Область техники
Настоящее изобретение имеет отношение к липопептиду, предназначенному для эффективного ингибирования ВИЧ, его производному или фармацевтической композиции, и их использованию в области биомедицины.
Уровень техники
Синдром приобретенного иммунодефицита (СПИД) является инфекционным заболеванием, которое в настоящее время наносит серьезный ущерб здоровью человека и развитию общества. Вирус иммунодефицита человека, вызывающий СПИД, подразделяется на два типа, т.е., ВИЧ-1 и ВИЧ-2. В мире около 36 миллионов людей, инфицированных вирусом ВИЧ, при этом ВИЧ-1 является основным патогеном (www.unСПИД.org). На сегодняшний день нет доступной эффективной вакцины против СПИД, и главную роль в лечении и предотвращении инфекции ВИЧ играют лекарственные средства, блокирующие репликацию вируса на разных стадиях. В настоящее время лекарственные средства, используемые при лечении в клинике, главным образом включают нуклеозидные ингибиторы обратной транскриптазы, ненуклеозидные ингибиторы обратной транскриптазы, ингибиторы протеаз, ингибиторы проникновения вирусов и ингибиторы интегразы (www.fda.gov). Высокоэффективный режим противовирусного лечения, получивший широкое распространение в клинике, т.е., так называемая “коктейльная” терапия, состоит главным образом из 3-4 ингибиторов обратной транскриптазы и ингибиторов протеаз. Вследствие персистенции инфекции ВИЧ, необходимо вводить лекарственные средства пациентам в течение длительного периода времени, что легко приводит к лекарственной устойчивости, которая существенно влияет на результат клинического лечения [1]. Соответственно, разработка новых анти-ВИЧ лекарственных средств является важной стратегией для предотвращения и контроля над СПИД.
В отличие от других типов лекарственных средств ингибиторы проникновения ВИЧ функционируют на ранних стадиях репликации вируса и действуют, блокируя вхождение вируса в клетку-мишень по принципу “отражение врага происходит за границей страны”, таким образом, ингибиторы проникновения ВИЧ имеют явные преимущества, как при лечении, так и при предотвращении инфицирования. Однако только два ингибитора проникновения ВИЧ в настоящее время одобрены для применения в клинике: первый является ингибитором слияния ВИЧ с мембраной энфувиртидом (также известным как T-20), который представляет собой полипептидный лекарственный препарат, имеющий 36 аминокислот, происходящих из белка слияния gp41 ВИЧ, и второй, маравирок, является антагонистом корецептора CCR5. Благодаря успешному созданию двух ингибиторов проникновения ВИЧ, добавляются новые средства для клинического лечения СПИД. К сожалению, необходимо введение T-20 в большой дозе каждый день (в виде подкожной инъекции по 90 мг два раза в день) из-за его относительно низкой активности, при этом T-20 легко приводит к лекарственной резистентности, а маравирок действует селективно против CCR5-тропного вируса и является неэффективным против CRCR4-тропного вируса [2].
Вход ВИЧ в клетки-мишени опосредуется поверхностным гликопротеином оболочки (Env), который образуется путем связывания поверхностной субъединицы gp120 с трансмембранной субъединицей gp41 через нековалентную связь и в естественном состоянии является тримерной структурой [3]. Прежде всего, поэтапное связывание gp120 с клеточным рецептором CD4 и корецептором (таким как CCR5 или CXCR4) запускает каскад изменения конформации gp120, затем экспонирует gp41 и активирует функцию мембранного слияния gp41. Gp41 структурно делится на три части: внемембранный участок, трансмембранный участок (TM) и внутримембранный участок, при этом внемембранный участок дополнительно включает несколько важных функциональных участков, таких как N-концевой участок слияния гидрофобного пептида (FP), участок N-концевого гептадного повтора (NHR), участок C-концевого гептадного повтора (CHR) и мембранно-проксимальный внешний участок (MPER) (Фигура 1). Еще в 1997 путем анализа кристаллической структуры комплекса происходящего от NHR полипептида N36 и происходящего от CHR полипептида C34 была обнаружена типичная структура в виде шести-α-спирального «пучка» (6-HB), в котором три NHR образуют расположенный в центре спиральный тример путем взаимодействия аминокислот в положениях a и d, при этом аминокислоты в положениях e и g «обнажаются» вокруг внешней поверхности центрального тримера и взаимодействуют с тремя CHR спиралями в a и d положении [4]. Три CHR спирали соответственно объединяются в бороздку, образованную тремя NHR спиралями в антипараллельной ориентации, как и в случае с трехтрубной шпилькообразной структурой. Исходя из 6-HB структуры, механизм слияния ВИЧ с мембраной становится хорошо понятным: подвергающийся воздействию пептид слияния gp41 сначала вставляется в мембрану клетки-мишени, затем CHR обратимо связывается с NHR, вирусная мембрана плотно подводится к мембране клетки-мишени, приводя к слиянию путем формирования стабильной 6-HB структуры, и таким образом, генетический материал ВИЧ в конечном итоге проникает в клетку-мишень. Структура 6-HB также указывает на то, что существует отдельный глубокий гидрофобный карман, образованный на C-конце спиралей NHR, в то время как три аминокислоты на N-конце CHR, т.е., так называемый карман-связывающий домен (PBD), вставляются в гидрофобный карман NHR, при этом взаимодействие между ними играет важную роль в стабилизации структуры 6-HB и, следовательно, является необходимым для ВИЧ инфицирования. В течение длительного времени гидрофобный карман NHR расценивался как важная мишень для лекарственных средств против ВИЧ, при этом мотив PBD CHR является ключом для создания анти-ВИЧ пептидных ингибиторов [5,6].
Предшествующие исследования показывают, что полипептиды, происходящие из CHR или NHR gp41, обладают значительной анти-ВИЧ активностью, главным образом конкурентно связываются с соответствующим NHR или CHR, чтобы предотвратить формирование вирусной 6-HB структуры, блокируя, таким образом, слияние вируса и клеточной мембраны [6]. В большинстве случаев антивирусная активность прототипа полипептида CHR является существенно более высокой, чем прототипа полипептида NHR. Лекарственное средство T-20 относится к одному из полипептидов CHR, и его последовательность показана на Фигуре 1, которая соответствует аминокислотной последовательности в положениях 127 - 162 gp41 из штамма HXB2 ВИЧ-1. Одна из структурных особенностей последовательности T-20 заключается в том, что у нее имеется гидрофобный, богатый триптофаном мотив (TRM: WASLWNWF) на C-конце, но при этом отсутствует последовательность PBD (WMEWDREI) на N-конце. При исследовании было обнаружено, что TRM T-20 опосредует связывание полипептида с липидами клеточной мембраны и поэтому считается, что он является липид-связывающим доменом (LBD), причем это свойство является важным для противовирусной активности T-20. Вследствие очевидных недостатков T-20 при клиническом применении, исследование и разработка нового поколения ингибиторов слияния ВИЧ с мембраной всегда является актуальной международной проблемой, однако большинство исследований основывается на полипептиде CHR C34, содержащем 34 аминокислоты, в качестве «матрицы», а об использовании T-20 в качестве «матрицы» сообщается редко. Это может объясняться тем, что: 1) C34 используется для анализа структуры 6-HB на начальном этапе и соответствует аминокислотной последовательности в положениях 117-150 gp41, которая, как считается, является коровой последовательностью CHR; 2) C34 содержит важную последовательность PBD на N-конце, и C34 обладает связывающей активностью и противовирусной активностью NHR, превышающей таковую у T-20; и 3) C34 обладает существенно повышенной ингибирующей активностью в отношении T-20-резистентных штаммов вируса. Все вновь созданные ингибиторы мембранного слияния ВИЧ, такие как T2635, SC35EK, SC29EK, сифувиртид (SFT), албувиртид (ABT), C34-Chol и тому подобные, получены путем оптимизации и/или модификации последовательности C34 [6,7], а также они обладают лучшей ингибирующей активностью и стабильностью, чем T-20.
Совсем недавнее открытие структуры “крючка M-T” полипептидов CHR предоставляет новый подход к созданию высокоактивных ингибиторов слияния ВИЧ с мембраной [8-10]. Эти исследования показывают, что вставка двух аминокислотных остатков (т.е., Met115 и Thr116), которые могут образовывать структуру “крючка M-T” снаружи PBD полипептида CHR, может существенно увеличить активность связывания с целевой последовательностью и противовирусную активность ингибиторов, в частности, увеличить активность ингибиторов против T-20-резистентных штаммов и значительно усилить генетический барьер в отношении лекарственной резистентности ингибиторов [11, 12]. Структура “крючка M-T” также делает возможным создание коротких пептидов, нацеленных на гидрофобный карман NHR, таких как MT-SC22EK с длиной 24 аминокислоты и HP23 и 2P23 с длиной 23 аминокислоты [13-15]. Эти короткие пептиды демонстрируют более высокую противовирусную активность и активность связывания с целевой последовательностью, чем другие полипептиды с длинной последовательностью. 2P23 не только является эффективным в отношении ВИЧ-1 и его T-20-резистентных штаммов, но также является очень эффективным в отношении ВИЧ-2 и вируса иммунодефицита обезьян (ВИО), и, таким образом, 2P23 представляет собой ингибитор слияния вируса с мембраной широкого спектра действия [13].
Липидный фундамент клеточной мембраны богат холестерином и сфингомиелином, а также многими трансмембранными белками и рецепторами (например, рецептор CD4 ВИЧ), и играет важную роль в проникновении вируса и инфицировании. С другой стороны, структура липидной бислойной мембраны оболочки вируса, происходящей из клеточной мембраны, также богата холестерином и сфингомиелином, и участвует в сохранении нормальной структуры и функции белков вирусной оболочки [16,17]. Во время проникновения ВИЧ в клетку-мишень липидный фундамент и липиды (например, холестерин и сфингомиелин), содержащиеся в нем, обеспечивают подходящую платформу для взаимодействия между gp120 вируса и клеточным рецептором CD4 или корецептором. Исследования показывают, что путем заякоривания ингибитора слияния вируса с мембраной (например, пептидов, протеинов, антител и так далее) на поверхности клеточной мембраны, локальная концентрация ингибитора на клеточной мембране может быть повышена, тем самым значительно увеличивая его противовирусную активность [18-20]. Фактически, ингибиторы слияния ВИЧ с мембраной на основе полипептида CHR, такие как T-20, T-1249 и сифувиртид, сами обладают способностью взаимодействовать с клеточной мембраной [21-23]. Peisajovich et al. показали важную роль TRM во взаимодействии между TRM T-20 и клеточной мембраной, продемонстрировав противовирусную функцию при помощи мутационного анализа аминокислотных остатков TRM T-20 и модификации липофильных функциональных групп на C-конце [24]. Экспрессия T-20 на поверхности клеточной мембраны с помощью метода рекомбинантного конструирования также может значительно увеличивать его ингибирующую активность против вируса [25, 26]. Последние исследования также показывают, что химическая модификация полипептида путем использования липидов, так называемые “липопептиды” могут увеличивать способность нацеливаться на клеточную мембрану и антивирусную активность полипептида, а также значительно улучшать стабильность и биологическое время полужизни полипептида [18-20, 27]. Исследования ингибиторов слияния мембран ВИЧ показывают, что увеличение активности полипептидов CHR зависит от C-концевой модификации, при этом N-концевая модификация подходит для полипептидов NHR, что согласуется со структурой 6-HB и механизмом слияния вируса с мембраной. Другими словами, C-концевое «заякоривание» способствует связыванию полипептида CHR с NHR вируса. В отличие от NHR полипептида, N-концевое «заякоривание» на клеточной мембране является более предпочтительным для связывания CHR вируса [19, 28, 29]. Аналогично дизайну немодифицированных полипептидов CHR, при создании липопептидов в качестве ингибиторов слияния ВИЧ с мембраной особое внимание обращается на использование C34, включающего PBD в качестве матрицы. Характерным примером является липопептид C34-Chol (см. Фигуру 1) разработанный Ingallinella et al. в 2009, он получен путем соединения холестерина с C-концом C34 посредством гибкого линкера и цистеина, и на основании результатов измерения противовирусной активности считается, что он является ингибитором слияния ВИЧ с мембраной, имеющим самую высокую активность, при этом время его метаболической полужизни у животных также является существенно увеличенным [20]. В лаборатории авторов изобретения три липидных соединения, пальмитиновая кислота (C16), холестерин и дигидросфингозин, используются для модификации коротких пептидов HP23 и HP23L, нацеленных на карманы NHR, соответственно, для получения группы липопептидов, имеющих высокую активность, при этом in vivo стабильность LP-11 также значительно повышается [18]. Недавно в лаборатории авторов изобретения получен липопептид, модифицированный пальмитиновой кислотой, LP-19, на основе короткого анти-ВИЧ пептида 2P23 широкого спектра действия, обладающий более высокой противовирусной активностью и высокрй специфичностью [30]. Эти достижения в исследованиях обеспечивают твердую теоретическую основу и намечают технические пути для разработки способов для проектирования новых ингибиторов слияния ВИЧ с мембраной.
Ссылки:
1. Flexner C. ВИЧ drug development: the next 25 years. Nat Rev Drug Discov 2007,6:959-966.
2. Este JA, Telenti A. ВИЧ entry inhibitors. Lancet 2007,370:81-88.
3. Eckert DM, Kim PS. Mechanisms of viral membrane fusion and its inhibition. Annu Rev Biochem 2001,70:777-810.
4. Chan DC, Fass D, Berger JM, Kim PS. Core structure of gp41 from the ВИЧ envelope glycoprotein. Cell 1997,89:263-273.
5. Chan DC, Chutkowski CT, Kim PS. Evidence that a prominent cavity in the coiled coil of ВИЧ type 1 gp41 is an attractive drug target. Proc Natl Acad Sci U S A 1998,95:15613-15617.
6. He Y. Synthesized peptide inhibitors of ВИЧ-1 gp41-dependent membrane fusion. Curr Pharm Des .2013,19:1800-1809.
7. Eggink D, Berkhout B, Sanders RW. Inhibition of ВИЧ-1 by fusion inhibitors. Curr Pharm Des 2010,16:3716-3728.
8. Chong H, Yao X, Sun J, Qiu Z, Zhang M, Waltersperger S, et al. The M-T hook structure is critical for design of ВИЧ-1 fusion inhibitors. J Biol Chem 2012,287:34558-34568.
9. Chong H, Qiu Z, Su Y, He Y. The N-Terminal T-T Motif of a Third-Generation ВИЧ-1 Fusion Inhibitor Is Not Required for Binding Affinity and Antiviral Activity. J Med Chem 2015,58:6378-6388.
10.Chong H, Yao X, Qiu Z, Qin B, Han R, Waltersperger S, et al. Discovery of critical residues for viral entry and inhibition through structural Insight of ВИЧ-1 fusion inhibitor CP621-652. J Biol Chem 2012,287:20281-20289.
11. Chong H, Yao X, Qiu Z, Sun J, Qiao Y, Zhang M, et al. The M-T hook structure increases the potency of ВИЧ-1 fusion inhibitor sifuvirtide and overcomes drug resistance. J Antimicrob Chemother 2014,69:6759.
12. Chong H, Qiu Z, Sun J, Qiao Y, Li X, He Y. Two M-T hook residues greatly improve the противовирусная активность and resistance profile of the ВИЧ-1 fusion inhibitor SC29EK. Retrovirology 2014,11:40.
13. Xiong S, Borrego P, Ding X, Zhu Y, Martins A, Chong H, et al. A helical short-peptide fusion inhibitor with highly potent activity against human immunodeficiency virus type 1 (ВИЧ-1), ВИЧ-2, and simian immunodeficiency virus. J Virol 2017,91:e01839-16.
14. Chong H, Qiu Z, Su Y, Yang L, He Y. Design of a highly potent ВИЧ-1 fusion inhibitor targeting the gp41 pocket. СПИД 2015,29:13-21.
15. Chong H, Yao X, Qiu Z, Sun J, Zhang M, Waltersperger S, et al. Short-peptide fusion inhibitors with high potency against wild-type and enfuvirtide-resistant ВИЧ-1. FASEB J 2013,27:1203-1213.
16. Brugger B, Glass B, Haberkant P, Leibrecht I, Wieland FT, Krausslich HG. The ВИЧ lipidome: a raft with an unusual composition. Proc Natl Acad Sci U S A 2006,103:2641-2646.
17. Ono A, Freed EO. Plasma membrane rafts play a critical role in ВИЧ-1 assembly and release. Proc Natl Acad Sci U S A 2001,98:13925-13930.
18. Chong H, Wu X, Su Y, He Y. Development of potent and long-acting ВИЧ-1 fusion inhibitors. СПИД 2016,30:1187-1196.
19. Wexler-Cohen Y, Shai Y. Membrane-anchored ВИЧ-1 N-heptad repeat peptides are highly potent cell fusion inhibitors via an altered mode of action. PLoS Pathog 2009,5:e1000509.
20. Ingallinella P, Bianchi E, Ladwa NA, Wang YJ, Hrin R, Veneziano M, et al. Addition of a cholesterol group to an ВИЧ-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc Natl Acad Sci U S A 2009,106:5801-5806.
21. Matos PM, Castanho MA, Santos NC. ВИЧ-1 fusion inhibitor peptides enfuvirtide and T-1249 interact with erythrocyte and lymphocyte membranes. PLoS One 2010,5:e9830.
22. Franquelim HG, Loura LM, Santos NC, Castanho MA. Sifuvirtide screens rigid membrane surfaces. establishment of a correlation between efficacy and membrane domain selectivity among ВИЧ fusion inhibitor peptides. J Am Chem Soc 2008,130:6215-6223.
23. Veiga AS, Santos NC, Loura LM, Fedorov A, Castanho MA. ВИЧ fusion inhibitor peptide T-1249 is able to insert или adsorb to lipidic bilayers. Putative correlation with improved efficiency. J Am Chem Soc 2004,126:14758-14763.
24. Peisajovich SG, Gallo SA, Blumenthal R, Shai Y. C-terminal octylation rescues an inactive T20 mutant: implications for the mechanism of ВИЧ/SIMIAN immunodeficiency virus-induced membrane fusion. J Biol Chem 2003,278:21012-21017.
25. Hildinger M, Dittmar MT, Schult-Dietrich P, Fehse B, Schnierle BS, Thaler S, et al. Membrane-anchored peptide inhibits human immunodeficiency virus entry. J Virol 2001,75:3038-3042.
26. Egelhofer M, Brandenburg G, Martinius H, Schult-Dietrich P, Melikyan G, Kunert R, et al. Inhibition of human immunodeficiency virus type 1 entry in cells expressing gp41-derived peptides. J Virol 2004,78:568-575.
27. Ashkenazi A, Viard M, Unger L, Blumenthal R, Shai Y. Sphingopeptides: дигидросфингозин-based fusion inhibitors against wild-type and enfuvirtide-resistant ВИЧ-1. FASEB J 2012,26:4628-4636.
28. Wexler-Cohen Y, Shai Y. Demonstrating the C-terminal boundary of the ВИЧ 1 fusion conformation in a dynamic ongoing fusion process and implication for fusion inhibition. FASEB J 2007,21:3677-3684.
29. Wexler-Cohen Y, Ashkenazi A, Viard M, Blumenthal R, Shai Y. Virus-cell and cell-cell fusion mediated by the ВИЧ-1 envelope glycoprotein is inhibited by short gp41 N-terminal membrane-anchored peptides lacking the critical pocket domain. FASEB J 2010,24:4196-4202.
30. Chong H,Xue J, Xiong S,Cong Z,Ding X,Zhu Y,Liu Z,Chen T,Feng Y,He L,Guo Y,Wei Q,Zhou Y,Qin C,He Y. Липопептид ВИЧ-1/2fusion inhibitor with highly potent in vitro, ex vivo and in vivo antiviral activity. J Virol 2017, 91: e00288-17.
Раскрытие настоящего изобретения
Техническая задача, подлежащая решению при помощи настоящего изобретения, заключается в определении того, как эффективно ингибировать ВИЧ.
Для решения вышеуказанной технической задачи настоящее изобретение предоставляет эффективный ингибитор слияния ВИЧ с мембраной. Эффективный ингибитор слияния ВИЧ с мембраной, предоставленный настоящим изобретением, представляет собой липопептид, обладающий сильной ингибирующей активностью в отношении ВИЧ, его фармацевтически приемлемую соль или его производное, при этом липопептид является следующим a) или b):
a) липопептид, образованный путем соединения полипептида, обладающего противовирусной активностью, с липофильным соединением, соединенным с карбоксильным концом полипептида;
b) липопептид, образованный связыванием обладающего противовирусной активностью полипептида с концевой защитной группой и липофильного соединения, связанного с карбоксильным концом полипептида, при этом концевая защитная группа является защитной амино-концевой группой и/или защитной карбокси-концевой группой;
в a) и b), полипептид представляет собой что-либо одно из P1 - P5;
P1 имеет последовательность, показанную в следующей Формуле I,
Формула I
X1X2X3X4X5X6X7X8X9X10X11X12X13X14X15X16X17X18X19X20X21X22X23X24X25X26X27X28
в Формуле I,
X1 - X28 каждый является аминокислотным остатком, X1 представляет собой W, L или Y, X2 представляет собой E или T, X3 представляет собой Q, A или S, X4 представляет собой K, N или L, X5 представляет собой I или L, X6 представляет собой E, D, K, R или A, X7 представляет собой E, D, K, R или A, X8 представляет собой L или I, X9 представляет собой L или I, X10 представляет собой K, R, E, D или A, X11 представляет собой K, R, E, D или A, X12 представляет собой A или S, X13 представляет собой E, D, K, R или A, X14 представляет собой E, D, K, R или A, X15 представляет собой Q, X16 представляет собой Q, X17 представляет собой K, R, E, D или A, X18 представляет собой K, R, E, D или A, X19 представляет собой N, X20 представляет собой E или D, и X21 представляет собой E, D, K, R или A, X22 представляет собой E, D, K, R или A, X23 представляет собой L или I, X24 представляет собой K, R, E, D или A, X25 представляет собой K, R, E, D или A, X26 представляет собой L или I, X27 представляет собой E или D, X28 представляет собой K или R;
P2 представляет собой полипептид, полученный делецией от 1 до 4 аминокислотных остатков на амино-конце P1 (т.е., от 1 до 4 из четырех аминокислотных остатков X1, X2, X3 и X4 в Формуле I);
P3 представляет собой полипептид, полученный делецией от 1 до 3 аминокислотных остатков на карбоксильном конце P1 (т.е., от 1 до 3 из трех аминокислотных остатков X26, X27 и X28 в Формуле I);
P4 представляет собой полипептид, полученный путем вставки остатка цистеина на карбоксильном конце P1;
P5 имеет последовательность, показанную в следующей Формуле II,
Формула II
X5X6X7X8X9X10X11X12X13X14X15X16X17X18X19X20X21X22X23X24X25
в Формуле II определения X5 - X25 являются такими же, как в Формуле I;
полипептид или липопептид, обладающий противовирусной активностью в отношении любого одного вируса, выбранного из группы, состоящей из следующего v1-v7:
v1: ВИЧ-1,
ВИЧ-2 и ВИО;
v2: ВИЧ-1 и ВИЧ-2;
v3: ВИЧ-1 и ВИО;
v4: ВИЧ-2 и ВИО;
v5: ВИЧ-1;
v6: ВИЧ-2; и
v7: ВИО.
Экспериментально подтверждено, что указанный выше P5 является коровой последовательностью липопептида настоящего изобретения. Противовирусная активность коровой последовательности существенно улучшается путем вставки от 1 до 4 аминокислотных остатков на ее N-конце и/или вставки от 1 до 3 аминокислотных остатков на ее C-конце.
В упомянутом выше липопептиде, его фармацевтически приемлемой соли, его производном, липопептид обладает более высокой противовирусной активностью, чем LP-19 и/или T-20 и/или C34-Chol.
P1 имеет последовательность, показанную в следующей последовательности::
X1X2X3X4IEELX9KKX12EEQQKKNEEELKKLEK;
P2 представляет собой P2-1, P2-2, P2-3 или P2-4, при этом
P2-1 имеет последовательность, как показано в следующей последовательности:
X2X3X4IEELX9KKX12EEQQKKNEEELKKLEK;
P2-2 имеет последовательность, как показано в следующей последовательности:
X3X4IEELX9KKX12EEQQKKNEEELKKLEK;
P2-3 имеет последовательность, как показано в следующей последовательности:
X4IEELX9KKX12EEQQKKNEEELKKLEK;
P2-4 имеет последовательность, как показано в следующей последовательности:
IEELX9KKX12EEQQKKNEEELKKLEK;
P3 имеет последовательность, как показано в следующей последовательности:
X1X2X3X4IEELX9KKX12EEQQKKNEEELKK;
P4 имеет последовательность, как показано в следующей последовательности:
X1X2X3X4IEELX9KKX12EEQQKKNEEELKKLEKC;
в P1, P2-1, P2-2, P2-3, P2-4, P3 и P4 определения X1, X2, X3, X4, X9 и X12 являются такими же, как в Формуле I.
В вышеупомянутом липопептиде, его фармацевтически приемлемой соли или его производном, исключая Xn (n представляет собой натуральное число от 1 до 28) в последовательностях полипептида, каждая из заглавных букв представляет собой обозначение аминокислоты, при этом аббревиатура аминокислоты имеет значение, хорошо известное в данной области техники, например: Y - тирозин, T - треонин, S – серин, L -лейцин, I – изолейцин, E – глутаминовая кислота, K - лизин, Q - глутамин, N - аспарагин, A – аланин и W - триптофан. Все аминокислоты в последовательностях полипептидов могут быть L-формой аминокислот, при этом одна или более (например, 2-5, 2-4 или 2-3) из аминокислот могут быть замещены D-формой аминокислоты (кислот), искусственно модифицированной аминокислотой(ами), редко встречающейся в природе аминокислотой(ами) и т.д. с целью улучшения биодоступности, устойчивости и/или противовирусной активности полипептидов, при этом D-форма аминокислоты относится к аминокислоте соответствующей аминокислоты L-формы, составляющей протеин; искусственно модифицированная аминокислота относится к обычной L-форме аминокислоты, которая составляет протеин, и модифицируется посредством метилирования, фосфорилирования и т.п.; и редкая аминокислота, встречающаяся в природе, включает необычные аминокислоты, составляющие протеин, и аминокислоту, не входящую в состав протеина, например, 5-гидроксилизин, метилгистидин, гамма аминомасляную кислоту, гомосерин и т.д.
В вышеупомянутом липопептиде, его фармацевтически приемлемой соли или его производном P1 представляет собой P-80/84/85/52, P-87/51 или P50, при этом P-80/84/85/52 является полипептидом, представленным последовательностью SEQ ID NO: 1 в перечне последовательностей (т.е., полипептид, представленный аминокислотными остатками в положениях 1 - 28 LP-80, LP-84, LP-85 или LP-52 на Фигуре 2), P-87/51 является полипептидом, представленным последовательностью SEQ ID NO: 2 в перечне последовательностей (т.е., полипептид, представленный аминокислотными остатками в положениях 1 - 28 LP-87 или LP-51 на Фигуре 2), и P50 является полипептидом, представленным последовательностью SEQ ID NO: 3 в перечне последовательностей (т.е., полипептид, представленный аминокислотными остатками в положениях 1-28 LP-50 на Фигуре 2). P2-1 представляет собой P-88/62, при этом P-88/62 является полипептидом, представленным последовательностью SEQ ID NO: 4 в перечне последовательностей (т.е., полипептид, представленный аминокислотными остатками в положениях 1 - 27 LP-88 или LP-62 на Фигуре 2). P2-2 представляет собой P63 или P60, при этом P63 является полипептидом, представленным последовательностью SEQ ID NO: 5 в перечне последовательностей (т.е., полипептид, представленный аминокислотными остатками в положениях 1 - 26 LP-63 на Фигуре 2), и P60 представляет собой полипептид, представленный последовательностью SEQ ID NO: 6 в перечне последовательностей (т.е., полипептид, представленный аминокислотными остатками в положениях 1 - 26 LP-60 на Фигуре 2). P2-3 представляет собой P-89/64, при этом P-89/64 является полипептидом, представленным последовательностью SEQ ID NO: 7 в перечне последовательностей (т.е., полипептид, представленный аминокислотными остатками в положениях 1 - 25 LP-89 или LP-64 на Фигуре 2). P2-4 представляет собой P-90/65 или P61, при этом P-90/65 является полипептидом, представленным последовательностью SEQ ID NO: 8 в перечне последовательностей (т.е., полипептид, представленный аминокислотными остатками в положениях 1 - 24 LP-90 или LP-65 на Фигуре 2); и P61 полипептид, представленный последовательностью SEQ ID NO: 9 в перечне последовательностей (т.е., полипептид, представленный аминокислотными остатками в положениях 1 - 24 LP-61 на Фигуре 2). P3 представляет собой P-91/55, при этом P-91/55 является полипептидом, представленным последовательностью SEQ ID NO: 10 в перечне последовательностей (т.е., полипептид, представленный аминокислотными остатками в положениях 1 - 25 LP-91 или LP-55 на Фигуре 2). P4 представляет собой P83 или P86, при этом P83 является полипептидом, представленным последовательностью SEQ ID NO: 11 в перечне последовательностей (т.е., полипептид, представленный аминокислотными остатками в положениях 1 - 29 LP-83 на Фигуре 2), и P86 является полипептидом, представленным последовательностью SEQ ID NO: 12 в перечне последовательностей (т.е., полипептид, представленный аминокислотными остатками в положениях 1 - 29 LP-86 на Фигуре 2).
В вышеупомянутом липопептиде, его фармацевтически приемлемой соли или его производном липофильное соединение может быть жирной кислотой, содержащей от 8 до 20 атомов углерода, холестерин (Chol), дигидросфингозин (DHS), витамин E (токоферол, Toc) и т.д.
В вышеупомянутом липопептиде, его фармацевтически приемлемой соли или его производном жирная кислота, содержащая от 8 до 20 атомов углерода, может быть пальмитиновой кислотой (также известной как гексадекановая кислота) (C16) или стеариновой кислотой (C18).
В вышеупомянутом липопептиде, его фармацевтически приемлемой соли или его производном липофильное соединение может быть связано с боковой цепью концевой аминокислоты или может непосредственно связываться c пептидной цепью. Модификация жирной кислотой, дигидросфингозином или витамином E в качестве липофильного соединения, связанного с C-концом, может осуществляться при помощи реакции амидирования аминогруппой лизина (Lys) боковой цепи на конце полипептида, и модификация холестерином может осуществляться путем «пересадки» холестерина на полипептидную цепь за счет реакции образования тиоэфира с высокой химической селективностью, которая протекает между тиоловой группой цистеина (Cys) боковой цепи на конце полипептида и холестерилом бромацетатом.
В упомянутом выше липопептиде, его фармацевтически приемлемой соли или его производном липопептид может быть каким-либо одним из числа следующих 12 липопептидов LP-80/84/85/52, LP-90/65, LP-87/51, LP-88/62, LP-50, LP-83, LP-91/55, LP-86, LP-63, LP-89/64, LP-60 и LP-61.
LP-80/84/85/52 представляет собой LP-80/84/85/52a или LP-80/84/85/52b, при этом LP-80/84/85/52a образуется путем связывания P-80/84/85/52 с липофильным соединением, связанным с карбоксильным концом P-80/84/85/52; LP-80/84/85/52b образуется путем связывания LP-80/84/ 85/52a с концевой защитной группой; в LP-80/84/85/52a и LP-80/84/85/52b липофильное соединение является стеариновой кислотой, дигидросфингозином, витамином E или пальмитиновой кислотой.
LP-90/65 представляет собой LP-90/65a или LP-90/65b, при этом LP-90/65a образуется путем связывания P-90/65 с липофильным соединением, связанным с карбоксильным концом P-90/65; LP-90/65b образуется путем связывания LP-90/65a с концевой защитной группой; в LP-90/65a и LP-90/65b липофильное соединение является стеариновой кислотой или пальмитиновой кислотой.
LP-87/51 представляет собой LP-87/51a или LP-87/51b, при этом LP-87/51a образуется путем связывания P-87/51 с липофильным соединением, связанным с карбоксильным концом P-87/51; LP-87/51b образуется путем связывания LP-87/51a с концевой защитной группой; в LP-87/51a и LP-87/51b, липофильное соединение является дигидросфингозином или пальмитиновой кислотой.
LP-88/62 представляет собой LP-88/62a или LP-88/62b, при этом LP-88/62a образуется путем связывания P-88/62 с липофильным соединением, связанным с карбоксильным концом P-88/62; LP-88/62b образуется путем связывания LP-88/62a с концевой защитной группой; в LP-88/62a и LP-88/62b липофильное соединение является стеариновой кислотой или пальмитиновой кислотой.
LP-50 представляет собой LP-50a или LP-50b, при этом LP-50a образуется путем связывания P-50 с пальмитиновой кислотой, соединенной с карбоксильным концом P-50; LP-50b образуется путем связывания LP-50a с концевой защитной группой.
LP-83 представляет собой LP-83a или LP-83b, при этом LP-83a образуется путем связывания P-83 с холестерином, соединенным с карбоксильным концом P-83; LP-83b образуется путем связывания LP-83a с концевой защитной группой.
LP-91/55 представляет собой LP-91/55a или LP-91/55b, при этом LP-91/55a образуется путем связывания P-91/55 с липофильным соединением, связанным с карбоксильным концом P-91/55; LP-91/55b образуется путем связывания LP-91/55a с концевой защитной группой; в LP-91/55a и LP-91/55b липофильное соединение является стеариновой кислотой или пальмитиновой кислотой.
LP-86 представляет собой LP-86a или LP-86b, при этом LP-86a образуется путем связывания P-86 с холестерином, соединенным с карбоксильным концом P-86; LP-86b образуется путем связывания LP-86a с концевой защитной группой. LP-63 представляет собой LP-63a или LP-63b, при этом LP-63a образуется путем связывания P-63 с пальмитиновой кислотой, соединенной с карбоксильным концом P-63; LP-63b образуется путем связывания LP-63a с концевой защитной группой. LP-89/64 представляет собой LP-89/64a или LP-89/64b, при этом LP-89/64a образуется путем связывания P-89/64 с липофильным соединением, связанным с карбоксильным концом P-89/64; LP-89/64b образуется путем связывания LP-89/64a с концевой защитной группой; в LP-89/64a и LP-89/64b липофильное соединение представляет собой стеариновую кислоту или пальмитиновую кислоту.
LP-60 представляет собой LP-60a или LP-60b, при этом LP-60a образуется путем связывания P-60 с пальмитиновой кислотой, соединенной с карбоксильным концом P-60; LP-60b образуется путем связывания LP-60a с концевой защитной группой. LP-61 представляет собой LP-61a или LP-61b, при этом LP-61a образуется путем связывания P-61 с пальмитиновой кислотой, соединенной с карбоксильным концом P-61; LP-61b образуется путем связывания LP-61a с концевой защитной группой.
В указанном выше липопептиде, его фармацевтически приемлемой соли или его производном липопептид настоящего изобретения может содержать N-концевую защитную группу на амино-конце, при этом N-концевая защитная группа может быть любой группой, выбранной из ацетила, аминогруппы, малеоила, сукцинила, трет-бутоксикарбонила, бензилоксигруппы, другой гидрофобной группы и группы высокомолекулярного носителя; липопептид настоящего изобретения может содержать C-концевую защитную группу на карбоксильном конце, при этом C-концевая защитная группа может быть любой группой, выбранной из числа аминогруппы, амидогруппы, карбоксильной группы, трет-бутоксикарбонила, другой гидрофобной группы и группы высокомолекулярного носителя.
Любой полипептид, выбранный из группы, состоящей из вышеуказанных P1 - P4, его фармацевтически приемлемая соль или его производное также включены в рамки настоящего изобретения.
Производное полипептида может быть, по меньшей мере, одним, выбранным из группы, состоящей из следующего 1) - 5):
1) производного, полученного путем связывания N-концевой защитной группы с амино-концом полипептида и/или путем связывания C-концевой защитной группы c карбоксильным концом полипептида;
2) производного, полученного путем связывания олигопептида или липофильного соединения с карбоксильным концом полипептида;
3) производного, полученного путем связывания олигопептида или липофильного соединения с амино-концом полипептида;
4) производного, полученного путем связывания олигопептида или липофильного соединения с двумя концами, как с карбоксильным концом, так и с амино-концом полипептида; и
5) производного, полученного путем модификации полипептида белком, полиэтиленгликолем или малеимидом.
Мультимер PM1 или PM2 также включено в рамки настоящего изобретения, при этом
PM1 представляет собой мультимер, образованный липопептидом, его фармацевтически приемлемой солью или его или производным; и
PM2 представляет собой мультимер, образованный полипептидом, его фармацевтически приемлемой солью или его или производным.
Следующая композиция также включено в рамки настоящего изобретения. Композиция, содержащая C1) и C2), причем
C1) представляет собой C11), C12) или/и C13), C11) представляет собой липопептид, производное, или его фармацевтически приемлемую соль, C12) представляет собой полипептид, его производное или его фармацевтически приемлемую соль, C13) представляет собой мультимер;
C2) является фармацевтически приемлемым носителем или адьювантом;
Композиция имеет, по меньшей мере, одну функцию из числа следующих функций F1)-F5):
F1) обладает противовирусной активностью;
F2) лечит и/или предотвращает и/или используется для адьюнктивного лечения болезни, вызванной вирусной инфекцией;
F3) ингибирует слияние вируса и клетки;
F4) ингибирует проникновение вируса в клетку; и
F5) ингибирует репликацию вируса;
в F1)-F5) вирус является одним вирусом, выбранным из группы, состоящей из следующего v1-v7:
v1: ВИЧ-1, ВИЧ-2 и ВИО;
v2: ВИЧ-1 и ВИЧ-2;
v3: ВИЧ-1 и ВИО;
v4: ВИЧ-2 и ВИО;
v5: ВИЧ-1;
v6: ВИЧ-2; и
v7: ВИО.
Применение C11), C12), C13) и/или C14) в производстве, по меньшей мере, одного продукта, выбранного из группы, состоящей из E1)-E5), также включено в рамки настоящего изобретения, при этом
C14) является композицией;
E1) является продуктом против вируса, таким как лекарственное средство или вакцина;
E2) является продуктом, таким как лекарственное средство или вакцина, для лечения и/или предотвращения и/или дополнительного (адьюнктивного) лечения болезни, вызванной вирусной инфекцией, такой как СПИД;
E3) является продуктом для ингибирования слияния вируса и клетки, таким как лекарственное средство или вакцина;
E4) является продуктом для ингибирования проникновения вируса в клетку, таким как лекарственное средство или вакцина; и
E5) является продуктом для ингибирования репликации вируса, таким как лекарственное средство или вакцина;
в E1)-E5) вирус является одним вирусом, выбранным из группы, состоящей из v1-v7:
v1: ВИЧ-1, ВИЧ-2 и ВИО;
v2: ВИЧ-1 и ВИЧ-2;
v3: ВИЧ-1 и ВИО;
v4: ВИЧ-2 и ВИО;
v5: ВИЧ-1;
v6: ВИЧ-2; и
v7: ВИО.
Настоящее изобретение предоставляет фармацевтическое соединение.
Фармацевтическое соединение, предоставленное настоящим изобретением, представляет собой C11), C12) или C13).
Вышеупомянутое фармацевтическое соединение имеет, по меньшей мере, один из следующих вариантов применения U1)-U5):
U1) применение против вируса
U2) применение для лечения и/или предотвращения и/или адьюнктивной терапии болезни, вызванной вирусной инфекцией (такой как СПИД);
U3) применение для ингибирования слияния вируса и клетки;
U4) применение для ингибирования проникновения вируса в клетку; и
U5) применение для ингибирования репликации вируса;
в U1)-U5) вирус является одним вирусом, выбранным из группы, состоящей из следующего v1-v7:
v1: ВИЧ-1, ВИЧ-2 и ВИО;
v2: ВИЧ-1 и ВИЧ-2;
v3: ВИЧ-1 и ВИО;
v4: ВИЧ-2 и ВИО;
v5: ВИЧ-1;
v6: ВИЧ-2; и
v7: ВИО.
Способ лечения или/и предотвращения инфекции, вызванной вирусом, у животного также включено в охраняемые рамки настоящего изобретения.
Способ лечения или/и предотвращения инфекции, вызванной вирусом, у животного включает введение животному C11), C12), C13) или/и C14) с целью ингибирования вирусной инфекции у животного, при этом
C14) является композицией; и
вирус является одним вирусом, выбранным из группы, состоящей из следующего v1-v7:
v1: ВИЧ-1, ВИЧ-2 и ВИО;
v2: ВИЧ-1 и ВИЧ-2;
v3: ВИЧ-1 и ВИО;
v4: ВИЧ-2 и ВИО;
v5: ВИЧ-1;
v6: ВИЧ-2; и
v7: ВИО.
Фармацевтически приемлемая соль липопептида и фармацевтически приемлемая соль полипептида включает согласно настоящему изобретению ацетат, лактобионат, бензолсульфонат, лаурат, бензоат, малат, бикарбонат, малеат, бисульфат, манделат, битартрат, мезилат, борат, метилбромид, бромид, метилнитрат, кальция эдетат, метилсульфат, камсилат, мукат, карбонат, напсилат, хлорид, нитрат, коавуланат, N-метилглюкамин, цитрат, аммониевую соль, дигидрохлорид, олеат, эдетат, оксалат, эдизилат, памоат, эмбонат, эстолат, пальмитат, эзилат, пантотенат, фумарат, фосфат/дифосфат, глюцептат, полигалактуронат, глюконат, салицилат, глютамат, стеарат, гликолиларсанилат, сульфат, гексилрезорцинат, субацетат, гидрабамин, сукцинат, гидробромид, таннат, гидрохлорид, тартрат, гидроксинафтоат, теоклат, иодид, тозилат, триэтиодид, лактат и валерат и т.д. В зависимости от применения фармацевтически приемлемая соль может быть образована из таких катионов как натрий, калий, алюминий, кальций, литий, магний, цинк и висмут, или может быть образована из таких оснований, как аммоний, этилендиамид, N-метил-глутамин, лизин, аргинин, орнитин, холин, N,N’-дибензилэтилен-диамин, хлорпрокаин, диэтаноламин, прокаин, диэтиламин, пиперазин, трис(гидроксиметиламинометан) и тетраметиламмоний гидроксид. Эти соли могут быть получены стандартными способами, например, путем взаимодействия свободной кислоты с органическим или неорганическим основанием. В присутствии основной группы (например, аминогруппы) кислая соль, такая как гидрохлорид, гидробромид, ацетат, памоат или тому подобное может использоваться как форма лекарственного средства; в присутствии кислотной группы (например, -COOH) или спиртовой группы фармацевтически приемлемый сложный эфир, такой как ацетат, малеат, пивалоилоксиметил, и сложный эфир, известный в литературе как улучшающий растворимость и гидролизуемость, может использоваться в форме с замедленным высвобождением лекарственного средства или пролекарства.
В настоящем изобретении противовирусная активность также может называться ингибирующей активностью в отношении вируса, в частности, ингибирующей слияние вируса и клетки и/или ингибирующей проникновение вируса в клетку и/или ингибирующей репликацию вируса. Значительный противовирусный эффект длительного действия проявляется у приматов, кроме человека (обезьян).
Липопептид или полипептид, его производное или его фармацевтически приемлемая соль, мультимер, композиция или фармацевтическое соединение, предоставленное настоящим изобретением, может использоваться для лечения инфекции ВИЧ (ВИЧ-1 и/или ВИЧ-2) и/или инфекции ВИО, включая различные стадии инфекции ВИЧ и/или инфекции ВИО, такие как начальная стадия, симпатическая стадия и бессимптомная стадия СПИД. Липопептид или полипептид, его производное или его фармацевтически приемлемая соль, мультимер, композиция или фармацевтическое соединение, предоставленное настоящим изобретением, также может использоваться для предотвращения инфекции ВИЧ (ВИЧ-1 и/или ВИЧ-2) и/или инфекции ВИО, включая профилактику до или после вызывающего подозрения внешнего воздействия, такого как переливание крови, трансплантация органа, обмен биологической жидкостью, укус, случайный укол иглой или воздействие на кровь пациента во время хирургической операции.
На практике липопептид или полипептид, его производное или его фармацевтически приемлемая соль, мультимер, композицию или фармацевтическое соединение согласно настоящему изобретению можно вводить пациенту в качестве медикамента или непосредственно или в смеси с подходящим носителем или эксципиентом с целью лечения и/или профилактики инфекции ВИЧ. Материал носителя согласно данному документу включает, но не ограничивается этим, водорастворимый материал для носителя (например, полиэтиленгликоль, поливинилпирролидон, органическую кислоту и т.д.), малорастворимый материал для носителя (например, этил целлюлоза, холетерин стеарат и т.д.), кишечнорастворимый материал для носителя (например, целлюлозы ацетатфталат, карбоксиметилцеллюлозу и т.д.), при этом предпочтительным является водорастворимый материал для носителя. Используя эти материалы, можно приготовить различные формы препаратов, включая, но не ограничиваясь этим, таблетки, капсулы, микропилюли, аэрозоль, пилюли, порошок, раствор, суспензию, эмульсию, гранулу, липосому, средство для трансдермальной доставки, буккальные таблетки, суппозитории, лиофилизированный порошок для инъекций и тому подобное, при этом суппозиторий может быть вагинальным суппозиторием, вагинальным кольцом или мазью, кремом или гелем, пригодным для вагинального применения. Препарат может иметь форму обычного препарата, форму препарата с замедленным высвобождением, препарата с контролируемым высвобождением и различных систем доставки с помощью частиц. Для создания стандартной лекарственной формы в виде таблеток может быть использован целый ряд носителей, известных в данной области техники. Примерами носителей являются, например, разбавитель и абсорбент, такой как крахмал, декстрин, сульфат кальция, лактоза, маннитол, хлорид натрия, глюкоза, мочевина, карбонат кальция, каолин, микрокристаллическая целлюлоза и силикат алюминия; увлажняющее вещество и связывающее вещество, такое как вода, глицерин, полиэтиленгликоль, этанол, пропанол, крахмальная суспензия, декстрин, сироп, мед, раствор глюкозы, гуммиарабик, желатиновый гель, натрий карбоксиметилцеллюлоза, шеллак, метилцеллюлоза, фосфат калия и поливинилпирролидон; разрыхлитель, такой как сухой крахмал, альгинат, агаровый порошок, крахмал бурых водорослей, натрия бикарбонат и лимонная кислота, карбонат кальция, полиоксиэтилен, сложный эфир сорбита и жирной кислоты, додецилсульфат натрия, метилцеллюлоза и этилцеллюлоза; ингибитор распадаемости, такой как сахароза, глицерил тристеарат, масло какао, гидрогенезированное масло и тому подобное; стимуляторы абсорбции, такие как соли четвертичного аммония и лаурил сульфат натрия; смазывающее вещество, такое как тальк, диоксид кремния, кукурузный крахмал, стеарат, борная кислота, жидкий парафин и полиэтиленгликоль. Таблетки, кроме того, могут быть созданы в виде таблеток, покрытых оболочкой, таких как, например, таблетки, покрытые сахарной оболочкой, таблетки с пленочным покрытием, таблетки, покрытые кишечнорастворимой оболочкой, или таблетки с двойным или многослойным покрытием. Для получения стандартной формы препарата в виде пилюли можно использовать большое разнообразие носителей, известных в данной области. Примерами носителей являются, например, разбавитель и адсорбирующее вещество, такое как глюкоза, лактоза, крахмал, масло какао, гидрогенезированное растительное масло, поливинилпирролидон, гелуцир, каолин и тальк; связующее вещество, такое как гуммиарабик, трагакантовая камедь, желатин, этанол, мед, жидкий сахар, рисовая паста и клейстер; разрыхлитель, такой как агаровый порошок, сухой крахмал, альгинат, натрий додецилсульфат, метилцеллюлоза и этилцеллюлоза. Для получения стандартной формы препарата в виде суппозитория можно использовать большое разнообразие носителей, известных в данной области. Примерами носителя являются, например, полиэтиленгликоль, лецитин, масло какао, высший спирт, сложный эфир высшего спирта, желатин и полусинтетический глицерид. Для получения стандартной формы препарата в виде препарата для инъекций, такого как раствор, эмульсия, лиофилизированный порошок и суспензия, могут использоваться все общепринятые разбавители, например, вода, этанол, полиэтиленгликоль, 1,3-пропандиол, этоксилированный изостеариловый спирт, полиоксилированный изостеариловый спирт, полиоксиэтиленовый эфир сорбита и жирной кислоты и т.д. Кроме того, для приготовления изотонических растворов для инъецируемых препаратов также можно использовать хлорид натрия, глюкозу или глицерин в соответствующем количестве, и также можно добавлять общепринятый сорастворитель, буфер, вещество, регулирующее pH. Кроме того, при необходимости к фармацевтическому препарату могут быть добавлены красящее вещество, консервирующее вещество, ароматизирующее вещество, вкусовая добавка, подсластитель и другие вещества.
Перечисленные выше формы препаратов могут вводиться с помощью инъекции, включая подкожную инъекцию, внутривенную инъекцию, внутримышечную инъекцию и внутрибрюшинную инъекцию, интрацистернальную инъекцию или инфузию и т.д., внутрипросветное введение, такое как трансректальное, вагинальное и сублингвальное введение, введение через дыхательные пути, такое как назальное введение; и введение через слизистую оболочку. Среди перечисленных способов введения введение с помощью инъекции является предпочтительным, а предпочтительным способом инъекции является подкожная инъекция.
Доза вводимого липопептида или полипептида, его производного, его фармацевтически приемлемой соли, мультимера, композиции или фармацевтического соединения настоящего изобретения зависит от различных факторов, например, природы и тяжести заболевания, которое необходимо предотвратить или лечить, пола, возраста, веса и индивидуального ответа пациента или животного, конкретного используемого активного вещества, способа введения и частоты введения и т.д. Вышеописанная доза может быть введена в виде единичной стандартной лекарственной формы или дозированной лекарственной формы, содержащей множество (например, две, три или четыре) структурно обособленных единиц.
Уровень отдельной терапевтически эффективной дозы для любого конкретного пациента будет зависеть от различных факторов, включая заболевание, которое необходимо лечить, и его тяжесть; активность конкретного используемого активного вещества; конкретную используемую композицию; возраст, вес, общее состояние здоровья, пол и диету пациента; время введения, способ введения и скорость экскреции конкретного используемого активного ингредиента; продолжительность лечения; лекарственное средство, используемое вместе с конкретным активным веществом, используемым в комбинации или одновременно; и подобных факторов, хорошо известных в области медицины. Например, считается широко распространенной практикой в данной области начинать с дозировки активного вещества с уровня ниже, чем требуется для достижения желательного терапевтического эффекта, и постепенно увеличивать дозировку до достижения желаемого эффекта. В общем, липопептид, его производное или его фармацевтически приемлемая соль, мультимер, композиция или фармацевтическое соединение настоящего изобретения может вводиться млекопитающему, в частности, человеку, в дозировке в пределах от 0,001 до 1000 мг/кг веса тела в день, например, в пределах от 0,01 и до 100 мг/кг веса тела в день, и например, в пределах от 0,1 и до 10 мг/кг веса тела в день и с частотой введения 1-2 раз/в день, 1 раз/2 дня, 1 раз/3 дня, 1 раз /4 дня, 1 раз /5 дней, 1 раз /6 дней или 1 раз/7 дней, предпочтительно 1 раз /1-2 дня или 1-2 раза / в неделю.
Липопептид или полипептид, его производное или его фармацевтически приемлемая соль, мультимер, композиция или фармацевтическое соединение настоящего изобретения могут использоваться непосредственно в отдельности для лечения или предотвращения ВИЧ у инфицированного пациента или могут использоваться в комбинации с одним или более анти-ВИЧ лекарственными средствами, или одновременно или с интервалами для достижения улучшенного общего терапевтического эффекта. Анти-ВИЧ лекарственные средства включают, но без ограничения этим, ингибиторы обратной транскриптазы, ингибиторы протеаз, ингибиторы проникновения, ингибиторы слияния, ингибиторы созревания и тому подобное. Вышеупомянутый ингибитор обратной транскриптазы может быть одним или более из нуклеозидных ингибиторов обратной транскриптазы, таким как, например, зидовудин (AZT), ламивудин (3TC), диданозин (ddI), зальцитабин (ddC), ставудин (d4T), тенофовир (TDF), абакавир (ABC), эмтрицитабин (FTC) и т.д., и также может быть одним или более из ненуклеозидных ингибиторов обратной транскриптазы, таким как, невирапин (NVP), эфавиренз (EFV), делавирдин (DLV), этравирин (ETR) и т.д. Упомянутые выше ингибиторы протеаз могут быть одним или более ингибиторами, выбранными из группы, состоящей из саквинавира (SQV-HGC), инидинавира (IDV), ритонавира (RTV), ампренавира (APV), лопинавира и ритонавира (LPV/RTV), нелфинавира (NFV), фосампренавира кальция (FPV), реатаза (ATV), презисты и тому подобного. Упомянутые выше ингибиторы слияния могут представлять собой один или более ингибиторов, выбранных из группы, состоящей из ралтегравира, долутегравира, элвитегравира и тому подобного. Упомянутые выше ингибиторы проникновения могут представлять собой один или более из числа маравирока, T-20, TAK-779, T2635, VIRIP (VIR-576), сифувиртила, албувиртида, растворимого белка CD4 и его аналога, антитела против корецептора CCR5 (например, PRO140), моноклонального антитела против gp120/gp41 (например, VRC01 и 10E8), моноклонального антитела против рецептора CD4 (например, TNX-355) и тому подобного.
Стратегия для дизайна липопептида настоящего изобретения заключается в том, что C-концевая TRM последовательность из 8 аминокислот (WASLWNWF) T-20 полипептида заменяется на липофильное соединение, такое как жирная кислота с длинной цепью (например, пальмитиновую кислоту или стеариновую кислоту), холестерин, дигидросфингозин или витамин E, чтобы получить липопептид, содержащий полипептидную последовательность, соответствующую первым 28 аминокислотам T-20, т.е., соответствующую аминокислотам в положениях 127-154 gp41 от ВИЧ-1 штамма HXB 2; далее, EE**KK аминокислотные остатки, которые способствуют образованию ионных пар, вводятся путем изменения аминокислоты на не-NHR связывающей поверхности (т.е., соответствующей аминокислоты в положениях b, c, и f, g) последовательности полипептида, и соответствующие аминокислотные остатки ВИЧ-2 и/или ВИО вводятся путем изменения аминокислоты на NHR связывающей поверхности (т.е., соответствующей аминокислоты в положениях a и d) последовательности полипептида. В дальнейшем C-концевая и/или N-концевая последовательность полученного липопептида укорачивается, чтобы получить набор липопептидов, имеющих меньше чем 28 аминокислот, т.е., содержащих от 24 до 27 аминокислот, и последовательность, соответствующая аминокислотам в положениях 5 - 25 T-20, т.е., соответствующая аминокислотам в положениях 131 - 151 gp41 от HXB2 штамма, определяется как являющаяся коровой последовательностью (т.е., P5 последовательностью) эффективных ВИЧ ингибиторов настоящего изобретения. Полипептиды настоящего патента имеют исключительные структурные характеристики последовательности, имеют химическую модификацию липофильным соединением, связанным с C-концом, и обладают значительно повышенной способностью связываться с целевой последовательностью, чрезвычайно сильной ингибирующей активностью против ВИЧ (ВИЧ-1 и/или ВИЧ-2) и/или ВИО и впечатляющей способностью ингибировать слияние клеток, вхождение вируса и инфицирование, опосредованное белком оболочки (Env) ВИЧ. Анти-ВИЧ активность липопептида настоящего изобретения выше, чем активность T-20 в несколько тысяч раз или даже десятки тысяч раз, и также значительно выше, чем активность анти-ВИЧ липопептида с более высокой активностью, такого как C34-Chol, LP-11, LP-19 и тому подобных. В то же самое время, липопептид настоящего изобретения имеет ряд преимуществ, таких как устойчивый длительный эффект, легкий синтез и низкая стоимость. Липопептид настоящего изобретения обладает очень сильной ингибирующей активностью в отношении различных ВИЧ-1 подтипов (таких как A, B, C, A/E и B/C подтипы), T-20 резистентных штаммов, штаммов ВИЧ-2 и вируса иммунодефицита обезьян (ВИО).
Описание чертежей
Фигура 1 показывает структуру и функцию гибридного белка gp41 ВИЧ и ингибитора слияния мембраны на основе полипептида. При этом, FP означает гибридный пептид gp41; NHR означает N-концевую последовательность повтора; CHR означает C-концевую последовательность повтора; и TM означает трансмембранный участок. Положение, указанное стрелкой, - это положение “крючка M-T” или положение мотива, богатого триптофаном (TRM). Последовательности N36 и N39 полипептида NHR представлены выше схематическим изображением gp41, и резистентные к T-20 сайты и гидрофобные карман-образующие сайты промаркированы соответственно, и последовательность CHR и последовательность ингибитора на основе CHR последовательности представлены ниже схематического изображения gp41, где последовательности M-T и PBD подчеркнуты, последовательность TRM показана курсивом, а измененные аминокислоты в последовательностях полипептидов настоящего изобретения показаны жирным шрифтом. Аминокислоты всех полипептидов или липопептидов на фигуре имеют модификацию ацетилированием на амино-конце (Ac-) и модификацию амидированием на карбоксильном конце (-NH2).
Фигура 2 показывает структуры последовательностей ингибиторов слияния ВИЧ с мембраной и их противовирусную активность. При этом, последовательность TRM T-20 показана курсивом, последовательности крючка M-T и PBD подчеркнуты. AHX в полипептидном линкерном рукаве относится к 6-аминокапроновой кислоте, AEEA означает 8-амино-3,6-диокси-октановую кислоту, PEG4, PEG8 и PEG12 относится к полиэтиленгликолям разной длины, при этом PEG4 является Fmoc-NH-PEG4-CH2CH2COOH, PEG 8 является Fmoc-NH-PEG8-CH2CH2COOH, и PEG 12 является Fmoc-NH-PEG12-CH2CH2COOH. C16 представляет пальмитиновую кислоту, C18 представляет стеариновую кислоту, Chol представляет холестерин, DHS представляет дигидросфингозин, Toc представляет собой витамин E, C12 представляет собой додекановую кислоту (лауриновую кислоту), и C8 представляет собой октановую кислоту (каприловую кислоту). Псевдовирусный NL4-3 представляет собой мутант gp41 с D36G. Эксперимент повторяют три раза и вычисляют среднее значение IC50. Некоторые из эффективных липопептидов показаны жирным шрифтом. “Слияние с клетками HXB2” представляет экспериментальные результаты ингибирования клеточного слияния опосредованного ВИЧ-1, “проникновение NL4-3” представляет экспериментальные результаты ингибирования входа в клетку, опосредованного псевдовирусом ВИЧ-1, и “репликация JRCSF” представляет результаты ингибирования репликации ВИЧ-1.
Фигура 3 показывает ингибирующий эффект ингибиторов слияния ВИЧ с мембраной на различных подтипах штамма ВИЧ-1. Эксперимент повторяют три раза и вычисляют среднее значение IC50.
Фигура 4 показывает ингибирующий эффект ингибиторов слияния мембраны с ВИЧ на резистентных к T-20 мутантных штаммах, штаммах ВИЧ-2 и штаммах ВИО. Резистентные к T-20 мутантные штаммы и штаммы ВИО являются псевдовирусами, и штаммы ВИЧ-2 являются инфекционным штаммом ROD. Эксперимент повторяют три раза и вычисляют среднее значение IC50.
Фигура 5 показывает противовирусную активность в сыворотке макаки после инъекции ингибиторов слияния ВИЧ с мембраной. На фигуре M248, M249, M250, M252, M253 и M254 - это порядковые номера макаки. На Фигуре 5, A показывает противовирусную активность в сыворотке макаки после инъекции T-20; B показывает противовирусную активность в сыворотке макаки после инъекции LP-19; C показывает противовирусную активность в сыворотке макаки после инъекции LP-51; D показывает противовирусную активность в сыворотке макаки после инъекции LP-52; E показывает противовирусную активность в сыворотке макаки после инъекции LP-80; F показывает результат сравнения противовирусной активности ингибиторов в сыворотке.
Фигура 6 показывает результаты анализа кругового дихроизма взаимодействия между ингибиторами слияния ВИЧ с мембраной и NHR. Структуры последовательностей инигибиторов являются такими же, как на фигуре 2 настоящего изобретения, при этом эффективные липопептиды настоящего изобретения показаны жирным шрифтом. Ингибиторы и N39 полипептид растворяют в фосфатно- солевом буферном растворе (PBS) при значении pH 7,2 до достижения конечной концентрации 10 мкM.
Фигура 7 показывает результаты анализа кругового дихроизма взаимодействия между NHR и T-20 или характерными липопептидами. На фигуре 7, A показывает результаты CD сканирования; B показывает результаты температурного сканирования.
Фигура 8 показывает результаты анализа вторичной структуры T-20 и самих характерных липопептидов. На фигуре 8, A и B показывают результаты CD сканирования и температурного сканирования ингибиторов при 10 мкM, соответственно; C и D показывают результаты CD сканирования и температурного сканирования ингибиторов при 20 мкM, соответственно; и E и F показывают результаты CD сканирования и температурного сканирования ингибиторов при 40 мкM, соответственно.
Фигура 9 показывает результаты фармакокинетического анализа LP-80 у крыс. На Фигуре 9, A показывает результаты обнаружения концентраций лекарств в сыворотке после введения LP-80; B показывает метаболические кинетические параметры LP-80, при этом T1/2 означает конечный период полувыведения, Cmax означает концентрацию пика, Tmax означает время выхода на пик, AUC (0-216h) означает площадь под кривой (0-216час), Vz означает кажущееся распределение по объему, CL означает выведение, MRT означает среднее время удержания, и Fabs – это абсолютная биодоступность.
Фигура 10 показывает терапевтический эффект LP-80 на модели инфекции обезьяны.
Оптимальный способ осуществления настоящего изобретения
Варианты осуществления настоящего изобретения далее будут подробно описаны со ссылкой на примеры, тем не менее, специалисту в данной области техники понятно, что следующие примеры предоставляются только для иллюстрации настоящего изобретения и не должны рассматриваться как ограничивающие объем настоящего изобретения. В том случае, когда условия не указаны в примерах, примеры осуществлялись при общепринятых условиях или условиях, рекомендованных производителями. Использованные в данной работе реактивы и приборы, производители которых не указаны, являются обычными коммерчески доступными продуктами. Аминокислоты во всех полипептидах в следующих примерах являются аминокислотами типа L.
Пример 1. Получение липопептидов
Структурная формула липопептидов, предоставленных в этом варианте осуществления, была следующая: Ac-X1X2X3X4X5X6X7X8X9X10X11X12X13X14X15X21X17X18X19X20X26X27X28Z-NH2, при этом X1-X28 представляли полипептидную последовательность, соответствующую аминокислотам в положениях 127 - 154 последовательности gp41 от штамма ВИЧ-1 HXB2 (YTSLIHSLIEESQNQQEKNEQELLELDK), при этом X1 соответствовал Y в положении 127, и X2 соответствовал T в положении 128, X3 соответствовал S в положении 129, ... X28 соответствовал K в положении 154. Новая последовательность, полученная посредством большого количества мутаций, была компонентом потенциальных ингибиторов. Характерные пептиды включали LP-50, LP-51, LP-52, LP-80, LP-83, LP-84, LP-85, LP-86 и LP-87 и т.д. Определения X1-X28 были те же самые, как определения в формуле I, Z представлял собой липофильное соединение, Ac был ацетильной группой, и NH2 был аминогруппой.
В этом примере были синтезированы липопептиды или полипептиды, показанные на фигуре 2, при этом амино-конец каждого липопептида или полипептида был соединен с ацетильной группой в качестве амино-концевой защитной группы, и карбоксильный конец был соединен с амино-группой в качестве карбокси-концевой защитной группы.
Причем, модификацию полипептида пальмитиновой кислотой (липопептиды, модифицированные пальмитиновой кислотой: LP-40, LP-41, LP-42, LP-43, LP-44, LP-45, LP-50, LP-51, LP-52, LP- 53, LP-54, LP-55, LP-56, LP-57, LP-58, LP-59, LP-60, LP-61, LP-62, LP-63, LP-64, LP-65, LP-66, LP-67, LP-68, LP-69, LP-70, LP-71, LP-72, LP-73, LP-74, LP-75, LP-11, LP-19, C34-C16), стеариновой кислотой (липопептиды, модифицированные стеариновой кислотой: LP-80, LP-88, LP-89, LP-90, LP-91, LP-92), дигидросфингозином (липопептиды, модифицированные дигидросфингозином: LP-84, LP-87), витамином E (липопептид, модифицированный витамином E: LP-85), додекановой кислотой (липопептид, модифицированный додекановой кислотой: LP-81) и октановой кислотой (липопептид, модифицированный октановой кислотой: LP-82) проводили, используя реакцию их амидирования при помощи боковой цепи аминогруппы лизина (Lys) на C-конце полипептида, см. ссылки 18 и 27, перечисленные в разделе «Уровень техники». В дальнейшем LP-52 и LP-80 были взяты в качестве примеров, чтобы проиллюстрировать синтез вышеперечисленных липопептидов.
Использованные химические реактивы, такие как амидная смола Ринка MBHA, различные аминокислоты Fmoc, пальмитоилхлорид, стеароилхлорид, сукцинат витамина E, D-эритро-дигидросфингозин, N,N'-дисукцинимидил карбонат, N,N'-диизопропилкарбодиимид (DIC), 1-гидроксибензотриазол (HOBt), трифторуксусная кислота (TFA), этандитиол (EDT), нингидрин, гексагидропиридин (PIPE), фенол, N,N'-диметилформамид (DMF), хроматографически чистый ацетонитрил и т.д. покупали у основных поставщиков химических реактивов и дополнительно не очищали перед использованием.
Синтез полипептида: синтез проводили в направлении от C-конца к N-концу с использованием амидной смолы Ринка MBHA (константа замещения 0,34 ммоль/г) в качестве исходного материала с использованием ручного метода Fmoc-твердофазного синтеза. Fmoc-защитную группу на смоле Ринка удаляли с помощью 25% гексагидропиридин/DMF (объемное отношение) и затем смолу «привили» с использованием 2 эквивалентов Fmoc-Lys(Dde)-OH/HOBt/DIC, чтобы ввести первый аминокислотный остаток на C-конце. После этого снова удалили N-концевую Fmoc-защитную группу с использованием 25% гексагидропиридин/DMF (объемное отношение), чтобы сделать N-конец свободной аминогруппой. Различные аминокислотные остатки были последовательно связаны. Использовали следующие материалы и их количества, как указано далее: Fmoc-Glu(OtBu)-OH (3экв.), Fmoc-Leu-OH (3экв.), Fmoc-Lys(Boc)-OH (3экв.), Fmoc-Lys(Boc)-OH (3экв.), Fmoc-Leu-OH (3экв.), Fmoc-Glu(OtBu)-OH (3экв.), Fmoc-Glu(OtBu)-OH (3экв.), Fmoc-Glu(OtBu)-OH (3экв.), Fmoc-Asn (Trt)-OH (3экв.), Fmoc-Lys(Boc)-OH (3экв.), Fmoc-Lys(Boc)-OH (3экв.), Fmoc-Gln(Trt)-OH (3экв.), Fmoc-Gln(Trt)-OH (3экв.), Fmoc-Glu(OtBu)-OH (3экв.), Fmoc-Glu(OtBu)-OH (3экв.), Fmoc-Ala-OH (3экв.), Fmoc-Lys(Boc)-OH (3экв.), Fmoc-Lys(Boc)-OH (3экв.), Fmoc-Leu-OH (3экв.), Fmoc-Leu-OH (3экв.), Fmoc-Glu(OtBu)-OH (3 экв.), Fmoc-Glu (OtBu) -OH (3 экв.), Fmoc-Ile-OH (3 экв.), Fmoc-Lys(Boc)-OH (3экв.), Fmoc-Gln(Trt)-OH (3экв.), Fmoc-Glu(OtBu)-OH (3экв.), Fmoc-Trp(Boc)-OH (3экв.). И наконец, N-конец был обработан посредством ацетилирования (3 эквивалентов Ac2O, 6 эквивалентов диизопропилэтиламина) для завершения синтеза основной цепи. Время реакции каждой стадии было, как указано далее: снятие защитных групп в течение 8 минут, дважды; «пересадка» обычных аминокислот в течение 60 минут.
После каждой стадии вышеописанной реакции требовалась промывка смолы с использованием DMF шесть раз или более, и реакцию контролировали с помощью теста Кайзера. Если реакция конденсации аминокислоты не была закончена, конденсацию повторяли еще раз до получения желаемого представляющего интерес пептидного сегмента.
Модификация полипептида: смолу обработали 2% раствором гидразин гидрат/DMF (объемное отношение), чтобы удалить защитную группу Dde боковой цепи C-концевого Lys, и затем смешивали с 3 эквивалентами пальмитоил хлорида или стеароил хлорида и 6 эквивалентами диизопропил этиламина, чтобы осуществить реакцию амидирования с использованием аминогруппы боковой цепи C-концевого Lys (60 минут), тем самым достигая модификации пальмитоилированием (LP-52) или стеароил-модификации (LP-80) C-концевого остатка Lys. Модификация полипептида дигидросфингозином (LP-84, LP-87) была осуществлена путем первого добавления N,N'-дисукцинимидил карбоната после того, как была удалена защитная группа Dde боковой цепи Lys, затем добавили дигидросфингозин и проводили реакцию в течение 48 часов. Модификацию полипептида витамином E (LP-85) проводили путем амидирования незащищенной аминогруппы боковой цепи Lys непосредственно сукцинатом витамина E.
Расщепление и снятие защитной боковой группы: после того, как синтез липопептида был завершен, смолу высушивали под вакуумом. Реактивы для расщепления (трифторуксусная кислота : 1,2-этандитиол : тиоанизол : фенол : H2O: триизопропилсилан = 68.5 : 10 : 10 : 5 : 3.5 : 1, об/об) добавили к высушенной смоле и расщепление проводили при 30°C в течение 3 часов, в результате представляющий интерес полипептид отщеплялся от смолы, и защитная группа боковой цепи была удалена. Затем проводили фильтрование. Фильтрат добавили к большому количеству охлажденного безводного диэтилового эфира, чтобы осадить полипептид, затем центрифугировали, полипептид промывали диэтиловым эфиром семь раз и высушивали с получением липопептида в виде сырого продукта.
Очистка и исследование характеристик липопептида: очистку сырого продукта липопептида осуществляли с помощью высокоэффективной обращено-фазовой жидкостной хроматографии с использованием колонки100 × 250 мм, содержащей обращено-фазовый силикагель C18 или C4 с размером частиц 10 мкм и диаметром пор 100 ангстрем (Å). Условия проведения хроматографии: проводили элюирование в линейном градиенте, при этом элюент состоял из подвижной фазы A и подвижной фазы B, подвижная фаза A представляла собой водный раствор, содержащий ацетат аммония 20 мM (pH 4,5) и 5% ацетонитрил, а подвижная фаза В представляла собой водный раствор 80% (концентрация в процентах от общего объема) ацетонитрила; скорость потока составляла 250 мл/мин; и длина волны детектирования в ультрафиолетовой области была 220 нм. После высушивания растворителя сублимацией был получен сырой продукт полипептида в рыхлом состоянии, химическая структура которого была охарактеризована MALDI-TOF масс-спектрометрией, и чистоту которого определяли с помощью аналитической высокоэффективной жидкостной хроматографии (C18-10 × 250 мм, скорость потока: 1 мл/мин). Результаты показали, что чистота синтезированных липопептидов составляла более 95%.
Способ синтеза модифицированных холестерином липопептидов (LP-83, LP-86, C34-Chol) проводили, руководствуясь ссылкой 18 и ссылкой 20, приведенных в разделе «Уровень техники». Сначала был синтезирован холестерил бромацетат согласно технологическому способу, описанному в литературных источниках, который затем «привили» на полипептидную цепь с помощью химически высоко-селективной реакции образования простого тиоэфира, которую проводили между тиоловой группой боковой цепи C-концевого цистеина (Cys) полипептида и холестерил бромацетатом, говоря иначе, после того, как был синтезирован сырой полипептидный продукт обычным образом, он был растворен в чистом DMSO, сюда добавили 1 эквивалент холестерил бромацетата, растворенного в небольшом количестве трифторуксусной кислоты (TFA) и затем добавили чистый диизопропилэтиламин (DIEA), чтобы отрегулировать pH до щелочного значения. После реакции проводили ВЭЖХ с обращенной фазой, и реакция заканчивалась в основном за 1 час. Липопептид был очищен и охарактеризован как описано выше, и чистота полученного липопептида составляла более 95%.
Пример 2. Идентификация эффективных ингибиторов слияния ВИЧ с мембранами
2.1 Экспериментальные материалы и методы
Каждый из липопептидов и полипептидов с фигуры 2 использовали в качестве тестируемых веществ, при этом их противовирусную активность устанавливали с помощью анализа ингибирования клеточного слияния, анализа ингибирования псевдовируса и анализа ингибирования репликации вируса согласно ссылке 18, указанной в разделе «Уровень техники». Конкретный метод состоял в следующем.
Анализ ингибирования ВИЧ-1 опосредованного слияния с клеткой: эффекторные клетки (HL2/3 клетки) и клетки-мишени (TZM-b1 клетки) были предоставлены AIDS Reagent Program Национального института здоровья (NIH) (каталожные номера: 1294 и 8129, соответственно). Оба вида клеток были адгезивными клетками, которые культивировали в среде для культивирования клеток DMEM, содержащей два антибиотика ампициллин/стрептомицин и 10% эмбриональную телячью сыворотку (FBS). Сначала в 96-луночный культуральный планшет добавили TZM-b1 (1×104 клеток/лунку) и культивировали в течение ночи при 37°C и 5% CO2. Тестируемые вещества в 3 раза разбавляли средой для культивирования клеток DMEM и смешивали с эффекторными клетками HL2/3 (3×104 клеток/лунку), затем добавили клетки-мишени TZM-b1 и культивировали далее в течение 6 часов до полного слияния. Затем определяли активность люциферазы (относительные единицы флуоресценции, RLU) при помощи набора для определения репортерного гена люциферазы от компании Promega согласно инструкциям. Вычисляли степень ингибирования каждого образца в каждой концентрации и вычисляли половинную эффективную ингибирующую концентрацию (значение IC50) при помощи программного обеспечения GraphPad Prism Software 2.01.
Анализ ингибирования опосредованного псевдовирусомом ВИЧ-1 проникновения в клетки: основные стадии включали: (1) получение псевдовируса ВИЧ-1: клетки 293T котрансфицировали плазмидой, экспрессирующей белок оболочки (Env) штамма ВИЧ-1 NL4-3 (т.е., рекомбинантной экспрессирующей плазмидой, полученной путем «вставки» гена, кодирующего белок оболочки (ENV) D36G мутанта штамма ВИЧ-1 NL4-3 из таблицы 2 ссылки 14 (раздел «Уровень техники»), в вектор pcDNA3.1(-)) и остов ВИЧ-1 плазмиды pSG3Δenv (предоставленный AIDS Reagent Program Национального института здоровья (NIH), каталожный номер: 11051) при помощи реагента для трансфекции клеток, инкубировали в инкубаторе для клеток при 37°C и 5% CO2 в течение 6 часов, затем среду заменили, а клетки инкубировали далее в течение 48 часов. Надосадочную жидкость от культуры клеток, содержащую частицы псевдовируса, пипетировали и фильтровали при помощи фильтра0,45 мкм, чтобы собрать супернатант, затем туда добавили 20% эмбриональную бычью сыворотку (FBS), конечный раствор перенесли в полипропиленовую пробирку и хранили при -80°C для последующего использования или непосредственно проводили титрование вируса. (2) Титрование псевдовируса ВИЧ-1: вирусы разбавляли в 5 раз в 96-луночном планшете, и 4 повтора лунки с градиентом 8 имели конечный объем 100 микролитров. Клетки TZM-bl трипсинизировали и подсчитывали, и клетки разбавляли полной средой DMEM до 1 × 105 клеток/мл; добавили 100 мкл раствора для разведения клеток (содержащего 15 мкг/мл DEAE-декстрана) на лунку и культивировали при 37°C и 5% CO2 в течение 48 часов. Затем 96-луночный планшет извлекали из инкубатора для культур клеток. Супернатант из лунок отбрасывали. К каждой лунке добавили 30 мкл клеточного лизата и через 10 минут добавили 100 мкл реагента для обнаружения люциферазы. 100 мкл жидкости из каждой лунки пипетировали и добавляли в соответствующий 96-луночный белый планшет, и значения люминесценции регистрировали с помощью микропланшетного фотометра. Титр вируса вычисляли с помощью метода Рида-Мюнча. (3) Анализ противовирусной активности: тестируемое вещество растворяли в DMSO, разбавляли раствором клеточной культуры в 3 раза и помещали в 96-луночный планшет в конечном объеме 50 мкл. 50 мкл среды DMEM использовали вместо тестируемого вещества в качестве отрицательного контроля. К каждой лунке добавили 100 мкл раствора клеток-мишеней TZM-bl (содержащий 15 мкг/мл DEAE-декстрана) в концентрации 1×105 клеток/мл, и затем 50 мкл (что соответствует 100 TCID50 на лунку) псевдовируса ВИЧ-1, полученного выше. После инкубации при 37°C и 5% CO2 в течение 48 часов, определяли относительные световые единицы (RLU) в каждой лунке, используя реагент для обнаружения люциферазы (Promega). Вычисляли степень ингибирования (%) и значение IC50.
Анализ ингибирования репликации ВИЧ-1: плазмида pYK-JRCSF для клонирования молекул, кодирующая штамм ВИЧ-1 JRCSF, была предоставлена СПИД Reagent Program Национального Института здоровья (каталожный номер: 2708). Клетки 293T трансфицировали pYK-JRCSF при помощи реагента для трансфекции и инкубировали в инкубаторе для клеток при 37°C и 5% CO2 в течение 6 часов, и затем среду заменили, а клетки инкубировали далее в течение 48 часов. Супернатант клеточной культуры, содержащий вирусные частицы JRCSF, осторожно пипетировали и фильтровали с помощью фильтра 0,45 мкм, чтобы собрать супернатант, затем сюда добавили 20% эмбриональную телячью сыворотку (FBS), конечный раствор перенесли в полипропиленовую пробирку и хранили при -80°C для последующего использования или непосредственно проводили титрование вируса. Титрование вируса было таким же, как вышеописанное титрование псевдовируса ВИЧ-1. Для обнаружения противовирусной активности тестируемое вещество растворяли в DMSO и разбавляли раствором клеточной культуры в 3 раза, помещали в 96-луночный планшет в конечном объеме 50 мкл. В качестве отрицательного контроля вместо тестируемого вещества использовали 50 мкл среды DMEM. К каждой лунке добавили 100 мкл раствора TZM-bl клеток-мишеней (содержащего 105 клеток/мл и 15 мкг/мл DEAE-декстрана) и затем добавили 50 мкл (100 TCID50 на лунку) вирусов, полученных выше. После инкубации при 37°C и 5% CO2 в течение 48 часов в каждой лунке определяли относительные световые единицы (RLU) с использованием реагента для обнаружения люциферазы (Promega). Вычисляли степень ингибирования (%) и значение IC50.
2.2 Экспериментальные результаты и анализы
2.2.1 Липопептид на основе T-20 (LP-40) обладал сильной противовирусной активностью.
Для проведения скрининга и идентификации эффективных ингибиторов слияния ВИЧ с мембраной в настоящем изобретении был разработан новый метод, согласно которому полипептидное лекарственное средство T-20, которое не содержит карман-связывающий домен (PBD) NHR, было использовано в качестве шаблона. Ингибирующую активность в качестве ингибитора слияния с клеткой, опосредованного ВИЧ-1, ингибитора проникновения псевдовируса и ингибитора репликации вируса оценивали при помощи трех методов анализа (фигура 2). Во-первых, посредством полного удаления 8 аминокислот на C-конце T-20 был синтезирован полипептид T20-TRM, который не содержал мотив TRM, и было установлено, что он не обладает значительной противовирусной активностью при высокой концентрации 2000 нM, что показало важную роль TRM в функции T-20. Затем, был синтезирован липопептид LP-40 посредством замещения TRM T-20 пальмитиновой кислотой (C16). Неожиданно было обнаружено, что LP-40 обладал существенно улучшенной противовирусной активностью по сравнению с T-20, и что его ингибирующая активность против HXB2-опосредованного слияния с клеткой, входа псевдовируса NL4-3 и репликации JRCSF была примерно в 59, 21 и 18 раз выше, чем у T-20, соответственно, таким образом, было продемонстрировано, что замена TRM T-20 липофильным соединением может значительно улучшить противовирусную активность полипептида, причем это может быть важной стратегией для создания ингибиторов слияния ВИЧ с мембраной.
2.2.2 Добавление линкерного рукава приводило к значительному снижению противовирусной активности LP-40.
Авторы настоящего изобретения ранее сконструировали набор высокоактивных анти-ВИЧ липопептидов, основанных на модификации короткого пептида (HP23 или 2P23), нацеленного на карман NHR, и обнаружили, что непосредственное присоединение липидного соединения C16, холестерина и дигидросфингозина к C-концу полипептида приводило к значительному снижению анти-ВИЧ активности полипептида, тогда как введение линкерного рукава между полипептидной последовательностью и соединением-модификатором приводило к существенному повышению активности полипептида, и противовирусная активность увеличивалась вместе с увеличением длины линкерного рукава (пожалуйста, см. ссылки 18 и 30, приведенные в разделе «Уровень техники»). Сконструированные в конечном итоге высокоактивные липопептиды, такие как LP-11 и LP-19, имели более длинный PEG8 линкерный рукав, давая основание полагать, что добавление линкерного рукава облегчало липопептидам преодоление стерического несоответствия для обеспечения функционирования. Для того, чтобы также улучшить активность LP-40, было разработано и синтезировано пять липопептидов LP-41, LP-42, LP-43, LP-44 и LP-45, к которым соответственно были добавлены AHX, AEEA, PEG4, PEG8 и PEG12 в качестве линкерного рукава (Фигура 2). Неожиданно, добавление линкерного рукава приводило к значительному снижению активности липопептидов, причем снижение активности было более значительным при увеличении длины линкерного рукава, что было прямо противоположно результатам в отношении липопептидов на основе HP23 и 2P23. Эти результаты наводят на мысль о том, что липопептиды на основе HP23 и 2P23 имеют разные механизмы действия по сравнению с липопептидами на основе T-20, и это может являться следствием их разных сайтов связывания на NHR.
2.2.3 Добавление ионных пар приводило к существенному увеличению противовирусной активности LP-40.
Кроме того, в настоящем изобретении была предпринята попытка стимулировать формирование спиральной структуры и противовирусную активность LP-40 путем введения ионных пар. В технологии согласно настоящему изобретению аминокислотные остатки EE**KK, которые способствуют формированию “структуры солевого мостика” были введены путем изменения аминокислот не на поверхности последовательности полипептида связывающей NHR (т.е., положения b, c и f, g). Как видно на фигуре 1, 11 аминокислот в полипептидной последовательности LP-40 были заменены на E или K, тем самым три пары EE**KK мотивов были введены в положениях i и i+4, и синтезированный липопептид был назван P-50. Ингибирующая активность LP-50 была определена с помощью трех антивирусных анализов, и были получены невероятные результаты! Как показано на фигуре 2, значения IC50 LP-50 для ингибирования слияния с клеткой псевдовируса и репликативного вируса были 21 пM, 7 пM и 23 пM, соответственно, и были больше в 151 раз, 1345 раз и 226 раз, чем значения для T-20, соответственно, и в 20 раз, 63 раза и 12 раз больше, чем значения у LP-40. Следовательно, введение ионных пар может увеличить устойчивость спиральной структуры липопептида путем формирования “структуры солевого мостика”, тем самым значительно увеличивая противовирусную активность липопептида. Это было подтверждено последующими анализами кругового дихроизма (см. экспериментальные результаты примера 7 ниже).
2.2.4 Добавление аминокислотных остатков ВИЧ-2/ВИО дополнительно улучшает активность LP-50.
Для того, чтобы дополнительно улучшить противовирусную активность LP-50, были предприняты дальнейшие попытки синтезировать липопептиды LP-51 и LP-52 путем введения соответствующих аминокислотных остатков, полученных от ВИЧ-2 и/или ВИО на поверхности полипептида, связывающей NHR, т.е., положения a и d, или смежные положения. Измененные аминокислоты показаны на фигуре 1. Последовательности полипептидов LP-51 и LP-52 сохранили только 10 исходных аминокислотных остатков gp41 и были менее, чем на 28% идентичны последовательности T-20. Результаты противовирусных анализов показали, что активность LP-51 была сравнима с активностью LP-50, и ингибирующая активность LP-52 против опосредованного ВИЧ-1 HBX2 слияния с клеткой, псевдовируса NL4-3 и репликативного вируса JRCSF была также улучшена примерно в 2 раза, в 2 раза и в 5 раз, соответственно. По сравнению с T-20 ингибирующая активность LP-52 против активности ВИЧ в трех системах анализа была улучшена в 1859 раз, 2353 раз и 1038 раз, соответственно. Следовательно, можно сделать вывод, что LP-50, LP-51 и LP-52 были чрезвычайно эффективными ингибиторами слияния ВИЧ с мембранами.
2.2.5 Идентификация коровой последовательности эффективных анти-ВИЧ липопептидов.
Последовательность полипептида вышеупомянутого ингибитора ВИЧ составляла 28 аминокислот в длину. Для того, чтобы идентифицировать ключевую последовательность и рассмотреть возможность создания липопептида, содержащего более короткую последовательность, в настоящем изобретении был впервые синтезирован усеченный на C-конце липопептид LP-53 на основе LP-40 и был синтезирован усеченный на C-конце липопептид LP-54 на основе LP-50, и было обнаружено, что его противовирусная дееспособность была заметно понижена (Фигура 2). В дополнение к этому, LP-55 и LP-56 были синтезированы с использованием LP-52 в качестве матрицы, при этом LP-56 содержал AEEA в качестве линкерного рукава для замены трех аминокислотных остатков (LEK) на C-конце. Противовирусный анализ показал, что хотя ингибирующая активность LP-55 и LP-56 против слияния HXB2 с клеткой существенно не изменилась, их ингибирующая активность против инфекции NL4-3 и JRCSF была уменьшена (примерно в 2 раза). Эти экспериментальные результаты показали, что три аминокислотных остатка (LEK) на C-конце липопептида играют важную роль в противовирусной активности.
Был также синтезирован набор усеченных на N-конце липопептидов (LP-60 - LP-68). Противовирусный анализ подтвердил, что активность двух усеченных липопептидов на основе LP-50, т.е., LP-60 и LP-61, была также значительно уменьшена, однако, неожиданно, активность усеченных липопептидов на основе LP-52, т.е., LP-62, LP-63 и LP-65, в значительной степени не изменилась, конкретнее, активность LP-65, содержащего только 24 аминокислоты, была равноценна активности LP-52, а активность, в частности, ингибирующая активность против слияния с клеткой LP-64, содержащего 25 аминокислот, была существенно понижена. В ходе исследований было обнаружено, что дальнейшее N-концевое укорочение приводило к значительному снижению активности липопептидов (LP-66, LP-67) или даже потере их противовирусной дееспособности (LP-68). LP-69 был синтезирован путем укорочения C-концевого LEK на основе LP-65. Хотя противовирусная активность LP-69 была значительно снижена, он все еще обладал сильной ингибирующей активностью против вирусов по сравнению с липопептидом, имеющим только 21 аминокислоту. Результаты этих исследований показали, что последовательность “IEELX9KKX12EEQQKKNEEELKK”, состоящая из 21 аминокислоты, была коровой последовательностью эффективных липопептидов настоящего изобретения, которая соответствовала аминокислотной последовательности в положениях 5 - 25 T-20, то есть, соответствовала аминокислотной последовательности в положениях 131 - 151 (IHSLIEESQNQQEKNEQELLE) gp41 от штамма ВИЧ-1 HXB2. Добавление WEQK (или LEAN или YTSL) к N-концу коровой последовательности или добавление LEK к C-концу может эффективно увеличить противовирусную активность; если аминокислотные мотивы сохранить на обоих концах (например, LP-52), то активность такого липопептида может быть дополнительно улучшена.
Результаты также показали, что противовирусная активность LP-57 была уменьшена примерно в 15 – 150 раз по сравнению с активностью LP-55, показывая, что три концевых аминокислоты LKK LP-55 были важны и не подходили для дальнейшего укорочения; противовирусная активность LP-66 была примерно в 54 – 158 раз ниже, чем LP-65, показывая, что первая аминокислота (Ile) LP-65 была чрезвычайно важна и не подходила для дальнейшего укорочения. В то же самое время, различие между двумя усеченными липопептидами LP-65 и LP-61 составляет только одну аминокислоту (S и A в положении 8, соответственно), но их активность отличается от 5 до 9 раз, показывая, что замена A на S была очень важной для эффективных липопетидов настоящего изобретения.
В то же самое время, для того, чтобы показать взаимоотношения между структурой последовательности и функцией эффективных противовирусных липопептидов, в настоящем изобретении был дополнительно спроектирован и синтезирован набор липопептидов, удлиненных на N-конце, (LP-70 - LP-75 на фигуре 2), при этом LP-74 содержал последовательность карман-связывающего домена (PBD), LP-75 содержал и последовательность PBD и последовательность, формирующую крючок M-T. Неожиданным образом, при удлинении полипептидной последовательности по N-концу противовирусная активность липопептидов была не увеличена, а уменьшена и, в частности, активность LP-74 и LP-75 была существенно уменьшена.
2.2.6 Производные эффективных анти-ВИЧ липопептидов и их противовирусная активность
Для выявления последовательности и структурной специфики эффективных анти-ВИЧ липопептидов в настоящем изобретении было продолжено конструирование и синтез липопептидов, модифицированных различными липофильными соединениями, включая жирные кислоты с разной длиной цепи, холестерин, дигидросфингозин и витамин E. Результаты противовирусных анализов показаны на фигуре 2. Ингибирующая активность LP-80, модифицированного стеариновой кислотой (C18), против NL4-3 вхождения и JRCSF репликации была даже больше, чем активность модифицированного C16 LP-52, однако ингибирующая активность LP-81, модифицированного (C12)-додекановой кислотой, и LP-82, модифицированного(C8)-октановой кислотой, была значительно понижена. Эти четыре липопептида имели ту же самую последовательность полипептида, но ингибирующая активность липопептидов определялась длиной цепи жирной кислоты. По этой причине жирные кислоты с длиной цепи C18 и C16 были более подходящими для модификации последовательности полипептида. Результаты противовирусных анализов также продемонстрировали, что липопептиды, модифицированные холестерином (LP-83 и LP-86), дигидросфингозином (LP-84 и LP-87) и витамином E (LP-85) также обладали сильным противовирусным действием. Кроме того, C18-модифицированные укороченные на N-конце липопептиды LP-88, LP-89 и LP-90 также обладали сильной противовирусной активностью. Интересно отметить, что LP-89 с 25 аминокислотами был менее активным, чем LP-90 с 24 аминокислотами. Это явление было сходно с аналогичным явлением с C16-модифицированными LP-64 и LP-65. Соответственно, N-концевой лизин (K) не был необходим для эффективного короткого липопептида на основе коровой последовательности. Что касается коровой последовательности из 21 аминокислоты, активность C16- и C18-модифицированных липопептидов (LP-69 и LP-92) была по существу эквивалентна.
Вместе с тем, в настоящем изобретении была определена противовирусная активность нескольких контрольных липопептидов, включая LP-11, LP-19, C34-Chol и C34-C16 (смотри фигуру 2). Видно, что контрольные липопептиды могут эффективно ингибировать опосредованное ВИЧ-1 слияние с клеткой, проникновение и репликацию, и их активность была существенно выше, чем активность T-20, но значительно ниже, чем активность некоторых эффективных липопептидов настоящего изобретения, таких как C16-модифицированные LP-52, LP-55 и LP-65 и C18-модифицированные LP-80, LP-90 и LP-91 и тому подобные.
Пример 3. Ингибирующая активность эффективных ингибиторов слияния ВИЧ с мембранами в отношении разных подтипов ВИЧ-1
СПИД вызывается, главным образом, ВИЧ-1, и вследствие изменчивости вирусов были получены многочисленные подтипы, включая A-D, F-H, J и K подтипы и тому подобные. Среди них A, B и C подтипы - это основные вирусы, вызывающие эпидемию СПИД в мире, тогда как рекомбинантные вирусы B/C и A/E - основные вирусы в Китае. Для того, чтобы дополнительно оценить активность эффективных ингибиторов слияния ВИЧ с мембранами, в настоящем изобретении была получена группа из 35 псевдовирусов ВИЧ-1, включающая типичные международные штаммы и штаммы ВИЧ-1, эпидемические в настоящее время в Китае, при этом штаммы включали 3 подтипа А штаммов, 8 подтипов B штаммов, 4 подтипа B’ штаммов, 7 подтипов C штаммов, 1 подтип G штамма, 1 рекомбинантный штамм A/C, 5 рекомбинантных штаммов A/E и 6 рекомбинантных штаммов B/C. Из числа экспрессирующих Env плазмид, использованных для получения псевдовируса, за исключением экспрессирующих Env плазмид, использованных для получения PVO, Du156 и CAP 210.2.00.E8, были получены от AIDS Reagent Program Национального института Здоровья в США, другие плазмиды хранились в лаборатории профессора Хе Йуксиан, Институт патогенной биологии, Китайской академии медицинских наук, см. ссылки 13, 14 и 18, упомянутые в разделе «Уровень техники» и статьи Chong et al. (Chong H, Yao X, Zhang C, Cai L, Cui S, Wang Y, He Y. Biophysical property and broad anti-ВИЧ activity of Albuvirtide, a 3-maleimimidoproprotionic acid-modified peptide fusion inhibitor. PLoS One, 2012; 7 (3): e 32599). Получение псевдовируса и противовирусный анализ были такими же, как в примере 2.1 (анализ ингибирования проникновения в клетку опосредованного псевдовирусом ВИЧ-1). Для сравнения и анализа в настоящем примере была определена ингибирующая активность 12 ингибиторов, включая T-20, LP-40, LP-50, LP-51, LP-52, LP-55, LP-65, LP-80, LP-85, LP-90 и контрольных липопептидов LP-19, C34-Chol, в отношении 35 псевдовирусов, описанных выше. Как показано на фигуре 3, средние значения IC50 T-20, LP-40, LP-50, LP-51, LP-52, LP-55, LP-65, LP-80, LP-85 и LP-90 для ингибирования различных типов псевдовирусов ВИЧ-1 составляли 41410 пМ, 6369 пМ, 41 пМ, 33 пМ, 16 пМ, 34 пМ, 52 пМ, 6 пМ, 44 пМ и 14 пМ, соответственно. Видно, что ингибирующая активность вновь синтезированных липопептидов настоящего изобретения против разных субтипов ВИЧ-1 была значительно выше, чем активность T-20, которая была выше в 7 раз, 1010 раз, 1255 раз, 2588 раз, 1218 раз, 796 раз, 6902 раз, 941 раз и 2958 раз, чем активность T-20. LP-80 среди них продемонстрировал самую сильную ингибирующую активность против вирусов, и среднее значения IC50 для 35 псевдовирусов было 6 пМ, и значения IC50 для многих штаммов были даже ниже, чем 1 пМ. Средние значения IC50 для контрольных LP-19 и C34-Chol в отношении ингибирования различных ВИЧ-1 псевдовирусов были 439 пМ и 66 пМ, соответственно, и их активность была ниже, чем активность LP-50, LP-51, LP-55, LP-65 и LP-85, и была значительно ниже, чем активность LP-52, LP-80 и LP-90. При сравнении IC50 значений LP-52 и LP-80, LP-65 и LP-90, было установлено, что противовирусная активность C18-модифицированных липопептидов превосходила активность C16-модифицированных липопептидов.
Пример 4. Ингибирующая активность эффективных ингибиторов слияния ВИЧ с мембраной в отношении штаммов, устойчивых к T-20
T-20 является единственным ингибитором слияния ВИЧ с мембраной, одобренным для использования в клинике в настоящее время, однако его активность не только ниже активности нового поколения полипептидов, но также легко вызывает мутации, приводящие к устойчивости к лекарственному средству, зачастую приводя к неэффективному клиническому лечению. Для того, чтобы в полной мере показать широкий противовирусный спектр и преимущества эффективных липопептидов настоящего изобретения, в этом примере были получены псевдовирусы NL4-3, несущие общий для T-20 сайт резистентной мутации NHR (как показано на фигуре 4, подпись названия штамма на фигуре 4 представляла собой название штамма в таблице 2 в ссылке 14, приведенной в разделе «Уровень техники»). Методы получения плазмид и псевдовируса и использованный метод анализа противовирусной активности были описаны в литературе, опубликованной авторами настоящего изобретения (см. ссылки 11, 12, 14 и 18, приведенные в разделе «Уровень техники») и в примерах 2 и 3 выше. Результаты представлены на фигуре 4. По сравнению с типичным мутантом NL4-3D36G сам дикий тип NL4-3WT продемонстрировал устойчивость к T-20, и соответствующие значения IC50 были 13,65 нМ и 152,23 нМ, соответственно. Однако лекарственная устойчивость штаммов, содержащих одиночные или двойные мутации T-20, была существенно повышена в несколько раз. Результаты показали, что чувствительность этих T-20 резистентных штаммов к LP-40 была улучшена. В результате проведенных экспериментов было обнаружено, что ингибирующая активность липопептида LP-50, созданного введением ионных пар на основе LP-40 для формирования структуры солевого мостика, против T-20 устойчивых штаммов была дополнительно улучшена в сотни и даже тысячи раз. Способность липопептидов LP-51, LP-52, LP-80 и LP-85, модифицированных ВИЧ-2/ВИО последовательностью, «побеждать» резистентные штаммы была сильно улучшена, при этом она была лучше от тысяч до десятков тысяч раз по сравнению с LP-40. При сравнении LP-52, LP-55 и LP-65, LP-80 и LP-90 было обнаружено, что хотя усеченный на C-конце липопептид LP-55 и усеченные на N-конце липопептиды LP-65 и LP-90, которые, как было показано в предыдущих анализах на противовирусную активность, являлись выдающимися, обладали ингибирующей активностью против большого количества штаммов ВИЧ-1, которая была даже сравнима с активностью LP-52 и LP-80, причем они были намного менее активными против T-20 резистентных мутантных штаммов, чем LP-52 и LP-80. Особого внимания заслуживает то, что характерные липопептиды как эффективные ингибиторы настоящего изобретения, включая LP-50, LP-51, LP-52, LP-55, LP-65, LP-80, LP-85 и LP-90, обладали значительно сниженной ингибирующей способностью в отношении большинства T-20 резистентных штаммов, однако они все же обладали сильной противовирусной активностью, в частности, активность LP-52 и LP-80 оставалась исключительно хорошей в этой области. Этот пример также показал, что последовательность NHR gp41 оставалась главной целью эффективных липопептидов настоящего изобретения.
Пример 5. Ингибирующая активность эффективных ингибиторов слияния ВИЧ с мембранами в отношении ВИЧ-2 и ВИО
Для того, чтобы дополнительно отразить противовирусные преимущества сильных липопептидов настоящего изобретения, в данном примере была определена их ингибирующая активность в отношении ВИЧ-2 и ВИО. Использованные методы анализа противовирусной активности были описаны в печатных материалах, опубликованных авторами настоящего изобретения (см. ссылки 13 и 30, перечисленные в разделе «Уровень техники»). Полученная методом плазмида для клонирования молекул ROD штамма ВИЧ-2 ROD (ВИЧ-2ROD) была любезно предоставлена профессором Нуно Тавеира из университета в г. Лиссабон, Португалия, и плазмиды, экспрессирующие оболочечные белки штаммов ВИО ВИОpbj (ВИОPBJ) и ВИО239 (pВИОpbj-Env и pВИО239, соответственно) были любезно предоставлены профессором Ксу Жианкинг из Фуданьского университета. Получение инфекционного ROD было таким же, как получение инфекционного JRCSF в предыдущем разделе 2.1, псевдовирусы ВИОpbj и ВИО239 были получены тем же методом, который описан в примерах 2 и 3 выше. Результаты показаны на фигуре 4, из которых видно, что ингибирующая активность T-20 против штаммов ВИЧ-2 и ВИО была чрезвычайно слабой, тогда как активность LP-40 была только немного улучшена. Однако, было признано, что установленные эффективные липопептиды, включая LP-50, LP-51, LP-52, LP-65, LP-80, LP-85 и LP-90, обладали чрезвычайно сильной ингибирующей активностью в отношении как ВИЧ-2, так и ВИО. Соответственно, эффективные липопептиды настоящего изобретения были не только высокоэффективными в отношении различных подтипов ВИЧ-1, но также высокоэффективными против T-20 резистентных штаммов ВИЧ-2 и ВИО и обладали чрезвычайно сильной противовирусной активностью широкого спектра действия. При сравнении LP-52, LP-55 и LP-65 было обнаружено, что усечение N-концевых аминокислот WEQK оказывало незначительное действие на ингибирующую активность против ВИЧ-2 и ВИО, в то время как усечение C-концевых аминокислот LEK значительно влияло на активность.
Пример 6. Противовирусная активность эффективных ингибиторов слияния ВИЧ с мембранами in vivo
Последние исследования показали, что ингибиторы слияния ВИЧ с мембранами на основе липопептидов не только обладали улучшенной противовирусной активностью, но также демонстрировали устойчивый метаболизм in vivo и, следовательно, они имели более длительное время полужизни. Для того, чтобы дополнительно продемонстрировать полезность применения и преимущества для создания лекарственных средств на основе эффективных липопептидов настоящего изобретения, в этом примере была проанализирована противовирусная активность липопептидов LP-51, LP-52 и LP-80 in vivo, и данные методы были описаны в литературе, опубликованной авторами изобретения (смотри ссылки 18 и 30, приведенные в разделе «Уровень техники»), при этом ингибитор инъецировали обезьянам подкожно, образцы крови собирали в разные моменты времени, и противовирусную активность ингибитора измеряли in vitro; с помощью данного метода могла быть изучена не только активность ингибитора in vivo, но также косвенным образом отражалась его устойчивость in vivo. В дополнение к трем эффективным липопептидам, описанным выше, этот пример включает два контроля, T-20 и LP-19, для сравнения и анализа. Конкретный метод представлял собой следующее: для эксперимента было отобрано 6 обезьян (макак-резус), половина самцов и половина самок, в возрасте 3-4 лет весом 3,4-4,7 кг. T-20, LP-19, LP-51, LP-52 или LP-80 (все растворяли в стерильной дистиллированной воде) вводили подкожно в дозе 3 мг/кг веса тела, и 0,4 мл образца венозной крови собирали перед инъекцией (0 час) и через 1, 2, 4, 6, 8, 12, 18, 24, 36, 48, 60 и 72 часа после инъекции, соответственно. Для LP-80 в дополнение к упомянутым выше временным точкам сбора образцов крови были добавлены четыре временные точки через 96, 120, 144 и 168 часов после инъекции. Сыворотку отделяли согласно общепринятому методу. Интервал между введением каждого ингибитора был более 2 недель, чтобы обеспечить отсутствие остатка предшествующего исследуемого вещества. Активность в сыворотке против штамма ВИЧ-1 NL4-3 (NL4-3D36G) измеряли согласно экспериментальному методу анализа на основе псевдовируса в вышеприведенных примерах. Сыворотку разбавляли в три раза. Экспериментальные результаты показаны на фигуре 5. Что касается подкожной инъекции T-20, пики ингибирования возникали через 2 и 4 часа после инъекции, при этом максимальное разведение сыворотки для ингибирования 50% инфекционности NL4-3 было 45 раз и 46 раз, соответственно (A на фигуре 5); что касается подкожной инъекции LP-19, пики ингибирования возникали через 6 и 8 часов после инъекции, при этом максимальное разведение сыворотки было 5396 раз и 4720 раз, соответственно (B на фигуре 5). Однако неожиданно оказалось, что при подкожной инъекции LP-51 или LP-52 пики ингибирования возникали через 4 и 6 часов после инъекции, при этом максимальное разведение сыворотки было 700482 раз и 584381 раз для LP-51, соответственно, и максимальное разведение сыворотки было 700802 раз и 669112 раз для LP-52 (C и D на фигуре 5); и при подкожной инъекции LP-80 пики ингибирования возникали через 6 и 8 часов после инъекции, максимальное разведение сыворотки было 491409 раз и 537206 раз, соответственно (E на фигуре 5). Видно, что пик ингибирования в сыворотке эффективных липопептидов мог быть 11678-15235-кратным пиком ингибирования в сыворотке T-20, и 100-130-кратным пиком ингибирования в сыворотке LP-19 (подсчитано согласно самым высоким значениям). Более впечатляющим результатом был длительный эффект in vivo трех липопептидов, LP-51, LP-52 и LP-80, они демонстрировали высокий пик ингибирования в сыворотке даже через 72 часа (3 дня) после инъекции, соответственно, пики ингибирования в сыворотке которых были 1122, 182 и 16157-кратными, соответственно. В частности, пик ингибирования LP-80 в сыворотке сохранялся 1980-кратным через 96 часов (4 дня) после инъекции, 211-кратным через 120 часов (5 дней) после инъекции и через 144 часов (6 дней) после инъекции, и пик ингибирования в сыворотке был таким же, как пик T-20 через 4 часа (46 раз) (F на фигуре 5). Следовательно, липопептиды настоящего изобретения были не только эффективными, но также их действие было пролонгированным.
Пример 7. Взаимодействие эффективных ингибиторов слияния ВИЧ с мембраной с целевыми последовательностями NHR
Для изучения механизма действия эффективных анти-ВИЧ липопептидов использовали анализ кругового дихроизма (CD), чтобы установить взаимодействие между ингибиторами и целевыми последовательностями NHR, включая вторичную структуру (α-спираль) и устойчивость спирали (Tm) образованных комплексов. Для измерения кругового дихроизма использовали спектрофотометр Jasco-815 от компании JASCO Inc., данный метод анализа можно найти в опубликованных авторами изобретения статьях (смотри ссылки 18 и 30, приведенные в разделе «Уровень техники»). Целевой полипептидной последовательностью, полученной от NHR, была N39 (см. фигуру 1) и ее последовательность была Ac-STMGAASMTLTVQARQLLSGIVQQQNNLLRAIEAQQHLL-NH2, которая соответствовала сайту-мишени на NHR, с которым связывается T-20. N39 и ингибитор по отдельности растворили в фосфатно-солевом буферном растворе (PBS) чтобы приготовить PBS раствор (pH 7,2) в концентрации 20 мкM. N39 смешали с ингибитором в объемном соотношении 1 : 1 (конечная концентрация 10 мкM для каждого), смешанный образец держали при 37°C в течение 30 минут, чтобы реакция прошла полностью, и затем содержание спирали и Tm значение полученного в результате комплекса измеряли при помощи спектрофотометра кругового дихроизма. Сканируемый прибором диапазон длин волн был 195-260 нм с интервалом длины волны 1 нм, значения, полученные путем трехкратного сканирования, усредняли. Исходя из CD сигналов, определяли взаимодействие между полипептидами и содержание спирали. Затем, образец для измерения CD сигнала перенесли в кювету для температурного сканирования, программа CD устройства была настроена на температурное сканирование с длиной волны детектирования 220 нм и диапазон сканирования 20-98°C, и осуществляли программу сканирования, чтобы получить кривую CD сигналов в зависимости от температуры, на основе которой вычисляли значение Tm. Исходя из Tm значения, судили о стабильности образованной спиральной структуры ингибитора и N39.
Результаты CD анализа показаны на фигуре 6. Видно, что T-20 может взаимодействовать с N39, при этом полученный комплекс имел содержание спирали 48,6% и значение Tm 43,9°C; однако, взаимодействие между T20-TRM и N39 было чрезвычайно слабым, и, таким образом, значение Tm не обнаруживалось прибором, дополнительно демонстрируя важную роль TRM в T-20. Взаимодействие между липопептидом LP-40 и N39 было существенно повышенным, и полученный комплекс имел содержание спирали 57,7% и значение Tm 51,3°C. Добавление линкерного рукава приводило к снижению содержания спирали, однако большинство из линкерных рукавов оказывали незначительное влияние на значение Tm, и только LP-45 с самым длинным линкерным рукавом (PEG12) имел существенно уменьшенное значение Tm. Неожиданно, введение ионной пары EE**KK значительно увеличивало устойчивость связывания липопептидов, что отражалось в том, что Tm значение LP-50/N39 комплекса было повышено до 63,3°C; и добавление ВИЧ-2/ВИО аминокислот могло дополнительно усилить способность к связыванию липопептидов, при этом значения Tm LP-51/N39 и LP-52/N39 комплексов были 72°C и 79,1°C, соответственно, которые были существенно повышенными по сравнению со значениями Tm T-20 и LP-40 комплексов, как показано на фигуре 7. Соответственно, три эффективных противовирусных липопептида были способны формировать экстремально устойчивую спиральную структуру с целевой последовательностью, в частности LP-52. Более того, LP-52/N39 комплекс также имел самое высокое содержание спирали (63,8%).
В этом примере было обнаружено, что C-концевое или N-концевое укорочение липопептидов может влиять на способность липопептидов к связыванию до различной степени, и некоторые липопептиды продемонстрировали уменьшение значения Tm, а некоторые продемонстрировали уменьшение, как содержания спирали, так и Tm значения. Значение Tm укороченных на C-конце липопептидов (LP-53 - LP-59) было существенно уменьшено, указывая на важную роль трех аминокислот (LEK) на C-конце в связывании липопептидов с NHR. Следует отметить, что значения Tm LP-55 и LP-56 с сильной противовирусной активностью также были значительно понижены (от 79, 1 до 63,1°C), однако значения Tm были намного выше, чем значения LP-53, LP-54, LP-57, LP- 58 и LP-59. В частности, значения Tm LP-58 и LP-59 не могли быть определены из-за низкого содержания спирали. Tm значения укороченных на N-конце липопептидов (LP-60 - LP-68) были также существенно снижены. N-концевое укорочение на основе LP-52 не оказывало влияния на устойчивость связывания липопептидов (LP-62 - LP-65), однако Tm значения соответствующих комплексов оставались больше, чем 70°C, показывая, что липопептиды по-прежнему обладали сильной способностью к связыванию, и это может быть причиной, почему они сохранили сильные противовирусные возможности. Было отмечено, что липопептид LP-65 с последовательностью из 24 аминокислот имел более высокое содержание спирали (63%) и Tm значение (72,1°C), тогда как дополнительное укорочение жестко влияло на способность к связыванию соответствующих липопептидов, таких как LP-66, LP-67 и LP-68, что согласовывалось с их противовирусной активностью. Для сравнения, эффект удаления 3 аминокислот (LEK) на C-конце на устойчивость связывания (Tm значение) был более существенным, чем эффект удаления 1-4 аминокислот (WEQK) на N-конце, показывая, что C-конец липопептидов играл более важную роль в связывании с мишенью. Однако коровая последовательность липопептида LP-69 с удалением аминокислот на обоих концах и C-конце и N-конце имела значительно уменьшенную устойчивость связывания и значение Tm 51°C, которое было ниже, чем значение LP-52 28,1°C.
Другое интересное явление заключалось в том, что удлиненные на N-конце липопептиды имели повышенное значение Tm, такое как показатель LP-70 - LP-75, которое не соответствовало их уменьшенной противовирусной активности. Следует отметить, что LP-74 и LP-75 содержали карман-связывающий домен (PBD) NHR и мотив крючка M-T, что делает невозможным полное соответстветствие N39.
Результаты этого примера также показали, что модифицированный стеариновой кислотой (C18) липопептид LP-80 также обладал сильной устойчивостью связывания с N39 (Tm значение = 79°C). Однако, липопептиды, модифицированные жирной кислотой с короткой длиной цепи, например, LP-81 модифицированный C12 жирной кислотой и LP-82 модифицированный C8 жирной кислотой, имели существенно уменьшенное содержание спирали и связывающую способность, Tm значения его были 74,1°C и 65,1°C, соответственно, и их противовирусная активность была снижена более существенно (см. фигуру 2). Липопептиды, модифицированные холестерином, LP-83 и LP-86, модифицированный витамином E липопептид LP-85, липопептиды, модифицированные дигидросфингозином LP-84 и LP-87, все имели сильное связывания, опосредованное стабильностью спирали, что согласовывалось с их противовирусной активностью. Аналогично, укороченные на N-конце липопептиды на основе LP-80 (LP-88, LP-89, LP-90) также обладали сильной связывающей способностью, и значение Tm было 76,5°C, 70°C и 71,1°C, соответственно. Однако, липопептид LP-91 с удаленным C-концевым LEK и коровая последовательность липопептида LP-92 с усечением на обоих концах имели существенно уменьшенную стабильность спирали, и Tm значение было 61°C и 55,1°C, соответственно.
Этот пример показал корреляцию между структурой последовательности, устойчивостью связывания и противовирусной активностью ингибиторов при помощи большого количества экспериментальных результатов и предоставил важную информацию для понимания механизма эффективных липопептидов настоящего изобретения. Хотя связывающая способность некоторых ингибиторов была иногда недостаточно сопоставима с их противовирусной активностью, в целом, эффективные липопептиды настоящего изобретения имели чрезвычайно высокие значения Tm. Этот пример также продемонстрировал, что противовирусная активность таких липопептидов зависела от их полипептидных последовательностей и также от свойств липофильных соединений.
Пример 8. Анализ вторичной структуры эффективных ингибиторов слияния ВИЧ с мембраной
Для исследования механизма действия эффективных анти-ВИЧ липопептидов использовали анализ кругового дихроизма (CD), чтобы проанализировать вторичные структурные характеристики T-20 и типичных липопептидов в растворе тем же способом, как в примере 7. При проведении анализа содержание α-спирали и значение Tm ингибиторов были измерены при концентрациях 10 мкM, 20 мкM и 40 мкM (раствор в PBS), соответственно. Результаты показаны на фигуре 8. T-20 продемонстрировал неправильную беспорядочную структуру при трех концентрациях, LP-40 продемонстрировал небольшое количество спиральной структуры при 20 мкM и 40 мкM, и четыре эффективных липопептида (LP-50, LP-51, LP-52, LP-80) продемонстрировали различные спиральные структуры, среди которых LP-80 имел более высокое содержание спирали и значение Tm. Следовательно, эффективные липопептиды настоящего изобретения сами могли формировать типичную спиральную структуру, которая значительно отличалась от T-20.
Пример 9. Фармакокинетический анализ эффективного липопептида LP-80 у крыс
Приведенные выше результаты исследования показали, что LP-80 – липопептид, обладающий более высокой противовирусной активностью in vivo, который был очень устойчивым среди эффективных липопептидов настоящего изобретения. В этом примере LP-80 использовали в качестве типичного представителя, для того, чтобы проанализировать его фармакокинетические свойства на SD крысах. В тесте использовали 12 крыс SD 5-8-недельного возраста весом 182-219 грамм, их разделили на 2 группы с внутривенным введением и подкожным введением, в каждой группе было 6 животных, половина самцов и половина самок. Доза LP-80 составляла 6 мг/кг веса тела (мг/кг), и LP-80 растворяли в стерильной дистиллированной воде. Образцы сыворотки у животных в каждой группе отбирали до введения и через 5 минут, 15 минут, 30 минут, 1 час, 2 час, 4 часа, 8 часов, 24 часа, 48 часов, 72 часа, 96 часов, 120 часов, 168 часов и 216 часов после введения. Концентрацию LP-80 в сыворотке крыс количественно определяли с помощью жидкостной хроматографии с тандемной масс спектрометрией (LC-MS/MS), и нижний предел был количественно определен как 1 нг/мл (нг/мл). Фармакокинетические параметры были вычислены с использованием модели некомпартментного анализа (NCA). Результаты экспериментов показаны на фигуре 9. Средний конечный период полувыведения (T1/2) LP-80 в группе с внутривенным введением и в группе с подкожной инъекцией был 6,04 часов и 6,28 часов, соответственно. Однако, следует отметить, что концентрация LP-80 в сыворотке через 3 дня (72 часа) после внутривенной и подкожной инъекции была 7,75 нг/мл и 6,86 нг/мл, соответственно, т.е., молярная концентрация 2021,12 пМ и 1789,02 пМ, и концентрация была в 1010,56 раз и 894,51 раз выше значения IC50 (2 пМ) LP-80 для ингибирования штаммов ВИЧ-1 NL4-3 и JRCSF, соответственно. Этот результат дополнительно подтвердил сильную и длительную противовирусную способность LP-80 согласно примеру 6 выше у макак с точки зрения фармакокинетики.
Пример 10. Оценка терапевтического эффекта липопептидов LP-80 на модели СПИДа у обезьян.
В этом примере был дополнительно изучен терапевтический эффект LP-80 на модели инфекции ВИЧ у обезьян, и что касается методики, ознакомьтесь, пожалуйста, с методом, использованным авторами изобретения для оценки LP-19 (т.е., ссылка 30 в разделе «Уровень техники»). В этом тесте использовали шесть взрослых макак-резус (пронумерованных от A до F, половина самцов и половина самок), и было установлено отсутствие антител к ВИО, герпесвирусу B и обезьяньему T-лимфотропному вирусу. ВИОЧ штамм SF162P3 был предоставлен AIDS Reagent Program Национального центра исследования здоровья (NIH) в Соединенных Штатах и был амплифицирован в мононуклеарных клетках периферической крови (PBMC) обезьян, и был определен показатель TCID50. Обезьянам внутривенно инокулировали 1,000 TCID50 вируса SF162P3 и периодически измеряли вирусную нагрузку в плазме у обезьян (число копий РНК/мл). На 197-ой день после инфицирования обезьян SF162P3 вводили LP-80 (растворенный в стерильной дистиллированной воде) с помощью подкожного введения, и LP-80 вводили в дозе 2 мг на килограмм веса тела (2мг/кг), один раз в день в течение 2 недель и затем один раз в 4 дня в течение 4 недель. Из крови обезьян, собранной в заранее установленные моменты времени, выделяли образцы плазмы и определяли вирусную нагрузку в плазме (число копий РНК/мл) с помощью количественной полимеразной цепной реакции с обратной транскриптазой в реальном времени (qRT-PCR). Плазменную РНК извлекали обычным способом и синтезировали образцы кДНК с помощью реакции обратной транскрипции. ПЦР-праймеры были направлены на gag477 ВИО (использовали прямой праймер GCAGAGGAGGAAATTACCCAGTAC, обратный праймер CAATTTTACCCAGGCATTTAATGTT и детекторный зонд FAM-ACCTGCCATTAAGCCCGA-MGB). Использовали ПЦР-машину PE ABI7500. Чувствительность анализа составляла 100 копий РНК на миллилитр образца плазмы.
Результаты экспериментов показаны на фигуре 10. Вирусная нагрузка у трех из шести обезьян на четвертый день после лечения уменьшилась до уровня ниже линии обнаружимого уровня; вирусная нагрузка у пяти обезьян не была обнаружена на восьмой день после лечения; и вирусная нагрузка у всех шести обезьян не была обнаружена на 14-ый день после лечения. Вирусную нагрузку у всех обезьян контролировали до уровня, ниже линии обнаружимого уровня в ходе дальнейшего лечения при помощи введения лекарственного средства один раз в 4 дня. Вирус не возвращался на 4-ый день после того, как было прекращено введение лекарственного средства; наблюдался возврат вируса у одной из обезьян (обезьяна A) на 10-ый день после того, как было прекращено введение; наблюдался возврат вируса у других 5 обезьян за исключением обезьяны C на 17-ый день после того, как было прекращено введение; и наблюдался возврат вирусной нагрузки у всех обезьян на 24-ый день после того, как было прекращено введение лекарственного средства. Результаты продемонстрировали сильный противовирусный терапевтический эффект LP-80.
Промышленная применимость
Эффективные липопептиды, их производные или фармацевтически приемлемые соли, мультимеры, композиции или фармацевтические соединения, предоставляемые настоящим изобретением, могут использоваться для лечения и/или предотвращения инфекции ВИЧ (ВИЧ-1 и/или ВИЧ-2) и/или ВИО. При применении на практике липопептиды, их производные или их фармацевтически приемлемые соли, мультимеры, композиции или фармацевтические соединения согласно настоящему изобретению могут непосредственно вводиться пациенту в качестве лекарственного средства или могут вводиться пациенту в смешанном с подходящим носителем или эксципиентом виде с целью лечения и/или профилактики инфекции ВИЧ.
--->
Перечень последовательностей
<110> Институт патогенной биологии, Китайская академия медицинских наук
<120> Липопептид для эффективного ингибирования ВИЧ, его производное,
его фармацевтическая композиция и их применение
<160> 12
<170> Патент в версии 3.5
<210> 1
<211> 28
<212> белок
<213> Искусственная последовательность
<220>
<223>
<400> 1
Trp Glu Gln Lys Ile Glu Glu Leu Leu Lys Lys Ala Glu Glu Gln Gln
1 5 10 15
Lys Lys Asn Glu Glu Glu Leu Lys Lys Leu Glu Lys
20 25
<210> 2
<211> 28
<212> белок
<213> Искусственная последовательность
<220>
<223>
<400> 2
Leu Glu Ala Asn Ile Glu Glu Leu Leu Lys Lys Ala Glu Glu Gln Gln
1 5 10 15
Lys Lys Asn Glu Glu Glu Leu Lys Lys Leu Glu Lys
20 25
<210> 3
<211> 28
<212> белок
<213> Искусственная последовательность
<220>
<223>
<400> 3
Tyr Thr Ser Leu Ile Glu Glu Leu Ile Lys Lys Ser Glu Glu Gln Gln
1 5 10 15
Lys Lys Asn Glu Glu Glu Leu Lys Lys Leu Glu Lys
20 25
<210> 4
<211> 27
<212> белок
<213> Искусственная последовательность
<220>
<223>
<400> 4
Glu Gln Lys Ile Glu Glu Leu Leu Lys Lys Ala Glu Glu Gln Gln Lys
1 5 10 15
Lys Asn Glu Glu Glu Leu Lys Lys Leu Glu Lys
20 25
<210> 5
<211> 26
<212> белок
<213> Искусственная последовательность
<220>
<223>
<400> 5
Gln Lys Ile Glu Glu Leu Leu Lys Lys Ala Glu Glu Gln Gln Lys Lys
1 5 10 15
Asn Glu Glu Glu Leu Lys Lys Leu Glu Lys
20 25
<210> 6
<211> 26
<212> белок
<213> Искусственная последовательность
<220>
<223>
<400> 6
Ser Leu Ile Glu Glu Leu Ile Lys Lys Ser Glu Glu Gln Gln Lys Lys
1 5 10 15
Asn Glu Glu Glu Leu Lys Lys Leu Glu Lys
20 25
<210> 7
<211> 25
<212> белок
<213> Искусственная последовательность
<220>
<223>
<400> 7
Lys Ile Glu Glu Leu Leu Lys Lys Ala Glu Glu Gln Gln Lys Lys Asn
1 5 10 15
Glu Glu Glu Leu Lys Lys Leu Glu Lys
20 25
<210> 8
<211> 24
<212> белок
<213> Искусственная последовательность
<220>
<223>
<400> 8
Ile Glu Glu Leu Leu Lys Lys Ala Glu Glu Gln Gln Lys Lys Asn Glu
1 5 10 15
Glu Glu Leu Lys Lys Leu Glu Lys
20
<210> 9
<211> 24
<212> белок
<213> Искусственная последовательность
<220>
<223>
<400> 9
Ile Glu Glu Leu Ile Lys Lys Ser Glu Glu Gln Gln Lys Lys Asn Glu
1 5 10 15
Glu Glu Leu Lys Lys Leu Glu Lys
20
<210> 10
<211> 25
<212> белок
<213> Искусственная последовательность
<220>
<223>
<400> 10
Trp Glu Gln Lys Ile Glu Glu Leu Leu Lys Lys Ala Glu Glu Gln Gln
1 5 10 15
Lys Lys Asn Glu Glu Glu Leu Lys Lys
20 25
<210> 11
<211> 29
<212> белок
<213> Искусственная последовательность
<220>
<223>
<400> 11
Trp Glu Gln Lys Ile Glu Glu Leu Leu Lys Lys Ala Glu Glu Gln Gln
1 5 10 15
Lys Lys Asn Glu Glu Glu Leu Lys Lys Leu Glu Lys Cys
20 25
<210> 12
<211> 29
<212> белок
<213> Искусственная последовательность
<220>
<223>
<400> 12
Leu Glu Ala Asn Ile Glu Glu Leu Leu Lys Lys Ala Glu Glu Gln Gln
1 5 10 15
Lys Lys Asn Glu Glu Glu Leu Lys Lys Leu Glu Lys Cys
20 25
<---
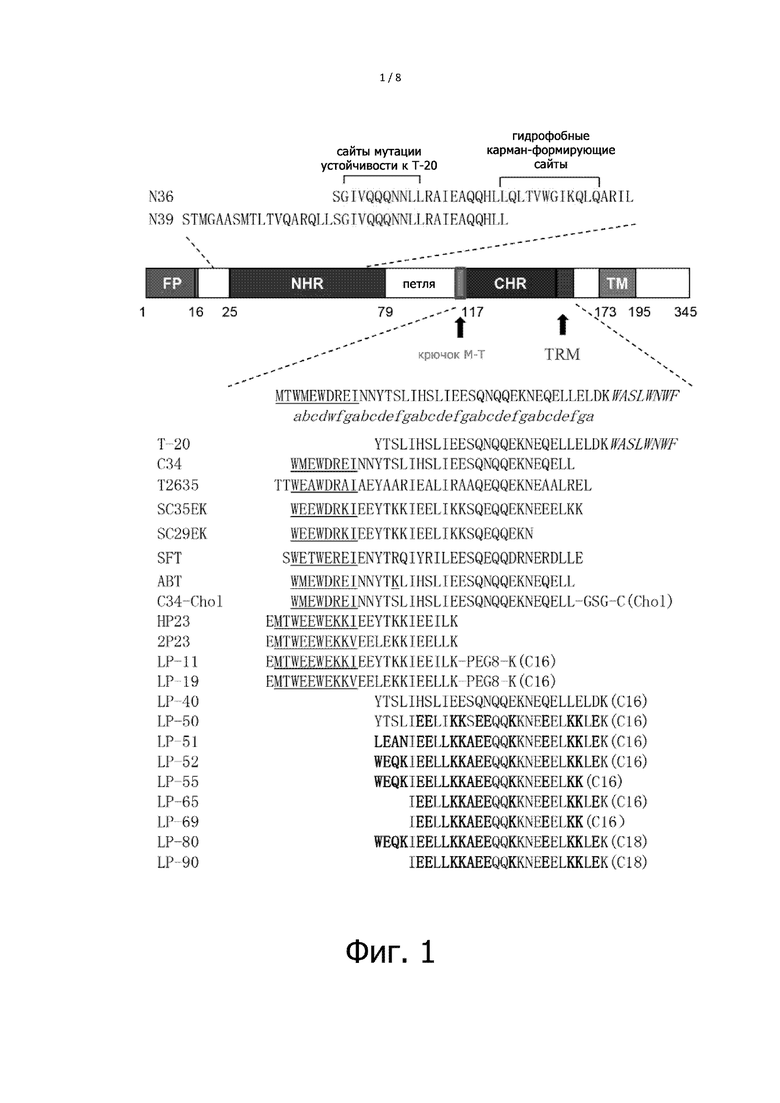
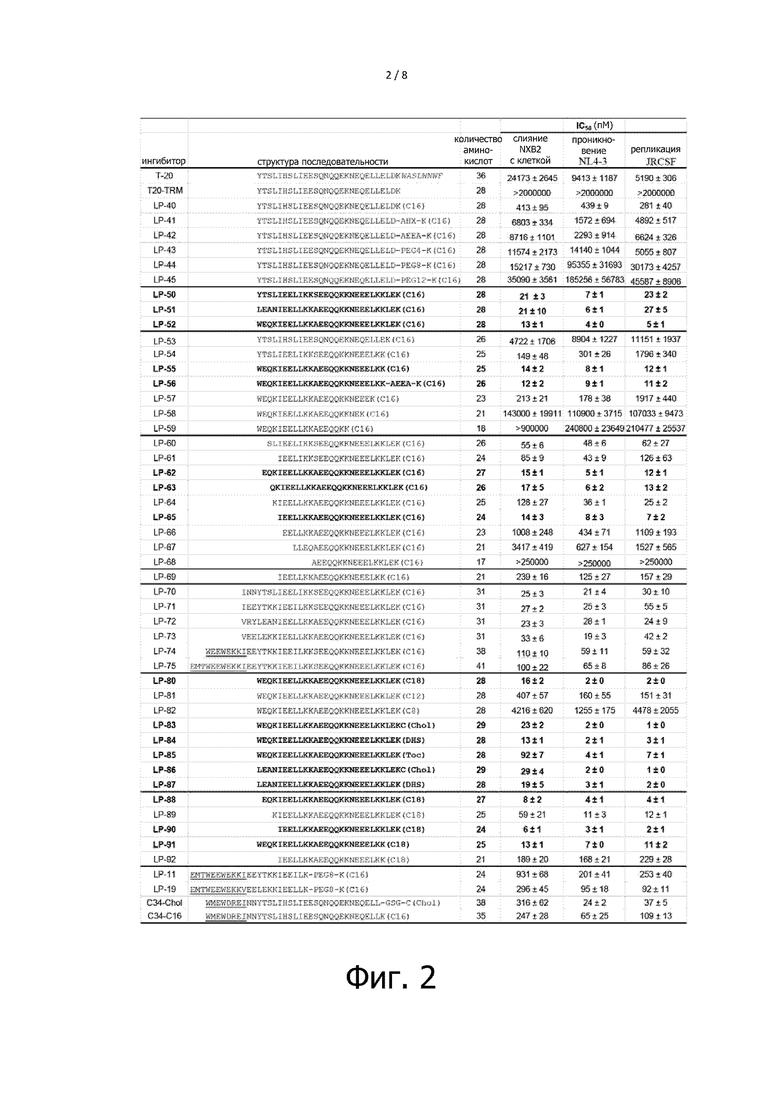
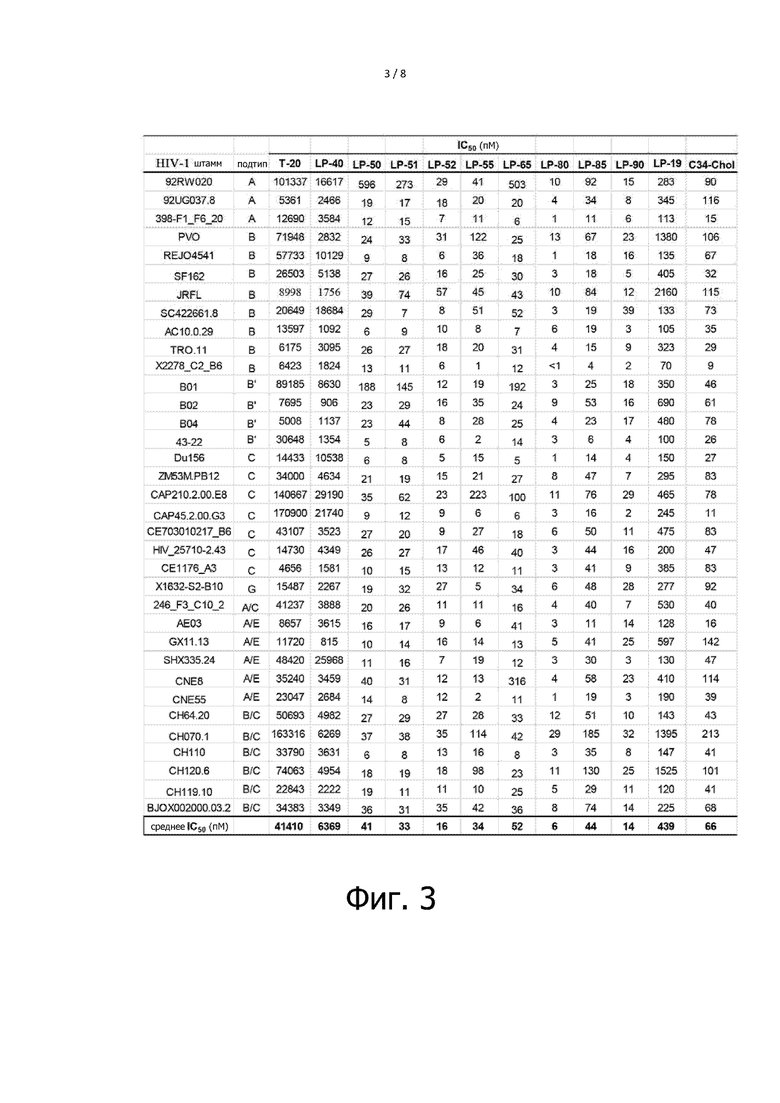
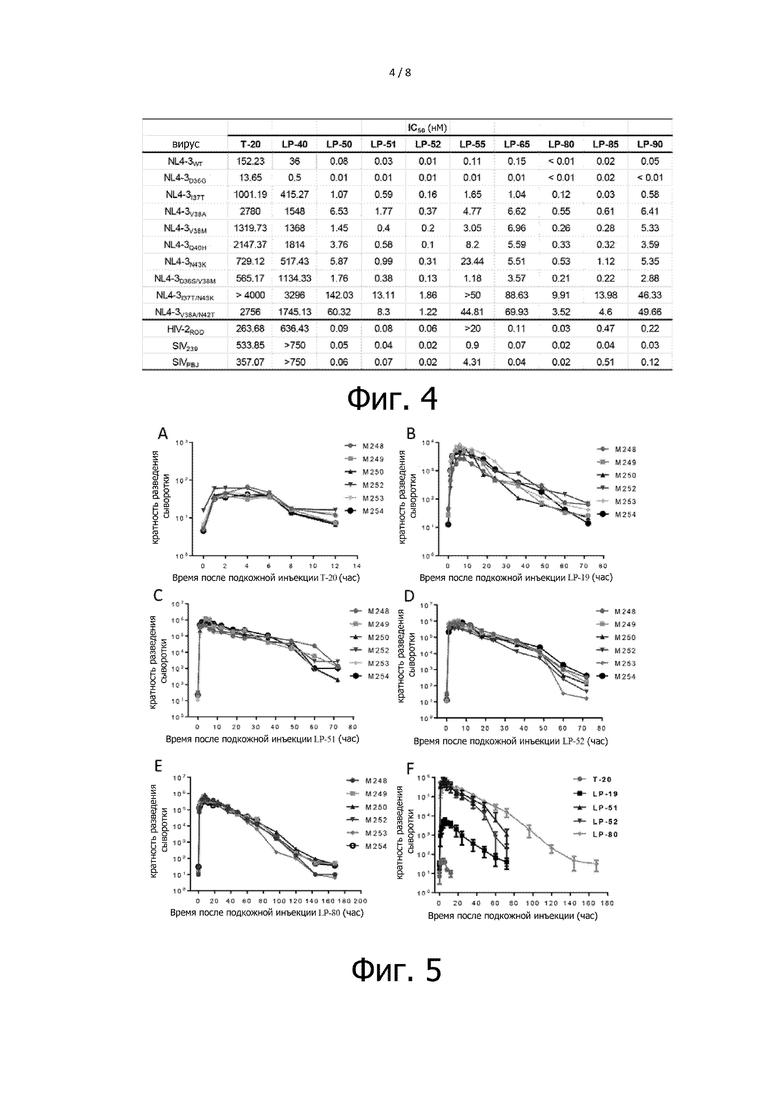
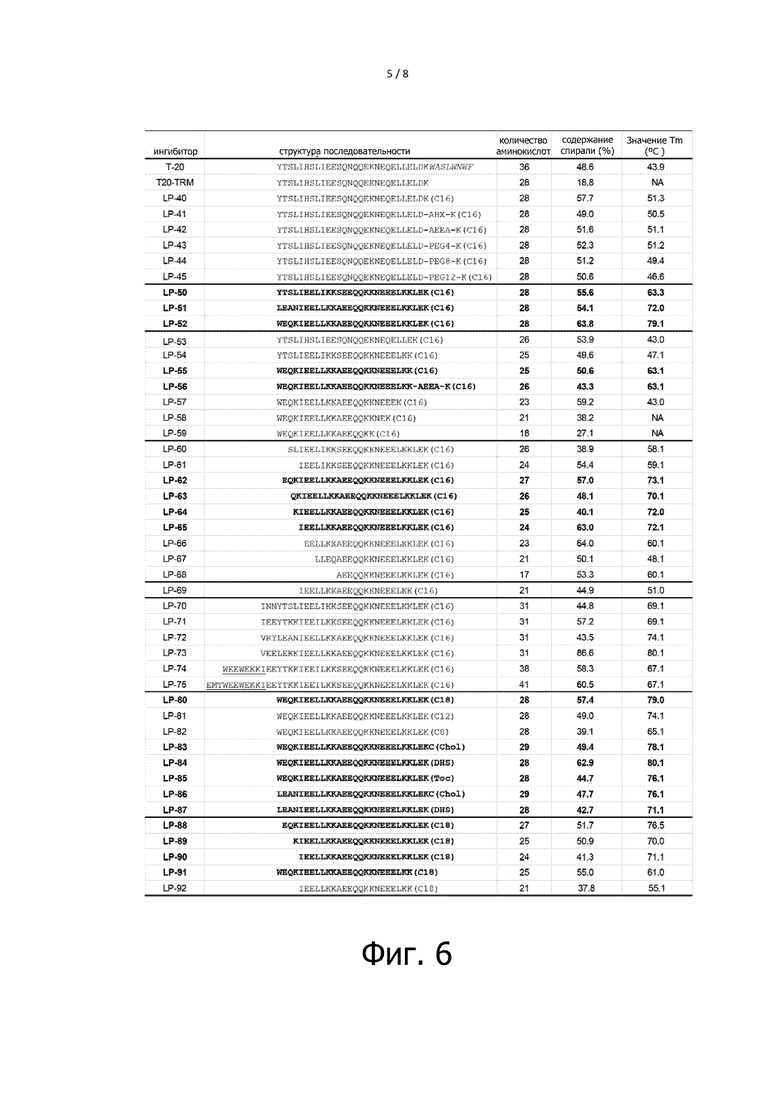
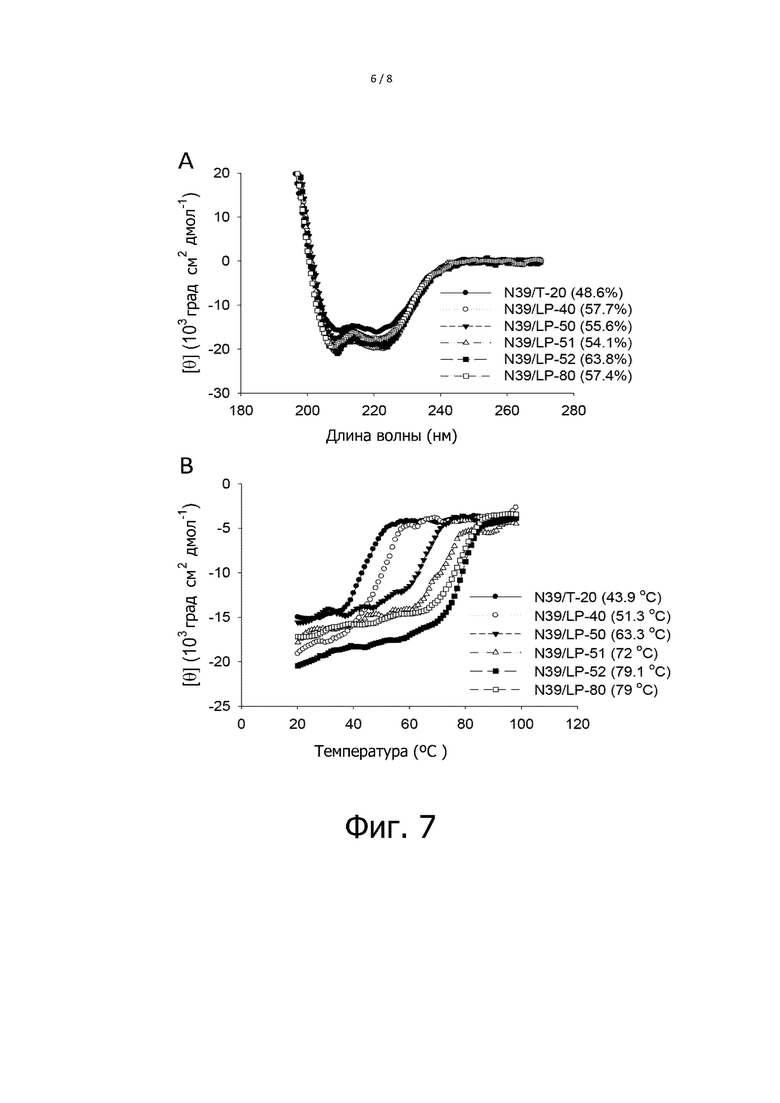
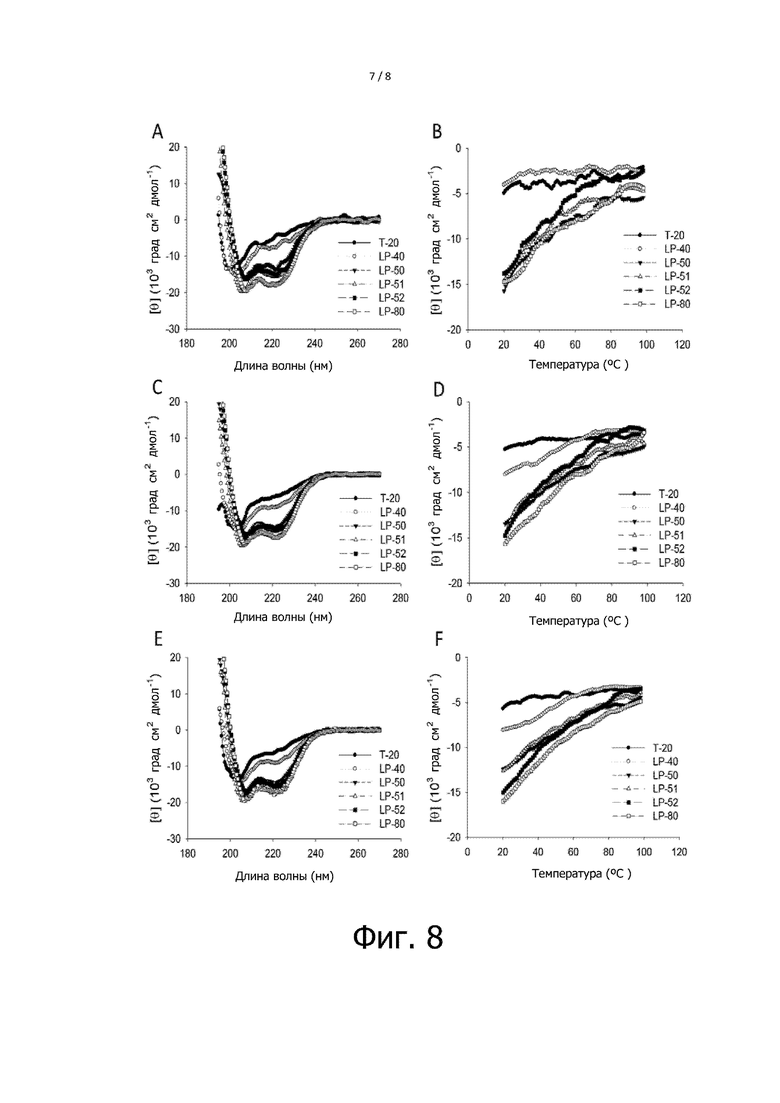
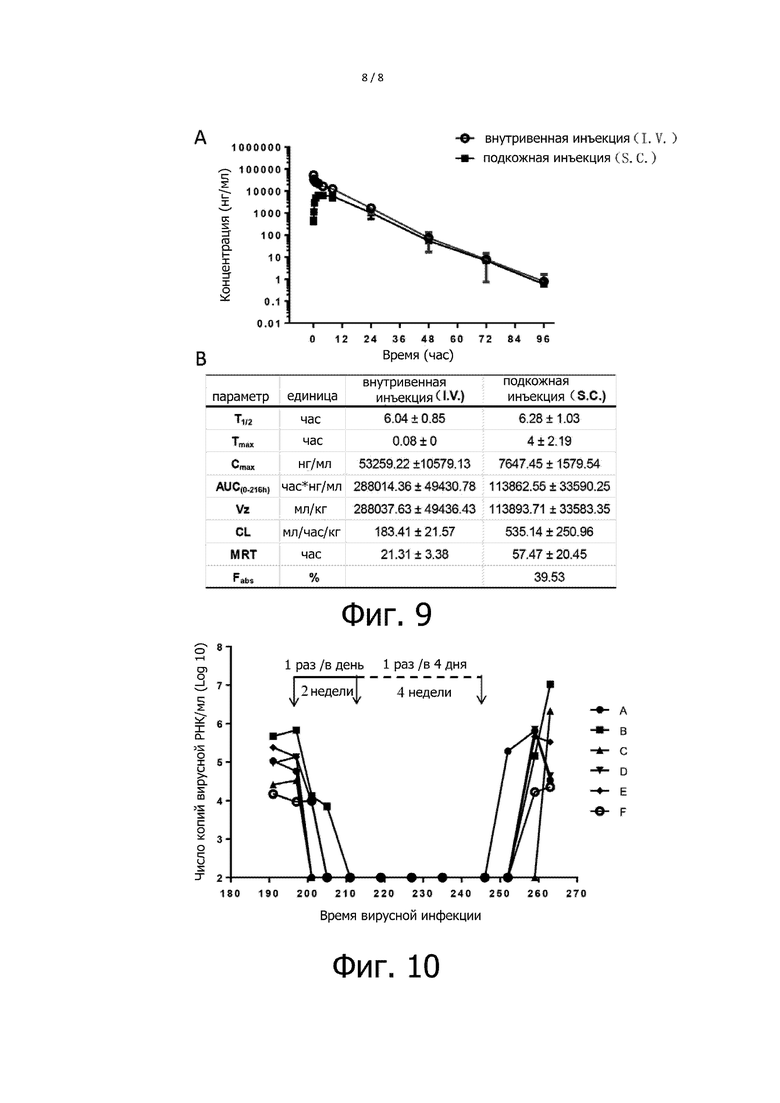
Настоящее изобретение имеет отношение к области биотехнологии, в частности к липопептиду, направленному на ингибирование ВИЧ, и может быть использовано в медицине. Изобретение позволяет получить как способный к эффективному ингибированию стадии проникновения ВИЧ в клетку млекопитающего (человека или примата) полипептид, так и соответствующий липопептид, образованный конъюгацией указанного полипептида с липофильным соединением, например жирной органической кислотой, или их производные. В силу механизма своего действия изобретение может применяться как для терапии ВИЧ-инфицированных больных, так и в качестве средства для профилактики инфицирования ВИЧ. В сравнении с известными аналогами, анти-ВИЧ активность липопептида настоящего изобретения выше, чем активность T-20 в несколько тысяч раз, а также существенно выше, чем активность известных анти-ВИЧ липопептидов, таких как C34-Chol, LP-19 или им подобных. 6 н. и 11 з.п. ф-лы, 10 ил., 10 пр.
1. Противовирусный липопептид, направленный против вируса иммунодефицита человека или примата, не являющегося человеком, или его фармацевтически приемлемая соль, отличающиеся тем, что липопептид представляет собой следующее a) или b):
a) липопептид, образованный соединением полипептида, обладающего противовирусной активностью, с липофильным соединением, соединенным с карбоксильным концом полипептида;
b) липопептид, образованный соединением полипептида, обладающего противовирусной активностью, с концевой защитной группой и липофильным соединением, соединенным с карбоксильным концом полипептида, при этом концевая защитная группа является защитной группой амино-конца и/или защитной группой карбокси-конца;
в a) и b),
причем липофильное соединение является жирной кислотой, содержащей от 8 до 20 атомов углерода, холестерином, дигидросфингозином или витамином Е,
где полипептид является любым одним из P1 - P5:
P1 имеет последовательность, показанную в следующей формуле I:
формула I
X1X2X3X4X5X6X7X8X9X10X11X12X13X14X15X16X17X18X19X20X21X22X23X24X25X26X27X28
причем в формуле I,
каждый из X1 - X28 является аминокислотным остатком, X1 представляет собой W, L или Y, X2 представляет собой E или T, X3 представляет собой Q, A или S, X4 представляет собой K, N или L, X5 представляет собой I или L, X6 представляет собой E, D, K, R или A, X7 представляет собой E, D, K, R или A, X8 представляет собой L или I, X9 представляет собой L или I, X10 представляет собой K, R, E, D или A, X11 представляет собой K, R, E, D или A, X12 представляет собой A или S, X13 представляет собой E, D, K, R или A, X14 представляет собой E, D, K, R или A, X15 представляет собой Q, X16 - Q, X17 представляет собой K, R, E, D или A, X18 представляет собой K, R, E, D или A, X19 представляет собой N, X20 представляет собой E или D, и X21 представляет собой E, D, K, R или A, X22 представляет собой E, D, K, R или A, X23 представляет собой L или I, X24 представляет собой K, R, E, D или A, X25 представляет собой K, R, E, D или A, X26 представляет собой L или I, X27 представляет собой E или D, X28 представляет собой K или R;
P2 представляет собой полипептид, полученный делецией от 1 до 4 остатков аминокислот на амино-конце P1;
P3 представляет собой полипептид, полученный делецией от 1 до 3 остатков аминокислот на карбоксильном конце P1;
P4 представляет собой полипептид, полученный добавлением остатка цистеина к карбоксильному концу P1;
P5 имеет последовательность, показанную в следующей формуле II:
формула II
X5X6X7X8X9X10X11X12X13X14X15X16X17X18X19X20X21X22X23X24X25,
причем в формуле II определения X5 - X25 являются такими же, как в формуле I;
и полипептид или липопептид обладает противовирусной активностью, направленной против указанного вируса иммунодефицита,
где необязательно один или более аминокислотных остатков полипептида указанного липопептида заменены L- или D-аминокислотами, искусственно модифицированными аминокислотами и/или редко встречающимися в природе аминокислотами.
2. Липопептид или его фармацевтически приемлемая соль по п. 1, антивирусная активность которого против вируса иммунодефицита человека или примата, не являющегося человеком, является активностью против одного из следующих v1-v7:
v1: ВИЧ-1, ВИЧ-2 и ВИО;
v2: ВИЧ-1 и ВИЧ-2;
v3: ВИЧ-1 и ВИО;
v4: ВИЧ-2 и ВИО;
v5: ВИЧ-1;
v6: ВИЧ-2; и
v7: ВИО.
3. Липопептид или его фармацевтически приемлемая соль по п. 1, отличающиеся тем, что
липопептид обладает противовирусной активностью, более высокой, чем активность LP-19, и/или T-20, и/или C34-Chol.
4. Липопептид или его фармацевтически приемлемая соль по любому из пп. 1-3, отличающиеся тем, что
P1 имеет последовательность, как показано в следующей последовательности:
X1X2X3X4IEELX9KKX12EEQQKKNEEELKKLEK;
P2 представляет собой P2-1, P2-2, P2-3 или P2-4, при этом
P2-1 имеет последовательность, как показано в следующей последовательности:
X2X3X4IEELX9KKX12EEQQKKNEEELKKLEK;
P2-2 имеет последовательность, как показано в следующей последовательности:
X3X4IEELX9KKX12EEQQKKNEEELKKLEK;
P2-3 имеет последовательность, как показано в следующей последовательности:
X4IEELX9KKX12EEQQKKNEEELKKLEK;
P2-4 имеет последовательность, как показано в следующей последовательности:
IEELX9KKX12EEQQKKNEEELKKLEK;
P3 имеет последовательность, как показано в следующей последовательности:
X1X2X3X4IEELX9KKX12EEQQKKNEEELKK;
P4 имеет последовательность, как показано в следующей последовательности:
X1X2X3X4IEELX9KKX12EEQQKKNEEELKKLEKC;
в P1, P2-1, P2-2, P2-3, P2-4, P3 и P4, определения X1, X2, X3, X4, X9 и X12 являются такими же, как в формуле I.
5. Липопептид или его фармацевтически приемлемая соль по п. 4, отличающиеся тем, что
P1 представляет собой P-80/84/85/52, P-87/51 или P50, при этом P-80/84/85/52 является полипептидом, представленным последовательностью SEQ ID NO: 1 в списке последовательностей, P-87/51 является полипептидом, представленным последовательностью SEQ ID NO: 2 в списке последовательностей, и P50 является полипептидом, представленным последовательностью SEQ ID NO: 3 в списке последовательностей;
P2-1 представляет собой P-88/62, при этом P-88/62 является полипептидом, представленным последовательностью SEQ ID NO: 4 в списке последовательностей;
P2-2 представляет собой P63 или P60, при этом P63 является полипептидом, представленным последовательностью SEQ ID NO: 5 в списке последовательностей, и P60 является полипептидом, представленным последовательностью SEQ ID NO: 6 в списке последовательностей;
P2-3 является P-89/64, при этом P-89/64 является полипептидом, представленным последовательностью SEQ ID NO: 7 в списке последовательностей;
P2-4 представляет собой P-90/65 или P61, при этом P-90/65 является полипептидом, представленным последовательностью SEQ ID NO: 8 в списке последовательностей; и P61 является полипептидом, представленным последовательностью SEQ ID NO: 9 в списке последовательностей;
P3 представляет собой P-91/55, при этом P-91/55 является полипептидом, представленным последовательностью SEQ ID NO: 10 в списке последовательностей; и
P4 представляет собой P83 или P86, при этом P83 является полипептидом, представленным последовательностью SEQ ID NO: 11 в списке последовательностей, и P86 является полипептидом, представленным последовательностью SEQ ID NO: 12 в списке последовательностей.
6. Липопептид или его фармацевтически приемлемая соль по п. 1, отличающиеся тем, что жирная кислота, содержащая от 8 до 20 атомов углерода, является стеариновой или пальмитиновой кислотой.
7. Липопептид или его фармацевтически приемлемая соль по п. 6, отличающиеся тем, что
липопептид является каким-либо из следующих 12 липопептидов LP-80/84/85/52, LP-90/65, LP-87/51, LP-88/62, LP-50, LP-83, LP-91/55, LP-86, LP-63, LP-89/64, LP-60 и LP-61;
LP-80/84/85/52 представляет собой LP-80/84/85/52a или LP-80/84/85/52b, при этом LP-80/84/85/52a формируют путем соединения полипептида, называемого P-80/84/85/52, с липофильным соединением, соединенным с карбоксильным концом P-80/ 84/85/52; LP-80/84/85/52b формируют путем соединения LP-80/84/ 85/52a с концевой защитной группой; в LP-80/84/85/52a и LP-80/84/85/52b P-80/84/85/52 представляет собой полипептид, представленный последовательностью SEQ ID NO: 1 в списке последовательностей, и липофильное соединение является стеариновой кислотой, дигидросфингозином или витамином E;
LP-90/65 представляет собой LP-90/65a или LP-90/65b, при этом LP-90/65a формируют путем соединения полипептида, называемого P-90/65, с липофильным соединением, соединенным с карбоксильным концом P-90/65; LP-90/65b формируют путем соединения LP-90/65a с концевой защитной группой; в LP-90/65a и LP-90/65b P-90/65 является полипептидом, представленным последовательностью SEQ ID NO: 8 в списке последовательностей, и липофильное соединение является стеариновой кислотой или пальмитиновой кислотой;
LP-87/51 представляет собой LP-87/51a или LP-87/51b, при этом LP-87/51a формируют путем соединения полипептида, называемого P-87/51, с липофильным соединением, соединенным с карбоксильным концом P-87/51; LP-87/51b формируют путем соединения LP-87/51a с концевой защитной группой; в LP-87/51a и LP-87/51b P-87/51 является полипептидом, представленным последовательностью SEQ ID NO: 2 в списке последовательностей, и липофильное соединение является дигидросфингозином или пальмитиновой кислотой;
LP-88/62 представляет собой LP-88/62a или LP-88/62b, при этом LP-88/62a формируют путем соединения полипептида, называемого P-88/62, с липофильным соединением, соединенным с карбоксильным концом P-88/62; LP-88/62b формируют путем соединения LP-88/62a с концевой защитной группой; в LP-88/62a и LP-88/62b P-88/62 является полипептидом, представленным последовательностью SEQ ID NO: 4 в списке последовательностей, и липофильное соединение является стеариновой кислотой или пальмитиновой кислотой;
LP-50 представляет собой LP-50a или LP-50b, при этом LP-50a формируют путем соединения полипептида, называемого P50, с пальмитиновой кислотой, соединенной с карбоксильным концом P50; LP-50b формируют путем соединения LP-50a с концевой защитной группой; в LP-50a и LP-50b P50 является полипептидом, представленным последовательностью SEQ ID NO: 3 в списке последовательностей;
LP-83 представляет собой LP-83a или LP-83b, при этом LP-83a формируют путем соединения полипептида, называемого P83, с холестерином, соединенным с карбоксильным концом P83; LP-83b формируют путем соединения LP-83a с концевой защитной группой; в LP-83a и LP-83b P83 является полипептидом, представленным последовательностью SEQ ID NO: 11 в списке последовательностей;
LP-91/55 представляет собой LP-91/55a или LP-91/55b, при этом LP-91/55a формируют путем соединения полипептида, называемого P-91/55, с липофильным соединением, соединенным с карбоксильным концом P-91/55; LP-91/55b формируют путем соединения LP-91/55a с концевой защитной группой; в LP-91/55a и LP-91/55b P-91/55 является полипептидом, представленным последовательностью SEQ ID NO: 10 в списке последовательностей, и липофильное соединение является стеариновой кислотой или пальмитиновой кислотой;
LP-86 представляет собой LP-86a или LP-86b, при этом LP-86a формируют путем соединения полипептида, называемого P86, с холестерином, соединенным с карбоксильным концом P86; LP-86b формируют путем соединения LP-86a с концевой защитной группой; в LP-86a и LP-86b P86 является полипептидом, представленным последовательностью SEQ ID NO: 12 в списке последовательностей;
LP-63 представляет собой LP-63a или LP-63b, при этом LP-63a формируют путем соединения полипептида, называемого P63, с пальмитиновой кислотой, соединенной с карбоксильным концом P63; LP-63b формируют путем соединения LP-63a с концевой защитной группой; в LP-63a и LP-63b P63 является полипептидом, представленным последовательностью SEQ ID NO: 5 в списке последовательностей;
LP-89/64 представляет собой LP-89/64a или LP-89/64b, при этом LP-89/64a формируют путем соединения полипептида, называемого P-89/64 с липофильным соединением, соединенным с карбоксильным концом P-89/64; LP-89/64b формируют путем соединения LP-89/64a с концевой защитной группой; в LP-89/64a и LP-89/64b P-89/64 является полипептидом, представленным последовательностью SEQ ID NO: 7 в списке последовательностей, и липофильное соединение является стеариновой кислотой или пальмитиновой кислотой;
LP-60 представляет собой LP-60a или LP-60b, при этом LP-60a формируют путем соединения полипептида, называемого P60, с пальмитиновой кислотой, соединенной с карбоксильным концом P60; LP-60b формируют путем соединения LP-60a с концевой защитной группой; в LP-60a и LP-60b P60 является полипептидом, представленным последовательностью SEQ ID NO: 6 в списке последовательностей; и
LP-61 представляет собой LP-61a или LP-61b, при этом LP-61a формируют путем соединения полипептида, называемого P61, с пальмитиновой кислотой, соединенной с карбоксильным концом P61; LP-61b формируют путем соединения LP-61a с концевой защитной группой; в LP-61a и LP-61b P61 является полипептидом, представленным последовательностью SEQ ID NO: 9 в списке последовательностей.
8. Антивирусный полипептид, его фармацевтически приемлемая соль или его производное, направленные против вируса иммунодефицита человека или примата, не являющегося человеком, отличающиеся тем, что полипептид является
любым одним из P1 - P5:
P1 имеет последовательность, показанную в следующей формуле I:
формула I
X1X2X3X4X5X6X7X8X9X10X11X12X13X14X15X16X17X18X19X20X21X22X23X24X25X26X27X28
причем в формуле I
каждый из X1 - X28 является аминокислотным остатком, X1 представляет собой W, L или Y, X2 представляет собой E или T, X3 представляет собой Q, A или S, X4 представляет собой K, N или L, X5 представляет собой I или L, X6 представляет собой E, D, K, R или A, X7 представляет собой E, D, K, R или A, X8 представляет собой L или I, X9 представляет собой L или I, X10 представляет собой K, R, E, D или A, X11 представляет собой K, R, E, D или A, X12 представляет собой A или S, X13 представляет собой E, D, K, R или A, X14 представляет собой E, D, K, R или A, X15 представляет собой Q, X16 - Q, X17 представляет собой K, R, E, D или A, X18 представляет собой K, R, E, D или A, X19 представляет собой N, X20 представляет собой E или D, и X21 представляет собой E, D, K, R или A, X22 представляет собой E, D, K, R или A, X23 представляет собой L или I, X24 представляет собой K, R, E, D или A, X25 представляет собой K, R, E, D или A, X26 представляет собой L или I, X27 представляет собой E или D, X28 представляет собой K или R;
P2 представляет собой полипептид, полученный делецией от 1 до 4 остатков аминокислот на амино-конце P1;
P3 представляет собой полипептид, полученный делецией от 1 до 3 остатков аминокислот на карбоксильном конце P1;
P4 представляет собой полипептид, полученный добавлением остатка цистеина к карбоксильному концу P1;
P5 имеет последовательность, показанную в следующей формуле II:
формула II
X5X6X7X8X9X10X11X12X13X14X15X16X17X18X19X20X21X22X23X24X25,
причем в формуле II определения X5 - X25 являются такими же, как в формуле I;
и указанный полипептид имеет противовирусную активность против указанного вируса иммунодефицита,
где указанное производное полипептида является по меньшей мере одним, выбранным из группы, состоящей из следующих 1) - 5):
1) производного, полученного путем соединения N-концевой защитной группы с амино-концом полипептида и/или путем соединения C-концевой защитной группы с карбоксильным концом полипептида;
2) производного, полученного путем соединения олигопептида или липофильного соединения с карбоксильным концом полипептида;
3) производного, полученного путем соединения олигопептида или липофильного соединения с амино-концом полипептида;
4) производного, полученного путем соединения олигопептида или липофильного соединения и с карбоксильным концом и с амино-концом полипептида; и
5) производного, полученного путем модификации полипептида протеином, полиэтиленгликолем или малеимидом.
9. Полипептид, его фармацевтически приемлемая соль или его производное по п.8, антивирусная активность которого против вируса иммунодефицита человека или примата, не являющегося человеком, является активностью против одного из следующих v1-v7:
v1: ВИЧ-1, ВИЧ-2 и ВИО;
v2: ВИЧ-1 и ВИЧ-2;
v3: ВИЧ-1 и ВИО;
v4: ВИЧ-2 и ВИО;
v5: ВИЧ-1;
v6: ВИЧ-2; и
v7: ВИО.
10. Композиция, содержащая C1) и C2), отличающаяся тем, что
C1) представляет собой C11) или/и C12), при этом C11) представляет собой липопептид или фармацевтически приемлемую соль по п. 1; C12) представляет собой полипептид, его производное или фармацевтически приемлемую соль по п. 8; C2) является фармацевтически приемлемым носителем или адъювантом;
причем композиция оказывает одно действие из лечения, предотвращения и лечения в качестве адъюнктивной (дополнительной) терапии болезни, обусловленной вирусной инфекцией, вызванной вирусом иммунодефицита человека или примата, не являющегося человеком.
11. Композиция по п. 10, где вирусная инфекция вызвана любым одним вирусом, выбранным из группы, состоящей из следующих v1-v7:
v1: ВИЧ-1, ВИЧ-2 и ВИО;
v2: ВИЧ-1 и ВИЧ-2;
v3: ВИЧ-1 и ВИО;
v4: ВИЧ-2 и ВИО;
v5: ВИЧ-1;
v6: ВИЧ-2; и
v7: ВИО.
12. Применение C11), C12) и/или C14) в производстве продукта для лечения, предотвращения или лечения в качестве адъюнктивной (дополнительной) терапии болезни, обусловленной вирусной инфекцией, вызванной вирусом иммунодефицита человека или примата, не являющегося человеком, где
C11) представляет собой липопептид или фармацевтически приемлемую соль по п. 1; C12) представляет собой полипептид, его производное или фармацевтически приемлемую соль по п. 8; а C14) представляет собой композицию по п. 10.
13. Применение по п. 12, в котором вирусная инфекция вызвана любым одним вирусом, выбранным из группы, состоящей из следующих v1-v7:
v1: ВИЧ-1, ВИЧ-2 и ВИО;
v2: ВИЧ-1 и ВИЧ-2;
v3: ВИЧ-1 и ВИО;
v4: ВИЧ-2 и ВИО;
v5: ВИЧ-1;
v6: ВИЧ-2; и
v7: ВИО.
14. Фармацевтическое соединение, отличающееся тем, что фармацевтическое соединение представляет собой C11) или C12), при этом
C11) представляет собой липопептид или фармацевтически приемлемую соль по п. 1; C12) представляет собой полипептид, его производное или фармацевтически приемлемую соль по п. 8, и это фармацевтическое соединение имеет применение для лечения, или предотвращения, или адъюнктивного лечения болезни, обусловленной вирусной инфекцией, вызванной вирусом иммунодефицита человека или примата, не являющегося человеком.
15. Фармацевтическое соединение по п. 14, где вирусная инфекция вызвана любым одним вирусом, выбранным из группы, состоящей из следующих v1-v7:
v1: ВИЧ-1, ВИЧ-2 и ВИО;
v2: ВИЧ-1 и ВИЧ-2;
v3: ВИЧ-1 и ВИО;
v4: ВИЧ-2 и ВИО;
v5: ВИЧ-1;
v6: ВИЧ-2; и
v7: ВИО.
16. Способ лечения или профилактики инфекции, вызванной вирусом, у животного, включающий введение животному C11), C12) или/и C14) с целью ингибирования вирусной инфекции у животного, согласно которому
C11) представляет собой липопептид или фармацевтически приемлемую соль по п. 1; C12) представляет собой полипептид, его производное или фармацевтически приемлемую соль по п. 8; а C14) представляет собой композицию по п. 10;
причем вирус является вирусом иммунодефицита человека или примата, не являющегося человеком.
17. Способ по п. 16, в котором вирус является любым одним вирусом, выбранным из группы, состоящей из следующих v1-v7:
v1: ВИЧ-1, ВИЧ-2 и ВИО;
v2: ВИЧ-1 и ВИЧ-2;
v3: ВИЧ-1 и ВИО;
v4: ВИЧ-2 и ВИО;
v5: ВИЧ-1;
v6: ВИЧ-2; и
v7: ВИО.
| INGALLINELLA P | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Светоэлектрический измеритель длин и площадей | 1919 |
|
SU106A1 |
| Паровоз для отопления неспекающейся каменноугольной мелочью | 1916 |
|
SU14A1 |
| Оправа для абонементных карточек, проездных билетов, личных удостоверений и проч. | 1925 |
|
SU5801A1 |
| LIU SH | |||
| Механический грохот | 1922 |
|
SU41A1 |
| Способ изготовления электрических сопротивлений посредством осаждения слоя проводника на поверхности изолятора | 1921 |
|
SU19A1 |
| Походная разборная печь для варки пищи и печения хлеба | 1920 |
|
SU11A1 |

