1. Введение
Настоящее изобретение относится к последовательностям пептида-усилителя, исходно происходящим от различных последовательностей белка оболочки ретровируса (gp41) и улучшающим фармакокинетические свойства любого корового полипептида, с которым они связаны. Настоящее изобретение частично основано на обнаружении того факта, что гибридные полипептиды, содержащие последовательности пептида-усилителя, присоединенные к коровому полипептиду, обладают улучшенными фармакокинетическими свойствами, такими как более продолжительное время полужизни. Настоящее изобретение также относится к новым антифузогенным и/или антивирусным пептидам, включая пептиды, содержащие вышеуказанные последовательности пептида-усилителя, и к способам использования таких пептидов. Настоящее изобретение, кроме того, относится к способам улучшения фармакокинетических свойств любого корового полипептида посредством присоединения к этому коровому полипептиду указанных последовательностей пептида-усилителя. Коровые полипептиды, используемые в настоящем изобретении, могут включать любые фармакологически приемлемые пептиды, которые могут быть использованы, например, в качестве терапевтического или профилактического реагента. В неограничивающем примере, иллюстрирующем вариант осуществления настоящего изобретения, было продемонстрировано, что гибридный полипептид, содержащий, например, коровый полипептид ВИЧ, присоединенный к последовательностям пептида-усилителя, является сильным нетоксичным ингибитором ВИЧ-1-, ВИЧ-2- или SIV-инфекции. Кроме того, последовательности пептида-усилителя настоящего изобретения были присоединены к коровому полипептиду респираторно-синцитиального вируса (RSV) и коровому полипептиду рецептора лютеинизирующего гормона (LH-RH). В каждом случае было обнаружено, что этот гибридный полипептид обладает улучшенными фармакокинетическими свойствами, а гибридный полипептид на основе RSV обладает значительной активностью против RSV.
2. Предпосылки создания изобретения
Полипептидные продукты широко используются в качестве терапевтических и/или профилактических агентов для предупреждения и лечения заболеваний. Многие из этих полипептидов способны регулировать биохимические или физиологические процессы, что приводит либо к предупреждению заболевания, либо к ослаблению симптомов, ассоциированных с этими заболеванием. Так, например, полипептиды, такие как вирусные или бактериальные полипептиды, могут быть с успехом использованы в качестве вакцин для предупреждения патологических состояний. Кроме того, пептиды могут быть с успехом использованы в качестве терапевтических агентов для лечения симптомов заболевания. Такие пептиды входят в состав белков различных категорий, таких как, например, гормоны, ферменты, иммуномодуляторы, белки сыворотки и цитокины.
Для того чтобы эти полипептиды оказывали присущее им биологическое и терапевтическое действие на сайты-мишени, они должны присутствовать в этих сайтах в соответствующих концентрациях. Кроме того, должна, в основном, сохраняться их структурная целостность. Следовательно, композиция полипептидов для их терапевтического использования в качестве лекарственных средств зависит от химической природы и характеристик этих полипептидов, таких как размер и структура, от конформационных требований, а часто от трудно предсказуемой стабильности и параметров растворимости. Фармакокинетические свойства любого конкретного терапевтического пептида зависят от биологической доступности, распределения и выведения указанного пептида.
Поскольку многие биологически активные вещества, такие как пептиды и белки, быстро разрушаются в организме, то для увеличения эффективности таких пептидов и для минимизации случаев возникновения опасных побочных эффектов и их тяжести в настоящее время крайне необходимо разработать эффективные системы для поддержания устойчивой концентрации пептида в кровотоке.
3. Краткое описание изобретения
Настоящее изобретение, главным образом, относится к последовательностям пептида-усилителя, исходно происходящим от различных последовательностей белка оболочки ретровируса (gp41), т.е. ВИЧ-1, ВИЧ-2 и SIV, и улучшающим фармакокинетические свойства любого корового полипептида, к которому они присоединены. Настоящее изобретение основано на обнаружении того факта, что в случае, когда описанные последовательности пептида-усилителя присоединены к любому коровому полипептиду, то полученный в результате этого гибридный полипептид обладает улучшенными фармакокинетическими свойствами, включая, например, более продолжительное время полужизни и более низкую скорость его выведения из кровотока, по сравнению с коровым полипептидом, взятым отдельно. Настоящее изобретение, кроме того, относится к указанным гибридным полипептидам и к коровым полипептидам, а также к новым пептидам, которые обладают антифузогенной активностью, антивирусной активностью и/или способностью модулировать внутриклеточные процессы, ассоциированные с биспиральными пептидными структурами. Такими пептидами являются пептиды, которые содержат последовательности пептида-усилителя.
Коровые полипептиды могут включать в себя любые пептиды, которые могут быть введены в живую систему, например любые пептиды, которые способны функционировать как терапевтические, профилактические или визуализирующие агенты, используемые для лечения или предупреждения заболеваний, или которые могут быть использованы в диагностических или прогностических методах, включая методы визуализации in vivo. Такими пептидами являются, например, факторы роста, гормоны, цитокины, ангиогенные факторы роста, внеклеточные полипептиды матрикса, лиганды рецепторов, агонисты, антагонисты или обратные агонисты, пептидные агенты для доставки, такие как визуализирующие агенты или цитотоксические агенты для доставки, либо полипептиды, которые обладают антифузогенной и/или антивирусной активностью, а также пептиды или полипептиды, которые функционируют как антигены или иммуногены, включая, например, вирусные и бактериальные полипептиды.
Настоящее изобретение, кроме того, относится к способам улучшения фармакокинетических свойств любого коревого полипептида посредством присоединения этого корового полипептида к последовательностям пептида-усилителя с образованием гибридных полипептидов.
Настоящее изобретение, кроме того, относится к способам использования описанных здесь пептидов, включая гибридные полипептиды, содержащие последовательности пептида-усилителя. Так, например, способами настоящего изобретения являются способы ослабления или ингибирования вирусной инфекции, например вируса ВИЧ-1, ВИЧ-2, RSV, кори, гриппа, парагриппа, Эпштейна-Барра и гепатита, и/или событий индуцированного вирусом слияния клеток. Эти последовательности пептида-усилителя настоящего изобретения, кроме того, могут быть использованы для увеличения времени полужизни in vitro или ex-vivo корового полипептида, к которому присоединены последовательности пептида-усилителя, например последовательности пептида-усилителя могут увеличивать время полужизни присоединенных коровых полипептидов в образцах клеточных культур, клеток или тканей.
Настоящее изобретение продемонстрировано в примерах, где показано, что гибридные полипептиды, содержащие коровый полипептид ВИЧ, присоединенный к последовательностям пептида-усилителя, обладают в значительной степени улучшенными фармакокинетическими свойствами и действуют как сильные нецитотоксические ингибиторы ВИЧ-1-, ВИЧ-2- и SIV-инфекции. Настоящее изобретение, кроме того, продемонстрировано в примерах, где показано, что гибридные полипептиды, содержащие коровый полипептид RSV или полипептид лютеинизирующего гормона, обладают в значительной степени улучшенными фармакокинетическими свойствами. Кроме того, гибридный полипептид на основе RSV обладает значительной активностью против RSV.
3.1. Определения
Пептиды, полипептиды и белки определены в настоящем описании как органические соединения, включающие две или несколько аминокислот, ковалентно связанных между собой, например, пептидными амидными связями. Пептиды, полипептиды и белки могут также включать неприродные аминокислоты и любые их модификации, а также дополнительные амино- и карбоксильные группы, описанные в настоящей заявке. Следовательно, термины "пептид", "полипептид" и "белок" используются здесь как взаимозаменяемые понятия.
Пептидные последовательности, определенные в настоящем описании, представлены однобуквенными символами, используемыми для обозначения нижеследующих аминокислотных остатков:
А (аланин)
R (аргинин)
N (аспарагин)
D (аспарагиновая кислота)
С (цистеин)
Q (глутамин)
Е (глутаминовая кислота)
G (глицин)
Н (гистидин)
I (изолейцин)
L (лейцин)
К (лизин)
М (метионин)
F (фенилаланин)
Р (пролин)
S (серин)
Т (треонин)
W (триптофан)
Y (тирозин)
V (валин)
Х (любая аминокислота)
"Последовательности пептида-усилителя" определены как пептиды, имеющие нижеследующие "консенсусные" аминокислотные последовательности: "WXXWXXXI", "WXXWXXX", "WXXWXX", "WXXWX", "WXXW", "WXXXWXWX", "XXXWXWX", "XXWXWX", "XWXWX", "WXWX", "WXXXWXW", "WXXXWX", "WXXXW", "IXXXWXXW", "XXXWXXW", "XXWXXW", "XWXXW", "XWXWXXXW", "XWXWXXX", "XWXWXX", "XWXWX", "XWXW", "WXWXXXW" или "XWXXXW", где Х и может быть любой аминокислотой, W означает триптофан, а I означает изолейцин. Как описано ниже, последовательности пептида-усилителя настоящего изобретения также представляют собой пептидные последовательности, которые в той или иной степени аналогичны консенсусным аминокислотным последовательностям, но которые содержат аминокислотные замены, вставки или делеции, не влияющие на способность этого пептида улучшать фармакокинетические свойства корового пептида, с которым он связан, по сравнению с фармакокинетическими свойствами указанного корового полипептида, взятого отдельно.
Используемый здесь термин "коровый полипептид" означает любой полипептид, который может быть введен в живую систему и, таким образом, представляет собой биологически активную молекулу, например любой полипептид, который может функционировать как фармакологически эффективный пептид для лечения или предупреждения заболевания.
Используемый здесь термин "гибридный полипептид" означает любой полипептид, содержащий амино-, карбокси- или амино- и карбоксиконцевую последовательность пептида-усилителя и коровый полипептид. Обычно последовательность пептида-усилителя присоединена непосредственно к коровому полипептиду. Следует отметить, что пептид-усилитель может быть также присоединен к промежуточной аминокислотной последовательности, локализованной между последовательностью пептида-усилителя и коровым пептидом.
Используемые здесь термины "антифузогенный" и "направленный против слияния мембран" относятся к способности пептида ингибировать или снижать число событий слияния между двумя или более структурами, например между клеточными мембранами или вирусными оболочками или пилями, по сравнению с числом событий слияния мембран, происходящих между структурами в отсутствие этого пептида.
Используемый здесь термин "антивирусный" означает способность пептида ингибировать вирусную инфекцию клеток, например, посредством слияния клеток или инфицирования свободной вирусной частицей. Такое инфицирование может включать в себя слияние мембран, как это происходит в случае вирусов с оболочкой, или другое событие слияния, происходящее между вирусной структурой и клеточной структурой, например слияние вирусных пилей и бактериальной мембраны во время конъюгации бактерий.
4. Краткое описание чертежей
Фиг.1. Гибридные полипептиды. Показаны последовательности пептида-усилителя, происходящие от предполагаемых N-концевой и С-концевой интерактивных областей и присоединенные к коровому полипептиду соответствующего вида. Консервативные последовательности пептида-усилителя заштрихованы. Следует отметить, что последовательности пептида-усилителя могут быть использованы либо как N-концевые, С-концевые, либо как N- и С-концевые добавления. Кроме того, последовательности пептида-усилителя могут быть добавлены к коровому полипептиду в прямой или обратной ориентации, отдельно или в любых возможных комбинациях, в целях улучшения фармакокинетических свойств данного пептида.
Фиг.2А. Последовательности пептида-усилителя, происходящие от различных последовательностей белка оболочки (gp41) и представляющие собой N-концевую интерактивную область, наблюдаемую во всех известных последовательностях изолятов ВИЧ-1, ВИЧ-2 и SIV. Конечная последовательность "WXXWXXXI" представляет собой "консенсусную" последовательность.
Фиг.2В. Варианты последовательности пептида-усилителя, происходящие от различных последовательностей белка оболочки (gp41) и представляющие собой С-концевую интерактивную область, наблюдаемую по всех известных последовательностях изолятов ВИЧ-1, ВИЧ-2 и SIV. Конечная последовательность "WXXXWXWX" представляет собой консенсусную последовательность.
Фиг.3. Сравнение титров ВИЧ-1 в тканях мыши SCID-HuPBMC, инфицированной ВИЧ-1 9320, как было измерено по уровням Р24 в анализах при культивировании вместе с НЧРВМС человека. На этой фигуре проиллюстрировано сравнение in vivo-ингибирования вирусной инфекции полипептидами Т20 и Т1249.
Фиг.4А-4В. Фармакокинетический профиль Т1249 в плазме по сравнению с фармакокинетическим профилем контрольного корового полипептида Т1387 у CD-крыс после внутривенной инъекции в течение периода времени вплоть до 2 часов (Фиг.4А) и 8 часов (Фиг.4В). Полипептид Т1387 представляет собой коровый полипептид, а полипептид Т1249 представляет собой коровый полипептид, присоединенный к последовательностям пептида-усилителя.
Фиг.5. Фармакокинетический профиль Т1249 в плазме по сравнению с профилем Т20-контроля у CD-крыс после внутривенного введения. Полипептид Т1249 представляет собой гибридный полипептид, содержащий коровый полипептид (Т1387), присоединенный к последовательностям пептида-усилителя. Т20: n=4; Т1249: n=3.
Фиг.6. Сравнение активности и цитотоксичности Т20/Т1249, направленной против ВИЧ-1/IIIb.
Фиг.7. Прямое связывание Т1249 с gp41-конструкцией М41Δ178. 125I-T1249 был очищен с помощью ВЭЖХ до максимальной специфической активности. Показано связывание до состояния насыщения с М41Δ178 (гибридный белок с эктодоменом gp41, не содержащий аминокислотной последовательности Т20), иммобилизованным на микротитрационных планшетах при 0,5 мг/мл.
Фиг.8. Кривая ассоциации/диссоциации Т1249 в зависимости от времени. Полученные результаты продемонстрировали, что 125I-T1249 и 125I-T20 имеют идентичные аффинности связывания, составляющие 1-2 нМ. Первоначальные скорости ассоциации и диссоциации для 125I-T1249 значительно ниже, чем скорости для 125I-T20. Диссоциацию связанного радиолиганда измеряли после присоединения немеченого пептида до конечной концентрации 10 мкм в 1/10 всего объема анализа.
Фиг.9. Сравнение связывания Т1249 с М41Δ178. Немеченые Т1249 и Т20 титровали в присутствии одной концентрации либо 125I-T1249, либо 125I-T20. Лиганд добавляли сразу после начала инкубирования немеченого пептида.
Фиг.10А-10В. Фармакокинетический профиль RSV-гибридных полипептидов Т1301 (10А) и Т1302 (10В) в плазме по сравнению с Т786 у крыс CD.
Фиг.11А. Анализ на снижение числа бляшек. Гибридный полипептид способен ингибировать RSV-инфекцию с IC50=2,6 мкг/мл.
Фиг.11В. Анализ на снижение числа бляшек продемонстрировал способность RSV-гибридных полипептидов Т1301, Т1302 и Т1303 ингибировать RSV-инфекцию.
Фиг.12А и 12В. Фармакокинетический профиль гибридного полипептида лютеинизирующего гормона Т1324 в плазме по сравнению с Т1323 у самцов CD-крыс. Полипептид Т1323 представляет собой коровый полипептид лютеинизирующего гормона, а полипептид Т1324 представляет собой гибридный полипептид, содержащий коровый полипептид, присоединенный к последовательностям пептида-усилителя.
Фиг.13. Последовательности гибридного полипептида, происходящие от различных коровых полипептидов. Последовательности корового полипептида заштрихованы. Не заштрихованные амино- и карбоксиконцевые последовательности представляют собой последовательности пептида-усилителя.
Фиг.14А-В. Спектр кругового дихроизма (CD) для Т1249 в растворе (забуференном фосфатом солевом растворе, рН 7) отдельно (10 мкм при 1°С, Фиг.14А) и в комбинации с пептидом из 45 остатков от связывающего домена HR1 др41 (Т1346); заштрихованные квадраты (n) представляет теоретический спектр CD, предсказанный для "неинтерактивной модели", а истинные спектры CD представлены заштрихованными кружками (l).
Фиг.15. Электрофорез в полиакриламидном геле продемонстрировал Т1249-защиту gp41-конструкции М41Δ178 от гидролиза протеиназной К; дорожка 1: маркер-затравка; дорожка 2: необработанный М41Δ178; дорожка 3: М41Δ178, инкубированный с протеиназой К; дорожка 4: необработанный Т1249; дорожка 5: Т1249, инкубированный с протеиназой К; дорожка 6: М41Δ178, инкубированный с Т1249; дорожка 7: инкубирование Т1249 и М41Δ178 перед добавлением протеиназы К.
Фиг.16А-С. Фармакокинетика полипептида Т1249 у крыс-альбиносов Sprague-Dawley; Фиг.16А: фaрмакокинетика полипептида Т1249 при введении разовой дозы путем непрерывной подкожной инфузии; Фиг.16В: фармакокинетика полипептида Т1249, введенного путем подкожной инъекции (s.c.) или путем внутривенной инъекции (i.v.); Фиг.16С: кинетический анализ Т1249 в лимфе и плазме после внутривенного введения.
Фиг.17А-В. Фармакокинетика полипептида Т1249 у собакоподобных обезьян; Фиг.17А: фармакокинетика полипептида Т1249 в плазме при введении разовой дозы 0,8 мг/кг Т1249 путем подкожной (s.c.), внутривенной (i.v.) или внутримышечной (i.m.) инъекции; Фиг.17В: фармакокинетика полипептида Т1249 в плазме, введенного путем подкожной инъекции Т1249 в трех различных дозах (0,4 мг/кг, 0,8 мг/кг и 1,6 мг/кг).
5. Подробное описание изобретения
Описанные здесь пептидные последовательности, называемые последовательностями пептида-усилителя, были получены из различных последовательностей белка оболочки (gp41) ретровируса, способных улучшать фармакокинетические свойства коровых полипептидов, к которым они присоединены. Такие последовательности пептида-усилителя могут быть использованы в способах улучшения фармакокинетических свойств любого корового полипептида посредством присоединения последовательностей пептида-усилителя к коровому полипептиду с образованием гибридного полипептида, обладающего улучшенными фармакокинетическими свойствами по сравнению с коровым полипептидом, взятым отдельно. Время полужизни корового пептида, к которому присоединена (или присоединены) последовательность (или последовательности) пептида-усилителя, может быть также увеличено in vitro. Так, например, присоединенные последовательности пептида-усилителя могут увеличивать время полужизни корового полипептида при их присутствии в клеточной культуре, в тканевой культуре или в образцах, взятых от пациента, таких как клетки, ткань или другие образцы.
Коровые полипептиды гибридных полипептидов настоящего изобретения включают любой пептид, который может быть введен в живую систему, например любой пептид, который может функционировать как терапевтический или профилактический агент, используемый для лечения или предупреждения заболевания, или как визуализирующий агент, используемый для визуализации структур in vivo.
В настоящей заявке также описаны пептиды, включая пептиды, содержащие последовательности пептида-усилителя, которые обладают антифузогенной и/или антивирусной активностью. Кроме того, в настоящей заявке описаны способы использования таких пептидов, включая способы ослабления или ингибирования вирусной инфекции и/или вирус-индуцированного слияния клеток.
5.1. Гибридные полипептиды
Гибридные полипептиды настоящего изобретения содержат, по крайней мере, одну последовательность пептида-усилителя и коровый полипептид. Гибридные полипептиды настоящего изобретения предпочтительно содержат, по крайней мере, две последовательности пептида-усилителя и коровый полипептид, при этом, по крайней мере, один пептид-усилитель присутствует в указанном гибридном полипептиде со стороны аминоконца по отношению к коровому полипептиду, и, по крайней мере, одна последовательность пептида-усилителя присутствует в указанном гибридном полипептиде со стороны карбоксиконца по отношению к коровому полипептиду.
Последовательности пептида-усилителя настоящего изобретения содержат пептидные последовательности, исходно происходящие от различных последовательностей белка оболочки (gp41) ретровируса, включая последовательности ВИЧ-1, ВИЧ-2 и SIV, и их специфические варианты или модификации, описанные ниже. Коровый полипептид может содержать любую пептидную последовательность, предпочтительно любую пептидную последовательность, которая может быть введена в живую систему, включая, например, пептиды, предназначенные для использования в терапевтических или профилактических целях либо в целях визуализации.
Обычно гибридный полипептид имеет длину, составляющую от около 10 до около 500 аминокислотных остатков, а предпочтительно от около 10 до около 100 аминокислотных остатков и наиболее предпочтительно от около 10 до около 40 аминокислот.
Не претендуя на какую-либо конкретную теорию, можно отметить, что структура белка оболочки построена, очевидно, таким образом, что предполагаемая область α-спирали, локализованная в С-концевой области белка, ассоциирована с областью "лейциновой молнии", локализованной в N-концевой области этого белка. Проводили сравнительный анализ первичных N-концевых и С-концевых последовательностей пептида-усилителя с областями gp41, наблюдаемыми во всех известных идентифицированных "консенсусных" аминокислотных последовательностях изолятов ВИЧ-1, ВИЧ-2 и SIV.
В частности, были идентифицированы нижеследующие "консенсусные" аминокислотные последовательности, представляющие собой консенсусные последовательности пептида-усилителя (ниже перечислены консенсусные последовательности в прямой и обратной ориентации, поскольку указанные последовательности пептида-усилителя могут быть использованы либо в прямой, либо в обратной ориентации): "WXXWXXXI", "WXXWXXX", "WXXWXX", "WXXWX", "WXXW", "WXXXWXWX", "XXXWXWX", "XXWXWX", "XWXWX", "WXWX", "WXXXWXW", "WXXXWX", "WXXXW", "IXXXWXXW", "XXXWXXW", "XXWXXW", "XWXXW", "XWXWXXXW", "XWXWXXX", "XWXWXX", "XWXWX", "XWXW", "WXWXXXW" или "XWXXXW", где Х может быть любой аминокислотой, W означает триптофан, а I означает изолейцин. Консенсусные аминокислотные последовательности в прямой ориентации показаны на Фиг.1 и 2.
Обычно длина последовательности пептида-усилителя составляет около 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 или 30 аминокислотных остатков, а предпочтительно от около 4 до около 20 остатков, более предпочтительно от около 4 до около 10 остатков, а наиболее предпочтительно от около 6 до около 8 остатков.
В предпочтительном варианте осуществления изобретения последовательности пептида-усилителя, которые могут быть использованы для улучшения фармакокинетических свойств полученных гибридных полипептидов, содержащих специфические последовательности пептида-усилителя, представлены на Фиг.2, 13 и ниже в Таблице 1. Наиболее предпочтительными последовательностями пептида-усилителя являются пептиды, содержащие нижеследующие аминокислотные последовательности: "WQEWEQKI" и WASLWEWF.
В качестве неограничивающих примеров, ниже в Таблице 1, перечислены аминокислотные последовательности, которые входят в предпочтительные варианты последовательностей пептида-усилителя настоящего изобретения. Следует отметить, что, хотя нижепривиденные последовательности представлены в прямой ориентации, однако, в объем настоящего изобретения также входят последовательности в обратной ориентации. Так, например, хотя нижеуказанная последовательность "WMEWDREI" пептида-усилителя приводится в прямой ориентации, однако, эта последовательность в обратной ориентации, то есть "IERDWEMW", также входит в объем настоящего изобретения.

В другом предпочтительном варианте осуществления изобретения конкретные последовательности настоящего изобретения содержат последовательности пептида-усилителя, показанные на Фиг.2, 13 и в Таблице 1 и имеющие консервативные аминокислотные замены в одном, двух или трех положениях, где указанные замены не влияют на способность последовательности пептида-усилителя улучшать фармакокинетические свойства гибридного полипептида по сравнению с его соответствующим коровым полипептидом.
Особенно предпочтительно, чтобы такие замены приводили к образованию пептида-усилителя, имеющего в своей последовательности одну из консенсусных последовательностей. Обычно такие замены осуществляют в аминокислотных остатках, соответствующих положениям "X", показанных в консенсусных аминокислотных последовательностях, представленных выше и на Фиг.1 и 2. Термин "консервативные замены" означает замены на аминокислотные остатки, имеющие такой же заряд и размер и/или такие же гидрофобные/гидрофильные свойства, как заменяемый аминокислотный остаток. Такие характеристики аминокислот хорошо известны специалистам.
Настоящее изобретение также относится к последовательностям пептида-усилителя, содержащим аминокислотные последовательности, представленные на Фиг.1, 2 и 13 и в Таблице 1, которые в той или иной степени являются аналогичными, но при этом указанные последовательности пептида-усилителя имеют одно или несколько аминокислотных добавлений (обычно не более чем примерно 15 аминокислотных остатков), делеций (например, усечения по амино- или [карбокси]-концу) или неконсервативных замен, которые, тем не менее, не влияют на способность полученного пептида-усилителя улучшать фармакокинетические свойства коровых полипептидов, к которым они присоединены, по сравнению с коровыми полипептидами без таких последовательностей пептида-усилителя.
Добавления обычно не превышают около 15 аминокислотных остатков и могут включать добавления около 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 или 15 следующих друг за другом аминокислотных остатков. Предпочтительно, чтобы полное число аминокислотных остатков, добавленных к исходному пептиду-усилителю, не превышало примерно 15 аминокислотных остатков, более предпочтительно, чтобы это число не превышало примерно десять аминокислотных остатков, а наиболее предпочтительно, чтобы это число не превышало примерно 5 аминокислотных остатков.
Предпочтительными делениями являются делеции, не превышающие в целом примерно 3 аминокислотных остатка (либо следующих друг за другом, либо не следующих друг за другом остатков), более предпочтительными являются делеции из 2 аминокислот, а наиболее предпочтительными являются делеции из одного аминокислотного остатка. Обычно делеции аминокислотных остатков соответствуют остаткам "X" консенсусных последовательностей пептида-усилителя.
Последовательности пептида-усилителя настоящего изобретения также содержат конкретные последовательности пептида-усилителя, показанные на Фиг.2, 13 и в Таблице 1 и имеющие одну, две или три неконсервативные аминокислотные замены, предпочтительно две таких замены, а наиболее предпочтительно одну такую замену. Термин "неконсервативные" замены означает замены на аминокислотные остатки, имеющие другой заряд, размер и/или гидрофобные/гидрофильные свойства по сравнению с заменяемым аминокислотным остатком. Такие свойства аминокислот хорошо известны специалистам.
Кроме того, замены аминокислот желательно не должны ограничиваться, а в некоторых вариантах осуществления изобретения предпочтительно не ограничиваются генетически кодируемыми аминокислотами. Действительно, эти пептиды могут содержать некодируемые генетически аминокислоты. Таким образом, помимо природных генетически кодируемых аминокислот, аминокислотные остатки в этих пептидах могут быть заменены на некодируемые в природе аминокислоты и синтетические аминокислоты.
Некоторыми наиболее часто встречающимися аминокислотами, которые могут быть использованы в качестве подходящих замен, являются, но не ограничиваются ими, β-аланин (β-Аlа) и другие омега-аминокислоты, такие как 3-аминопропионовая кислота, 2,3-диаминопропионовая кислота (Dpr), 4-аминомасляная кислота и т.п.; α-аминоизомасляная кислота (Aib); ε-аминогексановая кислота (Aha); δ-аминовалериановая кислота (Ava); N-метилглицин или саркозин (MeGly); орнитин (Оrn); цитруллин (Cit); трет-бутилаланин (t-BuA); трет-бутилглицин (t-BuG); N-метилизолейцин (MeIle); фенилглицин (Phg); циклогексилаланин (Cha); норлейцин (Nle); нафтилаланин (Nal); 4-хлорфенилаланин (Phe(4-Cl)); 2-фторфенилаланин (Phe(2-F)); 3-фторфенилаланин (Phe(3-F)); 4-фторфенилаланин (Phe(4-F)); пеницилламин (Pen); 1,2,3,4-тетрагидроизохинолин-3-карбоновая кислота (Tic); β-2-тиенилаланин (Thi) ; сульфоксид метионина (MSO); гомоаргенин (hArg); N-ацетиллизин (AcLys); 2,4-диаминомасляная кислота (Dbu); 2,3-ди-аминомасляная кислота (Dab); п-аминофенилаланин (Phe(pNH2)); N-метилвалин (MeVal); гомоцистеин (hCys); гомофенилаланин (hPhe); гомосерин (hSer); гидроксипролин (Hyp); гомопролин (hPro); N-метилированные аминокислоты и пептоиды (N-замещенные глицины).
Хотя в большинстве случаев аминокислоты пептида заменены L-энантиомерными аминокислотами, однако эти замены не ограничиваются L-энантиомерными аминокислотами. Таким образом, определение "мутированных" или "модифицированных" форм относится и к тем случаям, когда L-аминокислота заменена идентичной D-аминокислотой (например, L-Arg - D-Arg) или D-аминокислотой той же категории или подкатегории (например, L-Arg - D-Lys), и наоборот.
Следует отметить, что в настоящем изобретении также рассматриваются пептидные аналоги, где одна или несколько амидных связей необязательно заменены связью, не являющейся амидной, предпочтительно замещенным амидом или изостерой амида. Таким образом, хотя аминокислотные остатки в пептидах, обычно, называют аминокислотами, а предпочтительные варианты осуществления изобретения проиллюстрированы с использованием пептидов, однако в этой связи следует отметить, что в тех вариантах, где описаны не-амидные связи, используемый здесь термин "аминокислота" или "остаток" означает другие бифункциональные составляющие, несущие группы, которые по своей структуре аналогичны боковым цепям аминокислот. Кроме того, эти аминокислотные остатки могут быть блокированными или неблокированными.
Кроме того, одна или несколько амидных связей могут быть заменены пептидомиметиками или амид-имитирующими составляющими, которые, в основном, не влияют на структуру или активность пептидов. Подходящие амид-имитирующие составляющие описаны, например, Olson et al., 1993, J. Med. Chem. 36:3049.
Для улучшения фармакокинетических свойств корового полипептида последовательности пептида-усилителя могут быть использованы в виде либо N-концевых, либо С-концевых, либо в виде N-концевых и С-концевых добавлений. Хотя последовательности пептида-усилителя предпочтительно использовать в виде пары последовательностей, то есть предпочтительно, чтобы гибридные полипептиды содержали последовательность пептида-усилителя как у амино-, так и у карбоксиконца, однако гибридные полипептиды могут также содержать один пептид-усилитель, где указанный пептид присутствует либо у амино-, либо у карбоксиконца гибридного полипептида. Кроме того, эти пептиды-усилители, связанные с коровым полипептидом, могут быть использованы либо в прямой, либо в обратной ориентации, или в любой подходящей комбинации. Следует отметить, что все пептиды-усилители могут быть введены либо у N-конца, либо у С-конца корового полипептида. Более того, в N-, С- или N- и С-концевые положения гибридных полипептидов может быть введено множество последовательностей пептида-усилителя. Множественные последовательности пептида-усилителя могут быть связаны непосредственно друг с другом посредством тех же самых типов связей, которые используются для присоединения последовательности пептида-усилителя к коровому полипептиду (см. ниже). Кроме того, между одной или несколькими множественными последовательностями пептида-усилителя может также присутствовать промежуточная аминокислотная последовательность того же типа, что и описанная ниже. Эти множественные последовательности пептида-усилителя обычно содержат от 2 до около 10 отдельных последовательностей пептида-усилителя (одинаковых или различных аминокислотных последовательностей), а предпочтительно от около 2 до около 4 последовательностей.
Следует отметить, что указанный коровый полипептид обычно связан с пептидом усилителем посредством пептидной амидной связи, хотя для присоединения последовательностей пептида-усилителя к коровым полипептидами могут быть использованы и неамидные связи. Такие связи хорошо известны специалистам, и ими являются, например, любая из углерод-углеродных, сложноэфирных и химических связей, которая связывает последовательности пептида-усилителя настоящего изобретения с коровым пептидом.
Обычно последовательность пептида-усилителя связана непосредственно с коровым полипептидом. Последовательность пептида-усилителя может быть также присоединена к промежуточной аминокислотной последовательности, присутствующей между последовательностью пептида-усилителя и коровым полипептидом. Обычно размер промежуточной аминокислотной последовательности составляет от около 1 до около 50 аминокислотных остатков, а предпочтительно от около 1 до около 10 остатков. Некоторые типы связей, описанные для связывания пептида-усилителя с коровым полипептидом, могут быть использованы для связывания пептида-усилителя с промежуточным пептидом.
Как обсуждалось выше для последовательностей пептида-усилителя, коровая и промежуточная аминокислотные последовательности необязательно должны быть ограничены генетически кодируемыми аминокислотами, и они могут содержать любые модификации аминокислот и связей, описанные выше.
Амино- и/или карбоксиконцы полученного гибридного полипептида может содержать аминогруппу (-NH2-) или карбоксигруппу (-СООН) соответственно. Альтернативно аминоконец гибридного полипептида может, например, представлять собой гидрофобную группу, включая, но ограничиваясь ими, карбобензильную, данзильную, трет-бутоксикарбонильную, деканоильную, нафтоильную или другую углеводную группу; ацетильную группу; 9-флуоренилметоксикарбонильную (FMOC) группу; или модифицированный неприродный аминокислотный остаток. Альтернативно карбоксиконец гибридного полипептида может, например, представлять амидогруппу; трет-бутоксикарбонильную группу; или модифицированный неприродный аминокислотный остаток. В качестве неограничивающего примера могут служить амино- и/или карбоксиконцы полученного гибридного полипептида, которые могут содержать любые амино- и/или карбоксиконцевые модификации, проиллюстрированные в пептидах, представленных на Фиг.13 или в нижеследующей Таблице 2.
Обычно гибридный полипептид содержит аминокислотную последовательность, которая представляет собой неприродную аминокислотную последовательность. То есть обычно аминокислотная последовательность гибридного полипептида не состоит только из аминокислотной последовательности фрагмента эндогенного природного полипептида. Кроме того, гибридный полипептид не должен состоять только из полноразмерного природного полипептида.
Коровые полипептиды могут содержать любой полипептид, который может быть введен в живую систему, например любой полипептид, который может функционировать как фармакологически эффективный полипептид. Такие коровью полипептиды могут быть, например, использованы для лечения или предупреждения заболевания или в диагностических или прогностических методах, включая методы визуализации in vivo. Нижний предел размера корового полипептида обычно составляет около 4-6 аминокислотных остатков. Теоретически, не существует верхнего предела размера корового полипептида, и этот коровый полипептид может содержать любой природный полипептид или его фрагмент или любой модифицированный или синтетический полипептид. Однако в основном размеры корового полипептида составляют от около 4-6 аминокислот до около 494-500 аминокислот, предпочтительно от около 4 до около 94-100 аминокислотных остатков, а наиболее предпочтительно от около 4 до около 34-40 аминокислотных остатков.
Примерами возможных коровых полипептидов, которые приводятся лишь в иллюстративных целях, но не целях ограничения, являются, но не ограничиваются ими, факторы роста, цитокины, терапевтические полипептиды, гормоны, например инсулин и пептидные фрагменты гормонов, ингибиторы или стимуляторы цитокинов, пептидные факторы роста или факторы дифференцировки, интерлейкины, хемокины, интерфероны, колониестимулирующие факторы, ангиогенные факторы, лиганды рецепторов, агонисты, антагонисты или обратные агонисты, агенты для доставки пептидов, такие как визуализирующие агенты, или агенты для доставки цитотоксических молекул, и белки внеклеточного матрикса, такие как коллаген, ламинин, фибронектин и интегрин и др. Кроме того, в качестве коровых полипептидов могут быть использованы вирусные или бактериальные полипептиды, которые прямо или опосредованно функционируют как иммуногены или антигены, а поэтому они могут быть использованы для лечения или предупреждения патологических состояний.
Репрезентативные примеры гибридных полипептидов, содержащих коровые полипептиды, происходящие от последовательности вирусного белка, представлены на Фиг.13, где указанные последовательности корового полипептида заштрихованы. Коровыми полипептидами также являются, но не ограничиваются ими, полипептиды, описанные в Патенте США №5464933, в Патенте США №5656480 и в WO 96/19495, каждый из которых во всей своей полноте вводится в настоящее описание посредством ссылки.
Последовательностями корового полипептида могут, кроме того, быть, не ограничиваясь ими, последовательности полипептидов, представленных в нижепривиденной Таблице 2. Следует отметить, что пептидами, перечисленными в Таблице 2, помимо коровых полипептидов, являются гибридные полипептиды. Однако с учетом последовательностей пептида-усилителя очевидно, что эта последовательность коровых полипептидов присутствует как часть гибридных полипептидов.
Следует отметить, что пептиды, перечисленные в Таблице 2, также входят в объем настоящего изобретения. Как обсуждалось выше, те пептиды, представленные в Таблице 2, которые еще не содержат последовательностей пептида-усилителя (то есть не являются гибридными полипептидами), могут быть использованы в сочетании с последовательностями пептида-усилителя и рассматриваются в данном описании для генерирования гибридных полипептидов. Кроме того, коровые полипептиды и коровый полипептид гибридных полипептидов, показанные в Таблице 2 и на Фиг.13, могут быть использованы в сочетании с любыми последовательностями пептида-усилителя, описанными в настоящей заявке, для рутинного продуцирования других гибридных полипептидов, которые также входят в объем настоящего изобретения.
Следует отметить, что, хотя ряд полипептидов, перечисленных в Таблице 2 и на Фиг.13, приводится с модифицированными, например с блокированными, амино- и/или карбоксиконцами или с D-изомерными аминокислотами (остатки, указанные в скобках), однако, подразумевается, что любой полипептид, содержащий первичную аминокислотную последовательность, представленную в Таблице 2 и на Фиг.13, также считается частью настоящего изобретения.
Последовательности корового полипептида, per se, показанные в Таблице 2 и на Фиг.13, а также гибридные полипептиды, содержащие такие коровые полипептиды, могут обладать антивирусной и/или антифузогенной активностью и/или могут обладать способностью модулировать внутриклеточные процессы, ассоциированные с пептидными биспиральными структурами. Среди последовательностей коровых полипептидов имеются, например, последовательности, происходящие от отдельных последовательностей вирусного белка. Среди последовательностей коровых полипептидов имеются, например, такие аминокислотные последовательности, которые происходят от более чем одного вирусного белка (например, коровый полипептид, происходящий от ВИЧ-1, ВИЧ-2 и SIV).
Кроме того, такие коровые полипептиды могут иметь замены, делеции и/или инсерции аминокислот, обсуждаемые выше для последовательностей пептида-усилителя, при условии, что они не будут оказывать негативного воздействия на антивирусную и/или антифузогенную активность конкретного корового полипептида (как такового или в виде части гибридного полипептида).
Что касается делеции аминокислот, то предпочтительно, чтобы длина полученного корового полипептида составляла, по крайней мере, около 4-6 аминокислотных остатков. Что касается инсерции аминокислот, то предпочтительно, чтобы они составляли не более чем около 50 аминокислотных остатков, а более предпочтительно не более чем около 15 аминокислотных остатков. Также предпочтительно, чтобы инсерциями коровых полипептидов были амино- и/или карбоксиконцевые инсерции.
Среди таких амино- и/или карбоксиконцевых инсерций могут быть инсерции, которые включают аминокислотные последовательности, находящиеся у амино- и/или карбоксиконцов по отношению к эндогенной последовательности белка, от которого происходит данный коровый полипептид. Так, например, если коровый полипептид происходит от белка gp41, то такая инсерция должна быть амино- и/или карбоксиконцевой инсерцией, содержащейся в аминокислотной последовательности gp41, смежной с последовательностью корового полипептида gp41. Такие амино- и/или карбоксиконцевые инсерции могут, в основном, составлять около 1, 5, 10, 15, 20, 25, 30, 35, 40, 45 или 50 аминокислотных остатков со стороны амино- и/или карбоксиконца по отношению к исходному коровому полипептиду.
Гибридные полипептиды настоящего изобретения могут, кроме того, содержать дополнительные модификации, которые позволяют легко детектировать данный полипептид. Так, например, гибридные полипептиды могут быть помечены прямым или непрямым методом. Методы мечения пептидов хорошо известны специалистам, и такими методами являются, но не ограничиваются ими, методы радиоактивного, флуоресцентного или колориметрического мечения. Методы непрямого мечения также хорошо известны специалистам, и такими методами являются, но не ограничиваются ими, методы мечения с использованием биотина/стрептавидина и непрямого мечения антителом.
Настоящее изобретение также относится к ассоциации последовательностей пептида-усилителя с молекулами других типов, которые не являются пептидами. Так, например, последовательности пептида-усилителя могут быть присоединены к молекулам нуклеиновых кислот (например, ДНК или РНК) или к небольшой органической молекуле любого типа для улучшения фармакокинетических свойств указанных молекул.
5.2 Синтез пептидов
Полипептид-усилитель, коровый полипептид и гибридный полипептид могут быть синтезированы или получены методами, хорошо известными специалистам. См., например, работу Creighton, 1983, Proteins: Structures and Molecular Principles, W.H. Freeman and Co., NY, которая во всей своей полноте вводится в настоящее описание посредством ссылки. Гибридные полипептиды могут быть получены с использованием стандартного метода постадийного синтеза в растворе или твердофазного синтеза, конденсации фрагментов, F-MOC или Т-ВОС-химии (см., например, Chemical approaches to the Synthesis of Peptides and Proteins, Williams et al., Eds., 1997. CRC Press, Boca Raton Florida, и работы, цитированные в этой работе; Solid Phase Peptide Synthesis: A Practical Approach, Atherton & Sheppard, Eds., 1989, IRL Press, Oxford, England, и работы, цитированные в этой работе). Аналогичным образом, могут быть использованы амино- и/или карбоксиконцевые модификации.
Полипептид-усилитель, коровый полипептид и гибридный полипептид могут быть очищены известными методами, такими как нормальная и обращенно-фазовая жидкостная хроматография, с высоким разрешением, ионообменная хроматография, гель-электрофорез, аффинная хроматография, эксклюзионная хроматография, преципитация и т.п. Конкретные условия, используемые для очистки конкретного полипептида, зависят отчасти от стратегии синтеза и от таких факторов, как суммарный заряд, гидрофобность, гидрофильность, растворимость, стабильность и т.п., и могут быть выбраны самим специалистом.
Гибридные полипептиды, полипептиды-усилители и коровые полипептиды могут быть также получены с использованием техники рекомбинантных ДНК. В данном случае, нуклеотидные последовательности, кодирующие полипептиды настоящего изобретения, могут быть синтезированы и/или клонированы и экспрессированы методами, хорошо известными каждому специалисту. См., например, Sambrook et al., (1989), Molecular Cloning: A Laboratory Manual, vol.1-3, Cold Spring Harbor Press, NY.
ДНК-сегмент, кодирующий нужный полипептид, может быть получен различными методами молекулярной биологии, известными специалистам. Так, например, для генерирования ДНК-фрагмента, кодирующего нужный белок, может быть использована полимеразная цепная реакция (ПЦР). Альтернативно данный ДНК-фрагмент может быть получен из коммерческого источника.
ДНК, кодирующая нужные полипептиды, может быть сконструирована методами рекомбинантных ДНК в ряде векторных системах-хозяевах, которые также обеспечивают крупномасштабную репликацию данной ДНК. Эти векторы могут быть сконструированы так, чтобы они содержали элементы, необходимые для регуляции транскрипции и/или трансляции ДНК-последовательности, кодирующей гибридный полипептид.
Векторами, которые могут быть использованы в этих целях, являются, но не ограничиваются ими, векторы, происходящие от рекомбинантной бактериофаговой ДНК, плазмидной ДНК или космидной ДНК. Так, например, могут быть использованы плазмидные векторы, такие как pcDNA3, pBR322, pUC 19/18, pUC 118, 119 и векторы серии M13mp. Бактериофаговыми векторами могут быть λgt10, λgt11, λgt18-23, λZAP/R и бактериофаговые векторы серии EMBL. Космидными векторами, которые могут быть использованы, являются, но не ограничиваются ими, pJB8, pCV 103, pCV 107, pCV 108, pTM, pMCS, pNNL, pHSG274, COS202, COS203, pWE15, pWE16 и векторы типа харомиды 9.
Альтернативно могут быть сконструированы рекомбинантные векторы на основе вирусов, включая, но не ограничиваясь ими, векторы, полученные на основе таких вирусов, как вирус герпеса, ретровирусы, вирусы коровьей оспы, аденовирусы, аденоассоциированные вирусы или вирусы бычьей папилломы, либо вирусов растений, таких как вирус табачной мозаики и бакуловирус.
Для экспрессии биологически активных полипептидов в соответствующий экспрессирующий вектор, то есть в вектор, который содержит элементы, необходимые для транскрипции и трансляции встроенных кодирующих последовательностей, могут быть встроены нуклеотидные последовательности, кодирующие белок. Конструирование экспрессирующих векторов, имеющих последовательности, кодирующие гибридный полипептид и функционально присоединенные к транскрипционным/трансляционным регуляторным сигналам, может быть осуществлено методами, хорошо известными специалистам. Такими методами является техника рекомбинантных ДНК in vitro и методы синтеза. См., например, методы, описанные в работах Sambrook et al., (1989), Molecular Cloning, A Laboratory manual. Cold Spring Harbor Laboratory, и Ausubel et al., Current Protocols in Molecular Biology, Greene Publishing Associates and Wiley Interscience, N.Y., которые во всей своей полноте вводятся в настоящее описание посредством ссылки.
Молекула нуклеиновой кислоты, кодирующая нужный гибридный полипептид, полипептид-усилитель и коровый полипептид, может быть функционально присоединена к различным промоторным/энхансерным элементам. Для оптимизации экспрессии терапевтических количеств белка могут быть выбраны соответствующие промоторные/энхансерные элементы. Экспрессионные элементы этих векторов могут варьироваться по своей длине и специфичности. В зависимости от используемой системы хозяин/вектор может быть использован любой из подходящих элементов транскрипции и трансляции. Данный промотор может присутствовать в виде промотора, естественно ассоциированного с нужным геном. Альтернативно данная ДНК может находиться под контролем рекомбинантного или гетерологичного промотора, т.е. промотора, который обычно не ассоциируется с этим геном. Так, например, тканеспецифические промоторные/энхансерные элементы могут быть использованы для регуляции экспрессии встраиваемой ДНК в клетках определенного типа.
Примерами транскрипционных регуляторных областей, обладающих тканеспецифичностью, которые были описаны и которые могут быть использованы, являются, но не ограничиваются ими, регуляторная область гена эластазы I, которая является активной в ацинарных клетках поджелудочной железы (Swift et al., 1984, Cell 38:639-646; Ornitz et al., 1986, Cold Spring Harbor Symp. Quant. Biol.50:399-409; MacDonald, 1987, Hepatology 7:42S-51S); регуляторная область гена инсулина, которая является активной в бета-клетках поджелудочной железы (Hanahan, 1985, Nature 315:115-122); регуляторная область гена иммуноглобулина, которая является активной в лимфоидных клетках (Grosschedl et al., 1984, Cell 38:647-658; Adams et al., 1985, Nature 318:533-538; Alexander et al., 1987, Mol.Cell.Biol. 7:1436-1444): регуляторная область гена альбумина, которая является активной в печени (Pinkert et al., 1987, Genes and Devel. 1:268-276), регуляторная область гена альфа-фетопротеина, которая является активной в печени (Krumlauf et al., 1985, Mol.Cell.Biol. 5:1639-1648; Hammer et al., 1987, Science 235:53-58); регуляторная область гена альфа-1-антитрипсина, которая является активной в печени (Kelsey et al., 1987, Genes and Devel. 1:161-171); регуляторная область гена бета-глобина, которая является активной в миелоидных клетках (Magram et al., 1985, Nature 315:338-340; Kollias et al., 1986, Cell, 46:89-94); регуляторная область гена основного белка миелина, которая является активной в олигодендроцитах головного мозга (Readhead et al., 1987, Cell, 48:703-712); регуляторная область гена легкой цепи-2 миозина, которая является активной в скелетной мышце (Shani, 1985, Nature 314:283-286); и регуляторная область гена гонадотропного рилизинг-гормона, которая является активной в гипоталамусе (Mason et al., 1986, Science 234:1372-1378). Могут быть использованы промоторы, выделенные из генома вирусов, выращенных в клетках млекопитающих (например, промоторы вируса коровьей оспы 7,5К, SV40, HSV, аденовирусов MLP, MMTV, LTR и CMV), а также промоторы, продуцированные с помощью техники рекомбинантных ДНК или методами синтеза.
В некоторых случаях промоторные элементы могут быть конститутивными или индуцибельными и могут быть использованы при соответствующих условиях для осуществления высокого уровня экспрессии или осуществления регулируемой экспрессии нужной нуклеотидной последовательности. Экспрессия генов под контролем конститутивных промоторов не требует для своего индуцирования присутствия специфического субстрата, и эта экспрессия будет происходить при любых условиях клеточного роста. В противоположность этому, экспрессия генов, регулируемая индуцибельными промоторами, зависит от присутствия или отсутствия индуцирующего агента.
Для достаточной трансляции встроенных последовательностей, кодирующих белок, необходимы также специфические сигналы инициации. Такими сигналами являются инициирующий кодон ATG и смежные последовательности. В случаях, когда в соответствующие экспрессирующие векторы встроена вся кодирующая последовательность, включая инициирующий кодон и смежные последовательности, дополнительных сигналов регуляции трансляции не требуется. Однако в случае, когда встроена только часть кодирующей последовательности, необходимо присутствие экзогенных сигналов регуляции трансляции, включая инициирующий кодон ATG. Кроме того, для обеспечения трансляции всей вставки инициирующий кодон должен находиться в той же рамке считывания, что и последовательности, кодирующие белок. Эти экзогенные сигналы регуляции трансляции и инициирующие кодоны могут происходить от разных источников, как природных, так и синтетических. Эффективность экспрессии может быть повышена путем включения последовательностей ослабителей транскрипции, энхансерных элементов и т.п.
5.3. Использование последовательностей пептидов-усидителей, коровых полипептидов и гибридных полипептидов настоящего изобретения
Как обсуждалось выше, последовательности пептида-усилителя могут быть использованы для улучшения фармакокинетических свойств любого корового полипептида посредством присоединения указанного корового полипептида к последовательностям пептида-усилителя с образованием гибридных полипептидов. Такое улучшение фармакокинетических свойств наблюдается по сравнению с фармакокинетическими свойствами корового полипептида, взятого отдельно. Стандартные параметры фармакокинетических свойств и методы определения и характеризации фармакокинетических свойств такого агента, как полипептид, хорошо известны специалистам. Нелимитирующие примеры таких методов представлены в нижеописанных Примерах.
Последовательности пептида-усилителя настоящего изобретения, кроме того, могут быть использованы для увеличения in vitro или ex vivo времени полужизни корового полипептида, к которому могут быть присоединены последовательности пептида-усилителя. Так, например, последовательности пептида-усилителя могут увеличивать время полужизни присоединенных коровых полипептидов в том случае, когда полученные гибридные полипептиды присутствуют в клеточной культуре, тканевой культуре или в образцах, взятых от пациента (например, в образцах клеток, образцах тканей для биопсии или в других образцах, содержащих физиологические жидкости).
Коровые полипептиды и гибридные полипептиды настоящего изобретения могут быть также использованы в методах модуляции (например, снижения, ингибирования, устранения, стабилизации или усиления) фузогенных событий. Такие пептиды обладают предпочтительно антифузогенной или антивирусной активностью. Пептиды настоящего изобретения могут также обладать способностью модулировать внутриклеточные процессы, включая взаимодействия, ассоциированные с пептидой биспиральной структурой.
В конкретном варианте осуществления изобретения гибридные полипептиды и коровые полипептиды настоящего изобретения, которые обладают антивирусной активностью, могут быть использованы в активируемых методах. Такие активируемые методы могут быть использованы, например, против ретровирусов человека, а в частности, ВИЧ (вируса иммунодефицита человека), например ВИЧ-1 и ВИЧ-2, и против Т-лимфотропных вирусов человека (HTLV-I и HTLV-II), и нечеловеческих ретровирусов, таких как вирус лейкоза коров, вирусы саркомы и лейкоза кошек, вирусы иммунодефицита (SIV), саркомы и лейкоза обезьян, и вирусы прогрессирующей пневмонии овец.
Антивирусные способы настоящего изобретения могут быть также использованы против вирусов, не принадлежащих к ретровирусам, включая, но не ограничиваясь ими, респираторно-синцитиальный вирус (RSV), вирус собачьей чумы, вирус ньюкастлской болезни, вирус парагриппа человека, вирусы гриппа, вирусы кори, вирусы Эпштейна-Барра, вирусы гепатита В и вирусы Мейсона-Пфайзера.
Вышеуказанные вирусы являются вирусами с оболочкой. Антивирусные способы настоящего изобретения могут быть также использованы против вирусов без оболочки, включая, но не ограничиваясь ими, пикорнавирусы, такие как полиовирусы, вирус гепатита А, энтеровирус, эховирусы и вирусы Коксаки, паповавирусы, такие как вирус папилломы, парвовирусы, аденовирусы и реовирусы.
Другими типами антифузогенной активности, которые могут быть модулированы способами с использованием пептидов настоящего изобретения, являются, но не ограничиваются ими, модуляция обмена нейротрансмиттеров посредством слияния клеток и слияния сперматозоида с яйцеклеткой. Из внутриклеточных нарушений, включая взаимодействия, обусловленные биспиральной структурой пептидов, которые могут быть ослаблены способами с использованием пептидов настоящего изобретения, являются, например, нарушения, вызванные бактериальными токсинами.
Антифузогеннная или антивирусная активность данного корового полипептида или гибридного полипептида может быть определена рутинными методами посредством стандартных in vitro- и ex vivo-анализов и анализов с использованием животных с моделью данного заболевания, которые, с точки зрения противовирусной активности, могут быть специфичными или частично специфичными по отношению к нужному вирусу и которые хорошо известны специалистам.
Вышепривиденное описание, главным образом, относится к антивирусной и антифузогенной активности коровых и гибридных полипептидов настоящего изобретения. Гибридные полипептиды настоящего изобретения могут быть также использованы в любом методе, в котором предусматривается введение или использование корового полипептида, взятого отдельно. Использование гибридных полипептидов в таких методах является особенно предпочтительным в тех случаях, когда необходимо улучшение фармакокинетических свойств корового полипептида. Так, например, при лечении некоторых типов диабета используется инсулин. Следовательно, гибридный полипептид, содержащий инсулин или фрагмент инсулина в виде корового полипептида, может быть также использован в методах ослабления симптомов некоторых форм диабета, в которых используется и/или предусматривается использование инсулина.
Помимо вышеуказанных терапевтических методов, пептиды настоящего изобретения могут быть, кроме того, использованы в прогностических методах для предупреждения расстройств, включая, но не ограничиваясь ими, расстройства, ассоциированные с событиями слияния; внутриклеточные процессы, ассоциированные с биспиральными пептидными структурами; вирусные инфекции вызываемые слиянием клеток и/или слиянием вируса с клеткой. Так, например, коровые и гибридные полипептиды настоящего изобретения могут быть использованы в профилактических методах для предупреждения вирусной инфекции.
Гибридные полипептиды настоящего изобретения, кроме того, могут быть использованы в диагностических методах. Такими методами могут быть in vivo- или in vitro-методы. Любой диагностический метод, в котором может быть использован конкретный коровый полипептид, может быть осуществлен с использованием гибридного полипептида, содержащего данный коровый полипептид и модификацию или первичную аминокислотную последовательность, которая позволяет детектировать гибридный полипептид. Такие методы, по сравнению с диагностическими методами, могут иметь преимущества, заключающиеся в том, что увеличенное время полужизни гибридного полипептида по сравнению с коровым полипептидом, взятым отдельно, может способствовать повышению чувствительности диагностического метода, в котором он используется. Такими диагностическими методами являются, но не ограничиваются ими, методы визуализации, например методы визуализации in vivo. В неограничивающем примере метода визуализации структура, связывающая коровый полипептид гибридного полипептида, может быть детектирована путем связывания с гибридным полипептидом и визуализации (прямым или непрямым методом) этого связанного гибридного полипетида.
5.4. Фармацевтические композиции, дозы и способы введения
Пептиды настоящего изобретения могут быть введены методами, хорошо известными специалистам. Соответствующие агенты предпочтительно изготавливают и вводят системно. Описание способов получения готовых композиций и их введения можно найти в руководстве "Remington’s Pharmaceutical Sciences", последнее издание. Mack Publishing Co., Easton, PA. Подходящими способами введения могут быть пероральное введение, ректальное введение, вагинальное введение, внутрилегочное введение (например, путем ингаляции), чрезкожное введение, введение через слизистую и интестинальное введение; парентеральное введение, включая внутримышечные, подкожные, интрамедуллярные инъекции, а также внутриоболочечные, прямые интравентикулярные, внутривенные, внутрибрюшинные, интраназальные или офтальмические инъекции, и т.п. Для внутривенной инъекции агенты настоящего изобретения могут быть изготовлены в виде водных растворов, предпочтительно в забуференном физиологическом растворе, таком как раствор Хэнкса, раствор Рингера или забуференный физиологический раствор и т.п. Кроме того, для введения пептида настоящего изобретения могут быть использованы инфузионные насосы. Для введения через слизистую в данной композиции используются пенетранты, способствующие прохождению данного пептида через барьер. Такие пенетранты, в основном, известны специалистам.
В тех случаях, когда предпочтительным является внутриклеточное введение пептидов настоящего изобретения или других ингибирующих агентов, могут быть использованы методы, хорошо известные специалистам. Так, например, такими агентами могут быть агенты, инкапсулированные в липосомы или микросферы, которые затем вводят как описано выше. Липосомы представляют собой сферические липидные бислои с водной сердцевиной. В эту водную сердцевину вводятся все молекулы, присутствующие в водном растворе в процессе формирования липосомы. Содержимое липосом защищено от внешнего микроокружения, и, благодаря слиянию липосом с клеточными мембранами, оно может быть эффективно доставлено в цитоплазму клетки. Кроме того, если необходимо ввести небольшие молекулы, то благодаря гидрофобности этих липосом может быть осуществлено прямое внутриклеточное введение.
Нуклеотидные последовательности, кодирующие пептиды настоящего изобретения, предназначенные для внутриклеточного введения, могут быть экспрессированы в нужных клетках методами, хорошо известными специалистам. Так, например, экспрессирующие векторы, полученные на основе вирусов, таких как ретровирусы, вирусы коровьей оспы, аденоассоциированные вирусы, вирусы герпеса или вирусы коровьей папилломы, могут быть использованы для доставки и экспресии таких нуклеотидных последовательностей в нужную клеточную популяцию. Методы конструирования таких векторов и экспрессионных конструкций хорошо известны специалистам. См., например, Sambrook et al., 1989, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Press, Cold Spring Harbor, NY, и Ausubel et al., 1989, Current Protocols in Molecular Biology, Greene Publishing Associates and Wiley Interscience, NY.
Эффективные дозы вводимых пептидов настоящего изобретения могут быть определены методами, хорошо известными специалистам, в соответствии с такими параметрами, как биологическое время полужизни, биологическая доступность и токсичность. В особенно предпочтительных вариантах осуществления изобретения эффективная доза гибридного полипептида составляет в интервалах, которые могут быть определены самим специалистом, исходя из данных, полученных в результате рутинных in vitro- и in vivo-исследований, хорошо известных специалистам. Так, например, анализы клеточных in vitro-культур с антивирусной активностью, такие как анализы, описанные ниже в Разделе 7 для Т1249, позволяют получить данные, из которых каждый специалист может легко определить среднюю ингибирующую концентрацию (IC) пептида или полипептида, необходимую для блокирования определенного количества инфекционности вируса (например, 50%, IC50; или 90% IC90). Затем, с использованием фармакокинетических данных, таких как фармакокинетические данные, описанные ниже в Разделе 10 для Т1249 и полученные от одного или нескольких экспериментальных животных с моделью данного заболевания, специалистом могут быть выбраны соответствующие дозы так, чтобы получить минимальную концентрацию пептида в плазме (Cmin), которая равна определенной величине IC или превышает эту величину.
В качестве примера могут служить дозы полипептидов, составляющие, по крайней мере, от 0,1 мкг/кг массы тела и вплоть до 10 мг/кг массы тела. Более предпочтительная эффективная доза составляет от 0,1 до 100 мкг/кг массы тела. В качестве других примеров могут служить дозы пептидов настоящего изобретения, составляющие 1-5 мг, 1-10 мг, 1-30 мг, 1-50 мг, 1-75 мг, 1-100 мг, 1-125 мг, 1-150 мг, 1-200 мг или 1-250 мг пептида. Термин "терапевтически эффективная доза" означает количество соединения, достаточное для достижения ослабления симптомов или для продления жизни пациента. Токсичность и терапевтическая эффективность таких соединений может быть определена с помощью стандартных терапевтических процедур, проводимых на клеточных культурах или на экспериментальных животных, например, для определения LD50 (летальная доза для 50% популяции) и ED50 (терапевтически эффективная доза для 50% популяции). Отношение доз для токсического и терапевтического эффектов представляет собой терапевтический индекс, который может быть выражен как отношение LD50/ED50. При этом предпочтительными являются соединения, которые имеют высокий терапевтический индекс. Для установления интервалов доз для введения человеку могут быть использованы данные, полученные из анализов клеточных культур и исследований, проведенных на животных. Дозы таких соединений предпочтительно представляют собой такие количества этих соединений, чтобы их концентрации в кровотоке составляли ED50 при незначительной токсичности или ее отсутствии. Эти дозы могут варьироваться в указанных пределах в зависимости от используемой формы лекарственного средства и способа его введения. Для любого соединения, используемого в способе настоящего изобретения, терапевтически эффективная доза может быть первоначально оценена посредством анализа клеточных культур. Определенная доза может быть введена животным с моделью данного заболевания для достижения концентрации в плазме, которая составляет ингибирующую концентрацию IC50 (например, концентрация тестируемого соединения, которая приводит к полумаксимальному ингибированию события слияния, такому как полумаксимальное ингибирование вирусной инфекции, по отношению к числу событий слияния в отсутствие тестируемого соединения), определенную в клеточной культуре. Такие данные могут быть использованы для более точного определения эффективных доз для введения человеку. Концентрации в плазме могут быть измерены, например, с помощью высокоразрешающей жидкостной хроматографии (ВРЖХ) или любого биологического или иммунологического анализа, в котором могут быть определены уровни пептида.
Гибридные полипептиды настоящего изобретения могут быть введены в виде разовых или дробных доз, периодически или непрерывно. Так, например, полипептиды настоящего изобретения могут быть введены в виде разовой дозы, такой как разовая доза, вводимая подкожно, разовая доза, вводимая путем внутривенной инфузии, или разовая доза, вводимая перорально. Полипептиды настоящего изобретения могут быть также введены несколько раз в виде многократных доз, включая периодическое введение. Так, например, в некоторых вариантах осуществления изобретения полипептиды настоящего изобретения могут быть введены один раз в неделю, один раз в день, два раза в день (например, через каждые 12 часов), через каждые 6 часов, через каждые 4 часа, через каждые 2 часа или через 1 час. Полипептиды настоящего изобретения могут быть также введены в непрерывном режиме, таком как непрерывное подкожное или внутривенное введение с помощью инфузионного насоса или с помощью подкожного или другого имплантата, что приводит к непрерывной абсорбции указанных полипептидов в организме пациента.
Гибридные полипептиды настоящего изобретения могут быть также введены в комбинации, по крайней мере, с одним другим терапевтическим агентом. При этом могут быть использованы другие виды терапии, хотя они и не являются предпочтительными для ВИЧ-терапии (например, противораковая терапия), и эти виды терапии могут быть осуществлены одновременно или последовательно, включая циклическую терапию (то есть введение первого соединения в течение определенного периода времени с последующим введением второго антивирусного соединения в течение определенного периода времени, а затем повторения такого последовательного введения в целях минимизации возможности развития резистентности к одному из этих видов терапии).
В случае вирусных, например ретровирусных, инфекций может быть введено эффективное количество гибридного полипептида или его фармацевтически приемлемого производного в комбинации, по крайней мере, с одним, а предпочтительно, по крайней мере, с двумя другими антивирусными агентами.
Если в качестве примера взять ВИЧ-инфекцию, то такими антивирусными агентами могут быть, не ограничиваясь ими, DP-107 (Т21), DP-178 (T20), любой другой коровый полипептид, указанный в Таблице 2 и происходящий от ВИЧ-1 или ВИЧ-2, любой другой гибридный полипептид, коровый полипептид которого, по крайней мере, частично происходит от ВИЧ-1 или ВИЧ-2, цитокинов, например, pIFNα, pIFNβ, pIFNγ; ингибиторов обратной транскриптазы, включая нуклеозидные и ненуклеозидные ингибиторы, например AZT, 3ТС, D4T, ddI, адефовир, абакавир или другие дидезоксинуклеозиды или дидезоксифторнуклеозиды, или мезилат делавиридина, невирапин, эфазиренц; от ингибиторов кэппинга вирусной мРНК, таких как рибавирин; от ингибиторов ВИЧ-протеазы, таких как ритонавир, мезилат нелфинавира, ампренавир, саквинавир, мезилат саквинавира, индинавир или АВТ378, АВТ538 или МК639; от амфотерицина В в качестве липидсвязывающей молекулы с активностью против ВИЧ; и от кастаноспермина в качестве ингибитора процессинга гликопротеина.
Гибридные и/или коровые полипептиды настоящего изобретения, кроме того, могут быть использованы в профилактических целях для предупреждения заболевания. Гибридные и/или коровые полипептиды могут действовать непосредственно для предупреждения заболевания, либо, альтернативно, они могут быть использованы в качестве вакцин, которые способствуют вырабатыванию у хозяина антител против гибридных полипептидов настоящего изобретения, где указанные антитела затем служат для нейтрализации патогенных микроорганизмов, включая, например, ингибирование вирусной, бактериальной или паразитарной инфекции.
Для всех видов лечения, описанных выше, конкретная композиция, способ введения и доза могут быть выбраны лечащим врачом, исходя из состояния пациента (см., например, Fingl et al., 1975, The Pharmacological Basis of Therapeutics, Ch.1, p.1).
При этом следует отметить, что лечащий врач должен знать, как и когда должно быть завершено, прекращено или скорректировано введение лекарственного средства вследствие его токсичности или дисфункции органов. И наоборот, лечащий врач должен знать, как и когда лечение должно быть скорректировано в сторону увеличения вводимых доз, в том случае, если не достигается адекватного клинического ответа (с исключением токсичности). Величина вводимой дозы при лечении данного онкогенного расстройства может варьироваться в зависимости от тяжести заболевания пациента, подвергаемого лечению, и от способа введения. Доза и, вероятно, схема введения может также варьироваться в зависимости от возраста, массы тела и восприимчивости каждого отдельного пациента. Программа, аналогичная описанной выше, может быть использована в ветеринарии.
В объем настоящего изобретения входит использование фармацевтически приемлемых носителей для изготовления препаратов соединений настоящего изобретения, в дозах, подходящих для системного введения. При правильном выборе носителя и подходящего способа изготовления, композиции настоящего изобретения, в частности композиции, полученные в виде растворов, могут быть введены парентерально, например, путем подкожной инъекции, внутривенной инъекции, подкожной инфузии или внутривенной инфузии, например, с помощью насоса. Эти соединения могут быть легко изготовлены с использованием фармацевтически приемлемых носителей, хорошо известных специалистам, в дозах, подходящих для перорального введения. Такими носителями, подходящими для введения соединений настоящего изобретения, являются таблетки, драже, капсулы, жидкости, гели, сиропы, суспензии, взвеси и т.п., используемые для перорального введения пациентом.
Фармацевтическими композициями, подходящими для использования в настоящем изобретении, являются композиции, где указанные активные ингредиенты содержатся в количестве, эффективном для достижения нужной цели. Каждый специалист может самостоятельно определить эффективное количество активного ингредиента, особенно исходя из подробного описания, приведенного в настоящей заявке.
Помимо активных ингредиентов, эти фармацевтические композиции могут содержать подходящие фармацевтически приемлемые носители, включающие наполнители и вспомогательные добавки, облегчающие приготовление из этих активных соединений препаратов, которые могут быть использованы в фармацевтических целях. Препараты для перорального введения могут быть изготовлены в форме таблеток, драже, капсул или растворов. Для перорального введения пептидов могут быть использованы методы, описанные, например, в Emisphere Technologies, которые хорошо известны и обычно применяются специалистами.
Фармацевтические композиции настоящего изобретения могут быть изготовлены способами, известными по существу, например, способами простого смешивания, растворения, гранулирования, изготовления драже, растирания в порошок, сушки распылением, эмульгирования, инкапсулирования, захватывания или лиофилизации.
Фармацевтическими композициями для парентерального введения являются водные растворы активных соединений в водорастворимой форме. Кроме того, эмульсии и суспензии активных соединений могут быть получены в виде подходящих масляных смесей для инъекций. Подходящими липофильными растворителями или носителями являются жирные масла, такие как кунжутное масло, или синтетические сложные эфиры жирных кислот, такие как этилолеат или триглицериды, липосомы или другие вещества, используемые специалистами для приготовления липидных или липофильных эмульсий. Водные суспензии для инъекций могут содержать вещества, которые улучшают вязкость суспензии, такие как натрийсодержащая карбоксиметилцеллюлоза, сорбит или декстран. Для получения растворов с высокой концентрацией эти суспензии могут также содержать, но не обязательно, подходящие стабилизаторы или агенты, которые повышают растворимость соединений.
Фармацевтические препараты для перорального введения могут быть получены путем объединения активных соединений с твердым наполнителем, необязательного измельчения полученной смеси и приготовления смеси гранул после добавления подходящих добавок, если это необходимо, с получением сердцевины для таблеток или драже. Подходящими носителями являются, в частности, наполнители, такие как сахара, включая лактозу, сахарозу, трегалозу, маннит или сорбит; целлюлозные препараты, такие как, например, кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагакантовая камедь, метилцеллюлоза, гидроксипропилметилцеллюлоза, натрийсодержащая карбоксиметилцеллюлоза и/или поливинилпирролидон (ПВП). При необходимости могут быть добавлены дезинтегрирующие агенты, такие как поливинилпирролидон с поперечными связями, агар или альгиновая кислота или ее соли, такие как альгинат натрия.
Сердцевину драже покрывают подходящими покрытиями. Для этой цели могут быть использованы концентрированные растворы cахаров, которые могут содержать, но не обязательно, аравийскую камедь, тальк, поливинилпирролидон, гель карбопол, полиэтиленгликоль и/или двуокись титана, растворы лаков и подходящие органические растворители или смеси растворителей. Для идентификации или характеризации различных комбинаций доз активных соединений в покрытия таблеток или драже могут быть добавлены красители или пигменты.
Фармацевтическими препаратами, которые могут быть использованы для перорального введения, являются капсулы с устройством для выталкивания, изготовленные из желатина, а также мягкие герметичные капсулы, изготовленные из желатина и пластификатора, такого как глицерин или сорбит. Капсулы с устройством для выталкивания могут содержать активные ингредиенты в смеси с наполнителем, таким как лактоза, со связующими агентами, такими как крахмалы, и/или с замасливателями, такими как тальк или стеарат магния, и необязательно, со стабилизаторами. В мягких капсулах активные соединения могут быть растворены или суспендированы в подходящих жидкостях, таких как жирные масла, вазелиновое масло или жидкие полиэтиленгликоли. Кроме того, могут быть добавлены стабилизаторы.
В случаях, где необходимо усиление иммунного ответа у хозяина, гибридные полипептиды могут быть изготовлены с подходящим адъювантом для усиления этого иммунологического ответа. Такими адъювантами могут быть, но не ограничиваются ими, минеральные гели, такие как гидроксид алюминия; поверхностно-активные вещества, такие как лизолецитин, полиолы типа плюроников, полианионы; другие пептиды; масляные эмульсии; и потенциально подходящие адъюванты, такие как BCG и Corynebacterium parvum. Для введения описанных в настоящей заявке вакцинных композиций может быть использовано множество способов. Такими способами являются, но не ограничиваются ими, пероральное, чрескожное, внутримышечное, внутрилегочное, внутривенное, подкожное и интраназальное введение.
6. Пример: Идентификация консенсусных аминокислотных последовательностей, содержащих последовательности пептида-усилителя
Белок gp41 ретровируса содержит структурные домены, называемые областью α-спирали, расположенной в С-концевой области белка, и областью "лейциновой молнии", расположенной в N-концевой области белка. Сравнительный анализ участков первичных последовательностей пептида-усилителя, происходящих от gp41 (Фиг.2А и 2В) с gp41 для всех опубликованных в настоящее время идентифицированных консенсусных аминокислотных последовательностей изолятов ВИЧ-1, ВИЧ-2 и SIV, проиллюстрирован на Фиг.1.
Как подробно описано в нижепривиденных Примерах, такие последовательности представляют собой последовательности пептида-усилителя, в которых присоединение этих пептидных последовательностей к другим разнообразным коровым полипептидам приводит к улучшению фармакокинетических свойств полученных гибридных полипептидов.
7. Пример: Гибридные полипептиды, функционирующие как сильные ингибиторы ВИЧ-1-инфекции
Т1249, показанный на Фиг.13, представляет собой гибридный полипептид, содержащий последовательности пептида-усилителя, присоединенные к коровому полипептиду ВИЧ. Как продемонстрировано ниже, гибридный полипептид Т1249 обладает улучшенными фармакокинетическими свойствами и сильной активностью in vitro против изолятов ВИЧ-1, ВИЧ-2 и SIV, при этом повышенная активность против клинических изолятов ВИЧ-1 наблюдалась в in vitro-анализax на степень инфицирования НЧРВМС, а также в in vivo-исследованиях на ВИЧ-1-инфекцию у мышиной модели SCID-HuPBMC. В биологических анализах, описанных выше, активность Т1249 была сравнима с сильной антивирусной активностью полипептида Т20. Полипептид Т20, известный также как DP-178 и полученный из последовательности белка gp41 ВИЧ-1, был описан и заявлен в патенте США №5464933.
7.1. Материалы и методы
7.1.1. Синтез и очистка пептидов
Пептиды синтезировали с использованием экспресс-Мос-химии. В основном, если это не указано особо, эти пептиды содержали амидированные карбоксиконцы и ацетилированные аминоконцы. Очистку проводили с помощью обращенно-фазовой ВЭЖХ.
Т1249 (Ac-WQEWEQKITALLEQAQIQQEKNEYELQKLDKWASLWEWF-NH2) представляет собой пептид в 39 аминокислот (МW=5036,7), целиком состоящий из природных аминокислот и блокированный на своем аминоконце ацетильной группой, а на своем карбоксиконце - амидогруппой для повышения стабильности. Т1387 представляет собой пептид из 23 аминокислот, у которого отсутствует последовательность пептида-усилителя (Ac-TALLEQAQIQQEKNEYELQKLDK-NH2). Таким образом, Т1387 представляет собой коровый полипептид гибридного полипептида Т1249. Т1387 блокирован на своих карбокси- и аминоконцах так же, как и Т1249.
В частности, Т1249 синтезировали стандартными методами твердофазного синтеза. Идентичность главного пика на ВЭЖХ-хроматограмме, которая была подтверждена масс-спектроскопией, указывала на Т1249.
Т1249 был легко очищен с помощью обращенно-фазовой хроматографии на 6-дюймовой колонке, упакованной С18, 10 микрон, подложка 120 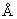 .
.
7.1.2. Вирус
Вирус ВИЧ-1LAI (Popovic M. et al., 1984, Science 224:497-508) размножали в клетках СЕМ, культивированных в RPMI-1640, содержащей 10% фетальную телячью сыворотку. Супернатант от инфицированных клеток СЕМ пропускали через 0,2-микронный фильтр и титр инфекционности оценивали путем проведения анализа на микроинфекционность с использованием клеточной линии АА5 для поддержания репликации вируса. Для этого 20 мкл серийно разведенного вируса добавляли к 20 мкл клеток СЕМ при концентрации 6 х 105/мл в 96-луночном микротитрационном планшете. Каждое разведение вируса тестировали в трех дубликатах. Клетки культивировали в течение 7 дней путем добавления свежей среды через день. Через 7 дней после инфицирования образцы супернатантов тестировали на репликацию вируса, о чем свидетельствовала активность обратной транскриптазы, высвобождаемой в супернатант. TCID50 вычисляли по формуле Рида и Мениша (Reed L.J. et al., 1938, Am. J. Hyg. 27:493-497).
7.1.3. Анализ на слияние клеток
Приблизительно 7×104 клеток Molt-4 инкубировали с 1 х 104 клеток СЕМ, хронически инфицированных вирусом ВИЧ-1LAI в 96-луночных планшетах для культивирования тканей в конечном объеме 100 мкл культуральной среды (RPMI-1640, содержащей 10% термоинактивированный FBS с добавлением 1% L-глутамина и 1% Pen-Strep), описанной ранее (Matthews T.J. et al., 1987, Proc. Natl. Acad. Sci., USA, 84:5424-5428). Пептидные ингибиторы добавляли в объеме 10 мкл, и клеточные смеси инкубировали в течение 24 ч при 37°С в 5% СО2. На этой стадии полинуклеарные гигантские клетки (синцитий, ширина в 5 клеток или более) подсчитывали под микроскопом при увеличении 10х и 40х, что позволяло визуализировать всю лунку в одном поле. Обработанные клетки сравнивали с инфицированными необработанными контрольными клетками, и результаты выражали как процент ингибирования от инфицированного контроля.
7.1.4. Анализы на инфекционность Magi-CCR-5
Приблизительно 1×106 клеток Magi-CCR-5 (полученных в рамках программы Национального института здравоохранения (NIH) по исследованиям СПИД’а (NIH AIDS) и стандартных реагентов, Отдел СПИД’а, Национальный институт артрита и инфекционных заболеваний (NIAID; Chackerian В. et al., 1997, J. Virol. 71:3932-3939) засевали в 48-луночный планшет для культивирования тканей (приблизительно 2×104 клеток/лунку в объеме 300 мкл/лунку селективной культуральной среды, состоящей из DMEM с добавлением 10% термоинактизированного FBS, 1% L-глутамина, 1% Pen-Strep, гигромицина В, генетицина и пуромицина) и оставляли на ночь для связывания при 37°С в 5% СО2. На следующий день конфлюентность клеток составляла приблизительно 30%. Среду для засевания удаляли и добавляли разведенный пептидный ингибитор в объемах 50 мкл/лунку (в необработанный контроль - только среду), а затем добавляли 100 мкл/лунку разведенного вируса (желательный вводимый титр вируса = 100-200 б.о.е./лунку). Наконец, в каждую лунку добавляли 250 мкл селективной культуральной среды, и планшет инкубировали в течение 2 дней при 37°С в 5% СО2. Фиксацию и окрашивание проводили в соответствии с протоколом, представленным Национальным институтом артрита и инфекционных заболеваний, и с использованием клеток MAGI-CCR5. Для этого среду удаляли из планшета, и в каждую лунку добавляли 500 мкл фиксатора. Планшеты оставляли для фиксации на 5 минут при комнатной температуре. Фиксатор удаляли, каждую лунку дважды промывали DPBS и в каждую лунку добавляли 200 мкл окрашивающего раствора. Затем планшет инкубировали при 37°С в 5% СО2 в течение 50 минут, окрашивающий раствор удаляли и каждую лунку дважды промывали DPBS. Планшет оставляли для сушки на воздухе, а затем синие клетки подсчитывали на микроскопе, при этом все лунки были пронумерованы. Обработанные лунки сравнивали с инфицированными необработанными контрольными клетками, и результаты выражали как процент ингибирования от инфицированного контроля.
7.1.5. Анализ на обратную транскриптазу
Микроанализ на обратную транскриптазу (RT) проводили адаптированным методом Goff et al. (Goff, S. et al., 1981, J.Virol. 38:239-248) и Willey et al. (Willey, R. et al., 1988, J.Virol. 62:139-147). Супернатанты от вирусных/клеточных культур доводили до объема 1% Тритон-Х100. 10 мкл каждого образца супернатанта/Тритона-Х-100 добавляли к 50 мкл смеси RT (75 мМ КCl, 2 мМ реагента Clevelands, 5 мМ MgCl2, 5 мкг/мл poly A, 0,25 единиц/мл oligo dT, 0,05% NР40, 50 мМ Трис-HCl, рН 7,8, 0,5 мкМ нерадиоактивного dTTP и 10 сКи/мл 32P-dTTP) в 96-луночном микротирационном планшете с U-образным дном и инкубировали при 37°С в течение 90 минут. После инкубирования 40 мкл реакционной смеси из каждой лунки переносили в аппарат для дот-блоттинга Schleicher & Schuell (S+S) в неполном вакууме, содержащий сетчатый 96-луночный фильтр-планшет (каталог Wallac №1450-423) и фильтр, предварительно насыщенный буфером 2x SSC (0,3 M NaCl и 0,003 М цитрат натрия). Каждую лунку промывали 4 раза, по крайней мере, 200 мкл 2x SSC в полном вакууме. Фильтр (Minifold) разбирали и сетчатую фильтровальную бумагу удаляли и 3 раза промывали 2х SSC. И наконец, фильтровальную мембрану дренировали на абсорбционной бумаге, оставляли на воздухе для сушки и помещали в пакеты, которые герметично запаивали. Образцы помещали в кассету с люминесцентным экраном, "стирали" нанесенный люминесцентный экран (по крайней мере, 8 минут) и закрывали. Экпонирование составляло 16 часов. Значения индексов пикселов (PIV), генерированных в формате представления данных об объеме, выбранном исходя из люминесцентной визуализации (Molecular Dynamic Phosphorimager) блотов, были использованы для определения поврежденной или ингибированной фракции (Fa) для всех доз ингибитора(тов) путем сравнения с необработанным инфицированным контролем (проанализированным с помощью данных об объеме ImageQuant, скорректированных с учетом фонового значения).
7.1.6. Анализ на инфицирование/нейтрализацию человеческих МКПК (НчРВМС)
Был использован прототип анализа, где в качестве клеточных линий использовали первичный изолят клеток МКПК, полученных из Банка крови Interstate и активированных в течение 2-3 дней ОКТЗ (0,5 мкг/мл) в комбинации с антителами против CD28 (0,1 мкг/мл). Эти клетки-мишени наносили полосами на среду для разделения лимфоцитов (LSM), промывали и замораживали. Клетки оттаивали, если это необходимо, и перед проведением анализа активировали как описано выше минимум 2-3 дня. В этом анализе с использованием 96-луночного планшета концентрация клеток составляла 2×106/мл в среде 5% IL-2, а конечный объем составлял 100 мкл. Исходные пептидные растворы приготавливали в DPBS (1 мг/мл). Разведения пептидов осуществляли в 20% FBS-RPMI-1640/полная среда с 5% IL-2.
7.1.7. HU-PBMC-SCID-модель ВИЧ-1-инфекции in vivo
Самкам мышей SCID (в возрасте 5-7 недель) внутрибрюшинно инъецировали 5-10 х 107 МКПК взрослого человека. Через две недели после восстановления мышей на день 0 инфицирововали внутрибрюшинно (i.p.) ВИЧ-1 9320 с 103 TCID50 (изолят А018, восприимчивый к АЗТ). Обработку пептидами осуществляли i.p. два раза в день, начиная со дня -1 и продолжая в течение 6 дней. Степень инфицирования клеток крови, спленоцитов, лимфоузлов и перитониальных клеток оценивали каждую неделю в течение последующих трех недель при количественном культивирования с бластами человеческих МКПК, затем животных умерщвляли путем обескровливания, и ткани собирали (на день 7, приблизительно через 12-18 часов после последней обработки лекарственным средством). Супернатанты от этой со-культуры оценивали на продуцирование антигена р24 ВИЧ-1 как показателя вирусной инфекции (наборы и протокол Immunotek Coulter).
7.1.8. Исследования фармакокинетики на крысах
Были использованы 250-300 г самцы крыс CD, которым вводили двойной шейный катетер, полученный от Charles River Laboratories. В один шейный катетер инъецировали пептиды в объеме 200 мкл пептидного раствора (приблизительно 3,75 мг/мл), при этом дозу концентрации раствора определяли методом Эдельхоха (Edeihoch, 1967, Biochemistry 6:1948-1954) и корректировали в расчете на массу животного так, чтобы каждое животное получало дозу 2,5 мг/кг. Через заранее определенные интервалы времени (через 0, 15, 30 мин и через 1, 2, 4, 6 и 8 часов) брали приблизительно 250-300 мкл крови и добавляли в пробирки Capiject с EDTA. Из осажденных клеток после центрифугирования удаляли плазму, и эту плазму либо замораживали, либо использовали непосредственно для флуоресцентного ВЭЖХ-анализа.
7.1.9. Флуоресцентный ВРЖХ-анализ образцов плазмы
100 мкл образцов плазмы добавляли к 900 мкл буфера для преципитации (ацетонитрил, 1,0% TFA, детергент), что приводило к осаждению большинства белков плазмы. После центрифугирования при 10000 об/мин в течение 10 минут 400 мкл супернатанта удаляли и добавляли к 600 мкл воды, пригодной для ВЭЖХ. Серийные разведения проводили в буфере для разведения, состоящем из 40% буфера для преципитации и 60% воды, пригодной для ВЭЖХ, в соответствии с нужными концентрациями пептида, присутствующего в каждом образце. Помимо разведений образца, серийные разведения или дозирование раствора осуществляли в буфере, а также в плазме и использовали для построения стандартной кривой, соответствующей площади пика для известной концентрации пептида. Эту кривую затем использовали для подсчета концентрации пептида в плазме с учетом всех проведенных разведений и количества пептида, введенного в колонку.
7.1.10. Протокол ХТТ
Для измерения цитотоксического/цитостатического действия пептидов осуществляли анализ ХТТ (Weislow, O.S. et al., 1989, J.Natl.Cancer Inst. 81: 577-586) в присутствии различных концентраций пептида для эффективного определения селективного индекса (SI). В этом анализе TC50 определяли путем инкубирования клеток в присутствии и отсутствие серийно разведенного пептида с последующим добавлением ХТТ. В выживших/метаболизирующих клетках ХТТ восстанавливался до растворимого ХТТ-формазана с коричневой окраской. Величину оптической плотности регистрировали, и для определения TC50 сравнивали значения, полученные в присутствии и отсутствие пептидов с использованием метода Karber (см., например, Lennette, E.H. et al., eds., 1969, Diagnonostic Procedures for Viral and Rickettsial Infections, American Public Health Association, Inc., fourth ed., pp.47-52). Клетки Molt 4, СЕМ (80000 клетка/лунку) и комбинацию клеток этих двух типов (70000 и 10000 соответственно) засевали и инкубировали с серийно разведенным пептидом в течение 24 часов в полном объеме 100 мкл. После инкубирования в каждую лунку добавляли 25 мкл рабочий раствор ХТТ (1 мг/мл ХТТ, 250 мкМ PMS в полной среде, содержащей 5% ДМСО) и планшеты инкубировали при 37°С. Развитие окраски регистрировали, и результаты использовали для вычисления величин, полученных от лунок, содержащих пептид, в виде процента от необработанных контрольных лунок.
7.2. Результаты
7.2.1. Антивирусная активность - анализы на слияние
Т1249 непосредственно сравнивали с Т20 в анализах на вирусопосредованное слияние клеток, проводимых с использованием хронически инфицированных клеток СЕМ, смешанных с неинфицированными клетками Molt-4, как показано ниже в Таблице 3. Ингибирование полипептидом Т1249 слияния клеток, опосредованного лабораторными изолятами, такими как IIIb, MN и RF, было сравнимым с ингибированием полипептидом Т20 и приблизительно в 2,5-5 раз превышало ингибирование слияния клеток полипептидом Т20. Т1249 был также более активным (в 3-28 раз), чем Т20, по отношению к нескольким синцитийиндуцированным клиническим изолятам, включая АЗТ-резистентый изолят (G691-2), предварительно АЗТ-обработанный изолят (G762-3) и изолят 9320 (используемый в исследованиях с моделью HuPBMC-SCID). Следует особо подчеркнуть, что Т1249 был в 800 раз более активным, чем Т20, по отношению к ВИЧ-2 NIHZ.
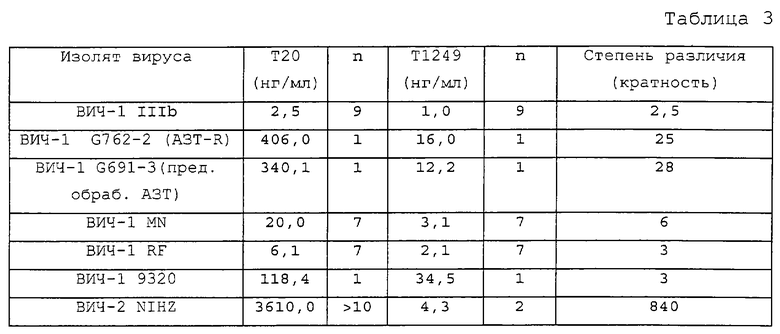
7.2.2. Антивирусная активность - анализы на степень инфицирования Magi-CCR-5
Анализы на инфицирование Magi-CCR-5 позволяют проводить непосредственные сравнения синцитиально и несинцитиально индуцирующих вирусных изолятов, а также проводить сравнения между лабораторными и клиническими изолятами. Этот анализ также позволяет непосредственно установить степень инфицирования вирусом (экспрессия ТАТ после инфицирования, трансактивация LTR, приводящая к продуцированию бета-галактозидазы) по сравнению с обычно используемыми непрямыми методами измерения инфекционности, такими как анализ на продуцирование антигена р24 или обратной транскриптазы. Анализы на степень инфицирования Magi-CCR-5 (см. Таблицу 4, ниже) выявили, что Т1249 является стабильно более эффективным, чем Т20 для всех тестируемых изолятов, исходя из вычислений параметров ингибирования, ЕС50 и Vn/Vo=0,1. Было обнаружено, что Т1249 является значительно более сильным ингибитором (в >25 раз) в отношении клинического изолята ВИЧ-1 301714, который является одним из наименее чувствительных изолятов для Т20. Кроме того, Т1249, по крайней мере, является в 100 раз более сильным ингибитором, чем Т20, в отношении изолята ВИЧ В670. Эти данные вместе с данными о слиянии позволяют предположить, что Т1249 является сильным пептидным ингибитором ВИЧ-1, ВИЧ-2 и SIV.

7.2.3. Антивирусная активность - анализы на степень инфицирования МКПК человека
Т1249 непосредственно сравнивали с Т20 в анализах на степень инфицирования МКПК человека (см. ниже Таблицу 5), которые представляют собой распознаваемую суррогатную in vitro-систему для определения предварительных концентраций лекарственного средства в плазме, необходимых для ингибирования вируса in vivo. Эти сравнения показали, что Т1249 является более сильным агонистом по отношению ко всем изолятам ВИЧ-1, протестированным в настоящее время, причем все величины Vn/Vo=0,1 (доза, необходимая для снижения титра вируса на один log) были снижены до субмикрограммовых концентраций. Многие из клинических изолятов, наименее чувствительных к Т20, обнаруживали в 10 раз или большую чувствительность к Т1249. Следует особо подчеркнуть, что ВИЧ-1 9320, то есть изолят, используемый для инфицирования мышиной модели HuPBMC-SCID, был в 46 раз менее чувствителен к Т20, чем к Т1249, что указывало на очень хорошую корреляцию с in vivo-результатами.

7.2.4. Антивирусная активность - Т20-резистентные лабораторные изоляты (LAB-изоляты)
Т1249 непосредственно сравнивали с Т20 в анализах на вирусопосредованное слияние клеток, проводимых с использованием хронически инфицированных клеток СЕМ, смешанных с неинфицированными клетками Molt-4 (см. ниже Таблицу 6). Т1249 обнаруживал почти в 200 раз большую активность против Т20-резистентного изолята, чем Т20.
В анализах Magi-CCR-5 (см. ниже Таблицу 7) Т1249 обнаруживал в 50000 раз более высокую активность, чем Т20, против Т20-резистентных изолятов, таких как pNL4-3 SM и pNL4-3 STM (Rimsky, L. & Mattews, Т., J. Virol. 72:986-933).

Т1249 непосредственно сравнивали с Т20 в анализах на степень инфицирования МКПК человека (см. ниже Таблицу 8) для оценки различия их активностей против резистентного изолята. Т1249 обнаруживал почти в 250 раз большую активность, чем Т20, против Т20-резистентного изолята pNL4-3 SM.

7.2.5. Антивирусная активность - HU-PBMC-SCID-модель in vivo
Антивирусную in vivo-активность Т1249 непосредственно сравнивали с активностью Т20 в мышиных моделях HuPBMC-SCID, инфицированных вирусом ВИЧ-1 9320 (Фиг.3). Через две недели после восстановления мышей с МКПК человека этих мышей на день 0 внутрибрюшинно (i.p.) инфицировали изолят от ВИЧ-1 9320 с TCID50 103 (изолят А018, восприимчивый к AZT), пассированный на МКПК. Обработку пептидами осуществляли i.p. два раза в день суточными дозами 67 мг/кг (Т20), 20 мг/кг (Т1249), 6,7 мг/кг (Т1249), 2,0 мг/кг (Т1249) и 0,67 мг/кг (Т1249) в течение 8 дней начиная со дня -1. Степень инфицирования клеток крови, спленоцитов, лимфоузлов и перитониальных клеток оценивали каждую неделю в течение последующих трех недель при количественном совместном культивировании с бластами человеческих МКПК, затем животных умерщвляли путем обескровливания, и ткани собирали (на 7-й день, приблизительно через 12-18 часов после последней обработки лекарственным средством). Супернатанты от этой со-культуры оценивали на продуцирование антигена р24 ВИЧ-1 как показателя вирусной инфекции. Инфекционный вирус не обнаруживался в тканях крови или лимфы Т20-обработанных животных, однако этот вирус был обнаружен в перитониальных промывках и в препаратах селезенки. Все компартменты были отрицательными на инфекционный вирус при дозе 6,7 мг/кг полипептида Т1249, что указывало на его более высокую активность, по крайней мере, в 10 раз превышающую активность Т20. При дозе 2,0 мг/кг Т1249 и в лимфе, и в селезенке не было обнаружено инфекционного вируса, причем в перитониальных промывках наблюдалось снижение в 2 log10 титра вируса, а в крови наблюдалось снижение 1 log10 титра вируса по сравнению с инфицированным контролем. При наименьшей дозе Т1249, то есть 0,67 мг/кг, перитониальные промывки и кровь были эквивалентны инфицированному контролю; однако в тканях лимфы и в тканях селезенки наблюдалось, по крайней мере, снижение 1 log10 титра инфекционного вируса. В целом, эти результаты показали, что при указанных условиях Т1249 обладает в 30-100 раз более сильной активностью против ВИЧ-1 9320 in vivo.
7.2.6. Исследования фармакокинетики на крысах
Для дополнительного определения фармакокинетического профиля Т1249 были использованы канюлированные крысы. Самцам крыс CD, 250-300 г, внутривенно (i.v.) вводили Т1249 и Т20 через шейный катетер (Фиг.4А-5). Полученные образцы плазмы оценивали с использованием флуоресцентной ВЭЖХ для оценки количества пептидов в экстрагированной плазме. Период полужизни в бета-фазе и полная площадь под кривой (AUC) для Т1249 были почти в три раза выше, чем для Т20 (Фиг.5).
7.2.7. Цитотоксичность
Как показано на Фиг.6, никакой цитотоксичности Т1249 in vitro обнаружено не было.
Кроме того, Т1249 не обнаруживал сильной токсичности (с летальным исходом через 24 часа) при дозе 167 мг/кг (самая высокая тестируемая доза), вводимой i.v. через шейную канюлю (0,3 мл в течение 2-3 минут).
7.2.8. Прямое связывание с конструкцией gp41 М41Δ178
T1249 подвергали радиоактивному мечению 125I, а ВЭЖХ-очистке до получения максимальной специфической активности. Т20 иодировали тем же способом. Связывание до состояния насыщения с М41Δ178 (гибридный белок с усеченным эктодоменом gp41, не содержащий аминокислотной последовательности Т20), иммобилизованным на микротитрационных планшетах при 0,5 мг/мкл, проиллюстрировано на Фиг.7. Неспецифическое связывание определяли как связывание радиолиганда в присутствии 1 мкМ немеченого пептида. Специфическое связывание представляло собой разность между общим и неспецифическим связыванием. Результаты показали, что 125I-T1249 и l25I-T20 имеют аналогичные аффинности связывания при 1-2 нМ. Графики обратной линейной зависимости Скэтчарда дают основание предположить, что каждый лиганд связывается с сайтами гомогенного класса.
Кинетику связывания 125I-T1249 и 125I-T20 определяли на сцинтилляционных микротитрационных планшетах, покрытых 0,5 мкг/мл М41Δ178. Время, затраченное на ассоциацию и диссоциацию, показано на Фиг.8. Диссоциацию связанного радиолиганда определяли после добавления немеченого пептида до конечной концентрации 10 мкМ в 1/10 полного объема анализа. Первоначальные скорости ассоциации и диссоциации для 125I-T1249 были значительно ниже, чем для 125I-T20. Характер диссоциации для обоих радиолигандов был неизменным в том случае, когда диссоциация была инициирована другим немеченым пептидом (т.е. 125I-T1249 с Т20).
Для дополнительной иллюстрации того факта, что оба лиганда конкурируют за связывание в одном и том же сайте-мишени, немеченые Т1249 и Т20 титровали в присутствии одной концентрации либо 125I-T1249, либо 125I-T20. Лиганд добавляли непосредственно после начала инкубирования немеченого пептида. Кривые конкурентного связывания показаны на Фиг.9 и позволяют предположить, что хотя оба этих лиганда имеют аналогичные аффинности, однако для обеспечения полной конкуренции за связывание с 125I-T1249 требуется более высокая концентрация либо немеченого Т20, либо Т1249.
7.2.9. Прямое связывание с HR1-областью gp41
Для измерения вторичной структуры Т1249 в растворе (забуференном фосфатом физиологическом растворе, рН 7), взятом отдельно и в комбинации с пептидом из 45 остатков (Т1346), происходящим от связывающей области HR1 (7-членный повтор 1) gp41, использовали спектроскопию кругового дихроизма (CD). На Фиг.14А показан CD-спектр одного Т1249 в растворе (10 мкМ, 1°С). Этот спектр является типичным спектром для пептидов с альфа-спиральной структурой. В частности, обратная свертка этого спектра с использованием сингулярного разложения с основной спектральной серией для 33 белков позволяет предсказать содержание спирали в Т1249 (взятого отдельно в растворе), составляющее 50%. На фиг.14В проиллюстрирован характерный CD-спектр Т1249, смешанного с Т1346. Заштрихованные квадраты (n) означают теоретический CD-спектр, предсказанный для "неинтерактивной модели", где высказана гипотеза о том, что эти пептиды не взаимодействуют в растворе. Фактический экспериментальный спектр (l) заметно отличается от указанного теоретического спектра "неинтерактивной модели", что указывает на то, что эти два пептида действительно взаимодействуют с образованием измеряемых структурных изменений, наблюдаемых в CD-спектре.
7.2.10. Защита от протеазы области связывания Т1249 в GP41
Восприимчивость химерного белка М41Δ178, описанного выше в разделе 7.2.8, к расщеплению протеиназой-К, определяли и анализировали с помощью электрофореза в полиакриламидном геле. Результаты проиллюстрированы на Фиг.15.
При отдельном инкубировании либо М41Δ178 (необработанного; Фиг.15, дорожка 2), либо Т1249 (необработанного; Фиг.15, дорожка 4) с протеиназой К (Фиг.15, дорожки 3 и 5 соответственно) оба они были расщеплены. Однако инкубирование Т1249 с М41Δ178 перед добавлением протеиназы-К (Фиг.15, дорожка 7) приводило к получению защищенного HR-1-фрагмента приблизительно в 6500 Дальтон. Секвенирование защищенного фрагмента показало, что он соответствует области первичной последовательности, локализованной в эктодомене gp41. Этот защищенный фрагмент включает растворимый пептид HR1 (Т1346), используемый в исследованиях CD, описанных выше в разделе 7.2.9, и, кроме того, содержит дополнительные семь аминокислотных остатков, локализованных на аминоконце. Эта защита может быть показателем связывания Т1249 со специфической последовательностью gp41, которая присутствует в конструкции М41Δ178.
8. Пример: Гибридные полипептиды на основе респираторно-синцитиального вируса
В этом примере описаны гибридные полипептиды с респираторно-синцитиальным вирусом (RSV), обладающие улучшенными фармакокинетическими свойствами. Кроме того, результаты представленные ниже, показали, что гибридные полипептиды, полученные на основе RSV, являются сильными ингибиторами RSV-инфекции.
8.1. Материалы и методы
8.1.1. Синтез и очистка пептидов
RSV-полипептиды были синтезированы с использованием стандартных химических экспресс-Мос-методов. В основном, если это не оговорено особо, эти пептиды содержат амидированные карбоксиконцы и ацетилированные аминоконцы. Очистку проводили с помощью обращенно-фазовой ВЭЖХ.
8.1.2. Анализ на снижение числа бляшек, образованных под действием респираторно-синцитиального вируса
Все необходимые разведения пептидов осуществляли в чистых стерильных 96-луночных планшетах ТС. Были собраны данные для всех одиннадцати разведений для каждого пептида и для одной контрольной лунки, не содержащей пептида. Интервал конечных концентраций пептида составлял от значения 50 мкг/мл или 100 мкг/мл и до значений, соответствующих всем одиннадцати двухкратным разведениям. RSV получали при концентрации 100 б.о.е./лунку в 100 мкл 3% ЕМЕМ, определенной с помощью известного титра RSV. Затем во все эти лунки добавляли вирус.
Из одного 96-луночного планшета с субконфлюэнтными клетками Нер2 удаляли среду. Материал из планшетов для разведения переносили на планшеты с клетками, начиная с ряда 1, а затем переносили ряд 12, ряд 11 и т.д. до тех пор, пока все ряды не были перенесены. Планшеты снова помещали в инкубатор на 48 часов.
Эти клетки проверяли на присутствие синцития в контрольных лунках. Затем среды удаляли, и в каждую лунку добавляли приблизительно 50 мкл 0,25% кристаллического фиолетового в метаноле. Для удаления избытка красителя лунки сразу промывали водой и оставляли для сушки. Число синцитиев в каждой лунке подсчитывали с помощью препаровальной лупы.
8.2. Результаты:
Фармакокинетические исследования гибридных RSV-пептидов Т1301 (AC-WQEWDEYDASISQVNEKINQALAYIREADELWAWF-NH2) и Т1302 (Ac-WQAWDEYDASISQVNEKINQALAYIREADELWAWF-NH2), содержащих последовательности пептида-усилителя, обнаружили значительно более продолжительное время полужизни по сравнению с коровым пептидом Т786 (Ac-VYPSDEYDASISQVNEEINQALAYIRKADELLENV-NH2), показанным на Фиг.10А-10В. Гибридные полипептиды Т1301, Т1302 и Т1303 (Ac-WQAWDEYDASISDVNEKINQALAYIREADELWEWF-NH2) также показали значительное увеличение времени полужизни по сравнению с коровым пептидом Т1476 (Ac-DEYDASISQVNEKINQALAYIREADEL-NH2).
Гибридные RSV-полипептиды Т1301, Т1302 и Т1303, а также полипептиды Т786 и Т1293 были протестированы на их способность ингибировать образование RSV-бляшек в клетках Нер2. Как показано на Фиг.11А и 11В, оба протестированных гибридных RSV-полипептида, а также коровый полипептид Т786, обладали способностью ингибировать RSV-инфекцию. Неожиданно было обнаружено, что гибридный полипептид Т1293 также является соединением, обладающим сильной активностью против RSV (Фиг.13).
9. Пример: Гибридные полипептиды, полученные на основе лютеинизирующего гормона
В этом примере описаны гибридные белки, полученные на основе лютеинизирующего гормона (LH) и обладающие улучшенными фармакокинетическими свойствами. Методами, описанными выше, синтезировали и очищали нижеследующие гибридные LH-пептиды: коровый пептид Т1323 (Ac-QHWSYGLRPG-NH2) и гибридный полипептид Т1324 (Ac-WQEWEQKIQHWSYGLRPGWASLWEWF-NH2), который содержит аминокислотную последовательность корового полипептида Т1323, присоединенную к пептиду-усилителю по амино- и карбоксиконцам. Как показано на Фиг.12А и 12В, гибридный пептид Т1324 обладал значительно более продолжительным временем полужизни по сравнению с коровым пептидом Т1323, у которого отсутствовали последовательности пептида-усилителя.
10. Пример: Фармакология гибридного полипептида Т1249
Т1249, показанный на Фиг.13, представляет собой гибридный полипептид, содержащий последовательность пептида-усилителя, присоединенную к коровому полипептиду, полученному из смеси вирусных последовательностей. Как продемонстрировано выше в Примере раздела 7, гибридный полипептид Т1249 обладает улучшенными фармакокинетическими свойствами и обладает сильной in vitro-, а также in vivo-активностью против ВИЧ-1. В представленном ниже Примере также описаны фармакологические свойства Т1249 как у грызунов, так и у приматов, используемых в качестве модели.
10.1. Материалы и методы
10.1.1. Введение разовой дозы грызунам
Т1249 вводили в виде разовой дозы крысам-альбиносам Sprague-Dawley путем непрерывной подкожной инфузии (SCI), подкожной инъекции (SС) или внутривенной инъекции (i.v.). Каждая обрабатываемая группа состояла из девяти крыс одного пола на группу. Этим группам вводили стерильные препараты порошкообразного лекарственного вещества Т1249 в дозе 0,5, 2,0 или 6,5 мг/кг посредством CSI. Одной группе давали 50 мМ карбонат-бикарбонат, рН 8,5, в качестве контроля. Эти пептиды вводили в течение 12 часов через поливинилхлоридный/полиэтиленовый катетер, хирургически имплантированный под кожу в заднюю область шеи. Двум группам вводили разовую дозу Т1249, составляющую 1,2 или 1,5 мг/кг, путем подкожной инъекции во внутрилопаточную область. Двум группам вводили разовую дозу Т1249, составляющую 1,5 или 5 мг/кг, путем внутривенной инъекции. Фактическое миллиграммовое количество Т1249 вычисляли исходя из содержания пептидов, которое было определено для введения каждой партии.
Заключительные этапы анализов включали осмотр клеток (два раза в день на летальность и серьезные поражения), клинические наблюдения, получение данных клинических лабораторных параметров, массы тела и аутопсии. Образцы крови брали методом рассеянной выборки в течение 12 часов у трех крыс каждого пола на группу через промежутки времени: 0,5, 1, 2, 4, 6, 8, 9, и 12 часов после введения дозы. Образцы оценивали с помощью анализа PcAb ECLIA (Blackburn, G. et al., 1991, Clin. Chem. 37:1534-1539; Deaver, D. 1995, Nature 377:758).
Для проведения анализа на фармакокинетические свойства Т1249 в плазме и лимфе у крыс Т1249 приготавливали в виде стерильного раствора в бикарбонатном буфере и вводили в разовой дозе путем внутривенной инъекции болюса в латеральную хвостовую вену в дозе 20 мк/кг. У животного брали кровь через вставленный шейный катетер. Образцы собирали сразу после взятия дозы через 5, 15 и 30 мин и 1, 2, 4 и 6 часов после введения лекарственного средства. Для анализа лимфатической жидкости образцы брали непосредственно перед введением дозы и через каждые 20 минут в течение первых шести часов после введения дозы. Лимфатическую жидкость собирали через катетер, установленный непосредственно в лимфатический проток грудной клетки, как было описано ранее (Kirkpatrick & Silver, 1970, The Journal of Surgical Research 10:147-158). Концентрации T1249 в плазме и в лимфатической жидкости определяли с использованием стандартного конкурентного анализа ELISA для T1249 (Hamilton, G., 1991, р.139, Immunochemistry of Solid-Phase Immunoassay, Butler, J., ed., CRC Press, Boston).
10.1.2. Введение разовой дозы приматам
Стерильные препараты порошкообразного лекарственного вещества T1249 вводили собакоподобным обезьянам в виде разовых доз посредством подкожной (s.c.), внутримышечной (i.m.) или внутривенной (i.v.) инъекции. В последующем перекрестном эксперименте одной группе животных, состоящей из двух животных каждого пола, вводили одну дозу болюса Т1249 внутривенно (0,8 мг/кг), внутримышечно (0,8 мг/кг) или подкожно (0,4, 0,8 и 1,6 мг/кг). Каждый день введения дозы сопровождался периодом промывки в течение, по крайней мере, трех дней. Непосредственно перед введением лиофилизованный Т1249 восстанавливали в стерильном забуференном фосфатом физиологическом растворе рН 7,4. Реальное миллиграммовое количество тестируемого препарата вычисляли исходя из содержания пептида, которое было определено для данной вводимой партии.
Заключительные этапы анализа включали осмотр клеток, физическое обследование и определение массы тела. Для i.v.-фазы этого исследования образцы крови собирали в гепаринизированные пробирки в следующие периоды времени: сразу после введения дозы и через 0,25, 0,5, 1,5, 3, 6, 12 и 24 часа после введения дозы. Для i.m.- и s.c.-фаз данного исследования образцы крови от каждого животного собирали в гепаринизированные пробирки в следующие периоды времени: через 0,5, 1, 2, 3, 6, 12 и 24 часа после введения дозы. Образцы плазмы получали в течение одного часа после сбора и быстрого замораживания в жидком азоте. Образцы оценивали с использованием анализа ELISA PcAb (Blackburn, G. et al., 1991, Clin. Chem. 37:1534-1539; Deaver, D. 1995, Nature 377:758).
10.1.3. Промежуточные фармакокинетические исследования
Шесть самцов собакоподобных обезьян случайно разделяли на три группы по два животных в каждой. Все дозы Т1249 в форме болюса вводили путем подкожной инъекции. Это исследование разделяли на две стадии. В стадии 1 животным в группах 1, 2 и 3 вводили стерильный препарат порошкообразного лекарственного вещества Т1249 (т.е. порошкообразный Т1249, растворенный в карбонате-бикарбонате, рН 8,5) два раза в день в течение четырех последующих дней (дни исследования 1-4) в дозах 0,2, 0,6 и 2,0 мг/кг/дозу соответственно. Стадию 1 и Стадию 2 разделял период десятидневной промывки. В Стадии 2 животным в группах 1, 2 и 3 вводили стерильный препарат готовой лекарственной композиции Т1249 (т.е. в водном растворе, рН 6,5 + маннит) два раза в день в течение четырех последующих дней (дни исследования 15-18) в дозах 0,2, 0,6 и 2,0 мг/кг/дозу соответственно.
Образцы крови для фармакокинетических анализов собирали в дни исследования 1 и 15 для оценки фармакокинетических параметров после введения разовых доз и в дни исследования 4 и 18 для оценки стационарных фармакокинетических параметров в плазме. Образцы крови собирали в следующие периоды времени: непосредственно перед введением дозы и через 0,5, 1,5, 3,0, 4,0, 6,0, 8,0 и 12,0 часов после введения дозы. Во время проведения Стадий 1 и 2, животных непрерывно наблюдали на клинические признаки и изменения массы тела.
10.2. Результаты
10.2.1. Фармакокинетика Т1249, введенного крысам
Крысиные модели использовали для проведения предварительной оценки фармакокинетики Т1249 в плазме и его распределения. У животных во всех дозовых группах не наблюдалось изменений массы тела, данных физического обследования, гематологии и клинической биохимии или данных макроскопической патологии, связанной с введением Т1249.
У крыс, которым вводили Т1249 путем непрерывной подкожной инфузии (c.s.i.), наблюдались постоянные концентрации пептидов в плазме через четыре часа после их введения. Как постоянная концентрация в плазме (Cpss), так и вычисленная площадь под кривой (AUC) зависимости концентрации в плазме от времени были прямо пропорциональны вводимой дозе, что указывало на то, что Т1249 обнаруживает линейную фармакокинетику в интервалах тестируемых доз от 0,5 до 6,5 мг/кг. Вычисленные фармакокинетические параметры и кривая зависимости концентрации в плазме от времени при с.s.i.-введении представлены в Таблице 9 и на Фиг.16А соответственно.
Введение Т1249 в виде болюса путем i.v.-инъекции приводило к линейной дозозависимой фармакокинетике в пределах тестируемых доз. В отличие от этого, введение Т1249 путем s.с.-инъекции не давало дозозависимой фармакокинетики в пределах исследуемых доз. Вычисленные фармакокинетические параметры и кривая зависимости концентрации в плазме от времени как для s.c.-, так и для i.v.-введения Т1249 представлены в Таблице 10 и на Фиг.16В соответственно.
Была определена биологическая доступность Т1249, вводимого крысам путем подкожной инъекции по сравнению с внутривенным введением. Результаты показаны ниже в Таблице 11. При низкой дозе (1,2 мг/кг) Т1249 обнаруживал относительную биологическую доступность (FR), составляющую 73% для подкожного введения. Относительная биологическая доступность Т1249 составляла 30% в случае, когда концентрация Т1249 при введении высокой дозы (15 мг/кг) введения превышала концентрацию, которая на 90 % (IС90) ингибирует ВИЧ-инфекцию, в течение всех 12 часов исследования при всех тестируемых дозах.

Кинетические данные для концентрации Т1249 в плазме и лимфе проиллюстрированы на Фиг.16С и систематизированы ниже в Таблице 12. Т1249 быстро проникал в лимфатическую систему и уравновешивался резервуаром лекарственного вещества в плазме в пределах приблизительно одного часа после введения. После установления равновесия между двумя компартментами уровни лекарственного средства в плазме и лимфе были сравнимыми в течение трех часов после введения дозы у четырех из пяти животных. Одно животное имело стабильно более низкие концентрации Т1249 в лимфе, чем другие животные, однако профиль элиминации этого лекарственного средства в лимфе этого животного не отличался от профиля других членов данной группы. Сравнение периода полужизни (t1/2) на фазе элиминации в плазме и лимфе позволяет предположить, что транспорт Т1249 между этими двумя компартментами представляет собой процесс, регулируемый диффузией. Через три часа наблюдалась вторая более быстрая фаза элиминации из лимфатической системы. Это отличие может быть обусловлено определенным механизмом (например, вследствие перераспределения или ускоренного разрушения пептида в лимфе) или другими факторами. Концентрация Т1249 в лимфатической жидкости через 6 часов после инъекции превышала IС50 для вирусной инфекционности стандартных лабораторных штаммов и первичных клинических изолятов ВИЧ-1.
Была также оценена степень проникновения Т1249 в цереброспинальную жидкость (ЦСЖ). Концентрации Т1249 были ниже пределов детекции (LOD; 2,0 нг Т1249/мл CSF) во всех интервалах времени измерения, что указывало на то, что Т1249 не проникает в центральную нервную систему после введения разовой дозы.
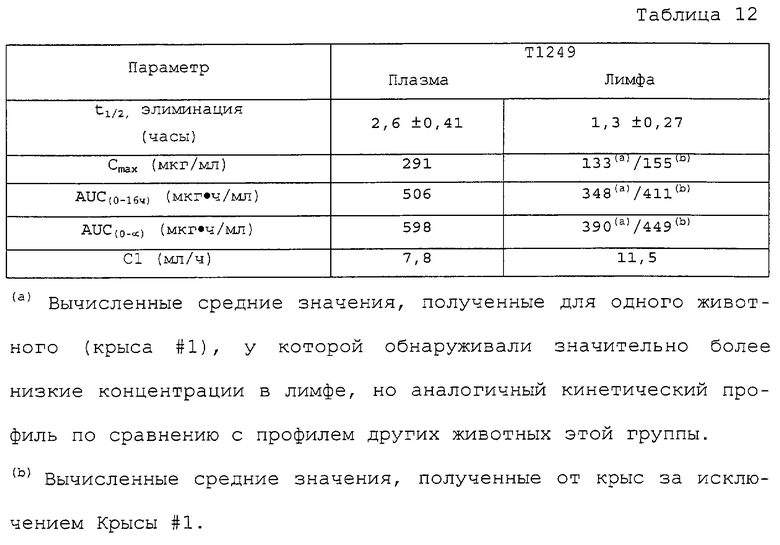
10.2.2. Фармакокинетика Т1249, введенного приматам
Для оценки зависимости между уровнем дозы и различными фармакокинетическими параметрами, ассоциированными с парентеральным введением Т1249, в качестве модели были использованы приматы. Концентрации Т1249 в плазме, превышающие 6,0 мкг/мл и достигаемые при всех способах введения и количественных уровнях (то есть уровнях, превышающих 0,5 мкг/мл), детектировали через 24 часа после s.c.- и i.v.-введения. Время выведения t1/2 было сравнимо во всех способах введения (5,4 часа, 4,8 часа и 5,6 часа для i.v.-, s.c.- и i.m.-введения соответственно). Концентрации Т1249 в плазме, которые превышали величины IC50 для лабораторных штаммов и клинических изолятов ВИЧ-1, наблюдали во все периоды времени измерений в течение 24-часового взятия образцов.
Сравнение данных, полученных при парентеральном введении 0,8 мг/кг Т1249 для всех способов введения (s.c., i.v. и i.m.), представлено на Фиг.17А. На Фиг.15В проиллюстрировано сравнение данных, полученных в результате s.с.-инъекции при трех различных дозах Т1249 (0,4 мг/кг, 0,8 мг/кг и 1,6 мг/кг). Вставка на Фиг.17В представляет кривую вычисленной AUC в зависимости от вводимой дозы.
Т1249 обнаруживает линейную фармакокинетику у собакоподобных обезьян после его s.с.-введения в интервалах вводимых доз, что указывает на то, что при этих интервалах доз отсутствует(ют) механизм(ы) насыщения клиренса. Суммарные фармакокинетические данные после парентерального введения Т1249 собакоподобным обезьянам показаны ниже в Таблице 13. Сравнение значений AUC плазмы показало, что по сравнению с внутривенным введением, биологическая доступность Т1249 при внутримышечном введении составляет приблизительно 64%, а при подкожном введении - 92%.
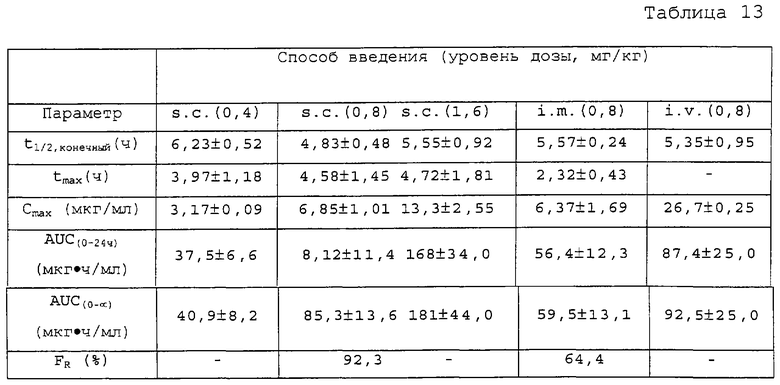
10.2.3. Промежуточные фармакокинетические исследования
Промежуточные фармакокинетические исследования осуществляли для сравнения фармакокинетических профилей в плазме для порошкообразных лекарственных веществ Т1249, используемых в неклинических испытаниях, описанных выше, с профилями готовой лекарственной композиции Т1249, которая была введена конкретному индивидууму или пациенту, например, для лечения ВИЧ-инфекции. В этом исследовании проводили одностороннее перекрестное сравнение на параллельных группах, которым вводили три уровня доз порошкообразного лекарственного вещества Т1249 и три уровня доз готового лекарственного продукта. Фармакокинетику в плазме оценивали после введения разовой дозы и после достижения стабильного состояния.
Введение Т1249 путем подкожной инъекции приводило к измеряемым уровням пептидов для всех дозовых групп. Кривые зависимости концентрации в плазме от времени были строго параллельны для всех дозовых групп после введения начальной дозы (дни 1 и 15) и оставались постоянными (дни 4 и 18) как для порошкообразного лекарственного вещества Т1249, так и для готового лекарственного продукта Т1249. Кроме того, величины AUC(0-12 ч) варьировались прямо пропорционально уровню дозы для обоих лекарственных препаратов. Вычисленные значения AUC(0-12 ч) для готового лекарственного продукта составляли от 43% до 80% для величин AUC(0-12 ч), вычисленных после введения разовой дозы лекарственного вещества, и от 36% до 71% при стационарном состоянии.
Порошкообразное лекарственное вещество Т1249 и готовый лекарственный продукт показали аналогичные фармакокинетические профили у собакоподобных обезьян после подкожного введения болюса при тестируемых уровнях и объемах доз. Прямое сравнение форм кривых зависимости концентрации в плазме от времени, полученных в настоящем исследовании, и форм кривых, полученных из предыдущих исследований, проводимых на собакоподобных обезьянах, дало основание предположить, что существует депо-эффект при введении 125I-T1249 путем подкожной инъекции. Это было подтверждено увеличением времени, при котором достигалась максимальная концентрация в плазме (tmax), и t1/2.
Эти результаты показали, что препарат порошкообразного лекарственного вещества, используемый в данной фармакологической программе, дает сравнимые величины AUC и других кинетических параметров по сравнению с величинами и параметрами, наблюдаемыми после введения готового лекарственного продукта. Эти наблюдения показали, что введение Т1249 в клинических условиях, в итоге, приводит к воздействию Т1249 на всех пациентов.
Объем настоящего изобретения не ограничивается конкретными вариантами его осуществления, описанными в настоящей заявке и представленными лишь в целях иллюстрации отдельных аспектов изобретения, а поэтому функционально эквивалентные методы и компоненты также входят в объем настоящего изобретения. Действительно, помимо модификаций, проиллюстрированных и описанных выше, в настоящее изобретение могут быть введены и различные другие модификации, которые являются очевидными из вышеизложенного описания и сопровождающих его чертежей. Такие модификации не должны выходить за рамки сформулированной ниже формулы изобретения.
ДОПОЛНЕНИЕ К ОПИСАНИЮ
Настоящее изобретение относится к коровым полипептидам, содержащим следующие аминокислотные последовательности:
YTSLIHSLIEESQNQQEKNEQELLELDK; LEENITALLEEAQIQQEKNMYELQKLNS;
LEANISQSLEQAQIQQEKNMYELQKLNS; NNYTSLIHSLIEESQNQQEKNEQELLEL;
DFLEENITALLEEAQIQQEKNMYELQKL; RYLEANISQSLEQAQIQQEKNMYELQKL;
RYLEANITALLEQAQIQQEKNEYELQKL; NNYTSLIHSLIEESQNQQEKNEQELLELDK;
TALLEQAQIQQEKNEYELQKLDK;
TALLEQAQIQQEKNEYELQKLDE;
TALLEQAQIQQEKNEYELQKLIE;
TALLEQAQIQQEKIEYELQKLDK;
TALLEQAQIQQEKIEYELQKLDE;
TALLEQAQIQQEKIEYELQKLIE;
TALLEQAQIQQEKIEYELQKLE;
TALLEQAQIQQEKIEYELQKLAK;
TALLEQAQIQQEKIEYELQKLAE;
TALLEQAQIQQEKAEYELQKLE;
TALLEQAQIQQEKNEYELQKLE;
TALLEQAQIQQEKGEYELQKLE;
TALLEQAQIQQEKAEYELQKLAK;
TALLEQAQIQQEKNEYELQKLAK;
TALLEQAQIQQEKGEYELQKLAK;
TALLEQAQIQQEKAEYELQKLAE;
TALLEQAQIQQEKNEYELQKLAE;
TALLEQAQIQQEKGEYELQKLAE;
DEFDASISQVNEKINQSLAFIRKSDELL;
DEYDASISQVNEKINQALAYIREADEL;
DEYDASISQVNEEINQALAYIRKADEL; DEFDESISQVNEKIEESLAFIRKSDELL;
DEFDESISQVNEKIEESLAFIRKSDEL; или QHWSYGLRPG.
В предпочтительном варианте осуществления коровый полипептид содержит аминокислотную последовательность TALLEQAQIQQEKNEYELQKLDK.
































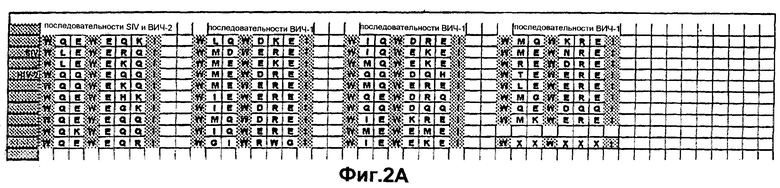
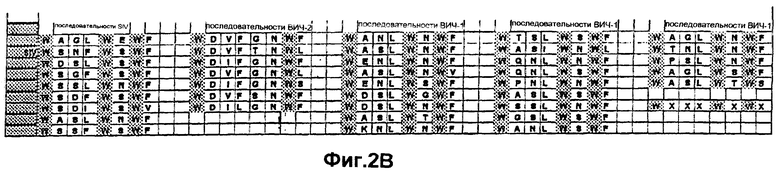

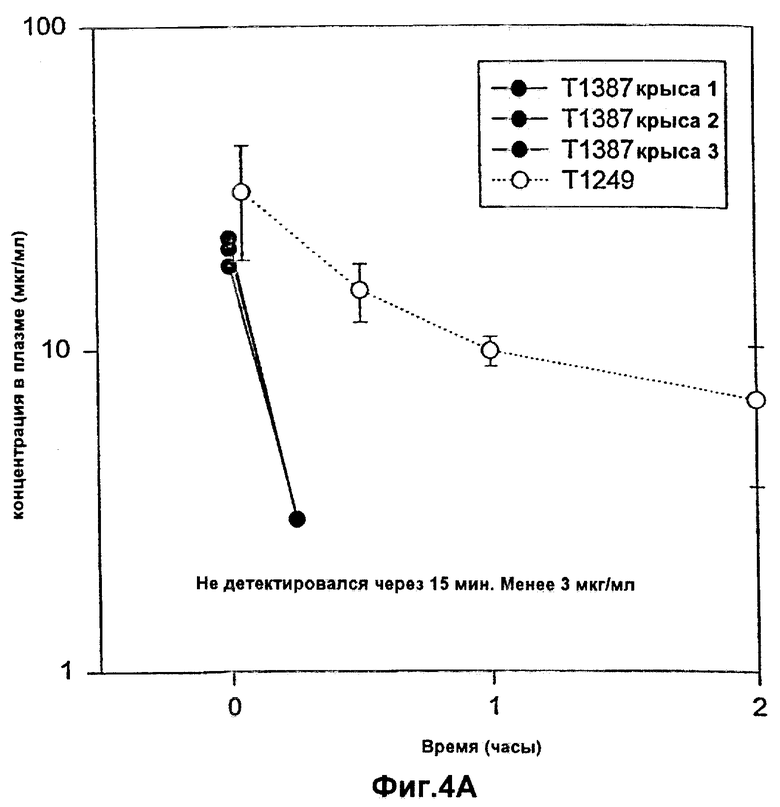


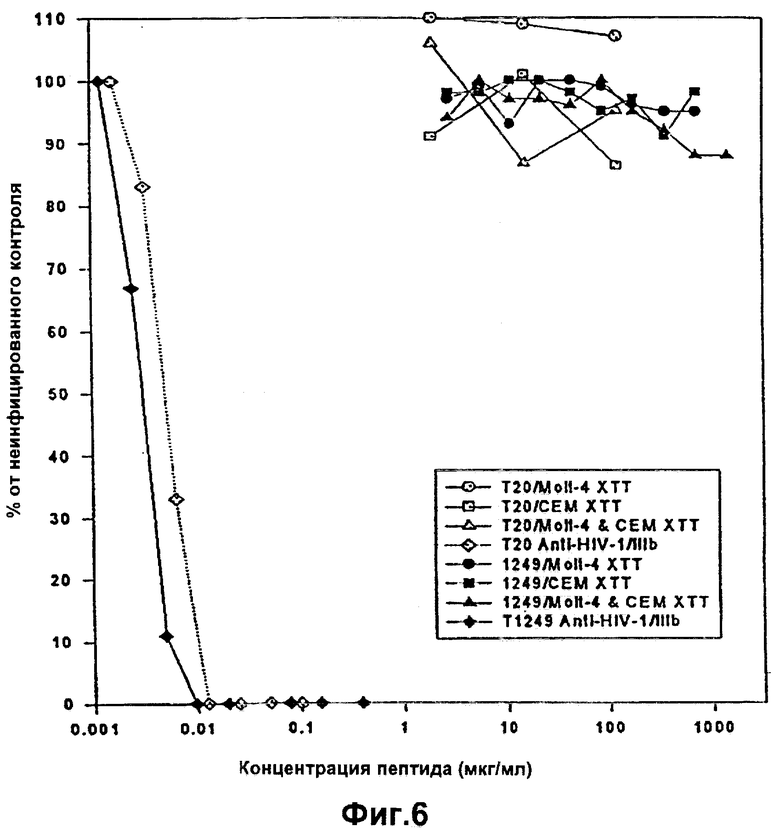


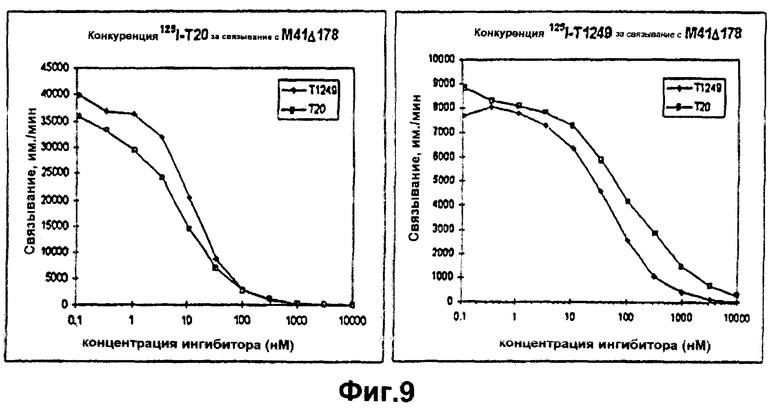
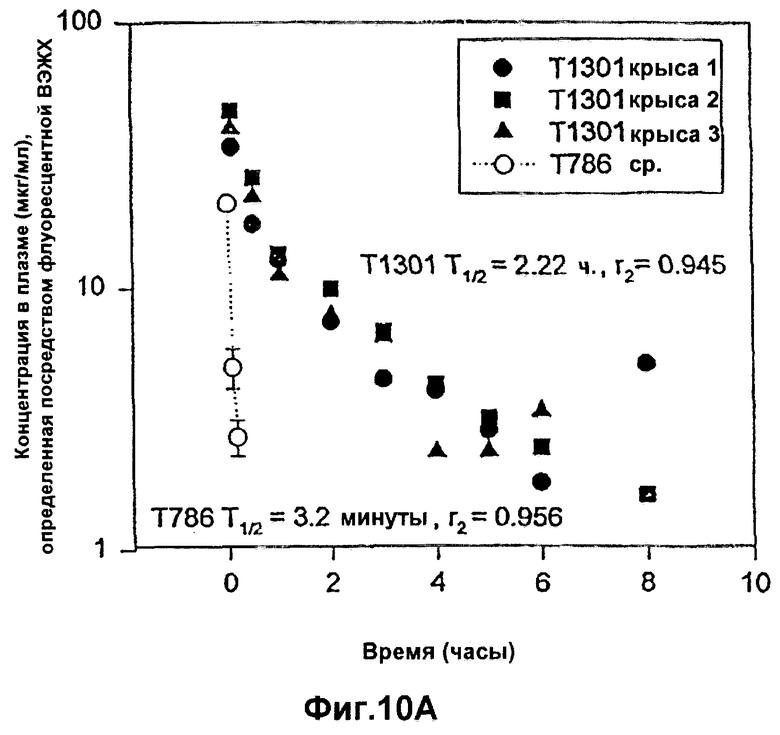
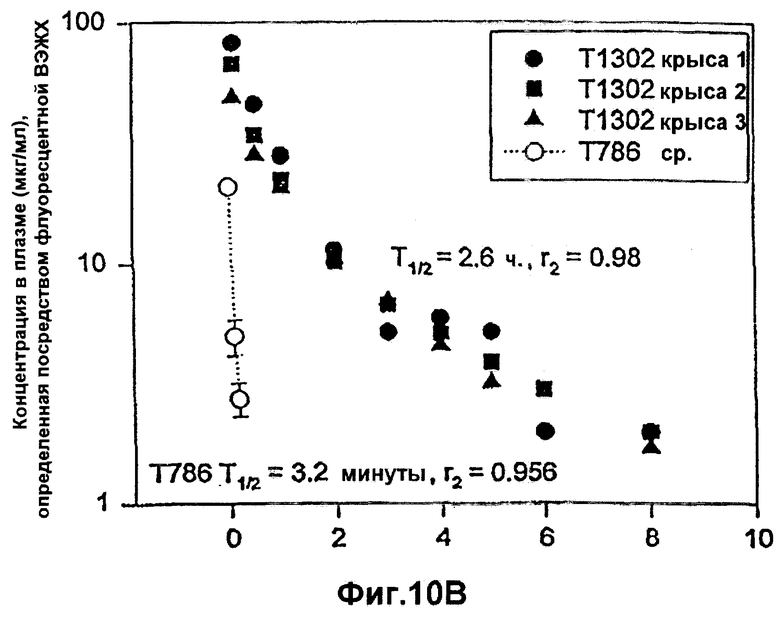


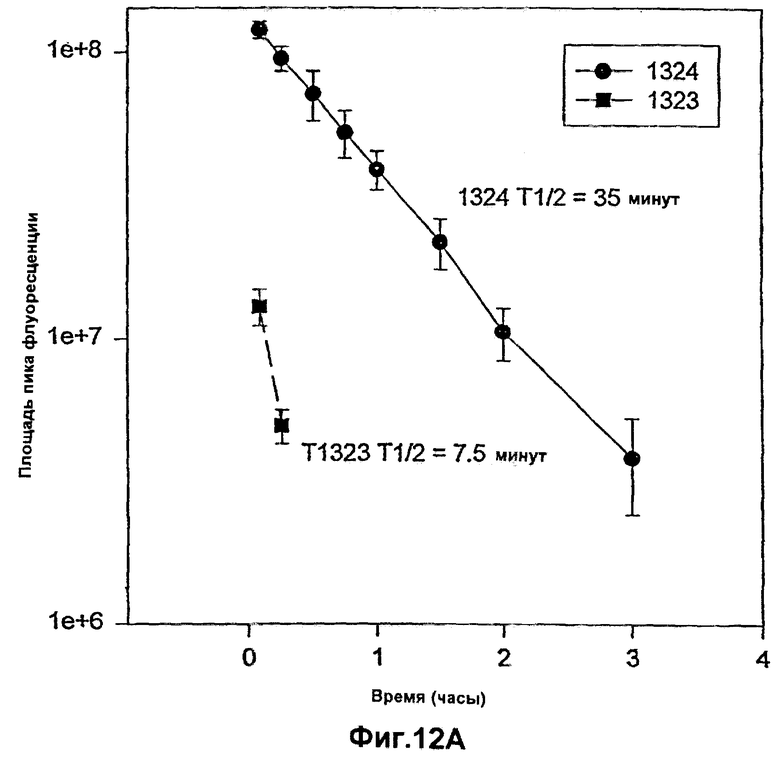
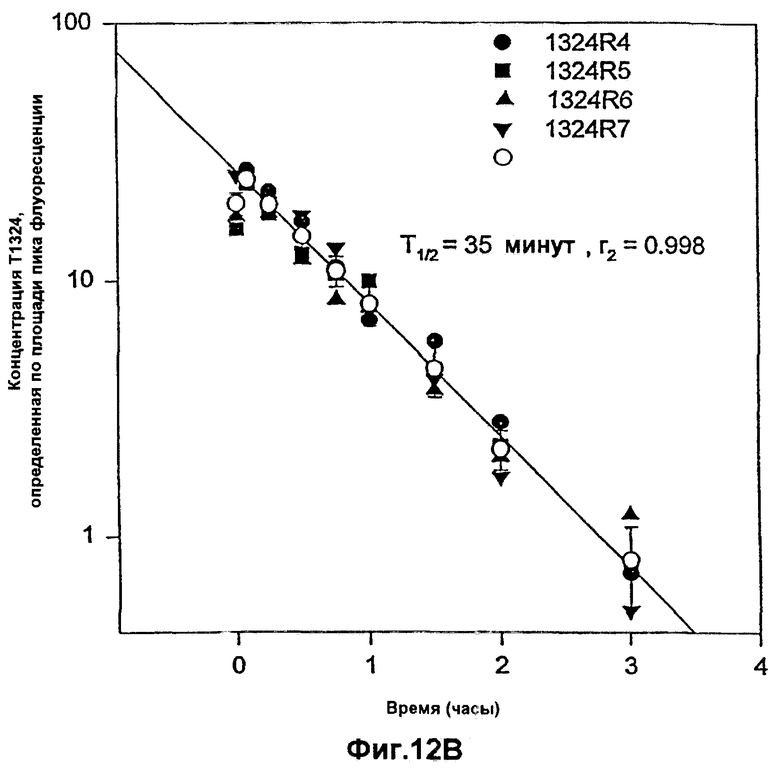


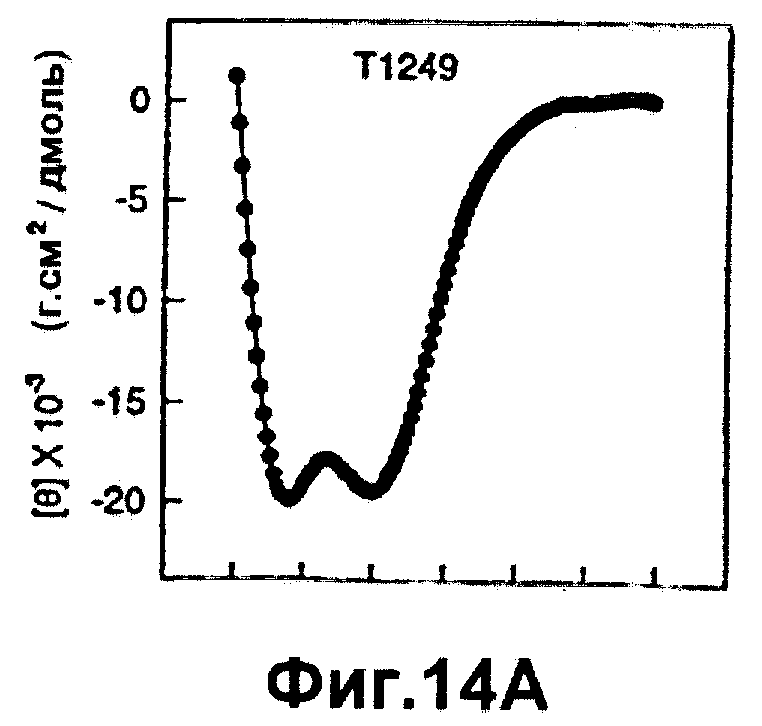

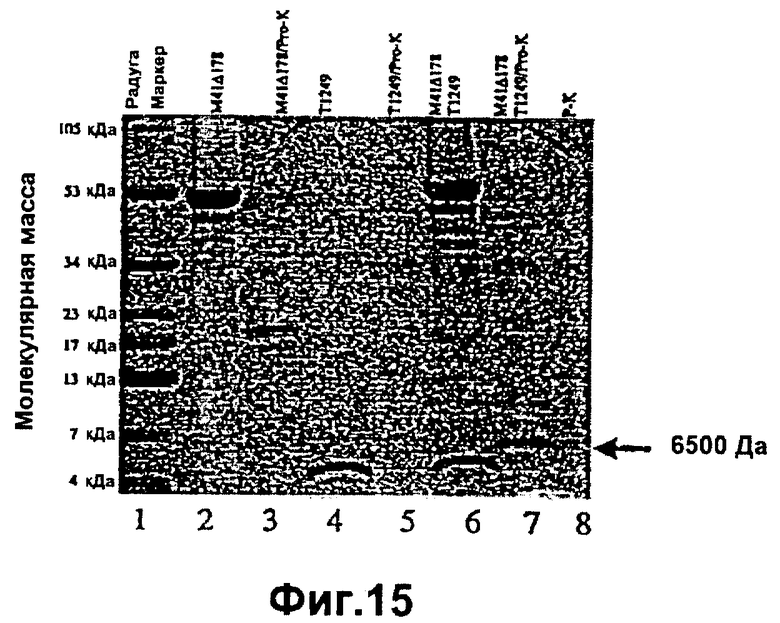

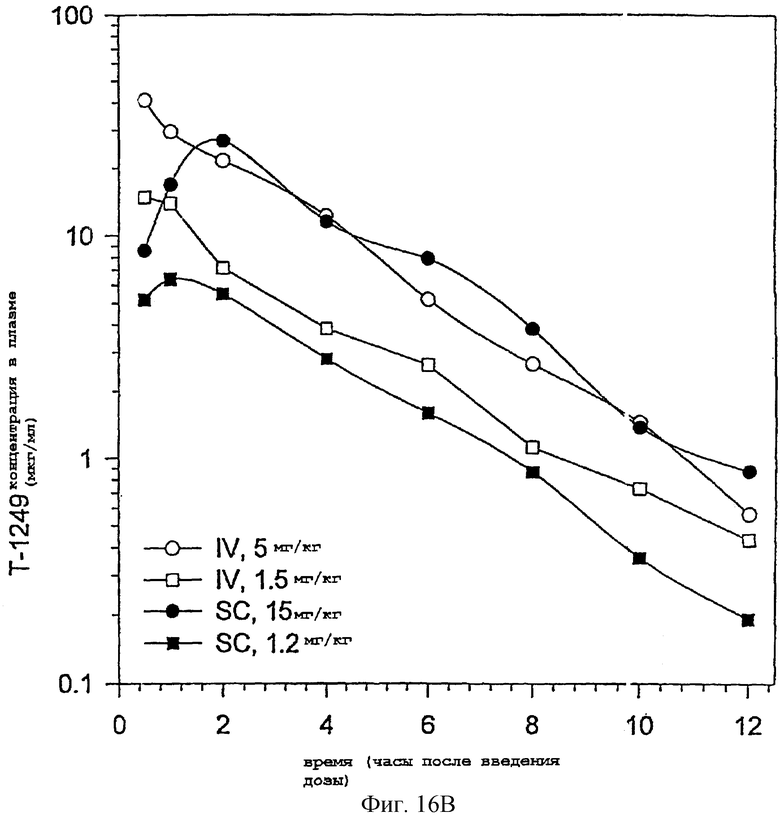
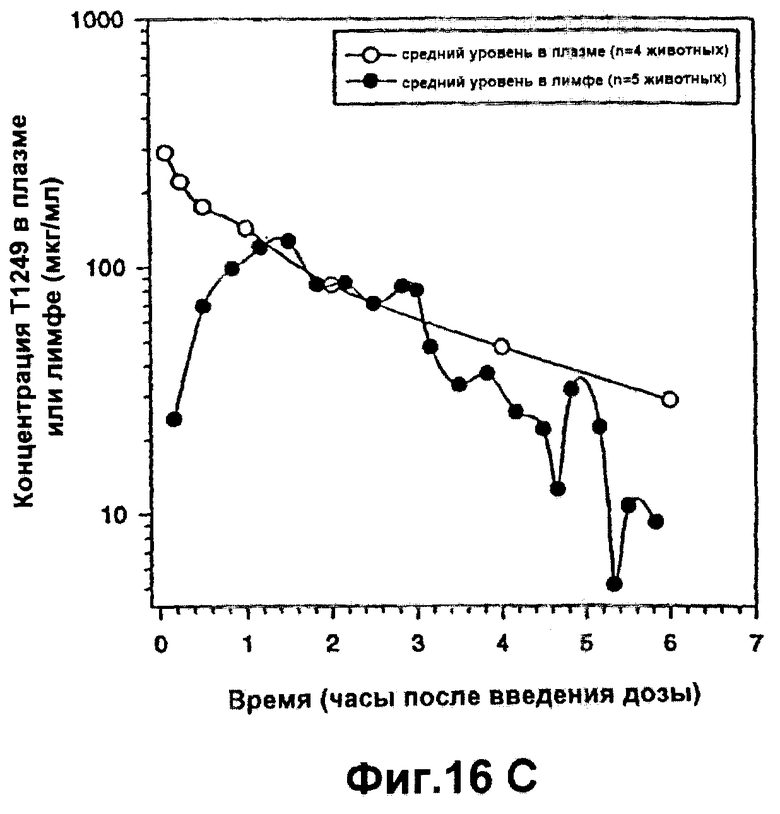
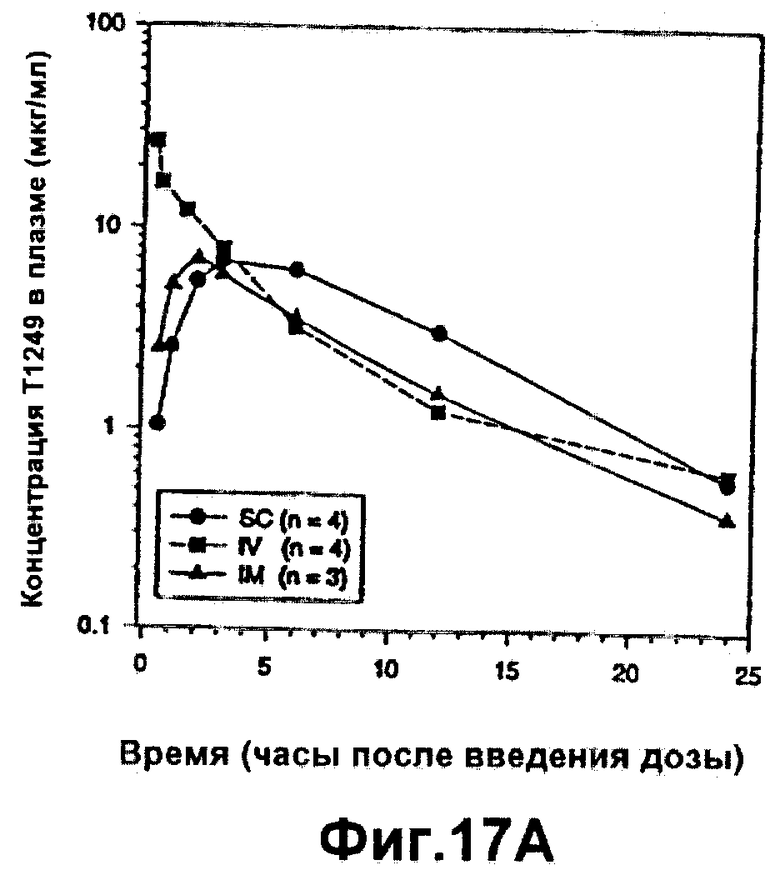
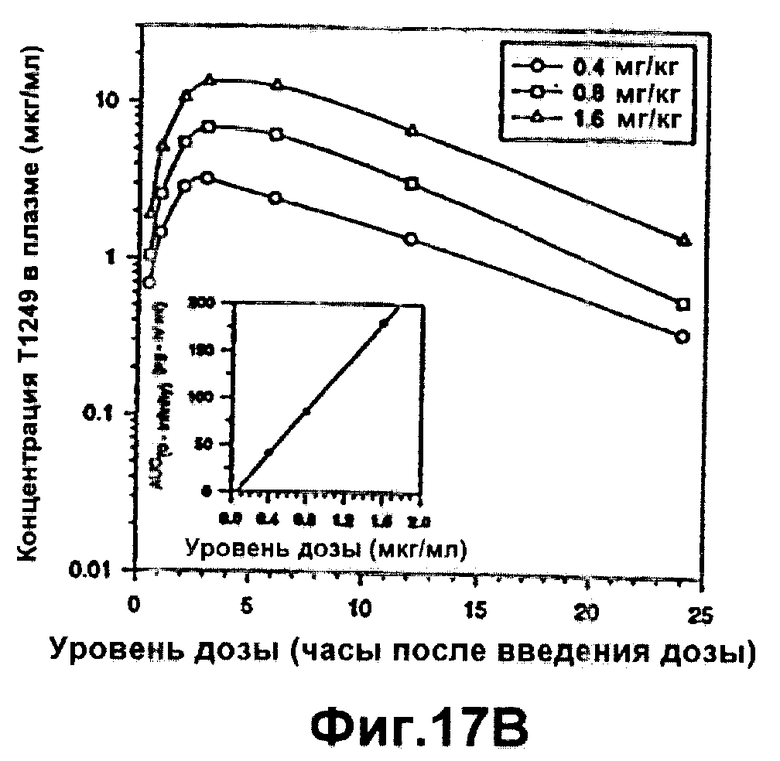
Настоящее изобретение относится к последовательностям пептида-усилителя, происходящим от различных последовательностей белка оболочки ретровируса (gp41), которые улучшают фармакокинетические свойства любого корового полипептида, к которому они присоединены, и основано на обнаружении того факта, что гибридные полипептиды, содержащие последовательности пептида-усилителя, присоединенные к коровому полипептиду, обладают улучшенными фармакокинетическими свойствами, такими как более продолжительное время полужизни. Настоящее изобретение, кроме того, относится к способу улучшения фармакокинетических свойств любого корового полипептида посредством присоединения указанных последовательностей пептида-усилителя к указанному коровому полипептиду. Коровыми полипептидами, используемыми для осуществления настоящего изобретения, могут быть любые фармакологически приемлемые пептиды, которые могут быть использованы, например, в качестве терапевтического или профилактического агента. Преимущество изобретения заключается в повышении эффективности биологически активных пептидов в кровотоке. 3 н. и 35 з.п.ф-лы, 13 табл., 26 ил.
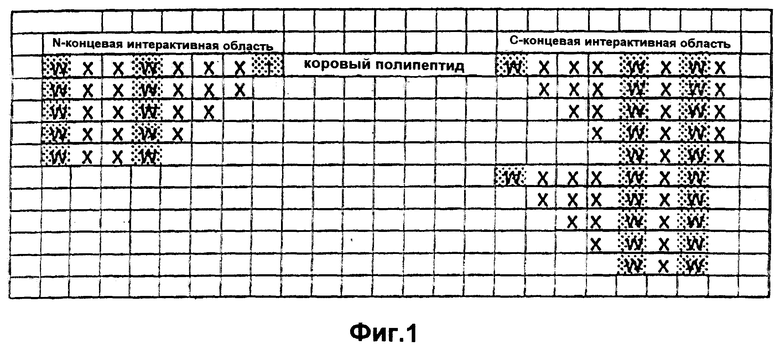
YTSLIHSLIEESQNQQEKNEQELLELDK; LEENITALLEEAQIQQEKNMYELQKLNS;
LEANISQSLEQAQIQQEKNMYELQKLNS; NNYTSLIHSLIEESQNQQEKNEQELLEL;
DFLEENITALLEEAQIQQEKNMYELQKL; RYLEANISQSLEQAQIQQEKNMYELQKL;
RYLEANITALLEQAQIQQEKNEYELQKL; NNYTSLIHSLIEESQNQQEKNEQELLELDK;
TALLEQAQIQQEKNEYELQKLDK;
TALLEQAQIQQEKNEYELQKLDE;
TALLEQAQIQQEKNEYELQKLIE;
TALLEQAQIQQEKIEYELQKLDK;
TALLEQAQIQQEKIEYELQKLDE;
TALLEQAQIQQEKIEYELQKLIE;
TALLEQAQIQQEKIEYELQKLE;
TALLEQAQIQQEKIEYELQKLAK;
TALLEQAQIQQEKIEYELQKLAE;
TALLEQAQIQQEKAEYELQKLE;
TALLEQAQIQQEKNEYELQKLE;
TALLEQAQIQQEKGEYELQKLE;
TALLEQAQIQQEKAEYELQKLAK;
TALLEQAQIQQEKNEYELQKLAK;
TALLEQAQIQQEKGEYELQKLAK;
TALLEQAQIQQEKAEYELQKLAE;
TALLEQAQIQQEKNEYELQKLAE;
TALLEQAQIQQEKGEYELQKLAE;
DEFDASISQVNEKINQSLAFIRKSDELL;
DEYDASISQVNEKINQALAYIREADEL;
DEYDASISQVNEEINQALAYIRKADEL; DEFDESISQVNEKIEESLAFIRKSDELL;
DEFDESISQVNEKIEESLAFIRKSDEL или
QHWSYGLRPG.
WQEWEQKITALLEQAQIQQEKNEYELQKLDKWASLWEWF
WQEWEQKITALLEQAQIQQEKIEYELQKLIEWEWF или
VYPSDEYDASISQVNEEINQALAYIRKADELLENV.
YTSLIHSLIEESQNQQEKNEQELLELDK;
LEENITALLEEAQIQQEKNMYELQKLNS;
LEANISQSLEQAQIQQEKNMYELQKLNS;
NNYTSLIHSLIEESQNQQEKNEQELLEL;
DFLEENITALLEEAQIQQEKNMYELQKL;
RYLEANISQSLEQAQIQQEKNMYELQKL;
RYLEANITALLEQAQOEKNEYELOKL;
NNYTSLIHSLIEESQNQQEKNEQELLELDK;
TALLEQAQIQQEKNEYELQKLDK;
TALLEQAQIQQEKNEYELQKLDE;
TALLEQAQIQQEKNEYELQKLIE;
TALLEQAQIQQEKIEYELQKLDK;
TALLEQAQIQQEKIEYELQKLDE;
TALLEQAQIQQEKIEYELQKLIE;
TALLEQAQIQQEKIEYELQKLE;
TALLEQAQIQQEKIEYELQKLAK;
TALLEQAQIQQEKIEYELQKLAE;
TALLEQAQIQQEKAEYELQKLE;
TALLEQAQIQQEKNEYELQKLE;
TALLEQAQIQQEKGEYELQKLE;
TALLEQAQIQQEKAEYELQKLAK;
TALLEQAQIQQEKNEYELQKLAK;
TALLEQAQIQQEKGEYELQKLAK;
TALLEQAQIQQEKAEYELQKLAE;
TALLEQAQIQQEKNEYELQKLAE;
TALLEQAQIQQEKGEYELQKLAE;
DEFDASISQVNEKINQSLAFIRKSDELL;
DEYDASISQVNEKINQALAYIREADEL;
DEYDASISQVNEEINQALAYIRKADEL;
DEFDESISQVNEKIEESLAFIRKSDELL;
DEFDESISQVNEKIEESLAFIRKSDEL или
QHWSYGLRPG.
YTSLIHSLIEESQNQQEKNEQELLELDK;
LEENITALLEEAQIQQEKNMYELQKLNS;
LEANISQSLEQAQIQQEKNMYELQKLNS;
NNYTSLIHSLIEESQNQQEKNEQELLEL;
DFLENNITALLEEAQIQQEKNMYELQKL;
RYLEANISQSLEQAQIQQEKNMYELQKL;
RYLEANITALLEQAQIQQEKNEYELQKL;
NNYTSLIHSLIEESQNQQEKNEQELLELDK;
TALLEQAQIQQEKNEYELQKLDK;
TALLEQAQIQQEKNEYELQKLDE;
TALLEQAQIQQEKNEYELQKLIE;
TALLEQAQIQQEKIEYELQKLDK;
TALLEQAQIQQEKIEYELQKLDE;
TALLEQAQIQQEKIEYELQKLIE;
TALLEQAQIQQEKIEYELQKLE;
TALLEQAQIQQEKIEYELQKLAK;
TALLEQAQIQQEKIEYELQKLAE;
TALLEQAQIQQEKAEYELQKLE;
TALLEQAQIQQEKNEYELQKLE;
TALLEQAQIQQEKGEYELQKLE;
TALLEQAQIQQEKAEYELQKLAK;
TALLEQAQIQQEKNEYELQKLAK;
TALLEQAQIQQEKGEYELQKLAK;
TALLEQAQIQQEKAEYELQKLAE;
TALLEQAQIQQEKNEYELQKLAE;
TALLEQAQIQQEKGEYELQKLAE;
DEFDASISQVNEKINQSLAFIRKSDELL;
DEYDASISQVNEKINQALAYIREADEL;
DEYDASISQVNEEINQALAYIRKADEL; DEFDESISQVNEKIEESLAFIRKSDELL;
DEFDESISQVNEKIEESLAFIRKSDEL или
QHWSYGLRPG.
| Способ получения циклопропилацетилена | 1972 |
|
SU578293A1 |
| Огнетушитель | 0 |
|
SU91A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |

