Настоящее изобретение относится к новым N-замещенным [(1'R), 2R, 3S, 4S, 5R, 6R, 9R, 11R, 12R, 14R]-11-(1'-гидроксипропил)-3-[(2,6-дидезокси-3-С-метил-3-О-метил-α-L-рибо-гексопиранозил)окси] -5-[(3,4,6-тридезокси-3-амино-β-D-ксилогексопиранозил)окси] -2,4,6,8,11,14-гексаметил-10,13,15-триоксацикло-[9.2.1.19.6]-пентадекан-1-онам с агонистическими мотилину свойствами и их солям, а также к содержащим эти соединения фармацевтическим композициям и способу получения этих соединений. Предлагаемые согласно изобретению соединения представляют собой производные N-дезметил-N-изопропилспироацеталя эритромицина А с суженным циклом.
Антибиотик эритромицин А, как известно, наряду со своим антибиотическим действием обладает также нежелательным для антибиотиков желудочно-кишечным побочным действием, в том числе сильное увеличение контракционной активности в желудочно-кишечной области со спазмами желудка и кишечника, тошнота, рвота и диарея.
Было осуществлено несколько попыток изменить эритромицин А так, чтобы получить производные, в которых антибиотическое действие практически отсутствует, однако достигается действие, влияющее на подвижность желудочно-кишечного тракта. Из европейской заявки на патент 0 550 895 известны производные N-дезметил-N-изопропилэритромицина А с суженным циклом, обладающие эффективными в отношении желудочно-кишечного тракта, агонистическими мотилину свойствами.
В основу настоящего изобретения положена задача получения новых, перорально эффективных производных эритромицина А с суженным циклом без антибиотического действия и с оказывающими благоприятное влияние на подвижность желудочно-кишечного тракта свойствами при улучшенном профиле действия.
В настоящее время показано, что новые производные N-дезметил-N-изопропил-спироацеталя эритромицина А с суженным циклом обладают селективными, агонистическими мотилину свойствами и стимулируют подвижность желудочно-кишечного тракта благоприятным образом, а также оказывают действия, усиливающие тонус нижнего эзофагусного сфинктера и тонус желудка. Благодаря их действию предлагаемые согласно изобретению вещества пригодны для лечения нарушений подвижности в желудочно-кишечном тракте и при этом отличаются хорошей совместимостью, хорошей оральной эффективностью и хорошей стабильностью.
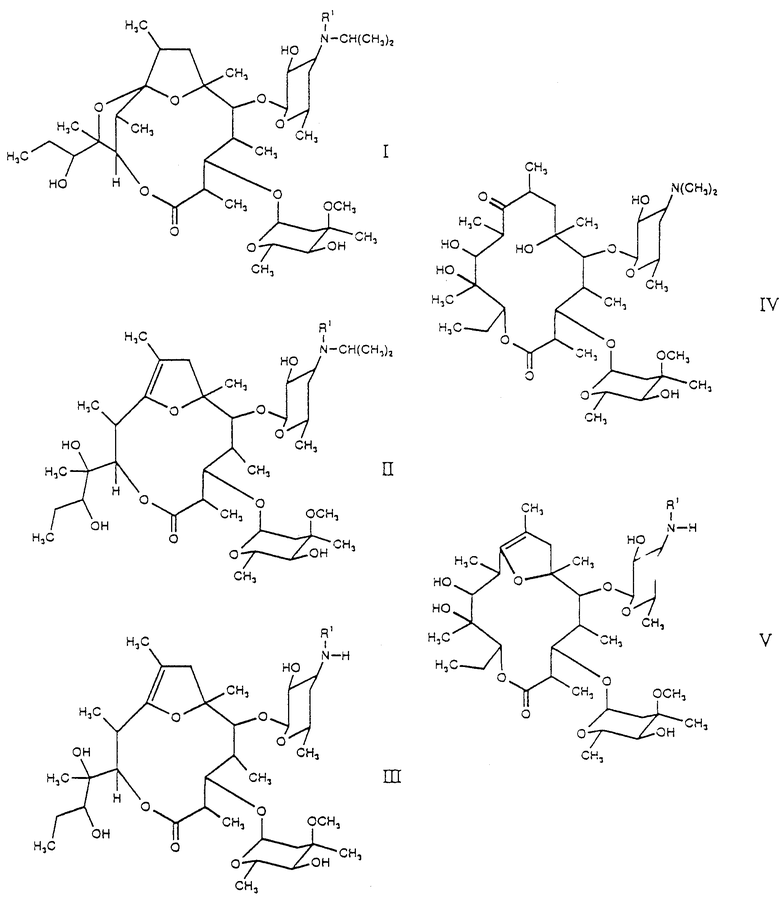
Настоящее изобретение поэтому относится к новым производным [(1'R),2R, 3S, 4S, 5R, 6R,9R,11R,12R,14R]-11-(1'-гидроксипропил)-2,4,6,8,11,14-гексаметил-10,13,15-триоксатрицикло-[9.2.1.19.6] -пентадекан-1-она общей формулы (I):
(см. формулу (I)),
где R' означает метил или водород,
и их стабильным и физиологически приемлемым солям.
В особенности благоприятным оказывается соединение формулы (I), где R1 означает метил.
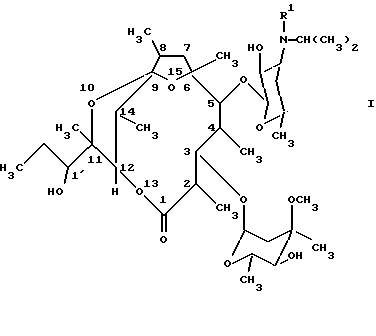
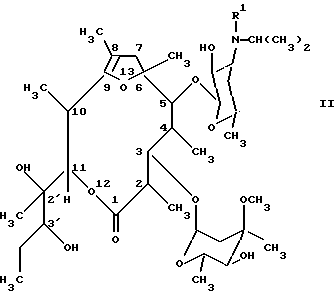
Соединения формулы (I) можно получать тем, что известным образом производные [2R(2'R, 3'R), 3S, 4S,5R,6R,10R,11R]-11-(2',3'-дигидроксипент-2'-ил)-2,4,6,8,10-пентаметил-12,13-диоксабицикло[8.2.1] тридец-8-ен-1-она общей формулы (II):
(см. формулу (II),
где R' имеет вышеуказанное значение,
путем обработки кислотой переводят в соединения формулы (I) и, при необходимости, в полученное соединение формулы (I), где R1 означает водород, вводят метильный остаток R1, или от полученного соединения формулы (I), где R1 означает метил, отщепляют метильный остаток R1, и, при необходимости, свободные соединения формулы (I) переводят в их стабильные соли или соли переводят в свободные соединения формулы (I).
Соединения формулы (I) получают из соединений формулы (II) путем катализируемой протонами внутримолекулярной спироциклизации. Спироциклизацию осуществляют известным образом путем обработки кислотой предпочтительно в водной среде, при низких значениях рН, например при значениях рН самое большее 3, целесообразнее при значениях рН от 1,5 до 3. В качестве кислот можно использовать инертные по отношению к остальным функциональным группам соединений формулы (I) и формулы (II) водорастворимые неорганические или органические кислоты. Целесообразно избегать снижения значения рН ниже 1, чтобы не протекали побочные реакции гидролиза. Пригодными реакционными средами являются, например, водный раствор соляной кислоты или водный раствор уксусной кислоты. Предпочтительно реакция циклизации протекает в водном растворе соляной кислоты при комнатной температуре.
Полученное соединение формулы (I), где R1 означает водород, при необходимости, можно дополнительно известным образом алкилировать с получением соответствующего N-метильного соединения. Алкилирование можно осуществлять известным образом путем взаимодействия с метилгалогенидом или как восстановительное алкилирование путем взаимодействия с формальдегидом в восстановительных условиях и можно проводить, например, в условиях, указанных для алкилирования соединений формулы (III).
От соединения формулы (I), где R1 означает метил, дополнительно можно отщеплять, при необходимости, метильный остаток R1. Деметилирование можно осуществлять известным образом путем обработки соединения галогеном, в особенности иодом и/или бромом, в инертном растворителе в присутствии пригодного основания. В качестве оснований пригодны, например, алкоголяты щелочных металлов, гидроксиды щелочных металлов и соли щелочных металлов слабых органических кислот.
Соединения формулы (I) можно известным образом выделять из реакционной смеси и очищать. Соли присоединения кислоты обычным образом можно переводить в свободные основания и, при желании, эти основания можно переводить известным образом в фармакологически приемлемые соли. Для избежания побочных реакций гидролиза целесообразно использовать для солеобразования только эквивалентные количества кислот.
В качестве фармакологически приемлемых солей соединений формулы (I) пригодны, например, их соли с неорганическими кислотами, как, например, угольная кислота, галогенводородные кислоты, в особенности соляная кислота, или с органическими кислотами, как, например, низшие алифатические моно- или дикарбоновые кислоты, как малеиновая, фумаровая, молочная, винная или уксусная кислоты.
В случае образующегося за счет реакции спироциклизации хирального центра, атома углерода в положении 8, могут получаться две эпимерные формы, так что возможны два изомера соединений формулы (I). Настоящее изобретение включает как смесь изомеров, так и чистые изомерные соединения формулы (I). При реакции циклизации образуется смесь изомеров. Чистые изомеры из этой смеси можно выделять известным образом путем обычных способов разделения, например путем хроматографического разделения.
Исходные соединения формулы (II) известны из ЕР-А-0 550 895 и их можно получать описанными там способами. Так, соединения формулы (II) можно получать способом, в котором в соединения общей формулы (III):
(см. формулу (III)),
где R1 имеет вышеуказанное значение, известным образом вводят изопропильный остаток.
Для введения изопропильного остатка соединения формулы (III) можно алкилировать известным образом. Предпочтительно алкилирование осуществляют как восстановительное алкилирование известным образом путем взаимодействия соединений формулы (III) с ацетоном в восстановительных условиях. Например, соединения формулы (III) можно вводить во взаимодействие с ацетоном в присутствии восстановителя, как, например, комплексное соединение гидрида бора, как натрийцианоборгидрид, натрийтриацетоксиборгидрид или натрийборгидрид. При желании, алкилирование, в особенности тех соединений формулы (III), где R1 означает метил, можно осуществлять также путем взаимодействия с изопропилгалогенидом, в особенности изопропилиодидом, или изопропилсульфатом, или сложным эфиром изопропилсульфокислоты. Целесообразно алкилирование осуществлять в инертном в условиях реакции органическом растворителе. Для восстановительного алкилирования может служить в качестве растворителя, например, избыток ацетона. Далее, в качестве растворителя пригодны также простые циклические эфиры, как тетрагидрофуран или диоксан, ароматические углеводороды, как толуол, или также низшие спирты. Алкилирование можно проводить при температурах от комнатной до температуры кипения растворителя. При алкилировании с помощью изопропильного производного, например изопропилгалогенида, как изопропилиодид, целесообразно работать в присутствии основания, как, например, карбонат щелочного металла или третичный органический амин.
При желании, в полученное соединение формулы (II), где R1 означает водород, можно вводить метильный остаток R1 или от полученного соединения формулы (II), где R1 означает метил, можно отщеплять метильный остаток R1. Такого рода метилирование и деметилирование можно осуществлять известным образом, например, в условиях, описываемых для введения или отщепления метильной группы в соединениях формулы (I).
Соединения формулы (III) можно получать известными способами, исходя из эритромицина А формулы (IV):
(см. формулу IV).
Так, эритромицин А сначала известным образом, например согласно способу DE-OS 21 54 032, можно моно- или дидеметилировать путем взаимодействия с галогеном, предпочтительно с иодом, в инертном растворителе в присутствии пригодного основания. В качестве оснований пригодны, например, алкоголяты, гидроксиды и карбонаты щелочных металлов и соли щелочных металлов слабых карбоновых кислот, как, например, ацетаты или пропионаты щелочных металлов. Можно использовать от 1 до 10 эквивалентов галогена в расчете на количество деметилируемого эритромицина. Для монодеметилирования в качестве оснований предпочтительно используют гидроксиды и/или соли щелочных металлов. Количество основания предпочтительно выбирают так, чтобы обеспечивалось значение рН в области 5-9. В качестве растворителей пригодны метанол, простые циклические эфиры, как диоксан или тетрагидрофуран, диметилформамид или смеси указанных растворителей с водой. Монодеметилирование целесообразно осуществлять при температурах от комнатной до 50oС. Реакцию можно ускорять путем облучения светом, например светом с длиной волны выше 290 нм от ртутной лампы низкого давления с фильтром из кварца или жаростойкого стекла (например, марки Пирекс®). Дидеметилирование (удаление двух метильных) групп предпочтительно осуществляют в абсолютном низшем спирте, например, как метанол, в присутствии соответствующего алкоголята щелочного металла при температуре от 0 до 10oС. При желании, для получения дидеметилированного продукта можно также исходить из уже монодеметилированного (с одной удаленной метильной группой) продукта.
Моно- или дидеметилированный эритромицин А известным образом путем мягкой обработки кислотой можно переводить в соответствующий моно- или дидеметилированный 8,9-ангидро-эритромицин-А-6,9-полукеталь общей формулы (V):
(см. формулу (V)),
где R1 означает водород или метил. Полукеталь можно получать, например, путем обработки органической кислотой, как лимонная, муравьиная или ледяная уксусная кислоты, или разбавленной неорганической кислотой при температурах от комнатной до примерно 50oС.
В соединениях формулы (V) известным образом путем внутримолекулярной транслактонизации можно осуществлять сужение 14-членного лактонового цикла эритромицинового скелета до 12-членного лактонового кольца с образованием соответствующих соединений формулы (III). Для этой цели соединения формулы (V) известным образом нагревают в низшем спирте в присутствии основания, например, при температурах от 40oС до 70oС, предпочтительно при температуре кипения реакционной смеси. В качестве оснований в особенности пригодны карбонаты щелочных металлов, также органические основания, как третичные амины, в особенности третичные низшие алкиламины. При этом сужении цикла конфигурация хиральных центров не изменяется.
Новые соединения формулы (I) и их физиологически приемлемые соли обладают представляющими интерес фармакологическими свойствами, в особенности агонистическими мотилину, стимулирующими подвижность желудочно-кишечного тракта свойствами. При этом они характеризуются благоприятным профилем действия с хорошей оральной активностью. Они лишены антибиотического действия и обладают высоким селективным сродством к рецепторам мотилина, в то время как в агонистически эффективных мотилину диапазонах доз не проявляют практически никакого релевантного сродства к другим рецепторам в желудочно-кишечном тракте, как адреналиновые, ацетилхолиновые, гистаминные, допаминные или серотониновые рецепторы.
Соединения характеризуются неожиданно хорошей переносимостью печенью, что позволяет их применять в течение продолжительного времени.
Для того чтобы обеспечить правильное переваривание употребленной пищи, в здоровом состоянии действуют одновременно автономная нервная система и гормоны желудочно-кишечного тракта, чтобы создать правильную сокращаемость желудочно-кишечного тракта не только после приема пищи, но и в случае пустого желудочно-кишечного тракта. Мотилин представляет собой известный желудочно-кишечный пептидный гормон, который стимулирует подвижность желудочно-кишечного тракта и индуцирует координированную подвижность во всем желудочно-кишечном тракте натощак, а также после приема пищи.
Соединения формулы (I) обладают мотилинообразным физиологическим действием тем, что они эффективны в качестве агонистов для рецепторов мотилина. Так, соединения формулы (I) оказывают ярковыраженные стимулирующие воздействия желудочно-кишечной области и в нижнем сфинктере пищевода. В особенности они вызывают ускорение опорожнения желудка, повышение тонуса желудка и продолжительное повышение тонуса в покое эзофагусного сфинктера. На основании своего мотилинообразного профиля действия вещества пригодны для лечения заболеваний, которые связаны с нарушениями подвижности в желудочно-кишечном тракте и/или оттоком пищевой кашицы из желудка в пищевод. Так, соединения формулы (I), например, показаны при парезе желудка различного происхождения, нарушениях тонуса желудка, нарушениях опорожнения желудка и гастроэзофагального оттока, диспепсии и послеоперационных нарушениях подвижности.
Эффективные в отношении желудочно-кишечного тракта свойства соединений формулы (I) можно подтвердить фармакологическими стандартными методами испытаний in vitro и in vivo.
Описание методов испытаний.
1. Определение способности испытуемых соединений связываться с рецепторами мотилина.
Сродство соединений формулы (I) к рецепторам мотилина определяют in vitro при использовании фракции тканевого гомогенизата из полости пазуха кролика. Определяют вытеснение радиоактивно маркированного иодированного мотилина из связи мотилин-рецептор испытуемыми соединениями.
Изучение связывания рецептора осуществляют согласно модификации метода Бормана и др. (Borman и др. Regulatory Peptides, 15, 143-153 (1986)). Для получения маркированного с помощью 125I-мотилина мотилин известным образом, например аналогично методу, описанному Bloom и др. (Scand. J. Gastroenterol. 11, 47-52 (1976)), ферментативно иодируют при использовании лактопероксидазы.
Для получения используемой в тесте фракции тканевого гомогенизата из полости пазуха кролика освобожденную от слизистых оболочек полость пазуха размельчают и гомогенизируют в десятикратном объеме холодного раствора буфера для гомогенизации (50 ммоль ТРИС-НСl-буфера; 250 ммоль сахарозы; 25 ммоль хлорида калия; 10 ммоль хлорида магния; рН 7,4) с добавкой ингибиторов (1 ммоль иодацетамида, 1 мкмоль пепстатина, 0,1 ммоль метилсульфонилфторида, 0,1 г/л ингибитора трипсина, 0,25 г/л бактрацина) с помощью гомогенизатора в течение 15 секунд с числом оборотов 1500 в минуту. Гомогенизат затем центрифугируют в течение 15 минут при 1000 g, полученный остаток промывают четырехкратно раствором буфера для гомогенизации и, наконец, снова суспендируют в 0,9%-ном растворе хлорида натрия (в объеме, соответствующем пятикратному массовому количеству полости пазуха). Таким образом полученная тканевая фракция, которая называется "сырая мембранная композиция", используется для испытания.
Для испытания на связывание 200 мкл сырой мембранной фракции (0,5-1 мг протеина) в 400 мкл буферного раствора А (50 ммоль ТРИС-НСl-буфера, 1/5% бычьего сывороточного альбумина, 10 ммоль хлорида магния, рН 8,0) вместе со 100 мкл иодированного мотилина, разбавленного в буферном растворе Б (10 ммоль ТРИС-НСl-буфера, 1% бычьего сывороточного альбумина, рН 8) (конечная концентрация 50 пмоль), инкубируют в течение 60 минут при температуре 30oС. Реакцию прекращают путем добавления 3,2 мл холодного буферного раствора Б, и связанный и несвязанный мотилин отделяют друг от друга путем центрифугирования (15 минут; 1000 g). Полученный после центрифугирования в виде осадка остаток промывают буферным раствором Б и подсчитывают число импульсов в гамма-счетчике. Изучение вытеснения осуществляют путем добавления возрастающих количеств испытуемого вещества в инкубационную среду. В качестве растворов испытуемых веществ используют водные растворы, которые получают путем пригодного разбавления 60•10-4 М водных основных (исходных) растворов. Труднорастворимые в воде испытуемые вещества сначала растворяют в 60%-ном этаноле, и этот раствор разбавляют таким количеством воды, чтобы в испытуемом растворе концентрация этанола не превышала 1,6 об.%. Из полученных результатов измерения определяют концентрацию ИK50 соответствующего тестируемого соединения, которая вызывает 50%-ное подавление специфического связывания иодированного мотилина с рецепторами мотилина. Из этой концентрации рассчитывают соответствующие пИК50-значения. Согласно настоящему методу для вещества примера 1 определяют пИК50-значение, равное 7,85.
2. Определение in vivo влияния веществ на тонус желудка.
Тонус желудка играет важную роль при опорожнении желудка. Повышенный тонус желудка способствует ускоренному опорожнению желудка.
Влияние веществ на тонус желудка определяют на гончих собаках с помощью баростата, который связан с пластиковым мешочком в желудке собаки и позволяет измерять объем и давление в желудке собаки. С помощью баростата определяют объем желудка при постоянном давлении в желудке или давление в желудке при постоянном объеме желудка. При повышении тонуса желудка при определенном давлении устанавливают уменьшенный объем желудка и повышенное давление при определенном объеме. В тест-модели, используемой для исследования повышения тонуса желудка, вызываемого веществами, определяют изменение объема желудка, вызываемое веществами, при постоянном давлении. Желудок подопытного животного расслабляется за счет поступления липидов, тонус желудка снижается, благодаря чему объем желудка соответственно увеличивается. В качестве меры действия веществ, повышающих тонус желудка, измеряют уменьшение увеличенного за счет введения липидов объема желудка в %, вызванное повышением тонуса желудка после введения вещества. В этой тест-модели вещество примера 1 при максимально переносимой дозе вызывает уменьшение увеличенного после введения липидов объема желудка на 69%.
На основании своего воздействия в желудочно-кишечном тракте соединения формулы (I) пригодны в гастроэнтерологии в качестве лекарственного средства для более крупных млекопитающих, в особенности людей, для профилактики и лечения нарушений подвижности желудочно-кишечного тракта.
Используемые дозы могут быть индивидуально различными и, естественно, могут изменяться в зависимости от рода излечиваемого состояния и формы введения. Например, парентерально вводимые лекарственные формы в общем содержат меньше активного вещества, чем орально вводимые препараты. В общем, для введения более крупным млекопитающим, в особенности людям, пригодны лекарственные формы с содержанием активного вещества от 1 до 100 мг на разовую дозу.
В качестве лекарственного средства соединения формулы (I) могут находиться вместе с обычными фармацевтическими вспомогательными веществами в галеновых композициях, как, например, таблетки, капсулы, суппозитории или растворы. Эти готовые лекарственные формы можно получать обычными способами с применением обычных твердых носителей, как, например, лактоза, крахмал или тальк, или жидких разбавителей, как, например, вода, жирные масла или жидкие парафины, и при использовании фармацевтически обычных вспомогательных веществ, например, как порофоры для таблеток, агенты растворения или консерванты.
Нижеследующие примеры должны подробнее пояснить изобретение, но никоим образом не ограничивая его объема притязаний.
Пример 1
[(1'R), 2R, 3S,4S,5R,6R,9R,11R,12R,14R]-11-(1'-Гидроксипропил)-3-[(2,6-дидезокси-3-С-метил-3-О-метил-α-L-рибо-гексопирано-зил)окси] -5-[(3,4,6-тридезокси-3-(N-метил-N-изопропиламино)β-D-ксилогексопиранозил)окси] -2,4,6,8,11,14-гексаметил-10,13,15-три-оксатрицикло[9.2.1.1. 9.6] -пентадекан-1-он(= смесь изомеров соединения формулы (I); R1=метил).
А) Получение N-дезметидэритромицина А
20 г Эритромицина А (27,2 ммоль) и 11,2 г (136,2 ммоль) ацетата натрия растворяют в 200 мл смеси метанола с водой в соотношении 8:2. Раствор нагревают до 47oС. Затем добавляют 6,9 г (136,2 ммоль) иода. рН-Значение поддерживают равным 8-9 путем добавления разбавленного водного раствора гидроксида натрия. Спустя 3 часа реакционную смесь с целью обработки выливают в смесь из 1 л воды и 20 мл раствора гидроксида аммония. Реакционную смесь экстрагируют этилацетатом, органический экстракт промывают содержащей гидроксид аммония водой и концентрируют. Получаемый после удаления растворителя сырой продукт перекристаллизовывают из смеси ацетона с раствором гидроксида аммония в соотношении 50:3. Т. пл. 143-148oС.
Б) Получение N-дезметид-8,9-ангидроэритромицин-А-6,9-полукеталя
(соединение формулы (V); R1=метил).
21 г Полученного на стадии А продукта растворяют в 110 мл ледяной уксусной кислоты, и раствор перемешивают в течение 1 часа при комнатной температуре. Затем с целью обработки реакционную смесь при охлаждении льдом прикапывают к 400 мл концентрированного раствора гидроксида аммония. Реакционную смесь экстрагируют этилацетатом, органический экстракт промывают водой, и растворитель удаляют. Получаемый в виде остатка сырой продукт перекристаллизовывают сначала из эфира и затем из метанола. Получают 14 г чистого продукта с т. пл. 145oС.
В) Получение [2R(2'R, 3'R), 3S,4S,5R,6R,10R,11R]-11-(2',3'-дигидроксипент-2'-ил)-3-[(2,6-дидезокси-3-С-метил-3-О-метил-α-L-рибо-гексопиранозил)окси] -5-[(3,4,6-тридезокси-3-метил-амино-β-D-ксило-гексопиранозил)окси] -2,4,6,8,10-пентаметил-12,13-диоксабицикло[8.2.1]тридец-8-ен-1-она
(соединение формулы (III); R1=метил).
9,4 г (13,4 ммоль) Полученного на стадии Б продукта вместе с 1,9 г (13,4 ммоль) карбоната калия в метаноле кипятят с обратным холодильником в течение 2,5 часов. С целью обработки реакционную смесь концентрируют, разбавляют водой и экстрагируют этилацетатом. Получаемый после удаления растворителя сырой продукт перекристаллизовывают из изопропанола. Получают 7,1 г чистого продукта с т. пл. 199-200oС; величина оптического вращения: [α]
Г) Получение [2R(2'R, 3'R), 3S, 4S,5S,6R,10R,11R]-11(2',3'-дигидроксипент-2'-ил)-3-[(2,6-дидезокси-3-С-метил-3-О-метил-α-L-рибо-гексопиранозил)окси]-5-[(3,4,6-тридезокси-3-(N-метил-N-изопропиламино)-β-D-ксилогексопиранозил)окси] -2,4,6,8,10-пентаметил-12,13-диоксабицикло[8.2.1]тридец-8-ен-1-она
(соединение формулы (III); R1=метил).
2 г (2,8 ммоль) Полученного на стадии В продукта растворяют в метаноле и значение рН раствора устанавливают равным 4 путем добавления разбавленной соляной кислоты. К раствору добавляют 2 г молекулярного сита (алюмосиликат кальция, диаметр пор 4  ), ацетон в избытке и 0,4 г (6,4 ммоль) цианоборгидрида натрия. Реакционную смесь перемешивают в течение 12 часов. С целью обработки отфильтровывают от молекулярного сита, фильтрат концентрируют, смешивают с водой и экстрагируют этилацетатом. Получаемый в виде остатка после концентрирования этилацетатного экстракта сырой продукт очищают путем колоночной хроматографии на силикагеле (элюирующее средство: смесь этилацетата с метанолом в соотношении 95:5). Получают 1,4 г очищенного продукта с т. пл. 130-134oС; величина оптического вращения [α]
), ацетон в избытке и 0,4 г (6,4 ммоль) цианоборгидрида натрия. Реакционную смесь перемешивают в течение 12 часов. С целью обработки отфильтровывают от молекулярного сита, фильтрат концентрируют, смешивают с водой и экстрагируют этилацетатом. Получаемый в виде остатка после концентрирования этилацетатного экстракта сырой продукт очищают путем колоночной хроматографии на силикагеле (элюирующее средство: смесь этилацетата с метанолом в соотношении 95:5). Получают 1,4 г очищенного продукта с т. пл. 130-134oС; величина оптического вращения [α]
Д) Получение целевого соединения
30 г Полученного на стадии Г продукта вносят в 2250 мл воды. При перемешивании к смеси прикапывают концентрированную соляную кислоту вплоть до достижения рН, равного 2-3. Затем реакционную смесь перемешивают в течение 7 часов при комнатной температуре. С целью обработки к реакционной смеси добавляют концентрированный раствор аммиака вплоть до достижения значения рН, равного 11. После этого реакционную смесь экстрагируют дихлорметаном. Органический экстракт концентрируют. Получаемый после концентрирования дихлорметанового экстракта сырой продукт очищают путем перекристаллизации из ацетонитрила. Получают 19,6 г целевого соединения с т. пл. 181-183oС; величина оптического вращения: [α]
Разделение изомеров
Изомеры разделяют путем полупрепаративной высокоэффективной жидкостной хроматографии (сокращенно ВЭЖХ) на готовой колонке с размерами 300 мм (высота) х 7,8 мм (внутренний диаметр) фирмы ВОТЕРС. Используют колонку с обращенной фазой "Symmetry-Prep®" С 18 (7 мкм). В качестве элюирующего средства служит смесь из 600 мл водного 0,05 М раствора КН2РО4 с рН-значением 6,0 (устанавливают с помощью 1 М раствора NaOH) и 400 мл ацетонитрила.
При времени удерживания 5,2 минуты получают 8R-изомер.
При времени удерживания 6,8 минуты получают 8S-изомер.
Пример 2
[(1'R), 2R, 3S, 4S,5R,6R,9R,11R,12R,14R]-11-(1'Гидроксипропил)-3-[(2,6-дидезокси-3-С-метил-3-О-метил-α-L-рибогексопиранозил)окси] -5-[(3,4,6-тридезокси-3-(N-изопропиламино)-β-D-ксилогексопиранозил)окси] -2,4,6,8,11,14-гексаметил-10,13,15-триоксатрицикло[9.2.1.1. 9.6] -пентадекан-1-он (= смесь изомеров соединения формулы (I); R1 = водород).
А) Получение (2R(2'R, 3'R), 3S, 4S,5R,6R,10R,11R]-11-(2'3'-дигидроксипент-2'-ил)-3-[(2,6-дидезокси-3-С-метил-3-О-метил-α-L-рибогексопиранозил)окси] -5-[(3,4,6-тридезокси-3-(N-изопропиламино)-β-D-ксилогексопиранозил)окси] -2,4,6,8,10-пентаметил-12,13-диоксабицикло[8.2.1]тридец-8-ен-1-она
Смесь из 7,3 г метилата натрия и 500 мл метанола в атмосфере азота охлаждают до 0oС. Затем прикапывают раствор 20 г полученного в примере 1 Г соединения формулы (II) (R1=метил) в 100 мл метанола. После этого порциями добавляют 34,1 г иода и реакционную смесь в течение 24-х часов выдерживают при температуре 0-5oС. С целью обработки реакционную смесь вносят в раствор 58 г тиосульфата натрия и 48 мл концентрированного раствора аммиака в 1,5 л воды. Водную фазу экстрагируют четырехкратно по 100 мл хлороформом. Объединенные органические фазы промывают один раз с помощью смеси из 5 мл концентрированного раствора аммиака и 100 мл воды, сушат над сульфатом натрия и концентрируют. Получаемый остаток очищают с помощью колоночной хроматографии на силикагеле. Получают 0,5 г очищенного продукта с т. пл. 147-155oС; величина оптического вращения: [α]
Б) Получение целевого соединения
1 г Вышеполученного продукта вводят во взаимодействие согласно описанному в примере 1 Д способу. Получают 0,47 г целевого соединения с т. пл. 201-209oС; величина оптического вращения: [α]
Пример I
[(1R'), 2R, 3S,4S,5R,6R,9R,11R,12R,14R]-11-(1'-Гидроксипропил)-3-[(2,6-дидезокси-3-С-метил-3-О-метил-α-L-рибогексопирано-зил)окси] -5-[(3,4,6-тридезокси-3-(N-метил-N-изопропиламино)β-D-ксилогексопиранозил)окси] -2,4,6,8,11,14-гексаметил-10,13,15-триоксатрицикло[9.2.1.1. 9.6]-пентадекан-1-он (= смесь изомеров соединения формулы (I); R1=метил) 20 мг, кукурузный крахмал 60 мг, лактоза 135 мг, желатин (в виде 10%-ного раствора) 6 мг.
Активное вещество, кукурузный крахмал и лактозу концентрируют с помощью 10%-ного раствора желатина. Пасту размельчают, и полученный гранулят наносят на пригодный металлический лист и высушивают при 45oС. Высушенный гранулят пропускают через машину для измельчения (дробилку) и в смесителе смешивают со следующими вспомогательными веществами:
Тальк - 5 мг
Стеарат магния - 5 мг
Кукурузный крахмал - 9 мг
и после этого прессуют в таблетки массой по 240 мг.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 11-АЦЕТИЛ-12,13-ДИОКСАБИЦИКЛО [8.2.1] ТРИДЕЦЕНОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ ЛЕКАРСТВЕННЫЕ СРЕДСТВА | 1999 |
|
RU2218346C2 |
| ПРОИЗВОДНЫЕ [2R,3R (2'R,3'R,), 6R,7S,8S,9R,10R] -3-(2',3'-ДИГИДРОКСИПЕНТ-2'-ИЛ)-2,6,8,10,12-ПЕНТАМЕТИЛ -4,13-ДИОКСАБИЦИКЛО [8,2,1]-ТРИДЕЦ-12-ЕН-5-ОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО | 1992 |
|
RU2107070C1 |
| Иммуномодулирующие азалиды на основе мочевины | 2021 |
|
RU2811591C1 |
| ПРОИЗВОДНЫЕ МОЧЕВИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1998 |
|
RU2198883C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4"-ЗАМЕЩЕННЫХ ПРОИЗВОДНЫХ 9-ДЕОКСО-9А-АЗА-9А-ГОМОЭРИТРОМИЦИНА А | 2002 |
|
RU2263117C2 |
| ПРОИЗВОДНЫЕ БЕНЗАЗЕПИНОН-N-УКСУСНОЙ КИСЛОТЫ, ЗАМЕЩЕННЫЕ ФОСФОНОВОЙ КИСЛОТОЙ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННЫЕ СРЕДСТВА, СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ | 1998 |
|
RU2211219C2 |
| ТВЕРДЫЕ СОЛИ БЕНЗАЗЕПИНОВЫХ СОЕДИНЕНИЙ И ИХ ПРИМЕНЕНИЕ ПРИ ПОЛУЧЕНИИ ФАРМАЦЕВТИЧЕСКИХ СОЕДИНЕНИЙ | 2003 |
|
RU2303041C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ У МЛЕКОПИТАЮЩИХ | 2004 |
|
RU2344121C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4''-ЗАМЕЩЕННЫХ ПРОИЗВОДНЫХ 9-ДЕОКСО-9А-АЗА-9А-ГОМОЭРИТРОМИЦИНА А | 2002 |
|
RU2233287C2 |
| СОЕДИНЕНИЯ С-МАННОЗИДА, ПРИМЕНИМЫЕ ДЛЯ ЛЕЧЕНИЯ ИНФЕКЦИЙ МОЧЕВЫВОДЯЩИХ ПУТЕЙ | 2019 |
|
RU2790228C2 |
Изобретение относится к производным [(1'R), 2R, 3S, 4S, 5R, 6R, 9R, 11R, 12R, 14R] -11-(1'-гидроксипропил)-2,4,6,8,11,14-гексаметил-10,13,15-триоксатрицикло [9.2.1.1. 9.6] -пентадекан-1-она общей формулы (I), где R1 означает метил или водород, и их стабильным и физиологически приемлемым солям. Описывается способ получения этих производных путем обработки соединений формулы (II) кислотой. Также описывается лекарственное средство для лечения и/или профилактики нарушений подвижности желудочно-кишечного тракта, гастроэзофагального оттока, диспепсии и/или нарушений опорожнения желудка, содержащее фармакологически эффективное количество соединения формулы (I). 3 c. и 1 з.п. ф-лы.

где R1 означает метил или водород,
или их стабильные и физиологически приемлемые соли.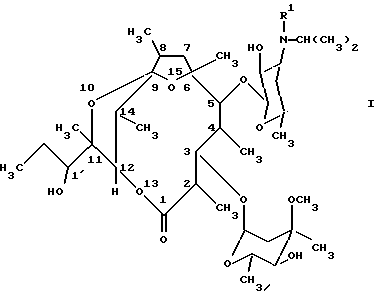
где R1 означает метил или водород,
или их стабильных и физиологически приемлемых солей, отличающийся тем, что производное [2R (2'R, 3'R), 3S, 4S, 5R, 6R, 10R, 11R] -11-(2', 3'-дигидроксипент-2'-ил)-2,4,6,8,10-пентаметил-12, 13-диоксабицикло[8.2.1] тридец-8-ен-1-она общей формулы (II)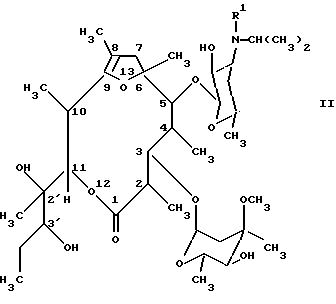
где R1 имеет вышеуказанное значение,
путем обработки кислотой переводят в соединение формулы (I) и, в случае необходимости, в полученное соединение формулы (I), где R1 означает водород, вводят метильный остаток R1 или от полученного соединения формулы (I), где R1 означает метил, отщепляют метильный остаток R1, и, в случае необходимости, свободные соединения формулы (I) переводят в их стабильные соли присоединения кислот или соли присоединения кислот переводят в свободные соединения формулы (I).
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Регулятор расхода газа | 1975 |
|
SU550895A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| EP 561413 A1, 22.09.1993 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| МАКРОЛИДНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2086560C1 |

