Перекрестная ссылка на родственные заявки
Данная заявка притязает на приоритет китайской заявки на патент № 01130985.7, поданной 29 августа 2001 г., в настоящее время китайский патент № ZL 01130985.7, заявки РСТ/CN02/99405, поданной 6 июня 2002 г., содержание которых полностью включено в данное описание изобретения в качестве ссылки.
Область техники
Настоящее изобретение относится к ингибитору слияния, который можно использовать для лечения ВИЧ-инфекции.
Предпосылки изобретения
1. ВИЧ и эпидемия СПИДа
Инфицирование вирусом иммунодефицита человека (ВИЧ), который является патогенным ретровирусом, может вызвать синдром приобретенного иммунодефицита (СПИД) (Barre-Sinossi, F. et al., 1983, Science 220:868-870). Несмотря на то что макрофаги, нейроны и другие клетки могут быть инфицированы ВИЧ (Maddon et al., 1986, Cell 47:333-49), главными клетками-мишенями для ВИЧ являются лимфоциты CD4+ (Dalgleish, A. et al., 1984, Nature 312:767-8), так как ВИЧ обладает сильным сродством к молекулам CD4 на поверхности клеток CD4+. ВИЧ-инфекция, попавшая в организм человека, разрушает так много лимфоцитов CD4+, что организм начинает утрачивать иммунную функцию, в результате чего больной СПИДом становится восприимчивым к разным инфекциям, нарушениям функции нервных клеток, опухолям и другим заболеваниям. Больные, у которых возникают указанные симптомы, в конце концов умирают (edited by Levy, J.A.: Acute HIV infection and susceptible cells, published in USA, 2000, Page 63-78).
Из-за тяжести симптомов и высокого коэффициента смертности эпидемическое распространение СПИДа стало одной из главных причин, угрожающих здоровью человека. В настоящее время число ВИЧ-инфицированных людей во всем мире составило в общей сложности 57900000. 21800000 человек умерли от СПИДа за последнее десятилетие. Установлено, что в 2000 г. были ВИЧ-инфицированы 5300000 человек. В Китае наблюдается быстрое распространение ВИЧ-инфекции. По оценке экспертов, в 2000 г. число людей с положительной реакцией на ВИЧ превысило 800000-1000000, причем в указанное число входят как взрослые, так и дети (отчет ВОЗ за 2000 г., UNAIDS and WHO).
В настоящее время обнаружены по крайней мере два типа ВИЧ: ВИЧ-1 (Gallo, R. et al., 1984, Science 224:500-503) и ВИЧ-2 (Clavel, F. et al., 1986, Science 223:343-346). Оба типа вируса обладают высокой генетической гетерогенностью. Только ВИЧ-1 имеет не менее 11 разных генотипов (подтипы A-J и О) (Jonassen, T.O. et al., 1997, Virol. 231:43-47). Подтип Е ВИЧ-1 распространен главным образом в Центральной Африке, Таиланде, Индии, Вьетнаме, Кампучии, Малайзии, Бирме, Китае и западном полушарии (отчет ВОЗ за 1996 г.). Подтипы ВИЧ, обнаруженные в Китае, представляют в основном подтипы В, Е или С (Yu, E.S. et al., 1996, American J. Public Health 86(8 Pt1): 1116-22).
Репродуктивный цикл ВИЧ включает несколько важных стадий. Сначала гликопротеин gp120 вирусной оболочки прикрепляется к мембране клетки-хозяина посредством специфического связывания с молекулой CD4, находящейся на поверхности лимфоцита Т4. При помощи корецептора хемокина оболочка вируса сливается с мембраной клетки-хозяина (Berger, E.A., et al., 1999, Ann. Rev. Immunol., 17:657-700). После слияния вирион ВИЧ, упакованный в нуклеокапсид, проникает в клетку-хозяина, при этом капсид разрушается и выделяется нуклеиновая кислота вируса. Обратная транскриптаза вируса катализирует транскрипцию одноцепочечной РНК ВИЧ в одноцепочечную ДНК, которая затем трансформируется в двухцепочечную ДНК в результате катализа клеточной полимеразой. Двухцепочечная ДНК может свободно существовать в цитоплазме или встраиваться в виде провируса в хромосомную ДНК хозяина в результате катализа вирусной интегразой, вследствие чего происходит латентное инфицирование ВИЧ (Roe, T. et al., 1997, J.Virol. 71(2):1334-40). Провирус, который нельзя вырезать из хромосомы хозяина, является очень устойчивым и репродуцируется вместе с репликацией хромосомы хозяина. После трансляции мРНК ВИЧ в полипротеин большой длины вирусные протеазы разрезают данный полипротеин и процессируют его с образованием структурных белков зрелого вируса (Xiang, Y. & Leis, J., 1997, J.Virol. 71(3):2083-91). Затем происходит сборка указанных структурных белков и нуклеиновых кислот ВИЧ с образованием новых вирусных гранул, которые высвобождаются из клетки путем баддинга (Kiss-Lazozlo, Hohn, T., 1996, Trends in Microbiology 4(12):480-5).
Основные стадии репликации ВИЧ можно кратко охарактеризовать следующим образом: 1) прикрепление к клеточной оболочке и проникновение в клетку-хозяин в результате процесса слияния; 2) обратная транскрипция и встраивание в хромосому; 3) трансляция и процессинг белка; 4) сборка и высвобождение вируса.
2. Лечение ВИЧ-инфекции
Несмотря на большие многолетние усилия, направленные на создание эффективных способов лечения и профилактики, до сих пор не существует действенной вакцины или способа лечения СПИДа.
Идеальная вакцина должна быть безвредной и способной индуцировать нейтрализующие антитела, а также устойчивые иммунные реакции в слизистой оболочке и крови (Levy, J.A., and Levy, J.A., 1988, Trens Med. Rev. 2:265-71). Многие вакцины против ВИЧ, создаваемые в настоящее время в мире, все еще находятся на стадиях экспериментов на животных. Хотя вакцины против белков gp160 и gp120 оболочки ВИЧ уже прошли первую, вторую или третью стадии клинических испытаний, результаты таких испытаний являются неутешительными. Многие вакцины, которые эффективно предотвращают инфицирование ВИЧ у лабораторных животных, оказываются неэффективными для человека (McElrath, M.J. et al., 1996, Pro. Natl. Acad. Sci. USA 93:3972-77). То, что ученые не добились значительных успехов в создании вакцины против ВИЧ, можно объяснить сложностью и вариабельностью генетических веществ ВИЧ (Bloom, B.R., 1996, Science 272:1888-1900).
Лекарственные средства против СПИДа, нашедшие признание во всем мире, можно классифицировать в две категории: ингибиторы обратной транскриптазы ВИЧ (Charles, C.J. et al., 1996, JAMA 276:146) и ингибиторы протеазы ВИЧ (Miles, S.A. et al., International AIDS Society USA 4(3):15). Лекарственные средства, относящиеся к обеим категориям, воздействуют на более поздние стадии репликации ВИЧ-инфекции - транскрипцию и сборку новых вирусов. Хорошо известная "смешанная терапия" представляет комбинированное лечение с использованием ингибиторов обоих типов (Lafeuillade, A. et al., 1997, J. Infect. Dis. 175:1051-55).
Ингибиторы обратной транскриптазы, включающие AZT, ddI, ddC, 3TC, d4Т и т.д., рано или поздно вызывают лекарственную устойчивость, в результате чего вирусы становятся менее восприимчивыми к указанным лекарственным средствам, поэтому эффективные ингибирующие концентрации лекарственных средств увеличиваются в несколько раз или даже в десятки раз (Vella, S. and Floridia, M., 1996, International AIDS Society USA 4(3): 15). Подобная лекарственная устойчивость обусловлена высокой частотой мутаций ВИЧ. В организме человека один вирус ВИЧ может ежедневно продуцировать 108-1010 новых вирусных гранул, при этом частота мутаций равна 3×105 в одном цикле репликации. Множество миссенс-мутаций, воздействующих на экспрессию аминокислот, может произойти в регуляторных генах и белках оболочки. В некоторых штаммах ВИЧ частота мутаций может охватывать до 40% аминокислотных последовательностей определенных генов (Myers, G. and Montaner. J.G., 1992, The Retroviridae vol.1, Plenum Press, New York 51-105). В результате этого ингибиторы обратной транскриптазы вызывают лекарственную устойчивость, облегчая пролиферацию устойчивых штаммов, которые появляются до и после мутаций наряду с контрольными чувствительными вирусными штаммами.
Кроме того, все ингибиторы обратной транскриптазы характеризуются специфической токсичностью в зависимости от дозы. Возникающие симптомы включают подавление функции спинного мозга, рвоту, нарушение функции печени, мышечную слабость, заболевания периферической нервной системы и воспаление поджелудочной железы. Многим субъектам приходится приостанавливать лечение из-за непереносимых побочных эффектов (Fischl, M.A., et al., 1987, N. Engl. J. Med. 317:185-91; Lenderking, W.R., et al., 1994, N. Engl. J. Med. 330:738-43).
Лекарственная устойчивость также является важной проблемой при использовании ингибиторов протеазы. Мутации гена вирусной протеазы вызывают лекарственную устойчивость при использовании всех ингибиторов протеазы, применяемых в настоящее время для лечения СПИДа (Condra, J.H. et al., 1995, Nature 374:569-71). Побочные эффекты, вызываемые ингибиторами протеазы, включают нарушение функции печени, расстройство желудочно-кишечного тракта, почечно-каменную болезнь, онемение тканей вокруг рта, нарушение липидного обмена и психическое расстройство (Deeks, et al., 1997, JAMA 277:145-53).
Большинство используемых в настоящее время лекарственных средств против ВИЧ являются высокотоксичными и вызывают лекарственную устойчивость. Поэтому по-прежнему существует непреодолимое препятствие при лечении ВИЧ-инфекции. Несомненно, существует острая нужда в новых лекарственных средствах для лечения ВИЧ-инфекции, характеризующихся большей эффективностью и меньшей токсичностью.
Необходимы новые лекарственные средства, направленно воздействующие на разные стадии цикла репликации ВИЧ. После фундаментального исследования ВИЧ и СПИДа недавно были созданы несколько лекарственных средств против СПИДа с новыми механизмами действия. Указанные лекарственные средства включают некоторые новые ингибиторы обратной транскриптазы ВИЧ и ингибиторы протеазы ВИЧ, а также некоторые новые лекарственные средства против ВИЧ, направленно воздействующие на другие указанные здесь мишени (De, C.E., 2000, Rev. Med. Virol. 10(4):255-77).
1) Абсорбенты вирусов, такие как лаурилсульфат натрия, сульфат декстрозы и гепарин, могут препятствовать сцеплению gp120 на оболочке ВИЧ с лимфоцитом благодаря воздействию полианионных групп. Однако указанные абсорбенты характеризуются плохой специфичностью и высокой токсичностью. Некоторые из них даже могут увеличивать вирусный потенциал (Baba, M. et al., 1988. Pro. Natl. Acad. Sci. USA, 85:6132-6).
2) Растворимые CD4 используют для предотвращения связывания gp120 с клетками-хозяевами. Некоторые рекомбинантные растворимые CD4 могут связывать вирусные гранулы до контактирования gp120 с молекулами CD4 на клеточной мембране и предотвращать распространение ВИЧ-инфекции. Однако указанные рекомбинантные растворимые CD4 не оказывают выраженного воздействия на штаммы ВИЧ-1, выделенные у некоторых пациентов. Кроме того, клинические эксперименты не позволили получить надежные данные, свидетельствующие об их антивирусной активности (Gomatos, P.J. et al., 1990, J.Immunol. 144:4183-9).
3) Хемокины и их аналоги, включающие RANTES, MIP-1α, MIP-1β, связывающийся с CCR5, и SDF, связывающийся с CXCR4, можно использовать для предотвращения проникновения ВИЧ в клетки-хозяева. Указанные средства могут не только конкурентно блокировать сцепление gp120 ВИЧ с клеточными корецепторами хемокина, но и ограничивать участки проникновения ВИЧ, подавляя экспрессию указанного корецептора на клетке. Самые последние блокаторы корецептора хемокина включают положительно заряженные короткие пептиды, такие как ALX40-4C и Т22, и соединения, такие как AMD3100, ТАК-779 и трихосантин.
4) Хотя растворимый CD4-IgG может подавлять репликацию ВИЧ in vitro, указанное вещество не демонстрирует надежную антивирусную активность при выполнении клинических испытаний.
5) Лекарственные средства, такие как 2,2'-дитио-бис-бензамиды (DIBA) и азадикарбонамид (ADA), могут блокировать сборку и распад вирусов в результате взаимодействия с цинксодержащим пальцеобразным сайтом NCp7.
6) Сегмент gp41 или его аналог можно использовать в качестве ингибитора слияния. Например, Т-20 способен блокировать проникновение вируса в клетку (Jiang, S. et al., 1993, Nature 365:113).
7) Ингибиторы транскриптазы вирусной мРНК, такие как CGP64222, фторхинолон К-12 и ЕМ2487.
8) Ингибиторы интегразы, такие как производные карбонила J [N,N'-бис-(2-(5-гидрокси-7-нафталинсульфоновая кислота)мочевины], могут предотвращать встраивание генома ВИЧ в геном лимфоцита хозяина (Maurer K., et al., 2000, Bioorg Chem 28(3): 140-155).
3. Ингибиторы слияния, блокирующие проникновение вируса в клетки
Многие биологические процессы включают слияние мембран. В эукариотических клетках постоянно происходит слияние клеточных мембран, включая эндоцитоз, секрецию, рециркуляцию мембранных компонентов и так далее (White, J. M., 1992, Science 258:917-24). Примеры слияния в некоторых конкретных клетках включают секрецию гормона регулируемого слияния, фермента и нервных медиаторов. Некоторые более известные примеры включают слияние зародышевых клеток и мышечных клеток.
В соответствии с одним вариантом осуществления настоящего изобретения лекарственное средство, препятствующее слиянию клеток или слиянию мембран, представляет вещество, которое ингибирует или подавляет слияние двух или большего числа биологических мембран. В соответствии с одним вариантом осуществления настоящего изобретения две или большее число биологических мембран являются клеточными или вирусными структурами, такими как клеточная мембрана и оболочка вируса. В соответствии с одним вариантом осуществления настоящего изобретения антивирусное средство представляет соединение, которое ингибирует инфицирование клеток вирусом, например, ингибирует слияние вируса с клеткой или слияние клеток. В соответствии с одним вариантом осуществления настоящего изобретения инфицирование представляет слияние мембран, например, при инфицировании клеток вирусом с оболочкой, и другие процессы, аналогичные слиянию вирусов с клетками, которые происходят во время бактериальной конъюгации.
И наконец, слияние мембран является главной стадией процесса воздействия вируса с оболочкой на клетки-хозяева и проникновения в указанные клетки (Weissenhorn, W. et al., 1997, Nature 387:426-30). Лекарственное средство против ВИЧ по настоящему изобретению, в частности фузонекс и его производные, представляет блокатор слияния, предотвращающий проникновение вирусов в клетки-хозяева.
Процесс слияния контролируется гликопротеинами в оболочке ВИЧ. Предшественником гликопротеинов является gp160, содержащий полисахаридные группы. В процессе репродукции вируса gp160 гидролизуется определенной протеазой с образованием двух субъединиц: gp120, находящегося снаружи оболочки, и gp41, являющегося трансмембранным белком. После гидролиза gp120 и gp41 все еще остаются связанными нековалентными связями и полимеризованными в виде тримеров снаружи вирусной гранулы. Трансмембранный белок gp41, эктодомен которого имеет высокоспиральную структуру, обладает очень эффективным механизмом инициации слияния мембран и известен как главная молекула, открывающая доступ в клетки благодаря ее непосредственному участию в процессе слияния клеточных мембран (Ferrer, M., et al., 1999, Nat. Struct. Biol. 6(10):953-60; Zhou, G., et al., 2000, 1:Bioorg. Med. Chem. 8(9):2219-27).
Анализ дифракции на кристалле продемонстрировал, что при слиянии вирусов с клетками ядро gp41 состоит из шести спиральных пучков, в которых N-концевая и С-концевая спирали образуют три "шпильки", которые крепят оболочку ВИЧ к клеточной мембране. Хотя тример gp41 может образовывать отверстие для слияния, которое облегчает проникновение вируса в клетку-хозяин (Chan, D.C. et al., 1997, Cell 89:263-73), он существует в виде неустойчивой естественной неслитой конформации на поверхности свободной вирусной гранулы, только что высвободившейся из инфицированных клеток. Сначала N-концевая спираль завертывается внутрь С-концевой спирали, благодаря чему скрывается N-концевая область слияния, затем после объединения gp120 на поверхности вируса с рецептором CD4 и корецептором хемокина на клеточной мембране происходит активируемое рецептором конформационное изменение gp41, в результате которого N-конец проникает с поверхности вируса в клеточную мембрану хозяина. В это время gр41 трансформируется из неустойчивой естественной неслитой конформации в промежуточную конформацию "прошпильки". Когда С-концевой и N-концевой пептиды gp41 связываются друг с другом, обнажается гидрофобное N-концевое ядро тримерной структуры и промежуточная "прошпилька" трансформируется в более энергостабилизированную конформацию "шпильки", и к этому времени оболочка вируса сливается с клеточной мембраной (Jones, P.L. et al., 1998, J. Biol. Chem. 273:404).
Впервые обнаруженный ингибитор слияния представляет пептид, содержащий 36 аминокислот и выделенный из С-конца (127-162) gp41, который получил название Т-20 и имеет нижеследующую последовательность:
X-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-Z
Структурное сходство Т-20 с С-концом gp41 позволяет ему конкурировать с С-концевым пептидом gp41 за связывание с N-концевой областью слияния. Т-20, находящийся на поверхности Т-клетки в очень низкой концентрации, может препятствовать слиянию gp41 ВИЧ с клеточной мембраной хозяина (значение IC50 находится в диапазоне нМ) (Jiang, S. et al., 1993, Nature 365:113; Wild, C.T. et al., 1994, Pro. Natl. Acad. Sci. USA 91:9770-74). В состоянии "прошпильки", которое сохраняется в течение многих минут, Т-20 эффективно препятствует связыванию С-концевого пептида gp41 с N-концевой областью слияния, блокируя таким образом образование "шпильки" между оболочной вируса и клеточной мембраной (Kliger, Y. and Shai, Y., 2000, J. Mol. Biol. 295:163-8).
Так как ингибитор слияния действует на клеточной мембране, не нужно обеспечивать его проникновение внутрь клеток для выполнения указанной функции. В отличие от этого все лекарственные средства против ВИЧ, применяемые в настоящее время в клинической практике, действуют на средней или поздней стадии инфицирования вирусом клеток-хозяев, из чего следует, что их нужно сначала ввести в клетки для подавления репродукции проникшего в клетку ВИЧ. Кроме того, высококонсервативная аминокислотная последовательность гидрофобного ядра gp41 позволяет предположить, что, по-видимому, вирус будет не в состоянии выработать лекарственную устойчивость к ингибиторам слияния. Результаты экспериментов in vitro показывают, что Т-20 может специфически блокировать проникновение ВИЧ в клетки. С другой стороны, на первой и второй стадиях клинических испытаний было установлено, что больные СПИДом хорошо переносят введение Т-20. Т-20 не оказывает токсического воздействия на спинной мозг, и большинство побочных эффектов выражены в низкой или средней степени. При суточной дозе, равной 200 мг, Т-20 может существенно уменьшить ВИЧ-потенциал у большинства субъектов, причем у 30% субъектов вирусный потенциал снижается ниже обнаруживаемого уровня (менее 400/мл). Кроме того, Т-20 эффективно воздействует на ВИЧ-инфицированных субъектов, у которых уже возникла лекарственная устойчивость. В научной литературе приведены данные об увеличении количества клеток CD4+ у некоторых субъектов после введения Т-20 (Kilby, J.M. et al., 1998, Nat. Med. 4:1302-1307). Некоторое беспокойство вызывал тот факт, что при длительном введении Т-20 может индуцировать образование специфических антител против Т-20, в результате чего у больных СПИДом появится устойчивость к Т-20. Тем не менее, при выполнении эксперимента в течение нескольких недель Т-20 сохранял антивирусную активность на протяжении всего времени (Pilcher, C.D. et al., 1999, AIDS 13(15): 2171-4).
По сравнению с ингибиторами обратной транскриптазы и ингибиторами протеазы ВИЧ, применяемыми в настоящее время в клинической практике, ингибитор слияния Т-20 обладает такими преимуществами, как более высокая эффективность, более низкая токсичность и отсутствие лекарственной устойчивости. Однако клиническая доза Т-20, равная 200 мг в сутки, является показателем плохой устойчивости указанного средства и низкого противодействия слиянию. Кроме того, из-за высокой дозы Т-20 вызывает некоторые местные реакции у некоторых субъектов (Kilby, J.M. et al., 1998, Nat. Med. 4:1302-1307).
Краткое изложение существа изобретения
Настоящее изобретение относится к ингибиторам слияния, обладающим высокой устойчивостью и высоким потенциалом лечения ВИЧ-инфекции. Ингибиторы слияния согласно настоящему изобретению можно использовать при комбинированном лечении СПИДа, при изготовлении лекарственных средств или в других случаях. По сравнению с Т-20 ингибиторы согласно настоящему изобретению обладают более высокой эффективностью, характеризуются более низкими дозами введения и, как следствие этого, более низкой токсичностью.
В соответствии с одним вариантом осуществления настоящего изобретения ингибитор слияния представляет специфический пептид, содержащий 36 аминокислотных остатков, к обоим концам которого добавлены концевые кэппирующие группы.
Настоящее изобретение относится к нижеследующим технологическим схемам.
В соответствии с предпочтительным вариантом осуществления настоящего изобретения ингибитор слияния представляет пептид, выделенный из трансмембранного гликопротеина gp41 ВИЧ. В соответствии с другим предпочтительным вариантом осуществления настоящего изобретения указанный пептид имеет аминокислотную последовательность, представленную в SEQ ID NO:1 и приведенную ниже:
X-SWETWEREIENYTKQIYKILEESQEQQDRNEKDLLE-Z (SEQ ID NO:1)
В указанном пептиде (именуемом также "фузонекс" в приводимом ниже описании изобретения):
Х представляет аминогруппу или -Х1-Х2, где Х1 означает иминогруппу и Х2 означает ацетильную группу, гидрофобную группу или макромолекулярную несущую группу, причем гидрофобная группа предпочтительно является карбобензоксигруппой, дансильной группой, трет-бутилоксикарбонильной группой или 9-флуоренилметилоксикарбонильной группой; макромолекулярная несущая группа является хелатным комплексом липид - жирная кислота, полиэтиленгликолем или углеводом;
Z представляет карбоксильную группу или -Z1-Z2, где Z1 означает карбонильную группу и Z2 означает аминогруппу, трет-бутилоксикарбонильную группу, гидрофобную группу или макромолекулярную несущую группу.
Используемые в данном описании изобретения однобуквенные коды, служащие для обозначения аминокислотных остатков, имеют нижеследующие значения:
В соответствии с предпочтительным вариантом осуществления настоящего изобретения Х2, или Z2, либо оба вместе означают гидрофобную группу. Гидрофобная группа представляет карбобензоксигруппу, дансильную группу, трет-бутилоксикарбонильную группу или 9-флуоренилметилоксикарбонильную группу.
В соответствии с альтернативным вариантом осуществления настоящего изобретения Х2, или Z2, либо оба вместе означают макромолекулярную несущую группу. Макромолекулярная несущая группа представляет хелатный комплекс липид - жирная кислота, полиэтиленгликоль или углевод.
В другом предпочтительном варианте осуществления настоящего изобретения вышеуказанный Х2 означает ацетильную группу и Z2 означает аминогруппу.
В соответствии с одним вариантом осуществления настоящего изобретения композиция, предназначенная для комбинированного лечения СПИДа, содержит указанный ингибитор и по крайней мере один компонент, выбранный из группы, включающей ингибиторы обратной транскриптазы, ингибиторы вирусной протеазы, ингибиторы гликозидазы, ингибиторы кэппирования вирусной мРНК, амфотерицин В, связующие молекулы кастаноспермина сложноэфирной связи, обладающие анти-ВИЧ активностью, гидроксимочевину, α-интерферон, β-интерферон и γ-интерферон.
В соответствии с предпочтительным вариантом осуществления настоящего изобретения ингибитор обратной транскриптазы является по крайней мере одним веществом, выбранным из группы, включающей AZT(3'-азид-2',3'-дидезоксицитидин), ddI (2',3'-дидезоксиинозин), ddC (2',3'-дидезоксицитидин), ddA (2',3'-дидезоксиаденозин), d4T (2',3'-дидезоксидидезокситимидин), 3ТС, невирапин, атевирапин, делавирдин, PMEA, PMPA и/или ловирид; ингибитор гликозидазы представляет SC-48334, или MDL-28574, либо оба вместе; ингибитор кэппирования вирусной мРНК представляет рибовирин.
Настоящее изобретение относится также к ингибитору, который вводят для лечения ВИЧ-инфекции в виде инъекции дозированной лекарственной формы для перорального введения, дозированной лекарственной формы для ректального введения или дозированной лекарственной формы для чрескожного введения.
Композицию, содержащую указанный ингибитор, можно также вводить для лечения ВИЧ-инфекции в виде инъекции, дозированной лекарственной формы для перорального введения, дозированной лекарственной формы для ректального введения или дозированной лекарственной формы для чрескожного введения.
Указанный ингибитор можно получить обычными способами, известными в данной области. Например, короткие пептиды можно синтезировать на определенном носителе или в растворе. Более длинные пептиды можно получить методами рекомбинантных ДНК или синтезировать в виде нескольких отдельных сегментов и затем соединить друг с другом. Нуклеотидную последовательность, кодирующую данный пептид, можно синтезировать и/или клонировать и экспрессировать методами, известными специалистам в данной области.
Одну или несколько пептидных связей ингибитора, соединяющих аминокислотные остатки, можно заменить непептидными связями, включающими (не ограничиваясь ими) имино, сложноэфирные, фталгидразидные, семикарбазидные, азосвязи и т.д. Реакции замены непептидных связей хорошо известны специалистам в данной области. Указанный пептид может быть синтезирован путем введения других химических групп в его аминоконец и/или карбоксильный конец для усиления устойчивости, биологической доступности и/или ингибирующей активности и т.д. Например, к аминоконцу можно добавить гидрофобные группы, такие как карбобензоксигруппа, дансильная группа или трет-бутилоксикарбонильная группа, и в аминоконец может быть также введен ацетил или 9-флуоренилметилоксикарбонил. К карбоксильному концу пептида можно добавить вышеуказанную гидрофобную группу, трет-бутилоксикарбонильную группу или аминогруппу. Кроме того, пептид можно синтезировать, изменяя его пространственную конфигурацию. Например, можно использовать D-изомеры одной или нескольких аминокислот пептида вместо обычных L-изомеров. По крайней мере один аминокислотный остаток пептида по настоящему изобретению можно заменить известным неприродным аминокислотным остатком, чтобы усилить устойчивость, биологическую доступность и/или ингибирующую слияние активность данного пептида.
Кроме того, любые вышеуказанные пептиды могут содержать непептидную макромолекулярную несущую группу, связанную с аминоконцом и/или карбоксильным концом при помощи ковалентной связи, которая включает (не ограничиваясь ими) хелатный комплекс липид - жирная кислота, полиэтиленгликоль или углевод.
Краткое описание чертежей
На фигуре 1 показана аминокислотная последовательность фузонекса (SEQ ID NO:1), которая представляет полипептид, состоящий из 36 аминокислот. Фузонекс выделен из аминокислотной последовательности (117-151) (SEQ ID NO:2) у С-концевого пептида gp41 ВИЧ-1 (подтип Е). На всех фигурах для обозначения аминокислот использованы вышеуказанные однобуквенные коды.
На фигуре 2 показан профиль элюирования в хроматографической колонке Superdex, на котором изображены характеристические пики фузонекса и комплекса фузонекса с N-концевым пептидом gp41 (35-70).
На фигуре 3 показаны спектры кругового дихроизма макромолекулярной вторичной структуры комплекса фузонекса с N-концевым пептидом gp41 (35-70).
На фигуре 4 показаны результаты сравнения устойчивости фузонекса и Т-20, где кривая с правой стороны (закрашенные кружки) представляет кривую плавления комплекса фузонекса с N-концевым пептидом gp41 и кривая с левой стороны (незакрашенные кружки) представляет кривую плавления комплекса Т-20 с N-концевым пептидом gp41. Как видно, в интервале температур 35-80°С фузонекс является более устойчивым, чем Т-20 при той же температуре.
На фигуре 5 показаны величины ингибирующей слияние активности фузонекса и Т-20. В данном эксперименте для количественного анализа ингибирующей слияние активности фузонекса (кружки) и Т-20 (квадратики) использована активность люциферазы. Слияние клеток было индуцировано gp160НХВ ВИЧ-1. Когда эффекторная клетка, экспрессирующая ген gp160НХВ и ген полимеразы Т7, сливается с клеткой-мишенью, экспрессирующей ген CD4 и ген-репортер люциферазы, происходит экспрессия гена люциферазы и возникает химическая флуоресценция. Результаты данного эксперимента свидетельствуют о 100% активности люциферазы в контрольной группе при отсутствии любого ингибитора слияния. При одинаковой концентрации фузонекс характеризуется лучшей ингибирующей активностью, чем Т-20, что установлено на основании сравнения их активности ингибирования слияния клеток, индуцированного gp160НХВ ВИЧ-1.
Подробное описание изобретения
На основании анализа трехмерной структуры gp41 получен новый ингибитор слияния, названный фузонексом, который является объектом настоящего изобретения. Ниже дано подробное описание антивирусного пептида по настоящему изобретению.
1. Фузонекс является полипептидом, состоящим из 36 аминокислотных остатков. Фузонекс (SEQ ID NO:1) выделен из сегмента (аминокислотная последовательность, содержащая остатки 117-152) (SEQ ID NO:2) С-концевого пептида эктодомена трансмембранного белка gp41 ВИЧ-1 (подтип Е), который имеет нижеследующую аминокислотную последовательность:
WIEWEREISNYTNQIYEILTESQNQQDRNEKDLLE (SEQ ID NO:2)
2. К N-концу добавлен S (серин), так как серин обычно добавляют к N-концу α-спирали для увеличения ее устойчивости.
3. I (изолейцин) заменен E (глутаминовой кислотой) в положении остатка 118, в результате чего возникает взаимодействие зарядов с R (аргинином) в положении остатка 122, что может увеличивать устойчивость α-спирали.
4. Е (глутаминовая кислота) заменен Т (треонином) в положении остатка 119 для закрытия гидрофобного кармана, включающего остатки W (триптофан) 117, W120 и W60.
5. S (серин) заменен Е (глутаминовой кислотой) в положении остатка 125, в результате чего возникает взаимодействие зарядов с К (лизином) в положении остатка 129, что может увеличивать устойчивость α-спирали.
6. N (аспарагин) заменен К (лизином) в положении остатка 129, в результате чего возникает взаимодействие зарядов с Е (глутаминовой кислотой) в положении остатка 125, что может увеличивать устойчивость α-спирали.
7. Е (глутаминовая кислота) заменен К (лизином) в положении остатка 133, в результате чего возникает взаимодействие зарядов с Е (глутаминовой кислотой) в положениях остатков 136 и 137, что может увеличивать устойчивость α-спирали.
8. Т (треонин) заменен Е (глутаминовой кислотой) в положении остатка 136, в результате чего возникает взаимодействие зарядов с К (лизином) в положении остатка 133, что может увеличивать устойчивость α-спирали.
9. N (аспарагин) заменен Е (глутаминовой кислотой) в положении остатка 140, в результате чего возникает взаимодействие зарядов с R (аргинином) в положении остатка 144, что может увеличивать устойчивость α-спирали.
После выполнения ряда вышеуказанных замен получают новый ингибитор слияния вируса "фузонекс", который имеет нижеследующую аминокислотную последовательность:
SWETWEREIENYTKQIYKILEESQEQQDRNEKDLLE (SEQ ID NO:1)
Настоящее изобретение относится к пептиду (фузонекс) с высоким потенциалом антивирусной активности. Фузонекс содержит 36 аминокислот и выделен из С-концевой аминокислотной последовательности (остатки 117-151) эктодомена трансмембранного гликопротеина gp41 ВИЧ-1 (подтип Е). Полипептид фузонекс по настоящему изобретению даже при очень низкой концентрации способен блокировать процесс слияния между вирусами и клетками и между вирус-инфицированными клетками и неинфицированными клетками. Блокируя доступ к клеткам, фузонекс способен предотвращать проникновение вирусов в клетки и распространение вирусов из инфицированных клеток в неинфицированные клетки.
Результаты анализа слияния вируса с клеткой показывают, что концентрация ингибирования слияния для фузонекса в 20 раз ниже аналогичного показателя для Т-20. Таким образом, пептид по настоящему изобретению более эффективно борется с ВИЧ-инфекцией и обладает более низкой токсичностью. Благодаря высокой устойчивости и высокой эффективности пептид по настоящему изобретению является гораздо лучшим ингибитором слияния вируса, чем Т-20.
Антивирусная активность фузонекса включает (не ограничиваясь ею) предотвращение распространения ВИЧ в неинфицированные клетки CD4+ и другие клетки. Кроме того, антивирусная активность ингибитора по настоящему изобретению не требует возбуждения какой-либо иммунной реакции у хозяина.
Ингибитор по настоящему изобретению может быть использован в любых областях биологии, относящихся к ингибированию слияния мембран, в том числе для предотвращения переноса ретровируса человека или животного кроме человека (в частности, ВИЧ) в неинфицированные клетки. В соответствии с предпочтительным вариантом осуществления настоящего изобретения ингибитор фузонекс и его производные используют в качестве ингибиторов переноса ретровируса (в частности, ВИЧ) в неинфицированные клетки человека или животного кроме человека.
Ингибитор по настоящему изобретению может также регулировать биологические процессы внутри клеток, которые относятся к спиралям вторичной структуры белка. В используемом здесь значении термин "регулировать" означает активирующее или подавляющее действие, производимое пептидом по настоящему изобретению на уровне определенной биологической активности внутри клеток (по сравнению с отсутствием пептида по настоящему изобретению).
Ингибитор по настоящему изобретению можно также использовать для идентификации соединений, способных ингибировать слияние вируса, антивирусных соединений и соединений, обладающих регуляторной активностью внутри клетки. Кроме того, фузонекс по настоящему изобретению можно также использовать для диагностики биологически специфических типов и/или подтипов вирусов.
Настоящее изобретение относится к новому ингибитору слияния фузонексу и его производным. Настоящее изобретение относится также к совместному введению фузонекса или его производных с другими лекарственными средствами, например, с другими антивирусными средствами, для лечения и/или профилактики вирусной инфекции, в частности, ВИЧ-инфекции. Указанные лекарственные средства могут иметь или не иметь такие же участки или механизмы действия, что и ингибиторы слияния вирусов. В результате совместного введения лекарственных средств может возникать комплексный или синергичный эффект.
В соответствии с одним вариантом осуществления настоящего изобретения фузонекс или его производные можно вводить вместе с другими лекарственными средствами в соответствии с нижеследующими схемами введения, которые включают (не ограничиваясь ими) одновременное введение, последовательное введение, периодическое введение и периодическое лечение (например, введение антивирусного соединения, затем введение второго антивирусного соединения через определенный период времени с повторением такой последовательности введения (то есть периода) для уменьшения возможной лекарственной устойчивости в процессе антивирусного лечения).
Комбинированное лечение фузонексом или его производными в сочетании с другими антивирусными средствами представляет новый метод лечения, который позволяет уменьшить эффективную дозу и, таким образом, токсичность указанных антивирусных препаратов. Кроме того, лекарственная композиция позволяют подавлять вирусную инфекцию в клетках-хозяевах при помощи разных механизмов, что не только увеличивает антивирусную эффективность, но и предотвращает возникновение устойчивости вирусов к любому отдельно используемому препарату. Поэтому повышается вероятность успешного лечения.
Настоящее изобретение относится также к лекарственным композициям и препаратам, предназначенным для лечения или профилактики вирусной (в частности, ВИЧ) инфекции. Лекарственная композиция содержит эффективную дозу фузонекса или его производных, по крайней мере одно другое лекарственное средство и/или фармацевтически приемлемый носитель.
Лекарственные средства, используемые вместе с фузонексом или его производными, включают любые средства, которые известны в данной области или находятся в процессе исследования. В соответствии с предпочтительным вариантом осуществления настоящего изобретения фузонекс или его производные вводят вместе с другим лекарственным средством, обладающим другим механизмом действия. Указанные лекарственные средства включают (не ограничиваясь ими) антивирусные средства, такие как цитокины rIFNα, rIFNβ... и rIFNγ; ингибиторы обратной транскриптазы, такие как AZT, 3TC, ddI, ddC, невирапин, атевирапин, делавирдин, PMEA, PMPA, ловирид и другие дидезоксирибонуклеозиды или фтордидезоксирибонуклеозиды; ингибиторы вирусной протеазы, такие как саквинарир, ритонавир, индинавир, нелфинавир и VX-478; гидроксимочевину; ингибиторы кэппирования вирусной мРНК, такие как вирусный рибовирин; амфотерицин В; связующие молекулы кастаноспермина сложноэфирной связи, обладающие анти-ВИЧ активностью, ингибитор процессинга гликопротеина; ингибиторы гликозидазы SC-48334 и MDL-28574; абсорбенты вирусов; блокатор рецепторов CD4; ингибитор корецепторов хемокина; нейтрализующее антитело; ингибиторы интегразы и другие ингибиторы слияния.
Таким образом, настоящее изобретение относится к усовершенствованной антивирусной терапии для борьбы с широкой вирусной (включая ВИЧ) инфекцией. Кроме того, настоящее изобретение относится к способу совместного введения лекарственных средств с целью усиления лечебного эффекта, включающему использование фузонекса, его производных, по крайней мере одного другого лекарственного средства и/или фармакологически приемлемого носителя. Комбинированная терапия позволяет предотвратить возникновение устойчивости вируса к каждому лекарственному средству, используемому отдельно, а также уменьшить токсичность лекарственного средства и повысить терапевтический индекс.
В используемом здесь значении термин "вирусная инфекция" означает болезнь, которая вызвана проникновением вируса в клетку. Когда вирус проникает в здоровую клетку, он использует репродуктивный механизм хозяина для собственной репродукции и в конце концов уничтожает клетку. После высвобождения из клетки вновь продуцированное поколение вирусов начинает инфицировать другие клетки. Гены некоторых вирусов могут также встраиваться в хромосомную ДНК хозяина в форме провируса, получившего название латентной инфекции. Провирус продуцируется вместе с репликацией хромосомы хозяина и может в любое время вызвать заболевание человека, будучи активированным разными внутренними и внешними факторами.
В используемом здесь значении термин "лечение или профилактика вирусной инфекции" означает подавление репликации и распространения вирусов, предотвращение саморазмножения вируса внутри хозяина и ослабление или устранение симптомов, вызываемых вирусной инфекцией. Критерии, определяющие эффективность лечения, включают снижение вирусного потенциала, уменьшение коэффициента смертности и/или коэффициента заболеваемости и т.д.
В используемом здесь значении термин "производные" означает любые пептиды, гомологичные фузонексу, которые представляют последовательность, гомолог, аналог или сегмент фузонекса или включают замену, вставку и/или делецию одной или нескольких аминокислот.
В используемом здесь значении термин "лекарственный препарат" означает любую молекулу, соединение или лекарственное средство, предназначенные для лечения вирусной инфекции или вызванных вирусом заболеваний, в частности антивирусные средства.
В используемом здесь значении термин "синергичное действие" означает совместное введение лекарственных средств, которое является более эффективным, чем аддитивное действие отдельно используемых двух или большего числа лекарственных препаратов, используемых для лечения или профилактики вирусной инфекции. Синергичное действие может усиливать эффективность антивирусных средств и позволяет избежать или ослабить устойчивость вируса к любому отдельно используемому препарату.
Пептид по настоящему изобретению представляет комплекс двух или большего числа аминокислот, связанных пептидными связями. Номенклатура пептидов определяется числом составляющих их аминокислот. Например, дипептид содержит два аминокислотных остатка, в то время как трипептид содержит три аминокислотных остатка и т.д. Пептид, состоящий из десяти или меньшего числа аминокислот, называется олигопептидом, и пептид, состоящий из более чем десяти аминокислот, называется полипептидом.
Ниже дано описание возможных применений фузонекса для лечения ВИЧ и других вирусных инфекций.
Фузонекс представляет полипептид, обладающий антивирусной активностью. Пептиды по настоящему изобретению включают фузонекс (пептид, содержащий 36 аминокислот и выделенный из gp41), его сегменты и/или аналоги. Указанные пептиды присутствуют также в других вирусах с оболочной. Пептиды по настоящему изобретению способны подавлять распространение ретровирусов, в частности ВИЧ, в организме человека и других млекопитающих.
Считается, что ВИЧ и другие вирусы непрерывно реплицируются в течение суток начиная с момента инфицирования. Таким образом, необходимо использовать антивирусные средства на разных стадиях развития вирусной инфекции. Настоящее изобретение относится также к совместному введению указанного пептида с другими антивирусными средствами для ингибирования слияния вируса с клеткой и межклеточного распространения вирусов.
Ниже дано описание совместного введения лекарственных средств, включающих фузонекс, для лечения ВИЧ-инфекции.
Фузонекс направленно воздействует на гликопротеин gp41 на поверхности вируса. Функциональный механизм фузонекса состоит в ингибировании слияния для предотвращения инфицирования новых клеток свободными вирусными гранулами и распространения вирусов из инфицированных клеток в неинфицированные клетки.
Считается, что фузонекс, вводимый вместе с одним или несколькими лекарственными средствами, направленно воздействующими на другие мишени, может оказывать аддитивное или синергичное действие. Настоящее изобретение относится к совместному введению лекарственных средств, в том числе фузонекса и его производных. При совместном введении лекарственных средств можно не только уменьшить эффективную дозу, но и снизить токсичность антивирусных средств. Кроме того, можно повысить эффективность благодаря использованию разных механизмов воздействия на вирусы. И наконец, при совместном введении лекарственных средств можно предотвратить или уменьшить вероятность возникновения лекарственной устойчивости.
Настоящее изобретение относится к способу лечения ВИЧ-инфекции у человека и других млекопитающих. Данный способ включает введение фузонекса или его производных в эффективной дозе и по крайней мере еще одного лекарственного средства, которое предпочтительно является другим антивирусным средством.
Настоящее изобретение относится к усовершенствованному способу лечения вирусной инфекции (в частности, ВИЧ-инфекции). Настоящее изобретение относится также к лекарственной композиции для лечения ВИЧ-инфекции, которая содержит фузонекс или его производное в эффективной дозе и по крайней мере одно другое антивирусное соединение. Фузонекс или его производное следует предпочтительно использовать с ингибиторами ретровирусов, ингибиторами вирусной протеазы, цитокинами, ингибиторами цитокинов и другими ингибиторами слияния вирусов. Совместное введение лекарственных средств позволяет получить более эффективные результаты при подавлении репликации и переноса вирусов.
Способы по настоящему изобретению включают введение фузонекса или его производного отдельно и совместно с другими антивирусными средствами. Фузонекс и по крайней мере одно другое лекарственное средство можно вводить одновременно (в виде смеси или раздельно) или последовательно (включая периодическое лечение). При периодическом лечении нуждающимся субъектам вводят одно антивирусное соединение в течение определенного периода времени и затем другое антивирусное соединение в течение последующего периода времени. Такую последовательность введения (а именно периодичность) повторяют для уменьшения токсичности лекарственных средств и снижения вероятности возникновения лекарственной устойчивости в процессе лечения.
Настоящее изобретение относится также к новому способу периодического лечения, в соответствии с которым сначала вводят пептид по настоящему изобретению, затем другое антивирусное средство и снова пептид по настоящему изобретению или другой ингибитор слияния вирусов. Таким образом, ингибитор по настоящему изобретению или его производное вводят вместе с другими антивирусными средствами.
Термин "совместное введение" означает не только использование двух или большего числа лекарственных препаратов в виде смеси, но и раздельное применение двух или большего числа лекарственных препаратов при одновременном введении, например, в разные вены. Термин "совместное введение" означает также последовательное введение лекарственных средств, то есть введение сначала первого лекарственного средства и затем второго лекарственного средства.
Предпочтительные антивирусные средства, используемые вместе с фузонексом, могут воздействовать на вирусы разными способами: они могут ингибировать обратную транскриптазу, ингибировать кэппирование вирусной мРНК, ингибировать протеазу ВИЧ, ингибировать гликозилирование белков, ингибировать интегразу и ингибировать слияние вирусов. Лекарственные средства, созданные на основе вышеуказанных способов воздействия, включают (не ограничиваясь ими) антивирусные средства, такие как цитокины, включая rIFNα, rIFNβ и rIFNγ; ингибиторы цитокинов; ингибиторы обратной транскриптазы, такие как AZT, 3ТC, ddI, ddC, d4T, невирапин, атевирапин, делавирдин, РМЕА, РМРА, ловирид и другие дидезоксирибонуклеозиды или фтордидезоксирибонуклеозиды; ингибиторы вирусной протеазы, такие как саквинавир, ритонавир, индинавир, нелфинавир и VX-478; ингибиторы гликозидазы, такие как SC-48334 и MDL-28574; ингибиторы кэппирования вирусной мРНК, такие как рибовирин; амфотерицин В; связующие молекулы кастаноспермина сложноэфирной связи, обладающие анти-ВИЧ активностью; гидроксимочевину; ингибиторы процессинга гликопротеинов; ингибиторы гликозидазы SC-48334 и MDL-28574; абсорбенты вирусов; блокаторы рецепторов CD4; ингибиторы корецепторов хемокина; нейтрализующие антитела; ингибиторы интегразы и другие ингибиторы слияния.
Ниже дано описание структуры полипептида фузонекса.
Пептид фузонекс является высокоэффективным ингибитором слияния, способным подавлять ВИЧ-инфекцию. Вполне вероятно, что указанный результат достигается благодаря связыванию фузонекса с gp41 на оболочке вируса и прерыванию процесса слияния вируса. Например, в процессе трансформации вирусного белка gp41 из естественной структуры в слитую структуру фузонекс может конкурировать за сайт связывания с gp41 вируса, препятствуя таким образом слиянию вирусов с клетками.
Фузонекс, пептид по настоящему изобретению, полученный из аминокислотных остатков 117-151 трансмембранного белка gp41 ВИЧ-1, состоит из 36 аминокислотных остатков. При считывании от аминоконца к карбоксильному концу фузонекс имеет нижеследующую аминокислотную последовательность:
X-SWETWEREIENYTKQIYKILEESQEQQDRNEKDLLE-Z (SEQ ID NO:1)
Кроме того, настоящее изобретение относится также к сегментам пептида фузонекс (SEQ ID NO:1), обладающим антивирусной активностью. Такие усеченные пептиды фузонекс могут содержать 4-36 аминокислот (то есть от тетрапептида до пептида, содержащего 36 аминокислот).
Х представляет аминогруппу или -Х1-Х2, где Х1 означает иминогруппу и Х2 означает гидрофобную группу, включающую (не ограничиваясь ими) карбобензокси, дансильную, трет-бутилоксикарбонильную группу, ацетил или 9-флуоренилметилоксикарбонил (FMOC) или ковалентно связанную макромолекулярную несущую группу, включающую (не ограничиваясь ими) хелатный комплекс липид - жирная кислота, полиэтиленгликоль и углевод.
Z представляет карбоксильную группу или -Z1-Z2, где Z1 означает карбонильную группу и Z2 означает аминогруппу, трет-бутилоксикарбонильную группу или макромолекулярную несущую группу, включающую (не ограничиваясь ими) хелатный комплекс липид - жирная кислота, полиэтиленгликоль и углевод.
Помимо фузонекса и усеченного фузонекса ингибиторами по настоящему изобретению могут быть также пептиды, содержащие SEQ ID NO:1, или пептиды, содержащие SEQ ID NO:1 с заменой, вставкой и/или делецией одной или нескольких аминокислот. Аминокислотная вставка может состоять из одного аминокислотного остатка или сегмента остатков 2-15 аминокислот. Фузонекс, его сегменты, аналоги и/или гомологи могут иметь одну или несколько вставок.
Кроме того, ингибиторы по настоящему изобретению включают гомологи фузонекса (SEQ ID NO:1), аналоги гомологов фузонекса и/или сегменты фузонекса, обладающие антивирусной активностью. Ингибиторами по настоящему изобретению являются фузонекс, сегменты фузонекса, аналоги фузонекса и гомологи усеченного фузонекса. Указанный усеченный фузонекс содержит пептидную последовательность фузонекса, из которой удалена одна или несколько аминокислот, в результате чего полученные пептиды имеют не менее 4-6 аминокислот. Усеченный фузонекс может содержать одну смежную часть или несколько несвязанных частей вышеуказанной пептидной последовательности.
В соответствии с одним вариантом осуществления настоящего изобретения заменяющие аминокислоты обладают защитными свойствами. Заменяющие защитные аминокислоты представляют аминокислоты, имеющие такие же заряды, размеры и/или гидрофобные характеристики, что и аминокислоты (одна или несколько), которые они заменяют в пептидной последовательности фузонекса (SEQ ID NO:1).
Ниже дано описание антивирусных средств, вводимых вместе с фузонексом.
Ингибиторы слияния по настоящему изобретению можно вводить вместе с другими лекарственными препаратами для усиления антивирусной эффективности. В соответствии с предпочтительным вариантом осуществления настоящего изобретения фузонекс вводят с другими антивирусными средствами, включающими (не ограничиваясь ими) лекарственные средства, воздействующие на другие мишени в процессе репликации вируса, такие как ингибиторы обратной транскриптазы, ингибиторы вирусной протеазы, ингибиторы гликозилирования и т.д.; антивирусные средства, воздействующие на другие мишени в процессе распространения вирусов; антивирусные средства, воздействующие на другие участки той же молекулы; и антивирусные средства, способные предотвращать или уменьшать возникновение лекарственной устойчивости. Механизмы воздействия и преимущества совместного введения должны быть известны научным работникам и технологам в данной области.
Ингибиторы по настоящему изобретению можно вводить вместе с ингибиторами ретровирусов, включающими (не ограничиваясь ими) нуклеозидные производные. Нуклеозидные производные являются усовершенствованными производными пуриновых и пиримидиновых нуклеозидов. Механизм воздействия указанных веществ направлен на предотвращение синтеза РНК и ДНК. Нуклеозидные производные при отсутствии любого 3'-заместителя, который может связываться с другими нуклеозидами, способны подавлять синтез кДНК, катализируемый обратной транскриптазой и, таким образом, прекращать репликацию вирусной ДНК. Именно поэтому они считаются лекарственными средствами против ВИЧ. Например, AZT и ddT, которые могут подавлять репликацию ВИЧ-1 in vivo и in vitro, признаны лечебными средствами против ВИЧ-инфекции и СПИДа. Однако применение таких лекарственных средств для лечения вышеуказанных заболеваний вызывает массовое размножение устойчивых к лекарственным средствам штаммов ВИЧ и многочисленные побочные эффекты.
Ингибиторы по настоящему изобретению можно вводить вместе с нуклеозидными производными и ненуклеозидными производными. Нуклеозидные производные включают (не ограничиваясь ими) 2',3'-дидезоксиаденозин (ddA); 2',3'-дидезоксигуанозин (ddG); 2',3'-дидезоксиинозин (ddI); 2',3'-дидезоксицитидин (ddC); 2',3'-дидезокситимидин (ddT); 2',3'-дидезоксидидезокситимидин (d4T) и 3'-азид-2',3'-дидезоксицитидин (AZT). В соответствии с одним вариантом осуществления настоящего изобретения нуклеозидные производные представляют галогеннуклеозиды, предпочтительно 2',3'-дидезокси-2'-фторнуклеотиды, включающие (не ограничиваясь ими) 2',3'-дидезокси-2'-фтораденозин; 2',3'-дидезокси-2'-фторинозин; 2',3'-дидезокси-2'-фтортимидин; 2',3'-дидезокси-2'-фторцитидин; и 2',3'-дидезокси-2',3'-дидегидро-2'-фторнуклеотиды, включающие (не ограничиваясь ими) 2',3'-дидезокси-2',3'-дидегидро-2'-фтортимидин (Fd4T). Более предпочтительно нуклеозидные производные представляют 2',3'-дидезокси-2'-фторнуклеотиды, имеющие фтористую связь в β-положении, которые включают (не ограничиваясь ими) 2',3'-дидезокси-2'β-фтораденозин (F-ddA), 2',3'-дидезокси-2'β-фторинозин (F-ddI) и 2',3'-дидезокси-2'β-фторцитидин (F-ddC). При совместном введении лекарственных средств может быть уменьшена доза нуклеозидных производных и, таким образом, снижена токсичность, а также лекарственная устойчивость вируса при сохранении антивирусной активности указанных лекарственных средств.
В соответствии с предпочтительным вариантом осуществления настоящего изобретения композиция, состоящая из антивирусных пептидов и нуклеотидных производных, включает фузонекс или его производные в эффективной дозе и AZT, ddC и/или d4T в эффективной дозе для лечения ВИЧ-инфекции. Более предпочтительная лекарственная композиция содержит (не ограничиваясь ими) фузонекс или его производные и ddT в эффективной дозе; и/или 3ТС, вирамун, рескриптор, сустива, ловирид, невирапин и атевирдин в эффективной дозе.
Фузонекс или его производные можно также вводить вместе с ингибиторами уридинфосфорилирующего фермента, включающими (не ограничиваясь ими) ациклоуридиновые соединения, в том числе бензилациклоуридин (BAU); бензоксибензилациклоуридин (BBAU); аметобензилациклоуридин (AMBAU); аметобензоксибензилациклоуридин (AMB-BAU); гидроксиметилбензилациклоуридин (HMBAU) и гидроксиметилбензоксибензилациклоуридин (HMBBAU).
Фузонекс или его производные можно также вводить вместе с цитокинами или ингибиторами цитокинов, которые включают (не ограничиваясь ими) rIFNα, rIFNβ,... и rIFNγ; ингибиторы TNFα, MNX-160, рекомбинантный αА-интерферон человека, рекомбинантный β-интерферон человека и рекомбинантный γ-интерферон человека. Более предпочтительное совместное введение лекарственных средств включает введение фузонекса или его производных с β-интерфероном в эффективной дозе.
Ингибиторы протеазы препятствуют созреванию вируса главным образом в период сборки или после сборки вируса (то есть во время баддинга вируса). Ингибиторы протеазы проявляют антивирусную активность как in vivo, так и in vitro. После введения ингибиторов протеазы у больных СПИДом наблюдается экспоненциальное снижение уровня ВИЧ и увеличение числа лимфоцитов CD4 (Deeks, et al., 1997, JAMA 277:145-53). Совместное введение ингибиторов вирусной протеазы и ингибитора слияния фузонекса может оказывать синергичное действие и способствовать достижению удовлетворительных клинических результатов. Настоящее изобретение относится к способу лечения ВИЧ-инфекции, который представляет совместное введение лекарственных средств с использованием фузонекса или его производных в эффективной дозе и ингибитора протеазы в эффективной дозе, причем указанное последним лекарственное средство включает (не ограничиваясь ими) индинавир, инвиразу, норвир, вирацепт и агенеразу.
Фузонекс или его производные можно также использовать вместе с лекарственными средствами против ВИЧ, нарушающими процессинг 5'-мРНК, такими как виразол. Механизм действия виразола до сих пор неизвестен, но предполагается, что он конкурирует с гуанином при образовании кэппирующей структуры мРНК и/или препятствует метилированию указанных молекул. Фузонекс и виразол, по-видимому, оказывают синергичное действие.
Кроме того, фузонекс или его производные можно вводить вместе с амфотерицином В. Амфотерицин В представляет полиен, являющийся противогрибковым антибиотиком, который может необратимо связываться со стеролом. Амфотерицин В и его формиат обладают ингибирующим действием против многих вирусов с липидной оболочкой, включая ВИЧ. Амфотерицин В является высокотоксичным для человека, в то время как его формиат гораздо менее токсичен. Таким образом, амфотерицин В или его формиат можно вводить вместе с фузонексом или его производными с достижением синергичного действия против ВИЧ, что позволяет клиницистам использовать амфотерицин В или его формиат в меньших дозах при сохранении антивирусной активности указанных веществ.
Фузонекс или его производные можно также вводить вместе с кастаноспермином, представляющим ингибитор процессинга гликопротеина, который является растительным алкалоидом, способным ингибировать процессинг гликопротеина. Оболочка ВИЧ содержит два гликопротеина большой длины gp120 и gp41. Гликозилирование белков играет важную роль во взаимодействии gp120 и CD4. Потомство вируса, синтезированное в присутствии кастаноспермина, характеризуется меньшей эффективностью по сравнению с исходным вирусом. Совместное введение фузонекса или его производных с кастаноспермином может оказывать синергичное действие.
Лечебное действие, достигаемое при совместном введении фузонекса или его производных и вышеуказанных антивирусных препаратов, можно оценить обычными методами, используемыми в данной области. Например, совместный эффект фузонекса и AZT можно определить при помощи целого ряда экспериментов in vitro, включающих ингибирование токсичности ВИЧ в отношении клеток, ингибирование образования синплазмы, ингибирование активности обратной транскриптазы или ингибирование способности вируса синтезировать РНК или белок и т.д.
Ниже дано описание дозированных лекарственных форм, используемых доз и способов введения. Сначала будут рассмотрены лекарственные композиции.
Лекарственные композиции по настоящему изобретению, содержащие фузонекс или его производные в сочетании по крайней мере с одним другим лекарственным средством, можно использовать для лечения или профилактики вирусной инфекции у человека или животного кроме человека. Совместное введение лекарственных средств по настоящему изобретению может создавать аддитивный/синергичный эффект.
Предпочтительная лекарственная композиция содержит фузонекс или его производные и по крайней мере одно другое антивирусное средство, такое как ингибиторы обратной транскриптазы, ингибиторы протеазы, ингибиторы процессинга мРНК, ингибиторы гликозилирования белка, абсорбенты вирусов, ингибиторы рецепторов CD4, ингибиторы корецепторов хемокинов, нейтрализующее антитело, ингибиторы интегразы и другие ингибиторы слияния, включающие (не ограничиваясь ими) аналоги нуклеозидов или терминаторы цепи; ингибиторы корецепторов хемокина AMD-3100 (Tremblay, C.L. et al., 2000, J. AIDS 1:25(2)99-10).
В соответствии с одним вариантом осуществления настоящего изобретения лекарственные средства, которые можно использовать вместе с фузонексом или его производными, включают (не ограничиваясь ими) 2-дезокси-D-глюкозу (2dGlc), дезоксинойиримицинациклогуанозин, виразол, рифадин, адамантанамин, рифабутин, ганцикловер (DHPG), фамцикловер, буцикловер (DHBG), фториодарацитозин, иодоксуридин, трифтортимидин, ара-А, ара-АМР, бромвинилдезоксиуридин, BV-арау, 1-b-D-гликоарабинофуранозид-Е-5-[2-бромвинил]урацил, адамантэтиламин, гидроксимочевину, фенилуксусный гептандион, диариламидин, (S)-(п-нитробензил)-6-тиоинозин и фосфоноформиат. Настоящее изобретение относится к лекарственной композиции, содержащей фузонекс или его производные в сочетании с любыми другими вышеуказанными соединениями.
Кроме того, пептиды по настоящему изобретению можно также использовать в качестве профилактического средства для субъектов, подверженных заражению ВИЧ, но еще не инфицированных. Примеры использования таких профилактических средств включают (не ограничиваясь ими) профилактику передачи вирусов от матери ребенку; профилактику ВИЧ-инфекции в других ситуациях, например, у медицинских работников, имеющих дело с ВИЧ-инфицированной кровью, продуктами крови и жидкостями организма, в случае неосторожного обращения. В указанных случаях пептиды по настоящему изобретению можно использовать в качестве профилактической вакцины. При вакцинации пептидом по настоящему изобретению в организме хозяина вырабатываются антитела, которые могут ингибировать ВИЧ-инфекцию и нейтрализовать вирусы ВИЧ.
Настоящее изобретение относится к схеме профилактической вакцинации, которая включает введение хозяину указанного пептида в эффективной концентрации для возбуждения иммунной реакции, достаточной для нейтрализации ВИЧ, например, для подавления клетками ВИЧ-инфекции. Вызванную иммунную реакцию можно обнаружить стандартными методами, хорошо известными специалистам в данной области. В соответствии с одним вариантом осуществления настоящего изобретения пептид, используемый в качестве вакцины, можно вводить при помощи внутримышечной инъекции.
Для увеличения иммунной реакции пептид по настоящему изобретению можно вводить вместе с некоторыми приемлемыми адъювантами, включающими (не ограничиваясь ими) минеральный гель, такой как гидроксид алюминия; поверхностно-активное вещество, такое как лизолецитин; многоатомный пуроновый спирт, полианион; другие пептиды; масляную эмульсию и другие добавки, приемлемые для организма человека, такие как Bacillus Calmette-Guerin (BCG) и мелкие булавовидные бактерии. Способы введения вышеуказанной вакцины включают (не ограничиваясь ими) пероральное, внутрикожное, внутримышечное, внутрибрюшинное, внутривенное, подкожное введение и через мышечную оболочку.
Ниже дано описание дозировки фузонекса.
При лечении острой вирусной инфекции у млекопитающих, таких как человек, фузонекс или его производные необходимо вводить в эффективной дозе, достаточной для подавления репликации вирусов. Эффективную дозу можно определить методами, известными специалистам в данной области, с использованием установочных параметров, таких как биологический полупериод существования, биологическая доступность, токсичность и т.д. Например, фузонекс можно постоянно вводить в виде инъекций в течение 4-52 недель в дозе 0,2-10,0 мг/кг в сутки. Предпочтительная доза равна 20-200 мг в сутки. Наиболее предпочтительная доза равна 30-80 мг в сутки при продолжительности лечения около 52 недель.
Промежуток между введением фузонекса или его производных составляет примерно от 2 суток до 1/4 суток, наиболее предпочтительно 1-1/2 суток. При наиболее эффективной дозировке максимальная концентрация фузонекса или его производных в плазме крови может достигать 1-10 мг/мл. Концентрацию фузонекса в крови можно определить следующим способом: получают стерильный инъекционный раствор 20% фузонекса в приемлемом буферном физиологическом растворе, производят постоянное введение указанного раствора и измеряют концентрацию в крови при помощи ВЭЖХ.
Эффективную дозу лекарственных средств, таких как антивирусные средства, вводимых вместе с фузонексом или его производными, определяют на основании рекомендуемой дозы используемых антивирусных средств, хорошо известных специалистам в данной области. Предпочтительная доза при совместном введении примерно на 10-50% меньше по сравнению с рекомендуемой в научной литературе дозой для раздельного введения. Специалисты в области медицины должны обращать особое внимание на дозы, при которых могут возникать токсические реакции. При нарушении функции костного мозга, печени и/или почки или при возникновении сильного взаимодействия между лекарственными средствами врач должен знать, каким образом и когда приостановить или прекратить введение лекарственных средств и уменьшить их дозу. В отличие от этого при отсутствии предполагаемых клинических результатов, врач должен знать, как увеличить дозу.
Эффективная лечебная доза означает такую дозу, при которой лекарственная композиция является достаточной для улучшения состояния субъекта или для увеличения продолжительности его жизни. Токсичность и терапевтическое действие лекарственных средств подобного типа можно определить стандартными фармакологическими методами культивирования клеток или выполнения экспериментов с использованием животных. Например, можно определить среднюю летальную дозу (LD50, дозу, при которой погибает 50% экспериментальной колонии) и среднюю эффективную дозу (ED50, дозу, при которой выздоравливает 50% экспериментальной колонии). Соотношение лечебной способности и токсичности представляет терапевтический индекс и может быть выражено как LD50/ED50. Чем выше терапевтический индекс, тем лучшими качествами обладает соединение. Статистические данные, полученные при выполнении экспериментов с клетками и животными, можно использовать для определения предельных доз для человека. Дозы соединений указанного типа наиболее предпочтительно находятся в пределах определенной лекарственной концентрации, повышающей количество гемоглобина в крови, то есть выше ED50, но при отсутствии или низкой токсичности, причем указанная доза может быть отрегулирована в зависимости от типа дозированной лекарственной формы и способа введения. На основании полученных данных можно точно определить дозу для человека. Концентрацию лекарственного средства в плазме крови можно измерить при помощи ВЭЖХ.
Ниже дано описание дозированных лекарственных форм и способов введения.
Нуждающимся субъектам можно вводить лекарственную композицию, содержащую только фузонекс или его производные, или лекарственную композицию, содержащую фузонекс или его производные в смеси с приемлемым носителем или наполнителем для получения дозы, необходимой для лечения вирусной инфекции, в частности ВИЧ-инфекции. Способы получения и введения лекарственных средств по данному изобретению хорошо известны специалистам в данной области.
Антивирусная активность пептидов по настоящему изобретению может характеризоваться специфичностью к подтипу вируса, то есть специфический пептид ингибирует только специфический вирус. Пептиды по настоящему изобретению наиболее восприимчивы к ВИЧ-1, и данное преимущество можно использовать в диагностических целях. Например, специфичность указанного пептида против ВИЧ-1 можно использовать для идентификации типа определенного выделенного вирусного штамма (ВИЧ-1 или ВИЧ-2). Например, выделенную неинфицированную клетку CD4+ инфицируют неизвестным вирусным штаммом в присутствии пептида фузонекса и культивируют данную клетку. Затем определяют активность ретровируса в клеточном супернатанте, и, если указанная активность полностью подавляется, вирусный штамм должен быть штаммом ВИЧ-1; если активность не подавляется или подавляется в незначительной степени, вирусный штамм, по-видимому, не является штаммом ВИЧ-1.
Настоящее изобретение относится также к использованию фармакологически приемлемого носителя для получения дозированной лекарственной формы для системного введения на основе пептидов и/или лекарственных композиций по настоящему изобретению. При использовании соответствующего носителя и препарата пептиды или композиции по настоящему изобретению, в частности композиции, полученные в виде раствора, можно вводить способами, не затрагивающими желудочно-кишечный тракт, которые включают (не ограничиваясь ими) внутривенные инъекции. Пептиды или композиции по настоящему изобретению можно также получить в виде раствора, предназначенного для перорального введения, используя фармакологически приемлемый носитель, хорошо известный в данной области. Соответствующие носители необходимы для получения пептидов или композиций в виде таблеток, пилюль, капсул, жидкости, геля, сиропов, взвесей, суспензий и других дозированных лекарственных форм.
Пептиды и лекарственные композиции по настоящему изобретению можно вводить способами, хорошо известными специалистам в данной области, которые включают актинальное или ректальное введение, введение через диализную мембрану или тонкий кишечник, введение, не затрагивающее желудочно-кишечный тракт, в виде внутримышечных, подкожных, интрамедуллярных, подоболочечных, интравентрикулярных, внутривенных, внутрибрюшинных, назальных или глазных инъекций; чрескожное, местное, вагинальное введение и т.д. Дозированные лекарственные формы включают (не ограничиваясь ими) таблетки, пастилки, порошки, суспензии, суппозитории, раствор, капсулы, глясе, пластыри и микромоторные препараты. Из лекарственных композиций по настоящему изобретению известными способами могут быть получены любые фармакологически приемлемые препараты при использовании одного или нескольких физиологически приемлемых носителей. Лекарственные композиции по настоящему изобретению могут содержать один или несколько наполнителей и адъювантов, облегчающих процесс обработки активных соединений. Тип препарата определяется способом введения. Для облегчения введения в виде инъекций пептиды или композиции по настоящему изобретению могут быть получены в виде раствора, например, физиологического раствора. Для введения через диализную мембрану необходимо использовать вещества, облегчающие проникновение препарата через барьеры, которые хорошо известны в данной области.
Дозированную лекарственную форму для перорального введения, содержащую пептиды и лекарственные композиции по настоящему изобретению, можно получить, измельчая активные компоненты вместе с твердыми наполнителями с образованием однородной смеси, используемой для получения гранул, из которых затем прессуют таблетки или сердцевину таблеток с сахарным покрытием; при необходимости к смеси может быть добавлен соответствующий адъювант. В качестве приемлемых наполнителей можно использовать сахар, такой как лактоза, сахароза, маннит или сорбиколан; фибринсодержащие продукты, такие как кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, глютин, трагакант, метилцеллюлоза, гидроксипропилметилцеллюлоза, натрийкарбоксиметилцеллюлоза и/или поливинилпирролидон. При необходимости можно использовать вещества, улучшающие распадаемость таблетки, такие как сшитый поливинилпирролидон, агар, альгиновая кислота или альгинат натрия в виде соли. На сердцевину таблеток может быть нанесено сахарное покрытие. Такое покрытие может быть получено из концентрированного сахарного раствора, содержащего аравийскую камедь, тальк, поливинилпирролидон, гель карбополь, полиэтиленгликоль, оксид титана, нитрат целлюлозы и соответствующий органический растворитель или комбинация растворителей. В таблетки или сахарное покрытие таблеток могут быть добавлены разные комбинации красящих веществ или съедобных пигментов для выделения или маркировки активного соединения.
Лекарственная композиция для перорального введения может быть получена в виде наполняемых твердых капсул или запаянных мягких капсул, изготовленных из глютина и пластификатора, такого как глицерин или сорбиновая кислота. Наполняемые капсулы содержат наполнитель, такой как лактоза, клейкое вещество, такое как крахмал, и/или смазывающее вещество, такое как тальк или стеарат. Кроме того, можно использовать стабилизирующую добавку для стабилизации активных компонентов. В мягких капсулах активное соединение может быть растворено или суспендировано в приемлемой жидкости, такой как нелетучее жидкое масло, жидкий олефин или текучий полиэтиленгликоль. Кроме того, можно также использовать стабилизирующую добавку. Все дозированные лекарственные формы для перорального введения должны быть удобны для использования нуждающимися субъектами. В случае актинального введения вышеуказанную композицию можно получить в виде удобной дозированной лекарственной формы, представляющей лепешку.
Для введения путем ингаляции пептиды или композиции по настоящему изобретению могут быть получены в форме аэрозоля при использовании баллона или ингалятора под высоким давлением или соответствующего пропеллента, такого как дихлордифторметан, трихлорфторметан, дихлортетрафторэтан, диоксид углерода или другие приемлемые газы. В случае аэрозоля, распыляемого под высоким давлением, дозирующее устройство может быть снабжено клапаном, высвобождающим требуемое количество препарата. Глютиновые капсулы и кассеты, используемые в качестве инсуффлятора или аспиратора, могут содержать смесь пептидов и соответствующего порошкообразного субстрата (такого как лактоза или крахмал).
Пептиды или композиции по настоящему изобретению можно получить в виде дозированной лекарственной формы для введения, не затрагивающего желудочно-кишечный тракт. Например, лекарственные вещества могут быть использованы для получения инъекционных препаратов, которые включают периодические инъекции или непрерывное внутривенное вливание. Препараты для инъекций могут быть получены в виде дозированных лекарственных форм. Например, они могут находиться в ампулах. Препараты в больших дозах также могут быть получены в виде дозированных лекарственных форм, таких как ампула или емкость для большой дозы, с добавлением консерванта. Композиции по настоящему изобретению могут иметь форму суспензии, раствора или эмульсии с использованием в качестве среды масла или воды и могут содержать некоторые добавки, такие как суспендирующие вещества, стабилизатор и/или диспергатор.
Лекарственные композиции, предназначенные для введения, не затрагивающего желудочно-кишечный тракт, могут представлять водный раствор активного вещества, то есть водорастворимую форму. Суспензию активного вещества можно также получить в виде соответствующей масляной суспензии для инъекций. Приемлемый олеофильный растворитель или носитель включает нелетучее жидкое масло, такое как кунжутное масло, синтезированный сложный эфир жирной кислоты, такой как этилолеат, или триглицерид, или липосому. Водная суспензия для инъекций может содержать вещество, увеличивающее вязкость суспензии, такое как натрийкарбоксиметилцеллюлоза, сорбиновый спирт и глюкозан. Вышеуказанная суспензия может также избирательно содержать приемлемый стабилизатор или вещество, повышающее растворимость соединения, для получения высококонцентрированного раствора. Активный компонент порошкообразного препарата для инъекций перед введением может быть растворен в приемлемом растворителе, таком как стерильная вода для инъекций, не содержащая пирогена.
Пептиды или композиции по настоящему изобретению могут быть также получены в виде дозированных лекарственных форм для ректального введения, таких как суппозитории или удерживающие клизмы. Указанные препараты могут быть получены с использованием быстро расплавляющегося субстрата, такого как масло какао или другие сложные эфиры глицерина.
Помимо вышеописанных дозированных лекарственных форм, пептиды или лекарственные композиции могут быть также получены в виде дозированных лекарственных форм пролонгированного действия, которые можно использовать в качестве подкожных или внутримышечных трансплантатов или внутримышечных инъекций. Поэтому пептиды и их производные или лекарственные композиции можно получить в сочетании с приемлемыми полимерами, гидрофобными веществами (например, масляная эмульсия), веществами для ионообменной хроматографии или их плохо растворимыми производными, такими как плохо растворимая соль.
Лекарственные носители для гидрофобных пептидов или композиций по настоящему изобретению представляют совместно растворимую систему органических полимеров и водной фазы, которая смешивается с водой и содержит бензиловый спирт и неполярное поверхностно-активное вещество. Такой совместно растворимой системой может быть совместно растворимая система VPD. VPD представляет раствор, содержащий 3% (мас./об.) бензилового спирта, 8% (мас./об.) неполярного поверхностно-активного вещества, являющегося мультиэтоксиэфиром, и 65% (мас./об.) полиэтиленгликоля 300 в абсолютном спирте; совместно растворимую систему VPD (VPD:5W) получают, растворяя VPD в воде в отношении 1:1 с 5% глюкозы. Совместно растворимая система подобного типа может лучше растворять гидрофобные вещества и является менее токсичной при системном введении. При сохранении показателей растворимости и токсичности пропорции совместно растворимой системы можно изменять в широких пределах. Кроме того, можно также изменять компоненты совместно растворимого носителя. Например, вместо мультиэтоксиэфира можно использовать другое неполярное поверхностно-активное вещество с низкой токсичностью; можно изменять пропорцию полиэтиленгликоля; вместо полиэтилена можно использовать другие биологически смешивающиеся полимеры, такие как поливинилпирролидон; вместо глюкозы можно использовать другой сахар или полиозу.
Лекарственные композиции могут также включать приемлемые носители или наполнители в твердой или гелеобразной фазе. Указанные носители или наполнители включают (не ограничиваясь ими) карбонат кальция, фосфат кальция, разные сахара, крахмал; производные целлюлозы, желатин или полимеры, такие как полиэтиленгликоль. Лекарственные композиции по настоящему изобретению могут также включать несколько активных компонентов в эффективной дозе для достижения требуемой лечебной цели. Способ определения эффективной дозы хорошо известен специалистам в данной области.
Ниже дано описание применения пептидов по настоящему изобретению.
Пептид, получивший название фузонекс (SEQ ID NO:1), обладает эффективной антивирусной активностью. Благодаря этому указанный пептид и его производные можно использовать в качестве ингибиторов ретровирусов, в частности ингибиторов ВИЧ, у человека или животных кроме человека, предотвращая таким образом распространение вируса в неинфицированные клетки.
Пептиды по настоящему изобретению можно использовать для подавления распространения ретровирусов, поражающих человека, которые включают (не ограничиваясь ими) штаммы ВИЧ-1 и ВИЧ-2 и Т-лимфоциты человека (HTLV-I и HTLV-II). Пептиды по настоящему изобретению можно также использовать для подавления распространения ретровирусов, поражающих животных кроме человека, которые включают (не ограничиваясь ими) вирус лейкоза крупного рогатого скота, вирус саркомы кошек, вирус лейкоза, вирус иммунодефицита обезьян и вирус прогрессирующей пневмонии овец.
Пептиды по настоящему изобретению, по-видимому, способны подавлять распространение других ретровирусов и/или неретровирусов, включающих (не ограничиваясь ими) респираторно-синцитиальный вирус человека, вирус чумы собак, вирус псевдочумы птиц, вирус парагриппа человека и вирус гриппа.
Кроме того, настоящее изобретение относится также к применению пептидов при совместном введении лекарственных средств для лечения заболеваний, вызываемых вышеуказанными ретровирусами и неретровирусами.
Ниже дано описание совместного введения лекарственных средств для подавления ВИЧ.
Настоящее изобретение относится к совместному введению фузонекса и других лекарственных средств. Совместное введение лекарственных средств может предотвращать образование синплазмы и репликацию ВИЧ, подавляя таким образом репродукцию ВИЧ у нуждающихся субъектов. Совместное введение лекарственных средств по настоящему изобретению можно также применять для ослабления или излечивания заболеваний, обусловленных ВИЧ-инфекцией. Например, антивирусный пептид фузонекс или его производные можно вводить вместе с противогрибковыми средствами, антибиотиками или другими антивирусными средствами для подавления HBV, EBV, CMV-инфекции и другой случайной инфекции (включая туберкулез).
Лучшим применением для фузонекса или его производных является подавление ВИЧ-инфекции. Эффективная доза для совместного введения может быть определена в зависимости от нижеследующих способов. Например, в зависимости от способов получения лекарственного средства в пригодном носителе и любых приемлемых способов его введения, включающих (не ограничиваясь ими) инъекции (такие как внутривенные, внутрибрюшинные, внутримышечные, подкожные инъекции и т.д.), всасывание через эпителий или слизистую оболочку, в частности через актинальную слизистую оболочку, эпителий прямой кишки или влагалища, слизистую оболочку носоглотки, слизистую оболочку тонкого кишечника и т.д., через костную ткань; чрескожное введение или другие фармакологически приемлемые способы введения.
По сравнению с лекарственными средствами, существующими в данной области, пептиды по настоящему изобретению обладают такими преимуществами, как значительно более высокая эффективность подавления слияния ВИЧ с клеточной мембраной, лучшая устойчивость и более высокий потенциал, уменьшение дозы при увеличении эффективности и сокращение побочных эффектов.
Примеры
Ниже приведены примеры, иллюстрирующие настоящее изобретение.
Пример 1. Получение и очистка фузонекса
Фузонекс синтезируют в биосистемном синтезаторе полипептидов типа 431А. Указанный пептид получают методом химического синтеза Fast-Moc и стандартным методом твердофазного синтеза с использованием аминокислот, защищенных 9-флуоренилметилоксикарбонилом (FMOC). Используемые реагенты включают TFA, воду, 5% анизилсульфида, 2,5% этилендисульфгидрата и 0,8 М кристаллического фенилфенола.
Чтобы продлить биологический полупериод существования фузонекса, его аминоконец ацетилируют и карбоксильный конец амидируют. Пептид автоматически отделяют от смолы в синтезаторе, при этом автоматически удаляют большие боковые группы. Отделенный от смолы сырой пептид фузонекс осаждают в течение 20 минут в четырехкратном объеме холодного простого эфира. Затем пептид центрифугируют, дважды промывают в холодном простом эфире и сушат в течение 24 часов.
Сырой пептид фузонекс очищают ВЭЖХ. Линейное элюирование выполняют в колонке С18 (15 мкм обычного наполнителя) в смеси вода/ацетонитрил, содержащей 0,1% TFA. Степень чистоты очищенного пептида фузонекса, измеренная при помощи аналитической ВЭЖХ, превышает 95%. И наконец, фузонекс проверяют путем секвенирования аминокислоты и сравнения электронных масс-спектров распыления. Молекулярная масса очищенного фузонекса равна 4641,24 Да.
Пример 2. Эксперимент по элюированию фузонекса
При выполнении эксперимента по элюированию фузонекса использован нижеследующий способ.
В хроматографической колонке Superdex 75 элюируют избыток фузонекса и комплекс фузонекса с N-концевым пептидом gp41 (35-70). В качестве элюента использован фосфатный буфер. Общий собранный объем для построения кривой равен 30 мл. На основании полученных результатов построен профиль элюирования (фигура 2), который показывает, что пик элюирования комплекса фузонекса с N-концевым пептидом gp41 (35-70) находится в положении, соответствующем 13,2 мл, и пик элюирования самого фузонекса находится в положении, соответствующем 17,3 мл.
Пример 3. Определение структуры комплекса фузонекса с N-концевым пептидом gp41
Для определения структуры комплекса фузонекса с N-концевым пептидом gp41 использованы спектры кругового дихроизма. Сначала образец разводят до 25 мкМ, рН 7,4, в 0,1 М NaCl/20 мМ фосфата калия и затем при помощи спектров кругового дихроизма (CD) определяют вторичную структуру комплекса фузонекса с N-концевым пептидом gp41. Анализ выполняют в устройстве для получения спектров кругового дихроизма Aviv 62A DS. Пептидный раствор, содержащий фузонекс и N-концевой пептид gp41 (35-70), измеряют в диапазоне длин волн 200-260 нм.
Спектры кругового дихроизма получены в следующих условиях: температура 20°С, циклическое изменение поляризованного по кругу света соответствует 1,5 нм с шагом изменения, равным 0,5 нм, постоянная времени равна 2,0 секунды и длина фотоэлемента равна 10 мм. Для коррекции полученных результатов с эталонными данными использован трехчленный полином. Температура образца поддерживается при помощи термоэлемента с диапазоном ошибок в пределах 1°С.
Результаты представлены в виде спектров кругового дихроизма (CD) (фигура 3). На фигуре 3 показано, что вторичная структура комплекса фузонекса с N-концевым пептидом gp41 (35-70) на 100% состоит из α-спиралей. В качестве единицы эллиптичности оси Y спектра использован градус (θ), при этом установлено, что эллиптичность θ равна примерно -40000/градус·см2·дмоль-1 (фигура 3) при длине волны 222 нм.
Пример 4. Испытание устойчивости фузонекса и Т-20
При помощи данного эксперимента определяют устойчивость полипептида, измеряя его кривую плавления при разных температурах. Указанный эксперимент выполняют в устройстве для определения спектров кругового дихроизма Aviv 62A DS. Пептидный раствор нагревают в интервале температур от 20 до 95°С.
Данный эксперимент позволяет исследовать устойчивость полипептида на основании его кривой плавления при разных температурах. Температура 50% плавления для комплекса фузонекса с N-концевым пептидом gp41 (кривая с правой стороны, изображенная закрашенными кружками) равна 70°С, в то время как температура 50% плавления для комплекса Т-20 с N-концевым пептидом gp41 (кривая с левой стороны, изображенная незакрашенными кружками) равна 51°С. При прочих равных условиях комплекс фузонекса плавится при более высокой температуре.
Полученные результаты показаны кривыми плавления (фигура 4) для комплекса фузонекса и комплекса Т-20. Как показано на фигуре 4, фузонекс обладает более высокой устойчивостью, чем Т-20.
Пример 5. Ингибирующая активность фузонекса в отношении инфекции ВИЧ-1
В данном рабочем примере рассмотрен анализ межклеточного слияния, результаты которого показывают, что фузонекс может очень эффективно блокировать вирусную инфекцию. При выполнении анализа слияния в одинаковых условиях IC50 фузонекса равно 1,2±0,2 нМ, в то время как IC50 Т-20 равно 23±6 нМ. 50% ингибирующая концентрация (IC50) фузонекса в 20 раз меньше аналогичного показателя Т-20. С учетом высокого потенциала фузонекса установлено, что указанный пептид более эффективно предотвращает слияние вирусов ВИЧ с клетками человека и ингибирует ВИЧ-индуцированное внутриклеточное слияние, блокируя таким образом проникновение вируса в неинфицированные клетки.
Ниже дано подробное описание указанного анализа.
Антивирусную активность фузонекса можно определить путем испытания указанного пептида in vitro. В настоящем изобретении для количественного определения способности фузонекса ингибировать образование синплазмы под действием gp160НХВ ВИЧ-1 использован эксперимент на основе люциферазы.
Данный эксперимент выполняют описанными ниже способами. Протомор Т7 встраивают в верхнюю последовательность pSP64 сайта клонирования для сборки плазмиды гена-репортера люциферазы Pst7luc. Сайт проникновения внутреннего рибозима (IRES) вируса энцефаломиокардита (CMCV) связывают с 5'-концом гена люциферазы и с нижней областью промотора Т7. Ген CD4, ген полимеразы Т7 и ген gp160НХВ ВИЧ-1 субклонируют в экспрессирующей плазмиде РМТ3 млекопитающего.
В 10 см чашку для культивирования переносят клетки 293Т методом на основе фосфата кальция. Эффекторные клетки трансфектируют равными количествами гена gp160НХВ и гена полимеразы Т7 и клетки-мишени трансфектируют pSP64 и геном CD4. Плазмида РМТ3, не содержащая гена gp160НХВ или CD4, служит в качестве отрицательного контрольного образца.
Клетки промывают несерозной модифицированной по способу Дульбекко средой Игла (DMEM) и добавляют преципитат ДНК. Клетки культивируют в течение 30-40 минут при 37°С в 9 мл/чашка несерозной DMEM. Затем клетки осторожно перемешивают, отсасывая их в чашке вверх и вниз пипеткой для переноса, центрифугируют и собирают осажденные клетки. Клетки в каждой чашке вновь суспендируют в 3-4 мл DMEM, содержащей 10% фетальной телячьей сыворотки (FBS), после чего эффекторные клетки смешивают с клетками-мишенями (по 45 мкл) на 96-луночном культуральном планшете с достижением хорошего распределения.
Фузонекс или Т-20 сначала растворяют в ДМСО и затем разводят до 20-кратного объема. Раствор, используемый в качестве маточного раствора, перед применением подвергают серийному разведению в культуральной среде. В каждую лунку добавляют 10 мкл культуральной среды или физиологического раствора с фосфатным буфером (PBS) и добавляют разведенный фузонекс или Т-20 в непрерывно увеличивающейся от нуля концентрации. Смесь клеток-мишений, эффекторных клеток и ингибитора слияния перемешивают и культивируют при 37°С в течение 8 часов. Прежде чем произвести анализ гена люциферазы, культуральные среды блоттируют в лунках, после чего клетки растворяют в 60 мкл цитолитического буфера в концентрации 1:1. В каждую лунку черного 96-луночного культурального планшета переносят 40 мкл лизата и добавляют 100 мкл флуоресцеина и кофермента А. Получают по три образца для каждой концентрации. Через две минуты после добавления люциферазного субстрата на черный 96-луночный культуральный планшет при помощи флуориметра Spex fluorolog-3 измеряют интенсивность химической флуоресценции при длине волны излучения 552 нм и ширине щели 7 мм. На основании трех результатов, полученных для каждой концентрации, высчитывают среднее и стандартное отклонение.
Результаты ингибирования, полученные в данном эксперименте, определяют при помощи простой формулы баланса сродства (% активности люциферазы = 100/(1+(С/IC50) и затем высчитывают значение IC50. Результаты представлены в виде профилей ингибирования фузонексом и Т-20 слияния вирусов (фигура 5). На фигуре показано, что 50% ингибирующая концентрация (IC50) фузонекса (кружки) составляет примерно 1/20 аналогичного показателя Т-20 (квадратики). Значение IC50 равно 1,2±0,2 нМ для фузонекса и 23±6 нМ для Т-20. Другими словами, потенциал ингибирования слияния фузонексом в 20 раз выше потенциала Т-20.
В заключение следует отметить, что фузонекс представляет соединение, кэппированное с обоих концов и имеющее специфическую последовательность, состоящую из 36 аминокислот. Данный ингибитор и содержащая его композиция обладают сильной ингибирующей активностью в отношении ВИЧ-инфекции и характеризуются лучшей устойчивостью и более высоким потенциалом. Поэтому указанный ингибитор может повысить эффективность лечения при использовании более низкой дозы, благодаря чему уменьшаются побочные эффекты.
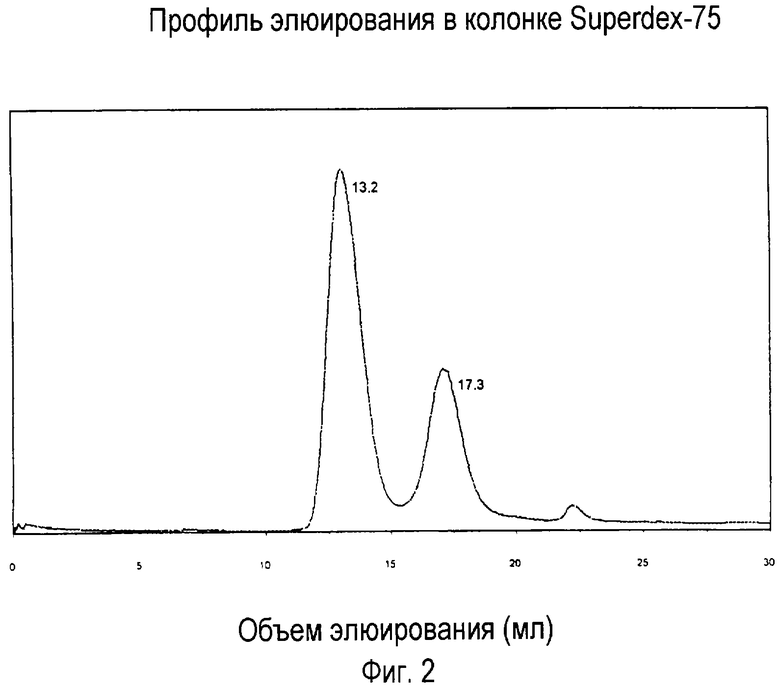
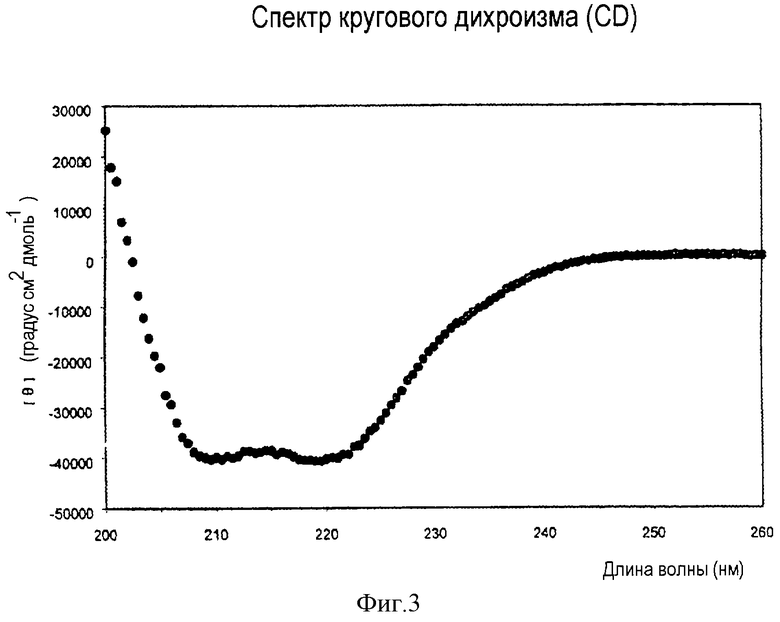
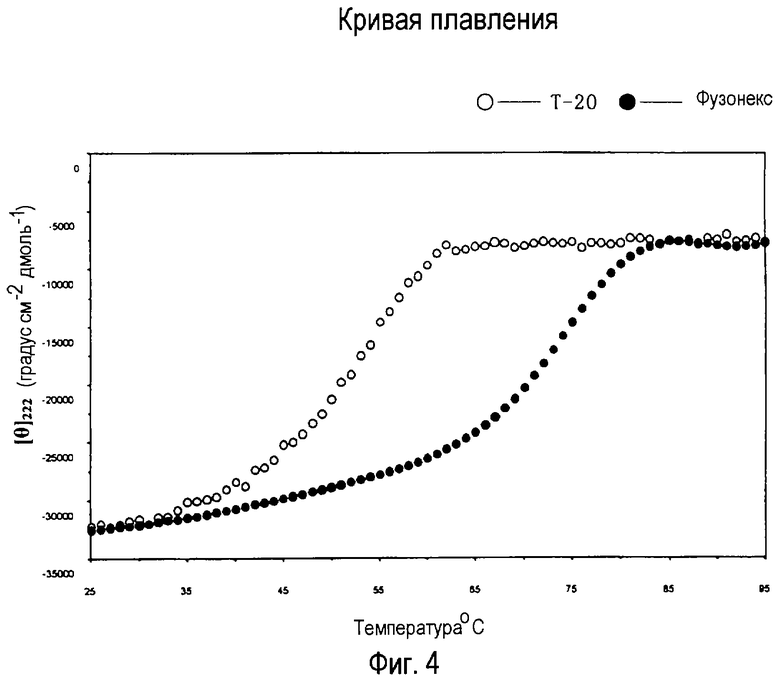

Изобретение относится к фармакологии и медицине и касается фармацевтического средства для лечения ВИЧ-инфекции и содержащее: ингибитор слияния для лечения ВИЧ-инфекции, содержащий аминокислотную последовательность:
X-SWETWEREIENYTKQIYKILEESQEQQDRNEKDLLE-Z (SEQ ID NO:1), в которой: Х представляет аминогруппу или -X1-Х2, где X1 означает иминогруппу и Х2 выбирают из группы, состоящей из ацетильной группы, гидрофобной группы и макромолекулярной несущей группы; Z представляет карбоксильную группу или -Z1-Z2, где Z1 означает карбонильную группу и Z2 выбирают из группы, состоящей из аминогруппы, трет-бутилоксикарбонильной группы, гидрофобной группы и макромолекулярной несущей группы. Указанный ингибитор обладает сильной ингибирующей активностью в отношении ВИЧ-инфекции. 5 н. и 3 з.п. ф-лы, 5 ил.
X-SWETWEREIENYTKQIYKILEESQEQQDRNEKDLLE-Z (SEQ ID NO:1),
в которой Х представляет аминогруппу или -X1-Х2, где X1 означает иминогруппу и Х2 выбирают из группы, состоящей из ацетильной группы, гидрофобной группы и макромолекулярной несущей группы;
Z представляет карбоксильную группу или -Z1-Z2, где Z1 означает карбонильную группу и Z2 выбирают из группы, состоящей из аминогруппы, трет-бутилоксикарбонильной группы, гидрофобной группы и макромолекулярной несущей группы.
| WO 9402505 A1, 03.02.1994 | |||
| FISCHER PB, KARISSON GB, et | |||
| Кровля из глиняных обожженных плит с арматурой из проволочной сетки | 1921 |
|
SU120A1 |
| Предохранительное устройство для паровых котлов, работающих на нефти | 1922 |
|
SU1996A1 |
| ROOKE R, at all | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

