Область техники, к которой относится изобретение
Данное изобретение относится к способам получения пирроло[2,3-d]пиримидиновых соединений и к промежуточным продуктам для их получения.
Уровень техники
Пирроло[2,3-d]пиримидиновые соединения являются сильнодействующими ингибиторами протеинкиназ, таких как фермент Янус киназы 3 (JAK 3), и являются поэтому полезными для лечения большого разнообразия иммунологических расстройств. Такие расстройства включают, но не ограничиваются этим, волчанку, рассеянный склероз, ревматоидный артрит, псориаз, диабет первого типа и осложнения при диабете, рак, астму, атопический дерматит, аутоиммунные тироидные расстройства, язвенный колит, болезнь Крона, болезнь Альцгеймера и лейкемию. Соединения являются полезными для лечения и предупреждения хронического или острого отторжения трансплантированного органа (аллотрансплантаты или ксенотрансплантаты).
Конкретные пирроло[2,3-d]пиримидиновые соединения, способы их применения, их синтез и промежуточные соединения ранее были описаны в патенте US 6627754, WO 02096909, WO 03048162 и опубликованной заявке US 2004/0102627A1, в основном принадлежащих заявителю данной заявки и включенных в описание изобретения в полном объеме. Патент US 6627754 описывает соединение 3-{4-метил-(7Н-пирроло[2,3-d]пиримидин-4-ил)амино}пиперидин-1-ил)-3-оксопропионитрил и его соль с лимонной кислотой как ингибиторы протеинкиназ (таких как фермент JAK 3), которые являются пригодными для лечения, в качестве иммуноподавляющих агентов для трансплантированных органов, ксенотрансплантации, волчанки, рассеянного склероза, ревматоидного артрита, псориаза, диабета первого типа и осложнений при диабете, рака, астмы, атопического дерматита, аутоиммунных тироидных расстройств, язвенного колита, болезни Крона, болезни Альцгеймера, лейкемии и их симптомов, где иммунное подавление может быть желательным. В опубликованной заявке US 2004/0102627A1 описан синтез промежуточных соединений, гидрохлорида цис-(1-бензил-4-метилпиперидин-3-ил)метиламина, которое используется в синтезе 3-{(3R,4R)-4-метил-(7Н-пирроло[2,3-d]пиримидин-4-ил)амино}пиперидин-1-ил)-3-оксопропионитрила и его соли с лимонной кислотой.
Сущность изобретения
Предшествующие синтезы пирролопиримидиновых соединений, описанных выше, и конкретно 3-{(3R,4R)-4-метил-(7Н-пирроло[2,3-d]пиримидин-4-ил)амино}пиперидин-1-ил)-3-оксопропионитрила, характеризовались низкими выходами и большой продолжительностью реакции сочетания 4-хлорпирролопиримидина с нуклеофилами, конкретно с (3R,4R)-(1-бензил-4-метилпиперидин-3-ил)метиламином и его солями. Было неожиданно обнаружено, что введение способной к удалению активирующей группы на пирролопиримидиновый центр с уходящей группой в положении 4 заметно повышает выходы и снижает продолжительности реакций с нуклеофилами, особенно с (3R,4R)-(1-бензил-4-метилпиперидин-3-ил)метиламином и его солями. Предварительно был описан синтез ключевого промежуточнго соединения, полезного в осуществлении одного аспекта вышеупомянутой последовательности синтеза 2,4-дихлор-7Н-пирроло[2,3-d]пиримидина (Saxena, S.K., et al., J. Med. Chem., 31, 1501, 1998; выход 26,5%; WO 04021979; выход 41%). Тем не менее синтез, описанный в литературе, обеспечивал низкие выходы целевого продукта. Было неожиданно обнаружено, что с помощью тщательного контролирования количеств фосфористого оксихлорида и триалкиламинового основания, используемых для реакции с исходным соединением, 7Н-пирроло[2,3-d]пиримидин-2,4-диолом, можно получить значительно увеличенные выходы желаемого продукта.
Прежние попытки асимметричной гидрогенизации пиридиниевых солей без закрепленных хиральных дополнений (Glorius, F., et al. Angew. Chem. Int. Ed., 43, 2859, 2004 и ссылки, приводимые там; см. также Legault, C.Y., et al., J. Am. Chem. Soc. 127, 8966, 2005; обе публикации включены в описание изобретения в полном объеме в качестве ссылки) обычно приводили к образованию пиперидина, но с низкими энантиомерными избытками (исключая ошибки). Было неожиданно обнаружено, что асимметричная гидрогенизация соответственно замещенного N-бензила или замещенных N-бензилпиридиниевых солей или соответственно замещенного N-бензила или замещенных N-бензилтетрагидропиридинов с определенными катализаторами асимметрической гидрогенизации приводит к получению энантиомерно обогащенных пиперидиновых производных, полезных в синтезе пирролопиримидиновых соединений, описанных выше, и конкретно 3-{(3R,4R)-4-метил-(7Н-пирроло[2,3-d]пиримидин-4-ил)амино}пиперидин-1-ил)-3-оксопропионитрила.
Как реализовано и в общих чертах описано в настоящем описании, это изобретение, в одном аспекте, относится к способам получения соединения формулы IVa
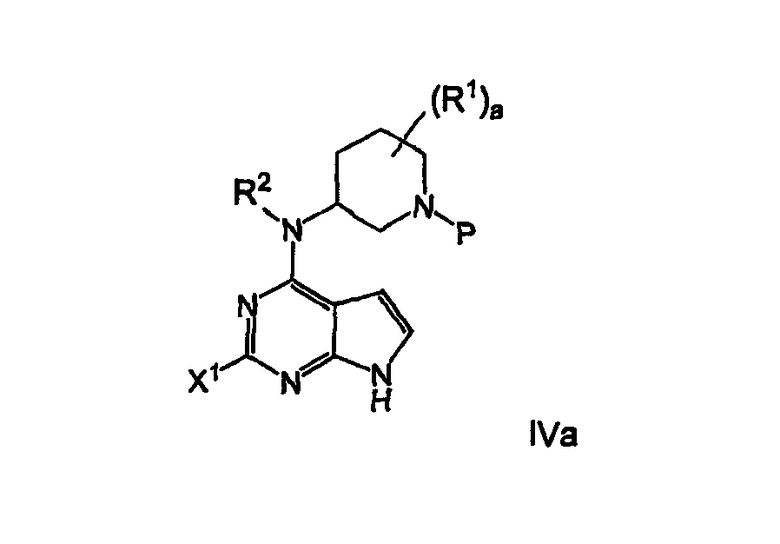
включающим стадию
сочетания активированного пирролопиримидинового соединения формулы IIa
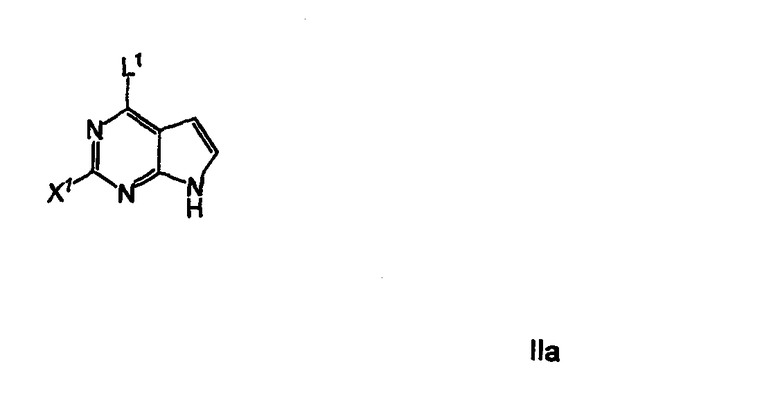
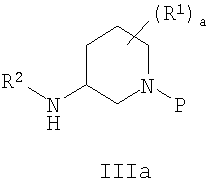
где L1 представляет собой уходящую группу, предпочтительно хлор, бром, йод или фтор, наиболее предпочтительно хлор, и Х1 представляет собой активирующую группу, предпочтительно хлор, бром, йод, фтор, CO2R', COCO2R', SO2Ar и COAr, где Ar представляет собой (С3-С12) ароматическую группу, необязательно включающую от 1 до 6 гетероатомов, выбранных из О, NR' и S, необязательно замещенных от 1 до 3 группами, выбранными из (С1-С6)алкила, галогена, нитро и циано, где R' выбирают из группы, состоящей из (С1-С6)алкила и бензила; наиболее предпочтительно, когда Х1 представляет собой хлор; с амином формулы IIIa или его солью

где R1 представляет собой водород или (С1-С6)алкил; а является целым числом от 0 до 4; R2 представляет собой водород или (С1-С6)алкил и Р представляет собой азотзащищающую группу, предпочтительно бензил; в присутствии основания, предпочтительно щелочи или триалкиламинового основания, более предпочтительно карбоната калия или карбоната натрия, наиболее предпочтительно карбоната калия, чтобы получить продукт сочетания формулы IVa.
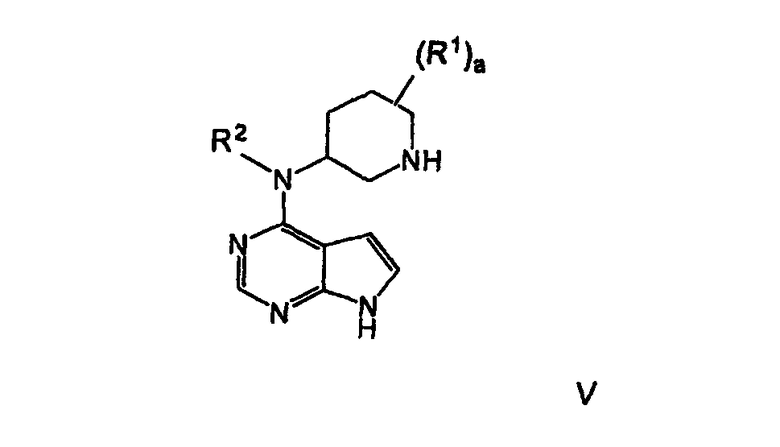
В другом варианте осуществления описанная выше стадия дополнительно включает удаление активирующей группы, предпочтительно хлора, и азотзащищающей группы, предпочтительно бензила, из соединения формулы IVa

последовательно, в любом порядке, или в одном реакторе, чтобы получить соединение формулы V
В другом варианте осуществления удаление активирующей группы, где активирующая группа представляет собой хлор, и защищающей группы, где защищающая группа является лабильной к гидрогенолизу, желательно бензилом или замещенным бензилом, выполняется в одном реакторе, в присутствии водорода или источника водорода и катализатора, желательно гидроксида палладия, палладия-на-угле и платины-на-угле, наиболее предпочтительно гидроксида палладия.
В еще одном варианте осуществления амин формулы IIIa или его соль являются IIIb или IIIc или рацемической или энантиомерно обогащенной смесью их.

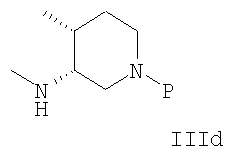
В еще одном аспекте осуществления амин формулы IIIa или его соль представляют собой IIId или IIIe или рацемическую или энантиомерно обогащенную смесь их.

В другом варианте осуществления амин формулы IIIa или его соль имеют формулу IIId, которая имеет 3R,4R-конфигурацию.

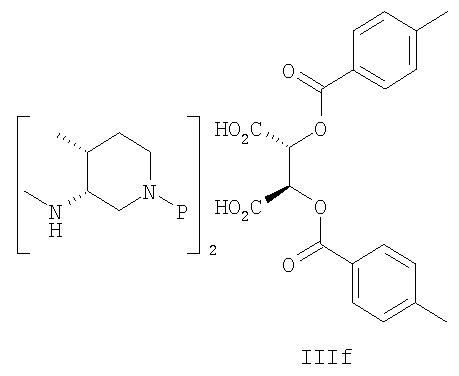
В другом варианте осуществления соль амина формулы IIId характеризуется 3R,4R-конфигурацией и имеет формулу IIIf.
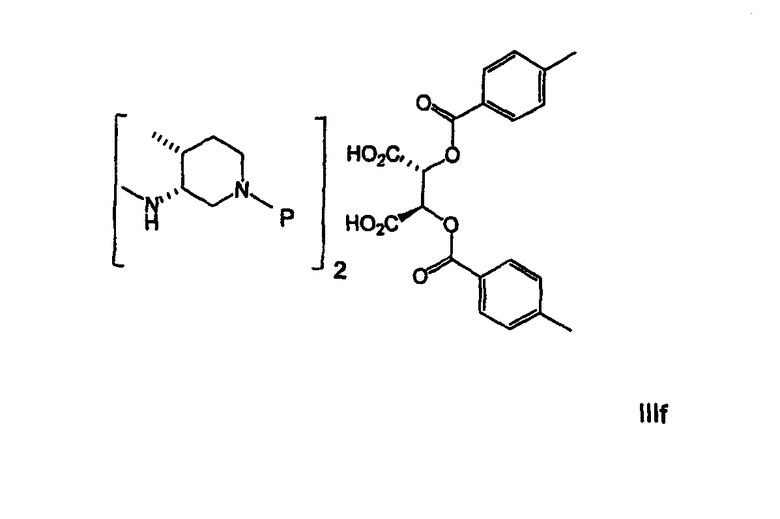
В еще одном варианте осуществления Р в формулах IIIb-f представляет собой защищающую группу, лабильную к гидрогенолизу, желательно бензил или замещенный бензил.
В другом варианте осуществления соединение формулы IVa имеет формулу Ivb
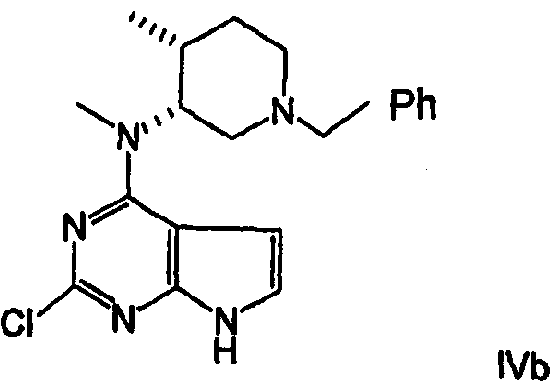
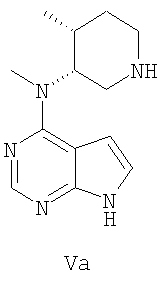
В другом варианте осуществления удаление хлора, в качестве активирующей группы, и N-бензильной защищающей группы из соединения формулы IVb выполняется в одном реакторе в присутствии водорода или источника водорода и катализатора гидрогенизации, желательно гидроксида палладия, палладия-на-угле, платины-на-угле, наиболее предпочтительно гидроксида палладия, чтобы получить соединение формулы Va.

В другом аспекте осуществления соединение формулы Va ацилируют ацилирующим агентом формулы VI
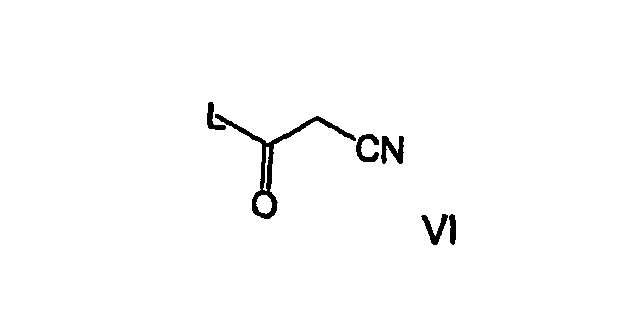
где L представляет собой уходящую группу, выбранную из группы, состоящей из -OCOR3, OR3, галогена, гидроксилсукцинимидила, сульфонатов, таких как тозил и мезил; где R3 представляет собой (С1-С6)алкил; предпочтительно, чтобы L представлял собой -OCOR3, где R3 представляет собой трет-бутил; необязательно в присутствии основания, чтобы получить соединение формулы Ia.

Дополнительные агенты сочетания и условия для образования амида между активированными и неактивированными производными карбоксильных кислот и аминами, которые являются подходящими для упомянутой выше стадии ацилирования, могут быть найдены в Benz, G. Comprehensive Organic Synthesis, B. Trost Ed., том 6, глава 2.3, таблицы 1-7, которые включаются в описание изобретения в полном объеме.
В другом варианте осуществления соединение формулы Ia реагирует с лимонной кислотой для получения соответствующей соли лимонной кислоты.
Еще одним вариантом осуществления данного изобретения является способ получения соединения формулы IVb

включающий стадию активированного пирролопиримидинового соединения формулы IIb

где L1 представляет собой уходящую группу, предпочтительно хлор, бром, йод или фтор, наиболее предпочтительно хлор, и Х2 представляет собой активирующую группу, предпочтительно бензил, SO2Ar, CO2R' и COAr; Ar представляет собой С3-С12 ароматическую группу, необязательно включающую от 1 до 6 гетероатомов, выбранных из О, NR' и S, необязательно замещенных от 1 до 3 группами, выбранными из (С1-С6)алкила, галогена, нитро и циано; где R' выбирают из группы, состоящей из
(С1-С6)алкила и бензила; предпочтительно Х2 представляет собой тозил (пара-толуолсульфонильную группу; -SO2C6H4CH3); с амином формулы IIIa или его солью,

где R1 представляет собой (С1-С6)алкил; а является целым числом от 0 до 4; R2 представляет собой водород или (С1-С6)алкил и Р представляет собой защищающую азот группу, предпочтительно бензил; в присутствии основания, предпочтительно щелочи или триалкиламинового основания, более предпочтительно карбоната калия или карбоната натрия, наиболее предпочтительно карбоната калия, чтобы получить продукт сочетания формулы IVb.
В другом варианте осуществления, когда Х2 представляет собой тозильную группу, тозильная группа и азотзащищающая группа удаляются из соединения формулы IVb последовательно в любом порядке или в одном реакторе, чтобы получить соединение формулы V

В другом варианте осуществления тозильная группа удаляется с помощью водного щелочного основания, предпочтительно водного гидроксида натрия.
В предпочтительном варианте осуществления амин формулы IIIa или его соль представляют собой соединение формулы IIIb или IIIc или их рацемическую или энантиомерную смесь.

В еще одном варианте осуществления амин формулы IIIa или его соль представляют собой соединение формулы IIId или IIIe или их смесь.
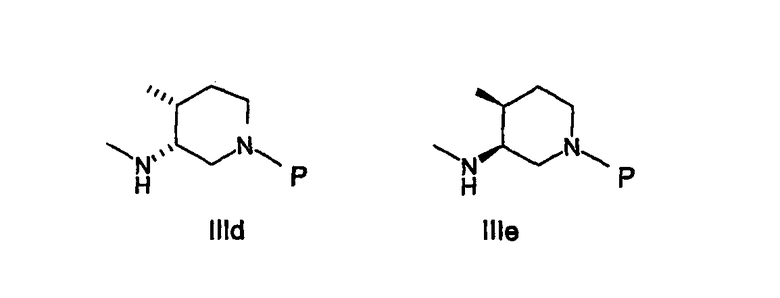
В другом варианте осуществления амин формулы IIIa или его соль представляют собой соединение формулы IIId, которая имеет 3R,4R-конфигурацию.

В другом варианте осуществления соль амина формулы IIId, которая имеет 3R,4R-конфигурацию, представляет собой соединение формулы IIIf.

В другом варианте осуществления Р в формулах IIIb-f представляет собой защищающую группу, лабильную к гидрогенолизу, желательно бензил.
В другом варианте осуществления формула IVb имеет формулу IVс
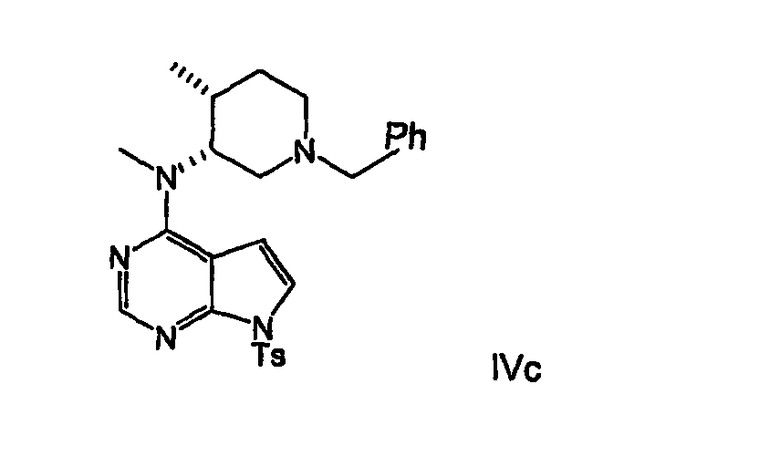
В другом варианте осуществления удаление тозильной группы и бензильной группы формулы IVс выполняется с помощью водного щелочного основания для удаления тозильной группы и с помощью водорода или источника водорода и катализатора, предпочтительно гидроксида палладия, палладия-на-угле, платины-на-угле, наиболее предпочтительно гидроксида палладия, необязательно в присутствии кислоты, предпочтительно уксусной кислоты, для удаления бензильной группы, чтобы получить соединение формулы Vа

Удаление тозильной группы и бензильной группы может быть выполнено в любом порядке.
Еще одним вариантом осуществления данного изобретения является способ синтеза 2,4-дихлор-7Н-пирроло[2,3-d]пиримидина, включающий стадии:
а) взаимодействие 7Н-пирроло[2,3-d]пиримидин-2,4-диола с фосфористым оксихлоридом в ароматическом растворителе, предпочтительно толуоле, для получения первого раствора при первой температуре, предпочтительно в пределах от около 0°С до около 50°С, наиболее предпочтительно около 25°С;
b) повышение первой температуры первого раствора до второй температуры, предпочтительно в пределах от около 40°С до около 100°С, наиболее предпочтительно около 75°С;
c) добавление к описанному первому раствору при второй температуре третичного аминового основания, предпочтительно диизопропилэтиламина, чтобы образовать второй раствор при второй температуре, и
d) повышение описанной второй температуры второго раствора до третьей температуры, предпочтительно в пределах от около 75°С до около 125°С, наиболее предпочтительно около 105°С, в течение первого промежутка времени, предпочтительно в пределах от около 1 час и до около 24 час, наиболее предпочтительно около 16 час,
где фосфористый оксихлорид присутствует в пределах от около 1,5 и до около 6 эквивалентов и третичное аминовое основание, предпочтительно диизопропилэтиламин, присутствует в пределах от около 1,0 и до около 8,0 эквивалентов, в обоих случаях относительно начального 7Н-пирроло[2,3-d]пиримидин-2,4-диола. Предпочтительно, чтобы фосфористый оксихлорид присутствовал в пределах от около 2,0 и до около 3 эквивалентов и чтобы третичное аминовое основание присутствовало в пределах от около 1,1 и до около 2 эквивалентов. Наиболее предпочтительно, чтобы фосфористый оксихлорид присутствовал около 3,0 эквивалентами и чтобы третичное аминовое основание присутствовало около 2,0 эквивалентами.
В другом варианте осуществления предложен способ получения энантиомерно обогащенных пиперидинов формулы Хс;

где R1 представляет собой водород или (С1-С6) алкил; а является целым числом от 0 до 4; R2 представляет собой водород или (С1-С6) алкил; и R'' выбирают из группы, состоящей из водорода, С1-С6 алкила и CF3 групп; b является целым числом от 0 до 4; включающий стадию
а) асимметричной гидрогенизации бензилпиридиниевой соли формулы Xd

где Х- выбирают из группы, состоящей из хлора, брома, фтора, йода, трифлата, тозилата или -BF4, предпочтительно брома, R''' представляет собой (С1-С6) алкил; в присутствии родиевого, иридиевого или рутениевого катализатора, предпочтительно бис(1,5-циклооктадиен)родий(I)трифторметансульфонат и хиральный фосфиновый лиганд, предпочтительно (R)-(-)-1-{(S)-2-(дифенилфосфино)ферроценил}этилди-трет-бутилфосфин; (R)-(-)-1-{(S)-2-(дициклогексилфосфино)ферроценил}этилди-трет-бутилфосфин, или (R)-(-)-1-{(S)-2-ди-(пара-трифторметилфенил)фосфино)ферроценил}этилди-трет-бутилфосфин, в присутствии водорода или источника водорода.
В другом предпочтительном варианте бензилпиридиниевая соль имеет формулу Хе

Предпочтительно, когда бензилпиридиниевая соль имеет формулу Хf
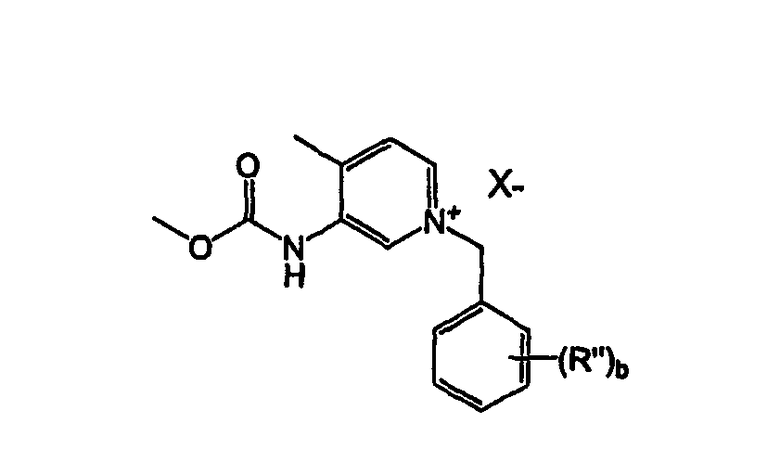
В другом варианте осуществления предложен способ получения энантиомерно обогащенных пиперидинов формулы Хg

где R1 представляет собой водород или (С1-С6) алкил; R2 представляет собой (С1-С6) алкил и R'' выбирают из группы, состоящей из водорода, С1-С6 алкила и CF3 групп; b является целым числом от 0 до 4; включающий стадию
а) асимметричной гидрогенизации тетрагидропиридина формулы Xh

где R''' представляет собой (С1-С6) алкил; в присутствии родиевого, иридиевого или рутениевого катализатора, предпочтительно бис(1,5-циклооктадиен)родий(I)трифторметансульфонат и хиральный фосфиновый лиганд, предпочтительно (R)-(-)-1-{(S)-2-(дифенилфосфино)ферроценил}этилди-трет-бутилфосфин; (R)-(-)-1-{(S)-2-(дициклогексилфосфино)ферроценил}этилди-трет-бутилфосфин или (R)-(-)-1-{(S)-2-ди-(пара-трифторметилфенил)фосфино)ферроценил}этилди-трет-бутилфосфин, в присутствии водорода или источника водорода.
Предпочтительно, когда тетрагидропиридин имеет формулу Xi

Данное изобретение может быть понято более легко с помощью обращения к следующему подробному описанию примерных осуществлений изобретения и к примерам, включенным сюда.
В этом описании и формуле изобретения, приведенной ниже, будут встречаться термины, которые должны быть определены следующим образом:
Термин «алкил» включает насыщенные моновалентные С1-С20 углеводородные радикалы, имеющие прямые, разветвленные или циклические части или их комбинации. Примеры подобных групп включают, но не ограничиваются, метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, циклопропил, циклобутил, циклопентил и циклогексил.
Термин «алкокси» включает О-алкильные группы, где алкил представляется, как определено выше. Примеры включают, но не ограничиваются, метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, трет-бутокси, циклопропокси, циклобутокси и циклопентокси.
Термин «галоген» включает фтор, хлор, бром или йод.
Термин «алкенил» включает моновалентные прямые, разветвленные или циклические углеводородные радикалы, содержащие одну или более двойных связей. Примеры включают, но не ограничиваются, этенил, пропенил, бутенил, изобутенил, циклопентенил и циклогексенил.
Термин «алкинил» включает моновалентные прямые или разветвленные углеводородные радикалы, содержащие одну или более тройных связей. Примеры включают, но не ограничиваются, этинил, пропинил, бутинил.
Термин «ацил» относится к -С(О)-части.
Термин «азотзащищающая группа» или «аминозащищающая группа», как использовалось в настоящем описании, будет понятен квалифицированному лицу, так как они включены в «Protective Groups in Organic Synthesis» 2nd Edition, T.W. Greene & P.G.M. Wutz, Wiley Interscience (1991), они конкретно нумеруются на страницах 218-222 этой ссылки, сущность этого документа включается в описание изобретения в полном объеме. Примеры подходящих защищающих групп включают, но не ограничиваются, бензил, карбобензилокси, трет-бутоксикарбонил (ВОС), 9-флуоренилметиленоксикарбонил и аллилоксикарбонил.
Термин «уходящая группа», как использовалось в настоящем описании, представляет группу, которая может быть заменена нуклеофилами. Примеры уходящих групп включают, но не ограничиваются, галоген, мезилокси, тозилокси и ангидридные остатки карбоновых кислот, такие как трет-бутоксикарбонилокси.
Термин «активирующая группа» относится к любой группе на пирролопиримидиновых фрагментах данного изобретения, которая увеличивает его реакционную способность по отношению к реакциям типа присоединения-удаления с нуклеофилами. Активирующие группы являются обычно электрон-акцепторными группами, такими как галогены, например хлор, сульфонаты, например тозилаты, мезилаты и подобные, сложные эфиры, карбонаты, карбоматы и ацилпроизводные. Также включенными являются бензилпроизводные.
Термин «кислоты», как принято, означает неорганические и органические кислоты. Неорганические кислоты включают, но не ограничиваются, соляную, бромистоводородную, фосфорную, метафосфорную, азотную, серную. Органические кислоты включают, но не ограничиваются, винную, уксусную, метансульфокислоту, трифторуксусную, лимонную, малеиновую, молочную, фумаровую, бензойную, янтарную, метансульфокислоту, щавелевую и пара-толуол сульфокислоты.
Термин «основания», как принято, означает неорганические и органические основания. Неорганические основания включают, но не ограничиваются, карбонат натрия, карбонат калия, гидроксид лития, гидроксид натрия, гидроксид калия, карбонат лития, карбонат натрия, карбонат калия, гидрид лития, гидрид калия, гидрид натрия, литийгидрокарбонат, натрийгидрокарбонат и калийгидрокарбонат. Органические основания включают, но не ограничиваются, металл-алкоксиды, металл-алкильные комплексы, пиридин, алкиламины, диалкиламины и триалкиламины. Примеры алкила, диалкила и триалкиламинов включают, но не ограничиваются, метиламин, диметиламин, триэтиламин и диизопропилэтиламин. Также включенными являются циклические амины, такие как 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU). Примеры металл-алкоксидов включают, но не ограничиваются, метоксид натрия, метоксид калия, трет-бутоксид натрия, трет-бутоксид калия и этоксид натрия. Металл-алкильные комплексы включают, но не ограничиваются, алкиллитиевые реагенты, такие как н-бутиллитий, см. бутиллитий и трет-бутиллитий. Также включенными в это определение являются реактивы Гриньяра, такие как фенилмагнийбромид и этилмагнийбромид.
Кислоты, которые используются для приготовления фармацевтически приемлемой кислотно-аддитивной соли вышеупомянутого соединения формулы Ia, которые формируют нетоксичные кислотно-аддитивные соли, т.е. соли, содержащие фармакологически приемлемые анионы, такие как гидрохлорид, гидробромид, гидроиодид, нитрат, сульфат, бисульфат, фосфат, кислый фосфат, ацетат, лактат, цитрат, кислый цитрат, тартрат, битартрат, сукцинат, малеат, фумарат, глюконат, сахарат, бензоат, метансульфонат, этансульфонат, бензолсульфонат, пара-толуолсульфонат и памоат [т.е. 1,1'-метилен-бис-(2-гидрокси-3-нафтоат)]соли.
Термин «третичный амин» относится к С1-С8 триалкиламинам. Примеры включают, но не ограничиваются, триэтиламин, диизопропилэтиламин и триметиламин. Также включенным в данное определение является пиридин.
Термин «около», как использовалось в настоящем описании, означает плюс или минус 10% для каждой из числовых единиц.
Термин «энантиомерный избыток», как использовалось в настоящем описании, означает избыток одного из двух энантиомеров по сравнению с другим, в основном в качестве процента. Поэтому энантиомерный избыток 90% соответствует присутствию 95% одного энантиомера и 5% другого в смеси при рассмотрении.
Термин «энантиомерно обогащенный» означает, что один энантиомер присутствует в избытке по отношению к другому. То есть, один энантиомер составляет более чем 50% смеси. Также включенными в это определение являются полные энантиомеры (т.е. 100%, исключая ошибку).
Термин «замещенный бензил» относится к бензильной группе, замещенной на фенильном кольце одной или более группами, выбранными из С1-С6 алкила, -СF3, OMe, NO2 и CN.
Термин «psi» относится к фунтам на квадратный дюйм.
Термин «(С3-С12) арил», необязательно, с замещенными от 1 до 6 гетероатомами включает, но не ограничивается, фенил, нафтил, пиридил, хинолинил, бензофуранил, бензотиофен, индол и подобные.
Термин «источник водорода» относится к любому источнику водорода, который может быть получен на месте. В отношении межфазной гидрогенизации муравьиная кислота и ее соли, такие как формиат аммония, формиаты щелочных металлов и тому подобное, могут быть использованы в качестве источников водорода.
Подробное описание изобретения.
Следующие реакционные схемы иллюстрируют приготовление соединений данного изобретения. Если не указывается иное описание, то все R группы, L, Х1, Х2 и Р в реакционных схемах и обсуждении, которое затем следует, являются такими, как определено выше.
Схема I

Соответственно замещенный 3-аминопиридин формулы VII может прореагировать с диалкилкарбонатом или алкилхлорформиатом, чтобы получить VIII. VIII можно затем восстановить в присутствии водорода, предпочтительно в атмосфере водорода под давлением, предпочтительно в пределах от около 70 до около 80 psi, и 5% родия-на-угле или родия-на-оксиде алюминия в присутствии кислоты, желательно уксусной кислоты, при повышенной температуре в пределах от около 50°С и до около 150°С, предпочтительно 70-80°С, чтобы получить цис-пиперидинилкарбаматы IXa и b. Пиперидинильный азот может быть затем защищен взаимодействием IXa и b с подходящей защищающей группой, чтобы получить Xa и b. (см. «Protective Groups in Organic Synthesis» 2nd Edition, T.W. Greene & P.G.M. Wutz, Wiley Interscience (1991) для примеров). Например, реагент, такой как Р-L, где Р представляет собой защищающую группу и L представляет собой уходящую группу, может реагировать, в присутствии основания, такого как карбонат калия или натрия, с пиперидильным азотом из IXa и b, чтобы получить N-защищенные цис-пиперидинилкарбаматы Xa и b. В качестве альтернативы альдегид РСНО, где Р представляет собой защищающую группу, может реагировать при условиях гидрогенизации с пиперидильным азотом. Реакционные условия могут быть определены с помощью одной из обычных практик в технике в зависимости от применявшейся защищающей группы. Карбаматная группа из Xa и b затем может быть восстановлена, используя подходящий восстанавливающий реагент, например алюмогидрид лития, в эфирном растворителе, таком как тетрагидрофуран, при пониженной температуре в пределах от около -78°С до около 70°С, обычно 70°С, чтобы получить IIIb и с (где R2 представляет собой метил). В качестве альтернативы карбаматная группа из Xa и b может быть расщеплена гидролизом, и получившийся в результате свободный амин затем может реагировать с разнообразными алкилирующими реагентами, например R2-L, где L представляет собой уходящую группу, в полярном апротонном растворителе, таком как диметилформамид (DMF), необязательно в присутствии основания, такого как триалкиламиновое основание, чтобы получить промежуточные продукты IIIb и с.
В качестве альтернативы либо бензильная, либо замещенная бензильная пиридиниевая соль формулы VIIIa, где Х- представляет собой хлор, бром, фтор, йод, трифлат, тозилат или -BF4, предпочтительно бром; и где R'' выбирают, независимо для каждой позиции, способной к замещению, из водорода, С1-С6 алкила или CF3 группы или подходящего замещенного тетрагидропиридинпроизводного, где R'' определен выше, может быть подвергнуто асимметрической гидрогенизации при условиях, таких как в присутствии родиевого, иридиевого и рутениевого катализатора и хирального фосфинового лиганда, включающего монофосфиновый и бифосфиновый лиганды. Примеры подходящих хиральных фосфиновых лигандов могут быть обнаружены в Chem. Rev. 2003, 103, 3029 в настоящем описании, включенном в описание изобретения посредством ссылки в полном объеме. Предпочтительными катализаторами являются те, которые образованы из бис(1,5-циклооктадиен)родий(I)трифторметансульфоната и хирального лиганда Josiphos типа. Эти предпочтительные лиганды могут быть коммерчески приобретены в Solvias (Базель, Швейцария). Предпочтительными реагентами и условиями для проведения преобразования являются бис(1,5-циклооктадиен)родий(I)трифторметансульфонат и (R)-(-)-1-{(S)-2-(дифенил, дициклогексил или ди-(пара-трифторметилфенил)фосфино)ферроценил}этилди-трет-бутилфосфины в присутствии герметичной водородной атмосферы под давлением, предпочтительно в пределах от около 50 и до около 200 psi, в растворителе, предпочтительно THF этанольной смеси, при повышенной температуре, желательно а пределах от около 50°С и до около 70°С; приводящие к энантимерно обогащенному пиперидину Ха (например, 50-70% (исключая ошибки) преобладание Ха над Хb). Таким образом полученный обогащенный Xa может быть произведен и позднее использован в хиральной разделяющей стадии ниже и может быть использован, чтобы присоединяться прямо к актвированному пирролопиримидину после образования IIb и с (т.е. образования энантиомерно обогащенного IIb).
Смеси R1 замещенных цис- и транс-3-аминопиперидинов (IIIa) могут быть синтезированы из подходящих замещенных N-защищенных-пиперидин-3-онов, приготовленных способом Iorio, M.A. and Damia, G.; Tetrahedron, Vol. 26: p.55-59 (1970) and Grieco, et al., Journal of the American Chemical Society, Vol. 107: p.1768 (1985). Обе публикации включены в описание изобретения в виде ссылки в полном объеме, измененном в части использования 5% метанола в качестве сорастворителя в реакции уксусной кислоты с R2-NH2, и триацетоксиборгидрида натрия в соответствии с процедурой, описанной в примере 1 согласно заявке US 2004/0053947, которая также включена в описание изобретения в виде ссылки в полном объеме. Пиперидины формулы IIIa могут быть использованы в любом из дальнейших взаимодействий с активированными пирролопиримидинами для получения продуктов соединения как смесей всех возможных диастереомеров.
Схема II

Цис-N-защищенные 3-аминопиперидины IIIb и с затем могут взаимодействовать с активированным пирролопиримидином формулы IIa в присутствии основания, такого как карбонат калия или натрия в полярном растворителе или смеси полярных растворителей, такой как вода или вода и ацетонитрил или DMSO (диметилсульфоксид), при повышенной температуре в пределах от около 50°С и до около 150°С, предпочтительно 100°С, чтобы получить продукт соединения IVa.
Схема III

В качестве альтернативы IIIb и с может быть сначала растворен с одним энантиомером ди-пара-толуоилвинной кислоты, предпочтительно L-энантиомером, или другими хиральными кислотами, и получившаяся в результате полная диастереомерная аминовая соль реагировала непосредственно или путем образования единственного энантиомерного свободного амина из аминовой соли реакцией с водным основанием, таким как гидроксид натрия, с активированным пирролопиримидином IIа в присутствии основания в растворителе или смеси полярных растворителей, таких как вода или вода и ацетонитрил или DMSO, при повышенной температуре в пределах от около 50°С и до около 150°С, предпочтительно 100°С, чтобы получить IVa, как единственный энантиомер. (Для альтернативных способов разделения см. Jacques, J, et al., «Enantiomers, Racemates and Resolutins», Wiley, New York, 1981; в настоящем описании включено в описание изобретения посредством ссылки в полном объеме).
Схема IV

Соединение IVа или как единственный энантиомер, или как смесь энантиомеров затем подвергается воздействию условий для удаления защиты, соответствующей конкретной азотзащищающей группы. Обычно эти условия для удаления защиты будут также способствовать удалению активирующей группы, чтобы образовать соединение формулы V. Например, когда Р представляет собой группу, лабильную к гидрогенолизу, и Х1 представляет собой хлор, защищающая группа и активирующая группа могут быть удалены с помощью водорода или источника водорода в присутствии катализатора. Предпочтительными условиями, которые могут быть применены, являются герметичная водородная атмосфера под давлением, обычно 50 psi, и водородный катализатор, такой как Pd(OH)2, в полярном растворителе, таком как вода, необязательно в присутствии кислоты, такой как уксусная кислота или HCl. В качестве альтернативы и защищающая группа, и активирующая группа могут быть удалены сначала использованием одного набора условий с последующим удалением оставшейся группы при втором наборе условий.
Схема V
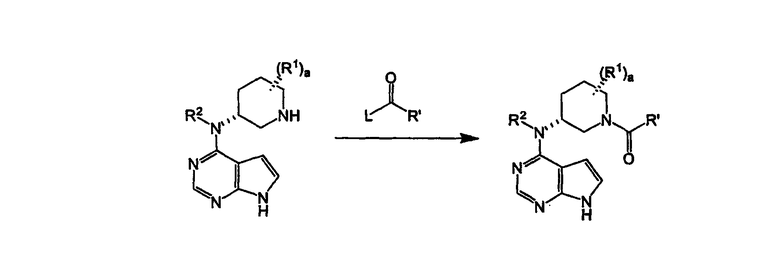
Пиперидильный азот из соединения формулы V может быть ацилирован, используя ацилирующий агент формулы VII, и L представляет собой уходящую группу, необязательно в присутствии основания, такого как триалкиламиновое основание, в полярном протонсодержащем или апротонном растворителе, таком как метиленхлорид, чтобы получить соединение формулы VII. Соединение формулы VII может быть преобразовано в фармацевтически приемлемую соль посредством воздействия органической кислоты, такой как лимонная, соляная и подобные. Необязательно, фармацевтически приемлемая соль соединения формулы VII может быть рекристаллизована из органического растворителя или смесей органических растворителей.
Схема VI
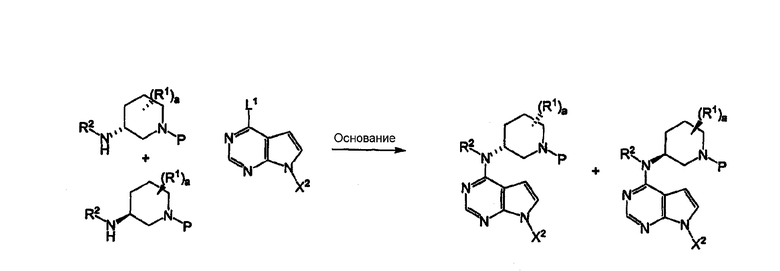
В качестве альтернативы активированный пироллопиримидин формулы IIb может быть присоединен к цис-N-защищенному 3-аминопиперидину IIIb и с, полученным в соответствии со способом, описанным в схеме 1, в присутствии основания в полярном растворителе и смеси полярных растворителей, таких как вода или вода и ацетонитрил, при повышенной температуре в пределах от около 50°С и до около 150°С, предпочтительно 100°С, чтобы получить продукт присоединения IVb как смесь энантиомеров.

В качестве альтернативы IIIb и с, произведенные в соответствии со способом, описанным в схеме 1, могут быть сначала растворены с одним энантиомером ди-пара-толуоилвинной кислоты, предпочтительно L-энантиомером, или другими хиральными кислотами, используя стандартные методы разделения, т.е. кристаллизацию единственной диастереомерной соли, и получившаяся в результате единственная диастериомерная аминовая соль реагировала непосредственно или сначала образуя единственный энантиомерный свободный амин из аминовой соли реакцией с водным основанием, таким как гидроксид натрия, с активированным пирролопиримидином IIb в присутствии основания, такого как карбонат натрия или калия, в растворителе или смеси полярных растворителей, таких как вода или вода и метанол, при повышенной температуре в пределах от около 50°С и до около 150°С, предпочтительно 100°С, чтобы получить IVb, как единственный энантиомер. (Для альтернативных способов разделения см. Jacques, J., et al., «Enantiomers, Racemates and Resolutins», Wiley, New York, 1981; в настоящем описании включено в описание изобретения посредством ссылки в полном объеме.)
Схема VIII
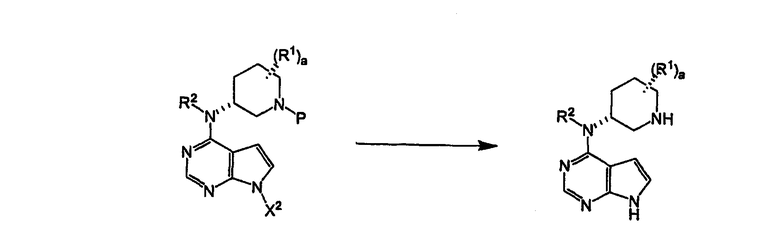
Продукт присоединения IVb затем подвергается воздействию условий удаления соответствующей конкретной азотзащищающей группы и условий для удаления тозильной активирующей группы. Когда защищающая группа является лабильной к гидрогенолизу, гидрогенизирующие условия, такие как герметичная водородная атмосфера под давлением от 10 до 100 psi, и катализатор гидрогенизации, такой как Pd(OH)2 в полярном растворителе или смеси полярных растворителей, такой как смесь изопропилового спирта и воды, необязательно в присутствии кислоты, такой как уксусная или соляная кислота, могут быть применены для удаления азотзащищающей группы с последующим применением щелочного основания, например, такого как водный гидроксид натрия, для удаления активирующей группы, когда активирующая группа является тозильной, чтобы получить соединение формулы V. Когда и активирующая группа, и защищающая группа являются лабильными к гидрогенолизу, обе могут быть удалены с помощью гидрогенизирующих условий в одном реакционном сосуде. В качестве альтернативы либо защищающая группа, либо активирующая группа могут быть сначала удалены, используя один ряд условий с последующим удалением остающейся группы при втором ряде условий.
Соединение формулы V, таким образом полученное, может в дальнейшем реагировать, как предварительно описывалось.
Экспериментальная часть
Пример 1
Приготовление обогащенного -(3R,4R)-(1-бензил-4-метилпиперидин-3-ил)метиламина с помощью асимметрической гидрогенизации
Стадия А. Для приготовления 1-бензил-3-метоксикарбониламино-4-метилпиридинийбромида, для очистки, высушивания, в продутую азотом колбу объемом 500 мл были добавлены метиловый сложный эфир (4-метилпиридин-3-ил)карбаминовой кислоты (25,0 г, 150 ммоль), толуол (250 мл) и бензилбромид (28,3 г, 165 ммоль). Реакция была нагрета до 110°С по меньшей мере 20 час. После охлаждения до пределов между 20-25°С проба была профильтрована и полученные в результате твердые вещества были отмыты толуолом (100 мл). После высушивания в вакууме, в течение по меньшей мере 12 час, в пределах между 40-50°С при слабом токе азота бромид метилового эфира (1-бензил-4-метилпиридин-3-ил)карбаминовой кислоты (48,6 г, 140 ммоль) был выделен с выходом 96,1% (чистота больше чем 95% по ЯМР).
В подходящий по размерам реакционный сосуд были добавлены 1-бензил-3-метоксикарбониламино-4-метилпиридинийбромид (150 мг, 0,446 моль), бис(1,5-циклооктадиен)родий(I)трифторметансульфонат (11 мг, 0,0223 ммоль, полученный от Strem Chemical Co. Newburyport, Massachusetts) и (R)-(-)-1-[(S)-2-(дифенилфосфино)ферроценил]этилди-трет-бутилфосфин (32 мг, 0,033 ммоль, полученный от Soolvias, Basel, Switzerland). Твердое вещество было продуто азотом (5× при 90 psi), затем были добавлены дегазированный THF (2 мл) и дегазированный этанол (1 мл). Смесь была продута азотом (5× при 90 psi), затем водородом (1× при 210 psi). Реакционная смесь была нагрета до 70°С, затем помещена под давление 200 psi с водородом. Через 48 час смесь была охлаждена до 30°С и продута азотом (5× при 90 psi). Аликвота была удалена для GCMS анализа.
Приготовление образца: 100 мкл аликвоты реакционной смеси разводятся в 1 мл МеОН. Добавляют 10 мкл триэтиламина. Смешивают. Отфильтровывают осадки. Анализируют на GC MS (колонна Cyclosil B, температурный градиент 140-240°С, 2°/мин) GC MS анализ: 84% цис-продукта 68%, исключая ошибку, 2% транс-продукта, 4% дебензилированного побочного продукта, 3% алкенового промежуточного продукта.
В качестве альтернативы хиральная жидкостная хроматография высокого давления (HPLC) может быть использована для анализа. Анализ выполняли на Agilent 1100 system: HPLC условия: Daicel Chiralcel OJ 4,6 мм × 250 мм аналитическая колонна, 5% этанол/гексаны, течение 1 мл/мин, 210 нм, 20 мин цикл.
Пример 2
Приготовление метилового эфира 1-бензил-4-метил-1,2,5,6-тетрагидропиридин 3-ил)карбаминовой кислоты. Бензилхлорид (2,1 мл, 18,3 ммоль) был добавлен к суспензии метилового эфира (4-метилпиридин-3-ил)карбаминовой кислоты (3,16 г, 19,0 ммоль) в толуоле (15 мл) при 80°С. Гомогенную реакционную смесь получили через 10 мин, и затем твердое вещество выпало в осадок через 30 мин. Суспензия перемешивалась при 80°С 16 час, затем охлаждалась до комнатной температуры. Твердое вещество было профильтровано и отмыто с толуолом. После высушивания продукт был выделен как серое твердое вещество (4,17 г, 78%).
К реакционной смеси 1-бензил-3-метоксикарбониламино-4-метилпиридинийхлорида (4,0 г, 13,0 ммоль) в EtOH (этаноле) (20 мл) был добавлен боргидрид натрия (0,645 г, 17,0 ммоль). Наблюдалось выделение газа, и реакция была экзотермической. Реакционную смесь перемешивали при комнатной температуре 16 час. Реакцию гасили водой Н2О (10 мл), фильтровали через Celite и слой Celite отмывали EtOH (2×10 мл). Объем растворителя уменьшался в вакууме. Полученную в результате смесь доводили до 9 рН с помощью 1 N гидроксида натрия, затем экстрагировали с помощью MTBE (метил-трет-бутил эфира) (2×15 мл). Комбинированные органические слои были отмыты Н2О (10 мл), высушены (сульфат натрия) и сконцентрированы до желтого масла (2,44 г, 72%).
Пример 3
Приготовление обогащенного (3R,4R)-(1-бензил-4-метилпиперидин-3-ил)метиламина с помощью асимметрической гидрогенизации метилового эфира 1-бензил-4-метил-1,2,5,6-тетрагидропиридин 3-ил)карбаминовой кислоты
В реакционный сосуд были добавлены метиловый эфир 1-бензил-4-метил-1,2,5,6-тетрагидропиридин-3-ил)карбаминовой кислоты (150 мг, 0,577 ммоль), бис(1,5-циклооктадиен)родий(I)трифторметансульфонат (13 мг, 0,0288 ммоль, полученный от Strem Chemical Co. Newburyport, Massachusetts) и (R)-(-)-1-[(S)-2-(дициклогексилфосфино)ферроценил]этилди-трет-бутилфосфин (32 мг, 0,033 ммоль, полученный от Soolvias, Basel, Switzerland). Твердое вещество было продуто азотом (5× при 90 psi), затем были добавлены дегазированный THF (2 мл) и дегазированный этанол (1 мл). Смесь была продута азотом (5× при 90 psi), затем водородом (1× при 210 psi). Реакционная смесь была нагрета до 70°С, затем помещена под давление 200 psi с водородом. Через 48 час смесь была охлаждена до 30°С и продута азотом (5× при 90 psi). Аликвота была удалена для GCMS анализа. GC MS анализ: 97% цис-продукта 66%, исключая ошибку, 2% транс-продукта. Такой же анализ с помощью GCMS или хиральной HPLC, как описывалось выше.
Пример 4
Синтез 3-{(3R,4R)-4-метил-(7Н-пиролло[2,3-d]пиримидин-4-ил)амино}пиперидин-1-ил)-3-оксопропионитрила
Приготовление бис-(3R,4R)-(1-бензил-4-метилпиперидин-3-ил)метиламина ди-пара-толуоил-L-винной кислоты
В чистую сухую продутую азотом колбу объемом 250 мл были загружены рацемический цис-(1-бензил-4-метилпиперидин-3-ил)метиламин-бис-гидрохлорид (20,0 г, 68,7 ммоль), ди-пара-толуоил-L-винная кислота (L-DPTT) (15,9 г, 41,2 ммоль) и метанол (100 мл). Раствор гидроксида натрия (5,5 г, 137,3 ммоль в воде (100 мл)) добавляли к реакционной смеси таким образом, чтобы поддерживать температуру ниже 30°С. Реакцию нагревали до пределов между 70-80°С и поддерживали при этой температуре по меньшей мере 60 мин. Реакцию охлаждали до 5-15°С в течение более по меньшей мере 4 час и поддерживали при данной температуре по меньшей мере 12 час. Твердое вещество отфильтровывали и промывали смесью 1:1 МеОН:вода (60 мл). Осадок с фильтра возвращали в 250-мл колбу, и загружали метанол (100 мл) и воду (100 мл). Реакцию охлаждали до 5-15°С в течение более по меньшей мере 4 час и поддерживали при данной температуре в течение по меньшей мере 12 час. Твердые вещества отфильтровывали и промывали смесью 1:1 МеОН:вода (60 мл). Влажный осадок отбирали до чистоты (99,4%, исключая ошибку), чтобы обеспечить отсутствие дополнительной необходимости ресуспензирования. После высушивания в вакууме при 40-50°С в течение по меньшей мере 24 час при слабом токе азота указанное в заголовке соединение (11,9 г, 28,9 ммоль) выделяли с выходом 42,1% (98,6% энантиомерного избытка, 0,63 транс-изомера с помощью GC (колонна Cyclosil B 30 м × I.D. 0,25 мм; входная температура 250°С, 2,0 мл/мин скорость течения; 15 мин цикл; 160°С изотермальный способ)).
Пример 5
Приготовление N-[(3R,4R)-1-бензил-4-метилпиперидин-3-ил]-2-хлор-N-метил-7Н-пиролло[2,3-d]пиримидин-4-амина (формула IVb)
В чистый сухой продутый азотом реактор объемом 500 мл были загружены 2,4-дихлор-7Н-пиролло[2,3-d]пиримидин, приготовленный, как описано ниже (20,0 г, 0,106 моль), бис-(3R,4R)-(1-бензил-4-метилпиперидин-3-ил)метиламин ди-пара-толуоил-L-винной кислоты (20 г, 0,106 моль), карбонат калия (44,6 г, 0,319 моль) и вода (200 мл). Реактор нагревали до 95-105°С минимум 10 час, затем охлаждали до 20-30°С и поддерживали минимум 3 час. Полученное в результате твердое вещество выделяли фильтрацией, отмывали водой (60 мл) и высушивали при 50°С с получением 39,5 г (100%) указанного в заголовке соединения.
Аналитически рассчитано для C20H24ClN5: С, 64,94; H, 6,54; N, 18,93. Найдено: С, 64,78; H, 6,65; N, 18,83.
1Н ЯМР (400 МГц, d6-ацетон): δ 10,80 (ушир.с, 1Н), 7,36 (д, J=7,0 Гц, 2Н), 7,30 (т, J=7,0 Гц, 2Н), 7,24-7,20 (м, 1Н), 7,13 (д, J=3,7 Гц, 1Н), 6,66 (ушир.с, 1Н), 5,15 (ушир.с, 1Н), 3,69 (ушир.с, 3Н), 3,54 (АВq, J=13,3 Гц, 1Н), 3,50 (АВq, J=13,3 Гц, 1Н), 2,92 (дд, J=12,0, 5,4 Гц, 1Н), 2,88-2,83 (м, 1Н), 2,77 (ушир.с, 1Н), 2,64-2,59 (м, 1Н), 2,29 (ушир.с, 1Н), 2,16 (ушир.с, 1Н), 1,75-1,69 (м, 2Н), 0,94 (д, J=6,6 Гц, 3Н).
13С ЯМР (400 МГц, d6-ДМСО, смесь изомеров): δ 158,0, 152,5, 151,8, 138,3, 129,1, 128,6, 128,1, 127,6, 126,8, 121,0, 102,3, 100,8, 62,5, 54,6, 53,1, 50,8, 35,3, 32,0, 30,9, 15,3.
Пример 6
Приготовление метил-[(3R,4R)-4-метилпиперидин-3-ил]-7Н-пиролло[2,3-d]пиримидин-4-ил)амина
В чистый сухой продутый азотом реактор гидрогенизации объемом 500 мл были загружены 20% по массе Pd(ОН)2/С (гидроксид палладия-на-угле) (5,0 г, с 50%-м содержанием воды), вода (200 мл) и N-[(3R,4R)-1-бензил-4-метилпиперидин-3-ил]-2-хлор-N-метил-7Н-пиролло[2,3-d]пиримидин-4-амин (50,0 г, 0,135 моль). Реактор продували три раза при 50 psi азотом и три раза при 50 psi водородом. Когда продувку закончили, реактор нагрели до 70-75°С и создавали давление 50 psi непрерывной подачей водорода. Поглощение водорода контролировали до тех пор, пока никакого водорода не подавалось минимум 1 час. Реактор охлаждали до 20-30°С и продували три раза при 50 psi азотом. Реакционную смесь отфильтровывали через гидрофильный Celite и перемещали в чистый сухой продутый водородом реактор объемом 500 мл для последующей обработки.
Пример 7
Приготовление 4-хлор-7-(толуол-4-сульфонил)-7Н-пиролло[2,3-d]пиримидина
В чистый сухой продутый азотом реактор загружали ацетон (87,5 мл), пара-толуолсульфонилхлорид (17,1 г, 0,09 моль) и 4-хлор-7Н-пиролло[2,3-d]пиримидин (25,0 г, 0,16 моль). Реактор охлаждали до температуры от -5,0°С до 5,0°С и добавляли 2,5 М гидроксид натрия (78,1 мл) таким образом, чтобы обеспечить температуру ниже 5,0°С. Реактор подогревали до температуры от 20 до 30°С и перемешивали минимум 5 час. Полученное в результате твердое вещество выделяли фильтрацией и отмывали смесью ацетон/вода (1:1, 25 мл каждого). После высушивания в течение минимум 12 час в вакууме при 40-50°С при слабом токе азота получали 44,9 г (90,1%) указанного в заголовке соединения.
Т.пл. 140,2-147,7°С.
Аналитически рассчитано для C13H10ClN3О2S: С, 50,73; H, 3,28; N, 13,65. Найдено: С, 50,50; H, 3,06; N, 13,63.
1Н ЯМР (400 МГц, d6-ДМСО): δ 8,79 (с, 1Н), 8,09 (д, J=4,2 Гц, 1Н), 8,01 (д, J=8,5 Гц, 2Н), 7,43 (д, J=8,5 Гц, 2Н), 6,92 (д, J=4,2 Гц, 1Н), 2,32 (с, 3Н).
13С ЯМР (400 МГц, d6-ДМСО): δ 153,2, 152,7, 151,2, 147,2, 134,3, 131,0, 129,3, 128,5, 119,9, 103,9, 21,8.
Пример 8
Приготовление [(3R,4R)-1-бензил-4-метилпиперидин-3-ил]метил-[7-(4-метилбензолсульфонил)-7Н-пиролло[2,3-d]пиримидин-4-ил]амина
В чистый сухой продутый азотом реактор загружали 4-хлор-7-(4-метилбензолсульфонил)-7Н-пиролло[2,3-d]пиримидин (25,12 г, 0,082 моль), бис-(3R,4R)-(1-бензил-4-метилпиперидин-3-ил)метиламин ди-пара-толуоил-L-винной кислоты (40,31 г, 0,041 моль), карбонат калия (34,2 г, 0,245 моль) и воду (125,6 мл). Смесь нагревали до 95-105°С в течение минимум 10 час, затем охлаждали до 45-55°С. Загружали ацетонитрил (25 мл) и суспензию поддерживали при 45-55°С в течение минимум 1 час. В дальнейшем смесь охлаждали до 20-30°С и перемешивали в течение минимум 5 час. Полученное в результате твердое вещество выделяли фильтрацией и отмывали водой (50 мл). После высушивания было выделено 32,8 г (82,1%) указанного в заголовке соединения.
Т.пл. 181,7-184,4°С.
Аналитически рассчитано для C27H31N5О2S: С, 66,231; H, 6,38; N, 14,3. Найдено: С, 66,04; H, 6,47; N, 14,44.
1Н ЯМР (400 МГц, CDCl3): δ 8,34 (с, 1Н), 8,06 (д, J=8,7 Гц, 2Н), 7,43 (д, J=4,2 Гц, 1Н), 7,30-7,29 (м, 6Н), 7,25-7,21 (м, 1Н), 6,67-6,66 (м, 1Н), 5,14 (ушир.с, 1Н), 3,56-3,44 (м, 5Н), 2,82-2,78 (м, 1Н), 2,73 (ушир.с, 1Н), 2,58-2,55 (м, 1Н), 2,38 (с, 3Н), 2,31 (ушир.с, 1Н), 2,12 (ушир.с, 1Н), 1,74 (ушир.с, 1Н), 1,69-1,61 (м, 1Н), 0,90 (д, J=7,0 Гц, 3Н)
13С ЯМР (400 МГц, CDCl3): δ 158,2, 152,9, 152,1, 145,5, 138,7, 135,4, 129,9, 129,1, 128,5, 128,4, 127,3, 120,8, 106,6, 104,9, 63,7, 55,5 (b), 53,0 (b), 51,8 (b), 35,9 (b), 32,8, 31,4, 21,7, 15,9 (b).
Пример 9
Приготовление [(3R,4R)-1-бензил-4-метилпиперидин-3-ил]метил-(7Н-пиролло[2,3-d]пиримидин-4-ил)амина
В чистый продутый азотом реактор загружали 50% раствор гидроксида натрия (210 мл) и (1-бензил-4-метилпиперидин-3-ил)метил-[7-(толуол-4-сульфонил)-7Н-пиролло[2,3-d]пиримидин-4-ил]амин (30,0 г, 0,061 моль). Смесь нагревали до 95-105°С в течение минимум 5 час, затем охлаждали до 70-90°С и добавляли воду (300 мл). Суспензию охлаждали до комнатной температуры в течение более чем 1,5 час и выдерживали при комнатной температуре 1 час. Твердое вещество выделяли фильтрованием и отмывали водой (120,0 мл) с получением 25,2 г указанного в заголовке влажного соединения.
1Н ЯМР (400 МГц, CD3OD): δ 8,05 (с, 1Н), 7,36-7,28 (м, 5Н), 7,24-7,20 (м, 1Н), 7,06 (д, J=3,7 Гц, 1Н), 6,62 (д, J=3,7 Гц, 1Н), 5,08 (ушир.с, 1Н), 3,58-3,52 (м, 5Н), 2,85 (дд, J=11,2, 7,0 Гц, 1Н), 2,68 (дд, J=11,2, 3,7 Гц, 1Н), 2,65-2,59 (м, 1Н), 2,45-2,39 (м, 1Н), 2,29-2,20 (м, 1Н), 1,90-1,81 (м, 1Н), 1,70-1,63 (м, 1Н), 0,98 (д, J=7,0 Гц, 3Н).
13С ЯМР (400 МГц, d6-ДМСО, смесь изомеров): δ 166,8, 164,4, 158,6, 155,1, 138,4, 137,9, 137,8, 136,6, 135,5, 135,3, 112,7, 110,0, 72,4, 64,3 (b), 62,4 (b), 60,3 (b), 44,5, 41,8, 40,9, 30,5, 24,8.
Пример 10
Приготовление метил-[(3R,4R)-4-метилпиперидин-3-ил]-7Н-пиролло[2,3-d]пиримидин-4-ил)амина
В чистый сухой продутый азотом реактор гидрогенизации объемом 2 л были загружены 20% по массе Pd(ОН)2/С (гидроксид палладия-на-угле) (24,0 г, с 50%-м содержанием воды), вода (160 мл), изопропанол (640 мл), (1-бензил-4-метилпиперидин-3-ил)метил-(7Н-пиролло[2,3-d]пиримидин-4-ил)амин (160,0 г, 0,48 моль) и уксусная кислота (28,65 г, 0,48 моль). Реактор продували 3 раза при 50 psi азотом и 3 раза при 50 psi водородом. Когда продувание было закончено, реактор нагревали до 45-55°С и создавали давление 50 psi непрерывной подачей водорода. Поглощение водорода контролировали до тех пор, пока никакого водорода не подавалось минимум 1 час. Реактор охлаждали до 20-30°С и продували 3 раза при 50 psi азотом. Реакционную смесь отфильтровывали через гидрофильный Celite и фильтрат отправляли в чистый сухой продутый азотом сосуд. Добавляли раствор гидроксида натрия (39,33 г) в воде (290 мл) и смесь перемещивали в течение минимум 1 час, затем нагревали до 75-90°С. Изопропанол удаляли дистилляцией. Реакционную смесь охлаждали до 20-30°С и добавляли 2-метилтетрагидрофуран (1,6 л). Водный слой был слит и 2-метилтетрагидрофуран заменили на толуол (1,6 л). Дистилляцию продолжали до тех пор пока, конечный объем не стал 800 мл. Суспензию охлаждали до 20-30°С и поддерживали в течение минимум 7 час. Полученное в результате твердое вещество выделяли фильтрованием и отмывали толуолом (480 мл). После высушивания в вакууме при температуре от 40 до 50°С в течение минимум 24 час при слабом токе азота было выделено 102,3 г (87,3%) указанного в заголовке соединения.
Т.пл. 158,6-159,8°С.
1Н ЯМР (400 МГц, CDCl3): δ 11,38 (ушир.с, 1Н), 8,30 (с, 1Н), 7,05 (д, J=3,5 Гц, 1Н), 6,54 (д, J=3,5 Гц, 1Н), 4,89-4,87 (м, 1Н), 3,39 (с, 3Н), 3,27 (дд, J=12,0, 9,3 Гц, 1Н), 3,04 (дд, J=12,0, 3,9 Гц, 1Н), 2,94 (тд, J=12,6, 3,1 Гц, 1Н), 2,84 (дт, J=12,6, 4,3 Гц, 1Н), 2,51-2,48 (м, 1Н), 2,12 (ушир.с, 2Н), 1,89 (ддт, J=13,7, 10,6, 4 Гц, 1Н), 1,62 (дкв., J=13,7, 4 Гц, 1Н), 1,07 (д, J=7,3 Гц, 3Н)
13С ЯМР (400 МГц, CDCl3): δ 157,9, 152,0, 151,0, 120,0, 103,0, 102,5, 56,3, 46,2 42,4, 34,7, 33,4, 32,4, 14,3.
Пример 11
Приготовление 3-{(3R,4R)-4-метил-3-[метил-(7Н-пиролло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-ил}-3-оксопропионитрила
В чистый сухой продутый азотом реактор объемом 1,0 л были загружены метил-(4-метилпиперидин-3-ил)-(7Н-пиролло[2,3-d]пиримидин-4-ил)амин (32,0 г, 0,130 моль), толуол (160 мл), этилцианацетат (88,53 г, 0,783 моль) и триэтиламин (26,4 г, 0,261 моль). Реакционную смесь нагревали до 100°С и поддерживали в течение 24 час. Реакционную смесь отмывали водой (160 мл). Были смешаны органический слой, сконцентрированный до объема 10 мл, и вода (20 мл). Остаточный толуол был удален дистилляцией, и смесь охладили до комнатной температуры. Ацетон (224 мл) был добавлен после добавления раствора лимонной кислоты (27,57 г, 0,144 моль) в воде (76 мл). Полученную в результате суспензию перемешивали 7 час. Твердое вещество отделили фильтрацией, отмыли ацетоном (96 мл) и высушили в вакууме с получением 42,85 г (65,3%) указанного в заголовке соединения.
Пример 12
Альтернативное приготовление 3-{(3R,4R)-4-метил-3-[метил-(7Н-пиролло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-ил}-3-оксопропионитрила с помощью хлорида кислоты
В чистый сухой продутый азотом реактор были загружены цианоуксусная кислота (2,30 г, 27,0 ммоль), метиленхлорид (20 мл), оксалилхлорид (3,36 г, 26,5 ммоль) и DMF (1 капля). Реакционную смесь перемешивали при комнатной температуре в течение минимум 40 мин. Реакционную смесь затем охлаждали до температуры от -15 до -10°С и поддерживали. В отдельный сухой продутый азотом реактор были добавлены метил-(4-метилпиперидин-3-ил)-(7Н-пиролло[2,3-d]пиримидин-4-ил)амин (1,3 г, 5,3 ммоль), DMF (7 мл) и триэтиламин (5,5 г, 54,0 ммоль). Реакционную смесь перемешивали до получения гомогенной смеси. Смесь затем медленно добавляли к кислотному хлориду, сохраняя температуру ниже 5°С. Реакционную смесь перемешивали при температуре от -10 до 5°С в течение 30 мин и затем нагревали до комнатной температуры, затем поддерживали в течение минимум 1 час. Добавляли этилацетат и раствор отмывали насыщенным бикарбонатом натрия (2×30 мл). Легколетучие компоненты удаляли дистилляцией, и остаток растворяли в ацетоне (27 мл) и воде (5 мл). Добавляли лимонную кислоту (1,02 г, 5,3 ммоль), и полученные в результате твердые вещества перемешивали в течение минимум 12 час. Твердые вещества отфильтровывали и отмывали ацетоном (3 мл) и водой (5 мл), затем высушивали в вакууме с получением 2,0 г (74%) указанного в заголовке соединения.
Пример 13
Приготовление 3-{(3R,4R)-4-метил-3-[метил-(7Н-пиролло[2,3-d]пиримидин-4-ил)амино]пиперидин-1-ил}-3-оксопропионитрильной соли лимонной кислоты
В чистый сухой продутый азотом реактор объемом 500 мл были загружены метил-(4-метилпиперидин-3-ил)-(7Н-пиролло[2,3-d]пиримидин-4-ил)амин (25,0 г, 0,102 моль) и метиленхлорид (250 мл). Смесь перемешивали при комнатной температуре в течение минимум 2,5 час. В чистый сухой продутый азотом реактор объемом 1 л были загружены цианоуксусная кислота (18,2 г, 0,214 моль), метиленхлорид (375 мл) и триэтиламин (30,1 мл, 0,214 моль). Смесь охлаждали до температуры от -15,0°С до -5,0°С более 1 час и добавляли триметилацетилхлорид (25,6 мл, 0,204 моль) таким образом, чтобы обеспечить температуру ниже 0°С. Реакцию поддерживали в течение минимум 2,5 час, затем добавляли раствор амина для обеспечения температуры ниже 0°С. После перемешивания в течение 1 час смесь нагревали до комнатной температуры и добавляли 1М гидроксид натрия (125 мл). Органический слой отмывали водой (125 мл). Раствор метиленхлорида заменяли ацетоном до объема 500 мл и до достижения температуры 55-65°С. Воду (75 мл) добавляли к смеси, сохраняя температуру 55-65°С. Загружали раствор лимонной кислоты (20,76 г, 0,107 моль) в воде (25,0), и смесь охлаждали до комнатной температуры. Реакционную смесь перемешивали в течение минимум 5 час, и затем полученные в результате твердые вещества выделяли фильтрацией и отмывали ацетоном (2×75 мл), который подавали на фильтр. Соль загружали в чистый сухой продутый азотом реактор объемом 1 л с 2В этанолом (190 мл) и водой (190 мл). Суспензию нагревали до 75-85°С в течение минимум 4 час. Смесь охлаждали до 20-30°С и перемешивали дополнительно 4 час. Твердые вещества выделяли фильтрацией и отмывали 2В этанолом (190 мл). После высушивания в вакуумном термошкафу при 50°С при слабом токе азота было выделено 34,6 г, (67,3%) указанного в заголовке соединения.
1Н ЯМР (500 МГц, d6-ДМСО): δ 8,14 (с, 1Н), 7,11 (д, J=3,6 Гц, 1Н), 6,57 (д, J=3,6 Гц, 1Н), 4,96 (кв., J=6,0 Гц, 1Н), 4,00-3,90 (м, 2Н), 3,80 (м, 2Н), 3,51 (м, 1Н), 3,32 (с, 3Н), 2,80 (Аbq, J=15,6 Гц, 2Н), 2,71 (Аbq, J=15,6 Гц, 2Н), 2,52-2,50 (м, 1Н), 2,45-2,41 (м, 1Н), 1,81 (м, 1Н), 1,69-1,65 (м, 1Н), 1,04 (д, J=6,9 Гц, 3Н).
Пример 15
Приготовление 2,4-дихлор-7Н-пиролло[2,3-d]пиримидина
В реактор добавили 7Н-пиролло[2,3-d]пиримидин-2,4-диол (10,0 г, 66,2 ммоль) и толуол (30 мл) при перемешивании. Добавили фосфороксихлорид (18,5 мл, 198,5 ммоль) и реактор нагрели до 70°С. Добавляли диизопропилэтиламин (23,0 мл, 132,3 ммоль) более чем 2,5 час для контроля экзотермы. После завершения основного добавления реактор нагрели до 106°С и реакционную смесь перемешивали при температуре 16 час. Смесь охладили до 25°С и медленно добавили в колбу, содержащую воду (230 мл) и этилацетат (120 мл) при комнатной температуре, затем перемешивали в течение ночи при комнатной температуре. После фильтрации через Celite слои были разделены, водный слой извлекали этилацетатом (3×75 мл). Органические слои объединили и отмыли рассолом (100 мл). Darco KBB (1,24 г) добавляли к органическим соединениям, затем фильтровали через Celite и высушивали над сульфатом натрия (10,0 г). Раствор концентрировали в вакууме с получением указанного в заголовке соединения (выход 52%).
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМБИНАЦИИ, ВКЛЮЧАЮЩИЕ АГОНИСТ РЕЦЕПТОРА S1P И ИНГИБИТОР КИНАЗЫ JAK3 | 2005 |
|
RU2415678C2 |
| Макрогетероциклические нуклеозидные производные и их аналоги, получение и применение | 2017 |
|
RU2731385C1 |
| ЗАМЕЩЕННЫЕ (2R,3R,5R)-3-ГИДРОКСИ-(5-ПИРИМИДИН-1-ИЛ)ТЕТРАГИДРОФУРАН-2-ИЛМЕТИЛ АРИЛ ФОСФОРАМИДАТЫ | 2013 |
|
RU2553996C1 |
| АЛКИЛ 2-{ [(2R,3S,5R)-5-(4-АМИНО-2-ОКСО-2Н-ПИРИМИДИН-1-ИЛ)- -ГИДРОКСИ-ТЕТРАГИДРО-ФУРАН-2-ИЛМЕТОКСИ]-ФЕНОКСИ-ФОСФОРИЛАМИНО} -ПРОПИОНАТЫ, НУКЛЕОЗИДНЫЕ ИНГИБИТОРЫ РНК-ПОЛИМЕРАЗЫ HCV NS5B, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2013 |
|
RU2534613C2 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |
| ПРОИЗВОДНЫЕ БЕНЗИЛФЕНИЛЦИКЛОГЕКСАНА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2008 |
|
RU2505521C2 |
| НОВЫЕ ПИПЕРИДИНИЛЬНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2017 |
|
RU2743163C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРОВ SGLT2 | 2009 |
|
RU2530494C2 |
| НОВЫЕ (ГЕТЕРО)АРИЛ-ЗАМЕЩЕННЫЕ ПИПЕРИДИНИЛЬНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2017 |
|
RU2742271C2 |
| НОВОЕ ФОСФОРАМИДАТНОЕ ПРОИЗВОДНОЕ НУКЛЕОЗИДА И ЕГО ПРИМЕНЕНИЕ | 2014 |
|
RU2621709C2 |
Данное изобретение относится к вариантам нового способа получения производных пирроло[2,3-d]пиримидина общей формулы V, обладающих свойствами ингибиторов протеинкиназ, включающим способ получения новых промежуточных соединений. Способ получения соединения формулы V


где X1 представляет собой активирующую группу, выбранную из хлора, брома, йода, R1 представляет собой (С1-С6)алкил; а является целым числом от 0 до 4; R2 представляет собой водород или (С1-С6)алкил, включает сочетание активированного пирролопиримидинового соединения формулы IIa:

где L1 представляет собой уходящую группу и X1 представляет собой активирующую группу, выбранную из хлора, брома, йода; с амином формулы IIIa или его солью, где R1 представляет собой (C1-С6)алкил; а является целым числом от 0 до 4; R2 представляет собой водород или (С1-С6)алкил и P представляет собой азотзащищающую группу, лабильную к гидрогенолизу, такую как бензил; в присутствии основания с получением соединения формулы IVa, и последующее удаление из полученного соединения формулы IVa активирующей группы X1 и азотзащищающей группы Р гидрогенолизом в присутствии водорода или источника водорода и катализатора в любом порядке. Способ позволяет повысить выход целевого продукта. 6 н. и 20 з.п. ф-лы.
1. Способ получения соединения формулы IVa
где X1 представляет собой активирующую группу, выбранную из хлора, брома, йода, R1 представляет собой (С1-С6)алкил; а является целым числом от 0 до 4; R2 представляет собой водород или (С1-С6)алкил и Р представляет собой азотзащищающую группу, лабильную к гидрогенолизу, такую как бензил;
включающий сочетание активированного пирролопиримидинового соединения формулы IIa:
где L1 представляет собой уходящую группу и X1 представляет
собой активирующую группу, выбранную из хлора, брома, йода; с амином формулы IIIa или его солью;
где R1 представляет собой (C1-С6)алкил; а является целым числом от 0 до 4; R2 представляет собой водород или (С1-С6)алкил и Р представляет собой азотзащищающую группу, лабильную к гидрогенолизу, такую как бензил; в присутствии основания с получением соединения формулы IVa.
2. Способ получения соединения формулы V
где R1 представляет собой (С1-С6)алкил; а является целым числом от 0 до 4; R2 представляет собой водород или (C1-С6)алкил;
включающий сочетание активированного пирролопиримидинового соединения формулы IIa
где L1 представляет собой уходящую группу и Х1 представляет собой активирующую группу, выбранную из хлора, брома, йода; с амином формулы IIIa или его солью;
где R1 представляет собой (С1-С6)алкил; а является целым числом от 0 до 4; R2 представляет собой водород или (С1-С6)алкил и Р представляет собой азотзащищающую группу, лабильную к гидрогенолизу, такую как бензил; в присутствии основания с получением соединения формулы IVa
где X1 представляет собой активирующую группу, выбранную из хлора, брома, йода, R1 представляет собой (С1-С6)алкил; а является целым числом от 0 до 4; R2 представляет собой водород или (С1-С6)алкил и Р представляет собой азотзащищающую группу, лабильную к гидрогенолизу, такую как бензил;
с последующим удалением из полученного соединения формулы IVa активирующей группы X1 и азотзащищающей группы Р гидрогенолизом в присутствии водорода или источника водорода и катализатора в любом порядке с получением соединения формулы V.
3. Способ по п.1, где уходящую группу L1 выбирают из группы, состоящей из хлора, брома, йода и фтора.
4. Способ по п.3, где L1 представляет собой хлор.
5. Способ по п.1, где X1 представляет собой хлор.
6. Способ по п.1, где основание выбирают из группы, состоящей из щелочного основания и триалкиламинового основания.
7. Способ по п.6, где основание представляет собой бикарбонат калия.
8. Способ по п.2, где X1 представляет собой хлор.
9. Способ по п.2, где катализатор выбирают из группы, состоящей из гидроксида палладия, палладия-на-угле и платины-на-угле.
10. Способ по п.1, где амин или его соль формулы IIIa являются соединением формулы IIIe или его солью
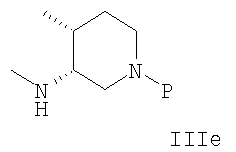
11. Способ по п.10, где амин формулы IIIe представляет собой аминовую соль формулы IIIf

12. Соединение формулы IVb
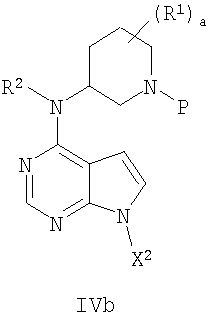
где X2 представляет собой активирующую группу, выбранную из хлора, брома, йода, бензила и тозила, R1 представляет собой (С1-С6)алкил; а является целым числом от 0 до 4; R2 представляет собой водород или (С1-С6)алкил, и Р представляет собой азотзащищающую группу, лабильную к гидрогенолизу, такую как бензил.
13. Способ получения соединения формулы IVb
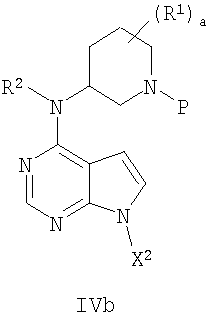
где X2 представляет собой активирующую группу, выбранную из хлора, брома, йода, бензила и тозила, R1 представляет собой (C1-С6)алкил; а является целым числом от 0 до 4; R2 представляет собой водород или (С1-С6)алкил, и Р представляет собой азотзащищающую группу, лабильную к гидрогенолизу, такую как бензил;
включающий сочетание активированного пирролопиримидинового соединения формулы IIb
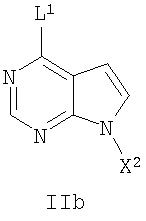
где L1 представляет собой уходящую группу, и X2 представляет собой активирующую группу, выбранную из хлора, брома, йода, бензила и тозила; с амином формулы IIIa или его солью,

где R1 представляет собой (С1-С6)алкил; а является целым числом от 0 до 4; R2 представляет собой водород или (С1-С6)алкил, и Р представляет собой азотзащищающую группу, лабильную к гидрогенолизу, такую как бензил; в присутствии основания с получением соединения формулы IVb.
14. Способ получения соединения формулы V
где R1 представляет собой (С1-С6)алкил; а является целым числом от 0 до 4; R2 представляет собой водород или (С1-С6)алкил;
включающий сочетание активированного пирролопиримидинового соединения формулы IIb
где L1 представляет собой уходящую группу, и X2 представляет собой активирующую группу, выбранную из хлора, брома, йода, бензила и тозила; с амином формулы IIIa или его солью,
где R1 представляет собой (С1-С6)алкил; а является целым числом от 0 до 4; R2 представляет собой водород или (С1-С6)алкил, и Р представляет собой азотзащищающую группу, лабильную к гидрогенолизу, такую как бензил; в присутствии основания с получением соединения формулы IVb
где X2 представляет собой активирующую группу, выбранную из хлора, брома, йода, бензила и тозила, R1 представляет собой (С1-С6)алкил; а является целым числом от 0 до 4; R2 представляет собой водород или (С1-С6)алкил, и Р представляет собой азотзащищающую группу, лабильную к гидрогенолизу, такую как бензил;
с последующим удалением из полученного соединения формулы IVb активирующей группы X2 и азотзащищающей группы Р гидрогенолизом в присутствии водорода или источника водорода, катализатора, и, необязательно, в присутствии кислоты, в любом порядке для получения соединения формулы V.
15. Способ по п.13, где основание выбирают из группы, состоящей из щелочных оснований и триалкиламиновых оснований.
16. Способ по п.15, где основание представляет собой карбонат калия.
17. Способ по п.13, где L1 представляет собой хлор.
18.Способ по п.14, где активирующая группа X2 представляет собой тозильную группу и удаляется щелочным основанием.
19. Способ по п.18, где щелочное основание представляет собой гидроксид натрия.
20. Способ по п.13, где амин формулы IIIa или его соль имеют формулу IIId или его соли
21. Способ по п.20, где амин формулы IIId представляет собой аминовую соль формулы IIIf
22. Способ по п.14, где активирующая группа X2 представляет собой тозильную группу и удаляется щелочным основанием, и активирующую группу Х2, и азотзащищающую группу Р удаляют с получением соединения формулы Va
23. Способ по п.22, где катализатор выбирают из группы, состоящей из гидроксида палладия, палладия-на-угле и платины-на-угле.
24. Способ по п.23, где катализатор представляет собой гидроксид палладия.
25. Способ по п.22, где кислота представляет собой уксусную кислоту.
26. Соединение формулы
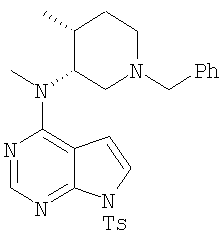 .
.
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| DAVID H | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Перекатываемый затвор для водоемов | 1922 |
|
SU2001A1 |
| NAVEEN K.SAXENA ET AL.: | |||

