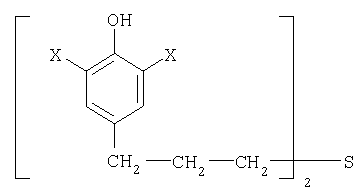
Изобретение относится к области аналитической химии, в частности к адсорбционному инверсионному вольтамперометрическому способу определения бутопрофида (БП) нового синтезированного вещества с выраженными антиоксидантными свойствами [Н.М.Сторожок, А.П.Крысин, Н.В.Гуреева. Антиоксидантное действие новых аналогов пробукола и их композиций с α-токоферолом //Вопросы медиц.химии. - 2001. - т.47, №5, с.517-525]. Соединение БП является производным тио-бис-фенола и представляет собой бис-[3-(3,5-ди-трет-бутил-4гидроксифенил)пропил]сульфид:
Применение природных и синтетических антиоксидантов (АО) является актуальным для стабилизации окисления ненасыщенных липидов, в качестве дополнительных средств неспецифической терапии многих заболеваний (злокачественных новообразований, ишемической болезни сердца и др.). В настоящее время выявлена способность БП блокировать накопление гидропероксидов. Вещество не обладает местным и общетоксическим действием, не оказывает влияние на эмбриогенез и развитие потомства [Т.Н.Орлова, Т.Г.Толстикова, И.В.Сорокина, М.П.Долгих, Т.В.Воевода. Фармакокинетика нового фенольного антиоксиданта СО-3. // Химико-фармацевтический журнал, 2000. - т.34, №9, с.9-10]. Это позволяет использовать БП не только в лечебной практике, но и в пищевой технологии, медицине, косметологии. Применение эффектов синергизма позволяет создавать высокоэффективные композиции АО [Н.М.Сторожок, А.П.Крысин, Н.В.Гуреева. Антиоксидантное действие новых аналогов пробукола и их композиций с α-токоферолом // Вопросы медиц. химии. - 2001. - т.47, №5, с.517-525]. Ингибирование окисления может осуществляться низким количеством ингибиторов (менее 10-4 моль/л), что особенно важно при стабилизации окисления лекарственных препаратов, пищевых продуктов, биологически активных добавок, косметических средств. Это, в свою очередь, предъявляет повышенные требования к контролю за качеством продуктов, лекарственных средств и совершенствованию методов определения АО.
В настоящее время практически отсутствуют методы определения БП. Известен метод высокоэффективной жидкостной хроматографии (ВЭЖХ) [Т.Н.Орлова, Т.Г.Толстикова, И.В.Сорокина, М.П.Долгих, Т.В.Воевода. Фармакокинетика нового фенольного антиоксиданта СО-3. // Химико-фармацевтический журнал, 2000. - т.34, №9, с.9-10] определение БП в биологическом материале после предшествующих совокупности операций по пробоподготовки (высушивание, гомогенизация, фильтрование и упаривание). Сухой остаток органической пробы растворяли в метаноле и анализировали методом ВЭЖХ на микроколоночном хроматографе Милихром (Россия). Колонка: Lichrosper С-18 (2×50 мм), элюент - метанол, скорость потока 0,1 мл/мин, объем пробы 2 мкл. Для увеличения чувствительности использовали ультрафиолетовое детектирование при длине волны 230 и 280 нм. Концентрацию БП в пробе определяли методами абсолютной калибровки. Для построения калибровочных прямых в интактные пробы вносили ПБ в диапазоне от 0,25 мг до 1 мг, после чего проводили процедуру пробоподготовки и определение ВЭЖХ, как описано выше. Несмотря на достаточно высокую чувствительность метода ВЭЖХ (1·10-6 г), длительность анализа с учетом времени пробоподготовки, а также высокая стоимость приборов существенно ограничивают его использование в экспрессном определении БП. Другие инструментальные методы количественного химического анализа (КХА) БП в литературе не описаны.
Одним из наиболее перспективных методов определения БП является вольтамперометрический (ВА) и в первую очередь такие его высокочувствительные варианты, как адсорбционная инверсионная дифференциальная вольтамперометрия (ИВА). Преимущество ИВА благодаря высокой чувствительности состоит в возможности работы с малыми по массе и объемами пробами в мутных и окрашенных средах. Пробоподготовка может быть существенно упрощена из-за необязательной дополнительной очистки определяемого вещества перед собственно электрохимическим анализом. Поэтому часто допустимы более простые методики предварительного выделения соединений из сложной многокомпонентной системы.
Информация о применении метода ВА для определения БП в литературе отсутствует. Однако имеются сведения об условиях вольтамперометрического окисления родственного пространственно-затрудненного 4,4-тио-бис-(6-трет-бутил-3-метилфенола) на фоне (C4H9)NOH в ацетонитриле при потенциале, равном 0,08 В, и концентрации 1·10-4 моль/л [Ю.В.Водзинский. Анодная вольтамперометрия органических соединений на графитовом электроде. Автореферат диссертации на соискание ученой степени доктора хим. наук. - 1975, Казань, 50. с]. Наиболее близким является метод анодной вольтамперометрии 4,4-тио-бис-фенолов (прототип) [Ю.В.Водзинский, А.А.Васильева, И.А.Коршунов. Анодная вольтамперометрия пространственно-затрудненных фенолов. // Журн. орг. химии. - 1969. - 39, №6, c.1196-1202]. Сущность методики состоит в электроокислении пространственно-затрудненных фенолов с использованием торцового графитового индикаторного электрода, пропитанного под вакуумом парафином. Потенциалы полуволн фенолов определяли в вводно-спиртовых буферных средах с рН 2 (смесь 0,04 моль/л борной, уксусной и фосфорной кислот с добавлением 0,2 моль/л водного раствора NaOH и 40% изопропилового спирта) в диапазоне потенциалов от 0,60 В до 0,75 В (электрод сравнения 0,5 моль/л CdSO4/Cd) и концентраций 1·10-5÷1·10-3 моль/л при скорости поляризации 10 мВ/с.
В данных условиях определение БП невозможно. Поэтому разработка экспериментальных и высокочувствительных методов определения БП продолжает представлять интерес.
Задачей заявляемого изобретения является повышение чувствительности, экспрессности и селективности определения БП методом инверсионной вольтамперометрии.
Поставленная задача достигается тем, что бутопрофид переводят из пробы в раствор и проводят вольтамперометрическое определение, отличающийся тем, что используют инверсионную вольтамперометрию, при этом накопление бутапрофида проводят с использованием индикаторного стеклоуглеродного электрода в перемешиваемом растворе в течение 30÷60 с при потенциале электролиза от -0,10 В до -0,05 В относительно насыщенного хлоридсеребряного электрода (нас.х.с.э.) на фоне 0,01÷0,02 моль/л гидроксида натрия, рН 10÷12 в присутствии 0,1% этилового спирта с последующей регистрацией катодных пиков в дифференциальном режиме съемки вольтамперограмм при скорости развертки потенциала 20÷50 мВ/с, и концентрацию бутопрофида определяют по высоте пика в диапазоне потенциалов от -0,55 В до -0,70 В методом добавок аттестованных смесей.
В прототипе количественное определение родственных БП соединений основано на реакции окисления в неводных или 40% растворах изопропилового спирта на графитовом торцовом электроде, импрегнированном парафином.
В предлагаемом способе впервые установлена способность БП восстанавливаться в щелочных средах на графитовых индикаторных электродах. Величина потенциала восстановления БП на графитовых электродах опрегонах графита, имеющих π-зонную структуру, а также возможностью взаимодействия адсорбата с адсорбентом. По-видимому, адсорбция плоскостью молекул органического соединения и структурное подобие материала электрода (стереоспецифическая адсорбция) благоприятствует переходу электронов при меньшем значении потенциала. Это способствует более легкому восстановлению БП на графитовых электродах, чем на ртутно-пленочном с серебряной подложкой. Для БП не удалось зарегистрировать пики восстановления в водных средах на ртутно-пленочном электроде. Наибольшая величина тока достигается на электроде из графита (Г), пропитанного полиэтиленом с парафином в вакууме или эпоксидной смолой, что обусловлено более упорядоченной структурой графита по сравнению с искусственными графитами (стеклоуглеродным, СУ). Однако Г электрод оказался неудобен в работе из-за плохой воспроизводимости аналитического сигнала, чем СУ электрод, особенно при определении БП в щелочных средах. СУ электрод обладает высокой химической и электрохимической устойчивостью, широкой областью рабочих потенциалов, а также простотой механического обновления поверхности. Для определения БП использовали «игольчатый» по форме СУ электрод после предварительного электрохимического модифицирования его поверхности. Такая обработка приводит к снижению нижней границы определяемых содержаний БП и улучшению воспроизводимости вольтамперометрических измерений. Графитовые СУ «игольчатые» электроды впервые использованы для определения БП.
В прототипе описано использование в качестве фонов буферных растворов рН 2, приготовленных из смеси 0,04 моль/л борной, уксусной и фосфорной кислот с добавлением 0,2 моль/л NaOH и 40% изопропилового спирта. Определение БП в этих условиях невозможно. Предлагаемый в заявляемом изобретении фон 0,01÷0,02 моль/л NaOH (рН 10÷12) в присутствии 0,1% этилового спирта позволяет регистрировать на вольтампе-рограмме пики восстановления в диапазоне потенциалов от -0,55 В до -0,70 В, высота которых линейно зависит от содержания БП в объеме раствора в интервале концентраций 5·10-7÷1·10-5 моль/л. Относительное стандартное отклонение (Sr) не превышает 0,02 для концентрации 5·10-7 моль/л и увеличивается до 0,07 с увеличением концентрации до 1·10-5 моль/л. По-видимому, это связано с ограниченной растворимостью БП в водно-спиртовых растворителях с увеличением концентрации. Абсолютной новизной является использование подобранных значений рН раствора NaOH, от чего зависит количественное определение БП. Оптимальный диапазон рН растворов фонового электролита 10÷12 определяется хорошей воспроизводимостью и фиксированием практически одного пика при регистрации вольтамперограмм. Использование рН меньше 10 приводит к регистрации дополнительного предпика при Еп, равном -0,45 В, и снижает селективность определения; при рН больше 12 снижается электрическая проводимость растворов и чувствительность. Применение 0,1% этилового спирта позволяет существенно облегчить удаление продуктов электродной реакции с поверхности электрода, от чего зависит стабильность и воспроизводимость во времени результатов.
Другой отличительный признак - установленные условия электрохимического накопления: потенциал электролиза от -0,10 В до -0,05 В. Опытные данные показали зависимость величины тока восстановления от Еэ (таблица). Величина тока увеличивалась и достигала максимального значения в области потенциалов от -0,10 В до -0,05 В. При Еэ больше -0,05 В и Еэ меньше -0,10 В уменьшалась величина тока; так, при Еэ больше -0,05 В ухудшалась воспроизводимость пиков. Использование предварительного электролиза при значениях потенциала от -0,01 В до -0,05 В позволяет регистрировать вольтамперограммы с четко выраженным максимумом. Это позволяет повысить точность и разрешающую способность способа и экспрессно определять концентрации меньше, чем 1·10-5 моль/л.
Время предварительного электролиза (τэ) выбирают в зависимости от концентрации определяемого вещества. Максимальное значение величины тока достигается при τэ, равном 30÷60 с. При τэ меньше 30 с снижается чувствительность и увеличивается ошибка определения, а при τэ больше 60 с снижается экспрессность. Исследование зависимости величины тока от времени выдержки электрода без наложения на него поляризующего напряжения свидетельствовало о достижении адсорбционного равновесия раствора за 30÷60 с. Величина тока при этом возрастала на 7÷9%. Дальнейшее увеличение времени выдержки электрода не влияло на характер аналитического сигнала.
Важным для определения БП является выбор скорости развертки потенциала. Оптимальной является скорость 20÷50 мВ/с. Увеличение скорости более 50 мВ/с увеличивает чувствительность, но при этом растет остаточный ток и уменьшается разрешающая способность способа. Использование скорости менее 20 мВ/с существенно снижает величину катодного тока и понижает чувствительность определения БП.
Нижняя граница определяемых содержаний БП зависит от режима съемки вольтамперных кривых. В дифференциальном режиме высокая точность определяется тем, что потенциал пика измеряется в точке, в которой круто падающая первая производная пересекает нулевую линию. Использование режима дифференцирования позволяет фиксировать четкие пики даже при концентрациях 5·10-7÷1·10--6 моль/л, что повышает чувствительность определения и позволяет регистрировать удобные для измерения поляризационные кривые с хорошей воспроизводимостью (Sr для указанного диапазона концентраций не превышает 0,02÷0,05). Для определения БП, его аналогов и родственных соединений он ранее не применялся. При линейном наложении потенциала в интегральном режиме (прототип) определение концентрации БП, на уровне 5·10-7÷1·10-6 моль/л практически невозможно из-за большого значения остаточного тока. Поэтому для количественного определения БП рекомендован дифференциальный режим съемки вольтамперограмм.
Таким образом, установленные условия впервые позволили количественно определять новый полифункциональный бисфенольный антиоксидант БП на основе реакции электровосстановления (в прототипе количественное определение родственного пространственно-затрудненного тио-бис-фенольного соединения проводят по волнам окисления). Для повышения чувствительности определения использовали предварительное адсорбционное концентрирование БП на поверхности СУ электрода. Предлагаемый вольтамперометрический способ позволил существенно улучшить метрологические характеристики анализа БП, повысить чувствительность определения. Предел обнаружения (Cmin, p), рассчитанный по трехсигмовому критерию Кайзера (n=13, р=0,95), равен 1,2·10-7 моль/л (Cmin, p в прототипе не приводится). Нижнюю границу определяемых содержаний БП находили опытным путем как минимальное содержание вещества, определяемого по данной методике при Sr, равном 0,02÷0,03 (Сн). Значение Сн, равное 5·10-7 моль/л, в 20 раз ниже по сравнению с прототипом. Определению не мешают вещества, присутствие которых возможно в виде примесей на стадии синтеза препарата БП [Н.М.Сторожок, А.П.Крысин, Н.В.Гуреева. Антиоксидантное действие новых аналогов пробукола и их композиций с α-токоферолом // Вопросы медиц. химии. - 2001. - т.47, №5, с.517-525].
Пример 1. Определение бутопрофида на уровне 5·10-7÷1·10-6 моль/л
В кварцевый стаканчик вместимостью 20,0÷25,0 мл вносят 10,0 мл раствора фонового электролита гидроксида натрия концентрации 0,01 моль/л с содержанием 0,1% этилового спирта и помещают в электролитическую ячейку вольтамперометрического анализатора. Опускают в раствор электроды: насыщенный хлоридсеребряный, стеклоуглеродный и вспомогательный платиновый. Удаляют из раствора кислород струей очищенного азота с содержанием кислорода менее 0,001% в течение трех минут. Не прекращая перемешивания, проводят процесс электронакопления при потенциале -0,10 В в течение 60 с. Отключают газ и фиксируют катодную дифференциальную вольтамперограмму в диапазоне потенциалов от -0,20 В до -1,00 В при скорости развертки напряжения 30 мВ/с. Отсутствие пиков свидетельствует о чистоте фона. Затем в стаканчик с фоновым раствором вносят аликвоту подготовленного раствора анализируемой пробы объемом 0,01÷0,06 мл, перемешивают раствор 10 с и проводят электрохимическое концентрированно осадка при Еэ, равном -0,10 В, и τэ, равном 60 с и вновь регистрируют вольтамперограмму в тех же условиях. Пик для указанной концентрации вещества регистрируют в диапазоне потенциалов от -0,55 В до -0,70 В (отн. нас. х.с.э.) при чувствительности прибора 1·10-10 А/мм. Содержание бутопрофида оценивают методом добавок аттестованных смесей, измеряя высоту катодных пиков.
Пример 2. Определение бутопрофида в техническом продукте
В кварцевый стаканчик вместимостью 20,0÷25,0 мл вносят 10,0 мл раствора фонового электролита гидроксида натрия концентрации 0,01 моль/л с содержанием 0,1% этилового спирта и помещают в электролитическую ячейку вольтамперометрического анализатора. Опускают в раствор электроды: насыщенный хлоридсеребряный, стеклоуглеродный и вспомогательный платиновый. Удаляют из раствора кислород струей очищенного азота с содержанием кислорода менее 0,001% в течение пяти минут. Не прекращая перемешивания, проводят процесс электронакопления при потенциале -0,05 В в течение 30 с. Отключают газ и фиксируют катодную дифференциальную вольтамперограмму в диапазоне потенциалов от -0,20 В до -1,00 В при скорости развертки напряжения 50 мВ/с. Отсутствие пиков свидетельствует о чистоте фона. Затем в стаканчик с фоновым раствором вносят аликвоту подготовленного спиртового раствора анализируемой пробы объемом 0,02 мл, перемешивают раствор 10 с и проводят электрохимическое накопление при Еэ , равном -0,05 В, и τэ, равном 30 с, и вновь снимают вольтамперограмму в тех же условиях. Катодный пик регистрируют в диапазоне потенциалов от -0,55 В; до -0,65 В (отн. нас. х.с.э.) при чувствительности прибора 5·10-10 А/мм. Содержание бутопрофида оценивают методом добавок аттестованных смесей, измеряя высоту катодных пиков. Время анализа одной пробы не превышает 15÷20 мин.
Предложенный способ прост, не требует больших трудозатрат, большого количества реактивов, отличается высокой селективностью, экспрессностью и чувствительностью и может быть применен в любой химической лаборатории, имеющей полярограф, особенно в настоящее время, когда налажен выпуск отечественной и зарубежной электроаппаратуры с компьютерным управлением и обработкой данных (анализаторы типа СТА, ТА, ВАМ и др.). Предложенный способ может быть использован для количественного определения бутопрофида в его лекарственных формах и фармакокинетических исследованиях, в анализе сложных биологических материалов, в пищевой промышленности, медицине, косметологии, для контроля сточных вод и воздушной зоны химико-фармацевтических предприятий.
Зависимость величины тока восстановления бутопрофида от потенциала электролиза (С=2,8·10-6 моль/л, w=30 мВ/с, τэ=30 с) мкА/мВ
мкА/мВ
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ СТРЕПТОМИЦИНА МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2005 |
|
RU2276354C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ГЕСПЕРИДИНА МЕТОДОМ ДИФФЕРЕНЦИАЛЬНОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2008 |
|
RU2381502C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СУРЬМЫ, ВИСМУТА, МЕДИ В ВОДНЫХ РАСТВОРАХ МЕТОДОМ АНОДНО-КАТОДНОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2010 |
|
RU2419786C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ СМЕСИ АФЛАТОКСИНОВ B1, B2, G1, G2 МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2015 |
|
RU2592049C1 |
| ВОЛЬТАМПЕРОМЕТРИЧЕСКИЙ СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ 4-ЙОДАНТИПИРИНА | 2001 |
|
RU2184370C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ЦИСТЕИНА В ВОДНЫХ РАСТВОРАХ МЕТОДОМ ЦИКЛИЧЕСКОЙ ВОЛЬТАМПЕРОМЕТРИИ НА ГРАФИТОВОМ ЭЛЕКТРОДЕ, МОДИФИЦИРОВАННОМ КОЛЛОИДНЫМИ ЧАСТИЦАМИ ЗОЛОТА | 2011 |
|
RU2463587C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ РТУТИ КАТОДНО-АНОДНОЙ ВОЛЬТАМПЕРОМЕТРИЕЙ | 2013 |
|
RU2533337C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ РЕНИЯ КИНЕТИЧЕСКИМ ИНВЕРСИОННО-ВОЛЬТАМПЕРОМЕТРИЧЕСКИМ МЕТОДОМ В ВОДНЫХ РАСТВОРАХ ПРИРОДНОГО И ТЕХНОГЕННОГО ПРОИСХОЖДЕНИЯ | 2012 |
|
RU2490625C1 |
| Вольтамперометрический способ определения дифениламина в продуктах выстрела | 2017 |
|
RU2657552C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ПЛАТИНЫ В РУДАХ МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2010 |
|
RU2426108C1 |
Изобретение относится к области аналитической химии. Технический результат изобретения: повышение чувствительности, экспрессности и селективности определения бутопрофида. Сущность: бутопрофид переводят из пробы в раствор и проводят инверсионное вольтамперометрическое определение бутопрофида с использованием стеклоуглеродного индикаторного электрода в перемешиваемом растворе в течение 30-60 с при потенциале электролиза от -0,10 В до -0,05 В относительно насыщенного хлоридсеребряного электрода на фоне 0,01÷0,02 моль/л гидроксида натрия (рН 10÷12) в присутствии 0,1% этилового спирта с последующей регистрацией катодных пиков в дифференциальном режиме съемки вольтамперограмм при скорости развертки потенциала 20÷50 мВ/с. Концентрацию бутопрофида определяют по высоте пика в диапазоне потенциалов от -0,55 В до -0,70 В методом добавок аттестованных смесей. 1 табл.
Способ количественного определения бутопрофида, включающий перевод вещества из пробы в раствор и вольтамперометрическое определение, отличающийся тем, что используют инверсионную вольтамперометрию, при этом накопление бутопрофида проводят с использованием индикаторного стеклоуглеродного электрода в перемешиваемом растворе в течение 30÷60 с при потенциале электролиза от -0,10 до -0,05 В относительно насыщенного хлоридсеребряного электрода на фоне 0,01÷0,02 моль/л гидроксида натрия, рН 10÷12 в присутствии 0,1%-ного этилового спирта с последующей регистрацией катодных пиков в дифференциальном режиме съемки вольтамперограмм при скорости развертки потенциала 20÷50 мВ/с и концентрацию бутопрофида определяют по высоте пика в диапазоне потенциалов от -0,55 до -0,70 В методом добавок аттестованных смесей.
| ВОДЗИНСКИЙ Ю.В | |||
| и др | |||
| Анодная вольтамперометрия пространственно-затрудненных фенолов // Журн | |||
| орг | |||
| химии | |||
| Приспособление к индикатору для определения момента вспышки в двигателях | 1925 |
|
SU1969A1 |
| Машина для изготовления проволочных гвоздей | 1922 |
|
SU39A1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ФЛАВОНОИДОВ МЕТОДОМ ДИФФЕРЕНЦИАЛЬНОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2002 |
|
RU2215288C2 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ВИТАМИНА E (α-ТОКОФЕРОЛ АЦЕТАТА) МЕТОДОМ ДИФФЕРЕНЦИАЛЬНОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2000 |
|
RU2180747C1 |
| US 6108570 A, 22.08.2000. | |||

