Область техники
Настоящее изобретение относится к гену, который кодирует полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, применимый для структурного анализа полисахарида, содержащего сульфатированную фукозу, а также для получения продуктов разрушения указанного полисахарида; к способу получения указанного полипептида методами генной инженерии; к полипептиду, полученному указанным способом.
Уровень техники
Полисахарид, содержащий сульфатированную фукозу, полученный из морских водорослей, который называют фукоидан, является смесью сульфатированных полисахаридов, в основном состоящей из фукозы, и который также содержит галактозу, глюкуроновую кислоту, ксилозу, маннозу, глюкозу и т.д. Типы и число таких составляющих сахаров изменяются в зависимости от вида морских водорослей, используемых в качестве исходного материала. Например, есть данные, в соответствии с которыми комерческий фукоидан, производимый Sigma, подразделяется более чем на 13 молекулярных разновидностей [Carbohydrate Research, 255, 213-224 (1994)]. Предварительно их классифицируют на два типа, в одном из которых уроновая кислота, по существу не содержится, а основным составляющим сахаром является фукоза, а другой содержит уроновую кислоту, но в качестве составляющих сахаров содержатся фукоза и манноза.
Относительно биологических активностей полисахарида, содержащего сульфатированную фукозу, сообщалось о различных активностях, таких как потенцирование макрофагальной активности, ингибирование метастазирования злокачественных опухолей и антикоагуляционная активность крови. Однако поскольку полисахарид, содержащий сульфатированную фукозу, представлен несколькими молекулярными разновидностями после выделения и очистки полисахарида, содержащего сульфатированную фукозу, необходимо исследовать, какая его молекулярная разновидность действительно обладает активностью. Однако стандартными методами невозможно провести разделение достаточно эффективно, и поэтому трудно получить большое количество продукта, применимого в качестве фармацевтического препарата. Кроме того, полисахарид, содержащий сульфатированную фукозу, является сульфатированным полисахаридом, имеющим большую молекулярную массу, и при применении его в качестве фармацевтического препарата в неизмененном виде, возникают проблемы с точки зрения антигенности, гомогенности, антикоагулянтной активности и т.д., вследствие чего считается необходимым расщепление полисахарида, содержащего сульфатированную фукозу, в определенной степени.
Способ, в котором полисахарид, содержащий сульфатированную фукозу, расщепляется ферментативно для получения низкомолекулярных продуктов, является выгодным, поскольку реакцию можно проводить в мягких условиях и получать однородные продукты расщепления соответственно субстратной специфичности фермента. Уже имеются данные, что awabi (морское ушко), гребешок, морской еж, морские микроорганизмы и т.д. продуцируют фермент, разрушающий полисахарид, содержащий сульфатированную фукозу. Однако такой фермент, как правило, содержится в живых организмах лишь в небольших количествах, и, кроме того, имеется множество ферментов, разрушающих полисахарид, содержащий сульфатированную фукозу, вследствие чего необходимо проводить различные стадии очистки для получения отдельного фермента. Более того, аминокислотные последовательности и генетические структуры данных ферментов полностью до сих пор не ясны.
Проблемы, решаемые изобретением
Целью настоящего изобретения является обеспечение гена, кодирующего полипептид, имеющий расщепляющую активность по отношению к полисахариду, содержащему сульфатированную фукозу, применимый для получения и структурного анализа полисахарида, содержащего сульфатированную фукозу, а также для получения расщепленного полисахарида, содержащего сульфатированную фукозу. Настоящее изобретение также призвано обеспечить полипептид, который можно получать методами генной инженерии, применяя указанный ген, и обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу.
Способы решения проблем
Авторы настоящего изобретения провели интенсивное исследование гена микроорганизмов, которые продуцируют ферменты, расщепляющие полисахарид, содержащий сульфатированную фукозу, для того чтобы выяснить аминокислотную последовательность полипептида, обладающего расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, и кодирующую его нуклеотидную последовательность. В результате они выяснили, что существует по два типа гена, кодирующего полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, полученный из бактерий, принадлежащих к родам Alteromonas и Flavobacterium соответственно, определили их полную нуклеотидную последовательность, выяснили аминокислотную последовательность указанного полипептида и разработали способ промышленно выгодного получения полипептида, обладающего расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, применяя указанный ген, таким образом осуществив настоящее изобретение.
Суть настоящего изобретения такова, что первый аспект настоящего изобретения относится к выделенному гену, содержащему последовательность ДНК, кодирующую полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, или имеющий функционально идентичную активность, как сказано выше.
Второй аспект настоящего изобретения относится к рекомбинантной ДНК, содержащей ген, описанный в первом аспекте настоящего изобретения.
Третий аспект настоящего изобретения относится к экспрессирующему вектору, где рекомбинантная ДНК, описанная во втором аспекте настоящего изобретения, встроена в микроорганизмы, животные клетки или растительные клетки, применяемые в качестве клеток-хозяев.
Четвертый аспект настоящего изобретения относится к трансформанту, который трансформирован экспрессирующим вектором, представленным в третьем аспекте настоящего изобретения.
Пятый аспект настоящего изобретения относится к способу получения полипептида, обладающего расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, или имеющего функционально идентичную активность, который характеризуется тем, что трансформант согласно четвертому аспекту настоящего изобретения инкубируют и полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, или имеющий функционально идентичную активность, выделяют из продукта инкубации.
Шестой аспект настоящего изобретения относится к полипептиду, имеющему аминокислотную последовательность, представленную в любой из SEQ ID NO: 1 до NO: 4 в списке последовательностей, и имеющему расщепляющую активность по отношению к полисахариду, содержащему сульфатированную фукозу, или к полипептиду, имеющему функционально идентичную ему активность.
Краткое пояснение чертежей
На фиг.1 показаны положения ORF-1 и ORF-2.
На фиг.2 показан коэффициент преципитации полисахарида, содержащего сульфатированную фукозу.
На фиг.3 показано положение fdlA.
На фиг.4 показано положение fdlB.
Фиг.5 представляет собой хроматограмму, полученную с использованием DEAE-сефарозы FF.
Осуществление изобретения
Настоящее изобретение будет конкретно проиллюстрировано ниже.
Настоящее изобретение относится к гену, кодирующему полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу. Примером полипептида, кодируемого указаннутым геном, является полипептид, имеющий эндо-расщепляющую активность по отношению к полисахариду, содержащему сульфатированную фукозу, полученный из бактерий рода Alteromonas, что показано далее в (1):
(1) действие на полисахарид, содержащий сульфатированную фукозу, имеющий следующие физико-химические свойства (в дальнейшем называемый "полисахарид-F, содержащий сульфатированную фукозу") и расщепление указанного полисахарида, содержащего сульфатированную фукозу:
(а) составляющие сахариды: по существу не содержащие уроновую кислоту и
(б) по существу не способный расщепляться под действием фукоиданазы, продуцируемой Flavobacterium sp. SA-0082 (FERM BP-5402).
Что касается полипептида, обладающего расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, существует фермент, эндо-расщепляющий полисахарид, содержащий сульфатированную фукозу, продуцируемый штаммом Alteromonas sp. SN-1009, и указанный фермент может быть получен способом, упомянутым в ссылочном примере 1-(3).
Полисахарид-F, содержащий сульфатированную фукозу, может быть получен, как описано в ссылочном примере 1.
Фукоиданаза, продуцируемая штаммом Flavobacterium sp. SA-0082 (FERM BP-5402), может быть получена, как описано в ссылочном примере 5.
Относительно других полипептидов, имеющих расщепляющую активность по отношению к полисахариду, содержащему сульфатированную фукозу, в пример можно привести полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, как показано ниже в (2), полученный из бактерий рода Flavobacterium:
(2) действие на полисахарид, содержащий сульфатированную фукозу, имеющий следующие физико-химические свойства (в дальнейшем называемый "полисахарид-U, содержащий сульфатированную фукозу") и расщепление указанного полисахарида, содержащего сульфатированную фукозу, посредством чего высвобождается одно из соединений нижеприведенных формул [I], [II], [III] и [IV].
(в) составляющие сахариды: содержащие уроновую кислоту и
(г) способный расщепляться под действием фукоиданазы, продуцируемой Flavobacterium sp. SA-0082 (FERM BP-5402).
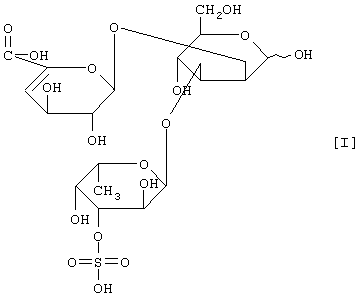
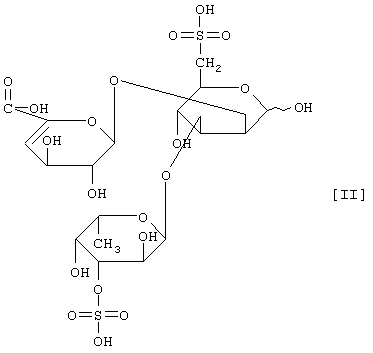
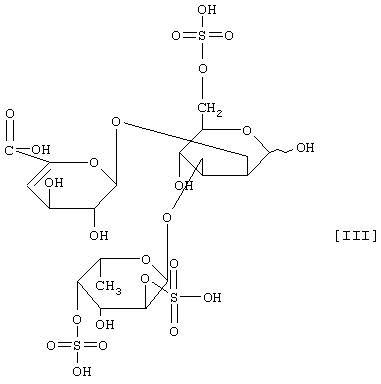
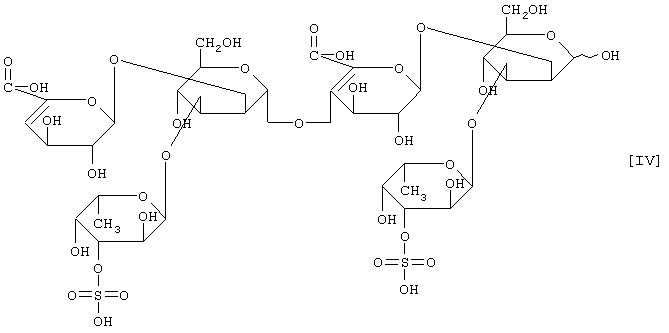
Примером полипептида, обладающего расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, является фукоиданаза, продуцируемая штаммом Flavobacterium sp. SA-802, и указанная фукоиданаза может быть получена способом, описанным в ссылочном примере 5.
В дальнейшем смесь полисахарида-F, содержащего сульфатированную фукозу, и полисахарида-U, содержащего сульфатированную фукозу, будет называться смесью полисахаридов, содержащих сульфатированную фукозу.
Полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, в настоящем изобретении означает не только фермент, расщепляющий полисахарид, содержащий сульфатированную фукозу, природного типа, но также полипептид, аминокислотная последовательность которого модифицирована делецией, замещением, вставкой, добавлением и т.д. аминокислот в аминокислотной последовательности природного типа до тех пор, пока данный полипептид будет иметь расщепляющую активность по отношению к полисахариду, содержащему сульфатированную фукозу.
В данном случае примером фермента природного типа, расщепляющего полисахарид, содержащий сульфатированную фукозу, могут служить ферменты, полученные из бактерий рода Alteromonas и бактерий рода Flavobacterium. Однако настоящее изобретение не ограничивается ими и, разумеется, охватывает ферменты, полученные из других бактерий, микроорганизмов, таких как дрожжи, плесневые грибы, Ascomycetes, Ваsidiomycetes и т.д., и из организмов, таких как растения и животные.
В настоящем изобретении значение термина полипептид, имеющий функционально идентичную активность, рассмотрено ниже.
В природных белках помимо полиморфизма и мутации гена, кодирующего указанный белок, возможны такие мутации как делеция, добавление, вставка, замещение и т.д. аминокислотных остатков аминокислотной последовательности являются результатом реакции модификации белка после продукции in vivo или в ходе очистки, но вопреки этому известно, что некоторые из них имеют по существу такую же физиологическую и биологическую активность, как и белок, не претерпевший мутаций. Таким образом, вещество, имеющее структурное различие, но не имеющее большого различия с точки зрения их функции, будет называться полипептидом, имеющим функционально идентичную активность.
Употребление данного термина справедливо и в случае, когда указанные мутации искусственно введены в аминокислотную последовательность белка, и в таком случае можно получить гораздо больше вариантов. Однако такой вариант может быть интерпретирован как полипептид, имеющий функционально идентичную активность, поскольку он проявляет по существу такую же физиологическую активность, как и полипептид без мутаций.
Например, считается, что остаток метионина, находящийся на N-конце белка, экспрессируемого Escherichia coli, во многих случаях удаляется действием метионинаминопептидазы, но в зависимости от типа белка продуцируются как продукт, содержащий, так и не содержащий метиониновый остаток. Однако наличие или отсутствие метионинового остатка обычно не влияет на активность белка. Также известно, что полипептид, в котором определенный цистеиновый остаток в аминокислотной последовательности человеческого интерлейкина-2 (IL-2) замещен серином, сохраняет активность интерлейкина-2 [Science, 224, 1431, (1984)].
Кроме того, когда продукцию белка выполняют методами генной инженерии, это часто делают для экспрессия гибридного белка. Например, N-конец белка, полученного из другого белка, присоединяют к N-концу желаемого белка для того, чтобы увеличить количество экспрессируемого желаемого белка, или присоединяют подходящую пептидную цепь и экспрессируют на N-конце или С-конце желаемого белка таким образом, что очистить желаемый белок становится легче, используя носитель, имеющий сродство к указанной присоединенной пептидной цепи.
Более того, часто замечается, что полипептид, в котором проведена, по крайней мере, одна делеция, добавление, вставка, замещение одного или более аминокислотных остатков в аминокислотной последовательности желаемого белка, имеет функционально идентичную активность с таковой желаемого белка. Такие полипептид и ген, который кодирует указанный полипептид, также охватываются настоящим изобретением независимо от того, является ли указанный полипептид природным и выделенным или искусственно полученным.
Известно, что обычно существует от 1 до 6 типов кодонов (комбинация из трех нуклеотидов), которые кодируют аминокислоту в гене для каждого типа аминокислоты. Соответственно ген, который кодирует аминокислотную последовательность, находится в избытке, что обусловлено особенностями аминокислотной последовательности. Ген никогда не присутствует в стабильном состоянии в природе, а наблюдаемые изменения в нуклеиновой кислоте являются нередкими. В некоторых случаях изменения, происходящие в гене, не влияют на аминокислотную последовательность, кодируемую им (что называется молчащей мутацией), и в таком случае продуцируются различные гены, кодирующие одну и ту же аминокислотную последовательность. Соответственно даже если выделить ген, который кодирует конкретную аминокислотную последовательность, возможно, что в течение серии пассажей в живых организмах, содержащих данный ген, производится множество типов генов, кодирующих одинаковую аминокислотную последовательность.
Далее, не трудно искусственно получить множество типов генов, кодирующих одинаковую аминокислотную последовательность при условии применения различных методов генной инженерии.
Например, в некоторых случаях продукция экспрессируемого белка является низкой, что обеспечивается методами генной инженерии, в которых используемый кодон родительского гена, кодирующего желаемый белок, встречается в клетке-хозяине с низкой частотой. В этом случае была сделана попытка увеличения экспрессии желаемого белка с помощью искусственного превращения кодона в такой, который часто встречается в клетках-хозяевах, без изменения кодируемой аминокислотной последовательности. И это, не говоря о том, что подобным образом может быть искусственно получено множество типов гена, кодирующего конкретную аминокислотную последовательность. Соответственно такой искусственно полученный и многообразный полинуклеотид также охватывается настоящим изобретением, поскольку кодирует аминокислотную последовательность, которая раскрыта в настоящем изобретении.
Кроме того, во многих случаях гены, кодирующие полипептиды, имеющие функционально идентичную активность, имеют гомологию. Поэтому ген, который способен к гибридизации в жестких условиях с геном, применяемым по настоящему изобретению, и кодирующий полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, также охватывается данным изобретением.
Ниже настоящее изобретение будет конкретно проиллюстрировано на примере штамма Alteromonas sp. SN-1009 и штамма Flavobacterium sp. SA-0082.
Штамм Alteromonas sp. SN-1009 депонирован в National Institute of Bioscience and Human-Technology, Agency of Industrial Science and Technology (1-3, Higashi 1-chome, Tsukuba, Ibaraki, 305-8566 JAPAN) 13 февраля 1996 года (дата первоначального поступления) под номером доступа FERM ВР-5747 в качестве международного поступления. Штамм Flavobacterium sp. SA-0082 депонирован в National Institute of Bioscience and Human-Technology, Agency of Industrial Science and Technology 29 марта 1995 года (дата первоначального поступления) под номером доступа FERM ВР-5402 в качестве международного поступления.
Для того чтобы получить ген, кодирующий полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, продуцируемому штаммом Alteromonas sp. SN-1009 или штаммом Flavobacterium sp. SA-0082, например, можно использовать метод гибридизации, метод ПЦР или их комбинацию. В данных методах необходим зонд, который способен гибридизоваться с указанным геном, или праймер, который пригоден для амплификации указанного гена или его части методом ПЦР, но поскольку совершенно не известны аминокислотная последовательность и структура гена полипептида, обладающего расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, продуцируемого этими штаммами, невозможно получить синтетический олигонуклеотид, который мог бы служить зондом или праймером. Поэтому в первую очередь исследуют частичную аминокислотную последовательность фермента, расщепляющего полисахарид, содержащий сульфатированную фукозу, продуцируемого указанными микроорганизмами, и получение синтетического олигонуклеотида, применимого в качестве зонда или праймера.
Сначала культивируют штамм Alteromonas sp. SN-1009 или штамм Flavobacterium sp. SA-0082 и затем каждый из продуцируемых ферментов, расщепляющих полисахарид, содержащий сульфатированную фукозу, выделяют из среды и очищают.
Далее получают информацию, касающуюся частичной аминокислотной последовательности каждого из выделенных ферментов, расщепляющих полисахарид, содержащий сульфатированную фукозу. Для того чтобы определить частичную аминокислотную последовательность, фермент, расщепляющий полисахарид, содержащий сульфатированную фукозу, например, подвергают анализу аминокислотной последовательности по Эдману в соответствии со стандартной методикой (например, можно использовать Protein Sequencer 476A, производимый Applied Biosys-tems), посредством чего может быть определена аминокислотная последовательность N-конца фермента, расщепляющего полисахарид, содержащий сульфатированную фукозу. Альтернативно, очищенный фермент, расщепляющий полисахарид, содержащий сульфатированную фукозу, подвергают ограниченному гидролизу, используя протеолитический фермент, имеющий высокую субстратную специфичность, такой как протеаза I Achromobacter, трипсин, обработанный N-тозил-L-фенилаланилхлорметилкетоном (ТРСК) и т.д., выделяют полученные пептидные фрагменты и очищают с помощью обратнофазовой ЖХВД, а очищенные пептидные фрагменты подвергают анализу аминокислотной последовательности, вследствие чего получают информацию об аминокислотной последовательности.
Полученная подобным способом информация, касающаяся частичной аминокислотной последовательности, конкретной для фермента, расщепляющего полисахарид, содержащий сульфатированную фукозу, выделяется и, основываясь на указанной информации, получают вырожденный олигонуклеотид со сконструированной и синтезированной последовательностью нуклеотидов. В то же время необходимо синтезировать длинный олигонуклеотид, имеющий низкую степень вырожденности, или, другими словами, олигонуклеотид, обладающий высокой специфичностью к гену, кодирующему полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу. Конструкция олигонулеотида является важным показателем для аналога гена, кодирующего полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу.
Далее, необходимо подобрать условия специфической гибридизации синтетического олигонуклеотида с геном, кодирующим полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, методом Саузерн-гибридизации.
Например, геномную ДНК штамма Alteromonas sp. SN-1009 или штамма Flavobacterium sp. SA-0082 полностью расщепляют подходящей рестриктазой, разделяют с помощью электрофореза в агарозном геле и подвергают блоттингу на нейлоновую мембрану или подобную стандартным методом. При проведении гибридизации нейлоновую мембрану сначала выдерживают при 65°С в течение нескольких часов в гибридизационном буфере, содержащем 6xSSC (1x SSC означает продукт, полученный путем растворения 8,77 г хлорида натрия и 4,41 г цитрата натрия в одном литре воды), 1% додецилсульфат натрия (SDS), 100 мкг/мл ДНК спермы лосося и 5х раствор Денхардта (содержащий 0,1% бычий сывороточный альбумин, поливинилпирролидон и фиколл), затем туда добавляют синтетический олигонуклеотид, меченный 32P и т.п., и смесь выдерживают при 42°С в течение ночи. Данную нейлоновую мембрану отмывают 1 хSSC, содержащим 0,1% SDS, при 42°С в течение 30 минут и подвергают авторадиографии для определения фрагментов ДНК, гибридизовавшихся с синтетическим олигонуклеотидным зондом. Для эффективности необходимо подобрать оптимальные условия, исследуя температуру инкубации, концентрацию соли в отмывающем буфере и т.д., принимая во внимание длину применяемого синтетического олигонуклеотида и комплементарность с геном, кодирующим полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу.
Для получения фрагментов ДНК, определяемых подобным образом, содержащих ген, кодирующий полипептид с расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, может быть использован способ, в котором фрагменты ДНК, соответствующие положению непосредственно определяемых полос, экстрагируют из геля и очищают, и библиотека, где фрагменты интегрируют в вектор из обычно используемой системы хозяин-вектор, и проводят вертикальную или горизонтальную гибридизацию в условиях, аналогичных таковым при Саузерн-гибридизации, для отбора и выделения клона, содержащего желаемые фрагменты ДНК. Альтернативно, геномную ДНК штамма Alteromonas sp. SN-1009 или штамма Flavobacterium sp. SA-0082 непосредственно расщепляют с помощью подходящей рестриктазы, создают библиотеку, где фрагменты интегрируют в вектор из обычно используемой системы хозяин-вектор и гибридизацию проводят аналогичным способом, вследствие чего отбирают и выделяют клон, содержащий желаемые фрагменты ДНК.
По отношению к системе хозяин-вектор, используемой здесь, может быть использована любая известная система, и ее примерами могут служить плазмидные векторы, такие как pUC18 и pUC19, при использовании в качестве хозяина Escherichia coli, или фаговой вектор, такой как фаг лямбда, хотя настоящее изобретение этим не ограничивается.
Относительно типа и управления такими системами хозяин-вектор могут быть использованы обычно применяемый тип и методика, упоминаемые, например, в "Molecular Cloning A Laboratory Manual", Second Edition, by J. Sambrook, et al., (published by Cold Spring Harbor Laboratory, 1989).
Когда можно выделить вектор, содержащий желаемые фрагменты ДНК, нуклеотидная последовательность желаемых фрагментов ДНК, встроенных в вектор, может быть определена в соответствии с традиционной методикой, такой как дидезокси-метод [Proceedings of the National Academy of Science, USA, 74, 5463 (1977)]. После сравнения определяемой нуклеотидной последовательности с данными N-концевого анализа, частичной аминокислотной последовательностью, молекулярной массой и т.д. фермента, расщепляющего полисахарид, содержащий сульфатированную фукозу, стало возможно узнать структуру гена в полученном фрагменте ДНК а также аминокислотную последовательность полипептида, который кодируется указанным геном.
Кроме того, может быть применен метод ПЦР как способ получения гена, кодирующего полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, используя олигонуклеотид, полученный на основе частичной аминокислотной последовательности указанного выше фермента, расщепляющего полисахарид, содержащий сульфатированную фукозу. Между тем, метод ПЦР с применением кассетной ДНК представляет собой метод, в котором фрагменты желаемого гена, применимые для гибридизационного метода, могут быть получены за короткое время с аминокислотной последовательности, содержащей небольшую информацию.
Например, геномную ДНК, выделенную стандартным методом из культуры клеток Flavobacterium sp. SA-0082, обрабатывают подходящей рестриктазой и лигируют с синтетической ДНК (кассетной ДНК), имеющей известную последовательность. Полученную конструкцию применяют как матрицу и ПЦР проводят, используя указанный геноспецифический олигонуклеотидный праймер, который сконструирован на основании информации об указанной выше частичной аминокислотной последовательности, и олигонуклеотидный праймер (кассетный праймер), комплементарный кассетной ДНК, посредством чего может быть проведена амплификация желаемых фрагментов ДНК. Относительно кассетной ДНК и кассетного праймера могут быть использованы, например, таковые, произведенные Takara Shuzo. Предпочтительно, чтобы кассетная ДНК содержала последовательность, соответствующую двум типам кассетных праймеров, и эффективнее, если сначала первую ПЦР проводят, используя праймер, находящийся далеко от сшивки по сайту рестрикции, и вторую ПЦР проводят, используя часть указанного реакционного раствора как матрицу и применяя внутренний праймер. Далее, относительно указанного геноспецифического олигонуклеотидного праймера, специфичность указанного гена становится выше, и возможность специфической амплификации желаемых фрагментов ДНК становится выше, когда конструктируют и параллельно синтезируют два типа праймеров, и верхний праймер применяется при первой ПЦР, а нижний праймер используется при второй ПЦР.
Однако поскольку нуклеотидная последовательность желаемого гена остается невыясненной, не всегда верно, что сайт рестрикции, используемый для сшивки кассетной ДНК, расположен в положении, подходящем для амплификации при ПЦР области, кодирующей частичную аминокислотную последовательность. Поэтому необходимо использовать различные типы рестриктаз кассетных ДНК. Кроме того, хотя ПЦР может проводиться в обычно применяемых условиях, как описано, например, в "PCR Technology" (edited by H.A.Eriich, published by Stockton Press, 1989), необходимо подобрать оптимальные условия для минимизации неспецифических полос амплификации с помощью выбора температуры отжига, числа циклов, концентрации ионов магния, концентрации термостабильной полимеразы и т.д., учитывая зависимость от длины используемого синтетического олигонуклеотида и комплементарности к гену, кодирующему полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу.
ПЦР-смесь подвергают электрофорезу, например, в агарозном геле, для подтверждения наличия амплифицированных фрагментов ДНК. Эти фрагменты экстрагируют и очищают стандартным способом и встраивают в обычно применяемый в клонировании вектор, такой как pUC18 или pUC19, и их нуклеотидная последовательность может быть проанализирована, например, с использованием дидезоксиметода. Альтернативно, выделенные амплифицированные фрагменты ДНК могут быть подвергнуты прямому определению нуклеотидной последовательности с использованием кассетного праймера, применяемого при ПЦР. Когда в результате получают последовательность, кодирующую уже установленную частичную аминокислотную последовательность фермента, расщепляющего полисахарид, содержащий сульфатированную фукозу, и дополняющую последовательность праймера, делают вывод о том, что может быть получен ген, кодирующий указанный фермент, или фрагменты гена, гомологичные ему.
Когда фрагменты ДНК, полученные с помощью Саузерн-гибридизации или метода ПЦР, являются частью гена, кодирующего желаемый фермент, проводят скрининг геномной библиотеки с помощью гибридизации, применяя указанные фрагменты ДНК в качестве зонда, или с помощью ПЦР, используя в качестве праймера олигонуклеотид, полученный на основании нуклеотидной последовательности указанных фрагментов ДНК, посредством чего могут быть получены фрагменты ДНК, содержащие полную длину гена, кодирующего желаемый фермент.
Далее, когда геномную ДНК штамма Alteromonas sp. SN-1009 или штамма Flavobacterium sp. SA-0082 анализируют с помощью Саузерн-гибридизации, применяя ген фермента, расщепляющего полисахарид, содержащий сульфатированную фукозу, полученный, как описано выше, или часть гена фермента, по положению определяемой полосы возможно получить информацию о размере рестрикционных фрагментов геномной ДНК штамма Alteromonas sp. SN-1009 или штамма Flavobacterium sp. SA-0082, содержащих ген, кодирующий полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу. Более того, можно сделать предположение о числе генов, кодирующих полипептид с расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, а также о числе генов, комплементарных им, и фрагменты ДНК, содержащие эти гены, могут быть выделены с помощью уже описанного метода.
Факт, что фрагменты ДНК, полученные таким образом, содержат ген, кодирующий желаемый фермент, может быть подтвержден созданием экспрессирующего вектора, содержащего указанный фрагмент ДНК, выделенный на конечном этапе, проведением трансформации клетки-хозяина указанным вектором, культивированием указанного трансформанта и измерением расщепляющей активности экспрессированного полипептида по отношению к полисахариду, содержащему сульфатированную фукозу.
В настоящем изобретении ген, имеющий аминокислотную последовательность, представленную в списке последовательностей SEQ ID NO: 1 и NO: 2, и имеющий нуклеотидную последовательность, кодирующую полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, был получен из штамма Alteromonas sp. SN-1009. Примеры нуклеотидной последовательности, кодирующей полипептид, имеющий аминокислотную последовательность, представленную SEQ ID NO: 1 и NO: 2, показаны соответственно в SEQ ID NO: 5 и NO: 6 в списке последовательностей.
Далее ген с нуклеотидной последовательностью, кодирующей полипептид, имеющий аминокислотную последовательность, представленную SEQ ID NO: 3 и NO: 4 в списке последовательностей, и обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, был выделен из штамма Flavobacterium sp. SA-0082. Примеры нуклеотидной последовательности, кодирующей полипептид с аминокислотной последовательностью SEQ ID NO: 3 и NO: 4, показаны в SEQ ID NO: 7 и NO: 8 в списке последовательностей соответственно.
Относительно способа получения гена, кодирующего полипептид, обладающего расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, или имеющего функционально идентичную активность с применением нуклеотидной последовательности гена по настоящему изобретению, может быть применен, например, следующий метод.
Во-первых, хромосомную ДНК, получаемую из источника, содержащего желаемый ген, или кДНК, полученную с мРНК с помощью обратной транскриптазы, вводят в клетку-хозяин стандартным методом при помощи лигирования с плазмидой или фаговым вектором для получения библиотеки. Библиотеку культивируют на чашке, а выросшие колонии или бляшки переносят на нитроцеллюлозу или нейлоновую мембрану и денатурируют для фиксирования ДНК на мембране. Данную мембрану инкубируют в гибридизационном буфере, содержащем зонд, меченный, например, 32P (относительно зонда, применяемого здесь, может быть использована любая аминокислотная последовательность, представленная SEQ ID NO: 1 до NO: 4 в списке последовательностей, или последовательность нуклеотидов, кодирующая его часть, и, например, может быть использована нуклеотидная последовательность, представленная любой из SEQ ID NO: 5 до NO: 8 в списке последовательностей или ее часть), вследствие чего формируют гибрид между зондом и ДНК на мембране. Например, мембрану с фиксированной ДНК подвергают гибридизации с зондом при температуре 65°С в течение 20 часов в гибридизационном буфере, содержащем 6х SSC, 1% додецилсульфат натрия (SDS), 100 мкг/мл ДНК спермы лосося и 5х раствор Денхардта. После гибридизации отмывают неспецифически адсорбировавшийся зонд и идентифицируют клон, который связался с зондом, например, посредством авторадиографии. Данную процедуру повторяют до тех пор, пока связавшийся с зондом клон не остается единственным. В клон, полученный подобным образом, встраивают ген, кодирующий желаемый полипептид.
Нуклеотидную последовательность полученного гена определяют, например, следующим способом и подтверждают факт, что полученный ген является геном, кодирующим полипептид, обладающий желаемой расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, или имеющий функционально идентичную активность.
Когда трансформантом является Escherichia coli, которую трансформируют плазмидой, определение нуклеотидной последовательности проводят путем ее культивирования в пробирке и тому подобном и выделения плазмиды стандартным методом. То есть вырезанный рестриктазой, встроенный фрагмент высвобождается и субклонируется в фаговый вектор М13, и нуклеотидную последовательность определяют дидезоксиметодом. Когда для рекомбинанта применяют фаговый вектор, последовательность нуклеотидов также может быть определена в основном этими же стадиями. Относительно основных экспериментальных методов от культивирования до определения нуклеотидной последовательности, существует описание, например, в "Molecular Cloning A Laboratory Manual", second edition, by J. Sambrook, et al., (published by Cold Spring Harbor Laboratory, 1989).
Для того чтобы подтвердить, является ли полученный ген геном, кодирующим полипептид, обладающий желаемой расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, или обладающий функционально идентичной активностью, определенную нуклеотидную последовательность или кодируемую ей аминокислотную последовательность сравнивают с нуклеотидной последовательностью, представленной в любой из SEQ ID NO: 5 до NO: 8 в списке последовательностей или сравнивают с аминокислотной последовательностью, представленной любой из SEQ ID NO: 1 до NO: 4 в списке последовательностей настоящего изобретения.
Если полученный ген не содержит всю область, кодирующую полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, или имеющий функционально идентичную активность, синтетическую библиотеку ДНК формируют на основании полученного гена, затем недостающую область амплифицируют с помощью ПЦР или фрагменты полученного гена применяют в качестве зонда для дальнейшего скрининга библиотеки ДНК или библиотеки кДНК, посредством чего может быть определена нуклеотидная последовательность всей области, кодирующей полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, или имеющий функционально идентичную активность.
Также возможно сконструировать праймер для ПЦР с нуклеотидной последовательности гена настоящего изобретения. При проведении ПЦР с применением указанного праймера можно определить фрагмент гена, имеющего высокую гомологию с геном настоящего изобретения, и также получить целиком весь ген.
Затем полученный ген экспрессируют и измеряют расщепляющую активность по отношению к полисахариду, содержащему сульфатированную фукозу, вследствие чего устанавливают функцию полученного гена.
Для продукции полипептида, обладающего расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, удобным является следующий метод, применяющий ген настоящего изобретения, кодирующий полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу.
Трансформацию клетки-хозяина проводят с применением вектора, содержащего ген, кодирующий полипептид, обладающий требуемой расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, и затем трансформант культивируют в обычно применяемых условиях, в результате чего может быть получен полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу. В то же время указанный полипептид получают в некоторых случаях в виде включения. В качестве клетки-хозяина могут быть использованы культивированные клетки микроорганизмов, животные клетки, растительные клетки и т.д.
Подходящим доказательством экспрессии является, например, измерение расщепляющей активности по отношению к полисахариду, содержащему сульфатированную фукозу. Измерение активности может быть выполнено, например, с применением экстракта рекомбинантных клеток Escherichia coli в качестве раствора фермента.
Когда установлена экспрессия полипептида, имеющего желаемую расщепляющую активность по отношению к полисахариду, содержащему сульфатированную фукозу, определяют оптимальные условия для состава среды, рН среды, температуры культивирования, количества и периода используемого индуктора, времени культивирования и т.д., при этом трансформантом, например, является Escherichia coli, вследствие чего возможна эффективная продукция полипептида, обладающего расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу.
Для очистки полипептида, обладающего расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, из культивированного трансформанта применяют стандартный метод. Когда полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, накапливается в клетках, как в случае, когда трансформантом является Escherichia coli, трансформант собирают посредством центрифугирования после завершения культивирования, разрушают обработкой ультразвуковыми волнами и центрифугируют для получения бесклеточного экстракта. Желаемый полипептид может быть очищен, когда данный экстракт подвергают стандартным методам очистки для белков, таким как высаливание, ионообменная гель-фильтрация, гидрофобная или аффинная хроматография. В некоторых применяемых системах хозяин-вектор экспрессируемый продукт может секретироваться трансформантом, и в этом случае супернатант может быть очищен подобным образом.
Когда продуцируемый трансформантом полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, вырабатывается внутри клеток, в клетках присутствуют одновременно различные ферменты, но поскольку их количество слишком мало по сравнению с количеством полипептида, обладающего расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, очистка представляется не слишком трудной. Кроме того, если выделить клетки, применяемые в качестве клеток-хозяев, количество получаемого из клеток-хозяев фермента, действующего на полисахарид, содержащий сульфатированную фукозу, заметно уменьшается. Далее, когда полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, секретируется из клеток, он оказывается вместе с компонентами
среды, но их можно легко отделить от полипептида, обладающего расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу.
Более того, когда, например, клеткой-хозяином является Escherichia coli, экспрессируемый продукт в некоторых случаях может вырабатываться как нерастворимое включение. В этом случае клетки собирают центрифугированием или подобным способом, после завершения культивирования разрушают обработкой ультразвуковыми волнами или подобным способом и центрифугируют, после чего собирают нерастворимые фракции, содержащие включения. После отмывки включений их растворяют, используя обычно применяемый растворяющий агент для белков, такой как мочевина или гуанидингидрохлорид, и, если необходимо, очищают при помощи различных хроматографических методов, таких как ионобменная гель-фильтрация, гидрофобная или аффинная хроматография, и подвергают восстановлению при помощи, например, диализа или дилюции, после чего может быть получен полипептид, сохраняющий активность и обладающий желаемой расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу. В случае, когда при необходимости далее очищают полученный образец с помощью различных хроматографических методов, возможно получить высокоочищенный полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу.
Между прочим, в случае продукции полипептида, имеющего функционально идентичную активность полипептиду, обладающему расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, могут быть использованы те же методы получения и очистки.
Настоящее изобретение обеспечивает первичную структуру и структуру гена, который кодирует полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу. Также возможно получение полипептида, обладающего расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, или полипептида, имеющего функционально идентичную активность, методами генной инженерии.
При применении в настоящем изобретении генно-инженерного метода получения стало возможно получить высокоочищенные полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, а также полипептид, имеющий функционально идентичную активность, с низкой стоимостью.
Таким образом, в способе получения фермента, обладающего расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, посредством культивирования микроорганизмов, принадлежащих штаммам Alteromonas sp. SN-1009 или Flavobacterium sp. SA-0082, продуцирующих фермент, расщепляющий полисахарид, содержащий сульфатированную фукозу, одновременно вырабатываются протеазы и другие расщепляющие полисахариды ферменты, и поэтому для изоляции желаемого фермента, расщепляющего полисахарид, содержащий сульфатированную фукозу, требуется отделение от других ферментов и очистка, которые являются очень проблематичными, и также необходимо для индуцирования продукции фермента добавление дорогостоящего полисахарида, содержащего сульфатированную фукозу, в процессе культивирования, вследствие чего продуцируется фермент, расщепляющий полисахарид, содержащий сульфатированную фукозу. Однако сейчас согласно настоящему изобретению возможно получить высокоочищенный полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, низкой стоимости.
ПРИМЕРЫ
Настоящее изобретение будет далее проиллюстрировано следующими примерами, хотя ими настоящее изобретение не ограничивается.
Ссылочный пример 1.
(1) 2 кг высушенных Kjellmaniella crassifolia измельчали с помощью Model M-2 (производство Nara Kikai Seisakusho). Полученный таким образом сухой порошок обрабатывали более чем 4,5-кратным количеством 80% этанола в течение 2 часов при температуре 80°С и затем фильтровали. Полученный остаток повторно отмывали 80% этанолом и фильтровали три раза описанным выше способом до получения, таким образом, 1,870 г остатка после отмывания этанолом. К данному остатку добавляли 36 л воды и смесь подвергали обработке при 100°С в течение 2 часов для получения экстракта. Концентрацию соли в экстракте доводили до уровня 400 мМ раствора хлорида натрия. Затем к нему добавляли 5% цетилпиридинхлорид до полного насыщения. После центрифугирования преципитат повторно отмывали 80% этанолом для полной элиминации цетилпиридинхлорида. Далее преципитат растворяли в 3 л 2 М хлорида натрия. После удаления центрифугированием нерастворимых веществ преципитат суспендировали в 100 мл DEAE-Cellulofine А-800, уравновешенной с помощью 2 М хлорида натрия. Суспензия перемешивали и затем отфильтровывали для удаления смолы. Фильтрат вносили в колонку, содержащую 100 мл DEAE-Cellulofine А-800, уравновешенную с помощью 2 М хлорида натрия, обессоливали прошедшую через нее фракцию и удаляли низкомолекулярные вещества с помощью ультрафильтрации (фильтр с порами, исключающими молекулярную массу 100000). Полученный таким образом преципитат удаляли центрифугированием. Супернатант высушивали вымораживанием до получения 82,2 г очищенной смеси, включающей полисахарид, содержащий сульфатированную фукозу, из Kjellmaniella crassifolia.
(2) 7 г указанной смеси, включающей полисахарид, содержащий сульфатированную фукозу, полученной из Kjellmaniella crassifolia, растворяли в 700 мл 20 мМ ацетата натрия (рН 6,0), содержащего 0,2 М хлорида кальция. Затем раствор вносили в колонку, содержащую 4000 мл DEAE-Sepharose FF, предварительно уравновешенную с помощью 20 мМ ацетата натрия (рН 6,0), содержащего 0,2 М хлорид кальция. Затем колонку полностью отмывали 20 мМ ацетатом натрия (рН 6,0), содержащим 0,2 М хлорида кальция, и проводили элюцию хлоридом натрия с линейным градиентом концентрации от 0 до 4 М. Затем собирали фракции, элюированные при концентрации хлорида натрия 0,9-1,5 М, и обессоливали при помощи ультрафильтрации с ультрафильтрационным фильтром, исключающим молекулярную массу 100000. После высушивания вымораживанием получали 4,7 г сухого препарата, включающего полисахарид-F, содержащий сульфатированную фукозу.
Также собирали фракции, элюированные при концентрации натрия хлорида 0,05-0,8 М, и обессоливали при помощи ультрафильтрации с ультрафильтрационным фильтром, исключающим молекулярную массу 100000. После высушивания вымораживанием получали 2,1 г сухого препарата, включающего полисахарид-U, содержащий сульфатированную фукозу.
(3) Штамм Alteromonas sp. SN-1009 (FERM BP-5747) высевали в 600 мл среды, включающей искусственно полученную морскую воду (рН 8,2, производство Jamarin Laboratory), содержащей 0,25% глюкозу, 1,0% пептонный бульон, и 0,05% экстракта дрожжей, стерилизовали (120°С, 20 минут) и пипетировали в 2 л Erienmeyer колбе. Затем штамм культивировали при 25°С в течение 25 часов до получения, таким образом, проросшей культуры. В 30 л колбу ферментера вносили 18 л среды, включающей искусственно полученную морскую воду (рН 8,0), 200 г пептона, 4 г экстракта дрожжей и 4 мл пеноудаляющего агента (КМ70 производство Shin-Etsu Chemical Co., Ltd) и стерилизовали при 120°С 20 минут. После охлаждения в среду добавляли 20 г полисахарида-F, содержащего сульфатированную фукозу, из Kjellmaniella crassifolia, полученного способом, описанным в справочном примере 1-(1), который отдельно был растворен в 2 л искусственно полученной морской воды, стерилизованной при 120°С в течение 15 минут, и вносили 600 мл вышеуказанной проросшей культуры для культивирования при 24°С в течение 20 часов при аэрации с уровнем 10 л в минуту и перемешивании 250 об/мин. После завершения культивирования культуральную среду центрифугировали, посредством чего получали клетки и культуральный супернатант.
При определении активности, применяя в качестве субстрата полисахарид-F, содержащий сульфатированную фукозу, способом, описанным в ссылочном примере 2, культуральный супернатант показал активность фермента, расщепляющего полисахарид, содержащий сульфатированную фукозу, равную 10 мЕ/мл среды.
Полученный культуральный супернатант концентрировали ультрафильтрацией с исключением частиц с молекулярной массой 10000 и сформированный таким образом преципитат элиминировали центрифугированием. Затем проводили высаливание с помощью 85% сульфата аммония. Полученный таким образом преципитат отбирали центрифугированием и полностью диализовали против 20 мМ Трис НСl буфера (рН 8,2), содержащего искусственно полученную морскую воду (Jamarin S), 10-кратно разведенную. Таким образом, получали 400 мл неочищенного фермента.
Полученный раствор неочищенного фермента адсорбировали на колонке с DEAE-Cellulofine A-800 (производство Seikagaku Kogyo), уравновешенной 20 мМ Трис НСl буфером (рН 8,2), содержащим 5 мМ азид натрия и искусственно полученную морскую воду (Jamarin S), 10-кратно разведенную. Затем адсорбированные вещества полностью промывали указанным буфером и элюировали в тот же буфер растворами, содержащими 100, 200, 300, 400 и 600 мМ хлорида натрия. Активные фракции смешивали.
При измерении активности способом, описанным в ссылочном примере 2, полученная активная фракция показала активность фермента 20400 мЕ (20,4 Е).
Полученные активные фракции концентрировали ультрафильтром с исключением частиц с молекулярной массой 10000 и подвергали ультрафильтрации с добавлением 20 мМ Трис НСl буфера (рН 8,2), содержащего 10 мМ хлорид кальция и 50 мМ хлорид натрия, вследствие чего происходила полная замена буфера.
Полученный раствор фермента наносили на колонку с DEAE-Sepharose FF, предварительно уравновешенную с помощью данного буфера, адсорбированные вещества хорошо промывали данным буфером, затем промывали данным буфером с концентрацией хлорида натрия 150 мМ и подвергали градиентной элюции хлоридом натрия, используя данный буфер, содержащий хлорид натрия в концентрации от 150 до 400 мМ.
Полученные активные фракции собирали, концентрировали посредством ультрафильтрации и подвергали гель-фильтрации, применяя Sephacryl S-200. В качестве элюента применяли 10 мМ Трис НСl буфер (рН 8,0), содержащий 5 мМ азида натрия и Jamarin S в 10-кратном разведении. Кроме того, с помощью указанной хроматографии была определена молекулярная масса, которая составила приблизительно 100000.
Полученные активные фракции собирали, тщательно диализовали против 20 мМ Трис НСl буфера (рН 8,2), содержащего 10 мМ хлорид кальция, 10 мМ хлорид калия и 4,2 М хлорид натрия, наносили на колонку с phenyl Sepharose CL-4B, предварительно уравновешенную таким же буфером с концентрацией хлорида натрия 4 М, и элюировали указанным буфером, содержащим 4, 3, 2, 1 М, 0,5 или 0,15 М хлорида натрия.
Полученные активные фракции собирали, концентрировали посредством ультрафильтрации и затем подвергали ультрафильтрации с добавлением 20 мМ Трис НСl буфера (рН 8,2), содержащего 10 мМ хлорид кальция, 10 мМ хлорид калия и 150 мМ хлорид натрия, за счет чего происходила полная замена буфера. Раствор фермента наносили на колонку с DEAE-Cellulofine A-800, предварительно уравновешенную тем же буфером, промывали указанным буфером и подвергали градиентной элюции с концентрацией хлорида натрия от 150 до 350 мМ.
Полученные активные фракции собирали, тщательно диализовали против указанного буфера, содержащего 50 мМ хлорид натрия, адсорбировали на колонке с DEAE-Cellulofine A-800, предварительно уравновешенной тем же буфером, содержащим 50 мМ хлорид натрия, промывали данным буфером и подвергали градиентной элюции с концентрацией хлорида натрия от 50 до 150 мМ. Собирали полученные активные фракции, содержащие очищенный фермент.
Далее определяли молекулярную массу указанного очищенного фермента с помощью SDS (додецилсульфат натрия) электрофореза в полиакриламидном геле, которая составила приблизительно 90000.
Ссылочный пример 2
Используя полученный согласно ссылочному примеру 1-(2) полисахарид-F, содержащий сульфатированную фукозу, эндорасщепляющую активность по отношению к полисахариду, содержащему сульфатированную фукозу, определяют следующим способом.
Собственно, готовят смесь, включающую 12 мкл 2,5% раствора полисахарида-F, содержащего сульфатированную фукозу, 6 мкл 1 М раствора хлорида кальция, 9 мкл 4 М раствора хлорида натрия, 60 мкл буфера (рН 7,5), содержащего 50 мМ уксусной кислоты, имидазол и Трис НС1 буфер, 21 мкл воды и 12 мкл раствора, подвергающегося определению расщепляющей активности, и проводят реакцию при 30°С в течение 3 часов. Затем реакционную смесь обрабатывают при 100°С в течение 10 минут и центрифугируют. Далее определяют степень расщепления в 100 мкл реакционной смеси с помощью ЖХВД.
В качестве контролей берут реакционную смесь, приготовленную из таких же компонентов, кроме раствора, подвергающегося определению расщепляющей активности, который заменяют буфером, входящим в данный раствор, и другую реакционную смесь, приготовленную из таких же компонентов, исключая раствор полисахарида-F, содержащего сульфатированную фукозу, который заменяют водой. Данные контроли также анализируют с помощью ЖХВД.
Количество фермента, которое может расщепить связи с фукозой в 10 мкмоль полисахарида-F, содержащего сульфатированную фукозу, за 1 минуту, берется за 1 Е. Связи с фукозой, расщепляемые подобным образом, рассчитывают согласно следующему уравнению:
Активность (Е/мл)={(12×2,5)/(100×MF)}×{(MF/M)-1}×{1/(180×0,01)}×1000
Для определения средней молекулярной массы продукта реакции анализируют меченые стандарты с известной молекулярной массой (STANDARD P-82, производство Showa Denko К.К.) с помощью ЖХВД в тех же условиях, как описано выше. Затем строится график зависимости молекулярной массы стандартов от времени прохождения колонки OHpak SB-806, который применяется в качестве калибровочной кривой для определения молекулярной массы указанного продукта ферментативной реакции.
Ссылочный пример 3
Используя полученный согласно ссылочному примеру 1-(2) полисахарид-U, содержащий сульфатированную фукозу, расщепляющую активность по отношению к полисахариду, содержащему сульфатированную фукозу, определяют следующим способом.
Готовят раствор, включающий 50 мкл 2,5% раствора полисахарида-U, содержащего сульфатированную фукозу, 10 мкл раствора, подвергающегося определению расщепляющей активности и 60 мкл 83 мМ фосфатного буфера (рН 7,5), содержащего 667 мМ хлорида натрия, и проводят реакцию при 37°С в течение 3 часов. Затем 105 мкл реакционной смеси смешивают с 2 мл воды при перемешивании и измеряют поглощение при 230 нм. В качестве контролей берутся реакционная смесь, приготовленная подобным образом, кроме внесения раствора, подвергающегося определению расщепляющей активности, который заменяют вышеуказанным буфером, содержащимся в этом растворе, и другая реакционная смесь, приготовленная подобным образом, исключая раствор полисахарида-U, содержащего сульфатированную фукозу, который заменяют водой, и также измеряют поглощение (АВ1 и АВ2).
Количество фермента, которое может специфически расщепить 1 мкмоль гликозидных связей между маннозой и уроновой кислотой за 1 минуту, принимают за 1 Е. Связи, расщепляемые подобным образом, определяют с помощью миллимолярного молекулярного коэффициента экстинкции ненасыщенной уроновой кислоты, полученной по реакции элиминации, 5,5. Активность фермента определяют согласно следующему уравнению:
Активность (Е/мл)=(AT-АВ1-АВ2)×2,105×120/5,5×105×0,01×180;
Ссылочный пример 4
(1) Молекулярные массы полисахарида-F, содержащего сульфатированную фукозу, и полисахарида-U, содержащего сульфатированную фукозу, определяют методом гель-фильтрации, применяя Sephacryl S-500. В результате распределение молекулярной массы каждого из них оказалось около 190000.
(2) На фигуре 2 показан коэффициент преципитации полисахарида-U, содержащего сульфатированную фукозу, и коэффициент преципитации полисахарида-F, содержащего сульфатированную фукозу, при различных концентрациях хлорида натрия в присутствии избытка цетилпиридинхлорида.
На фигуре 2 ордината показывает коэффициент преципитации (%), а абсцисса показывает концентрацию (М) хлорида натрия. Сплошная линия и открытый кружок отражают зависимость коэффициента преципитации полисахарида-U, содержащего сульфатированную фукозу, от различной концентрации хлорида натрия, а прерывистая линия и открытый треугольник отражают зависимость коэффициента преципитации полисахарида-F, содержащего сульфатированную фукозу, от различной концентрации хлорида натрия (М).
Коэффициент преципитации определяют при температуре раствора 37°С следующим образом.
Полисахарид-U, содержащий сульфатированную фукозу, и полисахарид-F, содержащий сульфатированную фукозу, каждый растворяют в воде и 4М хлориде натрия до 2% концентрации. Затем данные растворы смешивают в различных соотношениях для получения порций по 125 мкл растворов полисахарида-U, содержащего сульфатированную фукозу, и полисахарида-F, содержащего сульфатированную фукозу, при различных концентрациях хлорида натрия. Затем растворяют цетилпиридинхлорид в воде и 4 М хлориде натрия до концентрации 2,5% и полученные растворы смешивают в различных соотношениях для получения растворов, содержащих 1,25% цетилпиридинхлорида при различной концентрации хлорида натрия.
Необходимо более чем 3,2 объема 1,25% раствора цетилпиридинхлорида для полного осаждения полисахарида-U, содержащего сульфатированную фукозу, и полисахарида-F, содержащего сульфатированную фукозу, каждый из которых растворен в воде до 2% концентрации. К порциям по 125 мкл 2% растворов полисахарида-U, содержащего сульфатированную фукозу, и полисахарида-F, содержащего сульфатированную фукозу, содержащих различные концентрации хлорида натрия, добавляют по 400 мкл раствора цетилпиридинхлорида с различной концентрацией хлорида натрия. После полного перемешивания и выдерживания в течение 30 минут каждую смесь центрифугировали и содержание сахарида определяли фенолсернокислотным методом [Analytical Chemistry, 28, 350 (1956)] с последующим расчетом коэффициента преципитации для каждого из полисахаридов, содержащих сульфатированную фукозу, в каждой концентрации хлорида натрия.
(3) Далее, анализировали составляющие полисахарида-F, содержащего сульфатированную фукозу, следующим способом.
Сначала определяли содержание фукозы согласно методу, описанному в Journal of Biological Chemistry, 175, 595 (1948).
Сухой препарат полисахарида-F, содержащего сульфатированную фукозу, растворяли в 1 N соляной кислоте до получения 0,5% концентрации и обрабатывали при 110°С в течение 2 часов для гидролиза на составляющие моносахариды. Восстановленные концы моносахаридов, полученные при гидролизе, пиридил-(2)-аминировались с использованием GlycoTAG™ и GlycoTAG™ Reagent Kit (оба производства Takara Shuzo Co., Ltd.) и анализировали композиционное соотношение составляющих моносахаридов с помощью ЖХВД.
Далее, содержание уроновой кислоты определяли методом, описанным в Analytical Biochemistry, 4, 330 (1962).
Содержание серной кислоты определяли методом, описанным в Biochemical Journal, 84, 106 (1962).
Выяснилось, что составляющими сахаридами полисахарида-F, содержащего сульфатированную фукозу, являются фукоза и галактоза в молярном соотношении приблизительно 10:1. Ни уроновая кислота, ни какой-либо другой нейтральный сахарид, по существу, не входят в его состав. Молярное отношение фукозы к сульфату составляет приблизительно 1:2.
Составляющие полисахарида-U, содержащего сульфатированную фукозу, определяли вышеописанным способом. В результате выяснили, что составляющими сахаридами полисахарида-U, содержащего сульфатированную фукозу, являются фукоза, манноза, галактоза, глюкоза, рамноза, ксилоза и уроновая кислота и, по существу, не содержатся никакие другие нейтральные сахариды. Композиционное соотношение основных компонентов в молях выглядит следующим образом: фукоза : манноза : галактоза : уроновая кислота : сульфогруппа = приблизительно 10:7:4:5:20.
(4) При измерении высокоскоростным высокочувствительным поляриметром SEPA-300 (производство Horiba Seisakusho) высушенного вымораживанием препарата полисахарида-F, содержащего сульфатированную фукозу, удельное вращение составило -135°.
С другой стороны, удельное вращение для полисахарида-U, содержащего сульфатированную фукозу, составило -53,6°.
(5) Далее готовят раствор, включающий 16 мл 1% раствора полисахарида-F, содержащего сульфатированную фукозу, 12 мл 50 мМ фосфатного буфера (рН 8,0), 4 мл 4 М хлорида натрия и 8 мл 32 мЕ/мл раствора эндофукоиданазы, описанной в следующем ссылочном примере 5, и проводят реакцию при 25°С в течение 48 часов. В результате продукта расщепления не образуется.
Проводят реакцию между полисахаридом-U, содержащим сульфатированную фукозу, и фукоиданазой, описанной в ссылочном примере 5, при 25°С в течение 48 часов в вышеуказанных условиях. Установлено, что поглощение реакционной смеси при 230 нм увеличивается в ходе протекания реакции, таким образом, доказывая, что происходит расщепление полисахарида-U, содержащего сульфатированную фукозу, данным ферментом.
Ссылочный пример 5
Описанную в ссылочном примере 4 фукоиданазу получают следующим способом. На использование штамма, продуцирующего указанную фукоиданазу, не накладывают ограничение при условии наличия способности к продукции указанного фермента. В качестве частного примера можно указать штамм Flavobacterium sp. SA-0082 (FERM BP-5402).
Данный штамм был обнаружен и собран в морской воде Аоmori. Данный штамм был классифицирован как Flavobacterium sp. SA-0082 и депонирован в National Institute of Bioscience and Human-Technology, Agency of Industrial Science and Technology 29 марта, 1995 года (дата первоначального поступления) под номером доступа FERM BP-5402 в качестве международного поступления.
Питательные вещества, добавляемые в среду для культивирования данного штамма, могут быть произвольными при условии, что применяемый штамм может использовать их, продуцируя фукоиданазу. Подходящими примерами источника углерода являются фукоидан, порошок морских водорослей, альгиновая кислота, фукоза, глюкоза, маннит, глицерин, сахароза, мальтоза, лактоза и крахмал, а подходящими примерами источника азота являются экстракт дрожжей, пептон, casamino кислоты, жидкость вымоченной кукурузы, мясной экстракт, обезжиренные соевые бобы, сульфат аммония и хлорид аммония. Кроме того, среда может содержать неорганические вещества и соли металлов, такие как соли натрия, фосфаты, соли калия, соли магния и соли цинка.
Продукция фукоиданазы, происходящая при культивировании штамма, варьирует в зависимости от условий культивирования. В основном, предпочтительным является температурный интервал культивирования от 15 до 30°С и интервал значения рН среды от 5 до 9. Продукция фукоиданазы достигает максимума в случае культивирования штамма при аэрации и помешивании в течение 5 до 72 часов. Естественно, условия инкубации подбирают соответственно применяемому штамму, составу среды и т.д. для достижения максимальной продукции.
Фукоиданаза содержится как в клетках, так и в культуральном супернатанте.
Вышеуказанный штамм Flavobacterium sp. SA-0082 культивируют в подходящей среде, клетки собирают и разрушают обычно применяемым способом для разрушения клеток, таким как обработка ультразвуком. Таким образом, может быть получен бесклеточный экстракт.
Впоследствии экстракт подвергают очистке с помощью обычно применяемого в технике способа для получения препарата очищенного фермента. Например, очистка может проводиться высаливанием, ионообменной хроматографией, гидрофобной хроматографией, гель-фильтрацией или другим подобным способом для получения очищенной фукоиданазы.
Культуральный супернатант, получаемый удалением клеток из вышеуказанной культуральной среды, также содержит большое количество данного фермента (внеклеточного фермента), который может быть очищен теми же способами, которые применяли для очистки внутриклеточного фермента.
Ниже приведен пример проведения очистки фукоиданазы. Штамм Flavobacterium sp. SA-0082 (FERM BP-5402) помещают в 600 мл среды, включающей искусственно полученную морскую воду (рН 7,5, производство Jamarin Laboratory), содержащей 0,25% глюкозы, 1,0% пептона и 0,05% стерилизованного экстракта дрожжей (120°С, 20 минут) и пипетируют в 2 л Erienmeyer колбе. Затем штамм культивируют при 24°С в течение 24 часов до получения, таким образом, проросшей культуры. В 30 л колбу ферментера вносят 20 л среды, включающей искусственно полученную морскую воду (рН 7,5, производство Jamarin Laboratory), содержащей 0,25% глюкозы, 1,0% пептона и 0,05% экстракта дрожжей и 0,01% пеноудаляющего агента (КМ70 производство Shin-Etsu Chemical Co., Ltd) и стерилизуют при 120°С в течение 20 минут. После охлаждения в среду вносят 600 мл вышеуказанной проросшей культуры для культивирования при 24°С в течение 24 часов при аэрации с уровнем 10 л в минуту и помешивании 125 об/мин. После завершения культивирования культуральную среду центрифугируют, посредством чего собирают клетки.
Данные клетки суспендируют в 20 мМ ацетатфосфатном буфере (рН 7,5), содержащем 200 мМ хлорид натрия, разрушают обработкой ультразвуком и центрифугируют для получения клеточного экстракта. Активность фукоиданазы в данном клеточном экстракте оказалась равной 5 мЕ/мл среды при определении способом, описанном в ссылочном примере 3. В данном случае определение активности будет описано ниже.
К данному экстракту добавляют сульфат аммония до окончательного достижения 90% насыщения. После растворения при помешивании раствор центрифугируют, и полученный преципитат суспендируют в указанном выше буфере, в котором суспендировались клетки. Далее суспензию полностью диализуют против 20 мМ ацетатфосфатного буфера (рН 7,5), содержащего 50 мМ хлорида натрия. После удаления образующегося при диализе преципитата с помощью центрифугирования его наносят на DEAE-Sepharose FF колонку, предварительно уравновешенную с помощью 20 мМ ацетатфосфатного буфера (рН 7,5), содержащего 50 мМ хлорида натрия. Затем адсорбированные вещества хорошо промывают данным буфером и подвергают элюции с линейным градиентом концентрации хлорида натрия от 50 до 600 мМ. Активные фракции смешивают и добавляют хлорид натрия до конечной концентрации 4 М. Далее, раствор наносят на Phenyl Sepharose CL-4B колонку (производство Pharmacia), уравновешенную 20 мМ фосфатным буфером (рН 8,0), содержащим 4 М хлорида натрия. Затем, адсорбированное вещество тщательно отмывают данным буфером и подвергают элюции с линейным градиентом концентрации хлорида натрия от 4 до 1 М. Активные фракции смешивают и концентрируют с помощью ультрафильтрации. Далее смесь подвергают гель-фильтрации с применением Sephacryl S-300 колонки (производство Pharmacia), уравновешенной 10 мМ фосфатным буфером, содержащим 50 мМ хлорида натрия. Активные фракции смешивают. Молекулярная масса фермента, определенная по времени прохождения колонки Sephacryl S-300 составляет приблизительно 460000. Далее активную фракцию диализуют против 10 мМ фосфатного буфера (рН 7,0), содержащего 250 мМ хлорида натрия. Раствор фермента адсорбируют на Mono Q HR5/5 колонке (производство Pharmacia), уравновешенной 10 мМ фосфатным буфером (рН 7,0), содержащим 250 мМ хлорида натрия. Адсорбированное вещество тщательно отмывают данным буфером и подвергают элюции с линейным градиентом концентрации хлорида натрия от 250 до 450 мМ. Активные фракции смешивают, посредством чего получают раствор очищенного фермента. Таблица 1 суммирует вышеописанные стадии очистки. В данном случае количество белка определяют измерением поглощения раствора фермента при 280 нм. Расчет производят, принимая за единицу поглощение 1 мг/мл раствора белка.
Более того, фукоиданаза может быть очищена следующим способом. Штамм Flavobacterium sp. SA-0082 (FERM BP-5402) помещали в 600 мл среды, включающей искусственно полученную морскую воду (рН 7,5, производство Jamarin Laboratory), содержащей 0,1% глюкозы, 1,0% пептона и 0,05% экстракта дрожжей, которую стерилизовали при 120°С в течение 20 минут и пипетировали в 2 л Erlenmeyer колбе. Затем штамм культивировали при 24°С в течение 20 часов до получения вследствие этого проросшей культуры. В 30 л колбу ферментера вносили 20 л среды, включающей искусственно полученную морскую воду (рН 7,5, производство Jamarin Laboratory), содержащей 0,3% фукоидана, полученного из Kjellmaniella crassifolia, 0,5% пептона и 0,01% экстракта дрожжей и 0,01% пеноудаляющего агента (КМ70 производство Shin-Etsu Chemical Co., Ltd) и стерилизовали при 120°С в течение 20 минут. После охлаждения в среду вносили 600 мл вышеуказанной проросшей культуры для культивирования при 24°С в течение 20 часов при аэрации с уровнем 10 л в минуту и помешивании 125 об/мин. После завершения культивирования культуральную среду центрифугировали, вследствие чего получали клетки и культуральный супернатант. Клетки, полученные культивированием, суспендировали в 20 мМ ацетатфосфатном буфере (рН 7,5), содержащем 200 мМ хлорид натрия, разрушали обработкой ультразвуком и центрифугировали для получения клеточного экстракта. Активность фукоиданазы, определенная в данном клеточном экстракте, оказалась равной 20 мЕ/мл культуральной среды.
Отдельно культуральный супернатант концентрировали ультрафильтрацией (фильтр с исключением молекулярной массы 10000, производство Amicon) и проводили определение фукоиданазы. Активность составила 6 мЕ/мл культуральной среды.
К вышеуказанному концентрату культурального супернатанта добавляли сульфат аммония до полного установления 90% насыщения. После растворения при помешивании раствор центрифугировали и полученный преципитат суспендировали в описанном выше буфере, в котором суспендировались клетки. Далее суспензию полностью диализовали против 20 мМ ацетат-фосфатного буфера (рН 7,5), содержащего 50 мМ хлорида натрия. После удаления образующегося при диализе преципитата с помощью центрифугирования его наносили на DEAE-Sepharose FF колонку, предварительно уравновешенную с помощью 20 мМ ацетатфосфатного буфера (рН 7,5), содержащего 50 мМ хлорид натрия. Затем адсорбированные вещества хорошо промывали данным буфером и подвергали элюции с линейным градиентом концентрации хлорида натрия от 50 до 600 мМ. Активные фракции смешивали с добавлением хлорида натрия до конечной концентрации 4 М. Далее, раствор наносили на Phenyl Sepharose CL-4B колонку (производство Pharmacia), уравновешенную 20 мМ фосфатным буфером (рН 8,0), содержащим 4 М хлорид натрия. Затем адсорбированное вещество тщательно отмывали данным буфером и подвергали элюции с линейным градиентом концентрации хлорида натрия от 4 до 1 М. Активные фракции смешивали и концентрировали с помощью ультрафильтрации (производство Amicon). Далее смесь подвергали гель-фильтрации с использованием Sephacryl S-200 геля, уравновешенного 10 мМ фосфатным буфером, содержащим 50 мМ хлорид натрия. Активные фракции смешивали с добавлением хлорида натрия до конечной концентрации 3,5 М. Далее раствор адсорбировали на Phenyl Sepharose HP колонке, уравновешенной 10 мМ фосфатным буфером (рН 8.0), содержащим 3,5 М хлорид натрия. Затем адсорбированное вещество тщательно отмывали данным буфером и подвергали элюции с линейным градиентом концентрации хлорида натрия от 3,5 до 1,5 М. Активные фракции смешивали, посредством чего получали раствор очищенного фермента. Молекулярная масса фермента, определенная по времени прохождения колонки Sephacryl S-200, составила приблизительно 70000.
Пример 1
(1) Штамм Alteromonas sp. SN-1009 (FERM BP-5474), продуцирующий фермент, эндорасщепляющий полисахарид, содержащий сульфатированную фукозу, высевали в двухлитровую Еrlenmeyer колбу, с 500 мл среды, включающей искусственно полученную морскую воду (рН 8,0, производство Jamarin Laboratory), содержащую 0,25% глюкозу, 1,0% пептонный бульон и 0,05% экстракта дрожжей, стерилизовали (при 120°С в течение 20 минут) и затем культивировали при 25°С в течение 23 часов. После завершения культивирования среду центрифугировали для получения клеток, половину полученных клеток суспендировали в 10 мл буфера для экстракции [содержащего 50 мМ Трис НСl буфера (рН 8,0) и 100 мМ этилендиаминтетрауксусной кислоты (ЭДТА)], добавляли раствор (20 мг/мл лизоцима, растворенного в 1 мл буфера для экстракции) и смесь помещали на ледяную баню на 30 минут. После этого добавляли 10 мл раствора протеиназы К [содержащего 1 мг/мл протеиназы К, 50 мМ Трис НСl буфера (рН 8,0), 100 мМ (ЭДТА) и 1% SDS] и смесь выдерживали при 50°С в течение двух часов. Затем смесь охлаждали до комнатной температуры, добавляли равный объем фенола, насыщенного ТЕ буфером [содержащим 10 мМ Трис НСl буфера (рН 8,0) и 1 мМ ЭДТА], смесь осторожно помешивали в течение одного часа, и центрифугировали при 10000 об/мин в течение 20 минут и отбирали верхнюю фазу (в дальнейшем эта процедура будет называться фенольной экстракцией).
К данной верхней фазе добавляли равный объем смеси фенола и хлороформа в соотношении 1:1, насыщенной ТЕ буфером, затем раствор осторожно помешивали, центрифугировали при 10000 об/мин в течение 20 минут и отбирали верхнюю фазу (в дальнейшем эта процедура будет называться фенолхлороформной экстракцией). Фенолхлороформную экстракцию проводили повторно, добавляли хлорид натрия в водную фазу до концентрации 0,1 М, затем добавляли двукратный объем этанола для выделения ДНК, полученную ДНК накручивали на стеклянный капилляр, прополаскивали 80% этанолом и осторожно высушивали на воздухе. Данную геномную ДНК растворяли в 20 мл ТЕ буфера, содержащего растворенную рибонуклеазу А в концентрации 20 мкг/мл, и смесь выдерживали при 37°С в течение пяти часов для расщепления РНК. После фенольной экстракции и фенолхлороформной экстракции добавляли описанным выше способом этанол, выделяли ДНК и суспендировали в 5 мл ТЕ буфера. В результате проведенных операций получали приблизительно 20 мг геномной ДНК.
(2) Полученную в примере 1-(1) геномную ДНК (100 мкг) обрабатывали 10 единицами рестриктазы Sau3AI при 37°С в течение одной минуты сорока секунд для частичной деградации, затем проводили фенолхлороформную экстракцию и отбирали верхнюю фазу. К верхней фазе добавляли 0,1-кратный объем 3 М водного раствора ацетата натрия (рН 5,0) и 2,5-кратный объем этанола для осаждения ДНК и указанную ДНК прополаскивали 80% этанолом и осторожно высушивали на воздухе (в дальнейшем эта процедура будет называться осаждением этанолом). Полученный частично расщепленный продукт подвергали разделению по размеру с помощью градиентного ультрацентрифугирования с концентрацией хлорида натрия от 1,25 до 5 М и отбирали ДНК из фракций, содержащих фрагменты длиной 10-20 т.п.н., с помощью осаждения этанолом. Полученную частично расщепленную геномную ДНК (0,18 мкг) и 0,6 мкг λ, Blue Star BamHI линкера (производство Novagene) смешивали и лигировали с помощью DNA Ligation Kit (производство Takara Shuso) и ДНК упаковывали с помощью фага λ, применяя Gigapack II Gold Kit (производство Stratagene) для получения библиотеки геномной ДНК штамма Alteromonas sp. SN-1009.
(3) Очищенный белковый фермент (200 пмоль), обладающий расщепляющей активностью по отношению к полисахариду, содержащему эндосульфатированную фукозу, полученный в ссылочном примере 1-(3) из штамма Alteromonas sp. SN-1009, подвергали обессоливанию на колонке (Fast Desalting Column PC 3.2/10; производство Pharmacia), уравновешенной 20 мМ кислым углекислым аммонием, элюировали данным буфером и заменяли буфер. Элюат собирали в стеклянный стаканчик, концентрировали и выпаривали до сухого состояния, образец помещали вместе со стеклянным стаканчиком в стеклянную пробирку большего размера, куда вносили 10 мкл пиридина, 2 мкл 4-винилпиридина, 2 мкл три-N-бутилфосфина и 10 мкл воды, стеклянную пробирку плотно закрывали и проводили реакцию при 95°С в течение десяти минут для выполнения пиридилэтилирования. После завершения реакции стеклянный стаканчик вынимали и подвергали аэеотропной обработке водой несколько раз для удаления летучих компонентов.
К полученному пиридилэтилированному белковому ферменту, расщепляющему полисахарид, содержащий сульфатированную фукозу, добавляли 40 мкл 10 мМ Трис НСl буфера (рН 9,0), содержащего 8 М мочевину, 90 мкл 10 мМ Трис НСl буфера (рН 9,0) и 0,5 пмоль Achromobacter protease I (производство Таkara Shuzo), проводили обработку при 30°С в течение ночи и белковые фрагменты выделяли из полученного продукта обработки с помощью системы ЖХВД (Smart System; производство Pharmacia). Применяли колонку типа uRPC C2/C18 SC2.1/10 (производство Pharmacia) со скоростью элюции 100 мкл/мин. При элюции образца в качестве элюентов применяли 0,12% водный раствор трифторуксусной кислоты (элюент А) и ацетонитрил, содержащий 0,1% трифторуксусную кислоту (элюент В), и для выделения и очистки проводили элюцию образца с применением линейного градиента концентрации элюента В с начальной долей 0% и последующим возрастанием до 55% в течение 90 минут. Для каждой белковой фракции проводили анализ аминокислотной последовательности и определили частичные аминокислотные последовательности F27 (SEQ ID NO: 9), F34 (SEQ ID NO: 10), F47 (SEQ ID NO: 11) и F52 (SEQ ID NO: 12).
(4) Геномную ДНК (20 мкг), полученную в примере 1-(1), расщепляли рестриктазами BamHI, EcoRI, HindIII, PstI, SacI, SalI, SphI и Xbal, no 100 единиц при 37°С в течение четырех часов для каждой и затем экстрагировали с помощью фенолхлороформа. Продукт реакции выделяли посредством осаждения этанолом, каждые 10 мкг которого обрабатывали вышеуказанными рестриктазами по 50 единиц при 37°С в течение 16 часов для каждой, экстрагировали фенолхлороформом и продукт реакции также выделяли посредством осаждения этанолом. Каждые 5 мкг продукта реакции подвергали электорофорезу с 0,8% агарозным гелем и ДНК переносили на нейлоновый фильтр (торговое название: Hybond-N+; производство Amersham) применяя Саузерн блоттинг (cf. "Method for Studying Genes, II, pages 218-221, published by Tokyo Kagaku Dojin).
В качестве зонда для гибридизации синтезировали гибридный олигонуклеотид pFDA27 (SEQ ID NO: 13) на основе частичной аминокислотной последовательности F27 (SEQ ID NO: 9), определенной в примере 1-(3). Синтетический олигонуклеотид (20 пмоль) метили 32P, применяя Mega-Label Kit (производство Takara Shuzo).
Фильтр, подготовленный как указано выше, подвергали прегибридизации при 65°С в течение трех часов в буфере, содержащем 6 х SSC, 1% SDS, 100 мкг/мл ДНК спермы лосося и 5-кратный раствор Денхардта, куда добавляли меченый зонд до концентрации 0,5 пмоль/мл, и смесь подвергали гибридизации при 42°С в течение ночи. После завершения гибридизации фильтр отмывали 6 х SSC при комнатной температуре в течение десяти минут, 1 x SSC в 0,1% SDS при комнатной температуре в течение десяти минут, 1 х SSC в 0,1% SDS при 42°С в течение 30 минут. После удаления избытка воды фильтр помещали на экспозицию на 30 минут Imaging Plate (производство Fuji Photo Film) и анализировали полосы с помощью BAS 2000 Imaging Analyzer (производство Fuji Foto Film).
В результате были определены полосы гибридизации с зондом продукта, расщепленного BamHI, EcoRI и SalI, в положении 23 т.п.н. или более; в положении 4,8, 1,4 и 0,3 т.п.н. при расщеплении HindIII; в положении 23 т.п.н. или более и 3,6 т.п.н. при расщеплении PstI; в положении 23 т.п.н. или более и 9,8 т.п.н. при расщеплении SacI; в положении 23 т.п.н. или более, 4,9 т.п.н. и 3,0 т.п.н. при расщеплении SphI; и в положении 12, 5,2 и 3,5 т.п.н. при расщеплении Xbal.
Клон, содержащий ген фермента, расщепляющего полисахарид, содержащий сульфатированную фукозу, анализировали с помощью бляшечного гибридизационного метода согласно инструкциям для λ Blue Star Novagene из библиотеки геномной ДНК штамма Alteromonas sp. SN-1009, полученной в примере 1-(2). Затем этой фаговой библиотекой заражали клетки штамма Escherichia coli ER1647 и рассевали на четыре зоны чашки с L средой и диаметром 8,5 см для формирования приблизительно 500 бляшек на чашку. К данной чашке прикладывали нейлоновые фильтры (торговое название: Hybond-N+; производство Amersham), таким образом, что на первую зону приходилось 30 секунд, а на вторую приходилось около двух минут, вследствие чего фаг трансфецировался в каждые две зоны на чашке. Нейлоновые фильтры денатурировали в течение пяти минут на бумажном фильтре, погруженном в раствор, содержащий 0,5 М гидроксид натрия и 1,5 М хлорида натрия, затем нейтрализовали в течение пяти минут на бумажном фильтре, погруженном в 0,5 М Трис НСl буфер (рН 7,0) и 3,0 М хлорид натрия, и полоскали в 2 х SSC. Нейлоновые фильтры гибридизовали с синтетическим олигонуклеотидом pFDA27 (SEQ ID NO:13), отмывали и анализировали в условиях, аналогичных вышеуказанной Саузерн гибридизации, получая 18 позитивных сигналов. Бляшки, дающие позитивный сигнал, соскабливали с чашки и суспендировали в SM буфере, после чего формировали бляшки в новых чашках и соответственно повторяли вышепроделанные операции. В результате были выделены 14 фагов, дающих позитивный сигнал.
Каждым из полученных фагов инфицировали штамм Escherichia coli ВМ25.8, согласно инструкциям для λ Blue Star Novagene, отбирали колонии, резистентные к ампициллину, и фаг трансформировали в плазмиду. Колонии из каждого клона, полученного подобным образом, высевали на L среду (1% триптона, 0,5% экстракта дрожжей, 0,5% хлорида натрия), содержащей 100 мкг/мл ампициллина и инкубировали при 37°С в течение ночи и получали плазмидные ДНК способом щелочного лизиса. Штамм Escherichia coli JM109 (производство Takara Shuzo), трансформировали с помощью указанных плазмидных ДНК, колонии из каждого полученного клона, высевали на L среду, содержащую 100 мкг/мл ампициллина и инкубировали при 37°С в течение ночи, полученную среду опять подвергали щелочному лизису для получения каждой из плазмидных ДНК. Полученные плазмиды из каждого клона нумеровали соответственно pSFDA1, 2, 4, 5, 6, 7, 8, 10, 11, 12, 13, 14, 16 и pSFDA17.
Каждую плазмиду расщепляли рестриктазой NotI по обоим концам сайта клонирования Л, Blue Star вектора и анализировали посредством электрофореза в 0,3% агарозном геле, после чего выяснилось, что были встроены фрагменты длиной 8,4-17,4 т.п.н. Далее каждую плазмиду расщепляли рестриктазой HindIII, подвергали Саузерн блотингу, гибридизовали с синтетическим олигонуклеотидом pFDA27 (SEQ ID NO:13) и анализировали, после чего их разделили на шесть групп, состоящих из группы, дающей полосы приблизительно 4,8 т.п.н. (pSFDA1 и pSFDA17), второй группы, дающей полосы приблизительно 4,8 т.п.н. и 0,3 т.п.н. (pSFDA5, 6, 7 и pSFDA13), третьей группы, дающей полосу приблизительно 1,4 т.п.н. (pSFDA10), четвертой группы, дающей полосу приблизительно 0,3 т.п.н, и полосы других длин (pSFDA2 и pSFDA14), пятой группы, дающей полосу приблизительно 4,8 т.п.н. и полосы других длин (pSFDA16) и шестой группы, дающей полосу приблизительно 5,0 т.п.н. или более (pSFDA4, 8 и pSFDA12). Однако исключая pSFDA10, дающей полосу приблизительно 1,4 т.п.н., множество полос, имеющих одинаковые длины, определяли при окраске агарозного геля бромистым этидием, и поэтому было предположено, что в каждой плазмиде HindIII фрагменты длиной приблизительно 4,8 и 0,3 т.п.н., или один из них, или часть его встраивались в геномную ДНК в одинаковом положении. Также было предположено, что HindIII фрагменты длиной приблизительно 4,8 и 0,3 т.п.н., гибридизовавшиеся с pFDA27, локализуются в геноме в очень близких положениях. Соответственно полученные 14 плазмид предварительно могут быть разделены на pSFDA10, дающую полосу приблизительно 1,4 т.п.н., и другие плазмиды, и предположено, что они включают любой из фрагментов длиной около 4,8, 1,4 и 0,3 т.п.н. или оба фрагмента длиной 4,8 и 0,3 т.п.н., определенных с помощью Саузерн гибридизации геномной ДНК, расщепленной рестриктазой HindIII. Среди полученных плазмид плазмида pSFDA7, имеющая вставленный фрагмент длиной около 10,2 т.п.н. и содержащая оба HindIII фрагмента длиной 4,8 и 0,3 т.п.н., и плазмида pSFDA10, имеющая вставленный фрагмент длиной около 8,4 т.п.н. и содержащая HindIII фрагмент длиной 1,4 т.п.н., были обработаны различными типами рестриктаз и проанализированы с помощью электрофореза в агарозном геле для получения карты рестрикции. В то же время участок, который гибридизовался с синтетическим олигонуклеотидом pFDA27, субклонировали в плазмиду pUS119 и аналогичные плазмиды и анализировали нуклеотидную последовательность дидезоксиметодом. Последовательность в олигонуклеотиде pFDA27, в которой 14 оснований из 17 идентичны, обнаружили с помощью HindIII фрагмента из плазмиды pSFDA10 длиной около 1,4 т.п.н., но не обнаружили совпадения с последовательностью, кодирующей аминокислотную последовательность F27, и поэтому было предположено, что хотя указанный фрагмент и гибридизуется с олигонуклеотидом pFDA27, он не имеет отношения к гену, кодирующему фермент, эндорасщепляющий полисахарид, содержащий сульфатированную фукозу. С другой стороны, последовательность, кодирующая аминокислотную последовательность идентичную последовательности в олигонуклеотиде pFDA27, и, включая близлежащие последовательности, идентичную определенной в примере 1-(3) частичной аминокислотной последовательности F27 (SEQ ID NO: 9), была найдена из обоих HindIII фрагментов длиной около 4,8 и 0,3 т.п.н. плазмиды pSFDA7.
Кроме того, HindIII фрагменты длиной около 4,8 т.п.н. и 0,3 т.п.н. разделялись на рестрикционной карте отрезком около 3 т.п.н. или более и были больше длины указанного гена, кодирующего фермент, который предположительно имел молекулярную массу около 100000, как определено в ссылочном примере 1-(3) с помощью гель-фильтрации фермента, расщепляющего полисахарид, содержащий эндосульфатированную фукозу, выделенного из штамма Alteromonas sp. SN-1009, вследствие чего было предположено, что существуют, по крайней мере, два вида родственных генов.
Штамм Escherichia coli JM109, в который введена плазмида pSFDA7, будет называться как Escherichia coli JM109/pSFDA7. Далее, штамм Escherichia coli JM109, в который введена плазмида pSFDA7, называемый как Escherichia coli JM109/pSFDA7, был депонирован под номером доступа FERM Р-16362 в National Institute of Bioscience and Human-Technology, Agency of Industrial Science and Technology 1 августа 1997 года и был депонирован в качестве международного поступления в National Institute of Bioscience and Human-Technology, Agency of Industrial Science and Technology под номером доступа FERM BP-6340 (дата запроса для перевода в международное поступление: 6 мая 1998 года).
Относительно pSFDA7, более подробно нуклеотидную последовательность анализировали дидезоксиметодом, применяя метод удлинения затравки, и определили нуклеотидную последовательность длиной 8,3 т.п.н. во вставленном фрагменте длиной 10,2 т.п.н. pSFDA7, после чего были найдены две открытых рамки считывания - рамка считывания 1 длиной 2646 оснований (включая терминирующий кодон) (в дальнейшем называемая ORF-1) и другая рамка считывания 2 длиной 2445 оснований (включая терминирующий кодон) (в дальнейшем называемая ORF-2). HindIII фрагменты длиной 0,3 и 4,8 т.п.н., определяемых с помощью Саузерн гибридизации геномной ДНК, расщепленной рестриктазой HindIII, при более точном определении оказались длиной 140 и 4549 пар оснований соответственно и содержали часть или целиком всю длину ORF-1 и ORF-2 соответственно. В аминокислотной последовательности, кодируемой ORF-2, были найдены последовательности, имеющие высокую гомологию с частичными аминокислотными последовательностями F34 (SEQ ID NO: 10), F47 (SEQ ID NO: 11) и F52 (SEQ ID NO: 12), в дополнение к последовательности, идентичной частичной аминокислотной последовательности F27 (SEQ ID NO: 9) фермента, эндорасщепляющего полисахарид, содержащий сульфатированную фукозу. Соответственно предположили, что по существу ORF-2 кодирует фермент штамма Alteromonas sp. SN-1009, эндорасщепляющий полисахарид, содержащий сульфатированную фукозу. С другой стороны, в аминокислотной последовательности, кодируемой ORF-1, были найдены последовательности, имеющие высокую гомологию с частичными аминокислотными последовательностями F34 (SEQ ID NO: 10) и F52 (SEQ ID NO: 12), в дополнение к последовательности, идентичной частичной аминокислотной последовательности F27 (SEQ ID NO:9) фермента, эндорасщепляющего полисахарид, содержащий сульфатированную фукозу, хотя не было найдено последовательности, имеющей высокую гомологию с частичной аминокислотной последовательностью F47 (SEQ ID NO: 11). При сравнении аминокислотных последовательностей, кодируемых ORF-1 и ORF-2, была найдена вставочная последовательность из 67 аминокислотных остатков вблизи N-конца для ORF-1, которая не присутствовала в ORF-2, и аминокислотная последовательность после вставочной последовательности показала очень высокую гомологию, равную 70% или более, вследствие чего предположили, что ORF-1 также кодирует полипептид, эндорасщепляющий полисахарид, содержащий сульфатированную фукозу. Также были определены полные нуклеотидные последовательности гена (ORF-2), который, как предположили, кодирует полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, и гена (ORF-1), кодирующего новый полипептид, который, как предположили, обладает расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, и имеющего очень высокую гомологию с указанным геном.
Результат показан на фигуре 1. Таким образом, на фигуре 1 показаны положения ORF-1 и ORF-2. На фигуре 1 черная стрелка отражает область кодирования и направление ORF-1, в то время как стрелка со штриховкой отражает область кодирования и направление ORF-2. Нуклеотидная последовательность ORF-1 показана в SEQ ID NO:6 в списке последовательностей, а аминокислотная последовательность, кодируемая ORF-1, показана в SEQ ID NO: 2 в списке последовательностей. Нуклеотидная последовательность ORF-2 показана в SEQ ID NO: 5 в списке последовательностей, а аминокислотная последовательность, кодируемая ORF-2, показана в SEQ ID NO: 1 в списке последовательностей.
Как упоминалось ранее, ген (ORF-2), который предположительно кодирует фермент, расщепляющий полисахарид, содержащий эндосульфатированную фукозу, и ген (ORF-1), который, как предположили, кодирует новый полипептид, предположительно обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, были выделены и очищены согласно настоящему изобретению.
Пример 2
Для создания прямого вектора экспрессии гена (ORF-2), полученного в примере 1, предположительно, по существу, кодирующего фермент, эндорасщепляющий полисахарид, содержащий сульфатированную фукозу, инициирующий кодон ORF-2 сделали идентичным оптимизированному инициирующему кодону вектора экспрессии для создания плазмиды pEFDA-N, содержащей часть 5’-области гена ORF-2.
Сначала синтезировали синтетические ДНК: FDA-N1 (SEQ ID NO: 14) и FDA-N2 (SEQ ID NO: 15). FDA-N1 является синтетической ДНК 15 мер, содержащей последовательность из нуклеотидной последовательности номер 1-13 из SEQ ID NO: 5 в списке последовательностей, a FDA-N2 является синтетической ДНК 15 мер, содержащей последовательность, комплементарную последовательности из нуклеотидной последовательности номер 4-13 из SEQ ID NO: 5 в списке последовательностей.
Данные синтетические ДНК выдерживали при 70°С в течение 10 минут в растворе, содержащем 0,2 М Трис НСl буфер (рН 7,5) и 0,3 М хлорид натрия, постепенно охлаждали до комнатной температуры для образования двойной спирали, вследствие чего получали синтетический ДНК линкер FDA-N. Полученный FDA-N представлял собой синтетический отрезок ДНК, который содержал SnaBI сайт в нуклеотидной последовательности номер 8-13 из SEQ ID NO: 5 в списке последовательностей и был способен присоединяться по NcoI сайту около инициирующего кодона, а также по BamHI сайту непосредственно ниже SnaBI сайта.
Между тем, pET21d (производство Novagene), экспрессирующий вектор, использующий Т7 промотор, расщепляли по Ncol сайту, содержащему оптимизированный для экспрессии инициирующий кодон ниже Т7 промотора, и по BamHI сайту в поликлональном сайте. Данный продукт расщепления лигировали с уже приготовленным синтетическим ДНК линкером FDA-N с помощью DNA Ligation Kit (производство Takara Shuzo), затем трансформировали штамм Escherichia coli JM109 и отбирали колонии, выросшие на чашке с L средой, содержащей 100 мкг/мл ампициллина. Каждый трансформант высевали на L среду, содержащую 100 мкг/мл ампициллина, инкубировали при 37°С в течение ночи, и выделяли плазмидную ДНК из инкубированных клеток методом щелочного лизиса. После расщепления плазмидной ДНК SnaBI проводили электрофорез в 1% агрозном геле для выделения плазмиды, способной расщепляться SnaBI, затем устанавливали нуклеотидную последовательность вставленного фрагмента дидезоксиметодом и получали плазмиду pEFDA-N, в которой область от инициирующего кодона ORF-2 до SnaBI сайта располагалась между Ncol и BamHI сайтами pET21d.
Затем приблизительно 5 мкг плазмиды pSFDA7, полученной в примере 1, расщепляли при 37°С в течение двух часов 30 единицами SnaBI, разделяли с помощью электрофореза в 1% агарозном геле, и SnaBI фрагмент длиной 2,5 т.п.н., включающий приблизительно всю длину ORF-2 области, вырезали, выделяли и очищали. SnaBI фрагмент смешивали с продуктом расщепления рестриктазой SnaBI уже созданной плазмиды pEFDA-N и лигировали с помощью DNA Ligation Kit, затем проводили трансформацию штамма Escherichia coli JM109 и отбирали колонии, выросшие на чашке с L средой, содержащей 100 мкг/мл ампициллина. Плазмидные ДНК, полученные таким способом, расщепляли SnaBI, подвергали электрофорезу в 1% агарозном геле и выделяли плазмиду, содержащую высвобожденные SnaBI фрагменты длиной 2,5 т.п.н. Далее проводили дидезоксиметод для установления направления вставочного фрагмента и выделяли плазмиду, где ORF-2 область вставлена в том же направлении, что и Т7 промотор. Экспрессирующая плазмида, в которой содержится область от инициирующего кодона около NcoI сайта pET21d до полной длины ORF-2, была названа pEFDAII103.
Штамм Escherichia coli BL21 (DE3) (производство Novagene) трансформировали, применяя плазмиду pEFDAII103, полученную подобным способом. Полученный трансформант Escherichia coli BL21 (ВЕЗ)/pEFDAII103 вносили в 5 мл L среды, содержащей 100 мкг/мл ампициллина и 5 мМ хлорид кальция, подвергали посеву внутри среды при 37°С, добавляли IPTG до конечной концентрации 1 мМ на стадии, когда мутность, выражающаяся в O.D. 600, равнялась 0,8, и подвергали засеву внутри среды при 15°С в течение еще одной ночи. После завершения инкубации среду центрифугировали для получения клеток и клетки суспендировали в 1 мл буфера для разделения клеток [20 мМ Трис НСl буфер (рН 7,5), 10 мМ хлорида кальция, 10 мМ хлорида калия и 0,3 М хлорида натрия] и разрушали с помощью обработки ультразвуковыми волнами. Их центрифугировали, отбирали супернатант и использовали в качестве экстракта Escherichia coli.
В качестве контроля одновременно культивировали штамм Escherichia coli BL21 (DE3)/pET21d, трансформированный плазмидой pET21d, в тех же условиях и полученный экстракт Escherichia coli использовали для дальнейшего анализа.
Таким образом, сначала экстракты Escherichia coli анализировали с помощью SDS электрофореза в полиакриламидном геле, вследствие чего полосу с молекулярной массой 90000, не обнаруженную в экстракте Escherichia coli BL21 (DE3)/pET21d, обнаружили в экстракте Escherichia coli BL21 (DE3)/pEFDAII103. Данная молекулярная масса хорошо совпала с молекулярной массой полипептида (88210), рассчитанной из аминокислотной последовательности, которая способна кодироваться геном ORF-2 и которая показана в SEQ ID NO: 1 в списке последовательностей, и также совпала с молекулярной массой (приблизительно 90000), полученной при анализе фермента, эндорасщепляющего полисахарид, содержащий сульфатированную фукозу, выделенного из штамм Alteromonas sp. SN-1009 посредством SDS электрофореза в полиакриламидном геле. Соответственно было установлено, что трансформант Escherichia coli BL21 (DE3)/pEFDAII103 экспрессирует полипептид, кодируемый геном ORF-2.
Затем определяли эндорасщепляющую активность экстракта Escherichia coli по отношению к полисахариду, содержащему сульфатированную фукозу, способом, описанным в ссылочном примере 2.
В результате эндорасщепляющая активность по отношению к полисахариду, содержащему сульфатированную фукозу, определенная в экстракте Escherichia coli BL21 (DE3)/pEFDAII103 равнялась 520 мЕ/мл. Таким образом, было установлено, что полипептид, кодируемый геном ORF-2, имеет расщепляющую активность по отношению к полисахариду, содержащему сульфатированную фукозу, и что полипептид, кодируемый геном ORF-2, обладающий приблизительно 104 мЕ расщепляющей активности по отношению к полисахариду, содержащему сульфатированную фукозу, продуцируется в одном мл среды трансформанта Escherichia coli BL21 (DE3)/ pEFDAII103, содержащего ген настоящего изобретения.
Альтернативно, в экстракте Escherichia coli BL21 (DE3)/pET21d не было обнаружено эндорасщепляющей активности по отношению к полисахариду, содержащему сульфатированную фукозу.
Пример 3
Последовательности в ORF-1 и ORF-2 от инициирующего кодона до 15 основания полностью одинаковые, и в пределах указанной области располагается сайт рестрикции для рестриктазы SnaBI. Таким образом, вставочная последовательность в плазмиде pEFDA-N, созданной в примере 2, полностью одинакова в ORF-1 и в ORF-2, и поэтому плазмида pEFDA-N может быть использована для создания экспрессирующего вектора ORF-1.
Сначала около 5 мкг плазмиды p3FDA7, полученной в примере 1, обрабатывали при 37°С в течение 2 часов 30 единицами SnaBI, разделяли с помощью электрофореза в 1% агарозном геле и полученный SnaBI фрагмент длиной около 3,2 т.п.н., включающий почти всю длину ORF-1 области, вырезали, выделяли и очищали. После SnaBI фрагмент смешивали с продуктом расщепления плазмиды pEFDA-N рестриктазой SnaBI и лигировали с помощью DNA Ligation Kit и штамм Escherichia coli JM109 трансформировали данной смесью. Выделяли колонии, выросшие на чашке с L средой, содержащей 100 мкг/мл ампициллина.
Плазмидные ДНК получали способом, описанном в примере 2, расщепляли рестриктазой SnaBI, подвергали электрофорезу в 1% агарозном геле и выделяли плазмиду, содержащую SnaBI фрагмент длиной 3,2 т.п.н. Далее проводили определение направления вставочного фрагмента дидезоксиметодом и отбирали плазмиду, в которой ген ORF-1 был вставлен в одинаковом направлении с Т7 промотором. Полученная экспрессирующая плазмида, в которую вставлена полная длина ORF-1 до инициирующего кодона в NcoI сайте плазмиды pET21d, была названа pEFDAI103.
Используя плазмиду pEFDAI103, полученную подобным образом, экспрессию полипептида, кодируемого геном ORF-1, и расщепляющую активность полипептида по отношению к полисахариду, содержащему сульфатированную фукозу, устанавливали способом, описанным в примере 2.
Таким образом, сначала штамм Escherichia coli BL21 (DE3) трансформировали с помощью плазмиды pEFDAI103. Полученный трансформант Escherichia coli BL21 (DE3)/pEFDAI103 высевали на 5 мл L среды, содержащей 100 мкг/мл ампициллина и 5 мМ хлорида кальция и инкубировали при 37°С на шейкере. После добавления IPTG до конечной концентрации 1 мМ при мутности, выражающейся в O.D. 600, равной 0,8, далее культуру инкубировали при 15°С в течение еще одной ночи. После завершения инкубации среду центрифугировали для получения клеток. Затем клетки суспендировали в 1 мл буфера для разделения клеток и разрушали с помощью обработки ультразвуковыми волнами. Данные клетки центрифугировали для отбора супернатанта, используемого в качестве экстракта Escherichia coli.
Escherichia coli экстракт анализировали посредством SDS электрофореза в полиакриламидном геле, вследствие чего в экстракте Escherichia coli BL21 (DE3)/pEFDAI103 была получена полоса, соответствующая молекулярной массе 100000, которая не обнаруживалась в экстракте Escherichia coli BL21 (DE3)/pET21d. Данная молекулярная масса хорошо совпала с молекулярной массой (94910) полипептида, рассчитанной из аминокислотной последовательности, которая способна кодироваться геном ORF-1 и которая показана SEQ ID NO: 2 в списке последовательностей, и было установлено, что трансформант Escherichia coli BL21 (DE3)/pEFDAI103 экспрессирует полипептид, кодируемый геном ORF-1.
Затем определяли эндорасщепляющую активность экстракта Escherichia coli по отношению к полисахариду, содержащему сульфатированную фукозу, способом, описанным в ссылочном примере 2.
В результате эндорасщепляющая активность по отношению к полисахариду, содержащему сульфатированную фукозу, определенная в экстракте Escherichia coli BL21 (DE3)/pEFDAI103, равнялась 35,8 мЕ/мл. Таким образом, было установлено, что полипептид, кодируемый геном ORF-1, имеет расщепляющую активность по отношению к полисахариду, содержащему сульфатированную фукозу, и что в одном мл среды трансформанта Escherichia coli BL21 (DЕ3)/pEFDAI103, содержащего ген настоящего изобретения, продуцируется полипептид, кодируемый геном ORF-1, обладающий приблизительно 7,2 мЕ расщепляющей активности по отношению к полисахариду, содержащему сульфатированную фукозу.
С другой стороны, в экстракте Escherichia coli BL21 (DE3)/pET21d не было обнаружено эндорасщепляющей активности по отношению к полисахариду, содержащему сульфатированную фукозу.
Пример 4
(1) Штамм Flavobacterium sp. SA-0082 (FERM BP-5402), продуцирующий фукоиданазу, инкубировали при 25°С в течение 23 часов в двухлитровой Erlenmeyer колбе с 500 мл среды, включающей искусственно полученную морскую воду (производство Jamarin Laboratory), содержащей 0,25% глюкозу, 1,0% пептонный бульон и 0,05% экстракта дрожжей, и стерилизовали (при 120°С в течение 20 минут). После завершения инкубации среду центрифугировали для получения клеток, половину полученных клеток суспендировали в 10 мл буфера для экстракции [содержащего 50 мМ Трис НСl буфера (рН 8,0) и 100 мМ этилендиаминтетрауксусной кислоты (ЭДТА)], добавляли раствор 20 мг/мл лизоцима, растворенного в 1 мл буфера для экстракции, и смесь помещали на ледяную баню на 30 минут. После этого добавляли 10 мл раствора протеиназы К [содержащего 1 мг/мл протеиназы К, 50 мМ Трис НСl буфера (рН 8,0), 100 мМ ЭДТА и 1% SDS] и смесь выдерживали при 50°С в течение двух часов. Затем смесь охлаждали до комнатной температуры, добавляли равный объем фенола, насыщенного ТЕ буфером [содержащим 10 мМ Трис НСl буфера (рН 8,0) и 1 мМ ЭДТА], смесь осторожно помешивали в течение одного часа, центрифугировали при 10000 об/мин в течение 20 минут и отбирали верхнюю фазу. К данной верхней фазе добавляли равный объем смеси фенола и хлороформа в соотношении 1:1, насыщенной ТЕ буфером. Затем раствор осторожно помешивали и центрифугировали при 10000 об/мин в течение 20 минут. Отбирали полученную верхнюю фазу. Фенолхлороформную экстракцию проводили повторно, добавляли хлорид натрия в водную фазу до концентрации 0,1 М, затем добавляли двукратный объем этанола для выделения ДНК. Полученную ДНК накручивали на стеклянный капилляр, прополаскивали 80% этанолом и осторожно высушивали на воздухе. Данную геномную ДНК растворяли в 20 мл ТЕ буфера, содержащего растворенную рибонуклеазу А в концентрации 20 мкг/мл, и смесь выдерживали при 37°С в течение пяти часов для расщепления РНК. После фенольной экстракции и фенолхлороформной экстракции туда добавляли этанол, выделяли ДНК описанным выше способом и ДНК суспендировали в 5 мл ТЕ буфера. В результате проведенных операций получали приблизительно 20 мг геномной ДНК.
(2) Полученную в примере 4-(1) геномную ДНК (100 мкг) обрабатывали 10 единицами рестриктазы Sau3AI при 37°С в течение 1 минуты 40 секунд для частичной деградации, затем проводили фенолхлороформную экстракцию для отбора верхней фазы. К верхней фазе добавляли 0,1-кратный объем трехмолярного водного раствора ацетата натрия (рН 5,0) и 2,5-кратный объем этанола для осаждения ДНК, и с помощью центрифугирования получали преципитат, прополаскивали 80% этанолом и осторожно высушивали на воздухе. Полученный частично расщепленный продукт подвергали разделению по размеру с помощью ультрацентрифугирования по градиенту плотности с концентрацией хлорида натрия от 1,25 до 5 М, и отбирали ДНК из фракций, содержащих фрагменты длиной 10-20 т.п.н., с помощью осаждения этанолом. Полученную частично расщепленную геномную ДНК (0,2 мкг) и 0,6 мкг λ Blue Star BamHI линкера (производство Novagene) смешивали и лигировали с помощью DNA Ligation Kit (производство Takara Shuso) и ДНК упаковывали с помощью фага λ, применяя Gigapack II Gold Kit (производство Stratagene) для получения библиотеки геномной ДНК штамма Flavobacterium sp. SA-0082.
(3) Очищенную белковую фукоиданазу (200 пмоль) из штамма Flavobacterium sp. SA-0082, полученную в ссылочном примере 5, подвергали обессоливанию на колонке (Fast Desalting Column PC 3.2/10; производство Pharmacia), уравновешенной 20 мМ кислым углекислым аммонием, и элюировали данным буфером до замены буфера. После элюат собирали в стеклянный стаканчик, концентрировали и выпаривали до сухого состояния, образец помещали вместе со стеклянным стаканчиком в стеклянную пробирку большего размера, куда вносили 10 мкл пиридина, 2 мкл 4-винилпиридина, 2 мкл три-N-бутилфосфина и 10 мкл воды. Стеклянную пробирку плотно закрывали и проводили реакцию при 95°С в течение 10 минут для выполнения пиридилэтилирования. После завершения реакции стеклянный стаканчик вынимали и подвергали несколько раз азеотропной обработке водой для удаления летучих компонентов.
К полученной пиридилэтилированной белковой фукоиданазе добавляли 40 мкл 10 мМ Трис НСl буфера (рН 9,0), содержащего 8 М мочевину, 90 мкл 10 мМ Трис НСl буфера (рН 9,0) и 0,5 пмоль Achromobacter protease I (производство Таkara Shuzo), обработку проводили при 30°С в течение ночи и выделяли белковые фрагменты из полученного продукта обработки с помощью системы ЖХВД (Smart System; производство Pharmacia). Применяли колонку типа uRPC C2/C18 SC2.1/10 (производство Pharmacia) со скоростью элюции 100 мкл/мин. При элюции образца в качестве элюентов применяли 0,12% водный раствор трифторуксусной кислоты (элюент А) и ацетонитрил, содержащий 0,1% трифторуксусную кислоту (элюент В). При нанесении образца начальная доля элюента В составляла 0%, и элюцию проводили с применением линейного градиента концентрации с последующим возрастанием доли элюента В до 55% в течение 80 минут. В области пиков элюции, где доля элюента В составила 27% или более, были получены фракции L27, L31 и L36. После этого фракции, где доля элюента В составляла 17-27% и разделение было недостаточным, собирали, концентрировали, наносили на эту же систему ЖХВД и подвергали элюции с линейным градиентом концентрации, при этом доля элюента В возрастала от 15 до 40% в течение 87 минут, давая элюированные фракции LR8, LR9, LR14 и LR16. Для каждой белковой фракции проводили анализ аминокислотной последовательности и были определены частичные аминокислотные последовательности L27 (SEQ ID NO: 16), L36 (SEQ ID NO: 17), LR9 (SEQ ID NO: 18), LR14 (SEQ ID NO: 19) и LR16 (SEQ ID NO: 20).
(4) Геномную ДНК (2,4 мкг), полученную в примере 4-(1), обрабатывали при 37°С в течение трех часов рестриктазами EcoRI, HindIII, Munl, Spel, Xbal, и Sau3AI no 30 единиц для каждой с последующей экстракцией фенолхлороформом. Продукт реакции выделяли посредством осаждения этанолом. Каждые 5 мкг продукта реакции смешивали с EcoRI кассетой для продуктов расщепления EcoRI и Munl; с HindIII кассетой для продукта расщепления HindIII; с Xbal кассетой для продуктов расщепления Spel и Xbal; и Sau3AI кассетой для продукта расщепления Sau3AI, где кассету использовали в каждом случае в количестве 20 нг (в случае Sau3AI кассеты 200 нг) (кассеты, произведенные Takara Shuzo), и лигировали с помощью DNA Ligation Kit (производство Takara Shuzo). Каждый из продуктов реакции выделяли посредством осаждения этанолом и растворяли в 10 мкл воды, давая матричную ДНК для ПЦР с применением кассетной ДНК.
Между тем, с аминокислотной последовательности 1-6 из частичной аминокислотной последовательности LR14 (SEQ ID NO: 19), установленной в примере 4-(3), синтезировали комбинированный олигонуклеотид pL14F17 (SEQ ID NO: 21) в указанном аминокислотной последовательностью направлении, а с аминокислотной последовательности 3-11 из той же частичной аминокислотной последовательности синтезировали комбинированный олигонуклеотид pL14F26 (SEQ ID NO: 22) аналогичным способом.
Каждую реакционную смесь получали при смешивании 1 мкл уже подготовленной матричной ДНК, 20 пмоль кассетного праймера С1 (производство Takara Shuzo), 100 пмоль комбинированного олигонуклеотида pL14Fl7 (SEQ ID NO: 21) и стерилизованной воды, давая объем смеси 22 мкл, и нагревали при 94°С в течение 10 минут с последующим быстрым охлаждением. К каждому раствору до объема 100 мкл добавляли 10 мкл 10-кратного Ex Taq буфера (производство Takara Shuzo), 16 мкл раствора смешанных dNTP по 1,25 мМ каждого, 2,5 единицы Takara EX Taq (производство Takara Shuzo) и стерилизованной воды с последующим наслаиванием минерального масла. Данные смеси подвергали реакции амплификации, применяя автоматический амплификатор генов (DNA Thermal Cycler 480; производство Takara Shuzo). В ходе амплификации проводили 25 циклов, каждый из которых включал в себя денатурацию при 94°С в течение 0,5 минуты, отжиг при 45°С в течение двух минут и реакцию синтеза при 72°С в течение трех минут, и наконец, смеси выдерживали при 72°С в течение семи минут для завершения реакции.
После этого проводили вторую ПЦР, используя каждую матричную ДНК, полученную 10-кратным разведением реакционной смеси первой ПЦР стерилизованной водой и нагреванием до 94°С в течение 10 минут с последующим быстрым охлаждением. Таким образом, смешивали 10 мкл термически обработанной смеси первой ПЦР, 20 пмоль кассетного праймера С2 (производство Takara Shuzo), 100 пмоль комбинированного олигонуклеотида pL14F26 (SEQ ID NO: 22), 10 мкл 10-кратного Ex Taq буфера для амплификации (производство Takara Shuzo), 16 мкл раствора смешанных dNTP по 1,25 мМ каждого, 2,5 единицы Takara EX Taq и стерилизованную воду до объема 100 мкл с последующим наслаиванием минерального масла. Каждую смесь подвергали амплификации из 25 циклов, каждый из которых включал в себя денатурацию при 94°С в течение 0,5 минуты, отжиг при 55°С в течение двух минут и реакцию синтеза при 12°С в течение трех минут, и наконец, смеси выдерживали при 72°С в течение семи минут для завершения реакции. В то же время проводили ПЦР для реакционных смесей, содержащих только кассетный праймер С2 и содержащих только комбинированный олигонуклеотид pL14F26, что являлось контролем неспецифических продуктов амплификации для каждого праймера.
После анализа реакционного раствора с помощью элекрофореза в агарозном геле в реакционных растворах определили множество полос амплификации, которые сравнивали с контрольной реакцией на неспецифический продукт амплификации. Исходя из данного выделили и очистили полосу приблизительно 0,7 т.п.н. из раствора второй ПЦР, где использовали в качестве матрицы ДНК, полученную из продукта расщепления Munl и кассеты EcoRI, для которой свойственны относительно низкий фон и строго одна полоса амплификации. Данный фрагмент ДНК смешивали с рТ7bluе Т-вектором (производство Novagene) и лигировали с помощью DNA Ligation Kit. Штамм Escherichia coli JM109 трансформировали полученным продуктом сшивки и отделяли белые колонии, выросшие на чашке с L средой, содержащей 100 мкг/мл ампициллина, 0,04% X-Gal и 1 мМ IPTG. После каждый трансформант высевали на L среду, содержащую 100 мкг/мл ампициллина, и инкубировали при 37°С в течение ночи, из проинкубировавшихся клеток при помощи щелочного лизиса получали плазмидную ДНК, и выделенная плазмида, включающая вставку из полосы длиной приблизительно 0,7 т.п.н., была названа pT7-Mun. Вставочную последовательность данной плазмиды pT7-Mun подвергали определению нуклеотидной последовательности дидезоксиметодом, вследствие чего, следуя последовательности pL14F26, использованной во второй ПЦР, были найдены область, кодирующая аминокислотную последовательность из 12 аминокислот, и далее частичная аминокислотная последовательность LR14 (SEQ ID N0:19). Далее, ниже была найдена область, кодирующая аминокислотную последовательность, имеющую очень высокую гомологию с частичной аминокислотной последовательностью L27 (SEQ ID NO: 16). Исходя из данных находок очевидно, что фрагмент ДНК длиной приблизительно 0,7 т.п.н. является частью гена фукоиданазы. Последовательность от праймера pL14F26 0,7 т.п.н. фрагмента ДНК до точки лигирования с EcoRI кассетой показана в SEQ ID NO: 23.
(5) Каждые 20 мкг геномной ДНК, полученной в примере 4-(1), обрабатывали рестриктазами BamHI, EcoRI, HindIII, PstI, SacI, SalI, SphI или Xbal no 100 единиц каждой при 37°С в течение четырех часов и затем экстрагировали с помощью фенолхлороформа. Продукт реакции выделяли посредством осаждения этанолом, каждые 10 мкг которого повторно обрабатывали вышеуказанными рестриктазами по 50 единиц при 37°С в течение 16 часов для каждой, экстрагировали фенолхлороформом и продукт реакции также выделяли посредством осаждения этанолом. Каждые 5 мкг продукта реакции подвергали электорофореэу в 0,8% агарозном геле и ДНК переносили на нейлоновый фильтр (Hybond-N+; производство Amersham), применяя Саузерн блоттинг.
Кроме того, около 4 мг плазмиды pT7-Mun, полученной в примере 4-(4), обрабатывали BamHI и SphI и высвободившийся фрагмент длиной приблизительно 0,7 т.п.н., содержащий часть гена фукоиданазы, выделяли и очищали. Фрагмент ДНК метили 32P, используя BcaBEST Labeling Kit (производство Takara Shuzo) для получения ДНК зонда для гибридизации.
Фильтр, подготовленный как указано выше, подвергали прегибридизации при 65°С в течение одного часов в буфере, содержащем 6 х SSC, 1% SDS, 100 мкг/мл ДНК спермы лосося и 5-кратный раствор Денхардта, куда добавляли меченый зонд до концентрации 1000000 cpm/ml, и смесь подвергали гибридизации при 60°С в течение ночи. После завершения гибридизации фильтр отмывали 6 х SSC при комнатной температуре десять минут, 0,2 х SSC в 0,1% SDS при комнатной температуре десять минут, 0,2 х SSC в 0,1% SDS при комнатной температуре пять минут, 0,2 х SSC в 0,1% SDS при 45°С в течение 30 минут, для удаления излишка воды, фильтр закладывали на экспозицию на 30 минут Imaging Plate (производство Fuji Photo Film) и анализировали с помощью BAS 2000 Imaging Analyzer (производство Fuji Photo Film).
В результате в продуктах расщепления BamHI, SalI/SphI и SalI определяли интенсивную полосу гибридизации с зондом в положении 23 т.п.н. или более; в продукте расщепления EcoRI определяли две подобные полосы в положениях приблизительно 8 и 3 т.п.н.; в продукте расщепления HindIII определяли две подобные полосы в положениях приблизительно 11 и 4 т.п.н.; в продукте расщепления PstI определяли две подобные полосы в положениях приблизительно 12 и 2,5 т.п.н.; в продукте расщепления SacI определяли две подобные полосы в положениях приблизительно 10 и 9,5 т.п.н.; и в продукте расщепления XbaI определяли одну полосу в положении приблизительно 6 т.п.н. Соответственно было твердо предположено, что существует два типа гена фукоиданазы в геномной ДНК Flavobacterium sp. SA-0082.
(6) Клоны, содержащие ген фукоиданазы, анализировали с помощью бляшечного гибридизационного метода согласно инструкциям для λ Blue Star Novagene из библиотеки геномной ДНК штамма ДНК Flavobacterium sp. SA-0082, полученной в примере 4-(2).
Сначала фаговой библиотекой заражали клетки штамма Escherichia coli ER1647 и формировались приблизительно 300 бляшек на чашку на пяти чашках с L средой и диаметром 8,5 см. К каждой чашке прикладывали нейлоновый фильтр (Нуbond-N+; производство Amersham) в течение 30 секунд, вследствие чего происходила трансфекция фага. Нейлоновые фильтры денатурировали в течение пяти минут в растворе, содержащем 0,5 М гидроксид натрия и 1,5 М хлорида натрия, затем нейтрализовали в течение пяти минут в 0,5 М Трис НСl буфере (рН 7,0) и 3,0 М хлориде натрия, и полоскали в 2 х SSC и высушивали на воздухе. После обработки ультрафиолетом для фиксирования ДНК нейлоновые фильтры гибридизовали, отмывали и анализировали в условиях, аналогичных описанным в примере 4-(5), с помощью Саузерн гибридизации, вследствие чего были получены 36 позитивных сигналов. Бляшки, дающие позитивный сигнал, соскабливали с чашки и суспендировали в SM буфере для получения фага. Для некоторых из полученных растворов фага опять формировали бляшки в новых чашках, и соответственно повторяли выше проделанные операции, в результате были выделены девять фагов, дающих позитивный сигнал.
Согласно инструкциям для λ Blue Star Novagene каждым из полученных фагов инфицировали штамм Escherichia coli ВМ25,8, высевали на чашку с L средой, содержащей 100 мкг/мл ампициллина, и отбирали колонии, резистентные к ампициллину, в результате чего фаг преобразовывали в плазмиду. Колонии из каждого полученного клона высевали на L среду, содержащую 100 мкг/мл ампициллина, инкубировали при 37°С в течение ночи и выделяли из среды плазмидные ДНК способом щелочного лизиса. Штамм Escherichia coli JM109 (производство Takara Shuzo) трансформировали с помощью указанных плазмидных ДНК, колонии из каждого полученного клона высевали на L среду, содержащую 100 мкг/мл ампициллина, и инкубировали при 37°С в течение ночи, полученную среду опять подвергали щелочному лизису для получения плазмидных ДНК. Плазмиды из каждого полученного клона нумеровали соответственно pSFLA1, 5, 10, 11, 12, 13, 15, 17 и pSFLA18.
Каждую плазмиду обрабатывали подходящей комбинацией рестриктаз KpnI, SacI и XbaI, подвергали электрофорезу в агарозном геле и затем подвергали Саузерн гибридизации как описано выше, вследствие чего для каждого вставочного фрагмента получали и анализировали рестрикционную карту. В результате, после выдерживания агарозного геля в бромистом этидии в каждой дорожке определяли множество полос, при этом в каждой имелись полосы одинакового размера, после чего было установлено, что они разделяются на две группы - одна группа, состоящая из pSFLA1, 10, 12, 15 и pSFLA18, где имелись две полосы, гибридизующиеся с вставочным фрагментом длиной приблизительно 0,7 т.п.н. плазмиды рТ7-Мun, обработанной SacI, и вторая группа, состоящая из pSFLA5, 11, 13 и pSFLA17, где имелись две полосы, гибридизующиеся с вставочным фрагментом длиной приблизительно 0,7 т.п.н. плазмиды pT7-Mun, при обработке KpnI. Далее было выяснено, что три плазмиды - pSFLA5, pSFLA10 и pSFLA17 - при обработке рестриктазой ХbаI дают фрагмент длиной приблизительно 6 т.п.н., предполагая, что полоса длиной приблизительно 6 т.п.н. в продукте расщепления XbaI, которую определяли с помощью Саузерн гибридизации в примере 4-(5), обусловлена двумя частично совпадающими полосами, и стало очевидно, что существуют, по крайней мере, два типа гена фукоиданазы в геномной ДНК штамма Flavobacterium sp. SA-0082.
Таким образом, были выделены pSFLA10 и pSFLA17 из каждой группы плазмид, и далее проведен анализ вставочных фрагментов. Приблизительно 3 мкг каждой плазмиды обрабатывали рестриктазой Xbal и высвобожденный фрагмент длиной приблизительно 6 т.п.н. выделяли и очищали. Каждый из данных XbaI фрагментов смешивали с продуктом расщепления XbaI, pHSG399 (производство Takara Shuzo), который являлся вектором устойчивости к хлорамфениколу, и лигировали с помощью DNA Ligation Kit (производство Takara Shuzo). Затем штамм Escherichia coli JM109 трансформировали данным продуктом, и выделяли белые колонии, выросшие на чашке с L средой, содержащей 30 мкг/мл хлорамфеникола, 0,004% X-Gal и 1 мМ IPTG. Каждый из трансформантов высевали на L среду, содержащую 30 мкг/мл хлорамфеникола и инкубировали при 37°С в течение ночи. Затем выделяли плазмидную ДНК из инкубированных клеток с помощью щелочного лизиса и анализировали на агарозном геле электрофорезе после обработки рестриктазами. В результате были получены плазмиды рН10Х6-1 и рН10Х6-2, где полученная из pSFLA10 ДНК длиной приблизительно 6 т.п.н. была вставлена в обратных друг другу направлениях. Аналогично были получены плазмиды рН17Х6-7 и рН17Х6-11, где содержали ДНК длиной приблизительно 6 т.п.н., полученную из pSFLAl7. Штамм Escherichia coli JM109, в который введена плазмида рН10Х6-1, был назван Escherichia coli JM109/pH10X6-l, и был депонирован под номером доступа FERM Р-16659 в National Institute of Bioscience and Human-Technology, Agency of Industrial and Technology, MITI 24 февраля 1998 года и был депонирован в качестве международного поступления под номером доступа FERM BP-6341 в той же организации (дата запроса для перевода в международное поступление: 6 мая 1998 года). Штамм Escherichia coli JM109 в который введена плазмида рН17Х6-7, был назван Escherichia coli JM109/pH17X6-7 и был депонирован под номером доступа FERM P-16660 в National Institute of Bioscience and Human-Technology, Agency of Industrial and Technology, MITI 24 февраля 1998 года и был депонирован в качестве международного поступления под номером доступа FERM BP-6342 в той же организации (дата запроса для перевода в международное поступление: 6 мая 1998 года). Далее данные плазмиды подвергали точному определению нуклеотидной последовательности с помощью непосредственного метода удлинения затравки или с помощью субклонирования после расщепления рестриктазой, после чего были получены рамка считывания размером 2094 оснований (включая терминирующий кодон), из фрагмента ДНК длиной приблизительно 6 т.п.н., полученного из плазмиды pSFLA10, и рамка считывания размером 2115 оснований (включая терминирующий кодон), из фрагмента ДНК длиной приблизительно 6 т.п.н., полученного из плазмиды pSFLA17, и в аминокислотных последовательностях, кодируемых данными рамками считывания, были найдены области, имеющие очень высокую гомологию с частичной аминокислотной последовательностью, определенной в примере 4-(3).
Рамка считывания, происходящая из плазмиды pSFLA10, была названа fdlA, а рамка считывания, происходящая из плазмиды pSFLA17, была названа fdlB.
Результат показан на фиг.3 и на фиг.4. Таким образом, на фиг.3 показано положение fdlA в плазмиде pSFLA10, а на фиг.4 показано положение fdlB в плазмиде pSFLA17. Черной стрелкой на фиг.3 показана кодирующая область и направление fdlA, а черной стрелкой на фиг.4 показана кодирующая область и направление fdlB.
В кодируемой fdlA аминокислотной последовательности были найдены последовательности, идентичные частичной аминокислотной последовательности L36 (SEQ ID NO: 17) и LR14 (SEQ ID NO: 19) фукоиданазы, определенных в примере 4-(3), и также были найдены последовательности, имеющие очень высокую гомологию с частичной аминокислотной последовательностью L27 (SEQ ID NO: 16), LR9 (SEQ ID NO: 18) и LR16 (SEQ ID NO: 20) фукоиданазы, определенных в примере 4-(3). Далее, в белковой фракции, названной LR8 в примере 4-(3), были определены две аминокислотных производных из каждого цикла с помощью анализа аминокислотной последовательности, вследствие чего аминокислотная последовательность не была безусловно определена. Однако при изучении последовательностей в двух положениях в аминокислотной последовательности, кодируемой fdlA, могла быть объяснима аминокислотная производная из каждого цикла, поэтому стало очевидно, что в указанной фракции существует два белка, каждый из которых содержит одну из последовательностей из двух положений. Эти последовательности были названы LR8-1 (SEQ ID NO: 24) и LR8-2 (SEQ ID NO: 25). Также, кодируемые fdlA аминокислотные последовательности оказались идентичными или высокогомологичными со всеми аминокислотными последовательностями, установленными с помощью анализа частичной аминокислотной последовательностью фукоиданазы в примере 4-(3), и поэтому вероятно, что они, по существу, кодируют фукоиданазу штамма Flavobacterium sp. SA-0082.
Тем временем, в кодируемых геном fdlB аминокислотных последовательностях были найдены последовательности, идентичные частичным аминокислотным последовательностям LR14 (SEQ ID NO: 19) и LR8-1 (SEQ ID NO: 24) фукоиданазы, и также были найдены последовательности, имеющие высокую гомологию с частичными аминокислотными последовательностями L27 (SEQ ID NO: 16), LR16 (SEQ ID NO: 20) и LR8-2 (SEQ ID NO: 25) фукоиданазы, хотя не было найдено последовательности, имеющей высокую гомологию с частичными аминокислотными последовательностями L36 (SEQ ID NO: 17) или LR9 (SEQ ID NO: 18). Однако при сравнении fdlA и fdlB была найдена гомология нуклеотидной последовательности и аминокислотной последовательности, составляющая около 67% и около 56% соответственно, вследствие чего вероятно, что fdlB также кодирует полипептид, обладающий расщепляющей активностью по отношению к фукоидану. Также, были определены полная нуклеотидная последовательность гена fdlA, который предположительно кодирует полипептид, обладающий расщепляющей активностью по отношению к фукоидану, и гена fdlB, имеющего очень высокую гомологию с данным геном. Нуклеотидная последовательность гена fdlA показана в SEQ ID NO: 7 в списке последовательностей, а аминокислотная последовательность, кодируемая fdlA, показана в SEQ ID NO: 3 в списке последовательностей. Далее, нуклеотидная последовательность гена fdlB показана SEQ ID NO: 8 в списке последовательностей, а аминокислотная последовательность, кодируемая fdlB, показана SEQ ID NO:4 в списке последовательностей.
Также согласно настоящему изобретению выделены и очищены ген (fdlA), который, как предполагают, по существу, кодирует фукоиданазу, и ген (fdlB), который, как предполагают, имеет гомологию с данным геном и кодирует новый полипептид, предположительно обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу.
Пример 5
Сначала конструировали непосредственный экспрессирующий вектор гена (fdlA), полученного в примере 4 и предположительно, по существу, кодирующего фукоиданазу.
В плазмиду рН10Х6-1, полученную в примере 4, был вставлен выделенный из pSFLA10 Xbal фрагмент длиной приблизительно 6 т.п.н. в таком направлении, что fdlA ген располагался навстречу lac-промотору вектора. Данную плазмиду рН10Х6-1 расщепляли рестриктазой SacI по сайту, локализованному в fdlA гене, затем обрабатывали рестриктазой BspHI по положению инициирующего кодона в fdlA гене, разделяли с помощью электрофореза в 1% агарозном геле, и фрагменты ДНК приблизительно 400 пар оснований, кодирующие N-концевую область fdlA гена, вырезали, выделяли и очищали.
С другой стороны, плазмиду pET21d (производство Novagene), которая являлась экспрессирующим вектором, использующим Т7 промотор, разрезали по NcoI сайту, содержащему оптимизированный для экспрессии инициирующий кодон и расположенному ниже Т7 промотора, и также по SacI сайту, локализованному в поликлональном сайте, и затем вставляли BspHI-
SacI фрагменты длиной около 400 пар оснований, кодирующие N-концевую область уже полученного fdlA гена, вследствие чего создавали плазмиду pSFLA10-N.
Затем плазмиду р10Х6-1 обрабатывали HindIII для расщепления по HindIII сайтам, происходящим из вектора и внутренней области fdlA гена, и фрагменты ДНК длиной около 2 т.п.н., кодирующие С-концевую область fdlA гена, включая терминирующий кодон, вырезали, выделяли и очищали. Фрагмент лигировали с продуктом расщепления HindIII ранее полученной плазмиды pEFLA10-N и выделяли плазмиду, в которой указанные HindIII фрагменты длиной 2 т.п.н. были вставлены в направлении, при котором восстанавливалась полностью длина fdlA гена. Экспрессирующая плазмида, в которую вставлена от инициирующего кодона, расположенного в Ncol сайте плазмиды pET21d, полностью длина fdlA гена и которая получена подобным образом, была названа pEFLA10.
Затем штамм Escherichia coli BL21 (DE3) (производство Novagene) трансформировали, применяя плазмиду pEFLA10, полученную подобным способом. Полученный трансформант Escherichia coli BL21 (DE3)/pEFLA10 вносили в 5 мл L среды, содержащей 100 мкг/мл ампициллина, подвергали посеву внутри среды при 37°С. Когда мутность культуры, выражающаяся в O.D. 600, достигала 0,8, к культуре добавляли IPTG до конечной концентрации 1 мМ и далее культуру подвергали засеву внутри среды при 15°С в течение еще одной ночи. После завершения инкубации среду центрифугировали для получения клеток и клетки суспендировали в 0,5 мл буфера для разделения клеток [20 мМ натрийфосфатный буфер (рН 7,0) и 0,3 М хлорида натрия] и разрушали с помощью обработки ультразвуковыми волнами. Часть суспензии отбирали и использовали в качестве лизата Escherichia coli. Суспензию центрифугировали с удалением нерастворимых веществ для получения экстракта Escherichia coli.
В качестве контроля одновременно культивировали штамм Escherichia coli BL21 (DE3)/pET21d, трансформированный плазмидой pET21d, в тех же условиях, для получения лизата и экстракта Escherichia coli. Их использовали для дальнейшего анализа.
Таким образом, сначала лизат Escherichia coli анализировали с помощью SDS полиакриламидного гель-электрофореза, вследствие чего полоса с молекулярной массой около 76000 (не обнаруженная в лизате Escherichia coli BL21 (DE3)/pET21d), была обнаружена в лизате Escherichia coli BL21 (DE3)/pEFLA10. Данная молекулярная масса хорошо совпала с молекулярной массой полипептида (75740), рассчитанной из аминокислотной последовательности, которая способна кодироваться геном fdlA и которая показана в SEQ ID NO: 3 в списке последовательностей, и также хорошо совпала с молекулярной массой (приблизительно 70000), полученной при анализе с помощью гель-фильтрации хорошо очищенной фукоиданазы, выделенной из штамма Flavobacterium sp. SA-0082, описанной в ссылочном примере 5. Таким образом, было установлено, что трансформант Escherichia coli BL21 (DE3) /pEFLA10 экспрессирует полипептид, кодируемый геном fdlA.
Затем была определена расщепляющая активность экстракта Escherichia coli по отношению к полисахариду, содержащему сульфатированную фукозу, способом, описанным в ссылочном примере 3.
В результате расщепляющая активность по отношению к фукоидану, определенная в экстракте Escherichia coli BL21 (DE3)/pEFLA10, равнялась 2300 мЕ/мл. Таким образом, было установлено, что полипептид, кодируемый геном fdlA, имеет расщепляющую активность по отношению к фукоидану и что полипептид, кодируемый геном fdlA, обладающий приблизительно 230 мЕ расщепляющей активности по отношению к полисахариду, содержащему сульфатированную фукозу, продуцируется в одном мл среды трансформанта Escherichia coli BL21 (DE3)/pEFLA10, содержащем ген настоящего изобретения.
Альтернативно, в экстракте Escherichia coli BL21 (DE3)/pET21d не было обнаружено расщепляющей активности по отношению к фукоидану.
Пример 6
Создавался прямой экспрессирующий вектор гена fdlB, который является геном, полученным в примере 4, кодирующим новый белок, предположительно имеющий расщепляющую активность по отношению к полисахариду, содержащему сульфатированную фукозу. Опознаваемая рестриктазой BspHI последовательность находилась в данном fdlB гене в положении инициирующего кодона, но поскольку последовательность была подвержена метилированию благодаря влиянию вышерасположенной последовательности плазмида, полученная из используемого в качестве хозяина штамма Escherichia coli JM109, не способна расщепляться рестриктазой BspHI напрямую. Поэтому создание вектора проводилось с применением амплифицированных ДНК фрагментов посредством ПЦР.
В плазмиду рН17Х6-7, полученную в примере 4, вставлялись XbaI фрагменты длиной приблизительно 6 т.п.н. из плазмиды pSFLA17 в том же направлении, что и lac-промотор вектора и fdlB ген. Сначала рН17Х6-7 расщеплялась по КрnI сайту, происходящему из вектора и внутренней области fdlB гена, и сшивалась для создания плазмиды рН17Х6-7К. Используя плазмиду рН17Х6-7К в качестве матрицы, проводилась ПЦР с применением пары синтетических ДНК праймеров, один из которых являлся синтетическим ДНК праймером 17X6F4 (SEQ ID NO:26) и содержал последовательность и направление подобные последовательности, локализованный в вышерасположенной области от кодирующего региона fdlB, и М13 праймер М4 (производство Takara Shuzo), способный к отжигу на векторе. При проведении ПЦР цикл, состоящий из денатурации при 94°С в течение 0,5 минуты, отжига праймера при 55°С в течение 0,5 минуты и реакции синтеза при 72°С в течение 1 минуты, проводился 25 раз в том же составе реакционной смеси, что и в примере 4-(4), и наконец, смесь выдерживалась при 72°С в течение семи минут для завершения реакции. Реакционная смесь подвергалась экстракции фенолхлороформом, и затем амплифицированные ДНК фрагменты длиной около 1 т.п.н. получались с помощью осаждения этанолом. Фрагмент разрезался по BspHI сайту, расположенному в области инициирующего кодона fdlB гена и также по MflI сайту в пределах этой же кодирующей области, разделялся с помощью 3% агарозного гель-электрофореза, выделялся и очищался, в результате получая BspHI-MflI фрагменты длиной приблизительно 450 пар оснований, кодирующих N-концевую область fdlB гена.
С другой стороны, как и в примере 5, плазмида pET21d, являющаяся экспрессирующим вектором, использующим Т7 промотор, разрезалась по NcoI сайту и BamHI сайту, локализованному в поликлональном сайте, и вставлялись ранее полученные BspHI-MflI фрагменты длиной приблизительно 450 пар оснований, кодирующие N-концевую область fdlB гена, для создания плазмиды pEFLA17-N.
Затем плазмида рН17Х6-7 разрезалась по NspV сайту, расположенному в fdlB гене, и по EcoRI сайту, расположенному ниже fd1 гена, и высвобождались NspV-EcoRI фрагменты длиной около 2 т.п.н., кодирующие С-концевую область, включая терминирующий кодон fdlB гена, выделялись и очищались. Фрагмент вставлялся между NspV и EcoRI сайтами ранее созданной плазмиды pEFLA17-N. Созданная таким образом экспрессирующая плазмида, в которой полная область fdlB гена вставлена от инициирующего кодона в NcoI сайте pET21d, была названа pEFLA17.
Используя полученную выше плазмиду pEFLA17, экспрессия полипептида, кодируемого fdlB геном, и расщепляющая активность полипептида по отношению к фукоидану были установлены тем же способом, что и в примере 3.
Таким образом, штамм Escherichia coli BL21 (DE3) трансформировался с помощью плазмиды pEFLA17. Полученный трансформант Escherichia coli BL21 (DE3)/pEFLA17 вносился в 5 мл L среды, содержащей 100 мкг/мл ампициллина, подвергался посеву внутри среды при 37°С. Когда мутность культуры, выражающаяся в O.D. 600, достигала 0,8, к культуре добавлялся IPTG до конечной концентрации 1 мМ, и далее культура подвергалась засеву внутри среды при 15°С в течение еще одной ночи. После завершения инкубации среда центрифугировалась для получения клеток и клетки суспендировались в 0,5 мл буфера для разделения клеток и разрушались с помощью обработки ультразвуковыми волнами. Часть суспензии отбиралась и использовалась в качестве лизата Escherichia coli. Суспензия центрифугировалась с удалением нерастворимых веществ для получения экстракта Escherichia coli.
Таким образом, сначала лизат Escherichia coli анализировался с помощью SDS полиакриламидного гель электрофореза, вследствие чего полоса с молекулярной массой около 77000 (не обнаруженная в лизате Escherichia coli BL21 (DЕ3)/pET21d) была обнаружена в лизате Escherichia coli BL21 (DЕ3)/pEFLA17. Данная молекулярная масса хорошо совпала с молекулярной массой полипептида (76929), рассчитанной из аминокислотной последовательности, которая способна кодироваться геном fdlB и которая показана в SEQ ID NO: 4. Таким образом, было установлено, что трансформант Escherichia coli BL21 (DЕ3)/pEFLA17 экспрессирует полипептид, кодируемый геном fdlB.
Затем была определена расщепляющая активность экстракта Escherichia coli по отношению к фукоидану способом, описанным в ссылочном примере 3.
В результате расщепляющая активность по отношению к фукоидану, определенная в экстракте Escherichia coli BL21 (DЕ3)/pEFLAl7 равнялась 480 мЕ/мл. Таким образом, было установлено, что полипептид, кодируемый геном fdlB, имеет расщепляющую активность по отношению к полисахариду, содержащему сульфатированную фукозу, и что полипептид, кодируемый геном fdlB и обладающий приблизительно 48 мЕ расщепляющей активности по отношению к полисахариду, содержащему сульфатированную фукозу, продуцируется в одном мл среды трансформанта Escherichia coli BL21 (DЕ3)/pEFLAl7, содержащем ген настоящего изобретения.
Наоборот, в экстракте Escherichia coli BL21 (DE3)/pET21d не было обнаружено расщепляющей активности по отношению к фукоидану.
Пример 7
Исследовалось действие полученных в примере 5 и в примере 6 полипептидов, имеющих расщепляющую активность по отношению к полисахариду, содержащему сульфатированную фукозу, на полисахарид-U, содержащий сульфатированную фукозу.
Таким образом, 10 мкл раствора полипептида, обладающего расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, полученного в примере 5 либо в примере 6, добавлялось к смеси, состоящей из 50 мкл 100 мМ фосфатного буфера (рН 7,5), 50 мкл 2,5% полисахарида-U, содержащего сульфатированную фукоэу и 10 мкл 4 М хлорида натрия, и смесь выдерживалась при 25°С. Отбирались порции из реакционной смеси на стадиях после 16, 40 и 65 часов от начала реакции и продукты реакции анализировались с помощью ЖХВД. Условия проведения ЖХВД были следующие:
Относительно продуктов реакции вещества, имеющие следующие структуры, к настоящему моменту являются известными, и поэтому они использовались в качестве стандартов. В данном случае вещества, имеющие следующие формулы от [I] до [IV], получались следующим способом и использовались.
Высушенные Kjellmaniella crassifolia измельчались с помощью дезинтегратора типа М-2 (производство Nara Kikai Seisakusho), обрабатывались 10-кратным объемом 85% метанола в течение 2 часов при температуре 70°С и затем фильтровались. К полученному остатку добавлялась вода в 20-кратном объеме, и смесь обрабатывалась в течение трех часов при температуре 100°С и фильтровалась до получения экстракта. Концентрация соли в экстракте доводилась до уровня 400 мМ раствора натрия хлорида, затем добавлялся 5% цетилпиридинхлорид до полного насыщения и смесь центрифугировалась. После центрифугирования преципитат хорошо отмывался этанолом для полной элиминации цетилпиридинхлорида, обессоливался и удалялись низкомолекулярные вещества с помощью ультрафильтрации (фильтр с порами, исключающими молекулярную массу 100000) (производство Amicon) и полученный вследствие этого преципитат удалялся центрифугированием. Супернатант высушивался вымораживанием для получения очищенного фукоидана из Kjellmaniella crassifolia. Выход по массе к высушенным Kjellmaniella crassifolia составил около 4%.
После этого, смешивались 600 мл 5% раствора указанного очищенного фукоидана из Kjellmaniella crassifolia, 750 мл 100 мМ фосфатного буфера (рН 8,0), 150 мл 4 М хлорида натрия и 3,43 мл раствора фукоиданазы (1750 мЕ/мл), упоминаемой в ссылочном примере 5, и проводилась реакция при 25°С в течение 144 часов. Реакционная смесь диализовалась с помощью проницаемого фильтра, имеющего размер пор 3500, для сбора фракций с молекулярной массой 3500 или менее. Фракции обессоливались с помощью Micro Acilyzer G3 (производства Asahi Kasei) и подвергались ионообменной хроматографии на колонке (4 см × 25 см) с DEAE-Sepharose FF, уравновешенной с помощью 10 мМ ацетата аммония. Элюция проводилась с помощью градиента концентрации ацетата аммония и элюенты собирались каждые 50 мл. Фигура 5 представляет собой профиль элюции указанной хроматографии. В результате указанной хроматографии были получены девять фракций (a)-(i), среди которых фракции (а), (b), (с) и (f) подвергались структурному анализу.
Затем проводился структурный анализ с помощью стандартной методики для фракций (а), (b), (с) и (f), и после установления, что фракции (а), (b), (с) и (f) являются соединениями, представленными следующими формулами [I], [II], [III] и [IV] соответственно, каждая фракция применялась в качестве стандарта для анализа вышеописанных продуктов реакции.
В данном случае, номера фракций для фракций вышепроведенной ионообменной хроматографии с применением DEAE-Sepharose FF колонки, следующие: 42-43 для (а), 84-91 для (b), 51-52 для (с) и 62-63 для (f).
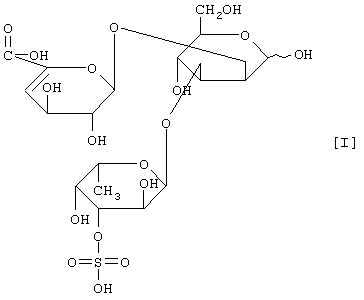
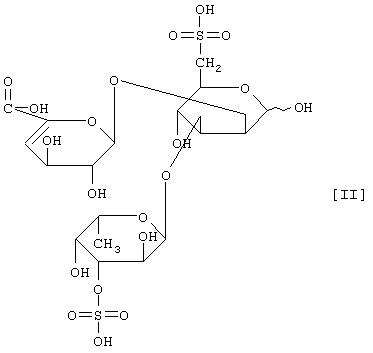
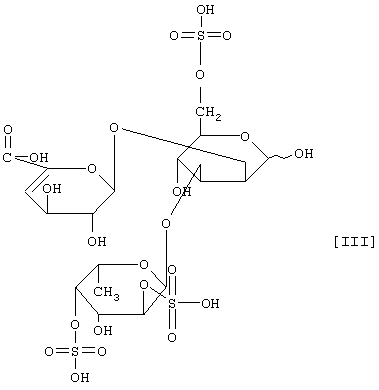
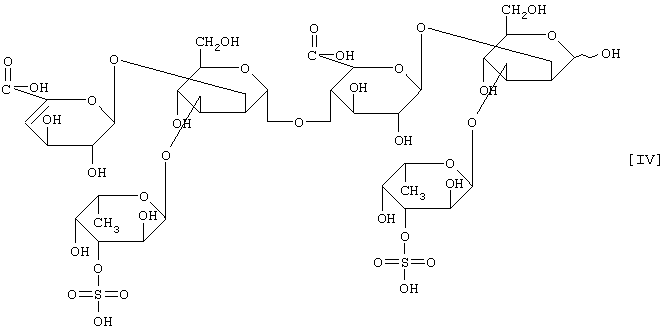
В результате анализа продуктов реакции при применении полученного в примере 5 полипептида, обладающего расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, получались только продукты [I], [II] и [III], а при применении полученного в примере 6 полипептида, обладающего расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, продукты реакции содержали большое количество продукта [IV], в дополнение вышеупомянутым продуктам [I], [II] и [III], и даже в случае дальнейшего проведения реакции продукт [IV] не разлагается до продукта [I].
Таким образом, было найдено, что полипептид, кодируемый геном fdlA полученный в примере 5, взаимодействует с полисахаридом-U, содержащим сульфатированную фукозу, посредством чего высвобождаются в качестве конечных единиц расщепления вышеуказанные три сахарида [I], [II] и [III], a в случае полипептида, кодируемого геном fdlB полученном в примере 6, установлено взаимодействие с полисахаридом-U, содержащим сульфатированную фукозу, с высвобождением в качестве конечной единицы расщепления гексозы, такой как вышеуказанный продукт [IV].
Пример 8
В примере 5 и примере 6 было установлено, что каждым рекомбинантом Escherichia coli, содержащим fdlA и fdlB ген, продуцируется полипептид, кодируемый каждым из указанных генов соответственно, что расщепляющая активность по отношению к полисахариду, содержащему сульфатированную фукозу, определялось в экстракте указанной Escherichia coli, и что полипептид, кодируемый геном fdlA и fdlB, обладает расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу. Однако при анализе каждого из экстрактов Escherichia coli с помощью SDS полиакриламидного гель-электрофореза установлено, что продуцируется только небольшое количество рекомбинантного полипептида по сравнению с лизатом Escherichia coli, и предположено, что только часть продуцируемого полипептида экспрессируется в виде растворимого полипептида в активной форме в каждом случае.
Фукоиданаза из штамма Flavobacterium sp. SA-0082 является ферментом, секретируемым во внеклеточное пространство, и предположительно экспрессируется в виде предшественника, в котором сигналы секреции для проникновения сквозь мембрану находятся на N-конце. Исходя из вышесказанного исследовалась N-концевая аминокислотная последовательность, кодируемая генами fdlA и fdlB, и было предположено, что области от инициирующего метионина до 25-го и 24-го остатка аланина являются сигналами секреции. Поскольку считалось, что наличие подобных гидрофобных сигналов секреции имеет отношение к растворимости каждого из рекомбинантных полипептидов в примерах 5 и 6, были созданы соответственно экспрессирующие плазмиды, в которых сигналы секреции в генах fdlA и fdlB были удалены.
(1) Сначала для того чтобы ввести сайт рестрикции BamHI в вышерасположенный 26-й остаток глутамина от инициирующего метионина гена fdlA, были спроектированы и синтезированы праймер FDL-Q-Bam (SEQ ID NO: 27) и праймер 10X6R4 (SEQ ID NO: 27). FDL-Q-Bam является синтетической ДНК 28 мер, в которой BamHI сайт ориентирован навстречу последовательности из последовательности нуклеотидов с номерами 76-95 в SEQ ID NO: 7 в списке последовательностей, а праймер 10X6R4 является синтетической ДНК 21 мер, которая комплементарна последовательности из последовательности нуклеотидов с номерами 1471-1491 в SEQ ID NO: 7 в списке последовательностей.
Проводилась ПЦР с комбинацией праймеров FDL-Q-Bam и 10X6R4, применяя в качестве матрицы плазмиду pEFLA10, полученную в примере 5. В ПЦР цикл, состоящий из денатурации при 94°С в течение 0,5 минуты, отжига праймера при 50°С в течение одной минуты и реакции синтеза при 72°С в течение двух минут, повторялся 25 раз в том же составе реакционной смеси, что и в примере 4-(4), и наконец, смесь выдерживалась при 72°С в течение семи минут для завершения реакции. Реакционная смесь для разделения подвергалась агарозному гель электрофорезу, после этого выделялись и очищались амплифицированные ДНК фрагменты длиной приблизительно 1,4 т.п.н. Затем плазмидная ДНК, в которой указанный фрагмент был вставлен в рТ7 Blue T-Vector (производство Novagene), получалась способом, описанным в примере 4-(4), и устанавливалась нуклеотидная последовательность. Полученная плазмида расщеплялась по BamHI сайту, расположенному в праймере FDL-Q-Bam и по SnaBI сайту в кодирующей области fdlA и разделялась с помощью 5% полиакриламидного гель-электрофореза, полученные BamHI-SnaBI фрагменты длиной около 480 пар оснований, кодирующие N-концевую область 26-го остатка глутамина от инициирующего метионина fdlA, и затем вырезались, выделялись и очищались.
С другой стороны, рЕТ21а (производство Novagene), которая является экспрессирующим вектором, использующим Т7 промотор из примера 5, расщеплялась по HindIII сайту в поликлональном сайте, и затем HindIII ДНК фрагмент длиной около 2 т.п.н., который кодирует С-концевую область, включая терминирующий кодон fdlA гена и полученный в примере 5, вставлялся в том же направлении, что и Т7 промотор и fdlA ген, вследствие чего получалась плазмида pEFDLA-C. Данная pEFDLA-C расщеплялась по BamHI сайту, происходящему из поликлонального сайта, и по SnaBI сайту в кодирующей области fdlA, и затем уже полученные BamHI-SnaBI фрагменты длиной около 480 пар оснований, кодирующие N-концевую область 26-го остатка глутамина от инициирующего метионина fdlA, вставлялись, вследствие чего получалась плазмида pEFDLA101.
Данная плазмида pEFDLA101 кодирует полипептид, в котором ниже Т7 промотора последовательность 26-го остатка глутамина (SEQ ID NO: 3) в списке последовательностей соединялась после N-концевой последовательности из 14 остатков
(SEQ ID NO: 29), полученной из рЕТ21а.
(2) последовательность из 20 оснований после 26-го глутаминового остатка гена fdlA, использующая для конструирования праймер FDL-Q-Bam, идентична последовательности (последовательность нуклеотидов с номерами 73-92 в SEQ ID NO: 8 списка последовательности) после 25-го остатка глутамина гена fdlB. Соответственно применяемый в примере 8-(1) праймер FDL-Q-Bam пригоден к использованию непосредственно для создания fdlB экспрессирующего вектора, и с помощью способа описанного в примере 8-(1) был создан fdlB экспрессирующий вектор pEFDLB101.
Сначала, проводя ПЦР с применением праймера FDL-Q-Bam, получалась синтетическая ДНК 17X6R1 (SEQ ID NO: 30) 20 мер комплементарная последовательность из последовательности нуклеотидов с номерами 1489-1508 из SEQ ID NO: 8 списка последовательностей. Используя в качестве матрицы pEFLB17 полученную в примере 6, проводилось ПЦР с комбинацией праймеров FDL-Q-Bam и 17X6R1, и амплифицированный ДНК фрагмент длиной около 1,4 т.п.н. выделялся и очищался. Фрагмент вставлялся в рТ7 Blue T-Vector для получения плазмидной ДНК, и устанавливалась ее нуклеотидная последовательность.
После этого полученная плазмида расщеплялась по BamHI сайту, находящемуся в праймере FDL-Q-Bam и по NspV сайту в кодирующей области fdlB, затем разделялась с помощью 5% полиакриламидного гель электрофореза, вследствие чего BamHI-NspV фрагменты длиной приблизительно 210 пар оснований, кодирующие N-концевую область 25-го остатка глутамина от инициирующего метионина fdlB и далее, вырезались, выделялись и очищались.
С другой стороны, полученная в примере 4 плазмида рН17Х6-7 расщеплялась по EcoRI сайту и высвобождающийся EcoRI фрагмент длиной приблизительно 2,6 т.п.н., включая полную длину гена fdlB, вырезался, выделялся и очищался. Фрагмент вставлялся по EcoRI сайту в поликлональном сайте плазмиды рЕТ21а, таким образом вводя Т7 промотор и fdlB ген в том же направлении, вследствие чего создавалась плазмида pEFDLB-W. Данная плазмида pEFDLB-W расщеплялась по BamHI сайту, происходящему из поликлонального сайта, и по NspV сайту в кодирующей области гена fdlB, затем ранее полученный BamHI-NspV фрагмент длиной приблизительно 210 пар оснований, кодирующий N-концевую область 25-го остатка глутамина от инициирующего метионина гена fdlB и далее, вставлялся для получения плазмиды pEFDLB101.
Данная плазмида pEFDLB101 кодирует полипептид, в котором ниже Т7 промотора последовательность от 25-го остатка глутамина в SEQ ID NO: 4 списка последовательностей и далее присоединялась после N-концевой лидирующей последовательности (SEQ ID NO: 29) из 14 остатков, которая идентична плазмиде pEFDLA101, полученной в примере 8-(1).
(3) Проводились инкубация рекомбинанта и получение клеточного экстракта с помощью способа, описанного в примере 5, используя плазмиды pEFDLA101 и pEFDLB101, полученные в примерах 8-(1) и (2), после чего проводилось сравнение с количеством экспрессии и расщепляющей активностью по отношению к фукоидану для каждого рекомбинантного полипептида плазмид pEFLA10 и pEFLA17, полученных в примерах 5 и 6.
Таким образом штамм Escherichia coli BL21 (DE3) трансформировался плазмидами pEFLA10, pEFLA17, pEFDLA101 и pEFDLB101. Каждый из полученных трансформантов высевался на 5 мл L среды, содержащей 100 мкг/мл ампициллина, подвергался посеву внутри среды при 37°С. Когда мутность культуры, выражающаяся в O.D. 600, достигала 0,8, к культуре добавлялся IPTG до конечной концентрации 1 мМ, и далее культура инкубировалась при 15°С в течение еще одной ночи. После завершения инкубации среда центрифугировалась для получения клеток, и клетки суспендировались в 0,5 мл буфера для разделения клеток и разрушались с помощью обработки ультразвуковыми волнами. Часть суспензии отбиралась и использовалась в качестве лизата Escherichia coli. Суспензия далее центрифугировалась с удалением нерастворимых веществ для получения экстракта Escherichia coli.
Каждый из лизатов и экстрактов Escherichia coli анализировался с помощью SDS полиакриламидного гель электрофореза с последующим окрашиванием Coomassie Brilliant Blue no стандартной методике, вследствие чего обнаруживалась экспрессия большого количества полипептида, обладающего ожидаемой молекулярной массой, во всех лизатах Escherichia coli. В то же время, количество экспрессии оценивалось посредством сравнения с количеством сывороточного бычьего альбумина с помощью электрофореза, и определялась расщепляющая активность по отношению к фукоидану в каждом экстракте с помощью методики, описанной в ссылочном примере 3. Данные результаты представлены в таблице 2.
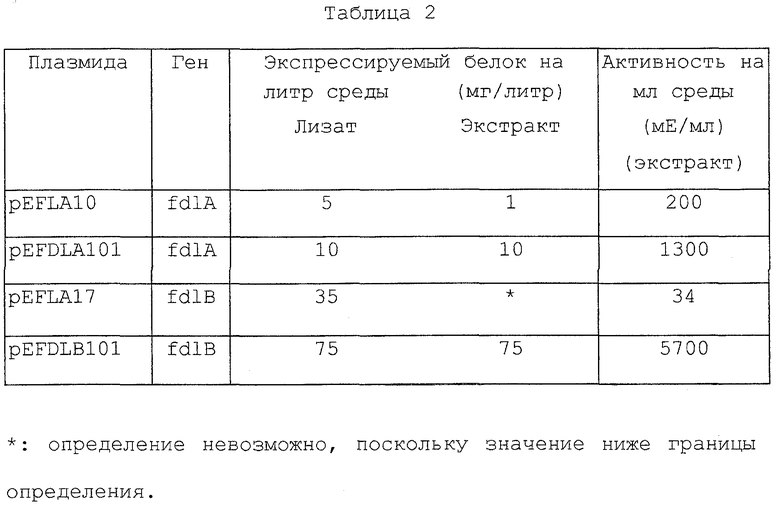
Как показано в таблице 2, относительно количества экспрессии желаемого полипептида, содержащегося в лизатах pEFDLA101 (fdlA) и pEFDLB101 (fdlB), где конструирование проводилось способом добавления N-концевой лидерной последовательности вместо последовательности, предположительно являющейся сигналом секреции, выявилось двукратное увеличение продукции для каждого из генов по сравнению с непосредственно экспрессирующими плазмидами pEFLA10 (fdlA) и pEFLA17 (fd1), содержащими предполагаемые сигналы секреции.
Также было выяснено, что в случае наличия сигналов секреции количество желаемого полипептида, находящегося в экстракте, очень мало по сравнению с лизатом и большое количество экспрессируемого полипептида находится в нерастворимом состоянии, а в случае отсутствия сигналов секреции желаемый полипептид содержится в экстракте приблизительно в таком же количестве, как и в лизате, и большее количество экспрессируемого полипептида находится в растворимом состоянии. Далее, активность в экстракте сильно возрастает соответственно количеству желаемого полипептида.
Пример 9
Исследовалось взаимодействие полипептида, обладающего расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, полученного в примере 2 и примере 3 на полисахарид-F, содержащий сульфатированную фукозу.
Таким образом, раствор полипептида (100 мкл), полученного в примере 2 или в примере 3, обладающего 9,7 мЕ/мл расщепляющей активности по отношению к полисахариду, содержащему сульфатированную фукозу, добавлялось к смеси, состоящей из 500 мкл 50 мМ имидазольного НСl буфера (рН 7,5), 50 мкл 2,5% раствора полисахарида-F, содержащего сульфатированную фукозу, описанного в ссылочном примере 1-(2), 50 мкл 1 М хлорида кальция, 75 мкл 4 М хлорида натрия и дистиллированной воды до общего объема 1 мл. После реакции при 25°С в течение 18 часов продукты реакции анализировались с помощью ЖХВД. Условия проведения ЖХВД были следующие:
Результатом анализа продукта реакции, полученного воздействием полипептида, кодируемого геном настоящего изобретения, являлось наличие во всех продуктах реакции, образующихся с помощью полипептида, обладающего расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, полученного в примере 2 и примере 3, двух пиков на 8,62 и 9,30 минутах, выраженных временем прохождения колонки при ЖХВД.
В данном случае, когда полученный в примере 2 полипептид, обладающий расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, сравнивался с полученным в примере 3 полипептидом, обладающим расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, выход веществ на 9,30 минуте в случае полипептида, полученного в примере 3, гораздо больше, чем в случае полипептида, полученного в примере 2.
Преимущества изобретения
Согласно настоящему изобретению были впервые установлены аминокислотная последовательность и нуклеотидная последовательность полипептида, обладающего расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, и в то же время предложен индустриально выгодный способ для производства полипептида, обладающего расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, методами генной инженерии.
Согласно настоящему изобретению нет необходимости добавлять полисахарид, содержащий сульфатированную фукозу, в среду для индукции продукции фермента, обладающего расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, поскольку его продуктивность высока. Кроме того, не существует такой протеазы и других ферментов, расщепляющих полисахариды, которые одновременно легко продуцировались и очищались. Поскольку предложены аминокислотная последовательность и нуклеотидная последовательность полипептида, обладающего расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, стало возможно получить антитела к полипептиду, обладающему расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, основываясь на аминокислотной последовательности, и также получить зонд и праймер, которые специфичны для нуклеотидной последовательности полипептида, обладающего расщепляющей активностью по отношению к полисахариду, содержащему сульфатированную фукозу, основываясь на нуклеотидной последовательности.
Список последовательностей





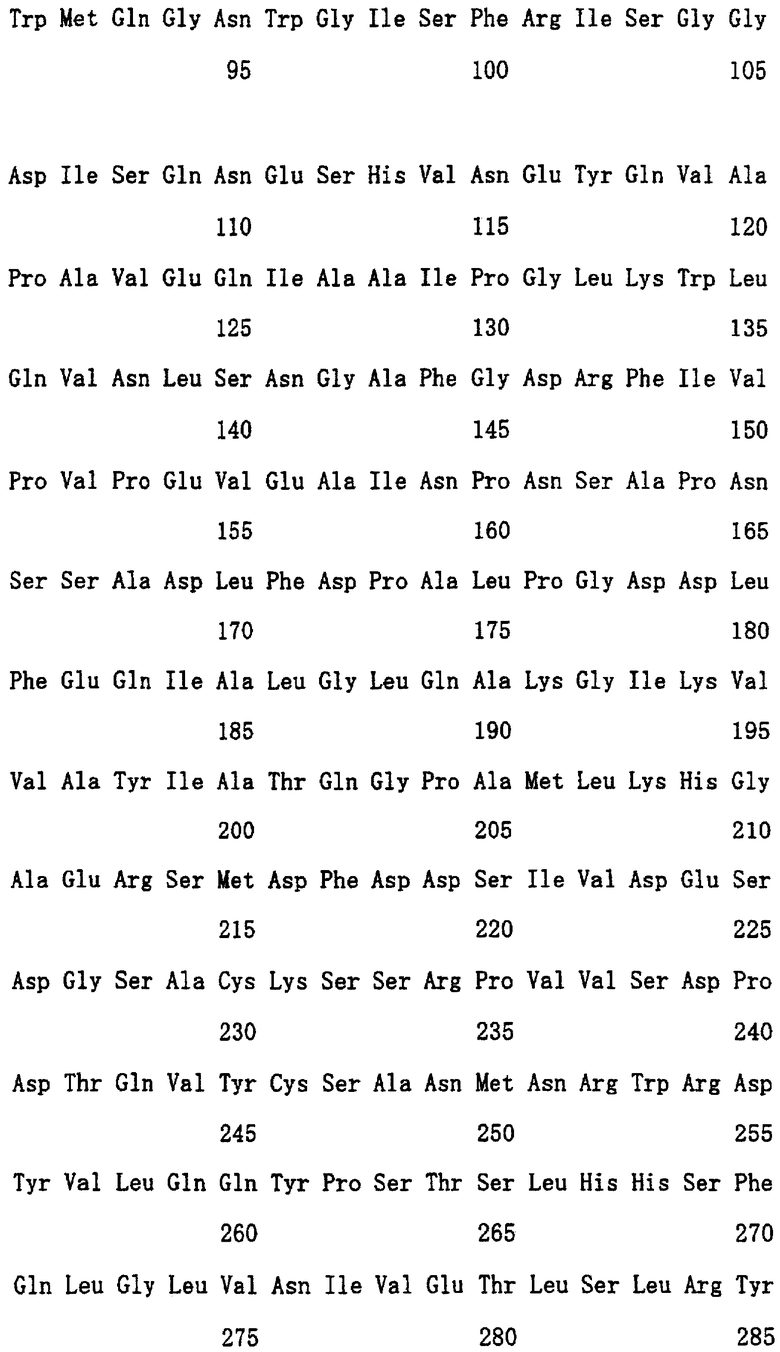


























| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ДИПЕПТИДА (ВАРИАНТЫ) | 2005 |
|
RU2316596C2 |
| ФЕРМЕНТАТИВНОЕ ПОЛУЧЕНИЕ 1-БУТАНОЛА | 2006 |
|
RU2429295C2 |
| СПОСОБ ПРОДУКЦИИ БЕЛКА | 2006 |
|
RU2435863C2 |
| ПОЛИНУКЛЕОТИД, КОДИРУЮЩИЙ ФОСФОЕНОЛПИРУВАТКАРБОКСИКИНАЗУ, И ЗОНД, ПРЕДНАЗНАЧЕННЫЙ ДЛЯ ЕГО ПОЛУЧЕНИЯ | 2000 |
|
RU2262532C2 |
| СПОСОБ ПРОДУКЦИИ СУЛЬФАТИРОВАННОГО ПОЛИСАХАРИДА И СПОСОБ ПРОДУКЦИИ PAPS | 2021 |
|
RU2811941C1 |
| КЛЕТКА НИТЧАТЫХ ГРИБОВ С ДЕФИЦИТОМ ПРОТЕАЗ И СПОСОБЫ ЕЕ ПРИМЕНЕНИЯ | 2013 |
|
RU2645252C2 |
| СПОСОБ ПОЛУЧЕНИЯ L-ОРНИТИНА С ИСПОЛЬЗОВАНИЕМ БАКТЕРИЙ, СВЕРХЭКСПРЕССИРУЮЩИХ LYSE | 2011 |
|
RU2571932C2 |
| МУТАНТЫ СИНТЕТАЗЫ АЦЕТООКСИКИСЛОТ, УСТОЙЧИВЫЕ К ОБРАТНОЙ СВЯЗИ | 2004 |
|
RU2325439C2 |
| ФЕРМЕНТАТИВНОЕ ПОЛУЧЕНИЕ ЧЕТЫРЕХУГЛЕРОДНЫХ СПИРТОВ | 2006 |
|
RU2394913C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2,4-ДИГИДРОКСИБУТИРАТА | 2013 |
|
RU2645260C2 |
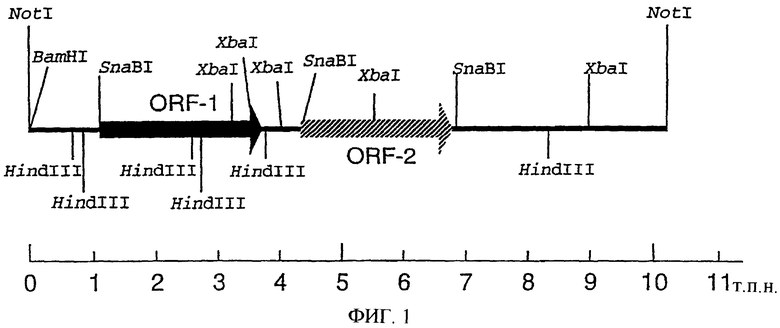
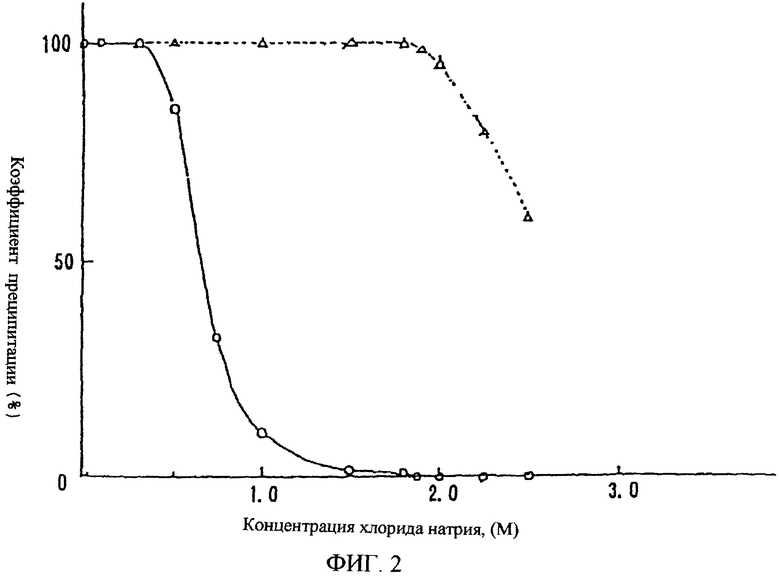
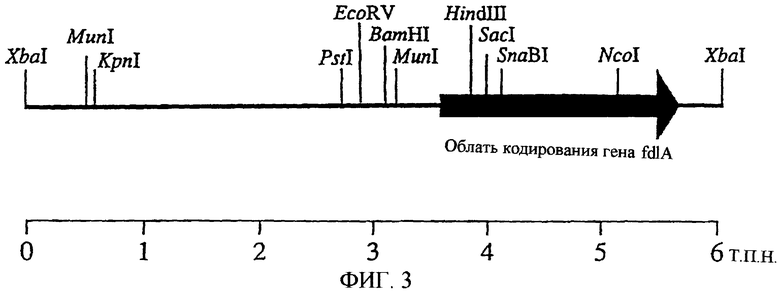
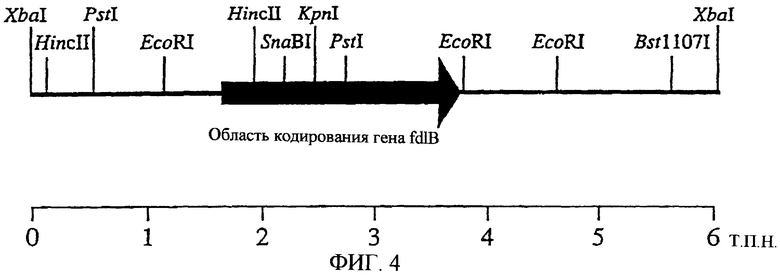
Изобретение относится к области генной инженерии и биотехнологии и может быть использовано в медико-биологической и фармацевтической промышленности. Из клеток штамма р. Alteromonas, продуцирующего фермент, который расщепляет полисахарид, включающий сульфатированную фукозу, выделен ген, кодирующий указанный фермент. Установлено наличие двух открытых рамок считывания (ORF-1 и ORF-2) и определены их последовательности. Путем экспрессии этих последовательностей в E.coli получены активные рекомбинантные формы фермента, способного эффективно гидролизовать полисахарид, содержащий сульфатированную фукозу, который не расщепляется под действием фукоиданазы, продуцируемой Flavobacterium sp. SA-0082 (FERM ВР-5402). Применение изобретения обеспечивает возможность получения больших количеств качественного сырья для фармацевтических препаратов. 5 с. и 1 з.п.ф-лы, 5 ил., 2 табл.
а) нуклеотидной последовательности, которая в соответствии с генетическим кодом кодирует аминокислотную последовательность, представленную SEQ ID NO: 1 или SEQ ID NO: 2;
б) нуклеотидной последовательности, способной гибридизоваться в условиях применения раствора, содержащего 6х SSC, 1% ДСН, 5х раствор Денхардта и 100 мкг/мл ДНК молок лосося, при 65°С с последовательностью, комплементарной последовательности ДНК, представленной SEQ ID NO: 5 или SEQ ID NO: 6.
(а) составляющий сахарид, по существу, не содержит уроновую кислоту; (б) по существу, не способен расщепляться под действием фукоиданазы, продуцируемой Flavobacterium sp. SA-0082 (FERM ВР-5402).
Приоритет по пунктам:
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| KEN SAKAI et al | |||
| Biosci | |||
| Biotech | |||
| Biochem | |||
| Способ получения молочной кислоты | 1922 |
|
SU60A1 |

