Изобретение относится к области аналитической химии, изучающей возможность определения анавидина методом инверсионной вольтамперометрии. Анавидин относится к катионоактивным полимерным соединениям и представляет собой полигексаметиленгуанидин фосфат (ПГМГ) с общей формулой:
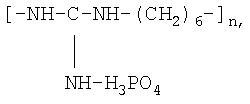
Анавидин является производным гуанидина, что обеспечивает его высокие бактерицидные и фунгицидные свойства. Обладая высокими антисептическими характеристиками, анавидин находит широкое применение в хирургии в качестве антимикробных тканей и перевязочных средств, для дезинфекции воздуха, обеззараживания воды, а также для придания антимикробных свойств одежде медицинского персонала и пациентов. В этой связи определение микроколичеств анавидина важно для контроля фармакокинетического действия препарата, оценки его токсичности и контроля качества воды и воздуха рабочей зоны.
Это в свою очередь повышает требования к современным методам количественного определения полимерных гуанидиновых соединений. Данные методы должны обладать высокой чувствительностью, достаточной селективностью, малыми временными затратами на анализ доступностью используемых реактивов, чтобы обеспечить широкое распространение в лабораториях данных методик.
Такие варианты электрохимических методов как вольтамперометрические (ВА) отвечают современным требованиям, предъявляемым к контролю за качеством фармпрепаратов, пищевых продуктов, продуктов сельского хозяйства.
Применение инверсии в вольтамперометрических методах позволяет достигнуть минимально определяемой концентрации вещества на уровне 10-10÷10-11 моль/л или 10-4÷10-5 г/л.
Возможности использования серийных компьютеризированных вольтамперометрических анализаторов, характеризующихся высокой экспрессностью, селективностью и возможностью многоэлементного анализа, часто без предварительной пробоподготовки, позволяют использовать ВА методы для серийных анализов во многих лабораториях производствах как контролирующие методы. Информация о применении электрохимических методов, в частности вольтамперометрических, для определения анавидина отсутствует.
Применение электрохимических методов количественного химического анализа для прямого определения анавидина не отмечено в литературе. Однако анавидин как представитель группы гуанидинов обладает достаточной электрохимической активностью.
При рН 9÷9,5 в растворе 30% сульфата аммония (каталит) и 20% серной кислоты (аналит) при подачи тока 300 А при температуре (18-21)°С в 2 электрохимической ячейке, где катодом служила амальгамированная медь, наблюдается восстановление нитрогуанидина до аминогуанидина (1) [Т.Duraiswamy Balakrishnan, К.Shrivara Udupa, G.Srinivasan Subramanian, and H. Venkatakrishna Udupa Electrolytic Production of Aminoguanidine Bicarbonate. / Ind. Eng. Chem. Process Des. Develop., Vol.10, No.4, 1971, 495-497].

В литературе встречаются данные о косвенном определении гуанидина вольтамперометрическими методами [Dengbai Luo, Jingui Lan, Chun Zhou, and Chenxia Luo Polarographic Behavior of Co(II)-BSA or -HSA Complex in the Presence of a Guanidine Modifier./ Anal. Chem. 2003, 75, 6346-6350]. На фоне 0,2 М NaOH при потенциале -1,73 В относительно каломельного электрода регистрировали адсорбционный пик комплекса кобальта (2+) и гуанидина хлорида. В данном эксперименте в качестве индикаторного электрода использовался ртутно-капающий электрод. Содержание гуанидина хлорида составляло 0,1-0,2 моль/л. В предлагаемых условиях гуанидин служит комплексообразователем.
В качестве прототипа был выбран способ капиллярного электрофореза для определения полигексаметиленгуанидина [Руднев А. В., Джераян Т.Г. Определение полигексаметиленгуанидина методом капиллярного электрофореза // Журнал аналитической химии, 2006, Т. 61, №10, с.1086-1089]. С помощью кварцевого капилляра с внешним полиимидным защитным покрытием при рабочей длине волны ультрафиолетового спектра, равной 200 нм, при рабочем напряжении 20 кВ, которому соответствует ток 20÷50 мкА, были получены электрофореграммы полигексаметиленгуанидина гидрохлорида и его олигомеров. Предел обнаружения полигексаметиленгуанидина в предложенном способе составляет до 2·10-3 г/л.
В данных условиях определение анавидина на уровне n·10-4 г/л невозможно.
Задачей заявляемого изобретения является установление рабочих условий вольтамперометрического определения анавидина, а также повышение чувствительности и экспрессности анализа.
Для определения анавидина использовали ртутно-пленочный электрод. Использование данного вида электродов при электрохимическом восстановлении полимера обусловлено получением воспроизводимых аналитических сигналов. А сочетание таких свойств электрода как адсорбционные способности и возможности электрохимического накопления позволяет существенно снизить границу определяемых содержаний, увеличить экспрессность.
Поставленная задача достигается тем, что способ количественного определения полигексаметиленгуанидина (ПГМГ) (анавидина) включает перевод анавидина из пробы в раствор путем экстракции и вольтамперометрического определение с использованием ртутно-пленочного (РП) электрода в качестве индикаторного. При этом используется накопление в течение 60÷90 с при потенциале электролиза - (0,15÷0,3) В относительно 1 М хлоридсеребрянного электрода на фоне 0,01 М гидрооксида натрия с последующей регистрацией катодных пиков при скорости развертки потенциала (80-100) мВ/с. Концентрацию анавидина определяют по высоте пика в диапазоне потенциалов от - 0,8 до - 0,95 В методом добавок аттестованных смесей (фиг.1).
В предлагаемом способе установлена способность анавидина к электрохимическому восстановлению на ртутных электродах. Абсолютной новизной является экспериментально подобранный фоновый электролит 0,01 М NaOH. Применение указанных в заявляемом изобретении фонового электролита впервые позволило определять анавидин с пределом обнаружения, рассчитанным по 3-сигмовому критерию, до 0,83·10-3 мг/л (Сmin,p). Минимально определяемая концентрация (Сн) 0,96·10-3 мг/л на фоне 0,01 М NaOH (фиг.2).
Другим отличительным признаком являются установленные условия электрохимического накопления: потенциал электролиза Еэ=-(0,15÷0,3) В. Опытные данные показали зависимость тока окисления анавидина от Еэ (фиг.3). Величина анодного тока увеличивалась примерно в 2,4 раза и достигала максимального значения в области потенциалов - (0,15÷0,3) В. При Еэ=-(0,15÷0,3) В уменьшалась величина тока восстановления анавидина. Использование предварительного электролиза при значениях потенциала - (0,15÷0,3) В позволяет регистрировать вольтамперограммы с четко выраженным максимумом. Это позволяет повысить точность и селективность способа и экспрессно определять концентрации анавидина меньше, чем 1·10-3 мг/л.
Оптимальное время предварительного электролиза (τэ) составляет 60÷90 с. При τэ меньше 60 с снижается чувствительность определения и увеличивается ошибка определения, а при τэ больше 90 с снижается экспрессность; величина тока достигала максимального значения при τэ, равном 60÷90 с.
Важным для определения анавидина методом адсорбционной ВА является выбор скорости развертки потенциала. Оптимальной является скорость (80÷100) мВ/с. Использование скорости менее (80÷100) мВ/с снижает величину анодного тока и понижает чувствительность определения (фиг.3).
Установленные условия проведения электродного процесса впервые позволили количественно определять анавидин на основе реакции электровосстановления. Для повышения чувствительности определения использовали предварительное концентрирование вещества на поверхности РП электрода. Предлагаемый вольтамперометрический способ позволил количественно определять анавидин в диапазоне концентраций полимера от 1·10-3 мг/л до 1,0 мг/л.
Измерения проводили на компьютеризированных вольтамперометрических анализаторах СТА, ВАМ (ООО «ИТМ», г.Томск).
Определению не мешают вещества, присутствие которых возможно в биологических объектах: водорастворимые витамины групп В (B1, В2, Вc, В6), РР, аскорбиновая и мочевая кислоты в соизмеримых количествах. Состав матрицы исследуемых вод практически не оказывает влияния на ток восстановления анавидина, поэтому не требовалось предварительное выделение полимера из матрицы до проведения собственно электрохимического анализа.
Пример 1. Определение содержания анавидна на уровне
(0,1÷10-3) мг/л.
В кварцевый стаканчик емкостью 20 мл наливают 10 мл раствора 0,1 М NaOH. Удаляют из раствора кислород струей очищенного азота с содержанием кислорода менее 0,001% в течение пяти минут. Не прекращая перемешивания, проводят электролиз раствора при условии: Еэ=-0,2 В, τэ=60 с. Отключают газ и фиксируют катодную вольтамперограмму при скорости развертки потенциала 100 мВ/с, начиная с потенциала Енач=-0,20 В. Отсутствие пиков свидетельствует о чистоте фона. Затем добавляют несколько капель объемом 0,01 мл аттестованной смеси анавидина 0,1-0,01 мг/л, перемешивают раствор 10 с и проводят электрохимическое концентрирование осадка при Еэ=-0,20 В, τэ=60 с. Съемку вольтампероной кривой начинают с потенциала -0,20 В. Пик для указанной концентрации вещества регистрируют в диапазоне потенциалов от -0,80 до -0,95 В (х.с.э.) при чувствительности прибора (0,5÷1)·10-9 А/мм. Массовую концентрацию анавидна в пробе оценивают методом добавок аттестованных смесей, измеряя высоту анодных пиков по формуле (1). Время единичного анализа не превышает 15 минут.

где: Xi - содержание компонента в анализируемой пробе, г/л;
CAC - концентрация аттестованной смеси (АС) анавидина, из которой делается добавка к анализируемой пробе, г/л;
VAC - объем добавки АС компонента, мл;
I1 - величина максимального катодного тока компонента в анализируемой пробе, А;
I2 - величина максимального катодного тока компонента в пробе с добавкой АС, А;
V - объем анализируемой пробы, мл;
Vпр - объем раствора подготовленной пробы, мл;
Vал - объем аликвоты раствора пробы, взятой для ВА измерения, мл.
Пример 2. Определение содержания анавидина в сточных водах
В кварцевый стаканчик емкостью 20 мл наливают 10 мл раствора 0,1 М NaOH. Удаляют из раствора кислород струей очищенного азота с содержанием кислорода менее 0,001% в течение пяти минут. Не прекращая перемешивания, проводят электролиз раствора при условии: Еэ=-0,2 В, τэ=60 с. Отключают газ и фиксируют катодную вольтамперограмму при скорости развертки потенциала 100 мВ/с, начиная с потенциала Eнач=-0,20 В. Отсутствие пиков свидетельствует о чистоте фона.
При анализе сточных вод, содержащих следовые количества анавидна, берут объем пробы 3 мл и вносят в электрохимическую ячейку, содержащую 7 мл фонового электролита. Электронакопление и регистрацию аналитического сигнала проводят в тех же условиях. Катодный пик анавидина фиксируют в диапазоне потенциалов от -0,85 до -0,95 в на РП электроде при чувствительности прибора (1÷5)·10-8 А/мм. Массовую концентрацию анавидна в пробе оценивают методом добавок аттестованных смесей, измеряя высоту анодных пиков по формуле (1). Время анализа одной пробы с учетом пробоподготовки менее 20 минут.
Таким образом, впервые установлена способность количественного химического анализа анавидина по пикам востановления его на РП электроде.
Анализ характеристик количественного химического определения анавидна по предлагаемому способу свидетельствуют о существенном повышении чувствительности определения (на 2 порядка по сравнению с методом капиллярного элетртрофореза) [Руднев А.В., Джераян Т.Г. Определение полигексаметиленгуанидина методом капиллярного электрофореза // Журнал аналитической химии, 2006, Т.61, №10, с.1086-1089]. Предел обнаружения и нижняя граница определяемых содержаний соответственно равны 0,83·10-3 мг/л и 0,96·10-3 мг/л. Значительно сократилось время проведения анализа. Предлагаемые условия позволяют контролировать сточные воды, воздушные зоны предприятий и химических лабораторий на уровне 0,5 ПДК (и меньше) анавидина.
Предложенный способ прост, не требует большого количества реактивов и трудозатрат и может быть применен в любой химической лаборатории, имеющей полярограф, особенно в настоящее время, когда налажен выпуск отечественной и зарубежной электроаппаратуры с компьютерным управлением и обработкой данных (анализаторы типа СТА, ТА и др.) Предложенный способ может быть использован в фармакокинетических и фармацевтических исследованиях, в токсикологическом и техническом анализе соединений группы гуанидинов, для контроля сточных вод и воздушной зоны вводно-очистных предприятий, а также для разработки методик анализа анавидина и родственных ему соединений в сложных многокомпонентных биосистемах (кровь, моча, плазма и др.) и пищевых продуктах.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ СТРЕПТОМИЦИНА МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2005 |
|
RU2276354C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СУРЬМЫ, ВИСМУТА, МЕДИ В ВОДНЫХ РАСТВОРАХ МЕТОДОМ АНОДНО-КАТОДНОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2010 |
|
RU2419786C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ БУТОПРОФИДА МЕТОДОМ ВОЛЬТАМПЕРОМЕТРИИ | 2005 |
|
RU2289127C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ РЕНИЯ КИНЕТИЧЕСКИМ ИНВЕРСИОННО-ВОЛЬТАМПЕРОМЕТРИЧЕСКИМ МЕТОДОМ В ВОДНЫХ РАСТВОРАХ ПРИРОДНОГО И ТЕХНОГЕННОГО ПРОИСХОЖДЕНИЯ | 2012 |
|
RU2490625C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ПЛАТИНЫ В ВОДНЫХ РАСТВОРАХ МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ ПО ПИКАМ СЕЛЕКТИВНОГО ЭЛЕКТРООКИСЛЕНИЯ ВИСМУТА ИЗ ИНТЕРМЕТАЛЛИЧЕСКОГО СОЕДИНЕНИЯ PtBi | 2009 |
|
RU2390011C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ГЕСПЕРИДИНА МЕТОДОМ ДИФФЕРЕНЦИАЛЬНОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2008 |
|
RU2381502C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ РЕНИЯ КИНЕТИЧЕСКИМ ИНВЕРСИОННО-ВОЛЬТАМПЕРОМЕТРИЧЕСКИМ МЕТОДОМ В ПОРОДАХ И РУДАХ | 2012 |
|
RU2506580C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ПЛАТИНЫ В РУДАХ МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2010 |
|
RU2426108C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ПЛАТИНЫ В РУДАХ ПО ПИКУ СЕЛЕКТИВНОГО ЭЛЕКТРООКИСЛЕНИЯ Сu ИЗ ИНТЕРМЕТАЛЛИЧЕСКОГО СОЕДИНЕНИЯ PtCu МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2012 |
|
RU2498289C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ЛЕВОМИЦЕТИНА В ПИЩЕВЫХ ПРОДУКТАХ И ФАРМПРЕПАРАТАХ | 2000 |
|
RU2180748C1 |



Изобретение относится к области аналитической химии, изучающей возможность определения анавидина методом инверсионной вольтамперометрии. Поставленная задача достигается тем, что способ количественного определения полигексаметиленгуанидина (ПГМГ) (анавидина) включает перевод анавидина из пробы в раствор путем экстракции и вольтамперометрического определение с использованием ртутно-пленочного (РП) электрода в качестве индикаторного. При этом используется накопление в течение 60÷90 с при потенциале электролиза - (0,15÷0,3) В относительно 1 М хлоридсеребрянного электрода на фоне 0,01 М гидрооксида натрия с последующей регистрацией катодных пиков при скорости развертки потенциала (80÷100) мВ/с. Концентрацию анавидина определяют по высоте пика в диапазоне потенциалов от -0,8 до -0,95 В методом добавок аттестованных смесей. Предел обнаружения равен 0,83·10-3 мг/л. Предложенный способ может быть использован в фармакокинетических и фармацевтических исследованиях, в токсикологическом и техническом анализе соединений группы гуанидинов, для контроля сточных вод и воздушной зоны вводно-очистных предприятий, а также для разработки методик анализа анавидина и родственных ему соединений в сложных многокомпонентных биосистемах (кровь, моча, плазма и др.) и пищевых продуктах. 3 ил.
Способ количественного определения анавидина методом инверсионной вольтамперометрии, включающий перевод анавидина из пробы в раствор и вольтамперометрическое определение с использованием индикаторного ртутно-пленочного электрода, при этом накопление анавидина в перемешиваемом растворе проводят в течение 60÷90 с при потенциале электролиза - (0,15÷0,3) В относительно 1М хлоридсеребряного электрода на фонах 0,01 М раствора гидрооксида натрия с последующей регистрацией катодных пиков при скорости развертки потенциала (80-100) мВ/с и концентрацию анавидна определяют по высоте пика в диапазоне потенциалов от -0,8 до -0,95В методом добавок аттестованных смесей.
| Dengbai Luo, Chun Zhou and Chenxia Luo Polarographic Behavior of Co(II)-BSA or -HAS Complex in Presence of a Guanidme Modifier., Anal | |||
| Chem., 2003, 75, p.p.6346-6350 | |||
| Руднев А.В., Джераян Т.Г | |||
| Определение полигексаметиленгуанидина методом капиллярного электрофореза | |||
| Журнал аналитической химии, 2006, т.61, №10, с.1086-1089 | |||
| СПОСОБ ИНВЕРСИОННОГО ВОЛЬТАМПЕРОМЕТРИЧЕСКОГО ОПРЕДЕЛЕНИЯ ГИДРАЗИНА | 2002 |
|
RU2219536C1 |

