Настоящее изобретение относится к способу получения энантиомерно и диастереомерно обогащенных циклобутанаминов и -амидов путем осуществления реакции (а) циклопропилкарбонитрила с получением циклопропилкарбальдегида (b) с последующим осуществлением реакции с получением циклобутанона или (d') с последующим осуществлением реакции с получением енамида; (с) с последующим осуществлением реакции с получением энантиомерно и диастереомерно обогащенного циклобутанамина или (d) с последующим осуществлением реакции с получением енамида, и (е) с получением энантиомерно и диастереомерно обогащенного циклобутанамида с получением (f) энантиомерно и диастереомерно обогащенного циклобутанамина, и (g) с последующим осуществлением реакции с получением энантиомерно и диастереомерно обогащенного циклобутанамида.
Настоящее изобретение относится к способу получения энантиомерно и диастереомерно обогащенных циклобутанаминов и -амидов в соответствии со следующей схемой 1.
Способ получения соединений формулы (VII) в соответствии со схемой 1 был описан в WO 2013/143811 и WO 2015/003951. Настоящее изобретение относится к улучшенным способам получения соединений формулы (VII), в частности, к стадиям способа (а), (b), (с), (d), (d'), (е), (f) и (g).
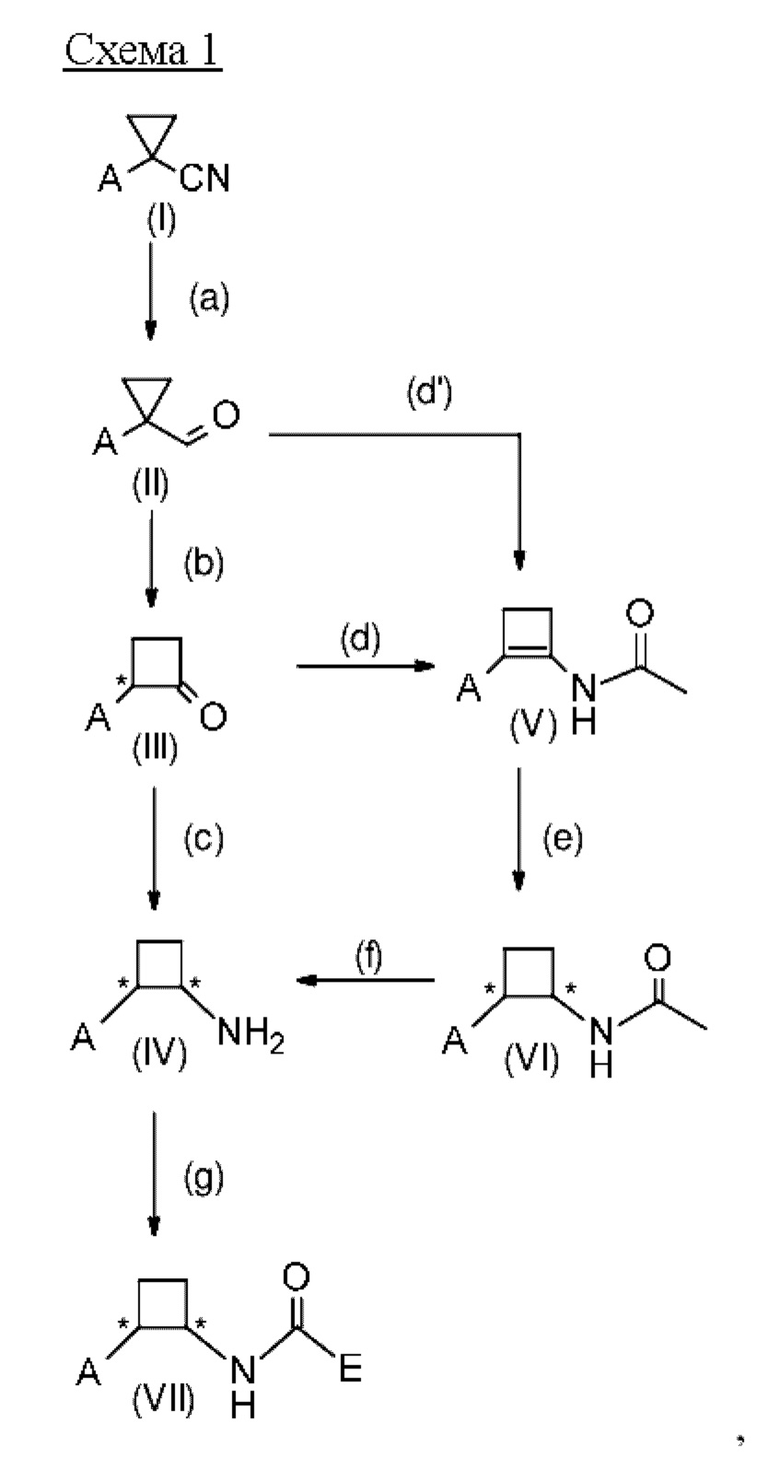
где А выбран из арила, гетероарила, С1-С6алкила, С2-С6алкенила, С2-С6алкинила и С3-С7циклоалкила, где арил, гетероарил, С1-С6алкил, С2-С6алкенил, С2-С6алкинил и С3-С7циклоалкил являются незамещенными или замещенными одним или несколькими заместителями, независимо выбранными из галогена, циано, С1-С6алкила, С1-С6галогеналкила, С1-С6алкокси, С1-С6галогеналкокси, С2-С6алкенила, С2-С6алкинила, С1-С6алкилсульфанила, С1-С6галогеналкилсульфанила, С1-С6алкилсульфинила, С1-С6галогеналкилсульфинила, С1-С6алкилсульфонила, С1-С6галогеналкилсульфонила, С2-С6галогеналкенила и С2-С6галогеналкинила; и где Е выбран из арила, гетероарила, водорода, С1-С6алкила, С2-С6алкенила, С2-С6алкинила и С3-С7циклоалкила, где арил, гетероарил, С1-С6алкил, С2-С6алкенил, С2-С6алкинил и С3-С7циклоалкил являются незамещенными или замещенными одним или несколькими заместителями, независимо выбранными из галогена, циано, С1-С6алкила, С1-С6галогеналкила, С1-С6алкокси, C1-С6галогеналкокси, С2-С6алкенила, С2-С6алкинила, С1-С6алкилсульфанила, С1-С6галогеналкилсульфанила, С1-С6алкилсульфинила, С1-С6галогеналкилсульфинила, С1-С6алкилсульфонила, С1-С6галогеналкилсульфонила, С2-С6галогеналкенила и С2-С6галогеналкинила, и где звездочка * обозначает стереоцентр.
Предпочтительно А и Е на схеме 1 выбраны из арила и гетероарила, где арил и гетероарил являются незамещенными или замещенными одним или несколькими заместителями, независимо выбранными из галогена, циано, С1-С6алкила, С1-С6галогеналкила, C1-С6алкокси и С1-С6галогеналкокси. Более предпочтительно, А представляет собой фенил, и Е представляет собой гетероарил, причем фенил и гетероарил являются незамещенными или замещенными одним или несколькими заместителями, независимо выбранными из галогена, циано, С1-С6алкила, С1-С6галогеналкила, C1-С6алкокси и С1-С6галогеналкокси. Еще более предпочтительно, А представляет собой фенил, замещенный одним или двумя галогенами, и Е представляет собой пиридил, замещенный одним С1-С6галогеналкилом, в частности, А представляет собой фенил, замещенный двумя атомами хлора, и Е представляет собой 3-пиридил, замещенный трифторметилом, более конкретно, А представляет собой 2,4-дихлорфенил, и Е представляет собой (2-трифторметил)-пирид-3-ил.
В первом аспекте настоящего изобретения предусмотрен способ (а) получения соединения формулы (II), предусматривающий восстановление нитрильного фрагмента соединения формулы (I) до альдегида, причем восстановление нитрильного фрагмента соединения формулы (I) осуществляют путем частичного гидрирования до соответствующего промежуточного имина с использованием Н2 и катализатора на основе металла или гидрида металла с последующим последовательным гидролизом с получением соединения формулы (II), где А определен выше на схеме 1. В частности, катализатор на основе металла выбран из Pd/C (палладия на угле) (a1) и никеля Ренея (Ra/Ni) (а2), гидрида металла, выбранного из DIBAL (гидрида диизобутилалюминия) (а3) и REDAL (бис(2-метоксиэтокси)алюминийгидрида натрия) (а4).
Типичные параметры реакции для реакции (а) являются следующими.
(a1) Преобразование соединения формулы (I) в соединение формулы (II) с применением Pd/C
Преобразование соединения формулы (I) в соединение формулы (II), как правило, осуществляют в присутствии Н2, катализатора, например, палладия на подходящих подложке или материале-носителе, необязательно растворителя, такого как спирт или углеводород, в частности, МеОН (метанол), толуол или метилциклогексан, органической или неорганической кислоты, в частности, HCl или трифторуксусной кислоты и воды.
Предпочтительные параметры реакции для способа (a1) в соответствии со схемой 1 являются следующими.
Реакцию (a1) предпочтительно осуществляют при температуре от 10°С до 150°С, более предпочтительно от 0°С до 50°С, еще более предпочтительно при приблизительно 5°С.
Давление газообразного водорода в реакционном сосуде может составлять от 0,1 до 100 бар, предпочтительно от 0,1 до 20 бар, более предпочтительно от 0,5 до 3 бар.
В реакционную смесь загружают предпочтительно 1-35 моль эквивалентов воды относительно соединения формулы (I), более предпочтительно от 2 до 16 моль эквивалентов и еще более предпочтительно от 3 до 6 моль эквивалентов.
В реакционную смесь загружают предпочтительно 1-50 моль эквивалентов кислоты относительно соединения формулы (I), более предпочтительно, от 2-30 моль эквивалентов и еще более предпочтительно от 4-20 моль эквивалентов.
Палладий на подходящих подложке или материале-носителе. В частности, добавляют от 0,001 до 1,0 моль эквивалента Pd/C относительно соединения формулы (I). Более конкретно, добавляют от 0,001 до 0,01 моль эквивалента Pd/C относительно соединения формулы (I).
Предпочтительные условия являются такими, чтобы концентрация соединения формулы (I) составляла 1-30% по весу от всей реакционной смеси.
(а2) Преобразование соединения формулы (I) в соединение формулы (II) с применением никеля Ренея (Ra/Ni)
Преобразование соединения формулы (I) в соединение формулы (II), как правило, достигается в присутствии Н2, катализатора на основе никеля Ренея, органической кислоты, в частности, уксусной кислоты или трифторуксусной кислоты, необязательно добавки, деактивирующей катализатор, в частности, пиридина или хинолина, и воды.
Предпочтительные параметры реакции для способа (а2) в соответствии со схемой 1 являются следующими.
Реакцию (а2) предпочтительно осуществляют при температуре от -10°С до 150°С, более предпочтительно от 0°С до 50°С, еще более предпочтительно при приблизительно 25°С.
Давление газообразного водорода в реакционном сосуде может составлять от 0,1 до 100 бар, предпочтительно от 0,1 до 20 бар, более предпочтительно от 0,5 до 3 бар.
В реакционную смесь загружают предпочтительно 1-60 моль эквивалентов воды относительно соединения формулы (I), более предпочтительно от 2 до 40 моль эквивалентов и еще более предпочтительно от 5 до 12 моль эквивалентов.
В реакционную смесь загружают предпочтительно 1-66 моль эквивалентов кислоты относительно соединения формулы (I).
В реакционную смесь необязательно загружают 0,1-25 моль эквивалентов добавки, деактивирующей катализатор, относительно соединения формулы (I).
Ra/Ni. В частности, добавляют от 0,01 до 1,0 моль эквивалента катализатора на основе Ra/Ni относительно соединения формулы (I), предпочтительно 0,1-0,5 мольных эквивалента.
Предпочтительные условия являются такими, чтобы концентрация соединения формулы (I) составляла 1-30% по весу реакционной смеси.
(а3) Преобразование соединения формулы (I) в соединение формулы (II) с применением DIBAL
Преобразование соединения формулы (I) в соединение формулы (II), как правило, достигается в присутствии DIBAL и органического растворителя с последующей кислотной обработкой.
Подходящий растворитель для реакции (а3) может быть выбран из следующих классов растворителей: простые эфиры или хлорированные и отличные от хлорированных углеводороды, или ароматические вещества. Подходящие растворители включают без ограничения дихлорметан, тетрагидрофуран, гексаны, гептан, ксилолы, толуол и циклогексан. Предпочтительными растворителями являются толуол и о-ксилол.
Предпочтительные параметры реакции для способа (а3) в соответствии со схемой 1 являются следующими.
Реакцию (а3) предпочтительно осуществляют при температуре от -20°С до 100°С, более предпочтительно от -10°С до 20°С, еще более предпочтительно при приблизительно 0°С.
В реакционную смесь загружают предпочтительно 1-2 моль эквивалентов DIBAL относительно соединения формулы (I).
Предпочтительные условия реакции являются такими, чтобы концентрация соединения формулы (I) составляла 1-30% по весу реакционной смеси.
(а4) Преобразование соединения формулы (I) в соединение формулы (II) с применением REDAL
Преобразование соединения формулы (I) в соединение формулы (II) достигается в присутствии REDAL и органического растворителя, предпочтительно с последующей кислотной обработкой.
Подходящий растворитель может быть выбран из следующих классов растворителей: простые эфиры или отличные от хлорированных углеводороды, или ароматические вещества. Подходящие растворители включают без ограничения THF (тетрагидрофуран), ксилолы и толуол. Предпочтительными растворителями являются толуол и THF.
Предпочтительные параметры реакции для способа (а4) в соответствии со схемой 1 являются следующими.
Реакцию (а4) предпочтительно осуществляют при температуре от 20°С до 100°С, более предпочтительно от -10°С до 25°С, еще более предпочтительно от 0 до 25°С.
В реакционную смесь загружают предпочтительно 0,5-1,2 моль эквивалента REDAL относительно соединения формулы (I).
Предпочтительные условия являются такими, чтобы концентрация соединения формулы (I) составляла 1-35% по весу реакционной смеси.
Следует понимать, что любой из предпочтительных признаков для каждого из способов (a1), (а2), (а3) и (а4) может быть объединен с одним или несколькими другими предпочтительными признаками способов (a1), (а2), (а3) и (а4) соответственно.
Во втором аспекте настоящего изобретения предусмотрен способ (b) получения соединения формулы (III), предусматривающий осуществление реакции соединения формулы (II) в присутствии подходящей кислоты Льюиса, и где А определен выше на схеме 1. В частности, кислота Льюиса выбрана из AlCl3 и GaCl3, предпочтительно AlCl3. Предпочтительно способ (b) осуществляют в инертных условиях, таких как в атмосфере газообразного азота, и в подходящем растворителе.
Специалисту в данной области техники будет понятно, что реакцию (b) в соответствии со схемой 1 осуществляют в подходящем растворителе. Подходящий растворитель может быть выбран из следующих классов растворителей: хлорированные углеводороды или хлорированные ароматические вещества. Подходящие растворители включают без ограничения дихлорметан, хлорбензол или дихлорбензол. Предпочтительные растворители представляют собой хлорированные ароматические растворители, в частности, дихлорбензол и хлорбензол.
Предпочтительные параметры реакции для способа (b) в соответствии со схемой 1 являются следующими.
Реакцию (b) предпочтительно осуществляют при температуре от 0°С до 100°С, более предпочтительно от 20°С до 80°С, еще более предпочтительно от 50 до 60°С.
В реакционную смесь загружают предпочтительно 1,0-1,5 моль эквивалента AlCl3 или GaCl3 относительно соединения формулы (II).
Концентрация соединения формулы (II) составляет предпочтительно 1-40% по весу реакционной смеси.
Было показано, что условия способа для способа (b) являются особенно эффективными, то есть они приводят к более высоким значениям выхода соединений формулы (III).
Следует понимать, что любой из предпочтительных признаков для способа (b) может быть объединен с одним или несколькими другими предпочтительными признаками для способа (b).
В третьем аспекте настоящего изобретения предусмотрен способ (с) получения энантиомерно и диастереомерно обогащенного циклобутанамина, предусматривающий осуществление реакции соединения формулы (III) с аммониевой солью и Н2 в присутствии хирального катализатора на основе переходного металла, и где А определен выше на схеме 1. В реакционную смесь необязательно добавляют одну или несколько добавок.
Хиральный катализатор на основе переходного металла содержит переходный металл, выбранный из Ru, Rh, Ir и Pd, и хиральный лиганд.
Хиральные лиганды известны из уровня техники и могут применяться в настоящем изобретении, примеры приведены в "Catalytic asymmetric synthesis", Iwao Ojima, third Edition, Wiley-VCH 2010, и в литературных источниках, приведенных в нем; типичные классы, которые известны специалисту в данной области техники, включают без ограничения TADDOL (α,α,α',α'-тетраарил-2,2-дизамещенный 1,3-диоксолан-4,5-диметанол), DUPHOS (фосфолановый лиганд), BOX (бис(оксазолиновый) лиганд), BINAP (2,2'-бис(дифенилфосфино)-1,1'-бинафтил), BINOL (1,1'-би-2-нафтол), DIOP (2,3-о-изопропилиден-2,3-дигидрокси-1,4-бис(дифенилфосфино)бутан), WALPHOS, TANIAPHOS, MANDYPHOS, CHENPHOS, JOSIPHOS, BIPHEMP, MeO-BIPHEP, SEGPHOS, CHIRAPHOS, PPHOS, TUNEPHOS и SYNPHOS.
Предпочтительно хиральный лиганд представляет собой лиганд, содержащий бидентатный фосфор, общей формулы (VIII):
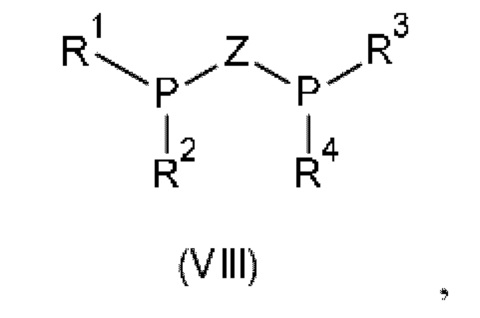
где Z представляет собой линкерную группу, и R1, R2, R3 и R4 независимо выбраны из арила, гетероарила, С1-С6алкила и С1-С6циклоалкила, каждый из которых является незамещенным или замещенным. Типичные заместители для R1, R2, R3 и R4 выбраны из С1-С6алкила, С1-С6алкокси, С1-С6галогеналкила и галогена. Предпочтительно линкерная группа Z выбрана из (R или S)-1,1'-бинафтила, (R или S)-4,4'-би-1,3-бензодиоксола, (R или S)-2,2',6,6'-тетраметокси-3,3'-бипиридина, (R или S)-6,6'-диметокси-1,1'-бифенила, (R или S)-4,4',6,6'-тетраметокси-1,1'-бифенила, 2,2'-бис-[(R или S)-сх-(диметиламино)бензил]ферроцена, ферроценилметила, ферроцена, бензола и этила. Более предпочтительно, бидентатный лиганд формулы (VIII) выбран из классов лигандов В IN АР, WALPHOS, JOSIPHOS, TANIAPHOS, MANDYPHOS, CHENPHOS, MeO-BIPHEP, PPHOS, DUPHOS, TUNEPHOS, SYNPHOS и SEPGPHOS. Подходящие хиральные лиганды в соответствии с настоящим изобретением включают без ограничения
(R)-2,2'-бис(дифенилфосфино)-1,1'-бинафтил,
(S)-2,2'-бис(дифенилфосфино)-1,1'-бинафтил,
(R)-2,2'-бис(ди-п-толилфосфино)-1,1'-бинафтил,
(S)-2,2'-бис(ди-п-толилфосфино)-1,1'-бинафтил,
(R)-2,2'-бис[ди(3,5-ксилил)фосфино]-1,1'-бинафтил,
(S)-2,2'-бис[ди(3,5-ксилил)фосфино]-1,1'-бинафтил,
(R)-5,5'-бис(дифенилфосфино)-4,4'-би-1,3-бензодиоксол,
(S)-5,5'-бис(дифенилфосфино)-4,4'-би-1,3-бензодиоксол,
(R)-5,5'-бис(ди[3,5-ксилил]фосфино)-4,4'-би-1,3-бензодиоксол,
(S)-5,5'-бис(ди[3,5-ксилил]фосфино)-4,4'-би-1,3-бензодиоксол,
(R)-1,13-бис(дифенилфосфино)-7,8-дигидро-6H-дибензо[f,h][1,5]диоксин,
(S)-1,13-бис(дифенилфосфино)-7,8-дигидро-6Н-дибензо[f,h][1,5]диоксин,
(R)-2,2',6,6'-тетраметокси-4,4'-бис(дифенилфосфино)-3,3'-бипиридин,
(S)-2,2',6,6'-тетраметокси-4,4'-бис(дифенилфосфино)-3,3'-бипиридин,
(R)-2,2',6,6'-тетраметокси-4,4'-бис(ди(3,5-ксилил)фосфино)-3,3'-бипиридин,
(S)-2,2',6,6'-тетраметокси-4,4'-бис(ди(3,5-ксилил)фосфино)-3,3'-бипиридин,
(R)-2,2'-бис(дифенилфосфино)-6,6'-диметокси-1,1'-бифенил,
(S)-2,2'-бис(дифенилфосфино)-6,6'-диметокси-1,1'-бифенил,
(R)-бис(дифенилфосфино)-4,4',6,6'-тетраметокси-1,1'-бифенил,
(S)-бис(дифенилфосфино)-4,4',6,6'-тетраметокси-1,1'-бифенил,
(R)-6,6'-бис(дифенилфосфино)-2,2',3,3'-тетрагидро-5,5'-би-1,4-бензодиоксин,
(S)-6,6'-бис(дифенилфосфино)-2,2',3,3'-тетрагидро-5,5'-би-1,4-бензодиоксин,
(R)-5,5'-бис(дифенилфосфино)-2,2,2',2'-тетрафтор-4,4'-би-1,3-бензодиоксол,
(S)-5,5'-бис(дифенилфосфино)-2,2,2',2'-тетрафтор-4,4'-би-1,3-бензодиоксол,
(R)-(-)-5,5'-бис[ди(3,5-ди-трет-бутил-4-метоксифенил)фосфино]-4,4'-би-1,3-бензодиоксол,
(S)-(+)-5,5'-бис[ди(3,5-ди-трет-бутил-4-метоксифенил)фосфино]-4,4'-би-1,3-бензодиоксол,
(S,S)-Fc-1,1'-бис[бис(3,5-диметилфенил)фосфино]-2,2'-бис[(S,S)С-(N,N-диметиламино)фенилметил]ферроцен,
(R,R)-Fc-1,1'-бис[бис(3,5-диметилфенил)фосфино]-2,2'-бис[(R,R)С-(N,N-диметиламино)фенилметил]ферроцен,
(R)-2,2'-бис[бис(3,5-диизопропил-4-диметиламинофенил)фосфино]-6,6'-диметокси-1,1'-бифенил,
(S)-2,2'-бис[бис(3,5-диизопропил-4-диметиламинофенил)фосфино]-6,6'-диметокси-1,1'-бифенил,
(S)-2,2'-бис[бис(3,4,5-триметоксифенил)фосфино]-4,4',5,5',6,6'-гексаметокси-1,1'-бифенил,
(R)-2,2'-бис[бис(3,4,5-триметоксифенил)фосфино]-4,4',5,5',6,6'-гексаметокси-1,1'-бифенил,
(R)-1-дифенилфосфино-2-[(R)-(N,N-диметиламино)-[2-(дифенилфосфино)фенил]метил]ферроцен,
(S)-1-дифенилфосфино-2-[(S)-(N,N-диметиламино)-[2-(дифенилфосфино)фенил]метил]ферроцен,
(R)-(+)-2,2'-бис(дифенилфосфино)-5,5',6,6',7,7',8,8'-октагидро-1,1'-бинафтил,
(S)-(-)-2,2'-бис(дифенилфосфино)-5,5',6,6',7,7',8,8'-октагидро-1,1'-бинафтил,
(R)-1-[(R)-1-[бис[3,5-бис(трифторметил)фенил]фосфино]этил]-2-[2-(дифенилфосфино)фенил]ферроцен,
(S)-1-[(S)-1-[бис[3,5-бис(трифторметил)фенил]фосфино]этил]-2-[2-(дифенилфосфино)фенил]ферроцен,
(R)-2,2'-бис[бис(3,5-ди-трет-бутил-4-метоксифенил)фосфино]-6,6'-диметокси-1,1'-бифенил,
(S)-2,2'-бис[бис(3,5-ди-трет-бутил-4-метоксифенил)фосфино]-6,6'-диметокси-1,1'-бифенил,
(R)-2,2'-бис[ди-3,5-ксилилфосфино]-6,6'-диметокси-1,1'-бифенил,
(S)-2,2'-бис[ди-3,5-ксилилфосфино]-6,6'-диметокси-1,1'-бифенил,
(R)-1-[(R)-1-(дифенилфосфино)этил]-2-[2-(дифенилфосфино)фенил]ферроцен,
(S)-1-[(S)-1-(дифенилфосфино)этил]-2-[2-(дифенилфосфино)фенил]ферроцен,
(R)-2,2'-бис(ди-p-диметиламинофенилфосфино)-6,6'-диметокси-1,1'-бифенил,
(S)-2,2'-бис(ди-p-диметиламинофенилфосфино)-6,6'-диметокси-1,1'-бифенил,
(R)-2,2'-бис[бис(3,5-ди-трет-бутилфенил)фосфино]-6,6'-диметокси-1,1'-бифенил,
(S)-2,2'-бис[бис(3,5-ди-трет-бутилфенил)фосфино]-6,6'-диметокси-1,1'-бифенил,
(R)-1-[(R)-1-[ди(3,5-ксилил)фосфино]этил]-2-[2-[ди(3,5-ксилил)фосфино]фенил]ферроцен,
(S)-1-[(R)-1-[ди(3,5-ксилил)фосфино]этил]-2-[2-[ди(3,5-ксилил)фосфино]фенил]ферроцен,
(R)-2,2'-бис(ди-м-диметиламинофенилфосфино)-6,6'-диметокси-1,1'-бифенил,
(S)-2,2'-бис(ди-м-диметиламинофенилфосфино)-6,6'-диметокси-1,1'-бифенил,
(R)-2,2'-бис[бис(3,5-диизопропил-4-диметоксифенил)фосфино]-6,6'-диметокси-1,1'-бифенил,
(S)-2,2'-бис[бис(3,5-диизопропил-4-диметоксифенил)фосфино]-6,6'-диметокси-1,1'-бифенил,
(-)-1,2-бис[(2S',5S)-2,5-диизопропилфосфолано]бензол,
(+)-1,2-бис[(2R,5R)-2,5-диизопропилфосфолано]бензол,
(R)-2,2'-бис(ди-п-толилфосфино)-6,6'-диметокси-1,1'-бифенил,
(S)-2,2'-бис(ди-п-толилфосфино)-6,6'-диметокси-1,1'-бифенил,
(R)-2,2'-бис(ди-6-метокси-2-нафталинилфосфино)-6,6'-диметокси-1,1'-бифенил,
(S)-2,2'-бис(ди-6-метокси-2-нафталинилфосфино)-6,6'-диметокси-1,1'-бифенил,
(R)-2,2'-бис[бис(3,5-диизопропилфенил)фосфино]-6,6'-диметокси-1,1'-бифенил,
(S)-2,2'-бис[бис(3,5-диизопропилфенил)фосфино]-6,6'-диметокси-1,1'-бифенил,
1-дициклогексилфосфино-1'-[(S)P-[(S)Fc-2-[(R)C-1-(диметиламино)этил]ферроценил]фенилфосфино]ферроцен,
1-дициклогексилфосфино-1'-[(R)P-[(R)Fc-2-[(S)C-1-(диметиламино)этил]ферроценил]фенилфосфино]ферроцен.
Предпочтительные хиральные лиганды выбраны из
(R)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила,
(S)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила,
(R)-2,2'-бис(ди-n-толилфосфино)-1,1'-бинафтила,
(S)-2,2'-бис(ди-п-толилфосфино)-1,1'-бинафтила,
(R)-2,2'-бис[ди(3,5-ксилил)фосфино]-1,1'-бинафтила,
(S)-2,2'-бис[ди(3,5-ксилил)фосфино]-1,1'-бинафтила,
(R)-5,5'-бис(дифенилфосфино)-4,4'-би-1,3-бензодиоксола,
(S)-5,5'-бис(дифенилфосфино)-4,4'-би-1,3-бензодиоксола,
(R)-5,5'-бис(ди[3,5-ксилил]фосфино)-4,4'-би-1,3-бензодиоксола,
(S)-5,5'-бис(ди[3,5-ксилил]фосфино)-4,4'-би-1,3-бензодиоксола,
(R)-5,5'-бис(ди[3,5-ди-трет-бутил-4-метоксифенил]фосфино)-4,4'-би-1,3-бензодиоксола,
(S)-5,5'-бис(ди[3,5-ди-трет-бутил-4-метоксифенил]фосфино)-4,4'-би-1,3-бензодиоксола,
(R)-1,13-бис(дифенилфосфино)-7,8-дигидро-6Н-дибензо[f,h][1,5]диоксина,
(S)-1,13-бис(дифенилфосфино)-7,8-дигидро-6Н-дибензо[f,h][1,5]диоксина,
(R)-2,2',6,6'-тетраметокси-4,4'-бис(дифенилфосфино)-3,3'-бипиридина,
(S)-2,2',6,6'-тетраметокси-4,4'-бис(дифенилфосфино)-3,3'-бипиридина,
(R)-2,2',6,6'-тетраметокси-4,4'-бис(ди[3,5-ксилил]фосфино)-3,3'-бипиридина,
(S)-2,2',6,6'-тетраметокси-4,4'-бис(ди[3,5-ксилил]фосфино)-3,3'-бипиридина,
(R)-2,2'-бис(дифенилфосфино)-6,6'-диметокси-1,1'-бифенила,
(S)-2,2'-бис(дифенилфосфино)-6,6'-диметокси-1,1'-бифенила,
(R)-бис(дифенилфосфино)-4,4',6,6'-тетраметокси-1,1'-бифенила,
(S)-бис(дифенилфосфино)-4,4',6,6'-тетраметокси-1,1'-бифенила,
(R)-6,6'-бис(дифенилфосфино)-2,2',3,3'-тетрагидро-5,5'-би-1,4-бензодиоксина,
(S)-6,6'-бис(дифенилфосфино)-2,2',3,3'-тетрагидро-5,5'-би-1,4-бензодиоксина,
(R)-(+)-2,2'-бис(дифенилфосфино)-5,5',6,6',7,7',8,8'-октагидро-1,1'-бинафтила,
(S)-(-)-2,2'-бис(дифенилфосфино)-5,5',6,6',7,7',8,8'-октагидро-1,1'-бинафтила,
(R)-5,5'-бис(дифенилфосфино)-2,2,2',2'-тетрафтор-4,4'-би-1,3-бензодиоксола,
(S)-5,5'-бис(дифенилфосфино)-2,2,2',2'-тетрафтор-4,4'-би-1,3-бензодиоксола,
(R)-1-[(R)-1-[ди(3,5-ксилил)фосфино]этил]-2-[2-[ди(3,5-ксилил)фосфино]фенил]ферроцена,
(S)-1-[(S)-1-[ди(3,5-ксилил)фосфино]этил]-2-[2-[ди(3,5-ксилил)фосфино]фенил]ферроцена,
(R)-1-[(R)-1-[бис[3,5-бис(трифторметил)фенил]фосфино]этил]-2-[2-(дифенилфосфино)фенил]ферроцена и
(S)-1-[(S)-1-[бис[3,5-бис(трифторметил)фенил]фосфино]этил]-2-[2-(дифенилфосфино)фенил]ферроцена.
Более предпочтительно, хиральный лиганд выбран из
(R)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила,
(R)-2,2'-бис(ди-и-толилфосфино)-1,1'-бинафтила,
(R)-2,2'-бис[ди(3,5-ксилил)фосфино]-1,1'-бинафтила,
(R)-5,5'-бис(дифенилфосфино)-4,4'-би-1,3-бензодиоксола,
(R)-5,5'-бис(ди[3,5-ксилил]фосфино)-4,4'-би-1,3-бензодиоксола,
(R)-5,5'-бис[ди(3,5-ди-трет-бутил-4-метоксифенил)фосфино]-4,4'-би-1,3-бензодиоксола,
(S)-1,13-бис(дифенилфосфино)-7,8-дигидро-6Н-дибензо[f,h][1,5]диоксина,
(R)-2,2',6,6'-тетраметокси-4,4'-бис(ди(3,5-ксилил)фосфино)-3,3'-бипиридина,
(R)-2,2'-бис(дифенилфосфино)-6,6'-диметокси-1,1'-бифенила,
(R)-2,2'-бис(дифенилфосфино)-4,4',6,6'-тетраметокси-1,1'-бифенила,
(R)-6,6'-бис(дифенилфосфино)-2,2',3,3'-тетрагидро-5,5'-би-1,4-бензодиоксина,
(R)-(+)-2,2'-бис(дифенилфосфино)-5,5',6,6',7,7',8,8'-октагидро-1,1'-бинафтила,
(R)-(+)-2,2'-бис(ди-3,5-ксилилфосфино)-5,5',6,6',7,7',8,8'-октагидро-1,1'-бинафтила,
(R)-5,5'-бис(дифенилфосфино)-2,2,2',2'-тетрафтор-4,4'-би-1,3-бензодиоксола,
(S)-1-[(S)-1-[ди(3,5-ксилил)фосфино]этил]-2-[2-[ди(3,5-ксилил)фосфино]фенил]ферроцена
и
(S)-1-[(5)-1-[бис[3,5-бис(трифторметил)фенил]фосфино]этил]-2-[2-(дифенилфосфино)фенил]ферроцена.
Еще более предпочтительно хиральный лиганд представляет собой (R)-2,2'-бис[ди(3,5-ксилил)фосфино]-1,1'-бинафтил.
Катализатор на основе переходного металла может состоять из предварительно образованного комплекса. Такие предварительно образованные комплексы обычно могут быть образованы путем осуществления реакции хирального лиганда с подходящим содержащим переходный металл соединением-предшественником.
Полученные таким образом комплексы затем можно применять в качестве катализатора для способа (с) в соответствии со схемой 1. Содержащие переходный металл соединения-предшественники содержат по меньшей мере переходные металлы, выбранные из Ru, Rh, Ir и Pd, и, как правило, содержат лиганды, которые легко замещаются хиральным лигандом, или они могут содержать лиганд, который легко удаляется путем гидрирования. Предпочтительными катализаторами на основе переходного металла являются комплексы на основе Ru.
Подходящие содержащие переходный металл соединения-предшественники включают без ограничения RuCl3, RuCl3⋅nH2O, [RuCl2(η6-бензол)]2, [RuCl2(η6-цимол)]2, [RuCl2(η6-мезитилен)]2, [RuCl2(η6-гексаметилбензол)]2, [RuCl2(η6-бензол)]2, [Rul2(η6-бензол)]2, транс-RuCl2(DMSO)4, RuCl2(PPh3)3, RuCl2(COD) (в котором
СОВ=1,5-циклооктадиен), Ru(COD)(метилаллил)2, Ru(COD)(трифторацетат)2, [Ir(COD)Cl]2,
Rh(COD)Cl, Rh(COD)2BF4, Rh(COD)2(OTf)2, Ru(COD)(OAc)2. Предпочтительные содержащие переходный металл соединения-предшественники выбраны из [RuCl2(η6-бензол)]2, [RuCl2(η6-цимол)]2, RuCl2(COD), Ru(COD)(метилаллил)2 и Ru(COD)(трифторацетат)2.
Примеры предварительно образованных комплексов катализаторов на основе Ru включают без ограничения [RuCl(п-цимол)((S)-DM-SEGPHOS)]Cl, [RuCl(п-цимол)((R)-DM-SEGPHOS)]Cl, [NH2Me2] [(RuCl((R)-xylbinap))2(u-Cl)3], [NH2Me2] [(RuCl((S)-xylbinap))2(u-Cl)3], Ru(OAc)2[(R)-binap], Ru(OAc)2[(S)-binap], Ru(OAc)2[(R)-xylbinap], Ru(OAc)2[(S)-xylbinap], RuCl2[(R)-xylbinap][(R)-daipen], RuCl2[(S)-xylbinap][(S)-daipen], RuCl2[(R)-xylbinap] [(R,R)-dpen], RuCl2[(S)-xylbinap] [(S,S-dpen]. Предпочтительные предварительно образованные комплексы катализаторов на основе Ru выбраны из RuCl(п-цимол)((R)-xylbinap)]Cl, [NH2Me2][(RuCl((R)-xylbinap))2(u-Cl)3], Ru(OAc)2[(R)-xylbinap], [NH2Me2][(RuCl((R)-H8-binap))2(u-Cl)3], [RuCl(п-цимол)((R)-H8-binap)]Cl, Ru(OAc)2[(R)-H8-binap], [NH2Me2][(RuCl((R)-H8-xylbinap))2(u-Cl)3], [RuCl(п-цимол)((R)-Н8-xylbinap)]Cl, Ru(OAc)2[(R)-H8-xylbinap], [NH2Me2][(RuCl((S)-1-[(S)-1-[бис[3,5-бис(трифторметил)фенил]фосфино]этил]-2-[2-(дифенилфосфино)фенил]ферроцен))2(u-Cl)3], [RuCl(п-цимол)((S)-1-[(S)-1-[бис [3,5-бис(трифторметил)фенил]фосфино]этил]-2-[2-(дифенилфосфино)фенил]ферроцен)]Cl и (S)-1-[(S)-1-[бис[3,5-бис(трифторметил)фенил]фосфино]этил]-2-[2-(дифенилфосфино)фенил]ферроцен]. Более предпочтительно предварительно образованные комплексы катализаторов на основе Ru выбраны из [NH2Me2][(RuCl((R)-xylbinap))2(u-Cl)3] и Ru(OAc)2[(R)-xylbinap].
Как указанно выше, в способе (с) на схеме 1 также требуется присутствие аммониевой соли. Аммониевые соли могут быть образованы in situ путем добавления аммиака и подходящей кислоты. В частности, кислоты выбраны из соединений формулы
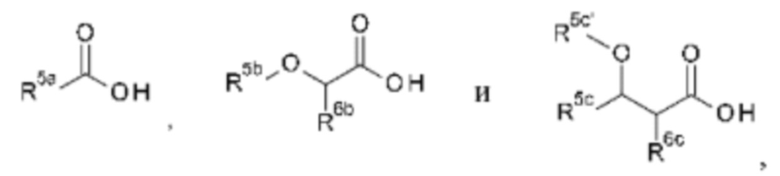
где R5a выбран из C1-С6алкила, -(С0-С3алкил)-С3-С6циклоалкила, -(С0-С3алкил)-С3-С6гетероциклоалкила, -(C0-C3алкил)-NR5a1R5a2, -(С0-С3алкил)арила и -(С0-С3алкил)гетероарила, где С1-С6алкил, -(С0-С3алкил)-С3-С6циклоалкил, -(С0-С3алкил)-С3-С6гетероциклоалкил, -(С0-С3алкил)арил и - (С0-С3алкил)гетероарил являются незамещенными или замещенными одним или несколькими заместителями, независимо выбранными из галогена, ОН, С1-С6алкила, С1-С6галогеналкила, С1-С6алкокси и С1-С6 галогеналкокси; R5a1 и R5a2 независимо выбраны из водорода и C1-С6алкила;
R5b и R5c' выбраны из водорода, С1-С6алкила, -(С0-С3алкил)-С3-С6циклоалкила, -(С0-С3алкил)-С(=О)ОН, -(С0-С3алкил)арила и -(С1-С3алкил)гетероарила, где С1-С6алкил, -(С0-С3алкил)-С3-С6циклоалкил, -(С0-С3алкил)арил и -(С0-С3алкил)гетероарил являются незамещенными или замещенными одним или несколькими заместителями, независимо выбранными из галогена, С1-С6алкила, C1-С6галогеналкила, С1-С6алкокси и С1-С6галогеналкокси;
R5c, R6b и R6c выбраны из водорода, галогена, С1-С6алкила, -(С0-С3алкил)-С3-С6циклоалкила, -(С0-С3алкил)арила и -(С3-С3алкил)гетероарила, где С1-С6алкил, -(С0-С3алкил)-С3-С6циклоалкил, -(С0-С3алкил)арил и -(С0-С3алкил)гетероарил являются незамещенными или замещенными одним или двумя заместителями, независимо выбранными из галогена, С1-С6алкила, С1-С6 галогеналкила, С1-С6алкокси и C1-С6галогеналкокси.
В частности, кислоты выбраны из соединений формулы
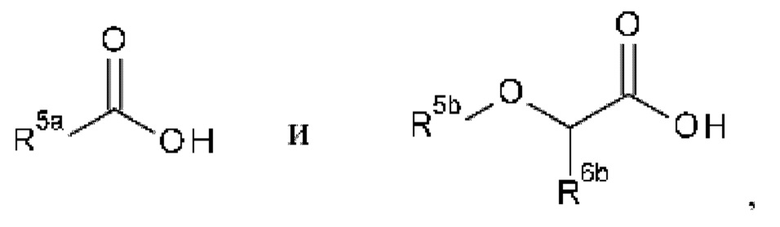
где R5a выбран из С1-С6алкила, -(С0-С3алкил)-С3-С6циклоалкила, -(С0-С3алкил)-С3-С6гетероциклоалкила, -(С0-С3алкил)арила и-(С0-С3алкил)гетероарила, где С1-С6алкил, -(С0-С3алкил)-С3-С6циклоалкил, -(С0-С3алкил)-С3-С6гетероциклоалкил, -(C0-С3алкил)арил и -(С0-С3алкил)гетероарил являются незамещенными или замещенными одним или несколькими заместителями, независимо выбранными из галогена, ОН, С1-С6алкила, С1-С6галогеналкила, C1-С6алкокси и C1-С6галогеналкокси;
R5b выбран из С1-С6алкила, -(С0-С3алкил)-С3-С6циклоалкила, -(С0-С3алкил)арила и -(С0-С3алкил)гетероарила, где С1-С6алкил, -(С0-С3алкил)-С3-С6циклоалкил, -(C0-С3алкил)арил и -(С0-С3алкил)гетероарил являются незамещенными или замещенными одним или несколькими заместителями, независимо выбранными из галогена, C1-С6алкила, С1-С6галогеналкила, С1-С6алкокси и С1-С6галогеналкокси;
R6b выбран из водорода, галогена, С1-С6алкила, С3-С6циклоалкила, арила и гетероарила, где С1-С6алкил, С3-С6циклоалкил, арил и гетероарил являются незамещенными или замещенными одним или двумя заместителями, независимо выбранными из галогена, С1-С6алкила, С1-С6галогеналкила, С1-С6алкокси и C1-С6галогеналкокси.
Более конкретно, кислоты выбраны из уксусной кислоты, метоксиуксусной кислоты, 2-метоксипропионовой кислоты, тетрагидрофуран-2-карбоновой кислоты, о-метоксибензойной кислоты, п-метоксибензойной кислоты, феноксиуксусной кислоты, 2-фуранкарбоновой кислоты, п-хлорфеноксиуксусной кислоты, салициловой кислоты, 4-трет-бутилфеноксиуксусной кислоты, уксусной кислоты и хлор уксусной кислоты, еще более конкретно феноксиуксусной кислоты.
В реакционную смесь также можно добавлять аммониевые соли в предварительно образованном состоянии. В частности, аммониевые соли выбраны из ацетата аммония, 2-метоксипропионата аммония, тетрагидрофуран-2-карбоксилага аммония, о-метоксибензоата аммония, п-метоксибензоата аммония, хлорацетата аммония, метоксиацетата аммония, феноксиацетата аммония, п-хлорфеноксиацетага аммония, 4-трет-б утилфе но кс и ацетат а аммония, фуран-2-карбоксилата аммония и салицилата аммония, более конкретно феноксиацетата аммония.
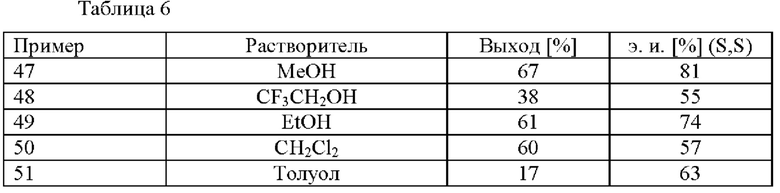
Специалисту в данной области техники будет понятно, что способ (с) в соответствии со схемой 1 осуществляют в подходящем растворителе. Подходящий растворитель может быть выбран из следующих классов растворителей: спирты, простые эфиры, сложные эфиры, хлорированные или отличные от хлорированных углеводороды, и ароматические вещества. Подходящие растворители включают без ограничения метанол, этанол, 2,2,2-трифторэтан-1-ол, дихлорметан и толуол. Предпочтительными растворителями являются спирты, в частности метанол.
Способ (с) в соответствии со схемой 1 осуществляют в присутствии газообразного водорода (Н2). Давление газообразного водорода в реакционном сосуде может составлять от 1 до 500 бар, в частности, от 1 до 250 бар, более конкретно от 1 до 50 бар, еще более конкретно от 5 до 30 бар.
Как правило, температура реакции (с) в соответствии со схемой 1 составляет от 0 до 150°С, предпочтительно от 30 до 120°С, более предпочтительно от 50 до 100°С, еще более предпочтительно от 70 до 90°С.
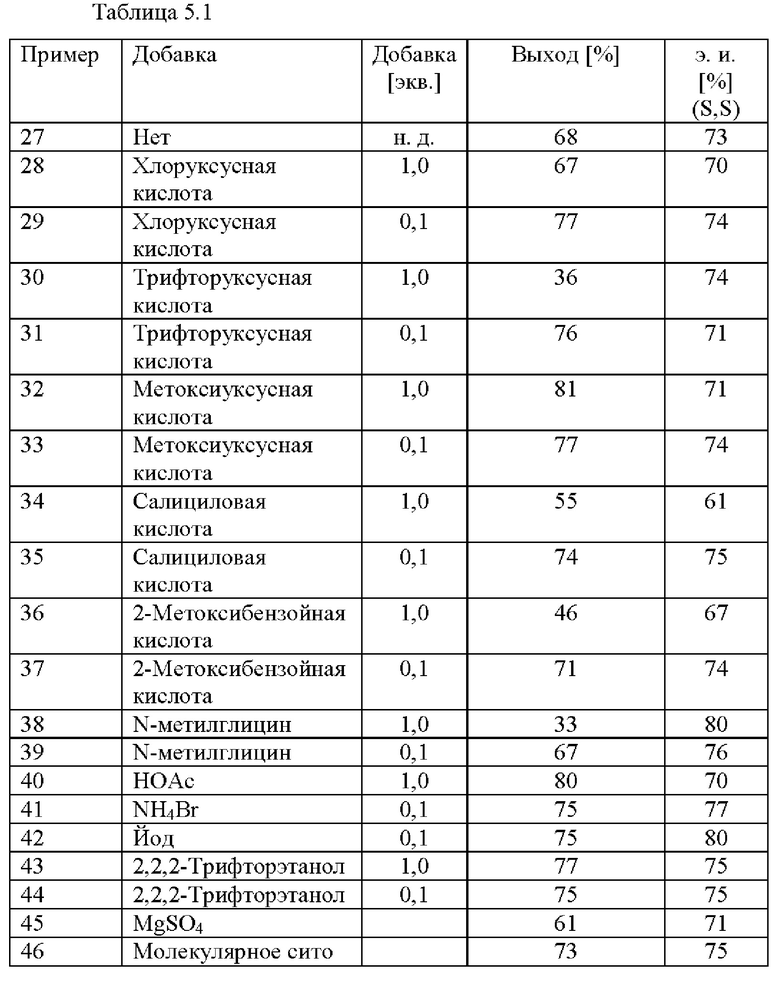
Также могут быть добавлены добавки в способ (с) в соответствии со схемой 1. Подходящие добавки могут быть выбраны из аммониевых солей, аммиака, кислот, высушивающих средств, таких как молекулярные сита и сульфат магния, йод и спирты. Примеры добавок, которые можно добавлять, включают без ограничения хлоруксусную кислоту, трифторуксусную кислоту, метоксиуксусную кислоту, салициловую кислоту, феноксиуксусную кислоту, фуран-2-карбоновую кислоту, 2-метоксибензойную кислоту, N-метилглицин, уксусную кислоту, бромид аммония, хлорид аммония, йод, 2,2,2-трифторэтанол, сульфат магния, материалы, представляющие собой молекулярные сита. Предпочтительными добавками являются аммониевые соли или кислоты, например, галогениды аммония, такие как NH4Cl, или карбоновые кислоты, такие как феноксиуксусная кислота.
Специалисту в данной области техники будет понятно, что рН реакционной смеси может влиять на результат реакции. Как правило, рН реакционной смеси составляет от 2 до 7, предпочтительно от 4 до 7 и еще более предпочтительно от 6 до 7.
Предпочтительные параметры способа для реакции (с) в соответствии со схемой 1 являются следующими.
Катализатор на основе переходного металла добавляют в реакционную смесь в количестве от 0,0001 до 0,1 моль эквивалента относительно соединения формулы (III). В частности, добавляют от 0,0005 до 0,01 моль эквивалента относительно соединения формулы (III).
Катализаторы на основе переходного металла могут быть предварительно образованы или могут быть образованы in situ путем добавления в реакционную смесь хиральных лигандов и содержащих переходный металл предшественников. Специалисту в данной области техники хорошо известно образование in situ катализаторов на основе переходных металлов, например, раскрытых в ЕР 1153908 А2.
В реакционную смесь загружают предпочтительно (i) 1-10 моль эквивалентов предварительно образованной аммониевой соли относительно соединения формулы (III) или (ii) загружают 1-10 моль эквивалентов аммиака относительно соединения формулы (III) и соответствующую кислоту с образованием аммониевой соли in situ.
Концентрация соединения формулы (III) составляет предпочтительно 1-30% по весу реакционной смеси.
Необязательно в реакционную смесь добавляют 0,01-2 моль эквивалентов одной или нескольких добавок относительно соединения формулы (III).
Предпочтительно в способе (с) предусмотрены цис-стереоизомеры соединения формулы (IV) с э. и. 50% или больше, предпочтительно 75% или больше, более предпочтительно 80% или больше. Более предпочтительно, в способе (с) предусмотрены цис-стереоизомеры соединений формулы (IV) с э. и. (S,S) 50% или больше, предпочтительно 75% или больше, более предпочтительно 80% или больше.
Следует понимать, что любой из предпочтительных признаков для способа (с) может быть объединен с одним или несколькими другими предпочтительными признаками способа (с).
В четвертом аспекте настоящего изобретения предусмотрен способ (d) получения соединения формулы (V), который предусматривает осуществление реакции соединения формулы (III) с ацетонитрилом в присутствии кислоты Льюиса или кислоты Бренстеда и подходящей добавки, и где А определен выше для схемы 1.
Способ (d) в соответствии со схемой 1 осуществляют либо в ацетонитриле, либо в подходящем растворителе. Подходящий растворитель может быть выбран из следующих классов растворителей: хлорированные углеводороды или ароматические вещества. Подходящие растворители включают без ограничения дихлорметан, хлорбензол или дихлорбензол. Предпочтительные растворители представляют собой хлорированные ароматические вещества, в частности 1,2-дихлорбензол.
Подходящие кислоты Льюиса или кислоты Бренстеда включают без ограничения AlCl3, GaCl3, FeCl3, BF3⋅Et2O, HBF3⋅Et2O, трифлатную кислоту, нонафлатную кислоту (перфторбутансульфоновую кислоту), MeSO3H и реактив Итона (7,7 вес.% пентаоксида фосфора в метансульфоновой кислоте).
Подходящие добавки могут представлять собой хлорангидриды, ангидриды или сложные эфиры. Примеры добавок, которые можно добавлять, включают без ограничения ацетилхлорид, изопропенилацетат, 4-метоксибензоилхлорид и п-анисовый ангидрид.
Предпочтительные параметры реакции для способа (d) в соответствии со схемой 1 являются следующими.
Для реакции (d) предпочтительной является температура от 0°С до 100°С, более предпочтительной от 20°С до 80°С, еще более предпочтительной приблизительно 50°С.
В реакционную смесь загружают предпочтительно 1,0-2,5 моль эквивалента кислоты Льюиса или кислоты Бренстеда относительно соединения формулы (III).
В реакционную смесь загружают предпочтительно 1-10 моль эквивалентов ацетонитрила относительно соединения формулы (III).
В реакционную смесь загружают предпочтительно 0,25-2,0 моль эквивалента добавки относительно соединения формулы (III).
Концентрация соединения формулы (III) составляет предпочтительно 1-30% по весу реакционной смеси.
Следует понимать, что любой из предпочтительных признаков для способа (d) может быть объединен с одним или несколькими другими предпочтительными признаками способа (d).
В пятом аспекте настоящего изобретения соединение формулы (V) может быть получено с помощью способа (d'), который является комбинацией способов (b) и (d) с тем отличием, что соединение (III) не выделяют, а непосредственно преобразуют в соединение (V). Преобразование соединения (II) в соединение (III) достигается в присутствии кислоты Льюиса, как описано во втором аспекте настоящего изобретения. Последовательное преобразование соединения (III) в соединение (V) достигается путем добавления ацетонитрила и добавки, описанной в четвертом аспекте настоящего изобретения. Предпочтительно способ (d') осуществляют в инертных условиях, таких как в атмосфере газообразного азота, и в подходящем растворителе.
Специалисту в данной области техники будет понятно, что способы (d) и (d') в соответствии со схемой 1 осуществляют в подходящем растворителе. Подходящий растворитель может быть выбран из следующих классов растворителей: хлорированные углеводороды или ароматические вещества. Подходящие растворители включают без ограничения дихлорэтан, хлорбензол или дихлорбензол. Предпочтительные растворители представляют собой хлорированные ароматические вещества, в частности, 1,2-дихлорбензол.
Подходящий катализатор/кислота Льюиса включает без ограничения AlCl3 и GaCl3, предпочтительно AlCl3.
Подходящие добавки могут представлять собой хлорангидриды, ангидриды или сложные эфиры. Примеры добавок, которые можно добавлять, включают без ограничения ацетилхлорид, изопропенилацетат, 4-метоксибензоилхлорид или п-анисовый ангидрид.
Предпочтительные параметры реакции для способа (d') в соответствии со схемой 1 соответствуют предпочтительным условиям реакции, уже описанным для преобразования соединения (II) в соединение (III) (стадия b) и преобразования соединения (III) в соединение (V) (стадия d) (второй и четвертый аспект настоящего изобретения).
Следует понимать, что любой из предпочтительных признаков для способа (d') может быть объединен с одним или несколькими другими предпочтительными признаками способов (d') и (d).
В шестом аспекте настоящего изобретения предусмотрен способ (е) получения соединения формулы (VI), который предусматривает осуществление реакции соединения формулы (V) с Н2 в присутствии хирального или энантиообогащенного катализатора по аналогии к тому, что было раскрыто в WO 2015/003951.
Хиральный катализатор на основе переходного металла содержит переходный металл, выбранный из Ru и Rh, и хиральный лиганд.
Хиральные лиганды известны из уровня техники и могут применяться в настоящем изобретении, примеры приведены в "Catalytic asymmetric synthesis", Iwao Ojima, third Edition, Wiley-VCH 2010, и в литературных источниках, приведенных в нем; типичные классы, которые известны специалисту в данной области техники, включают без ограничения TADDOL (α,α,α',α'-тетраарил-2,2-дизамещенный 1,3-диоксолан-4,5-диметанол), DUPHOS (фосфолановый лиганд), BOX (бис(оксазолиновый) лиганд), BIN АР (2,2'-бис(дифенилфосфино)-1,1'-бинафтил), BINOL (1,1'-би-2-нафтол), DIOP (2,3-о-изопропилиден-2,3-дигидрокси-1,4-бис(дифенилфосфино)бутан), WALPHOS, TANIAPHOS, MANDYPHOS, CHENPHOS, JOSIPHOS, BIPHEMP, MeO-BIPHEP, SEGPHOS, CHIRAPHOS, PPHOS, QUINOX-P, NORPHOS, TUNEPHOS и SYNPHOS.
Предпочтительно хиральный лиганд представляет собой лиганд, содержащий бидентатный фосфор, общей формулы (IX):
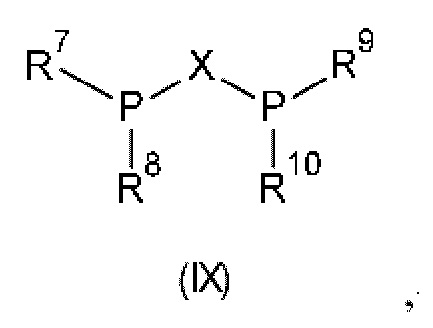
где X представляет собой линкерную группу, и R7, R8, R9 и R10 независимо выбраны из арила, гетероарила, С1-С6алккт и С1-С6циклоалкила, каждый из которых является незамещенным или замещенным. Предпочтительно линкерная группа X выбрана из (R или S)-1,1'-бинафтила, (R или S)-4,4'-би-1,3-бензодиоксола, (R или S)-2,2',6,6'-тетраметокси-3,3'-бипиридина, (R или S)-6,6'-диметокси-1,1'-бифенила, (R или S)-4,4',6,6' тетраметокси-1,1'-бифенила, 2,2'-бис-[(R или S)-cx-(диметиламино)бензил]ферроцена, ферроценилметила, ферроцена, бензола и этила. Более предпочтительно, бидентатный лиганд формулы (IX) выбран из классов лигандов В IN АР, MANDYPHOS, JOSIPHOS, MeO-BIPHEP, TANIAPHOS, CHENPHOS, QUINOX-P и DUPHOS.
Подходящие хиральные лиганды в соответствии с настоящим изобретением включают без ограничения
(R)-1-[(S)-2-(ди-трет-бутилфосфино)ферроценил]этил-ди-о-толилфосфин,
(R,R)Fc-1,1'-бис[бис(4-метокси-3,5-диметилфенил)фосфино]-2,2'-бис[(S,S)C-(N,N-диметиламино)фенилметил]ферроцен,
(R)-1-[(S)-2-циклогексилфосфино)ферроценил]этил-бис(2-метилфенил)-фосфин,
(S)-1-дифенилфосфино-2-[(S)-(N,N-диметиламино)-[2-(дифенилфосфино)фенил]метил]ферроцен,
(R,R)-(-)-2,3-бис(трет-бутилметилфосфино)хиноксалин,
(2S,3S)-(-)-2,3-бис(дифенилфосфино)бицикло[2.2.1]гепт-5-ен,
(S)-2,2'-бис(диизопропилфосфино)-6,6'-диметокси-1,1'-бифенил,
(2S,4S)-(-)-2,4-бис(дифенилфосфино)пентан,
1,1'-бис[(R)P-[(R)Fc-2-[(,S0c-l-(диметиламино)этил]ферроценил]фенилфосфино]ферроцен,
1-дициклогексилфосфино-1'-(S)Р-[(S)Fc-2-[(R)С-1-(диметиламино)этил] ферроценил]фенилфосфино] ферроцен,
(R)-1-[(S)-2-(дифенилфосфино)ферроценил]этил-ди(3,5-ксилил)фосфин,
(R)Fc-1-[(S)Р-трет-бутилфосфиноил]-2-[(S)-1-(дифенилфосфино)этил]ферроцен,
(S)-1-[(R)-2-(дифенилфосфино)ферроценил]этил-ди-трет-бутилфосфин,
аддукт этанола и (S)-1-[(R)-2-(дифенилфосфино)ферроценил]этил-дициклогексилфосфина,
(R)-2,2'-бис(дифенилфосфино)-6,6'-диметокси-1,1'-бифенил,
(R)-(+)-1,2-бис(дифенилфосфино)пропан,
(R)-1-[(S)-2-[ди(1-нафтил)фосфино]ферроценил]этил-ди-трет-бутилфосфин,
(S)-1-[(R)-2-[ди(2-фурил)фосфино]ферроценил]этил-ди-трет-бутилфосфин,
(R)-1-[(S)-2-(ди-трет-бутилфосфино)ферроценил]этилдифенилфосфин,
(S)-1-[(R)-2-(дифенилфосфино)ферроценил]этил-ди-трет-бутилфосфин,
(R)-1-[(S)-2-(дициклогексилфосфино)ферроценил]этил-дициклогексилфосфин,
(S)-1-[(R)-2-[бис(4-метокси-3,5-диметилфенил)фосфино]ферроценил]этил-дициклогексилфосфин,
(S)-1-[(R)-2-[бис(4-метокси-3,5-диметилфенил)фосфино]ферроценил]этил-ди-трет-бутилфосфин,
(S)-(+)-2,2'-бис[ди(3,5-ксилил)фосфино]-1,1'-бинафтил,
(S)-2,2'-бис[ди-3,5-ксилилфосфино]-6,6'-диметокси-1,1'-бифенил,
(-)-1,2-бис((2S,5S)-2,5-диэтилфосфолано)бензол,
(S,S)Fc-1,1'-бис[бис(4-метокси-3,5-диметилфенил)фосфино]-2,2'-бис[(R,R)С-(N,N-диметиламино)фенилметил] ферроцен,
(R)-1-дифенилфосфино-2-[(R)-(N,N-диметиламино)-[2-(дифенилфосфино)фенил]метил]ферроцен,
(R)-2,2'-бис[бис(3,5-ди-трет-бутил-4-метоксифенил)фосфино]-6,6'-диметокси-1,1'-бифенил,
(S)-1-[(R)-2-(дифенилфосфино)ферроценил]этил-ди(3,5-ксилил)фосфин,
(S,S)-(-)-2,3-бис(трет-бутилметилфосфино)хиноксалин,
(2S,3S)-(+)-2,3-бис(дифенилфосфино)бутан,
(S)-2,2'-бис(дифенилфосфино)-6,6'-диметокси-1,1'-бифенил,
(S)-(+)-1,2-бис(дифенилфосфино)пропан,
(S,S)-(+)-1,2-бис(трет-бутилметилфосфино)бензол,
(-)-1,2-бис[(2S,5S)-2,5-диметилфосфолано]бензол,
(+)-1,2-бис[(2S,5S)-2,5-диэтилфосфолано]бензол,
(-)-1,2-бис((2R,5R)-2,5-диэтилфосфолано)этан.
Предпочтительные хиральные лиганды в комбинации с Rh выбраны из
(R)-1-[(S)-2-(ди-трет-бутилфосфино)ферроценил]этил-ди-о-толилфосфина,
(R)-1-[(S)-2-циклогексилфосфино)ферроценил]этил-бис(2-метилфенил)фосфина
и
(R)-1-[(S)-2-(ди-трет-бутилфосфино)ферроценил]этилдифенилфосфина.
Предпочтительные хиральные лиганды в комбинации с Ru выбраны из
(S)-(+)-2,2'-бис[ди(3,5-ксилил)фосфино]-1,1'-бинафтила,
(-)-1,2-бис((2S,5S)-2,5-диэтилфосфолано)бензола,
(S,S)-(-)-2,3-бис(трет-бутилметилфосфино)хиноксалина,
(S,S)-(+)-1,2-бис(трет-бутилметилфосфино)бензола,
(-)-1,2-бис[(2S,5S)-2,5-диметилфосфолано]бензола,
(+)-1,2-бис [(2S,5S)-2,5-диэтилфосфолано] бензола,
(S)-1-[(R)-2-[бис(4-метокси-3,5-диметилфенил)фосфино]ферроценил]этил-ди-трет-бутилфосфина и
(S)-1-[(R)-2-[бис(4-метокси-3,5-диметилфенил)фосфино]ферроценил]этил-дициклогексилфосфина.
Катализатор на основе переходного металла может состоять из предварительно образованного комплекса. Такие предварительно образованные комплексы обычно могут быть образованы путем осуществления реакции хирального лиганда с подходящим содержащим переходный металл соединением-предшественником. Полученные таким образом комплексы затем можно применять в качестве катализатора для способа (е) в соответствии со схемой 1. Содержащие переходный металл соединения-предшественники содержат по меньшей мере переходные металлы Ru и Rh и, как правило, содержат лиганды, которые легко замещаются хиральным лигандом, или они могут содержать лиганд, который легко удаляется путем гидрирования.
Подходящие содержащие переходный металл соединения-предшественники включают без ограничения [Rh(COD)2]O3SCF3, [Rh(COD)2]BF4, [Rh(COD)2]BARF (в котором BARF = тетракис[3,5-бис(трифторметил)фенил]борат), [Rh(nbd)2]BF4 (в котором nbd = норборнадиен), бис(2-металлил)(COD)рутений, бис(СОВ)тетра[μ-трифторацетато]дирутения(II) гидрат и [Ru(COD)(2-металлил)2].
Примеры предварительно образованных комплексов катализаторов на основе Rh включают без ограничения
[Rh(COD)((R)-1-[(S)-2-(ди-трет-бутилфосфино)ферроценил]этил-ди-o-толилфосфин)]BF4,
[Rh(COD)((R)-1-[(S)-2-циклогексилфосфино)ферроценил]этил-бис(2-метилфенил)фосфин]BF4,
[Rh(COD)((R)-1-[(S)-2-(ди-трет-бутилфосфино)ферроценил]этилдифенилфосфин)]BF4,
[Rh(COD)((R)-1-[(5)-2-(ди-трет-бутилфосфино)ферроценил]этил-ди-о-толилфосфин)]O3SCF3,
[Rh(COD)((R)-1-[(S)-2-циклогексилфосфино)ферроценил]этил-бис(2-метилфенил)фосфин)]O3SCF3 и
[Rh(COD)((R)-1-[(S)-2-(ди-трет-бутилфосфино)ферроценил]этилдифенилфосфин)]O3SCF3.
Предпочтительные параметры реакции для способа (е) в соответствии со схемой 1 являются следующими.
Специалисту в данной области техники будет понятно, что способ (е) в соответствии со схемой 1 осуществляют в подходящем растворителе. Подходящий растворитель может быть выбран из следующих классов растворителей: спирты, простые эфиры, сложные эфиры, хлорированные или отличные от хлорированных углеводороды, или ароматические вещества. Предпочтительные растворители выбраны из метанола, изопропанола и 2,2,2-трифторэтан-1-ола, если применяют катализатор на основе Rh. Предпочтительными растворителями являются спирты, в частности, трифторэтанол и изопропанол. Предпочтительные растворители выбраны из спиртов, в частности, изопропанола, если применяют катализатор на основе Ru.
Способ (е) в соответствии со схемой 1 осуществляют в присутствии газообразного водорода (Н2). Давление газообразного водорода в реакционном сосуде может составлять от 1 до 500 бар, в частности, от 1 до 250 бар, более конкретно от 1 до 50 бар, еще более конкретно от 10 до 50 бар.
Как правило, температура в способе (е) в соответствии со схемой 1 составляет от 0 до 150°С, предпочтительно от 30 до 120°С, более предпочтительно от 20 до 100°С, еще более предпочтительно от 25 до 60°С.
Количество катализатора, которое добавляют, составляет от 0,0001 до 0,1 моль эквивалента катализатора на основе переходного металла относительно соединения формулы (V). В частности, добавляют от 0,0001 до 0,005 моль эквивалента катализатора на основе переходного металла относительно соединения формулы (V). Катализаторы на основе переходного металла могут быть предварительно образованы или могут быть образованы in situ путем добавления в реакционную смесь хиральных лигандов и содержащих переходной металл предшественников.
Концентрация соединения формулы (V) составляет предпочтительно 1-30% по весу реакционной смеси.
В данном способе (е) предусмотрены энантиомерно и диастереомерно обогащенные амиды формулы (VI) с э. и. (энантиомерным избытком) 75% или больше, предпочтительно 80% или больше. Предпочтительно в способе (е) предусмотрены цис-энантиомеры соединений формулы (IV) с э. и. 75% или больше, более предпочтительно 80% или больше.
Следует понимать, что любой из предпочтительных признаков для способа (е) может быть объединен с одним или несколькими другими предпочтительными признаками способа (е).
В седьмом аспекте настоящего изобретения предусмотрен способ (f) получения соединения формулы (IV), который предусматривает осуществление реакции соединения формулы (VI) в условиях кислой или основной среды, предпочтительно в условиях кислой среды.
Подходящие кислоты включают без ограничения HCl, H2SO4, Н3РО4, метансульфоновую кислоту, п-толуолсульфоновую кислоту и трифторметансульфоновую кислоту.
Специалисту в данной области техники будет понятно, что способ (f) в соответствии со схемой 1 осуществляют в подходящем растворителе. Подходящий растворитель может быть выбран из следующих классов растворителей: вода, спирты, простые эфиры, сложные эфиры, хлорированные или отличные от хлорированных углеводороды, или ароматические вещества. Подходящие растворители включают без ограничения воду и толуол. Предпочтительный растворитель представляет собой воду.
Типичные параметры реакции для способа (f) в соответствии со схемой 1 являются следующими.
Предпочтительными условиями для деацетилирования являются температура от 0°С до 200°С, более конкретно от 100°С до 180°С, в частности, при температуре возврата флегмы для соответствующей системы.
Кислота. Было подтверждено, что применение 1,0-7,0 моль эквивалента относительно соединения формулы (VI) является благоприятным.
Реакционный объем. Типичные условия являются такими, чтобы концентрация соединения формулы (VI) составляла 1-30% по весу реакционной смеси.
Следует понимать, что любой из предпочтительных признаков для способа (f) может быть объединен с одним или несколькими другими предпочтительными признаками способа (f).
В восьмом аспекте настоящего изобретения предусмотрен способ (g) получения соединения формулы (VII), предусматривавший осуществление реакции соединения формулы (IV) с соединением формулы (X),
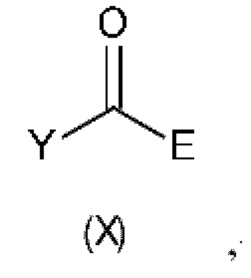
где Y представляет собой подходящую уходящую группу, такую как ОН, OR или галоген, предпочтительно хлор, R представляет собой С1-С6алкил, и А и Е определены ранее для схемы 1.
Реакцию соединения формулы (IV) с соединением формулы (X) осуществляют в типичных условиях для образования амидной связи, которые являются известными специалисту в данной области техники. Например, реакция (g) включает добавление подходящего основания. Подходящие основания включают без ограничения Et3N, NaHCO3 и NaOH.
Специалисту в данной области техники будет понятно, что реакцию (g) в соответствии со схемой 1 осуществляют в подходящем растворителе. Подходящий растворитель может быть выбран из следующих классов растворителей: простые эфиры, сложные эфиры, хлорированные или отличные от хлорированных углеводороды, или ароматические вещества. Подходящие растворители включают без ограничения толуол, ксилол, THF, MeTHF (метилтетрагидрофуран) и ацетонитрил.
Предпочтительные параметры реакции для способа (g) в соответствии со схемой 1 являются следующими.
Реакцию (g) предпочтительно осуществляют при температуре от 0°С до 200°С, более предпочтительно от 100°С до 180°С, еще более предпочтительно от 40 до 60°С.
Предпочтительно добавляют 1,0-3,0 моль эквивалента основания относительно соединения формулы (IV).
Предпочтительно в реакционную смесь добавляют 0-20 моль эквивалентов воды относительно соединения формулы (IV).
Концентрация соединения формулы (IV) составляет предпочтительно 1-30% по весу реакционной смеси.
Следует понимать, что любой из предпочтительных признаков для способа (g) может быть объединен с одним или несколькими другими предпочтительными признаками для способа (g).
Определения
Термин "алкил", используемый в данном документе отдельно или как часть химической группы, представляет собой углеводороды с прямой или разветвленной цепью, предпочтительно с 1-6 атомами углерода, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил, 1-метилбутил, 2-метилбутил, 3-метилбутил, 1,2-диметилпропил, 1,1-диметилпропил, 2,2-диметилпропил, 1-этилпропил, гексил, 1-метилпентил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 1,2-диметилпропил, 1,3-диметилбутил, 1,4-диметилбутил, 2,3-диметилбутил, 1,1-диметилбутил, 2,2-диметилбутил, 3,3-диметилбутил, 1,1,2-триметилпропил, 1,2,2-триметилпропил, 1-этилбутил и 2-этилбутил. Предпочтительными являются алкильные группы с 1-4 атомами углерода, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил или трет-бутил. Термин "алкенил", отдельно или как часть химической группы, представляет собой углеводороды с прямой или разветвленной цепью, предпочтительно с 2-6 атомами углерода и по меньшей мере одной двойной связью, например, винил, 2-пропенил, 2-бутенил, 3-бутенил, 1-метил-2-пропенил, 2-метил-2-пропенил, 2-пентенил, 3-пентенил, 4-пентенил, 1-метил-2-бутенил, 2-метил-2-бутенил, 3-метил-2-бутенил, 1-метил-3-бутенил, 2-метил-3-бутенил, 3-метил-3-бутенил, 1,1-диметил-2-пропенил, 1,2-диметил-2-пропенил, 1-этил-2-пропенил, 2-гексенил, 3-гексенил, 4-гексенил, 5-гексенил, 1-метил-2-пентенил, 2-метил-2-пентенил, 3-метил-2-пентенил, 4-метил-2-пентенил, 3-метил-3-пентенил, 4-метил-3-пентенил, 1-метил-4-пентенил, 2-метил-4-пентенил, 3-метил-4-пентенил, 4-метил-4-пентенил, 1,1-диметил-2-бутенил, 1,1-диметил-3-бутенил, 1,2-диметил-2-бутенил, 1,2-диметил-3-бутенил, 1,3-диметил-2-бутенил, 2,2-диметил-3-бутенил, 2,3-диметил-2-бутенил, 2,3-диметил-3-бутенил, 1-этил-2-бутенил, 1-этил-3-бутенил, 2-этил-2-бутенил, 2-этил-3-бутенил, 1,1,2-триметил-2-пропенил, 1-этил-1-метил-2-пропенил и 1-этил-2-метил-2-пропенил. Предпочтительными являются алкенильные группы с 2-4 атомами углерода, например, 2-пропенил, 2-бутенил или 1-метил-2-пропенил.
Термин "алкинил", отдельно или как часть химической группы, представляет собой углеводороды с прямой или разветвленной цепью, предпочтительно с 2-6 атомами углерода и по меньшей мере одной тройной связью, например, 2-пропинил, 2-бутинил, 3-бутинил, 1-метил-2- пропинил, 2-пентинил, 3-пентинил, 4-пентинил, 1-метил-3-бутинил, 2-метил-3-бутинил, 1-метил-2-бутинил, 1,1-диметил-2-пропинил, 1-этил-2-пропинил, 2-гексинил, 3-гексинил, 4-гексинил, 5-гексинил, 1-метил-2-пентинил, 1-метил-3-пентинил, 1-метил-4-пентинил, 2-метил-3-пентинил, 2-метил-4-пентинил, 3-метил-4-пентинил, 4-метил-2-пентинил, 1,1-диметил-3-бутинил, 1,2-диметил-3-бутинил, 2,2-диметил-3-бутинил, 1-этил-3-бутинил, 2-этил-3-бутинил, 1-этил-1-метил-2- пропинил и 2,5-гексадиинил. Предпочтительными являются алкинилы с 2-4 атомами углерода, например, этинил, 2-пропинил или 2-бутинил-2-пропенил.
Термин "циклоалкил", отдельно или как часть химической группы, представляет собой насыщенные или частично ненасыщенные моно-, би- или трициклические углеводороды, предпочтительно с 3-10 атомами углерода, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, бицикло[2.2.1]гептил, бицикло[2.2.2]октил или адамантил.
Термин "гетероциклил", отдельно или как часть химической группы, представляет собой насыщенные или частично ненасыщенные моно-, би- или трициклические углеводороды, предпочтительно с 3-10 атомами углерода, в которых по меньшей мере один атом углерода заменен на гетероатом, выбранный из О, N и S, например, тетрагидрофуран, пирролидин, тетрагидротиофен.
Термин "арил" представляет собой моно-, би- или полициклическую ароматическую систему с предпочтительно 6-14, более предпочтительно 6-10 атомами углерода в кольце, например, фенил, нафтил, антрил, фенантренил, предпочтительно фенил. "Арил" также представляет собой полициклические системы, например, тетрагидронафтил, инденил, инданил, флуоренил, бифенил. Арилалкилы являются примерами замещенных арилов, которые могут быть дополнительно замещены одинаковыми или различными заместителями как в арильной, так и в алкильной части. Бензил и 1-фенилэтил являются примерами таких арилалкилов.
Термин "гетероарил" представляет собой гетероароматические группы, а именно полностью ненасыщенные ароматические гетероциклический группы, которые подпадают под приведенное выше определение гетероциклов. "Гетероарилы" с 5-7-членными кольцами с 1-3, предпочтительно 1-2 одинаковыми или различными гетеро атомами, выбранными из N, О и S. Примерами "гетероарилов" являются фур ил, тиенил, пиразолил, имидазолил, 1,2,3- и 1,2,4-триазолил, изоксазолил, тиазолил, изотиазолил, 1,2,3-, 1,3,4-, 1,2,4- и 1,2,5-оксадиазолил, азепинил, пирролил, пиридил, пиридазинил, пиримидинил, пиразинил, 1,3,5-, 1,2,4- и 1,2,3-триазинил, 1,2,4-, 1,3,2-, 1,3,6- и 1,2,6-оксазинил, оксепинил, тиепинил, 1,2,4-триазолонил и 1,2,4-диазепинил.
Термин "галоген" или "гало" представляет собой фтор, хлор, бром или йод, в частности, фтор, хлор или бром. Химические группы, которые замещены с помощью галогена, например, галогеналкил, галогенциклоалкил, галогеналкилокси, галогеналкилсульфанил, галогеналкилсульфинил или галогеналкилсульфонил являются замещенными с помощью галогена в количестве от одного до максимального числа заместителей. Если "алкил", "алкенил" или "алкинил" являются замещенными с помощью галогена, атомы галогена могут быть одинаковыми или различными и могут быть связаны с одним атомом углерода или различными атомами углерода.
Термин "энантиомерно обогащенный" означает, что один из энантиомеров соединения присутствует в избытке по сравнению с другим энантиомером. Данный избыток будет далее в данном документе называться энантиомерным избытком или э. и. Э. и. может быть определен с помощью хиральной GC или HPLC-анализа. Э. и. равняется разнице между количеством энантиомеров, разделенной на сумму количеств энантиомеров, коэффициент которых может быть выражен в процентах после умножения на 100.
Экспериментальная часть
Примеры
Следующие примеры предназначены для иллюстрации настоящего изобретения и не должны рассматриваться как ограничивающие его.
Оборудование
Способы HPLC
Способ 1
Устройство для HPLC: Thermo Electron Corporation; SpectraSystem P200, SpectraSystemASlOOO и SpectraSystem UV1000.
Колонка: CHIRALPAK IA-3, 3 мкм, 4,6 мм × 100 мм.
Температура: комнатная температура (к. т.)
Подвижная фаза: EtOH + 0,1 диэтиламина/МеОН+0,1% диэтиламина (50/50).
Расход: 1,0 мл/мин.
Градиент: изократический.
Обнаружение: 223 нм.
Размер образца: 10 мкл.
Rt (время удерживания) (1S,2S)-2-(2,4-дихлорфенил)циклобутанамина: 4,8 мин.
Rt (1R,2R)-2-(2,4-дихлорфенил)циклобутанамина: 3,0 мин.
Способ 2
Устройство для HPLC: Agilent Technologies 1260 Infinity.
Колонка: Agilent XDB-C18, 1,8 мкм, 4,6 × 50 мм.
Температура: к. т.
Подвижная фаза: А: Ацетонитрил; В: МеОН; С: H2O+0,1% Н3РО4.
Расход: 1,5 мл/мин.
Градиент:
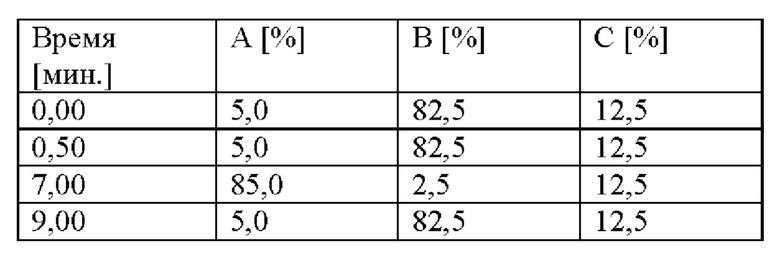
Обнаружение: 220 нм.
Размер образца: 1 мкл.
Rt цис-изомера 2-(2,4-дихлорфенил)циклобутанамина: 2,8 мин.
Способ 3
Устройство для HPLC: Agilent Technologies 1200 Series.
Колонка: Phenomenex Kinetex XB C18, 2,6 мкм, 4,6 × 150 мм
Температура: 40°C.
Подвижная фаза: A: H2O+0,1% (об./об.) Н3РО4; В: Ацетонитрил.
Расход: 1,0 мл/мин.
Градиент:
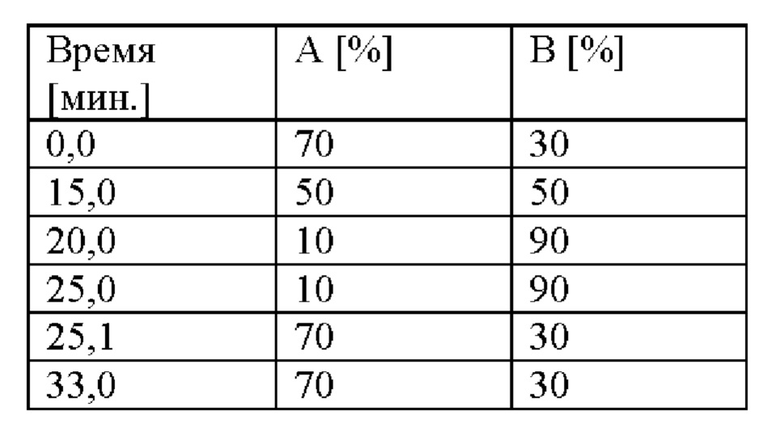
Обнаружение: 223 нм.
Размер образца: 5 мкл.
Rt цис-изомеров N-[2-(2,4-дихлорфенил)циклобутил]ацетамида: 9,2 мин.
Способ 4
Устройство для HPLC: Agilent Technologies 1200 Series.
Колонка: CHIRALPAK IA-3, 3 мкм, 4,6 мм × 100 мм.
Температура: к. т.
Подвижная фаза: н-гексан/этанол (98/2).
Расход: 1,0 мл/мин.
Градиент: изократический.
Обнаружение: 223 нм.
Размер образца: 5 мкл.
Rt N-[(1S,2S)-2-(2,4-дихлорфенил)циклобутил]ацетамида: 12,4
мин. Rt N-[(1R,2R)-2-(2,4-дихлорфенил)циклобутил]ацетамида: 15,6 мин.
Первый аспект
Реакция (a1) в соответствии со схемой 1
Пример 1.1
Добавляли 1-(2,4-дихлорфенил)циклопропанкарбонитрил (1,0 г, 4,61 ммоль) и трифторуксусную кислоту (6 мл) в высушенную пламенем двугорлую колбу объемом 50 мл, которая была оснащена вакуумным насосом, двойным баллоном давления и магнитной мешалкой. Реакционную колбу вакуумировали и в вакуум дважды запускали азот. Затем в реакционную колбу добавляли 5% палладий на торфяном угле с 50% воды (50 мг, 0,013 ммоль) и колбу продували азотом, после чего присоединяли баллон с водородом. Реакционную смесь перемешивали при к. т. (комнатная температура) в течение 2-4 ч. Водород высвобождали и реакционную колбу продували азотом, когда степень превращения составила >98%. Реакционную массу фильтровали в атмосфере азота. Отгоняли трифторуксусную кислоту при пониженном давлении и остаток разбавляли с помощью МТВЕ и промывали водой. Фазы разделяли и нижнюю водную фазу дважды экстрагировали с помощью МТВЕ. Объединенные органические слои промывали водой и концентрировали при пониженном давлении с получением 1-(2,4-дихлорфенил)циклопропанкарбальдегида (1,1 г) в виде масла.
1Н ЯМР (400МГц, DMSO-d6) δ 8,75 (s, 1 Н), 7,60 (d, J=1,90 Гц, 1 Н), 7,46-7,27 (m, 2 Н), 1,80-1,61 (m, 2 Н), 1,47-1,36 (m, 2 Н).
Пример 1.2
Добавляли 1-(2,4-дихлорфенил)циклопропанкарбонитрил (9,49 г, 44,0 ммоль), трифторуксусную кислоту (70 г), воду (4,7 г) и 5% палладий на углероде с 50% воды (300 мг, 0,15 мол. %) в реактор объемом 100 мл, который был оснащен вакуумным насосом, входом для азота и клапаном, соединенным с входом для водорода. Реактор вакуумировали и вакуум трижды заменяли азотом. Эту же процедуру повторяли с H2 и затем реакционную смесь перемешивали в течение 2 часов при 5°С и давлении Н2 1,5 бар. После того, как превращение было завершено, водород высвобождали и колбу продували азотом. Реакционную смесь фильтровали через слой целита (промывали с помощью TFA) и фильтрат концентрировали при пониженном давлении, разбавляли толуолом и дважды промывали водой. Полученный органический слой концентрировали при пониженном давлении с получением 1-(2,4-дихлорфенил)циклопропанкарбальдегида (4,2 г) в виде прозрачного коричневого масла.
1Н ЯМР (400МГц, DMSO-d6) δ 8,75 (s, 1 Н), 7,60 (d, J=1,90 Гц, 1 Н), 7,46-7,27 (m, 2 Н), 1,80-1,61 (m, 2 Н), 1,47-1,36 (m, 2 Н).
Реакция (а2) в соответствии со схемой 1
Пример 2.1
Добавляли 1-(2,4-дихлорфенил)циклопропанкарбонитрил (1,0 г, 4,64 ммоль), уксусную кислоту (17,5 мл) и воду (5 мл) в высушенную пламенем двугорлую колбу объемом 50 мл, которая была оснащена вакуумным насосом, двойным баллоном давления и магнитной мешалкой. Реакционную колбу вакуумировали и в вакуум дважды запускали азот. Затем добавляли никель Ренея (концентрация 50% вес./вес.) с 50% воды (201 мг, 0,71 ммоль) и колбу продували азотом, после чего присоединяли баллон с водородом. Реакционную смесь перемешивали при к. т. в течение 6-10 ч. Водород высвобождали и реакционную колбу продували с азотом, когда степень превращения составила >98%. Реакционную массу фильтровали через слой целита в атмосфере азота. Фильтрат подкисляли с помощью концентрированного водного раствора HCl до рН 1,0 и разбавляли водой и этилацетатом. Органический слой экстрагировали 15% водным раствором карбоната натрия и затем водой. Растворитель удаляли при пониженном давлении с получением 1-(2,4-дихлорфенил)циклопропанкарбальдегида (0,81 г) в виде масла.
1Н ЯМР (400МГц, DMSO-d6) δ 8,75 (s, 1 Н), 7,60 (d, J=1,90 Гц, 1 Н), 7,46-7,27 (m, 2 Н), 1,80-1,61 (m, 2 Н), 1,47-1,36 (m, 2 Н).
Пример 2.2
Добавляли DCP-нитрил (9,50 г, 44,0 ммоль), трифторуксусную кислоту (70 г), воду (7,0 г) и никель Ренея (520 мг, 20 мол. %) в реактор объемом 100 мл, который был оснащен соединением с вакуумным насосом, входом для азота и клапаном, соединенным с входом для Н2. Реактор вакуумировали и вакуум трижды заменяли азотом. Затем применяли атмосферу Н2 с парциальным давлением 0,5 бар и реакционную смесь перемешивали при 20°С. После того, как превращение было завершено, Н2 высвобождали и колбу продували азотом. Реакционную смесь фильтровали через слой целита (промывали с помощью TFA) и фильтрат концентрировали при пониженном давлении, разбавляли толуолом и промывали водой. Полученный органический слой концентрировали при пониженном давлении с получением DCP-альдегида (7,4 г) в виде желтоватого масла.
1Н ЯМР (400МГц, DMSO-d6) δ 8,75 (s, 1 Н), 7,60 (d, J=1,90 Гц, 1 Н), 7,46-7,27 (m, 2 Н), 1,80-1,61 (m, 2 Н), 1,47-1,36 (m, 2 Н).
Реакция (а3) в соответствии со схемой 1
Пример 3
Добавляли 1,2 М раствор DIBAL в толуоле (75,2 г, 105 ммоль) в раствор 1-(2,4-дихлорфенил)циклопропанкарбонитрила (20 г, 91,5 ммоль) в толуоле (40 г) таким образом, что температуру поддерживали в пределах от -5°С до 0°С. После периода перемешивания длительностью 30 минут после добавления в реакционную смесь добавляли 2 М водный раствор HCl при -20°С. Затем обеспечивали достижение реакционной смесью к. т. Реакционную смесь разбавляли этилацетатом и экстрагировали водой. Фазы разделяли и водную фазу дважды экстрагировали этилацетатом. Объединенные органические фазы экстрагировали водой и солевым раствором, высушивали над Na2SO4, фильтровали и растворители удаляли при пониженном давлении. Выделяли 1-(2,4-дихлорфенил)циклопропанкарбальдегид (20,1 г) в виде желтого масла.
1Н ЯМР (400МГц, CDCl3) δ 9,05 (s, 1H), 7,45 (m, 1H), 7,20 (m, 2Н), 1,70 (m, 2Н), 1,40 (m, 2Н).
Реакция (а4) в соответствии со схемой 1
Пример 4.1
Дозировали 60% раствор REDAL в толуоле (87,6 г, 260 ммоль) в течение 4 ч. в раствор 1-(2,4-дихлорфенил)циклопропанкарбонитрила (84,8 г, 400 ммоль) в толуоле (212 г) при температуре 0°С.После периода перемешивания длительностью 2 часа после добавления при 0°С реакционную смесь дозировали в 50% водный раствор уксусной кислоты (384 г, 3,2 моль) таким образом, чтобы поддерживать температуру реакционной смеси ниже <25°С. Затем в смесь дозировали 32% водный раствор HCl (137 г, 1,2 моль). Фазы разделяли и органическую фазу дважды экстрагировали водой (75 г), после чего растворитель органической фазы удаляли при пониженном давлении. Выделяли 1-(2,4-дихлорфенил)циклопропанкарбальдегид (81,0 г) в виде оранжевого масла.
1Н ЯМР (400МГц, DMSO-d6) δ 8,75 (s, 1 Н), 7,60 (d, J=1,90 Гц, 1 Н), 7,46-7,27 (m, 2 Н), 1,80-1,61 (m, 2 Н), 1,47-1,36 (m, 2 Н).
Пример 4.2
Дозировали 70% раствор REDAL в толуоле (97,3 г, 0,34 моль) в течение 80 минут в раствор 1-(2,4-дихлорфенил)циклопропанкарбонитрила (120,0 г, 0,54 ммоль) в толуоле (182 г) при температуре 20-25°С. После периода перемешивания длительностью 1 ч. после добавления при 20-25°С реакционную смесь дозировали в смесь, состоящую из 50% водного раствора уксусной кислоты (266,0 г, 2,19 моль) и 35% водного раствора HCl (160,0 г, 1,54 моль), с поддержанием температуры реакционной смеси ниже 25°С. Фазы разделяли и органическую фазу дважды экстрагировали водой (60 г), после чего растворитель удаляли из органической фазы при пониженном давлении. Выделяли 1-(2,4-дихлорфенил)циклопропанкарбальдегид (118,4 г) в виде темно-оранжевого масла.
1Н ЯМР (400МГц, DMSO-d6) δ 8,75 (s, 1 Н), 7,60 (d, J=1,90 Гц, 1 Н), 7,46-7,27 (m, 2 Н), 1,80-1,61 (m, 2 Н), 1,47-1,36 (m, 2 Н).
Второй аспект
Реакция (b) в соответствии со схемой 1
Пример 5.1
В высушенную пламенем двугорлую колбу объемом 10 мл, оснащенную термометром и барботером, содержащую 1-(2,4-дихлорфенил)циклопропанкарбальдегид (1,4 г, 6,0 ммоль), в атмосфере аргона добавляли хлорбензол (6 мл) с последующим добавлением безводного AlCl3 (1,2 г, 9,0 ммоль). Полученную суспензию нагревали в течение 1 ч. при 45°С.
После охлаждения до к. т. смесь выливали в холодный 1 н. HCl и разбавляли этилацетатом. Водную фазу дважды экстрагировали дихлорметаном. Объединенную органическую фазу один раз промывали с помощью 1 и. HCl и один раз солевым раствором, затем высушивали с помощью твердого Na2SO4, фильтровали и концентрировали в вакууме. Выделяли 2-(2,4-дихлорфенил)циклобутанон (1,35 г) в виде коричневого масла.
1Н ЯМР (400МГц, CDCl3) δ 7,40 (d, J=2,2 Гц, 1H), 7,30 (d, J=8,1 Гц, 1H), 7,21 (dd, J=2,2 и 8,4 Гц, 1H), 4,76 (m, 1H), 3,25 (m, 1H), 3,10 (m, 1H), 2,63 (m, 1H), 2,12 (m, 1H).
Пример 5.2
Добавляли раствор 1-(2,4-дихлорфенил)циклопропанкарбальдегида (109,9 г, 0,5 моль) в 1,2-дихлорбензоле (64,8 г, 0,44 моль) в течение 90 минут в суспензию AlCl3 (87,6 г, 0,65 моль) в 1,2-дихлорбензоле (100,0 г, 0,68 моль) при Ti=50-55°С; суспензию продували легким потоком азота во время дозирования и периода перемешивания после добавления. По сути полное превращение было достигнуто после периода перемешивания длительностью 1 ч. после добавления при Ti=50-55°С. Реакционную смесь охлаждали до к. т. и затем дозировали в 14% водный раствор HCl (352,7 г, 1,34 моль) таким образом, что Ti поддерживали ниже 40°С. Смесь перемешивали в течение 45 минут, после чего фазы разделяли. Затем органическую фазу дважды экстрагировали водой (83,3 г, 4,6 моль и 80,0 г, 4,4 моль), после чего органическую фазу концентрировали при пониженном давлении. Выделяли 2-(2,4-дихлорфенил)циклобутанон (110,6 г) в виде коричневого масла.
1Н ЯМР (400МГц, CDCl3) δ 7,40 (d, J=2,2 Гц, 1Н), 7,30 (d, J=8,1 Гц, 1H), 7,21 (dd, J=2,2 и 8,4 Гц, 1H), 4,76 (m, 1H), 3,25 (m, 1H), 3,10 (m, 1H), 2,63 (m, 1H), 2,12 (m, 1H).
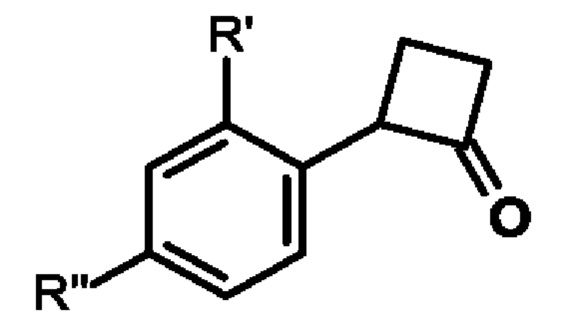
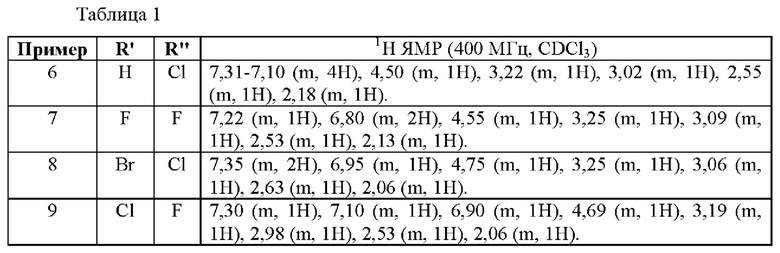
Примеры 6-9
Следующие соединения получали образом, аналогичным примеру 5.
Третий аспект
Реакция (с) в соответствии со схемой 1
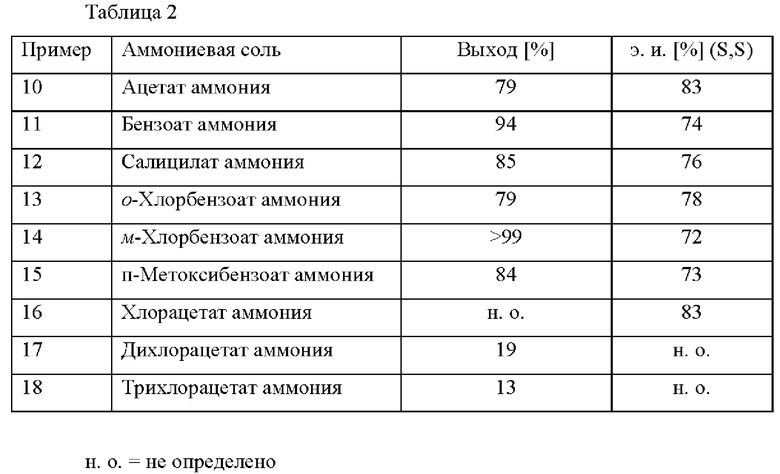
Примеры 10-18
В автоклав под давлением загружали 2-(2,4-дихлорфенил)циклобутанон (0,5 ммоль), [RuCl(п-цимол)((S)-DM-SEGPHOS)]Cl (0,005 ммоль), соответствующую соль аммония (0,5 ммоль, см. таблицу 2) и МеОН (5 мл). Применяли атмосферу водорода под давлением 30 бар и реакционную смесь нагревали до 80°С. После периода перемешивания длительностью 22 ч. реакционную смесь охлаждали до к. т. и анализировали с помощью хиральной HPLC (способ 1) и 1Н ЯМР.
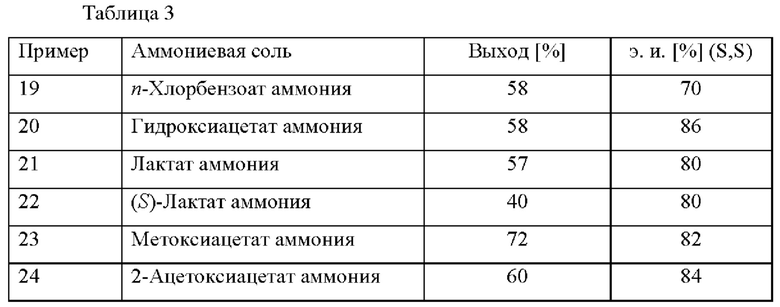
Примеры 19-24
В автоклав под давлением загружали 2-(2,4-дихлорфенил)циклобутанон (0,5 ммоль), [RuCl(п-цимол)((S)-DM-SEGPHOS)]Cl (0,005 ммоль), соответствующую соль аммония (0,6 ммоль, см. таблицу 3) и МеОН (2,5 мл). Применяли атмосферу водорода под давлением 30 бар и реакционную смесь нагревали до 80°С. Реакционную смесь охлаждали до к. т. после периода перемешивания длительностью 18,5 ч. и добавляли H2O (20 мл) и HCl (1 М, 2 мл). Смесь экстрагировали с помощью МТВЕ (20 мл) и органический слой экстрагировали водным раствором HCl (2 мл 1 М HCl и 20 мл H2O). Повышали основность водной фазы с применением водного раствора NaOH (5 М, 2 мл) и ее экстрагировали с помощью МТВЕ (3 × 20 мл). Объединенные органические слои высушивали над Na2SO4, фильтровали и растворитель удаляли при пониженном давлении. Остаток анализировали с помощью хиральной HPLC (способ 1) и 1H ЯМР.
Примеры 25-26
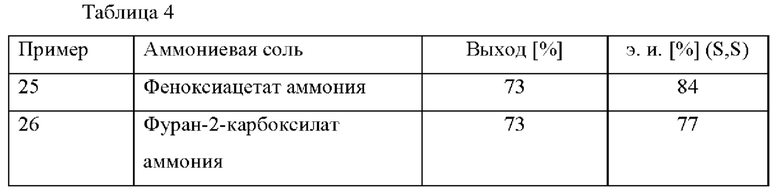
Добавляли 7 н. раствор аммиака в МеОН (108 ммоль) в смесь соответствующей кислоты (110 ммоль) в МеОН (столько же, чтобы получить 10% раствор аммониевой соли) при к. т. Смесь перемешивали в течение приблизительно 10 минут, и она была готова к применению на стадии прямого асимметричного восстановительного аминирования.
Прямое асимметрическое восстановительное аминирование
В автоклав под давлением загружали [NH2Me2][(RuCl((R)-xylbinap))2(μ-Cl)3] (0,135 ммоль), полученный ранее 10% раствор соответствующей аммониевой соли в МеОН (108 ммоль, см. таблицу 4) и МеОН (44 г). Реакционную смесь нагревали до 80°С и применяли атмосферу водорода под давлением 30 бар. Затем в автоклав добавляли раствор 2-(2,4-дихлорфенил)циклобутанона (624 ммоль) в МеОН (20 г) в течение 4 ч. Реакционную смесь охлаждали до к. т. после периода перемешивания длительностью 2 ч. после добавления. Затем повышали основность реакционной смеси и ее анализировали с помощью хиральной (способ 1) и ахиральной (способ 2) HPLC.
Примеры 27-46
Различные добавки тестировали на скрининговой платформе. В реакционный сосуд загружали 2-(2,4-дихлорфенил)циклобутанон (2,0 ммоль), [NH2Me2] [(RuCl((R)-xylbinap))2(u-Cl)3] (0,25 мол. %), ацетат аммония (2,4 ммоль), соответствующую добавку (см. таблицу 5) и МеОН (2,5 мл). Применяли атмосферу водорода под давлением 30 бар и реакционную смесь нагревали до 80°С. После периода перемешивания длительностью 16 ч. реакционную смесь охлаждали до к. т. и анализировали с помощью хиральной (способ 1) и ахиральной (способ 2) HPLC.
Примеры 46.1, 46.2
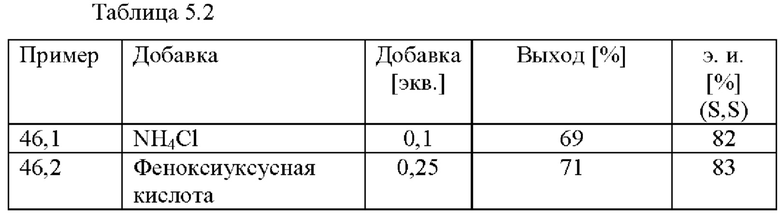
В автоклав под давлением загружали 2-(2,4-дихлорфенил)циклобутанон (93,0 ммоль), [NH2Me2][(RuCl((R)-xylbinap))2(u-Cl)3] (0,1 мол. %), феноксиацетат аммония (158,4 ммоль), соответствующую добавку (см. таблицу 5.2) и МеОН (4,1 моль). Применяли атмосферу водорода под давлением 30 бар и реакционную смесь нагревали до 80°С. После периода перемешивания длительностью 17 ч. реакционную смесь охлаждали до к. т. и анализировали с помощью хиральной (способ 1) и ахиральной (способ 2) HPLC.
Примеры 47-51
В автоклав под давлением загружали 2-(2,4-дихлорфенил)циклобутанон (0,5 ммоль), [RuCl(п-цимол)((S)-DM-SEGPHOS)]Cl (0,005 ммоль), ацетат аммония (0,6 ммоль) и соответствующий растворитель (5,0 мл). Применяли атмосферу водорода под давлением 30 бар и реакционную смесь нагревали до 80°С. Реакционную смесь охлаждали до к. т. после периода перемешивания длительностью 19 ч. и добавляли H2O (20 мл) и 1 М водный раствор HCl (2 мл). Смесь экстрагировали с помощью МТВЕ (20 мл) и органический слой экстрагировали водным раствором HCl (2 мл 1 М HCl и 20 мл H2O). Повышали основность водной фазы с применением 5 М водного раствора NaOH (2 мл) и ее экстрагировали с помощью МТВЕ (3 × 20 мл). Объединенные органические слои высушивали над Na2SO4, фильтровали и растворитель удаляли при пониженном давлении. Остаток анализировали с помощью хиральной HPLC (способ 1) и 1H ЯМР.
Пример 52
В автоклав под давлением загружали 2-(2,4-дихлорфенил)циклобутанон (1,4 ммоль), [RuCl(п-цимол)((S)-DM-SEGPHOS)]Cl (0,014 ммоль), метахлорбензоат аммония (1,4 ммоль) и МеОН (13,8 мл). Применяли атмосферу водорода под давлением 5 бар и реакционную смесь нагревали до 80°С. Реакционную смесь охлаждали до к. т. после периода перемешивания длительностью 16 ч. Добавляли H2O (50 мл) и 1 М водный раствор HCl (5 мл). Смесь экстрагировали с помощью МТВЕ (50 мл) и органический слой экстрагировали водным раствором HCl (5 мл 1 М HCl и 20 мл H2O). Повышали основность водной фазы с применением 5 М водного раствора NaOH (5 мл) и ее экстрагировали с помощью МТВЕ (3 × 50 мл). Объединенные органические слои высушивали над Na2SO4, фильтровали и растворитель удаляли при пониженном давлении. Остаток анализировали с помощью хиральной HPLC (способ 1) и 1Н ЯМР.
Пример 53
В автоклав под давлением загружали 2-(2,4-дихлорфенил)циклобутанон (0,5 ммоль), [RuCl(п-цимол)((S)-DM-SEGPHOS)]Cl (0,005 ммоль), метахлорбензоат аммония (0,5 ммоль) и МеОН (5 мл). Применяли атмосферу водорода под давлением 10 бар и реакционную смесь нагревали до 80°С. Реакционную смесь охлаждали до к. т. после периода перемешивания длительностью 17 ч. и анализировали с помощью хиральной HPLC (способ 1) и 1H ЯМР.
Пример 54
В автоклав под давлением загружали 2-(2,4-дихлорфенил)циклобутанон (0,46 ммоль), [RuCl(п-цимол)((S)-DM-SEGPHOS)]Cl (0,0046 ммоль), метахлорбензоат аммония (0,55 ммоль) и МеОН (2,3 мл). Применяли атмосферу водорода под давлением 10 бар и реакционную смесь нагревали до 80°С. Реакционную смесь охлаждали до к. т. после периода перемешивания длительностью 19 ч. Добавляли H2O (20 мл) и 1 М водный раствор HCl (2 мл). Смесь экстрагировали с помощью МТВЕ (20 мл) и органический слой экстрагировали водным раствором HCl (2 мл 1 М HCl и 20 мл H2O). Повышали основность водной фазы с применением 5 М водного раствора NaOH (2 мл) и ее экстрагировали с помощью МТВЕ (3 × 20 мл). Объединенные органические слои высушивали над Na2SO4, фильтровали и растворитель удаляли при пониженном давлении. Остаток анализировали с помощью хиральной HPLC (способ 1) и 1Н ЯМР.
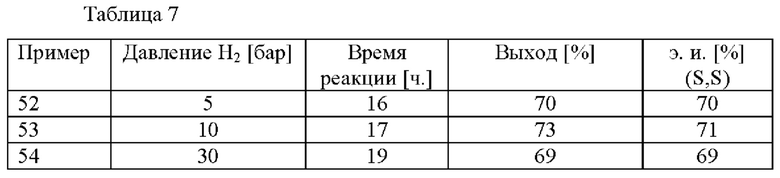
Результаты примеров 52-54 обобщены в таблице 7.
Примеры 55-56
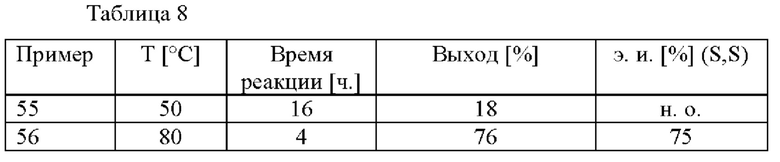
В автоклав под давлением загружали 2-(2,4-дихлорфенил)циклобутанон (4,0 ммоль), Ru(OAc)2[(R)-xylbinap] (0,02 ммоль), ацетат аммония (4,8 ммоль), уксусную кислоту (6,4 ммоль) и МеОН (10 мл). Применяли атмосферу водорода под давлением 30 бар и реакционную смесь нагревали до 80°С. После соответствующего периода перемешивания реакционную смесь охлаждали до к. т. Смесь разбавляли с помощью МТВЕ и воды и добавляли 1 М раствор HCl. Органическую фазу дважды экстрагировали 1 М раствором HCl. Повышали основность объединенных водных фаз с применением водного 5 М раствора NaOH и их три раза экстрагировали с помощью МТВЕ. Объединенные органические слои высушивали над Na2SO4, фильтровали и растворитель удаляли при пониженном давлении. Остаток анализировали с помощью хиральной HPLC (способ 1) и 1H ЯМР.
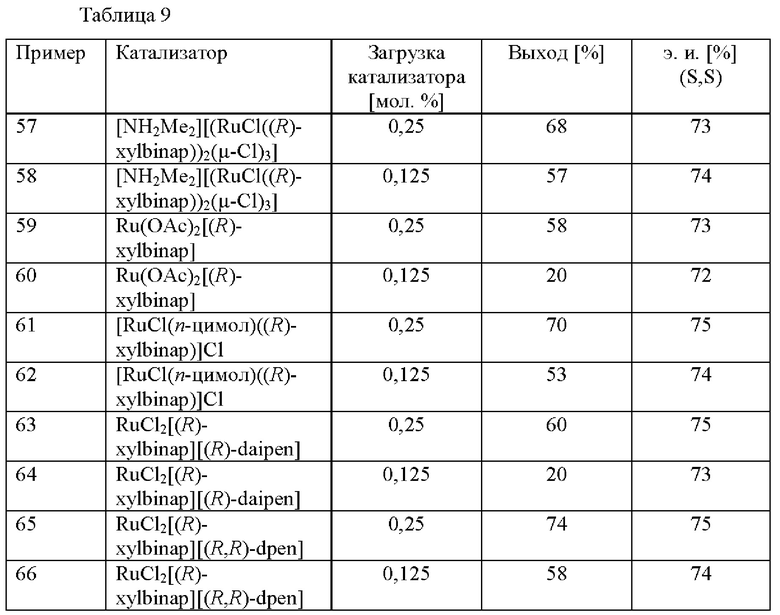
Примеры 57-66
Различные предварительно образованные катализаторы на основе Rh тестировали на скрининговой платформе. В реакционный сосуд загружали 2-(2,4-дихлорфенил)циклобутанон (2,0 ммоль), соответствующий катализатор (см. таблицу 9), ацетат аммония (2,4 ммоль) и МеОН (2,5 мл). Применяли атмосферу водорода под давлением 30 бар и реакционную смесь нагревали до 80°С. После периода перемешивания длительностью 16 ч. реакционную смесь охлаждали до к. т. и анализировали с помощью хиральной (способ 1) и ахиральной (способ 2) HPLC.
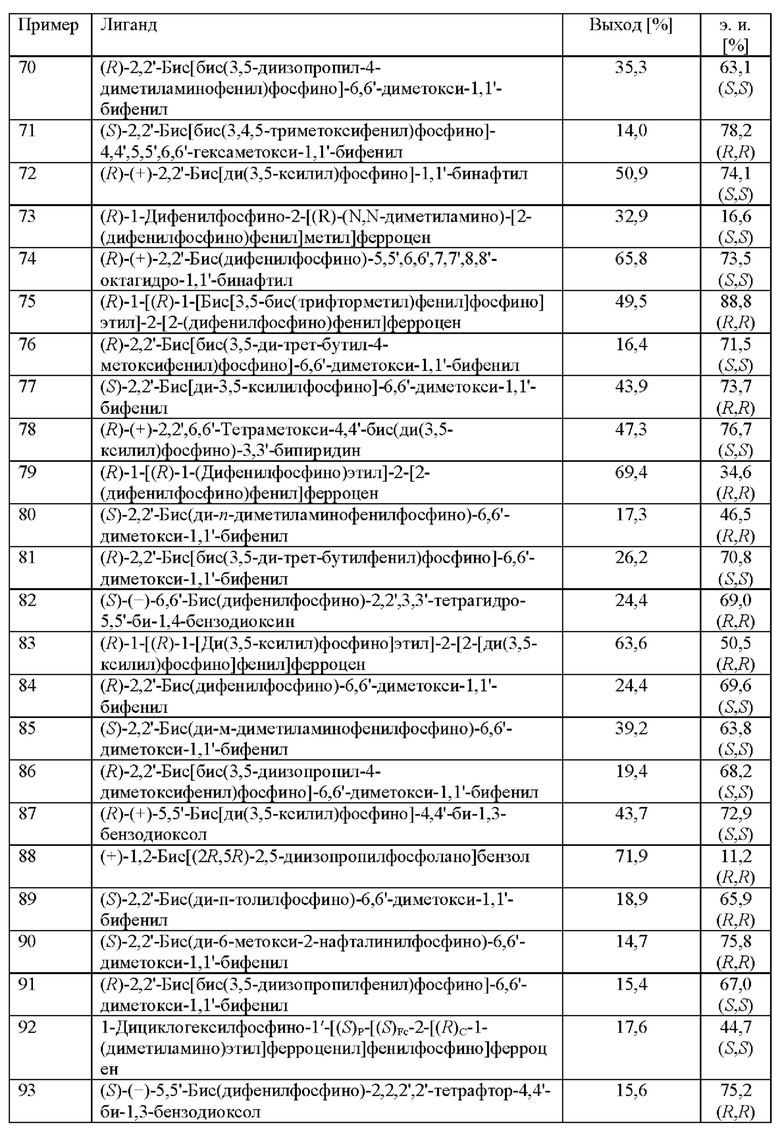
Примеры 67-93
Различные хиральные лиганды в комбинации с рутением тестировали на скрининговой платформе. Растворяли [Ru(Ме-аллил)2(cod)] и соответствующий лиганд (см. таблицу 10) в ацетоне (2,5 мл) и затем перемешивали в течение 1 ч. при 25°С. Смесь выпаривали до сухого состояния, добавляли МеОН и переносили раствор катализатора (0,25 мол. %) в автоклав вместе с 2-(2,4-дихлорфенил)циклобутаноном (2,0 ммоль), ацетатом аммония (2,4 ммоль) и МеОН (2,5 мл). Применяли атмосферу водорода под давлением 30 бар и реакционную смесь нагревали до 80°С. После периода перемешивания длительностью 16 ч. реакционную смесь охлаждали до к. т. и анализировали с помощью хиральной (способ 1) и ахиральной (способ 2) HPLC.

Пример 94
В реактор высокого давления загружали раствор метоксиацетата аммония в МеОН (323 г, 301 ммоль), метанол (123 г), диацетато-((R)-(+)-2,2'-бис[ди(3,5-ксилил)фосфино]-1,1'-бинафтил)рутений (0,72 г, 0,75 ммоль). Реактор герметично закрывали и создавали 3 перепада давления до N2 (6 бар) без перемешивания, затем 3 перепада давления до Н2 (6 бар) при перемешивании и смесь нагревали до 55°С. Давление повышали до 10 бар с применением водорода и реактор нагревали до 80°С. При 80°С давление увеличивалось до 30 бар. Добавляли раствор 2-(2,4-дихлорфенил)циклобутанона в МеОН (110 г, 249 ммоль) в реактор в течение 4 ч. Реакционную смесь перемешивали в течение 8 часов, пока поглощение водорода не прекратилось. Давление понижали и создавали 3 перепада давления до N2 (6 бар). Затем реакционную массу концентрировали в вакууме (60°С, 90 мбар) с получением коричневого масла. К неочищенному продукту добавляли воду (450 мл) и этилацетат (300 мл). Добавляли 32% водный раствор HCl (37,5 г) для достижения рН 1 и обеспечивали разделение фаз. Нижнюю водную фазу удаляли и верхнюю органическую фазу промывали 1 М водным раствором HCl (50 мл) и обеспечивали разделение фаз. Нижнюю водную фазу объединяли с предыдущей водной фазой и верхнюю органическую фазу отбрасывали. К объединенным водным фазам добавляли толуол (100 мл) и двухфазную систему охлаждали до 10°С. Добавляли 30% водный раствор NaOH (80 г) для достижения рН 10, с поддержанием температуры ниже 20°С. Обеспечивали разделение фаз. Нижнюю водную фазу удаляли и дважды промывали толуолом (2 × 100 мл). Объединенные органические слои и слои толуола промывали водой (100 мл) и концентрировали при пониженном давлении с получением (1S,2S)-2-(2,4-дихлорфенил)циклобутанамина (45,5 г) в виде масла.
1Н ЯМР (400МГц, DMSO-d6) δ 7,54 (m, 1H), 7,41(d, J=1 Гц, 2H); 3,85-3,75 (m, 2Н), 2,40-2,24 (m, 2Н), 2,11-2,02 (m, 1H), 1,61-1,53 (m, 1 Н), 1,15 (br s, 2H).
Примеры 95-98
Следующие соединения в таблице 11 получали образом, аналогичным примерам 10-94.
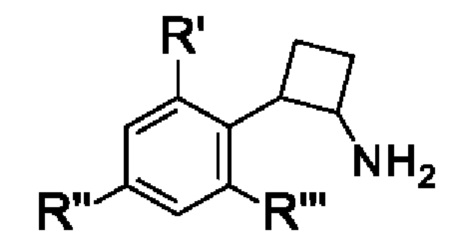
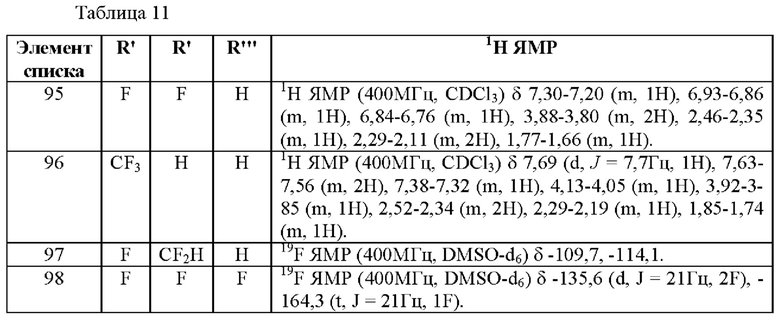
Четвертый аспект
Реакция (d) в соответствии со схемой 1
Пример 99
В раствор 2-(2,4-дихлорфенил)циклобутанона (5,00 г, 23,2 ммоль) в хлорбензоле (23,2 мл) добавляли при к. т. AlCl3 (4,65 г, 34,9 ммоль), CH3CN (6,08 мл, 116 ммоль) и AcCl (2,48 мл, 34,9 ммоль). Наблюдали слабый экзотермический эффект при добавлении AcCl. Полученную желтоватую суспензию нагревали при 50°С в течение 1 ч. После охлаждения до к. т. реакционную смесь добавляли по каплям в холодный (0°С) 4 н. водный раствор NaOH (116 мл) и 50 мл толуола. Во время добавления образовывался бледно-желтый осадок. После фильтрации получали N-[2-(2,4-дихлорфенил)циклобутен-1-ил]ацетамид (3,56 г) в виде белого твердого вещества.
1H ЯМР (400МГц, DMSO-d6) δ 9,30 (bs, 1H); 7,51 (s, 1H), 7,47 (m, 2Н), 2,85 (m, 2Н), 2,68 (m, 2Н), 1,92 (s, 3Н).
Примеры 100-107
Следующие соединения в таблице 12 получали образом, аналогичным примеру 99.
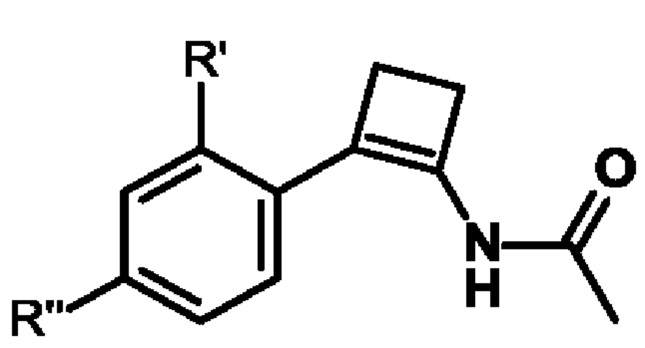

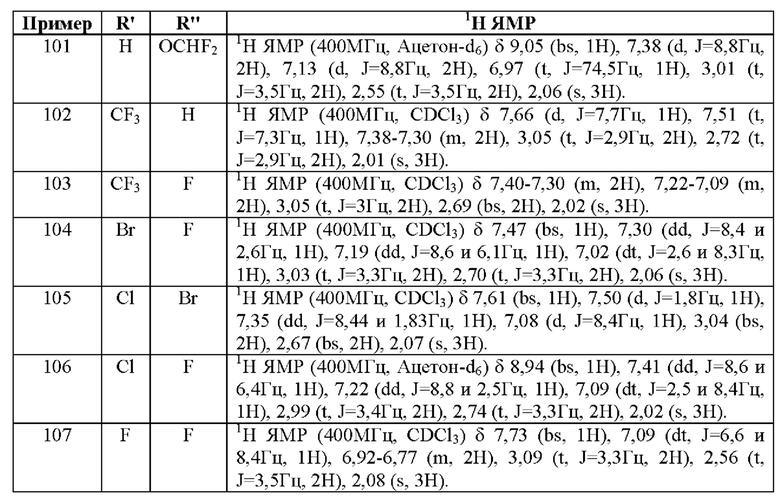
Примеры 108-119
N-[2-(2,4-Дихлорфенил)циклобутен-1-ил]ацетамид (соединение формулы (VI)) может быть получен аналогично примеру 99 с применением параметров реакции, 5 указанных в примерах 108-119.
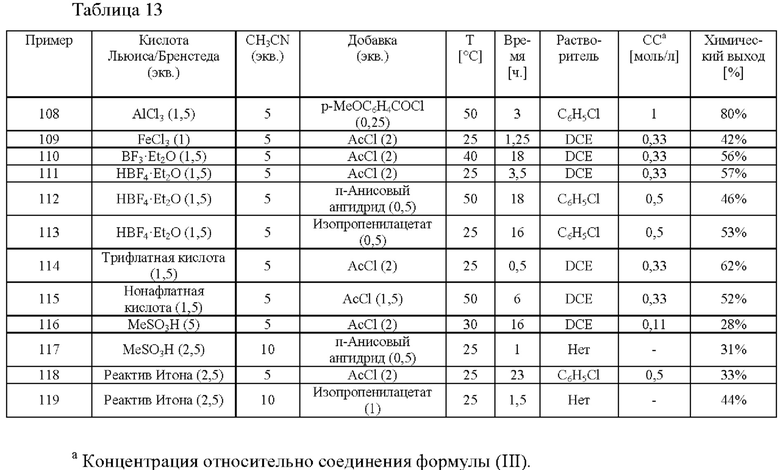
Пятый аспект
Реакция (d') в соответствии со схемой 1
Пример 120
В раствор 1-(2,4-дихлорфенил)циклопропанкарбальдегида (2,1 г, 9,3 ммоль) в хлорбензоле (9 мл) добавляли при к. т. AlCl3 (1,9 г, 14 ммоль). Полученную желтоватую суспензию нагревали при 60°С в течение 30 мин. После охлаждения до 35°С добавляли CH3CN (4,38 мл, 83,7 ммоль) и AcCl (1,70 мл, 23,3 ммоль). Наблюдали слабый экзотермический эффект при добавлении AcCl. Реакционную смесь нагревали до 50°С в течение 30 мин, затем после охлаждения до 5°С реакционную смесь добавляли по каплям в холодный (0°С) 4 н. водный раствор NaOH (46 мл) и 50 мл толуола. Во время добавления образовывался бледно-желтый осадок. Внутреннюю температуру поддерживали в пределах от 0°С до 5°С. Двухфазную суспензию переносили в делительную воронку объемом 250 мл, содержащую 200 мл толуола. Водную фазу дважды экстрагировали толуолом (250 и 50 мл), объединенные органические фазы один раз промывали 1 н. водным раствором NaOH (50 мл) и водой (2 × 50 мл; пока рН водной фазы не становился нейтральным). Затем объединенные органические слои высушивали над твердым Na2SO4, фильтровали и концентрировали при пониженном давлении. Получали N-[2-(2,4-дихлорфенил)циклобутен-1-ил]ацетамид (2,3 г) в виде желтого твердого вещества.
1Н ЯМР (400МГц, DMSO-d6) δ 9,30 (bs, 1H), 7,51 (s, 1H), 7,47 (m, 2H), 2,85 (m, 2H), 2,68 (m, 2H), 1,92 (s, 3Н).
Примеры 121-125
Следующие соединения в таблице 14 получали образом, аналогичным примеру 120.
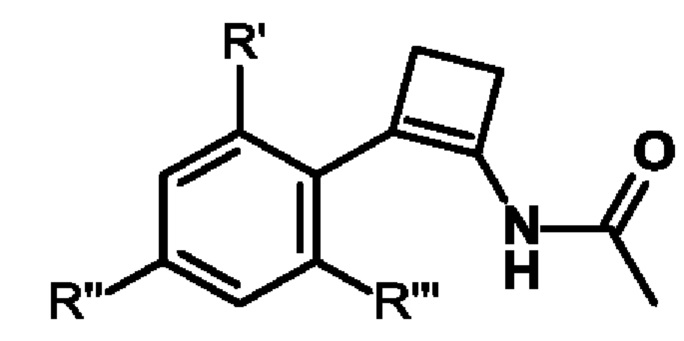


Шестой аспект
Реакция (е) в соответствии со схемой 1
Примеры 126-131
Предварительное образование катализатора
В стерильной камере с перчатками в сосуд загружали предкатализатор бис(COD)родия(I) трифторметансульфонат (0,01 ммоль) и (R)-1-[(S)-2-(ди-трет-бутилфосфино)ферроценил]этил-ди-о-толилфосфин (0,011 ммоль). Затем добавляли соответствующий растворитель (10,0 мл) и раствор перемешивали в течение 90 минут при к. т.
Асимметричное гидрирование
В автоклав под давлением загружали N-[2-(2,4-дихлорфенил)циклобутен-1-ил]ацетамид (0,97 ммоль), соответствующий растворитель (1,50 мл) и ранее полученный раствор катализатора (1,00 мл). Применяли атмосферу водорода под давлением 10 бар и реакционную смесь нагревали до 50°С. Реакционную смесь охлаждали до к. т.после периода перемешивания длительностью 2 ч. при 50°С.
Реакционную смесь концентрировали при пониженном давлении и анализировали с помощью количественного 1H-ЯМР и хиральной HPLC (способ 4).
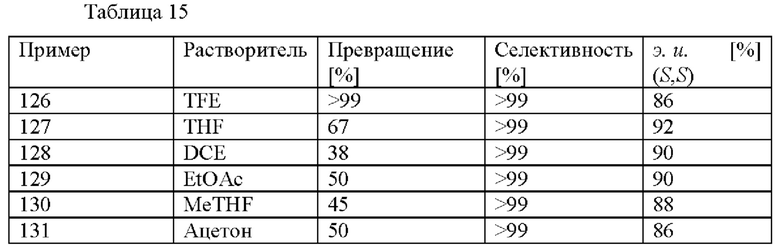
TFE: 2,2,2-тетрафторэтанол; DCE: 1,2-дихлорэтан. Термин "селективность", используемый в таблице выше, означает селективность реакции в отношении соединений формулы (VI).
Примеры 132-133
Предварительное образование катализатора
В стерильной камере с перчатками в сосуд загружали предкатализатор бис(COD)родия(1) трифторметансульфонат (0,01 ммоль) и (R)-1-[(S)-2-(ди-трет-бутилфосфино)ферроценил]этил-ди-о-толилфосфин (0,011 ммоль). Затем добавляли МеОН (5,00 мл) и раствор перемешивали в течение 30 минут при к. т.
Асимметричное гидрирование
В автоклав под давлением загружали N-[2-(2,4-дихлорфенил)циклобутен-1-ил]ацетамид (0,98 ммоль), МеОН (4,00 мл) и ранее полученный раствор катализатора (1,00 мл). Применяли атмосферу водорода под соответствующим давлением и реакционную смесь нагревали до 50°С. Реакционную смесь охлаждали до к. т. после периода перемешивания длительностью 2 ч. при 50°С.
Реакционную смесь концентрировали при пониженном давлении и анализировали с помощью количественного 1H-ЯМР и хиральной HPLC (способ 4).
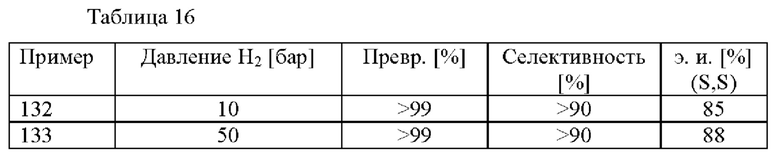
Термин "селективность", используемый в таблице выше, означает селективность реакции в отношении соединений формулы (VI).
Примеры 134-135
Предварительное образование катализатора
В стерильной камере с перчатками в сосуд загружали предкатализатор бис(COD)родия(1) трифторметансульфонат (0,01 ммоль) и (R)-1-[(S)-2-(ди-трет-бутилфосфино)ферроценил]этил-ди-о-толилфосфин (0,011 ммоль). Затем добавляли МеОН (5,00 мл) и раствор перемешивали в течение 30 минут при к. т.
Асимметричное гидрирование
В автоклав под давлением загружали N-[2-(2,4-дихлорфенил)циклобутен-1-ил]ацетамид (0,98 ммоль), МеОН (4,00 мл) и ранее полученный раствор катализатора (1,00 мл). Применяли атмосферу водорода под давлением 50 бар и реакционную смесь нагревали до температуры, указанной в таблице. Реакционную смесь охлаждали до к. т. после периода перемешивания длительностью 2 ч. при температуре, указанной в таблице.
Реакционную смесь концентрировали при пониженном давлении и анализировали с помощью количественного 1H-ЯМР и хиральной HPLC (способ 4).
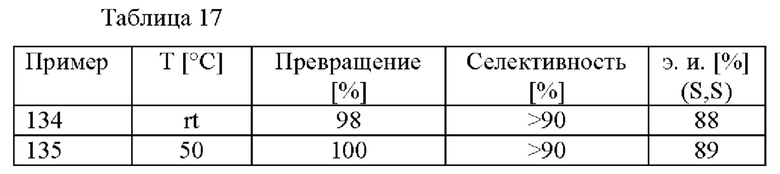
Термин "селективность", используемый в таблице выше, означает селективность реакции в отношении соединений формулы (VI).
Примеры 136-139
Предварительное образование катализатора
В стерильной камере с перчатками в сосуд загружали соответствующий предкатализатор (0,01 ммоль) и (R)-1-[(S)-2-(ди-трет-бутилфосфино)ферроценил]этил-ди-отолилфосфин (0,011 ммоль). Затем добавляли соответствующий растворитель (10,0 мл) и раствор перемешивали в течение 90 минут при к. т.
Асимметричное гидрирование
В автоклав под давлением загружали N-[2-(2,4-дихлорфенил)циклобутен-1-ил]ацетамид (0,97 ммоль), соответствующий растворитель (1,50 мл) и ранее полученный раствор катализатора (1,00 мл). Применяли атмосферу водорода под давлением 10 бар и реакционную смесь нагревали до 50°С. Реакционную смесь охлаждали до к. т. после периода перемешивания длительностью 2 ч. при 50°С.
Реакционную смесь концентрировали при пониженном давлении и анализировали с помощью количественного 1H-ЯМР и хиральной HPLC (способ 4).
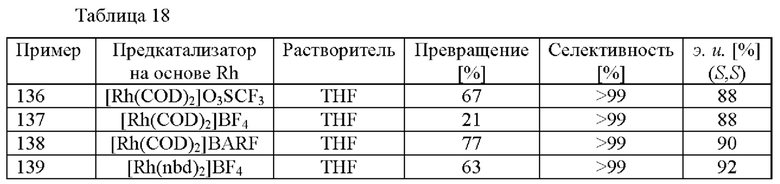
Термин "селективность", используемый в таблице выше, означает селективность реакции в отношении соединений формулы (VI).
Примеры 140-149
Различные предварительно образованные катализаторы на основе Rh тестировали на скрининговой платформе. В реакционный сосуд загружали N-[2-(2,4-дихлорфенил)циклобудет-1-ил]ацетамид (31,7 мкмоль), соответствующий катализатор (0,158 мкмоль) и соответствующий растворитель (500 мкл). Применяли атмосферу водорода под давлением 50 бар и реакционную смесь нагревали до 50°С. После периода перемешивания длительностью 16 ч. реакционную смесь охлаждали до к. т. и анализировали с помощью ахиральной (способ 3) и хиральной HPLC (способ 4).
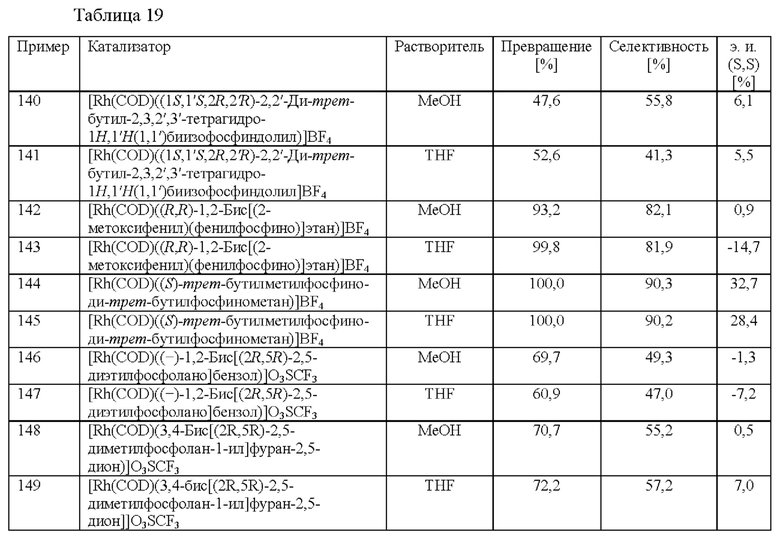
Термин "селективность", используемый в таблице выше, означает селективность реакции в отношении соединений формулы (VI).
Примеры 150-166
Предварительное образование катализатора
В стерильной камере с перчатками в сосуд загружали [Rh(COD)2]O3SCF3 (0,158 мкмоль) и соответствующий хиральный лиганд (0,190 мкмоль). Затем добавляли дихлорэтан и раствор перемешивали в течение 30 минут при к. т., после чего растворитель выпаривали при пониженном давлении.
Асимметричное гидрирование
Добавляли N-[2-(2,4-дихлорфенил)циклобутен-1-ил]ацетамид (31,7 мкмоль) и МеОН (500 мкл) к ранее полученному катализатору. Применяли атмосферу водорода под давлением 50 бар и реакционную смесь нагревали до 50°С. Реакционную смесь охлаждали до к. т.после периода перемешивания длительностью 16 ч. при 50°С.
Реакционную смесь концентрировали при пониженном давлении и анализировали с помощью ахиральной (способ 3) и хиральной HPLC (способ 4).
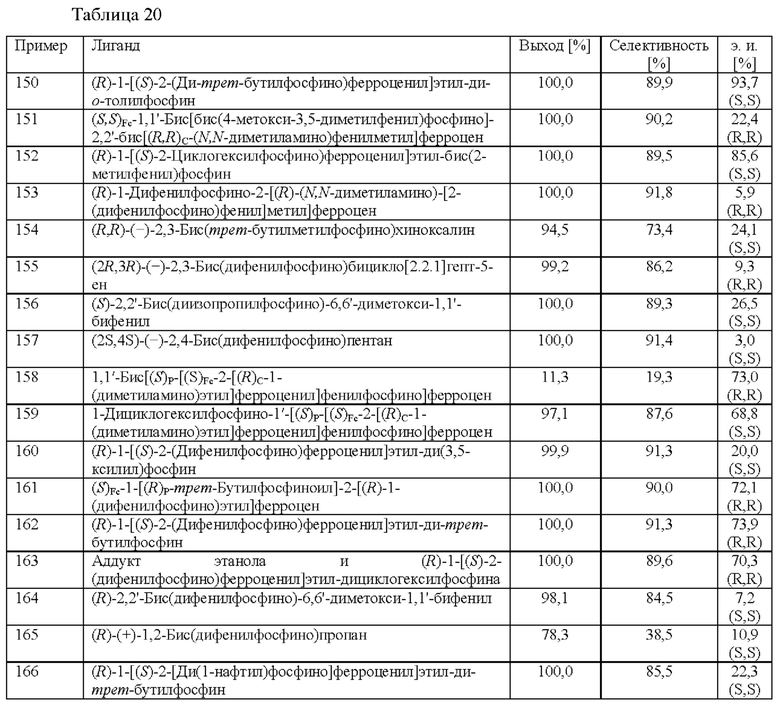
Термин "селективность", используемый в таблице выше, означает селективность реакции в отношении соединений формулы (VI).
Примеры 167-172
Предварительное образование катализатора
В стерильной камере с перчатками в сосуд загружали [Rh(COD)2]O3SCF3 (4,52 мг, 0,0096 ммоль) и соответствующий хиральный лиганд (0,01208 ммоль). Затем добавляли трифторэтанол, дегазированный с помощью аргона (10,0 мл), и раствор перемешивали в течение 60 минут при к. т.
Асимметричное гидрирование
В автоклав под давлением загружали N-[2-(2,4-дихлорфенил)циклобутен-1-ил]ацетамид (250 мг, 0,966 ммоль), трифторэтанол (0,50 мл) и ранее полученный раствор катализатора (2,00 мл). Применяли атмосферу водорода под давлением 10 бар и реакционную смесь нагревали до 50°С. Реакционную смесь охлаждали до к. т. после периода перемешивания длительностью 2 ч. при 50°С.
Реакционную смесь концентрировали при пониженном давлении и анализировали с помощью ахиральной (способ 3) и хиральной HPLC (способ 4).
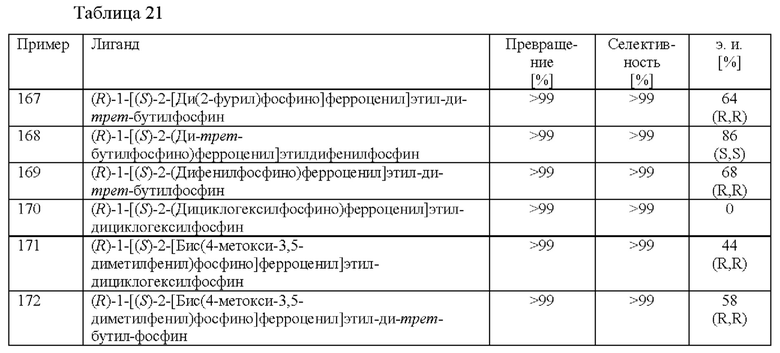
Термин "селективность", используемый в таблице выше, означает селективность реакции в отношении соединений формулы (VI).
Пример 173
Перемешивали смесь Rh(COD)2OTf (1,3 кг, 0,5 мол. %) и (R)-1-[(S)-2-(ди-трет-бутилфосфино)ферроценил]этил-ди-о-толилфосфина (2,1 кг, 0,7 мол. %) в TFE (174 кг, 1,74 кмоль) в течение 90 минут при 50°С, после чего ее добавляли в суспензию N-[2-(2,4-дихлорфенил)циклобутен-1-ил]ацетамида (142 кг, 0,55 кмоль) в IPA (изопропиловый спирт, 866 кг, 14,4 кмоль). Гидрирование осуществляли при 50°С и давлении водорода 10 бар до достижения полного превращения. Затем реакционный раствор (1176 кг; э. и. (S,S) 91%) переносили на следующую стадию.
1H ЯМР (400МГц, DMSO-d6) δ 8,1 (bs, 1Н), 7,6 (d, J=4 Гц, 1H), 7,5 (m, 2H), 4,1 (m, 1H), 4,0 (m, 1H), 3,3 (s, 3H), 2,9 (квинтет, J=8 Гц, 1H), 2,5 (m, 1H), 2,2 (m, 1H), 1,8 (m, 1H).
Реакция (e) в соответствии со схемой 1 с применением катализаторов на основе Ru
Примеры 174-180
Предварительное образование катализатора:
В стерильной камере с перчатками в сосуд загружали предкатализатор бис(2-металлил)(COD)рутений (3,72 мг) и (S,S)-(-)-2,3-бис(трет-бутилметилфосфино)хиноксалин (4,26 мг). Затем добавляли дихлорметан, дегазированный с помощью аргона (3,0 мл), с последующим добавлением метансульфоновой кислоты (1,2 мг). Раствор перемешивали в течение 30 минут при к. т.
Асимметричное гидрирование
В автоклав под давлением загружали N-[2-(2,4-дихлорфенил)циклобутен-1-ил]ацетамид (0,25 г, 0,97 ммоль), соответствующий растворитель, дегазированный с помощью аргона (2,0 мл), и часть ранее полученного раствора катализатора (0,5 мл). Применяли атмосферу водорода под давлением 50 бар и реакционную смесь нагревали до 50°С. Реакционную смесь охлаждали до к. т. после периода перемешивания длительностью 2 ч. при 50°С.
Реакционную смесь концентрировали при пониженном давлении и анализировали с помощью количественного 1Н-ЯМР и хиральной HPLC (способ 4).
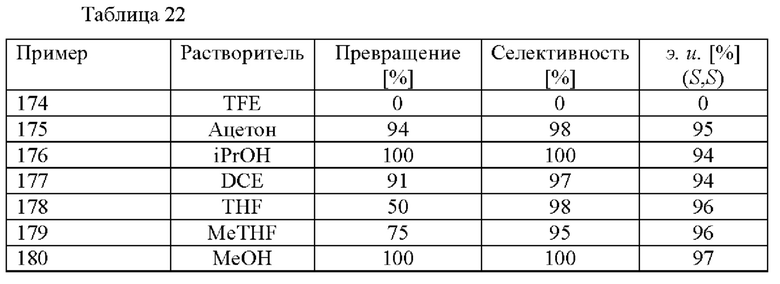
Термин "селективность", используемый в таблице выше, означает селективность реакции в отношении соединений формулы (VI).
Примеры 181-182
Предварительное образование катализатора:
В стерильной камере с перчатками в сосуд загружали бис(2-металлил)(COD)рутений (6,2 мг) и (S,S)-(-)-2,3-бис(трет-бутилметилфосфино)хиноксалин (7,1 мг). Затем добавляли дихлорметан, дегазированный с помощью аргона (4,0 мл), с последующим добавлением комплекса фторборной кислоты с диэтиловым эфиром (0,24 мл, 0,081 ммоль/мл). Раствор перемешивали в течение 30 минут при к. т. Объем в сосуде уменьшали путем барботирования аргона через раствор (конечный объем: 1 мл), после чего добавляли дегазированный МеОН (9 мл).
Асимметричное гидрирование
В автоклав под давлением загружали N-[2-(2,4-дихлорфенил)циклобутен-1-ил]ацетамид (0,5 г, 1,933 ммоль), МеОН, дегазированный с помощью аргона (4,0 мл), и часть ранее полученного раствора катализатора (1,0 мл). Применяли атмосферу водорода под соответствующим давлением и реакционную смесь нагревали до 50°С. Реакционную смесь охлаждали до к. т. после периода перемешивания длительностью 2 ч. при 50°С.
Реакционную смесь концентрировали при пониженном давлении и анализировали с помощью количественного 1Н-ЯМР и хиральной HPLC (способ 4).
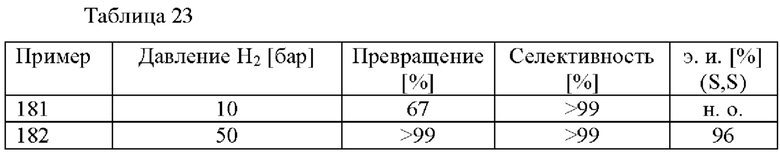
Термин "селективность", используемый в таблице выше, означает селективность реакции в отношении соединений формулы (VI).
н. о. = не определено.
Пример 183
Предварительное образование катализатора:
В сосуд загружали бис(COD)тетра[μ-трифторацетато]дирутения(II) гидрат (1,40 мг, 0,0015 ммоль), (S,S)-(-)-2,3-бис(трет-бутилметилфосфино)хиноксалин (1,10 мг, 0,0032 ммоль) и N-[2-(2,4-дихлорфенил)циклобутен-1-ил]ацетамид (0,40 г, 1,53 ммоль). Затем в сосудах создавали инертную атмосферу с помощью аргона и добавляли МеОН, дегазированный с помощью аргона (5,0 мл). Применяли атмосферу водорода под давлением 50 бар и реакционную смесь нагревали до 50°С. Реакционную смесь охлаждали до к. т. после периода перемешивания длительностью 2,5 ч. при 50°С.
Реакционную смесь концентрировали при пониженном давлении и анализировали с помощью количественного 1Н-ЯМР и хиральной HPLC (способ 4).

Термин "селективность", используемый в таблице выше, означает селективность реакции в отношении соединений формулы (VI).
Примеры 184-199
Предварительное образование катализатора
В стерильной камере с перчатками в сосуд загружали соответствующий предшественник катализатора (0,158 мкмоль; когда применяли [Ru(COD)(2-металлил)2] в качестве предшественника катализатора, в сосуд также загружали HBF4, (0,316 мкмоль)) и соответствующий хиральный лиганд (0,19 мкмоль). Затем добавляли дихлорэтан и раствор перемешивали в течение 30 минут при к. т., после чего растворитель выпаривали при пониженном давлении.
Асимметричное гидрирование
Добавляли N-[2-(2,4-дихлорфенил)циклобутен-1-ил]ацетамид (31,7 мкмоль) и МеОН (500 мкл) к ранее полученному катализатору. Применяли атмосферу водорода под давлением 50 бар и реакционную смесь нагревали до 50°С. Реакционную смесь охлаждали до к. т. после периода перемешивания длительностью 16 ч. при 50°С.
Реакционную смесь концентрировали при пониженном давлении и анализировали с помощью ахиральной (способ 3) и хиральной HPLC (способ 4).
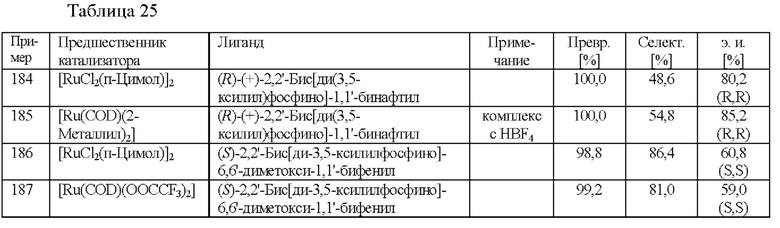
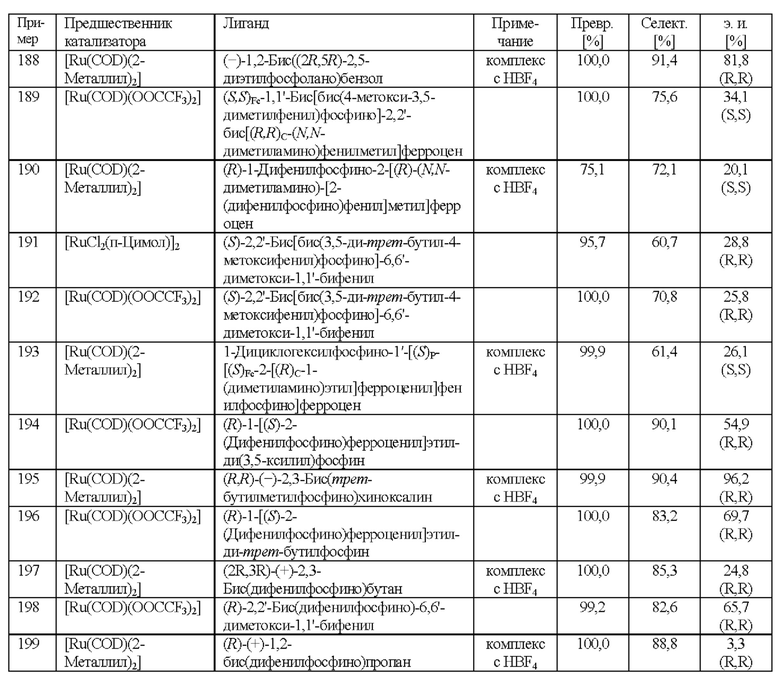
Термин "селективность", используемый в таблице выше, означает селективность реакции в отношении соединений формулы (VI).
Примеры 200-206
Предварительное образование катализатора
В стерильной камере с перчатками в сосуд загружали бис(2-металлил)(COD)рутений (3,10 мг, 0,0097 ммоль), соответствующий хиральный лиганд (0,0107 ммоль) и комплекс фторборной кислоты с диэтиловым эфиром (1,72 мг, 0,0106 ммоль). Затем добавляли дихлорметан, дегазированный с помощью аргона (2,5 мл), и раствор перемешивали в течение 30 минут при к. т.
Асимметричное гидрирование
В автоклав под давлением загружали N-[2-(2,4-дихлорфенил)циклобутен-1-ил]ацетамид (250 мг, 0,97 ммоль), МеОН, дегазированный с помощью аргона (2,0 мл), и ранее полученный раствор катализатора (0,5 мл). Применяли атмосферу водорода под давлением 50 бар и реакционную смесь нагревали до 50°С. Реакционную смесь охлаждали до к. т. после периода перемешивания длительностью 2 ч. при 50°С.
Реакционную смесь концентрировали при пониженном давлении и анализировали с помощью ахиральной (способ 3) и хиральной HPLC (способ 4).
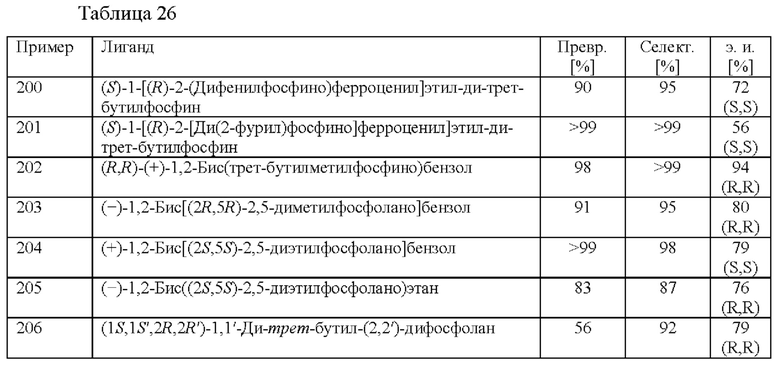
Термин "селективность", используемый в таблице выше, означает селективность реакции в отношении соединений формулы (VI).
Примеры 207-208
Предварительное образование катализатора
В стерильной камере с перчатками в сосуд загружали бис(2-металлил)(COD)рутений (3,72 мг, 0,0116 ммоль) и соответствующий хиральный лиганд (0,0127 ммоль). Затем добавляли дихлорметан, дегазированный с помощью аргона (3,00 мл), и 0,081 М раствор метансульфоновой кислоты в дихлорметане (0,16 мл, 0,0130 ммоль) и раствор перемешивали в течение 30 минут при к. т.
Асимметричное гидрирование
В автоклав под давлением загружали N-[2-(2,4-дихлорфенил)циклобутен-1-ил]ацетамид (250 мг, 0,97 ммоль), МеОН, дегазированный с помощью аргона (2,0 мл), и ранее полученный раствор катализатора (0,5 мл). Применяли атмосферу водорода под давлением 50 бар и реакционную смесь нагревали до 50°С. Реакционную смесь охлаждали до к. т. после периода перемешивания длительностью 2 ч. при 50°С.
Реакционную смесь концентрировали при пониженном давлении и анализировали с помощью ахиральной и хиральной HPLC.

Термин "селективность", используемый в таблице выше, означает селективность реакции в отношении соединений формулы (VI).
Седьмой аспект
Реакция (f) в соответствии со схемой 1
Пример 209
Реакционную смесь, состоящую из N-[(1S,2S)-2-(2,4-дихлорфенил)циклобутил]ацетамида в изопропаноле/трифторэтаноле (428 г, 153 ммоль) и суспензии Aerosil-200 (23 мг) в изопропаноле (0,5 г), нагревали до Та=130°С. Часть изопропанола и трифторэтанола (примерно 220 г) удаляли при температуре 82°С, измеренной в верхней части оборудования для перегонки, после чего добавляли 50% водный раствор Н3РО4 (150 г, 765 ммоль). Удаляли большее количество изопропанола и трифторэтанола до тех пор, пока температура в верхней части ректификационной колонны не достигла 100°С. Затем реакционную массу перемешивали в при температуре возврата флегмы до достижения превращения >95% (время реакции: приблизительно 20 часов).
Добавляли воду (145 г, 8,1 моль) в реакционную смесь после того, как ее охлаждали до Ti=60°С. Реакционную смесь дважды экстрагировали толуолом (первый: 169 г, 1,8 моль; второй: 86 г, 0,9 моль), после чего регулировали рН водного раствора до 8,0-8,5 с применением 25% водного раствора аммиака (117,5 г, 1,7 моль). Основную водную фазу экстрагировали толуолом (169 г, 1,8 моль). Затем органический слой концентрировали при пониженном давлении и фильтровали с получением 25,3% раствора (1S,2S)-2-(2,4-дихлорфенил)циклобутанамина в толуоле (136 г).
1H ЯМР (400МГц, CDCl3) δ 7,54 (t, J=4 Гц, 1H), 7,41 (d, J=4 Гц, 2Н); 3,86-3,75 (m, 2Н), 2,41-2,24 (m, 2Н), 2,13-2,00 (m, 1Н), 1,60-1,53 (m, 1 Н).
Пример 210
Реакционную смесь, состоящую из N-[(1S,2S)-2-(2,4-дихлорфенил)циклобутил]ацетамида (10 г, 35,8 ммоль), метансульфоновой кислоты (6,9 г, 71,5 ммоль) и воды (6,4 г, 358 ммоль), нагревали до 110°С до достижения температуры возврата флегмы. Реакционную массу перемешивали при температуре возврата флегмы в течение 21 ч., после чего ее охлаждали до к. т. В реакционную смесь добавляли воду (40 мл) и затем ее дважды экстрагировали толуолом (первый: 50 мл; второй: 30 мл). Регулировали рН водного раствора до 8,9 с применением 25% водного раствора аммиака (9,6 г, 141 моль). Основную водную фазу экстрагировали толуолом (50 мл) с получением 11,9% раствора (1S,2S)-2-(2,4-дихлорфенил)циклобутанамина в толуоле (59,8 г).
Пример 211
Добавляли N-[(1S,2S)-2-(2,4-дихлорфенил)циклобутил]ацетамид (5,0 г, 17,9 ммоль) в 60% водный раствор H2SO4 (17,5 г, 107 ммоль). Реакционную смесь нагревали до 128°С до достижения температуры возврата флегмы. Реакционную массу охлаждали до 60°С после периода перемешивания длительностью 21 ч. при температуре возврата флегмы. В реакционную смесь добавляли воду (75 мл) и затем ее дважды экстрагировали толуолом (первый: 25 мл; второй: 25 мл). Регулировали рН водного раствора до 8,8 с применением 25% водного раствора аммиака (5,8 г, 85,1 моль). Основную водную фазу экстрагировали толуолом (100 мл). Затем органический слой концентрировали при пониженном давлении с получением 15,4% раствора (1S,2S)-2-(2,4-дихлорфенил)циклобутанамина в толуоле (17,3 г).
Восьмой аспект
Реакция (g) в соответствии со схемой 1
Пример 212
Добавляли раствор (1S,2S)-2-(2,4-дихлорфенил)циклобутанамина в толуоле (339 г, 0,40 моль) к твердому NaHCO3 (47 г, 0,56 моль). Затем в реакционную смесь добавляли воду (140 г, 7,79 моль) и смесь нагревали до Ti=50°С. Затем в реакционную смесь добавляли раствор 2-(трифторметил)пиридин-3-карбонилхлорида в толуоле (247 г, 0,42 моль) в течение 53 минут при Ti=50°С. После достижения полного превращения реакционную смесь нагревали до Ti=70°С и перемешивали в течение 20 минут при этой же температуре. После разделения фаз органическую фазу экстрагировали водой (201 г, 11,1 моль) при Ti=80°С. После разделения фаз органическую фазу концентрировали до примерно 35% раствора МСН (140 г, 1,4 моль) и затем ее добавляли в течение 20 минут к концентрированной органической фазе при Ti=80°С. Затем реакционную смесь охлаждали до Ti=5°С в течение 2,5 ч., при этом затравку добавляли при Ti=72°С (кристаллизация осуществлялась также без введения затравки). Реакционную смесь перемешивали в течение 30 минут до тех пор, пока реакционная смесь не достигла Ti=5°С, после чего суспензию фильтровали, промывали с помощью МСН (200 г, 2,0 моль) и высушивали при повышенной температуре при пониженном давлении для выделения N-[(1S,2S)-2-(2,4-дихлорфенил)циклобутил]-2-(трифторметил)пиридин-3-карбоксамида (141,6 г) в виде ангидрида.
FT-IR 3282, 3077, 2981, 2952, 1650, 1593, 1543, 1473, 1353, 1187, 1138, 1074, 1066, 1054 см-1
Растворяли ангидрид N-[(1S,2S)-2-(2,4-дихлорфенил)циклобутил]-2-(трифторметил)пиридин-3-карбоксамида (80 г) в смеси ацетона (240 г, 4,1 моль) и воды (80 г, 4,4 моль) при Ti=55°С. Затем смесь охлаждали до Ti=8°С и добавляли кристаллы затравки при Ti=29°С. В реакционную смесь добавляли воду (86 г, 4,8 моль) в течение 60 минут после того, как реакционная смесь достигла температуры 8°С. Реакционную смесь перемешивали в течение 30 минут после добавления другой аликвоты воды (174 г, 9,7 моль) в течение 1 ч. Затем добавляли конечную аликвоту воды (340 г, 18,9 моль) и суспензию перемешивали в течение 80 минут. Суспензию фильтровали и осадок на фильтре промывали водой (2 × 80 г, 4 раза), после чего его сушили при пониженном давлении и температуре 35°С с получением моногидрата N-[(1S,2S)-2-(2,4-дихлорфенил)циклобутил]-2-(трифторметил)пиридин-3-карбоксамида (94,6 г).
1H ЯМР (400МГц, CDCl3) δ 8,65 (dd, J=4,6 Гц, J=1,2 Гц, 1H), 7,60-7,58 (m, 1H), 7,47-7,44 (m, 1H), 7,41-7,40 (m, 1H), 7,33-7,25 (m, 2Н), 5,54 (br d, J=7,8 Гц, 1H), 5,03 (quin, J=7,3 Гц, 1Н), 4,24 (q, J=7,8 Гц, 1H), 2,65-2,56 (m, 1H), 2,44-2,28 (m, 2Н), 2,10-2,01 (m, 1 Н).
FT-IR 3403, 3232, 3079, 2948, 1660, 1645, 1593, 1575, 1471, 1326, 1186, 1126, 1076, 1054 см-1.
Пример 213
Охлаждали двухфазную смесь (1S,2S)-2-(2,4-дихлорфенил)циклобутанамина (20,0 г, 87,3 ммоль) в толуоле (20 г) и воды (40 г) до 0°С и добавляли кристаллы затравки продукта в форме гидрата (1,53 г). Раствор 2-(трифторметил)пиридин-3-карбонилхлорида (19,7 г, 91.6 ммоль) в толуоле (60 г) параллельно дозировали в 30% водн. NaOH (14,0 г, 105 ммоль) в течение 2 ч. таким образом, чтобы поддерживать рН в диапазоне 7-9. После окончания дозирования реакционную смесь перемешивали в течение дополнительно 3 ч. Полученную густую суспензию фильтровали, осадок на фильтре промывали водой (2 × 25 г) и высушивали при давлении 150 мбар в течение 16 ч. с получением N-[(1S,2S)-2-(2,4-дихлорфенил)циклобутил]-2-(трифторметил)пиридин-3-карбоксамида (36,4 г, содержащего 7% воды) в виде моногидрата.
FT-IR 3403, 3232, 3079, 2948, 1660, 1645, 1593, 1575, 1471, 1326, 1186, 1126, 1076, 1054 см-1.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЙ СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ, ИСПОЛЬЗУЕМЫХПРИ ПОЛУЧЕНИИИНГИБИТОРОВ NEP | 2011 |
|
RU2573824C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОПТИЧЕСКИ АКТИВНЫХ АМИНОФОСФИНИЛБУТАНОВЫХ КИСЛОТ | 2007 |
|
RU2442787C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВОРИКОНАЗОЛА И ЕГО АНАЛОГОВ | 2013 |
|
RU2619928C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПРОПИОНОВОЙ КИСЛОТЫ | 2011 |
|
RU2575345C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПРОПИОНОВОЙ КИСЛОТЫ | 2010 |
|
RU2544989C2 |
| СПОСОБ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 5-БИФЕНИЛ-4-ИЛ-2-МЕТИЛПЕНТАНОВОЙ КИСЛОТЫ | 2009 |
|
RU2513521C2 |
| СПОСОБ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 5-БИФЕНИЛ-4-ИЛ-2-МЕТИЛПЕНТАНОВОЙ КИСЛОТЫ | 2013 |
|
RU2636936C2 |
| СПОСОБ ПОЛУЧЕНИЯ БИАРИЛЗАМЕЩЕННОЙ 4-АМИНОМАСЛЯНОЙ КИСЛОТЫ ИЛИ ЕЕ ПРОИЗВОДНЫХ И ИХ ПРИМЕНЕНИЕ В ИЗГОТОВЛЕНИИ ИНГИБИТОРОВ НЭП | 2007 |
|
RU2469019C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФЕНИЛЗАМЕЩЕННОГО ГЕТЕРОЦИКЛИЧЕСКОГО ПРОИЗВОДНОГО ПОСРЕДСТВОМ СОЧЕТАНИЯ С ИСПОЛЬЗОВАНИЕМ ПЕРЕХОДНОГО МЕТАЛЛА В КАЧЕСТВЕ КАТАЛИЗАТОРА | 2010 |
|
RU2510393C9 |
| ФУНГИЦИДНЫЕ ФЕНИЛПИРИМИДИНИЛАМИНО ПРОИЗВОДНЫЕ | 2008 |
|
RU2459819C2 |
Изобретение относится к способу получения энантиомерно и диастереомерно обогащенных циклобутанамидов формулы (VII). Способ предусматривает (a) восстановление нитрильного фрагмента соединения формулы (I) до альдегида, где восстановление нитрильного фрагмента соединения формулы (I) осуществляют путем частичного гидрирования до соответствующего промежуточного имина с использованием Н2 и катализатора на основе металла с последующим последовательным гидролизом с получением соединения формулы (II). Затем способ предусматривает (b) осуществление реакции соединения формулы (II) в присутствии кислоты Льюиса с получением соединения формулы (III). Затем способ предусматривает (c) осуществление реакции соединения формулы (III) с аммониевой солью и Н2 в присутствии хирального катализатора на основе переходного металла с получением энантиомерно и диастереомерно обогащенного амина формулы (IV) и последующее осуществление реакции амина формулы (IV) с соединением формулы (X) с образованием энантиомерно и диастереомерно обогащенного амида формулы (VII). В указанных формулах А выбран из арила и гетероарила, где арил и гетероарил являются незамещенными или замещенными одним или несколькими заместителями, независимо выбранными из галогена, циано, C1-С6алкила, C1-С6галогеналкила, C1-С6алкокси и C1-С6галогеналкокси; Y представляет собой подходящую уходящую группу, такую как ОН, OR или галоген, предпочтительно хлор, R представляет собой C1-С6алкил; Е выбран из арила и гетероарила, где арил и гетероарил являются незамещенными или замещенными одним или несколькими заместителями, независимо выбранными из галогена, циано, C1-С6алкила, C1-С6галогеналкила, C1-С6алкокси и C1-С6галогеналкокси; * обозначает стереоцентр; арил представляет собой моно-, би- или полициклическую ароматическую систему с 6-14 атомами углерода в кольце, гетероарил представляет собой 5-7-членные кольца с 1-3 одинаковыми или различными гетероатомами, выбранными из N, O и S. Предлагаемый способ позволяет улучшить существующие способы получения энантиомерно и диастереомерно обогащенных циклобутанамидов формулы (VII). Изобретение относится также к способам получения соединения формулы (III) и способу получения соединения формулы (V). 4 н. и 10 з.п. ф-лы, 27 табл., 213 пр.
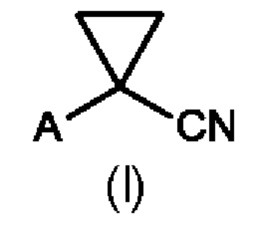
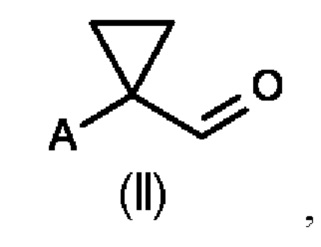
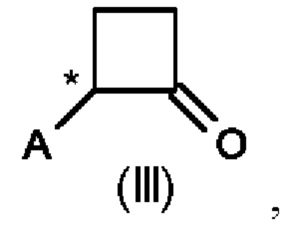
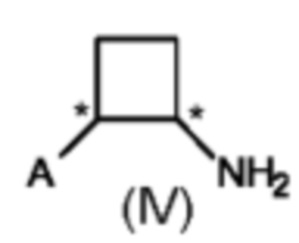
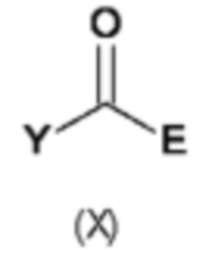
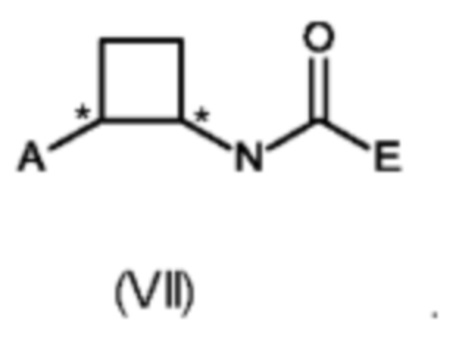
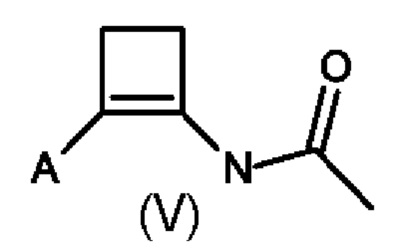
1. Способ получения энантиомерно и диастереомерно обогащенных циклобутанамидов формулы (VII), предусматривающий
(a) восстановление нитрильного фрагмента соединения формулы (I) до альдегида
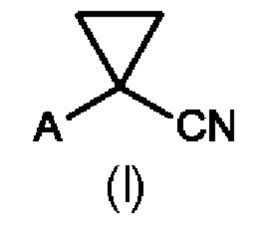 ,
,
где А выбран из арила и гетероарила, где арил и гетероарил являются незамещенными или замещенными одним или несколькими заместителями, независимо выбранными из галогена, циано, C1-С6алкила, C1-С6галогеналкила, C1-С6алкокси и C1-С6галогеналкокси;
где восстановление нитрильного фрагмента соединения формулы (I) осуществляют путем частичного гидрирования до соответствующего промежуточного имина с использованием Н2 и катализатора на основе металла с последующим последовательным гидролизом с получением соединения формулы (II)
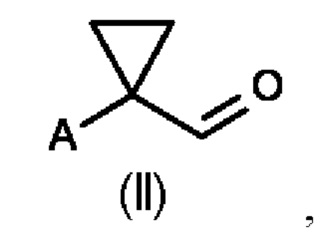 затем
затем
(b) осуществление реакции соединения формулы (II) в присутствии кислоты Льюиса с получением соединения формулы (III)
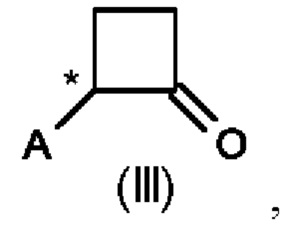
где * обозначает стереоцентр, затем
(c) осуществление реакции соединения формулы (III) с аммониевой солью и Н2 в присутствии хирального катализатора на основе переходного металла с получением энантиомерно и диастереомерно обогащенного амина формулы (IV)
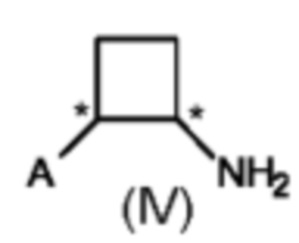 ,
,
где * обозначает стереоцентр, и затем
последующее осуществление реакции амина формулы (IV) с соединением формулы (X)
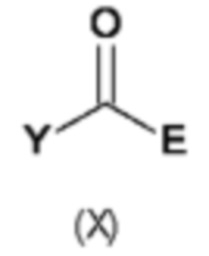 ,
,
где Y представляет собой подходящую уходящую группу, такую как ОН, OR или галоген, предпочтительно хлор, R представляет собой C1-С6алкил и Е выбран из арила и гетероарила, где арил и гетероарил являются незамещенными или замещенными одним или несколькими заместителями, независимо выбранными из галогена, циано, C1-С6алкила, C1-С6галогеналкила, C1-С6алкокси и C1-С6галогеналкокси;
с образованием энантиомерно и диастереомерно обогащенного амида формулы (VII)
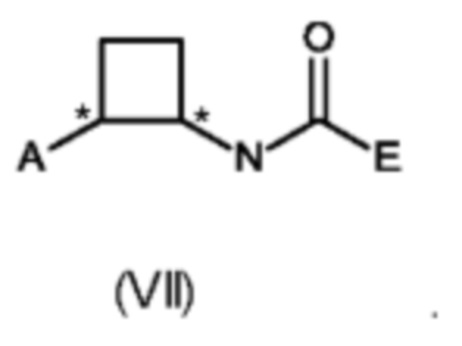 ,
,
где арил представляет собой моно-, би- или полициклическую ароматическую систему с 6-14 атомами углерода в кольце,
гетероарил представляет собой 5-7-членные кольца с 1-3 одинаковыми или различными гетероатомами, выбранными из N, O и S.
2. Способ по п. 1, где А представляет собой фенил и Е представляет собой гетероарил, где фенил и гетероарил являются незамещенными или замещенными одним или несколькими заместителями, независимо выбранными из галогена, циано, C1-С6алкила, C1-С6 галогеналкила, C1-С6алкокси и C1-С6галогеналкокси.
3. Способ по любому из пп. 1, 2, где соединение формулы (II) вводят в реакцию на стадии (b) в присутствии кислоты Льюиса, выбранной из AlCl3 и GaCl3, предпочтительно AlCl3.
4. Способ по п. 3, где на стадии (b) добавляют 1,0-1,5 моль эквивалента AlCl3 или GaCl3 относительно соединения формулы (II).
5. Способ по любому из пп. 1-4, где хиральный катализатор на основе переходного металла на стадии (с) содержит переходный металл, выбранный из Ru, Rh, Ir и Pd, предпочтительно Ru, и хиральный лиганд с бидентатным фосфором общей формулы (VIII)
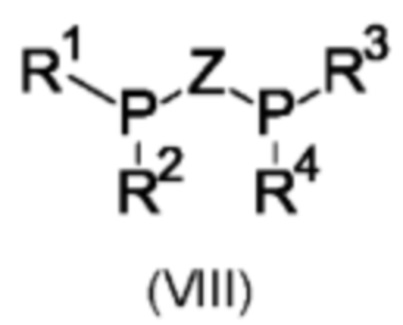 ,
,
где Z представляет собой линкерную группу и R1, R2, R3 и R4 независимо выбраны из арила, гетероарила, C1-С6алкила и C1-С6циклоалкила, каждый из которых является незамещенным или замещенным одним или несколькими заместителями, независимо выбранными из C1-С6алкила, C1-С6алкокси, C1-С6галогеналкила и галогена.
6. Способ по п. 5, где линкерная группа Z выбрана из (R и S)-1,1'-бинафтила, (R и S)-4,4'-бис-1,3-бензодиоксола, (R и S)-2,2',6,6'-тетраметокси-3,3'-бипиридина, (R и S)-6,6'-диметокси-1,1'-бифенила, (R и S)-4,4',6,6'-тетраметокси-1,1'-бифенила, 2,2'-бис-[(R)-cx-(диметиламино)бензил]ферроцена, ферроценилметила, ферроцена, бензола и этила, предпочтительно (R и S)-1,1'-бинафтила.
7. Способ по п. 5 или 6, где хиральный лиганд выбран из
(R)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила,
(R)-2,2'-бис(ди-n-толилфосфино)-1,1'-бинафтила,
(R)-2,2'-бис[ди(3,5-ксилил)фосфино]-1,1'-бинафтила,
(R)-5,5'-бис(дифенилфосфино)-4,4'-би-1,3-бензодиоксола,
(R)-5,5'-бис(ди[3,5-ксилил]фосфино)-4,4'-би-1,3-бензодиоксола,
(R)-5,5'-бис[ди(3,5-ди-трет-бутил-4-метоксифенил)фосфино]-4,4'-би-1,3-бензодиоксола,
(S)-1,13-бис(дифенилфосфино)-7,8-дигидро-6Н-дибензо[f,h][1,5]диоксина,
(R)-2,2',6,6'-тетраметокси-4,4'-бис(ди(3,5-ксилил)фосфино)-3,3'-бипиридина,
(R)-2,2'-бис(дифенилфосфино)-6,6'-диметокси-1,1'-бифенила,
(R)-2,2'-бис(дифенилфосфино)-4,4',6,6'-тетраметокси-1,1'-бифенила,
(R)-6,6'-бис(дифенилфосфино)-2,2',3,3'-тетрагидро-5,5'-би-1,4-бензодиоксина,
(R)-(+)-2,2'-бис(дифенилфосфино)-5,5',6,6',7,7',8,8'-октагидро-1,1'-бинафтила,
(R)-(+)-2,2'-бис(ди-3,5-ксилилфосфино)-5,5',6,6',7,7',8,8'-октагидро-1,1'-бинафтила,
(R)-5,5'-бис(дифенилфосфино)-2,2,2',2'-тетрафтор-4,4'-би-1,3-бензодиоксола,
(S)-1-[(S)-1-[ди(3,5-ксилил)фосфино]этил]-2-[2-[ди(3,5-ксилил)фосфино]фенил]ферроцена
и
(S)-1-[(S)-1-[бис[3,5-бис(трифторметил)фенил]фосфино]этил]-2-[2-(дифенилфосфино)фенил]ферроцена.
8. Способ по п. 5, где хиральный катализатор на основе переходного металла выбран из [RuCl(п-цимол)((S)-DM-SEGPHOS)]Cl, [RuCl(п-цимол)((R)-DM-SEGPHOS)]Cl, [NH2Me2][(RuCl((R)-xylbinap))2(u-Cl)3], [NH2Me2][(RuCl((S)-xylbinap))2(u-Cl)3], Ru(OAc)2[(R)-binap], Ru(OAc)2[(S)-binap], Ru(OAc)2[(R)-xylbinap], Ru(OAc)2[(S)-xylbinap], RuCl2[(R)-xylbinap][(R)-daipen], RuCl2[(S)-xylbinap][(S)-daipen], RuCl2[(R)-xylbinap][(R,R)-dpen] и RuCl2[(S)-xylbinap][(S,S)-dpen].
9. Способ получения соединения формулы (III)
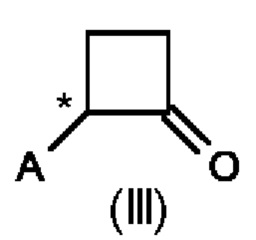 ,
,
где А определен в любом из пп. 1, 2 и * обозначает стереоцентр,
предусматривающий осуществление реакции соединения формулы (II)
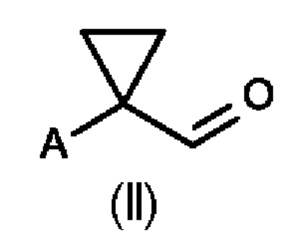
с кислотой Льюиса.
10. Способ получения соединения формулы (III)
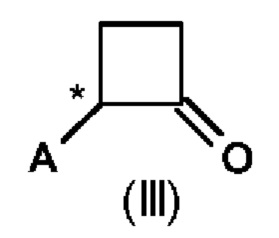 ,
,
где А определен в любом из пп. 1, 2 и * обозначает стереоцентр,
предусматривающий осуществление реакции соединения формулы (II)
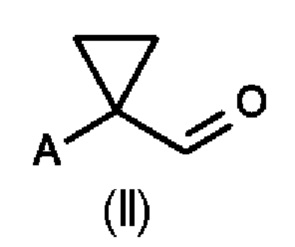
с кислотой Льюиса, выбранной из AlCl3 и GaCl3, предпочтительно AlCl3.
11. Способ по п. 10, где добавляют 1,0-1,5 моль эквивалента AlCl3 или GaCl3 относительно соединения формулы (II).
12. Способ по любому из пп. 9-11, где соединение формулы (III)
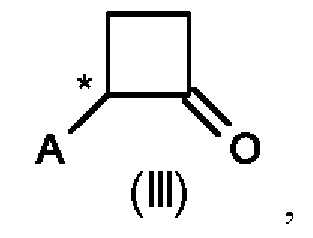
где А определен в любом из пп. 1, 2 и * обозначает стереоцентр,
дополнительно вводят в реакцию с аммониевой солью и Н2 в присутствии хирального катализатора на основе переходного металла с получением энантиомерно и диастереомерно обогащенного амина формулы (IV)
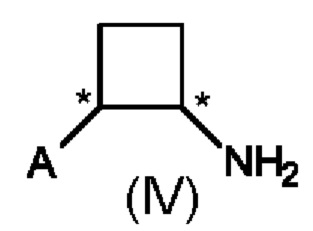 ,
,
где * обозначает стереоцентр, и затем
дополнительно осуществляют реакцию амина формулы (IV) с соединением формулы (X)
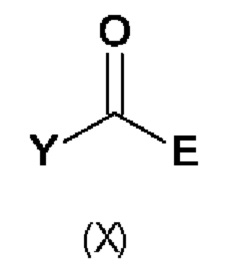 ,
,
где Y представляет собой подходящую уходящую группу, такую как ОН, OR или галоген, предпочтительно хлор, R представляет собой C1-С6алкил и Е выбран из арила и гетероарила, где арил и гетероарил являются незамещенными или замещенными одним или несколькими заместителями, независимо выбранными из галогена, циано, C1-С6алкила, C1-С6галогеналкила, C1-С6алкокси и C1-С6галогеналкокси;
с образованием энантиомерно и диастереомерно обогащенного амида формулы (VII)
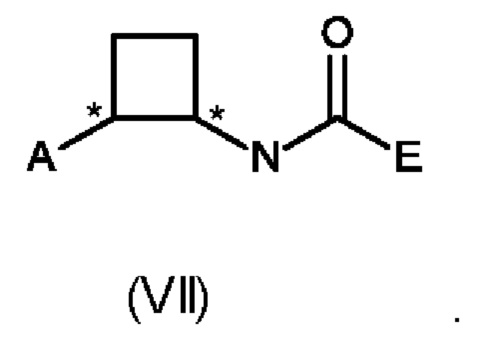
13. Способ по п. 12, где хиральный катализатор на основе переходного металла определен в любом из пп. 5-8.
14. Способ получения соединения формулы (V)
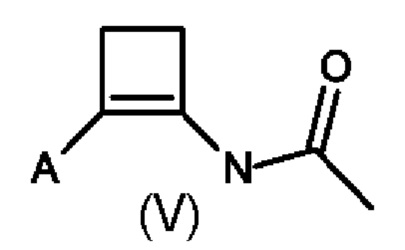 ,
,
где А определен в любом из пп. 1, 2, который предусматривает осуществление реакции соединения формулы (II)
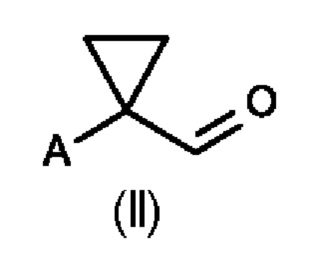
в присутствии кислоты Льюиса, выбранной из AlCl3 и GaCl3, предпочтительно AlCl3, с получением соединения формулы (III)
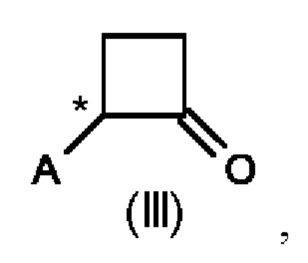
и последующее осуществление реакции соединения формулы (III) с ацетонитрилом и подходящей добавкой, где добавка выбрана из хлорангидридов, ангидридов или сложных эфиров, предпочтительно добавка выбрана из ацетилхлорида, изопропенилацетата, 4-метоксибензоилхлорида и п-анисового ангидрида.
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| M | |||
| TRUONG ET AL., First synthesis of (1S,2S)- and (1R,2R)-1-amino-2-isopropylcyclobutanecarboxylic acids by asymmetric Strecker reaction from 2-substituted cyclobutanones, TETRAHEDRON: ASYMMETRY, 2003, vol | |||
| Паровоз для отопления неспекающейся каменноугольной мелочью | 1916 |
|
SU14A1 |
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| ЭЛЕКТРИЧЕСКИЙ ПЕРЕНОСНЫЙ ФОНАРЬ | 1924 |
|
SU1063A1 |
| M | |||
| YAMASHITA ET AL., A new synthesis of | |||

