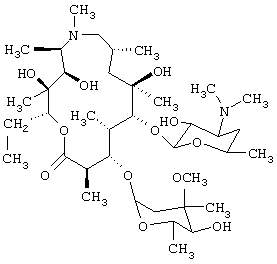
Изобретение относится к области аналитической химии, в частности к инверсионному вольтамперометрическому способу определения антибиотика (АБ) азитромицина дигидрата, который представляет собой 9-Deoxo-9a-aza-9a-methyl-9a-homoerythromycin A dihydrate (C38H72N2O12·2Н2О):
Азитромицина дигидрат(азитромицин) относится к полусинтетическим антибиотикам второго поколения и обладает высокой микробиологической и клинической эффективностью в лечении ряда тяжелых инфекций дыхательных путей, кожи и мягких тканей, некоторых урогенитальных инфекций. Определение макроколичеств азитромицина важно для оценки фармакологического действия и эффективности антибактериальной терапии, идентификации действующих веществ в лекарственных формах, а также его метаболитов в биологических матрицах. Это в свою очередь предъявляет повышенные требования к контролю за качеством лекарственных средств и совершенствованию методов количественного определения антибиотиков группы макролидов. Важной проблемой остается разработка новых, более чувствительных и селективных методов их анализа.
В настоящее время имеется сравнительно небольшое число работ по определению азитромицина. Для количественного определения применяли микробиологический метод [Krichhoff R.M., Laufen Н., Schacke G., Kirchhoff G., Gallo E. Determination of azithromycin in gastric biopsy samples./Int.J.Clin Parmacol Ther 1999 Jul; 37(7):361-4], а также хроматографические методы анализа (тонкослойная хроматография (ТСХ), высокоэффективная жидкостная хроматография (ВЭЖХ) [Фармакопейная статья на препарат сумамед капсулы 250 мг от 11 июня 1997 г НД 42-1205-97].
Авторы отмечают мешающее действие растворителей, которые могут перекрывать пик азитромицина и снижать точность анализа. Для увеличения чувствительности определения методом ВЭЖХ использовали химическую ионизационную масс-спектрометрию и электрохимическое детектирование для фармакокинетического исследования содержания антибиотика в сыворотке крови у детей [Founda H.G., Schneider R.P. (HPLC) - atmospheric pressure chemical ionization mass spectrometry: correlation with a standard HPLC - electrochemical method./J.Chromatogr A 1998 Jul 3; 812(1-2):287-93]. Несмотря на достаточно высокую чувствительность вариантов ВЭЖХ (от 1,3· 10-7 моль/л до 3,2· 10-5 моль/л азитромицина), длительность анализа с учетом времени пробоподготовки, очистки, экстракции, хроматографического разделения, а также высокая стоимость приборов существенно ограничивают его использование в контрольно-аналитических лабораториях для экспрессного количественного определения антибиотика. Применение ВЭЖХ за рубежом в анализе лекарственных средств, по-видимому, связано прежде всего с интенсивной разработкой теоретических основ метода, созданием и промышленным выпуском необходимых сорбентов и аппаратуры.
Электрохимические методы, и, в первую очередь, такие его высокочувствительные варианты, как инверсионные вольтамперометрические (ВА), отвечают современным требованиям к контролю разнообразных и сложных по составу систем: лекарственных и фармацевтических препаратов, природных объектов, пищевых продуктов. В инверсионных вариантах ВА минимально определяемая концентрация органических веществ может достигать 10-10-10-11 моль/л. Методы ВА позволяют проводить серийные анализы с использованием современных компьютеризированных вольтамперометрических анализаторов с высокой экспрессностью, селективностью и, зачастую, без предварительной пробоподготовки с одновременным определением нескольких веществ из одной навески, что существенно упрощает анализ. Имеются ограниченные сведения о применении электрохимических методов, в том числе и вольтамперометрических для определения азитромицина.
Наиболее близким является метод циклической вольтамперометрии (ЦВА), используемый для детектирования антибиотика после предварительного его выделения из пробы методом ВЭЖХ [Shepard R.M., Duthu G.S, Ferraina R.A., Mullins M.A. High-performance liquid chromatographic assay with electrochemical detection for azithromycin in seum and tissues./J.Chromatogr 1991 Apr 19; 565(1-2), 321-337] (прототип). Циклическое ВА определение азитромицина проводили после растворения препарата в подвижной фазе (по ВЭЖХ условиям), которая служила фоном: смесь 0,02 М ацетата аммония - 0,02 М перхлората натрия - ацетонитрил - метанол в соотношении 22:23:45:10 соответственно, с использованием рабочего СУ электрода. Сигнал в виде анодной волны фиксировали при значениях E1/2 0,95 В при рН 11 относительно хлоридсеребряного электрода при скорости изменения потенциала 5 мВ/с. Минимально определяемая концентрация азитромицина ~3· 10-8 моль/л. Для улучшения воспроизводимости и снижения пассивации электрода требовалась очистка электрода 6 М азотной кислотой и последующая настройка детектора.
В данных условиях определение азитромицина на уровне n· 10-10 моль/л невозможно.
Задачей заявляемого изобретения является повышение чувствительности и экспрессности определения азитромицина методом адсорбционной инверсионной дифференциальной вольтамперометрии.
Поставленная задача достигается тем, что способ количественного определения азитромицина дигидрата (азитромицина) включает перевод азитромицина из пробы в раствор и вольтамперометрическое определение с использованием индикаторного стеклоуглеродного электрода, отличающийся тем, что используют инверсионную вольтамперометрию, при этом накопление азитромицина в перемешиваемом растворе проводят в течение при потенциале 30-60 с при потенциале электролиза (0,10-0,25) В относительно насыщенного хлоридсеребряного электрода на фонах 0,2н. раствора гидрофосфата натрия или буферного раствора Бриттона-Роббинсона, рН 8,0-9,0 в присутствии 0,1%-ного этилового спирта с последующей регистрацией анодных пиков в дифференциальном режиме съемки вольтамперограмм при скорости развертки потенциала 20-30 мВ/с и концентрацию азитромицина определяют по высоте пика в диапазоне потенциалов от 0,70 до 0,85 В методом добавок аттестованных смесей.
В предлагаемом способе впервые установлена способность азитромицина окисляться на различных типах графитовых электродов. В качестве индикаторных применяли СУ и графитовый (Г) электрод, пропитанный полиэтиленом и парафином в вакууме (в прототипе применяли только СУ торцевой электрод). Использование таких электродов обусловлено высокой химической и электрохимической устойчивостью графита, широкой областью рабочих потенциалов, как в водных, так и в неводных средах, а также простотой механического обновления поверхности и требованиями техники безопасности. Способность к окислению азитромицина зависит от материала электрода и состояния его поверхности. Максимальное значение регистрируемого тока с использованием Г электрода выше (примерно на 10-15%), однако из-за большого остаточного тока он оказался менее удобным в работе, чем СУ. Для определения азитромицина использовали “игольчатые” по форме индикаторные электроды, после предварительного электрохимического модифицирования их поверхности. Это приводит к снижению нижней границы определяемых содержаний, улучшению воспроизводимости вольтамперометрических измерений и экспрессности анализа. “Игольчатые” СУ и Г электроды впервые использованы для идентификации антибиотиков группы макролидов на примере определения азитромицина.
Реакция электроокисления азитромицина является электрофильной. Роль электрофильного реагента с электронным дефицитом выполняет анод, а субстрата органическая молекула, реакционная способность которой определяется электронной плотностью связей, атомов и групп в молекуле и сильно зависит от рН среды. Предварительные исследования показали, что электроокисление азитромицина осложнено как адсорбцией, так, возможно, и дополнительными химическими стадиями и представляет сложный диффузионно-контролируемый электродный процесс с участием более одного электрона.
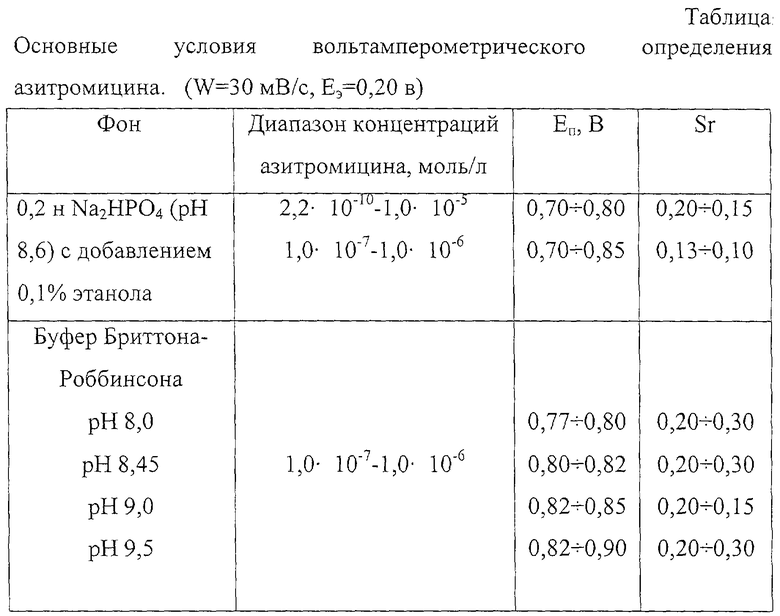
В прототипе описано использование в качестве фона смеси 0,02 М ацетата аммония - 0,02 М перхлората натрия - ацетонитрил - метанол в соотношении 22:23:45:10 соответственно, рН 11. Определение азитромицина в этих условиях затруднено. Молекула азитромицина содержит два основных радикала и азитромицин очень чувствителен к изменению рН среды. С увеличением рН раствора буфера Бриттона-Роббинсона от 8,0 до 9,0 потенциал пика окисления азитромицина смещается в более положительную область потенциалов от 0,774 до 0,850 В. По-видимому, это связано с тем, что у основных радикалов молекул макролидных антибиотиков, в частности эритромицина и азитромицина, рН составляет 8,4-8,8 [Страчунский Л.С., Козлов С.Н. Макролиды в современной клинической практике. - Смоленск: Русич, 1998. - С.166-197]. Поэтому практически при рН больше 9,0 молекула азитромицина практически находится преимущественно в неионизированном состоянии и окисляется при более положительном потенциале. Кроме того, при рН больше 9,0 может регистрироваться дополнительный пик с ЕП 0,90 В, что снижает разрешающую способность способа. При рН меньше 8,0 молекула азитромицина находится в ионизированном состоянии и поэтому окисляется при менее положительном потенциале. Значения рН 8,0-9,0 являются оптимальными для количественного химического определения азитромицина. В кислой среде при рН меньше 6 пик окисления не регистрируется, по-видимому, происходит распад молекул макролидов [Мелентьева Г.А. Фармацевтическая химия. - М.: Медицина, 1976, 828 с.]. Предлагаемые в заявленном изобретении фоны 0,2н. (что соответствует рН 8,6) гидрофосфат натрия (Na2HPO4) или раствор буфера Бриттона-Роббинсона (рН 8,0-9,0) в присутствии 0,1%ного (по объему) этилового спирта позволяет определять азитромицин на уровне 10-10 моль/л с хорошей воспроизводимостью. Относительное стандартное отклонение (Sr) не превышает 0,20 и 0,25 при регистрации анодных пиков соответственно на фонах 0,2н. Na2HPO4 и раствора буфера Бриттона-Роббинсона рН 8,0-9,0 для диапазона концентраций 2,2· 10-10-3,0· 10-9 моль/л и Sr равно 0,13 для диапазона концентрации 1· 10-6-1· 10-7 моль/л (таблица).
Фоны 0,2н. Na2HPO4 и растворы буфера Бриттона-Роббинсона подобраны экспериментально. Абсолютной новизной является экспериментально установленный диапазон рН от 8,0 до 9,0 и использование спиртового водно-органического растворителя. Присутствие 0,1%-ного по объему этилового спирта способствует лучшему удалению антибиотика с поверхности электрода и приводит к стабилизации и воспроизводимости анодных пиков азитромицина. Применение указанных в заявляемом изобретении фоновых электролитов впервые позволило снизить предел обнаружения азитромицина, рассчитанного по 3-сигмовому критерию, до 1,84· 10-10 моль/л (Cmin,p). Минимально определяемая концентрация (Сн) 2,2· 10-10 моль/л на фоне 0,2н. Na2HPO4 и 1,0· 10-9 моль/л на фоне раствора буфера Бриттона-Роббинсона рН 8,0-9,0. Минимально определяемая концентрация азитромицина на указанных фонах в прототипе ~3· 10-8 моль/л. Авторами статьи (прототип) отмечена низкая воспроизводимость волн окисления азитромицина и высокая пассивация индикаторного электрода. Значения Sr в прототипе не указаны. Кроме того, в прототипе использована высокотоксичная смесь ацетонитрила - метанола, что ограничивает ее применение в массовых анализах.
Другим отличительным признаком являются установленные условия электрохимического накопления: потенциал электролиза Еэ=(0,10-0,25)В. Опытные данные показали зависимость тока окисления азитромицина от Еэ (чертеж). Величина анодного тока увеличивалась примерно в 2,4 раза и достигала максимального значения в области потенциалов (0,10-0,25)В. При Еэ=(0,10-0,25) В уменьшалась величина тока окисления антибиотика. Использование предварительного электролиза при значениях потенциала (0,10-0,25) В позволяет регистрировать вольтамперограммы с четко выраженным максимумом.
Это позволяет повысить точность и селективность способа и экспрессно определять концентрации азитромицина меньше чем 1· 10-8 моль/л.
Оптимальное время предварительного электролиза (τ э) составляет 30-60 с. При τ э меньше 30 с снижается чувствительность определения и увеличивается ошибка определения, а при τ э больше 60 с снижается экспрессность; величина тока достигала максимального значения при τ э равном 30-60 с.
Важным для определения азитромицина методом дифференциальной ВА является выбор скорости развертки потенциала. Оптимальной является скорость 20-30 мВ/с. Увеличение скорости развертки потенциала более 30 мВ/с увеличивает чувствительность, но при этом растет остаточный ток и уменьшается разрешающая способность метода. Использование скорости менее 30 мВ/с снижает величину анодного тока и понижает чувствительность определения. Использование скорости 5 мВ/с (прототип) не позволяет определять азитромицина на уровне (10-9-10-10) моль/л.
Нижняя граница определяемых содержаний азитромицина зависит от режима съемки вольтамперных кривых. Использование режима дифференцирования позволяет фиксировать четкие пики даже при очень низких концентрациях (10-9-10-10) моль/л, что повышает чувствительность определения. Для определения азитромицина он ранее не применялся. При линейном наложении потенциала в интегральном режиме определение концентраций азитромицина на уровне (10-8-1010) моль/л практически невозможно из-за большого значения остаточного тока, что приводит к регистрации нечеткой волны окисления, а минимально определяемая концентрация составляет 1· 10-7 моль/л. Поэтому для количественного химического анализа рекомендован дифференциальный режим съемки вольтамперограмм. В прототипе использован вариант циклической вольтамперометрии.
Установленные условия проведения электродного процесса впервые позволили количественно определять азитромицин на основе реакции электроокисления. Для повышения чувствительности определения использовали предварительное концентрирование антибиотика на поверхности СУ электрода. Предлагаемый вольтамперометрический способ позволил существенно улучшить метрологические характеристики анализа азитромицина; повысить чувствительность определения (Сmin,р=1,84· 10-10 моль/л, Сн=2,2· 10-10 моль/л), что на 2 порядка ниже по сравнению с прототипом. Диапазон определяемых концентраций азитромицина от 2,2· 10-10 моль/л до 1,0· 10-4 моль/л.
Измерения проводили на компьютеризированных вольтамперометрических анализаторах СТА, ВАМ (ООО “ИТМ”, г.Томск), а также универсальном полярографе типа ПУ.
Определению не мешают вещества, присутствие которых возможно в биологических объектах: водорастворимые витамины групп В (b1, В2, Вс, В6), РР, аскорбиновая и мочевая кислоты в соизмеримых количествах. Состав матрицы лекарственных препаратов “Сумамед” капсулы, таблетки практически не оказывают влияния на ток окисления азитромицина, поэтому не требовалось предварительное выделение антибиотика из матрицы до проведения собственно электрохимического анализа.
Пример 1. Определение содержания азитромицина на уровне (10-9-10-10) моль/л.
В кварцевый стаканчик емкостью 20 мл наливают 9,9 мл раствора 0,2н. Na2HPO4 и 0,1 этанола. Удаляют из раствора кислород струей очищенного азота с содержанием кислорода менее 0,001% в течение пяти минут. Не прекращая перемешивания, проводят электролиз раствора при условии: Еэ=0,20 В, τ з=60 с. Отключают газ и фиксируют анодную дифференциальную вольтамперограмму при скорости развертки потенциала 30 мВ/с, начиная с потенциала Енач=0,20 В. Отсутствие пиков свидетельствует о чистоте фона. Затем добавляют несколько капель объемом 0,01 мл аттестованной смеси азитромицина (2-4)· 10-7-10-6 моль/л, перемешивают раствор 10 с и проводят электрохимическое концентрирование осадка при Еэ=0,20 В, τ э=60 с. Съемку вольтамперной кривой начинают с потенциала 0,20 В. Пик для указанной концентрации вещества регистрируют в диапазоне потенциалов от 0,70 до 0,75 В (нас.х.с.э.) при чувствительности прибора (0,5-1)· 10-9 А/мм. Время единичного анализа не превышает 10 минут.
Пример 2. Определение содержания азитромицина в таблетках “Сумамед”
В стаканчик для вольтамперометрических измерений вносят 10 мл 0,2н. Na2HPO4 или 10 мл раствора буфера Бриттона-Роббинсона, рН 8,0-9,0 и удаляют кислород из раствора пропусканием газообразного азота в течение пяти минут. Не прекращая перемешивание, проводят электролиз раствора при условии Еэ=0,15 В, τ э=30 с. Отключают газ и регистрируют вольтамперограмму фона в диапазоне потенциалов от 0,20 до 1,00 В (нас.х.с.э.). Отсутствие пиков на вольтамперограмме в области потенциалов 0,70-0,85 В свидетельствует о чистоте фонового электролита и полярографического стаканчика.
При анализе таблеток, содержащих азитромицин, измельчают в ступке 3-6 таблеток без оболочки. Берут навеску пробы 0,200 г, взвешенную с точностью до 0,001 г, вносят в колбу вместимостью 100 мл, растворяют навеску пробы в 50 мл 96% этилового спирта и доводят до метки бидистиллированной водой. Через 3-5 минут раствор отфильтровывают через бумажный фильтр. Затем 0,01 мл полученного фильтрата вносят в кварцевый стаканчик с фоновым раствором. Электронакопление и регистрацию аналитического сигнала проводят в тех же условиях. Анодный пик азитромицина фиксируют в диапазоне потенциалов от 0,75 до 0,85 В на СУ электроде при чувствительности прибора (1-5)· 10-8 А/мм в дифференциальном режиме съемки вольтамперограмм. Массовую концентрацию азитромицина в пробе оценивают методом добавок аттестованных смесей, измеряя высоту анодных пиков. Время анализа одной пробы с учетом пробоподготовки менее 30 минут.
Таким образом, впервые установлена способность количественного химического анализа азитромицина по пикам окисления его на СУ “игольчатом” электрохимически модифицированном электроде (в прототипе количественное определение азитромицина проводят по волнам окисления с использованием торцевого СУ электрода).
Анализ характеристик количественного химического определения азитромицина по предлагаемому способу свидетельствует о существенном повышении чувствительности определения (на 2 порядка по сравнению с прототипом и более чем на 4 порядка по сравнению с методом ВЭЖХ) [Founda H.G., Schneider R.P. (HPLC) - atmospheric pressure chemical ionization mass spectrometry: correlation with a standard HPLC - electrochemical method./J. Chromatogr A 1998 Jul 3; 812(1-2):287-93]. Предел обнаружения и нижняя граница определяемых содержаний соответственно равны 1,84· 10-10 моль/л и 2,2· 10-10 моль/л. Значительно сократилось время проведения анализа. Условия, используемые в прототипе, не позволяют контролировать сточные воды, воздушные зоны предприятий и химических лабораторий на уровне нанограммовых содержаний (и меньше) азитромицина.
Предложенный способ прост, не требует большого количества реактивов и трудозатрат и может быть применен в любой химической лаборатории, имеющей полярограф, особенно в настоящее время, когда налажен выпуск отечественной и зарубежной электроаппаратуры с компьютерным управлением и обработкой данных (анализаторы типа СТА, ТА и др.) Предложенный способ может быть использован в фармакокинетических и фармацевтических исследованиях, в токсикологическом и техническом анализе антибиотиков группы макролидов, для контроля сточных вод и воздушной зоны химико-фармацевтических предприятий, а также для разработки методик анализа азитромицина и родственных ему соединений в сложных многокомпонентных биосистемах (кровь, моча, плазма и др.) и пищевых продуктах.
Изобретение относится к области аналитической химии и касается количественного определения антибиотика группы макролидов азитромицина дигидрата (азитромицин). Сущность способа: азитромицин переводят из пробы в раствор и проводят вольтамперометрическое определение, для чего осуществляют накопление антибиотика в перемешиваемом растворе в течение 30-60 с при потенциале электролиза (0,10-0,25) В относительно насыщенного хлоридсеребряного электрода на фонах 0,2н. раствора гидрофосфата натрия или буферного раствора Бриттона-Роббинсона, рН 8,0-9,0, в присутствии 0,1%-ного этанола с последующей регистрацией анодных пиков в дифференциальном режиме съемки вольтамперограмм при скорости развертки потенциала 20-30 мВ/с. Концентрацию азитромицина определяют по высоте пика в диапазоне потенциалов от 0,70 до 0,85 В методом добавок аттестованных смесей. Способ обладает высокой чувствительностью, предел обнаружения равен 1,86·10-10 моль/л. 1 табл., 1 ил.
Способ количественного определения азитромицина дигидрата (азитромицина), включающий перевод азитромицина из пробы в раствор и вольтамперометрическое определение с использованием индикаторного стеклоуглеродного электрода, отличающийся тем, что используют инверсионную вольтамперометрию, при этом накопление азитромицина в перемешиваемом растворе проводят в течение 30-60 с при потенциале электролиза (0,10-0,25) В относительно насыщенного хлоридсеребряного электрода на фонах 0,2 н раствора гидрофосфата натрия или буферного раствора Бриттона-Роббинсона, рН 8,0-9,0, в присутствии 0,1%-ного этилового спирта с последующей регистрацией анодных пиков в дифференциальном режиме съемки вольтамперограмм при скорости развертки потенциала 20-30 мВ/с и концентрацию азитромицина определяют по высоте пика в диапазоне потенциалов 0,70 - 0,85 В методом добавок аттестованных смесей.
| SHEPARD RM | |||
| et al | |||
| High-performance liquid chromatographic assay with electrochemical detection for azithromycin in serum and tissues | |||
| J.Chromatogr | |||
| Циркуль-угломер | 1920 |
|
SU1991A1 |
| СПОСОБ ЛЕЧЕНИЯ БАКТЕРИАЛЬНОЙ ИНФЕКЦИИ | 1995 |
|
RU2128998C1 |
| ЛЕКАРСТВЕННЫЕ ФОРМЫ АЗИТРОМИЦИНА С КОНТРОЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ | 1995 |
|
RU2130311C1 |
| ИВАНОВСКАЯ Е.А | |||
| и др | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Химико-фармацевтический журнал | |||
| - М.: Медицина, 1995, №3, с.57-58. | |||

