По данной заявке испрашивается приоритет в соответствии с предварительной заявкой на патент США №60/157850, поданной 6 октября 1999 года, которая специально включена сюда в качестве ссылки.
Настоящее изобретение относится к новым бис-тетрагидро-фуранбензодиоксолилсульфонамидным соединениям, содержащим их композициям и способам их получения. Также изобретение относится к применению настоящих соединений в качестве фармацевтически активных соединений для лечения и профилактики ретровирусных инфекций, особенно ВИЧ-инфекций, и в особенности для резистентных ко многим, лекарственным препаратам ВИЧ-инфекций млекопитающих.
Резистентность ВИЧ к ингибиторам является наиболее важной причиной безуспешной терапии. Половина пациентов, получающих комбинированную терапию против ВИЧ, не полностью восприимчивы к лечению главным образом из-за реэистентности вируса к одному или нескольким применяемым препаратам. Боле того, было показано, что резистентный вирус переносится вновь инфицированным лицам, приводя к сильному ограничению выбора терапии и для данных, не получавших ранее препаратов пациентов. Следовательно, существует необходимость в данной области получения новых соединений для лечения ретровирусной инфекции, в особенности для лечения СПИД. Необходимость в данной области является особенно острой относительно соединений, которые активны не только в отношении вируса дикого типа, но также по отношению к все чаще встречающимся резистентным вирусам. Кроме того, ингибиторы протеазы обычно вводятся больным СПИД в сочетании с другими препаратами, активными по отношению к ВИЧ, такими как НИОТ и/или ННИОТ. Данное обстоятельство является причиной высокой лекарственной нагрузки для пациента. Одним из способов снижения данной лекарственной нагрузки является поиск препаратов, активных по отношению к ВИЧ, таких как ингибиторы протеазы с высокой биодоступностью, т.е. благоприятным фармакокинетическим и метаболическим профилем, так чтобы суточная доза могла быть минимальной. Другой важной характеристикой хорошего ингибитора протеазы и, в общем, препаратов, активных по отношению к ВИЧ, является минимальное связывание ингибитора протеазы белками плазмы или даже полное отсутствие влияния на его эффективность.
Несколько опубликованных заявок на выдачу патента относятся к ингибиторам ВИЧ- протеазы. Например, WO 95/06030 относится к ингибиторам протеазы со структурой, содержащей гидроксиэтиламиносульфонамидное ядро. Также Ghosh et al. (Bioorganic & Medicinal Chemistry Letters, 8, 1998, 687-690) раскрывает гидроксиэтиламиносульфонамидные ингибиторы ВИЧ-протеазы.
Соединения согласно настоящему изобретению являются неожиданно эффективными ингибиторами ВИЧ в отношении их активности против широкого спектра мутантов ВИЧ и в отношении их биодоступности. Дополнительные объекты и преимущества будут изложены частично в нижеследующем описании и частично будут понятны из описания или могут быть изучены при практическом применении изобретения. Объекты и преимущества согласно изобретению будут реализованы и достигнуты с помощью элементов и сочетаний, подробно указанных в формуле изобретения.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
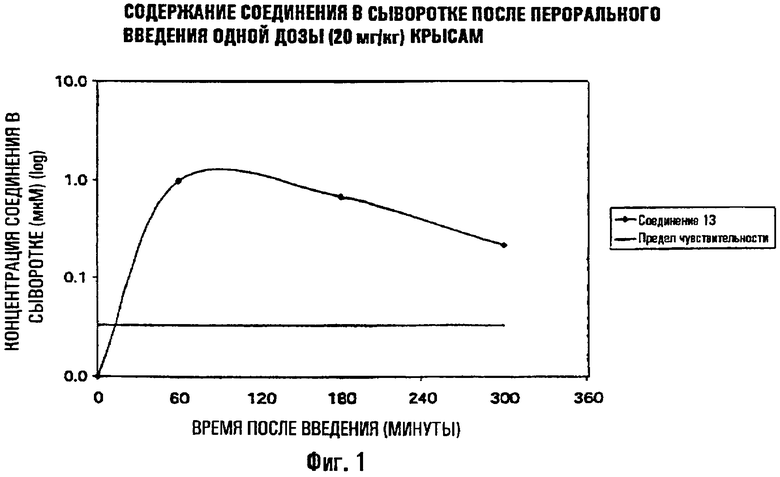
На фиг.1 показана концентрация соединения 13 в сыворотке после однократного введения пероральной дозы как функция времени.
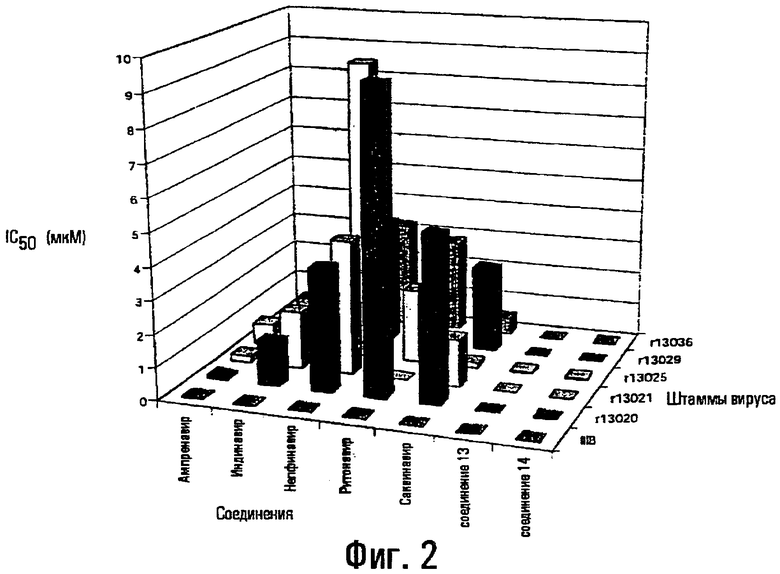
На фиг.2 показано сравнение активности соединений 13 и 14 согласно изобретению и нескольких коммерчески доступных противовирусных соединений по отношению к небольшой панели вирусных штаммов.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Для более полного понимания описанного здесь изобретения приводится нижеследующее подробное описание.
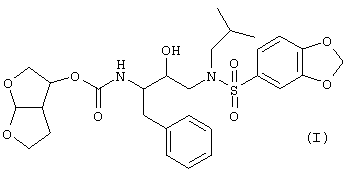
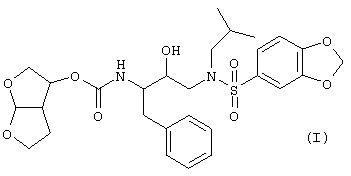
Настоящее изобретение относится к соединениям, представленным формулой
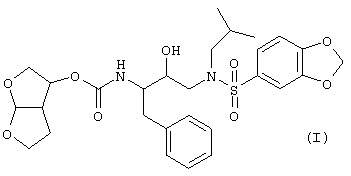
и его N-оксидам, солям, сложным эфирам, стереоизомерным формам, рацемическим смесям, пролекарствам и метаболитам. Молекулярная структура, изображенная выше, называется гексагидрофуро[2,3-b]фуран-3-ил-N-{3-[(1,3-бензодиоксол-5-илсульфонил)(изобутил)амино]-1-бензил-2-гидроксипропил}- карбамат.
Данное изобретение также предусматривает кватернизацию атомов азота настоящих соединений. Основной атом азота может быть кватернизован с помощью любого агента, известного специалисту в данной области, включая, например, низшие алкилгалогениды, диалкилсульфаты, длинноцепочечные галогениды и аралкилгалогениды. С помощью подобной кватернизации могут быть получены водо- или жирорастворимые или дисперсные продукты.
Термин "пролекарство", используемый в данном тексте, означает фармакологически приемлемые производные, такие как сложные эфиры, амиды и фосфаты, такие, что полученный продукт в результате биотрансформации производного представляет собой активное лекарственное вещество, как представленное формулой (I). В настоящее описание включена ссылка Goodman и Gilman (The Pharmacological Basis of Therapeutics, 8th ed, McGraw-Hill, Int. Ed. 1992, "Biotransformation of Drugs", p 13-15), в которой описаны пролекарства в общем смысле. Типичные примеры пролекарств описаны, например, в WO 99/33795, WO 99/33815, WO 99/33793 и WO 99/33792, включенных сюда в качестве ссылки.
Пролекарства характеризуются превосходной растворимостью в воде, повышенной биодоступностью и легко метаболизируются в активные ингибиторы in vivo.
Для применения в терапии приемлемы такие соли соединений формулы (I), в которых противоион является фармацевтически или физиологически приемлемым. Однако также могут применяться соли, содержащие фармацевтически неприемлемый противоион, например, при получении или очистке фармацевтически приемлемого соединения, представленного формулой (I). Все соли, фармацевтически приемлемые или неприемлемые, входят в объем настоящего изобретения.
Фармацевтически приемлемые или физиологически переносимые аддитивные соли, которые способны образовывать соединения согласно настоящему изобретению, могут быть легко получены с помощью подходящих кислот, таких как, например, неорганические кислоты, такие как галогеноводородные кислоты, например хлористоводородная или бромистоводородная кислота, серная, азотная, фосфорная или подобные кислоты; или органические кислоты, такие как, например, уксусная, пропановая, гидроксиуксусная, молочная, пировиноградная, щавелевая, малоновая, янтарная, малеиновая, фумаровая, яблочная, винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламовая, салициловая, п-аминосалициловая, памовая и подобные кислоты.
И наоборот, указанные солевые формы могут быть преобразованы в фирму свободного основания обработкой подходящим основанием.
Термин "соли" также включает в себя гидраты и сольваты, которые могут образоваться соединениями по настоящему изобретению. Примерами таких форм являются, например, гидраты, алкоголяты и тому подобные.
N-оксидные формы согласно настоящему изобретению означают соединения, представленные формулой (I), где один или несколько атомов азота окислены до так называемого N-оксида.
Настоящие соединения также могут существовать в таутомерных формах. Хотя подобные формы прямо не указаны в вышеприведенной формуле, подразумевается, что они включены в объем настоящего изобретения.
Термин "стереохимически изомерные формы соединений согласно настоящему изобретению", применяемый здесь ранее, определяет все возможные соединения, состоящие из тех же атомов, связанных в той же последовательности связями, но имеющие различные пространственные структуры, не являющиеся взаимозаменяемыми, которые соединения согласно настоящему изобретению могут принимать. Если не обозначено или не указано иначе, химическое обозначение соединения включает смесь всех стереохимически возможных изомерных форм, которые указанное соединение может принимать. Указанная смесь может включать все диастереомеры и/или энантиомеры основной молекулярной структуры указанного соединения. Все стереохимически изомерные формы соединений согласно настоящему изобретению как в чистом виде, так и в виде примеси друг к другу, включены в объем настоящего изобретения.
Чистые стереоизомерные формы соединений и промежуточных продуктов, как упоминалось здесь, определяют как изомеры, по существу свободные от других энантиомерных или диастереомерных форм этой же основной молекулярной структуры указанных соединений и промежуточных продуктов. В частности, термин "стереоизомерно чистый" относится к соединениям или промежуточным продуктам, содержащим избыток стереоизомера, по крайней мере, 80% (т.е. минимум 90% одного изомера и максимум 10% других возможных изомеров) до избытка стереоизомера 100% (т.е. 100% одного изомера и отсутствие других), особенно соединениям или промежуточным продуктам, имеющим избыток стереоизомера от 90% до 100%, в особенности имеющим избыток стереоизомера от 94% до 100% и в наибольшей степени имеющим избыток стереоизомера от 97% до 100%. Термины "энантиомерно чистый" и "диастереомерно чистый" следует понимать одинаково, и тогда, соответственно, избыток энантиомера означает избыток стереоизомера в данной смеси.
Чистые стереоизомерные формы соединений и промежуточных продуктов согласно данному изобретению могут быть получены с помощью известных в данной области способов. Например, энантиомеры могут быть отделены друг от друга с помощью селективной кристаллизации их диастереомерных солей с оптически активными кислотами. Альтернативно энантиомеры могут быть разделены с помощью хроматографии, применяя хиральные неподвижные фазы. Указанные чистые стереохимически изомерные формы могут также быть получены из соответствующих чистых стереохимически изомерных форм подходящих исходных веществ, обеспечивая стереоспецифичность проведения реакции. Предпочтительно, если требуется конкретный стереоизомер, указанное соединение будет синтезироваться с помощью стереоспецифических способов получения. В данных способах преимущественно будут применяться энантиомерно чистые исходные вещества.
Специалисту в данной области понятно, что соединения, представленные формулой (I), содержат 5 хиральных центров и, таким образом, существуют в стереоизомерных формах. Данные 5 центров хиральности указаны с помощью номеров со звездочками (*1, *2, *3, *4 и *5) на фигуре ниже.
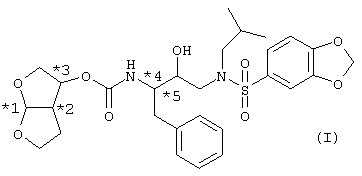
Абсолютная конфигурация каждого центра асимметрии может быть указана с помощью стереохимических обозначений R и S, данная R и S конфигурация соответствует правилам, описанным в Pure Appl. Chem. 1976, 45, 11-30. Предпочтительной конфигурацией бис-тетрагидрофуранового кольца является такая, в которой атом углерода *1 находится в R-конфигурации, атом углерода *2 находится в S-конфигурации и атом углерода *3 находится в R-конфигурации, в которой атом углерода *1 находится в S-конфигурации, атом углерода *2 находится в R-конфигурации и атом углерода *3 находится в S-конфигурации. Атом углерода *4 предпочтительно находится в S-конфигурации и атом углерода *5 находится в R-конфигурации.
Для соединения, представленного основной структурой (I), существуют следующие 32 энантиомерные формы, указанные в таблице A. Хиральные атомы углерода обозначены, как указано на фигуре выше.
Соединения l и w являются предпочтительными энантиомерными чистыми формами, особенно соединение 1.
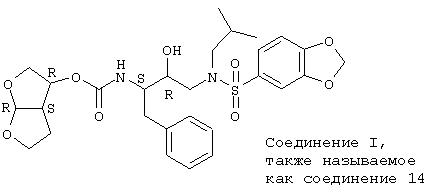
Применяемый в дальнейшем термин "соединения формулы (I)" или "настоящие соединения" или подобный термин включают в себя соединение, представленное выше, его N-оксиды, соли, сложные эфиры, стереоизомерные формы, рацемические смеси, пролекарства и метаболиты, так же как и его кватернизованные азотные производные.
Настоящие соединения, таким образом, могут применяться у животных, предпочтительно млекопитающих и особенно человека в качестве лекарственного средства отдельно, в смесях одного с другим или в форме фармацевтических препаратов.
Кроме того, настоящее изобретение относится к фармацевтическим препаратам, которые содержат в качестве активных составляющих эффективную дозу, по крайней мере, одного из соединений формулы (I) и/или их физиологически переносимую соль в дополнение к обычным фармацевтически безопасным наполнителям и вспомогательным веществам. Фармацевтические препараты обычно содержат от 0,1% до 90% по массе соединения формулы (I) и/или его физиологически переносимой соли. Фармацевтические препараты могут быть получены способами, известными специалистам в данной области. С данной целью, по крайней мере, одно из соединений формулы (I) и/или его физиологически переносимые соли вместе с одним или несколькими твердыми или жидкими фармацевтическими наполнителями и/или вспомогательными веществами и, если необходимо, в комбинации с другими фармацевтически активными соединениями вводят в подходящую для введения форму или дозированную форму, которая может далее быть использована в качестве фармацевтического препарата в медицине или ветеринарии.
Фармацевтические препараты, содержащие соединение согласно изобретению и/или его физиологически переносимые соли, может вводиться орально, парентерально, например внутривенно, ректально, в виде ингаляций или местно, предпочтительный способ введения будет зависеть от конкретного случая, например особенности течения подвергаемого лечению заболевания. Предпочтительным является пероральное введение.
Специалист в данной области легко определит на основе опыта вспомогательные вещества, подходящие для желаемого фармацевтического препарата. Кроме растворителей, гелеобразующих веществ, основ для суппозиториев, вспомогательных веществ для таблеток и других носителей активного соединения также применяют антиоксиданты, диспергирующие агенты, эмульгаторы, противовспенивающие агенты, ароматизаторы, консерванты, солюбилизаторы, вещества для создания депо, буферные вещества или красители.
Соединения согласно настоящему изобретению вследствие своих антиретровирусных свойств, в частности активности против ВИЧ, особенно активности по отношению к ВИЧ-1, применимы для лечения ВИЧ-инфицированных лиц и для профилактических мероприятий у данных лиц. В общем, соединения согласно настоящему изобретению могут быть пригодны для лечения теплокровных животных, инфицированных вирусами, существование которых опосредовано или зависит от фермента протеазы. Состояния, профилактика или лечение которых проводится соединениями согласно настоящему изобретению, особенно состояния, связанные с ВИЧ и другими патогенными ретровирусами, включают СПИД, СПИД-ассоциированный комплекс (ARC), прогрессирующую генерализованную лимфоаденопатию (PGL), равно как и хронические заболевания ЦНС, вызванные ретровирусами, такие как, например, ВИЧ- опосредованную деменцию и рассеянный склероз.
Соединения согласно настоящему изобретению или любая их подгруппа, следовательно, может быть использована в качестве лекарственного средства для вышеупомянутых состояний. Указанное применение в качестве лекарственного препарата или способа лечения включает систематическое введение ВИЧ-инфицированным пациентам эффективного количества для коррекции состояний, связанных с ВИЧ и другими патогенными ретровирусами, особенно ВИЧ-1. Следовательно, соединения согласно настоящему изобретению могут применяться для получения лекарственных препаратов, подходящих для лечения состояний, связанных с ВИЧ и другими патогенными ретровирусами, в частности лекарственных препаратов с ингибирующей активностью по отношению к ретровирусной протеазе.
Также комбинация, включающая соединение с антиретровирусной активностью, и соединение согласно настоящему изобретению может иметь применение в качестве лекарственного препарата. Таким образом, настоящее изобретение также относится к продукту, содержащему (а) соединение согласно настоящему изобретению и (б) другое соединение с антиретровирусной активностью, в виде комбинированного препарата для одновременного, раздельного или последовательного применения при лечении ретровирусной инфекции, особенно ретровирусов, резистентных ко многим лекарственным препаратам. Таким образом, для борьбы или лечения ВИЧ-инфекций или инфекции или заболевания, связанных с ВИЧ-инфекциями, таких как синдром приобретенного иммунодефицита (СПИД) или СПИД-ассоциированный комплекс (ARC), соединения согласно данному изобретению могут сочетанно вводиться в комбинации, например, со связывающими ингибиторами, такими как, например, декстрансульфат, сурамин, полианионы, растворимый CD4; гибридными ингибиторами, такими как, например, Т20, Т1249, SHC-C; ингибиторами, связывающими корецептор, такими как, например, AMD 3100 (Bicyclams), ТАК 779; ингибиторами обратной транскриптазы, такими как, например, фоскарнет и пролекарства; нуклеозидами - ингибиторами обратной транскриптазы, такими как, например, AZT, 3TC, DDC, DDI, D4T, Abacavir, FTC, DAPD, dOTC; нуклеотидными ИОТ, такими как, например, РМЕА, РМРА; ННИОТ, такими как, например, невирапин, делавирдин, ифавиренц, 8 и 9-С1 TIBO (тивирапин), ловирид, ТМС-125, ТМС-120, МКС-442, UC 781, каправирин, DPC 961, DPC963, DPC082, DPC083, каланолид A, SJ-3366, TSAO, 4"-деаминированный TSAO; ингибиторами РНК-азы Н, такими как, например, SP1093V, PD126338; ингибиторами тирозинаминотрансферазы, такими как, например, RO-5-3335, K12, К37; ингибиторами интегразы, такими как, например, L 708906, L 731988; ингибиторами протеазы, такими как, например, ампренавир, ритонавир, нелфинавир, саквинавир, индинавир, лопинавир, BMS 232632, DPC 681, DPC 684, типранавир, AG1776, DMP 450, L 756425, PD178390; ингибиторами гликозилирования, такими как, например, кастаноспермин, дезоксиноджиримицин.
Комбинации могут обеспечивать синергический эффект, посредством чего инфективность вируса и связанные с ней симптомы могут быть предупреждены, существенно ослаблены или полностью устранены. Соединения согласно настоящему изобретению также могут вводиться в сочетании с иммуномодуляторами (например, бропиримином, антителами против человеческого альфа-интерферона, IL-2, метионинэнкефалином, альфа-интерфероном и налтрексоном) или с антибиотиками (например, пентамидиноизотиоратом) для ослабления, борьбы или уничтожения ВИЧ-инфекции и ее симптомов.
При получении формы для перорального введения соединения согласно настоящему изобретению или их соли смешивают с подходящими добавками, такими как наполнители, стабилизаторы или инертные разбавители, и придают с помощью обычных способов желаемые формы введения, такие как таблетки, таблетки в оболочке, твердые капсулы, водные, спиртовые или масляные растворы. Примерами подходящих инертных носителей являются аравийская камедь, магнезия, карбонат магния, фосфат калия, лактоза, глюкоза или крахмал, в частности кукурузный крахмал. В данном случае препарат может быть получен как в виде сухих, так и влажных гранул. Подходящими масляными наполнителями или растворителями являются растительные или животные масла, такие как растительное масло или рыбий жир. Подходящими растворителями для водных или спиртовых растворов являются вода, этанол, сахарный сироп или их смеси. Полиэтиленгликоли и полипропиленгликоли также являются пригодными в качестве дополнительных вспомогательных веществ для других форм введения.
Для чрескожного или внутривенного введения активные соединения, если необходимо вместе с принятыми для этого веществами, такими как солюбилизаторы, эмульгаторы или дополнительные вспомогательные вещества, вводят в форму раствора, суспензии или эмульсии. Соединения, представленные формулой (I), и их физиологически переносимые соли также могут быть лиофилизованы и полученные лиофилизаты могут применяться, например, для получения инъекционных или инфузионных препаратов. Подходящими растворителями являются, например, вода, физиологический солевой раствор или спирты, например этанол, пропанол, глицерин, кроме того, также растворы сахаров, такие как растворы глюкозы или маннит, или альтернативно смеси различных вышеупомянутых растворителей.
Подходящими фармацевтическими формами для введения в виде аэрозолей или спреев являются, например, растворы, суспензии или эмульсии соединений, представленных формулой (I), или их физиологически переносимых солей в фармацевтически приемлемом растворителе, таком как этанол или вода или смесь подобных растворителей. Если необходимо, препарат также может дополнительно содержать другие фармацевтические вспомогательные вещества, такие как поверхностно-активные вещества, эмульгаторы и стабилизаторы, так как и пропеллент. Подобный препарат обычно содержит активное соединение в концентрации приблизительно от 0,1 до 50%, в частности приблизительно от 0,3 до 3% по массе.
Для увеличения растворимости и/или стабильности соединений, представленных формулой (I), в фармацевтических препаратах может быть желательным применение α-, β- или γ-циклодекстринов или их производных. Также сорастворители, такие как спирты, могут повысить растворимость и/или стабильность соединений, представленных формулой (I), в фармацевтических препаратах. При получении водных составов добавление солей рассматриваемых соединений является, несомненно, более подходящим вследствие их более высокой водной растворимости.
Подходящими циклодекстринами являются α-, β- или γ-циклодекстрины (ЦД) или их эфиры или смешанные эфиры, в которых одна или несколько гидроксигрупп ангидроглюкозных остатков циклодекстрина замещены такими группами, как C1-6-алкил, особенно метил, этил или изопропил, например, случайным образом метилированный β-ЦД; гидрокси-С1-6-алкил, особенно гидроксиэтил, гидроксипропил или гидроксибутил; карбокси-C1-6-алкил, особенно карбоксиметил или карбоксиэтил; C1-6-алкилкарбонил, особенно ацетил; C1-6-алкилоксикарбонил-C1-6-алкил или карбокси-C1-6-алкилокси-C1-6-алкил, особенно карбоксиметоксипропил или карбоксиэтоксипропил; C1-6-алкил- карбонилокси-C1-6-алкил, особенно 2-ацетилоксипропил. Особенно заслуживают внимания в качестве комплексообразователей и/или солюбилизаторов β-ЦД, статистически метилированный β-ЦД, 2,6-диметил-β-ЦД, 2-гидроксиэтил-β-ЦД, 2-гидроксиэтил-γ-ЦД, 2-гидроксипропил-γ-ЦД и (2-карбоксиметокси)пропил-β-ЦД и особенно 2-гидроксипропил-β-ЦД (2-ГП-β-ЦД).
Термин "смешанный эфир" обозначает производные циклодекстринов, в которых, по крайней мере, две гидроксигруппы циклодекстрина этерифицированы разными группами, такими как, например, гидроксипропил и гидроксиэтил.
Интересный способ приготовления комбинации соединений по настоящему изобретению с циклодекстрином или его производным описан в ЕР-А- 721, 331. Хотя описанные там препараты содержат соединения с противогрибковой активностью, они в равной степени интересны для получения противоретровирусных соединений согласно настоящему изобретению. Препараты, описанные там, являются особенно подходящими для перорального введения и содержат в качестве активного компонента соединение с противогрибковой активностью, значительное количество циклодекстрина или его производного в качестве солюбилизатора, водную кислую среду в качестве жидкой фазы носителя и спиртовой сорастворитель, что значительно упрощает получение препарата. Вкус указанных препаратов также может быть улучшен с помощью фармацевтически приемлемых подсластителей и/или ароматизаторов.
Другие подходящие способы увеличения растворимости соединений согласно настоящему изобретению в фармацевтических препаратах описаны в WO-94/05263, РСТ- заявка РСТ/ЕР 98/01773, ЕР-А-499299 и WO 97/44014, включенных здесь в качестве ссылки.
Более подробно, соединения по настоящему изобретению могут быть применены для получения фармацевтического препарата, содержащего терапевтически эффективное количество частиц, состоящих из твердой дисперсной системы, включающей (а) соединение формулы (I) и (б) один или несколько фармацевтически приемлемых водорастворимых полимеров.
Термин "твердая дисперсная система" означает систему в твердой фазе (в отличие от жидкой и газообразной фаз), включающую, по крайней мере, два компонента, где один из компонентов диспергирован более или менее равномерно по отношению к другому компоненту или компонентам. Когда указанная дисперсная система компонентов представляет собой систему, химически и физически однородную или совершенно гомогенную или состоящую из одной фазы, как определено в термодинамике, подобную твердую дисперсную систему относят к "твердому раствору". Твердые растворы являются предпочтительными физическими системами, поскольку их компоненты обычно являются легко биодоступными для организмов, в которые они вводятся.
Термин "твердая дисперсная система" также включает дисперсные системы, которые менее гомогенны по сравнению с твердыми растворами. Подобные дисперсные системы не являются химически и физически однородными или содержат более одной фазы.
Водорастворимый полимер, входящий в частицы, представляет собой просто полимер, обладающий кажущейся вязкостью от 1 до 100 мПа·с при растворении в 2% водном растворе при 20°С раствора.
Предпочтительными водорастворимыми полимерами являются гидроксипропилметилцеллюлозы или ГПМЦ. ГПМЦ, имеющая степень замещения метоксигруппами примерно от 0,8 до 2,5 и молярное замещение гидроксипропильными группами примерно от 0,05 до 3,0, обычно являются водорастворимыми. Степень замещения метоксигруппами относится к среднему числу метилэфирных групп на глюкозный остаток молекулы целлюлозы. Молярное замещение гидроксипропильными группами относится к среднему количеству молей пропиленоксида, прореагировавшего с каждым ангидроглюкозным остатком молекулы целлюлозы.
Частицы, описанные здесь выше, могут быть получены с помощью первоначального получения твердой дисперсной системы соединений и затем необязательного растирания или перемалывания данной дисперсной системы. Существуют различные способы получения твердых дисперсных систем, включающие экструзию из расплава, осушение аэрозоля, выпаривание раствора, предпочтительной является экструзия из расплава.
Кроме того, может быть удобным преобразовать противоретровирусные соединения по настоящему изобретению в форму наночастиц, имеющих на своей поверхности адсорбированный модификатор поверхности в достаточном количестве для сохранения эффективного среднего размера частицы менее чем 1000 нм. Предполагается, что применяемые поверхностные модификаторы включают такие, которые связаны физически с поверхностью противоретровирусного соединения, но не имеют химической связи с противоретровирусным соединением.
Подходящие поверхностные модификаторы предпочтительно могут быть выбраны из известных органических и неорганических фармацевтических наполнителей. Подобные наполнители включают различные полимеры, низкомолекулярные олигомеры, природные соединения и поверхностно-активные вещества. Предпочтительные поверхностные модификаторы включают неионные и анионные поверхностно-активные вещества.
Другой интересный способ преобразования соединений по настоящему изобретению включает получение фармацевтического препарата, в котором противоретровирусное соединение встроено в гидрофильные полимеры, и наносят данную смесь в качестве пленочного покрытия на множество мелких шариков, таким образом получая соединение с хорошей биодоступностью, которое может быть легко получено и которое является подходящим для получения фармацевтических дозированных форм для перорального введения.
Указанные шарики включают в себя (а) центральное округлое или сферическое ядро, (б) покрывающую пленку из гидрофильного полимера и противоретровирусного агента и (в) водоизолирующий полимерный слой.
Вещества, подходящие в качестве ядер шариков, многочисленны при условии, что указанные вещества являются фармацевтически приемлемыми и имеют соответствующий размер и стабильность. Примерами подобных веществ являются полимеры, неорганические вещества, органические вещества и сахариды и их производные.
Вводимая доза соединений согласно настоящему изобретению или их физиологически переносимой соли (солей) зависит от конкретного случая и, как обычно, адаптирована к условиям каждого конкретного случая для достижения оптимального эффекта. Таким образом, она зависит, разумеется, от частоты введения и от эффективности и продолжительности действия применяемых соединений в каждом случае для лечения или профилактики, но и от вида и тяжести инфекционного процесса и симптомов, пола, возраста, массы и индивидуальной реакции нуждающегося в лечении человека или животного и от того, предназначена ли терапия для острой ситуации или является профилактической. Обычно суточная доза соединения формулы (I) в случае введения пациенту массой приблизительно 75 кг составляет от 1 мг до 1 г, предпочтительно от 3 мг до 0,5 г.
Доза может вводиться в виде отдельной дозы или быть разделена на несколько, например две, три или четыре отдельные дозы.
Органический синтез гексагидрофуро [2,3-b]фуран-3-ил-N-{3-[(1,3-бензодиоксол-5-илсульфонил)(изобутил)амино]-1-бензил-2-гидроксипропил}карбамата
Синтез гексагидрофуро[2,3-b]фуран-3-ил-N-{3-[(1,3-бензо- диоксол-5-илсульфонил)(изобутил)амино]-1-бензил-2-гидрокси- пропил}карбамата проводят посредством взаимодействия бис-тетрагидрофуранового кольца и соответствующего бензодиоксолиламина, как изложено ниже.
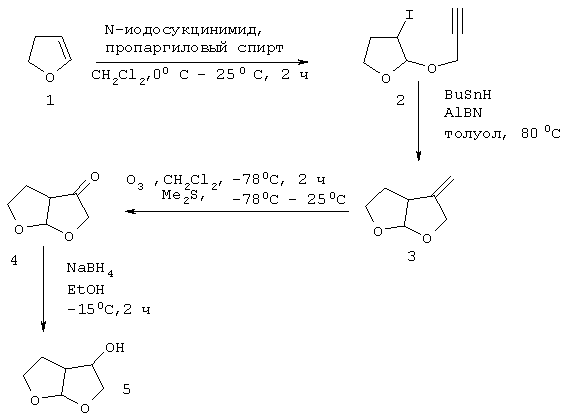
1) Синтез гексагидрофуро[2,3-b]фуран-3-ола 5
Рацемический синтез бис- тетрагидрофурана (бис-ТГФ) 5 проводят, как показано на схеме 1, согласно процессу по Ghosh et al., J. Med. Chem. 39: 3278-3290 (1996). Проводят реакцию коммерческого 2,3-дигидрофурана с N-йодсукцинимидом и пропаргиловым спиртом в метиленхлориде при 0-25°С в течение 2 часов с получением йодэфира 2 (выход 88%). Радикальная циклизация йодэфира 2 с трибутилтингидридом в толуоле при 80°С в присутствии каталитического количества 2,2'-азобисизобутиронитрила (AIBN) позволяет получить бициклический ацетат 3. Озонолитическое расщепление приводит к получению кетона 4. Восстановление полученного кетона с помощью боргидрида натрия в этаноле при -15°С приводит к получению рацемического эндоспирта 5 (см. схему 1).
Схема 1
2) Синтез аминоспирта 8 N-{3-[(1,3-бензодиоксол-5- илсульфонил)(изобутил)амино]-1-бензил-2-гидроксипропил}-амина
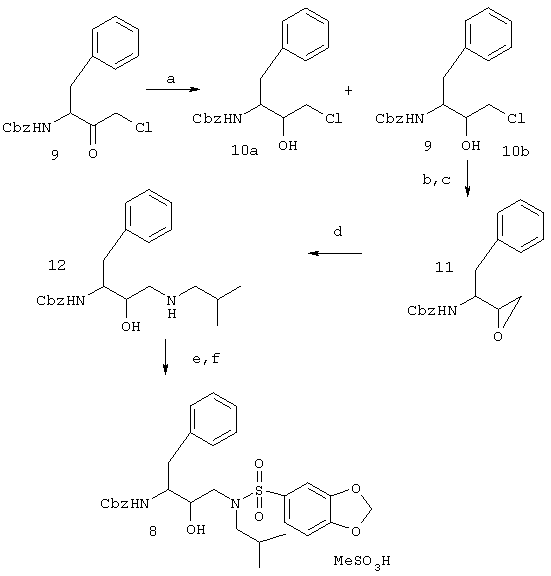
Восстановление ацилхлорида 9 с помощью NaBH4 в смеси метанол: тетрагидрофуран 1:1 (стадия а, схемы 2 ниже) приводит к получению рацематов 10а и 10b, которые разделяют и проводят взаимодействие соответствующего энантиомера с КОН в этаноле (стадии Ь, с) для получения эпоксида 11 согласно опубликованным способам (Getman, et al., J Med. Chem. 36: 288-291 (1993), Luly et al., J Org. Chem. 52(8): 1487-1492 (1987)). Эпоксид обрабатывали избытком изоамиламина в 2-пропаноле при кипении в колбе с обратным холодильником (стадия d) для получения аминоспирта 12. Дминоспирт 12 далее обрабатывают 1,3-бензодиоксол-5-илсульфонилхлоридом (стадия е) с получением аминоспирта 8 с карбобензокси (Cbz)-защищенным амином. Гидрогенизация Cbz-группы с помощью 10% Pd/C и Н2 в метаноле (стадия f) приводит к получению свободного аминоспирта 8. Данные стадии проводят согласно широко известным опубликованным способам (Vazquez et al., J. Med. Chem. 38: 581-584 (1995), схема 2).
Схема 2
3) Синтез гексагидрофуро[2,3-b]фуран-3-ил-N-{3-[(1,3- бензодиоксол-5-илсульфонил)(изобутил)амино]-1-бензил-2-гидроксипропил}карбамата
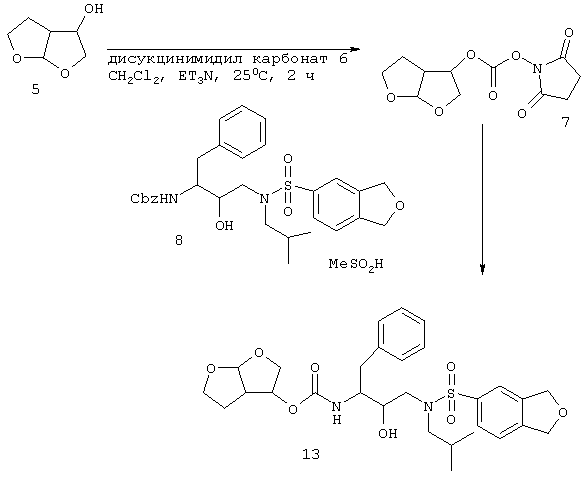
Проводят взаимодействие бис-тетрагидрофуранового лиганда 5 с дисукцинимидилкарбонатом 6 и триэтиламином в метиленхлориде с получением карбоната 7, который смешивают in situ с амином 8. Данное присоединение приводит к конечному соединению 13 (схема 3). Соединение 13 является смесью 2 диастереомеров, находящихся в стереоизомерных формах, указанных для соединений 1 и w в таблице А. Данная смесь может быть разделена с помощью хорошо известных в данной области способов разделения.
Альтернативно чистые энантиомерные формы, соответствующие соединению 1 и w, здесь и далее обозначаемые как соединения 14 и 15, могут быть получены с помощью разделения рацемического бис-ТГФ 5 посредством стадии ферментного расщепления, как описано в Tetrahedron Letters, 36, 4, (1995), 505-508, включенного здесь в качестве ссылки. Энантиомерные чистые промежуточные продукты бис-ТГФ далее могут быть подвергнуты реакциям, аналогичным описанным выше, с получением энантиомерно чистого соединения 14 и 15.
Схема 3
Экспериментальная часть
К перемешиваемому раствору (500 мг, 3,84 ммоль) (3R,3aS,6aR)-3-гидроксигексагидрофуро[2,3-b]-фурана 5 (схема 1) в СН2Сl2 (50 мл) при 25°С добавляют дисукцинимидилкарбонат 6 (1,08 г, 4,23 ммоль) и триэтиламин (0,77 г, 7,68 ммоль). Полученную смесь перемешивают в течение 6 ч при 25°С и добавляют амин 8 (схема 2, 2,42 г, 5,76 ммоль). Полученный раствор промывают водой и сушат над безводным Na2SO4. Упаривание растворителя при пониженном давлении приводит к получению остатка, который очищают с помощью хроматографии (CH2Cl2/MeOH: 98/2) с получением 1,36 г (62%) ингибитора 13 (схема 3) согласно настоящему изобретению в виде белого твердого вещества.
Спектр 1H-ЯМР в CDCl3 соединения 13 представлял собой следующее: 7,4-7,1 (ушир. м, 7Н), 6,9 (д, J=8,1 Гц, 1Н), 6,1 (с, 2Н), 5,7 (д, J=5,1 Гц, 1Н), 5 (д, J=6,7 Гц, 1Н), 5,1-4,8 (ушир. м, 1Н), 4-3,4 (ушир. м, 7Н), 3,25-2,6 (ушир. м, 6Н), 2,35-1,2 (ушир. м, 4Н), 1,17-0,7 (ушир. м, 6Н). Также спектр 13С-ЯМР в СDСl3 представлял собой следующее: 151 (СО), 148-138 (С-O), 132-129,4-129,34-128,55-126,67-126,59-123,1 (Аr-С), 109,16-108,36-107,52-102,36 (СН-O), 73,43-72,58-70,73-69,47-58,87-53,78-45,04-36-35-27, 27-25,76-20,1-19,85. Масс-спектр показал ожидаемый ион (m/z) 577, соответствующий М+ + Н.
Полученное соединение 13 и соединение 14 далее тестировали на биологическую и противовирусную активность в нескольких тестах, как описано ниже. Неожиданно обнаружили, что данные соединения обладают большей эффективностью и большей активностью как ингибиторы протеазы по сравнению с ранее известными соединениями.
Противовирусный тест:
Соединение 13 и соединение 14 далее тестировали на противовирусную активность в клеточном тесте. Тест показал, что данные соединения обнаруживают значительную активность против ВИЧ по отношению к дикому типу лабораторного штамма ВИЧ. Клеточный тест проводили согласно следующей методике.
Экспериментальный клеточный тест:
ВИЧ- или ложно-инфицированные клетки МТ4 инкубировали в течение 5 дней в присутствии различных концентраций ингибитора. По окончании периода инкубации все ВИЧ- инфицированные клетки погибали при репликации вируса в контрольных культурах в отсутствие ингибитора. Жизнеспособность клеток оценивали с помощью измерения концентрации МТТ, желтого водорастворимого тетразольного красителя, преобразовывающегося в пурпурный водонерастворимый формазан в митохондриях только живых клеток. После растворения полученных кристаллов формазана с помощью изопропанола проводили измерение поглощения раствора при 540 нм. Величины напрямую коррелировали с количеством живых клеток, оставшихся в культуре после завершения пяти дней инкубации. Ингибирующую активность соединения отслеживали с помощью вирус- инфицированных клеток и выражали как IC50 и IС90. Данные величины представляют собой количество соединения, необходимого для защиты 50% и 90%, соответственно, клеток от цитопатогенного эффекта вируса. Токсичность соединения оценивали с помощью ложно- инфицированных клеток и выражали как CC50, представляющую собой концентрацию соединения, необходимую для ингибирования роста клеток на 50%. Индекс селективности (SI) (отношение CC50/IC50) отражает селективность активности ингибитора по отношению к ВИЧ.
Результаты клеточного теста:
Соединение 13 показало IC50, равную 1,1 нМ, и IС90, равную 2,4 нМ (представляющие собой средние значения для 12 определений), по отношению к штамму ВИЧ-1 LAI. CC50 для соединения 13 составило 15,3 мкМ и SI для него равен 13900. Соединение 14 показало IC50, равную 0,8 нМ, по отношению к штамму ВИЧ-1 LAI. CC50 для соединения 14 составило более чем 100 мкМ.
Тест на связывание белков;
Известно, что белки сыворотки человека, такие как альбумин (HSA) и кислый гликопротеин альфа-1 (AAG), связываются со многими лекарственными препаратами, приводя к возможному снижению эффективности данных соединений. Для определения наличия нежелательного эффекта данного связывания соединения 13 оценивали анти-ВИЧ активность соединения в присутствии физиологических концентраций HSA или AAG, таким образом оценивая эффект связывания ингибитора с данными белками.
Результаты:
В обычном эксперименте HAS в концентрации 45 мг/мл не оказывал влияния на эффективность соединения 13. AAG в концентрации 2 мг/мл снижал эффективность соединения 13 в два - четыре раза.
Противовирусный спектр:
Вследствие увеличения появления штаммов ВИЧ, устойчивых к лекарственным препаратам, соединение 13 и соединение 14 оценивали на их эффективность по отношению к штаммам ВИЧ, несущим различные мутации. Данные мутации связаны с резистентностью к ингибиторам протеазы и выражаются наличием у вирусов различной степени фенотипической перекрестной резистентности к пяти из имеющихся в наличии коммерчески доступным лекарственным препаратам (саквинавир, ритонавир, нелфинавир, индинавир и ампренавир).
Результаты:
В таблице 1 представлены результаты данного тестирования в виде значений IC50 в мкМ. Соединения 13 и 14 являются эффективными ингибиторами даже данных резистентных вирусов в низких концентрациях, являющимися более низкими, чем достигаемые уровни в плазме. Фигура 2 представляет сравнение fold резистентности различных штаммов вируса к коммерчески доступным ингибиторам протеазы и соединения 13. Штаммы неожиданно показали повышенную чувствительность к соединению 13 по сравнению с препаратами саквинавир (SAQ), ритонавир (RIT), индинавир (IND), нелфинавир (NEL) и ампренавир (ДМР).
Активность соединений 13 и 14 и 5
коммерческих ингибиторов протеазы по отношению к штаммам
ВИЧ, резистентным к ингибиторам протеазы
Биодоступность:
Далее измеряли абсорбцию соединения 13 при пероральном введении на крысах для определения биодоступности. Соединение вводили через желудочный зонд (by gavage) крысам в виде отдельной дозы 20 мг/кг в ПЭГ 400. Животных забивали в различные моменты времени после введения, брали цельную кровь и получали сыворотку обычным способом. Концентрацию соединения в сыворотке определяли с помощью титрования активности образца по отношению к ВИЧ согласно описанной выше методике.
Результата определения биодоступности:
Результаты представлены в таблице 2 и графически проиллюстрированы с помощью фигуры 1. Концентрация соединения 13 в сыворотке достигает 1 мкМ через 1 час после перорального введения и еще превышает IC50 соединения, активного по отношению к штаммам, мультирезистентным к ингибиторам протеазы, вплоть до 3 часов после введения. Следовательно, соединение 13 обладает значительным преимуществом, выражающимся в терапевтическом преимуществе. Данная неожиданно высокая биодоступность в плазме является особенно важной в отношении резистентных вирусов.
Концентрация соединения в сыворотке после перорального введения
(минуты)
сыворотке (мкМ)
Настоящее изобретение может быть воплощено в других конкретных формах без отклонения от его сути или основных характеристик. Описанные воплощения рассматриваются во всех аспектах исключительно как иллюстративные, но не ограничивающие. Объем настоящего изобретения, таким образом, определяется приложенной формулой изобретения, а не вышеизложенным описанием. Все изменения, которые подпадают под значение и объем эквивалентности формулы изобретения, входят в объем притязаний.
Далее приводятся данные ЯМР спектров энантиомерного соединения 14:
1H-NMR (в СDСl3): 7.35-7.2 (m, 6H), 7.17 (d, J=1.7Hz, 1H), 6.85 (d, J= 8.1Hz, 1H), 6.1 (s, 2H), 5.65 (d, J=4.96Hz, 1H), 5 (d, J=6.7Hz, 1H), 4.9-5 (m, 1H), 3.9 (dd, J=9.5 Hz и J=6.6Hz, 1H), 3.8-3.6 (m, 5H), 3.6 (dd, J=9.5Hz, и J=6.6Hz, 1H), 3.1 (dd, J=15.1 и J=8.6Hz, 1H), 3.1-2.85 (m, 4H), 2.8 (d, J=13.4Hz и J=6.7Hz, 1H), 2 (br.s, 1H), 1.85-1.8 (m, 1H), 1.65-1.55 (br.s, 2H), 0.9 (d, J=6.6Hz, 3H), 0.85 (d, J=6.6Hz, 3H).
13C-NMR (в СDСl3): 155.6 (СО), 151.6-148.4 (С-O), 137.5-131.5-129.4128.6-126.7-123.1 (Ar-C), 109.2 (СН-O)-108.4-107.5 (CH-Ar), 102.4 (O-СН2-O), 73.4-72.6-70.7-69.5-58.8-55.3-53.7-45-35.3-27.3-25.9-20.1-19.8.
Масс-спектроскопия дала неожиданный ион (m/z) 577, соответствующий M++Н.
Изобретение относится к новым бис-тетрагидрофуранбензодиоксолилсульфонамидным соединениям формулы I
его солям, стереоизомерам и рацематам, являющимся эффективными ингибиторами протеазы. Изобретение также относится к фармацевтическим препаратам, способам ингибирования ретровирусных протеаз, в частности протеаз резистентных ретровирусов ко многим лекарственным препаратам, способам лечения и профилактики инфекции или заболевания, связанного с ретровирусной инфекцией, у млекопитающих и способам ингибирования репликации ретровируса. Технический результат - получение новых производных бис-тетрагидрофуранбензодиоксолилсульфонамидов, обладающих ценными фармацевтическими свойствами. 5 н. и 11 з.п. ф-лы, 2 ил.,3 табл.
его соль, стереоизомерная форма, рацемическая смесь.
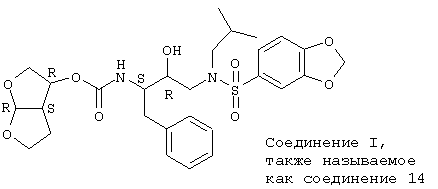
его соль.
| WO 9405639 A1, 17.03.1994 | |||
| WO 9506030 A1, 02.03.1995 | |||
| WO9633184 A1, 24.10.1996 | |||
| ПРОИЗВОДНЫЕ СУКЦИНОИЛАМИНОГИДРОКСИЭТИЛАМИНОСУЛЬФОНАМИДА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ РЕТРОВИРУСНОЙ ПРОТЕАЗЫ, СПОСОБ ЛЕЧЕНИЯ РЕТРОВИРУСНОЙ ИНФЕКЦИИ, СПОСОБ ЛЕЧЕНИЯ ВИЧ-ИНФЕКЦИИ | 1993 |
|
RU2130016C1 |
| RU 95106823 A1, 20.11.1996. | |||

