Настоящее изобретение касается способа получения оксирана посредством взаимодействия олефина с пероксидом водорода в присутствии катализатора и разбавителя. Более конкретно изобретение касается способа получения 1,2-эпоксипропана (или оксида пропилена) посредством взаимодействия пропилена с пероксидом водорода.
Известен способ получения оксида пропилена путем эпоксидирования пропилена с помощью пероксида водорода в присутствии катализатора типа TS-1, как описано, например, в заявке на патент ЕР 0230949.
Применяемый пероксид водорода, обычно, хорошо очищен от органических примесей. Таким образом, исходные растворы пероксида водорода (Н2О2), полученные в результате экстрагирования из смеси, полученной путем окисления по меньшей мере одного алкилантрагидрохинона, обычно проходят одну или несколько стадий промывки, экстрагирования и/или дистилляции перед продажей и/или применением в способах синтеза. Это относится, в частности, к растворам Н2О2, используемым для получения оксиранов.
Заявка на патент ЕР 549013 касается объединенного способа окисления органических веществ и получения Н2O2 способом с использованием алкилантрахинонов (АО), в котором в качестве растворителя, экстрагирующего H2O2 из хинонового улавливателя, применяют смесь вода/спирт, используемую при окислении органического вещества. Заявитель констатировал, что указанный способ имеет ряд недостатков:
- недостаточная гибкость всего способа в целом, связанная с тем, что каждая стадия синтеза (АО и окисление) зависит от другой стадии;
- ограничение содержания спирта с смесях вода/спирт, обусловленное условиями экстрагирования, которое отрицательно сказывается на выходе H2O2 в способе реакции эпоксидирования;
- трудности, возникающие при разделении фаз в способе экстрагирования смесью вода/спирт;
- прохождение значительного количества метанола в хиноновый улавливатель, в результате чего, учитывая низкую температуру вспышки метанола, возникает реальная опасность взрыва парообразной фазы на стадии окисления при синтезе H2O2;
- значительное экстрагированное количество хинонов в смеси вода/спирт, которое снижает рентабельность промышленной установки; и
- загрязнение хинонового улавливателя побочными продуктами реакции окисления.
Кроме того, в известных реакциях эпоксидирования обычно применяют пропилен, имеющий относительно высокую степень чистоты для того, в частности, чтобы избежать побочных реакций окисления примесей, главным образом, в целях повышения эффективности и безопасности. Действительно, пропан является основной примесью пропилена, и в патенте BE 1001884 указано, что пероксид водорода может окислять алкан в присутствии TS-1.
Кроме того, продуктом окисления пропана является изопропанол. Ознакомившись с патентом BE 1001884, специалист в данной области может прийти к выводу, что в непрерывном способе получения оксида пропилена с рециркуляцией органического разбавителя, используемого в реакции (обычно, метанола), и/или в непрерывном или периодическом способе с использованием источника пропилена, богатого пропаном, изопропанол аккумулируется в растворителе и, наконец, превращается в ацетон, который трудно отделить от указанного разбавителя. В присутствии пероксида водорода этот ацетон может образовывать взрывоопасные и, кроме того, нерастворимые в органической среде пероксиды, что еще больше увеличивает опасность взрыва в результате их осаждения. Такой вывод может быть сделан применительно к любому окисленному алкану в присутствии пероксидированного соединения и TS-1 и, следовательно, к любому источнику олефина (рециркулируемому или нерециркулируемому), богатому алканом (алканами), который может использоваться в реакции эпоксидирования.
Так, в патентах US 5599955 и 5599956 описано использование в основном очищенного пропилена, т.е. со степенью чистоты по меньшей мере 90% и предпочтительно по меньшей мере 98%, в котором основной примесью является пропан.
Однако различные способы синтеза пропилена (и олефинов в целом) обычно приводят к значительному содержанию пропана (или, более широко, алкана(ов)), которое даже выше, чем содержание пропилена, что предполагает осуществление соответствующих способов разделения и/или очистки. Упомянутые выше патенты US 5599955 и 5599956 иллюстрируют эту проблему.
Кроме того, в различных промышленных способах, в которых используется олефин, осуществляют рециркуляцию не преобразованной фракции последнего, обычно обогащенной алканом(алканами). В таких способах, следовательно, может также потребоваться разделение компонентов перед указанной рециркуляцией. Примерами таких способов могут служить полимеризация олефинов и их эпоксидирование.
Предметом настоящего изобретения является способ получения оксирана, в котором устранен по меньшей мере один из перечисленных выше недостатков и получен более высокий выход, а также улучшенная избирательность по сравнению с той, которую получают при использовании очищенного экстракта.
Таким образом, изобретение касается способа получения оксирана путем взаимодействия олефина с пероксидом водорода в присутствии катализатора и органического разбавителя, в котором пероксид водорода является водным раствором пероксида водорода, экстрагированным с помощью в основном очищенной воды из смеси, полученной в результате окисления по меньшей мере одного алкилантрагидрохинона без последующей обработки промывкой и/или очисткой.
Заявителем действительно было неожиданно установлено, что использование в реакции эпоксидирования раствора Н2О2, полученного экстракцией с помощью воды, а не смеси вода/спирт, позволяет повысить выход указанного H2O2. Кроме того, использование неочищенного экстракта позволяет выиграть в избирательности по сравнению с использованием очищенного экстракта.
Способы получения пероксида водорода с использованием алкилантрахинона(ов) или способы АО хорошо известны и широко описаны в литературе (см., например, "Ullmann Encyclopedia of Industrial Chemistry, Fifth Edition, 1989, том 3, стр. 447-57''). Они заключаются в том, чтобы подвергнуть гидрированию рабочий раствор по меньшей мере одного алкилантрахинона и/или по меньшей мере одного тетрагидроалкилантрахинона в разбавителе для получения одного или нескольких алкилантрагидрохинонов и/или алкилтетрагидроантрагидрохинонов. Рабочий раствор, прошедший стадию гидрирования, затем подвергают окислению кислородом, воздухом или воздухом, обогащенным кислородом, для получения пероксида водорода и повторного образования алкилантрахинонов и/или алкилтетрагидроантрахинонов. Полученный пероксид водорода затем отделяют от рабочего раствора на стадии экстрагирования. Согласно настоящему изобретению отделение происходит с помощью в основном очищенной воды. Полученный на стадии экстрагирования рабочий раствор направляют на стадию гидрирования для возобновления цикла получения пероксида водорода.
Под алкилантрахинонами подразумевают, например, 9,10-антрахиноны, замещенные по меньшей мере одной боковой алкильной цепью, линейной или разветвленной, алифатического типа, содержащей по меньшей мере один атом углерода. Обычно такие алкильные цепи содержат меньше 9 атомов углерода и предпочтительно меньше 6 атомов углерода. Примерами таких алкилантрахинонов являются 2-этилантрахинон, 2-изопропилантрахинон, - 2-втор- и 2-трет-бутилантрахиноны, 1,3-, 2,3-, 1,4- и 2,7-диметилантрахиноны, 2-изо- и 2-трет-амилантрахиноны и смеси указанных хинонов.
Под в основном очищенной водой подразумевают воду, содержащую менее 3 мас.% органических разбавителей, в частности, спирта(ов), предпочтительно менее 0,1%, даже менее 0,001% упомянутых разбавителей. Вода, применяемая для экстрагирования, может, тем не менее, преимущественно содержать неорганические вещества из расчета минимум 0,001 мас.%, предпочтительно 0,005%, даже минимум 0,01%. Содержание неорганических веществ не превышает, тем не менее, 1 мас.%, предпочтительно 0,5%, даже 0,1%. Этими неорганическими веществами преимущественно являются вещества, регулирующие рН, такие как кислоты и, в частности, сильные кислоты, такие как азотная кислота, фосфорная кислота или соли указанных кислот. Этими неорганическими веществами могут также преимущественно являться вещества, стабилизирующие Н2O2, такие как соли щелочных или щелочноземельных металлов и, в частности, натрия, такая как пирофосфат натрия. Экстрагирующий раствор может, таким образом, содержать катионы металлов (таких, как щелочные или щелочноземельные металлы, например натрий) и/или анионы, такие как фосфаты, нитраты... в малых количествах, обычно меньше 10 г/л, но больше 0,01 г/л.
Раствор Н2O2, полученный в результате экстрагирования, или исходный раствор H2O2 содержит обычно меньше 50 мас.% Н2О2, чаще меньше 40% H2O2. Обычно он содержит больше 5 мас.% Н2О2, чаще всего больше 10%, в частности, больше 20%, даже больше 30%. Перед использованием в реакции эпоксидирования он не подвергается дальнейшей обработке промывкой и/или очисткой. Следовательно, он содержит органические примеси (продукты распада хинонового улавливателя) и неорганические примеси (катионы и анионы, введенные с экстрагирующей водой, а также те, которые уже присутствовали в смеси, полученной в результате окисления одного или нескольких алкилантрагидрохинонов). Экстрагированный раствор может таким образом содержать органические примеси, выраженные СОТ (концентрацией всего органического углерода), определяемой в соответствии со стандартом ISO 8245 из расчета по меньшей мере 0,001 г/л, даже по меньшей мере 0,01 г/л или даже по меньшей мере 0,1 г/л, но не больше 10 г/л, даже 1 г/л или даже 0,2 г/л. Он может также содержать катионы металлов (таких как щелочные или щелочноземельные металлы, например натрий) и/или анионы, такие как фосфаты, нитраты... в незначительных количествах, обычно меньших или равных 10 г/л, но больших или равных 0,01 г/л.
Исходный раствор Н2О2 перед его использованием в реакции эпоксидирования может быть разбавлен водой или любым другим жидким растворителем или разбавителем, не оказывающим негативного влияния на реакцию эпоксидирования. Обычно водный раствор, используемый для эпоксидирования, содержит по меньшей мере 5 мас.%, чаще всего по меньшей мере 10 мас.% Н2O2, в частности по меньшей мере 20 мас.%. Он содержит чаще всего максимально 50 мас.% пероксидированного соединения, в частности 40 мас.%.
Оксиран, который может быть получен способом согласно изобретению является органическим соединением, содержащим группу общей формулы
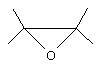
Оксиран обычно содержит от 3 до 10 атомов углерода, предпочтительно от 3 до 6 атомов углерода. Оксиран, который преимущественно может быть получен способом согласно изобретению, является 1,2-эпоксипропаном.
Олефины, которые приемлемы для способа согласно изобретению, обычно содержат от 3 до 10 атомов углерода и предпочтительно от 3 до 6 атомов углерода. Особенно подходят пропилен и бутилен. Пропилен является предпочтительным.
Катализаторы, используемые в способе согласно изобретению, преимущественно содержат цеолит, а именно твердое вещество, содержащее диоксид кремния, имеющий кристаллическую микропористую структуру. Цеолит преимущественно свободен от алюминия. Предпочтительно он содержит титан.
Цеолит, используемый в способе согласно изобретению, может иметь кристаллическую структуру типа ZSM-5, ZSM-11, МСМ-41 или типа бета-цеолита. Хорошо подходят цеолиты типа ZSM-5. Предпочтительными являются те, инфракрасная полоса поглощения которых составляет приблизительно 950-960 см-1.
Особенно подходящими цеолитами являются силикалиты, содержащие титан. Эффективными являются те, которые имеют формулу xTiO2(1-х)SiO2, где значение х составляет от 0,0001 до 0,5, предпочтительно от 0,001 до 0,05. Вещества этого типа, известные под названием TS-1 и имеющие кристаллическую структуру типа ZSM-5, дают особенно хорошие результаты.
Реакционная среда согласно изобретению обычно содержит жидкую фазу и газообразную фазу.
Органические разбавители, используемые в способе согласно изобретению, могут являться органическими производными, такими как алифатические спирты, содержащие от 1 до 4 атомов углерода. В качестве примера можно назвать метанол. Содержание разбавителя в жидкой фазе реакционной среды преимущественно больше 35 мас.%, предпочтительно больше 60%, даже 75%. Содержание разбавителя в жидкой фазе реакционной среды обычно тем не менее меньше 99 мас.%, предпочтительно меньше 95%.
В предпочтительном варианте осуществления способа согласно изобретению оксиран, полученный в реакционной среде, можно отделить жидкофазной экстракцией с помощью такого растворителя, как описан в заявке на патент WO 99/14208 настоящего заявителя.
Способ согласно изобретению может быть непрерывным или периодическим. При непрерывном способе не вступивший в реакцию олефин может быть рециркулирован в реактор.
В реактор, в котором осуществляют способ согласно изобретению, может вводиться раствор, получаемый непосредственно на стадии водной экстракции способа АО. В этом случае установка для осуществления способа согласно изобретению включает в себя также установку для получения раствора Н2О2 способом АО. Такая установка и способ, для осуществления которого она предназначена, являются также предметом настоящего изобретения.
В качестве альтернативы возможно хранение и/или транспортировка раствора перед его введением в реактор, как в случае с очищенными растворами, применяемыми в настоящее время.
В способе согласно изобретению газ, не имеющий негативного влияния на реакцию эпоксидирования, также может вводиться в реактор. Действительно, в заявке на патент WO 99/48883 заявитель констатировал, что введение газообразного соединения в реакционную среду с расходом, достаточным для того, чтобы перемещать полученный оксиран и выводить его из реактора одновременно с газообразным соединением, позволяет сократить продолжительность контакта полученного оксирана с реакционной средой эпоксидирования. Таким образом устраняется возможность образования побочных продуктов и улучшается избирательность эпоксидирования.
В предпочтительном варианте осуществления способа согласно изобретению газообразную фазу вводят в реактор с таким расходом, что он позволяет не только перемещать по меньшей мере часть полученного оксирана, но также способствовать циркуляции жидкой фазы в реакторе, особенно в том случае, если это реактор циркуляционного типа. В этом случае газообразная фаза обычно вводится в реактор в таком количестве, что молярное отношение расхода указанной газообразной фазы к расходу вводимого Н2O2 равно по меньшей мере 5, в частности по меньшей мере 8, при этом обычными являются значения, равные по меньшей мере 10. Молярное отношение таких расходов обычно меньше или равно 100, в частности 60, при этом обычными являются значения, меньшие или равные 40 и даже 20.
В способе согласно изобретению можно использовать реактор любого типа, в частности реактор циркуляционного типа. Хорошо подходят циркуляционные реакторы с барботажным сифоном, в которых циркуляция жидкости, а также, возможно, катализатора достигается барботированием газа в одном из колен. Такой тип реактора описан в вышеупомянутой заявке на патент WO 99/48883.
В способе согласно изобретению следует поддерживать значение рН жидкой фазы во время взаимодействия олефина с Н2O2, равным по меньшей мере 4,8, в частности по меньшей мере 5. Преимущественно значение рН меньше или равно 6,5, в частности 6. Хорошие результаты получают, когда рН равно 4,8-6,5, предпочтительно 5-6. Регулировать рН жидкой фазы во время реакции эпоксидирования можно посредством введения основания. Такое основание может выбираться среди водорастворимых оснований. Речь может идти о сильных основаниях. В качестве примеров можно назвать сильные основания NaOH и КОН. Речь также может идти о слабых основаниях. Слабые основания могут быть неорганическими. В качестве примеров можно назвать слабые неорганические основания NH4OH, Nа2СО3, NаНСО3, Na2HPO4, К2СО3, Li2СО3, КНСО3, LiНСО3, К2НРO4. Слабые основания могут также быть органическими. Приемлемыми слабыми органическими основаниями могут быть соли щелочных и щелочноземельных металлов с карбоновыми кислотами, содержащими предпочтительно от 1 до 10 атомов углерода. В качестве примера можно назвать ацетат натрия. Слабые основания дают хорошие результаты. Предпочтительными являются слабые органические основания. Наиболее предпочтительным является ацетат натрия.
Молярное соотношение между количеством используемого олефина и количеством используемого H2O2 обычно больше или равно 0,1, в частности больше или равно 1 и предпочтительно больше 5. Такое молярное соотношение чаще всего меньше или равно 100, в частности меньше или равно 50 и предпочтительно меньше или равно 25.
В способе согласно изобретению, когда он осуществляется непрерывно и в присутствии цеолита, используемое количество Н2О2 обычно составляет по меньшей мере 0,005 моль в час и на грамм цеолита, в частности по меньшей мере 0,01 моль в час и на грамм цеолита. Обычно количество Н2О2 меньше или равно 2,5 моль в час и на грамм цеолита и, в частности, меньше или равно 1 моль в час и на грамм цеолита. Предпочтительным является количество H2O2, которое больше или равно 0,03 моль в час и на грамм цеолита и меньше или равно 0,1 моль в час и на грамм цеолита.
Взаимодействие между олефином и Н2О2 может осуществляться в присутствии соли, такой как соль металла или соль аммония. Металл может быть выбран среди щелочных или щелочноземельных металлов, таких как литий, натрий, калий, цезий, магний, кальций, стронций и барий. Соли металлов предпочтительно являются галогенидами, оксидами, гидроксидами, карбонатами, сульфатами, фосфатами и солями органических кислот, такими как ацетаты. Галогенидами обычно являются фториды, хлориды, бромиды и йодиды. Предпочтительными являются хлориды. Солью, преимущественно используемой в способе согласно настоящему изобретению, является предпочтительно галогенид щелочного металла и преимущественно хлорид натрия. Используемое количество соли металла выражается как содержание ионов металлов или аммония, происходящих из соли, по отношению к количеству катализатора, выраженному в миллимолях (ммоль) металла или аммония на грамм цеолита. Такое содержание может быть больше или равно 10-4 ммоль/г цеолита и меньше или равно 10 ммоль/г цеолита. Преимущественно содержание соли металла больше или равно 10-3 ммоль/г цеолита и меньше или равно 1 ммоль/г цеолита. Предпочтительным является содержание, которое больше или равно 10-2 ммоль/г цеолита и меньше или равно 0,5 ммоль/г цеолита.
Температура реакции между олефином и Н2О2 преимущественно выше 35°С, что препятствует постепенной дезактивации катализатора. Предпочтительной является температура реакции, выше или равная 40°С и преимущественно выше или равная 45°С. Наиболее предпочтительной является температура выше или равная 50°С. Тем не менее температура реакции обычно ниже 100°С и предпочтительно ниже 80°С. Температура, при которой олефин взаимодействует с H2O2, обычно составляет от 40°С до 100°С и предпочтительно от 45°С до 80°С.
В способе согласно изобретению взаимодействие между олефином и Н2О2 может происходить при атмосферном давлении. Оно может также происходить под давлением. Обычно такое давление не превышает 40 бар. На практике приемлемым является давление 20 бар.
В соответствии с наиболее предпочтительным способом согласно изобретению олефин взаимодействует с пероксидом водорода в присутствии катализатора и органического разбавителя в жидкой фазе в реакторе, в который вводят лероксид водорода и органический разбавитель, а также текучее вещество, содержащее олефин и алкан(ы), содержание которых составляет по меньшей мере 10% объема. Предпочтительно содержание алкана в текучем веществе выше 10% объема.
Этот вариант является предпочтительным, поскольку в нем применяются различные источники олефинов, неочищенных от алканов, для получения оксиранов и поскольку в нем в способе реакции окисления алкана пероксидом водорода неожиданно уменьшается количество получаемого спирта и кетона в присутствии олефина, и это происходит с учетом коэффициента разбавления. Следовательно, опасность осаждения взрывоопасных пероксидов значительно меньше, чем можно было предположить теоретически, и может, таким образом, легко контролироваться в установке промышленного уровня.
Одно из основных преимуществ предпочтительного варианта заключается в том, что в реактор вводят текучее вещество, содержание одного или нескольких алканов в котором составляет по меньшей мере 10% объема. Содержание алкана(ов) в указанном текучем веществе может в некоторых случаях быть по меньшей мере равным 20% объема, даже 30%. Можно также использовать текучие вещества, содержание алкана(ов) в которых составляет по меньшей мере 50% объема. Не рекомендуется, напротив, использовать текучие вещества, содержание алкана(ов) в которых составляет больше 95% объема, и даже предпочтительно использовать текучие вещества, содержание алкана(ов) в которых не превышает 85%.
Содержание олефина в текучем веществе обычно составляет более 50% объема, в частности по меньшей мере 60% объема и предпочтительно по меньшей мере 70% объема. Количество водорода, вводимого в реактор эпоксидирования, чаще всего меньше 5% объема текучего вещества и предпочтительно равно 0%. Количество кислорода, вводимого в реактор эпоксидирования, обычно меньше 10% объема текучего вещества.
Один или несколько алканов, содержащихся в текучем веществе согласно настоящему изобретению, обычно содержат от 3 до 10 атомов углерода и предпочтительно от 3 до 6 атомов углерода. Предпочтительно алкан является линейным и не содержит, в частности, ароматических веществ. В том случае, если олефин согласно изобретению является пропиленом, один или несколько алканов состоят в основном из пропана. Предпочтительно алкан не применяется в качестве органического разбавителя в реакции эпоксидирования и отличается от органического разбавителя.
Способ согласно предпочтительному варианту может быть непрерывным или периодическим. Если он непрерывен, текучее вещество может рециркулироваться в реактор после окончания взаимодействия олефина с пероксидом водорода.
В первом случае осуществления предпочтительного варианта способа согласно изобретению способ осуществляется непрерывно, и текучее вещество, вводимое в реактор в начале процесса, содержит алкан(ы) в количестве, составляющем меньше 10% объема. В процессе осуществления способа текучее вещество рециркулируется в реактор после взаимодействия олефина с пероксидом водорода так, что рециркулируемое текучее вещество постепенно обогащается алканом. Содержание алкана в текучем веществе достигает тогда по меньшей мере 10% объема.
Во втором случае осуществления предпочтительного варианта способа согласно изобретению способ является непрерывным или периодическим, и текучее вещество, вводимое в реактор в начале осуществления способа, уже содержит количество алкана(ов), составляющее по меньшей мере 10% объема.
Предпочтительно текучим веществом (содержащим олефин и один или несколько алканов), вводимым в реактор, является газ. В этом случае особая форма осуществления предпочтительного варианта способа согласно изобретению заключается в том, что расход указанного газа, вводимого в реактор, таков, что позволяет не только перемещать по меньшей мере часть полученного оксирана, но также вызывать циркуляцию жидкой фазы в реакторе, в частности, если последний является реактором циркуляционного типа. В этом случае расход подаваемого газа обычно такой, что молярное отношение расхода указанного газа к расходу вводимого пероксидированного соединения равно по меньшей мере 5, в частности по меньшей мере 8, при этом обычными являются значения, равные 10. Молярное отношение таких расходов обычно меньше или равно 100, в частности 60, при этом обычными являются значения, меньшие или равные 40 и даже 20.
В предпочтительном варианте способа согласно изобретению при его непрерывном осуществлении предпочтительным является количество пероксида водорода, которое больше или равно 0,03 моль в час и на грамм цеолита и меньше или равно 0,25 моль в час и на грамм цеолита.
В предпочтительном варианте способа согласно изобретению водный раствор пероксида водорода чаще всего максимально содержит 70 мас.% пероксидированного соединения, в частности 50 мас.%.
Изобретение касается также другого способа получения оксирана, в соответствии с которым в реакторе в жидкой фазе происходит взаимодействие олефина с пероксидом водорода в присутствии катализатора и органического разбавителя, и в соответствии с которым в реактор вводят пероксид водорода и органический разбавитель, а также текучее вещество, содержащее олефин и по меньшей мере 10% объема алкана(ов).
Этот другой способ согласно изобретению соответствует предпочтительному варианту, описанному выше, когда его осуществляют как таковой, не комбинируя с первым способом согласно изобретению, в котором используют водный раствор пероксида водорода, экстрагированный с помощью в основном очищенной воды из смеси, полученной при окислении по меньшей мере одного алкилантрагидрохинона, без последующей обработки промывкой и/или очисткой.
Условия, в которых может быть осуществлен этот другой способ, идентичны условиям первого способа, за исключением использования исходного раствора пероксида водорода.
Пример 1 (согласно изобретению) и 2С (сравнительный)
В реакторе непрерывного действия, содержащем 5,25 г TS-1, поддерживается 35°С при атмосферном давлении, и в него вводят 0,57 моль Н2О2/час в виде 40 мас.% водного раствора, 475 мл метанола/час, а также 250 IN/I (или 11,2 моль/час) пропилена. Выходящие жидкие и газообразные фазы подвергают анализу с целью определения пропорций различных органических веществ, а также выход Н2О2.
Ниже в таблице показаны результаты опытов, в которых использовали свежий катализатор TS-1, полученный способами, известными из литературы.
Наблюдают известным образом постепенное снижение активности катализатора, на которую не влияет качество используемого Н2О2. Присутствие исходного H2O2 благоприятно влияет только на избирательность.
Следует отметить соответствующее содержание анионов и катионов в этих растворах Н2О2 (таблица):
Примеры 3 (согласно изобретению) и 4С (сравнительный)
В таблице показаны результаты опытов, сходных по всем позициям с описанным в примерах 1 и 2С во время цикла, следующего за восстановлением катализатора. Катализатор восстанавливается в течение 7 часов с помощью проходящего по нему воздуха, нагретого до 300°С.
Подтверждается, что активность осталась идентичной, с учетом погрешностей, допустимых при измерениях, и что отклонение избирательности сохраняется.
Примеры 5С (сравнительный) и 6 (согласно изобретению)
Раствор синтеза Н2O2, полученный после окисления хинонового улавливателя/гидрохинонов, экстрагировали с помощью смеси метанол/вода, в которой содержание метанола составляет 52 мас.%. Этот водный экстракт затем использовали в опыте по эпоксидированию пропилена (пример 5С) и полученные результаты сравнивали с результатами аналогичного опыта, осуществленного с исходным Н2О2 в количестве 40 мас.% в воде, полученной экстракцией из того же улавливателя с помощью в основном очищенной воды (пример 6). Этот улавливатель содержит 11,8 г/кг H2O2.
Экстракция с помощью смеси вода/спирт осуществлялась в 4 стадии. Первая экстракция осуществлялась путем обработки 14331 г улавливателя (содержащего в целом 169,1 г Н2О2) с помощью 1511 г смеси метанол/вода. Фаза метанол-вода имеет большую плотность, чем исходный органический раствор, и относительно быстро осаждается (приблизительно в течение 15 мин), образуя 1085 г экстракта. Концентрация Н2О2 в ней, определяемая йодометрией, составляет 3,18 моль Н2О2/кг, что соответствует 3,45 моль или 117,4 г Н2O2 (=69% от целого).
Вторая экстракция осуществлялась с помощью 1522 г той же смеси метанол/вода. Отделение было менее очевидным. Осаждение происходило очень медленно: потребовалось больше 1 часа для того, чтобы приступить к разделению фаз. В противоположность первой экстракции фаза метанол-вода на этот раз имела меньшую плотность и содержала 1215 г экстракта. Концентрация Н2О2 в ней равна 0,833 моль/кг, что соответствует 1,012 моль или 34,4 г Н2O2. Таким образом, на двух стадиях экстракции было собрано 90% всего Н2О2.
Третья экстракция осуществлялась с помощью 1511 г той же смеси метанол/вода. Отделение было также затруднено, и было собрано 1446 г фазы метанол-вода. Концентрация Н2O2 в ней равна 0,244 моль/кг, что соответствует 0,353 моль или 12,0 г H2O2 (т.е. на 3 стадиях экстракции было собрано 96,9% всего Н2O2).
Наконец, четвертая стадия экстракции осуществлялась с помощью 1517 г той же смеси метанол/вода. Отделение было также затруднено, и было собрано 1497 г фазы метанол-вода. Концентрация Н2О2 в ней равна 0,071 моль/кг, что соответствует 0,106 моль или 3,6 г Н2О2 (т.е. на 4 стадиях экстракции было собрано 99,0% всего H2O2).
Все четыре экстракта затем смешали, получив раствор метанол/вода, содержащий 0,94 моль Н2О2/кг (контроль осуществлялся титрованием). Содержание метанола, определяемое с помощью CPV, составляет около 437 г/кг.
Содержание "полезных" хинонов (= которые могут применяться для получения Н2О2), утраченное в этой фазе, составляет 0,020 г/кг экстракта.
Кроме того, имеется явный переход части метанола в хиноновый улавливатель, как это показывает расхождение между массами используемых смесей метанол/вода и собранных экстрактов (в частности, на 1 и 2 стадиях экстракции). Содержание метанола в хиноновом улавливателе, определяемое с помощью CPV, действительно близко к 6 мас.%.
Опыты по эпоксидированию пропилена (Ре) проводились на установке с барботажным сифоном в следующих условиях: Т: 55°С; расход Ре: 75 л/час при нормальных условиях; H2O2: 0,17 моль Н2О2/час; концентрация H2O2 в контуре при нулевой конверсии: 1,0 моль/кг; катализатор: 0,53 г TS-1.
Что касается примера 5, ввод смеси всех четырех экстрактов метанол/вода, содержащих Н2O2, в установку с барботажным сифоном привел бы после отгонки к образованию среды, "бедной" метанолом (концентрация <440 г/кг). Поэтому было введено дополнительное количество метанола, так чтобы поддерживать его концентрацию в контуре, равную приблизительно 440 г/кг, что соответствует содержанию метанола в исходном H2O2, использованном в ссылочном опыте (пример 6).
Полученные результаты изложены ниже в таблице.
Примеры 7-9
Оксид пропилена был получен в реакторе с барботажным сифоном, таком, как описан в заявке WO 99/48883, взаимодействием пропилена с пероксидом водорода в присутствии метанола и катализатора TS-1, применяемого в виде шариков, диаметром 0,5 мм.
Опыты проводились при температуре 55°С с непрерывной подачей 0,17 моль/час пероксида водорода. Общий расход газа составляет 75 л/час при нормальных условиях (или 3,3 моль/час). Исходная концентрация H2O2 в контуре при отсутствии конверсии составляла 1,5 моль/кг, при этом количество используемого катализатора составляло 4,5 г шариков, содержащих 1,5 г TS-1.
В примере 1 использовали смесь, содержащую 75% пропилена "polymer grade" (98% пропилена и 0,3% пропана) и 25% пропана (молярные %); в примере 2 использовали 100% пропилена "polymer grade" и в примере 3 использовали смесь, содержащую 75% пропилена "polymer grade" и 25% азота.
Полученные результаты указаны ниже в таблице.
Избирательность в отношении оксида пропилена приведена в молярном соотношении, выраженном в процентах, между количеством полученного оксида пропилена, разделенного на сумму всех органических веществ, полученных в опыте из примера С3.
Количество изопропанола, полученное в опыте 1, измеренное через 5 часов, составляет 0,007 ммоль/час. В опытах 2 и 3 следов изопропанола не обнаружено.
Пример 10
Опыт в условиях, идентичных условиям, описанным выше в примерах с 7 по 9, был осуществлен с использованием чистого пропана. Количество полученного изопропанола, измеренное через 5 часов, составляет 0,11 ммоль/час, т.е. имеет коэффициент 16 по отношению к количеству из примера 1. Получено также 0,04 ммоль/час ацетона. Конверсия H2O2 очень незначительна, т.е. составляет 1% через 5 часов.
| название | год | авторы | номер документа |
|---|---|---|---|
| ВОДНЫЙ РАСТВОР ПЕРОКСИДА ВОДОРОДА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ИСПОЛЬЗОВАНИЕ | 2008 |
|
RU2468990C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОКСИРАНА В ПРИСУТСТВИИ КАТАЛИЗАТОРА В ВИДЕ ЧАСТИЦ | 2001 |
|
RU2272032C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОКСИРАНА, ВКЛЮЧАЮЩИЙ ВЫДЕЛЕНИЕ ОКСИРАНА ИЗ РЕАКЦИОННОЙ СМЕСИ | 2001 |
|
RU2259362C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОКСИРАНА ПРИ ПОМОЩИ ПЕРОКСИДНОГО СОЕДИНЕНИЯ | 2001 |
|
RU2256656C2 |
| СПОСОБ ЭПОКСИДИРОВАНИЯ С - С ОЛЕФИНОВ | 1996 |
|
RU2168504C2 |
| ПОЛУЧЕНИЕ СОЕДИНЕНИЙ ОКСИРАНА | 2001 |
|
RU2282625C2 |
| СПОСОБ ЭПОКСИДИРОВАНИЯ ОЛЕФИНА | 1996 |
|
RU2162466C2 |
| СПОСОБ ЭПОКСИДИРОВАНИЯ ОЛЕФИНОВ | 2002 |
|
RU2290399C2 |
| СПОСОБ КАТАЛИТИЧЕСКОГО ЭПОКСИДИРОВАНИЯ ПРОПЕНА | 2002 |
|
RU2303035C2 |
| ПОЛУЧЕНИЕ СОЕДИНЕНИЙ ОКСИРАНА | 2001 |
|
RU2282624C2 |
Использование: получение оксиранов. Сущность: проводят взаимодействие олефина с пероксидом водорода в присутствии катализатора и органического разбавителя. При этом пероксид водорода является водным раствором пероксида водорода, экстрагированным с помощью в основном очищенной воды из смеси, полученной в результате окисления по меньшей мере одного алкилантрагидрохинона без последующей обработки промывкой и/или очисткой. Технический результат: повышение выхода и селективности по оксирану. 3 с. и 14 з.п. ф-лы, 5 табл.
| DE 19623611 А, 18.12.1997 | |||
| ЕР 549013 А1, 30.06.1993 | |||
| Станок для обработки оптических поверхностей деталей | 1977 |
|
SU709339A1 |
| Электромагнитный способ контроляфЕРРОМАгНиТНыХ издЕлий | 1977 |
|
SU819683A1 |
| СПОСОБ ЭПОКСИДИРОВАНИЯ ОЛЕФИНА | 1996 |
|
RU2140415C1 |

