ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к области синтеза оптически активных соединений. В нем описан способ синтеза пароксетина с высокой стереоспецифичностью.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
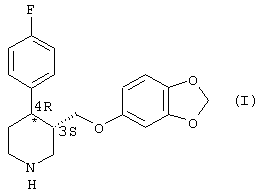
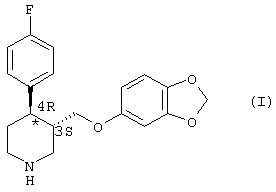
Пароксетин - это соединение, которое широко применяется при лечении депрессии. Структурная формула этого соединения имеет следующий вид (I):

Молекула формулы (I) имеет два хиральных центра в кольце пиперидина в положении 3 и 4 соответственно. Из четырех возможных изомеров фармакологически активным является только изомер, имеющий абсолютную конфигурацию 3S, 4R, известный как 4R-транс-4-(п-фторофенил-3-{[3,4-(метилендиокси)фенокси]метил}-пиперидин. Поэтому способы получения пароксетина должны приводить к получению структуры 4-(п-фторофенил-3-{[3,4-(метилендиокси)фенокси]метил}пиперидина исключительно в указанной выше конформации - 3S, 4R (или 4R-транс).
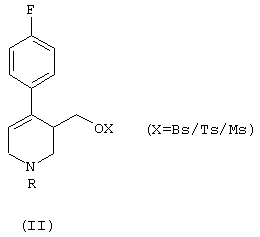
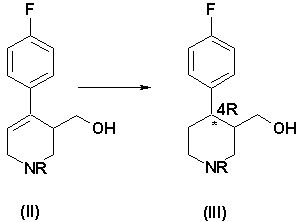
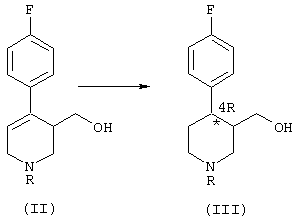
Некоторые способы синтеза пароксетина, известные в этой области, основаны на образовании промежуточного соединения формулы (II):
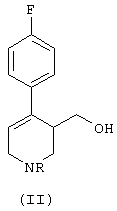
где R означает алкильную группу. Исходя из этого промежуточного соединения продукт формулы (I) получают путем: (i) восстановления двойной связи пиперидина, (ii) алкилирования кислорода гидроксиметильной группы и (iii) удаления связанной с азотом алкильной группы R. Для получения продукта (I) в фармакологически активной конформации необходимо выделить соответствующие изомеры из рацемической смеси и обработать их так, чтобы получить производное (I) в нужной конформации.
Например, в патентной заявке WO-A-9636636 раскрыт синтез 4-арилпиперидина, в котором производное формулы (II) разделяют на два оптических изомера кристаллизацией с оптически активными солями. Затем эти два оптических изомера по отдельности превращают в пароксетин. Следовательно, этот способ требует отдельных и независимых путей синтеза для обработки изомеров и поэтому его трудно довести до коммерческого масштаба.
Согласно другому способу (J. Labelled Compounds Radiopharm., 1993, 8, 785) производное формулы (II) гидрируют и алкилируют, как в описанной выше схеме, после чего выделяют диастереоизомеры хроматографией, а энантиомеры разделяют кристаллизацией с L-(+)-винной кислотой. Наконец, (-)-транс-изомер превращается в пароксетин с помощью N-деалкилирования. В этом случае требуются два отдельных цикла разделения изомеров, что приводит к значительной потере продукта в виде нежелательного изомера. Поэтому данный процесс тоже трудно применим для производства в коммерческом масштабе.
Способ, раскрытый в патентной заявке WO-A-9322284, основан на стереоспецифичности реакций, катализируемых эстеразами. В этом случае фермент вызывает образование транс-карбоксильного предшественника, (+)-транс- и (-)-транс-формы которого разделяют стандартными методами. Последняя форма затем подвергается восстановлению и алкилированию с образованием пароксетина. Преимуществом этого способа является высокая стереоспецифичность, а его недостатками - стоимость и нестабильность фермента. Указанные выше реакции обычно протекают медленно и должны проводиться с соблюдением точных значений рН и температуры.
Таким образом, известные в этой области способы разделения вызывают значительную потерю продукта в виде изомеров с нежелательной конфигурацией или требуют отдельных циклов обработки для превращения этих изомеров. В частности, известные в этой области способы влекут за собой разделение рацемических смесей, в которых право- и левовращающие компоненты присутствуют в примерно равных пропорциях, а это приводит к тому, что почти половина разделенного продукта идет в отходы или по отдельности превращается в нужную форму.
Следовательно, ощущается потребность в разработке высокостереоспецифичных способов синтеза пароксетина, с помощью которых можно получить требуемые изомеры с высоким выходом. Особенно настоятельна потребность в способах, которые не включают циклов разделения изомеров и не требуют раздельных методов обработки разделенных изомеров.
ПЕРЕЧЕНЬ ФИГУР
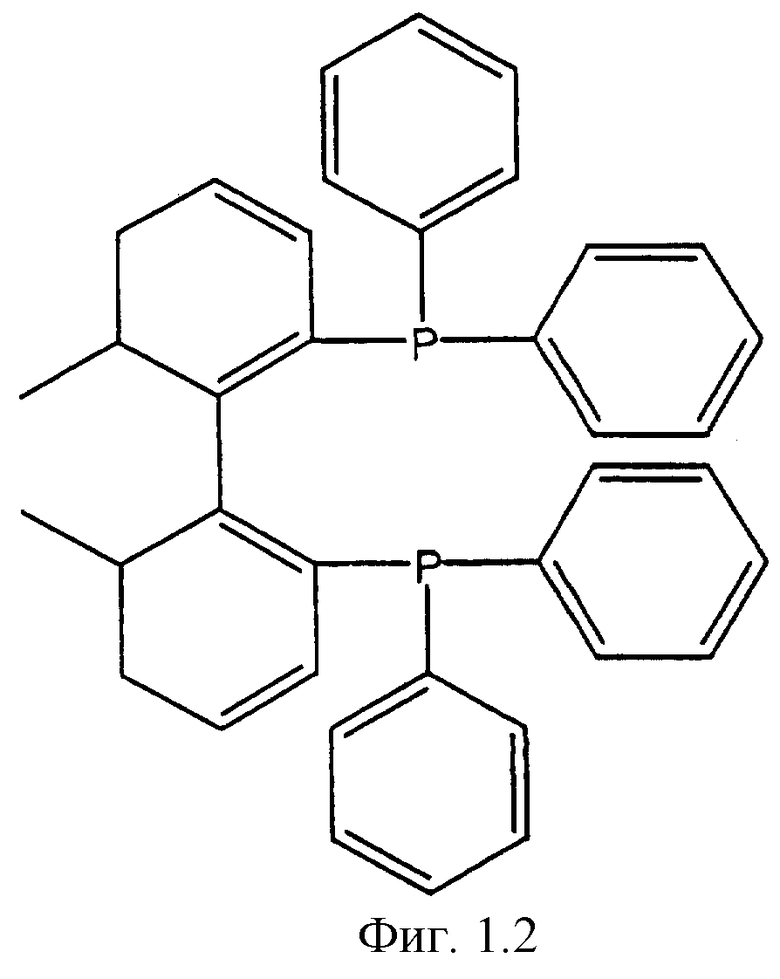
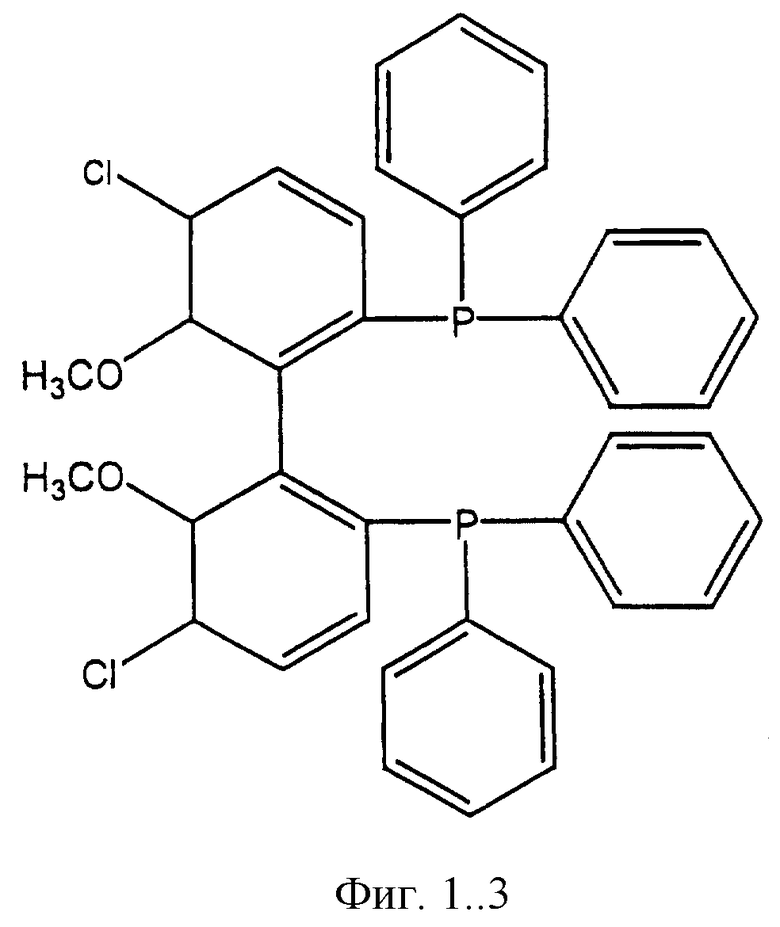
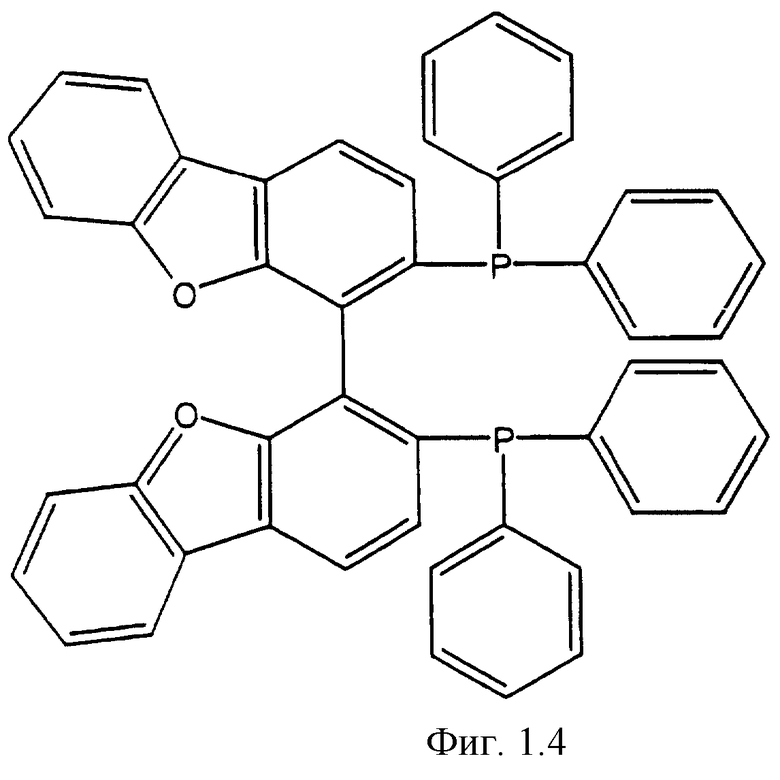
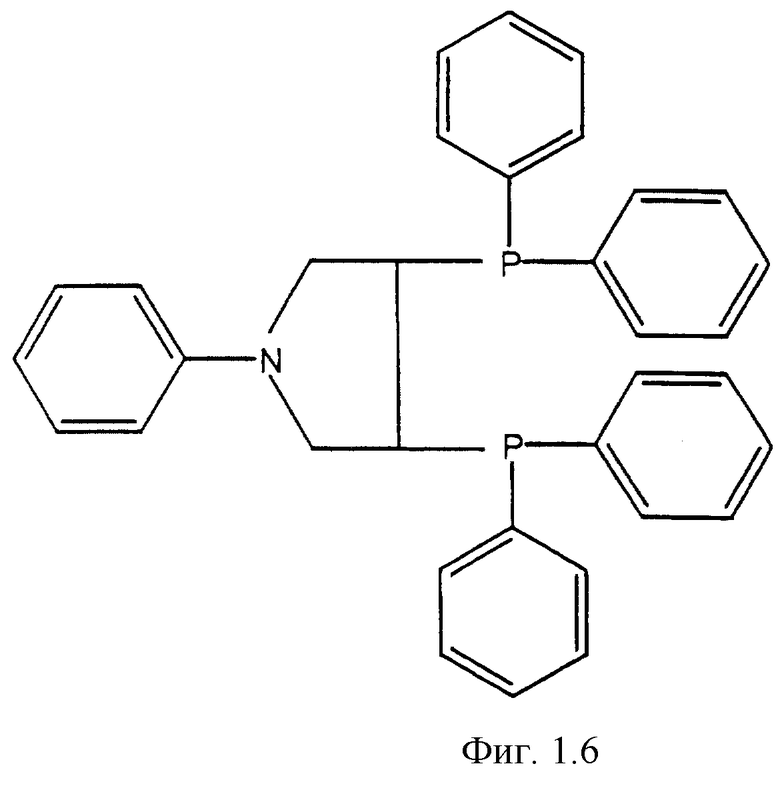
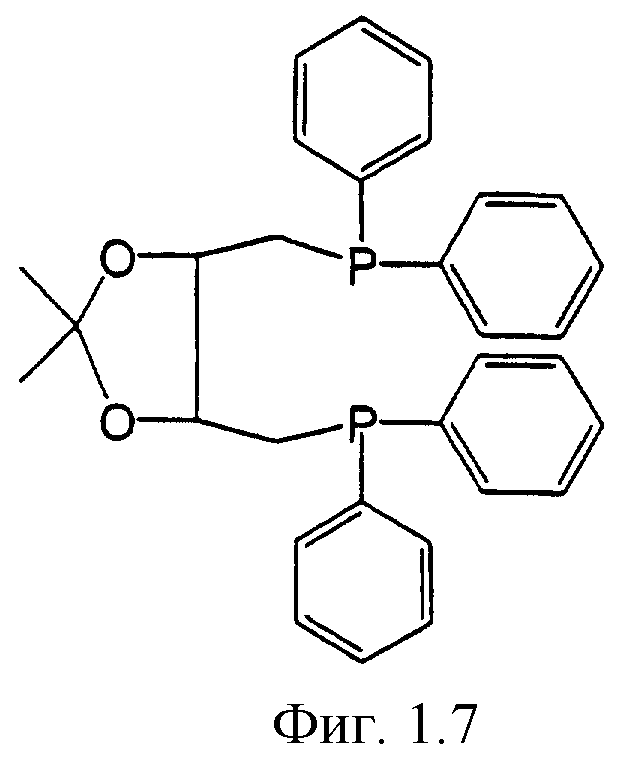
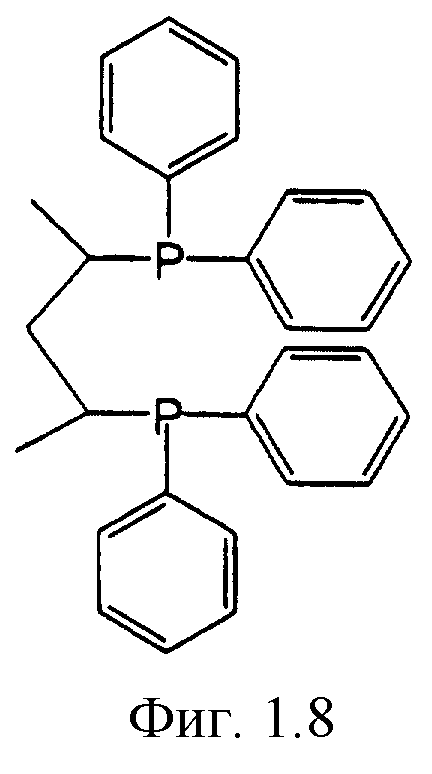
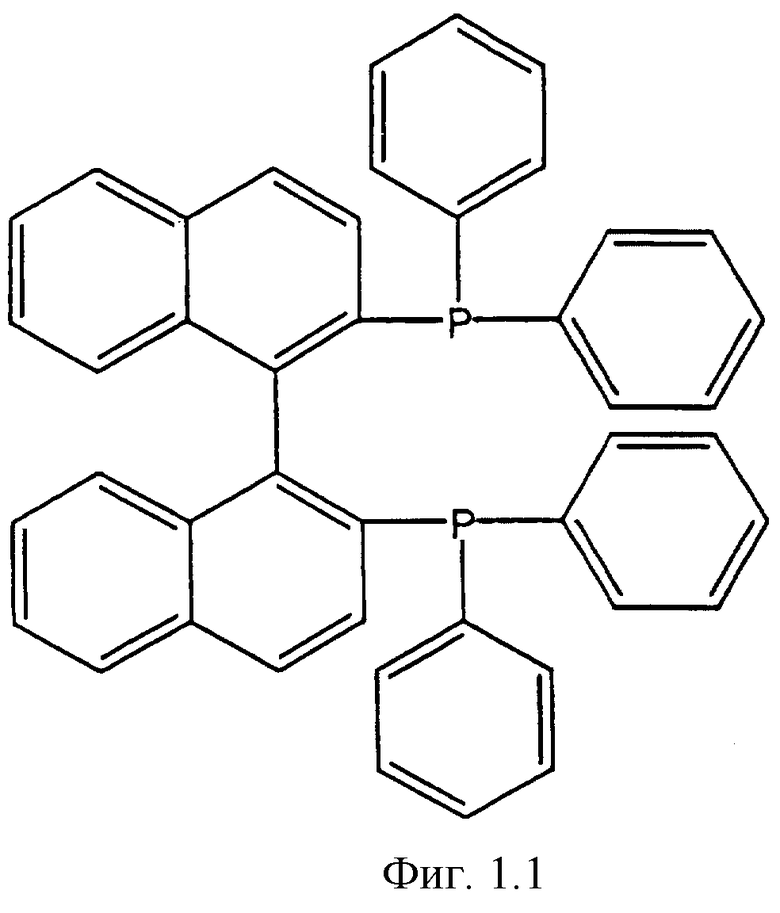
Фигуры 1.1, 1.2, 1.3, 1.4, 1.5, 1.6, 1.7 и 1.8. Примеры хиральных дифосфиновых лигандов.
Фигура 1.1. BINAP.
Фигура 1.2. BIPHEMP.
Фигура 1.3. (5,5'-дихлоро-6,6'-диметоксидифенил-2,2'-диил)-бис-(дифенилфосфин).
Фигура 1.4. (бис-4,4'-дибензофуран-3,3'-ил)-бис-дифенилфосфин.
Фигура 1.5. 4,4'-бис-дифенилфосфин-2,2',5,5'-тетраметил-3,3'-дитиофен.
Фигура 1.6. PYRPHOS.
Фигура 1.7. DIOP.
Фигура 1.8. BDPP.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является обеспечение способа синтеза пароксетина.
Способ включает следующие стадии:
а) гидрирование соединения формулы (II), в котором R означает группу, выбранную из (C1-C5)алкила, (C1-C5)карбоксиалкила, необязательно замещенного фенила и необязательно замещенного бензила, причем реакция гидрирования катализируется комплексным соединением переходного металла с хиральными дифосфиновыми лигандами, что приводит к образованию обогащенного 4R-энантиомером соединения формулы (III):
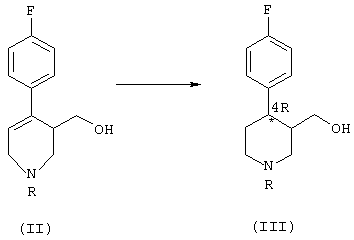
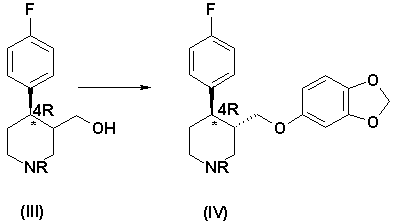
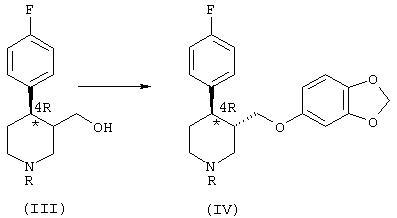
б) реакция соединения формулы (III) с реагентом, способным преобразовать ОН-группу в уходящую группу, с последующим нуклеофильным замещением ее сезамол(3,4-метилендиоксифенолом), что приводит к получению обогащенного 4R-энантиомером производного формулы (IV):
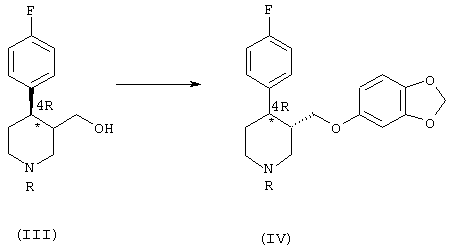
в) N-деалкилирование соединения (IV), что ведет к образованию пароксетина (I):
В формуле (II) R предпочтительно означает этильную группу. Если R представляет собой необязательно замещенный фенил или бензил, то он предпочтительно замещен (C1-C5)алкилом.
Полученное на стадии а соединение формулы (III) содержит два асимметричных центра в кольце пиперидина, в положении 3 и 4, соответственно. Этот продукт получают в виде смеси обогащенных 4R-энантиомерами цис-(IIIa) и транс- (IIIb) изомеров, в которой каждый из этих изомеров (цис- и транс-) преимущественно находится в форме, имеющей абсолютную конфигурацию R по атому углерода в положении 4 пиперидинового кольца.
Термин "обогащенный 4R-энантиомерами" означает, что общее содержание этих энантиомеров (энантиомерная чистота, сокращенно эн.ч.) превышает 80%. Содержание энантиомеров расчитывают, как описано в J. March, "Advanced Organic Chemistry", 3rd Ed., Chapter 4, p. 107 (John Wiley & Sons) (1985).
В реакции гидрирования (стадия а) применяются катализаторы, принадлежащие к классу комплексных соединений переходных металлов с хиральными дифосфиновыми лигандами. Эти катализаторы ответственны за стереоспецифичность реакции и позволяют получить обогащенное 4R-энантиомером производное формулы (III). К этому классу принадлежат все комплексные соединения переходных металлов с хиральными дифосфиновыми лигандами. В этих комплексах атом металла координирован с хиральными лигандами, образуя хиральный комплекс, способный катализировать с высокой стереоспецифичностью гидрирования двойной связи. Предпочтительными переходными металлами являются рутений, родий и иридий.
К числу хиральных дифосфиновых лигандов относятся соединения, представленные формулами, указанными на Фиг.1.
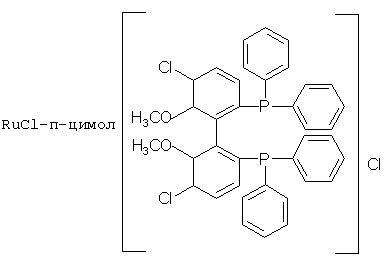
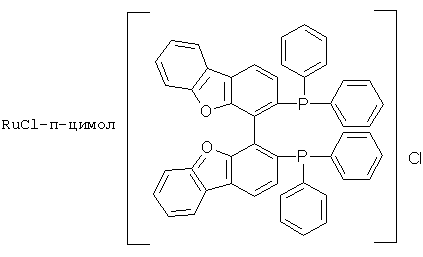
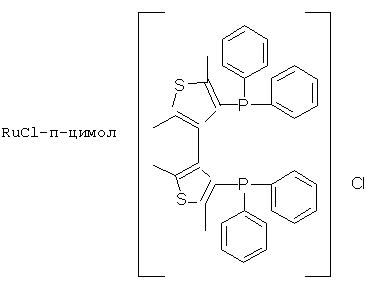
К числу комплексных соединений переходных металлов с хиральными дифосфиновыми лигандами относятся соединения формулы RuX1(L)m[BINAP]Yn, описанные в патентной заявке ЕР-А-366390, или соединения формулы, описанные в ЕР-А-245959. Предпочтительными комплексными соединениями являются {RuCl (п-цимол) [BINAP]}Cl, RuHCl [BINAP]2, Ru2Cl4 [BINAP]2Net3, Ru [BINAP] (OAc)2, Ru [BINAP] (CF3CO2)2 и соединения, имеющие формулы:
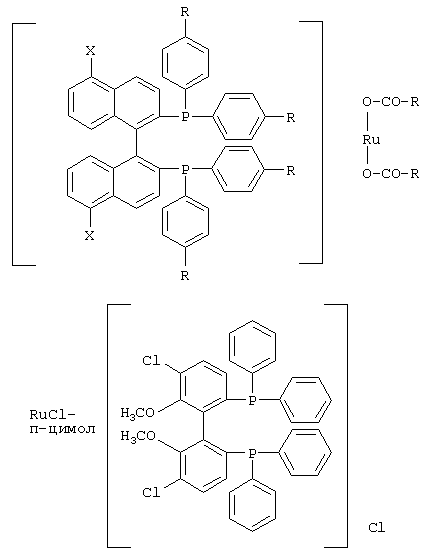
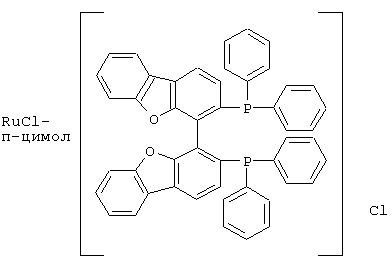
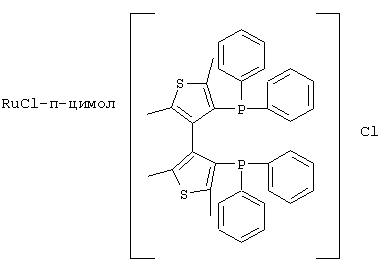
и соединения формулы {Ru(п-цимол)Х[BINAP]}+Х-, где Х означает атом галогена, например соединение {Ru(п-цимол)Cl[BINAP]}+Cl-.
Как отмечалось выше, все эти комплексные соединения хиральны - у каждого данного комплекса в обогащенном 4R-энантиомером продукте образуется только одна из этих хиральных форм -(S)BINAP или (R)BINAP, которую невозможно определить a priori, но можно легко установить, проведя предварительный опыт по гидрированию соединения (II) с помощью лиганда в одной из энантиомерных форм, например (S)BINAP, и проверяя, будет ли гидрированный продукт обогащен 4R- или 4S-формой - при синтезе пароксетина следует использовать тот энантиомерный лиганд, который дает 4R-обогащенную форму.
Комплексные соединения переходных металлов с хиральными дифосфиновыми лигандами можно применять сами по себе или на носителе, например, на полимерной матрице.
Гидрирование обычно проводится в спиртовых и/или галогенизированых растворителях при давлении от 1 до 150 атм и температуре от 60 до 150°С, предпочтительно от 5 до 15 атм и от 100 до 130°С, и наиболее предпочтительно при 10 атм и 120°С. К таким растворителям, не ограничивающим выбор, относятся, например, этанол, метанол, н-пропанол, изопропанол, н-бутанол, изобутанол, циклогексанол, дихлорометан, дихлороэтан, трихлороэтан и четыреххлористый углерод.
В более конкретном воплощении настоящего изобретения можно получить гидрированный продукт (III), практически свободный от 4S-формы, - это выражение означает, что каждый из изомеров (IIIa) и (IIIb) более чем на 95% состоит из энантиомера 4R, и суммарное содержание этого энантиомера (IIIa)+(IIIb) также ≥95%. Это воплощение наиболее предпочтительно, поскольку в нем сведены к минимуму или практически устранены потери продукта в виде нежелательного энантиомера, что усиливает простоту, избирательность и экономическую выгодность этого способа. Условия реакции практически не отличаются от тех, что описаны выше: давление от 1 до 150 атм и температура от 60 до 150°С, предпочтительно от 5 до 15 атм и от 100 до 130°С, и наиболее предпочтительно при 10 атм и 120°С. Однако для получения указанной выше энантиомерной чистоты не менее 95% растворитель необходимо выбирать из числа дихлорометана, н-пропанола, изопропанола, изобутанола, циклогексанола и их смесей, а комплексное соединение следует выбирать из числа {RuCl(п-цимол)[(S)-BINAP]}С1, RuHCl[(S)-BINAP]2/RU2Cl4[(S)-BINAP]2(NЕt3). Когда реакцию гидрирования катализирует {RuCl(п-цимол)[(S)-BINAP]}Сl в присутствии изопропанола как растворителя, дополнительным преимуществом служит то, что высокий выход указанного энантиомера сопровождается чрезвычайно хорошей скоростью превращения продукта (II) в (III) [100% за 5 ч реакции], что является еще одним важным достоинством этого способа.
Соединение (III), полученное на стадии а, используется в виде смеси цис- и транс-изомеров (IIIa+IIIb) в следующей реакции на стадии б. На этой стадии сначала требуется трансформировать ОН-группу в уходящую группу посредством реакции с соответствующим реагентом. К предпочтительным реагентам для этой реакции относятся тозилхлорид, метансульфонилхлорид, бензолсульфонилхлорид. Этот реагент добавляют к соединению (III) при температуре между -20 и +25°С (предпочтительно 0°С-5°С) в присутствии инертного растворителя (ароматического или алифатического углеводорода такого как толуол) и основного соединения (органического основания типа триэтиламина или неорганического основания или основной соли). Эта реакция ведет к образованию активированного промежуточного соединения формулы:
Этот продукт при добавлении сезамол(3,4-метилендиоксифенола) в щелочной среде превращается в обогащенное 4R-энантиомером соединение формулы (IV), преимущественно в виде трансизомера. Сезамол добавляют в виде жидкой смеси со спиртовым растворителем, предпочтительно 4-метилпентан-2-олом, в соотношении 1:1 относительно бензолсульфонилхлорида или метансульфонилхлорида или тозилхлорида. Эту смесь лучше всего прокипятить с обратным холодильником, предпочтительно в течение 2-4 часов, получая обогащенный 4R-энантиомером трансдиастереоизомер (IV) и небольшое количество цисдиастереоизомера, который удаляется путем кристаллизации.
Продукт (IV) получают преимущественно в виде транс-изомера, причем ранее достигнутое обогащение 4R-энантиомером почти полностью сохраняется, как показывают результаты анализа методом ВЭЖХ, представленные в экспериментальной части.
Стадия в заключается в элиминировании алкильной группы R, связанной с атомом азота. Реакцию проводят методами, известными в этой области. В предпочтительном воплощении продукт (IV) смешивают с фенилхлороформиатом в CH2Cl2 и проводят реакцию в течение 1-6 часов при комнатной температуре. При этом получают карбамидное производное следующей формулы:

которое затем подвергают гидролизу.
В конце стадии в получают соединение формулы (V)
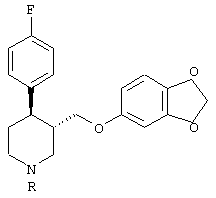
которое обогащено 4R-энантиомером (пароксетин). Небольшую примесь изомера, имеющего конфигурацию 4S, удаляют путем кристаллизации. Таким образом получают чистый пароксетин (I).
Соединения формулы (II), используемые в качестве реагентов на стадии а описанного здесь способа, можно легко получить различными методами, известными в этой области, например, при реакции 1-метил-4-фторостирола с формальдегидом и гидрохлоридом этиламина, как описано в WO-A-9636636.
С другой стороны, соединения формулы (II) можно получить, как показано на следующей схеме:
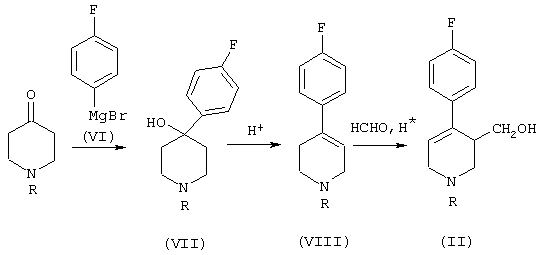
Проводят реакцию между 4-фторобромобензолом и магнием, получая 4-фторофенилмагний бромид (VI), к которому добавляют 1-алкил-4-пиперидон. При этом получают 1-алкил-4-(п-фторофенил)-4-гидроксипиперидин (VII), который дегидрируют в положении 3-4 пиперидинового кольца путем кипячения с обратным холодильником в присутствии Н2SO4, получая 1-алкил-4-(п-фторофенил)-1,2,5,6-тетрагидропиридин (VIII). Наконец, этот продукт превращают в продукт формулы (II), добавляя в ту же реакционную среду водный раствор формальдегида (реакция Принса).
В способе настоящего изобретения каталитическое гидрирование а сильно повышает процент энантиомеров, идущих на образование пароксетина (4R), а реакция б дает возможность использовать цис- и трансформы соединения (III). По этой причине количество отходов продукта в виде нежелательных изомеров очень низкое, вследствие чего повышается выход пароксетина.
Кроме того, преимущество настоящего изобретения заключается в том, что устраняется применение раздельных путей синтеза для цис- и трансизомеров, как того требуют существующие способы, известные в этой области.
Другое преимущество настоящего изобретения состоит в том, что высокий выход указанных энантиомеров достигается при умеренном давлении (5-15 атм) - в этих условиях данный способ можно легко довести до коммерческих размеров.
Еще одно преимущество настоящего изобретения заключается в том, что высокий выход энантиомеров достигается и при низких соотношениях катализатор/субстрат, а именно, от 1/200 до 1/1000. Применение малых количеств катализатора способствует снижению расходов.
Нижеследующие примеры приводятся для иллюстрации, а не для ограничения настоящего изобретения.
Сведения, подтверждающие возможность осуществления изобретения
ПРИМЕРЫ
А. Синтез 1-этил-4-(п-фторофенил)-4-гидроксипиперидина (VII) [R=Et]
0,9 М раствор 4-фторофенилмагний бромида (1,0 экв.; 0,31 моль), полученный обычным способом из магния и 4-фторобромобензола в THF, титровали и охлаждали до 0°С, затем по каплям на протяжении 45 минут добавляли прозрачный раствор коммерческого 1-этил-4-пиперидона (1 экв., 0,31 моль) в THF (60 мл). По окончании добавления поддерживали реакцию при комнатной температуре в течение 1 часа.
Реакционную смесь охлаждали до 5°С и добавляли 360 мл 20% раствора NH4Cl. После этого разделяли фазы и органическую фазу упаривали досуха. Водную фазу экстрагировали двумя порциями по 250 мл толуола. Остаток упаривали, а толуоловые фазы объединяли и промывали 420 мл 20% раствора NH4Cl.
Органическую фазу концентрировали и охлаждали. Образовавшийся осадок фильтровали, получая 40,9 г бледно-желтого твердого продукта. Анализ его методом ВЭЖХ (колонка Symmetry Shield RP8, элюент - ацетонитрил:буфер=90:10 (0,025 М КН2РO4, рН 2,5 с помощью Н3РO4), скорость 1 мл/мин, УФ-детектор при 215 нм) показал содержание 98% и чистоту >98% с выходом 58%.
Продукт реакции использовали без дальнейшей очистки в следующей реакции.
Неочищенный продукт реакции анализировали методами ГЖХ/масс-спектроскопии (МС) и 1H-ЯМР, на основании чего были установлены следующие характеристики соединения (VII): 1Н-ЯМР (CDCl3), δ (ppm): 7,55-7,44 (2Н), m, ароматический Н); 7, 01-(2Н, m, ароматический Н); 4,4-3,6 (1H, br s, OH); 3-1,75 (8H, br m, H пиперидинового кольца); 2,59 (2Н, q, J=7,3 Гц, СН2 этиловой группы); 1,18 (3Н, t, J=7,3 Гц, СН3 этиловой группы). МС, m/z (%): 223 (M+, 19), 208 (85), 190 (43), 122 (34), 109 (26), 95 (35), 94 (20), 84 (100), 71 (16), 57 (26), 56 (26).
В. Синтез 1-этил-4-(п-фторофенил)-3-гидроксиметил-1,2,3,6-тетрагидропиридина (II) [R=Et]
К раствору соединения (VII) [R=Et] (10,0 г, 44,8 ммоль) в разбавленной Н2SО4 (40 мл воды и 29,7 г Н2SO4) добавляли 4,0 г (49,3 ммоль) формальдегида (37% раствор в воде) и кипятили с обратным холодильником на магнитной мешалке в течение 6 часов. Затем эту смесь охлаждали до комнатной температуры, подщелачивали с помощью 60,9 мл 30% водного раствора NaOH и несколько раз подвергали экстракции. Объединенные органические экстракты промывали один раз водой и концентрировали.
Остаток в виде желто-оранжевого вязкого масла разбавляли 75 мл изопропанола и добавляли газообразный НС1, чтобы выпал осадок гидрохлорида соединения (II). Образовавшуюся суспензию фильтровали и промывали изопропанолом, получая твердое вещество, которое растворяли в воде и добавляли 30% NaOH до достижения рН 12. Эту смесь экстрагировали толуолом и упаривали органическую фазу. При этом получали 6,32 г соединения (II), имеющего чистоту согласно ВЭЖХ > 96% (колонка Symmetry Shield RP8, элюент-ацетонитрил:буфер=90:10 (0,025 М КН2РO4, рН 2,5 с помощью Н3РO4), скорость 1 мл/мин, УФ-детектор при 215 нм). Выход соединения (II) составил 60%.
Соединение (II) имело следующие характеристики:
т.пл. 58-60°С; 1H-ЯМР (CDCl3), δ (ppm): 7,37-7,29 (2Н, m, ароматический Н); 7,01 (2Н, t, J=8,6 Гц, ароматический Н); 6,07 (1Н, d, J=3 Гц, олефиновый Н); 5,2 (1Н, br s, ОН); 3,89 и 2,60 (7Н, m); 2,53 (2Н, q, J=7 Гц, СН2 этиловой группы); 1,16 (3Н, t, J=7 Гц, СН3). МС, m/z (%): 235 (M+, 29), 204 (100), 202 (36), 176 (21), 160 (21), 149 (25), 135 (37), 133 (39), 109 (85), 84 (17), 56 (68).
С. Синтез 4R-цис- и 4Н-транс-4- (п-фторофенил) -3-гидроксиметил-1-этилпиперидина (IIIa и IIIb) [R=Et] в присутствии {RuCl(п-цимол)[(S)-BINAP]}Сl-
Тетрагидропиридин (II) [R=Et] (35,5 г; 150,9 ммоль) гидрировали в 300 мл изопропанола при рабочем давлении 10 атм при 120°С в присутствии {RuCl(п-цимол)[(S)-BINAP]}С1-, который предварительно получали, смешивая раствор (S)-BINAP (0,378 ммоль; 0,235 г) в 21 мл CH2Cl2:MeOH 1:1 с [RuCl2 (п-цимол)]2 (0,188 ммоль; 0,115 г) и подвергая эту смесь кипячению с обратным холодильником в течение 2 часов. Реакцию контролировали, отбирая образцы на анализ методом ГЖХ (капиллярная колонка АТ-35), и останавливали через 2 часа при 120°С. Эту смесь охлаждали до комнатной температуры, фильтровали на целите (Celite) и концентрировали при пониженном давлении, получая 33,2 г продукта (выход 88%).
Неочищенный продукт анализировали методом ВЭЖХ (Chiradex β-cyclodextrin Merck, элюент - метанол:буфер = 15:85 (1,38 г/л NaH2PO4, pH 6 с помощью Na2HPO4), скорость 1 мл/мин, УФ-детектор при 215 нм) и определяли содержание 4R-энантиомеров (энантиомерную чистоту, эн.ч.) в цис- и транс-диастереоизомерах. Были получены следующие результаты:
транс, эн.ч. ≥ 99%; цис, эн.ч. ≥ 99%.
Соотношение цис/транс = 55:45, общая эн.ч. ≥ 99%.
Для дальнейшего анализа цис- и трансдиастереоизомеры разделяли и исследовали после очистки на силикагеле методом ЖХ при среднем давлении.
4R-цисдиастереоизомер: т.пл. 90-92°С; 1H-ЯМР (СDСl3), δ (ppm): 7,44-7,20 (2Н, m, ароматический Н); 7,15-6,90 (2Н, m, ароматический Н); 5,8-4,5 (1Н, br s, ОН); 3,9-1,6 (10Н, m, пиперидиновое кольцо + гексациклический СН2); 2,25 (2Н, q, J=7,2 Гц, СН2 этиловой группы); 1,13 (3Н, t, J=7,2 Гц, СН3 этиловой группы). МС, m/z (%): 237 (M+, 23), 222 (43), 206 (17), 133 (11), 114 (29), 109 (20), 84 (16), 72 (17), 58 (100).
4R-трансдиастереоизомер: т.пл. 90-92°С; 1H-ЯМР (СDС13), δ (ppm): 7,3-7,1 (2Н, m, ароматический Н); 7,1-6,85 (2Н, m, ароматический Н); 3,5-3,0 (4Н, m, пиперидиновое кольцо); 2,7-2,2 (4Н, m, пиперидиновое кольцо + гексациклический СН2); 2,15-1,65 (5Н, m, пиперидиновое кольцо + СН2 этиловой группы); 1,14 (3Н, t, J=7,2 гц, СН3 этиловой группы). МС, m/z (%): 237 (М+, 34), 222 (76), 206 (16), 133 (14), 114 (34), 109 (27), 84 (21), 72 (22), 58 (100).
D. Синтез 4R-цис- и 4R-транс-4-(п-фторофенил)-3-гидроксиметил-1-этилпиперидина (IIIa и IIIb) [R=Et] в присутствии Ru2Cl4[(S)-BINAP]2(NEt3)
Тетрагидропиридин (II) [R=Et] (5,0 г; 21,4 ммоль) гидрировали в 40 мл изопропанола при рабочем давлении 10 атм при 120°С в присутствии RugCl4[(S)-BINAP]2(NEt3), который предварительно получали, добавляя (S)-BINAP и Еt3N (0,032 мл) в суспензию [RuCl2(COD)]n (14,9 мг; 0,0531 ммоль) в 2 мл толуола, подвергая эту смесь кипячению с обратным холодильником в течение 12 часов и упаривая досуха. Реакцию останавливали через 5 часов при 120°С и смесь охлаждали до комнатной температуры. После фильтрования на целите (Celite) и упаривания при пониженном давлении получали 4,3 г продукта (выход 84%).
Содержание энантиомеров в диастереоизомерах (IIIa) и (IIIb) определяли методом ВЭЖХ (Chiradex β-cyclodextrin Merck, элюент - метанол: буфер = 15:85 (1,38 г/л NaH2PO4, pH 6 с помощью Na2HPO4), скорость 1 мл/мин, УФ-детектор при 215 нм). Были получены следующие результаты:
транс, эн.ч. ≥ 99%; цис, эн.ч. ≥ 98%.
Соотношение цис/транс = 50:50, общая эн.ч. ≥ 99%.
Е. Синтез 4R-циc- и 4R-транс-4-(п-фторофенил)-3-гидроксиметил-1-этилпиперидина (IIIa и IIIb) [R=Et] в присутствии RuHCl[(S)-BINAP]2
Тетрагидропиридин (II) [R=Et] (5,0 г; 21,4 ммоль) гидрировали в 40 мл изопропанола при рабочем давлении 10 атм при 120°С в присутствии RuHCl[(S)-BINAP]2, который предварительно получали, добавляя (S)-BINAP (74,0 мг; 0,120 ммоль) и Et3N (0,017 мл) в раствор [RuCl2(COD)]n (14,9 мг; 0,0531 ммоль) в 3 мл EtOH, подвергая эту смесь кипячению с обратным холодильником в течение 6 часов и упаривая досуха. Реакцию останавливали через 5 часов при 120°С и смесь охлаждали до комнатной температуры. После фильтрования на целите (Celite) и упаривания при пониженном давлении получили 4,2 г продукта (выход 83%).
Содержание энантиомеров в диастереоизомерах (IIIa) и (IIIb) определяли методом ВЭЖХ (Chiradex β-cyclodextrin Merck, элюент - метанол: буфер = 15:85 (1,38 г/л NaH2PO4, pH 6 с помощью Na2HPO4), скорость 1 мл/мин, УФ-детектор при 215 нм). Были получены следующие результаты:
транс, эн.ч. ≥ 99%; цис, эн.ч. ≥ 99%.
Соотношение цис/транс=50:50, общая эн.ч. ≥ 99%.
F. Синтез 4R-цис- и 4R-транс-4-(п-фторофенил)-3-гидроксиметил-1-этилпиперидина (IIIa и IIIb) [R=Et] в присутствии Ru[(S)-BINAP](ОАс)2
Тетрагидропиридин (II) [R=Et] (5,0 г; 21,4 ммоль) гидрировали в 40 мл изопропанола при рабочем давлении 10 атм при 120°С в присутствии Ru[(S)-BINAP](ОАс)2 (48,0 мг; 0,057 ммоль), который предварительно получали методом, описанным в Inorg. Chem. 27, 1988, 566-569, исходя из [RuCl2(COD)]n и (S)-BINAP. Реакцию останавливали через 18 часов при 120°С и смесь охлаждали до комнатной температуры. После фильтрования на целите (Celite) и упаривания при пониженном давлении получали 3,6 г продукта (выход - 71%).
Содержание энантиомеров в диастереоизомерах (IIIa) и (IIIb) определяли методом ВЭЖХ (Chiradex β-cyclodextrin Merck, элюент - метанол: буфер = 15:85 (1,38 г/л NaH2PO4, pH 6 с помощью Na2HPO4), скорость 1 мл/мин, УФ-детектор при 215 нм). Были получены следующие результаты:
транс, эн.ч. ≥ 43%; цис, эн.ч. ≥ 94%.
Соотношение цис/транс = 10:90, общая эн.ч. ≥ 89%.
G. Синтез 4R-цис- и 4R-транс-4-(п-фторофенил)-3-гидроксиметил-1-этилпиперидина (IIIa и IIIb) [R=Et] в присутствии Ru[(S)-BINAP] (СF3СO2)2
Тетрагидропиридин (II) [R=Et] (5,0 г; 21,4 ммоль) гидрировали в 40 мл изопропанола при рабочем давлении 10 атм при 120°С в присутствии Ru[(S)-BINAP] (СF3СO2) 2 (50,0 мг; 0,031 ммоль), который предварительно получали, растворяя Ru[(S)-BINAP](OAc)2 в CH2Cl2, добавляя трифторуксусную кислоту, перемешивая в течение 12 часов и упаривая, а затем растворяя в толуоле и гексане и подвергая кристаллизации. Реакцию останавливали через 20 часов и смесь охлаждали до комнатной температуры. После фильтрования на целите (Celite) и упаривания при пониженном давлении получили 3,7 г продукта (выход - 73%).
Содержание энантиомеров в диастереоизомерах (IIIa) и (IIIb) определяли методом ВЭЖХ (Chiradex β-cyclodextrin Merck, элюент - метанол: буфер = 15:85 (1,38 г/л NaH2PO4, pH 6 с помощью Na2HPO4), скорость 1 мл/мин, УФ-детектор при 215 нм). Были получены следующие результаты:
транс, эн.ч. ≥ 69%; цис, эн.ч. ≥ 93%.
Соотношение цис/транс = 34:66, общая эн.ч. ≥ 85%.
Полученные при энантиоселективном гидрировании результаты суммированы в таблице 1.
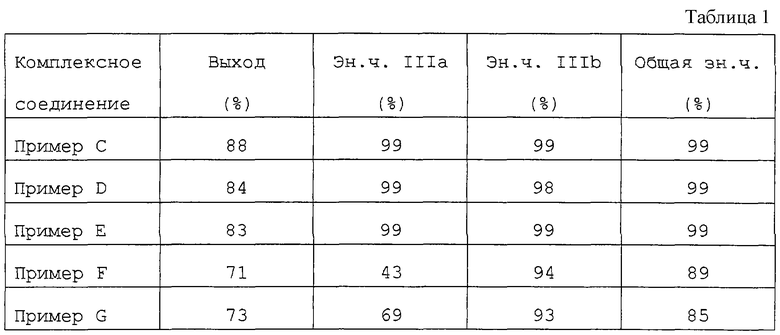
Из таблицы видно, что общая эн.ч. никогда не была меньше 85% во всех опытах, в частности в примерах С, D, Е, когда применялись предпочтительные лиганды и растворители, общие эн.ч. и удельные эн.ч. по IIIa и IIIb составили 98-99%.
Пример С был повторен с другими спиртовыми или галогенизированными растворителями, чтобы проверить влияние растворителей на энантиоселективность. Гидрирование проводилось при температуре 120°С и реакцию останавливали через 5 часов. Результаты суммированы в следующей таблице 2.
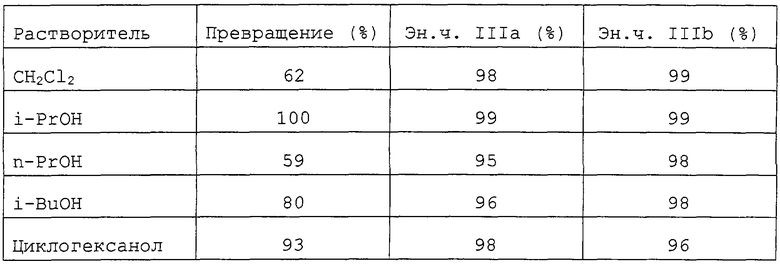
Как показывает таблица, все растворители давали величину эн.ч. не менее 95%. В случае i-PrOH наблюдалась очень высокая скорость превращения (100% за 5 часов) наряду со значением эн.ч. в 99%.
Н. Синтез 4R-цис- и 4R-транс-4-(п-фторофенил)-3-гидроксиметил-1-этил-3-(3,4-метилендиоксифеноксиметил)пиперидина (IV) [R=Et]
240 мл растворенной в толуоле смеси энантиомеров (IIIa) и (IIIb) (40/0 г; 169 ммоль), полученных согласно предыдущим примерам, смешивали с триэтиламином (1,7 экв; 286 ммоль; 21,0 мл) при комнатной температуре и с перемешиванием, после чего по каплям на протяжении 1 часа добавляли раствор метансульфонилхлорида (1,2 экв; 202 ммоль; 15,6 мл) в толуоле (40 мл).
Смесь перемешивали при 25°С в течение 3 часов и фильтровали. Жидкую фазу промывали водой и образовавшуюся органическую фазу концентрировали при пониженном давлении. Остаток (47,2 г) растворяли в толуоле (240 мл) и обрабатывали сначала раствором сезамола (1,0 экв относительно смеси (IIIa) и (IIIb); 169 ммоль; 23,3 г) в 4-метилпентан-2-оле (100 мл), а затем водным раствором 10 М NaOH (1,2 экв относительно смеси (IIIa) и (IIIb); 201 ммоль; 20,3 мл). Образовалась гетерогенная смесь, которую кипятили с обратным холодильником в течение 3 часов. Затем реакционную смесь промывали 3 раза водой до нейтральной реакции и отделяли органическую фазу. Водную фазу опять экстрагировали толуолом. Органические фазы объединяли и концентрировали при пониженном давлении. Анализ методом ГЖХ (капиллярная колонка SE-30) вязкого масляного остатка (55,0 г) показал присутствие цис-и трансдиастереоизомеров (IV) в соотношении 10:90.
Остаток растворяли в изопропаноле и добавляли газообразный НС1. Выпадал осадок, состоящий только из 4R-трансдиастереоизомера (гидрохлорида) с выходом 74% (125 ммоль; 49, 1 г).
Гидрохлорид затем обратно превращали в свободное основание путем растворения в воде, подщелачивания с помощью 30% NaOH и экстракции водной фазы толуолом. Органическую фазу упаривали, получая 4R-транс-диастереоизомер в виде свободного основания (44,1 г). Выход - 99%.
Энантиомерная чистота этого диастереоизомера, определенная методом ВЭЖХ (Chiradex β-cyclodextrin Merck, элюент - метанол:буфер=40:60 (1% триэтиламин, рН 4,1 с помощью АсОН), скорость 1 мл/мин, УФ-детектор при 290 нм), составила > 99%.
ЯМР-анализ проводили на спектрометре АМХ-600 фирмы Bruker, включая запись 1H и 13С спектров, 1Н-1Н COSY, корреляцию гетероядерного смещения, корреляцию гетероядерного смещения дальнего порядка и тесты NOESY, что позволило правильно установить стереохимию двух продуктов реакции. Свободное основание 4R-транс-диастереоизомера - бледно-желтый сироп - имело следующие спектральные характеристики: 1Н-ЯМР (СDСl3), δ (ppm): 7,16 (2Н, m, Н фторофенила); 6,96 (2Н, m, Н фторофенила); 6,62 (1Н, d, J=8,8 Гц, Н сезамола); 6,34 (1Н, d, J=2,4 Гц, Н сезамола); 6,13 (1Н, dd, J=8,8 и 2,4 Гц, Н сезамола); 5,87 (2Н, s, O-CH2-O); 3,58 (1Н, dd, J=9,5 и 2,8 Гц, СН2-O); 3,45 (1Н, dd, J=9,5 и 6,8 Гц, СН2-O); 3,31 (1Н, m, CH-N); 2,45 (3Н, m, СН2 этиловой группы + СН пиперидина); 2,20 (1Н, m, СН пиперидина); 2,1-1,7 (4Н, m, СН пиперидина); 1,17 (3Н, t, J=7,2 гц, СН3). МС, m/z (%): 357 {М+,2), 220 (13), 205 (16), 137 (7), 109 (14), 98 (10), 82 (12), 72 (100), 58 (23).
I. Синтез пароксетина гидрохлорида (I)
Раствор 4R-транс-4-(п-фторофенил)-3-гидроксиметил-1-этил-3-(3,4-метилендиоксифеноксиметил)пиперидина (IV) [R=Et] (26,5 г; 74,1 ммоль) в дихлорометане (135 мл) охлаждали до 0°С и добавляли по каплям на протяжении 15 минут раствор фенилхлороформиата (22,6 г; 144 ммоль) в дихлорометане (22 мл). Прозрачный желтый раствор держали при комнатной температуре в течение 3 часов, а затем промывали 150 мл 1М NaOH и двумя порциями по 150 мл 6М НС1. Органическую фазу упаривали досуха и растворяли в толуоле (190 мл). Затем эту смесь фильтровали, добавляли 19,2 г (343 ммоль) твердого КОН и кипятили с обратным холодильником в течение 2 часов. После этого смесь охлаждали до комнатной температуры и добавляли 150 мл воды. Органическую фазу отделяли, а водную фазу экстрагировали двумя порциями по 100 мл воды и упаривали досуха.
Остаток растворяли в изопропаноле (85 мл) и добавляли 37% НС1. Выпавший осадок пароксетина гидрохлорида отфильтровывали, промывали и высушивали. Было получено 21,9 г (59,3 ммоль) пароксетина гидрохлорида. Этот продукт при анализе методом ВЭЖХ (колонка Symmetry Shield RP8, элюент - ацетонитрил:буфер=70:30 (0,025 М КН2РO4, рН 2,5 с помощью Н3РО4), скорость 1 мл/мин, УФ-детектор при 290 нм) показал содержание 99% и чистоту 99,9% с выходом 80%. Результаты спектроскопии соответствовали данным литературы.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРРОЛО[2,3-d]ПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СИНТЕЗ | 2006 |
|
RU2384583C2 |
| СПОСОБ ПОЛУЧЕНИЯ ДИАРИЛЦИКЛОАЛКИЛПРОИЗВОДНЫХ | 2005 |
|
RU2414459C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭНАНТИОМЕРНО И ДИАСТЕРЕОМЕРНО ОБОГАЩЕННЫХ ЦИКЛОБУТАНАМИНОВ И -АМИДОВ | 2018 |
|
RU2793738C2 |
| НОВЫЙ СПОСОБ ПОЛУЧЕНИЯ ФЕНИЛАМИДА 5-(4-ФТОРФЕНИЛ)-1-[2-(2R, 4R)-4-ГИДРОКСИ-6-ОКСОТЕТРАГИДРОПИРАН-2-ИЛ)ЭТИЛ]-2-ИЗОПРОПИЛ-4- ФЕНИЛ-1-H-ПИ РРОЛ-3- КАРБОНОВОЙ КИСЛОТЫ | 2001 |
|
RU2244714C1 |
| СПОСОБ ПОЛУЧЕНИЯ (S)-ЭНАНТИОМЕРОВ | 1994 |
|
RU2114103C1 |
| СПОСОБ ПОЛУЧЕНИЯ (S)-3-(АМИНОМЕТИЛ)-5-МЕТИЛГЕКСАНОВОЙ КИСЛОТЫ | 2015 |
|
RU2643373C2 |
| СПОСОБ ГИДРОГЕНИЗАЦИИ ИМИНОВ И СПОСОБ ПОЛУЧЕНИЯ АМИНА | 1995 |
|
RU2150464C1 |
| ПРОИЗВОДНЫЕ 2-ОКСО-1-ПИРРОЛИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2001 |
|
RU2292336C2 |
| СПОСОБ ЭНАНТИОСЕЛЕКТИВНОГО ГИДРИРОВАНИЯ АМИНОСПИРТОВ | 2003 |
|
RU2340594C2 |
| ПРОИЗВОДНЫЕ 2-ОКСО-1-ПИРРОЛИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ | 2001 |
|
RU2355680C2 |

Изобретение относится к способу получения пароксетина, включающий следующие стадии: а) гидрогенизация соединения формулы (II), в котором R означает группу, выбранную из (C1-C5) алкила, (С1-С5)карбоксиалкила, необязательно замещенного фенила и необязательно замещенного бензила, причем реакция гидрогенизации катализируется комплексным соединением переходного металла с хиральными дифосфиновыми лигандами, где переходный металл выбран из группы, состоящей из рутения, иридия и родия, и процесс гидрирования проводят при давлении от 1 до 150 атм, в спиртовом или галогенизированном растворителе или их смеси при температуре от 60 до 150°С, что приводит к образованию обогащенного 4R-энантиомером соединения формулы (III):
б) реакция соединения формулы (III) с реагентом, способным преобразовать ОН-группу в уходящую группу, выбранным из ряда тозилхлорида, метансульфонилхлорида, бензолсульфонилхлорида, который добавляют к соединению (III) при температуре между -20 и +25°С в присутствии инертного растворителя и основного соединения, с последующим нуклеофильным замещением ее сезамол(3,4-метилендиоксифенолом), добавлением его в виде смеси со спиртовым растворителем в присутствии основания, и образовавшуюся смесь нагревают в течение 2-4 часов с получением обогащенного 4R-энантиомером производного формулы (IV):
в) N-деалкилирование соединения (IV) с образованием пароксетина. Технический результат – этот способ обладает высокой стереоспецифичностью и приводит к образованию промежуточных соединений, обогащенных требуемыми изомерными компонентами, которые превращаются с количественным выходом. 1 н. и 7 з.п.ф-лы, 8 ил., 2 табл.
б) реакция соединения формулы (III) с реагентом, способным преобразовать ОН-группу в уходящую группу, выбранным из ряда тозилхлорида, метансульфонилхлорида, бензолсульфонилхлорида, который добавляют к соединению (III) при температуре между -20°С и +25°С в присутствии инертного растворителя и основного соединения с последующим нуклеофильным замещением ее сезамол(3,4-метилендиоксифенолом) при добавлении его в виде смеси со спиртовым растворителем в присутствии основания и нагревают образовавшуюся смесь в течение 2-4 ч с получением обогащенного 4R-энантиомером производного формулы (IV)
в) N-деалкилирование соединения (IV) с образованием пароксетина (I)
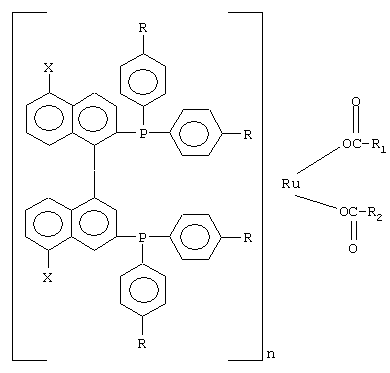
где Х представляет собой водород, аминогруппу, ацетиламиногруппу или сульфогруппу;
R представляет собой водород или (C1-С4)алкильную группу;
R1 и R2 каждый представляет собой (C1-С9)алкильную группу, галогенированную (C1-C4)алкильную группу; фенильную группу, фенильную группу, замещенную (C1-C4)алкильной группой, (C1-C4)-α-аминоалкильную группу или (С7-С10)-α-аминофенилалкильную группу, или R1 и R2 вместе образуют (C1-C4)алкиленовую группу;
n означает 1 или 2;
или соединений формулы
{RuX(п-цимол)[BINAP]}Х-,
где Х означает атом галогена,
или соединений формул
RuHCl [BINAP]2,
Ru2Cl4[BINAP]2 (NEt3),
Ru[BINAP](OAc)2,
Ru[BINAP](СF3СО2)2
или соединений, соответствующих одной из следующих формул:
| АНГИДРАТ ПАРОКСЕТИН ГИДРОХЛОРИДА, СОЛЬВАТЫ ПАРОКСЕТИН ГИДРОХЛОРИДА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1996 |
|
RU2125052C1 |
| WO 9636636 A, 21.11.1996 | |||
| WO 9322284 A, 11.11.1993 | |||
| WO 9209552 A, 11.06.1996. | |||

