Изобретение относится к области аналитической химии, в частности к вольтамперометрическому способу определения лекарственного препарата 4-йодантипирина (1-фенил-2,3-диметил-4-йод-5-пиразолон; J-АП), обладающего антивирусной активностью против вирусов Коксаки А и В, вазикулярного стоматита, гриппа и др. Препарат J-АП находит все большее применение для лечения и профилактики вирусного клещевого энцефалита, особенно в настоящее время, когда производство иммунных серопрепаратов, выделяемых из крови доноров (человеческий иммуноглобулин), ограничено и является достаточно дорогостоящим. Количественное определение J-АП является актуальным в биофармацевтических исследованиях и в оценке эффективности лечения такого опасного заболевания как клещевой энцефалит, а также заболеваниях глаз, периревмокардита, панкреатитов и др. В связи с этим аналитическая практика предъявляет повышенные требования к методам контроля и определения противовирусных лекарственных средств.
Главной проблемой остается разработка новых, более чувствительных, точных и селективных методов их анализа.
В настоящее время практически отсутствуют сведения по определению J-АП [Харламов В. Т., Инкин А.А., Сысоева Е.С. Определение 4-йодантипирина кулонометрическим титрованием. Аналитическая химия. - 1975, т. ХХХ, 9, с. 1845-1846] однако, имеется небольшое число работ по определению родственного соединения антипирина и его производных [Levine J., Weber J.D Determination of antipyrine in combinations with other drugs / J. Pharmac. Sci. - 1965, т. 54, 4, 636-638 р. , Пассет Б.В., Антипов М.А. Практикум по техническому анализу и контролю производства химико-фармацевтических препаратов и антибиотиков. - М.: Медицина. 1981, 272 с.], нашедших применение в медицинской практике.
Разработаны химические методы, основанные на цветных реакциях, характерных для пиразольного кольца, входящего в молекулу [Пассет Б.В., Антипов М. А. Практикум по техническому анализу и контролю производства химико-фармацевтических препаратов и антибиотиков. - М.: Медицина. 1981, 272 с.; Пентюк А. А. , Истошин В.М., Мусин Р.А., Лычик Г.З., Луцюк Н.В., Илика В.Г. Способ определения 4-аминоантипирина в биологических жидкостях. Авт. свид. 4760330/00-14(22)891120] . Полученные продукты окрашены в различные цвета в зависимости от характера окисления и условий проведения реакции. Такие методы просты и доступны, не требуют применения специальной аппаратуры и чаще всего используются для качественного обнаружения.
Описан титриметрический метод определения антипирина гипоброматом калия в присутствии бромида калия в кислой среде. Для количественного определения антипирина использовался метод амперометрического титрования раствором хлорида йода. Чувствительность этих методов не велика ≤1•10-5 моль/л [Мискиджьян С.П., Кравченюк Л.П. Полярография лекарственных препаратов. Киев: Вища школа, 1976, 232 с.].
Предложен метод кулонометрического титрования радиоактивного 4-йодантипирина в одноименном радиофармацевтическом препарате генерированном бромом [Харламов В. Т., Инкин А.А., Сысоева Е.С. Определение 4-йодантипирина кулонометрическим титрованием. Аналитическая химия. - 1975, т.ХХХ, 9, с.1845-1846] . Анализ затруднялся присутствием антипирина, являющимся исходным веществом при получении радиоактивного препарата. Поэтому для повышения селективности определения использовали предварительное выделение 4-йодантипирина методом хроматографии на бумаге. Методика анализа связана со значительными трудностями при количественной оценке хроматограмм, длительна (более 20 ч) и имеет невысокую чувствительность определения 3,2•10-5 моль/л. Многие же задачи химического анализа связаны с определением следов органических веществ, часто находящихся в пробах на уровне миллиардных долей и даже ниже и поэтому эти методы не нашли широкого применения для количественного определения антипирина и его производных.
Инверсионно-вольтамперометрические и вольтамперометрические методы отвечают современным требованиям анализа сложных по составу систем и при соблюдении требований ультрамикроанализа вполне реально определение 10-8-10-10 моль/л. Однако большинство опубликованных работ по анализу этими методами посвящено определению металлов, идентификация органических лекарственных препаратов становится с каждым годом все более серьезной проблемой. Практически отсутствует информация о применении вольтамперометрических методов для определения антипирина и его производных, в том числе йодантипирина. Полярографическому исследованию антипирина посвящены единичные работы [Харламов В. Т., Инкин А.А., Сысоева Е.С. Определение 4-йодантипирина кулонометрическим титрованием. Аналитическая химия. - 1975, т. XXX. 9, с. 1845-1846, Пассет Б. В. , Антипов М.А. Практикум по техническому анализу и контролю производства химико-фармацевтических препаратов и антибиотиков. - М. : Медицина. 1981, 272 с.]. Для определения йодантипирина метод полярографии также не применялся.
Наиболее близким является полярографический метод определения антипирина [Пассет Б. В. , Антипов М.А. Практикум по техническому анализу и контролю производства химико-фармацевтических препаратов и антибиотиков. - М.: Медицина. 1981, 272 с.] после предварительного его нитрозирования растворами нитрита натрия в серной кислоте (прототип). К 1 мл 0,1 н. раствора антипирина прибавляют 1 мл 0,1 н. раствора серной кислоты и 1 мл 0,1 н. раствора нитрита натрия. Избыток образующейся азотистой кислоты нейтрализуют 0,1 н. раствором щелочи, добавляют 0,5 мл 1%-ного раствора желатина и полученный 4-нитрозоантипирин восстанавливают на ртутном капельном электроде. Полученный результат пересчитывают на содержание антипирина и концентрацию его определяют методом калибровочных кривых. Предел обнаружения не указан. Минимально определяемая концентрация антипирина этим методом практически больше n•10-5 моль/л. В данных условиях определение J-АП на уровне 10-9-10-7 моль/л невозможно.
Задачей заявляемого изобретения является повышение чувствительности и экспрессности определения 4-йодантипирина методом анодной адсорбционной вольтамперометрии (ВА).
Поставленная задача достигается тем, что 4-йодантипирин электрохимически концентрируют на стеклоуглеродном (СУ) или пирографитовом (ПГ) электроде с последующей регистрацией анодных вольтамперограмм. Новым в способе является то, что проводят накопление йодантипирина в перемешиваемом растворе в течение 40-60 с при потенциалах электролиза Еэ=(-0,15-0,05) В на фонах: 0,1 н. раствора лимоннокислого натрия, или 0,2 н. раствора гидрофосфата натрия, или 0,2 н. раствора дигидрофосфата калия в присутствии 10%-ного этилового или изопропилового спирта с последующей регистрацией анодных пиков в дифференциальном режиме съемки вольтамперограмм при скорости развертки потенциала 30-50 мВ/с. Концентрацию 4-йодантипирина определяют по высоте анодного пика в диапазоне потенциалов от 1,00 до 1,25 В относительно насыщенного хлоридсеребряного электрода (нас.х.э).
В прототипе количественное определение антипирина основано на реакции восстановления и возможно только после предварительного нитрозирования с образованием полярографически активного производного 4-нитрозоантипирина, т. к. антипирин не восстанавливается на ртутном капельном электроде.
В предлагаемом способе впервые установлена способность антипирина и его производных окисляться на различных типах графитовых электродов. В качестве индикаторных применяли графитовый (Г) электрод, пропитанный полиэтиленом с парафином в вакууме или эпоксидной смолой, ПГ и СУ (в прототипе применяли ртутный капельный электрод). Использование таких электродов обусловлено высокой химической и электрохимической устойчивостью графита, широкой областью рабочих потенциалов как в водных, так и в неводных средах, а также простотой механического обновления поверхности и требованиями техники безопасности. Ртуть токсична и может вредно влиять на чувствительные биологические материалы.
Величина потенциалов окисления лекарственных веществ определяется строением, структурой и степенью адсорбируемости на гексагонах графита, имеющих π-зонную структуру. По легкости окисления на СУ электроде вещества можно расположить в ряд:
4-аминоантипирин 0,82 В, 4-йодатипирин 1,07 В, антипирин 1,09 В, 4-бромантипирин 1,12 В на фоне 0,2 н. Na2HPO4 и концентрации 3•10-6 моль/л.
Максимальное значение регистрируемого тока с использованием графитового электрода несколько выше (≈ на 15-20%), однако из-за большого остаточного тока он оказался менее удобным в работе, чем СУ и ПГ электроды. Для определения J-АП, антипирина и его производных использовали "игольчатые" по форме индикаторные электроды после предварительного электрохимического модифицирования их поверхности. Такая обработка приводит к снижению нижней границы определяемых содержаний J-АП и улучшению воспроизводимости вольтамперометрических измерений. Графитовые (СУ, ПГ) "игольчатые" электроды впервые использованы для определения J-АП, антипирина и его производных.
Реакция электроокисления J-АП является электрофильной. Роль электрофильного реагента с электронным дефицитом выполняет анод, а субстрата - органическая молекула, реакционная способность которой определяется электронной плотностью связей, атомов и групп в молекуле. Предварительные исследования показали, что электроокисление J-АП представляет сложный диффузионно-контролируемый электродный процесс, протекающий с участием одного электрона, осложненный как адсорбцией, так, возможно, и дополнительными химическими стадиями.
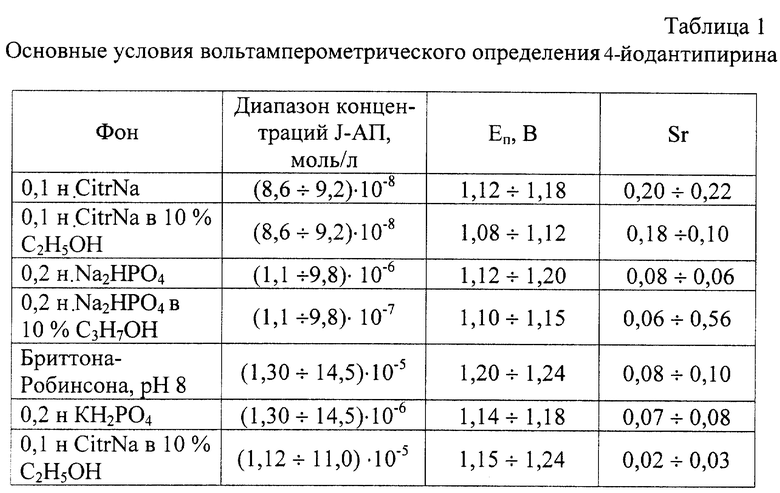
В прототипе описано использование в качестве фонов 0,1 н. раствора серной кислоты в присутствии 1%-ного желатина. Определение J-АП в этих условиях затруднено, и, по-видимому, связанно с меньшей устойчивостью J-АП в водных растворах в присутствии сильных окислителей, что делает невозможным проведение анализа на этих фонах. Предлагаемые в заявляемом изобретении фоны 0,1 н. лимоннокислый натрий (CitrNa), или 0,2 н. гидрофосфат натрия (Na2HPO4), или 0,2 н. дигидрофосфат калия (КН2PO4) в присутствии 10%-ного (по объему) этилового или изопропилового спирта позволяет определять J-АП на уровне 10-8 моль/л с хорошей воспроизводимостью. Относительное стандартное отклонение не превышает 0,03 для концентрации 10-5-10-4 моль/л и равно 0,18 для концентрации 8,6•10-8 моль/л. Фоны 0,1 н. CitrNa, 0,2 н. Na2HPО4 и 0,2 н. КН2PO4 подобраны экспериментально. Абсолютной новизной является экспериментально подобранный фон 0,1 н. CitrNa и использование водно-органических растворителей, от чего зависит количественное определение J-АП. Использование 0,1 н. CitrNa, или 0,2 н. Na2HPO4, или 0,2 н. КН2РO4 в присутствии 10%-ного по объему этилового или изопропилового спирта способствует хорошей растворимости определяемого вещества, приводит к стабилизации и воспроизводимости пиков J-АП, особенно при анализе сложных по составу биологических систем (плазмы крови, мочи). В табл.1 представлены основные условия вольтамперометрического определения J-АП в неводных и водно-органических средах. Наиболее широкий диапазон определяемых концентраций J-АП установлен для (10%-ного) этанольного раствора и составляет (8,6•10-8 - 9,9•10-5) моль/л. Для интервала концентраций J-АП (10-6-10-5) моль/л минимальное значение Sr наблюдалось в 10%-ном этаноле (Sr<0,05), максимальное - в пропиленкарбонате (Sr<0,15). Применение водно-органических сред позволяет существенно облегчить протекание процесса и, следовательно, снизить предел обнаружения определяемого вещества (Cmin) до 3,3•10-9 моль/л на фоне 0,1 н. CitrNa в 10% этанольном растворе.
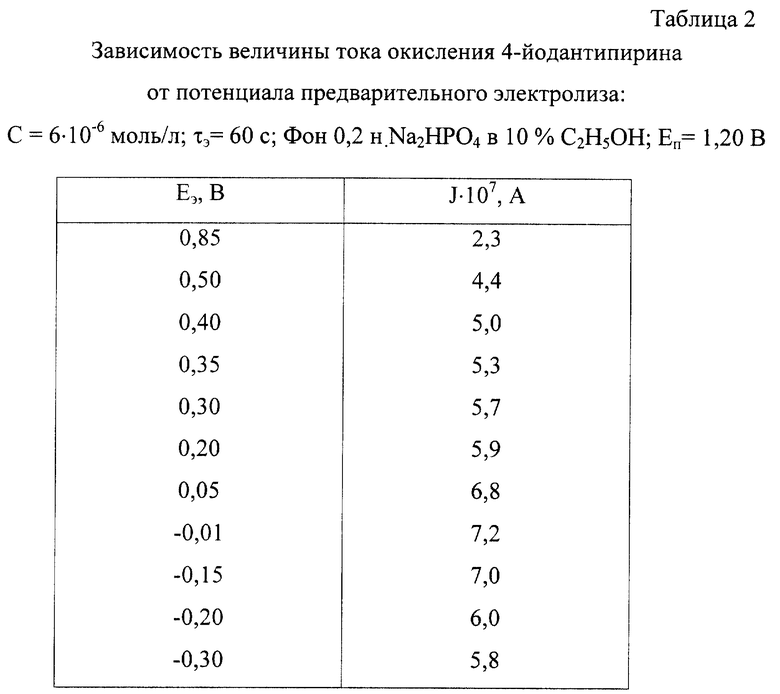
Другим отличительным признаком являются установленные условия электрохимического накопления: потенциал электролиза Еэ=(-0,15-0,05) В. Опытные данные показали зависимость тока окисления J-АП от Еэ (табл.2). Величина анодного тока увеличивалась и достигала максимального значения в области потенциалов (-0,15-0,05) В. При 0,05<Еэ<-0,15 В уменьшилась величина тока окисления органического вещества. Использование предварительного электролиза при значениях потенциала (-0,15±0,05) В позволяет регистрировать вольтамперограммы с четко выраженным максимумом. Это позволяет повысить точность и разрешающую способность способа и экспрессно определить концентрации меньше, чем 1•10-7 моль/л J-АП в сложных по составу смесях.
Время предварительного электролиза (τэ) выбирают в зависимости от концентрации определяемого вещества. Максимальное значение величины тока окисления достигается при τэ равном 40-60 с. При τэ<40 с снижается чувствительность определения и увеличивается ошибка определения, а при τэ>60 снижается экспрессность; величина тока достигала максимального значения при τэ равном 40-60 с. Исследования зависимости величины тока от времени выдержки электрода без наложения на него поляризующего напряжения свидетельствовало о достижении адсорбционного равновесия в растворе за 40-60 с. Величина тока при этом возрастала на 10-12%. Дальнейшее увеличение времени выдержки электрода не влияло на характер аналитического сигнала.
Важным для определения J-АП является выбор скорости развертки потенциала. Оптимальной является скорость 30-50 мВ/с. Увеличение скорости более 50 мВ/с увеличивает чувствительность, но при этом растет остаточный ток и уменьшается разрешающая способность способа. Использование скорости менее 30 мВ/с существенно снижает величину анодного тока и понижается чувствительность определения J-АП.
Нижняя граница определяемых содержаний J-АП зависит от режима съемки вольтамперных кривых. В дифференциальном режиме высокая точность определяется тем, что потенциал пика измеряется в точке, в которой круто падающая первая производная пересекает нулевую линию. Использование режима дифференцирования позволяет фиксировать четкие пики даже при очень низких концентрациях 10-8-10-7 моль/л, что повышает чувствительность определения. Для определения J-АП, антипирина и его производных он ранее не применялся. При линейном наложении потенциала в интегральном режиме определение концентрации J-АП на уровне n•10-8 моль/л практически не возможно из-за большого значения остаточного тока. Поэтому для количественного определения J-АП рекомендован дифференциальный режим съемки вольтамперограмм.
Таким образом, установленные условия впервые позволили количественно определять J-АП на основе реакции электроокисления. Для повышения чувствительности определения использовали предварительное концентрирование J-АП на поверхности графитовых электродов. Предлагаемый вольтамперомстрический способ позволил существенно улучшить метрологические характеристики анализа J-АП; повысить чувствительность определения (Cmin.p= 3,3•10-9 моль/л, Сn=8,6•10-8 моль/л), что на 3-4 порядка ниже по сравнению с прототипом [Пассет Б. В. , Антипов М.А. Практикум по техническому анализу и контролю производства химико-фармацевтических препаратов и антибиотиков. - М.: Медицина. 1981, 272 с.] и известным методом кулонометрического титрования J-АП [Харламов В. Т., Инкин А.А., Сысоева Е.С. Определение 4-йодантипирина кулонометрическим титрованием. Аналитическая химия. - 1975, т. ХХХ, 9, с. 1845-1846] , время анализа не превышает 15-20 мин, против 20-25 ч. Определению не мешают вещества, присутствие которых возможно в биологических объектах: аскорбиновая и мочевая кислоты, витамины В), Bs, Вс, Вб, PP.
Пример 1. Определение 4-йодантипирина на уровне 10-8-10-7 моль/л. В кварцевый стаканчик емкостью 20 мл наливают 9 мл 0,1 н. лимоннокислого натрия и 1 мл этилового спирта. Удаляют из раствора кислород струей очищенного азота с содержанием кислорода менее 0,001% в течение трех минут. Не прекращая перемешивания, проводят электролиз раствора при условии: Еэ=-0,15 В, τэ= 60 c. Отключают газ и фиксируют анодную дифференциальную вольтамперограмму при скорости развертки потенциала 30 мВ/с, начиная с потенциала Енач=0,75 В. Отсутствие пиков свидетельствует о чистоте фона. Затем добавляют несколько капель объемом 0,01 мл аттестованной смеси 4-йодантипирина (1-2)•(10-5-10-4 моль/л, перемешивают раствор 10 с и проводят электрохимическое концентрирование осадка при Еэ=-0,15 В и τэ= 60 c. Съемку вольтамперной кривой начинают с потенциала 0,75 В. Пик для указанной концентрации вещества регистрируют в диапазоне потенциалов от 1,00 до 1,20 В (нас.х.с.э.) при чувствительности прибора (0,5-1)(10-9 А/мм. Время единичного анализа не превышает 10 мин.
Пример 2. Определение 4-йодантипирина в лекарственных формах и плазме крови.
При анализе таблеток, содержащих 4-йодантипирин, измельчают в ступке 5 таблеток, берут навеску пробы 0,06-0,09 г, взвешенную с точностью до 0,001 г, вносят в стаканчик вместимостью 25 мл, растворяют навеску пробы в 10 мл этилового спирта (диметилформамида или изопропилового спирта) и доводят до метки этим же раствором. 0,2 мл полученного раствора вносят в кварцевый стаканчик вместимостью (25,0-30,0) мл и добавляют 10 мл 0,2 н. раствора Na2HPО4 и 1 мл этилового спирта. Удаляют из раствора кислород струей очищенного азота с содержанием кислорода менее 0,001% в течение трех минут и затем проводят вольтамперометрические измерения при условиях: Еэ=0,05 В, τэ= 40 c, скорость развертки потенциала 50 мВ/с. Анодный пик 4-йодантипирина регистрируют в диапазоне потенциалов от 1,10-1,25 В на стеклоуглеродном или пирографитовом электроде при чувствительности прибора (1-5)•10-8 А/мм в дифференциальном режиме съемки вольтамперограмм. Содержание 4-йодантипирина оценивают методом добавок аттестованных смесей, измеряя высоту анодных пиков. Время анализа одной пробы не превышает 10 мин.
Для анализа препарата 4-йодантипирина в плазме берут 1 - 2 мл пробы анализируемой плазмы, вносят в кварцевый стаканчик вместимостью 25,0 мл 8 мл 0,2 н. Na2HPО4 и 1 мл изопропилового спирта, удаляют из раствора кислород газообразным азотом в течение 5 мин, отключают газ и далее вольтамперометрические измерения проводят согласно описанному выше примеру 2.
По предлагаемому способу проведена метрологическая аттестация методики количественного химического анализа проб лекарственных препаратов на содержание массовой концентрации 4-йодантипирина методом адсорбционной инверсионной вольтамперометрии. Метрологические исследования проведены согласно требованиям ГОСТ Р 8.563-96 "Методики выполнения измерений" и МИ 2336-95 "Характеристики погрешности результатов количественного химического анализа. Алгоритм оценивания". Характеристика случайной составляющей погрешности (показатель воспроизводимости) оценена по результатам анализов 5 проб по две параллельных. При этом варьировали условия проведения анализов (время, приборы, температура, оператор и т.д.), чтобы учесть все возможные факторы.
Оценку характеристики систематической составляющей проводили по МИ 2336-95 с использованием образцов для контроля и методом добавок аттестованной смеси определяемого вещества в пробу. Образцы для контроля готовили из реальных рабочих проб анализируемых объектов, в которых препарат отсутствовал, с добавкой в них определяемого вещества. Обобщение расчетных данных по определению показателей сходимости и воспроизводимости находится в пределах от 0,28 до 0,94% при определении массовой концентрации 4-йодантипирина 101-151 мг/таблетку.
Таким образом, впервые установлена способность количественного химического анализа 4-йодантипирина по пикам окисления его на стационарных СУ и ПГ электродах (в прототипе количественное определение родственного соединения антипирина проводят по волнам восстановления на ртутном капельном электроде. Определение 4-йодантипирина в прототипе не описано). Анализ характеристик количественного химического определения 4-йодантипирина по предлагаемому способу свидетельствует о существенном улучшении метрологических характеристик анализа по сравнению с прототипом. Значительно повысилась чувствительность определения (более чем на 2-3 порядка) и воспроизводимость (σвоспр.<1%).
Предел обнаружения и нижняя граница определяемых содержаний соответственно равна 3,3•10-9 моль/л и 8,6•10-8 моль/л. Время проведения анализа сократилось более чем в три раза. Условия, используемые в прототипе, не позволяют анализировать биосистемы (плазма, лимфа, кровь), а также контролировать сточные воды на уровне микрограммовых содержаний (и меньше) йодантипирина.
Предложенный способ прост, исключает использование токсичной ртути, не требует больших трудозатрат, большого количества реактивов и может быть применен в любой химической лаборатории, имеющей современные компьютеризированные анализаторы типа СТА, ТА или полярограф. Предложенный способ может быть использован для определения 4-йодантипирина в фармацевтических исследованиях, в анализе сложных многокомпонентных биосистем (кровь, моча и др.), в токсикологическом и техническом анализе лекарственных средств на основе антипирина и его производных, для контроля сточных вод и воздушной зоны химико-фармацевтических предприятий.

Изобретение относится к вольтамперометрическому способу определения лекарственного препарата 4-йодантипирина (1-фенил-2,3-диметил-4-йод-5-пиразолон; J-АП), обладающего антивирусностной активностью, предложенный способ может быть использован для определения 4-йодантипирина в фармацевтических исследованиях, в анализе сложных многокомпонентных биосистемах (кровь, моча и др. ), в токсикологическом и техническом анализе лекарственных средств на основе антипирина, для контроля сточных вод и воздушной зоны химико-фармацевтических предприятий. Техническим результатом изобретения является повышение чувствительности и экспрессности определения 4-йодантипирина методом адсорбционной вольтамперометрии (ВА). Сущность изобретения: 4-йодантипирин электрохимически концентрируют на стеклоуглеродном или пирографитовом электроде в перемешиваемом растворе в течение 40-60 с при потенциалах электролиза Еэ=(-0,15-0,05) В на фонах: 0,1 н. раствора лимоннокислого натрия, или 0,2 н. раствора гидрофосфата натрия, или 0,2 н. раствора дигидрофосфата калия в присутствии 10%-ного этилового или изопропилового спирта с последующей регистрацией анодных пиков в дифференциальном режиме съемки вольтамперограмм при скорости развертки потенциала 30-50 мВ/с. Концентрацию 4-йодантипирина определяют по высоте анодного пика в диапазоне потенциалов от 1,00 до 1,25 В относительно насыщенного хлоридсеребряного электрода. 3 табл.
Способ количественного определения 4-йодантипирина методом адсорбционнной инверсионной вольтамперометрии, заключающийся в том, что 4-йодантипирин переводят из пробы в раствор и проводят вольтамперометрическое определение, отличающийся тем, что проводят накопление 4-йодантипирина в перемешиваемом растворе в течение 40-60 с при потенциалах электролиза (-0,15-0,05) В относительно насыщенного хлоридсеребряного электрода на фонах 0,1 н. раствора лимоннокислого натрия, или 0,2 н. раствора гидрофосфата натрия, или 0,2 н. раствора дигидрофосфата калия в присутствии 10%-ного этилового или изопропилового спирта с последующей регистрацией анодных пиков в дифференциальном режиме съемки вольтамперограмм при скорости развертки потенциала 30-50 мВ/с и концентрацию определяют по высоте пика в диапазоне потенциалов от 1,00 до 1,25 В методом добавок аттестованных смесей.
| ПАССЕТ Б.В., АНТИПОВ М.А | |||
| Практикум по техническому анализу и контролю производства химико-фармацевтических препаратов и антибиотиков | |||
| - М.: Медицина, 1981, с | |||
| Паровоз с приспособлением для автоматического регулирования подвода и распределения топлива в его топке | 1919 |
|
SU272A1 |
| ХАРЛАМОВ В.Т | |||
| и др | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Аналитическая химия, - 1975, т | |||
| ХХХ, № 9, с | |||
| Устройство для нейтрализирования статических зарядов, образующихся при выделке и обработке тканей и т.п. изделий из диэлектриков | 1922 |
|
SU1845A1 |
| Способ определения 4-аминоантипирина в биологических жидкостях | 1989 |
|
SU1705745A1 |
| Способ количественного определения амидопирина и антипирина при их совместном присутствии в смеси | 1977 |
|
SU636189A1 |
| US 3988430 A, 26.10.1976. | |||
