По данной заявке испрашивается приоритет на основании предварительной заявки США №60/462613, поданной 14 апреля 2003 г.
Область изобретения
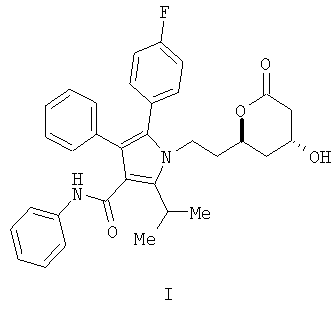
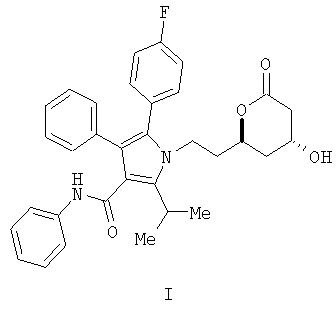
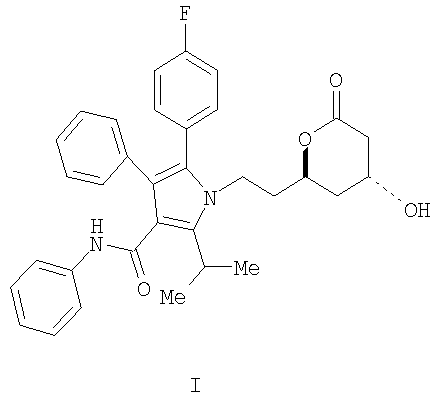
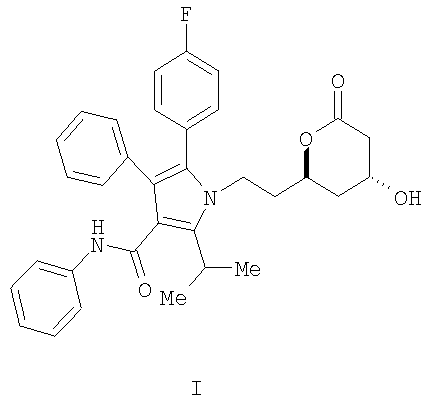
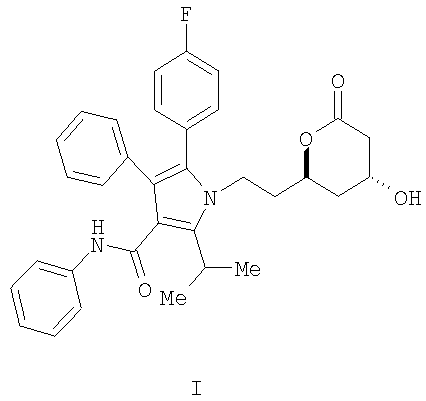
Описывается способ получения фениламида 5-(4-фторфенил)-1-[2-((2R,4R)-4-гидрокси-6-оксотетрагидропиран-2-ил)этил]-2-изопропил-4-фенил-1H-пиррол-3-карбоновой кислоты, ключевого промежуточного соединения в синтезе аторвастатина кальция.
Предпосылки создания изобретения
Фениламид 5-(4-фторфенил)-1-[2-((2R,4R)-4-гидрокси-6-оксотетрагидропиран-2-ил)этил]-2-изопропил-4-фенил-1Н-пиррол-3-карбоновой кислоты (I) является ключевым промежуточным соединением в синтезе аторвастатин кальция (Lipitor®), известного также под химическим наименованием тригидрат кальциевой соли (2:1) [R-(R*,R*)]-2-(4-фторфенил)-β,δ-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1Н-пиррол-1-гептановой кислоты. Аторватстин кальция ингибирует 3-гидрокси-3-метилглютарил-кофермент А редуктазу (HMG-CoA редуктаза) и таким образом является полезным в качестве гиполипидемического и/или гипохолестеринемического агента.
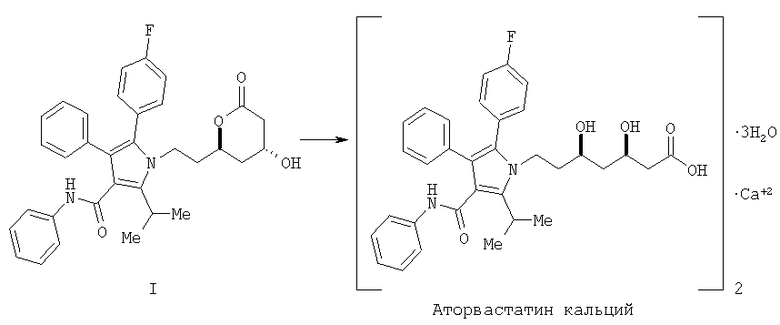
Аторвастатин кальций
Выдан ряд патентов, раскрывающих подходы к получению аторвастатин кальция, также как и различных аналогов, через такие промежуточные продукты как соединение (I). Данные патенты включают: патенты Соединенных Штатов 4681893; 5273995; 5003080; 5097045; 5103024; 5124482; 5149837; 5155251; 5216174; 5245047; 5248793; 5280126; 5397792; 5342952; 5298627; 5446054; 5470981; 5489690; 5489691; 5510488; 5998633 и 6087511; 5969156; 6121461; 5273995; 6476235; заявку Соединенных Штатов 60/401707 (поданную 6 августа 2002 г.).
Существующие подходы к получению ключевого промежуточного соединения (I) обнаружили некоторые недостатки. Например, один из подходов основывался на использовании дорогостоящего хирального сырья (этилового эфира (R)-4-циано-3-гидроксимасляной кислоты) и на низкотемпературном диастереоселективном восстановлении бораном.
Схема 1 обобщает альтернативный подход, описанный в патенте США 6476235. Гидрирование сложного β,δ-дикетоэфира 2 в присутствии хирального рутениевого катализатора протекало в кислой среде, давая диол 3 с низкими выходами и диастереоселективностью син:анти 1:1 в отношении хиральных центров С-3 и С-5.
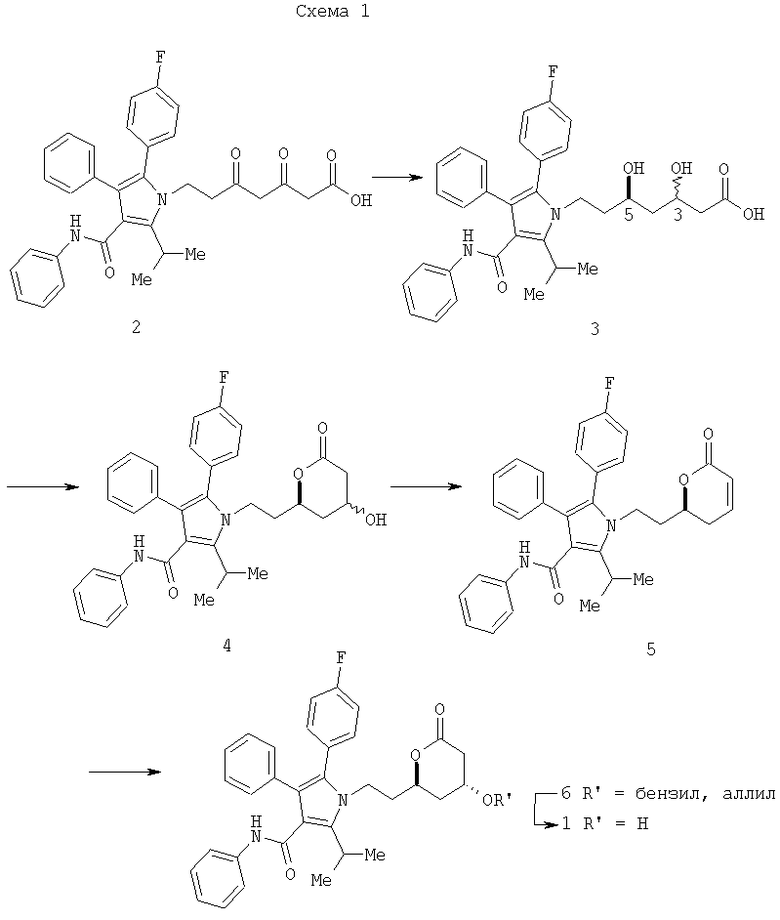
В качестве начальной стадии известны способы асимметрического гидрирования кетонов, описанные выше, для преобразования соединения 2 в 3. Однако сложность реакции увеличивается в случае 1,3,5-трикарбонильных систем, таких как соединение 2, и часто результатом являются низкий выход и плохие стереоселективности. В действительности исследования Saburi [Tetrahedron, 1997, 1993; 49) и Carpentier (Eur. J. Org. Chem. 1999; 3421) независимо продемонстрировали диастерео- и/или энантиоселективности от низких до умеренных для асимметрических способов гидрирования сложного дикетоэфира. Далее тот факт, что процессы, описанные в литературе, требуют гидрирования при высоком давлении и продолжительного времени реакций, делают данные способы в общем непрактичными и неадаптируемыми к крупномасштабным процессам производства, где критическими факторами являются безопасность, эффективность и стоимость.
Обращаясь снова к Схеме 1, ряд дополнительных преобразований необходим для того, чтобы возвращать исходную стереохимию центру С-3 в диоле 3 для получения ключевого промежуточного соединения (I). Данные стадии включают: (а) внутримолекулярную циклизацию соединения 3 для получения лактона 4; (b) отщепление воды в лактоне 4 с использованием кислоты для получения α,β-ненасыщенного лактона 5; (с) селективное присоединение с одной стороны плоскости молекулы аллилового или бензилового спирта к α,β-ненасыщенному лактону 5 по Михаэлю для получения насыщенного лактона 6 и удаление аллильной или бензильной части в лактоне 6 с помощью гидрогенолиза для получения ключевого промежуточного соединения (I).
В результате все еще остается потребность в подходе к получению ключевого промежуточного соединения (I), который являлся бы эффективным, недорогим, протекал с минимумом преобразований и проходил с хорошим выходом и высокими уровнями диастереоселективности.
Краткое описание изобретения
Данные и другие потребности удовлетворяются настоящим изобретением, которое направлено на способ получения соединения формулы (I)
включающий:
(а) контактирование в растворителе необязательно в присутствии кислоты Льюиса соединения формулы (II) с соединением, в котором М означает SiCl3, SiMe3, В(ОН2), CuLi, MgBr, ZnBr, InBr, SnR3, где R3 означает (C1-C6)алкил, с получением соединения (III)
(b) превращение соединения (III) в сложный акрилоиловый эфир формулы (IV) в присутствии основания с использованием соединения 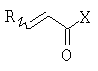 , в котором X означает Cl, Br, I, или соединения
, в котором X означает Cl, Br, I, или соединения 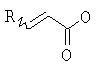 , и R означает Н, (C1-C6)алкил или фенил, или в эквивалент активированного акрилоилового сложного эфира
, и R означает Н, (C1-C6)алкил или фенил, или в эквивалент активированного акрилоилового сложного эфира
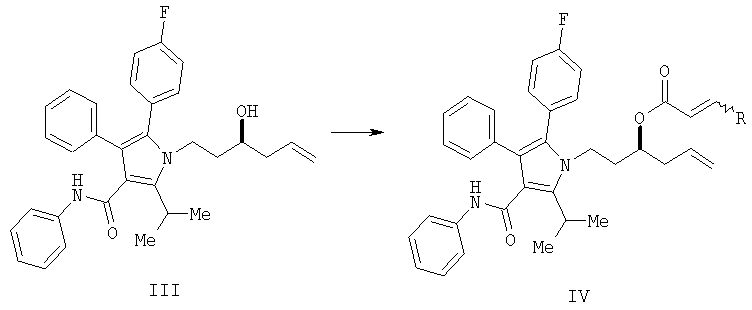
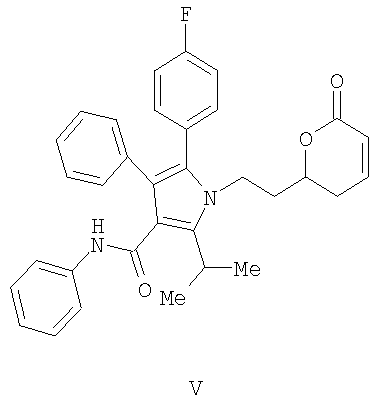
(с) контактирование в растворителе сложного акрилоилового эфира (IV) с катализатором с получением 5,6-дигидропиран-2-она (V)
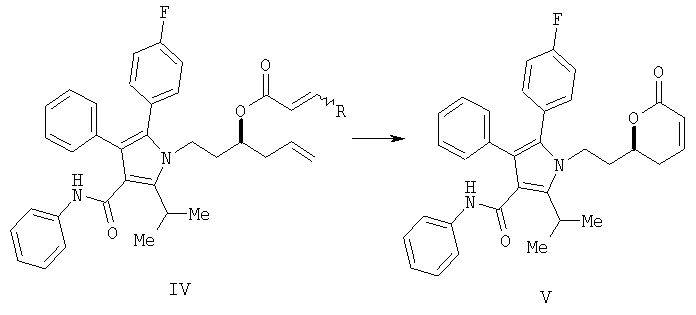
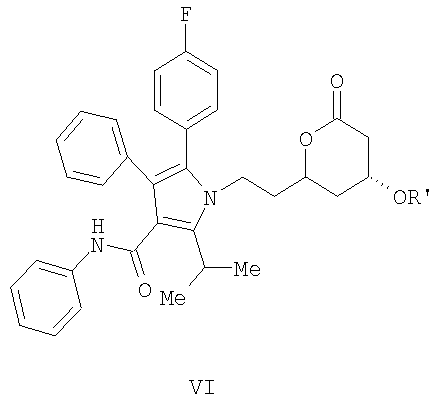
(d) превращение соединения формулы (V) в соединение формулы (VI) с помощью селективного 1,4-присоединения аллилового или бензилового спирта с одной стороны плоскости молекулы
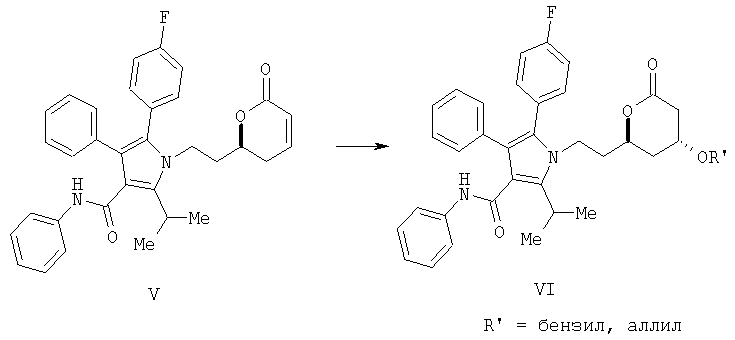
и
(е) удаление аллильной или бензильной части в соединении формулы (VI) с помощью гидрогенолиза с получением соединения формулы I
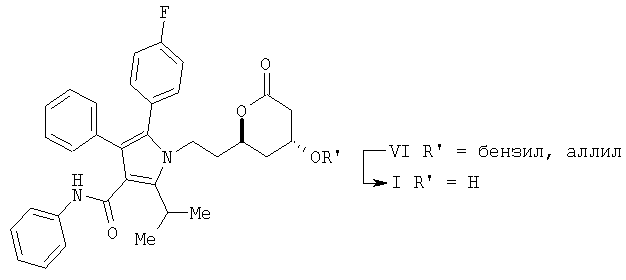
Описывается также способ получения соединения формулы (I)
включающий:
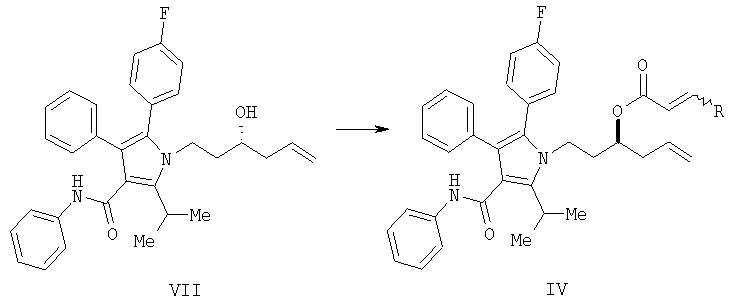
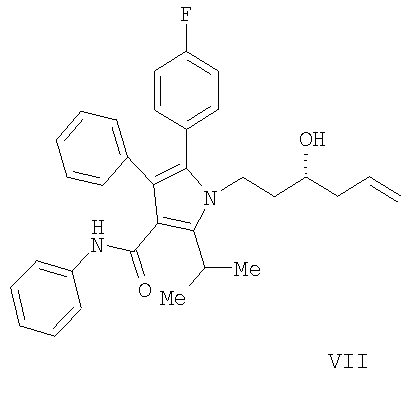
(а) контактирование в растворителе необязательно в присутствии кислоты Льюиса соединения формулы (II) с соединением  , в котором М означает SiCl3, SiMe3, B(OH)2, CuLi, MgBr, ZnBr, InBr, SnR3, где R3 означает (C1-C6)алкил, с получением соединения формулы (VII)
, в котором М означает SiCl3, SiMe3, B(OH)2, CuLi, MgBr, ZnBr, InBr, SnR3, где R3 означает (C1-C6)алкил, с получением соединения формулы (VII)
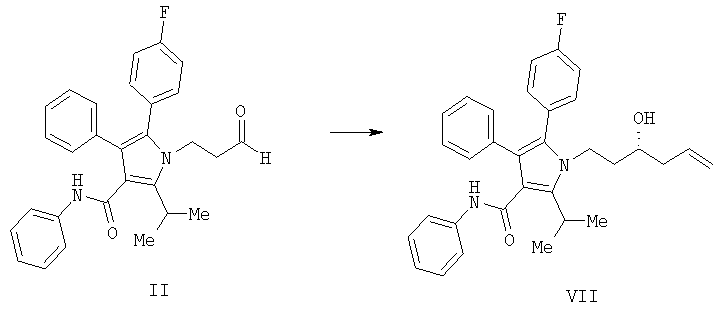
(b) превращение соединения формулы (VII) с сопутствующей стереохимической инверсией центра гомоаллилового спирта в сложный акрилоиловый эфир формулы (IV) по реакции Мицунобу в присутствии акриловой кислоты или аналога акриловой кислоты формулы 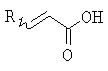 , в которой R означает Н, (C1-C6)алкил или фенил, в присутствии основания
, в которой R означает Н, (C1-C6)алкил или фенил, в присутствии основания
(с) контактирование в растворителе сложного акрилоилового эфира (IV) с катализатором с получением 5,6-дигидропиран-2-она V
(d) превращение соединения формулы (V) в соединение формулы (VI) с помощью селективного 1,4 присоединения аллилового или бензилового спирта с одной стороны плоскости молекулы
и
(е) удаление аллильной или бензильной части в соединении формулы (VI) с помощью гидрогенолиза с получением соединения формулы I
Далее описывается способ получения соединения формулы (I)
включающий:
(а) контактирование в растворителе необязательно в присутствии кислоты Льюиса соединения формулы (II) с соединением , в котором М означает SiCl3, SiMe3, В(ОН)2, CuLi, MgBr, ZnBr, InBr, SnR3, где R3 означает (C1-C6)алкил, с получением соединения формулы (VIII)
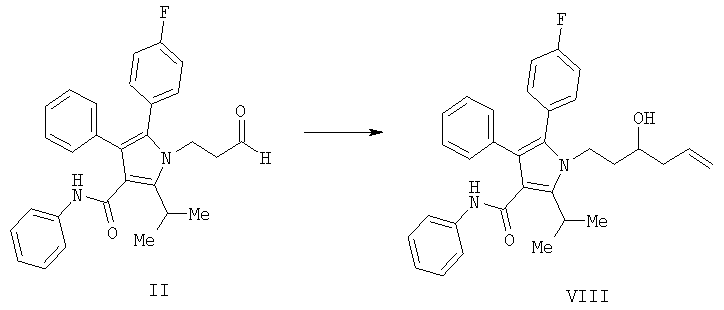
(b) выделение желаемого энантиомера (III) из энантиомерной смеси
(с) превращение соединения формулы (III) в сложный акрилоиловый эфир формулы (IV) в присутствии основания с использованием соединения , в котором Х означает Cl, Br, I, или соединения , и R означает Н, (C1-C6)алкил или фенил, или в эквивалент активированного акрилоилового сложного эфира
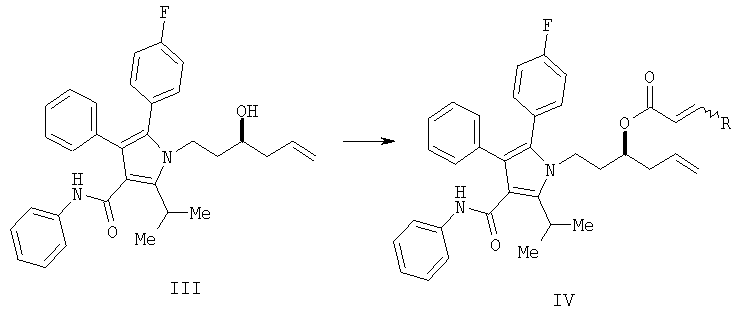
(d) контактирование в растворителе сложного акрилоилового эфира (IV) с катализатором с получением 5,6-дигидропиран-2-она V
(е) превращение соединения формулы (V) в соединение формулы (VI) с помощью селективного 1,4-присоединения аллилового или бензилового спирта с одной стороны плоскости молекулы
и
(f) удаление аллильной или бензильной части в соединении формулы (VI) с помощью гидрогенолиза с получением соединения формулы I
Описывается также способ получения соединения формулы (III)
включающий:
(а) контактирование соединения формулы (II) с алленилбороновым сложным эфиром с получением соединения формулы (XI)
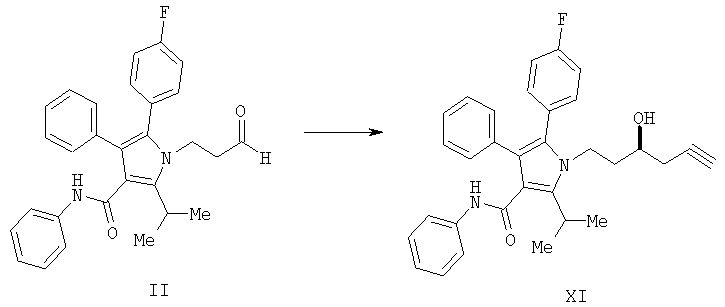
и
(b) гидрирование соединения формулы (XI) с получением соединения формулы III
Предоставляется также способ получения соединения формулы
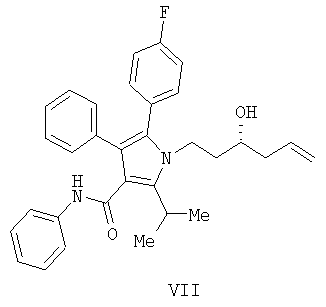
включающий:
(а) контактирование соединения формулы (II) с алленилбороновым сложным эфиром с получением соединения формулы (XII)
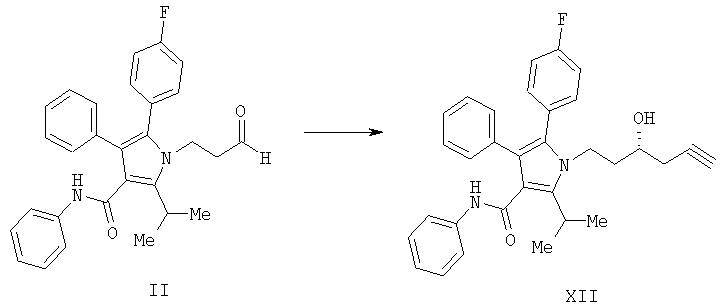
и
(b) гидрирование соединения формулы (XII) с получением соединения формулы (VII)
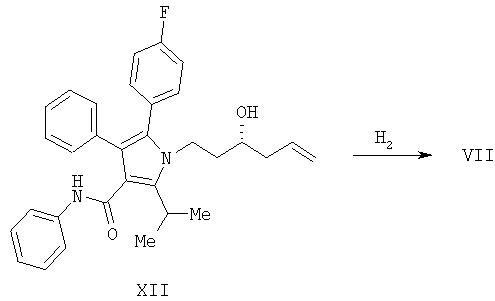
Предоставляется также способ получения соединения формулы
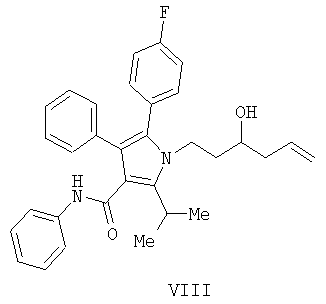
включающий:
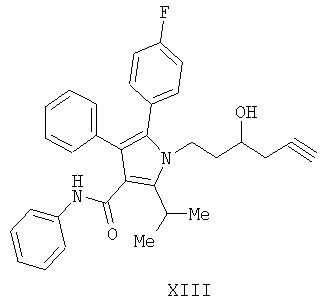
(а) контактирование соединения формулы (II) с алленилбороновой кислотой или алленилбороновым сложным эфиром с получением соединения формулы (XIII)
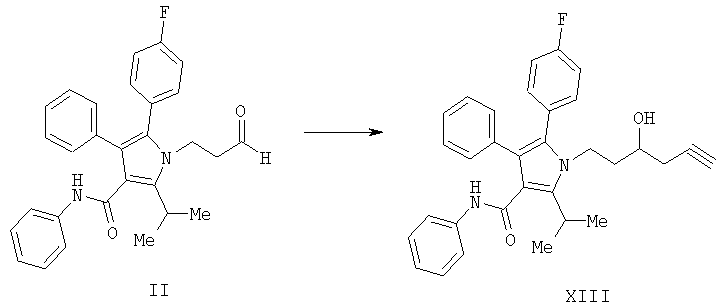
и
(b) гидрирование соединения формулы (XIII) с получением соединения формулы VII
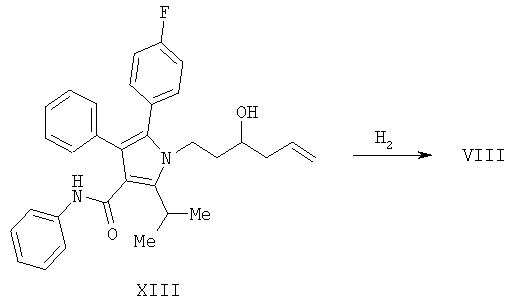
Предоставляется также соединение формулы III
Предоставляется также соединение формулы VIII
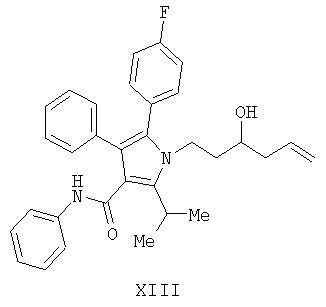
Предоставляется также соединение формулы VII
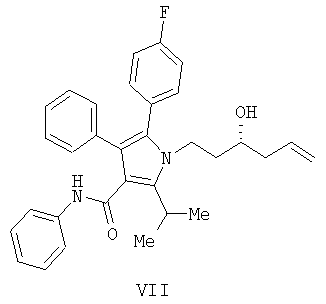
Предоставляется также соединение формулы IV
Предоставляется также соединение формулы IX
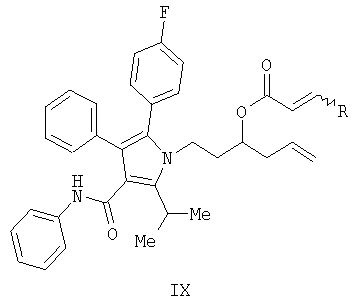
Предоставляется также соединение формулы X
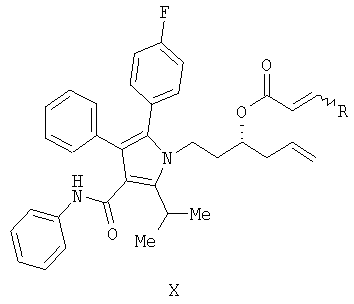
Предоставляется также соединение формулы XI
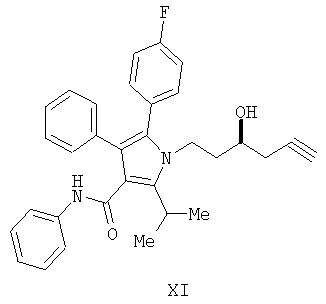
Предоставляется также соединение формулы XII
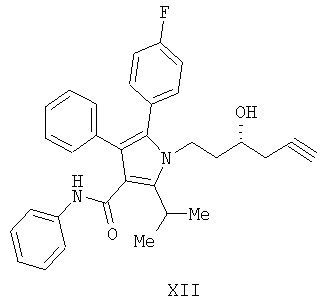
Предоставляется также соединение формулы XIII
Как описано в данном изобретении, на удивление и неожиданно, авторы обнаружили, что 5,6-дигидропиран-2-он (V) может быть удобно получен из акрилоилового сложного эфира (IV) с помощью мягкой и эффективной одностадийной реакции замыкания кольца в присутствии гомогенного катализатора. Реакция протекает с хорошими выходами при температурах ниже приблизительно 60°С и атмосферном давлении. Способ изобретения является, таким образом, более безопасным и более эффективным в крупных масштабах, чем ранее известные способы, потому что он не требует обязательного использования специализированного оборудования высокого давления. В дополнение к этому требуется минимальное число преобразований для введения С-3 гидроксигруппы и общее число стадий, необходимых для превращения соединения (II) в ключевое промежуточное соединение (I) сводится до минимума. Более того, способ изобретения не требует использования дорогого хирального сырья (этилового эфира (R)-4-циано-3-гидроксимасляной кислоты) и низкотемпературного диастереоселективного восстановления бораном, как это было необходимо в ранее известных способах получения ключевого промежуточного соединения (I).
Подробное описание изобретения
Определения
"(C1-С6)алкил" означает как прямые, так и разветвленные группы; но ссылка на индивидуальный радикал, такой как "пропил", охватывает радикал только с прямой цепью, а изомер, с разветвленной цепью, такой как "изопропил", называется конкретно. Таким образом, (C1-С6)алкилом может быть метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, пентил, 3-пентил или гексил.
Термин "приблизительно", используемый в данном описании в отношении качества, условия или количества, означает величину, указываемую по отношению к качеству, условию или количеству, которая является почти точной, или приблизительной, или больше, или меньше указанной.
Если для конкретной величины, указанной в данном описании (относящейся, например, к температуре реакции, времени, концентрации или стехиометрии), даются пределы или интервалы, определяемые двумя граничными точками, имеется в виду, что данный интервал охватывает конечные значения и все действительные величины как дробные, так и целые между конечными значениями.
Способ изобретения
Соединения, получаемые с помощью способа изобретения, описанного в данном описании, могут иметь один или более хиральных центров и могут существовать и использоваться или выделяться в оптически активной и рацемических формах. Таким образом, следует понимать, что способы настоящего изобретения могут давать любые рацемические или оптически активные формы, или их смеси, описанные в данном изобретении. Следует также понимать, что продукты способа изобретения могут быть выделены в виде рацемических, энантиомерных или диастереомерных форм, или их смесей. Очистка и методы определения характеристик таких продуктов известны обычным специалистам в данной области и включают приемы перекристаллизации, методы хирального хроматографического разделения, а также другие методы.
Способ изобретения, раскрытый в данном описании, обобщен на Схеме 2. Хотя она изображает синтез желаемого хирального ряда, последовательность реакций, показанных на Схеме 2, может быть при необходимости модифицирована (т.е. путем использования хиральных и нехиральных вспомогательных агентов, кислот Льюиса, или лигандов, в зависимости от типа реакции) для получения как хиральных, так и нехиральных продуктов
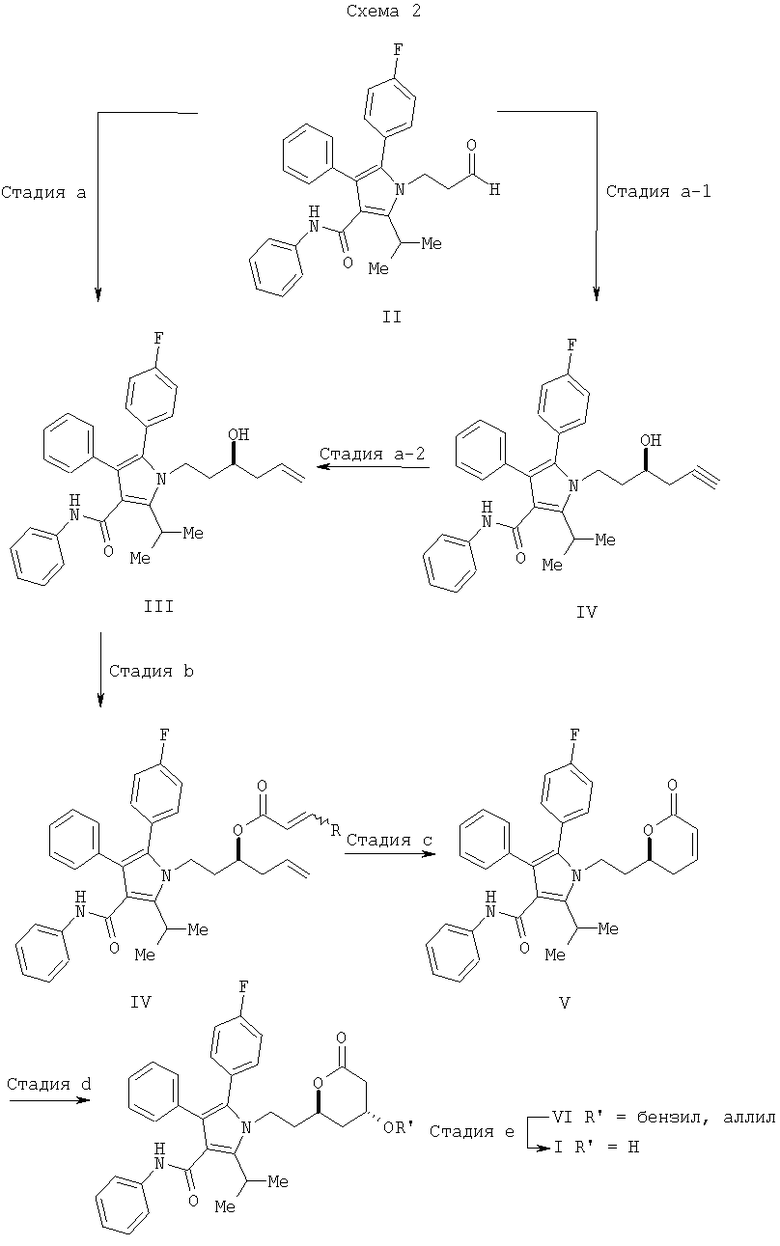
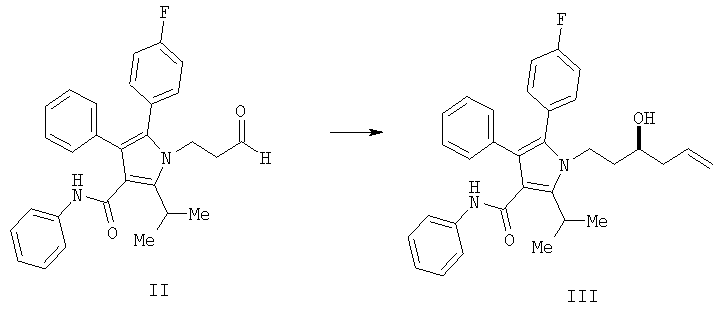
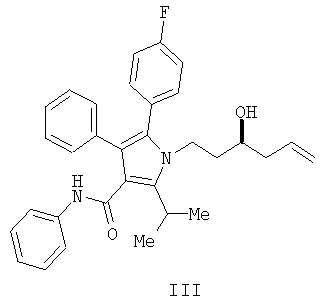
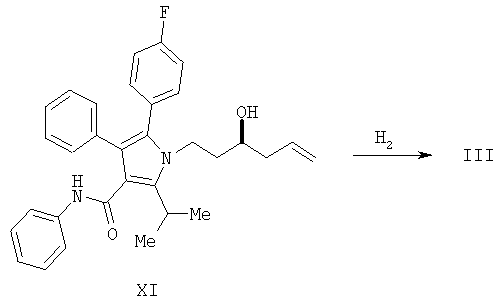
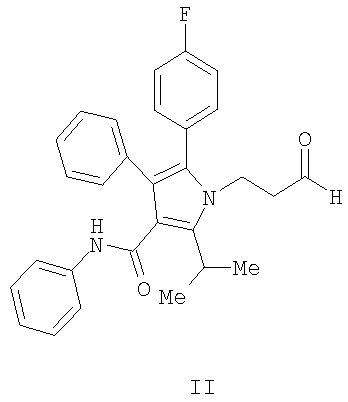
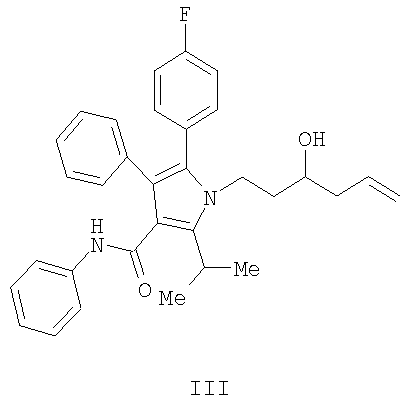
Способ изобретения начинается со стадии (а) или (а-1)/(а-2). На стадии (а) аллилирование альдегида (II) дает гомоаллиловый спирт III. На стадии (а-1)/(а-2) присоединение алленилборонового сложного эфира к альдегиду (II) дает гомопропаргиловый спирт XI. Гидрирование гомопропаргилового спирта (XI) на стадии (а-2) дает гомоаллиловый спирт III.
На стадии (b) гидроксильной группе в соединении (III) дают возможность реагировать с акрилоилхлоридом с получением акрилоилового эфира IV. На стадии (с) реакция замыкания кольца дает ключевое промежуточное соединение V. На стадии (d) С-3 гидроксильная группа, защищенная в виде соответствующего бензилового или аллилового эфира, стереоселективно вводится в соединение V. Удаление защитной группы и гидрогенолиз дают соединение I.
Последовательность синтеза, раскрытая на Схеме 2, описывается более подробно в следующих разделах.
Стадия (а)
На стадии (а) способа изобретения альдегид (II) подвергается аллилированию с использованием соединения 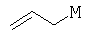 , в котором М означает SiCl3, SiMe3, В(ОН)2, CuLi, MgBr, ZnBr, InBr, SnR3, где R3 означает (C1-С6)алкил, с получением гомоаллилового спирта III. Методы выполнения аллилирования альдегидов хорошо известны и широко доступны квалифицированным специалистам и обычно основываются на использовании реактива Гриньяра (т.е. аллилмагнийбромида) или эквивалента реактива Гриньяра, такого как аллилцинк, аллилборан (такой как аллилдигидроксиборан), аллилбороновый сложный эфир, аллилкупрат, аллилолово (такой как аллил-три-н-бутилстаннан), аллилсилан (такой как аллилтрихлорсилан или аллилтриметилсилан) или аллилиндиевый реагент. Способы получения и использования данных реагентов хорошо известны квалифицированным специалистам в данной области на основе сообщений в химической литературе. Многие также коммерчески доступны.
, в котором М означает SiCl3, SiMe3, В(ОН)2, CuLi, MgBr, ZnBr, InBr, SnR3, где R3 означает (C1-С6)алкил, с получением гомоаллилового спирта III. Методы выполнения аллилирования альдегидов хорошо известны и широко доступны квалифицированным специалистам и обычно основываются на использовании реактива Гриньяра (т.е. аллилмагнийбромида) или эквивалента реактива Гриньяра, такого как аллилцинк, аллилборан (такой как аллилдигидроксиборан), аллилбороновый сложный эфир, аллилкупрат, аллилолово (такой как аллил-три-н-бутилстаннан), аллилсилан (такой как аллилтрихлорсилан или аллилтриметилсилан) или аллилиндиевый реагент. Способы получения и использования данных реагентов хорошо известны квалифицированным специалистам в данной области на основе сообщений в химической литературе. Многие также коммерчески доступны.
Кислота Льюиса необязательно может использоваться для опосредования асимметрического индуцирования и/или опосредования реакции аллилирования. Использование кислот Льюиса хорошо известно в органическом синтезе (см. Hisashi Yamamoto, Lewis acids in Organic Synthesis (2002)). В случае нехирального воплощения способа изобретения нехиральная кислота Льюиса может использоваться для катализирования процесса аллилирования, как изображено на Схеме 3, с получением гомоаллилового спирта (VIII) в виде рацемической смеси. В данной серии реакций желаемый энантиомер (III) может быть выделен с использованием методов, доступных специалисту в данной области, например, с помощью хроматографического разделения с использованием хиральной стационарной фазы или расщепления рацемической формы с помощью установившихся приемов перекристаллизации.
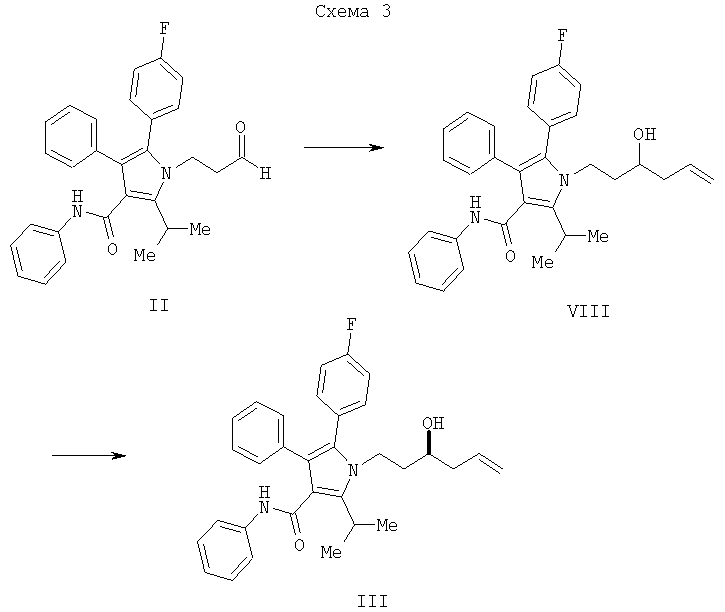
Согласно еще одному воплощению стадии (а) Схемы 2 хиральная кислота Льюиса может использоваться для контроля энантиоселективности, а также для опосредования процесса. Согласно одному из воплощений способа изобретения использовали кислоту Льюиса, генерированную на месте, полученную из трибромида бора и (S,S)-1,2-диамино-1,2-дифенилэтан-бис-толуолсульфонамида, с получением 94,4% энантиомерного избытка желаемого S изомера, как показано на Схеме 2.
Согласно еще одному воплощению стадии (а) Схемы 2, изображенному на Схеме 4, противоположный энантиомер (VII) может быть синтезирован с помощью подбора соответствующей хиральной кислоты Льюиса. В данном варианте реакции соединение (VII) свободно превращается в предпочтительный энантиомер (III) в условиях, доступных специалистам в данной области. Например, реакция соединения (VII) типа Мицунобу в присутствии трифенилфосфина, трибутилфосфина или подобного, диэтилазодикарбоксилата или эквивалентного реагента, такого как диизопропилазодикарбоксилат или 1,1'-(азодикарбонил)дипиперидин, и карбоновой кислоты, такой как бензойная, муравьиная или уксусная кислота, дает сложный эфир IIIa. Эфир IIIa может быть свободно превращен в гомоаллиловый спирт (III) в условиях восстановления или гидролиза, доступных специалистам в данной области. Альтернативно, в качестве кислотного компонента системы реагента Мицунобу может быть использована акриловая кислота с получением сложного гомоаллилового эфира (III) в одном сосуде.
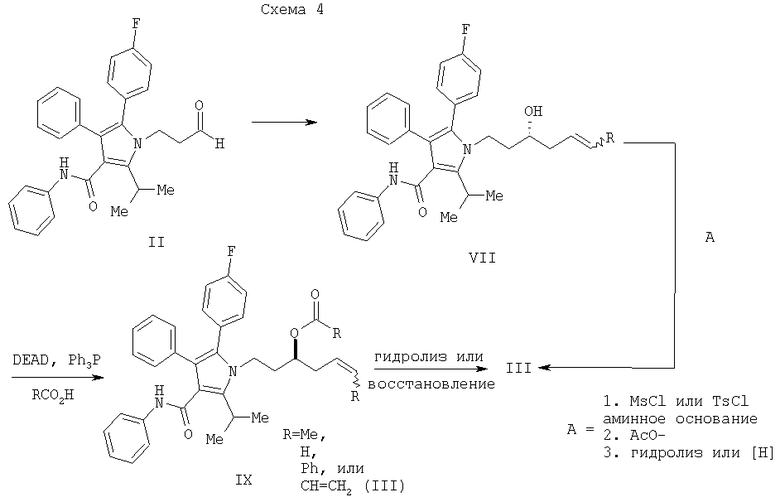
Альтернативный способ превращения соединения (VII) в соединение (III) также изображен на Схеме 4 и предусматривает превращение спиртовой части в соединении (VII) в уходящую группу, такую как мезилат или тозилат, например, с помощью мезилирования или тозилирования или подобных, с последующим нуклеофильным замещением соответствующим кислородным нуклеофилом, таким как ацетат, с получением сложного эфира. Восстановление или гидролиз сложного эфира дает соединение III. Методики выполнения данной последовательности преобразований легко доступны специалистам в данной области.
Следует заметить, что в некоторых случаях кислота Льюиса не является обязательным компонентом реакции, как в случае, когда используется аллилтрихлорсилан в присутствии аминоспирта или диамина (см. Kinnaird, et al., J. Am. Chem. Soc. 2002, 124, 7920). Стоит также отметить, что реакция протекает в присутствии основания Льюиса, когда используется аллилтрихлорсилан (см. Denmark, et al., J. Am. Chem. Soc., 2001, 123, 9488).
В одном из воплощений стадии (а) способа изобретения стехиометрия компонентов реакции аллилирования в конкретном случае составляет приблизительно:
1,0 эквивалент альдегида;
1,05-1,5 эквиалента кислоты Льюиса и
1,05-1,5 эквивалента аллильного реагента Гриньяра или эквивалента аллильного реагента Гриньяра.
Согласно еще одному из воплощений способа изобретения стехиометрия в реакции аллилирования в конкретном случае составляет приблизительно:
1,0 эквивалент альдегида;
1,05-1,3 эквивалента кислоты Льюиса и
1,05-1,3 эквивалента аллильного реагента Гриньяра или эквивалента аллильного реагента Гриньяра.
Согласно еще одному из воплощений способа изобретения стехиометрия реакции аллилирования в конкретном случае составляет приблизительно:
1,0 эквивалент альдегида;
1,05-1,2 эквиалента кислоты Льюиса и
1,05-1,2 эквивалента аллильного реагента Гриньяра или эквивалента аллильного реагента Гриньяра.
Согласно одному из воплощений способа изобретения концентрация альдегида в дихлорметане конкретно составляет приблизительно 0,05-0,125 мМ.
Согласно еще одному из воплощений способа изобретения концентрация альдегида в дихлорметане составляет в конкретном случае приблизительно 0,075-0,10 мМ.
Согласно еще одному воплощению способа изобретения концентрация альдегида в дихлорметане составляет в конкретном случае приблизительно 0,08-0,09 мМ.
Температура реакции аллилирования в конкретном случае находится в интервале приблизительно от -78°С до приблизительно комнатной температуры, или 25°С.
Время, требуемое для реакции аллилирования, составляет в конкретном случае интервал приблизительно от 12 до приблизительно 24 часов или до тех пор, пока общепринятые аналитические приемы, такие как ТСХ или ВЭЖХ, не покажут, что реакция завершилась.
В общем временные и температурные параметры реакции аллилирования будут зависеть в некоторой степени от реакционной концентрации и стехиометрии. Квалифицированный специалист может легко выбрать параметры реакции так, как это необходимо для оптимизации выходов в реакции, на основе проведения опыта за опытом.
При типичной методике с использованием хиральной кислоты Льюиса, генерируемой на месте, (S,S)-1,2-диамино-1,2-дифенилэтан-бис-толуолсульфонамид растворяют в полярном непротонном растворителе. Полярные непротонные растворители, полезные на первой стадии способа изобретения, включают, например, дихлорметан, хлороформ, 1,1,1-трихлорэтан, 1,1,2-трихлорэтан и подобные. Обычно используют дихлорметан. Смесь хирального вспомогательного агента в растворителе затем охлаждают до 0°С и по каплям добавляют BBr3 со скоростью, достаточной для поддержания температуры реакционной массы при 0°С. Полученную смесь перемешивают при 0°С в течение 10 минут, затем оставляют нагреваться до комнатной температуры, перемешивают в течение дополнительных 40 минут и затем концентрируют в вакууме. Остаток переносят в растворитель, такой как дихлорметан, и повторно концентрируют в вакууме для удаления избытка трибромида бора. Остаток затем растворяют в дихлорметане и полученную смесь охлаждают до 0°С. К данной охлажденной реакционной смеси добавляют аллилметаллический реагент, такой как трибутилстаннан, затем полученную смесь нагревают до температуры окружающей среды и перемешивают в течение приблизительно 1-4 часов. Смесь охлаждают до -78°С и по каплям добавляют альдегид (II), растворенный в дихлорметане. Смесь затем перемешивают в течение дополнительных 12-24 часов. Обычная обработка и очистка дают желаемый продукт.
Альтернатива Стадии (а): Стадии (а-1) и (а-2)
Альтернатива стадии (а) изображена на стадии (а-1) и стадии (а-2) и включает присоединение алленилборонового эфира к альдегиду (II) с получением гомопропаргилового спирта XI с последующим гидрированием.
Стадия (а-1)
Реакция алленилбороновых сложных эфиров с альдегидами и, более конкретно, использование в энантиоселективном синтезе хиральных алленилбороновых эфиров, хорошо известны специалисту в данной области (см. R.Haruta, M.Ishiguro, N.Ikeda, and H.Yamamoto. J. Am. Chem. Soc. 1982, 104, 7667; N.Ikeda, and H.Yamamoto. J. Am. Chem. Soc. 1986, 108, 483; E.J. Corey, C.-M.Yu, and D.-H.Lee. J. Am. Chem. Soc. 1990, 112, 878).
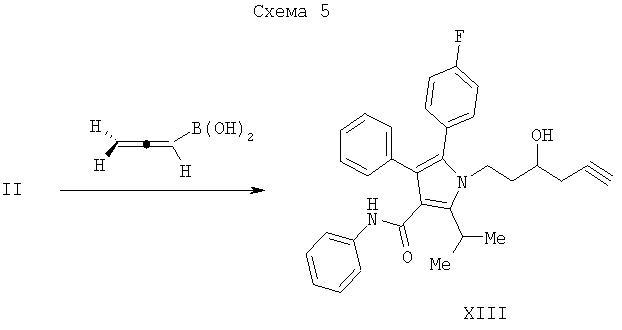
В нехиральном варианте обработка альдегида (II) алленилбороновой кислотой, полученной, как описано N.Ikeda, and H.Yamamoto. J. Am. Chem. Soc. 1986, 108, дает гомопропаргиловый спирт XIII, как изображено на Схеме 5.
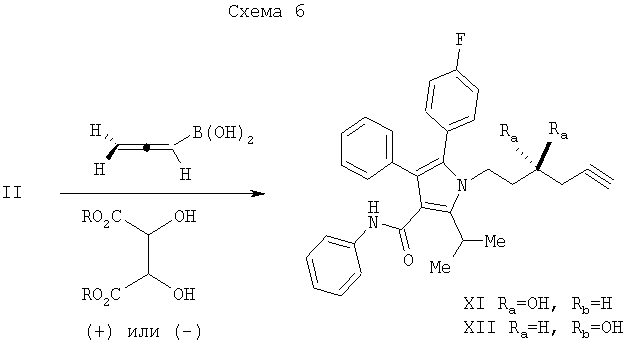
В хиральном варианте, в зависимости от используемого хирального вспомогательного агента, может образовываться или гомопропаргиловая кислота (XI), или (XII), как показано на Схеме 6. Например, как описано R.Haruta, M.Ishiguro, N.Ikeda, and H.Yamamoto. J. Am. Chem. Soc. 1982, 104, 7667; или N.Ikeda and H.Yamamoto. J. Am. Chem. Soc. 1986, 108, 483, присоединение хирального алленилборонового эфира, полученного из алленилбороновой кислоты с использованием (+)-диалкилтартрата, такого как диэтил, диизопропил, дициклопентил, диментил, дициклододецил или ди-2,4-диметил-3-пентилтартрата, дает гомопропаргиловую кислоту XI. Использование (-)-диалкилтартрата дает гомопропаргиловую кислоту XII. Другие варианты подхода известны специалистам в данной области и включают, например, методику, описанную E.J.Corey, C.-M.Yu, and D.-H.Lee. J. Am. Chem. Soc. 1990, 112, 878.
Согласно типичной методике алленилбороновая кислота может быть объединена с (+)-диэтилтартратом в тетрагидрофуране, как описано N.Ikeda and Н.Yamamoto. J. Am. Chem. Soc. 1986, 108. Тетрагидрофуран можно удалять в вакууме, оставляя алленилбороновый сложный эфир, который можно использовать без дальнейшей очистки. Альдегид (II) можно добавлять к раствору алленилборонового эфира в толуоле или подобном при температуре приблизительно от -80 до -10°С. Общепринятая обработка (экстракция диэтиловым эфиром, сушка над сульфатом магния и концентрированно в вакууме) и очистка (хроматография на колонке с силикагелем) дают гомопропаргиловый спирт XI. Аналогичная методика, за исключением использования (-)-диэтилтартрата, будет давать гомопропаргиловый спирт XII.
Стадия (а-2)
Гидрирование гомопропаргилового спирта (XI) дает гомоаллиловый спирт III. Условия проведения гидрирования хорошо известны специалистам в данной области, и процесс может быть осуществлен в гетерогенных условиях или гомогенных условиях. Для данного преобразования обычно используют гетерогенный катализатор, известный как катализатор Линдлара, который представляет собой модифицированный свинцом катализатор палладий-СаСО3 (см. H.Lindlar and R.Dubuis. Org. Synth. 1973, V, 880).
Стадия (b)
На стадии (b) способа изобретения гомоаллиловый спирт (III) превращают в сложный акрилоиловый эфир (IV) по реакции с соединением 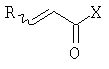 , в котором Х означает Cl, Br, I, или соединения
, в котором Х означает Cl, Br, I, или соединения 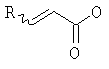 , и R означает Н, (C1-C6)алкил или фенил, или по реакции с эквивалентом активированного акрилоилового сложного эфира в присутствии основания. "Эквивалент активированного акрилоилового сложного эфира" означает акрилоиловый смешанный ангидрид, в котором Х означает стерически затрудненный фрагмент, такой как
, и R означает Н, (C1-C6)алкил или фенил, или по реакции с эквивалентом активированного акрилоилового сложного эфира в присутствии основания. "Эквивалент активированного акрилоилового сложного эфира" означает акрилоиловый смешанный ангидрид, в котором Х означает стерически затрудненный фрагмент, такой как 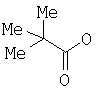 . Данный термин означает также акрилоиловый смешанный ангидрид, получаемый из хлорформиата или из карбонилдиимидазола. Реакция спирта с хлорангидридом кислоты, ангидридом или смешанным ангидридом хорошо известна в данной области (см., например, Junzo Otera, Esterification: Methods, Reactions, and Applications, Wiley-VCH, Weinheim, 2003). Обычно реакция требует использования аминного основания, такого как триэтиламин, диизопропилэтиламин, DBU, или DBN, или подобного, в присутствии каталитического количества 4-(диметиламино)пиридина (DMAP). Данное превращение протекает гладко без защиты амидного азота. Также могут быть использованы альтернативные методики, основанные на использовании карбодиимидных реагентов сочетания.
. Данный термин означает также акрилоиловый смешанный ангидрид, получаемый из хлорформиата или из карбонилдиимидазола. Реакция спирта с хлорангидридом кислоты, ангидридом или смешанным ангидридом хорошо известна в данной области (см., например, Junzo Otera, Esterification: Methods, Reactions, and Applications, Wiley-VCH, Weinheim, 2003). Обычно реакция требует использования аминного основания, такого как триэтиламин, диизопропилэтиламин, DBU, или DBN, или подобного, в присутствии каталитического количества 4-(диметиламино)пиридина (DMAP). Данное превращение протекает гладко без защиты амидного азота. Также могут быть использованы альтернативные методики, основанные на использовании карбодиимидных реагентов сочетания.
Согласно одному воплощению способа изобретения стехиометрия реакционных компонентов в реакции сложной этерификации составляет в конкретном случае приблизительно:
1,0 эквивалент гомоаллилового спирта;
1,05-1,5 эквивалента акрилоилхлорида;
1,05-1,5 эквивалента аминного основания и
0,1-0,5 эквивалента DMAP.
Согласно еще одному воплощению способа изобретения стехиометрия реакции в конкретном случае составляет приблизительно:
1,0 эквивалент гомоаллилового спирта;
1,1-1,4 эквивалента акрилоилхлорида;
1,1-1,4 эквивалента аминного основания и
0,15-0,4 эквивалента DMAP.
Согласно еще одному воплощению способа изобретения стехиометрия реакции в конкретном случае составляет приблизительно:
1,0 эквивалент гомоаллилового спирта;
1,15-1,3 эквивалента акрилоилхлорида;
1,15-1,3 эквивалента аминного основания и
0,2-0,3 эквивалента DMAP.
Согласно одному воплощению способа изобретения концентрация акрилатного сложного эфира в дихлорметане в конкретном случае составляет приблизительно 0,01-0,05 мМ.
Согласно еще одному из воплощений способа изобретения концентрация акрилатного сложного эфира в дихлорметане в конкретном случае составляет приблизительно 0,015-0,045 мМ.
Согласно еще одному воплощению способа изобретения концентрация альдегида в дихлорметане в конкретном случае составляет приблизительно 0,02-0,04 мМ.
Температура реакции этерификации в конкретном случае находится в интервале приблизительно комнатной температуры или приблизительно от -5°С до приблизительно 20°С.
Время, требуемое для реакции, составляет в конкретном случае интервал приблизительно от 4 до приблизительно 24 часов, или до тех пор, пока общепринятые аналитические приемы, такие как ТСХ или ВЭЖХ, не покажут, что реакция завершилась.
В общем временные и температурные параметры реакции зависят в некоторой степени от реакционной концентрации и стехиометрии. Квалифицированный специалист может легко выбрать параметры реакции так, как это необходимо для оптимизации реакционных выходов, на основе проведения опыта за опытом.
В типичной методике фениламид 5-(4-фторфенил)-1-(3-гидроксигекс-5-енил)-2-изопропил-4-фенил-1Н-пиррол-3-карбоновой кислоты (III) растворяют в полярном непротонном растворителе, таком как дихлорметан. Реакционную смесь охлаждают до -5°С и добавляют аминное основание, такое как триэтиламин, наряду с каталитическим количеством 4-(диметиламино)пиридина (DMAP). К данной охлажденной реакционной смеси медленно добавляют акрилоилхлорид, растворенный в дихлорметане. При необходимости может быть дополнительно добавлен триэтиламин и/или DMAP для доведения реакции до конца. Реакционную смесь гасят, обрабатывают и очищают в общепринятых условиях с получением соединения IV.
Стадия (с)
На стадии (с) способа изобретения акрилоиловый сложный эфир (IV) подвергается реакции замыкания кольца в присутствии гомогенного металлорганического катализатора с получением 5,6-дигидропиран-2-она IV. Для выполнения реакций замыкания кольца доступным является ряд металлических катализаторов, включающих, например, промышленно доступный дихлорид бис(трициклогексилфосфин)бензилиденрутения (IV) А ("катализатор Груббса") в присутствии или отсутствии Ti(O-iPr)4 (G.C.Fu and R.H.Grubbs, J. Am. Chem. Soc., 1992, 114, 5426; см. также A.K.Ghosh and H.Lei, J. Org. Chem., 2000, 65, 4779 и ссылки, цитированные там; Grubbs, R.H. and Chang, S., Tetrahedron Lett., 1998, 54, 4413; Cossy, J., Pradaux, F. and BouzBouz, S., Org. Lett., 2001, 3, 2233; Held, С., Frohlich, R. and Metz, P., Ang. Chem. Int. Ed. Eng., 2001, 40, 1058; Reddy, M.V., Rearick, J.P., Hoch, N. and Ramachandran, P.V., Org. Lett., 2001, 3, 19; P.V.Ramachandran, M.V.Reddy and H.C.Brown, J. Indian. Chem. Soc., 1999, 76, 739; Greer Р.В. and Donaldson W.A. Tetrahedron Lett., 2000, 41, 3801; Ghosh A. and Wang Y. Tetrahedron Lett., 2000, 41, 2319; Ghosh A. and Bilcer G. Tetrahedron Lett., 2000, 41, 1003; Ramachandran P.V., Reddy M.V. and Brown H.C., Ghosh A. and Wang Y. Tetrahedron Lett., 2000, 41, 583; Ghosh A. and Liu С. Chem. Commun., 1999, 1743; Ghosh А.К., Capiello J. and Shin D. Ghosh A. and Wang Y. Tetrahedron Lett., 1998, 39, 4651; Reddy M.V., Yucel A., Ramachandran P.V. J. Org. Chem., 2001, 66, 2512).
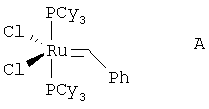
Альтернативным катализатором для использования в реакции способа изобретения является В.
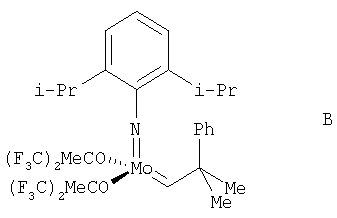
(см., например, Schrock R.R., Murdzek J.S., Bazan G.C., Robbins J., DiMare M. and O'Regan M.B. J. Am. Chem. Soc. 1990, 112, 3875; Bazan С., Khosravi E., Schrock R.R., Feast W.J., Gibson V.C., O'Regan M.B., Thomas J.K., Davis W.M. J. Am. Chem. Soc., 1990, 112, 8378; Bazan С., Oskam J.H., Cho H.N., Park L.Y., Schrock R.R. J. Am. Chem. Soc., 1991, 113, 6899).
Дополнительным альтернативным реакционным подходом является генерирование катализатора на месте, как предложено Morgan J.P. and Grabbs R.H. Org. Lett., 2000, 2, 3153; Huang J., Stevens E.D., Nolan S.P., Petersen J.L. J. Am. Chem. Soc., 1999, 121, 2674; Furstner A., Thiel O., Ackerman L., Schanz H.-J. and Nolan S.P. J. Org. Chem., 2000, 65, 2204. Такие катализаторы включают, например,
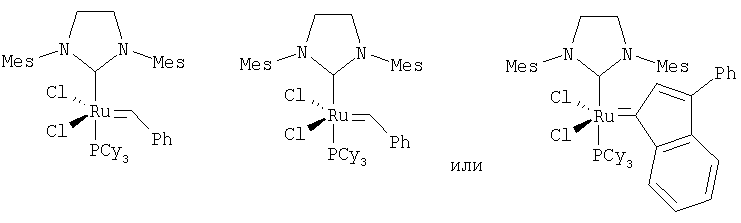
или подобные.
Согласно одному воплощению способа изобретения стехиометрия реакционных компонентов в конкретном случае составляет приблизительно:
1,0 эквивалент акрилатного эфира и
0,025-0,075 эквивалента катализатора.
Согласно еще одному воплощению способа изобретения стехиометрия реакции в конкретном случае составляет приблизительно:
1,0 эквивалент акрилатного эфира и
0,04-0,06 эквивалента катализатора.
Согласно еще одному воплощению способа изобретения стехиометрия реакции в конкретном случае составляет приблизительно:
1,0 эквивалент акрилатного эфира и
0,045-0,55 эквивалента катализатора.
Согласно одному воплощению способа изобретения концентрация акрилатного сложного эфира в дихлорметане в конкретном случае составляет приблизительно 0,05-0,125 мМ.
Согласно еще одному из воплощений способа изобретения концентрация акрилатного сложного эфира в дихлорметане в конкретном случае составляет приблизительно 0,08-0,11 мМ.
Согласно еще одному воплощению способа изобретения концентрация акрилатного сложного эфира в дихлорметане в конкретном случае составляет приблизительно 0,09-0,10 мМ.
Температура реакции в конкретном случае находится в интервале приблизительно от 25°С до приблизительно 50°С.
Время, требуемое для реакции, составляет в конкретном случае интервал приблизительно от 4 до приблизительно 24 часов или до тех пор, пока общепринятые аналитические приемы, такие как ТСХ или ГХ, не покажут, что реакция завершилась.
В общем временные и температурные параметры реакции зависят в некоторой степени от реакционной концентрации и стехиометрии. Квалифицированный специалист в данной области может легко выбрать реакционные параметры так, как это необходимо для оптимизации реакционных выходов, на основе проведения опыта за опытом.
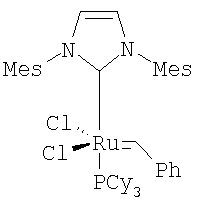
В типичной методике соединение (IV) растворяют в дихлорметане. Смесь дегазируют в вакууме, затем продувают азотом. Смесь затем нагревают до кипения с обратным холодильником, добавляют катализатор Груббса 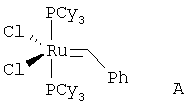 (CAS #1246047-72-3) в дегазированном дихлорметане. Смесь оставляют перемешиваться при кипении с обратным холодильником в течение приблизительно 12-24 часов. Обработка и очистка с помощью общепринятых методик дают соединение V.
(CAS #1246047-72-3) в дегазированном дихлорметане. Смесь оставляют перемешиваться при кипении с обратным холодильником в течение приблизительно 12-24 часов. Обработка и очистка с помощью общепринятых методик дают соединение V.
Выгоды от подхода к получению соединения (V) с помощью данной реакции замыкания кольца, особенно когда используется гомогенный катализатор, включают следующее:
- Необходимость меньших количеств катализатора вследствие обычно многократных оборотов гомогенных катализаторов, увеличивающих эффективность и снижающих общую стоимость превращения;
- Возможность проведения реакции в промышленном масштабе в минимальном количестве растворителя, снижая таким образом расходы на переработку отходов и нагрузку на окружающую среду;
- Возможность проведения реакции при комнатной температуре и атмосферном давлении, снижая таким образом потребность в использовании специализированного оборудования, работающего при высоком давлении в промышленном масштабе, и упрощая методики обработки, и
- Общее снижение числа стадий синтеза, необходимых для стереоселективного получения соединения.
Стадии (d)
Стадии (d) и (е) способа изобретения раскрыты в патенте США 6476235 (соответствующем заявке USSN 10/015558, выдан 22 июля 2002 г.).
В соответствии с методикой, раскрытой в патенте США 6476235, соединение формулы (12)
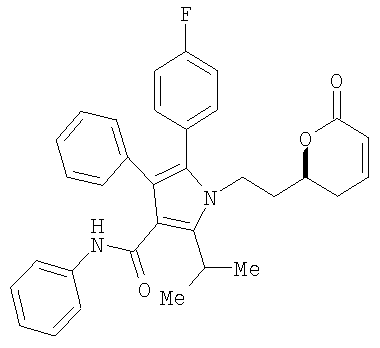
реагирует с соединением НО-М в спирте формулы (17) НОСН2-Арил или (17а) НО-Аллил, где М представляет натрий, литий, калий, цинк, магний, медь, кальций или алюминий, или с соединением формулы (16) МОСН2-Арил или соединением (16а) МО-Аллил, где М принимает значения, указанные выше, необязательно в сорастворителе, таком как, например, нуклеофильный растворитель, например, ацетон, тетрагидрофуран, 1,2-диметоксиэтан и подобные, с последующим добавлением водорода в присутствии катализатора, такого как, например, Pd(OH)2/C, Pd/C, PD/Al2O3 и подобные, в присутствии кислоты, такой как, например, хлористоводородная кислота, бромистоводородная кислота, серная кислота и подобные, с получением соединения формулы (13)
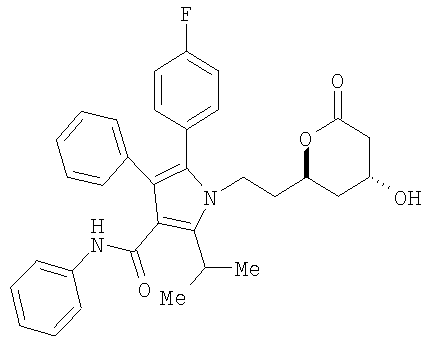
которое является соединением формулы (I) настоящего изобретения и является удобным предшественником аторвастатина.
Предпочтительно реакцию проводят с гидрохлоридом натрия в бензиловом спирте с последующим гидрированием в присутствии Pd(OH)2/C и серной кислоты.
ПРИМЕРЫ
Следующие примеры предназначены для иллюстрации различных воплощений изобретения и никоим образом не для ограничения его объема.
ПРИМЕР 1
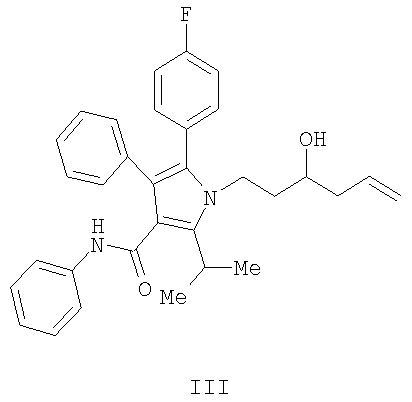
Получение фениламида 5-(4-фторфенил)-1-(3-гидроксигекс-5-енил)-2-изопропил-4-фенил-1Н-пиррол-3-карбоновой кислоты (III)
В колбу загружали 1,25 г (2,4 ммол., 1,14 экв.) (S,S)-1,2-диамино-1,2-дифенилэтан-бис-толуолсульфонамида и затем 20 мл СН2Cl2. Полученную смесь охлаждали до 0°С и по каплям добавляли 2,0 мл (2,33 ммол., 1,1 экв.) BBr3. Реакционную смесь перемешивали при 0°С в течение 10 минут, затем оставляли нагреваться до температуры окружающей среды и перемешивали в течение дополнительных 40 минут. Реакционную смесь концентрировали в вакууме, переносили в 8 мл CH2Cl2 и концентрировали в вакууме. К реакционной смеси снова добавляли 20 мл CH2Cl2 и полученный раствор охлаждали до 0°С. К охлажденной реакционной смеси добавляли 0,75 мл (2,31 ммол., 1,1 экв.) аллилтрибутилстаннана, затем смесь нагревали до температуры окружающей среды и перемешивали в течение двух часов. Реакционную смесь охлаждали до -78°С и по каплям добавляли 0,96 г (2,1 ммол., 1,0 экв.) альдегида (II), растворенного в 2,5 мл CH2Cl2. Спустя три часа добавляли дополнительно 0,5 г альдегида, растворенного в 2,5 мл CH2Cl2, и смесь перемешивали на протяжении ночи. Реакцию гасили добавлением 10 мл фосфатного буфера с рН 6,2. Органический слой промывали 10 мл насыщенного водного хлорида натрия и затем концентрировали. Полученную смесь растворяли в 10 мл СН2Cl2 и разбавляли 40 мл гептана. Хиральный вспомогательный диамин выделяли с выходом 97%. Фильтрат перемешивали с 20 мл 33% водного KF для удаления солей олова. Органический слой сушили над сульфатом магния и концентрировали с последующим растворением в 50 мл этилацетата, фильтрованием и снова концентрированием. Данную процедуру повторяли с дополнительными 12 мл этилацетата и окончательным концентрированием, получая 0,98 г (выход 95%) масла.
ЖХ-МС API-ES отрицательная ионизация М 496.3 и М-1 495.3; ЖХ-МС API-ES положительная ионизация М 496.3 и М+1 497.3.
ПРИМЕР 2
Получение 1-{2-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]этил}бут-3-енилового эфира акриловой кислоты (IV)
В колбу загружали 0,98 г (1,98 ммол., 1 экв.) фениламида 5- (4-фторфенил)-1-(3-гидроксигекс-5-енил)-2-изопропил-4-фенил-1H-пиррол-3-карбоновой кислоты (III) и 10 мл CH2Cl2. Реакционную смесь охлаждали до -5°С и добавляли 0,33 мл (2,38 ммол., 1,240 экв.) триэтиламина и 0,048 г (0,396 ммол., 0,2 экв.) 4-(диметиламино)пиридина. К охлажденной реакционной смеси медленно добавляли 0,19 мл (2,38 ммол., 1,2 экв.) акрилоилхлорида, растворенного в 10 мл CH2Cl2. К реакционной смеси добавляли дополнительно 0,33 мл триэтиламина и 0,048 г 4-(диметиламино)пиридина с последующим добавлением 0,19 мл акрилоилхлорида, растворенного в 3 мл СН2Cl2. Реакцию гасили 20 мл водного NaHCO3. Органический слой промывали 20 мл водного NaHCO3, затем насыщенным водным NaCl, сушили над MgSO4 и концентрировали в вакууме, получая 0,9 г (88% выход) соединения (IV) в виде оранжевого твердого вещества.
ВЭЖХ время удерживания 17,0 минут, длина волны 254 нм. Ацетонитрил:вода в./0,1% муравьиная кислота 60:40 (0-5 мин), 100:0 (15-22 мин), 60:40 (25 мин), YMC ODS-AQ S5; 120 A; 4,6×250 мм; объемный расход 1 мл/мин и температура колонки 30°С.
ПРИМЕР 3
Получение фениламида 5-(4-фторфенил)-2-иэопропил-1-[2-(6-оксо-3,6-дигидро-2Н-пиран-2-ил)этил]-4-фенил-1Н-пиррол-3-карбоновой кислоты (V)
В колбу загружали 0,9 г (0,8 ммол., 1 экв.) акрилатного сложного эфира в 45 мл CH2Cl2. Смесь дегазировали один раз в вакууме с последующей продувкой азотом. Реакционную смесь нагревали до кипения с обратным холодильником. К реакционной смеси добавляли 0,035 г (0,04 ммол., 0,05 экв.) катализатора Груббса (CAS #1246047-72-3) в 5 мл дегазированного растворителя. Реакционную смесь оставляли перемешиваться при кипении с обратным холодильником в течение 19 часов. Смесь концентрировали и подвергали флэш-хроматографии на силикагеле, элюируя градиентом смеси от 10% этилацетат/гептан до 40% этилацетат/гептан. После конденсации подходящей фракции выделяли 0,3 г белого твердого вещества (72% выход).
ВЭЖХ время удерживания 13,3 минуты, длина волны 254 нм. Ацетонитрил:вода в./0,1% муравьиная кислота 60:40 (0-5 мин), 100:0 (15-22 мин), 60:40 (25 мин), YMC ODS-AQ S5; 120 A; 4,6×250 мм; объемный расход 1 мл/мин и температура колонки 30°С.
Анализ хиральной ВЭЖХ гексан:изопропанол 90:10 Chirapak AD; 4,6×250 мм; объемный расход 1 мл/мин и температура колонки 30°С.
(S) время удерживания 16,6 мин.
(R) время удерживания 19,1 мин.
Соотношение 97,22:2,78
94,4% энантиомерный избыток.
На все патенты и патентные документы включены в данное описание ссылки, как если бы каждая была включена индивидуально. Изобретение описано со ссылкой на различные, конкретные и предпочтительные воплощения и технологические приемы. Однако следует понимать, что могут быть произведены многие варианты и модификации, сохраняя при этом сущность и объем изобретения.
Изобретение относится к новому способу получения фениламида 5-(4-фторфенил)-1-[2-((2R,4R)-4-гидрокси-6-оксотетрагидропиран-2-ил)этил]-2-изопропил-4-фенил-1Н-пиррол-3-карбоновой кислоты (I)
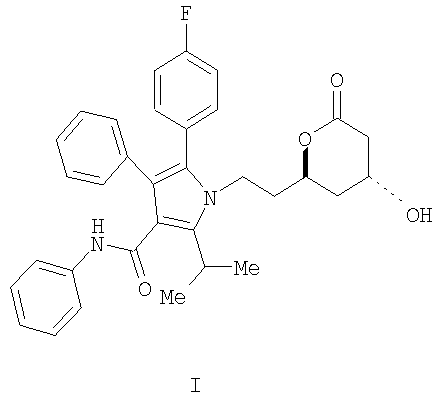
ключевого промежуточного соединения в синтезе аторвастатин кальция, который является гиполипидемическим и/или гипохолестеринемическим агентом. Изобретение также относится к способам получения промежуточного продукта и к новым промежуточным продуктам. 7 н. и 4 з.п. ф-лы.
включающий
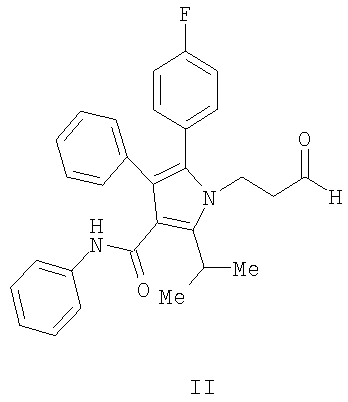
(а) контактирование в растворителе необязательно в присутствии хиральной кислоты Льюиса соединения формулы (II)
с соединением  , в котором М означает SiCl3, SiMe3,
, в котором М означает SiCl3, SiMe3,
B(OH)2, CuLi, MgBr, ZnBr, InBr, SnR3, где R3 означает (C1-C6)алкил, с получением соединения формулы (III)
 ;
;
(b) превращение соединения формулы (III), представленной выше, в сложный акрилоиловый эфир формулы (IV)
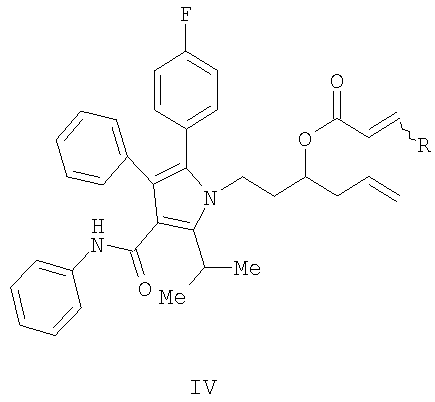
в присутствии основания с использованием соединения 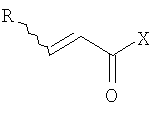 , в котором X означает Cl, Br, I, или
, в котором X означает Cl, Br, I, или 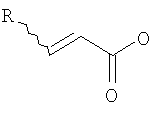 , и R означает Н, (С1-С6)алкил или фенил, или эквивалент активированного акрилоилового сложного эфира;
, и R означает Н, (С1-С6)алкил или фенил, или эквивалент активированного акрилоилового сложного эфира;
(с) контактирование в растворителе сложного акрилоилового эфира формулы (IV), представленной выше, с гомогенным металлорганическим катализатором с получением 5,6-дигидропиран-2-она формулы (V)
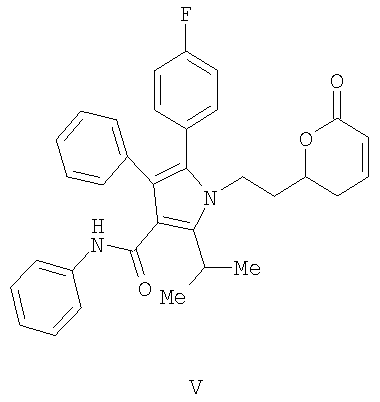
(d) превращение соединения формулы (V), представленной выше, в соединение формулы (VI)
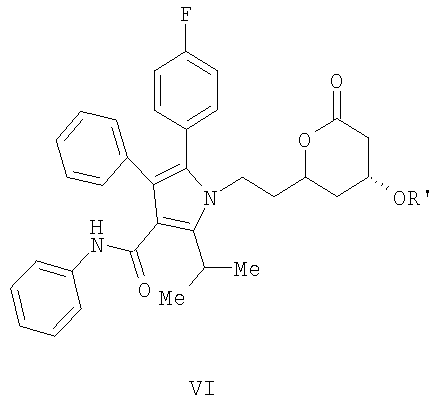
где R' означает бензил, аллил, с помощью селективного
1,4-присоединения аллилового или бензилового спирта с одной стороны плоскости молекулы; и
(е) удаление аллильной или бензильной части в соединении формулы (VI) с помощью гидрогенолиза с получением соединения формулы I.
соединение представляет собой аллил-три-н-бутилстаннан, аллилтриметилсилан, аллилтрихлорсилан, аллилмагнийбромид или аллилцинкбромид, необязательно используемый в присутствии аминоспирта, или диамина, или основания Льюиса.
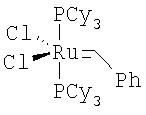 ,
, 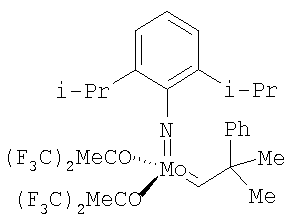 ,
, 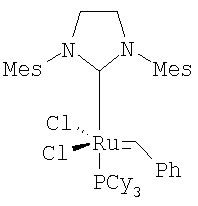 ,
,
 ,
, 
или бензилиден[1,3-бис(2,4,6-триметилфенил)-2-имидазолидинилиден]дихлор(трициклогексилфосфин)рутений.
включающий
(а) контактирование в растворителе необязательно в присутствии хиральной кислоты Льюиса соединения формулы (II)
с соединением  , в котором М означает SiCl3, SiMe3, B(OH)2, CuLi, MgBr, ZnBr, InBr, SnR3, где R3 означает (С1-С6)алкил, с получением соединения формулы (VII)
, в котором М означает SiCl3, SiMe3, B(OH)2, CuLi, MgBr, ZnBr, InBr, SnR3, где R3 означает (С1-С6)алкил, с получением соединения формулы (VII)
(b) превращение соединения формулы (VII), приведенной выше, с сопутствующей стереохимической инверсией центра гомоаллилового спирта в сложный акрилоиловый эфир формулы (IV)
по реакции Мицунобу в присутствии акриловой кислоты или аналога
акриловой кислоты формулы 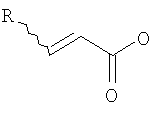 , в которой R означает Н, (С1-С6)алкил или фенил, в присутствии основания;
, в которой R означает Н, (С1-С6)алкил или фенил, в присутствии основания;
(с) контактирование в растворителе сложного акрилоилового эфира формулы (IV), приведенной выше, с гомогенным металлорганическим катализатором с получением 5,6-дигидропиран-2-она формулы (V)
 ;
;
(d) превращение соединения формулы (V), приведенной выше, в соединение формулы (VI)
где R' означает бензил, аллил, с помощью селективного 1,4-присоединения аллилового или бензилового спирта с одной стороны плоскости молекулы; и
(е) удаление аллильной или бензильной части в соединении формулы (VI) с помощью гидрогенолиза с получением соединения формулы I.
включающий
(а) контактирование в растворителе необязательно в присутствии нехиральной кислоты Льюиса соединения формулы (II)
с соединением , где М представляет SiCl3, SiMe3, В(ОН)2, CuLi, MgBr, ZnBr, InBr, SnR3,
где R3 означает (C1-C6) алкил, с получением соединения формулы (VIII)
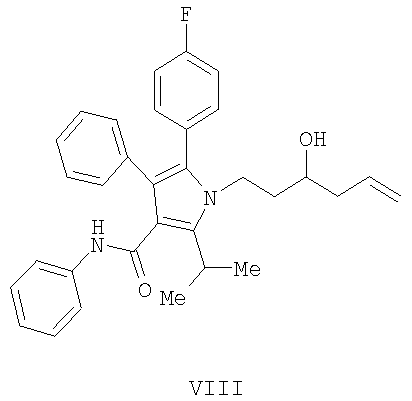
с последующим выделением соединения формулы (III) с помощью хроматографического разделения или расщепления.
включающий
(а) контактирование соединения формулы (II)
с алленилбороновым сложным эфиром в присутствии хирального вспомогательного агента с получением соединения формулы (XI)
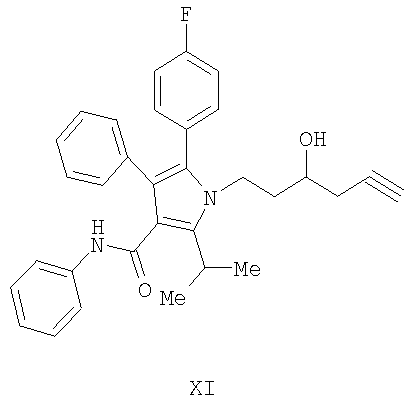
(b) гидрирование соединения формулы (XI), приведенной выше, с получением соединения III.
включающий
(а) контактирование соединения (II)
с алленилбороновым сложным эфиром в присутствии хирального вспомогательного агента с получением соединения формулы (XII)
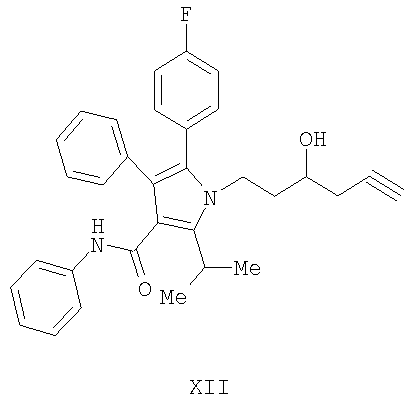
(b) гидрирование соединения формулы (XII), приведенной выше, с получением соединения формулы (VII)
(с) превращение соединения формулы (VII), представленной выше, с сопутствующей стереохимической инверсией центра гомоаллилового спирта в сложный акрилоиловый эфир формулы (IV)
по реакции Мицунобу в присутствии акриловой кислоты или аналога акриловой кислоты формулы 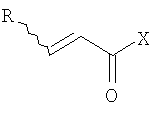 , в которой R означает Н, (С1-С6)алкил или фенил, в присутствии основания;
, в которой R означает Н, (С1-С6)алкил или фенил, в присутствии основания;
(d) контактирование в растворителе сложного акрилоилового эфира формулы (IV), приведенной выше, с гомогенным металлорганическим катализатором с получением 5,6-дигидропиран-2-она формулы (V)
(е) превращение соединения формулы (V), представленной выше, в соединение формулы (VI)
где R' означает бензил, аллил, с помощью селективного
1,4-присоединения аллилового или бензилового спирта с одной стороны плоскости молекулы; и
(f) удаление аллильной или бензильной части в соединении формулы (VI) с помощью гидрогенолиза с получением соединения формулы I.
включающий
(а) контактирование соединения (II)
с алленилбороновой кислотой или алленилбороновым сложным эфиром с получением соединения формулы (XIII)
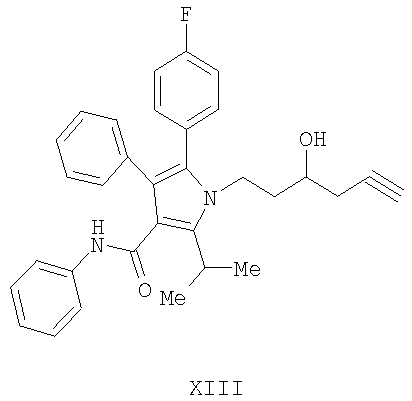
(b) гидрирование соединения формулы (XIII) с получением
соединения формулы (VIII), с последующим выделением соединения формулы (III) с помощью хроматографического разделения или расщепления.
где R представляет Н, (С1-С6)алкил или фенил.
| US 6476235 В2, 05.11.2002 | |||
| 1-[W-(N,N-ЗАМЕЩЕННЫЕ АМИНО) АЛКИЛ]-2-(2*991-АЦИЛЭТЕНИЛ) ПИРРОЛЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ОБЛАДАЮЩИЕ АНТИАРИТМИЧЕСКОЙ И ПРОТИВОИШЕМИЧЕСКОЙ АКТИВНОСТЬЮ | 1993 |
|
RU2088573C1 |
| RU 97117590 А, 10.07.1999 | |||
| RU 95119850 А, 20.09.1997 | |||
| RU 2000103212 А, 27.03.2002. | |||

