Настоящее изобретение относится к способу получения диарилциклоалкилпроизводных общей формулы (I). Кроме того, данное изобретение относится к новым промежуточным продуктам, которые образуются способом по настоящему изобретению, к способу получения промежуточных продуктов для соединений общей формулы (I) и к способу отделения смеси цис/транс-изомеров от исходных веществ, применяемых при получении соединений общей формулы (I)
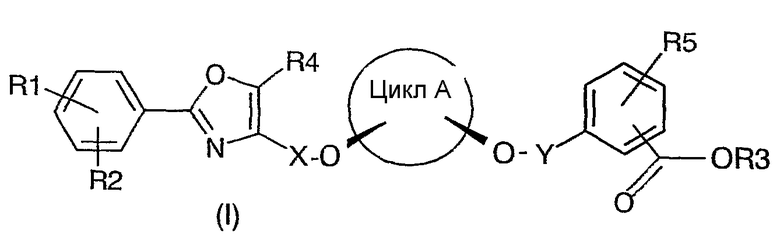
Соединения формулы (I) представляют собой активаторы рецепторов, активируемых пролифератором пероксисом (PPAR-активаторы), и уже известны из документа WO 03/020269. Из описанных в документе WO 03/020269 PPAR-активаторов эффективными PPAR-активаторами оказались те, которые проявляют способность к цис-замещению X- и Y-содержащих заместителей в центральном цикле A. В частности, это относится к соединениям, в которых цикл A представляет собой циклогексил‚ предпочтительно цис-1,3-циклогексил.
При синтезе или выделении требуемых целевых соединений формулы (I) трудность представляют, главным образом, два фактора. Одним из них является цис/транс-изомерия заместителей цикла A. Так как вместе с соединениями формулы (I) соответствующие цис-изомеры эффективных PPAR-активаторов имеются в виде соответствующих транс-изомеров, то целесообразно уже к началу синтеза на соответствующих промежуточных стадиях отделять соответствующие транс-изомеры цикла A, чтобы избежать нежелательного снижения выхода продукта. В качестве другого фактора при рассмотрении соответствующего цис-изомера цикла A следует учитывать то, что в большинстве промежуточных продуктов, а также в целевых соединениях формулы (I) имеются два асимметрических атома углерода и цикл A замещен двумя различными радикалами (X, Y). Следовательно, при присоединении к циклу A, например, X-содержащих заместителей необходимо учитывать, что при эквимолярном превращении образуется рацемическая смесь, поскольку данный заместитель принципиально может присоединяться к обеим функциональным группам цикла A. Если данное обстоятельство не учитывают, то получают соединения формулы (I) в виде рацемической смеси.
Хотя описанным в документе WO 03/020269 способом получения PPAR-активаторов принципиально можно получать соединения формулы (I) в энантиомерно чистой форме, однако описанный в данном документе способ обладает отдельными значимыми недостатками: применение и утилизация ядовитых соединений олова и фторида цезия; применение и утилизация йодсодержащих соединений; рацемический синтез, то есть после отделения ненужного энантиомера хиральной хроматографией по меньшей мере половина дорогостоящих исходных веществ теряется в качестве отходов, при этом хиральную хроматографию необходимо дополнительно сочетать с нехиральной хроматографией; половина продукта и, соответственно, половина применяемых ценных исходных веществ теряется при разделении рацемата с обоими энантиомерами; "ложный" энантиомер не может быть использован как вторичное сырье и должен быть утилизирован в качестве отхода; применение гидрида натрия в качестве основания и N,N-диметилформамида в качестве растворителя (возможно экзотермическое разложение).
Для получения избытка энантиомера или энантиомерно чистого соединения формулы (I) по способу, описанному в документе WO 03/020269, необходимо в обязательном порядке применять хиральную хроматографию. В частности, при крупномасштабном производстве большие издержки, связанные с применением хиральной хроматографии, оказываются главным недостатком данного способа.
Альтернативный способ получения PPAR-активаторов, которые относятся к PPAR-активаторам, описанным в документе WO 03/020269, описывается в международной заявке 10308350.2. В данном способе, который ограничивается только цис-1,3-двузамещенными производными циклогексана, алкилируют сначала цис-1,3-циклогександиол с защитной группой (бензил или силил) или уже с одним из двух заместителей целевого соединения, причем образуется рацемическая смесь соответствующего моноалкилированного соединения в цис-форме. В свою очередь, данный промежуточный продукт приводят во взаимодействие с донором ацильных групп, после чего его также в виде рацемата моноалкилированного и моноацилированного промежуточного продукта разделяют ферментативным расщеплением сложного эфира с последующей хроматографией на две фракции, из которых соответственно могут быть синтезированы раздельно проводимыми реакциями оба энантиомера целевого соединения. Рацемический моноалкилированный промежуточный продукт альтернативным образом может быть разделен ферментативной этерификацией с последующей хроматографией на две фракции, из которых, в свою очередь, могут быть синтезированы в двух отдельных загрузках обе энантиомерные формы целевого соединения. Недостатком данного способа является, в частности, то, что, несмотря на отсутствие хиральной хроматографии, вначале получают рацемический промежуточный продукт, из которого безальтернативно получают обе энантиомерные формы целевого соединения. При использовании варианта синтеза с предварительно введенной защитной группой бензилсодержащие защитные группы необходимо удалять гидрированием. При таком гидрировании первый заместитель целевого соединения, уже присоединенный на соответствующей промежуточной стадии, также может удаляться в известной степени, что ведет к снижению выхода продукта. Силилсодержащие защитные группы удаляют фторидом, но это также ведет к другим побочным реакциям с остальными заместителями соответствующих промежуточных соединений, и, следовательно, этого необходимо избегать.
Применение ферментов для разделения рацемических смесей различных соединений (исходных веществ или промежуточных продуктов) было уже многократно описано в литературе. Однако отыскание приемлемого фермента для энантиоселективного разделения соответствующей рацемической смеси, подлежащей разделению, представляет собой трудную задачу.
Так, Hirata et al., Chirality 9: 250-253 (1997), описывают гидролиз цис- и транс-1,3-диацетоксициклогексанов до ацилоксициклогексанола в присутствии культивированных растительных клеток маршанции разнообразной (Marchantia polymorpha). При этом необходимо культивировать растительные клетки, а соответствующие ферменты неизвестны. Избыток энантиомера при гидролизе мезо-цис-1,3-диацетоксициклогексана составляет при этом только 15% для (1R,3S)-1-ацетоксициклогексан-3-ола. Транс-1,3-диацетоксициклогексан превращается в (1R, 3R)-3-ацетоксициклогексан-1-ол (выход 60%) с 27% избытка энантиомера и циклогексан-1,3-диол (выход 70%). Поэтому данный способ не подходит для получения приемлемого избытка энантиомера или энантиомерно чистого цис-1S-ацилоксициклогексан-3R-ола.
K. Laumen et al., J. Chem. Soc., Chem. Common., (1986) 1298 - 1299, описали ферментативный гидролиз цис-1,4-диацетоксициклопент-2-ена в присутствии липаз, таких, как Pseudomonas species или Mucor miehei. При степени превращения приблизительно 50% образуется моноацетилированный энантиомер с энантиомерной чистотой от 95 до 97%. Энантиомерная чистота может быть повышена перекристаллизацией до значения более 99%.
Лежащая в основе данного изобретения задача состоит в том, чтобы разработать новый способ получения PPAR-активаторов общей формулы (I), который не обладает недостатками известных способов предшествующего уровня техники. В частности, необходимо разработать способ, которым можно получать PPAR-активаторы общей формулы (I) с приемлемым избытком энантиомера, то есть с более высокой энантиоселективностью, без необходимости применять хиральную хроматографию.
Решение задачи представляет собой способ получения соединения общей формулы (I), содержащий следующие стадии, причем:
а1) соединение формулы (IX) превращают в соединение формулы (V) посредством действия воды в присутствии фермента, обеспечивающего приемлемый избыток энантиомера соединения (V), или
a2) соединение формулы (X) превращают в соединение (V) посредством действия по меньшей мере одного донора ацильных групп в присутствии фермента, обеспечивающего приемлемый избыток энантиомера соединения (V);
b) соединение (V) в присутствии кислотного катализатора действием соединения, которое может образовывать защитную группу Z3, стабильную в щелочной среде и лабильную в кислой среде, превращают в соединение формулы (VIII) и
c) соединение (VIII) в присутствии нуклеофильного агента превращают в соединение формулы (II);
d) соединение (II) в присутствии основания B1 превращают действием соединения формулы (VI) в соединение формулы (IIIa) или действием соединения формулы (VII) в соединение формулы (IIIb)

e) соединение (IIIa) превращают в соединение формулы (IVa) или соединение (IIIb) превращают в соединение формулы (IVb), причем соответствующее превращение осуществляют действием спирта в присутствии кислотного катализатора;
f) соединение (IVa) посредством действия соединения (VII) или соединение (IVb) посредством действия соединения (VI) превращают в соединение формулы (Ia) в присутствии основания B1 и
g) при необходимости, соединение (Ia) гидролизуют или, если R3 представляет собой H, подвергают гидрогенолизу до соединения (Ia);
причем соединения (IX) и (X) представляют собой чистый цис-изомер или смесь цис/транс-изомеров соответственно;
при этом переменные и заместители имеют следующие значения соответственно:
цикл A представляет собой C3-C8циклоалкил или C3-C8циклоалкенил, причем в циклолалкильных или циклоалкенильных циклах один или несколько атомов углерода могут быть замещены атомами кислорода;
R1, R2, R4 и R5 представляют собой независимо друг от друга H, F, Cl, Br, OH, NO2, CF3, OCF3, C1-C6алкил или -O-(C1-C6алкил);
R3 представляет собой H, C1-C6алкил или бензил, которые, при необходимости, могут быть замещены F, Cl, Br, OH, NO2, CF3, OCF3, C1-C6алкилом или -O-(C1-C6алкилом);
R6 представляет собой C1-C6алкил или бензил, которые, при необходимости, могут быть замещены F, Cl, Br, OH, NO2, CF3, OCF3, C1-C6алкилом или -O-(C1-C6алкилом);
X представляет собой C1-C6алкил, причем в алкильной группе один или несколько атомов углерода могут быть замещены атомами кислорода;
Y представляет собой C1-C6алкил, причем в алкильной группе один или несколько атомов углерода могут быть замещены атомами кислорода;
Z1 и Z2 представляют собой независимо друг от друга защитные группы, стабильные в кислой среде;
Z3 представляет собой защитную группу, стабильную в щелочной среде и лабильную в кислой среде;
Z4 и Z5 представляют собой независимо друг от друга уходящие группы;
B1 представляет собой третичный алкоголят щелочноземельного металла, третичный алкоголят щелочного металла, амид щелочноземельного металла, амид щелочного металла, силазид щелочноземельного металла, силазид щелочного металла или гидрид щелочного металла.
Соединения, упомянутые в приведенных ранее стадиях способа, показаны на представленной далее схеме I, служащей для пояснения способа по настоящему изобретению.

Стадии способа, приведенные на схеме I, далее поясняются более подробно.
В соединениях (с I по VIII), приведенных на схеме I, касательно обоих заместителей, связанных с циклом A (в соответствующих соединениях), имеет место замещение в цис-положении упомянутыми обоими заместителями относительно цикла A. Например, речь может идти о замещении в цис-1,2-, цис-1,3- или цис-1,4-положении. При этом предпочтительным является замещение в цис-1,2- и цис-1,3-положении и более предпочтительным является замещение в цис-1,3-положении. В частности, предпочтительным является замещение в цис-1,3-положении циклогексильного цикла A. Для упрощения цикл A или также заместители X и Y далее обозначаются просто как радикалы (алкил или алкенил), даже если - в зависимости от рассматриваемого варианта - в случае цикла A под обозначением подразумевается алкан или алкен (цикл A как основной фрагмент формулы (I)) и соответственно алкилен или алкенилен.
Под приемлемым избытком энантиомера (высокой энантиоселективностью) следует понимать энантиомерную чистоту (ee) больше 50%, предпочтительно больше 90%, более предпочтительно больше 95%, еще более предпочтительно больше 98%, наиболее предпочтительно больше 99% и особо предпочтительно больше 99,5%.
Предпочтительно стадию a1) и/или a2) проводят в присутствии липазы B из Candida antarctica.
Способ по настоящему изобретению по сравнению со способами по предшествующему уровню техники обладает тем преимуществом, что посредством использования соответствующих ферментов хиральная структура вводится уже к началу процесса с соответствующими предшественниками, вследствие чего упомянутые предшественники уже обладают в отношении энантиоселективности приемлемым, при необходимости даже крайне высоким, избытком энантиомера (энантиомерная чистота более 99%). Следовательно, возможно также получать требуемые энантиомеры соединений (I) энантиоселективно с приемлемым, при необходимости даже крайне высоким, избытком энантиомера (энантиомерная чистота более 99%). Вследствие этого по сравнению с известными способами по предшествующему уровню техники не наблюдается снижение выхода продукта до 50% и также не требуется разделять рацемические смеси цис-энантиомеров соответствующих промежуточных продуктов для того, чтобы получить требуемый энантиомер соединений (I) с приемлемым избытком энантиомера.
Неожиданным образом, благодаря стабильной в щелочной среде защитной группе Z3, несмотря на двойную стадию алкилирования, сохраняется имеющаяся уже к началу синтеза хиральная структура (энантиомерная чистота предшественников отчасти более 99%) вплоть до получения требуемого хирального PPAR-активатора формулы (I), энантиомерная чистота которого, таким образом, также превосходит 99%. Кроме того, применяемые в способе по настоящему изобретению ферменты предоставляют возможность использовать применяемые исходные вещества не только в виде соответствующего чистого цис-изомера, но также и в виде смеси цис/транс-изомеров, при этом не ухудшая энантиомерную чистоту промежуточных продуктов и соответственно целевого соединения. При применении в способе по настоящему изобретению в качестве исходных соединений смеси цис/транс-изомеров соответствующие исходные соединения в транс-форме, благодаря применяемой технике защитных групп, можно без проблем удалять при очистке промежуточных продуктов, например экстракцией. Вследствие этого не требуется в обязательном порядке дополнительная стадия очистки, например хроматографией.
По сравнению с вариантом синтеза, изложенным в документе WO 03/020269, могут быть отмечены, в частности, следующие преимущества: при выборе приемлемой липазы ферментативной десимметризацией можно получать почти чистый энантиомер (чистота более 99%) соединения (V), который в качестве исходного вещества является важным элементом для стереоселективного синтеза PPAR-активаторов формулы (I) с оптической чистотой более 99%; стереохимическая структура сохраняется неожиданным образом благодаря соответствующей технике защитных групп вплоть до получения требуемого хирального PPAR-активатора, так что больше не требуется утилизировать в качестве отхода половину ценных исходных веществ; дорогостоящее разделение рацемата, например хиральной хроматографией, может больше не проводиться; применение и утилизация ядовитых соединений олова, йодсодержащих соединений и фторида цезия больше не являются необходимыми; применение гидрида натрия в качестве основания и N,N-диметилформамида в качестве растворителя больше не является необходимым; применение хроматографии требуется только лишь в незначительной степени, если вообще требуется.
В частности, при использовании липазы B из Candida antarctica, применяемой на стадии а1) по настоящему изобретению, в количестве более 90% уже в растворе может быть достигнута энантиомерная чистота более 99%, вследствие чего перекристаллизация не требуется.
Способом по настоящему изобретению можно получать соединения общей формулы (I):
где:
цикл A представляет собой C3-C8циклоалкил или C3-C8циклоалкенил, причем в циклолалкильных или циклоалкенильных циклах один или несколько атомов углерода могут быть замещены атомами кислорода;
R1, R2, R4 и R5 представляют собой независимо друг от друга H, F, Cl, Br, OH, NO2, CF3, OCF3, C1-C6алкил или -O-(C1-C6алкил);
R3 представляет собой H, C1-C6алкил или бензил, которые, при необходимости, могут быть замещены F, Cl, Br, OH, NO2, CF3, OCF3, C1-C6алкилом или -O-(C1-C6алкилом);
X представляет собой C1-C6алкил, причем в алкильной группе один или несколько атомов углерода могут быть замещены атомами кислорода;
Y представляет собой C1-C6алкил, причем в алкильной группе один или несколько атомов углерода могут быть замещены атомами кислорода.
Предпочтительно способом по настоящему изобретению можно получать соединения общей формулы (I), где:
цикл A представляет собой циклопентил, циклогексил или циклогептил;
R1, R2, R4 и R5 представляют собой независимо друг от друга H, F, Cl, Br, OH, NO2, CF3, OCF3, C1-C6алкил или O-(C1-C6алкил);
R3 представляет собой H или C1-C6алкил или бензил;
X и Y представляют собой независимо друг от друга C1-C6алкил.
Более предпочтительно способом по настоящему изобретению можно получать соединения общей формулы (I), где:
цикл A представляет собой циклогексил, причем X-содержащий и Y-содержащий заместители формулы (I) стоят в цис-1,3-положении циклогексильного фрагмента;
X и Y представляют собой метил.
Еще более предпочтительно способом по настоящему изобретению можно получать соединения общей формулы (I), где:
цикл A представляет собой циклогексил, причем X-содержащий и Y-содержащий заместители формулы (I) стоят в цис-1,3-положении циклогексильного фрагмента и атом углерода цикла A, связанный с Y-содержащим заместителем, имеет R-конфигурацию;
X и Y представляют собой метил.
Особо предпочтительно способом по настоящему изобретению можно получать соединения общей формулы (I), где:
цикл A представляет собой циклогексил, причем X-содержащий и Y-содержащий заместители формулы (I) стоят в цис-1,3-положении циклогексильного фрагмента и атом углерода цикла A, связанный с Y-содержащим заместителем, имеет R-конфигурацию;
X и Y представляют собой метил;
R1, R2 и R4 представляют собой независимо друг от друга H, F, Cl, C1-C3алкил или -O-(C1-C3алкил);
R5 представляет собой H или C1-C3алкил.
Приведенное на схеме I соединение (IX), которое по настоящему изобретению может применяться в качестве исходного вещества, в свою очередь, может быть получено:
i) превращением соединения (X) посредством действия по меньшей мере одного донора ацильных групп в присутствии фермента, обеспечивающего получение преимущественно цис-изомера соединения (IX), и отделением транс-изомеров соединений формулы (V), в случае их образования в качестве побочных продуктов, или
ii) превращением соединения (X) посредством действия по меньшей мере одного донора ацильных групп.
Стадию i) проводят предпочтительно в присутствии липазы поджелудочной железы свиньи, липазы из Burkholderia cepacia, липазы из Burkholderia species, липазы из Pseudomonas cepacia или липазы из Pseudomonas species.
Далее в качестве примера представлен способ по настоящему изобретению с учетом схемы I в пределах типично принимаемой последовательности реакций, включая предварительные стадии.
Отсылка на конкретный пример в следующем далее тексте служит только для пояснения последовательности реакций, описанной в качестве примера, и не означает, что способ по настоящему изобретению ограничивается данным конкретным примером.
Стадия a)
В способе по настоящему изобретению на стадии a) получают соединения (V), причем имеется несколько вариантов. Соединения (V) уже давно известны из литературных источников. Например, K. Dimroth et al., Ber. (1942), 75B, 322-6, описали моноацетат ацилоксициклогексанола. Кроме того, в процитированной ранее публикации T. Hirata и др. описано возможное выделение различных цис-1S-ацилоксициклогексан-3R-олов хиральной хроматографией. Главной проблемой при этом следует считать выделение соединения (V) в виде отдельного энантиомера, что на практике представляет очень большую трудность, так как для этого должна применяться преимущественно хиральная хроматография.
а) Ферментативное ацилирование
В качестве альтернативы хиральной хроматографии может быть представлено получение отдельных энантиомеров соединений (V) ферментативным ацилированием соединений (X). Соединения (X) могут использоваться или в виде смеси цис/транс-изомеров, или в виде чистых цис-изомеров и в такой форме продаются различными производителями(например, фирмами Merck, Fluka или Aldrich). Недостатком использования соединений (X) в виде чистых цис-изомеров является то, что изомеры сначала должны быть выделены из соответствующих смесей цис/транс-изомеров, и соответственно то, что чистые цис-изомеры стоят дороже. Для разделения смесей цис/транс-изомеров соединений (X) может использоваться, например, в случае 1,3-циклогександиола, кристаллизация в виде комплекса цис-1,3-циклогександиола с медью (W. Rigby, J. Chem. Soc. (1949), 1586; R. Sillanpää et al., Polyhetron 21 (2002), 1133-1138).
Исходя из соединений (X), можно проводить ферментативное ацилирование в пределах способа по настоящему изобретению в присутствии различных ферментов (например, липаз) с донором ацильных групп. При этом могут использоваться отдельные доноры ацильных групп или смесь нескольких доноров ацильных групп. Реакцию можно осуществлять без дополнительного органического растворителя (пример 1) или с дополнительным органическим растворителем (пример 2). В качестве органического растворителя принципиально могут быть приняты во внимание все традиционные органические растворители, такие как толуол, хлорированные углеводороды или простые эфиры, например метил-трет-бутиловый эфир. Однако реакция не может быть проведена в воде. В качестве доноров ацильных групп приемлемыми являются все химические соединения, которые могут образовывать стабильные в кислой среде защитные группы Z1 или Z2. В качестве примера могут быть приведены сложные эфиры карбоновых кислот. Предпочтительно приемлемыми являются виниловые сложные эфиры, такие как винилацетат, изопропенилацетат, виниллаурат или винилбутират, особо предпочтительными являются винилацетат или изопропенилацетат.
Для ферментативного ацилирования не может быть использован любой фермент. Более того, должен применяться такой фермент, с которым можно получать, прямо или косвенно, требуемое целевое соединение с приемлемым избытком энантиомера. Кроме того, от используемого фермента зависит, образуется ли из соединений формулы (X) соединения (V) прямым путем или сначала (при косвенном пути) образуется соединение (IX), которое, в свою очередь, затем должно быть превращено в соединение (V). Если используется липаза, которую выделяют из фракции B микроорганизма Candida antarctica (далее упоминается как липаза B из Candida antarctica, выделение фракций по EP-A 287634), то при использовании, в качестве исходной, смеси цис/транс-изомеров соединения (X) образуется преимущественно моноацильное соединение (V) в цис-форме, в то время как диацильное соединение (IX) образуется в качестве побочного продукта. Тогда как моноацильное соединение (V) в транс-форме при применении липазы B из Candida antarctica не образуется, поскольку соответствующее исходное соединение (X) в транс-форме или не вступает во взаимодействие, или превращается в соответствующее диацильное соединение (IX).
Прямое превращение (ферментативное ацилирование) соединения формулы (X) в соединение формулы (V) проводят предпочтительно в присутствии липазы B из Candida antarctica. Особо предпочтительно такое превращение проводят в присутствии липазы, выбранной из ферментов Chirazyme L2 lyo., Chirazyme L2 c.f. C2 или Chirazyme L2 c.f. C3. Соответствие упомянутых ферментов (по их коммерческим названиям) c надлежащими номерами доступа банка генов Национального центра биотехнологической информации (NCBI) может быть выяснено из таблицы 1, приведенной в примере 1.
Получающаяся при таком превращении реакционная смесь соединений формул (IX) и (V) может быть, при необходимости, разделена экстракцией, перегонкой или хроматографией. Однако для достижения избытка энантиомера разделение на данной стадии не требуется в обязательном порядке, так как образующееся в качестве побочного продукта соединение (IX) на следующей далее стадии b) может быть и без защитной группы Z3. Вследствие этого на стадии c) способа по настоящему изобретению снимают двойную защиту побочного продукта (IX) и, при необходимости, удаляют экстракцией при обработке соединения (II). Подобное соображение может быть принято во внимание в отношении непревращенного исходного соединения (X), чтобы провести разделение или уже на данной стадии экстракцией, перегонкой или хроматографией, или при обработке соединений (II) или (IX).
Вместо липазы B из Candida antarctica при ферментативном ацилировании могут применяться также липаза поджелудочной железы свиньи, липаза из Burkholderia cepacia, липаза из Burkholderia species, липаза из Pseudomonas cepacia или липаза из Pseudomonas species. При применении упомянутых липаз из исходного соединения (X) образуются как соединения (IX), так и соединения (V). Однако моноацильные соединения (V) не находятся в требуемой транс-форме, в то время как образовавшиеся при этом соединения (IX) неожиданным образом находятся преимущественно в цис-форме диацильных соединений. Упомянутые диацильные соединения (IX) в цис-форме могут быть превращены, как поясняется далее, ферментативной десимметризацией (ферментативным гидролизом) в требуемые цис-энантиомеры соединения (V).
Превращение соединения (V) ферментативным ацилированием в соединение (IX), которое находится преимущественно в виде цис-изомера, проводят предпочтительно в присутствии липазы, выбранной из липазы поджелудочной железы свиньи, липазы из Burkholderia cepacia, липазы из Burkholderia species, липазы из Pseudomonas cepacia или липазы из Pseudomonas species. Более предпочтительно липазу выбирают из липазы поджелудочной железы свиньи, липазы из Burkholderia cepacia, липазы из Burkholderia species или липазы из Pseudomonas cepacia. Наиболее предпочтительно липазу выбирают из ферментов Chirazyme L1 lyo, Chirazyme L1 c.f., Chirazyme L7 lyo или липазы PS. Особо предпочтительно липазу выбирают из ферментов Chirazyme L1 lyo., Chirazyme I1 c.f. или Chirazyme I7 lyo. Соответствие коммерческих названий упомянутых ферментов c номерами доступа банка генов NCBI может быть выяснено из таблицы 1.
Если используется исходное вещество (X) в виде смеси цис/транс изомеров, то для достижения избытка энантиомера, разумеется, требуется отделение соединения (V) в транс-форме, которое образуется при данном варианте синтеза в виде нежелательного побочного продукта, от соединения (IX) экстракцией, перегонкой или, при необходимости, хроматографией. Однако данная стадия обработки отпадает при применении соединения (X) в виде чистого цис-изомера. Возможное отделение побочного продукта (V) осуществляют предпочтительно экстракцией. Так как заключительную ферментативную десимметризацию соединения (IX) осуществляют с другим ферментом и, к тому же, в водной среде, необходимо заранее удалить примененный при ферментативном ацилировании фермент, например фильтрованием. Фермент удаляют предпочтительно перед отделением моноацильного соединения (соединение (V) в транс-форме).
Химическое ацилирование/ферментативная десимметризация
Другую возможную отправную точку для получения соединений (V) представляют соединения (IX), которые также в виде смеси цис/транс-изомеров или в виде чистых цис-изомеров продаются различными производителями (например, фирмами Merck, Fluka или Aldrich). Разделение смесей цис/транс-изомеров можно осуществлять, например, в случае цис-1,3-диацетоксициклогексана, перегонкой обоих изомеров, температуры кипения которых различаются на 1°C. Однако вследствие обычно небольшой разницы температур кипения данный метод является затратным и дорогостоящим. Соединения (IX) могут быть получены, как было упомянуто ранее, ферментативным ацилированием соединений (X). Альтернативно соединения формулы (X) также можно прямо превращать действием упомянутых ранее доноров ацильных групп (в присутствии ферментов) в соединения (IX). Данная реакция известна уже давно и называется химическим ацилированием, которое протекает, однако, нестереоселективно (примеры 3 и 4). Химическое ацилирование можно осуществлять, например, действием смеси уксусный ангидрид/4-диметиламинопиридин (4-DMAP), триэтиламина (TEA) в дихлорметане. Химическое ацилирование можно осуществлять действием отдельного донора ацильных групп или смеси доноров ацильных групп, предпочтительно действием отдельного донора ацильных групп, так что заместители Z1 и Z2 в соединении (IX) имеют одинаковое значение.
Соединения (IX) могут быть превращены в соединение (V) посредством действия воды в присутствии фермента, обеспечивающего приемлемый избыток энантиомера соединения (V). В качестве фермента применяют предпочтительно липазу B из Candida antarctica. Особо предпочтительно такое превращение проводят в присутствии липазы, выбранной из ферментов Chirazyme L2 lyo., Chirazyme L2 c.f. C2 или Chirazyme L2 c.f. C3. Данную реакцию необходимо проводить в водном растворе, заключительное применение органических растворителей в данном случае неприемлемо. Неожиданным образом диацильное соединение (IX) в транс-форме не видоизменяется липазой B из Candida antarctica.
Таким образом, в обоих упомянутых способах (ферментативном ацилировании и химической десимметризации), а также возможной комбинации обоих способов (ферментативного ацилирования с заключительной ферментативной десимметризацией) могут использоваться соединения (X) в виде смеси цис/транс изомеров для получения избытка энантиомера или энантиомерно чистого моноацильного соединения (V) в цис-форме. Данный способ является более экономичным, чем применение чистых изомеров. Таким образом, получение энантиомерно чистых соединений (V) является другим объектом данного изобретения. Под энантиомерно чистыми соединениями в пределах данного изобретения следует понимать соединения, чистота которых составляет более 98% (ee >98%), предпочтительно более 99% (ee >99%), особо предпочтительно более 99,5% (ee >99,5%).
Большим преимуществом применения липазы B из Candida antarctica следует считать то, что независимо от того, применяется ли отдельный донор ацильных групп или смесь доноров ацильных групп, соединение формулы (V) всегда образуется в энантиомерно чистой форме. В соединениях (IX), (V) и (XIII) защитные группы Z1 и Z2 независимо друг от друга представляют собой защитные группы, стабильные в кислой среде. Предпочтительно защитные группы Z1 и Z2 имеют одинаковые значения. Z1 и Z2 представляют собой предпочтительно -C(O)-R, где R представляет собой при необходимости замещенный алкил или арил, например C1-C6алкил или фенил. Z1 и Z2 независимо друг от друга представляют собой более предпочтительно -C(O)-(C1-C3алкил) и особо предпочтительно -C(O)-CH3. Липаза B из Candida antarctica может применяться как в неиммобилизованной форме (Chirazym L2), так и в иммобилизованной форме (c.f., c.f.C2, c.f.C3, производитель: Roche Diagnostics).
Липаза B из Candida antarctica поставляется также другими производителями, такими, как, например, Novozym (Novozym 435 в виде иммобилизата). К тому же, альтернативно может применяться также растворимая липаза B из Candida antarctica, такая, как, например, Novozym CALB L или Novozym 525 F после иммобилизации фермента.
Описанные ранее способы разделения смесей цис/транс-изомеров соединений (X) или (IX) и соответственно способы получения избытка энантиомера соединения (V) в цис-форме или энантиомерно чистого соединения (V) в цис-форме находят применение по настоящему изобретению, в частности, в том, что избыток энантиомера соединения (V) в цис-форме или энантиомерно чистое соединение (V) в цис-форме переводят соответствующей техникой защитных групп и последующим алкилированием в требуемое целевое соединение (I) (избыток энантиомера или энантиомерно чистое соединение), причем не требуется ни хиральная, ни нехиральная хроматография.
Все попытки по осуществлению селективного алкилирования энантиомерно чистого соединения (V) до сих пор не удавались, так как была отмечена меж- и внутримолекулярная миграция ацильной группы в неустранимых щелочных условиях алкилирования (ацил представляет собой, например, ацетил, бензоил). Поэтому пытаются применять технику защитных групп, стабильных в щелочных условиях, используя в качестве защитных групп такие, как, например, тетрагидропиранил, метоксиизопропил, так что сохраняется хиральная структура, образовавшаяся в соединении (V) вследствие ферментативной десимметризации, несмотря на щелочные условия алкилирования. Целенаправленным последовательным алкилированием с использованием техники защитных групп может быть достигнуто, в качестве второго объекта данного изобретения, получение требуемого стереоизомера PPAR-активатора, неожиданным образом, без потери хиральной структуры.
Стадия b)
Соединение (V) в присутствии кислотного катализатора действием соединения, которое может образовывать группу Z3, стабильную в щелочной среде и лабильную в кислой среде, превращают в соединение формулы (VIII). В качестве кислотных катализаторов могут применяться, например, неорганические кислоты, толуолсульфокислота, пириридин-пара-толуолсульфонат или кислотный ионообменник, такой как Amberlyst H15. С этой целью предпочтительно используют пириридин-пара-толуолсульфонат. Защитная группа Z3, имеющаяся в соединении (VIII), представляет собой защитную группу, стабильную в щелочной среде и лабильную в кислой среде. При этом речь идет предпочтительно об ацетальной или кетальной защитной группе. Z3 представляет собой более предпочтительно тетрагидропиранил или метоксиизопропил, особо предпочтительно тетрагидропиранил. В качестве соединения, которое может образовывать защитную группу, стабильную в щелочной среде и лабильную в кислой среде, предпочтительно приемлемым является дигидро-2H-пиран. Один эквивалент соединения (V) приводят во взаимодействие с соединением, образующим защитную группу Z3, стабильную в щелочной среде и лабильную в кислой среде, в количестве от 1 до 10 эквивалентов, предпочтительно от 1,1 до 1,4 эквивалента. Кислотный катализатор используют, как правило, в количестве от 0,01 до 1 эквивалента, предпочтительно от 0,05 до 0,1 эквивалента. Температура реакции составляет обычно от 20 до 80°C, предпочтительно от 50 до 60°C. Стадию b), как и все другие стадии по настоящему способу, осуществляют обычно при нормальном давлении. В качестве растворителя на стадии b) приемлемыми являются органические растворители, такие, как, например, хлорированные углеводороды, сложные эфиры карбоновых кислот, такие как этилацетат, амиды карбоновых кислот, такие как N-метилпирролидон, простые эфиры, такие как диэтиловый эфир или метил-трет-бутиловый эфир, ароматические углеводороды, такие как хлорбензол или толуол. Aльтернативно в качестве растворителя может применяться также и сам 3,4-дигидро-2H-пиран. Предпочтительным растворителем является толуол. Тогда как вода или спирты не являются приемлемыми растворителями, поскольку они взаимодействуют, например, с 3,4-дигидро-2H-пираном с образованием соответствующих ацеталей. Образующееся на данной стадии соединение (VIII) может быть очищено перегонкой, однако оно может использоваться на следующей стадии без дальнейшей очистки.
Стадия c)
Соединение (VIII) в присутствии нуклеофильного агента превращают в соединение (II). В данной реакции, называемой деацилированием, в качестве нуклеофильного агента могут использоваться, например, алкоголяты щелочных или щелочноземельных металлов, предпочтительно этилат натрия. На один эквивалент соединения (VIII) применяют от 0,1 до 10 эквивалентов нуклеофильного агента, каталитические количества составляют предпочтительно от 0,1 до 0,3 эквивалента. Температура реакции составляет обычно от 10 до 80°C, предпочтительно от 15 до 25°C. Данная стадия деацилирования может осуществляться во всех органических растворителях, которые не взаимодействуют с нуклеофильным агентом (этилатом натрия), например, в ароматических углеводородах, спиртах, хлорированных углеводородах. При этом толуол является предпочтительным растворителем, так как на предшествующей стадии можно осуществлять экстракцию толуолом, поэтому при деацилировании замена растворителя не требуется, при этом алкилирование на следующей стадии d) также можно осуществлять в толуоле. Соединение (II) может быть очищено перегонкой, которая, однако, не требуется в обязательном порядке.
Стадия d)
Соединение (II) превращают в присутствии основания B1 действием соединения формулы (VI) в соединение формулы (IIIa) или действием соединения формулы (VII) в соединение формулы (IIIb). В качестве основания B1 приемлемыми являются третичные алкоголяты щелочноземельных металлов, третичные алкоголяты щелочных металлов, амиды щелочноземельных металлов, амиды щелочных металлов, силазиды щелочноземельных металлов, силазиды щелочных металлов или гидриды щелочных металлов. Тогда как первичные или вторичные алкоголяты приемлемыми не являются. Предпочтительными основаниями B1 являются трет-бутилат калия (KOtBu), третичный изопентилат, диизопропиламид лития или бис-(триметилсилил)амид калия. Особо предпочтительными основаниями являются трет-бутилат калия или бис-(триметилсилил)амид калия. В качестве растворителя приемлемыми являются апротонные органические растворители, например, простые эфиры (диэтиловый эфир, метил-трет-бутиловый эфир), амиды карбоновых кислот (N-метилпирролидон), ароматические углеводороды (хлорбензол или толуол), при этом предпочтительным растворителем является толуол. Реакцию осуществляют в нормальном порядке при температуре от 20 до 80°C, предпочтительно при температуре от 50 до 60°C. При этом 1 эквивалент соединения (II) приводят во взаимодействие с алкилирующим агентом (соединения (VI) или (VII)) в нормальном порядке в количестве от 1 до 3 эквивалентов, предпочтительно от 1,1 до 1,3 эквивалента. Основание B1 применяют в количестве от 1 до 3 эквивалентов, предпочтительно от 1,5 до 2 эквивалентов.
Алкилирующие агенты формул (VI) или (VII) имеются в продаже или могут быть получены известными из литературных источников способами. Z4 и Z5 представляют собой независимо друг от друга уходящие группы. При этом могут применяться все традиционные уходящие группы, предпочтительно хлор или бром. Способы получения соединений формулы (VI) приведены в документе WO 03/020269 или в международной заявке 10308350.2 или в издании The Chemistry of Heterocyclic Compounds (Ed.: A. Weissberger, E. C. Taylor): Oxazoles (Ed.: I.J. Turchi), b). Methoden der Organischen Chemie, Houben-Weyl 4. Auflage, Hetarene III, Teilband 1; c) I. Simit, E. Chindris, Arch. Pharm. 1971, 303, 425; d). Y. Goto, M. Yamazaki, M. Hamana, Chem. Pharm. Bull. 1971, 19 (10), 2050-2057]. Соединения формулы (VII) описаны также в обеих упомянутых ранее заявках, а также в документах WO 00/64888 (изобутиловый сложный эфир) и WO 00/64876 (метиловый сложный эфир). Кроме того, упомянутые соединения могут быть получены радикальным галогенированием боковых цепочек (см. литературный обзор R.C. Larock, Comprehensive Organic Transformations, S. 313, 1989 VCH Publishers, Inc.) или из спиртов, или получаемых из них производных (см. литературный обзор R.C. Larock, Comprehensive Organic Transformations, S. 353-363., 1989 VCH Publishers, Inc.). Кроме того, известно (см. J. Chem. Soc. 1925, 127, 2275-2297; J. Chem. Soc. 1922, 121, 2202-2215) получение различных 2-бромметилбензоилбромидов радикальным бромированием, которые затем последующим взаимодействием со спиртами переводят в эфиры бромметилбензойной кислоты, принадлежащие к группе алкилирующих агентов формулы III.
Решение о применении для алкилирования на стадии d) соединения (VI) или соединения (VII) зависит от требуемого энантиомера целевого соединения (I). Предпочтительно соединения (II) приводят во взаимодействие с соединением формулы (VI), в частности, если цикл A представляет собой цис-1,3-циклогексил.
Стадия e)
Соединение (IIIa) превращают в соединение (IVa) или соединение (IIIb) превращают в соединение (IVb), причем соответствующее превращение осуществляют действием спирта в присутствии кислотного катализатора. В качестве кислотных катализаторов приемлемыми являются те же соединения, которые уже были упомянуты на стадии b), причем выбор кислотного катализатора на стадиях b) и e) осуществляют независимо друг от друга. В качестве спиртов приемлемыми являются предпочтительно первичные спирты, в частности метанол. Данную стадию осуществляют при температуре от 20 до 80°C, предпочтительно при температуре от 45 до 55°C. В качестве растворителя приемлемыми являются упомянутые уже на стадии d) апротонные органические растворители, предпочтительно толуол. Выбор растворителя на стадиях d) и e) можно осуществлять независимо друг от друга. Один эквивалент соединения (III) приводят во взаимодействие с кислотой в количестве от 0,01 до 10 эквивалентов, предпочтительно в количестве 0,05 эквивалента, например соляной кислоты. Применяемые спирты могут использоваться при этом в количестве от 1 до 3 эквивалентов. Образующееся на данной стадии соединение (IV) может быть очищено перегонкой. Если полученное соединение является кристаллическим, то предпочтительной является очистка перекристаллизацией, менее предпочтительна очистка хроматографией, так как для этого требуется повышенный расход растворителя.
Стадия f)
На стадии f) соединение (IVa) посредством действия соединения (VII) или соединение (IVb) посредством действия соединения (VI) превращают в соединение (Ia) в присутствии основания B1. Выбор основания B1 осуществляют при этом независимо от стадии d), однако используют предпочтительно те же соединения, что и на стадии d). Принципиально возможно также применять тот же растворитель, что и на стадии d), причем выбор растворителя осуществляют также независимо от стадии d). Предпочтительно в качестве растворителя на данной стадии второго алкилирования наряду с толуолом может быть выбран хлорбензол, причем хлорбензол в данном случае более предпочтителен, чем толуол, ввиду большей степени превращения по сравнению с толуолом. Применяемые соотношения количеств исходных соединений, алкилирующего агента, а также основания B1 соответствуют принятым на стадии d). Реакцию осуществляют обычно при температуре от -30 до +20°C, предпочтительно при температуре от -5 до +5°C.
Стадия q)
Данная стадия требуется только тогда, когда в целевом соединении формулы (I) остаток R3 представляет собой водород, то есть когда требуемый PPAR-активатор должен быть в форме свободной кислоты. В ином случае полученное на стадии f) соединение (Ia) соответствует соединению (I). Однако, если это не так, то соединение (Ia) гидролизом или гидрогенолизом переводят в соединение (I). Гидролиз может осуществляться традиционными способами как в щелочной среде (R6 представляет собой предпочтительно налкил) или в кислой среде (R6 представляет собой предпочтительно трет-бутил). Если R6 представляет собой бензильный остаток, то соединение (I) получают предпочтительно гидрогенолизом по известному специалисту в данной области техники способу. В случае щелочного омыления используют гидроксиды металлов, такие как, например, гидроксиды щелочных или щелочноземельных металлов, в количестве от 1 до 10 эквивалентов по отношению к омыляемому соединению. В качестве растворителя приемлемыми являются вода, спирты или другие органические растворители, такие как, например, простые эфиры (диэтиловый эфир, метил-трет-бутиловый эфир), амиды карбоновых кислот (N-метилпирролидон) или ароматические углеводороды. Предпочтительно применяют трет-бутанол. Температура реакции составляет от 20 до 100°C, предпочтительно от 65 до 75°C. Затем протонированием карбоксильной группы, например, органическими или неорганическими кислотами, предпочтительно соляной кислотой, переводят требуемый хиральный PPAR-активатор формулы (I) с энантиомерной чистотой более 99% в форму свободной кислоты. При необходимости, может быть осуществлена перекристаллизация из органических растворителей, таких, как, например, ароматические растворители, предпочтительно толуол, этилацетат или н-бутилацетат, или, при необходимости, из эфиров карбоновых кислот, алкиловых эфиров или алкиловых спиртов. Такая перекристаллизация может осуществляться также по окончании стадии e).
При осуществлении всех стадий от b) до g) в одном и том же растворителе предпочтительно применяют толуол.
В предпочтительном варианте осуществления способ по настоящему изобретению содержит следующие стадии, причем:
a) соединение формулы (IX) в присутствии липазы B из Candida antarctica действием воды превращают в соединение формулы (V);
b) соединение (V) в присутствии кислотного катализатора действием соединения, которое может образовывать защитную группу Z3, стабильную в щелочной среде и лабильную в кислой среде, превращают в соединение формулы (VIII) и
c) соединение (VIII) в присутствии нуклеофильного агента превращают в соединение формулы (II);
d) соединение (II) в присутствии основания B1 действием соединения формулы (VI) превращают в соединение формулы (IIIa);
e) соединение (IIIa) превращают в соединение формулы (IVa), причем превращение осуществляют действием спирта в присутствии кислотного катализатора;
f) соединение (IVa) посредством действия соединения (VII) превращают в соединение формулы (Ia) в присутствии основания B1 и
g) при необходимости, соединение (Ia) гидролизуют или подвергают гидрогенолизу до соединения (Ia), если R3 представляет собой H.
Данный предпочтительный вариант осуществления способа по настоящему изобретению является приемлемым, в частности, для получения соединений общей формулы (I), в которых цикл A представляет собой циклогексил, причем X-содержащий и Y-содержащий заместители формулы (I) стоят в цис-1,3-положении циклогексильного фрагмента и атом углерода цикла A, связанный с Y-содержащим заместителем, имеет R-конфигурацию.
Альтернативно по данному предпочтительному варианту осуществления соединение (V) может быть получено из соединения (X) ферментативным ацилированием с соответствующей липазой, как было описано ранее, однако соединение (IX) является предпочтительным в качестве исходного соединения. В свою очередь, соединение (IX) может быть получено из соединения (X) ферментативным ацилированием с соответствующей липазой или предпочтительно химическим ацилированием.
Типично принимаемая последовательность реакций, включая предварительные стадии, представлена далее в качестве примера для иллюстрации предпочтительного способа по настоящему изобретению. По данной последовательности реакций могут быть получены, в частности, соединения формулы (I), в которых цикл A представляет собой циклогексил, X-содержащий и Y-содержащий заместители расположены в цис-1,3-положении цикла A и атом углерода цикла A, связанный с Y-содержащим заместителем, имеет R-конфигурацию. Описанные на соответствующих стадиях синтеза условия способа, например, касательно применяемых кислот, оснований или растворителя, также действительны в отношении остальных соединений формулы (I), в которых цикл A не ограничен производными цис-1,3-циклогексана.
Следующей далее схемой (II) дополнительно поясняется данная типичная последовательность реакций получения данной выборки PPAR-активаторов. При этом исходят из изомерно чистого цис-1,3-циклогександиола (X-i-цис) (фирма Clariant) или смеси цис/транс-изомеров (X-i) (фирма Acros). Цис-1,3-циклогександиол может быть получен, при необходимости, известными специалисту в данной области техники способами из смеси изомеров цис/транс-циклогександиола, например хроматографией. 1,3-циклогександиол может быть превращен ферментативным ацилированием прямо в цис-1S-ацилоксициклогексан-3R-ол (V-i) или химическим ацилированием через 1,3-диацилоксициклогексан (IX-i) в качестве промежуточной стадии. Как при химическом, так и при ферментативном ацилировании используют предпочтительно донор ацильных групп только из числа ранее описанных, так что стабильные в кислой среде защитные группы Z1 и Z2 в формуле (IX-i) имеют то же самое значение. 1,3-диацилоксициклогексан также имеется в продаже в виде цис-изомера (IX-i-цис) или в виде смеси цис/транс-изомеров (фирма Clariant). При этом цис-1,3-диацилоксициклогексан или смесь цис/транс-изомеров (IX-i) ферментативной десимметризацией (ферментативным гидролизом), как было описано ранее, превращают в энантиомерно почти чистый цис-1S-ацилоксициклогексан-3R-ол (ee >99%) (V-i). После этого цис-1S-ацилоксициклогексан-3R-ол действием, например 3,4-дигидро-2H-пирана или метоксипропена в присутствии кислотных катализаторов, таких, как, например, неорганические кислоты, толуолсульфокислота, пиридин-пара-толуолсульфонат или кислотный ионобменник, ацеталируют до цис-1S-ацилокси-3R-O-тетрагидропиранилциклогексана (VIII-i, где Z3 представляет собой тетрагидропиранил). Затем после деацилирования нуклеофильными агентами, такими, как, например, органические амины или неорганические гидроксиды или алкоголяты щелочных и щелочноземельных металлов, до цис-O-тетрагидропиранилциклогексан-3S-ола (II-i) осуществляют алкилирование оксазолгалогенидом (VI-i, где Z4 представляет собой галогенид) в присутствии одного из упомянутых неорганических органических оснований до цис-1S-O-оксалил-3R-O-тетрагидропиранилциклогексана (IIIa-i). Здесь следует указать на то, что на следующей далее схеме в формулах (IIIa-i, IVa-i, VI-i, VII-i, Ia-i, а также I-i) обе переменные X и Y в целях наглядности представлены метильными группами. Однако это не является ограничением, так как данная последовательность реакций может быть приведена с любыми другими значениями переменных X и Y. Цис-1S-O-оксалил-3R-O-тетрагидропиранил-циклогексан (IIIa-i) посредством действия первичных спиртов, таких, как, например, метанол, этанол, в присутствии кислотных катализаторов, таких, как, например, неорганические кислоты, толуолсульфокислота, пиридин-пара-толуолсульфонат, Amberlyst-H15, переводят в цис-3S-оксазилциклогексан-1R-ол (IVa-i), который, например, алкилируют алкиловым эфиром бромметилбензойной кислоты (VII-i, где Z4 представляет собой Br) в щелочной среде (соединение формулы (Ia-i)) и затем гидролизуют до соединения формулы (I-i), если R3 представляет собой H. Если в требуемом целевом соединении и R3, и R6 представляют собой C1-C6алкил или бензил, то стадию гидролиза проводить не требуется, так как в этом случае соединение (Ia-i) соответствует соединению (I-i). Гидролиз может осуществляться как в кислой, так и в щелочной среде. Щелочной гидролиз является приемлемым предпочтительно для случая, когда R6 представляет собой налкил, причем омыление до требуемых стереоизомеров PPAR-активаторов осуществляют действием гидроксидов металлов, таких, как, например, гидроксиды щелочных или щелочноземельных металлов, в соответствующих растворителях, таких, как, например, вода, спирты, органические растворители, а карбоксильную группу переводят в H-форму протонированием. Если R6 представляет собой трет-бутил, то гидролиз предпочтительно осуществляют в кислой среде. Альтернативно соединение (I-i) может быть получено гидрогенолизом соединения (Ia-i). Гидрогенолиз целесообразно применять, в частности, тогда, когда R6 представляет собой бензил и требуется соответствующее соединение (I-i), где R3 представляет собой H.

Как явствует из варианта осуществления стадии a) способа по настоящему изобретению, разработка способа получения соединения формулы (V) является другим объектом данного изобретения,

причем:
a1) соединение формулы (IX) в присутствии липазы B из Candida antarctica действием воды превращают в соединение формулы (V);

a2) соединение формулы (X) в присутствии липазы B из Candida antarctica действием по меньшей мере одного донора ацильных групп превращают в соединение формулы (V);

соединения (IX) и (X) представляют собой чистый цис-изомер или смесь цис/транс-изомеров соответственно;
при необходимости, выделяют транс-изомер соединения (IX) по завершении стадии a1) или транс-изомер соединения (X) по завершении стадии a2);
при этом переменные и заместители имеют следующие значения соответственно:
цикл A представляет собой C3-C8циклоалкил или C3-C8циклоалкенил, причем в циклолалкильных или циклоалкенильных циклах один или несколько атомов углерода могут быть замещены атомами кислорода;
Z1 и Z2 представляют собой независимо друг от друга защитные группы, стабильные в кислой среде.
Предпочтительно в соединениях (V) цикл A представляет собой цис-1,3-циклогексил, причем атом углерода цикла A, связанный с группой OH, имеет R-конфигурацию, а Z1 и Z2 представляют собой -C(O)-(C1-C3алкил).
Другим объектом данного изобретения, с учетом варианта осуществления стадии a), является также способ разделения смеси цис/транс-изомеров соединения формулы (X), отличающийся тем, что:
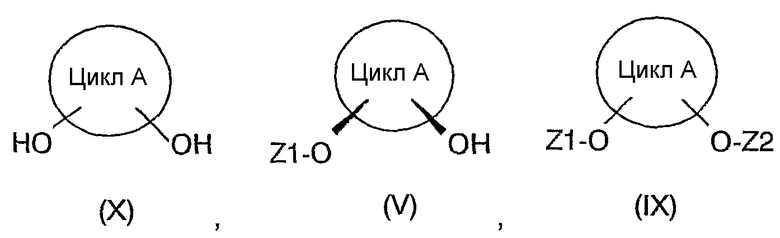
i) соединение (X) превращают в соединение формулы (IX) посредством действия по меньшей мере одного донора ацильных групп и соединение (IX) превращают в соединение формулы (V) посредством действия воды в присутствии фермента, обеспечивающего соответствующий избыток энантиомера соединения (V), и затем полученное таким образом соединение (V) отделяют от непрореагировавшего транс-изомера соединения (IX) и других побочных продуктов, в случае их образования, хроматографией, экстракцией или перегонкой, или
ii) соединение (X) превращают в соединение (V) посредством действия по меньшей мере одного донора ацильных групп в присутствии фермента, обеспечивающего соответствующий избыток энантиомера соединения (V), и затем полученное таким образом соединение (V) отделяют от непрореагировавшего транс-изомера соединения (X) и других побочных продуктов, в случае их образования, хроматографией, экстракцией или перегонкой, или
iii) соединение (X) превращают в соединение (IX) посредством действия по меньшей мере одного донора ацильных групп в присутствии фермента, обеспечивающего получение преимущественно цис-изомера соединения (IX), и затем цис-изомер соединения (IX) отделяют от образовавшегося при этом транс-изомера соединения (V) и других побочных продуктов, в случае их образования, хроматографией, экстракцией или перегонкой;
причем выделенные фракции соединений (IX) и (V) в присутствии нуклеофильного агента отщеплением защитной группы Z1 и/или Z2 превращают, при необходимости, в соответствующее соединение (X);
при этом переменные и заместители имеют следующие значения соответственно:
цикл A представляет собой C3-C8циклоалкил или C3-C8циклоалкенил, причем в циклолалкильных или циклоалкенильных циклах один или несколько атомов углерода могут быть замещены атомами кислорода;
Z1 и Z2 представляют собой независимо друг от друга защитные группы, стабильные в кислой среде.
В качестве возможно образующихся побочных продуктов, упоминаемых в вариантах от i) до iii) способа, могут быть приняты во внимание, в зависимости от выбранного варианта способа, например, транс-изомер соединения (IX), транс-изомер соединения (V) или цис-изомер соединения (IX). Упомянутые, возможно образовавшиеся в зависимости от хода реакции, другие побочные продукты, а также возможно оставшийся исходный продукт могут быть отделены от соответствующих образовавшихся главных фракций способами, известными специалисту в данной области техники, осуществляемыми в зависимости от обстоятельств в виде дополнительных стадий хроматографии, экстракции или перегонки.
Предпочтительно в данном способе разделения соответствующее превращение осуществляют по вариантам i) и/или ii) в присутствии липазы B из Candida antarctica и/или по варианту iii) в присутствии липазы поджелудочной железы свиньи, липазы из Burkholderia species, липазы из Pseudomonas cepacia или липазы из Pseudomonas species.
Особо предпочтительными данные способы разделения смесей цис/транс-изомеров являются тогда, когда цикл A представляет собой цис-1,3-циклогексил, причем атом углерода цикла A, связанный с группой OH, имеет R-конфигурацию, а Z1 и Z2 представляют собой -C(O)-(C1-C3алкил).
Другим объектом данного изобретения являются соединения формулы (IIIa), получающиеся в качестве промежуточных продуктов на стадии d) способа по настоящему изобретению.
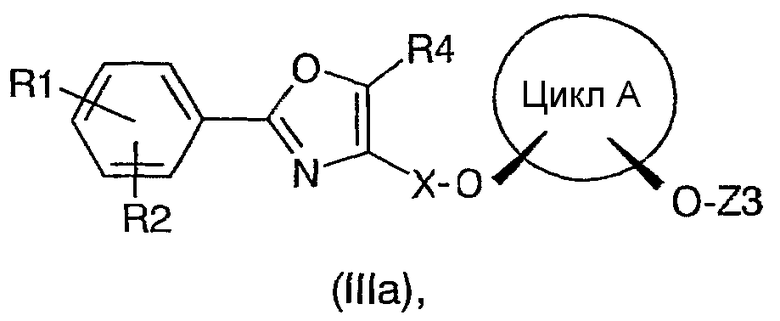
где:
R1, R2 и R4 представляют собой независимо друг от друга H, F, Cl, Br, OH, NO2, CF3, OCF3, C1-C6алкил или -O-(C1-C6алкил);
X представляет собой C1-C6алкил, причем в алкильной группе один или несколько атомов углерода могут быть замещены атомами кислорода;
цикл A представляет собой C3-C8циклоалкил или C3-C8циклоалкенил, причем в циклолалкильных или циклоалкенильных циклах один или несколько атомов углерода могут быть замещены атомами кислорода;
Z3 представляет собой защитную группу, стабильную в щелочной среде и лабильную в кислой среде.
В предпочтительных соединениях формулы (IIIa) следующие обозначения означают, что:
R1, R2 и R4 представляют собой независимо друг от друга H, F, Cl, Br, OH, NO2, CF3, OCF3, C1-C6алкил или -O-(C1-C6алкил);
X представляет собой C1-C3алкил;
цикл A представляет собой циклопентил, циклогексил или циклогептил;
Z3 представляет собой защитную группу, стабильную в щелочной среде и лабильную в кислой среде.
В более предпочтительных соединениях формулы (IIIa) следующие обозначения означают, что:
цикл A представляет собой циклогексил, причем X-содержащий и Z3-содержащий заместители стоят в цис-1,3-положении циклогексильного фрагмента;
R1, R2 и R4 представляют собой независимо друг от друга H, F, Cl, C1-C3алкил или -O-(C1-C3алкил);
Z3 представляет собой тетрагидропиранил;
X представляет собой метил.
В особо предпочтительных соединениях формулы (IIIa) следующие обозначения означают, что:
цикл A представляет собой циклогексил, причем X-содержащий и Z3-содержащий заместители стоят в цис-1,3-положении циклогексильного фрагмента и атом углерода цикла A, связанный с заместителем O-Z3, имеет R-конфигурацию;
R1, R2 и R4 представляют собой независимо друг от друга H, F, Cl, C1-C3алкил или -O-(C1-C3алкил);
Z3 представляет собой тетрагидропиранил;
X представляет собой метил.
Другим объектом данного изобретения являются соединения формулы (IIIb), получающиеся в качестве промежуточных продуктов на стадии d) способа по настоящему изобретению.

где:
R5 представляет собой независимо друг от друга H, F, Cl, Br, OH, NO2, CF3, OCF3, C1-C6алкил или -O-(C1-C6алкил);
R6 представляет собой C1-C6алкил или бензил, которые при необходимости могут быть замещены F, Cl, Br, OH, NO2, CF3, OCF3, C1-C6алкилом или -O-(C1-C6алкилом);
Y представляет собой C1-C6алкил, причем в алкильной группе один или несколько атомов углерода могут быть замещены атомами кислорода;
цикл A представляет собой C3-C8циклоалкил или C3-C8циклоалкенил, причем в циклолалкильных или циклоалкенильных циклах один или несколько атомов углерода могут быть замещены атомами кислорода;
Z3 представляет собой защитную группу, стабильную в щелочной среде и лабильную в кислой среде.
В предпочтительных соединениях формулы (IIIb) следующие обозначения означают, что:
цикл A представляет собой циклогексил, причем X-содержащий и Z3-содержащий заместители стоят в цис-1,3-положении циклогексильного фрагмента и атом углерода цикла A, связанный с заместителем O-Z3, имеет R-конфигурацию;
R5 представляет собой H, F, Cl, C1-C3алкил или -O-(C1-C3алкил);
R6 представляет собой C1-C6алкил или бензил;
Z3 представляет собой тетрагидропиранил;
Y представляет собой метил.
Другим объектом данного изобретения являются соединения формулы (VIII), получающиеся в качестве промежуточных продуктов на стадии b) способа по настоящему изобретению.

где:
цикл A представляет собой C3-C8циклоалкил или C3-C8циклоалкенил, причем в циклолалкильных или циклоалкенильных циклах один или несколько атомов углерода могут быть замещены атомами кислорода;
Z1 представляет собой защитную группу, стабильную в кислой среде;
Z3 представляет собой защитную группу, стабильную в щелочной среде и лабильную в кислой среде.
В предпочтительных соединениях формулы (VIII) следующие обозначения означают, что:
цикл A представляет собой циклопентил, циклогексил или циклогептил;
Z1 представляет собой -C(O)-(C1-C3алкил);
Z3 представляет собой тетрагидропиранил или метоксиизопропил.
В более предпочтительных соединениях формулы (VIII) следующие обозначения означают, что:
цикл A представляет собой циклогексил, причем Z1-содержащий и Z3-содержащий заместители стоят в цис-1,3-положении циклогексильного фрагмента;
Z1 представляет собой -C(O)CH3;
Z3 представляет собой тетрагидропиранил.
В особо предпочтительных соединениях формулы (VIII) следующие обозначения означают, что:
цикл A представляет собой циклогексил, причем Z1-содержащий и Z3-содержащий заместители стоят в цис-1,3-положении циклогексильного фрагмента и атом углерода цикла A, связанный с заместителем O-Z3, имеет R-конфигурацию;
Z1 представляет собой -C(O)CH3;
Z3 представляет собой тетрагидропиранил.
Как отмечено ранее, соединения формул (IIIa) и (IIIb) могут быть получены в соответствии со стадиями от a) до d) способа по настоящему изобретению. В связи с этим имеют силу те же самые варианты осуществления, что и в случае получения соединений формулы (I). Это действительно также в отношении соединений формулы (VIII), которые могут быть получены в соответствии со стадиями от a) до b) способа по настоящему изобретению. Следовательно, все упомянутые промежуточные соединения являются также приемлемыми, при необходимости, в качестве исходных соединений для синтеза PPAR-активаторов общей формулы (I).
Приведенные далее примеры предназначены для пояснения настоящего способ, однако, без наложения на него ограничений.
Примеры
ГХ анализ
Определение степени превращения: колонка Supelco BetaDex325 (30 м×0,25 мм×0,25 мкм); газ-носитель H2 (1,2 мл/мин); температура испарителя 250°C; ПИД; температура термостата 130°C, изотермический режим; усиление 100:1.
Время удерживания: 10,9 мин для цис-1-ацетоксициклогексан-3-ола, 14,6 мин для цис-1,3-диацетоксициклогексана, 12,6 и 12,8 мин для транс-1,3-диацетоксициклогексана.
Для определения избытка энантиомера цис-1S-ацетоксициклогексан-3R-ола проводят дериватизацию ангидридом гептафтормасляной кислоты: 2 мг пробы растворяют в 0,1 мл ангидрида гептафтормасляной кислоты и реакционную смесь выдерживают в течение 10 минут при 70°C. Пробу в токе азота упаривают до сухого состояния и снова растворяют в 0,5 мл метиленхлорида. Время удерживания: 24,4 мин для производного цис-1S-ацетоксициклогексан-3R-ола с гептафтормасляной кислотой; 25,2 мин для производного цис-1R-ацетоксициклогексан-3S-ола с гептафтормасляной кислотой;
колонка Supelco BetaDex325 (30 м×0,25 мм×0,25 мкм); газ-носитель He (1,5 мл/мин); температура испарителя 230°C; ПИД; температура термостата 100°C, изотермический режим; усиление 50:1.
Пример 1. Взаимодействие смеси цис/транс-изомеров циклогексан-1,3-диола с винилацетатом
4 мг смеси цис/транс-циклогексан-1,3-диола с соотношением 60:40 растворяют в 1 мл винилацетата и прибавляют 5 мг фермента согласно таблице 1. Смесь выдерживают при 25°C и через 21 или 45 ч отбирают пробу.
С ферментами Chirazyme L2 lyo., Chirazyme L2 c.f. C2 и Chirazyme L2 c.f. C3 получают предпочтительно моноацетат в цис-форме. Кроме того, ферменты Chirazyme L7 lyo., Chirazyme L1 lyo., Chirazyme L1 c.f., липаза PS и липаза AH неожиданным образом катализировали образование диацетата в цис-форме из цис-циклогексан-1,3-диола при одновременно высокой степени превращения, тогда как транс-изомер преимущественно превращается в моноацетат в транс-форме (таблица 2).
Образующаяся при применении соответствующего фермента смесь моноацетата и диацетата может быть разделена, например, экстракцией или перегонкой. Таким образом удается также выделить цис-1,3-диацетоксициклогексан из имеющейся в продаже смеси циc/транс-изомеров циклогексан-1,3-диола.
Пример 2. Превращение смеси цис/транс-изомеров циклогексан-1,3-диола в 1,3-диацетоксициклогексан действием винилацетата
30 г смеси цис/транс-циклогексан-1,3-диола с соотношением 60:40 растворяют в 900 мл метил-трет-бутилового эфира, прибавляют 100 мл винилацетата и 1,7 г липазы (Chirazyme L1 lyo.) (20°C). Через 24 ч отфильтровывают протеин и удаляют растворитель. Маслянистый остаток растворяют в 1000 мл циклогексана и промывают водой четыре раза по 150 мл. Органический слой удаляют в ротационном испарителе. Получают 10 г цис-1,3-диацетоксициклогексана в виде маслянистого продукта (выход 23% в пересчете на смесь цис/транс-изомеров, 95% в цис-форме).
Пример 3. Химическое ацилирование смеси цис/транс-изомеров циклогексан-1,3-диола до 1,3-диацетоксициклогексана с заключительной ферментативной десимметризацией до цис-1S-ацетоксициклогексан-3R-ола
Химическое ацилирование
Цис/транс-циклогексан-1,3-диол превращают в цис/транс-1,3-диацетоксициклогексан простыми способами ацилирования ацетангидридом или ацетилхлоридом, которые, например, известны из издания Organikum, Seite 405-7, 16.Auflage 1986 VEB Deutscher Verlag der Wissenschaften (Berlin).
Ферментативная десимметризация
21 г смеси цис/транс-1,3-диацетоксициклогексана с соотношением 96:4 и 0,2 г фермента Chirazyme L2 lyo. при 25°C вносят в 150 мл 0,1 M буферного раствора фосфата калия (pH=7,0). Значение pH поддерживают равным 7,0 добавлением 10 M раствора KOH и реакционную смесь перемешивают в течение 5 часов. Затем реакционную смесь экстрагируют метиленхлоридом три раза по 100 мл и растворитель удаляют в ротационном растворителе. Получают 15,8 г цис-1S-ацетоксициклогексан-3R-ола (выход 83% в пересчете на смесь цис/транс-изомеров, ee >99%). Идентификацию абсолютной конфигурации осуществляют по результатам хиральной ВЭЖХ на дальнейшей стадии синтеза цис-3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексан-1R-ола (пример 8) по сравнению с известным стандартным веществом.
Приведенная далее схема предназначена для пояснения способа получения по настоящему изобретению соединений формулы (I) на конкретном примере (10).
Схема III

Пример 4. Химическое ацилирование цис-циклогексан-1,3-диола до цис-1,3-диацетоксициклогексана с заключительной ферментативной десимметризацией до цис-1S-ацетоксициклогексан-3R-ола
Химическое ацилирование
Дис-циклогексан-1,3-диол превращают в цис-1,3-диацетоксициклогексан простыми способами ацилирования ацетангидридом или ацетилхлоридом, которые, например, известны из издания Organikum, Seite 405-7, 16. Auflage 1986 VEB Deutscher Verlag der Wissenschaften (Berlin).
Ферментативная десимметризация
166 г цис-1,3-диацетоксициклогексана и 1,6 г фермента Chirazyme L2 lyo. вносят в 1 л 0,1 М буферного раствора фосфата калия (pH=6,8), постоянное значение pH поддерживают добавлением 10 M раствора KOH. Реакционную смесь выдерживают в течение 6 часов при 35°C и затем нагревают при 75°C в течение 1 часа. Далее реакционную смесь охлаждают до 8°C и оставляют стоять в течение ночи. Протеин отфильтровывают и реакционную смесь экстрагируют толуолом два раза по 500 мл соответственно. Растворитель удаляют в ротационном испарителе. Получают 121,3 г цис-1S-ацетоксициклогексан-3R-ола в виде бесцветного маслянистого продукта (выход 87%, ee >99%).
Пример 5. Введение тетрагидропиранильной (THP) защитной группы с получением цис-1S-ацетокси-3R-(O-тетрагидропиранил)циклогексана
25 г (138 ммоль) цис-1S-ацетоксициклогексан-3R-ола с чистотой 87% (ее >99%) нагревают с 15,6 г (166 ммоль) 3,4-дигидро-2H-пирана в присутствии 1,76 г (0,05 г-экв) пиридин-пара-толуолсульфоната в 125 мл толуола в течение часа при перемешивании и температуре от 50 до 60°C. Контроль хода реакции с количественным определением степени превращения осуществляют ГХ анализом. Реакционную смесь охлаждают и отфильтровывают выпавший в осадок пириридин-пара-толуолсульфонат. Удаление пиридин-пара-толуолсульфоната фильтрованием не является обязательно необходимым, так как при заключительном деацетилировании избыток введенного метилата натрия нейтрализует кислую соль пиридин-пара-толуолсульфоната, вследствие чего влияние на деацетилирование не оказывается. Полученный фильтрат без дальнейшей очистки подвергают заключительному деацетилированию до цис-3R-(O-тетрагидропиранил)циклогексан-1S-ола. После концентрирования фильтрата получают маслянистый продукт, причем в качестве главного компонента получают цис-1S-ацетокси-3R-(O-тетра-гидропиранил)циклогексан с молекулярной массой 242,32 (C13H22O4); MS (EI): 241 (M-H+).
Пример 6. Деацетилирование до цис-3R-(O-тетрагидропиранил)циклогексан-1S-ола
Толуольный раствор цис-1S-ацетокси-3R-(O-тетрагидропиранил)циклогексана из примера 5 смешивают с 115 мл метанола и приводят во взаимодействие с 7,48 г (0,3 г-экв) 30%-го раствора метилата натрия в метаноле. Количественный контроль деацетилирования может проводиться ГХ анализом. Деацетилирование может быть осуществлено по способу A) или B):
Способ A)
Приблизительно через 2 часа перемешивания при комнатной температуре реакционную смесь смешивают с 600 мл воды. После отделения водного слоя толуольный слой экстрагируют водой три раза по 600 мл соответственно. Водные слои объединяют, насыщают NaCl и экстрагируют толуолом три раза по 600 мл соответственно. Образующиеся по ходу реакции прочие производные цис-1,3-циклогександиола, то есть исходные вещества, побочные продукты или предшественники, которые не соответствуют примеру 6 или соответствуют побочному продукту, образованному с 2 защитными THP-группами, остаются в виде водорастворимых соединений в водном слое. Органические слои объединяют и концентрируют в вакууме. Цис-3-(O-тетрагидропиранил)циклогексан-1S-ол получают в виде бесцветного маслянистого продукта с чистотой более 97% и выходом более 96% в пересчете на цис-1S-ацетоксициклогексан-3R-ол. Полученный таким образом цис-3R-(O-тетрагидропиранил)циклогексан-1S-ол имеет молекулярную массу 200,28 (C11H20O3); MS (EI): 199 (M-H+).
Способ B)
Приблизительно через 2 часа перемешивания при комнатной температуре реакционную смесь смешивают с 200 мл воды и двухфазную смесь перемешивают в течение часа. Избыток 3,4-дигидро-2H-пирана, метанола, толуола и воды отгоняют при температуре от 65°C и давлении от 300 до 150 мбар. Вязкий остаток обрабатывают 90 мл толуола и 14 мл воды и смешивают с 25 г хлорида натрия. После перемешивания данной двухфазной смеси слои разделяют и водный слой дополнительно экстрагируют 30 мл толуола.
Образующиеся по ходу реакции прочие производные цис-1,3-циклогександиола, то есть исходные вещества, побочные продукты или предшественники, которые не соответствуют примеру 5 или соответствуют побочному продукту, образованному с 2 THP-защитными группами, остаются в виде водорастворимых соединений в водном слое. Органические слои объединяют и концентрируют в вакууме. Цис-3-(O-тетрагидропиранил)циклогексан-1S-ол получают в виде бесцветного маслянистого продукта с чистотой более 95% и выходом более 95% в пересчете на цис-1S-ацетоксициклогексан-3R-ол. Полученный таким образом цис-3R-(O-тетрагидропиранил)циклогексан-1S-ол имеет молекулярную массу 200,28 (C11H20O3); MS (EI): 199 (M-H+).
Пример 7. Алкилирование 4-(хлорметил)-2-(4-фторфенил)оксазолом с получением цис-1S-(2-(4-фторфенил)оксазол-4-илметокси)-3R-O-тетрагидропиранилциклогексана
1,5 г (7,2 ммоль) цис-3R-(O-тетрагидропиранил)циклогексан-1S-ола нагревают в 40 мл толуола с 1,17 г (10,1 ммоль) трет-бутилата калия при перемешивании в течение 20 мин при 55°C. К реакционной смеси прибавляют по каплям при 55°C толуольный раствор 1,74 г (7,9 ммоль) 4-(хлорметил)-2-(4-фторфенил)оксазола в 26 мл толуола. По окончании прибавления реакционную смесь перемешивают в течение приблизительно 5 часов при 55°C. Контроль за ходом реакции осуществляют методом ВЭЖХ. Затем к реакционной смеси при интенсивном перемешивании прибавляют 20 мл воды. После концентрирования объединенных органических слоев получают коричневый маслянистый продукт цис-1-(2-(4-фторфенил)оксазол-4-илметокси)-3-O-тетрагидропиранилциклогексана с чистотой 74% и молекулярной массой 375,44 (C21H26FNO4); MS (EI): 375.
Объединенные органические слои могут быть использованы без последующей очистки для заключительного отщепления тетрагидропиранильной защитной группы с получением цис-3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексан-1R-ола.
Пример 8. Удаление тетрагидропиранильной защитной группы с получением цис-3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексан-1R-ола
2,3 г (4,53 ммоль) цис-3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексан-1R-ола с чистотой 74% перемешивают в 9 мл толуола с 3 мл метанола в присутствии 113 мг (0,1 г-экв) пиридин-пара-толуолсульфоната в течение 6 часов при 55°C. Затем реакционную смесь экстрагируют 10 мл водного раствора NaCl. При этом в водном слое остается возможно имеющийся 1,3-циклогександиол, который образуется из возможно образовавшегося в примере 5 побочного продукта, имеющего 2 защитные THP-группы. Органический слой концентрируют в вакууме. Полученный коричневый высоковязкий маслянистый продукт смешивают с небольшим количеством диизопропилового эфира. Через несколько часов кристаллизуют 44% цис-3-(2-(4-фторфенил)оксазол-4-илметокси)циклогексанола с ее >99% и чистотой 99%. После концентрирования маточного раствора кристаллизуют последующие 24% цис-3-(2-(4-фторфенил)оксазол-4-илметокси)циклогексанола с ее >99%, однако с чистотой 89%. Хотя дальнейшим концентрированием маточного раствора можно получить дополнительное количество кристаллического цис-3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексан-1R-ола, но с каждым последующим концентрированием маточного раствора также увеличивается содержание примесей в кристаллизованном продукте. Простое хроматографирование смесью этилацетат/толуол полученного в виде коричневого маслянистого продукта цис-3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексан-1R-ола альтернативно приводит к существенно лучшему выходу. После хроматографирования и концентрирования в вакууме получают цис-3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексан-1R-ол в виде белых кристаллов с ee >99%. Полученный цис-3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексан-1R-ол имеет молекулярную массу 291,32 (C16H18FNO3); MS (TOF MS ES+): 291,9 (M+H+).
Пример 9. Алкилирование метиловым эфиром 2-(бромметил)-6-метилбензойной кислоты с получением метилового эфира цис-2-(3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойной кислоты
5 г (17,2 ммоль) цис-3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексан-1R-ола растворяют в 175 мл хлорбензола и при 0°C смешивают с 3,54 г (30,9 ммоль) трет-бутилата калия. Альтернативно цис-3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексан-1R-ол и трет-бутилат калия могут быть взяты в виде твердых веществ и смешаны при 0°C с 175 мл хлорбензола. В течение 20 мин при перемешивании и температуре 0°C по каплям прибавляют 9,54 г (18,9 ммоль) метилового эфира 2-бромметил-6-метилбензойной кислоты в виде 50%-го раствора в циклогексане. После перемешивания при 0°C в течение ночи реакционную смесь экстрагируют водой три раза по 40 мл соответственно. Органический слой сушат над сульфатом магния и концентрируют в вакууме. Получают коричневый маслянистый продукт с содержанием от 80 до 90% метилового эфира цис-2-(3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойной кислоты, который без дальнейшей очистки может быть использован для последующего омыления. Полученный метиловый эфир цис-2-(3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойной кислоты имеет молекулярную массу 453,52 (C26H28FNO5); MS (ESI): 454 (M+H+).
Пример 10. Омыление и перевод цис-2-(3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойной кислоты в форму свободной кислоты протонированием карбоксильной группы
Пример 10a)
7,79 г (15,1 ммоль) метилового эфира цис-2-(3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойной кислоты с чистотой 88% перемешивают в течение ночи при 50°C с 2,99 г (45,3 ммоль) 85%-го гидроксида калия в 78 мл трет-бутанола. Затем трижды прибавляют по 78 мл воды и соответственно отгоняют в вакууме приблизительно 80 мл растворителя до получения мутного желтоватого раствора продукта. Данный раствор экстрагируют метил-трет-бутиловым эфиром три раза по 24 мл соответственно. После отделения органического слоя водный раствор смешивают с 50 мл ацетона и подкисляют 2 M раствором соляной кислоты до pH=4-5. Раствор перемешивают при температуре 0-5°C, при этом образуется цис-2-(3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойная кислота в виде белого кристаллического твердого вещества (чистота более 95%), который может быть отделен фильтрованием. Цис-2-(3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойная кислота, полученная в виде белого кристаллического твердого вещества с выходом более 80%, избытком энантиомера более 99% и чистотой более 95%, для дальнейшей очистки может быть перекристаллизована из толуола. Вместо толуола для перекристаллизации могут использоваться также другие растворители, такие, как, например, н-бутилацетат, спирты и водно-спиртовые смеси.
Полученная цис-2-(3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексил-1R-оксиметил)метилбензойная кислота имеет молекулярную массу 439,18 (C25H26FNO5); MS (TOF MS ES+): 440,2 (M+H+).
Пример 10b)
11,88 г (18,9 ммоль) метилового эфира цис-2-(3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойной кислоты с чистотой 72% перемешивают в течение ночи при 70°C с 6,23 г (94,4 ммоль) 85%-го гидроксида калия и 6 мл воды в 113 мл трет-бутанола. Затем трижды прибавляют по 85 мл воды и соответственно отгоняют в вакууме приблизительно 80 мл растворителя. Остаток экстрагируют метилизобутилкетоном три раза по 36 мл соответственно. Объединенные водные слои подкисляют 35 мл 2 M раствора HCl до pH=8 и смешивают с 21 мл н-бутилацетата. Добавлением еще 10,5 мл 2 M раствора HCl 2-фазную смесь подкисляют до pH=4 и нагревают до 90°C. Слои разделяют в предварительно нагретой делительной воронке. Органический слой нагревают с активированным углем до 90°C и фильтруют. Фильтрат охлаждают до комнатной температуры, при этом выкристаллизовывается цис-2-(3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойная кислота в виде белого твердого вещества (чистота более 90%). Цис-2-(3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойная кислоту, полученную с выходом 75% и избытком энантиомера более 99%, для дальнейшей очистки перекристаллизовывают из н-бутилацетата, при этом достигается чистота более 97% и избыток энантиомера более 99%. Полученная цис-2-(3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексил-1R-оксиметил)метилбензойная кислота имеет молекулярную массу 439,18 (C25H26FNO5); MS (TOF MS ES+): 440,2 (M+H+).
В приведенных далее примерах с 11 по 14 показывается синтез другого соединения общей формулы (I).
Пример 11. Алкилирование 4-(хлорметил)-2-(3-метоксифенил)-5-метилоксазолом с получением цис-1S-(2-(3-метоксифенил)-5-метилоксазол-4-илметокси)-3R-O-тетрагидропиранилциклогексана
Суспензию 13,3 г (115 ммоль) трет-бутилата калия в 140 мл толуола нагревают при перемешивании до 55°C. Раствор 8,58 г (40,2 ммоль) цис-3R-(O-тетрагидропиранил)циклогексан-1S-ола (пример 6) в 88,5 мл толуола прибавляют по каплям в течение 15 мин при перемешивании и температуре 55°C. По окончании прибавления смесь перемешивают в течение 15 мин при 55°C. Затем 52,24 г (40 ммоль) 4-(хлорметил)-2-(3-метоксифенил)-5-метилоксазола в толуоле (18,2%-й раствор) прибавляют по каплям в течение 45 мин при 55°C. Далее перемешивают при 55°C в течение часа. Затем реакционную смесь смешивают с 60 мл воды и 30 мл насыщенного раствора хлорида натрия и охлаждают до комнатной температуры при перемешивании. Слои разделяют и органический слой концентрируют в ротационном испарителе. Маслянистый остаток очищают хроматографией на кизельгуре смесью толуол/этилацетат в соотношении 3:1. После концентрирования в вакууме получают маслянистый продукт цис-1-(2-(3-метоксифенил)-5-метилоксазол-4-илметокси)-3-O-тетрагидропиранилциклогексана с молекулярной массой 401,5 (C23H31NO5); MS (EI): 401 (M+).
Толуольный раствор цис-1-(2-(3-метокси-фенил)-5-метилоксазол-4-илметокси)-3-O-тетрагидропиранилциклогексана может быть также использован без дальнейшей очистки при заключительном отщеплении тетрагидропиранильной защитной группы с получением цис-3S-(2-(3-метоксифенил)-5-метилоксазол-4-илметокси)циклогексан-1R-ола, так как не требуется смена растворителя.
Пример 12. Удаление тетрагидропиранильной защитной группы с получением цис-3S-(2-(3-метоксифенил)-5-метилоксазол-4-илметокси)циклогексан-1R-ола
8,04 г (20 ммоль) цис-1-(2-(3-метоксифенил)-5-метилоксазол-4-илметокси)-3-O-тетрагидропиранилциклогексана в толуоле смешивают с 13 мл метанола и 0,3 мл 30%-го раствора соляной кислоты и затем нагревают в течение приблизительно 2 часов при 55°C. Далее реакционную смесь смешивают с 25 мл воды. После разделения слоев органический слой концентрируют в вакууме при 40°C. Получают 6,03 г (выход 95%) коричневого маслянистого продукта, который растворяют в 10 мл толуола и очищают колоночной хроматографией смесью толуол/этилацетат в соотношении 1:2. После удаления органического растворителя получают 3,25 г (69,8%) цис-3S-(2-(3-метоксифенил)-5-метилоксазол-4-илметокси)циклогексан-1R-ола в виде коричневого вязкого маслянистого продукта с молекулярной массой 317,39 (C18H23NO4); MS (EI): 317 (M+).
Пример 13. Алкилирование метиловым эфиром 2-(бромметил)-6-метилбензойной кислоты с получением метилового эфира цис-2-(3S-(2-(3-метоксифенил)-5-метилоксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойной кислоты
59 г (89,3 ммоль) цис-3S-(2-(3-метоксифенил)-5-метилоксазол-4-илметокси)циклогексан-1R-ола растворяют в 825 мл хлорбензола и при 0°C смешивают с 17,7 г (158 ммоль) трет-бутилата калия. В течение 20 мин при перемешивании и температуре 0°C по каплям прибавляют 9,54 г (18,9 ммоль) метилового эфира 2-бромметил-6-метилбензойной кислоты в виде 50%-го раствора в циклогексане. После перемешивания при 0°C в течение ночи реакционную смесь смешивают с 350 мл насыщенного раствора NaCl и перемешивают. Органический слой отделяют и концентрируют в вакууме. Получают коричневый маслянистый продукт с содержанием 80% метилового эфира цис-2-(3S-(2-(3-метоксифенил)-5-метилоксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойной кислоты, который может быть использован для последующего омыления без дальнейшей очистки или перед дальнейшим превращением очищен хроматографией на кизельгуре смесью гептан/этилацетат. -Метиловый эфир цис-2-(3S-(2-(3-метоксифенил)-5-метилоксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойной кислоты, полученный с выходом 64%, имеет молекулярную массу 479,58 (C28H33NO6); MS (TOF MS ES+): 480,2 (M+H+).
Пример 14. Омыление и перевод цис-2-(3S-(2-(3-метоксифенил)-5-метилоксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойной кислоты в форму свободной кислоты протонированием карбоксильной группы
Пример 14a)
7 г (14,6 ммоль) метилового эфира цис-2-(3S-(2-(3-метоксифенил)-5-метилоксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойной кислоты с чистотой 96,5% нагревают с 7 мл 33%-го раствора гидроксида натрия в 70 мл этанола с обратным холодильником в течение 25 ч. Затем реакционную смесь полностью упаривают в вакууме. Маслянистый остаток растворяют в 71 мл воды и затем экстрагируют метил-трет-бутиловым эфиром два раза по 24 мл соответственно. Водную фазу продукта смешивают с 70 мл дихлорметана и перемешивают в течение 5 мин. Добавлением 11,9 мл 30%-го раствора соляной кислоты 2-фазную смесь подкисляют до pH=1. Слои разделяют, и водный слой еще раз экстрагируют дихлорметаном. Собранные органические слои сушат над сульфатом магния и концентрируют в вакууме. Оставшийся высоковязкий маслянистый продукт растворяют в 50 мл диизопропилового эфира. После внесения в раствор затравки 10 мг цис-2-(3S-(2-(3-метоксифенил)-5-метилоксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойной кислоты через 30 мин начинается кристаллизация, которая может быть улучшена многочасовым охлаждением при 0-5°C. Кристаллы отсасывают и сушат в вакууме при 40°C. Цис-2-(3S-(2-(3-метоксифенил)-5-метилоксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойная кислота, полученная с выходом 72,1%, избытком энантиомера более 99% и чистотой более 99%, для дальнейшей очистки может быть перекристаллизована из бутилацетата.
Полученная цис-2-(3S-(2-(3-метоксифенил)-5-метилоксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойная кислота имеет молекулярную массу 465,55 (C27H31NO6); MS (ES+): 466,3 (M+H+).
Пример 14b)
Омыление 7,9 г (16,5 ммоль) метилового эфира цис-2-(3S-(2-(3-метоксифенил)-5-метилоксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойной кислоты осуществляют непосредственно после алкилирования цис-3S-(2-(3-метоксифенил)-5-метилоксазол-4-илметокси)циклогексан-1R-ола метиловым эфиром 2-бромметил-6-метилбензойной кислоты в присутствии трет-бутилата калия, как описано в примере 13, в 750 мл хлорбензола без обработки водой добавлением к реакционной смеси 7,62 г (115 ммоль) 85%-го КОН и нагревании при 80°С в течение 3 часов. После охлаждения до комнатной температуры реакционную смесь экстрагируют 30 мл воды. Затем хлорбензольный раствор еще раз экстрагируют 80 мл воды. Объединенные водные слои смешивают с 40 мл метиленхлорида и подкисляют 10 мл 2 М раствора HCl до рН=2. После интенсивного перемешивания слои разделяют и органический слой концентрируют в вакууме. Остаток растворяют диизопропиловым эфиром и обрабатывают аналогично предыдущему примеру 14а.
В приведенных далее примерах с 15 по 19, а также на схеме IV показывается синтез еще одного соединения общей формулы (I).
Схема IV

В данном примерном варианте синтеза соединение примера 6 действием 4-(хлорметил)-5-метил-2-п-толилоксазол в присутствии, например, оснований щелочных или щелочноземельных металлов, предпочтительно в присутствии трет-бутилата калия или бис-(триметилсилил)амида калия, превращают в соединение примера 15. Затем соединение примера 15 действием метанола в присутствии кислотных катализаторов, таких, как, например, неорганические кислоты, толуолсульфокислота, пириридин-пара-толуолсульфонат, Amberlyst-H15, предпочтительно с неорганическими кислотами, переводят в соединение примера 16. 2-бромметил-6-метилбензоилбромид взаимодействует со спиртами, такими, как метанол (Mе-OH), трет-бутанол (tBu-OH) или бензиловый спирт (Bn-OH) с образованием соединения примера 17a, соединения примера 17b или соединения примера 17c. Соединение примера 16 алкилируют соединением примера 17a, соединением примера 17b или соединением примера 17c в присутствии, например, оснований щелочных или щелочноземельных металлов, предпочтительно в присутствии трет-бутилата калия, с получением соединения примера 18a, соединения примера 18b или соединения примера 18c. Соединение примера 18a омыляют действием гидроксидов металлов, таких, как, например, гидроксиды щелочных или щелочноземельных металлов, предпочтительно гидроксид натрия и калия, в приемлемом растворителе, таком, как вода, спирты или органические растворители, предпочтительно в этаноле или изопропаноле. Протонированием, например, органическими или неорганическими кислотами, предпочтительно неорганическими кислотами, выделяют требуемый хиральный PPAR-активатор примера 19. Соединение примера 19 получают также из соединения примера 18b расщеплением сложного эфира с кислотными катализаторами, такими, как, например, неорганические кислоты или трифторуксусная кислота (ТФУ), предпочтительно соляная или трифторуксусная кислота. Соединение примера 18c переводят в соединение примера 19 гидрогенолизом с гетерогенным катализатором, предпочтительно с катализатором из благородного металла и более предпочтительно с палладиевым катализатором. Соединение примера 19 выделяют с оптической чистотой более 99%, причем для очистки не требуется ни хиральная, ни нехиральная хроматография. Очистку осуществляют перекристаллизацией соединения примера 16 и PPAR-активатора примера 19, например, из органического растворителя, предпочтительно из диизопропилового эфира.
Пример 15. Алкилирование цис-3R-(O-тетрагидропиранил)циклогексан-1S-ола 4-(хлорметил)-5-метил-2-п-толилоксазолом с получением цис-1S-(2-п-толил-5-метилоксазол-4-илметокси)-3R-O-тетрагидропиранилциклогексана
К суспензии 130 г (652 ммоль) бис-(триметилсилил)амида калия в 1,5 л трет-бутилметилового эфира прибавляют при перемешивании раствор 110 г (550 ммоль) цис-3R-(O-тетрагидропиранил)циклогексан-1S-ола (пример 6) в 500 мл трет-бутилметилового эфира и 97 мл толуола. Реакционную смесь перемешивают в течение 10 минут при комнатной температуре перед прибавлением раствора 111 г (501 ммоль) 4-(хлорметил)-5-метил-2-п-толилоксазола в 1,5 л трет-бутилметилового эфира. Реакционную смесь перемешивают до окончания превращения (контроль методом ВЭЖХ) при 35°C. Аликвоту реакционной смеси промывают водой, сушат над сульфатом магния и затем полностью упаривают в вакууме. Полученный таким образом цис-1S-(2-п-толил-5-метилоксазол-4-илметокси)-3R-O-тетрагидропиранилциклогексан имеет молекулярную массу 385,51 (C23H31NO4); MS (ESI): 302 [M-THP+H]+.
Пример 16. Отщепление защитной THP-группы от цис-3S-(2-п-толил-5-метилоксазол-4-илметокси)-3R-O-тетрагидропиранилциклогексана с получением цис-3S-(2-п-толил-5-метилоксазол-4-илметокси)циклогексан-1R-ола
К раствору цис-3S-(2-п-толил-5-метилоксазол-4-илметокси)-3R-O-тетрагидропиранилциклогексана в трет-бутилметиловом эфире и толуоле из примера 15 прибавляют 100 мл метанола и затем 101 мл концентрированной соляной кислоты. Реакционную смесь перемешивают при 25°C. По окончании превращения прибавляют 1,0 л воды и 101 г (1,2 моль) гидрокарбоната натрия и смесь интенсивно перемешивают. После фильтрования водный слой отделяют и органический слой промывают водой три раза по 800 мл соответственно. Растворитель отгоняют в вакууме и получившийся маслянистый остаток растворяют в 450 мл диизопропилового эфира. Смесь перемешивают при комнатной температуре. Выпавший в осадок продукт отфильтровывают и промывают небольшим количеством диизопропилового эфира. Получают цис-3S-(2-п-толил-5-метилоксазол-4-илметокси)циклогексан-1R-ол с выходом 56% и молекулярной массой 301,39 (C18H23NO3); MS (ESI): 302 [M+H]+.
Пример 17a. Синтез метилового эфира 2-бромметил-6-метилбензойной кислоты из 2-бромметил-6-метилбензоилбромида
К 400 мл метанола прибавляют раствор 200 г (240 ммоль) 2-бромметил-6-метилбензоилбромида (35%-й раствор в гептане). После перемешивания в течение часа при комнатной температуре прибавляют 400 мл н-гептана. Раствор последовательно промывают 600 мл насыщенного раствора гидрокарбоната натрия и водой три раза по 200 мл соответственно. Затем растворитель полностью удаляют в вакууме. Получают метиловый эфир 2-бромметил-6-метилбензойной кислоты в виде бесцветного маслянистого продукта с выходом 98,8%, чистотой 80% (в пересчете на выход) и молекулярной массой 241,99 (C10H11BrO2); MS (ESI): 243,1 [M+H]+.
Пример 17b. Синтез трет-бутилового эфира 2-бромметил-6-метилбензойной кислоты
К смеси 15 мл н-гептана и 25 мл трет-бутанола прибавляют раствор 20 г (24 ммоль) 2-бромметил-6-метилбензоилбромида (35%-й раствор в гептане). После перемешивания в течение часа при 40°C прибавляют 400 мл н-гептана. Раствор последовательно промывают 60 мл насыщенного раствора гидрокарбоната натрия и водой четыре раза по 20 мл соответственно. Затем растворитель полностью удаляют в вакууме. При количественном превращении получают трет-бутиловый эфир 2-бромметил-6-метилбензойной кислоты в виде почти бесцветного маслянистого продукта с чистотой 93% и молекулярной массой 284,04 (C13H17BrO2); MS (ESI): 302,1 [M+NH4]+.
Пример 17c. Синтез бензилового эфира 2-бромметил-6-метилбензойной кислоты
К раствору 136 г (233 ммоль) 2-бромметил-6-метилбензоилбромида (35%-й раствор в гептане) прибавляют при охлаждении льдом 24,2 г (221 ммоль) бензилового спирта. Затем перемешивают в течение часа при комнатной температуре. После прибавления 500 мл трет-бутилметилового эфира органический раствор последовательно промывают 410 мл насыщенного раствора гидрокарбоната натрия и водой четыре раза по 300 мл соответственно. Затем растворитель полностью удаляют в вакууме. Получают 74 г бензилового эфира 2-бромметил-6-метилбензойной кислоты в виде бесцветного маслянистого продукта с чистотой 80%. Полученный таким образом бензиловый эфир 2-бромметил-6-метилбензойной кислоты имеет молекулярную массу 318,02 (C16H15BrO2); MS (ESI): 341,0 [M+Na]+.
Пример 18a. Алкилирование цис-3S-(2-п-толил-5-метилоксазол-4-илметокси)циклогексан-1R-ола метиловым эфиром 2-бромметил-6-метилбензойной кислоты с получением метилового эфира 2-метил-6-[(1R,3S)-3-(5-метил-2-п-толилоксазол-4-илметокси)циклогексилоксиметил]бензойной кислоты
50 г (166 ммоль) цис-3S-(2-п-толил-5-метилоксазол-4-илметокси)циклогексан-1R-ола и 30 г (262 ммоль) трет-бутилата калия растворяют в 500 мл ТГФ и смесь перемешивают в течение 30 минут при комнатной температуре. После прибавления 500 мл трет-бутилметилового эфира прибавляют раствор 51 г (210 ммоль) метилового эфира 2-бромметил-6-метилбензойной кислоты в 310 мл трет-бутилметилового эфира. Реакционную смесь перемешивают в течение 1 часа при температуре в интервале от 25 до 30°C, затем реакцию останавливают прибавлением 500 мл раствора соляной кислоты (0,5 M). После отделения водного слоя органический слой промывают водой три раза по 300 мл соответственно. Затем растворитель полностью удаляют в вакууме. Получают 74 г метилового эфира 2-метил-6-[(1R,3S)-3-(5-метил-2-п-толилоксазол-4-илметокси)циклогексилоксиметил]бензойной кислоты в виде желтоватого маслянистого продукта с чистотой 79% и молекулярной массой 463,57 (C28H33NO5); MS (ESI): 464,37 [M+H]+.
Пример 18b. Алкилирование цис-3S-(2-п-толил-5-метилоксазол-4-илметокси)циклогексан-1R-ола трет-бутиловым эфиром 2-бромметил-6-метилбензойной кислоты с получением трет-бутилового эфира 2-метил-6-[(1R,3S)-3-(5-метил-2-п-толилоксазол-4-илметокси)циклогексилоксиметил]бензойной кислоты
100 г (332 ммоль) цис-3S-(2-п-толил-5-метилоксазол-4-илметокси)циклогексан-1R-ола и 60 г (524 ммоль) трет-бутилата калия растворяют в 1,0 л ТГФ и смесь перемешивают в течение 30 минут при комнатной температуре. После прибавления 1 л трет-бутилметилового эфира прибавляют раствор 120 г (524 ммоль) метилового эфира 2-бромметил-6-метилбензойной кислоты в 850 мл трет-бутилметилового эфира. Реакционную смесь перемешивают в течение 30 минут при температуре 40°C. Затем органическую фазу последовательно промывают 1,0 л воды, 250 мл раствора соляной кислоты (0,5 M) и водным раствором хлорида натрия (1%-й раствор) четыре раза по 500 мл соответственно. После полного удаления в вакууме растворителя получают 195 г трет-бутилового эфира 2-метил-6-[(1R,3S)-3-(5-метил-2-п-толилоксазол-4-илметокси)циклогексилоксиметил]бензойной кислоты в виде желтого маслянистого продукта с чистотой 86%. Полученный таким образом трет-бутиловый эфир 2-метил-6-[(1R,3S)-3-(5-метил-2-п-толилоксазол-4-илметокси)циклогексилоксиметил]бензойной кислоты имеет молекулярную массу 505,65 (C31H39NO5); MS (ESI): 506,39 [M+H]+.
Пример 18c. Алкилирование цис-3S-(2-п-толил-5-метилоксазол-4-илметокси)циклогексан-1R-ола бензиловым эфиром 2-бромметил-6-метилбензойной кислоты с получением бензилового эфира 2-метил-6-[(1R,3S)-3-(5-метил-2-п-толилоксазол-4-илметокси)циклогексилоксиметил]бензойной кислоты
К раствору 30 г (100 ммоль) цис-3S-(2-п-толил-5-метилоксазол-4-илметокси)циклогексан-1R-ола в 300 мл ТГФ прибавляют 18 г (157 ммоль) трет-бутилата калия и смесь перемешивают в течение 30 минут при комнатной температуре. После прибавления 44,5 г (112 ммоль) бензилового эфира 2-бромметил-6-метилбензойной кислоты (чистота 80%) реакционную смесь перемешивают в течение 30 минут при 30°C. Затем реакционную смесь выливают в раствор 15 мл соляной кислоты (32%-й раствор) в 300 мл воды. После разделения слоев органический слой полностью упаривают в вакууме и остаток растворяют в 500 мл трет-бутилметилового эфира. Раствор промывают водой четыре раза по 300 мл соответственно. Растворитель удаляют в вакууме и остаток очищают хроматографией на кизельгуре. Получают бензиловый эфир метил-6-[(1R,3S)-3-(5-метил-2-п-толилоксазол-4-илметокси)циклогексилоксиметил]бензойной кислоты в виде желтого маслянистого продукта с выходом 90% и молекулярной массой 539,68 (C34H37NO5); MS (ESI): 540,41 [M+H]+.
Пример 19. Синтез 2-метил-6-[(1R,3S)-3-(5-метил-2-п-толилоксазол-4-илметокси)циклогексилоксиметил]бензойной кислоты расщеплением сложного эфира
a: из метилового эфира метил-6-[(1R,3S)-3-(5-метил-2-п-толилоксазол-4-илметокси)циклогексилоксиметил]бензойной кислоты: 97,1 г (165 ммоль) метилового эфира метил-6-[(1 R,3S)-3-(5-метил-2-п-толил-оксазол-4-илметокси)циклогексилоксиметил]бензойной кислоты растворяют в смеси 640 мл изопропанола и 80 мл воды. Прибавляют порциями 47,7 г (809 ммоль) гидроксида калия. Реакционную смесь перемешивают в течение 24 часов при 95°C. Затем растворитель полностью удаляют в вакууме и остаток растворяют в 800 мл трет-бутилметилового эфира и 800 мл воды. Смесь подкисляют прибавлением раствора соляной кислоты (2 M). После разделения слоев органический слой промывают водой четыре раза по 200 мл соответственно. Органический слой концентрируют в вакууме и остаток смешивают с небольшим количеством диизопропилового эфира. Твердое вещество отфильтровывают и высушивают в вакууме. Получают 2-метил-6-[(1R,3S)-3-(5-метил-2-п-толилоксазол-4-илметокси)циклогексилоксиметил]бензойную кислоту в виде кристаллического твердого вещества с выходом 57%.
b: из трет-бутилового эфира метил-6-[(1R,3S)-3-(5-метил-2-п-толилоксазол-4-илметокси)циклогексилоксиметил]бензойной кислоты: 200 г (396 ммоль) трет-бутилового эфира метил-6-[(1R,3S)-3-(5-метил-2-п-толилоксазол-4-илметокси)циклогексилоксиметил]бензойной кислоты растворяют в н-гептане. Прибавляют 300 г (2,6 моль) трифторуксусной кислоты. Реакционную смесь перемешивают в течение 24 часов при комнатной температуре и затем полностью упаривают. Остаток растворяют в 1,0 л диизопропилового эфира и промывают водой четыре раза по 500 мл соответственно. Затем прибавляют 1,0 л воды и добавлением раствора гидроксида натрия (4 M) устанавливают щелочную реакцию раствора. После разделения слоев водный слой промывают диизопропиловым эфиром два раза по 400 мл соответственно. После прибавления 1,6 л диизопропилового эфира смесь подкисляют раствором соляной кислоты (2 M). После разделения слоев органический слой промывают водой четыре раза по 500 мл соответственно и затем упаривают приблизительно до одной третьей объема. 2-метил-6-[(1R,3S)-3-(5-метил-2-п-толилоксазол-4-илметокси)циклогексилоксиметил]бензойную кислоту, выпадающую в виде кристаллического твердого вещества, отфильтровывают. После высушивания в вакууме получают 2-метил-6-[(1R,3S)-3-(5-метил-2-п-толилоксазол-4-илметокси)циклогексилоксиметил]бензойную кислоту с выходом 66%.
c: из бензилового эфира метил-6-[(1R,3S)-3-(5-метил-2-п-толилоксазол-4-илметокси)циклогексилоксиметил]бензойной кислоты: 15 г (27,8 ммоль) бензилового эфира метил-6-[(1R,3S)-3-(5-метил-2-п-толилоксазол-4-илметокси)циклогексилоксиметил]бензойной кислоты растворяют в 300 мл ТГФ. Прибавляют 700 мг палладия на угле (5%-й). Затем при нормальном давлении и комнатной температуре гидрируют до прекращения расходования водорода. Реакционную смесь отфильтровывают и затем органическую фазу полностью упаривают в вакууме. Остаток обрабатывают 150 мл трет-бутилметилового эфира, 150 мл воды и 150 мл насыщенного раствора гидрокарбоната натрия. После разделения слоев водный слой промывают 150 мл трет-бутилметилового эфира. Затем прибавляют 150 мл диизопропилового эфира и подкисляют концентрированной соляной кислотой. После разделения слоев органический слой промывают водой четыре раза по 100 мл соответственно и затем полностью упаривают в вакууме. Остаток смешивают с небольшим количеством диизопропилового эфира. Твердое вещество отфильтровывают и высушивают в вакууме. Получают 2-метил-6-[(1R,3S)-3-(5-метил-2-п-толилоксазол-4-илметокси)циклогексилоксиметил]бензойную кислоту в виде кристаллического вещества с выходом 42%.
Полученная различными вариантами 2-метил-6-[(1R,3S)-3-(5-метил-2-п-толилоксазол-4-илметокси)циклогексилоксиметил]бензойная кислота имеет молекулярную массу 449,55 (C27H31NO5); MS (ESI): 450,29 [M+H]+.
В приведенных далее примерах 20 и 21, а также на схеме V показывается синтез еще одного соединения общей формулы (I).
Схема V
Пример 20. Алкилирование цис-3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексан-1R-ола трет-бутиловым эфиром 2-бромметил-6-метилбензойной кислоты с получением цис-2-(3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойной кислоты
К раствору 5,1 г (17,5 ммоль) цис-3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексан-1R-ола (пример 8) в 50 мл ТГФ прибавляют 3,31 г (28 ммоль) трет-бутилата калия. Смесь перемешивают в течение 30 минут при комнатной температуре. Добавляют 6,83 г (22,8 ммоль) трет-бутилового эфира 2-бромметил-6-метил-бензойной кислоты (чистота 95%) (пример 17b). Реакционную смесь перемешивают в течение 1 часа при комнатной температуре. Затем прибавляют 100 мл воды и 17,5 раствора соляной кислоты (2 M). После разделения слоев органический слой промывают водой четыре раза по 100 мл соответственно и полностью упаривают в вакууме. Получают 10,6 г трет-бутилового эфира цис-2-(3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойной кислоты в виде желтоватого маслянистого продукта с чистотой 82%. Полученный таким образом трет-бутиловый эфир цис-2-(3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойной кислоты имеет молекулярную массу 495,59 (C29H34FNO5); MS (ESI): 440,19 [M-трет-бутил+H+H]+.
Пример 21. Отщепление трет-бутилового эфира от трет-бутилового эфира цис-2-(3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойной кислоты с получением цис-2-(3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойной кислоты
10,6 г (17,5 ммоль) трет-бутилового эфира цис-2-(3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойной кислоты (чистота 82%) растворяют в 20 мл дихлорметана. После прибавления 10 мл (128 ммоль) трифторуксусной кислоты реакционную смесь перемешивают в течение 16 часов при комнатной температуре. Затем реакционную смесь полностью упаривают. Остаток растворяют в 100 мл диизопропилового эфира и промывают водой шесть раз по 50 мл соответственно. Затем прибавляют 100 мл воды и добавлением раствора гидроксида натрия (2 M) устанавливают щелочную реакцию раствора. После разделения слоев водный слой промывают диизопропиловым эфиром три раза по 50 мл соответственно. Затем прибавляют 150 мл диизопропилового эфира и подкисляют раствором соляной кислоты (2 M). После разделения слоев органический слой промывают водой три раза по 50 мл соответственно и затем полностью упаривают. Остаток перекристаллизовывают из диизопропилового эфира. Выделяют 2,9 г цис-2-(3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойной кислоты в виде кристаллов. Полученная таким образом цис-2-(3S-(2-(4-фторфенил)оксазол-4-илметокси)циклогексил-1R-оксиметил)-6-метилбензойная кислота имеет молекулярную массу 439,48 (C25H26FNO5); MS (ESI): 438,31 [M -H]-.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ЭНАНТИОМЕРНЫХ ФОРМ ПРОИЗВОДНЫХ 1,3-ЦИКЛОГЕКСАНДИОЛА В ЦИС-КОНФИГУРАЦИИ | 2004 |
|
RU2372319C2 |
| ДИАРИЛЦИКЛОАЛКИЛЬНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ PPAR-АКТИВАТОРОВ | 2002 |
|
RU2330846C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ ПРОСТОГО АМИНОЦИКЛОГЕКСИЛОВОГО ЭФИРА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2008 |
|
RU2478617C2 |
| СОЕДИНЕНИЯ ПРОСТОГО АМИНОЦИКЛОГЕКСИЛОВОГО ЭФИРА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2003 |
|
RU2330017C2 |
| НОВЫЕ ЦИКЛИЧЕСКИЕ УГЛЕВОДОРОДНЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ | 2008 |
|
RU2524949C2 |
| (ГЕТЕРО)АРИЛЦИКЛОПРОПИЛАМИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ LSD1 | 2012 |
|
RU2668952C2 |
| ПРОИЗВОДНЫЕ ОКСОПИПЕРИДИНА, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ В ТЕРАПИИ | 2005 |
|
RU2376298C2 |
| ИМИДАЗОПИРАЗИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗ | 2004 |
|
RU2405784C2 |
| КОСМЕТИЧЕСКИЕ КОМПОЗИЦИИ | 2005 |
|
RU2395272C2 |
| ПОЗИТИВНЫЕ АЛЛОСТЕРИЧЕСКИЕ МОДУЛЯТОРЫ (РАМ) | 2010 |
|
RU2561920C2 |
Изобретение относится к способу получения соединения общей формулы (I), включающему стадии согласно следующей схеме:
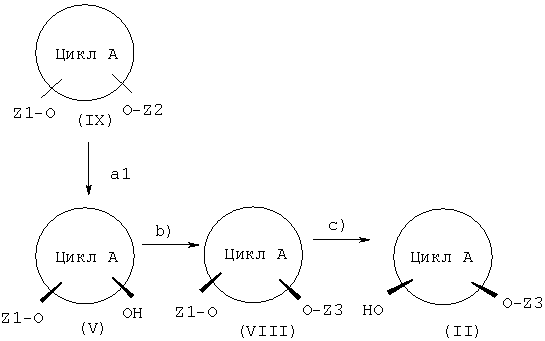


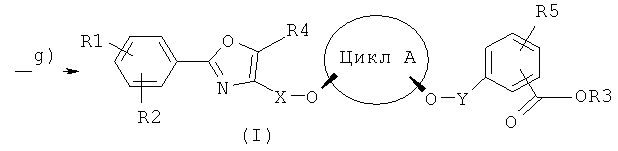

причем на отдельных стадиях: а1) соединение формулы (IX) превращают в соединение формулы (V) в присутствии фермента, выбранного из липазы В из Candida antarctica, b) соединение формулы (V) в присутствии кислотного катализатора посредством действия соединения, которое может образовывать защитную группу Z3, стабильную в щелочной среде и лабильную в кислой среде, превращают в соединение формулы (VIII) и с) соединение формулы (VIII) в присутствии нуклеофильного агента превращают в соединение формулы (II); d) соединение формулы (II) в присутствии основания В1 превращают посредством действия соединения формулы (VI) в соединение формулы (IIIа); е) соединение формулы (IIIа) превращают в соединение формулы (IVa), причем соответствующее превращение осуществляют посредством действия спирта в присутствии кислотного катализатора; f) соединение (IVa) посредством действия соединения (VII) превращают в соединение формулы (Iа) в присутствии основания В1 и g) при необходимости, соединение (Iа) гидролизуют или подвергают гидрогенолизу до соединения (I), если R3 представляет собой Н; причем соединения (IX) представляет собой чистый цис-изомер или смесь цис/транс-изомеров соответственно; при этом переменные и заместители имеют следующие значения соответственно: цикл А представляет собой С3-С8циклоалкил, R1, R2, R4 и R5 представляют собой независимо друг от друга Н, F, Cl, Вr, C1-С6алкил или -O-(С1-С6алкил); R3 представляет собой Н, C1-С6алкил; R6 представляет собой C1-С6алкил или бензил; Х представляет собой C1-С6алкил; Y представляет собой C1-С6алкил; Z1 представляет собой защитную группу, стабильную в кислой среде; Z2 представляет собой защитную группу, стабильную в кислой среде; Z3 представляет собой защитную группу, стабильную в щелочной среде и лабильную в кислой среде; Z4 представляет собой уходящую группу; Z5 представляет собой уходящую группу; В1 представляет собой третичный алкоголят щелочноземельного металла, третичный алкоголят щелочного металла, амид щелочноземельного металла, амид щелочного металла, силазид щелочноземельного металла, силазид щелочного металла или гидрид щелочного металла. Изобретение также относится к способу получения соединения общей формулы (I), включающему стадии согласно следующей схеме:



причем на отдельных стадиях: а2) соединение формулы (X) посредством по меньшей мере одного донора ацильных групп в присутствии фермента, выбранного из липазы В из Candida antarctica, превращают в соединение (V); стадии b) - g) описаны выше, причем соединение (X) представляет собой чистый цис-изомер или смесь цис/транс-изомеров соответственно. Изобретение также относится к соединению общей формулы (IIIа), где цикл А представляет собой циклогексил, причем Х-содержащий и Z3-содержащий заместители стоят в цис-1,3-положении циклогексильного фрагмента; R1, R2 и R4 представляют собой независимо друг от друга Н, F, Сl, C1-С3алкил или -С(O)-(С1-С6алкил); Z3 представляет собой тетрагидропиранил; Х представляет собой метил и к соединению общей формулы (VIII), где цикл А представляет собой циклогексил, причем Z1-содержащий и Z3-содержащий заместители стоят в цис-1,3-положении циклогексильного фрагмента; Z1 представляет собой -С(O)СН3; Z3 представляет собой тетрагидропиранил. 4 н. и 10 з.п. ф-лы, 2 табл.
1. Способ получения соединения общей формулы (I), включающий стадии согласно следующей схеме:
причем на отдельных стадиях:
а1) соединение формулы (IX) превращают в соединение формулы (V) в присутствии фермента, выбранного из липазы В из Candida antarctica,
b) соединение формулы (V) в присутствии кислотного катализатора посредством действия соединения, которое может образовывать защитную группу Z3, стабильную в щелочной среде и лабильную в кислой среде, превращают в соединение формулы (VIII) и
c) соединение формулы (VIII) в присутствии нуклеофильного агента превращают в соединение формулы (II);
d) соединение формулы (II) в присутствии основания В1 превращают посредством действия соединения формулы (VI) в соединение формулы (IIIa);


e) соединение формулы (IIIа) превращают в соединение формулы (IVa), причем соответствующее превращение осуществляют посредством действия спирта в присутствии кислотного катализатора;
f) соединение (IVa) посредством действия соединения (VII) превращают в соединение формулы (Iа) в присутствии основания В1 и
g) при необходимости, соединение (Iа) гидролизуют или подвергают гидрогенолизу до соединения (I), если R3 представляет собой Н;
причем соединения (IX) представляет собой чистый цис-изомер или смесь цис/транс-изомеров соответственно;
при этом переменные и заместители имеют следующие значения соответственно:
цикл А представляет собой С3-С8циклоалкил,
R1, R2, R4 и R5 представляют собой независимо друг от друга Н, F, Cl, Вr, С1-С6алкил или -O-(С1-С6алкил);
R3 представляет собой Н, C1-С6алкил;
R6 представляет собой C1-С6алкил или бензил;
Х представляет собой C1-С6алкил;
Y представляет собой C1-С6алкил;
Z1 представляет собой защитную группу, стабильную в кислой среде;
Z2 представляет собой защитную группу, стабильную в кислой среде;
Z3 представляет собой защитную группу, стабильную в щелочной среде и лабильную в кислой среде;
Z4 представляет собой уходящую группу;
Z5 представляет собой уходящую группу;
В1 представляет собой третичный алкоголят щелочноземельного металла, третичный алкоголят щелочного металла, амид щелочноземельного металла, амид щелочного металла, силазид щелочноземельного металла, силазид щелочного металла или гидрид щелочного металла.
2. Способ по п.1, отличающийся тем, что Z1 представляет собой -С(O)-СН3 и/или Z3 представляет собой тетрагидропиранил или метоксиизопропил.
3. Способ по п.1 или 2, в котором:
цикл А представляет собой циклопентил, циклогексил или циклогептил;
4. Способ по п.3, в котором:
цикл А представляет собой циклогексил, причем Х-содержащий и Y-содержащий заместители формулы (I) стоят в цис-1,3-положении циклогексильного фрагмента;
Х и Y представляют собой метил.
5. Способ по п.1 или 2, в котором:
цикл А представляет собой циклогексил, причем Х-содержащий и Y-содержащий заместители формулы (I) стоят в цис-1,3-положении циклогексильного фрагмента и атом углерода цикла А, связанный с Y-содержащим заместителем, имеет R-конфигурацию.
6. Способ по п.1 или 2, отличающийся тем, что соединения общей формулы (I) присутствует с энантиомерной чистотой больше чем 99%.
7. Способ получения соединения общей формулы (I), включающий стадии согласно следующей схеме:

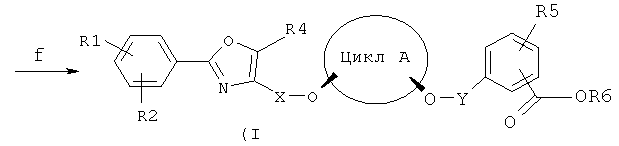
причем на отдельных стадиях:
а2) соединение формулы (X) посредством по меньшей мере одного донора ацильных групп в присутствии фермента, выбранного из липазы В из Candida antarctica, превращают в соединение (V);
b) соединение формулы (V) в присутствии кислотного катализатора посредством действия соединения, которое может образовывать защитную группу Z3, стабильную в щелочной среде и лабильную в кислой среде, превращают в соединение формулы (VIII) и
c) соединение формулы (VIII) в присутствии нуклеофильного агента превращают в соединение формулы (II);
d) соединение формулы (II) в присутствии основания В1 превращают посредством действия соединения формулы (VI) в соединение формулы (IIIа);


e) соединение формулы (IIIа) превращают в соединение формулы (IVa), причем соответствующее превращение осуществляют посредством действия спирта в присутствии кислотного катализатора;
f) соединение (IVa) посредством действия соединения (VII) превращают в соединение формулы (Iа) в присутствии основания В1 и
g) при необходимости, соединение (Iа) гидролизуют или подвергают гидрогенолизу до соединения (I), если R3 представляет собой Н;
причем соединение (X) представляет собой чистый цис-изомер или смесь цис/транс-изомеров соответственно;
при этом переменные и заместители имеют следующие значения соответственно:
цикл А представляет собой С3-С8циклоалкил;
R1, R2, R4 и R5 представляют собой независимо друг от друга Н, F, Cl, Вr, C1-С6алкил или -(O)-(С1-С6алкил);
R3 представляет собой Н, C1-С6алкил;
R6 представляет собой C1-С6алкил или бензил;
Х представляет собой C1-С6алкил;
Y представляет собой C1-С6алкил;
Z1 представляет собой защитную группу, стабильную в кислой среде;
Z3 представляет собой защитную группу, стабильную в щелочной среде и лабильную в кислой среде;
Z4 представляет собой уходящую группу;
Z5 представляет собой уходящую группу;
В1 представляет собой третичный алкоголят щелочноземельного металла, третичный алкоголят щелочного металла, амид щелочноземельного металла, амид щелочного металла, силазид щелочноземельного металла, силазид щелочного металла или гидрид щелочного металла.
8. Способ по п.7, отличающийся тем, что Z1 представляет собой -С(O)-СН3 и/или Z3 представляет собой тетрагидропиранил или метоксиизопропил.
9. Способ по п.7 или 8, в котором:
цикл А представляет собой циклопентил, циклогексил или циклогептил.
10. Способ по п.9, в котором:
цикл А представляет собой циклогексил, причем Х-содержащий и Y-содержащий заместители формулы (I) стоят в цис-1,3-положении циклогексильного фрагмента;
Х и Y представляют собой метил.
11. Способ по п.7 или 8, в котором:
цикл А представляет собой циклогексил, причем Х-содержащий и Y-содержащий заместители формулы (I) стоят в цис-1,3-положении циклогексильного фрагмента и атом углерода цикла А, связанный с Y-содержащим заместителем, имеет R-конфигурацию.
12. Способ по п.1 или 2, отличающийся тем, что соединения общей формулы (I) присутствует с энантиомерной чистотой больше, чем 99%.
13. Соединение общей формулы (IIIа)

где цикл А представляет собой циклогексил, причем Х-содержащий и Z3-содержащий заместители стоят в цис-1,3-положении циклогексильного фрагмента;
R1, R2 и R4 представляют собой независимо друг от друга Н, F, Cl, C1-С3алкил или -С(O)-(С1-С6алкил);
Z3 представляет собой тетрагидропиранил;
Х представляет собой метил.
14. Соединение общей формулы (VIII)
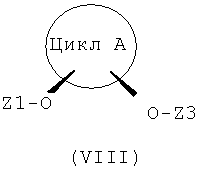
где цикл А представляет собой циклогексил, причем Z1-содержащий и Z3-содержащий заместители стоят в цис-1,3-положении циклогексильного фрагмента;
Z1 представляет собой -С(O)СН3;
Z3 представляет собой тетрагидропиранил.
| Nicolosi G | |||
| et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Способ получения транс-4-(транс-4-н-алкилциклогексил)-1-алкоксициклогексанов | 1990 |
|
SU1816753A1 |
| WO 03020269 A1, 13.03.2003. | |||

