Данной заявкой истребуется приоритет в соответствии с предварительной заявкой с регистрационным номером 60/133418, поданной 11 мая 1999 г., содержание которой включено сюда в качестве ссылки.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к системам и способам достижения оптимизированных режимов дозирования ЕРО для желаемого фармакодинамического/фармакокинетического ответа.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Эритропоэтин (ЕРО) является основным фактором, ответственным за регуляцию продукции красных кровяных клеток в обычных условиях и за ускоренное восстановление массы красных кровяных клеток после кровотечения. ЕРО является гликопротеидным гормоном с молекулярной массой 30 кДа и в значительной степени гликозилирован, что служит для защиты молекулы ЕРО от быстрой деградации in vivo. Концентрации ЕРО в сыворотке человека обычно находятся в пределах от 6 до 32 Ед/л (1), и сообщалось, что время полужизни (t1/2) ЕРО находится в пределах от 2 до 13 часов с объемом распределения, близким к объему плазмы. Как и ожидалось для крупного сиалогликопротеида, менее 10% ЕРО экскретируется с мочой (смотри, например, Lappin et al., 1966, Clin. Lab. Haem. 18:137-145).
Основным местом синтеза ЕРО во взрослых организмах является почка; хотя печень и костный мозг также были включены в рассмотрение, данные остаются недоказанными. Первичным стимулом для повышенного синтеза ЕРО является тканевая гипоксия, которая возникает в результате пониженного доступа кислорода в ткани. Гипоксия может быть результатом потери больших количеств крови, разрушения эритроцитов радиацией или воздействия больших высот. Кроме того, гипоксию вызывают различные формы анемии, так как эритроциты ответственны за транспорт кислорода в организме. В нормальном состоянии повышенный уровень ЕРО стимулирует продукцию новых красных кровяных клеток, тем самым повышая уровень кислорода и снижая или устраняя состояние гипоксии.
Основная функция ЕРО состоит в том, что он действует синергетически с другими факторами роста, стимулируя пролиферацию и дифференцировку клеток-предшественников эритроцитов в костном мозге, приводя к ретикулоцитозу и повышенному количеству эритроцитов в крови, процессу, также известному как эритропоэз (фигура 1). В ходе эритропоэза дифференцировка клеток эритроидного ряда у человека происходит в течение двухнедельного промежутка времени. Самым ранним предшественником является BFU-E (Burst-Forming Unit-Erythroid, бурст-образующая эритроидная единица), которая невелика по размеру и не обладает отличительными гистологическими характеристиками. Стадией после BFU-E является CFU-E (Colony Forming Unit-Erythroid, колониеобразующая эритроидная единица), которая крупнее, чем BFU-E, и непосредственно предшествует стадии, на которой начинается продукция гемоглобина. Клетками, которые начинают продуцировать гемоглобин, являются юные эритроциты, которые не только начинают продуцировать гемоглобин, но также начинают конденсировать свои ядра, в конечном итоге становясь зрелыми эритробластами. Зрелые эритробласты меньше, чем юные эритроциты, и имеют плотно упакованное ядро, которое вытесняется по мере того, как клетки становятся ретикулоцитами. Ретикулоциты называются так потому, что эти клетки содержат ретикулярные сети полирибосом, и, по мере того как ретикулоциты теряют свои полирибосомы, они становятся зрелыми красными кровяными клетками (RBC).
До последнего времени возможность получения ЕРО была очень ограничена. Несмотря на то, что белок присутствует в моче человека, экскретируемые уровни слишком малы, чтобы сделать мочу практическим источником ЕРО для терапевтического использования. Идентификация, клонирование, экспрессия генов, кодирующих ЕРО, и технологии очистки ЕРО, которые описаны, например, в патентах США №№4703008, 5389541, 5441868, 5614184, 5688679, 5888774, 5888772 и 5856298, сделали ЕРО легко доступными для терапевтических применений. Описание очистки рекомбинантного ЕРО (rHuEPO) из культуральной среды, которая поддерживала рост клеток млекопитающих, содержащих рекомбинантные ЕРО-плазмиды, включено, например, в патент США №4667016. Рекомбинантный ЕРО имеет аминокислотную последовательность, идентичную последовательности эритропоэтина мочи человека, при этом оба эритропоэтина неразличимы в химических, физических и иммунологических тестах. Экспрессия и извлечение биологически активного рекомбинантного ЕРО из хозяйских клеток млекопитающих, содержащих ген ЕРО в рекомбинантных плазмидах, сделали доступными количества ЕРО, пригодные для терапевтических применений. Кроме того, сведения о последовательности гена и доступность больших количеств очищенного белка привели к лучшему пониманию способа действия этого белка.
Биологическая активность белка зависит от его структуры. В частности, первичная структура белка, т.е. его аминокислотная последовательность, обеспечивает информацию, которая делает возможным образование полипептидом вторичной (например, α-спираль или β-складка) и третичной (общая трехмерная укладка) структур во время и после синтеза. Кроме того, биологическая активность белка определяется не только его структурой, но также модификациями, возникающими после того, как белок был транслирован. Действительно, многие белки клеточной поверхности и секреторные белки модифицированы одной или несколькими олигосахаридными группами. Указанная модификация, известная как гликозилирование, может коренным образом влиять на физические свойства белков и может быть важной для стабильности, секреции и субклеточной локализации белка. Правильное гликозилирование может быть существенным для биологической активности.
И ЕРО, полученный из мочи человека, и рекомбинантный ЕРО (экспрессированный в клетках млекопитающих), имеющий последовательность аминокислот 1-165 ЕРО человека, содержат три N-связанных и одну O-связанную олигосахаридные цепи, которые вместе составляют примерно 40% общей молекулярной массы гликопротеида. Было показано, что олигосахаридные цепи модифицированы концевыми остатками сиаловой кислоты. Ферментативная обработка гликозилированного ЕРО для удаления всех остатков сиаловой кислоты в результате приводит к потере активности in vivo, но не влияет на его активность in vitro (Lowy et al., 1960, Nature 185: 102; Goldwasser et al., 1974, J. Biol. Chem. 249: 4202). Такое поведение объяснили быстрым выведением асиалоэритропоэтина из циркулирующей крови при взаимодействии с белком печени, связывающим асиалогликопротеиды (Morrell et al., 1968, J. Biol. Chem. 243: 155; Briggs et al., 1974, Am. J. Physiol. 227: 1385; и Ashwell et al., 1978, Methods of Enzymol. 50: 287). Таким образом, ЕРО обладает биологической активностью in vivo только в том случае, когда он сиалирован, чтобы избежать связывания со связывающим белком печени.
Продукция дефицитного (или неэффективного) ЕРО по отношению к уровню гемоглобина связана с некоторыми формами анемии. Такие анемии включают анемию при почечной недостаточности и конечной стадии заболевания почек, анемию при хронических заболеваниях (хронические инфекции и ревматоидный артрит), аутоиммунном заболевании, синдроме приобретенного иммунодефицита (СПИД) и злокачественной опухоли. Многие из этих состояний связаны с образованием фактора, который, как показано, является ингибитором активности ЕРО. Другие анемии, очевидно, независимы от ЕРО и включают апластическую анемию, железодефицитную анемию, талассемию, мегалобластную анемию, истинную эритроцитарную аплазию и синдромы миелодисплазии.
Измерение уровней ЕРО в сыворотке человека имеет клиническую важность. Определение уровней ЕРО в сыворотке пациентов может быть полезным для того, чтобы отличить такие анемии и полицитемии, которые связаны с пониженными или повышенными уровнями ЕРО, от тех, которые не связаны. Кроме того, обнаружение несоответствующего норме низкого уровня ЕРО в сыворотке позволяет заключить, что для пациента с анемией полезным может быть лечение экзогенным ЕРО.
Эпоэтин-альфа оценили в клинических испытаниях на пациентах с нормальным здоровьем, а также пациентах с различными анемичными состояниями. Эпоэтин-альфа индуцирует быстрый гематологический ответ у здоровых добровольцев, при условии, что достигается адекватное потребление железа, чтобы обеспечить повышенный синтез гемоглобина. В большинстве испытаний исследовали безопасность и эффективность лечения анемии, связанной с почечной недостаточностью. Кроме того, эпоэтин-альфа можно использовать для корректировки анемии у других групп пациентов, включая анемию, связанную с химиотерапией злокачественных опухолей препаратами платины, анемию, связанную с лечением зидовудином больных СПИД, и анемию, связанную с применением других лекарственных препаратов, таких как цисплатин. Введение эпоэтина-альфа также имеет много других потенциальных терапевтических применений: введение эпоэтина-альфа увеличивает способность к аутологичному донорству крови у пациентов, которым запланировано проведение операции, и ослабляет снижение гематокрита, часто наблюдаемое у необработанных аутологичных доноров; введение эпоэтина-альфа увеличивает восстановление красных кровяных клеток после аллогенного - но не аутологичного - трансплантата костного мозга; и показано, что введение эпоэтина-альфа улучшает качество жизни у людей, страдающих ревматоидным артритом.
Альтернативным применением ЕРО является повышение спортивных данных атлетов, за счет того что он вызывает увеличение гематокрита у атлета. Увеличение гематокрита увеличивает объем кислорода, транспортируемого из легких в тренируемые скелетные мышцы. Так как синтез ЕРО осуществляется биоинженерным способом, инъекции атлетов ЕРО, известные также как допинг крови, стали популярными в спорте вообще и, в частности, в велоспорте (Scheen, A.J., 1998. Rev. Med. Liege 53(8): 499-502).
Ныне существует ряд недостатков, связанных со стандартным режимом дозирования ЕРО, вводимого пациентам. При особых показаниях, таких как злокачественная опухоль, пациентов лечат введением 150 ME ЕРО/кг три раза в неделю. Таким образом, остается важная задача изменить принятый в настоящее время режим дозирования на более подходящую схему и режим дозирования. Предполагается, что менее частое введение улучшит переносимость и будет более удобным. Кроме того, стандартные режимы дозирования могут не вызывать максимальный физиологический ответ у пациентов; и стандартные режимы дозирования могут быть не самыми экономически эффективными.
Кроме того, существует ряд недостатков, связанных с путем введения ЕРО: регулярное внутривенное введение неудобно для пациента; внутривенное введение неосуществимо для индивидуумов, страдающих некоторыми состояниями, а именно для пациентов при продолжительном амбулаторном перитонеальном диализе или для не диализуемых пациентов с ограниченным сосудистым доступом; быстрая доставка дозы rHuEPO посредством внутривенного введения приводит к более низкой биодоступности rHuEPO в течение более продолжительных периодов времени и может быть не так эффективна для стимулирования продукции эритроцитов, как требуется.
Следовательно, по всем детально описанным выше причинам необходим лучший путь введения и способы определения эффективной дозы и режима дозирования для введения ЕРО.
Поэтому одним из аспектов данного изобретения является разработка фармакокинетической/фармакодинамической (ФК/ФД) модели для характеристики и предсказания ответов на rHuEPO, тем самым для идентификации наиболее эффективных, экономически эффективных и/или подходящих режимов лечения пациентов. В конкретном варианте данного изобретения предполагается введение ЕРО один раз в неделю или один раз каждые две недели. Другой аспект данного изобретения обеспечивает методологию оценки фармакокинетических и фармакодинамических параметров ЕРО после введения при двух или нескольких режимах дозирования для сравнения клинических последствий наряду с параметрами переносимости и безопасности, проводимого между режимами дозирования ЕРО. Также рассматриваются связанные бизнес-способы и компьютерные системы.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ.
Конкретный вариант данного изобретение может включать способ достижения оптимизированных режимов дозирования ЕРО для желаемого фармакодинамического ответа, который может включать в себя выбор одного или нескольких режимов дозирования, затем использование ФК/ФД-модели для определения фармакодинамического профиля одного или нескольких режимов дозирования ЕРО и, наконец, выбор одного из режимов дозирования ЕРО для введения, чтобы достичь желаемого фармакодинамического (ФД) ответа на основе профиля ЕРО. В дополнительном варианте ФД-ответ может включать в себя один или несколько параметров из группы, состоящей из количества ретикулоцитов, количества эритроцитов и уровня гемоглобина.
Альтернативным вариантом данного изобретения также может быть способ достижения оптимизированных режимов дозирования ЕРО для желаемого фармакодинамического ответа, который включает в себя сначала выбор одного или нескольких желаемых фармакодинамических ответов, затем использование ФК/ФД-модели для определения режима дозирования ЕРО, который обеспечивает желаемые ответы, и, наконец, выбор одного из режимов дозирования ЕРО для введения, позволяющего достичь желаемого фармакодинамического ответа. В дополнительном варианте ФД-ответ может включать в себя один или несколько параметров из группы, состоящей из количества ретикулоцитов, количества эритроцитов и уровня гемоглобина.
Дополнительный предпочтительный вариант данного изобретения может включать компьютерную программу, которую можно использовать для достижения оптимизированных режимов дозирования для желаемого фармакодинамического ответа. Компьютерная программа может включать в себя машинный код. В дополнительном варианте, машинный код описывает ФК/ФД-модель для ЕРО и дает возможность пользователю выбрать один или несколько желаемых фармакодинамических ответов. Затем машинный код использует ФК/ФД-модель для определения режимов дозирования ЕРО, которые будут обеспечивать желаемые фармакодинамические ответы. Режим дозирования ЕРО можно вводить еженедельно или один раз каждые две недели, исходя из массы тела, дозы. Предпочтительно еженедельная доза ЕРО может включать в себя введение ЕРО в дозе 40000 МЕ/кг, и режим дозирования ЕРО один раз каждые две недели может включать в себя введение ЕРО в дозе примерно от 80000 до 120000 МЕ/кг. В дополнительном варианте ФД-ответ может включать в себя один или несколько параметров из группы, состоящей из количества ретикулоцитов, количества эритроцитов и уровня гемоглобина.
Альтернативный предпочтительный вариант данного изобретения может включать компьютерную программу для достижения оптимизированных режимов дозирования для желаемого фармакодинамического ответа. В дополнительном варианте компьютерная программа включает в себя машинный код. Машинный код дает возможность пользователю выбрать один или несколько режимов дозирования ЕРО. Затем машинный код использует ФК/ФД-модель для определения фармакодинамического ответа на основании выбранных режимов дозирования ЕРО.
Предпочтительный вариант данного изобретения может включать компьютерную программу для определения оптимизированных режимов дозирования для желаемого фармакокинетического ответа, включающую в себя этапы выбора одного или нескольких режимов дозирования ЕРО, использование ФК/ФД-модели для определения фармакокинетического ответа режимов дозирования ЕРО, и затем выбор желаемого режима дозирования ЕРО на основании фармакокинетического профиля, в особом варианте на основании одного или нескольких ЕРО или ЕРО-подобных соединений. В дополнительном варианте фармакокинетический ответ может включать в себя уровни ЕРО в сыворотке, биодоступность и пороговые уровни ЕРО.
Следующий вариант данного изобретения может включать способ достижения оптимизированных режимов дозирования ЕРО для желаемого фармакокинетического ответа, который включает в себя этапы сначала выбора одного или нескольких желаемых фармакокинетических ответов, затем использования ФК/ФД-модели для определения режима дозирования ЕРО, который обеспечивает один или несколько желаемых фармакокинетических ответов, и, наконец, выбора режима дозирования, который обеспечивает желаемые фармакокинетические ответы.
Дополнительный вариант данного изобретения может включать компьютерную программу для достижения оптимизированных режимов дозирования для желаемого фармакокинетического ответа, которая включает в себя машинный код, который описывает ФК/ФД-модель для ЕРО. В дополнительном варианте машинный код дает возможность пользователю выбрать один или несколько фармакокинетических ответов и затем использует ФК/ФД-модель для определения режимов дозирования ЕРО, которые обеспечивают желаемые фармакокинетические ответы.
Альтернативный предпочтительный вариант данного изобретения может включать компьютерную программу для достижения оптимизированных режимов дозирования для желаемого фармакокинетического ответа. В дополнительном варианте компьютерная программа включает в себя машинный код. Машинный код дает возможность пользователю выбрать один или несколько режимов дозирования ЕРО. Затем машинный код использует ФК/ФД-модель для определения фармакокинетического ответа на основании выбранных режимов дозирования ЕРО. Можно рассчитывать на использование одного или нескольких ЕРО или ЕРО-подобных соединений.
Другой предпочтительный вариант данного изобретения включает в себя ряд способов, включая бизнес-способ предоставления потребителю режима дозирования ЕРО, который включает в себя первую дозу ЕРО с последующей второй дозой ЕРО для пациента. Вторую дозу ЕРО предпочтительно вводят пациенту во временной точке после первой дозы, которая совпадает с ФД-профилем, полученным от первой дозы ЕРО. ФД-профиль может включать количество продуцируемых клеток-предшественников относительно времени, концентрацию ретикулоцитов относительно времени, количество продуцируемых эритроцитов относительно времени и концентрацию гемоглобина относительно времени. Наиболее предпочтительно ФД-профиль будет представлять собой профиль ретикулоцитов для данного режима. Вторую дозу ЕРО предпочтительно вводят так, чтобы она совпала с профилем ретикулоцитов, т.е. когда продукция ретикулоцитов достигает пика. Вторая доза ЕРО способствует созреванию юных эритроцитов в циркулирующей крови до зрелых эритроцитов.
Дополнительный вариант данного изобретения включает бизнес-способ предоставления пациенту режима дозирования ЕРО, который включает в себя первую дозу ЕРО с последующей второй дозой ЕРО для пациента. Вторую дозу вводят пациенту в момент времени после первой дозы, который совпадает с профилем ретикулоцитов пациента. Вторую дозу можно вводить в пределах от 6 до 10 дней после первой дозы. Предпочтительно вторую дозу будут вводить через 7 дней после первой дозы ЕРО.
Бизнес-способ согласно данной заявке относится к коммерческому и другим применениям методологий данного изобретения. В одном аспекте биснез-способ включает маркетинг, реализацию или лицензирование данных методологий в контексте предоставления потребителям, т.е. пациентам, практикующим медикам, поставщикам медицинским услуг и распространителям и производителям фармацевтических препаратов, режимов дозирования ЕРО, предоставляемых данным изобретением. Указанные режимы включают режимы дозирования ЕРО один раз в неделю и один раз каждые две недели.
Другой предпочтительный вариант данного изобретения обеспечивает способ создания фармакокинетической модели подкожного (п/к) введения ЕРО пациентам. Указанный способ может включать в себя получение фармакокинетических данных от пациентов, выбор уравнения, основанного на ФК-данных, собранных от пациентов, и подгонку уравнения в соответствии с ФК-данными. Кроме того, получение ФК-данных может включать в себя нормализацию значений концентрации ЕРО в сыворотке по собранным ФК-данным и создание временных профилей концентрации ЕРО в сыворотке, основанных на нормализованных данных. В следующем варианте ФК-данные могут быть нормализованы посредством получения сначала значений исходного уровня концентрации ЕРО в сыворотке по ФК-данным посредством усреднения значений концентрации ЕРО в сыворотке до введения дозы во множестве временных точек; затем получения значений концентрации ЕРО в сыворотке после п/к-введения ЕРО; затем получения нормализованных значений концентрации ЕРО в сыворотке вычитанием значений концентрации ЕРО перед введением дозы из значений концентрации ЕРО в сыворотке; и, наконец, подсчета средних нормализованных значений концентрации ЕРО в сыворотке в каждой временной точке.
В дополнительном варианте данного изобретения ФК-уравнение может включать в себя выбор уравнения Михаэлиса-Ментен. Можно сделать подгонку ФК-данных и ФК-уравнения с использованием, например, компьютерной программы ADAPT II и оценку параметров можно получить посредством использования наименьших квадратов посредством метода максимального подобия и обобщенной модели наименьших квадратов. В следующем варианте параметры можно выбрать из группы, состоящей из Vmax, Кm, Vd, Fr, τ (более низкие дозы) и τ (более высокие дозы).
Следующий вариант данного изобретения обеспечивает способ расчета биодоступности ЕРО после п/к введения ЕРО. Способ может включать в себя получение ФК-данных, расчет площади под кривой концентрации ЕРО в сыворотке (AUC) против дозы, нормализацию AUC по дозе и, наконец, получение уравнения посредством выполнения линейной регрессии ФК-данных.
Другой предпочтительный вариант данного изобретения обеспечивает способ создания фармакодинамической (ФД) модели после п/к введения ЕРО. Данный способ может включать в себя нормализацию концентраций ЕРО в сыворотке, получение ФД-данных, выбор ФД-модели, получение уравнения на основе ФД-модели и подгонку ФД-данных и ФД-уравнений. В дополнительном варианте нормализация концентраций ЕРО в сыворотке может включать в себя получение исходной концентрации ЕРО в сыворотке (Сbs) для каждой группы доз усреднением значений концентрации ЕРО в сыворотке перед введением дозы во множестве временных точек для каждой группы доз и затем корректировку Сbs добавлением Сbs к концентрации ЕРО в сыворотке, предсказанной ФК-моделью, и при этом скорректированная Сbs может быть использована как вынуждающая функция для ФД-анализа.
В следующем варианте ФД-данные могут быть получены посредством определения среднего количества клеток-предшественников, ретикулоцитов и эритроцитов и концентрации гемоглобина перед введением дозы, и затем получения профилей среднего количества ретикулоцитов, среднего количества эритроцитов и средней концентрации гемоглобина против времени в соответствии с дозой ЕРО.
В дополнительном варианте ФД-модель может включать в себя модель потери и продукции клеток. ФД-данные можно подогнать к уравнению модели, используя, например, компьютерную программу ADAPT II, и затем можно получить как оцениваемые, так и фиксированные параметры благодаря использованию наименьших квадратов посредством метода максимального подобия и обобщенной модели наименьших квадратов. Кроме того, оцениваемые параметры могут включать Ks, SC50 и ТР, тогда как фиксированные параметры могут включать RL, RBCL, Hb и пороговую величину.
Следующий предпочтительный вариант данного изобретения может обеспечивать способ предсказания ФД-ответа у пациента после разных п/к доз ЕРО. Кроме того, данный способ может включать в себя выбор дозы и режима дозирования и затем определение ФД-ответа, основанного на такой конкретной дозе и режиме дозирования, посредством ФК/ФД-модели. В дополнительном варианте ФД-ответ может включать в себя один или несколько параметров из группы, состоящей из количества ретикулоцитов, количества красных кровяных клеток и уровня гемоглобина.
Данное изобретение может быть направлено на удовлетворение потребностей пациентов, у которых может продуцироваться дефицитный или неэффективный в отношении уровня гемоглобина ЕРО, что может быть связано с некоторыми формами анемии. Такие формы могут включать, но не ограничиваются этим, анемию, связанную с конечной стадией заболевания почек или анемию, связанную с почечной недостаточностью, анемию, связанную с химиотерапией злокачественных опухолей препаратами платины, анемию, связанную с лекарственной терапией СПИД, при которой лекарственные средства могут включать цисплатин и зидовудин. Пациенты также могут подвергаться аутологичной трансфузии перед операцией, восстанавливаться после аллогенного трансплантата костного мозга, страдать ревматоидным артритом или относиться к атлетам или иным категориям людей, нуждающихся или желающих увеличить количество эритроцитов и/или содержание гемоглобина.
ФК/ФД-модель согласно данному изобретению имеет много потенциальных терапевтических применений. Например, врач может использовать данную систему ФК/ФД-моделирования, чтобы определить оптимальный режим дозирования ЕРО для введения пациенту, нуждающемуся в повышенном количестве эритроцитов и/или гемоглобина. В частности, врач мог бы иметь выбор либо определения режима дозирования ЕРО на основании желаемого фармакодинамического результата, либо определения фармакодинамического ответа на основании конкретного режима дозирования ЕРО.
КРАТКОЕ ОПИСАНИЕ ФИГУР
Фигура 1: Процесс эритропоэза.
Фигура 2: Профили концентрации rHuEPO против времени после внутривенного введения пяти указанных уровней доз. Данные для доз 150 и 300 МЕ/кг представляют собой средние величины, полученные для шести здоровых субъектов исследования, тогда как другие дозы представляют данные от одного субъекта исследования. Кружки представляют данные, скорректированные относительно концентраций ЕРО исходного уровня, тогда как сплошные линии получены из подгонки данных и уравнений 1, 2 и 3, приведенных ниже.
Фигура 3: Фармакокинетические параметры для внутривенных и подкожных доз ЕРО.
Фигура 4: Схематичное представление фармакокинетической модели согласно данному изобретению, используемой для анализа профилей rHuEPO (CEPO) против времени. Определения используемых символов даны в разделе определений детального описания изобретения ниже.
Фигура 5А: Профили концентрации rHuEPO против времени после подкожного введения доз, равных 300, 450, 600 и 900 МЕ/кг. Точки ввода данных для каждой дозы представляют собой средние значения для пяти здоровых субъектов исследования. Данные скорректированы относительно концентраций ЕРО исходного уровня, тогда как сплошная линия получена из подгонки данных и уравнений 1,2 и 3.
Фигура 5В: Профили концентрации rHuEPO против времени после подкожного введения доз, равных 1200, 1350, 1800 и 2400 МЕ/кг. Точки ввода данных для каждой дозы представляют собой средние значения для пяти здоровых субъектов. Данные скорректированы относительно концентраций ЕРО исходного уровня, тогда как сплошная линия получена из подгонки данных и уравнений 1,2 и 3.
Фигура 6: Площадь под кривой временной зависимости концентрация rHuEPO в сыворотке (AUC) против дозы после подкожного введения восьми уровней доз, указанных на фигурах 4А и 4В. AUC рассчитывали методом Spline.
Фигура 7: Биодоступность (F) rHuEPO против дозы после подкожного ведения восьми указанных уровней доз. Значения F получали из первичных подгонок фармакокинетических данных к модели, как объясняется в тексте. Линейная регрессия давала r2 0,9713, наклон 0,00024952 и точку пересечения 0,3884.
Фигура 8: Значения биодоступности при подкожном введении rHuEPO.
Фигура 9: Профили концентрации rHuEPO в сыворотке против времени в ходе режимов многократного дозирования 150 МЕ/кг т.р.н. (вверху) и 600 МЕ/кг/неделю (внизу). Закрашенные кружки представляют средние данные, в то время как линии представляют значения, предсказанные в модели.
Фигура 10: Схематичное представление фармакодинамической модели, используемой для анализа ретикулоцитов, эритроцитов и концентраций гемоглобина. Определения используемых символов даны в разделе определений детального описания изобретения ниже.
Фигура 11: Профили среднего количества ретикулоцитов против времени для восьми указанных уровней подкожных доз rHuEPO.
Фигура 12А: Профили количества ретикулоцитов против времени после подкожного введения доз, равных 300, 450, 600 и 900 МЕ/кг. Данные для каждой дозы представляют собой средние значения для пяти здоровых субъектов исследования. Символы показывают экспериментальные данные, тогда как сплошные линии получали из подгонки данных и уравнений 4, 5, 6 и 7, представленных ниже.
Фигура 12В: Профили количества ретикулоцитов против времени после подкожного введения доз, равных 1200, 1350, 1800 и 2400 МЕ/кг. Данные для каждой дозы представляют собой средние значения для пяти здоровых субъектов. Символы означают экспериментальные данные, тогда как сплошные линии получали из подгонки данных и уравнений 4, 5, 6 и 7.
Фигура 13: Оцениваемые и задаваемые фармакодинамические параметры для воздействий ЕРО при подкожном введении.
Фигура 14: Профили концентрации гемоглобина против времени после однократного подкожного введения восьми указанных уровней доз rHuEPO. Закрашенные кружки представляют средние данные, тогда как сплошные линии означают предсказания в модели.
Фигура 15: Ответы, выраженные в количестве ретикулоцитов, эритроцитов и гемоглобина, после многократного подкожного дозирования 150 МЕ/кг т.р.н. rHuEPO. Закрашенные кружки представляют измеренные данные, и сплошные линии представляют предсказания в модели.
Фигура 16: Ответы, выраженные в количестве ретикулоцитов, эритроцитов и гемоглобина, после многократного дозирования 600 МЕ/кг/неделю rHuEPO. Закрашенные кружки представляют измеренные данные, и сплошные линии представляют предсказания в модели.
Фигура 17: Краткое описание клинических фармакокинетических исследований эпоэтина-альфа, которые внесли вклад в фармакокинетические и фармакодинамические данные для субъектов исследования в клинических исследованиях EPO-PHI-358, EPO-PHI-359, EPO-PHI-370 и EPO-PHI-373.
Фигура 18А: Краткое описание биофармацевтического исследования в ходе клинического исследования EPO-PHI-373.
Фигура 18В: Краткое описание биофармацевтического исследования в ходе клинического исследования EPO-PHI-370.
Фигура 18С: Краткое описание биофармацевтического исследования в ходе клинического исследования EPO-PHI-358.
Фигура 18D: Краткое описание биофармацевтического исследования в ходе клинического исследования EPO-PHI-359.
Фигура 19: Сводные фармакокинетические данные для клинических исследований EPO-PHI-358, EPO-PHI-359, EPO-PHI-370 и EPO-PHI-373.
Фигура 20: Краткое описание аналитических способов для клинических исследований EPO-PHI-358, EPO-PHI-359, EPO-PHI-370 и EPO-PHI-373.
Фигура 21: Средние±3D демографические и исходные параметры для субъектов, включенных в клинические исследования EPO-PHI-358 и EPO-PHI-359.
Фигура 22: Временные профили средней концентрации эпоэтина-альфа в сыворотке (не корректировали относительно ЕРО исходного уровня) для субъектов клинического исследования EPO-PHI-358.
Фигура 23: Временные профили средней концентрации эпоэтина-альфа в сыворотке (не корректировали относительно ЕРО исходного уровня) для субъектов клинического исследования EPO-PHI-359.
Фигура 24: Средние±3D (%CV) фармакокинетические параметры (клинические исследования EPO-PHI-358 и EPO-PHI-359).
Фигура 25: Взаимосвязь между средним значением±SD Сmax и дозой для субъектов, получавших однократный или многократный п/к режим дозирования в клинических исследованиях EPO-PHI-358 и EPO-PHI-359.
Фигура 26: Взаимосвязь между средним значением±SD CL/F и дозой для субъектов, получавших однократный или многократный п/к режим дозирования в клинических исследованиях EPO-PHI-358 и EPO-PHI-359.
Фигура 27: Временные профили среднего процента ретикулоцитов для четырехнедельного периода исследования (клинические исследования EPO-PHI-358 и EPO-PHI-359).
Фигура 28: Среднее изменение±SD в количестве гемоглобина относительно профилей исходного уровня после введения эпоэтина-альфа 150 МЕ/кг т.р.н. (N=5) и 600 МЕ/кг р.н. (N=5) в течение четырех недель (клинические исследования EPO-PHI-358 и EPO-PHI-359).
Фигура 29: Демографические данные субъектов клинического исследования EPO-PHI-370.
Фигура 30: Временные профили средней концентрации в сыворотке эпоэтина-альфа (не корректировали относительно исходного уровня ЕРО) у здоровых субъектов после получения 150 МЕ/кг т.р.н. (N=24) или 40000 ME р.н. (N=22) в течение четвертой недели дозирования (клиническое исследование EPO-PHI-370).
Фигура 31: Средние±SD (%CV) фармакокинетические параметры (клиническое исследование EPO-PHI-370).
Фигура 32: Профиль среднего изменения процента ретикулоцитов относительно исходного уровня.
Фигура 33: Профиль среднего изменения гемоглобина (г/дл) относительно исходного уровня.
Фигура 34: Средние±SD (%CV) фармакокинетические параметры, скорректированные относительно значения исходного уровня (клиническое исследование EPO-PHI-370).
Фигура 35: Средние±SD демографические данные субъектов клинического исследования EPO-PHI-373.
Фигура 36: Временные профили средней концентрации в сыворотке эпоэтина-альфа (не корректировали относительно исходного уровня ЕРО) у здоровых субъектов после получения 150 МЕ/кг т.р.н. (N=17) или 40000 ME р.н. (N=17) в течение четвертой недели дозирования (клиническое исследование EPO-PHI-373).
Фигура 37: Средние±SD (%CV) фармакокинетические параметры, скорректированные относительно значения исходного уровня (клиническое исследование EPO-PHI-373).
Фигура 38: Профиль среднего изменения процента ретикулоцитов относительно исходного уровня для субъектов клинического исследования EPO-PHI-373.
Фигура 39: Профиль среднего изменения гемоглобина (г/дл) относительно исходного уровня для субъектов клинического исследования EPO-PHI-373.
Фигура 40: Профиль среднего изменения общего количества красных кровяных клеток (×102/л) относительно исходного уровня для субъектов клинического исследования EPO-PHI-373.
Фигура 41: Средние±SD (%CV) фармакокинетические параметры, скорректированные относительно значения исходного уровня для субъектов клинического исследования EPO-PHI-373.
Фигура 42: Средние + SD (%CV) фармакокинетические параметры, скорректированные относительно значения исходного уровня для субъектов клинических исследований EPO-PHI-358, ЕРО-PHI-359, EPO-PHI-370 и EPO-PHI-373.
Фигура 43: Средняя AUC изменений процента ретикулоцитов как функция AUC (дни 0-29) эпоэтина-альфа (клинические исследования EPO-PHI-358 и EPO-PHI-359).
Фигура 44: Средняя AUC изменений процента ретикулоцитов как функция AUC (дни 0-29) эпоэтина-альфа (клинические исследования EPO-PHI-370 и EPO-PHI-373).
Фигура 45: Профиль среднего изменения уровня гемоглобина (г/дл) относительно исходного уровня для субъектов клинического исследования EPO-PHI-373.
Фигура 46: Профиль среднего изменения уровня гемоглобина (г/дл) относительно исходного уровня для субъектов клинического исследования EPO-PHI-370.
Фигура 47: Демографические и исходные характеристики 34 субъектов, которые прошли полное исследование ЕРО (EPO-PHI-373). 18 субъектов составляли часть группы 1, которым вводили режим дозирования ЕРО 150 МЕ/кг т.р.н., и 18 субъектов составляли часть группы 2, которым вводили ЕРО в дозе 40000 р.н. Демографические характеристики включают пол, возраст (годы), массу (кг), рост (см) и расу.
Фигура 48: Концентрации эпоэтина-альфа в сыворотке, не скорректированные относительно концентраций эндогенного эритропоэтина до введения дозы для субъектов группы 1 (150 МЕ/кг т.р.н), обозначенные треугольниками, и группы 2 (40000 ME р.н.), обозначенные кружками.
Фигура 49: Данные концентрации эпоэтина-альфа в сыворотке, скорректированные относительно концентраций эндогенного эритропоэтина до введения дозы для субъектов группы 1 (150 МЕ/кг т.р.н), обозначенные треугольниками, и группы 2 (40000 ME р.н.), обозначенные кружками.
Фигура 50: Средние (SD)[%CV] значения фармакокинетических параметров по данным для индивидуальных субъектов в группе 1 (150МЕ/кг т.р.н.) и группе 2 (40000 ME р.н.).
Фигура 51: Сводные данные среднего изменения (SD) процента ретикулоцитов относительно исходного уровня по дням исследования для эффективной популяции для всех субъектов в группах дозирования 1 (150 МЕ/кг т.р.н.) и группе 2 (40000 ME р.н.) для клинического исследования EPO-PHI-373.
Фигура 52: Профиль среднего изменения (SD) процента ретикулоцитов относительно исходного уровня по дням исследования для эффективной популяции для всех субъектов. Не закрашенные кружки представляют группу 1 (150 МЕ/кг т.р.н.) и закрашенные кружки представляют группу 2 (40000 ME р.н.). Полученные параметры перечислены на фигуре 51.
Фигура 53: Сводные данные среднего изменения (SD) гемоглобина (г/дл) относительно исходного уровня по дням исследования для эффективной популяции для всех субъектов в группе 1 (150 МЕ/кг т.р.н.) и группе 2 (40000 ME р.н.) для клинического исследования EPO-PHI-373.
Фигура 54: Профиль среднего изменения (SD) гемоглобина (г/дл) относительно исходного уровня ко дню исследования для эффективной популяции для всех субъектов. Не закрашенные кружки представляют группу 1 (150 МЕ/кг т.р.н.) и закрашенные кружки представляют группу 2 (40000 ME р.н.). Полученные параметры перечислены на фигуре 53.
Фигура 55: Сводные данные среднего изменения (SD) количества эритроцитов (×102/л) относительно исходного уровня по дням исследования для эффективной популяции для всех субъектов клинического исследования EPO-PHI-373.
Фигура 56: Профиль среднего изменения (SD) количества эритроцитов (×102/л) относительно исходного уровня по дням исследования для эффективной популяции для всех субъектов. Не закрашенные кружки представляют группу 1 (150 МЕ/кг т.р.н.) и закрашенные кружки представляют группу 2 (40000 ME р.н.). Полученные параметры перечислены на фигуре 55.
Фигура 57: Средние значения фармакодинамических параметров, скорректированные относительно исходного значения. %CV означает коэффициент вариации в процентах. Примечания представляют собой следующее: а AUC% ретикулоцитов в течение периода исследования, равного одному месяцу, скорректированного относительно исходного значения до введения дозы; b AUC гемоглобина в течение периода исследования, равного одному месяцу, скорректированного относительно исходного значения до введения дозы; с AUC концентрации красных кровяных клеток в течение периода исследования, равного одному месяцу, скорректированной относительно исходного значения до введения дозы; d отношения среднего значения параметра для всех субъектов при 40000 ME р.н. к значению при 150 МЕ/кг т.р.н.; е включая все субъекты женского пола в обеих группах лечения; f включая все субъекты мужского пола в обеих группах лечения; g статистически различны (р<0,05) между субъектами мужского и женского пола.
Фигура 58: Нежелательные явления, появляющиеся при лечении, по предпочтительному наименованию для индивидуальных субъектов клинического исследования EPO-PHI-373.
Фигура 59: Средние изменения количества железа, ферритина, насыщения трансферрина и других химических параметров сыворотки относительно исходного уровня, как по группам лечения, так и по дням исследования. В группе 1 вводили 150 ME ЕРО/кг т.р.н., и в группе 2 вводили 40000 МЕ/кг р.н. в клиническом исследовании EPO-PHI-373.
Фигура 60: Профиль среднего изменения количества железа, ферритина (нг/мл) относительно исходного уровня, как по группам лечения, так и по дням исследования для клинического исследования EPO-PHI-373. Группа 1 (150 МЕ/кг т.р.н) указана не закрашенными кружками и группа 2 (40000 МЕ/кг р.н.) указана закрашенными кружками. Параметры, полученные для ферритина, перечислены на фигуре 59.
Фигура 61: Сводные данные средних изменений измерений показателей жизненно важных функций относительно исходного уровня для индивидуумов в группе 1 (150 МЕ/кг т.р.н.) в клиническом исследовании EPO-PHI-373.
Фигура 62: Схематичное представление модели стимулирующего действия rHuEPO на эритропоэз.
Фигура 63: Фармакокинетические параметры после дозирования rHuEPO EPREX® и PROLEASE®.
Фигура 64: Фармакодинамические параметры после дозирования rHuEPO. Данные по ретикулоцитам для субъектов мужского и женского пола анализировали отдельно. Эти параметры могут отражать некоторые незначительные фармакодинамические различия по полу.
Фигура 65: Профиль фармакодинамических параметров после введения 600 МЕ/кг/т.р.н. EPREX® в течение 4 недель. Показаны профили “средняя концентрация rHuEPO - время”, “концентрация ретикулоцитов - время” и “концентрация эритроцитов - время”. Субъекты мужского пола обозначены закрашенными кружками, тогда как субъекты женского пола обозначены не закрашенными кружками.
Фигура 66: Профиль фармакодинамических параметров после введения 150 МЕ/кг/т.р.н. EPREX® в течение 1 месяца. Показаны профили “средняя концентрация rHuEPO - время”, “концентрация ретикулоцитов - время” и “концентрация эритроцитов - время”. Субъекты мужского пола обозначены закрашенными кружками, тогда как субъекты женского пола обозначены не закрашенными кружками.
Фигура 67: Профиль фармакодинамических параметров после введения PROLEASE® в однократной дозе 2400 МЕ/кг/мес. Показаны профили “средняя концентрация rHuEPO - время”, “концентрация ретикулоцитов - время” и “концентрация эритроцитов - время”. Субъекты мужского пола обозначены закрашенными кружками, тогда как субъекты женского пола обозначены не закрашенными кружками.
Фигура 68: Моделирование уровней гемоглобина после введения разных доз/режимов rHuEPO.
Фигура 69: Моделирование временных профилей ответов, выраженных в концентрации гемоглобина и эритроцитов, для режима дозирования ЕРО 600 МЕ/кг/нед. в течение 24 недель в сравнении с введением суммарной дозы 40000 МЕ/нед. rHuEPO субъектам с массой тела 50, 70 и 90 кг.
Фигура 70А: Моделирование временных профилей ответов, выраженных в концентрации ретикулоцитов, эритроцитов и гемоглобина, у больных злокачественной опухолью для режима дозирования ЕРО 150 МЕ/кг/т.р.н. в течение 12 недель.
Фигура 70В: Моделирование временных профилей ответов, выраженных в концентрации ретикулоцитов, эритроцитов и гемоглобина, у больных злокачественной опухолью для режима дозирования ЕРО 300 МЕ/кг/т.р.н. в течение 12 недель.
Фигура 7ОС: Моделирование временных профилей ответов, выраженных в концентрации ретикулоцитов, эритроцитов и гемоглобина, у больных злокачественной опухолью для режима дозирования ЕРО 450 МЕ/кг/т.р.н. в течение 12 недель.
Фигура 70D: Моделирование временных профилей ответов, выраженных в концентрации ретикулоцитов, эритроцитов и гемоглобина, у больных злокачественной опухолью для режима дозирования ЕРО 600 МЕ/кг/т.р.н. в течение 12 недель.
Фигура 70Е: Моделирование временных профилей ответов, выраженных в концентрации ретикулоцитов, эритроцитов и гемоглобина, у больных злокачественной опухолью для режима дозирования ЕРО 900 МЕ/кг/т.р.н. в течение 12 недель.
Фигура 71: Временные профили средней концентрации гемоглобина по дням для иммуносупрессированных (IS) и не иммуносупрессированных собак. Закрашенные кружки представляют ЕРО, введенный в дозе 50 МЕ/кг/день IS-собакам, закрашенные квадраты представляют ЕРО, введенный в дозе 50 МЕ/кг/день т.р.н. не-IS-собакам, не закрашенный треугольник представляет ЕРО, введенный в дозе 600 МЕ/кг/нед. IS-собакам, закрашенные треугольники представляют ЕРО, введенный в дозе 600 МЕ/кг/нед. не-IS-собакам, закрашенные ромбы представляют контроль в виде солевого раствора для IS-собак, и заштрихованные восьмиугольники представляют контроль в виде солевого раствора для не-IS-собак.
Фигура 72: Временные профили средней концентрации эритроцитов по дням для иммуносупрессированных и не иммуносупрессированных собак. Закрашенные кружки представляют ЕРО, введенный в дозе 50 МЕ/кг/день IS-собакам, закрашенные квадраты представляют ЕРО, введенный в дозе 50 МЕ/кг/день т.р.н. не-IS-собакам, не закрашенный треугольник представляет ЕРО, введенный в дозе 600 МЕ/кг/нед. IS-собакам, закрашенные треугольники представляют ЕРО, введенный в дозе 600 МЕ/кг/нед. не-IS-собакам, закрашенные ромбы представляют контроль в виде солевого раствора для IS-собак, и заштрихованные восьмиугольники представляют контроль в виде солевого раствора для не-IS-собак.
Фигура 73: ФК/ФД-модель для rHuEPO у обезьян.
Фигура 74: Подгонки временных профилей концентрации rHuEPO после введения трех однократных внутривенных доз и шести однократных подкожных доз EPREX®. Полученные параметры перечислены на фигуре 75.
Фигура 75: ФК-параметры у обезьян.
Фигура 76А: Временные профили средней концентрации ретикулоцитов для режимов дозирования 400 МЕ/кг, 1000 МЕ/кг и 2400 МЕ/кг.
Фигура 76В: Временные профили средней концентрации ретикулоцитов для режимов дозирования 5000 МЕ/кг, 20000 МЕ/кг и 40000 МЕ/кг.
Фигура 77: ФД-параметры у обезьян.
Фигура 78А: Временные профили средней концентрации эритроцитов для режимов дозирования 400 МЕ/кг, 1000 МЕ/кг и 2400 МЕ/кг.
Фигура 78В: Временные профили средней концентрации эритроцитов для режимов дозирования 5000 МЕ/кг, 20000 МЕ/кг и 40000 МЕ/кг.
Фигура 79А: Временные профили средней концентрации гемоглобина для режимов дозирования 400 МЕ/кг, 1000 МЕ/кг и 2400 МЕ/кг.
Фигура 79В: Временные профили средней концентрации гемоглобина для режимов дозирования 5000 МЕ/кг, 20000 МЕ/кг и 40000 МЕ/кг.
Фигура 80: ФД-параметры у человека.
Фигура 81: ФК/ФД-модель для rHuEPO у человека.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ.
Следует понимать, что данное изобретение не ограничено описанными здесь конкретными методологией, протоколами и реагентами и т.д., так как они могут варьировать. Также должно быть понятно, что используемая здесь терминология применяется только в целях описания конкретных вариантов и не означает ограничение рамок данного изобретения. Необходимо отметить, что когда здесь или в прилагаемой формуле изобретения используются формы в единственном числе, они включают указания на множественное число, если контекст четко не предписывает другое понимание.
Если не оговорено особо, все используемые здесь технические и научные термины имеют те же значения, которые обычно подразумеваются специалистом в данной области, к которому данное изобретение относится. Описаны предпочтительные способы, устройства и материалы, хотя любые способы и материалы, сходные или аналогичные описанным здесь способам и материалам, можно использовать в практике или тестировании согласно данному изобретению. Все используемые при экспертизе данной заявки материалы включены здесь в виде ссылок в полном объеме.
ОПРЕДЕЛЕНИЯ
а.m. До полудня, утро
AUC Площадь под кривой концентрации против времени
AUC(0-168) AUC концентрации эпоэтина-альфа в сыворотке от 0 времени до 168 часов
AUC(RETI) AUC изменения% ретикулоцитов от исходного уровня AUC(НЕМО) AUC изменения гемоглобина от исходного уровня
AUC(RBC) AUC изменения общего количества красных кровяных клеток от исходного уровня
BUN Азот мочевины крови
°С Градусы по шкале Цельсия
Сmах Максимальная концентрация
Cmin Минимальная концентрация до введения дозы AUC от 0 времени до 168 часов
CL/F Клиренс/биодоступность
см сантиметры
CRF Индивидуальная регистрационная карта (ИРК)
CV% Коэффициент вариации, относительное стандартное отклонение
д. День
D Доза (количество)
Дл Децилитр
г Грамм
час Часы
Hb Гемоглобин
Hct Гематокрит
HIV Вирус иммунодефицита человека
IRB Этический комитет
I Механизм отрицательной обратной связи
IV Внутривенно
кг Килограмм
л Литр
LDH Лактатдегидрогеназа
LOQ Предел количественного анализа
мкг Микрограмм
мг Миллиграмм
мин Минута
мл Миллилитр
N/A Не применимый/доступный
нг Нанограмм
нм Нанометр
ЯМР Ядерный магнитный резонанс
No. Номер
NS Статистически незначимый
ОТС Безрецептурный препарат
р.н. Один раз в неделю
QC Контроль качества
r Коэффициент корреляции
r2 Коэффициент смешанной корреляции
RBC Красные кровяные клетки
REF Ссылка
RETI Ретикулоцит
РИА Радиоиммуноанализ
rHuEPO Рекомбинантный эритропоэтин человека
RWJPRI Фармацевтический научно-исследовательский институт Роберта Вуда Джонсона
п/к Подкожно
SD Стандартное отклонение
SE Стандартная ошибка
SEM Стандартная ошибка среднего
SGOT Сывороточная трансаминаза глутаминовой-щавелевоуксусной кислоты (AST)
SGPT Сывороточная трансаминаза глутаминовой-пировиноградной кислоты (ALT)
tmax Время максимальной концентрации
т.р.н Три раза в неделю
TIBC Общая способность связывать железо
t1/2 Период полувыведения
WBC Белые кровяные клетки
WHOART Терминология нежелательных реакций Всемирной организации здравоохранения
ЕРО по данному здесь определению относится к любой молекуле, которая специфично стимулирует терминальную дифференцировку эритроцитов из гематопоэтических стволовых клеток и стимулирует продукцию гемоглобина. В качестве примера, но не ограничивая рамки данного изобретения, молекулы ЕРО могут включать небольшие органические или неорганические молекулы, синтетические или природные полипептиды из аминокислот, очищенный белок из рекомбинантных или природных систем экспрессии, или синтетические или природные последовательности нуклеиновых кислот, или любые химические производные вышеупомянутого. Конкретные примеры эритропоэтина включают эпоэтин-альфа (EPREX®, ERYPO®), новый белок, стимулирующий эритропоэз (NESP) (гипергликозилированный аналог рекомбинантного эритропоэтина человека (эпоэтина), описанный в заявке на выдачу европейского патента ЕР640619), гибридные белки аналога эритропоэтина человека - сывороточного альбумина человека, описанные в международной заявке на патент WО 9966054, мутанты эритропоэтина, описанные в международной заявке на патент WO 9938890, эритропоэтин-омега, который можно получить из фрагмента рестрикции Ара I гена эритропоэтина человека, описанный в патенте США №5688679, измененный гликозилированный эритропоэтин человека, описанный в международной заявке на патент WО 9911781, ПЭГ-конъюгированные аналоги эритропоэтина, описанные в WO 9805363 или патенте США 5643575. Конкретные примеры клеточных линий, модифицированных для экспрессии эндогенного эритропоэтина человека, описаны в международных заявках на патент WO 9905268 и WO 9412650. Предпочтительной формой ЕРО является очищенный рекомбинантный ЕРО. Например, очищенный рекомбинантный ЕРО распространяют под торговыми марками EPREX®, PROLEASE® или ERYPO®. В частности, эпоэтин-альфа (EPREX®, ERYPO®) представляет собой стерильно чистый, бесцветный, водный раствор для инъекций, который поставляется в предварительно заполненных одноразовых шприцах, содержащих либо 4000, либо 10000 ME эпоэтина-альфа (рекомбинантный эритропоэтин человека) и 2,5 мг/мл сывороточного альбумина человека в 0,4 мл (4000 МЕ-шприц) или 1,0 мл (10000 МЕ-шприц) фосфатного буфера.
ЕРО также включает такие белки, которые обладают биологической активностью эритропоэтина человека, а также аналоги эритропоэтина, изоформы эритропоэтина, миметики эритропоэтина, фрагменты эритропоэтина, гибридные белки эритропоэтина, агонисты рецептора эритропоэтина, почечный эритропоэтин, олигомеры и мультимеры указанного выше, гомологи указанного выше и мутантные формы указанного выше, независимо от биологической активности таковых, и, кроме того, независимо от способа их синтеза или их производства, включающего, но не ограниченного, способами получения, например, ЕРО-подобных белков природного происхождения, рекомбинантных, синтетических, трансгенных и получаемых при активации генов. Такие ЕРО и методики получения описаны, например, в патентах США №5716644, 5674534, 5916597, 6048524, 5994127, 5955422, 5856298, 5756349, 5621080, 5547933 и 5441868.
Подкожное (п/к) введение ЕРО согласно данному здесь определению относится к доставке желаемой дозы ЕРО посредством устройства доставки препарата. Устройство доставки препарата может проникать сквозь эпидермис индивидуума, которого необходимо лечить, и в результате приводит к введению желаемой дозы ЕРО в ткани индивидуума. Устройство доставки согласно данному изобретению может включать, но не ограничено этим, любые традиционные подкожные шприцы с иглами, пневматические инъекторы без игл, безыгольные инъекторы или безыгольные инъекторы под давлением газа (смотри, например, патенты США №5730723, 5891086, 5957886 и 5851198).
Пороговый уровень согласно данному здесь определению относится к концентрации ЕРО в сыворотке на таком уровне, выше которого концентрации ЕРО, поддерживаемые в сыворотке, будут способствовать дифференцировке юных эритроцитов в зрелые эритроциты. Напротив, концентрации ЕРО в сыворотке, поддерживаемые ниже порогового уровня, не будут приводить к созреванию ретикулоцитов в эритроциты.
Пациент, как здесь описано, включает индивидуумов, которые требуют режима лечения вследствие состояния болезни или хотят увеличить гематокрит, количество эритроцитов или кислородную емкость.
Инфраструктурный домен, согласно данному здесь описанию, включает операционные аспекты системы/ которые в значительной степени прозрачны для пользователя системой. Примеры инфраструктурных доменных элементов включают функции, которые осуществляют взаимосовмещаемость, поддержку транзакций, поддержку структур данных, и функции безопасности и т.д.
Бизнес-домен согласно данному здесь описанию включает все операции и логическую схему, касающиеся действительных функциональных возможностей системы, когда это относится к реальным бизнес-моделям и способам, выполняемым системой. Например, если бизнес-модель предоставляла режим дозирования ЕРО и реализацию дозированного ЕРО, логическая схема бизнес-домена включала все аспекты ведения бизнеса. Аспекты включали производство, закупку и реализацию ЕРО, проверку и установление кредита клиентам и т.д. Важно отметить, что бизнес-домен не включает поддержку операций, связанных с инфраструктурой.
Предпочтительный вариант данного изобретения описывает режим дозирования, при котором ЕРО вводят примерно в дозе 40000 МЕ/кг один раз в неделю в течение двух недель подряд. Первая доза ЕРО способствует продукции ретикулоцитов из клеток предшественников эритроцитов. Вторую дозу ЕРО вводят так, чтобы она совпала с фармакодинамическим профилем ретикулоцитов у пациента. Вторую дозу будут вводить через 6-10 дней после начальной дозы, и предпочтительно в то время, когда концентрация ретикулоцитов достигнет пика после первой дозы ЕРО.
Указанный аспект данного изобретения относится к стимуляции пролиферации предшественников эритроидных клеток посредством ЕРО и высвобождения ретикулоцитов в циркуляцию крови. После введения однократной дозы ретикулоциты достигали пика в период времени от 6 до 12 дней и возвращались к уровням до введения дозы в период времени до 15 дней. Продолжительность жизни клеток в стадии ретикулоцитов составляет примерно от 1 до 2 дней в циркулирующей крови. Поэтому специалист в данной области будет ожидать, что юные эритроциты появятся в циркулирующей крови примерно от 7 до 14 дней после дозирования. Предполагается, что ЕРО необходим для созревания юных эритроцитов в зрелые эритроциты в течение от 7 до 14 дней после начала терапии ЕРО. Зрелые эритроциты у здоровых субъектов имеют среднюю продолжительность жизни 120 дней. Продолжительность жизни зрелых эритроцитов может быть короче у пациентов с хронической анемией или при других патологических состояниях.
Другой предпочтительный вариант данного изобретения включает в себя режим дозирования, при котором ЕРО вводят в виде цикла дозирования, включающего два или несколько циклов, в которых ЕРО вводят один раз в неделю в течение двух недель подряд. Продолжительность времени между циклами дозирования предпочтительно совпадает с продолжительностью жизни эритроцитов. Продолжительность жизни эритроцитов обычно составляет 120 дней, однако время может варьировать вследствие состояния болезни или режима лечения. Поэтому предпочтительно следующий цикл ЕРО предпочтительно будет вводится через 60-120 дней после предыдущей дозы.
Предпочтительный вариант данного изобретения описывает фармакокинетическую модель концентраций ЕРО в сыворотке у здоровых добровольцев после внутривенного (в/в) и подкожного (п/к) дозирования, а также фармакодинамическую модель изменений количества ретикулоцитов, эритроцитов и концентраций Нb в крови, вызванных п/к введением ЕРО. Кроме того, конкретные примеры будут следовать использованию ФК/ФД-моделей согласно данному изобретению. Примеры включают: определение вероятных различий в ответах на уровне гемоглобина для различных режимов дозирования rHuEPO; оценку влияния массы тела субъекта на ожидаемый ответ на ведение терапии rHuEPO и исследование того, отвечают ли больные злокачественной опухолью на лечение rHuEPO в той же степени, как и здоровые добровольцы.
ФАРМАКОКИНЕТИЧЕСКАЯ МОДЕЛЬ И АНАЛИЗ
Моделирование, описывающее данные, и фармакокинетику при п/к введении rHuEPO получали на основе клинических исследований, проводимых Фармацевтическим научно-исследовательским институтом Роберта Вуда Джонсона (Robert Wood Johnson Pharmaceutical Research Institute, RWJPRI). Исследование состояло из двух сравнительных, открытых, рандомизированных, параллельных, плацебо-контролируемых исследований на здоровых добровольцах, которым rHuEPO вводили в виде восьми однократных п/к доз EPREX®: 300, 450, 600, 900, 1200, 1350, 1800, 2400 МЕ/кг и в виде режимов многократного дозирования: 150 МЕ/кг три раза в неделю в течение четырех недель и 600 МЕ/кг один раз в неделю в течение четырех недель. Каждая группа лечения состояла из 5 субъектов.
Измеренные концентрации rHuEPO после введения rHuEPO корректировали относительно значений исходного уровня, поскольку используемый радиоиммуноанализ не мог отличить эндогенный ЕРО от rHuEPO. Концентрацию ЕРО исходного уровня для каждого субъекта определяли усреднением значений до введения дозы (10, 20 и 30 мин). Полученное значение вычитали из значений после введения дозы в каждой временной точке, чтобы получить скорректированную концентрацию rHuEPO в сыворотке. Средние значения скорректированных концентраций для всех субъектов использовали для анализа данных. Какие-либо измерения ниже предела, выявляемого при анализе (7,8 МЕ/л), не использовали в качестве точки ввода данных. Данные по внутривенному введению также корректировали относительно уровней ЕРО исходного уровня.
Исходя из предварительного анализа данных по в/в ведению, полученных из литературы, было обнаружено, что адекватной является однокомпартментная модель. Сообщалось, что распределение rHuEPO не линейно, главным образом, в результате зависимого от дозы снижения клиренса (смотри, например, Macdougall et al., 1991. Clin. Pharmacokinet. 20: 99-113). Поэтому для того, чтобы описать распределение rHuEPO, использовали функцию Михаэлиса-Ментен. Подгоняли данные для концентраций rHuEPO при в/в введении (СEPO=Ap/Vd) против времени и следующее уравнение:
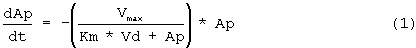
где Ар означает количество rHuEPO в организме, Vmax означает возможности процесса, Km означает константу аффинности или концентрацию rHuEPO в плазме, при которой скорость элиминации достигает половины Vmax, и Vd означает объем распределения.
Временные профили концентрации при в/в введении для разных доз показаны на фигуре 2. Для того чтобы описать кинетику rHuEPO, использовали однокомпартментную модель с нелинейным распределением. Примечательно, что двухкомпартментная модель может лучше подходить данным по в/в введению в ранний период; однако она будет давать большую сложность для подгонки данных в целом. Поэтому выбрали однокомпартментную модель, так как она давала приемлемые подгонки и удовлетворяла цели. Также, поскольку rHuEPO является белком с М.м. 30 кДа, можно ожидать, что он ограничен внутрисосудистым компартментом, таким образом, подтверждая выбор однокомпартментной модели.
Параметры, полученные подгонкой, перечислены на фигуре 3. Полученные значения Vd (0,0558 л/кг) и Vmax/Km (т.е. CL при низких дозах: 0,0066 л/час/кг) находятся в пределах оценок, сообщаемых в литературе (смотри, например, Macdougall et al. выше, и Lappin et al., 1996, Clin. Lab. Haem. 18: 137-1458). Высокое значение Km свидетельствует о том, что распределение ЕРО является только умеренно нелинейным, и дозозависимая элиминация будет становиться значительной только при высоких дозах. Исследования на крысах привели к предположению о том, что связывание rHuEPO с рецепторами в костном мозге и селезенке вносит вклад в насыщаемую элиминацию rHuEPO (смотри, например, Kato et al., 1997, J. Pharmacol. Exp. Ther. 283: 520-27).
Обнаружено, что фармакокинетика п/к-введенного ЕРО лучше всего характеризуется моделью двойной абсорбции с быстрым всасыванием нулевого порядка большей части лекарственного средства (87% абсорбированного средства) с последующим медленным всасыванием первого порядка небольшой части лекарственного средства (13% абсорбированного средства). Обнаружено, что биодоступность увеличивается с дозой (в диапазоне от 46 до 100%), в значительной степени внося вклад в нелинейность кинетики. Дифференциальные уравнения модели (фигура 4) выглядят следующим образом:
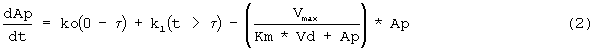
где
ko=0 когда t>τ

k1=0 когда t≤τ
и
k1=ka*F*Fr*Доза*e-(ka*(t-τ)) когда t>τ
Биодоступность rHuEPO была принята равной 100% при в/в введении. Биодоступность после п/к дозирования представлена параметром F. Соответственно количество rHuEPO, связанного с процессом первого порядка, описывается формулой F*Fr*Доза, тогда как количество, поглощаемое в ходе процесса нулевого порядка, описывается формулой F*(l-Fr)* Доза.
Все подгонки делали, используя компьютерную программу ADAPT II (смотри, например, Argenio et al., 1998. ADAPT II User's Guide, Biomedical Simulations Resource, University of Southern California, Los Angeles), хотя можно использовать любую подходящую компьютерную программу. Оценку параметров делали, используя подгонку наименьших квадратов способом максимального подобия, и используемая обобщенная дисперсионная модель наименьших квадратов задана уравнением
V(1)=Inter2*Y(1)сигма+0,0001, где V(1)=Ap/Vd
Была выполнена одновременная подгонка восьми однократных п/к доз и пяти уровней в/в доз. Во время начальных подгонок Vmax, Km, Vd, ka, Fr и параметры дисперсии сохраняли постоянными при всех дозах, тогда как τ и F давали возможность варьировать с изменением дозы. Результаты показали, что τ может быть установлено в виде единственного значения вплоть до дозы 1350 МЕ/кг, тогда как для двух последних доз оптимальным было более высокое значение τ. Обнаружено, что биодоступность F увеличивалась пропорционально дозе в пределах тестированных доз и была описана линейным уравнением (r2=0,9713) следующим образом:
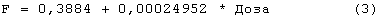
Для линейной регрессии значение F для дозы 450 МЕ/кг исключали, так как оно, по-видимому, является выбросом. Конечные подгонки выполняли, используя данную функцию, чтобы установить значения F по дозам, и фиксировали значение τ, равное 44 часам, для всех доз, за исключением двух последних, для которых τ устанавливали равным 60 часам. Это дало возможность описать все данные, используя единственный набор параметров для Vmax, Km, Vd, ka, Fr и параметров дисперсии. Данные многократного дозирования 600 МЕ/кг/неделю моделировали, используя те же параметры с F, установленным при 60%, так как это давало оптимальную подгонку. Для режима 150 ME/кг т.р.н. F задавали равным 25%. Кроме того, для указанной низкой дозы необходимо было увеличить Vd (0,1193 л/кг) и установить τ, равное 18 час.
На фигурах 5А и 5В показаны временные профили средней концентрации rHuEPO для разных однократных п/к доз. Визуальный просмотр данных по п/к введению четко свидетельствует о кинетике “флип-флоп”, так как конечный наклон гораздо более плоский по сравнению с моноэкспоненциальным снижением при в/в введении. Поэтому была задана константа скорости абсорбции первого порядка, чтобы охватить конечную фазу. Данные также показывают, что концентрации rHuEPO быстро достигают пиковой Сmax за один день, тем самым свидетельствуя, что также должен быть более быстрый процесс абсорбции. Указанная быстрая вогнутая кривая объясняется процессом поглощения нулевого порядка. Было обнаружено, что концевые наклоны по всем дозам параллельны, свидетельствуя о том, что единственное значение ka скорости абсорбции первого порядка может отвечать за эту фазу для всех доз. Часть дозы, связанная с медленным процессом абсорбции первого порядка, составляла только 13,1%. Таким образом, большая часть биодоступной дозы быстро поглощается в пределах 2-3 дней посредством компонента нулевого порядка. Значение ka скорости абсорбции первого порядка отвечает за медленное непрерывное высвобождение rHuEPO из места подкожного введения. Сходная модель абсорбции с двумя скоростями была использована для характеристики кинетики абсорбции другой макромолекулы - IL-10 после п/к дозирования (смотри, например, Radwanski et al., 1988. Pham. Res. 15: 1895-1902).
На фигуре 6 представлен график AUC против дозы для разных однократных п/к доз. Более значительное, чем пропорциональное, увеличение AUC с увеличением дозы свидетельствует о том, что либо CL, либо биодоступность, либо оба параметра изменяются с дозой. Было обнаружено, что элиминация rHuEPO является только умеренно нелинейной. С другой стороны, было обнаружено, что F увеличивается линейно с дозой (фигура 7) и оказывается главным фактором, ответственным за диспропорциональное увеличение AUC с дозой. В случае двух последних самых высоких доз в нелинейность также вносил вклад сниженный CL. На фигуре 8 перечислены значения F для всех доз, полученные деконволюцией и подгонкой предложенной модели к данным для индивидуальных доз. Оценки, полученные с использованием обоих способов, очень сходны, свидетельствуя таким образом, что фармакокинетическая модель согласно данному изобретению может адекватно объяснять нелинейность вследствие изменений значений F.
Причина неполной и нелинейной биодоступности rHuEPO, введенного п/к, не известна. Например, белок IL-10 проявляет только 42% биодоступности при п/к дозировании с потерей, предположительно являющейся результатом действия протеолитических ферментов (смотри, например, ту же ссылку). В свою очередь, эти ферменты могут насыщаться при более высоких концентрациях пептидных или белковых субстратов. Двойной процесс абсорбции может быть следствием роли лимфатических сосудов в контролировании доступа макромолекул после п/к дозирования. Фаза быстрой ранней абсорбции может быть вызвана прохождением основной части дозы в местные кровеносные сосуды, в то время как более поздняя фаза может быть связана с медленным поступлением через лимфатическую систему.
Моделирование зависимостей концентраций rHuEPO против времени для режимов многократного дозирования 150 МЕ/кг т.р.н. и 600 МЕ/кг/нед. показано на фигуре 9. Фармакокинетическая модель согласно данному изобретению в том случае, когда большинство параметров имеют фиксированное значение, полученное при подгонке однократных доз, точно описывает данные многократного дозирования. Для режима дозирования более низких многократных доз 150 МЕ/кг значение Vd пришлось увеличить. Это может быть следствием нелинейности, затрагивающей распределение в том случае, когда биодоступное количество так мало. Такое же изменение Vd также может обеспечить лучшую подгонку в/в дозы 10 МЕ/кг, когда максимальные концентрации находятся в таких же пределах.
ФАРМАКОДИНАМИЧЕСКАЯ МОДЕЛЬ И АНАЛИЗ
Данные, описывающее фармако динамику количества ретикулоцитов, эритроцитов и уровней Нb в крови, получали при клинических исследованиях в RWJPRI. Исследование состояло из двух сравнительных, открытых, рандомизированных, параллельных, плацебо-контролируемых исследований на здоровых добровольцах, которым rHuEPO вводили в виде восьми однократных п/к доз EPREX®:
300, 450, 600, 900, 1200, 1350, 1800, 2400 МЕ/кг и в виде режимов многократного дозирования: 150 МЕ/кг три раза в неделю в течение четырех недель и 600 МЕ/кг один раз в неделю в течение четырех недель. Каждая группа лечения состояла из 5 субъектов. Конечными измеряемыми параметрами были количество ретикулоцитов, эритроцитов в крови и количество гемоглобина.
Фармакодинамические данные описывали, используя модель продукции и потери клеток, которая описывает изменения количества клеток во времени после введения rHuEPO. Согласно данной модели предполагается, что все клетки, вовлеченные в процесс эритропоэза, продуцируются в режиме нулевого порядка (k0): они живут фиксированный период времени, по окончании которого они погибают, или превращаются в другие клетки. В результате потеря клеток происходит со скоростью, которая точно равна скорости, с которой они образовывались, за исключением того, что их элиминация замедляется к периоду времени, который равен продолжительности жизни клеток. Предполагается, что продолжительность жизни любого отдельного набора клеток постоянна относительно времени и является одной и той же для каждой клетки данного типа.
На фигуре 10 дано схематичное представление ФД-модели согласно данному изобретению. Указаны продолжительности жизни каждой из клеток - клетки предшественника (ТР), ретикулоцита (RL) и эритроцита (RBCL). Отделения отражают пулы эритроидных клеток-предшественников (Р), ретикулоцитов (R), эритроцитов (RBC) и гемоглобина (Нb) в крови. Стимуляция эритропоэза введением rHuEPO (Cp(t)) задается функцией Хилла (S(t)), действующей на продукцию клеток-предшественников в костном мозге.
Дифференциальные уравнения модели представлены нижеследующими уравнениями:

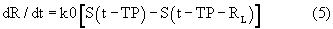
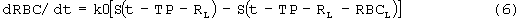
где
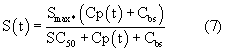
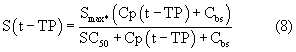
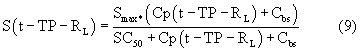
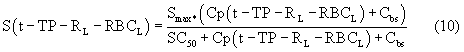
Smax означает максимально возможную стимуляцию продукции ретикулоцитов посредством rHuEPO, и SC50 означает концентрацию rHuEPO в плазме, которая вызывает полумаксимальную стимуляцию. Параметр Ks определяется как Ks=k0*Smax, и он означает максимально возможную скорость продукции клеток при стимуляции rHuEPO.
SC50 также использовали как пороговую концентрацию rHuEPO для образования RBC из ретикулоцитов. Предполагали, что когда концентрации rHuEPO падают ниже этого предела, ретикулоциты не превращаются в эритроциты. Для этого процесса функция стимуляции для уравнения модели (6) скорректирована следующим образом:
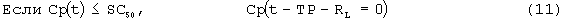
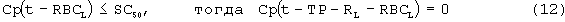
Условия исходного уровня (стационарные уровни) определяют следующим образом:
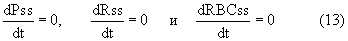
В результате начальное условие само определяет стационарные уровни.
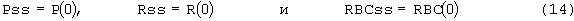
Стационарные условия для предшественников можно определить в явной форме следующим образом:

Так как условие исходного уровня для камеры предшественников неизвестно, значение 14*1010 клеток/л/час было установлено с использованием приведенного выше уравнения и оценок Кin из литературы (19).
Изменение уровней гемоглобина моделировали, просто используя фактор пропорциональности Hbcell, который представляет собой содержание гемоглобина на клетку (ретикулоцита или эритроцита).

Как изображено на фигуре 1, эритропоэз включает каскад событий. Камера предшественников в модели представляет все клетки в костном мозге, вовлеченные в этот процесс, которые в конечном итоге превращаются в ретикулоциты. Время ТР, таким образом, служит в качестве средней продолжительности времени, желаемого для того, чтобы самые ранние клетки-предшественники, стимулированные rHuEPO, подверглись каскаду процессов дифференцировки, чтобы, наконец, превратиться в ретикулоциты. Другими словами, оно отвечает за время задержки, наблюдаемое для ретикулоцитов, стимулированных rHuEPO. После того как ретикулоцит образован, он существует в течение времени, равного RL, и в этой точке он превращается в эритроцит. Предполагается, что основной путь, которым ретикулоцит может исчезнуть, заключается в превращении в эритроцит, за исключением подпорогового условия. Модель не объясняет случайное распределение клеток, такое как при кровотечении. Поэтому скорость продукции и элиминации всех этих клеток может быть представлена одной константой скорости нулевого порядка k0. После того как продуцируются эритроцит, он, в свою очередь, живет в течение периода RBCL дней, после которого он исчезает из крови.
Превращение ретикулоцитов в эритроциты частично происходит в костном мозге, а частично в крови, в течение периода, равного 72 часам (смотри, например, Jusko, 1994. Clin. Pharmacol. Ther. 56: 406-19). Ретикулоциты формируются в костном мозге и при диапедезе они просачиваются в циркуляцию, где эти незрелые клетки развиваются в течение периода 24-48 ч перед тем как трансформируются в эритроциты (смотри, например, Guyton, 1996. Textbook of Medical Physiology, W.B.Saunders, Philadelphia). Так как известно, что rHuEPO стимулирует преждевременное высвобождение ретикулоцитов из костного мозга, предположили, что вся продолжительность жизни этих новорожденных клеток, равная 72 часам, при стимуляции rHuEPO проходит в крови. Поэтому продолжительность жизни ретикулоцитов RL была установлена равной 72 часам. Продолжительность жизни эритроцитов RBCL была установлена равной 120 дням, и содержание Нb в клетке установлено равным 29,5 пг/клетку на основании значений из литературы (смотри, например, Jusko, выше и Guyton, выше). Как ретикулоциты, так и эритроциты, предположительно, вносят вклад в общее содержание Нb в крови.
Кроме того, модель продукции новых клеток (скорость Кs) и потери клеток использовали для получения оценок SC50 и Ks. Ретикулоциты и эритроциты, предположительно, имеют продолжительности жизни 3 дня (72 часа) и 120 дней (2880 часов). Время лаг-периода появления ретикулоцитов в крови оценили посредством введения камеры предшественников, представляющих клетки-предшественники. Получили значение Ks 0,3709×1010 клеток/л/час, дающее Smах, приблизительно равное 2, которое отражает среднюю максимальную стимуляцию эритропоэза. Полученное значение SC50 составляло 23 МЕ/л, свидетельствуя о том, что для поддержания стимуляции были достаточны низкие концентрации rHuEPO в сыворотке. Было обнаружено, что для превращения ретикулоцитов в эритроциты в крови важным является пороговая концентрация, предположительно равная SC50 rHuEPO. Однократные дозы rHuEPO вплоть до 900 МЕ/кг были неспособны поддерживать уровни rHuEPO выше указанного порогового значения во время продукции эритроцитов. Это объясняет отсутствие увеличения количества эритроцитов несмотря на стимуляцию продукции ретикулоцитов после введения четырех более низких однократных п/к доз, которые поэтому приводили только к очень небольшому изменению в Нb в течение короткого периода времени. Также идея порога объясняет лучшую способность уровней Hb к реагированию на многократные режимы дозирования rHuEPO, которые вызывают значительное повышение уровней Нb. Параллельный характер исследования давал вариабельность в ответах на разные дозы rHuEPO, но один набор параметров обеспечивал приемлемую характеристику ответов в пределах доз и режимов.
На фигуре 11 показан профиль среднего количества ретикулоцитов против времени для всех однократных п/к доз. Количество ретикулоцитов слегка увеличивается по сравнению с уровнями до введения дозы сразу в первой точке отбора образца. Этот уровень сохраняется 3-4 дня, после чего количества начинают монотонно возрастать вплоть до того момента, как достигается пик примерно около 200-300 часов. Затем количества начинают быстро снижаться с кажущимся временем полужизни 25 часов, достигая исходных уровней к 22 дню (528 часов).
Данные и модельные подгонки показаны на фигурах 12А и 12В. Оценки параметров, полученные подгонкой фармакодинамического уравнения к данным, представлены на фигуре 13. Получили Ks, равную 0,3709×1010, которая пересчитывается как 4,451×1010 клеток/день, полагая, что объем крови равен 5 л. Известно, что 1% всех эритроцитов у здоровых людей ежедневно разрушается и заменяется ретикулоцитами, давая скорость продукции эритроцитов (k0) 2-3×1011 клеток/день. Поэтому оцененная Smax равна 1,5-2,2, которая свидетельствует о том, что rHuEPO может давать максимально от 2,5 до 3,2-кратное увеличение скорости нулевого порядка продукции ретикулоцитов, относительно умеренная степень стимуляции которых является причиной медленного и ограниченного повышения в крови.
Полученное значение SC50, равное 22,58 МЕ/л, отражает концентрацию rHuEPO в сыворотке, необходимую для того, чтобы вызвать полумаксимальную стимуляцию. До тех пор пока концентрации rHuEPO в сыворотке поддерживаются выше этого уровня, количество клеток должно оставаться выше исходного уровня. Нормальные клетки-предшественники эритроцитов независимо от происхождения экспрессируют менее 1000 ЕРО-рецепторов на клеточной поверхности. Связывание ЕРО с таким рецептором вызывает события сигнальной трансдукции, которые, в конце концов, приводят к стимуляции дифференцировки и пролиферации предшественников эритроцитов в костном мозге (смотри, например, Lappin, выше). Кроме того, ЕРО ускоряет высвобождение ретикулоцитов из костного мозга, приводя к повышенному количеству ретикулоцитов и эритроцитов в сыворотке (там же). Незначительное увеличение уровней ретикулоцитов, наблюдаемое после нулевой временной точки, может быть вызвано ранним высвобождением незрелых ретикулоцитов из костного мозга, которое не объясняется моделью в указанных ранних временных точках.
Полученное низкое значение SC50, равное 22,58 МЕ/л, отражает тот факт, что имеется небольшое количество рецепторных мест на эритропоэтических клетках, которые могут быстро насыщаться так, что высокие дозы с быстрой доставкой могут приводить к значительной потери биодоступного rHuEPO впустую. Увеличение дозы или более медленная доставка способствует поддержанию уровней rHuEPO выше SC50 в течение более длительного времени, и поэтому имеет место увеличение степени и продолжительности стимуляции продукции ретикулоцитов. Эта концепция объясняет результаты двух недавних клинических исследований, которые показывают, что п/к-введенный rHuEPO более эффективен, чем в/в дозирование, для стимуляции продукции эритроцитов. Несмотря на более низкую биодоступность п/к дозы с пролонгированной абсорбцией приводят в результате к более эффективной стимуляции продукции эритроцитов (смотри Kaufmann et al., 1998. N. Engl. J. Med. 339: 578-83 и Besarab, et al., 1992. Am. Soc. Nephrol. 2: 1405-12).
На фигурах 12А и 12В показано, что фармакодинамические данные для нескольких уровней доз довольно варьируют, и это отражается в изменчивости степени стимуляции с увеличением дозы. Например, дозы 600 и 1200 МЕ/кг вызывают немного более высокие количества клеток по сравнению с дозами 900 и 1350 МЕ/кг. Однако характер возвращения к исходному уровню выглядит аналогичным при всех дозах. Во всяком случае, модели согласно данному изобретению улавливают общее направление ответов, учитывая изменчивую природу данных и тот факт, что один единственный набор параметров может адекватно описать фармакодинамические данные при всех дозах. Можно получить более хорошую подгонку данных при каждом уровне доз, давая возможность Ks и SC50 изменяться для каждой группы. Это было бы целесообразно, так как это были параллельные группы доз, но затем параметры надо было бы усреднять в целях обобщения.
На фигуре 14 показан ответ на уровне Нb против времени после 8 однократных п/к доз и имитационное моделирование. Очень небольшое изменение происходит, если вообще происходит, в уровнях Нb, и это может объясняться тем фактом, что концентрации rHuEPO понижаются близко к пороговому пределу к 7 дню, таким образом, препятствуя превращению новообразованных ретикулоцитов в эритроциты. Поэтому небольшое повышение уровней Нb вызвано только увеличением количества ретикулоцитов для доз до 900 МЕ/кг, после чего имеет место умеренное увеличение в количестве эритроцитов, также вносящее вклад в соответственно более высокие уровни Нb. Указанная концепция порога может объяснить дозозависимое увеличение в ответе ретикулоцитов без значительных увеличений ответов на уровне гематокрита или гемоглобина после однократных внутривенных доз до 1000 МЕ/кг у здоровых добровольцев, как сообщалось Flaharty et al., выше.
На фигурах 15 и 16 показан результат моделирования изменений в количестве ретикулоцитов, эритроцитов и Нb после многократного п/к дозирования rHuEPO. Охватываются фармакодинамические ответы для обоих режимов дозирования. Концентрации rHuEPO поддерживаются выше порога большую часть периода времени, давая ретикулоцитам возможность превратиться в эритроциты, что отражается в виде устойчивого увеличения уровней Нb после многократного дозирования, в противоположность однократным дозам. Подгонки разных однократных п/к доз и моделирование многократных п/к доз показывают, что поддержание концентраций rHuEPO выше SC50 при введении нескольких более малых доз имеет тенденцию обеспечивать постоянное увеличение Tib-уровней, в противоположность введению такой же общей дозы в виде однократной дозы. В последнем случае концентрации падают ниже SC50 намного быстрее, вызывая более слабый ответ на уровне ретикулоцитов и, что еще более важно, неэффективный ответ на уровне Нb.
В заключение, ФК/ФД-модели согласно данному изобретению демонстрируют важность дозы, режима дозирования и пути введения для контролирования ответов на rHuEPO и могут быть использованы в качестве полезного средства для расчета оптимальных доз rHuEPO и времени повторного введения для разных состояний. Кроме того, данное изобретение предоставляет основанную на компьютерных программах систему для того, чтобы адаптировать дозирование и режим лечения ЕРО так, чтобы пациент получал оптимальную пользу, на основе, например, повышенной продукции гемоглобина и ретикулоцитов, или чтобы предоставить режим дозирования, например, один раз в неделю или один раз каждые две недели, который подходит для нужд пациента.
Бизнес-способ
В отдельном варианте данного изобретения может быть осуществлен бизнес-способ, имеющий отношение к предоставлению режима дозирования ЕРО и реализации дозированного ЕРО. В конкретном варианте указанный способ может быть выполнен посредством компьютерных систем согласно данному изобретению. Например, пользователь (например, здоровый практикующий специалист, такой как врач или фармацевт) может получить доступ к компьютерным системам данного изобретения через компьютерный терминал и через Интернет или другими способами. Связь между пользователем и компьютерной системой предпочтительно защищена.
На практике пользователь может, например, ввести информацию, относящуюся к пациенту, такую как состояние болезни пациента, гематокрит, концентрацию гемоглобина и другие факторы, имеющие отношение к количеству ретикулоцитов и/или количеству эритроцитов пациента, а именно желаемое или оптимальное количество ретикулоцитов или эритроцитов. Затем компьютерная система может посредством использования резидентных компьютерных программ предоставить один или несколько подходящих режимов дозирования для пациента. Компьютерная программа через интерфейс пользователя также может предоставить калькуляцию цен и сравнение цен ЕРО и ЕРО-подобных лекарственных средств, вместе или отдельно от подходящих режимов дозирования для таких ЕРО или ЕРО-подобных лекарственных средств.
Компьютерной системой в соответствии с предпочтительным вариантом данного изобретения может быть, например, усовершенствованная средняя по производительности компьютерная система IBM AS/400. Однако специалистам в данной области будет понятно, что способы и аппарат данного изобретения в равной степени применимы к любой компьютерной системе, независимо от того, является ли компьютерная система усложненной многопользовательской вычислительной техникой или устройством для одного пользователя, таким как персональный компьютер или рабочая станция. Компьютерная система соответственно включает в себя процессор, основное запоминающее устройство, блок управления памятью, дополнительный интерфейс хранения данных, интерфейс терминала, которые все соединены между собой через системную шину. Обратите внимание, что в рамках данного изобретения могут быть произведены разные модификации, добавления или удаления в компьютерной системе, такие как добавление сверхоперативной памяти или других периферийных устройств.
Процессор выполняет расчет и контроль функций компьютерной системы и включает в себя подходящий центральный процессор (CPU). Процессор может содержать единственную интегральную схему, такой как микропроцессор, или может содержать любое подходящее количество устройств на интегральных схемах и/или монтажных плат, работающих в кооперации, чтобы выполнить функции процессора. Процессор соответствующим образом выполняет компьютерные программы ФК/ФД-моделирования согласно данному изобретению в пределах его оперативной памяти.
В предпочтительном варианте дополнительный интерфейс хранения данных позволяет компьютерной системе хранить и извлекать информацию из дополнительных устройств хранения, таких как магнитный диск (например, жесткие диски или гибкие диски) или оптическое устройство хранения (например, запоминающее устройство на компакт-диске, CD-ROM). Одним из подходящих устройств хранения является запоминающее устройство с прямым доступом (DASD). DASD может представлять собой дисковод гибких дисков, который считывает программы и данные с гибкого диска. Важно отметить, что наряду с тем, что данное изобретение было описано (и описание будет продолжено) в контексте полной функциональной компьютерной системы, специалистам в данной области будет понятно, что механизмы согласно данному изобретению поддаются распространению в виде программного продукта во многих формах, и что данное изобретение независимо от конкретного типа несущего сигнал носителя в равной мере применимо к фактическому осуществлению распространения. Примеры несущих сигнал носителей включают: носители перезаписываемого типа, такие как гибкие диски и компакт-диски (CD-ROM), и носители передающего типа, такие как каналы цифровой и аналоговой связи, включая каналы беспроволочных связей.
Компьютерные системы данного изобретения также могут включать в себя блок управления памятью, посредством применения отдельного процессора, который ответственен за перемещение запрашиваемой информации из оперативной памяти, и/или посредством дополнительного интерфейса хранения информации к главному процессору. Хотя в целях объяснения блок управления памятью описывается как отдельный объект, специалистам в данной области понятно, что на практике части функции, обеспечиваемой блоком управления памятью, могут в действительности возлагаться на схему, связанную с главным процессором, основным запоминающим устройством и/или дополнительным интерфейсом хранения информации.
Кроме того, компьютерные системы согласно данному изобретению могут включать в себя интерфейс терминала, который позволяет системным администраторам и компьютерным программистам устанавливать связь с компьютерной системой, обычно посредством программируемых рабочих станций. Следует понимать, что данное изобретение в равной степени относится к компьютерным системам, имеющим множественные процессоры и множественные системные шины. Сходным образом, хотя системная шина предпочтительного варианта обычно представляет собой постоянно замонтированную многоточечную шину, можно использовать любое средство связи, которое поддерживает двунаправленную передачу информации в связанное с компьютером окружение.
Основная память компьютерных систем согласно данному изобретению соответственно содержит одну или несколько компьютерных программ, имеющих отношение к ФК/ФД-моделированию введения ЕРО, и операционную систему. Компьютерная программа в памяти используется в ее самом широком смысле, и включает какую-либо и все формы компьютерных программ, включая исходный код, промежуточный код, машинный код и любое другое представление компьютерной программы. Термин “память” в используемом здесь смысле относится к любой ячейке запоминающего устройства в пространстве виртуальной памяти системы. Следует понимать, что части компьютерной программы и операционной системы могут быть загружены в кэш команд для главного процессора для выполнения, в то время как другие файлы хорошо могут быть сохранены в запоминающих устройствах на магнитных или оптических дисках. Кроме того, понятно, что основная память может включать в себя несовместимые ячейки памяти.
Другие цели, особенности и преимущества данного изобретения станут понятными из следующих конкретных примеров. Конкретные примеры, показывая конкретные варианты изобретения, представлены исключительно в иллюстративных целях. Соответственно данное изобретение также включает такие различные изменения и модификации в пределах сущности и в рамках изобретения, которые могут стать понятными специалистам в данной области на основании данного подробного описания.
Пример 1
ФАРМАКОКИНЕТИКА И БИОДОСТУПНОСТЬ ЕРО У ЧЕЛОВЕКА.
Нижеследующий пример данного изобретения представляет краткое изложение ФК/ФД-данных, которые свидетельствуют в пользу режима дозирования 40000 ME р.н. Данные получены как из литературы, так и из четырех клинических исследований, проведенных RWJPRI, Raritan, NJ. Три исследования были проведены под номером исследуемого нового лекарственного препарата ВВ-IND-2318, и одно исследование было проведено в Великобритании. Краткий обзор исследований приведен на фигуре 17.
Клинические фармакокинетические исследования, включенные в указанную техническую сводку, описаны на фигурах 18A-18D, а фармакокинетические данные этих исследований суммированы на фигуре 19. Аналитические способы, используемые для определения концентрации ЕРО в сыворотке, суммированы ниже и на фигуре 20.
Клиническое исследование EPO-PHI-373 (фигура 17) представляет данные для поддержки режима дозирования 40000 ME один раз в неделю. Клиническое исследование EPO-PHI-370 (фигура 17), которое имеет дизайн, сходный с клиническим исследованием EPO-PHI-373.
В клиническом исследовании EPO-PHI-370 были проблемы с рандомизацией, приводящие к флуктуациям уровней гемоглобина, и многие субъекты в начале имели дефицит железа. Ошибка в рандомизации в исследовательском центре привела в результате к неравному распределению субъектов мужского и женского пола в каждой группе лечения и способствовала расхождению в значениях среднего исходного уровня гемоглобина. Просмотр гематологических значений показал значительные колебания в гемоглобине для ряда субъектов между скрининговыми оценками и оценками исходного уровня. Дальнейшее исследование показало лабораторные несоответствия, относящиеся к калибровке оборудования. Чтобы соответствующим образом подтвердить заключения данного исследования, было решено, что будет проведено повторное исследование. Клиническое исследование EPO-PHI-373 было повтором клинического исследования EPO-PHI-370.
Клинические исследования EPO-PHI-358 (фигура 17) и ЕРО-PHI-359 (фигура 17) представляют собой пилотные исследования, проведенные для того, чтобы изучить пропорциональность доз ЕРО, и чтобы получить предварительную фармакокинетическую и фармакодинамическую информацию после разных однократных и многократных доз.
Общие аналитические способы для исследования in vivo
Краткое изложение
Разработаны и утверждены биоаналитические способы для поддержания клинической программы по ЕРО, проведенной RWJPRI, Raritan, NJ. Исходным способом, используемым для определения концентраций ЕРО в сыворотке человека, был радиоиммуноанализ (РИА) (исследование RWJPRI №DM 92001 и DM 96023). Указанный способ был успешно передан в PPD Development (PPD), Richmond VA, контрактно-исследовательскую организацию, которая проводила анализ поддерживающего клинического исследования (EPO-PHI-370). В PPD проведена полная сравнительная проверка способа РИА и твердофазного иммуноферментного анализа (ELISA) для анализа опорного исследования (EPO-PHI-373). Чтобы проверить совместимость РИА и ELISA, в RWJPRI был приготовлен набор из 16 объединенных образцов, полученных от людей - субъектов исследования, и проанализирован в PPD как ELISA, так и РИА. ELISA удобен в отношении доступности реагентов, времени анализа и расширенных пределов стандартной кривой без потери чувствительности.
Тип анализа
Радиоиммуноанализ использовали для количественной оценки ЕРО для поддержки клинических исследований EPO-PHI-358, ЕРО-PHI-359 (проведенных в RWJPRI) и EPO-PHI-370 (проведенных в PPD). ELISA использовали для количественной оценки ЕРО в клиническом исследовании EPO-PHI-373 (проводимом в PPD).
Метод РИА исходно разработали в лаборатории диагностических систем (Diagnostic Systems Laboratories, DSL), Webster, TX для количественного определения ЕРО, и результаты сравнили с результатами, полученными способом RWJPRI. Доступный для приобретения РИА представляет собой конкурентный способ с двойными антителами, который использует поликлональную антисыворотку кролика к ЕРО мочи человека в качестве первого антитела и 125I-меченый ЕРО мочи человека в качестве индикатора. Процедура следует основному принципу радиоиммуноанализа, в соответствии с которым существует конкуренция между радиоактивным и нерадиоактивным антигеном за фиксированное число мест связывания с антителами. Количество 125I-меченого ЕРО, связанного с антителом, обратно пропорционально концентрации ЕРО, присутствующего в сыворотке. Разделение свободного и связанного антигена легко и быстро достигается посредством использования ускоренной системы на основе полиэтиленгликоля с двойными антителами. Главной собственной модификацией набора DSL была замена ЕРО из мочи на рекомбинантный ЕРО человека в стандартах и меченых образцах для контроля качества. Стандартные концентрации, используемые в анализе, составляли 7,8; 15,6; 31,3; 50; 62,5; 100 и 125 мЕд/мл. Указанный точный способ был передан в PPD и там подтвержден.
Отделение иммунохимии PPD подтвердило пригодность ELISA для определения концентраций ЕРО в сыворотке человека. ELISA представляет собой прямой анализ на основе “сэндвича”, образованного двойными антителами, разработанный R&D Systems, Inc, Minneapolis, MN, для количественного определения концентраций ЕРО в плазме или сыворотке. Для захвата ЕРО использовали лунки планшета для микротитрования, предварительно покрытые моноклональным (мышиным) антителом, специфичным для r-HuEPO. Связанный ЕРО метили анти-ЕРО поликлональным (кроличьим) антителом и пероксидазой хрена (конъюгат). Оптический сигнал получали при добавлении тетраметилбензидина/забуференной перекиси водорода (субстрат). Количество вырабатываемой окраски прямо пропорционально концентрации ЕРО в образце или стандарте. Главной собственной модификацией набора P&D было использование собственного рекомбинантного ЕРО человека в стандартах и меченых образцах для контроля качества. Используемые в анализе стандартные концентрации составляли 7,8; 15,6; 31,3; 50; 62,5; 100 и 125 мЕд/мл.
Пределы стандартных кривых
В РИА, используемом для клинических исследований EPO-PHI-358, EPO-PHI-359 и EPO-PHI-370, пределы стандартной кривой составляли от 7,8 до 125 ми/мл при использовании аликвот образцов объемом 0,1 мл. Погрешность стандартной кривой в RWJPRI была ≤5,8%, а в PPD ≤10,8%. ELISA, используемый для анализа клинических образцов при клиническом исследовании EPO-PHI-373, показал пределы концентрации от 7,8 до 250 мЕд/мл, при использовании аликвот образца объемом 0,1 мл, с погрешностью ≤5,3%.
Нижний предел количественного анализа
Нижний предел количественного анализа (LLOQ), наименьшая измеряемая стандартная концентрация, которая может быть безошибочно и точно определена количественно, составляла 7,8 мЕд/мл как в РИА, так и в ELISA.
Контроли качества
Образцы для контроля качества (QCs) для обоих способов готовили в чистой сыворотке, отражая ожидаемые концентрации в исследовании. Анализируемые для QC концентрации в ходе проверки достоверности РИА в RWJPRI составляли 35, 60, 100, 300, 1000 и 2000 мЕд/мл. Анализируемые для QC концентрации в ходе проверки достоверности РИА в PPD составляли 100, 500, 2000 и 5000 мЕд/мл. Концентрации QC для ELISA составляли 7,8, 20, 100, 500, 2000 и 5000 мЕд/мл. Анализировали, по меньшей мере, три уровня QCs в сыворотке человека с исследуемыми образцами в ходе ежедневного анализа образцов.
В RWJPRI приготовили набор из 16 объединенных образцов сыворотки человека из предыдущего клинического исследования и слепые образцы отправили в PPD, чтобы проанализировать как в РИА, так и в ELISA. Относительное рассчитываемое различие в процентах (RPD) между значениями, полученными обоими способами, было определено равным 15,8% (для самого низкого уровня QC) или лучше.
Также была продемонстрирована линейность разведения, так как ожидаемые концентрации некоторых образцов были выше самой высокой концентрации стандартной кривой. Во время подтверждения РИА в RWJPRI линейность при разведении была установлена с использованием серийных от 1:30- до 1:256-кратных разведений QC-образцов. В PPD линейность разведения была продемонстрирована в ходе подтверждения РИА при разведениях QC-образцов от 1:20 до 1:200 и при разведениях от 1:20- до 1:100-кратных для образцов, полученных in vivo от субъекта с самыми высокими концентрациями ЕРО. В ходе подтверждения ELISA линейность разведения была установлена с использованием кратности разведении 1:50, 1:100 и 1:200 образца QC с концентрацией 5000 МЕ/мл.
Выход
Анализ выполняли без экстракции образца, поэтому оценка выхода продукта не требовалась.
Точность и погрешность
В РИА точность в пределах анализа (процент различий между измеренной концентрацией и заданной концентрацией) была от 102,7 до 127% в RWJPRI и от 95,5 до 100,3% в PPD. И точность между анализами была в пределах от 81,7 до 109,8% в RWJPRI и от 100 до 105,6% в PPD. В ELISA точность в пределах анализа составляла от 87,9% до 109,9%, и точность между анализами была в пределах от 83,6 до 116%.
Погрешность в пределах анализа РИА (процентный коэффициент вариации) составляла ≤8,2% в RWJPRI и ≤5,3% в PPD. Погрешность между анализами составляла ≤15,1% в RWJPRI. В ELISA погрешность между анализами составляла ≤10%, и погрешность внутри анализа составляла ≤12,9%.
Стабильность
Была показана стабильность ЕРО в сыворотке человека в течение 20 месяцев при -20°С, при комнатной температуре в течение 2 часов и в течение 4 циклов замораживания/оттаивания.
Анализ данных и критерии приемлемости
Использовали подтвержденную логистическую кривую по четырем параметрам для того, чтобы определить неизвестные концентрации ЕРО в образце сыворотки из стандартной кривой в обоих анализах. Данные РИА, выполненного в RWJPRI, предварительно обрабатывали, используя компьютерную программу Micromedic (ICN Biomedicals, Inc., Costa Mesa, CA). Данные РИА и ELISA, выполненных в PPD, предварительно обрабатывали, используя собственную базу данных RICORA PPD Development Oracle 7,3. Анализы РИА были приемлемы, когда QCs были в пределах 20% номинала, и различие между счетом в минуту повторов составляло ≤6%. Анализы ELISA были приемлемы, когда QCs были в пределах 20% как для точности, так и для погрешности.
Сводки индивидуальных исследований
Пробные пилотные исследования EPO-PHI-358 и EPO-PHI-359
Клиническое исследование EPO-PHI-358 и клиническое исследование EPO-PHI-359 представляли собой два пробных пилотных исследования, предназначенных для исследования дозовой пропорциональности после п/к введения. Вследствие сходства дизайна исследования данные от двух исследований объединили, чтобы сделать максимальными пределы доз фармакокинетического и фармакодинамического анализов.
Целью клинического исследования EPO-PHI-358 было определение фармакокинетических и фармакодинамических профилей ЕРО после однократной подкожной дозы ЕРО 450, 900, 1350 или 1800 МЕ/кг, и сравнение этих профилей с профилем обычного режима дозирования по 150 МЕ/кг т.р.н. × 4 недели. Целью клинического исследования EPO-PHI-359 было определение ФК-и ФД-профилей ЕРО после однократной п/к дозы ЕРО 300, 600, 1200 или 2400 МЕ/кг и сравнение этих профилей с профилем режима дозирования по 600 МЕ/кг р.н. × 4 недели. В каждом исследовании была группа, которой вводили плацебо.
Композиция ЕРО, используемая в этих двух исследованиях, представляла собой 40000 МЕ/мл раствора в фосфатном буфере без консервантов, содержащего 2,5 мг/мл сывороточного альбумина человека, 5,84 мг/мл хлорида натрия, 1,164 мг/мл дигидрата двузамещенного фосфата натрия и 2,225 мг/мл дигидрата однозамещенного фосфата натрия (Композиция №FD-22512-000-J-45; партия продукта 5C903J; дата производства - март 1995). Композиция плацебо, используемая в этих двух исследованиях, представляла собой раствор фосфатного буфера без консервантов, содержащий 2,5 мг/мл сывороточного альбумина человека, 5,84 мг/мл хлорида натрия, 1,164 мг/мл дигидрата двузамещенного фосфата натрия и 2,225 мг/мл дигидрата однозамещенного фосфата натрия (Композиция №FD-22512-000-ABX-45; партия продукта 5C902J; дата производства - март 1995).
Оба исследования представляли собой открытые, рандомизированные, плацебо-контролируемые исследования в параллельных группах и в одном центре исследования. Тридцать два здоровых субъекта были зарегистрированы в клиническом исследовании ЕРО-PHI-358, и 30 субъектов полностью прошли исследование и были включены в ФК/ФД-анализы. Тридцать субъектов было зарегистрировано в клиническом исследовании EPO-PHI-359. Демографические данные субъектов этих исследований перечислены на фигуре 21.
Проводили частый серийный сбор образцов крови для определения ЕРО в сыворотке в течение недель 1 и 4 в ходе режима дозирования 600 МЕ/кг р.н. и в течение недели 4 для режима дозирования 150 МЕ/кг т.р.н. Также еженедельно собирали образцы крови до введения дозы для определения ЕРО в сыворотке. Для групп, получавших однократные дозы, серийные образцы крови для определения ЕРО в сыворотке брали в течение 4-недельного периода исследования. Также собирали образцы крови для определения процента ретикулоцитов (%RETI), общего количества красных кровяных клеток (RBC) и гемоглобина (НЕМО) в крови в течение 4-недельного периода исследования.
Анализ образцов для определения ЕРО в сыворотке выполняли в RWJPRI, Raritan, NJ. Для определения концентраций ЕРО в сыворотке использовали процедуру на основе набора для РИА, произведенного DSL и модифицированного в RWJPRI. Этот способ и его подтверждение описаны в разделе 4. Фармакокинетические и фармакодинамические параметры суммированы посредством описательной статистики. Временные профили средней концентрации ЕРО в сыворотке для субъектов клинического исследования EPO-PHI-358 и EPO-PHI-359 показаны на фигурах 22 и 23, соответственно.
В то время как концентрации ЕРО после введения однократных доз снижаются до эндогенного уровня ЕРО к 15 дню, режим дозирования 150 МЕ/кг т.р.н. давал возможность поддерживать концентрации ЕРО в сыворотке выше эндогенного уровня ЕРО до введения дозы на протяжении периода лечения (фигура 22). Средняя эндогенная концентрация ЕРО до введения дозы для этой группы субъектов составляла 14±4 мМЕ/мл, и средние минимальные концентрации (скорректированные относительно ЕРО исходного уровня) перед первой, второй и третьей дозами в течение четвертой недели дозирования составляли 19±9, 48±18 и 52±25 мМЕ/мл, соответственно. Имело место накопление сывороточного ЕРО в ходе дозирования, так как Сmах после первой, второй и третьей доз в последнюю неделю дозирования была в пределах от 128 до 163, от 141 до 214 и от 152 до 334 мМЕ/мл, соответственно. Режим дозирования 600 МЕ/кг один раз в неделю поддерживал уровни ЕРО в сыворотке выше эндогенного уровня ЕРО до введения дозы до 5-6 дня недели дозирования (фигура 23). Средний эндогенный уровень ЕРО до введения дозы у этих субъектов составлял 13±3 мМЕ/мл, и средняя стационарная минимальная концентрация ЕРО (скорректированная относительно ЕРО исходного уровня) составляла 11±5 мМЕ/мл. При этом режиме дозирования достигалась более высокая Сmах, чем при режиме дозирования 150 МЕ/кг т.р.н., хотя минимальные концентрации до введения дозы были близки к эндогенному уровню ЕРО. Значения Сmах для режима дозирования 600 МЕ/кг р.н. в течение недель 1 и 4 были в пределах от 1203 до 2148 и от 920 до 1489 мМЕ/мл, соответственно. Значения средних±SD фармакокинетических и фармакодинамических параметров перечислены на фигуре 24.
Имела место линейная зависимость между средней Сmах и дозой с коэффициентом корреляции=0,982 (фигура 25), свидетельствуя о том, что скорость абсорбции ЕРО из места инъекции независима от дозы. С другой стороны, зависимость между AUC и дозой была нелинейной зависимостью, такой, при которой клиренс (CL/F) снижался с увеличением доз (фигура 26).
Временные профили среднего процента ретикулоцитов в течение 4-недельного периода исследования показаны на фигуре 27. Процент ретикулоцитов в крови достигает своих максимальных значений примерно на 10 день после введения лекарственного средства как при однократных дозах, так и при многократных дозах. В то время как процент ретикулоцитов в крови после введений однократных доз снижался к исходным значениям до введения дозы к 15 дню, процент ретикулоцитов в крови после двух многократных режимов дозирования (150 МЕ/кг т.р.н. и 600 МЕ/кг р.н.) поддерживался значительно выше исходных значений до введения дозы в течение периода вплоть до 30 дня. Это наблюдение не является неожиданным, так как нормальная продолжительность жизни клеток в стадии ретикулоцитов составляет около 3,5 дней в костном мозге и от 1 до 2 дней в циркулирующей крови (Hillman, выше). ЕРО вызывает свои биологические эффекты посредством связывания со специфичным рецептором клеточной поверхности на эритроидных клетках-предшественниках в костном мозге, являющихся его мишенью, колониеобразующих эритроидных единицах (CFU-E) и бурст-образующих эритроидных единицах (BFU-E) (Dessypris et al., 1984, Br. J. Haematol. 56: 295-306 и Wu et al., 1995, Cell, 83: 59-67). Указанные предшественники эритроидных клеток в конечном итоге созревают в ретикулоциты, которые затем высвобождаются в циркулирующую кровь. Данные указанных двух исследований показывают, что ретикулоциты, продуцируемые при стимуляции однократной дозой ЕРО, появлялись в циркулирующей крови примерно от 7 до 10 дня. При продолжительности жизни в циркулирующей крови от 1 до 2 дней следует ожидать, что ретикулоциты, продуцируемые в результате стимуляции однократной дозой, исчезнут из циркулирующей крови к 15 дню. Следовательно, для непрерывной продукции ретикулоцитов требуется, чтобы в сыворотке уровень ЕРО поддерживался постоянно (например, как после режима дозирования 150 МЕ/кг т.р.н.) или периодически (например, как после режима дозирования 600 МЕ/кг р.н.) выше эндогенной концентрации ЕРО.
Для данных при однократной дозе имела место тенденция увеличения средней AUC процента ретикулоцитов (AUC [некорректир. RETI]) по мере того, как увеличивается средняя AUC ЕРО [AUC(дни 0-29)] (концентрация ЕРО в сыворотке скорректирована относительно уровня ЕРО перед введением дозы, и значение AUC рассчитывали в течение 4-недельного периода). По сравнению с данными при сходных значениях AUC(дни 0-29) при однократных дозах данные при многократных дозах имеют более высокие средние значения AUC(некорректир. RETI). Следовательно, данные свидетельствуют о том, что многократное дозирование ЕРО более эффективно в стимулировании продукции ретикулоцитов, чем однократная доза.
Несмотря на связанное с AUC ЕРО увеличение продукции ретикулоцитов, не было видимого увеличения уровней гемоглобина после введения однократной дозы. Причина отсутствия увеличения уровня гемоглобина после однократного дозирования в настоящее время не известна. С другой стороны, оба режима многократного дозирования были способны вызывать неуклонный подъем уровней гемоглобина, и характер изменения гемоглобина от исходного уровня был сходным для двух режимов многократного дозирования (фигура 28).
В заключение, результаты данного исследования показывают, что фармакологический ответ на ЕРО является функцией дозы и режима дозирования. Скорость абсорбции ЕРО после п/к введения независима от дозы. Клиренс ЕРО зависел от дозы, снижаясь с увеличением дозы. Имела место тенденция увеличения ответа (АUC[некорректир. RETI]) с увеличением AUC (дни 0-29) для однократных доз. Для непрерывного фармакологического ответа (непрерывной продукции ретикулоцитов и продолжительного повышения уровня гемоглобина) требуется, чтобы концентрация ЕРО в сыворотке поддерживалась постоянно (например, как после режима дозирования 150 МЕ/кг т.р.н.) или периодически (например, как после режима дозирования 600 МЕ/кг р.н.) выше эндогенных уровней ЕРО.
Клиническое исследование EPO-PHI-370
Главной целью данного исследования было оценить фармакокинетический профиль ЕРО после введения 150 МЕ/кг т.р.н. или 40000 ME р.н. и продемонстрировать, что два режима дозирования дают сходные клинические выходы при использовании гемоглобина в качестве меры клинической эффективности. Дополнительные цели состояли в том, чтобы определить фармакодинамические профили ЕРО после введения 150 МЕ/кг т.р.н. или 40000 ME р.н. и сравнить параметры толерантности и безопасности при двух режимах дозирования ЕРО.
ЕРО, используемый в данном исследовании, готовили в виде стерильного, бесцветного, свободного от консервантов раствора, забуференного цитратом, во флаконах одноразового применения. ЕРО в концентрации 10000 МЕ/мл (формула №FD-22512-000 С-45, партия D000123) использовали для введения в группе 150 МЕ/кг т.р.н., и ЕРО в концентрации 40000 МЕ/мл (формула №FD-22512-000 AC-45, партия D000175) использовали для введения в группе 40000 ME р.н.
Данное исследование представляло собой проводимое в одном исследовательском центре открытое, рандомизированное исследование с параллельным дизайном, проводимое на 49 здоровых субъектах (49 зарегистрированных и проанализированных на безопасность; 46 прошли исследование полностью и проанализированы на ФК/ФД). Субъекты были случайным образом распределены в две группы лечения и получали ЕРО либо в виде стандартного режима при злокачественной опухоли (150 МЕ/кг, п/к, т.р.н.), либо в виде еженедельного фиксированного режима дозирования (40000, п/к, р.н.) в течение четырех недель. Образцы крови отбирали перед введением дозы в 1, 8 и 15 день и в течение недели 4 для определения концентраций ЕРО в сыворотке. Образцы крови также отбирали при исходном уровне (день 1) и в отдельных временных точках в течение 4-недельного периода исследования для определения процента ретикулоцитов, гемоглобина и суммарного количества красных кровяных клеток.
Из 46 субъектов, которые были проанализированы на ФК/ФД, 24 субъекта (9 мужского пола и 15 женского пола) были зарегистрированы в исследовании в группе с введением 150 МЕ/кг т.р.н., и 22 субъекта (14 мужского пола и 8 женского пола) зарегистрированы в исследовании в группе с введением 40000 ME р.н. Демографические данные и исходные уровни гемоглобина у этих субъектов перечислены на фигуре 29.
Анализ образцов на содержание ЕРО в сыворотке выполняли в PPD Development, Richmond, VA. Для определения концентраций ЕРО в сыворотке использовали процедуру на основе набора для РИА, произведенного DSL и модифицированного в RWJPRI.
Объем выборки в данном исследовании не был основан на статистическом анализе. Суммарная статистика фармакодинамических параметров была представлена по группам лечения и дням; рассчитывали среднее, стандартное отклонение, медиану, размах и стандартную ошибку. Чтобы сравнить ФД-профили ЕРО после 150 МЕ/кг т.р.н. и 40000 ME р.н., делали подгонку анализа дисперсионных моделей, подходящих для двух методических схем, к данным с использованием одного из интересующих ФД-параметров (лог-преобразованной AUC процента ретикулоцитов, гемоглобина и суммарного количества эритроцитов) в качестве зависимой переменной, и лечение, пол и взаимодействие пола и лечения в качестве неизменяемых эффектов. Так как было обнаружено, что эффект взаимодействия пола и лечения не значим для всех трех параметров, приведенные модели без элемента взаимодействия подгоняли к данным и тестировали влияние лечения и пола при 5% уровне, используя элемент остаточной ошибки. Отношение средних, полученных для введения 40000 МЕ/неделю, к средним, полученным для введения 150 МЕ/кг т.р.н., и отношение средних для субъектов женского пола к средним для субъектов мужского пола оценивали, используя геометрические среднеквадратичные отклонения, полученные из моделей ANOVA.
Временные профили средней концентрации ЕРО в сыворотке (не корректировали относительно эндогенного уровня ЕРО до введения дозы) для групп, получавших 150 МЕ/кг т.р.н. и 40000 ME р.н., в течение недели 4 периода исследования показаны на фигуре 30.
В течение недели 4 режима дозирования 150 МЕ/кг т.р.н. пиковые концентрации ЕРО в сыворотке (скорректированные относительно исходного уровня ЕРО) находились в пределах от 78 до 447 мМЕ/мл (средняя Cmах=191±100 мМЕ/мл) и снижались до минимальных концентраций от 7,3 до 88 мМЕ/мл (средняя минимальная концентрация Cmin=39±18 мМЕ/мл). В ходе недели 4 режима дозирования 40000 ME р.н. пиковые концентрации ЕРО в сыворотке (скорректированные относительно исходного уровня ЕРО) находились в пределах от 197 до 1992 мМЕ/мл (средняя Сmах=785±427 мМЕ/мл), которые достигались за время от 9 до 24 часов (среднее tmax=18±5 часов), затем снижались мультиэкспоненциально до минимальных уровней в пределах от значений ниже предела количественного анализа аналитического способа (7,8 мМЕ/мл) до 44 МЕ/мл (средняя минимальная концентрация на 29 день=19±10 мМЕ/мл) к концу недели дозирования на 29 день. Средняя Cmin в течение четырехнедельного периода исследования составляла 13±9 мМЕ/мл. Конечные фазы двух режимов дозирования выглядели параллельно со средними значениями времени полужизни 31,8±13,4 (N=13) и 39,3±7,1 (N=3) часа для режимов дозирования 150 МЕ/кг т.р.н. и 40000 ME р.н., соответственно.
Средние значения (SD) (%CV) фармакокинетических параметров перечислены на фигуре 31.
Биодоступность при режиме дозирования 400000 ME р.н. относительно биодоступности, полученной после режима дозирования 150 МЕ/кг т.р.н., рассчитывали, используя следующую формулу:
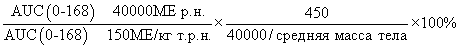
Среднюю массу тела рассчитывали, используя данные от субъектов, которые полностью прошли исследование.
Биодоступность ЕРО после режима дозирования 40000 ME р.н. относительно биодоступности после режима дозирования 150 МЕ/кг т.р.н. составляла 176%.
Линейные графики среднего изменения относительно исходного уровня против дня исследования для процента ретикулоцитов и концентраций гемоглобина представлены на фигурах 32 и 33, соответственно. Средние значения фармакодинамических параметров (скорректированные относительно исходного значения) представлены на фигуре 34. Динамические ответы при двух режимах дозирования были сходными, несмотря на тот факт, что AUC ЕРО в сыворотке для режима дозирования 40000 ME р.н. была больше, чем AUC ЕРО в сыворотке для режима дозирования 150 МЕ/кг т.р.н. Не было статистически значимых различий в AUC гемоглобина и AUC эритроцитов между двумя режимами дозирования, хотя AUC процента ретикулоцитов после режима дозирования 40000 ME р.н. была статистически больше (р<0,05), чем после режима дозирования 150 ME/кг т.р.н. Не было статистически значимых различий в AUC процента ретикулоцитов, AUC гемоглобина и AUC эритроцитов между субъектами мужского и женского пола.
Временные профили изменений в гемоглобине и суммарных красных кровяных клетках в течение одномесячного периода исследования были сходны при сравнении двух режимов дозирования, несмотря на различия в экспозиции (т.е. AUC[0-168]) EPO в сыворотке и несмотря на более высокую продукцию ретикулоцитов (измеренную по площади под кривой) для режима 40000 ME р.н. Площади под кривыми AUC гемоглобина и суммарных красных кровяных клеток в течение одномесячного периода исследования для двух режимов дозирования были сходны. В данном исследовании не было различий в фармакодинамических ответах субъектов мужского и женского пола.
Хотя данные этого исследования свидетельствуют о том, что ответы на уровне гемоглобина после режимов дозирования 150 МЕ/кг т.р.н и 40000 ME р.н. были сходны, и что два режима дозирования могут быть взаимозаменяемы, существовали проблемы с рандомизацией, приводящие к колебаниям уровней гемоглобина в начале исследования, и многие субъекты имели дефицит железа в начале (о чем свидетельствовали значения насыщения трансферрина). Ошибка в рандомизации в исследовательском центре привела в результате к неравному распределению субъектов мужского и женского пола в каждой группе лечения и внесла вклад в дисбаланс средних значений исходного уровня гемоглобина. Чтобы соответствующим образом подтвердить полученные в данном исследовании данные, было решено, что будет проведено повторное исследование (клиническое исследование ЕРО-373).
Клиническое исследование EPO-PHI-373
Главной целью данного исследования было оценить ФК-профиль ЕРО после введения 150 МЕ/кг т.р.н или 40000 ME р.н. и продемонстрировать, что два режима дозирования дают сходные клинические выходы. Дополнительные цели состояли в том, чтобы определить ФД-профили ЕРО после введения 150 МЕ/кг т.р.н или 40000 ME р.н. и сравнить параметры толерантности и безопасности при двух режимах дозирования ЕРО.
ЕРО, используемый в данном исследовании, готовили в виде стерильного, бесцветного, свободного от консервантов раствора, забуференного фосфатом, во флаконах одноразового применения. ЕРО в концентрации 10000 МЕ/мл (формула №FD-22512-000-T-45, партия 99KS077) использовали для введения в группе 150МЕ/кг т.р.н., и ЕРО в концентрации 40000 МЕ/мл (формула №FD-22512-000 АА-45, партия 99KS091) использовали для введения в группе 40000 МЕ р.н.
Данное исследование представляло собой проводимое в одном исследовательском центре открытое, рандомизированное исследование с параллельным дизайном, проводимое на 36 здоровых субъектах (36 зарегистрированных и проанализированных на безопасность; 34 прошли исследование полностью и проанализированы на ФК/ФД). Субъекты были случайным образом распределены в две группы лечения и получали ЕРО либо в виде стандартного режима при злокачественной опухоли (150 МЕ/кг, п/к, т.р.н.), либо в виде еженедельного фиксированного режима дозирования (40000, п/к, р.н.) в течение четырех недель. Образцы крови отбирали перед введением дозы в 1, 8 и 15 день и в течение недели 4 для определения концентраций ЕРО в сыворотке. Образцы крови также отбирали при исходном уровне (день 1) и в отдельных временных точках в течение 4-недельного периода исследования для определения процента ретикулоцитов, гемоглобина и суммарного количества красных кровяных клеток.
Из 34 субъектов, которые были проанализированы на ФК/ФД, 17 субъектов (девять мужского пола и восемь женского пола) были зарегистрированы в исследовании в группе с введением 150 МЕ/кг т.р.н., и 17 субъектов (девять мужского пола и девять женского пола) зарегистрированы в исследовании в группе с введением 40000 ME р.н. Демографические данные и исходные уровни гемоглобина у этих субъектов перечислены на фигуре 35.
Набор для ELISA, производства R&D Systems, Inc. (R&D), Minneapolis, MN, модифицированный в RWJPRI и перекрестно проверенный на достоверность с исходным РИА в PPD Development, Richmond, VA, использовали для определения концентраций ЕРО в сыворотке.
Объем выборки в данном исследовании не был основан на статистическом анализе. Суммарная статистика фармакодинамических параметров была представлена по группам лечения и дням; рассчитывали среднее, стандартное отклонение, медиану, размах и стандартную ошибку. Чтобы сравнить фармакодинамические профили ЕРО после введения 150 МЕ/кг т.р.н. и 40000 ME р.н., делали подгонку к данным анализа дисперсионных моделей, подходящих для двух методических схем, с использованием одного из интересующих фармакодинамических параметров (лог-преобразованной AUC процента ретикулоцитов, гемоглобина и суммарного количества красных кровяных клеток) в качестве зависимой переменной, и лечение, пол и взаимодействие пола и лечения в качестве фиксированных эффектов. Так как было обнаружено, что эффект взаимодействия пола и лечения не значим для всех трех параметров, приведенные модели без элемента взаимодействия подгоняли к данным и тестировали влияние лечения и пола при 5% уровне, используя элемент остаточной ошибки. Отношение средних, полученных при введении 40000 МЕ/неделю, к средним, полученным при введении 150 МЕ/кг т.р.н., и отношение средних для субъектов женского пола к средним для субъектов мужского пола оценили, используя геометрические среднеквадратичные отклонения, полученные из моделей ANOVA.
Временные профили средней концентрации ЕРО в сыворотке (не корректировали относительно эндогенного уровня ЕРО до введения дозы) для групп, получавших 150 МЕ/кг т.р.н. и 40000 ME р.н. в течение недели 4 периода исследования, показаны на фигуре 36.
В течение недели 4 режима дозирования 150 МЕ/кг т.р.н. концентрации ЕРО в сыворотке (скорректированные относительно исходного уровня ЕРО) находились в пределах от пиковых концентраций от 75 до 284 мМЕ/мл (средняя Cmах=143±54 мМЕ/мл) до значений минимального уровня, находящихся в пределах от значений ниже предела количественного анализа аналитического способа (7,8 мМЕ/мл) до 40 мМЕ/мл (средняя минимальная концентрация (Cmin)=18±9 мМЕ/мл). В ходе недели 4 режима дозирования 40000 ME р.н. концентрации ЕРО в сыворотке (скорректированные относительно исходного уровня ЕРО) достигали пиковых концентраций (средняя Cmах=861±445 мМЕ/мл) за время от 1 до 24 часов (среднее tmax=16±8 часов), затем снижались мультиэкспоненциально до значений минимального уровня в пределах от значений ниже предела количественного анализа аналитического способа (7,8 мМЕ/мл) до 5,9 МЕ/мл (средняя минимальная концентрация на 29 день=2,0±1,5 мМЕ/мл) к концу недели дозирования на 29 день. Средняя Сmin в течение четырехнедельного периода исследования для режима 40000 ME р.н. составляла 3,8±4,3 мМЕ/мл. Конечные фазы двух режимов дозирования выглядели параллельно со средними значениями времени полужизни 19,4±8,1 часа (n=9) и 15,0±6,1 часа (n=9) для режимов дозирования 150 МЕ/кг т.р.н. и 40000 ME р.н., соответственно.
Средние значения (SD) (%CV) фармакокинетических параметров перечислены на фигуре 37.
Биодоступность при режиме дозирования 400000 ME р.н. относительно биодоступности, полученной после режима дозирования 150 МЕ/кг т.р.н.. рассчитывали, используя следующую формулу:
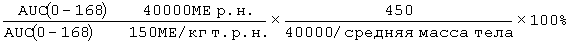
Среднюю массу тела рассчитывали, используя данные от субъектов, которые полностью прошли исследование.
Биодоступность ЕРО после режима дозирования 40000 ME р.н. относительно биодоступности после режима дозирования 150 МЕ/кг т.р.н. составляла 239%.
Линейные графики среднего изменения относительно исходного уровня против дня исследования для процента ретикулоцитов, концентраций гемоглобина и суммарного количества красных кровяных клеток представлены на фигурах 38, 39 и 40, соответственно.
Средние значения фармакодинамических параметров (скорректированные относительно исходного значения) представлены на фигуре 41. Динамические ответы при двух режимах дозирования были сходными, несмотря на тот факт, что AUC ЕРО в сыворотке для режима дозирования 40000 ME р.н. была больше, чем AUC ЕРО в сыворотке для режима дозирования 150 МЕ/кг т.р.н. Не было статистически значимых различий в AUC процента ретикулоцитов, AUC гемоглобина и AUC эритроцитов между двумя режимами дозирования. Не было статистически значимых различий (р>0,05) в AUC процента ретикулоцитов между субъектами мужского и женского пола. Однако AUC гемоглобина и AUC эритроцитов были статистически больше (р=0,038 и 0,042, соответственно) у женщин, чем у мужчин. Клинически эти различия были не значимы.
Временные профили изменений процента ретикулоцитов, гемоглобина и суммарных красных кровяных клеток в течение одномесячного периода исследования были сходны при сравнении двух режимов дозирования, несмотря на различия в экспозиции ЕРО в сыворотке (на основе AUC[0-168]). Кроме того, не было статистически значимых различий (р>0,05) в AUC процента ретикулоцитов, AUC гемоглобина и AUC суммарных красных кровяных клеток в течение одномесячного периода исследования между двумя режимами дозирования. В данном исследовании не было различий в фармакодинамических ответах между субъектами мужского и женского пола. Данные этого исследования свидетельствуют о том, что ответы на уровне гемоглобина после режимов дозирования 150 МЕ/кг т.р.н и 40000 ME р.н. были сходны, тем самым подтверждая, что два режима дозирования могут быть взаимозаменяемы.
В заключение, имело место ожидаемое различие в общей экспозиции ЕРО в сыворотке после режимов дозирования 150 МЕ/кг т.р.н и 40000 ME р.н. Ответы на уровне гемоглобина были сходными, тем самым подтверждая, что оба режима дозирования могут быть взаимозаменяемыми. Таким образом, данные исследования показали, что можно использовать режим дозирования ЕРО один раз в неделю. Указанный режим позволяет преодолеть недостатки, связанные с обычно используемыми режимами дозирования. В предпочтительном варианте предполагается доза 40000 ME на основании данных клинических исследований. Данное изобретение также предполагает режим дозирования один раз каждые две недели, например, при дозе ЕРО от 80000 до 120000 ME. В отдельном варианте используется доза ЕРО, вводимая один раз каждые две недели.
Результаты
Фармакокинетика после в/в введения
После в/в введения здоровым добровольцам или пациентам с нарушенной функцией почек r-HuEPO распределяется в объеме, сравнимом с объемом плазмы, и концентрации в плазме снижаются со средними значениями времени полужизни в пределах от 4 до 11,2 часов, которые, как сообщалось, укорачиваются после повторных введений (Macdougall et al., 1991, Clin. Pharmacokinet., 20: 99-113). Имели место пропорциональные дозе увеличения значений Сmах и AUC после однократных внутривенных доз от 50 до 1000 МЕ/кг у здоровых субъектов (Flaharty et al., 1990, Clin. Pharmacol. Ther., 47, 47: 557-64), и сообщалось, что клиренс (CL) независим от дозы после однократного болюсного внутривенного введения доз 10, 100 и 500 МЕ/кг здоровым субъектам, с соответствующими средними значениями 5,89±1,53, 7,02±1,14 и 6,88±1,19 мл/час/кг (Veng-Pendersen et al., 1995, J. Pharma. Sci., 84(6): 760-767). Однако также сообщалось, что клиренс выше после однократной внутривенной дозы 10 МЕ/кг (13,1 мл/час/кг), чем клиренс после однократных внутривенных доз 100 и 500 МЕ/кг у здоровых субъектов (соответственно CL=7,9 и 6,2 мл/час/кг) (Widness et al., 1996, J. Appl. Physiol., 80(1): 140-148).
Фармакокинетика после п/к введения
Сводка фармакокинетических параметров после п/к введения при клинических исследованиях EPO-PHI-358, EPO-PHI-359, ЕРО-PHI-370 и EPO-PHI-373 приведена на фигуре 42.
После однократного или еженедельного п/к введения концентрации ЕРО в сыворотке достигали максимального значения за время (tmax) в пределах от 9 до 36 часов. Среднее tmax было сходным для разных однократных доз и было в пределах от 15,6±5,8 до 28,8±7,8 часа. Существовала линейная зависимость между средним Сmах и дозой (корреляция=0,982), свидетельствуя о том, что скорость абсорбции ЕРО из места инъекции не зависела от дозы. С другой стороны, зависимость между AUC и дозой была нелинейной, такой что клиренс (CL/F) снижался с увеличением доз (Cheung et al., 1998, Clin. Pharmacol. Ther., 64: 412-423). Данные клинических исследований EPO-PHI-373 и EPO-PHI-370 свидетельствуют, что режим дозирования 150 МЕ/кг т.р.н. имеет более высокое значение CL/F, чем режим дозирования 40000 ME р.н., и данные клинических исследований EPO-PHI-358 и ЕРО-PHI-359 свидетельствуют, что режим дозирования 150 МЕ/кг т.р.н. имеет более высокое значение CL/F, чем режим дозирования 600 МЕ/кг р.н.
Как показано на фигуре 42, концентрация ЕРО снижается с более медленной скоростью после п/к введения, чем после в/в введения. Значения времени полужизни было в пределах от 15,9 до 221 часа после однократных п/к доз от 300 до 2400 МЕ/кг, и средние значения времени полужизни после режимов дозирования 150 МЕ/кг т.р.н. и 40000 ME р.н. составляли 19,4±8,1 и 15,0±6,1 часа, соответственно. Значения времени полужизни не зависели от дозы в пределах изученных доз. Более длительное время полужизни, наблюдаемое после п/к введения по сравнению с в/в введением, возможно, является отражением времени полупериода абсорбции из подкожных тканей.
В то время как концентрации ЕРО в сыворотке после введений однократных доз снижались до эндогенного уровня ЕРО к 15 дню (для более низких доз раньше), режим дозирования 150 МЕ/кг т.р.н. был способен поддерживать концентрации ЕРО в сыворотке выше эндогенного уровня ЕРО до введения дозы в течение периода лечения (Cheung, выше). Режимы еженедельного дозирования 600 МЕ/кг/мл и 40000 ME р.н. поддерживали уровни ЕРО в сыворотке выше эндогенного уровня ЕРО до введения дозы вплоть до 5-6 дня недели дозирования (Cheung, выше). При указанных режимах еженедельного дозирования достигалась более высокая Сmах, чем при режиме дозирования 150 МЕ/кг т.р.н., хотя минимальные концентрации перед дозами были ниже, чем эндогенный уровень ЕРО.
Фармакодинамика ЕРО после п/к введения
После однократных (от 300 до 2400 МЕ/кг) или многократных (150 МЕ/кг т.р.н., 600 МЕ/кг р.н. или 40000 МЕ/р.н.) п/к введений ЕРО процент ретикулоцитов начинал увеличиваться к 3-4 дню. Процент ретикулоцитов после введений однократной дозы достигал своих максимальных значений к 6-12 дню (Cheung, выше), тогда как процент ретикулоцитов после режимов введения многократных доз достигал пиковых значений во время в пределах от 8 дня до последней временной точки отбора образца крови на 29 день (Cheung, выше). Все режимы многократного дозирования стимулировали умеренное, но непрерывное увеличение процента ретикулоцитов (примерно от 2 до 7%), который поддерживался выше исходных значений до введения дозы в течение с 22 по 29 день, тогда как процент ретикулоцитов после введений однократных доз снижался до исходных значений к 15-22 дню (Cheung, выше).
Зависимость между средней AUC изменения (от исходного уровня) процента ретикулоцитов [AUC(RETI)] и средней AUC ЕРО (скорректированной относительно эндогенного уровня ЕРО до введения дозы) [AUC(дни 0-29)] показана на фигуре 43 для данных клинических исследований EPO-PHI-358, EPO-PHI-359 и на фигуре 44 для данных клинических исследований EPO-PHI-370 и EPO-PHI-373. Значения AUC как для изменения процента ретикулоцитов от исходного уровня, так и для ЕРО рассчитывали на протяжении четырехнедельного периода. Значения AUC(RETI) клинических исследований EPO-PHI-358 и EPO-PHI-359 нельзя было сравнить со значениями AUC(RETI) клинических исследований EPO-PHI-370 и ЕРО-PHI-373, так как временные точки отбора образцов для анализа ретикулоцитов были разными. Режим отбора образцов был менее частым в клиническом исследовании EPO-PHI-358 и EPO-PHI-359, и значения AUC(RETI) могли быть занижены. Данные клинических исследований EPO-PHI-358 и EPO-PHI-359 свидетельствуют о том, что для данных по однократным дозам имела место тенденция увеличения средней AUC(RETI) по мере увеличения средней AUC(дни 0-29) ЕРО. Для режимов многократного дозирования на основании данных клинических исследований EPO-PHI-370 и EPO-PHI-373 также имела место тенденция увеличения средней AUC(RETI) по мере увеличения средней AUC(дни 0-29) ЕРО.
Общее количество введенного ЕРО за один месяц для режима 150 МЕ/кг т.р.н. составляло 1800 МЕ/кг, и для режима 600 МЕ/кг р.н. составляло примерно 2400 МЕ/кг. Хотя при режиме 150 МЕ/кг т.р.н. значение AUC(дни 0-29) ЕРО было намного меньше, чем при режиме однократной дозы в 1800 МЕ/кг, при этих режимах средние значения AUC(RETI) были сходны. Подобным образом при режиме 600 МЕ/кг р.н. значение AUC(дни 0-29) ЕРО было намного меньше, чем при режиме однократной дозы в 2400 МЕ/кг, но при этих режимах средние значения AUC(RETI) также были сходны. Следовательно, ЕРО (на единицу AUC экспозиции) после многократного дозирования более эффективен для продуцирования ретикулоцитов, чем после однократной дозы.
Несмотря на связанное с AUC ЕРО увеличение продукции ретикулоцитов, не было явного увеличения количества гемоглобина после введения однократной дозы. С другой стороны, режимы многократного дозирования были способны обеспечить неуклонный подъем количества гемоглобина, и характеры увеличения количества гемоглобина были сходными при сравнении между режимами дозирования 150 МЕ/кг т.р.н. и 40000 ME р.н., как было показано в опорном клиническом исследовании EPO-PHI-373 (фигура 45) и поддерживающем клиническом исследовании EPO-PHI-370 (фигура 46), и были сходными при сравнении между режимами 150 МЕ/кг т.р.н. и 600 МЕ/кг р.н., как было показано в пилотных клинических исследованиях EPO-PHI-358 и EPO-PHI-359. Причины отсутствия ответа на уровне гемоглобина после однократного дозирования в настоящее время не известны. Существует два возможных объяснения этого факта: увеличение количества ретикулоцитов после однократных доз (максимальное увеличение процента ретикулоцитов после однократной дозы 2400 МЕ/кг=6,6%) может быть недостаточно продолжительным, чтобы привести к какому-либо существенному увеличению количества гемоглобина; предложена гипотеза, что выживаемость юных эритроцитов зависит от постоянного присутствия ЕРО в циркулирующей крови (Alfrey et al., 1997, The Lancet, 349: 1389-1390). После введения однократной дозы ретикулоциты достигают пика во время в пределах от 6 до 12 дней, и возвращаются к уровням до введения дозы во время до 15 дней. Продолжительность жизни клеток в циркулирующей крови в стадии ретикулоцитов составляет примерно от 1 до 2 дней (Hillman et al., 1967, Sem Haematol., 4(4): 327-336).
Поэтому следует ожидать появления юных эритроцитов в циркулирующей крови примерно от 7 до 14 дня после дозирования. К тому времени концентрации ЕРО после введения однократной дозы были на эндогенном уровне, что может в результате приводить к гибели юных эритроцитов из-за отсутствия достаточного количества ЕРО в циркулирующей крови для того, чтобы поддержать их выживание. С другой стороны, концентрации ЕРО сохранялись выше эндогенного уровня постоянно после режима дозирования т.р.н. или периодически после режима еженедельного дозирования, и поэтому поддерживалась выживаемость юных эритроцитов, приводя к непрерывному повышению количества гемоглобина в течение периода исследования.
Следовательно, чтобы поддержать выживаемость большого количества продуцируемых при фармакологическом воздействии юных эритроцитов, концентрации ЕРО в сыворотке должны быть выше эндогенного уровня.
Заключение
В заключение, данные указанных четырех исследований фазы 1 на здоровых субъектах показывают, что фармакокинетика и фармакодинамика ЕРО у людей нелинейны после п/к введения. Данные этих исследований также четко демонстрируют, что режим дозирования 150 МЕ/кг т.р.н. обеспечивает ответ на уровне гемоглобина, сходный с ответом при режиме дозирования 40000 ME в неделю, тем самым доказывая, что два режима дозирования могут быть взаимозаменяемыми. Соответственно ЕРО вводят в дозе примерно 40000 МЕ/кг один раз в неделю в течение двух недель подряд. Первая доза ЕРО способствует продукции ретикулоцитов из клеток-предшественников эритроцитов. Вторая доза ЕРО вводится так, чтобы она совпадала с фармакологическим профилем ретикулоцитов пациента. Вторая доза ЕРО будет вводиться через 6-10 дней после начальной дозы и предпочтительно в то время, когда концентрация ретикулоцитов достигнет пика после первой дозы ЕРО.
Пример 2
ОЦЕНКА ФК/ФД-ПРОФИЛЯ ЕРО ПОСЛЕ ВВЕДЕНИЯ 150 МЕ/КГ Т.Р.Н. И 40000 ME P.H.
При особых показаниях, таких как злокачественная опухоль, субъектов лечат 150 МЕ/кг эпоэтина-альфа т.р.н. Таким образом, остается важная задача изменить принятый в настоящее время график дозирования на более удобный (т.е. один раз в неделю или один раз каждые две недели) график и режим дозирования. Менее частое введение улучшит прием и будет удобным для пользователя.
Фармакокинетические и фармакодинамические профили режимов многократного дозирования эпоэтина-альфа были определены в предыдущих примерах (EPO-PHI-358 и EPO-PHI-359). Данные свидетельствуют о том, что режимы дозирования 150 МЕ/кг т.р.н. и 600 МЕ/кг/неделю давали сходные фармакодинамические ответы (например, повышение количества гемоглобина). Поэтому эпоэтин-альфа потенциально можно вводить в виде еженедельной дозы на килограмм. Поскольку 600 МЕ/кг эквивалентно 42000 ME на человека массой 70 кг, данное исследование было проведено, чтобы показать, что фиксированный режим дозирования 40000 ME в неделю обеспечивает сравнимый фармакодинамический ответ, как и принятый режим дозирования 150 МЕ/кг т.р.н.
Основной целью данного исследования было оценить фармакокинетический профиль эпоэтина-альфа после введения 150 МЕ/кг т.р.н. или 40000 ME р.н. и продемонстрировать, что два режима дозирования дают сходные клинические выходы.
Дополнительные цели состояли в том, чтобы определить фармакодинамические профили эпоэтина-альфа после введения 150 МЕ/кг т.р.н. или 40000 ME р.н. и сравнить параметры толерантности и безопасности при двух режимах дозирования эпоэтина-альфа.
Тридцать шесть здоровых взрослых добровольцев (18 мужчин и 18 женщин) были зарегистрированы в данном открытом, рандомизированном исследовании с параллельным дизайном, проводимом в одном исследовательском центре. Субъекты были в возрасте от 18 до 45 лет, с уровнями гемоглобина от 12,0 до 14,0 г/дл включительно для женщин, и от 13,0 до 14,0 г/дл включительно для мужчин. Субъектов тщательно отбирали на пригодность для исследования на основании критериев включения и исключения и случайным образом распределяли в одну из двух групп лечения. Группа 1 получала стандартный режим при злокачественной опухоли 150 МЕ/кг, п/к, ЕРО т.р.н в течение четырех недель. Группа 2 получала еженедельный фиксированный режим дозирования 40000 ME эпоэтина-альфа, п/к, р.н. в течение четырех недель.
Субъекты должны были в ходе исследования принимать ежедневно добавку железа перорально (две капсулы Ferro-Grad®, каждая из которых содержит 105 мг элемента железа).
Образцы крови отбирали при исходном уровне и в определенные временные точки в ходе исследования для определения концентраций эритропоэтина в сыворотке, полного анализа крови (СВС), включая процент ретикулоцитов, концентрации гемоглобина и значения гематокрита. Оценки безопасности были основаны на частоте и типе нежелательных явлений, появляющихся при лечении, изменениях клинических лабораторных тестов (гематология и химия), измерениях показателей жизненно важных функций и результатов объективного обследования. Кроме того, в ходе исследования следили за количеством железа в сыворотке, рассчитанным насыщением трансферрина и концентрациями ферритина.
Оценки безопасности были основаны на частоте и тяжести нежелательных явлений, появляющихся при лечении, и на изменениях результатов объективного обследования, измерений показателей жизненно важных функций и параметром клинического лабораторного анализа относительно данных, полученных перед исследованием.
В протокол вносили поправки после того, как все субъекты начали испытание. В поправке разъясняли критерии включения и исключения, сопутствующую терапию, лабораторные параметры, анализ мочи, измерения показателей жизненно важных функций и режим дозирования для группы 40000 ME р.н.
Три субъекта, случайно распределенных в группу 150 МЕ/кг т.р.н., и один субъект в группе 40000 ME р.н. имели значение критерия для включения в исследование по гемоглобину выше, чем предел, установленный в критериях включения/исключения (14,0 г/дл), но были включены в исследование. Субъекты 2012, 2015 и 2016 в группе 150 МЕ/кг т.р.н. имели концентрации гемоглобина при отборе 14,1 г/дл; субъект 2018 в группе 40000 ME р.н. имел концентрацию гемоглобина при отборе 14,2 г/дл. Четыре субъекта в группе 40000 ME р.н. имели значения ферритина при отборе ниже предельного значения 45 нг/мл, определенного в критериях исключения: субъект 1006 имел при отборе значение 40 нг/мл, субъект 1010 имел при отборе значение 43 нг/мл и субъекты 1015 и 2011 имели при отборе значения, равные 44 нг/мл. По одному субъекту в каждой группе лечения (субъект 2015 в группе 150 МЕ/кг и субъект 2014 в группе 40000 ME) имели при отборе значения насыщения трасферрина, равные 19%, немного ниже критерия исключения ≤20%. Субъект 2002 (40000 ME) весил на 0,1 кг больше максимального значения, допускаемого критериями протокола включения для людей с его ростом. Кроме того, субъект 2016 (150 МЕ/кг т.р.н.) принимал таблетки снотворного на основе трав за четыре дня до начала терапии эпоэтином-альфа.
Информация об исследуемых препаратах
Эпоэтин-альфа готовили в виде стерильного, бесцветного, свободного от консервантов раствора, забуференного фосфатом, поставляемого во флаконах одноразового применения. В исследовании использовали коммерческий продукт, и он имел торговый знак. Раствор эпоэтина-альфа в концентрации 10000 МЕ/мл имел формулу №FD 22512-000-T-45, партия №99KS077, и раствор эпоэтина-альфа в концентрации 40000 МЕ/мл имел формулу №FD 22512-000-АА-45, партия №99KS091.
Дозирование и введение
Субъекты были приняты к услугам исследователей, по меньшей мере, за 12 часов до введения исследуемого лекарственного препарата в 1 день. Субъекты голодали, по меньшей мере, в течение десяти часов перед дозированием в 1 день; вода была доступна по желанию. Субъекты были распределены случайным образом и получали один из двух исследуемых лекарственных средств следующим образом:
Для группы дозирования 1 объем инъекции для субъекта массой 70 кг составлял примерно 1,0 мл.
Для группы дозирования 2 объем инъекции составлял точно 1 мл.
Если в какой-либо момент во время фазы лечения концентрация гемоглобина у какого-либо субъекта становилась равной или превышала 18,0 г/дл, отбирали второй образец, чтобы подтвердить первое определение. В случае подтверждения делали флеботомию, чтобы снизить уровень гемоглобина; в начале удаляли одну единицу крови. Гемоглобин снова измеряли, после того как субъект стабилизировался. Если уровень гемоглобина еще составлял 18,0 г/дл или больше, удаляли дополнительно от 0,5 до одной единицы крови и измеряли гемоглобин. Любому пациенту после флеботомии прекращали терапию эпоэтином-альфа и подвергали его необходимым завершающим экспертизам и процедурам.
Композицию с концентрацией эпоэтина-альфа 10000 МЕ/мл использовали для группы 1 (стандартный режим при лечении злокачественной опухоли), и композицию с концентрацией эпоэтина-альфа 40000 МЕ/мл использовали для группы 1 (еженедельно фиксированный режим дозирования).
Сопутствующая терапия
Субъекты были проинструктированы по поводу того, чтобы не принимать лекарственных средств (рецептурных, безрецептурных, травяных или “природных”), начиная с двух недель до первой дозы исследуемого лекарственного препарата и впоследствии в течение всей продолжительности исследования. В случае головной боли или симптомах, подобных гриппу, мог быть введен парацетамол. Если становилось необходимым введение какого-либо медицинского средства, это записывалось в соответствующую индивидуальную регистрационную карту (CRF) и исходный документ.
Субъекты ежедневно получали добавку железа перорально в течение исследования (две капсулы Ferro-Grad®, каждая из которых содержит 105 мг элемента железа).
Оценки исследования
Время и график событий
Исследование было разделено на три фазы: отбор, лечение и завершение/досрочное прекращение. Субъекты оценивались по их пригодности во время периода отбора (процедуры, выполняемые в течение двух недель введения исследуемого лекарственного препарата). Субъекты были случайно распределены в одну из двух групп лечения и затем входили в фазу лечения. Субъекты были помещены в клинику, по меньшей мере, за 12 часов до введения исследуемого препарата в 1 день, и оставались в клинике, по меньшей мере, 24 часа после дозирования, пока не были выполнены все тесты. Субъекты голодали, по меньшей мере, десять часов перед дозированием в 1 день, но получали воду по желанию. Фаза лечения состояла из введения исследуемого препарата (дозирование в 1, 3, 5, 8, 10, 12, 15, 17, 19, 22, 24 и 26 день для группы 150 МЕ/кг т.р.н. и в 1, 8, 15 и 22 день для группы 40000 ME р.н.). На 22 день второй период госпитализации начинался с введения эпоэтина-альфа и продолжался в течение, по меньшей мере, 144 часов после дозирования. Оценки фармакокинетики, фармакодинамики и параметров безопасности выполняли для всех субъектов с периодическими интервалами в течение 28-дневной фазы лечения. Оценки и процедуры в фазе завершения исследования выполняли на 29 день, или при досрочном исключении из исследования.
Фармакокинетические оценки
Сбор и обработка образцов
Образцы венозной крови, по 2,5 мл каждый, собирали путем прямой венепункции в вакуумные пробирки для определения концентрации эритропоэтина в сыворотке для группы 150 МЕ/кг эпоэтина-альфа т.р.н. в следующих временных точках:
День 1: 30, 20 и 10 минут до начальной дозы исследуемого лекарственного препарата.
Дни 8 и 15: непосредственно перед дозированием.
Дни 22 и 24: непосредственно перед дозированием и в 0,5, 3, 6, 9, 12, 15, 18, 24, 30 и 36 час после дозирования.
День 26: непосредственно перед дозированием и в 0,5, 3, 6, 9, 12, 15, 18, 24, 36, 48 и 72 час после дозирования.
Образцы крови для определения концентрации эритропоэтина в сыворотке для группы 40000 ME р.н. (группа 2) собирали в следующих временных точках:
День 1: 30, 20 и 10 минут до начальной дозы исследуемого лекарственного препарата.
Дни 8 и 15: непосредственно перед дозированием.
Дни 22-28: непосредственно перед дозированием и в 0,5, 3, 6, 9, 12, 15, 18, 24, 30, 36, 48, 48,5, 51, 54, 57, 60, 63, 66, 72, 78, 84, 96, 96,5, 99, 102, 105, 108, 111, 114, 120, 126, 132, 144 и 168 час после введения эпоэтина-альфа на 22 день.
Образцам давали возможность свернуться при комнатной температуре примерно в течение 20 минут и затем центрифугировали в течение десяти минут при 1200 об/мин в центрифуге с охлаждением. Сыворотку отбирали в предварительно меченый флакон. Образцы замораживали при -20°С и хранили в замороженном виде при этой температуре вплоть до анализа.
Аналитические процедуры
Анализ образцов в отношении эпоэтина-альфа в сыворотке выполняли в PPD Development, Richmond, VA. Способ на основе набора для твердофазного иммуноферментного анализа (ELISA), произведенного R&D Systems, Inc., (R&D), Minneapolis, MN, и модифицированного RWJPRI, использовали для определения концентраций эритропоэтина в сыворотке. Доступный для приобретения ELISA представляет собой прямой анализ на основе “сэндвича”, образованного двойными антителами. Для захвата ЕРО используют лунки планшета для микротитрования, предварительно покрытые моноклональным мышиным антителом, специфичным для rHuEPO. Связанный ЕРО метят анти-ЕРО поликлональным (кроличьим) антителом и пероксидазой хрена (конъюгат). Оптический сигнал получают при добавлении субстрата. Главной собственной модификацией набора P&D было использование собственного рекомбинантного эритропоэтина человека в стандартах и меченых образцах для контроля качества.
Используемые в анализе стандартные концентрации составляли 7, 8; 15, 6; 31, 3; 50; 62, 5; 100; 125 и 250 мМЕ/мл. Чувствительность, определенная как наименьший стандарт, дающий допустимую погрешность, составляла 7,8 мМЕ/мл, и диапазон анализа был расширен до 5000 мМЕ/мл посредством разведении при контроле качества.
Фармакокинетические параметры
Измеряли концентрации эритропоэтина в сыворотке. Фармакокинетические параметры Сmах, tmax, Cmin, AUC(0-168), CL/F и t1/2 измеряли с помощью независимых от модели способов.
Анализ фармакодинамики
Фармакодинамические параметры включали измерение изменений от исходного уровня процента ретикулоцитов, эритроцитов и концентраций гемоглобина и их взаимосвязи с концентрациями эритропоэтина в сыворотке.
Оценки безопасности
Нежелательные явления
Нежелательные явления, появляющиеся при лечении, определяли как любые вредные и непредвиденные явления, наблюдаемые в ходе клинического исследования, которые были впервые выявлены, или усилены по тяжести или частоте, включая патологические изменения лабораторных показателей, которые требовали медицинского вмешательства, включая дополнительные диагностические процедуры или изменение терапии исследования.
Каждый субъект наблюдался по поводу нежелательных явлений на протяжении всего исследования, начиная с введения первой дозы. Сообщения о нежелательных явлениях идентифицировали по сообщениям добровольных субъектов исследования. Исследователь регистрировал в индивидуальной регистрационной карте (CRF) субъекта любые нежелательные явления, возникшие при лечении, независимо от их взаимосвязи с исследуемым препаратом. Нежелательные явления характеризовали по дате появления, тяжести (выраженное, умеренное или слабо выраженное), взаимосвязи с исследуемым препаратом (очень вероятно, вероятно, возможно, сомнительно или не связано), действию, предпринятому в отношении терапии исследования (не предпринимали, доза снижена, введение препарата временно остановлено, или введение препарата прекращено совсем) и было ли явление серьезным или нет. Также регистрировали информацию о сопутствующей терапии и последствиях.
Серьезные нежелательные явления определяли как явления, которые были летальными или непосредственно угрожали жизни, требовали осуществить или продолжить госпитализацию в стационаре, вызывали длительную или значительную потерю трудоспособности или недееспособность или были врожденными аномалиями, врожденными пороками, или передозировками. Исследователь получал инструкции о том, чтобы сообщать о всех серьезных нежелательных явлениях непосредственно в RWJPRI. Исследователь должен был собирать информацию о серьезных нежелательных явлениях вплоть до 35 дней после последней оценки на 29 день. Данные по безопасности также анализировали в отношении потенциальных серьезных нежелательных явлений; такими считали явления, которые были достаточно тяжелыми или тревожными, что требовали медицинского вмешательства.
Клинические лабораторные тесты
Анализы образцов для клинических лабораторных тестов выполняли в Havenfern Laboratories, Berks, UK. Образцы крови, включая образцы на гемоглобин, гематокрит, процент ретикулоцитов, эритроцитов (RBC), среднее гематокритное число (MCV), средний эритроцитарный гемоглобин (МСН), среднюю концентрацию эритроцитарного гемоглобина (МСНС) и тромбоциты, собирали во время отбора и в 1, 3, 5, 8, 10, 12, 15, 17, 19, 22, 24, 26 и 29 дни, по возможности от 8 до 10 часов утра. Полный анализ крови (СВС) при отборе также включал общее количество эритроцитов и общее количество лейкоцитов с дифференциацией.
Образцы для химического анализа сыворотки брали при отборе и в конце исследования (29 день); параметры включали глюкозу, кальций, натрий, калий, хлорид, фосфор, азот мочевины крови (BUN), общий билирубин, креатинин, суммарный белок, холестерин, альбумин, мочевую кислоту, щелочную фосфатазу, сывороточную трансаминазу глутаминовой-щавелево-уксусной кислоты (SGOT; аспартатаминотрансферазу (AST), сывороточную трансаминазу глутаминовой-пировиноградной кислоты (SGPT; аланинами-нотрансферазу (ALT) и лактатдегидрогеназу (LDH).
Образцы крови для определения параметров железа (сывороточное железо, общая железо-связывающая способность (TIBC) и уровни ферритина) брали во время отбора и на 8, 15, 22 и 29 день. Насыщение трансферрина рассчитывали в виде отношения железо/общая железосвязывающая способность.
Тестирование мочи выполняли с помощью измерительного стержня. Если проба на кровь или эстеразу лейкоцитов была позитивной (1+ или выше) и/или белок или нитрат присутствовал в следовых количествах или выше, образец мочи необходимо было послать в центральную лабораторию для микроскопического обследования.
Другие наблюдения по безопасности
Показатели жизненно важных функций
Измерения показателей жизненно важных функций - давления крови в положении сидя, скорости пульса и оральной температуры, регистрировали при отборочном визите, перед введением исследуемого препарата в 1, 8, 15 и 22 день и на 29 день (завершение исследования). Показания режима респирации и массы тела снимали при отборе и при завершении; размеры роста определяли только при отборочном визите.
Объективное обследование
Объективные исследования выполняли при отборочном визите и на 29 день.
Гарантия качества данных
Перед тем как был выбран центр исследования, исследователь, персонал центра исследования и ресурсы оценивались клиническим персоналом RWJPRI. Перед началом исследования протокол и заявление о согласии, основанном на получении информации, были рассмотрены и одобрены Этическим исследовательским комитетом (ЕС). Индивидуальные регистрационные карты проверялись для точности и полноты персоналом RWJPRI во время периодических визитов для мониторинга на месте.
Расхождения в данных решали с исследователем или назначенными должностными лицами. Данные вводили в базу данных RWJPRI и соответствующие компьютерные редакторские программы запускали для того, чтобы проверить точность базы данных.
Статистические способы
Объем выборки в данном исследовании не был основан на статистическом анализе. Поэтому анализ является описательным: не выполняли статистических тестов для фармакокинетических параметров. Суммарная статистика, включая среднее, стандартное отклонение, медиану и размах, была представлена по группам лечения для гематологии, химического состава сыворотки и измерений показателей жизненно важных функций.
Основные цели (т.е. оценить фармакокинетический профиль эпоэтина-альфа после введения 150 МЕ/кг т.р.н. или 40000 ME р.н., и показать, что два режима дозирования дают сходные клинические выходы при использовании гемоглобина в качестве меры клинической эффективности) были достигнуты описательным сравнением фармакокинетических параметров, полученных после введения исследуемого препарата.
Фармакокинетика
Следующие фармакокинетические параметры рассчитывали способами, независимыми от модели, с использованием компьютерной программы WinNonlin, версии 1.1 (Scientific Consulting, Incorporation, Apex, NC):
Пиковая концентрация в сыворотке (Сmах): максимальная концентрация в сыворотке, наблюдаемая в ходе четвертой недели периода дозирования для режима дозирования 150 МЕ/кг т.р.н. и режима дозирования 40000 ME р.н.
Время до Сmах (tmах): время, при котором имеет место Сmах. Показатель tmax не представляли для группы лечения эпоэтином-альфа т.р.н., так как Cmах достигалась случайным образом при любой из трех доз в ходе четвертой недели дозирования.
Средняя наименьшая концентрация до введения дозы (Cmin). Cmin для режима 150 МЕ/кг т.р.н. оценивали усреднением наименьших концентраций до введения дозы на 22, 24 и 26 день и концентрации через 72 часа после последней дозы на 26 день. Cmin для режима 40000 ME р.н. оценивали усреднением концентраций до введения дозы в 8, 15, 22 день и концентрации через 168 часов после последней дозы на 22 день.
Площадь под кривой “концентрация в сыворотке - время” от нулевого времени до времени отбора последнего образца крови AUC (0-168) в течение последней недели дозирования для эпоэтина-альфа 40000 ME р.н. и 150 МЕ/кг т.р.н. рассчитывали с использованием линейного варианта формулы трапеций.
Клиренс после п/к введения (CL/F): рассчитывали делением дозы (на кг) на AUC(0-168).
Период полуэлиминации на конечном этапе (t1/2): рассчитывали из отношения 0,693/константа скорости элиминации. Константу скорости элиминации определяли посредством линейной регрессии последовательных точек данных в конечном линейном районе логлинейного графика “концентрация - время”. В регрессии использовали минимально три точки ввода данных. Значения t1/2 не вносили в отчет для регрессии с коэффициентом корреляции (r) меньше 0,975 (или r2 <0,95).
Среднее, стандартное отклонение и коэффициент вариации фармакокинетических параметров рассчитывали для каждого лечения.
Фармакокинетические параметры рассчитывали, используя концентрации эритропоэтина в сыворотке, скорректированные относительно эндогенного уровня эритропоэтина до введения дозы. Значения концентрации в сыворотке после введения дозы корректировали относительно исходных концентраций эритропоэтина до введения дозы вычитанием из каждого значения средней исходной концентрации эритропоэтина, определенной для образцов, собранных за 30, 20 и 10 минут перед дозированием. Концентрации эритропоэтина в сыворотке до введения дозы не включали в расчет среднего значения, если они были ниже предела количественного анализа. Если значения концентрации для всех трех образцов от субъекта до введения дозы были ниже предела количественного анализа, то предел количественного анализа, 7,8 мМЕ/мл, принимали за среднюю исходную концентрацию эритропоэтина у данного субъекта. Фактическое время отбора крови (в часах относительно времени введения первой дозы) использовали при расчетах фармакокинетических параметров.
Биодоступность при режиме дозирования 400000 ME р.н. относительно биодоступности, полученной после режима дозирования 150 МЕ/кг т.р.н. рассчитывали, используя следующую формулу:
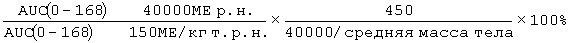
*Средняя масса тела субъектов в группе 40000 ME р.н., которые полностью прошли исследование.
Фармакодинамика
Суммарная статистика, включая среднее, стандартное отклонение, медиану, размах и стандартную ошибку, была представлена по группам лечения и дням для процента ретикулоцитов и концентраций гемоглобина. Расчеты были основаны на данных от субъектов, которые завершили исследование. Составляли сводки данных исходного уровня, дней 1, 3, 5, 8, 10, 12, 15, 17, 19, 22, 24, 26 и 29; выполняли “кадрирование”, чтобы включить результаты лабораторного тестирования в сводки, если субъект не имел данных, собранных в назначенный день (дни). Изменения относительно исходного уровня суммировали по группам лечения и дням. Построены линейные графики среднего изменения относительно значений исходного уровня против дня исследования для ретикулоцитов, гемоглобина и ферритина.
Фармакокинетика/Фармакодинамика
При расчете фармакодинамических параметров использовали номинальное время (в днях относительно первой дозы на 1 день), которое указано в протоколе. Способами, независимыми от модели, с помощью компьютерной программы WinNonlin, версии 1.1 (Scientific Consulting, Incorporation, Apex, NC) рассчитывали следующие фармакодинамические параметры:
Площадь под кривой “концентрация в сыворотке - время” процента ретикулоцитов от нулевого времени до 672 часов (29 день) после начала дозирования [AUC(RETI)] рассчитывали, используя линейный вариант формулы трапеции. AUC(RETI) оценивали, используя значения процента ретикулоцитов, скорректированные относительно значения процента ретикулоцитов до введения дозы. В качестве значения процента ретикулоцитов до введения дозы использовали среднее измерений в точках -10 минут и -30 минут до введения дозы. Для данных скорректированного процента ретикулоцитов, которые имели отрицательные значения, для оценки AUC(RETI) использовали нулевое значение.
Площадь под кривой “концентрация в сыворотке - время” гемоглобина от нулевого времени до 672 часов (29 день) после начала дозирования [AUC(HEMO)] рассчитывали, используя линейный вариант формулы трапеции. AUC(HEMO) оценивали, используя значения гемоглобина, скорректированные относительно значения гемоглобина до введения дозы. В качестве значения гемоглобина до введения дозы использовали среднее измерений в точках -10 минут и -30 минут до введения дозы. Для данных скорректированного значения гемоглобина, которые имели отрицательные значения, для оценки AUC(НЕМО) использовали нулевое значение.
Площадь под кривой “концентрация в сыворотке - время” суммарного количества красных кровяных клеток (RBC) от нулевого времени до 672 часов (29 день) после начала дозирования [AUC(RBC)] рассчитывали, используя линейный вариант формулы трапеции. AUC(RBC) оценивали, используя значения RBC, скорректированные относительно значения RBC до введения дозы. В качестве значения RBC до введения дозы использовали среднее измерений в точках -10 минут и -30 минут до введения дозы. Для данных скорректированного значения RBC, которые имели отрицательные значения, для оценки AUC(RBC) использовали нулевое значение.
Определяли отношения AUC(RETI), AUC(HEMO) и AUC(RBC) для режима 40000 ME р.н. к значениям этих показателей для режима 150 МЕ/кг т.р.н.
Безопасность
Оценки безопасности были основаны на типе и частоте нежелательных явлений, сообщенных субъектами, и изменениям объективного обследования, клинических лабораторных данных и показателей жизненно важных функций. Нежелательные явления, появляющиеся при лечении, классифицировали по системам организма, предпочтительному наименованию и включенному в исследование наименованию. Нежелательные явления кодировали в соответствии с терминологическим словарем нежелательных реакций Всемирной организации здравоохранения (WHOART), где включенный в исследование термин представляет собой описание, наиболее близкое терминологии исследователя, предпочтительный термин означает группу близких включенных в исследование терминов, и система организма представляет собой широкую категорию, включающую близкие предпочтительные термины. Нежелательные явления, появляющиеся при лечении, суммировали по системам организма и предпочтительным терминам и представили в виде распечаток данных индивидуальных субъектов.
Клинические лабораторные данные и показатели жизненно важных функций суммированы и представлены в виде распечаток данных индивидуальных субъектов.
Хранение данных
Протокол, отчет и исходные данные этого исследования хранятся в Управлении документооборотом, в отделении управления информацией RWJPRI. Данные можно найти в тетради проекта, где сохранены данные исследования метаболизма препарата DM00009.
Продолжительность исследования
Дозирование и сбор образцов сыворотки для обеих групп проводили в течение периода с 21 февраля 2000 г. по 29 марта 2000 г. Валидация анализа эритропоэтина в сыворотке проходила с 21 января 2000 г. по 3 февраля 2000 г. Образцы сыворотки анализировали в течение периода с 24 марта 2000 г. по 4 апреля 2000 г.
Результаты
Демографические и исходные параметры
Тридцать шесть здоровых взрослых людей (по 18 субъектов в группе) были зарегистрированы в данном исследовании и были случайным образом распределены в одну из групп лечения. В целом большинство субъектов (89%) были белой расы, и средний возраст составлял 26,5 лет (в пределах от 18 до 41 года). Средняя масса тела субъектов в группе 40000 ME р.н. была немного выше (70,3 кг) по сравнению с группой 150 МЕ/кг т.р.н. (66,8 кг), при этом остальные исходные и демографические параметры были очень сходны между двумя группами.
Демографические и исходные параметры 34 субъектов, которые завершили исследование, представлены на фигуре 47. Заметных отличий от общей популяции исследования не было.
Информация о завершении/исключении из исследования
Субъекты считались завершившими исследование, если они принимали участие на протяжении всего (29 дней) исследования. Кроме того, субъект должен принять все желаемые дозы исследуемого препарата, они должны были соглашаться на процедуры отбора образцов крови, и они должны были подвергнуться оценкам и процедурам на 29 день. Эффективная популяция включала субъектов, получавших все желаемые дозы, которые завершили исследование. Эффективную популяцию использовали для анализа фармакокинетических и фармакодинамических данных.
94% субъектов в обеих группах завершили исследование. Один субъект (1014) в группе введения эпоэтина-альфа в дозе 150 МЕ/кг прекратил исследование на 15 день вследствие нежелательного явления (постоянных головных болей), и один субъект (1006) в группе введения эпоэтина-альфа в дозе 40000 ME р.н. прекратил исследование на 10 день (по желанию субъекта). Указанных субъектов не включали в эффективную популяцию.
Аналитические результаты
Фармакокинетические результаты
Фигура 48 иллюстрирует временные профили средней концентрации эпоэтина-альфа в сыворотке (не скорректированные относительно эндогенного уровня эритропоэтина до введения дозы) для групп с режимами 150 МЕ/кг т.р.н. и 40000 ME р.н. в течение недели 4 периода исследования. Временные профили средней концентрации эритропоэтина в сыворотке, скорректированные относительно эндогенного уровня эритропоэтина до введения дозы показаны на фигуре 49.
Средние (SD) концентрации эндогенного эритропоэтина-альфа до введения дозы для субъектов в группах с режимами 150 МЕ/кг т.р.н. и 40000 ME р.н. составляли 8 (0,4) и 9 (2) мМЕ/мл, соответственно. В течение недели 4 при режиме дозирования 150 МЕ/кг т.р.н. концентрации эритропоэтина в сыворотке (скорректированные относительно исходного уровня эритропоэтина) были в пределах от пиковых концентраций от 75 до 284 мМЕ/мл [средняя (3D) Cmах=143 (54) мМЕ/мл] до значений минимального уровня, находящихся в пределах от значений ниже предела количественного анализа аналитического способа (7,8 мМЕ/мл) до 40 мМЕ/мл [средняя (3D) минимальная концентрация (Cmin)=18 (9) мМЕ/мл]. В ходе недели 4 режима дозирования 40000 ME р.н. концентрации эритропоэтина в сыворотке (скорректированные относительно исходного уровня эритропоэтина) достигали пиковых концентраций [средняя (3D) Сmах=861 (445) мМЕ/мл] за время от 1 до 24 часов [медианное tmax=15 (пределы 1-24) часам], затем снижались мультиэкспоненциально до значений минимальных уровней в пределах от значений ниже предела количественного анализа аналитического способа (7,8 мМЕ/мл) до 5,9 МЕ/мл [средняя (3D) минимальная концентрация на 29 день=2,0 (1,5) мМЕ/мл] к концу недели дозирования на 29 день. Средняя (3D) Cmin в течение четырехнедельного периода исследования для режима 40000 ME р.н. составляла 3,8 (4,3) мМЕ/мл. Конечные фазы двух режимов дозирования выглядели параллельно со средними (SD) значениями времени полужизни 19,4 (8,1) часа (n=9) и 15,0 (6,1) часов (n=9) для режимов дозирования 150 МЕ/кг т.р.н. и 40000 ME р.н., соответственно.
Средние значения (SD) (%CV) фармакокинетических параметров представлены на фигуре 50. Биодоступность эпоэтина-альфа при режиме дозирования 400000 ME р.н. относительно биодоступности при режиме дозирования 150 МЕ/кг т.р.н. составляла 239%.
Фармакодинамические результаты
Средние изменения относительно исходного уровня для процента ретикулоцитов, концентраций гемоглобина и значений количества красных кровяных клеток суммированы по группам лечения и дням исследования на фигурах 51, 53 и 55, соответственно. Среднее изменение относительно исходного уровня для фармакоди-намических результатов представлено только для субъектов, которые полностью прошли исследование.
Линейные графики среднего изменения относительно исходного уровня против дня исследования для процента ретикулоцитов, концентраций гемоглобина и значений для красных кровяных клеток представлены на фигурах 52, 54 и 56, соответственно.
Процент ретикулоцитов
В обеих группах среднее изменение процента ретикулоцитов увеличивалось до 10 дня и постепенно снижалось к 29 дню (фигуры 51 и 52).
Гемоглобин
Среднее значение гемоглобина на исходном уровне было эквивалентным в двух группах дозирования, 13,4 г/дл в группе 150 МЕ/кг т.р.н. и 13,5 г/дл в группе 40000 ME р.н. В обеих группах лечения среднее изменение от исходного уровня значений гемоглобина увеличивалось до 26 дня (фигуры 53 и 54). Среднее изменение относительно исходного уровня значения гемоглобина для группы 40000 ME р.н. было зеркальным отражением изменения в группе с режимом введения эпоэтина-альфа 150 МЕ/кг т.р.н. В общем, в обеих группах наблюдалось увеличение на 3,1 г/дл от исходного уровня до 29 дня.
Эритроциты
Среднее изменение от исходного уровня значений эритроцитов показано на фигурах 55 и 56. В обеих группах среднее изменение относительно исходного уровня для значений количества эритроцитов увеличивалось до 24 дня. Среднее изменение относительно исходного уровня значений количества эритроцитов для группы с режимом 40000 ME р.н. было зеркальным отражением изменения в группе с введением эпоэтина-альфа в режиме 150 МЕ/кг т.р.н. В общем, в обеих группах наблюдалось увеличение на 1,0×1012/л от исходного уровня до 29 дня.
Фармакокинетические/фармакодинамические результаты
Средние значения фармакодинамических параметров (скорректированные относительно исходного значения) представлены на фигуре 57. Динамические ответы при двух режимах дозирования были сходными, несмотря на тот факт, что AUC эритропоэтина в сыворотке для режима дозирования 40000 ME р.н. была больше, чем AUC эритропоэтина в сыворотке для режима дозирования 150 МЕ/кг т.р.н. Не было статистически значимых различий (р>0,05) в AUC% ретикулоцитов, AUC гемоглобина и AUC эритроцитов между двумя режимами дозирования. Не было статистически значимых различий (р>0,05) в AUC процента ретикулоцитов между субъектами мужского и женского пола. Однако AUC гемоглобина и AUC эритроцитов были статистически больше (р=0,038 и 0,042, соответственно) у женщин, чем у мужчин. Клинически эти различия были не значимы.
Результаты исследования безопасности
Степень экспозиции
Тридцать четыре субъекта (94%) из субъектов, принимавших участие в исследовании, получали все дозы исследуемого препарата (либо 150 МЕ/кг т.р.н. эпоэтина-альфа [12 доз], либо 40000 ME р.н. эпоэтина-альфа [четыре дозы]). Один субъект (1014), попавший в группу с режимом введения эпоэтина-альфа в дозе 150 МЕ/кг т.р.н., вышел из исследования вследствие нежелательного явления (постоянных головных болей) после получения шести доз исследуемого препарата. В группе 40000 ME р.н. один субъект (1006) по собственному выбору вышел из исследования после получения двух доз исследуемого препарата.
С одним исключением все субъекты ежедневно получали перорально добавку железа по протоколу; субъект 2008 (40000 ME р.н.) прекратил пероральный прием добавки железа на 19 день исследования. С одним исключением все женщины продолжали пользоваться способом контролирования беременности, которым они обычно пользовались до исследования (согласно протоколу); субъект 1010 (эпоэтин-альфа 40000 ME р.н.) прекратила пероральный прием средства контроля беременности за одиннадцать дней до начала исследования.
Нежелательные явления
Все нежелательные явления, возникшие при лечении, суммированы на фигуре 58. В целом, 13 (72%) из 18 субъектов, которым вводили эпоэтин-альфа в режиме 150 ME/кг т.р.н. имели нежелательное явление, по сравнению с 12 (67%) из 18 субъектов, которым вводили 40000 ME эпоэтина-альфа р.н. Большинство нежелательных явлений, возникающих при лечении, были умеренной тяжести с небольшими качественными различиями между двумя группами.
Наиболее часто заявляемыми нежелательными явлениями были боль (22% 150 МЕ/кг, 28% 40000 ME р.н.), головная боль (28% в обеих группах) и эритематозная сыпь. У пяти (28%) из 18 субъектов, получавших 150 ME эпоэтина-альфа/кг т.р.н,, выявлена эритематозная сыпь в месте введения канюли на предплечье, по сравнению с двумя (11%) из 18 субъектов, получавших 40000 ME эпоэтина-альфа р.н. Все явления, оцененные исследователем, не были связаны с терапией эпоэтином-альфа.
Сводка всех нежелательных явлений
Смертельные случаи, другие серьезные нежелательные явления и другие значительные нежелательные явления
В течение курса исследования не было смертельных исходов или серьезных нежелательных явлений. Один субъект (1014), получавший эпоэтин-альфа в режиме 150 МЕ/кг т.р.н. прекратил исследование из-за нежелательного явления (постоянной головной боли) на 15 день исследования. Головные боли были расценены исследователем как очень вероятно связанные с введением эпоэтина-альфа.
Субъект 2007 (150 МЕ/кг т.р.н., 28-летний белый мужчина, имел уровень гемоглобина 18,0 г/дл на 26 день. Повторная оценка гемоглобина после этого дня показала уровень 17,6 г/дл. На 29 день уровень гемоглобина у субъекта составлял 18,2 г/дл; повторная оценка на 31 день показала уровень гемоглобина 18,4 г/дл. Затем субъекту была сделана флеботомия, что детально изложено в протоколе; взято 450 мл крови. Субъект завершил исследование на 29 день, и впоследствии наблюдали за уровнями гемоглобина для безопасности; оценки на 32 и 39 день показали уровни гемоглобина 17,7 г/дл и 16,9 г/дл, соответственно.
Сопутствующие лекарственные средства, используемые во время исследования для лечения нежелательных явлений, включали: парацетамол от головной боли (субъекты 1014, 2004 [150 МЕ/кг] и 1011, 2008 [40000 ME]), зубной боли (субъект 2004 [150 МЕ/кг]), боли в шее (субъект 2110 [150 МЕ/кг]), периодической боли (субъект 1013 [150 МЕ/кг]), желудочного гриппа (субъект 1011 [40000 ME]), простуды (субъект 1013 [150 МЕ/кг]) и боли в месте введения канюли (субъект 1008 [150 МЕ/кг]); и физиологический раствор и хлорамфеникол (субъект 2004 [150 МЕ/кг]) для воспаления глаз.
Клиническая лабораторная оценка
Значения, полученные в лаборатории на протяжении времени исследования
Средние изменения от исходного уровня железа, ферритина и насыщения трансферрина суммированы на фигуре 59. Не было непротиворечивых примеров значений железа или насыщения трансферрина, которые бы свидетельствовали о том, что какое-либо из двух режимов лечения привело к клинически значимым отклонениям.
Картины, отражающие изменения ферритина, которые были сходны между группами, отражают ожидаемое использование запасов железа для продукции гемоглобина (фигура 60). Флуктуирующие уровни железа в сыворотке в обеих группах не считали клинически значимыми.
Как показано на фигуре 60, среднее изменение от исходного уровня значений ферритина уменьшалось до 8 дня и оставалось низким до 29 дня, свидетельствуя о продолжающемся эритропоэзе в течение этого периода. Не было заметных различий между группами в среднем изменении значений ферритина относительно исходного уровня.
Изменения у отдельных субъектов
В ходе исследования не было изменений у отдельных субъектов, регистрируемых как нежелательное явление.
Другие исследования безопасности
Показатели жизненно важных функций
Сводка средних изменений от исходного уровня в измерениях показателей жизненно важных функций по дням исследования по индивидуальным данным субъектов представлены на фигуре 61. Не было клинически значимых изменений в средних измерений показателей жизненно важных функций ни в какой из групп лечения, и не было значимых различий между группами.
У двух субъектов, которым вводили эпоэтин-альфа в дозе 150 МЕ/кг т.р.н., наблюдались значения систолического давления крови на уровне или выше верхнего предела 140 мм Hg; ни одно из этих событий не рассматривались исследователем как клинически значимое, и ни одно не было зарегистрировано как нежелательное событие.
Объективное обследование
Не было клинически значимых изменений от исходного уровня при объективном обследовании.
Заключения о безопасности
Эпоэтин-альфа, вводимый т.р.н. в дозе 150 МЕ/кг или р.н. в дозе 40000 ME, был безопасен и хорошо переносился здоровыми субъектами в данном исследовании. Ни в одной из групп лечения не было клинически значимых нежелательных явлений, возникающих при лечении. Большинство нежелательных явлений, возникающих при лечении, были умеренной тяжести с небольшими качественными различиями между двумя группами. Ни один из субъектов не умер во время исследования, и не сообщалось о серьезных нежелательных явлениях. Одному субъекту, которому вводили эпоэтин-альфа в дозе 150 МЕ/кг т.р.н., проводили флеботомию на 31 день исследования, из-за высоких уровней гемоглобина. Последующий мониторинг уровней гемоглобина у этого субъекта не выявил дальнейшего повышения уровней гемоглобина. Один субъект, получавший эпоэтин-альфа в дозе 150 МЕ/кг т.р.н., вышел из исследования вследствие нежелательного явления (постоянных головных болей). Не было отмечено клинически значимых изменений в значениях клинических лабораторных тестов, средних измерениях показателей жизненно важных функций, или объективном обследовании каждой группы; также не было видимых различий в результатах между группами.
Заключение и обсуждение
После введения эпоэтина-альфа в дозе 40000 ME р.н. значения Сmах были в шесть раз выше, а значения AUC(0-168)) - в три раза выше, чем значения при режиме дозирования 150 МЕ/кг т.р.н. Клиренс после режима дозирования 150 МЕ/кг т.р.н. был выше, чем после режима дозирования 40000 ME р.н. Временные профили изменений процента ретикулоцитов, гемоглобина и суммарных красных кровяных клеток в течение одномесячного периода исследования были сходными между двумя режимами дозирования, несмотря на различия в экспозиции эпоэтина-альфа в сыворотке [в пересчете на AUC(0-168)]. Кроме того, не было статистически значимых различий (р>0,05) в AUC процента ретикулоцитов, AUC гемоглобина и суммарных красных кровяных клеток в течение одномесячного периода исследования между двумя режимами дозирования. Хотя различия в AUC гемоглобина и AUC суммарных эритроцитов между субъектами мужского и женского пола были статистически значимыми, эти различия не считались клинически значимыми. Результаты данного исследования ясно свидетельствуют о том, что ответы на уровне гемоглобина после режимов дозирования 150 МЕ/кг т.р.н. и 40000 ME р.н. сходны.
Имело место ожидаемое различие в суммарной экспозиции эпоэтина-альфа в сыворотке после режимов дозирования 150 МЕ/кг т.р.н. и 40000 ME р.н. Ответы на уровне гемоглобина были сходными, свидетельствуя о том, что два режима дозирования могут быть взаимозаменяемыми.
Пример 3
СРАВНЕНИЕ ФК/ФД-ПАРАМЕТРОВ ПОСЛЕ ВВЕДЕНИЯ EPREX® И PROLEASE®
Фигура 62 является схематичным представлением модели стимулирующих эритропоэз эффектов rHuEpo. Эту модель использовали для оценки кинетических и динамических параметров ответов на rHuEpo после введения 8 однократных доз EPREX®, а также кинетических параметров после введения однократной дозы PROLEASE®.
Фармакокинетика
Фармакокинетику 600 МЕ/кг/нед. EPREX®, вводимого в течение 4 недель, моделировали, используя параметры, полученные из одновременной подгонки восьми однократных доз. Оценивали только значения Tau и Fr. Для режима при злокачественной опухоли INT-57 - 150 МЕ/кг/т.р.н. значения F, Tau и Vd были фиксированными, как показано на фигуре 63, основанными на предыдущих оценках более раннего исследования (ЕРО-358/359). Оценивали фармакокинетические параметры для однократной дозы PROLEASE® (2400 МЕ/кг) и эти параметры использовали для моделирования многократного режима дозирования 1800 МЕ/кг/месяц. Одни и те же наборы кинетических параметров использовали для характеристики профилей как для мужчин, так и для женщин, так как предварительные прогоны не показали значимых различий в оцениваемых параметрах на основании пола.
Фармакодинамика
Фиксировали кинетические параметры и использовали как вынуждающую функцию для управления динамическими ответами. Значения до введения дозы фиксировали как исходные уровни для ретикулоцитов, и средние 48-и 96-часовые значения фиксировали как исходные для RBC. Параметры продолжительности жизни, полученные на основании оценки однократной дозы EPREX®, фиксировали для дальнейших подгонок многократных доз. Smax и SC50 оценивали для ответа на уровне ретикулоцитов после применяемого в хирургии режима многократного дозирования 600 МЕ/кг/нед. Как сообщается на фигуре 64, эти параметры, по-видимому, не очень существенно изменяются, учитывая вариабельность в ответах. Различия могут просто отражать тот факт, что это было другое исследование фазы I, отличное от исследования однократной дозы, проводимое на новом наборе добровольцев. Кроме того, этот набор параметров, по-видимому, также хорошо характеризует режим при лечении злокачественной опухоли 150 МЕ/кг/т.р.н., на основании моделирований, показанных на фигурах 65 и 66. Данные по ретикулоцитам для мужчин и женщин анализировали отдельно, так как Smах и SC50, оцененные по данным для субъектов мужского пола, не описывали достаточно хорошо ответы у женщин. Как видно из фигуры 68, эти параметры отличались в том случае, когда оценивались отдельно для женщин, хотя было трудно судить, было ли различие существенным. Эти параметры могут отражать некоторые небольшие фармакодинамические различия, основанные на различии полов, так как все данные были получены в одном исследовании, в котором можно ожидать более низкую вариабельность.
Определенные параметры для EPREX® использовали для моделирования ответов, как для однократного, так и для двукратного режима дозирования PROLEASE®. Моделирование с использованием моделей согласно данному изобретению выполняли отдельно для мужчин и женщин, в соответствии с параметрами, определенными для соответствующих полов на основе оценок EPREX®. Кроме того, моделировали RBC-ответы на основании параметров, выработанных с использованием данных по ретикулоцитам, и эритроцитарный ответ, по-видимому, хорошо характеризуется во всех случаях как для EPREX®, так и для PROLEASE.
На основании моделирования, которое показано на фигурах 66 и 67, можно видеть, что один и тот же набор динамических параметров может хорошо описывать ответы для обеих композиций. Поэтому можно сделать вывод, что EPREX® и PROLEASA®,по-видимому, фармакодинамически эквивалентны. Модели согласно данному изобретению предсказывают, что различия в ответах между двумя композициями могут быть полностью учтены посредством измененной кинетики. Действительно, модели согласно данному изобретению можно использовать для сравнения ФК/ФД-характеристик новых форм и версий ЕРО и ЕРО-подобных соединений с соединениями, доступными в настоящее время, чтобы предоставить пациенту наиболее благоприятный режим лечения.
Пример 4
СРАВНЕНИЕ РАЗНЫХ РЕЖИМОВ ДОЗИРОВАНИЯ rHuEPO
Стандартными режимами дозирования для хронического введения rHuEPO являются 150 МЕ/кг/т.р.н. и 600 МЕ/кг/р.н. Экономия затрат и дополнительные удобства могут иметь место в том случае, если терапия пациента включает менее частое дозирование. Поэтому оценивали различия в ответах на уровне гемоглобина при разных режимах дозирования rHuEPO, используя модели согласно данному изобретению, чтобы охарактеризовать и предсказать ответы на rHuEPO, которые предлагают наиболее эффективные режимы лечения.
Использовали фармакодинамическую модель продукции/потери наряду с фармакокинетической моделью двойной абсорбции согласно данному изобретению. Параметры, полученные на основе подгонки динамики rHuEPO у здоровых добровольцев (фигура 3 и фигура 13), использовали для моделирования разных режимов дозирования. Концентрация ЕРО исходного уровня, равная 40 МЕ/л, была фиксированной при моделировании во всех случаях. Для всех моделирований использовали программу ADAPT II.
На фигуре 68 показаны смоделированные профили ответа на уровне гемоглобина против времени для нескольких разных доз и режимов дозирования rHuEPO. Все режимы давали непрерывное повышение концентраций Нb вплоть до того, как достигается стационарное состояние примерно к 126 дню (3024 час). Доза 600 МЕ/кг/нед., по-видимому, дает максимальное увеличение уровней Hb. Эта доза и режим могут сохранять концентрации rHuEPO выше порогового значения, равного 23 МЕ/л, в течение максимальных периодов времени, вызывая постоянное увеличение количества клеток, в конце концов, продуцирующих более высокие стационарные уровни Нb. Такая же суммарная доза, равная 1200 МЕ/кг, даваемая каждые 2 недели, дает ответы, которые значительно ниже, поскольку концентрации rHuEPO падают ниже порогового уровня до того, как большая часть ретикулоцитов превратится в эритроциты. Кроме того, последующие дозы rHuEPO также не даются достаточно скоро, чтобы повысить концентрации выше порогового уровня, как это происходило бы при еженедельном дозировании.
Сходный аргумент может быть приведен при сравнении режима 450 МЕ/кг/нед. с дозированием 900 МЕ/кг каждые 2 недели. Дозирование 150 МЕ/кг т.р.н. эквивалентно режиму дозирования 450 МЕ/кг/нед. в пересчете доставляемой суммарной дозы, но динамические профили после дозирования трижды в неделю давали немного лучший Нb-ответ. Как и ожидалось, режим дозирования 900 МЕ/кг/10 дней дает профиль стационарного ответа (увеличение 56%) лучше, чем 1200 МЕ/кг/2 нед. (увеличение 48%), но ниже, чем режим 600 МЕ/кг/нед. (увеличение 71%).
В том случае, когда лечение продолжается в течение достаточно продолжительного периода времени, соответствующие достигнутые стационарные ответы, по-видимому, отличаются для разных доз и режимов. Однако эти различия в стационарном ответе не так очевидны при кратковременном режиме лечения, который вызывает только слабое повышение уровней Hb (например, увеличение на 1 единицу), за исключением лечения 600 МЕ/кг/неделю, которое вызывает соответственно более высокий Нb-ответ по сравнению с другими режимами. Моделирование с использованием моделей согласно данному изобретению показывает, что когда время для повторного введения уменьшается, можно достичь более высокого и более стабильного увеличения уровня Нb при одной и той же дозе. Частое дозирование помогает сохранять концентрации rHuEPO выше порогового уровня, способствуя образованию RBC из ретикулоцитов. Эритроциты, имеющие в 40 раз большую продолжительность жизни, чем ретикулоциты, сохраняются в крови в течение более длительного периода времени, приводя в результате к стабильному увеличению уровней Нb. Также видно, что изменение ответа, наблюдаемое при более частом введении, также зависит от выбранной дозы. Переключение с режима введения через неделю на режим еженедельного дозирования влияет на дозу 600 МЕ/кг больше, чем на дозу 450 МЕ/кг. Несмотря на то, что дозирование трижды в неделю лучше в отношении достигаемого стационарного ответа, оно может быть непредпочтительным по сравнению с еженедельным дозированием той же самой суммарной дозы, поскольку степень, до которой улучшается ответ, достаточно мала по сравнению с неудобствами более частого дозирования. Поэтому для тестированных режимов дозирования показано, что еженедельная доза 600 ME ЕРО/кг обеспечивает желаемый ФК/ФД-ответ.
В действительности модели согласно данному изобретению могут предоставить любой желаемый режим дозирования, такой как от режима менее чем ежедневного до менее чем еженедельного, менее чем один раз каждые две, три или четыре недели, в зависимости от используемого ЕРО и желаемого ФК/ФД-ответа. Таким образом, модели согласно данному изобретению не ограничены применением какого-либо конкретного типа ЕРО или какого-либо конкретного типа режима дозирования, и могут быть модифицированы и использованы для любого типа ЕРО.
Пример 5
ЭФФЕКТЫ ДОЗИРОВАНИЯ 40000 ME rHuEPO/НЕДЕЛЮ ПО ОТНОШЕНИЮ К МАССЕ ТЕЛА ПАЦИЕНТА
Необходимость повторного введения rHuEPO является причиной того, что дозирование на основе массы тела неудобно и занимает много времени. Переход от такой практики к дозированию определенного количества независимо от массы тела субъекта, облегчило бы клиническое применение rHuEPO. Поэтому выполняли моделирование с помощью моделей согласно данному изобретению в попытке выявить степень изменений в ожидаемых профилях ответов на уровне RBC и гемоглобина с изменением массы тела. Планировалось использовать это моделирование для того, чтобы понять, допустимо ли такое изменение способа дозирования с теоретической точки зрения.
Использовали фармакодинамическую модель продукции/потери наряду с фармакокинетической моделью двойной абсорбции согласно данному изобретению. Для моделирования разных режимов дозирования использовали параметры, полученные из подгонки к динамическим параметрам rHuEPO у здоровых добровольцев (фигура 3 и фигура 13). Концентрация исходного уровня ЕРО, равная 40 МЕ/л, была фиксированной при моделировании во всех случаях. Для всех моделирований использовали программу ADAPT II.
Влияние массы тела субъекта на ответ, предполагаемый для поддержания терапии rHuEPO, изображено на фигуре 69. На фигуре 69 показаны смоделированные профили RBC- и Hb-ответов против времени для режима 600 МЕ/кг/нед. в течение 24 недель (4032 часа) по сравнению с введением суммарной дозы 40000 МЕ/нед. субъектам с массой тела 50, 70 и 90 кг. Наблюдалось постоянное повышение и RBC и Hb в ходе введения rHuEPO. Доза 40000 МЕ/нед. сходна с режимом дозирования 600 МЕ/кг/нед., предполагая, что большинство субъектов имеют массу 70 кг.
Изменение массы тела влияет на объем распределения (Vd). RHuEPO имеет объем распределения 0,0558 л/кг, который очень близок объему плазмы. По мере увеличения массы тела Vd (л) увеличивается, приводя к тому, что достигаемые максимальные концентрации становятся ниже. Изменение массы тела также влияет на параметр клиренса Vmax (МЕ/час/кг). Имеет место увеличение Vmax (МЕ/час) с увеличением массы тела, приводящее к увеличению клиренса (Vmax/(Km+серo)) rHuEPO. поскольку и Vmах и Vd затронуты в одинаковой степени, константа скорости элиминации (т.е. k при более низких концентрациях) остается неизменной. Во всяком случае, различия в массе тела, по-видимому, не влияют на кинетику в значительной степени, поскольку конечный наклон после п/к введения в действительности регулируется кинетикой абсорбции, и при очень низкой SС50 конечный наклон главным образом регулирует степень ответа.
Моделирование показало, что стационарные уровни количества RBC и количества Нb незначительно отличаются на основании массы тела. Однако продолжительность времени, необходимого для достижения стационарного состояния, та же самая, и поэтому Нb-ответы в раннее периоды времени, такие как 4 недели, не очень сильно отличаются (16,46; 16,33 и 16,19 г/дл для субъектов массой 50, 70 и 90 кг по сравнению с 16,35 г/дл для дозирования 600 МЕ/кг/нед.).
Поэтому данные, полученные из моделей согласно данному изобретению, показывают, что различия в массе тела в пределах от 50 до 90 кг не вносят существенного вклада в кинетику и динамику rHuEPO. Поэтому дозирование, основанное на массе тела, может быть неоптимальным, не обязательным, и изменение дозирования на основе массы тела на стандартный режим 40000 МЕ/нед. независимо от массы тела приемлемо и удобно. Подразумевается, что дозы 40000 и 650 ME не являются абсолютным и предполагают пределы значений дозы, которая может быть введена пациенту, и дать такой же или сходный эффект. В действительности выражение “приблизительно” 40000 или 650 ME предполагает пределы значений, которые обеспечивают такой же или сходный эффект у пациента, и в конкретном варианте предполагает пределы от +/-1 до 20% значения ME.
Пример 6
ОЦЕНКА ДИНАМИКИ rHuEPO У БОЛЬНЫХ ЗЛОКАЧЕСТВЕННОЙ ОПУХОЛЬЮ
Пациенты со злокачественной опухолью, подвергающиеся химиотерапии, часто имеют анемию. Предполагают, что одним из факторов, ответственных за состояние анемии у этих пациентов, является неадекватная продукция эндогенного ЕРО, и показано, что введение rHuEPO в дозе 150 МЕ/кг три раза в неделю (т.р.н.) оказывается целебным для коррекции анемии. Хотя указанный режим дает адекватные ответы и наиболее широко используется, необходимое при этом частое дозирование делает его неудобным, и это может быть не самый лучший режим. Поэтому искали оптимальную дозу rHuEPO, которую можно вводить в еженедельном режиме, чтобы получить сравнимое увеличение уровней гемоглобина, как и при используемом в настоящее время режиме введения три раза в неделю. Использовали ФК/ФД-модели согласно данному изобретению с использованием данных от нормальных субъектов, чтобы количественно сравнить ответы у больных злокачественной опухолью и объяснить возможные причины различий в динамике, если они существуют. Исследование также предоставляет благоприятную возможность для валидации моделей согласно данному изобретению, так что эти модели могут с большой вероятностью использоваться в целях предсказания в будущем.
Данные о больных злокачественной опухолью получали из RWJPRI. Это было открытое, рандомизированное, контролируемое исследование в параллельных группах и в нескольких центрах исследования, проведенное на 150 больных злокачественной опухолью с анемией, имеющих твердые опухоли и получающих химиотерапию содержащими платину препаратами (цисплатин или карбоплатин). Разные уровни доз PROCRIT® (эпоэтин-альфа, Amgen), которые включали еженедельные п/к дозы, равные 300, 450, 600, 900 МЕ/кг, и п/к дозу, равную 150 МЕ/кг т.р.н., вводили 5 группам субъектов (25 пациентов в группе) в течение 12-недельного периода. Контрольная группа не получала лечения. Требовались пациенты 18-летнего возраста или старше, имеющие Hb≤10 г/дл, скорректированные количества ретикулоцитов ≤3%, тромбоцитов ≥25000 клеток/мм3, креатина ≤2,0 мг/мл, отрицательный анализ стула на скрытую кровь, без симптомов гемолиза, и нормальными уровнями фолата и витамина B12 в сыворотке. Также в исследование включали только таких субъектов, которым не требовалось переливание крови в течение месяца до рандомизации, которые не имели дефицита по железу. Количество гемоглобина измеряли в качестве основной конечной точки фармакодинамики.
Параметры, полученные на основе подгонок кинетических и динамических данных для нормальных добровольцев, использовали для моделирования ответов после введения разных доз и режимов rHuEPO больным злокачественной опухолью. Использовали фармакодинамическую модель продукции/потери наряду с кинетической моделью двойной абсорбции согласно данному изобретению. Параметры, полученные на основе подгонок динамики rHuEPO у здоровых добровольцев (фигура 3 и фигура 13) вместе с концентрацией ЕРО исходного уровня, равной 40 МЕ/л, использовали в качестве исходной точки для моделирования разных режимов дозирования, и исследовали влияние изменения различных параметров на ответы. Для всех случаев моделирования использовали программу ADAPT II.
На фигурах 70А-70Е показаны данные по гемоглобину и моделирование ответов на уровне ретикулоцитов, RBC и Hb для больных злокачественной опухолью с анемией, которым давали разные дозы и режимы rHuEPO. Как видно на фигурах, в общем, пациенты, по-видимому, отвечали на терапию положительно с постоянным увеличением концентраций Hb. Имеет место только очень слабый ответ, связанный с режимом дозирования 300 ME/кг. С увеличением еженедельной дозы, по-видимому, увеличивается степень ответа. Однако самая высокая доза 900 МЕ/кг/неделю не давала значительно более высокого увеличения уровней Hb, по сравнению с дозой 600 МЕ/кг/нед. Данные показывают, что режим еженедельного введения 600 МЕ/кг дает ответы немного лучше, чем режим 150 МЕ/кг т.р.н. Также на фигурах можно увидеть, что моделирование с использованием параметров, полученных для здоровых субъектов, предсказывает ответы, которые являются более высокими, по сравнению с ответами, реально наблюдаемыми у больных злокачественной опухолью. Причину таких измененных профилей ответа могут объяснять различия в кинетике и/или динамике rHuEPO у больных злокачественной опухолью, по сравнению со здоровыми субъектами. Поэтому моделирование выполняли, изменяя выбранные параметры в фармакодинамической модели, чтобы объяснить эти различия.
Сообщается, что некоторыми возможными причинами анемии при хроническом заболевании являются эритроидная гипоплазия костного мозга, сниженная выживаемость RBC и пониженный ретикулоцитоз (смотри, например, Abels, 1992, Semin. Oncol. 19:29-35). Существует мнение, что противораковая лекарственная терапия также является одной из основных причин анемии у этих пациентов (смотри, например, Matsumoto, et al., 1990, Br. J. Pharmacol. 75: 463-68). Снижение значения Ks на 1/3, свидетельствующее о пониженной скорости собственной продукции клеток, и/или более низкое значение Smax может объяснять пониженные ответы, как видно при моделировании. Пациенты с злокачественной опухолью имеют исходные концентрации ЕРО, которые выше, чем в норме, но неудовлетворительно низкие для степени анемии (смотри, например. Case et al., 1993, J. Natl. Cancer Inst. 85: 801-806 и Miller, et al., 1990, N. Engi. J. Med. 322:1689-99. Сообщается, что исходные концентрации ЕРО находятся в пределах от значений ниже 40 до значений более 500 Ед/л (смотри, например, Ludwig, et al., 1994, Blood 84: 1056-63, Case et al., выше, Abels, выше, и Miller et al., выше) в зависимости от тяжести и типа анемии, связанной с злокачественной опухолью и химиотерапией. Исходные уровни ЕРО выше 500 МЕ/л сообщались, чтобы показать невосприимчивость к терапии rHuEPO (смотри, например, Ludwig et al., выше).
Увеличение исходных концентраций ЕРО в модели до 70 МЕ/л, предсказывающее пониженную чувствительность системы к ЕРО, сдвигало временные профили ответов вниз и давало лучшую подгонку к данным. Поскольку дозирование делали каждую неделю, концентрации оставались намного выше порогового уровня, давая возможность для превращения большинства ретикулоцитов в эритроциты. Поэтому изменение порогового уровня не способствовало существенному сдвигу кривых, что свидетельствует о том, что процесс превращения ретикулоциты - эритроциты не может быть значительно затронут у этих пациентов. В нормальных условиях эритроциты живут в течение периода, равного 2880 часам, и согласно модели продукции/потери соответствующий стационарный уровень достигается в течение одной продолжительности жизни после начала продукции новых эритроцитов. Любое уменьшение продолжительности жизни RBC вызвало бы достижение этого стационарного уровня раньше, и он был бы ниже у этих пациентов. Такая возможность не была показана, поскольку дозирование не проводили так долго, чтобы было достаточным для того, чтобы обеспечить достижение соответствующего стационарного уровня в обычных условиях исследования. Как отмечено выше, другой возможной причиной сниженных ответов у больных злокачественной опухолью могли быть различия в фармакокинетике rHuEPO. У здоровых добровольцев rHuEPO подвергается кинетике “флип-флоп” после п/к введения, что является причиной того, что концентрации остаются выше исходного уровня в течение продолжительных периодов времени, обеспечивая непрерывную стимуляцию продукции новых клеток. Предположили, что медленное поглощение первого порядка через лимфатические сосуды вносит вклад в этот феномен, и любые изменения физиологического функционирования лимфатических сосудов вследствие состояния болезни и химиотерапии может снизить медленную доставку, приводя к более низким ответам.
Хотя режим еженедельного введения 600 МЕ/кг требует более высоких суммарных доз, по сравнению с режимом 150 МЕ/кг т.р.н., он вызывает лучшие Нb-ответы у больных злокачественной опухолью и является более удобным режимом дозирования для терапии на основе поддержания rHuEPO. Поэтому изменение применяемого в настоящее время режима на режим 600 МЕ/кг/нед. может быть предпочтительным согласно моделям данного изобретения. Разработанная ФК/ФД-модель может объяснять различия в ответах вследствие таких патологических состояний, как злокачественная опухоль, и моделирование предсказывает, что более низкое значение Ks и/или более высокие исходные уровни ЕРО с изменениями или без изменений фармакокинетики rHuEPO могут быть ответственны за ослабленные ответы, наблюдаемые у этих пациентов.
При обращении к предыдущему детальному описанию и конкретным примерам специалист в данной области поймет, что систему ФК/ФД-моделирования согласно данному изобретению можно использовать во многих ситуациях. Например, врач может нуждаться в корректировке режима дозирования ЕРО, чтобы достичь желаемого фармакокинетического ответа у пациента, такого как концентрация ЕРО в сыворотке. Врач может использовать системы согласно данному изобретению, чтобы достичь этого результата. В альтернативном случае врачу может потребоваться конкретный фармакодинамический ответ у пациента, такой как конкретное увеличение уровней гемоглобина. Врач может использовать системы, заявленные в данном изобретении, чтобы определить, какой режим дозирования ЕРО будет способен обеспечить достижение желаемого результата. В другом аспекте врачу может потребоваться определить, какой тип фармакокинетического и фармакодинамического выхода будет результатом конкретного режима дозирования ЕРО. И снова врач сможет использовать ФК/ФД-модели, заявленные в данном изобретении, чтобы осуществить такое определение.
Пример 7
ИММУНОГЕННОСТЬ ЕРО У СОБАК В ХОДЕ ОДНОМЕСЯЧНЫХ РЕЖИМОВ ДОЗИРОВАНИЯ
Данное исследование было спланировано для того, чтобы оценить иммуногенность композиций ЕРО у иммуносупрессированных и не иммуносупрессированных гончих собак. Исследовали фармакодинамические и фармакокинетические профили композиций ЕРО.
Согласование с официальными инстанциями
Качественная лабораторная практика (GLP): Данное исследование не проводилось при строгом соответствии с постановлением по GLP управления по санитарному надзору за качеством пищевых продуктов и медикаментов США для неклинических лабораторных исследований (21 CRF, часть 58), но было выполнено согласно протоколу и соответствующим стандартным рабочим процедурам Oread.
Уход за животными и использование: Исследования на животных проводили согласно “Руководству по уходу и использованию лабораторных животных” NRC (пересмотренное в 1996 г.) и “Акту о благосостоянии лабораторных животных” USDA, опубликованному 24 августа 1966 г. L.89-544, и последующим поправкам. Oread Met/PK является объектом, аккредитованным AAALAC.
Идентификация и источник
Композиции:
EPREX®, одна 2000 ME/мл и одна 10000 ME/мл
Физиологический раствор в качестве контроля
Хранение
Композиции ЕРО хранили в холодильнике (~ 4°С), защищенными от света, когда не использовали в исследовании. Неиспользованные композиции возвращали в RWJPRI после дозирования или уничтожали.
Исследуемые животные
Вид: собака
Порода: гончая
Пол: самцы
Источник: Harlan Sprague Dawley, Inc.
Indianapolis, Indiana 46299
Возраст при дозировании: 8-9 месяцев
Масса объекта при первом
дозировании: 9-12 кг
Способ идентификации: татуировка, нанесенная поставщиком
Количество в исследовании: 18 (N=3 собаки/группу)
Содержание
Собак содержали в группе по группам лечения в конурах в комнате для содержания собак, перед введением дозы собаки привыкали к обращению и отбору образцов. Карантин продолжался, по меньшей мере, 5 дней до введения дозы. К концу карантинного периода состояние здоровья всех животных подтверждалось исследовательским персоналом. В течение периода исследования/отбора собаки оставались в группах содержания, за исключением случаев, необходимых по состоянию здоровья. Конуру метили номером животного и номером протокола.
Условия окружающей среды
В комнате для животных поддерживали 23±3°С при относительной влажности 50±15% и 12-часовым циклом свет/темнота. Воздух в комнате меняли, по меньшей мере, 10 раз в час.
Питание и вода
Во время исследования собаки имели доступ к сертифицированному питанию для собак Purina® №5007 и воде по желанию. Результаты анализа пищи (сертификат анализа, предоставленный продавцом) и анализа воды (растворенное сухое вещество, содержание микроорганизмов, выбранных элементов, тяжелых металлов и хлорированных углеводородов) сохраняли в файле исходных данных. Обоснованно надеялись на отсутствие примесей в пище или воде на таких уровнях, которые были бы достаточными, чтобы повлиять на результаты исследования.
Объяснение выбора дозы и вида
Доза была выбрана на основании существующих данных, полученных для композиций, оцениваемых в предыдущих исследованиях. Данное исследование проводили на иммуносупрессированных и не иммуносупрессированных гончих собаках, чтобы оценить иммуногенность композиций ЕРО и ФК/ФД-профили двух режимов дозирования. Количество собак, которое было использовано, было минимальным количеством, необходимым, чтобы предоставить научно обоснованные результаты. В распоряжении не было приемлемых моделей in vitro. Специально разводимых гончих собак обычно используют для проведения фармакокинетических, фармакодинамических и токсикологических исследований, удовлетворяющих обязательным требованиям.
Дизайн исследования
Краткое изложение сути
Гончих собак (N=3 собаки/группу, 6 групп) случайным образом распределяли по группам лечения. В день -2 трем группам собак вводили однократную пероральную дозу циклоспорина (25 мг/кг). После этого три группы собак получали ежедневную поддерживающую дозу циклоспорина (10 мг/кг). У иммуносупрессироанных и не иммуносупрессированных собак исследовали два режима дозирования EPREX® и наполнителя. Все композиции и наполнитель вводили подкожно (п/к) либо ежедневно, либо еженедельно. В назначенное время в течение четырехнедельного периода собирали образцы крови. Ежедневно осматривали место инъекции и массу тела получали еженедельно. Собак умерщвляли по соображениям гуманности или дарили другому исследовательскому институту после последних отборов проб.
Подготовка и введение доз тестируемых композиций
День 1
Тестируемые композиции сначала вводили собакам в 1 день (смотри таблицу 2 ниже). Все композиции вводили в объеме, указанном в следующей таблице.
Дозу набирали в шприц, снабженный иглой соответствующего калибра. П/к дозу вводили в район спины. Места введения дозы подстригали перед дозированием и метили несмываемыми чернилами.
Обследование, отбор образцов и обработка данных
Собак и места инъекций осматривали ежедневно. Любое аномальное проявление или поведение отмечали и оценивали. Массу тела регистрировали один раз в неделю.
В назначенные основные временные точки (смотри ниже) примерно 2 мл крови отбирали из яремной вены в гепаринизированные вакуумные контейнеры Vacutainers® (Becton Dickinson, Franklin Lakes, NJ). В случае неудачи взятия крови из яремной вены кровь отбирали из головной вены и отмечали. Первичный сбор крови, полученной перед введением циклоспорина, использовали для сбора плазмы. Кровь помещали на лед, центрифугировали (1500 × g, 10 мин, ~ 4°С) и собирали плазму. Плазму замораживали при -20°С для перевозки.
При вторичном сборе крови примерно 2 мл крови собранной с использованием Vacutainer®, содержащих ЭДТА, получали утром и помещали на лед. Второй сбор хранили при ~ 4°С в виде цельной крови и использовали для измерений ретикулоцитов, гемоглобина и суммарных красных кровяных клеток. Временные точки первичного отбора крови:
Группы 1 и 2 (ежедневно EPREX®): до введения дозы, 1, 3, 8, 12, 16 и 24 часа в 1 день и 28 день, и перед введением дозы на 3, 7, 14, 21 и 24 день.
Группы 3-6 (еженедельно EPREX® или физиологический раствор): до введения дозы, 1, 3, 8, 12, 24, 48, 72 и 96 час в 1 день и 22 день, и перед введением дозы на 7 и 14 день.
Временные точки вторичного отбора крови:
Все группы: до введения дозы на 1, 3, 7, 10, 14, 17, 21, 24 и 28 день.
По мере необходимости в ходе исследования собирали дополнительные образцы сыворотки, чтобы оценить функцию почек и печени. В пределах 24 часов последнего сбора образцов собак умерщвляли по соображениям гуманности большой внутривенной дозой раствора барбитурата для эвтаназии или дарили другому исследовательскому объекту.
Анализ образцов
Собранную цельную кровь (на ЭДТА) анализировали на ретикулоциты, гемоглобин и суммарные красные кровяные клетки.
Результаты:
Результаты данного исследования представлены на фигурах 71 и 72. Результаты данного исследования показывают, что динамические ответы (характер повышения уровня гемоглобина и эритроцитов) в течение 4-недельного периода исследования сходны после режимов дозирования 50 МЕ/кг/день и 600 МЕ/кг/неделю у иммуносупрессированных или не иммуносупрессированных собак.
Пример 8
ФК/ФД-МОДЕЛИРОВАНИЕ РЕКОМБИНАНТНОГО ЕРО ЧЕЛОВЕКА ПОСЛЕ ВВЕДЕНИЯ ТРЕХ В/В И ШЕСТИ П/К ДОЗ САМЦАМ МАКАК-КРАБОЕДОВ
Цели
Целью данной исследование было использование ФК/ФД-модели согласно данному изобретению для того, чтобы охарактеризовать профили фармакокинетики (ФК) и фармакодинамики (ФД) rHuEPO в показателях повышенного количества ритикулоцитов, эритроцитов и гемоглобина в крови после в/в введения трех однократных доз и п/к введения шести однократных доз rHuEPO (EPREX®) самцам макак-крабоедов.
Способы
Данные получали из двух исследований, выполненных RWJPRI (No. исследований Sbi 0876-49 и 0875-49 и No. исследований RWJPRI DM99146 и DM99124, декабрь 1999). Одно исследование представляло собой исследование в параллельных группах, выполненное на 12 самцах макак-крабоедов (No. исследований Sbi 0876-49 и No. исследований RWJPRI DM99146, декабрь 1999). Обезьян делили на 4 группы, одна группа была контрольной, тогда как трем другим внутривенно инъецировали 500, 2000 и 4000 МЕ/кг EPREX®. Образцы крови отбирали перед введением дозы и вплоть до 48 часов для измерения концентраций rHuEPO. Другое исследование представляло собой исследование в параллельных группах, выполненное на 21 самце макак-крабоедов, которые были разделены на 7 групп по 3 обезьяны в группе (No. исследований Sbi 0875-49 и No. исследований RWJPRI DM99124, декабрь 1999). Контрольная группа подкожно получала физиологический раствор, тогда как остальным шести группам вводили 400, 1000, 2400, 5000, 20000 и 40000 МЕ/кг EPREX® подкожно. Животные были распределены так, чтобы было однородное распределение по массе тела в группах. Образцы крови отбирали до введения дозы и в разное время после введения вплоть до 28 дня для определения концентрации rHuEPO, а также количества ретикулоцитов, эритроцитов и гемоглобина. Для этого анализа использовали средние данные.
Модель
Схематичное представление ФК/ФД-модели изображено на фигуре 73.
Фармакокинетика
Была выбрана 2-компартментная модель для объяснения многоэкспоненциальности в кинетических профилях при в/в введении. Бескомпартментный анализ показал нелинейность в кинетике, которую моделировали с использованием фармакокинетической функции Михаэлиса-Ментен. Использовали кинетическую модель двойной абсорбции с быстрым поглощением нулевого порядка части дозы с последующим медленным поглощением первого порядка оставшейся части, чтобы охарактеризовать абсорбцию rHuEPO при п/к введении. Одновременно подгоняли данную модель для шести однократных п/к доз, а также трех в/в доз, чтобы получить общий набор параметров для характеристики всех данных.
Дифференциальные уравнения, используемые для моделирования внутривенной кинетики, представляли собой следующее:

Данные п/к введения моделировали с помощью следующих уравнении:
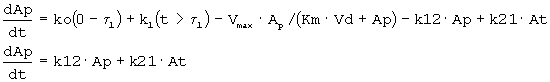
где ko=0 при t>τ1
 k1=0 при t≤τ1
k1=0 при t≤τ1
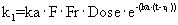 при t>τ1
при t>τ1
Ар представляет собой количество препарата в плазме, тогда как At представляет препарат в периферической камере (т.е. в ткани). Микроконстанты k12 и k21 являются скоростями первого порядка переноса между центральным (плазма) и периферическим компартментами. Vmах и Km являются константами михаэлиса-ментен, отражающими возможности процесса и концентрацию, при которой достигается половина vmax. Fr является частью дозы, связанной с траекторией абсорбции первого порядка k1. Период времени (tau 1) для поглощения нулевого порядка (ko) фиксировали при значении 10 часов на основании данных и исходных прогонов. Одна скорость абсорбции первого порядка ka могла описать все дозы, за исключением наименьшей дозы (400 ме/кг) для которой определяли отдельное значение ka. Биодоступность f, по-видимому, изменяется с дозой, и была определена для двух наименьших доз и фиксировалась на уровне 100% для остальных доз.
Фармакодинамическую модель цепного процесса созревания (фигура 73) с двумя камерами клеток предшественников, имеющих разные продолжительности жизни, использовали для моделирования фармакодинамики eprex®. предположили, что стимуляция продукции происходит при определенных скоростях продукции в обеих камерах предшественников. k0 представляет скорость продукции клеток нулевого порядка, в то время как tr и тrbc означают продолжительности жизни ретикулоцитов и красных кровяных клеток.
предполагали, что концентрации rhuepo исходного уровня были равны нулю, и поэтому исходный уровень ретикулоцитов был задан k0 tr. Для определения целей использовали следующие дифференциальные уравнения:
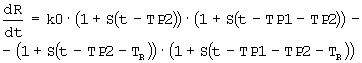
где функция стимуляции задана уравнением Хилла, при фиксированном значении γ, равном 1.
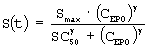
Указанное выше уравнение подгоняли к количеству ретикуло-цитов после введения шести уровней доз EPREX®, чтобы получить один набор динамических параметров, характеризующих данные по всем дозам. Параметры кинетической модели были фиксированными, и их использовали как вынуждающую функцию для динамики. Количества ретикулоцитов до введения дозы фиксировали как стационарные исходные значения.
Динамические параметры, полученные при подгонках, проведенных для ретикулоцитов, использовали, чтобы смоделировать количества RBC и уровни гемоглобина при всех дозах. Количество RBC при 48 часах использовали в качестве исходного уровня, в то время как содержание гемоглобина в клетке было фиксированным для каждой группы и полученным из отношения количества гемоглобина до введения дозы к суммарному количеству клеток до введения дозы (RBC+ ретикулоциты) для данной группы.
В целях моделирования использовали следующие дифференциальные уравнения:
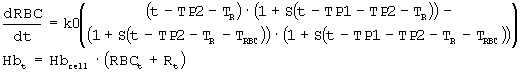
Результаты
На фигуре 74 показаны подгонки временных профилей концентрации rHuEPO после введения трех однократных внутривенных доз и шести однократных п/к доз EPREX®. Полученные параметры перечислены на фигуре 75. Двухкомпартментная кинетическая модель с нелинейным распределением могла адекватно фиксировать многофазные кинетические профили при в/в введении, хотя конечная фаза для наименьшей дозы была немного завышена. Определили высокое значение Km, которое свидетельствует о том, что нелинейность в распределении умеренная и будет заметной только при высоких дозах. Основной объем распределения Vd был определен до 57 мл/кг, что близко к объему крови. Для п/к введения биодоступность в значительной степени увеличивалась с увеличением дозы, при наименьшей дозе проявляя биодоступность 26,8% и при следующей большей дозе -73%. Наименьшая доза имела несколько отличное значение ka, по сравнению с остальными дозами. На основе оценок параметров можно сделать вывод, что основная часть биодоступной дозы следует медленной траектории первого порядка. Путь поглощения нулевого порядка, по-видимому, является быстрым и отвечает на меньшую фракцию (35,5%) биодоступной дозы.
Подгонки для ретикулоцитов показаны на фигурах 76а, 76b, и на фигуре 77 перечислены определенные фармакодинамические параметры. Время запаздывания, которое объясняется второй камерой предшественников ТР2, было небольшим (~ 15 час). Оцененная продолжительность жизни ретикулоцитов была близка к 6 дням. Smax, которое означает максимально возможное увеличение скорости продукции, составляло 3,133, в то время как было определено высокое значение SC50 равное 842,5 МЕ/л. На фигурах 78а и 78b показано моделирование количества RBC, и фигуры 79а и 79b представляют собой модели ответа на уровне гемоглобина.
На фигуре 80 показаны фармакодинамические параметры, полученные после введения EPREX® здоровым людям. Фармакокинетическая модель была упрощена от 2-компартментной до 1-компартментной модели на основании временных профилей концентрации при в/в введении. Фармакодинамическая модель была расширена применением регуляции продукции клеток эндогенными уровнями ЕРО, а также был включен дополнительный компонент, отвечающий за природный механизм обратной связи в организме (фигура 81). Это было сделано на основе предположения, что ретикулоциты вызывают ингибирование по принципу обратной связи своей собственного продукции посредством снижения скорости продукции самых ранних клеток, представленных камерой Р1. Также полагали, что требуется время ТРО часов для того, чтобы такое ингибирование возымело действие. Ингибирование моделировали, используя функцию Хилла. Сравнение оценок фармакодинамических параметров для обезьян и человека показало, что параметры продолжительности жизни в камере очень сходны между этими видами. Значения Smax и SС50, по-видимому, отличаются между видами, но это может быть следствием дополнительной сложности, связанной с обратной регуляцией и исходными концентрациями Еро в модели эффектов rHuEPO на ретикулоциты человека.
Обсуждение
Фармакокинетика: При в/в введении, кинетика следует биэкспоненциальному снижению, которое фиксировалось двухкомпартментной моделью с нелинейным распределением. Первоначальным местом действия rHuEPO является костный мозг, который представляет собой высоко перфузируемую ткань, и таким образом периферическая камера в модели может только представлять некоторое неспецифичное связывание rHuEpo. Конечная фаза для наименьшей дозы была завышена, что может быть следствием несоответствия измерений концентрации в более поздних временных точках, или возможно другим характером изменения кинетики в пределах этой дозы. Подобно человеку, оцениваемое значение Vd было очень близко объему крови у обезьян, и кинетики были умеренно нелинейными, о чем свидетельствует высокое значение Km.
После п/к введения пиковые концентрации rHuEPO достигались за один день, и rHuEPO оставался в циркуляции в течение более длительного периода времени по сравнению с rHuEPO после в/в введения, вследствие наличия явления “флип-флоп” с ka, регулирующей конечную фазу. Характер этих данных удовлетворительно описывали с использованием многофазной модели абсорбции. Начальные концентрации для самой высокой п/к дозы были немного завышены. Однако это должно быть допустимым, учитывая тот факт, что использовали один единственный набор параметров, чтобы описать все уровни доз. Кинетическую модель двойной абсорбции, предложенную авторами изобретения, можно использовать для того, чтобы объяснить разные пути абсорбции препарата из места п/к введения. Быстрое поглощение нулевого порядка части дозы можно объяснить прямым поступлением в кровь в месте подкожного введения через кровеносные сосуды. С другой стороны, другая часть дозы может, предположительно, поступать в лимфатические сосуды и подвергаться медленному процессу абсорбции первого порядка из лимфы в кровь. Это также объяснило бы 10-часовой лаг-период для начала абсорбции первого порядка. Биодоступность увеличивалась с увеличением дозы и составляла 100% для доз 2400 МЕ/кг и выше. Та же самая модель двойной абсорбции хорошо характеризовала кинетику при п/к введении у человека, и показала сходную тенденцию увеличения биодоступности с увеличением дозы. Однако в отличие от обезьян модель предсказывала, что большая часть дозы абсорбируется у человека посредством пути нулевого порядка.
Фармакодинамика: Количества ретикулоцитов начинали возрастать в пределах 48 часов и достигали пика примерно через 10 дней, после чего они начинали падать и возвращались к исходным уровням к 20 дню. Модель цепного созревания, предложенная авторами данного изобретения, очевидно, хорошо характеризует данные. Модели на фигурах 78а, 78b, 79а и 79b показывают, что динамику ретикулоцитов можно легко использовать для предсказания изменения количеств RBC, а также содержания гемоглобина.
Точный способ действия эритропоэтина еще не полностью понятен. Предполагалось, что первичное действие rHuEPO состоит в стимуляции пролиферации ранних клеток-предшественников. Однако существует доказательство, полученное в исследованиях на экспериментальных животных, что эритропоэтин также действует на дифференцированные эритробласты (Krantz et al., Erythropoietin and the regulation of erythropoesis, 1970, The University of Chicago Press). Размышления этой научной школы привели к предположению о том, что rHuEPO действует на зрелые эритробласты, вызывая ранний 24-часовой ответ ретикулоцитов с последующим макроцитозом вследствие дополнительного влияния на нормобласты. На основании этой теории авторы разработали механистическую модель цепного созревания при стимуляции rHuEPO, происходящего в двух популяциях клеток-предшественников, которые могут представлять собой эритробласты и ранние клетки-предшественники.
Известно, что эритробласты подвергаются 2-5 делениям со средним временем созревания или оборота от 11 до 48 часов в зависимости от вида (там же, Aplen et al., 1959, Ann. N.Y. Acad. Sci., 77: 753, Osgood, E., 1954, Blood, 9: 1141 и Flidner et al., 1959, Acta. Haematol., 22: 65). Модель авторов данного изобретения предсказывает, что существует 15-часовой лаг-период перед тем, как заново продуцированные ретикулоциты действительно высвободятся в циркуляцию, и это отражает время созревания эритробластов.
Оцененная продолжительность жизни ретикулоцитов составляет 6 дней. У человека нормальная продолжительность жизни клеток в стадии ретикулоцитов составляет около 3,5 дней в костном мозге и 1-2 дня в крови (Hillman et al., 1967, Sem. Haematol., 4(4): 327). Однако в моделях тяжелой анемии на животных было показано, что пул ретикулоцитов в костном мозге сдвигается в циркуляцию (там же, и Bessis et al., 1973, Living blood cells and their ultrastructure, Verlag New York-Heidelberg-Berlin). Эти перемещенные ретикулоциты костного мозга занимают на 3 дня больше, чем нормальные ретикулоциты, чтобы продуцировать эритроциты. Поэтому авторы могли ожидать, что средняя продолжительность жизни ретикулоцитов, определенная посредством модели, предложенной авторами, отражает сумму времени созревания в костном мозге и крови.
В литературе сообщалось, что у человека в среднем требуется 5 дней для того, чтобы эритроидные предшественники образовали ретикулоциты в костном мозге (Krantz et al., выше). В действительности это время отражает сумму времени, которую клетка расходует в камерах Р1 и Р2, которая была оценена как 85 часов.
В заключение, кинетика и динамика rHuEPO, по-видимому, довольно сходны у разных видов (обезьяна и человек), и модели, заявленные в данном изобретении, могут хорошо аппроксимировать кинетику и динамику эффектов rHuEPO и дают реалистические оценки параметров времени созревания клеток.
Различные модификации и вариации описанных примеров и систем согласно изобретению без выхода за рамки и без отклонения от сути изобретения будут очевидны для специалистов в данной области. Несмотря на то, что изобретение описано в связи с конкретными предпочтительными вариантами, следует понимать, что изобретение, как заявлено, не должно быть незаконно ограничено такими конкретными вариантами. В действительности различные модификации описанных способов выполнения изобретения, которые очевидны для специалистов в связанных областях, имеются ввиду в рамках следующей формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТЕРАПЕВТИЧЕСКИЕ СПОСОБЫ ДЛЯ ЛЕЧЕНИЯ СУБЪЕКТОВ РЕКОМБИНАНТНЫМ ЭРИТРОПОЭТИНОМ, ИМЕЮЩИМ ВЫСОКУЮ АКТИВНОСТЬ И УМЕНЬШЕННЫЕ ПОБОЧНЫЕ ДЕЙСТВИЯ | 2001 |
|
RU2282460C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ Эритропоэтина | 2000 |
|
RU2225221C2 |
| Способ лечения анемии с применением композиции ЕРО длительного действия | 2016 |
|
RU2673548C1 |
| КОМПОЗИЦИИ ИНСУЛИНОВ ДЛИТЕЛЬНОГО ДЕЙСТВИЯ | 2011 |
|
RU2564104C2 |
| ГИБРИДНЫЙ БЕЛОК НА ОСНОВЕ РЕКОМБИНАНТНОГО ЭРИТРОПОЭТИНА ЧЕЛОВЕКА, ОБЛАДАЮЩИЙ ПРОЛОНГИРОВАННЫМ ДЕЙСТВИЕМ (ВАРИАНТЫ), И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2013 |
|
RU2515914C1 |
| КОМПОЗИЦИИ ИНСУЛИНОВ ДЛИТЕЛЬНОГО ДЕЙСТВИЯ | 2015 |
|
RU2642662C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ АНЕМИИ | 2014 |
|
RU2705206C2 |
| РЕКОМБИНАНТНЫЙ СЛИТЫЙ ЧЕЛОВЕЧЕСКИЙ БЕЛОК EPO-FC С ПРОДЛЕННЫМ ВРЕМЕНЕМ ПОЛУЖИЗНИ И ПОВЫШЕННОЙ ЭРИТРОПОЭТИЧЕСКОЙ АКТИВНОСТЬЮ IN VIVO (ВАРИАНТЫ), ДИМЕРНАЯ БЕЛКОВАЯ КОНСТРУКЦИЯ, ДИМЕРНЫЙ БЕЛОК, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПОСЛЕДОВАТЕЛЬНОСТЬ НУКЛЕИНОВОЙ КИСЛОТЫ (ВАРИАНТЫ), ВЕКТОР ЭКСПРЕССИИ, КЛЕТКА, СПОСОБ ПОЛУЧЕНИЯ БЕЛКА И СПОСОБ СТИМУЛЯЦИИ ЭРИТРОПОЭЗА У МЛЕКОПИТАЮЩЕГО | 2007 |
|
RU2433181C2 |
| ПРИМЕНЕНИЕ СТАБИЛИЗАТОРОВ HIF-АЛЬФА ДЛЯ УСИЛЕНИЯ ЭРИТРОПОЭЗА | 2004 |
|
RU2414214C2 |
| ПРИМЕНЕНИЕ ЭРИТРОПОЭТИНА В ВОССТАНОВЛЕНИИ ПОСЛЕ ИНСУЛЬТА | 2004 |
|
RU2341284C2 |
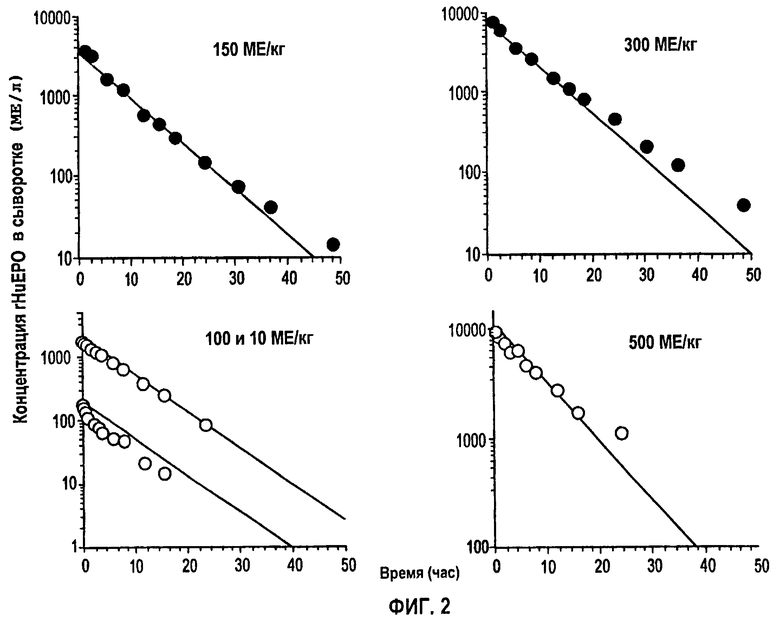
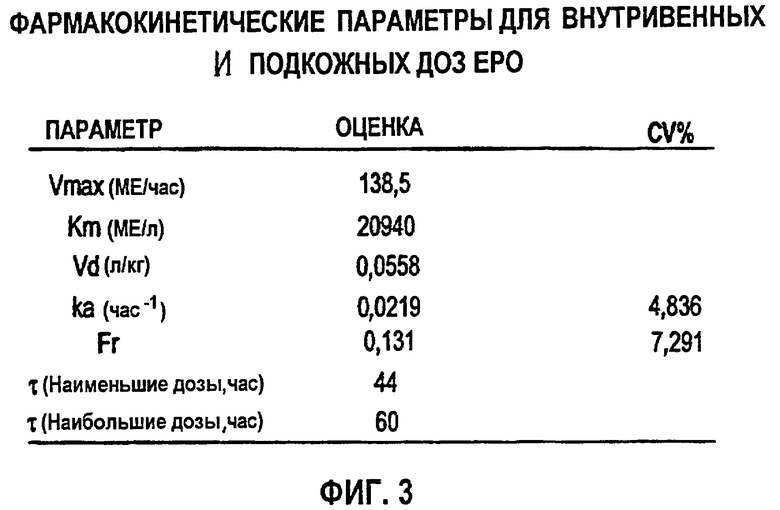
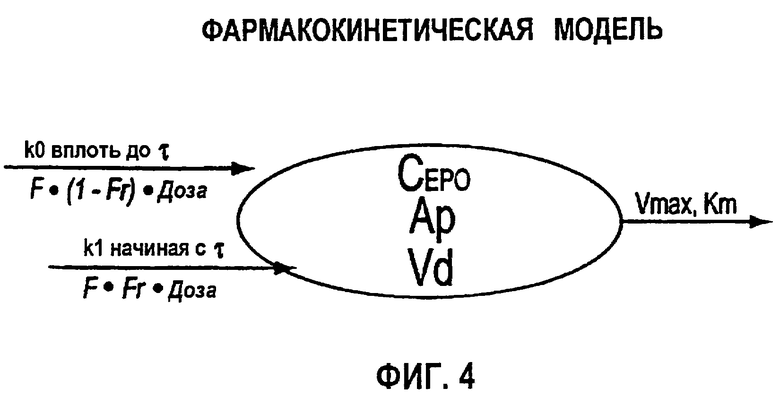
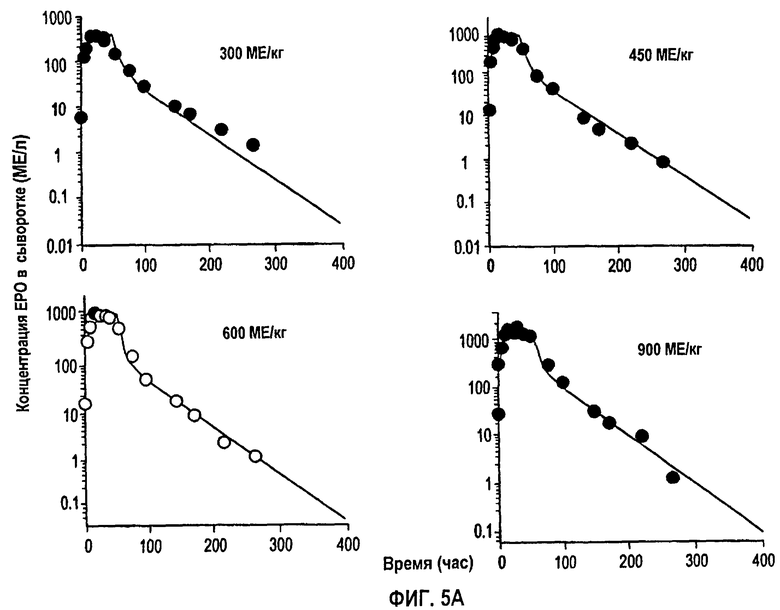
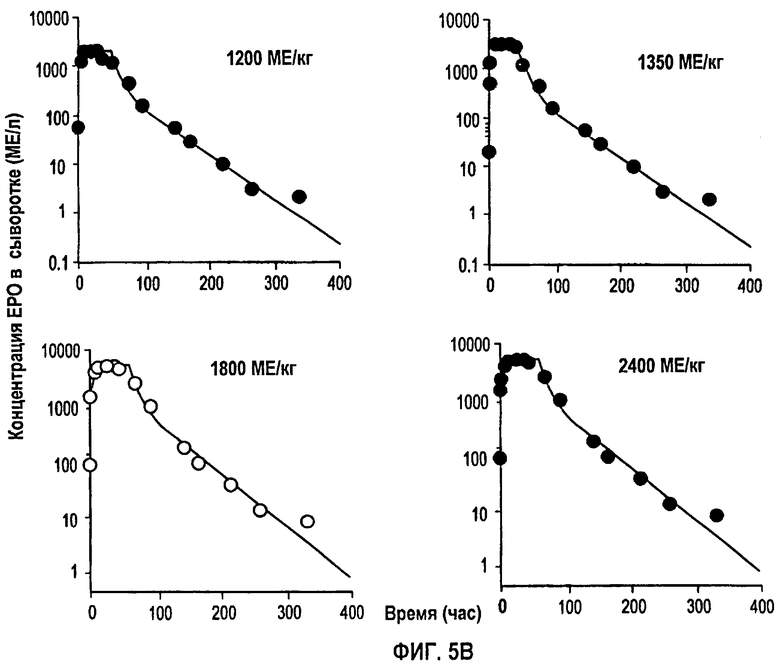
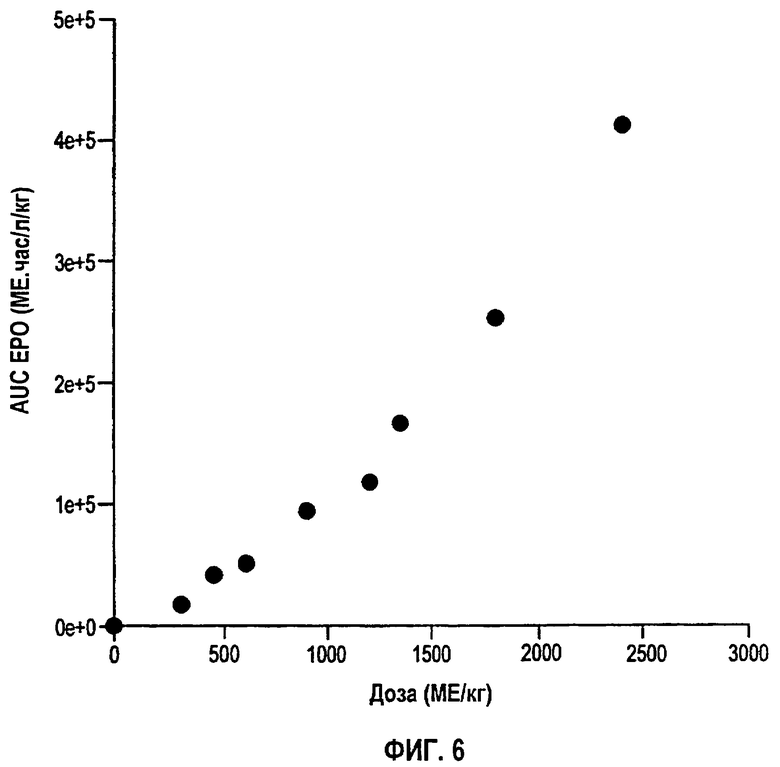
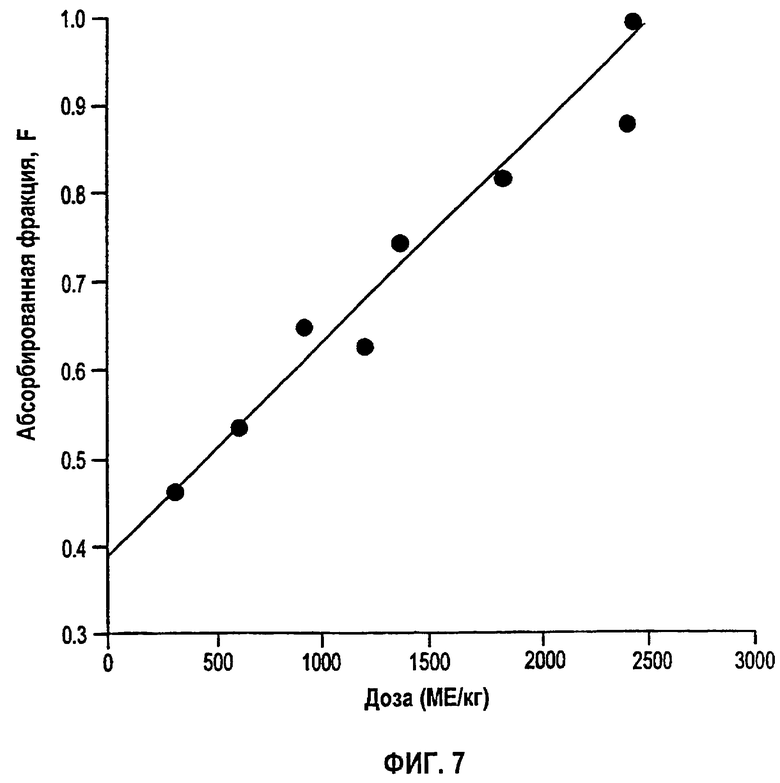
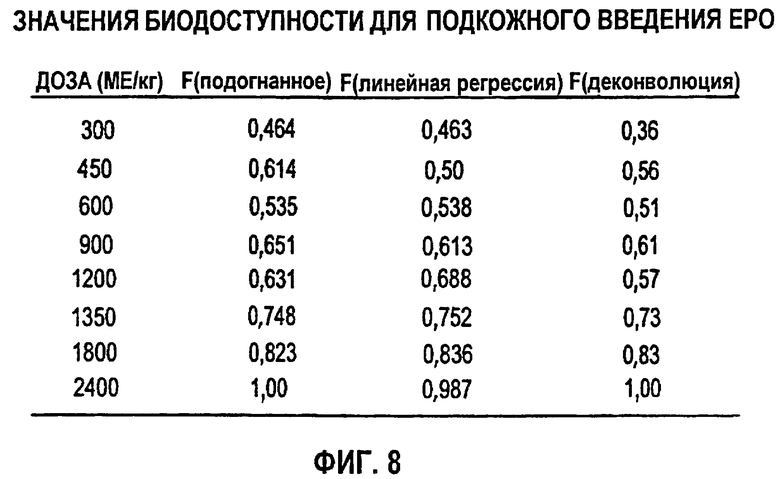
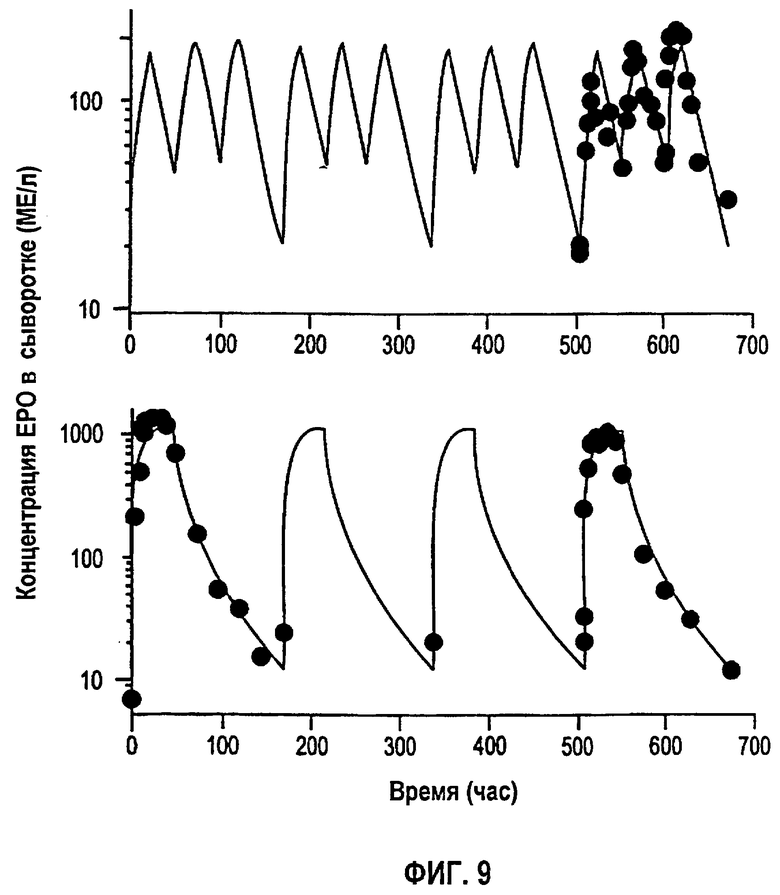
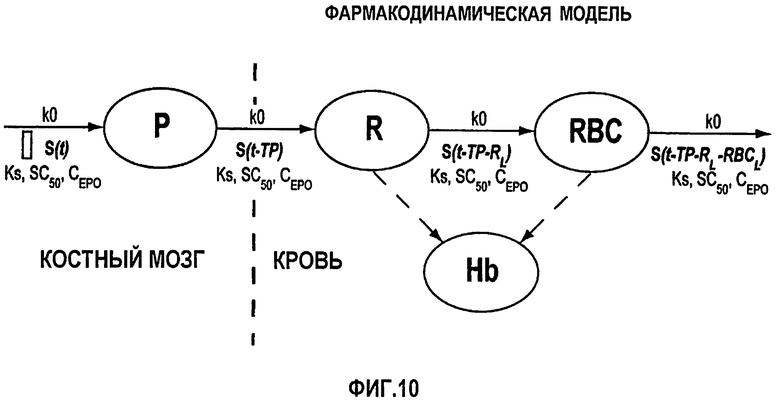
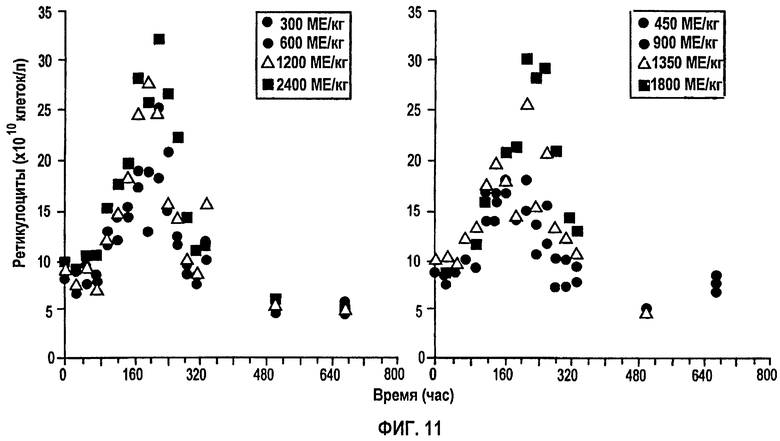
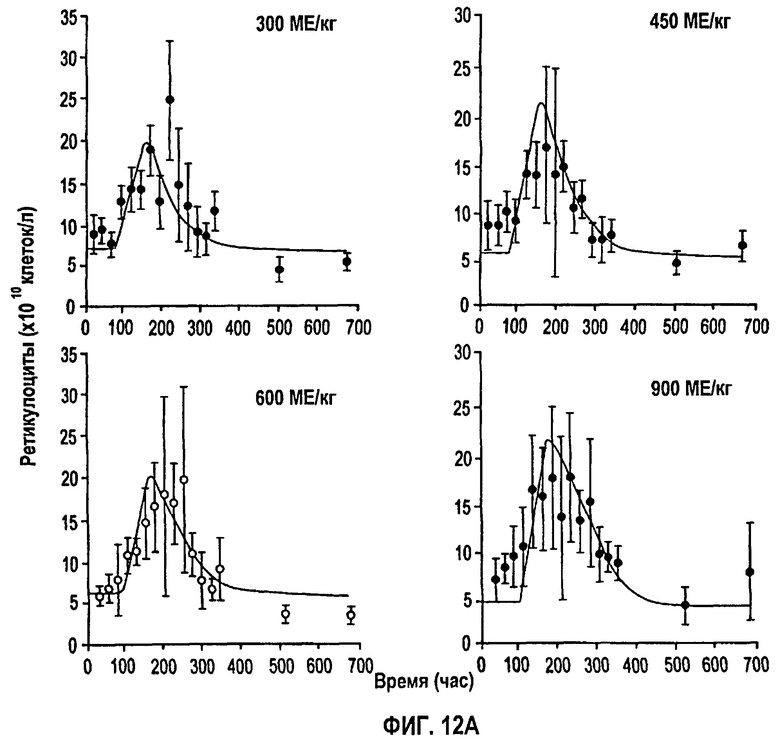
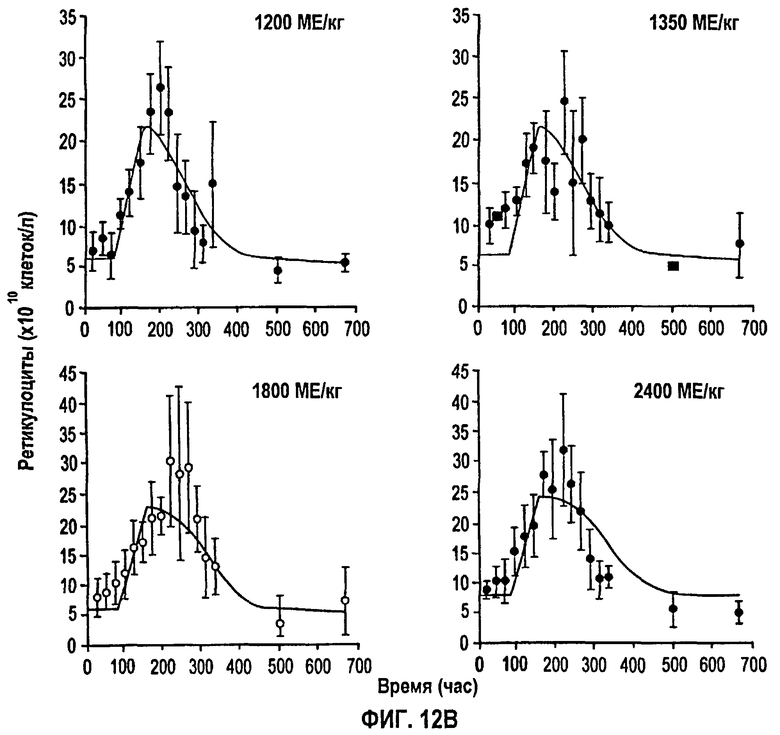

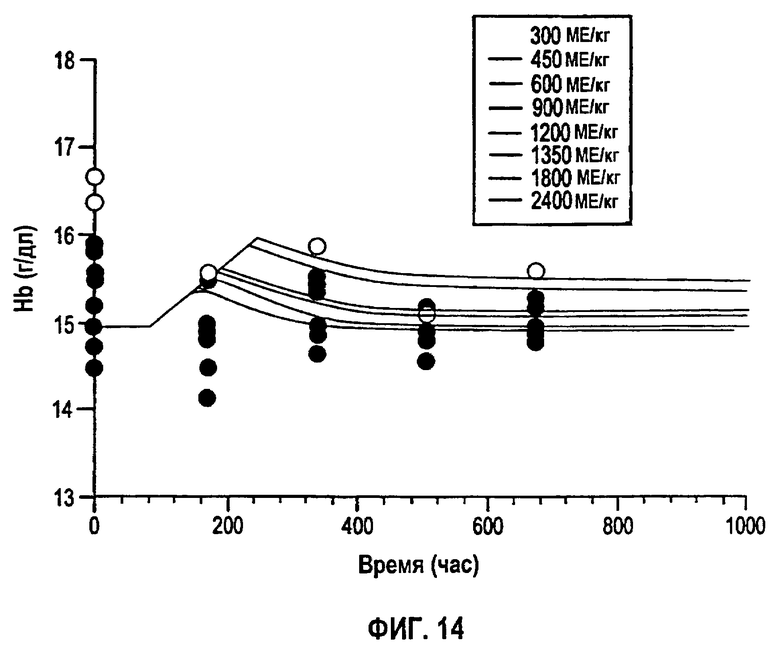
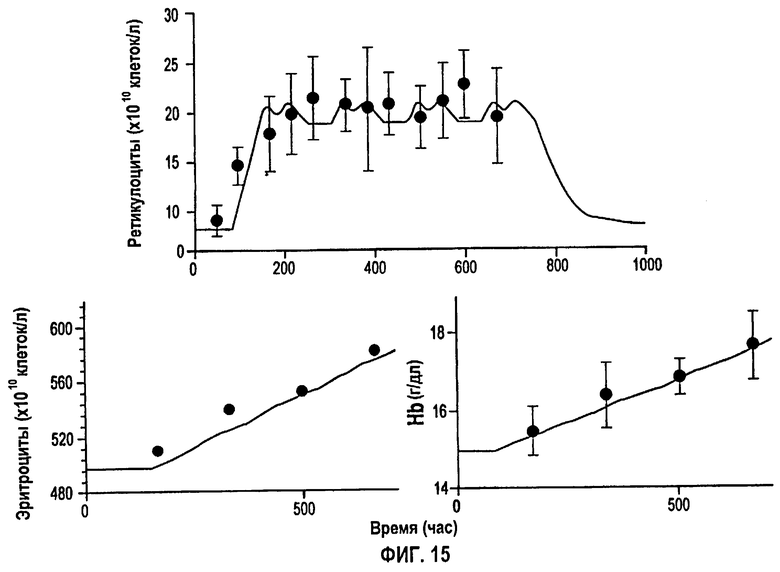
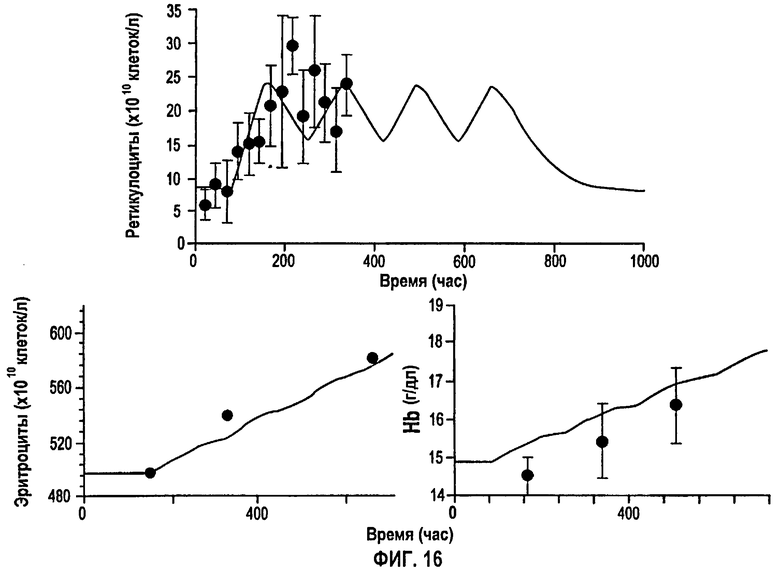
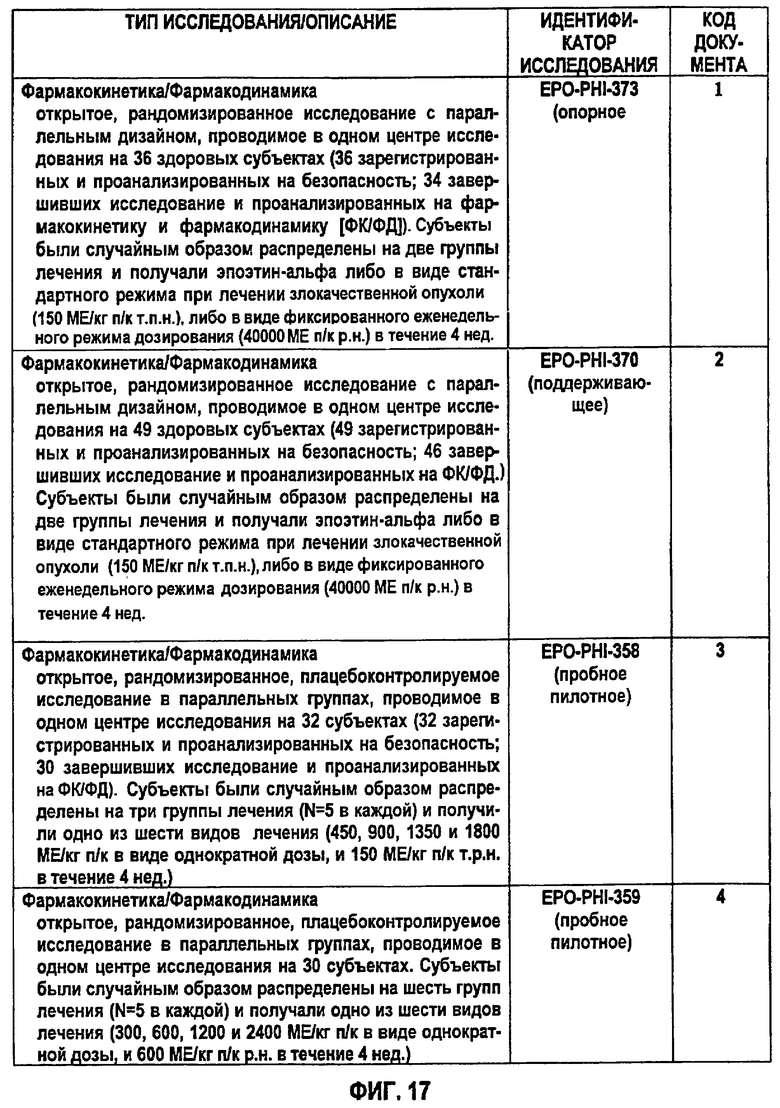
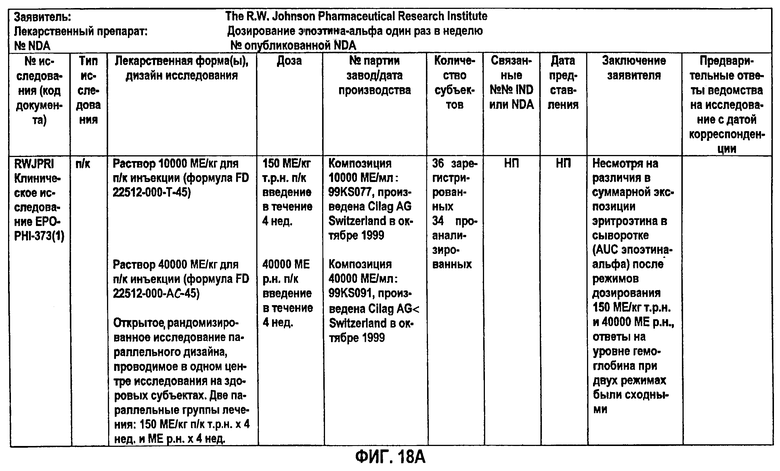
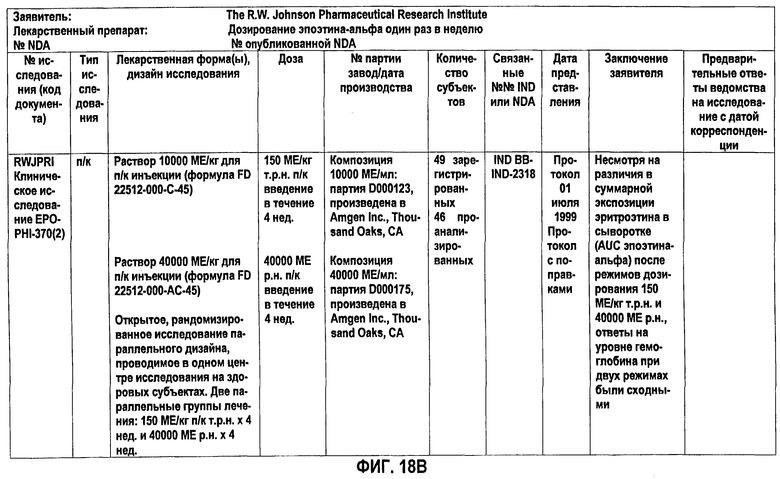
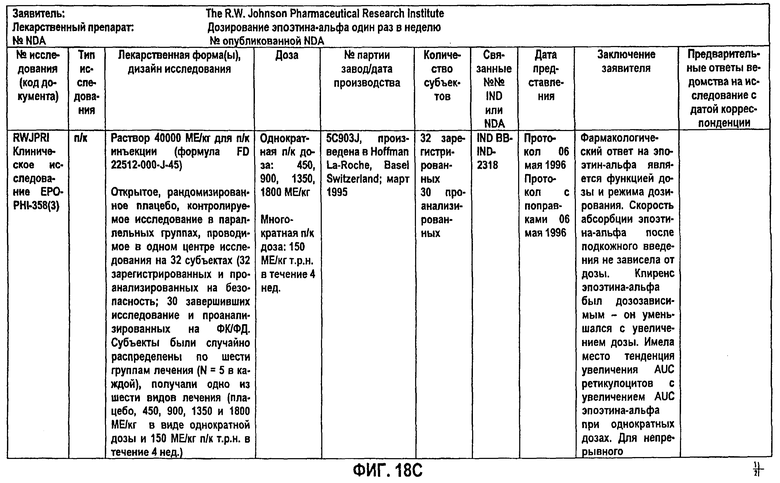
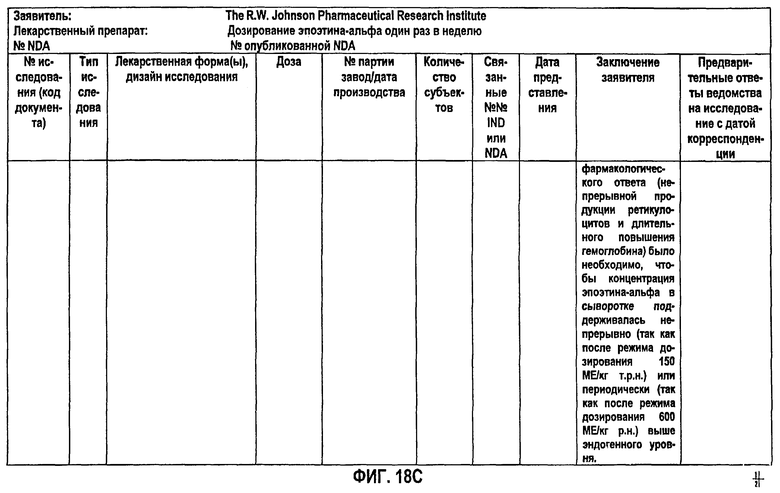
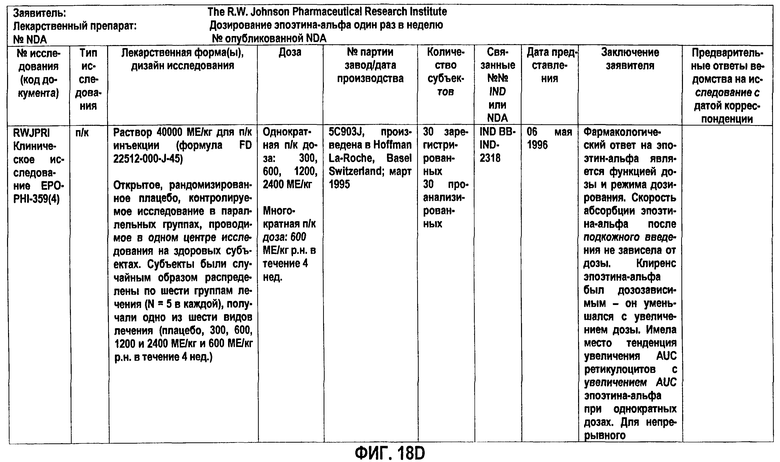
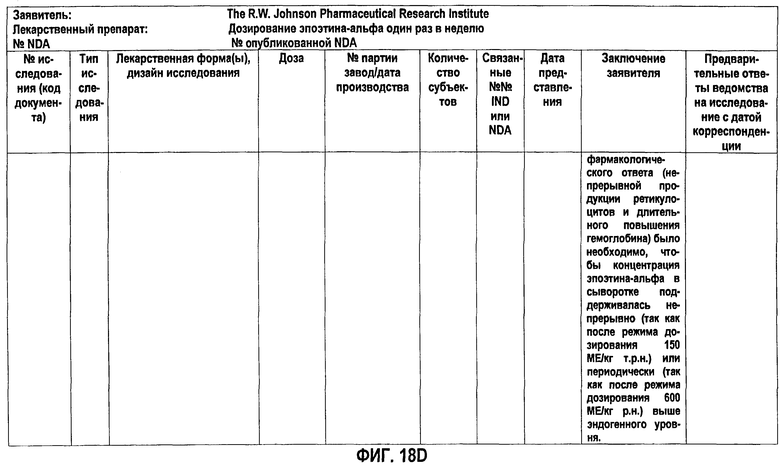
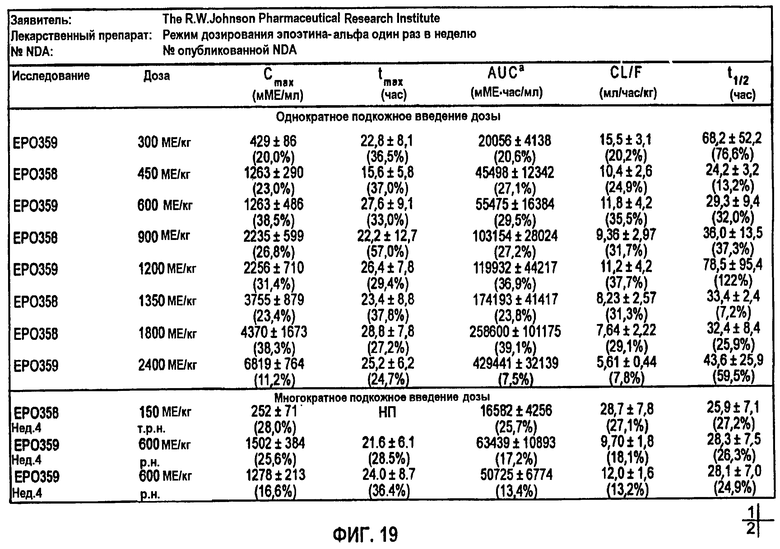
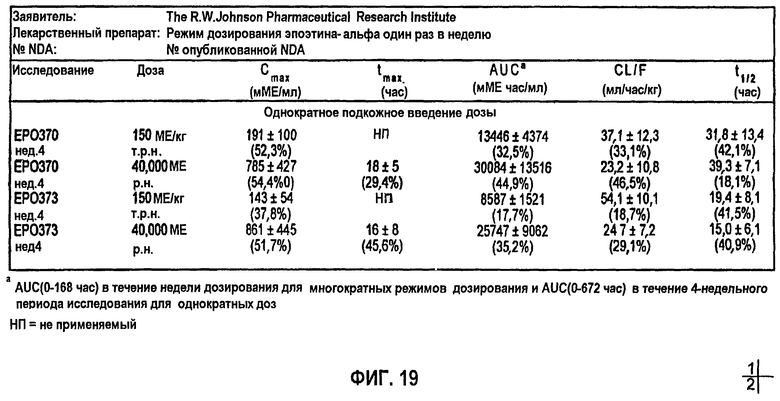


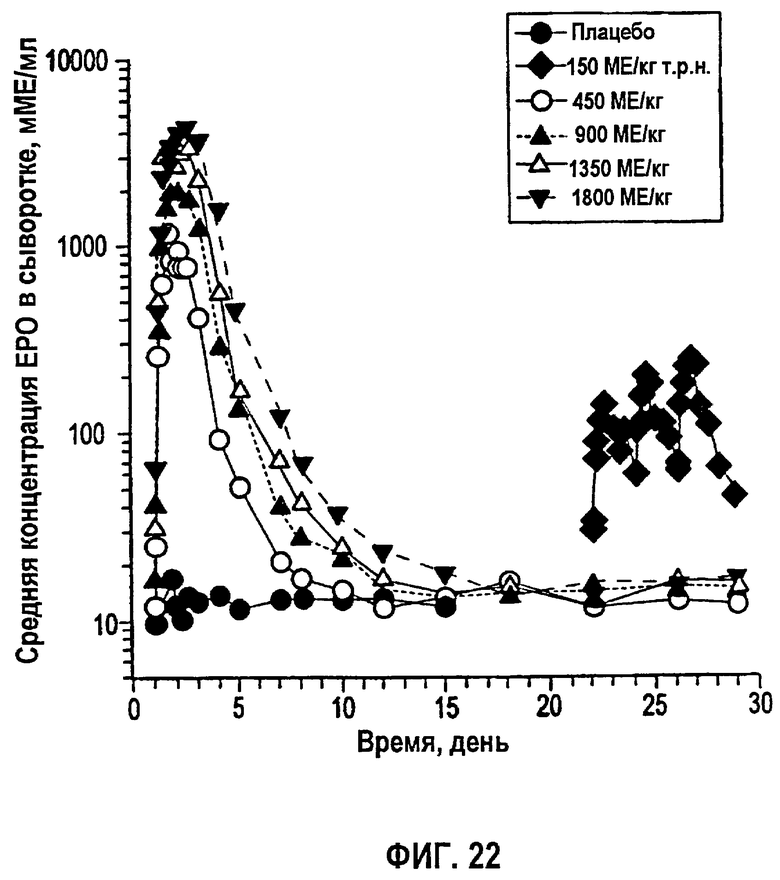
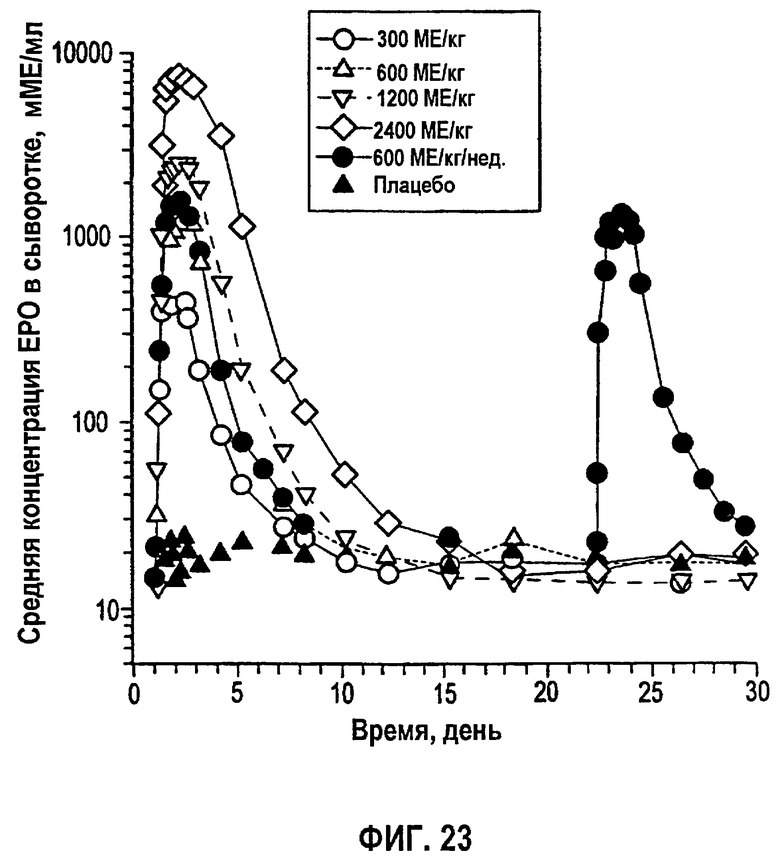
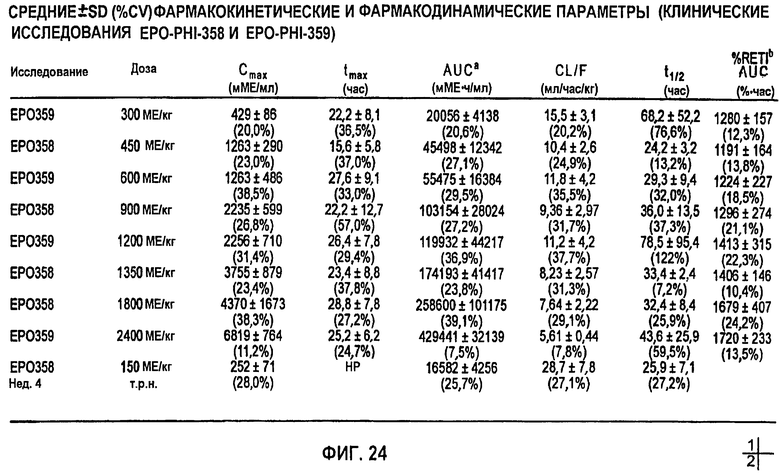
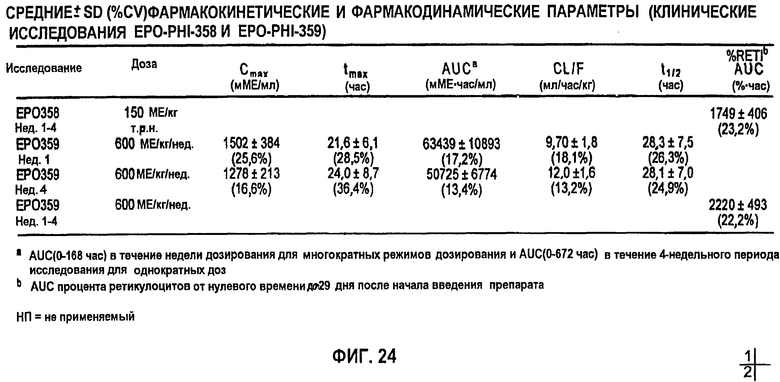
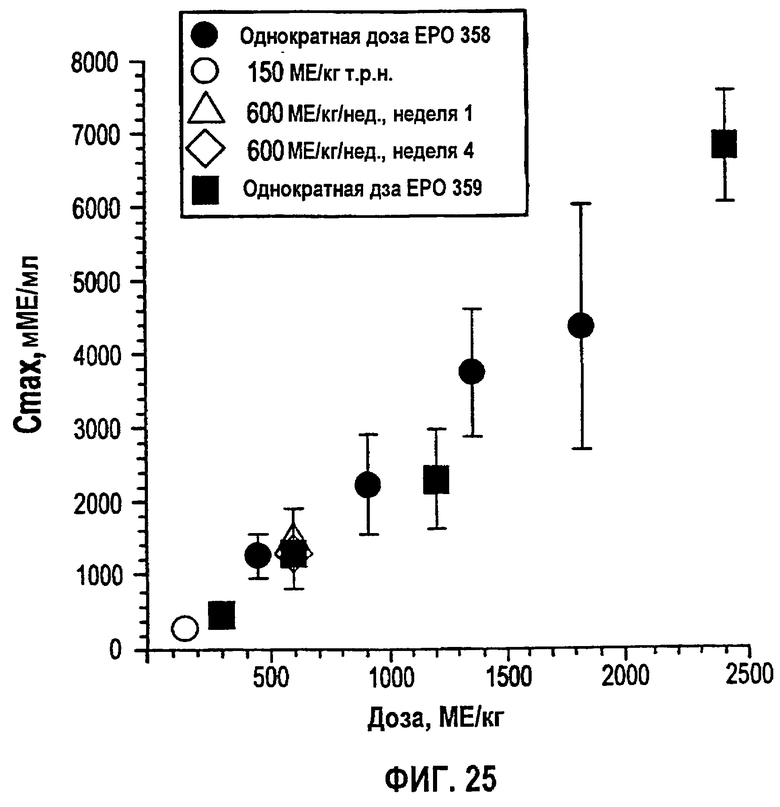
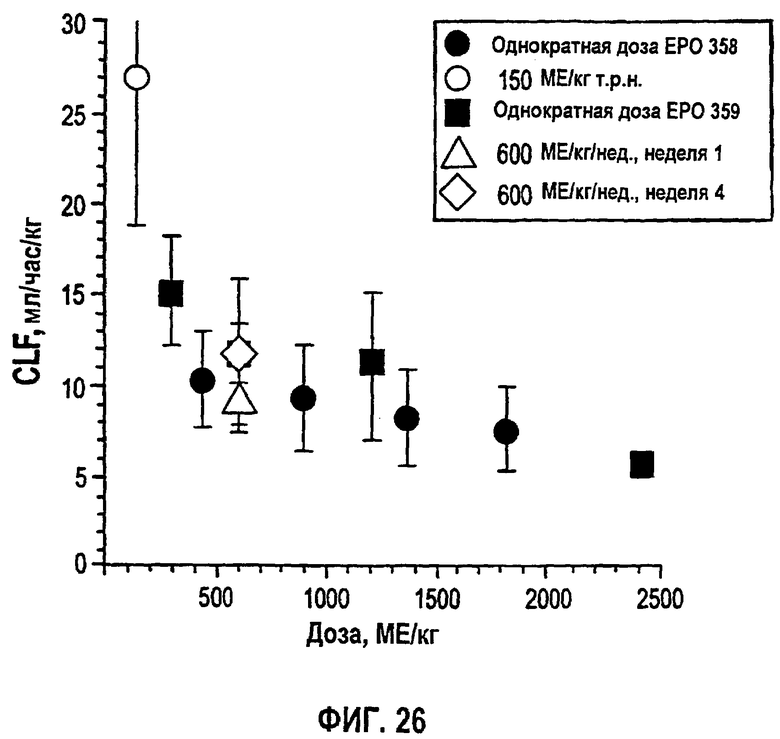
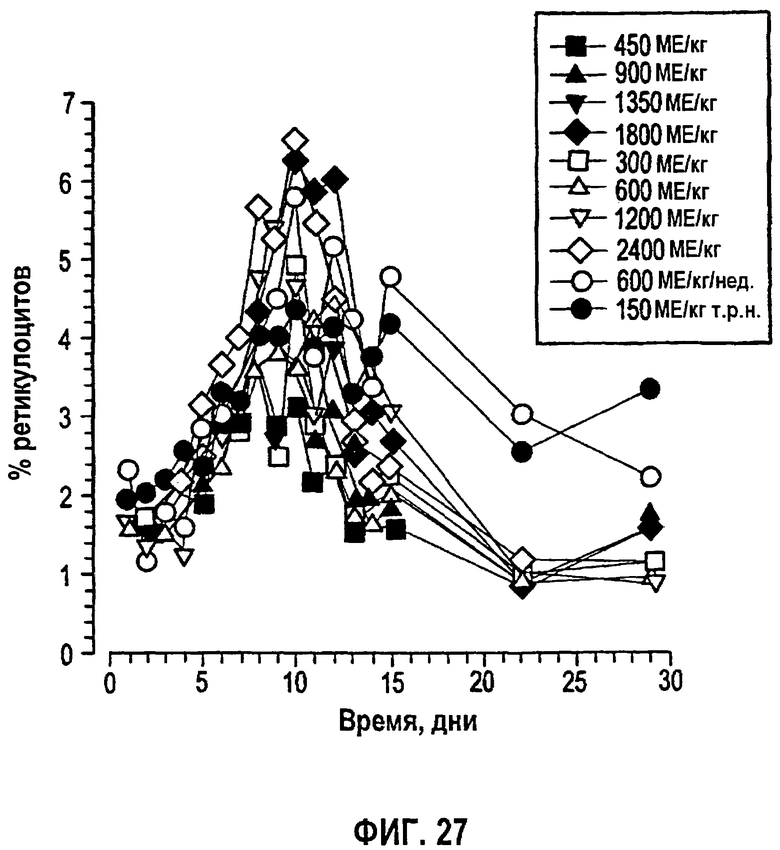
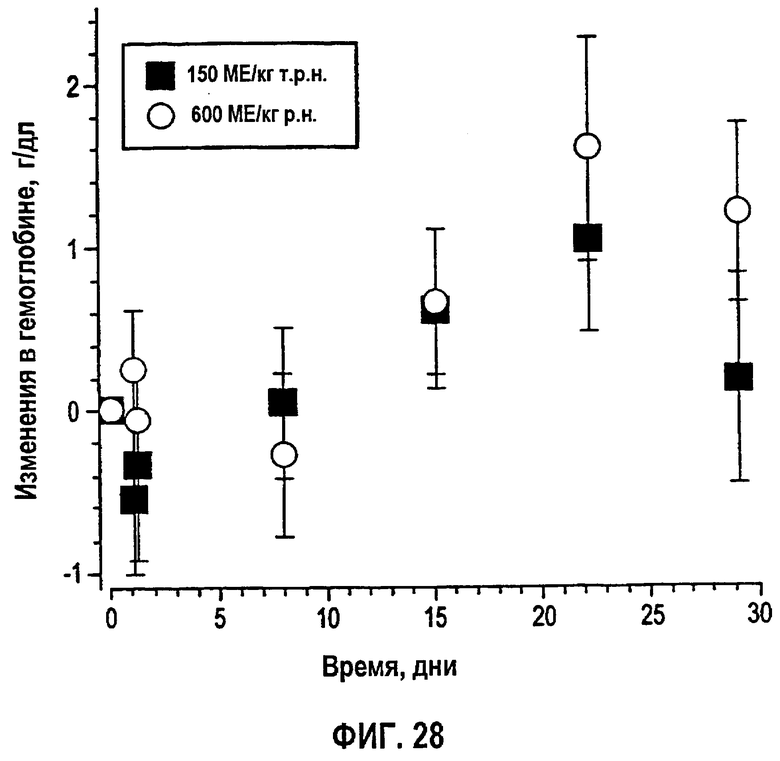
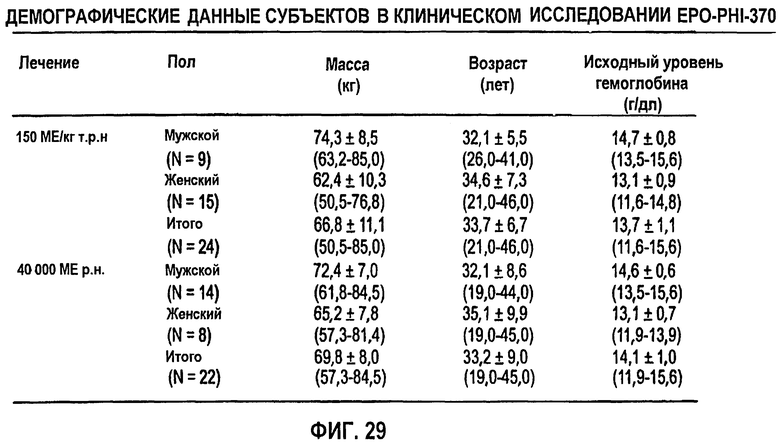
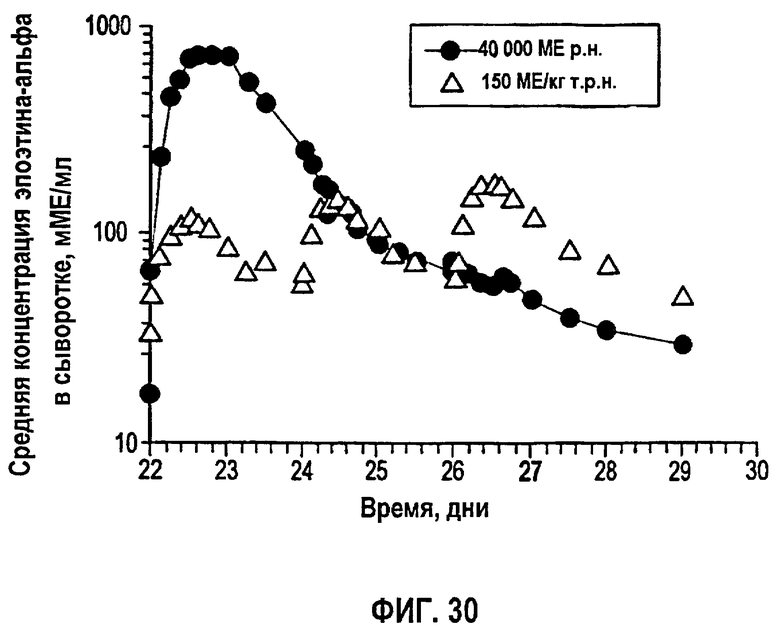
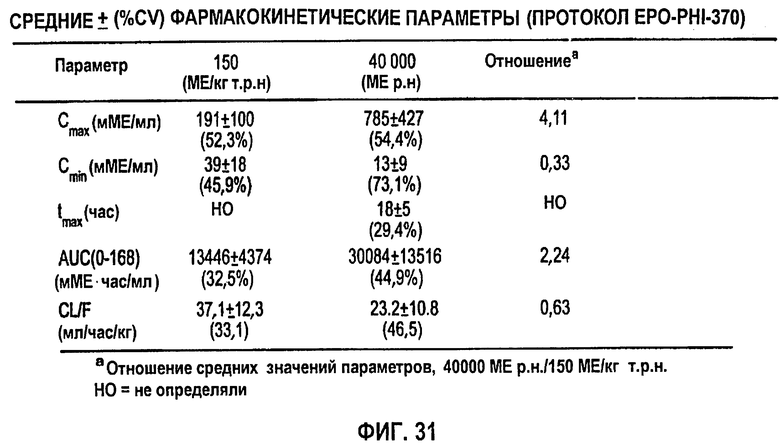
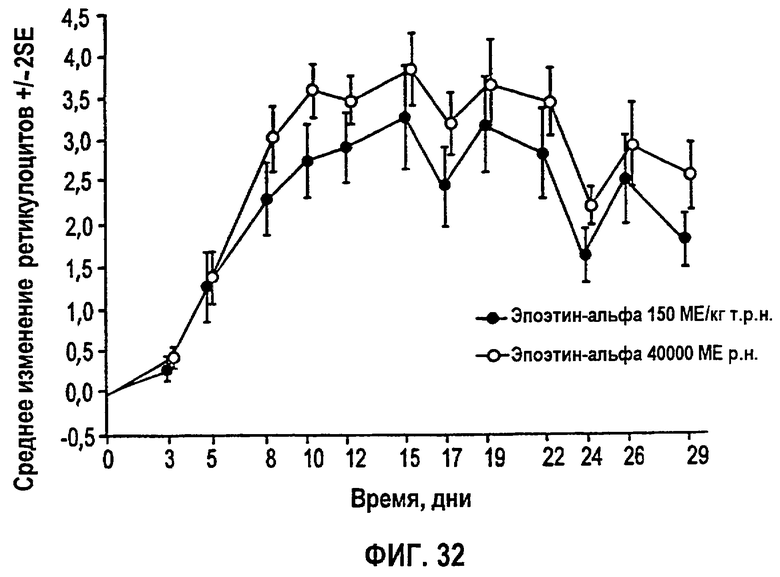
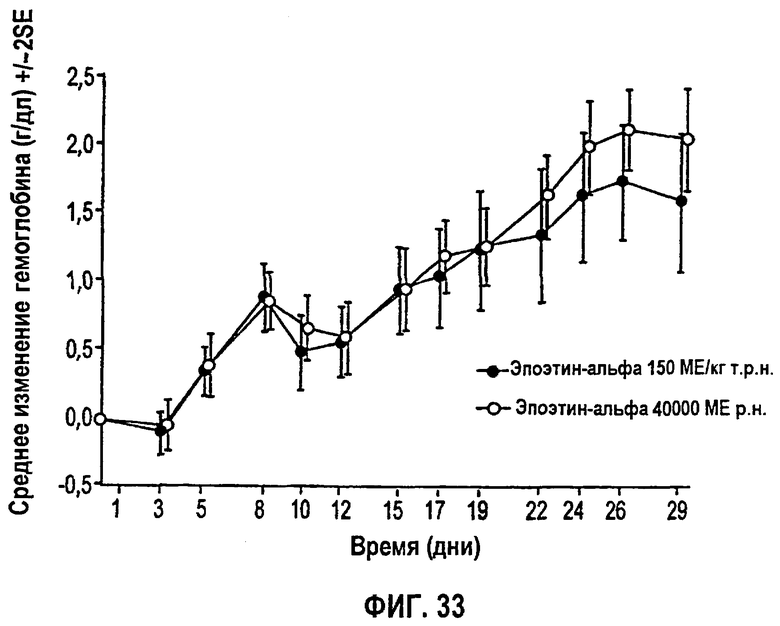
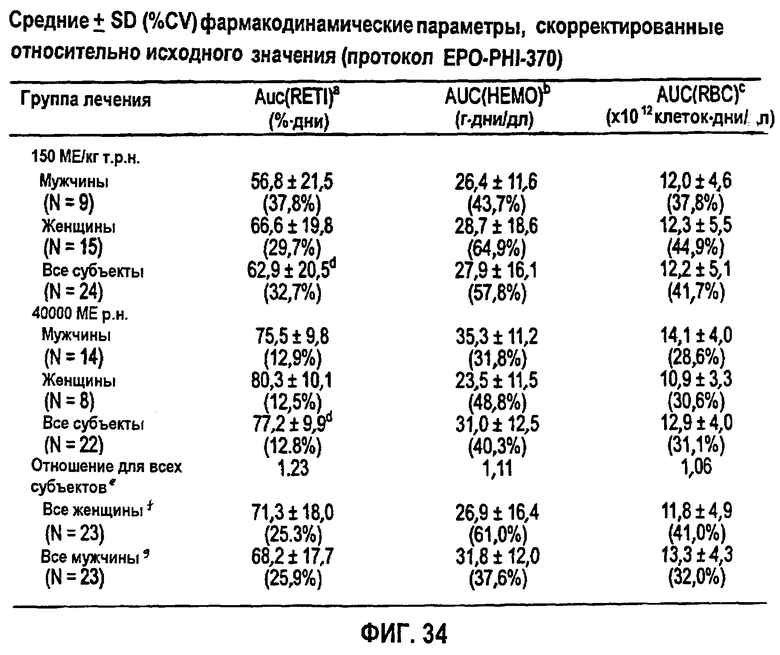
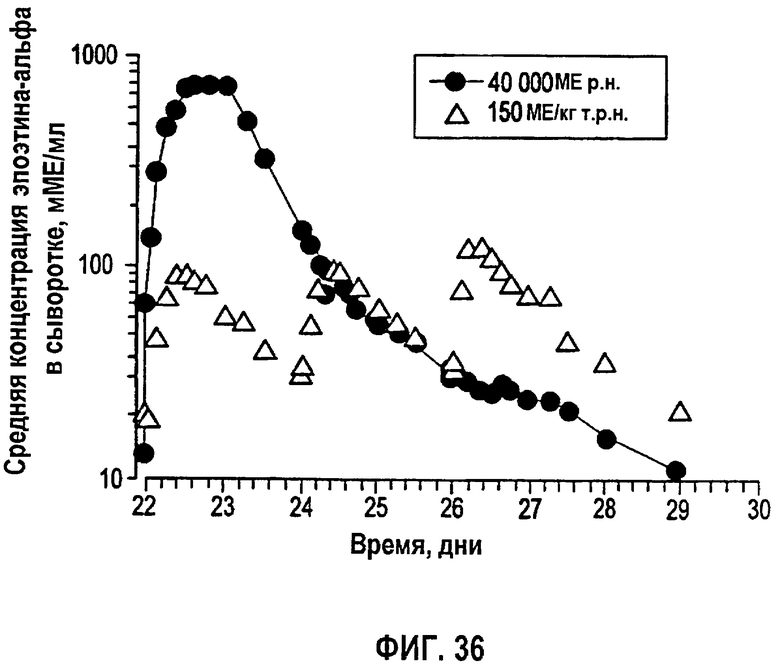
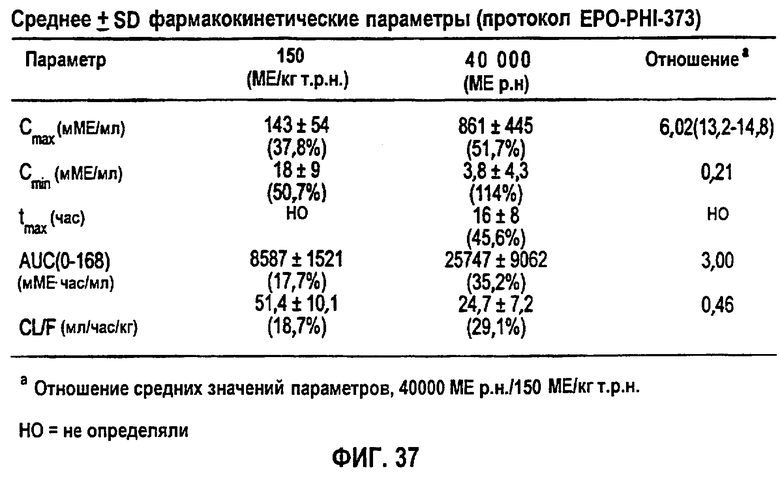
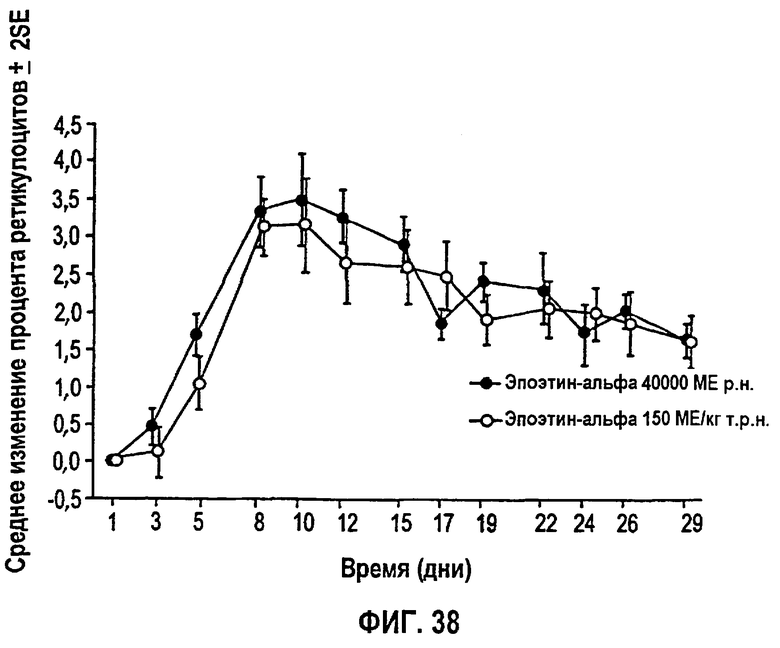
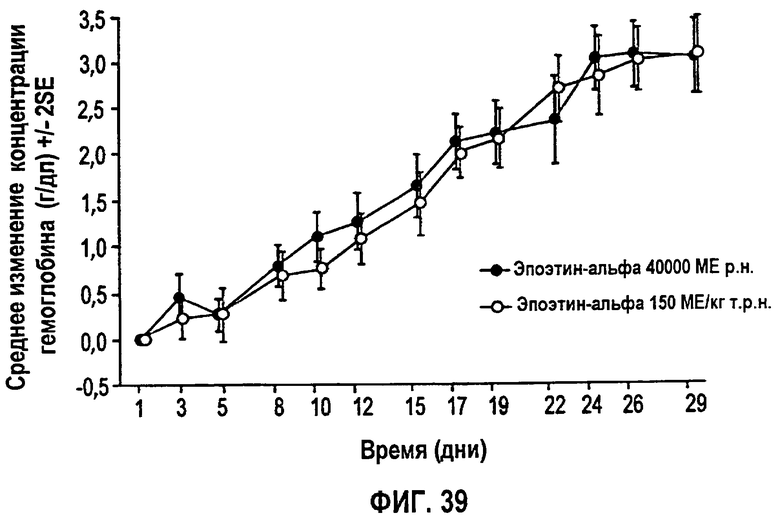
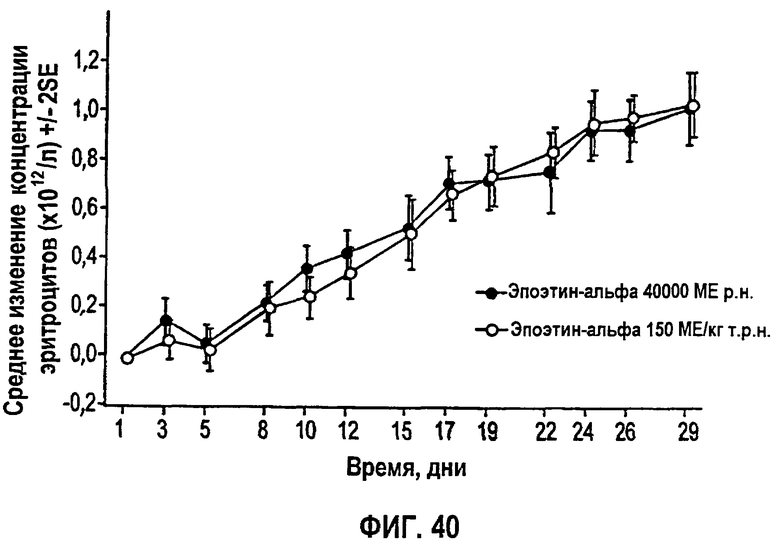
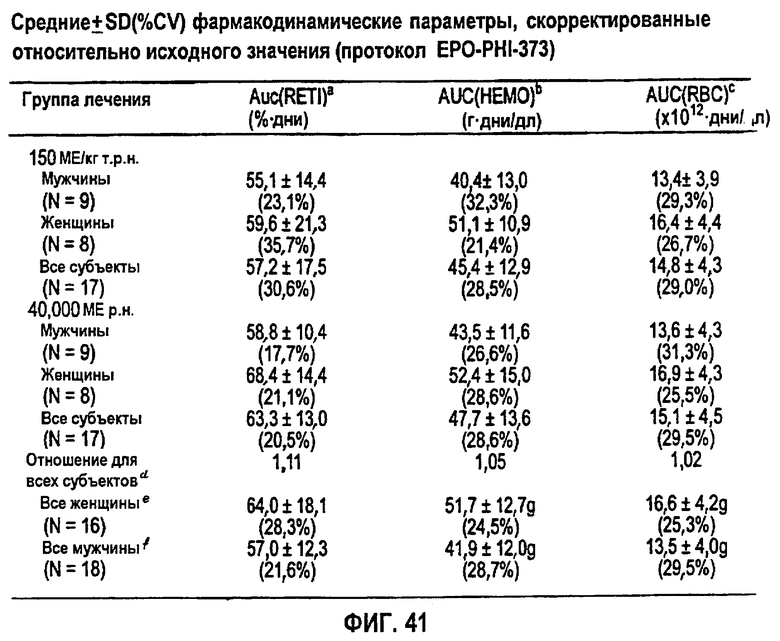
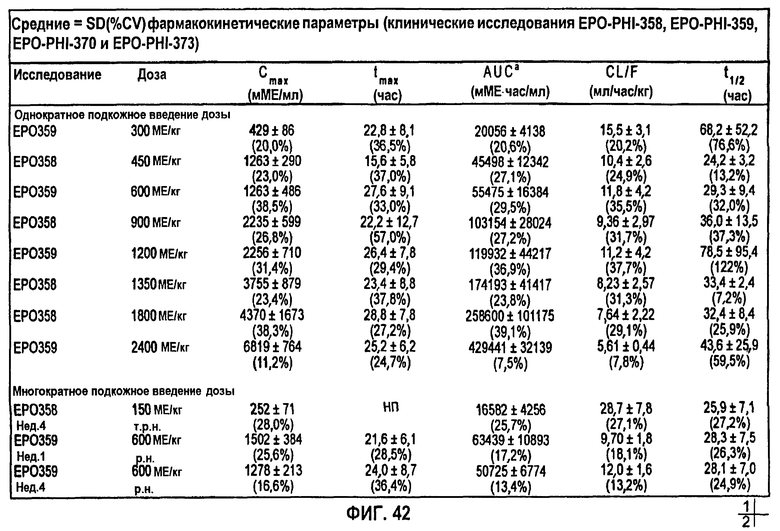
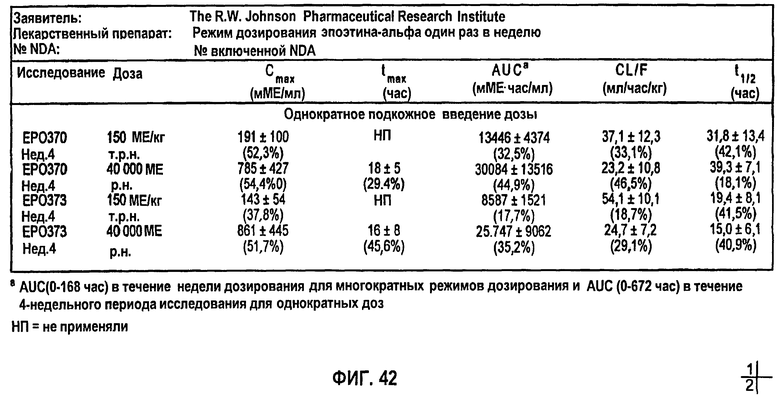
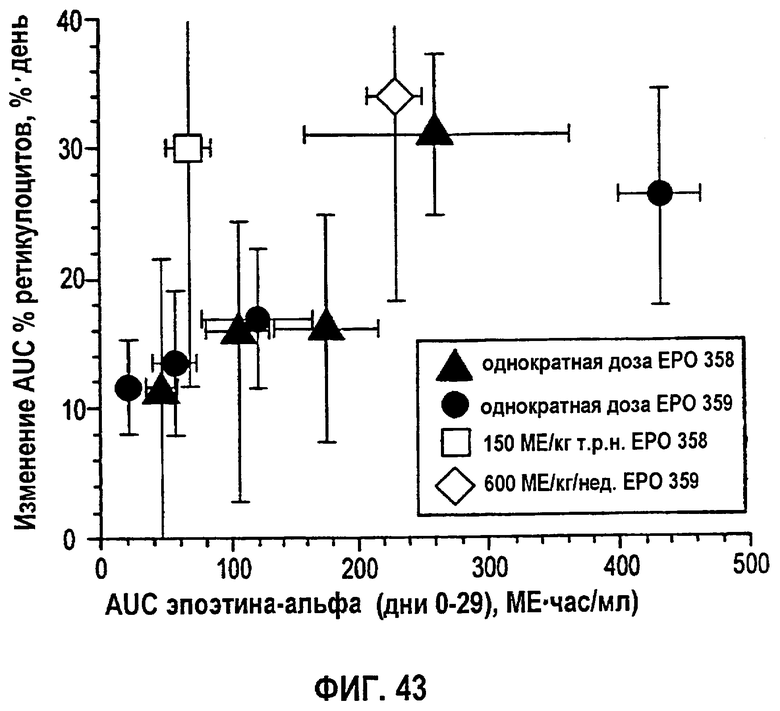
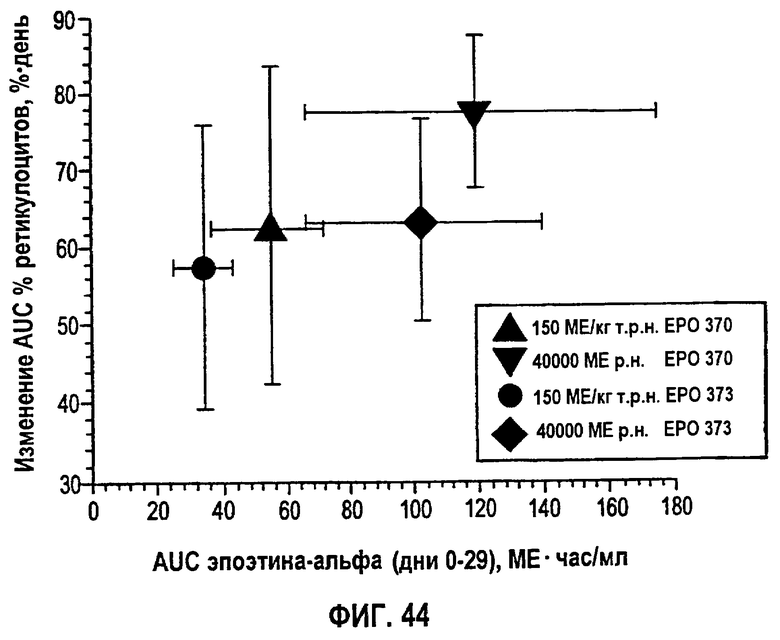
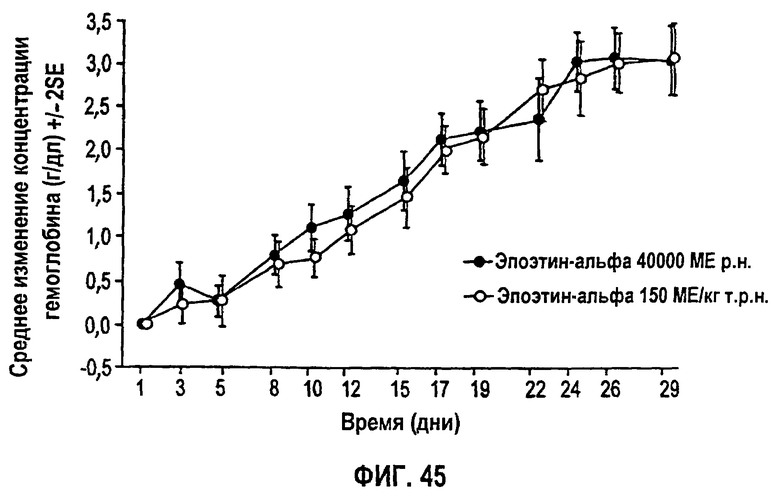
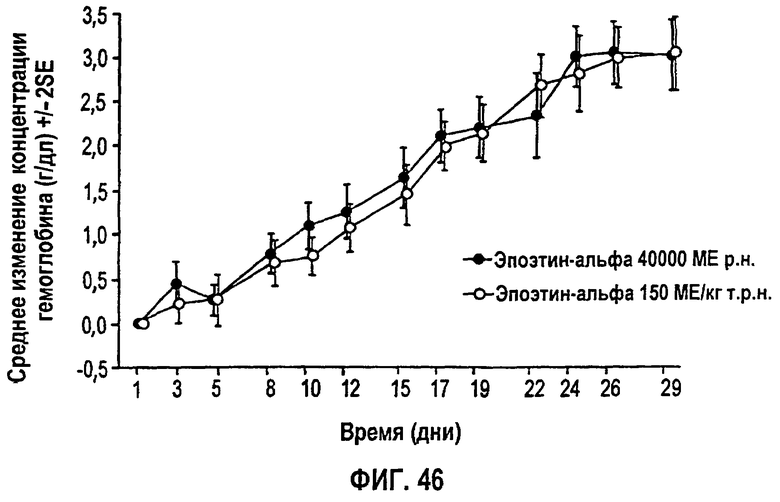
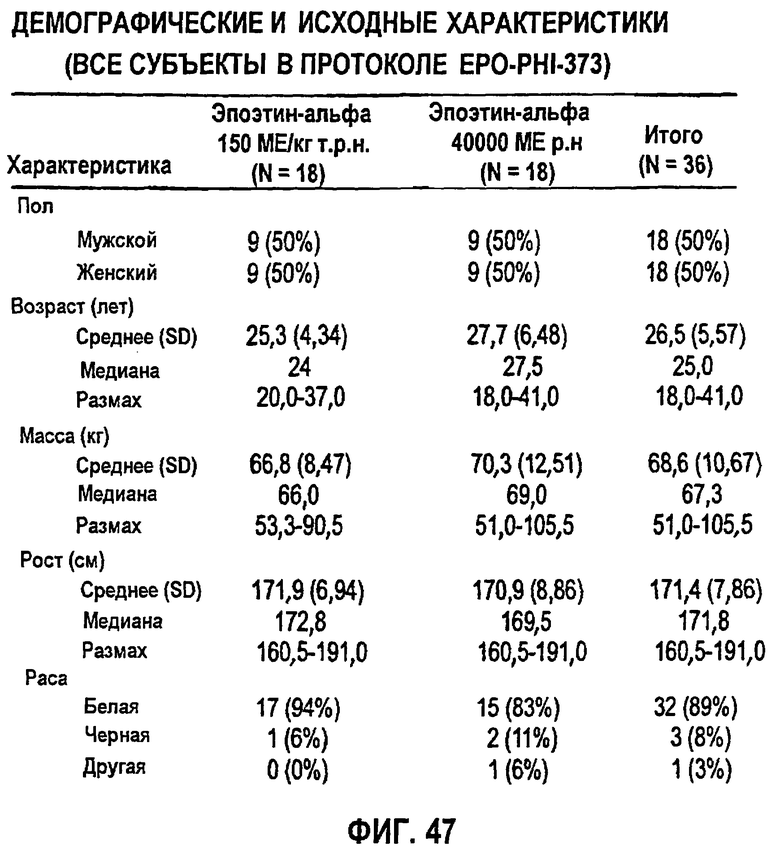
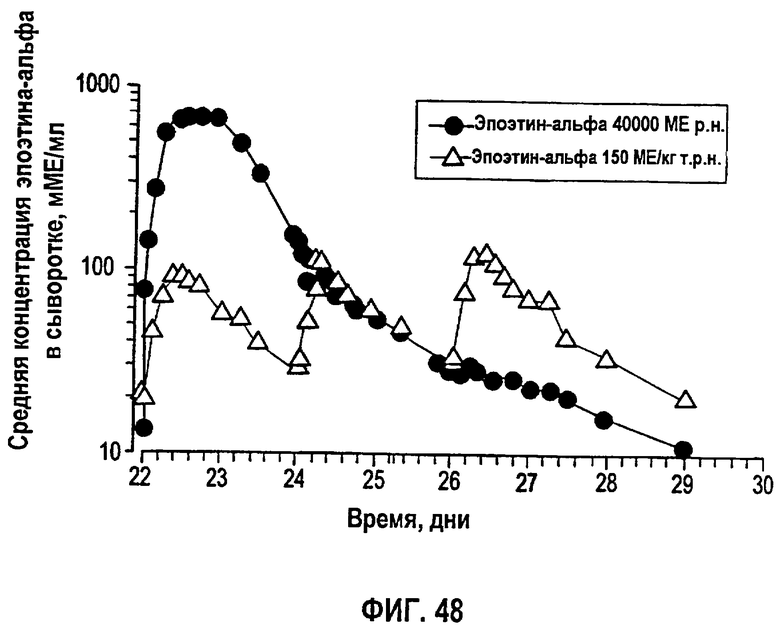
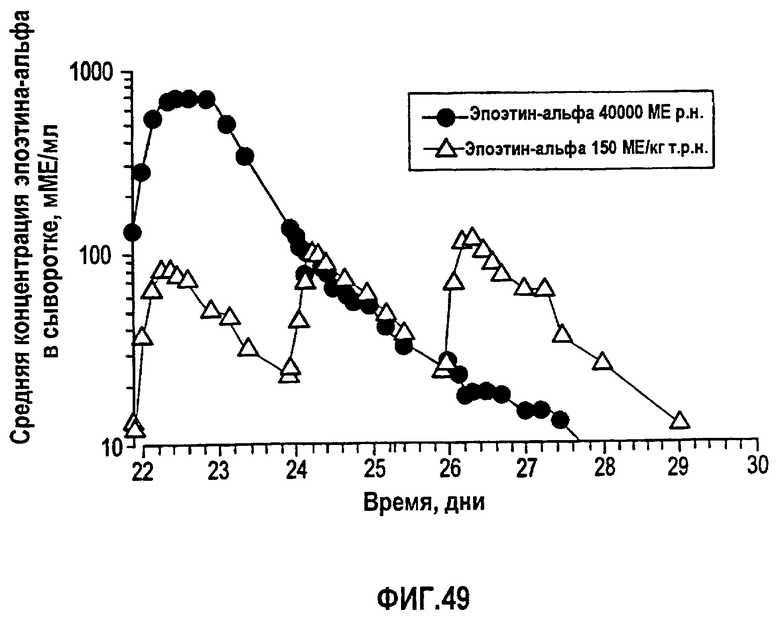
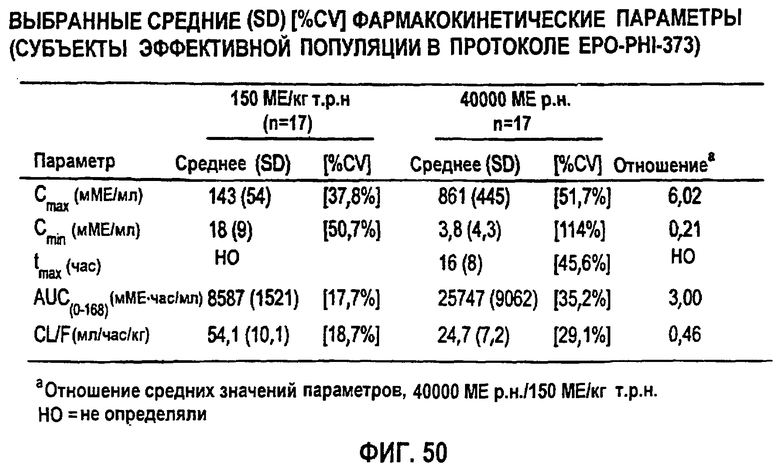
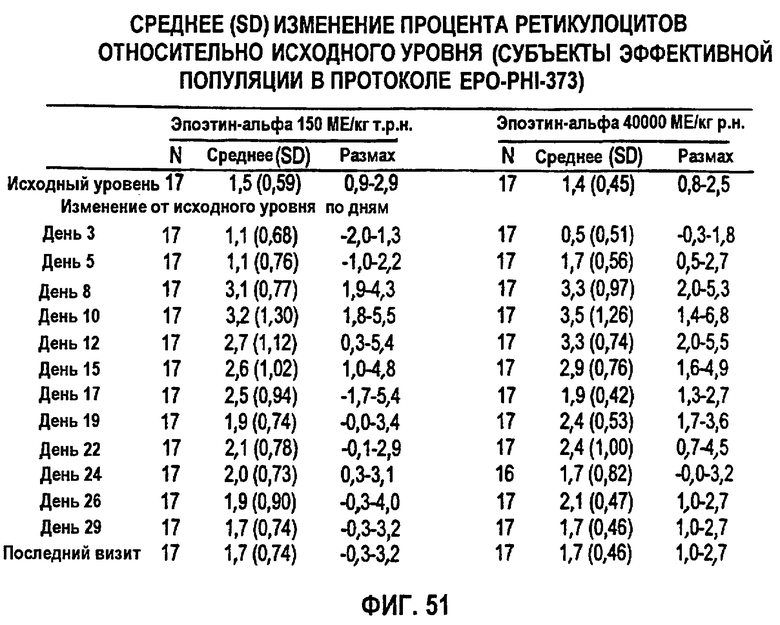
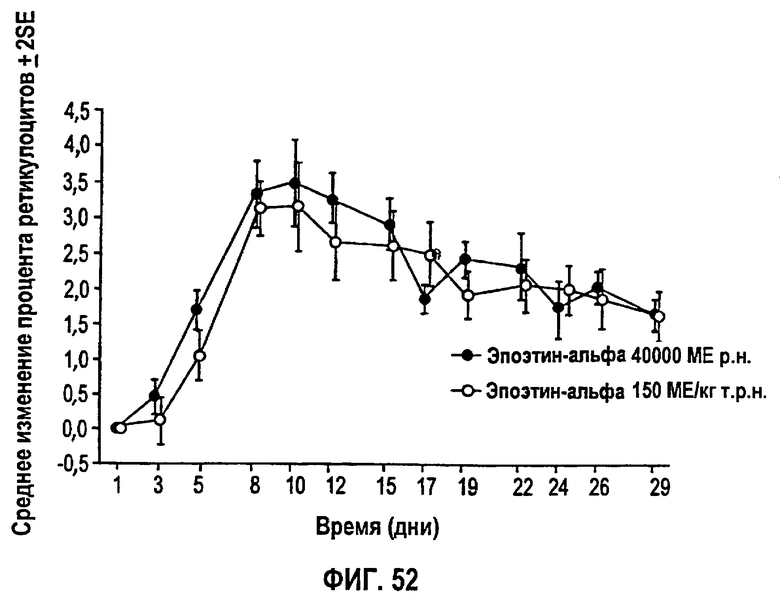
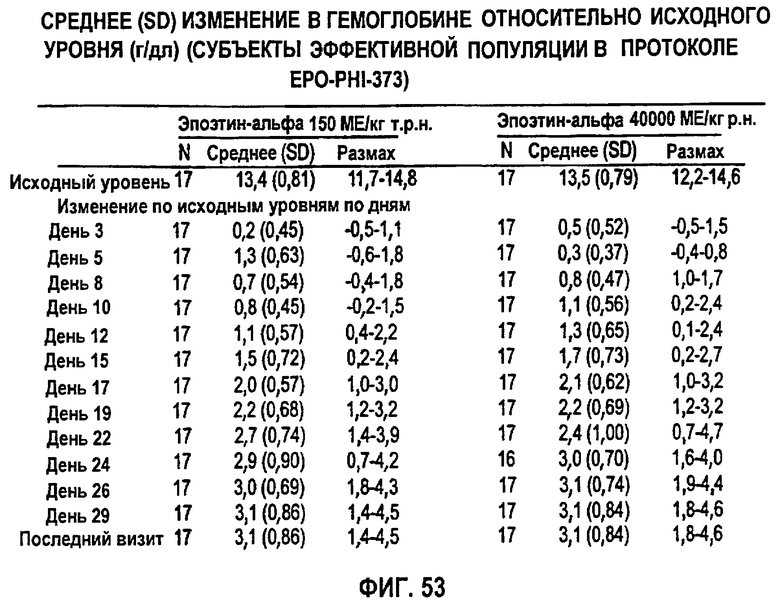
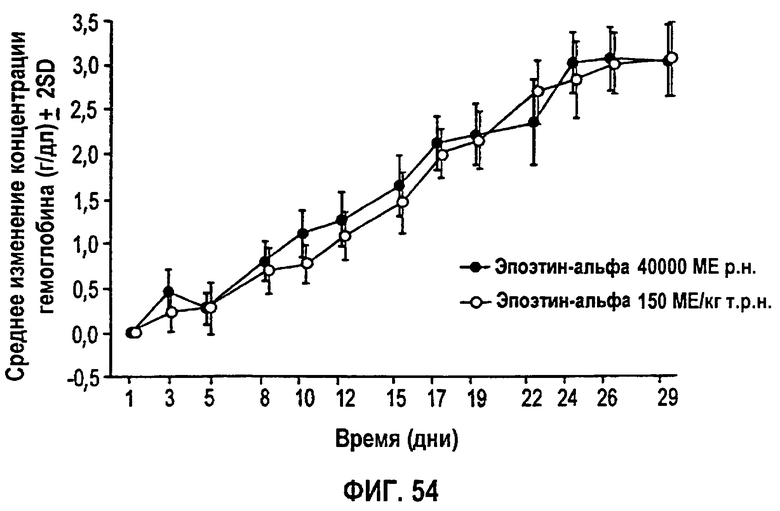
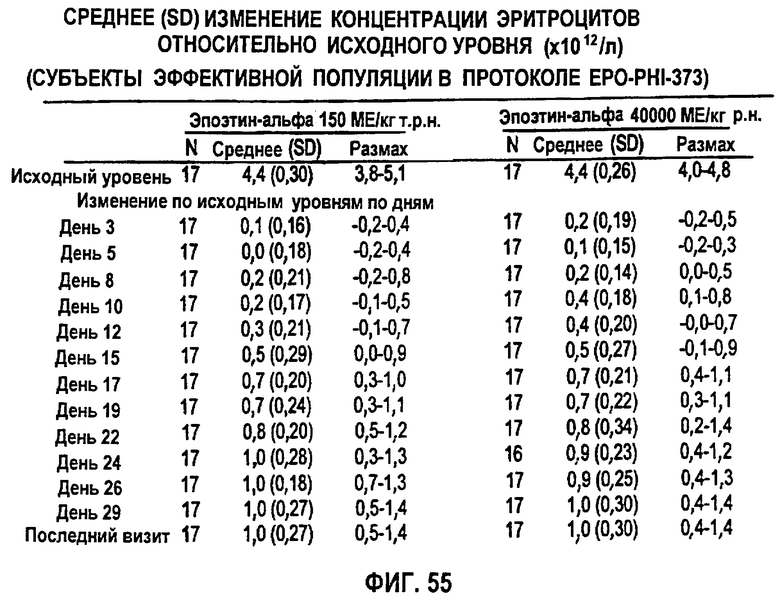
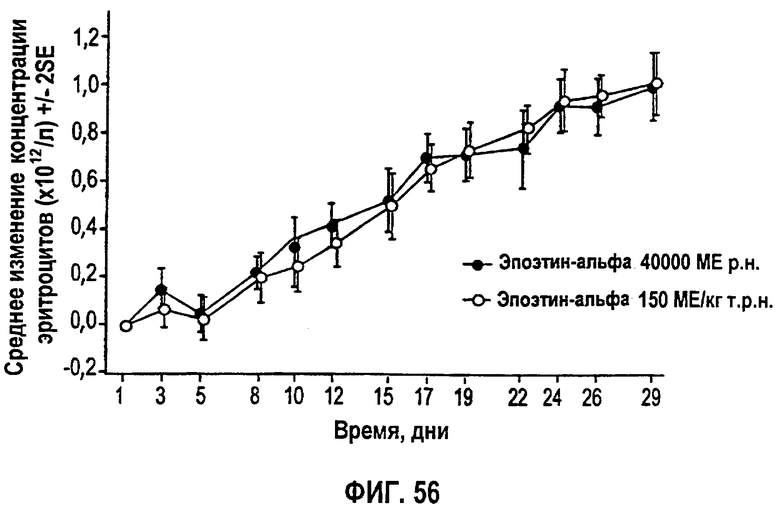
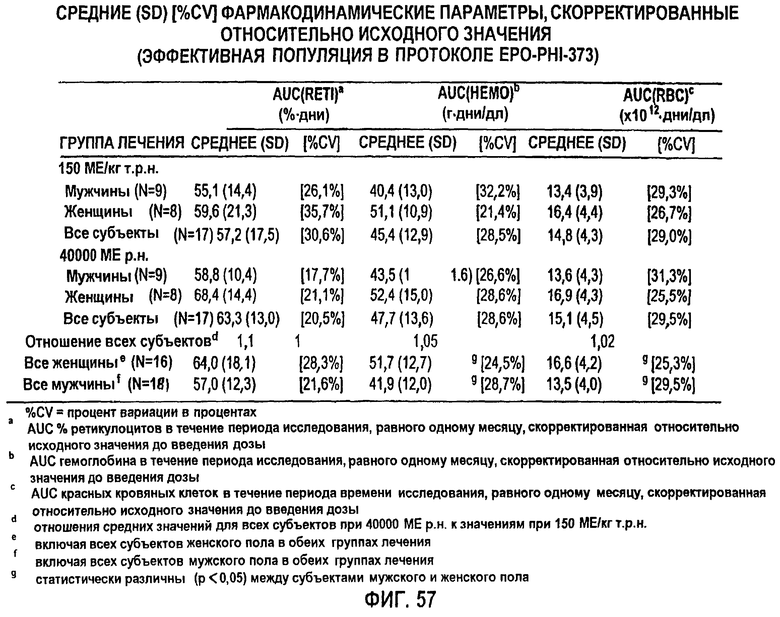

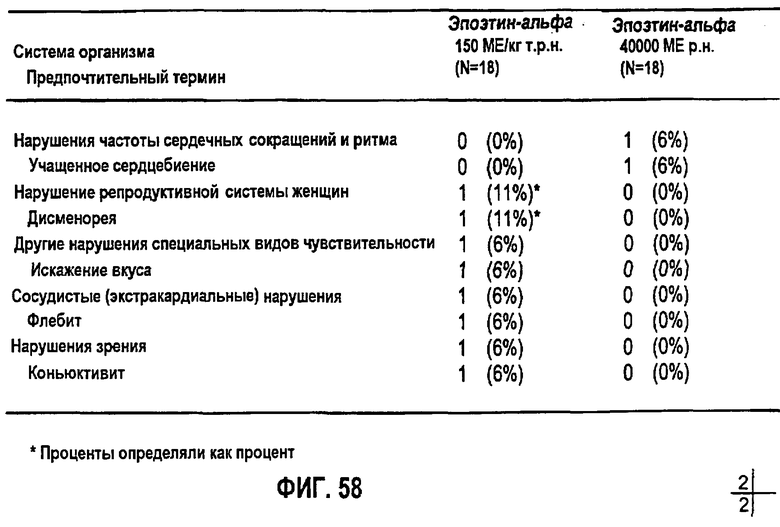
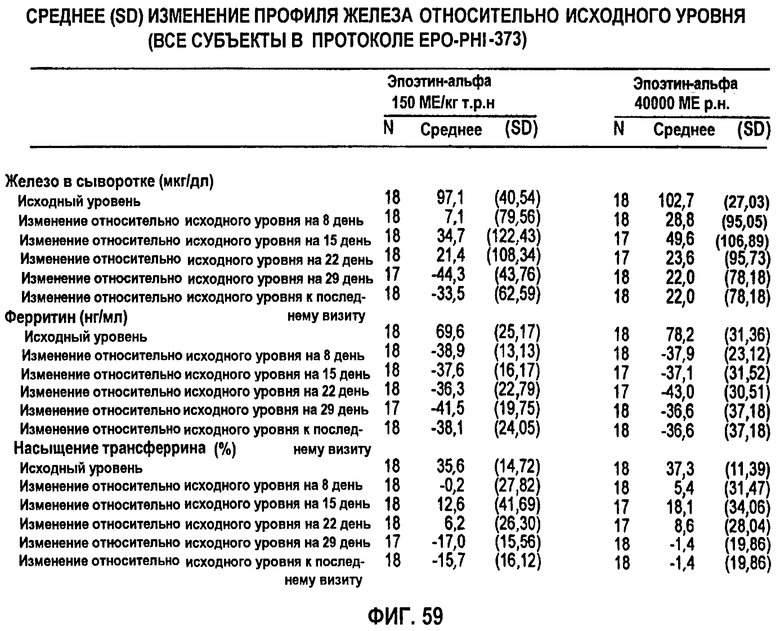
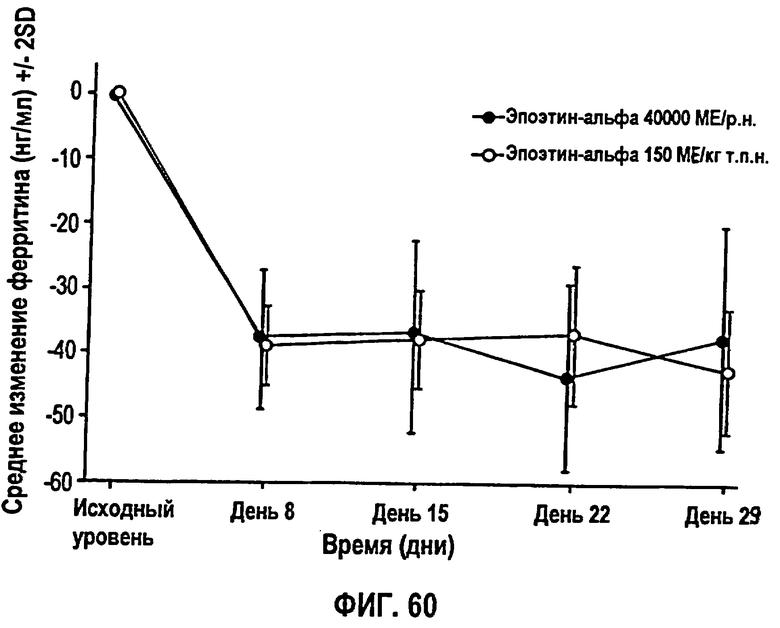

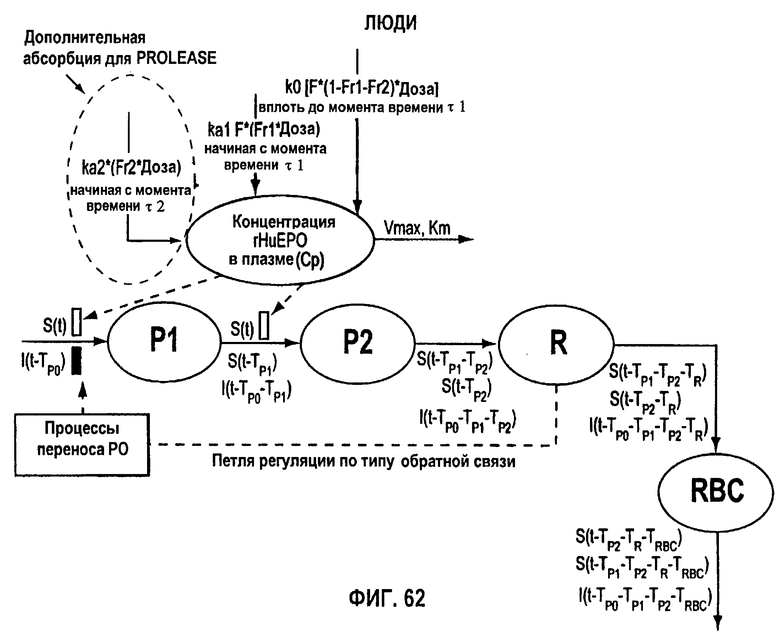
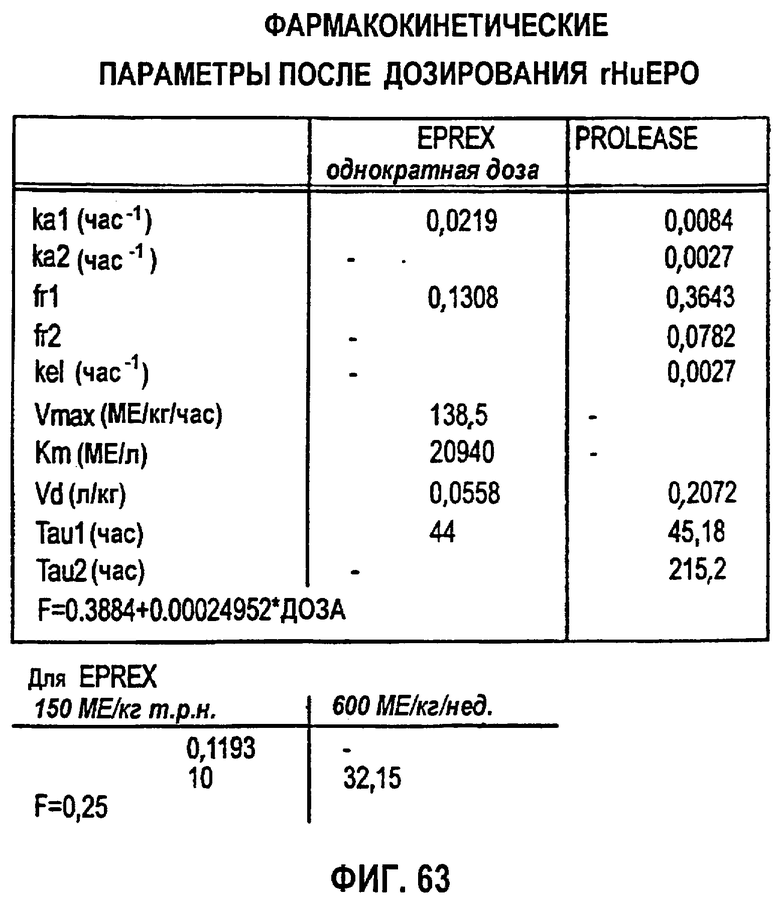

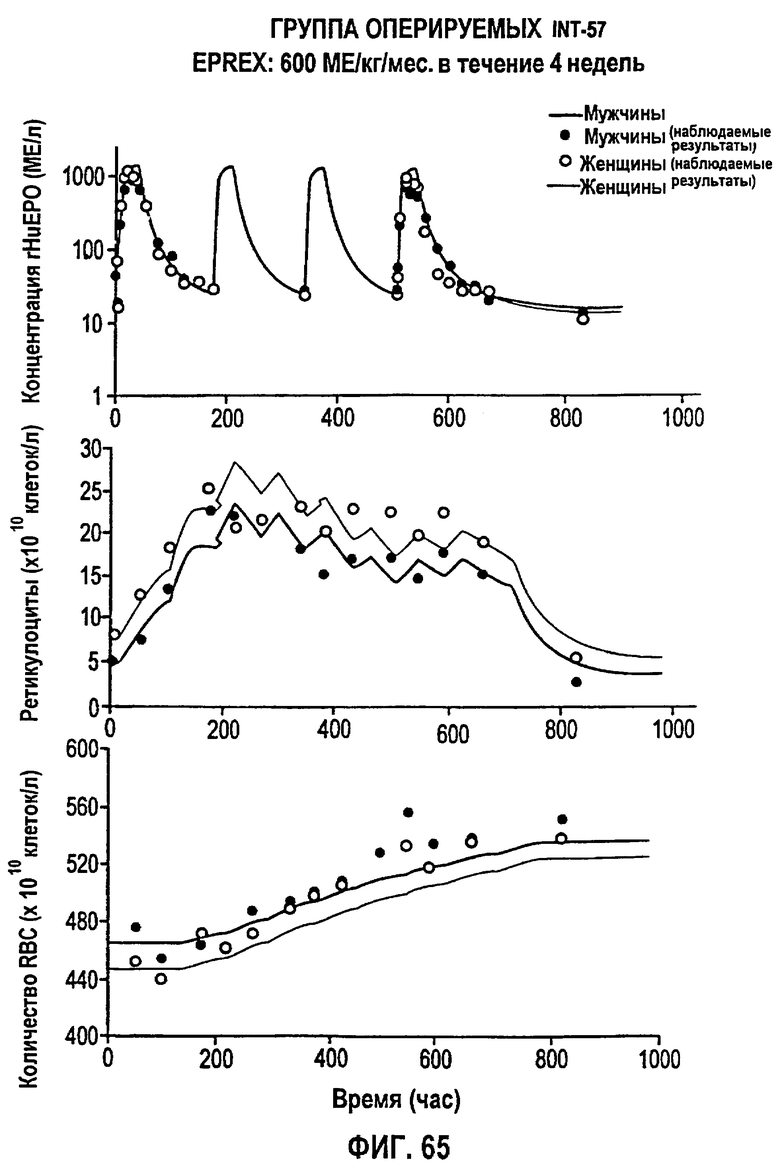
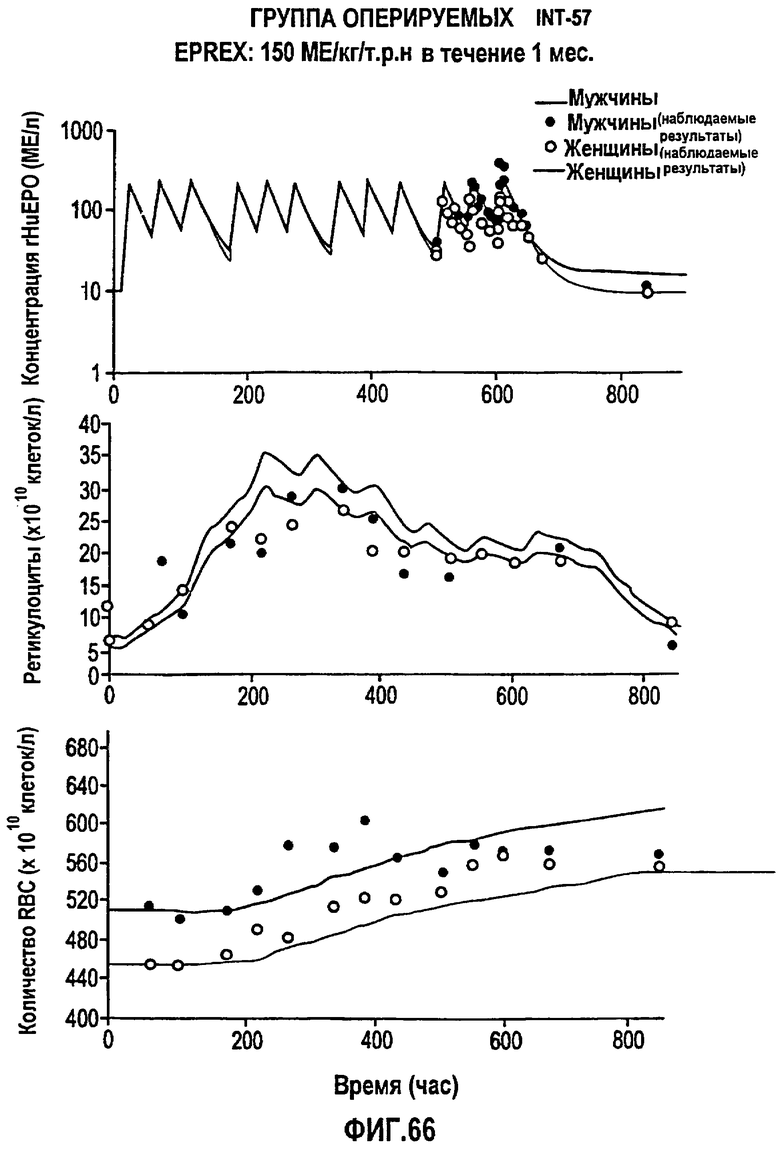
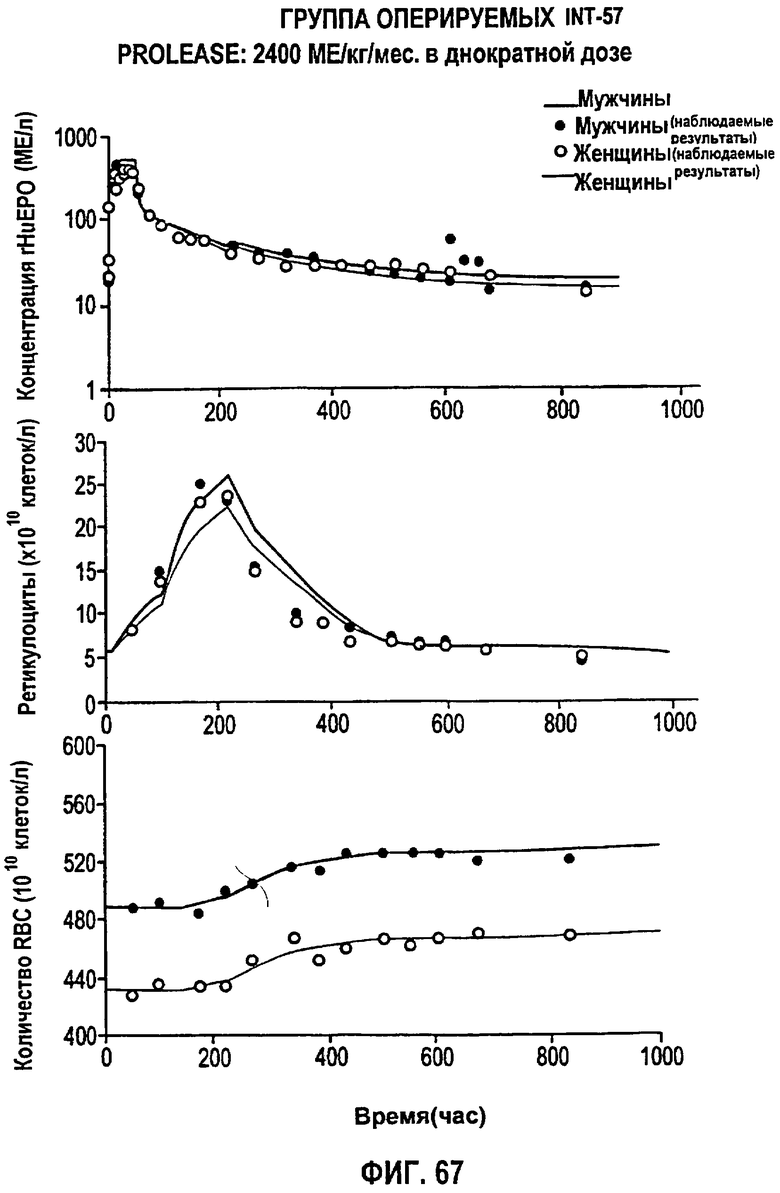
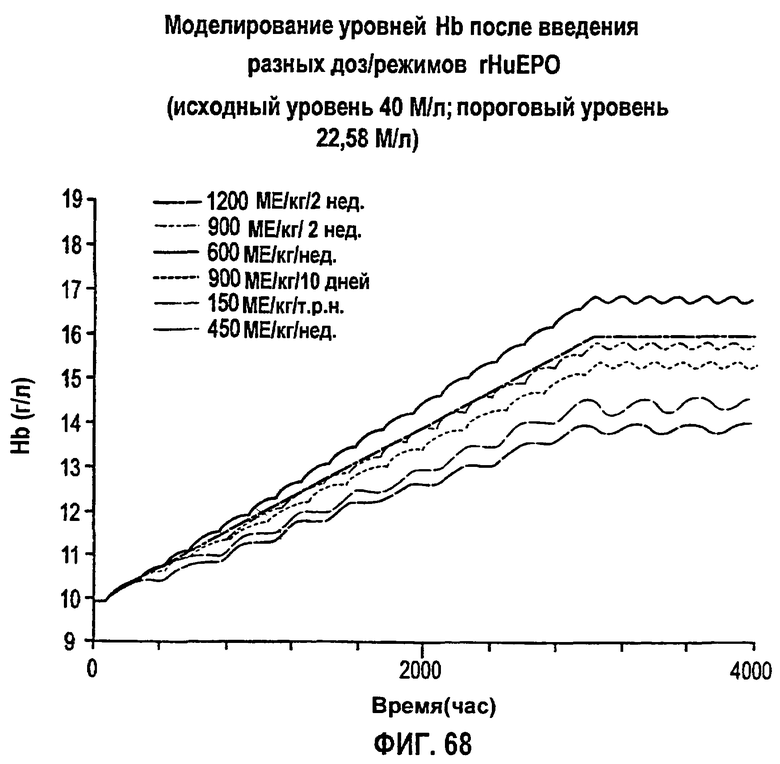
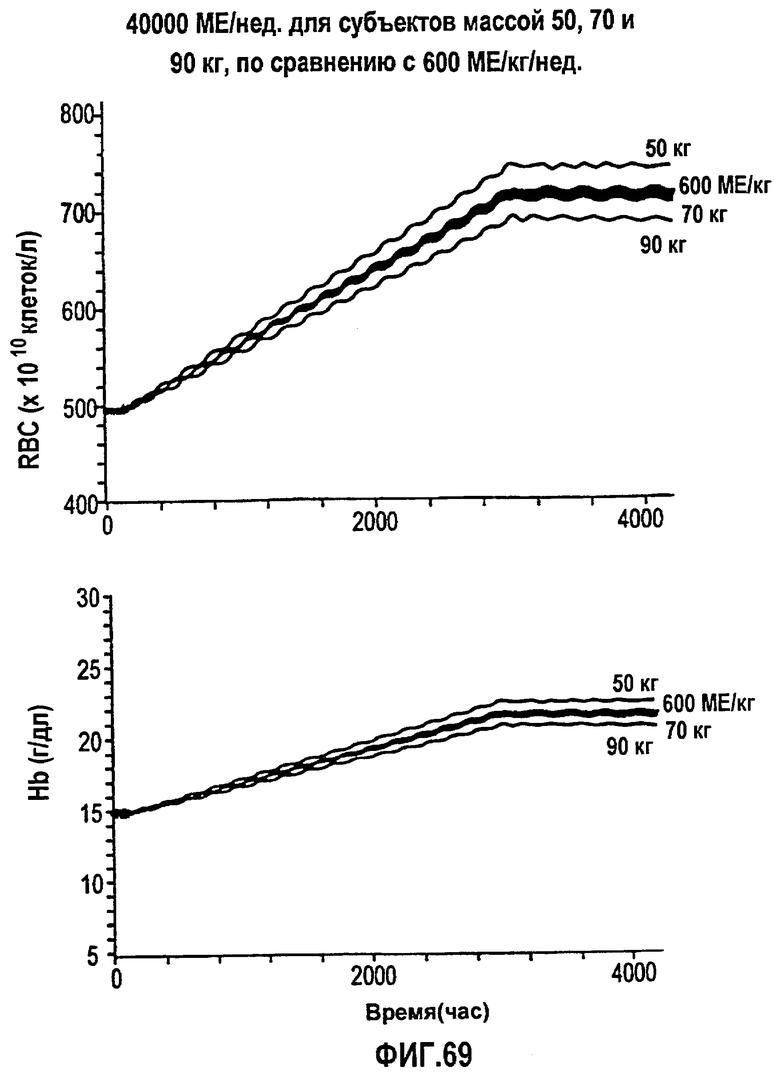
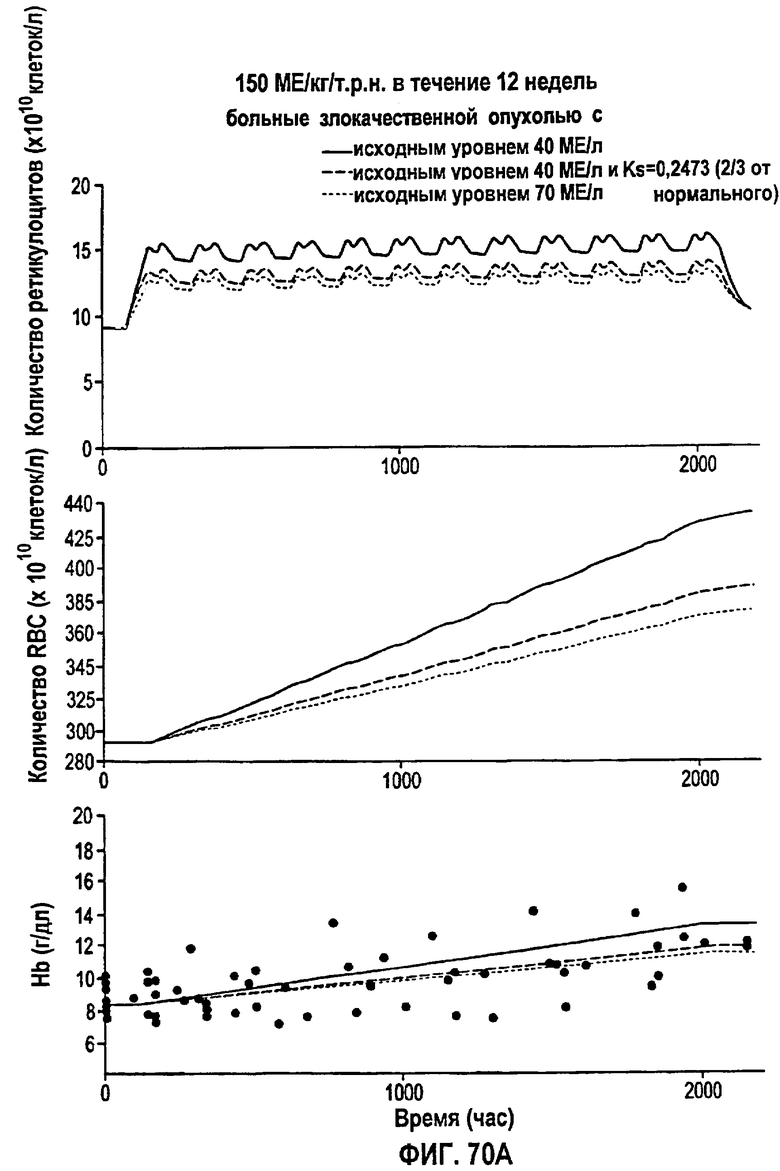
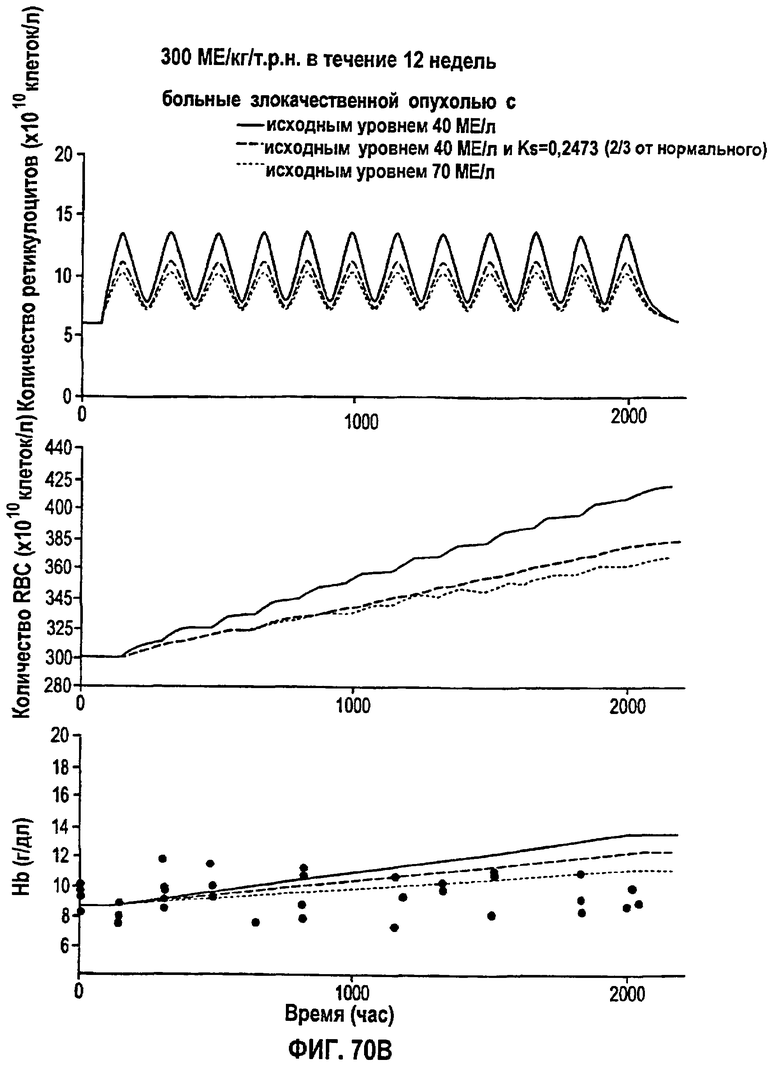
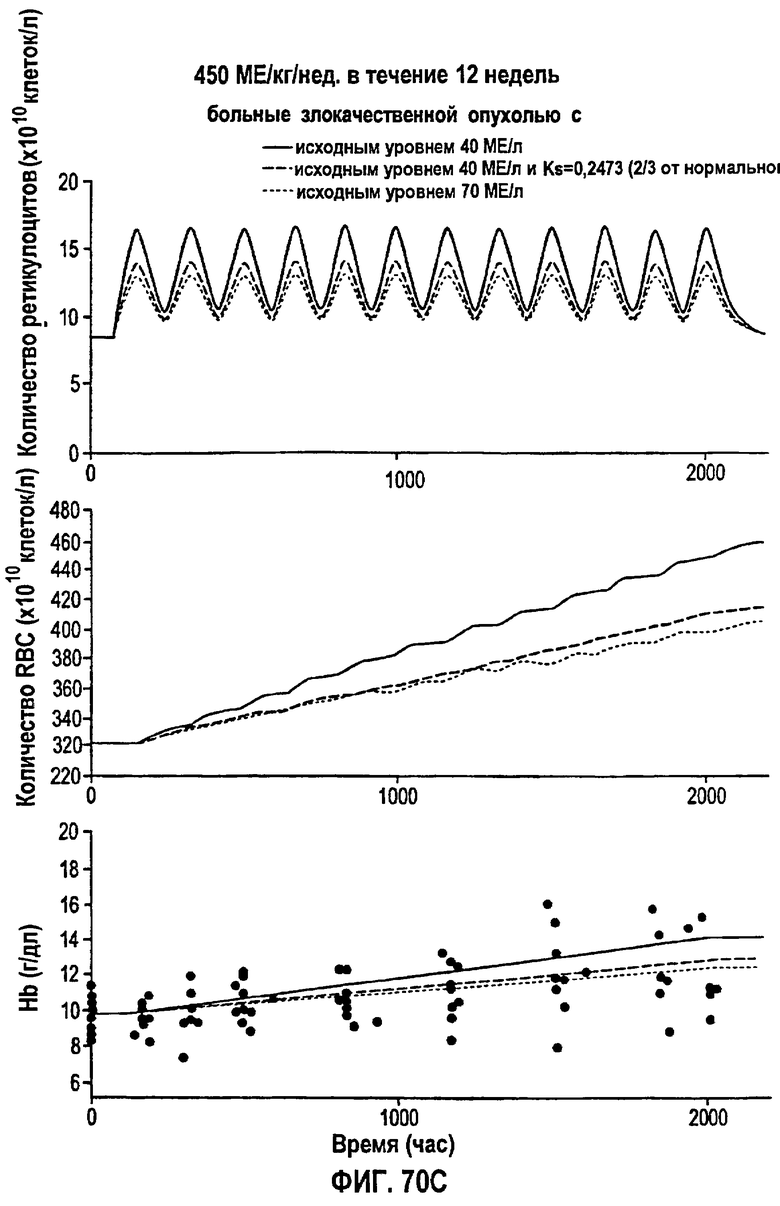
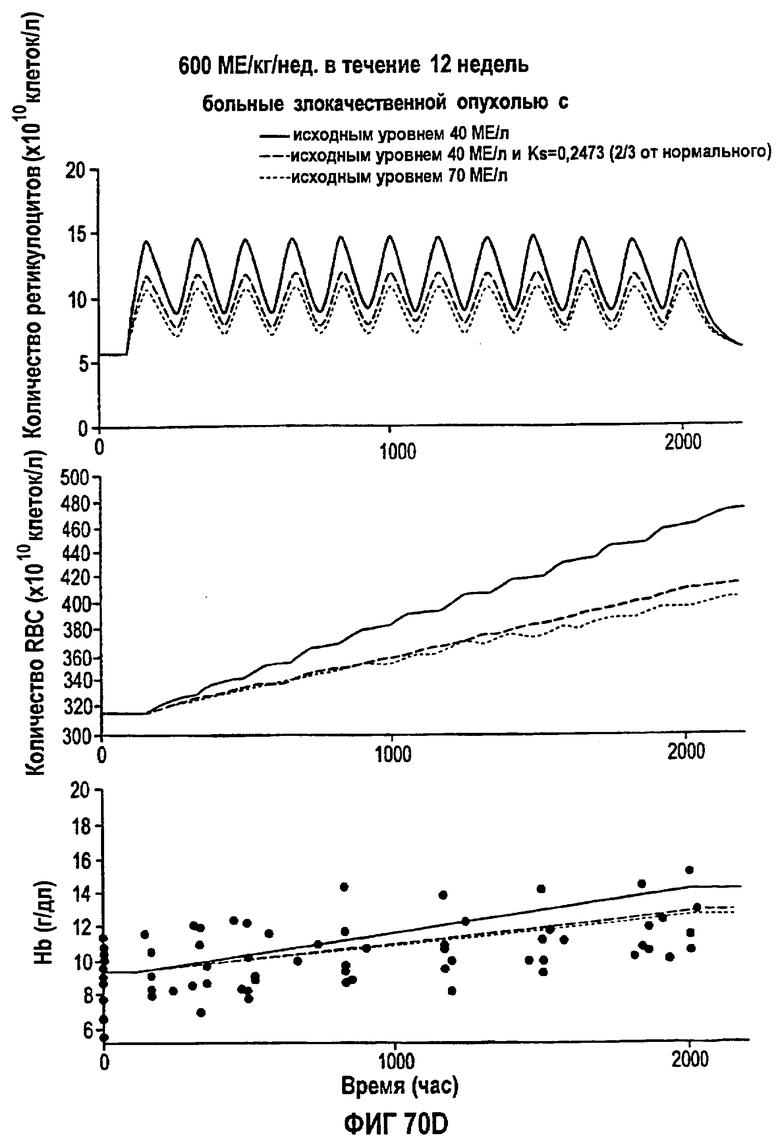
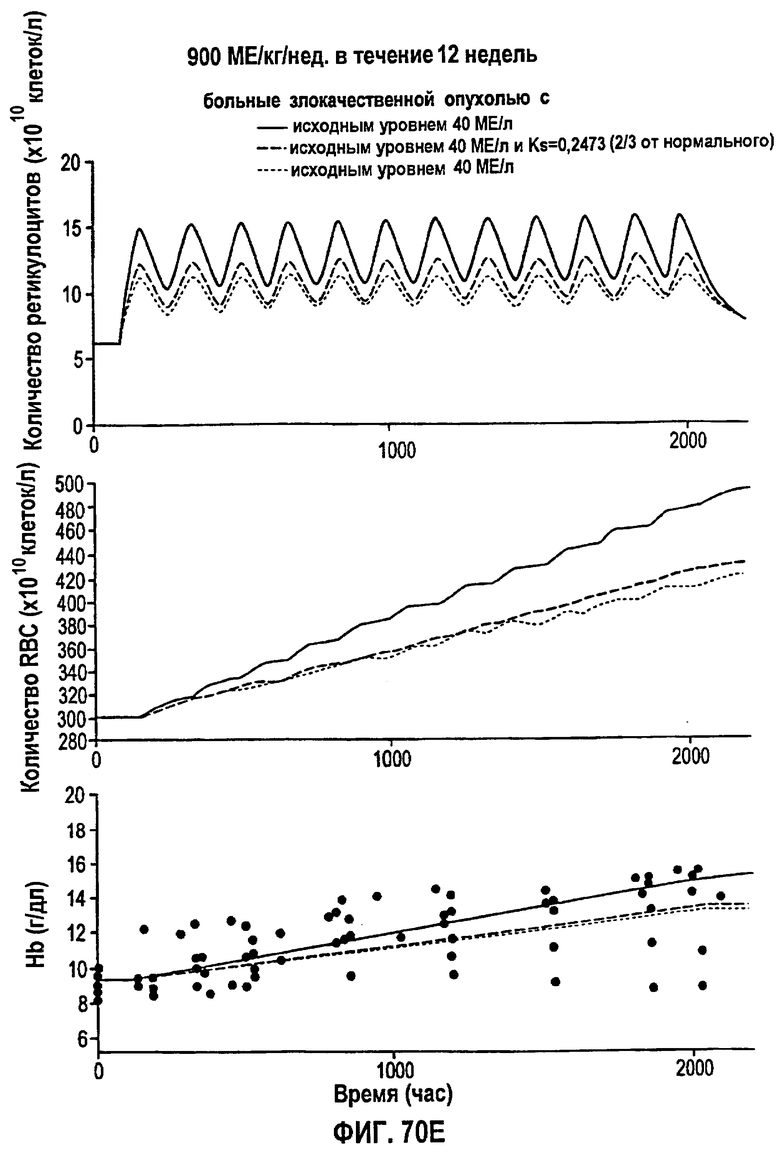
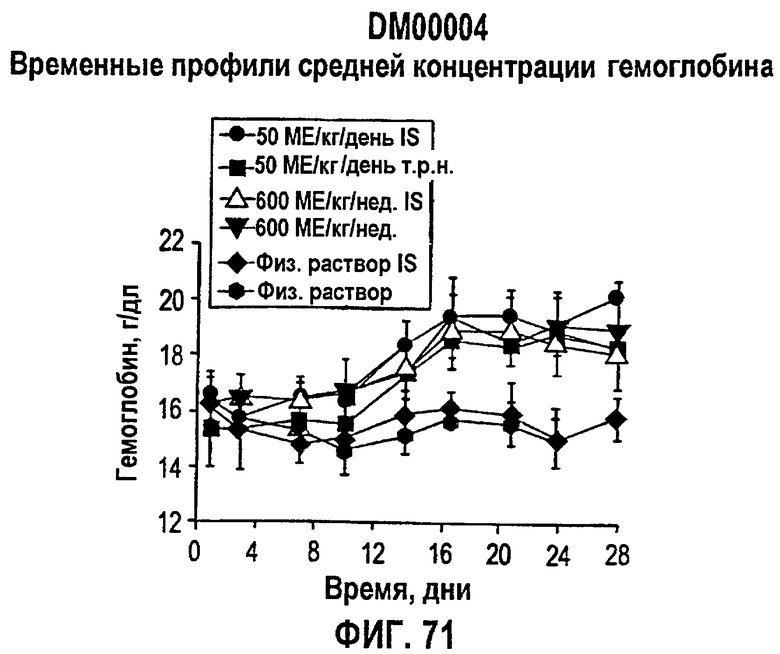
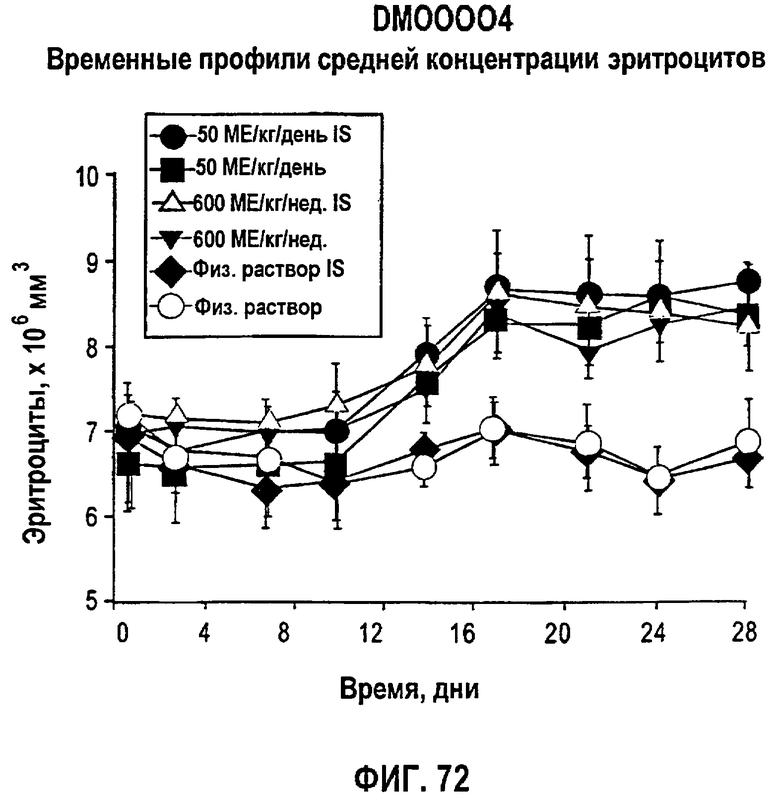
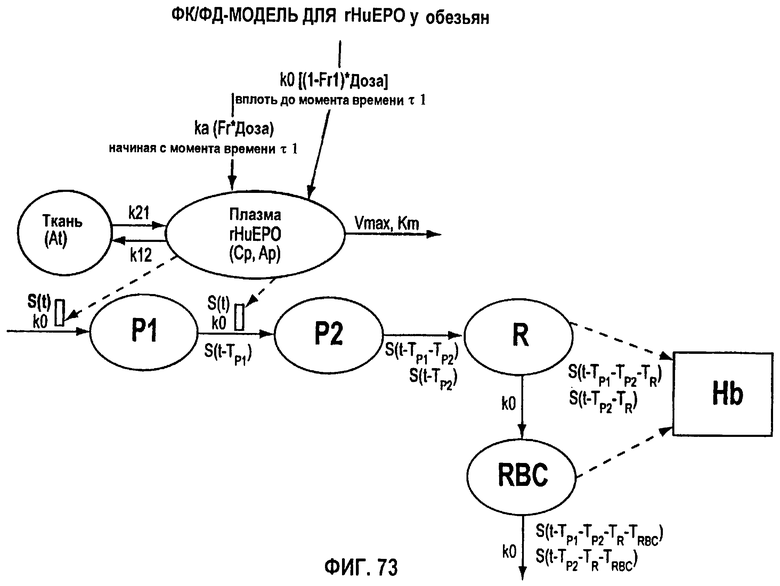
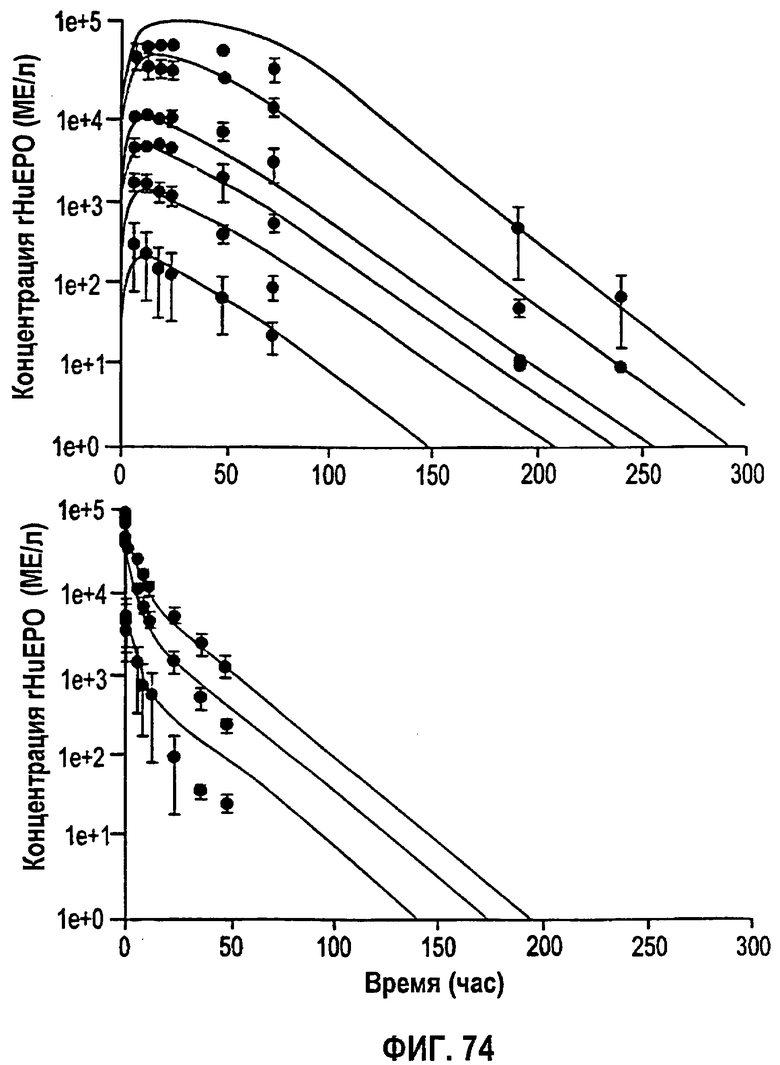
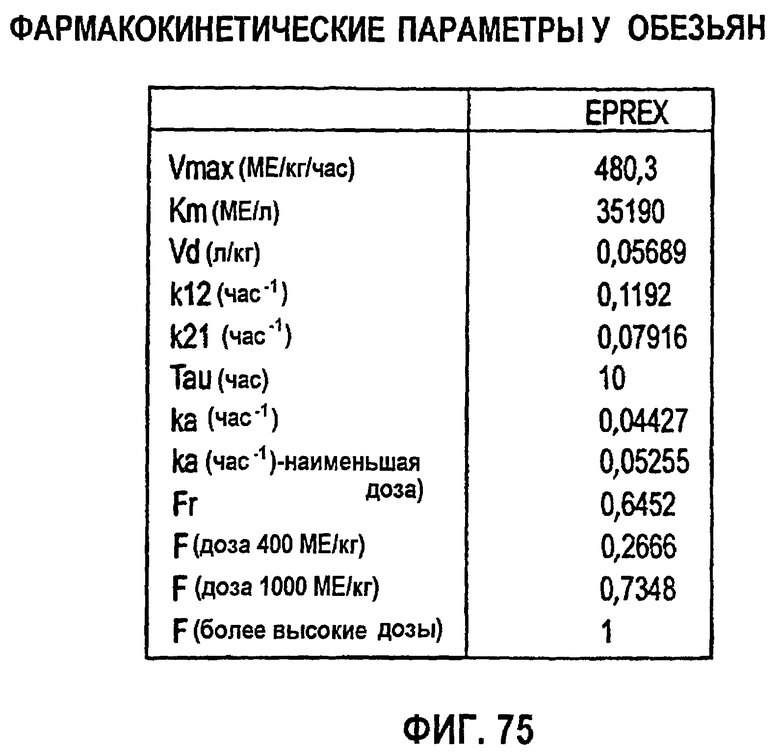
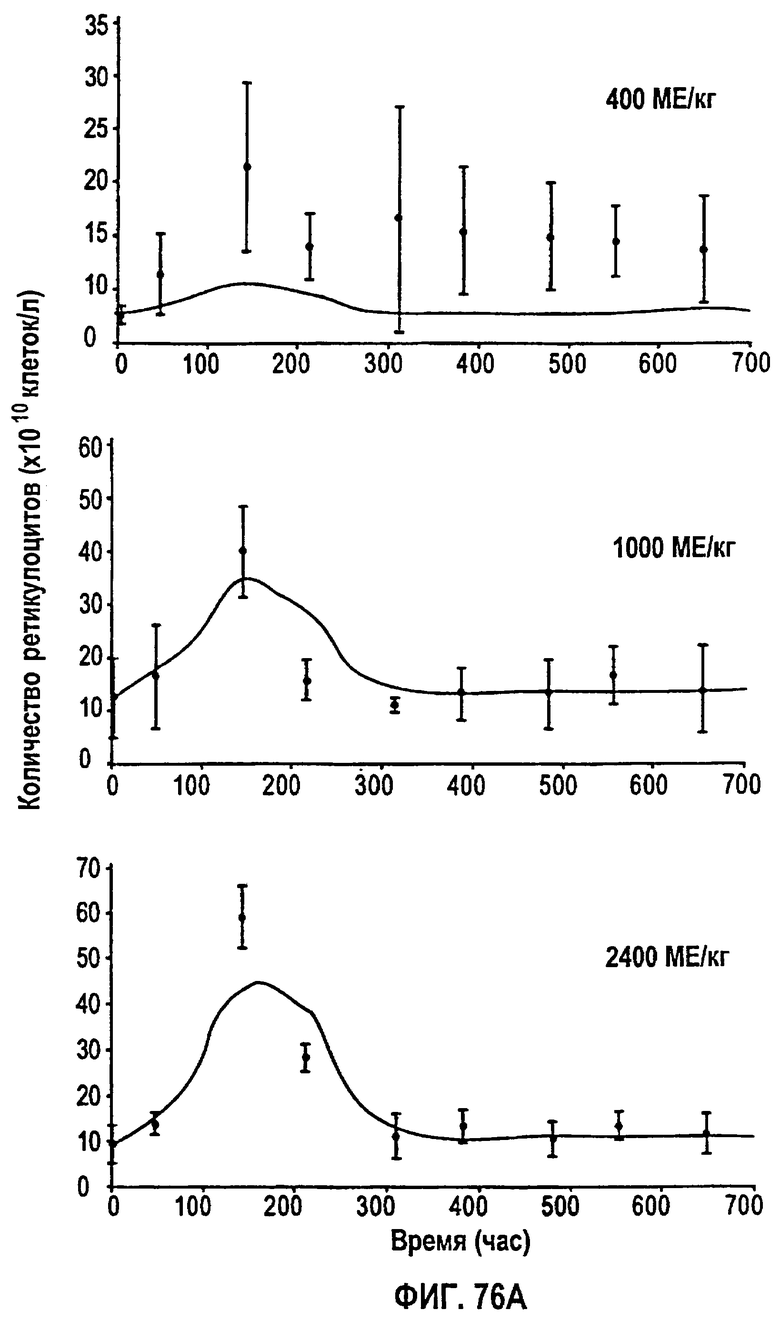
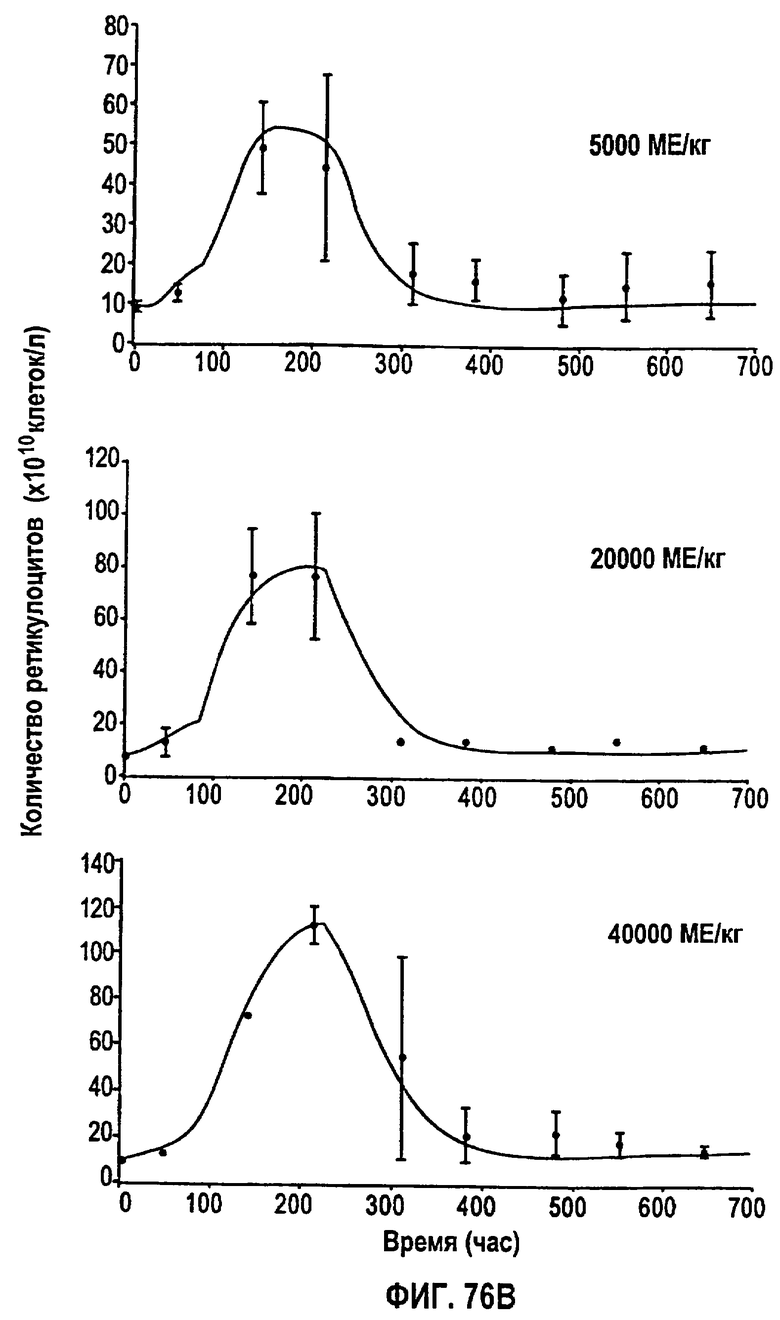

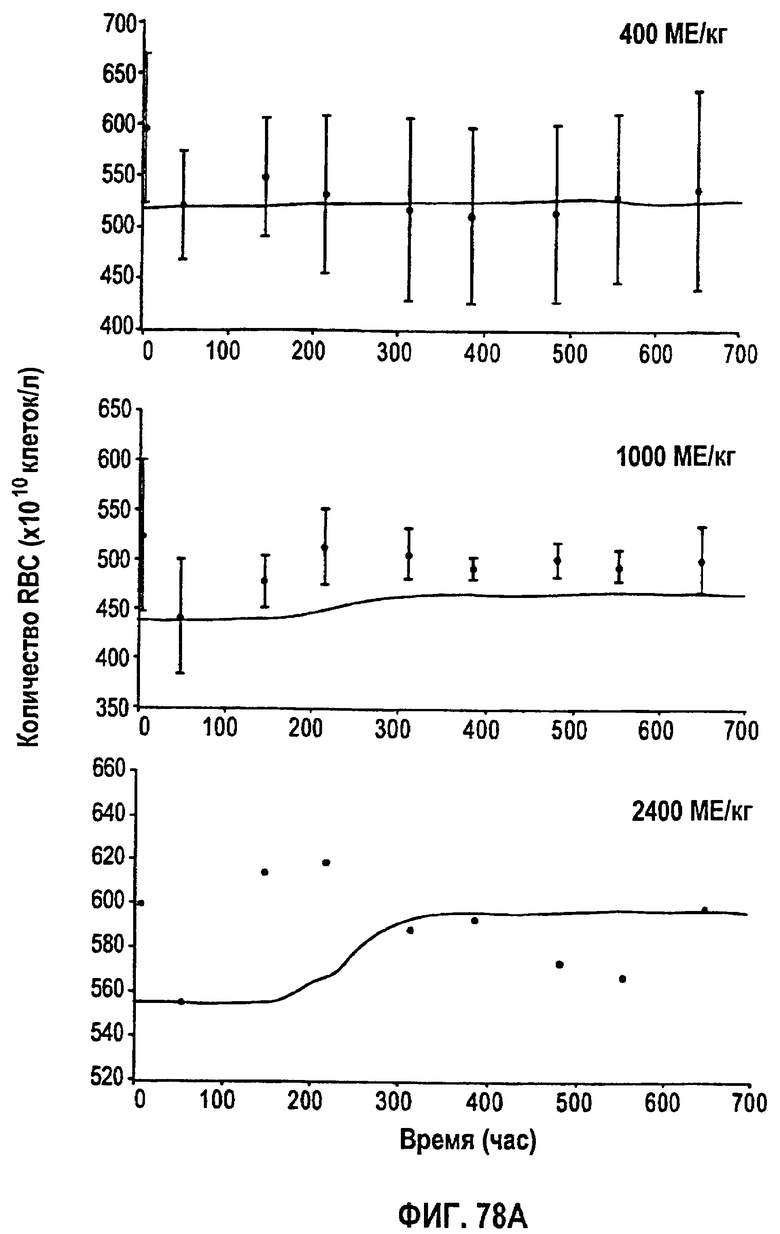
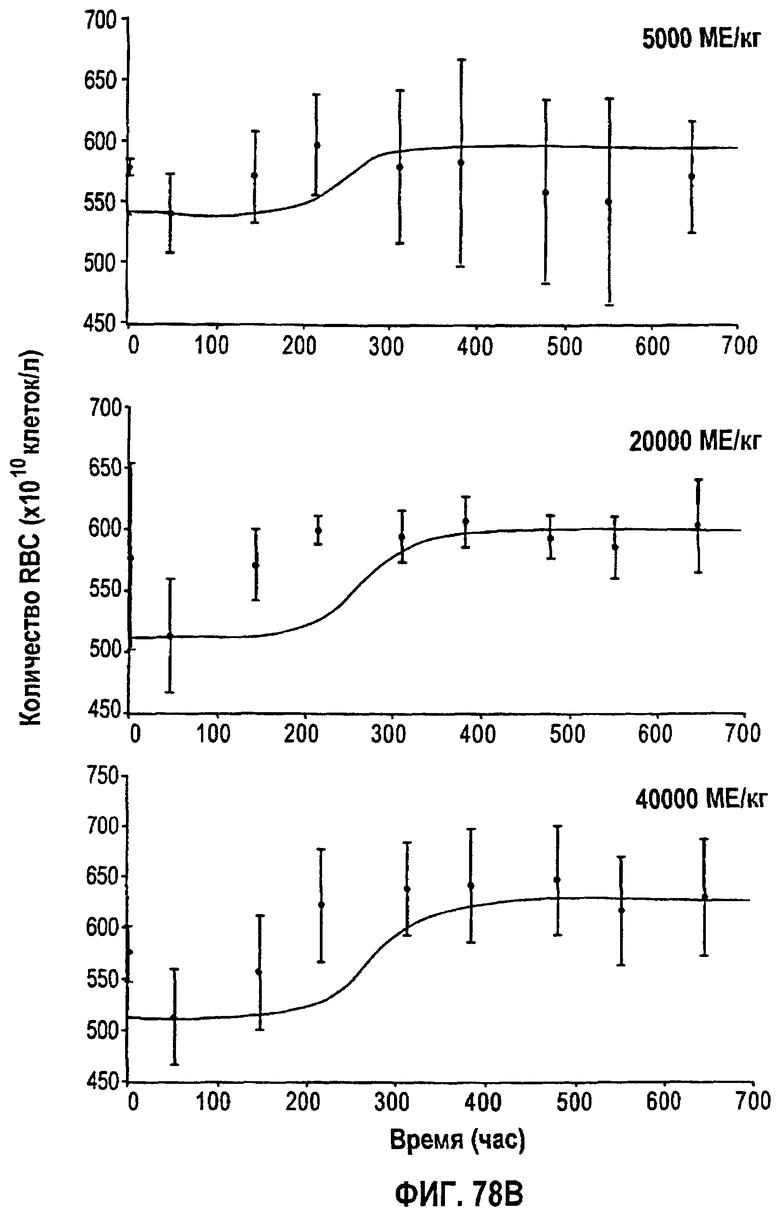
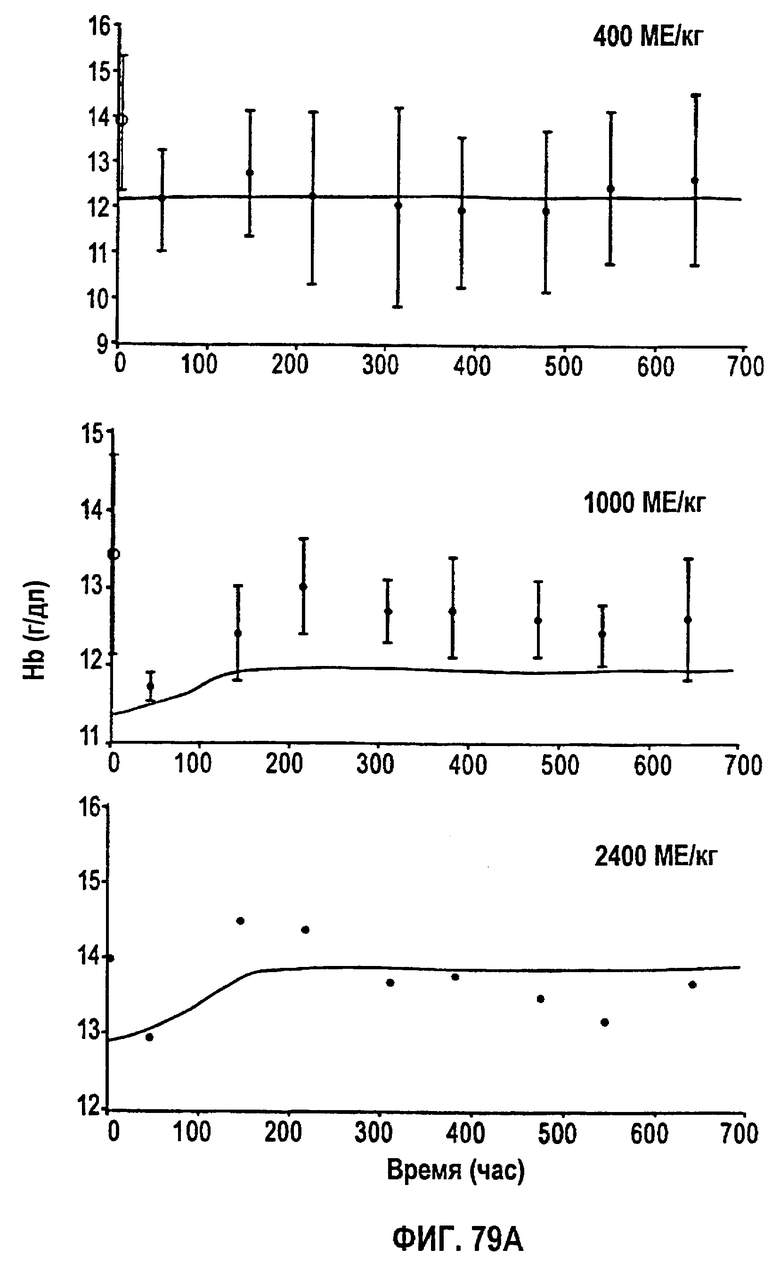
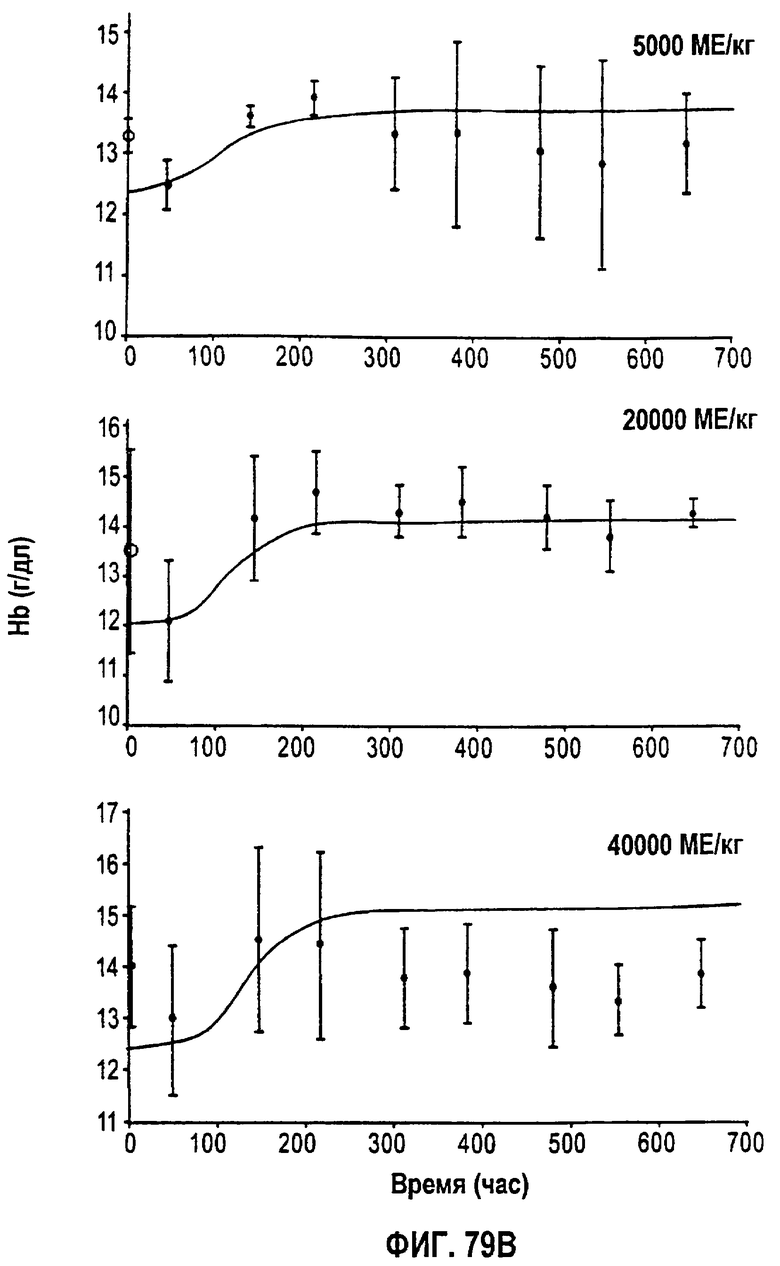
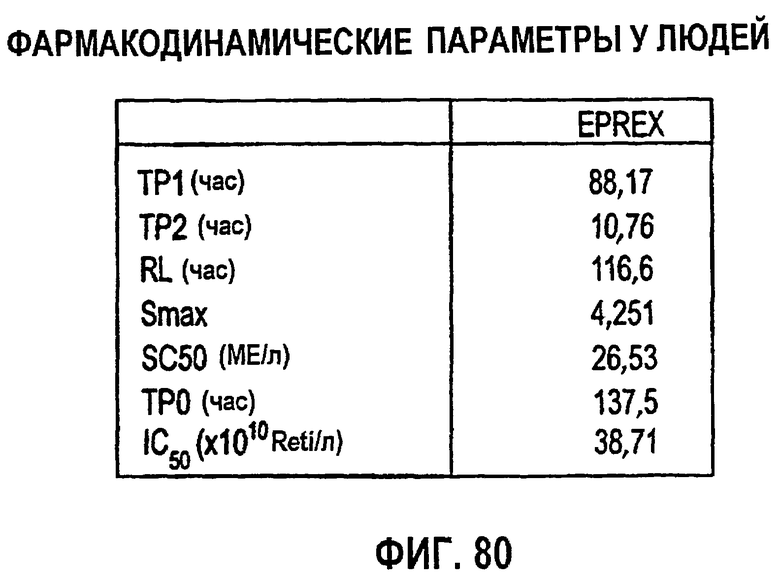
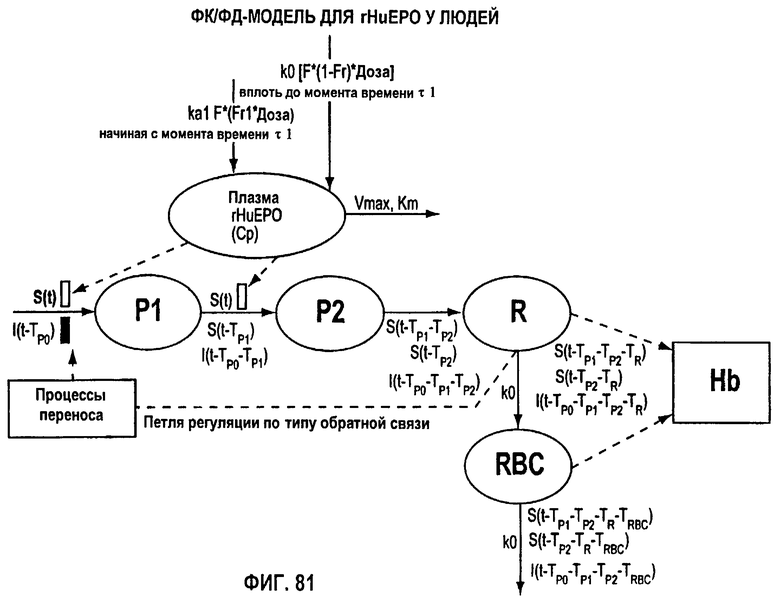
Изобретение относится к медицине, в частности к фармакологии, и касается определения оптимизированных режимов дозирования эритропоэтина (ЕРО). Для этого предлагается система выбора одного или нескольких режимов дозирования путем использования ФК/ФД - модели для определения ФК/ФД профиля режимов. Затем осуществляют отбор такого режима, который обеспечивает концентрацию ЕРО в сыворотке, превышающую ее до введения в течение 5-30 суток между введениями ЕРО. Изобретение позволяет оптимально использовать ЕРО для коррекции изменений показателей крови и лечения заболеваний, с ними связанных. 15 н. и 35 з.п. ф-лы, 81 ил., 2 табл.
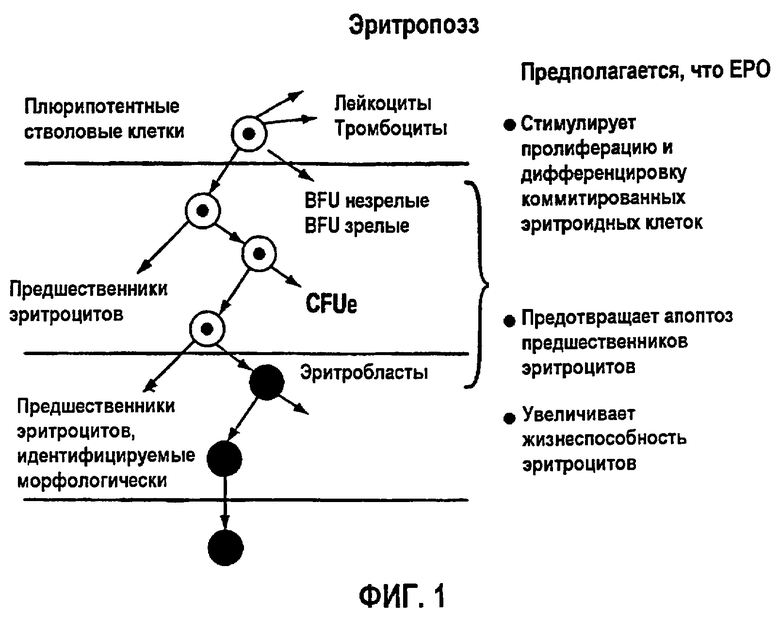
(a) выбор одного или нескольких режимов дозирования ЕРО;
(b) применение фармакокинетической/фармакодинамической модели для определения фармакодинамического профиля указанных одного или нескольких режимов дозирования ЕРО; и
(c) отбор указанных одного или нескольких режимов дозирования ЕРО, которые обеспечивают указанный желаемый фармакодинамический ответ, на основании указанного фармакодинамического профиля, причем указанные один или несколько режимов дозирования ЕРО позволяют поддерживать, по меньшей мере, концентрацию ЕРО в сыворотке, превышающую содержание до введения в течение приблизительно 5-30 суток между введениями ЕРО.
(а) выбор одного или нескольких желаемых фармакодинамических ответов;
(b) применение фармакокинетической/фармакодинамической модели для определения режимов дозирования ЕРО, которые обеспечивают указанный один или несколько фармакодинамических ответов; и
(c) отбор одного или нескольких режимов дозирования ЕРО, которые обеспечивают указанные желаемые фармакодинамические ответы, причем указанные один или несколько режимов дозирования ЕРО позволяют поддерживать, по меньшей мере, концентрацию ЕРО в сыворотке, превышающую содержание до введения в течение приблизительно 5-30 суток между введениями ЕРО.
(a) отбор дозы и режимов дозирования ЕРО, причем указанные один или несколько режимов дозирования ЕРО позволяют поддерживать, по меньшей мере, концентрацию ЕРО в сыворотке, превышающую содержание до введения в течение приблизительно 5-30 суток между введениями ЕРО; и
(b) определение указанного фармакодинамического ответа на основании указанной дозы и режимов дозирования.
(a) отбор одного или более желаемых фармакодинамических ответов;
(b) применение фармакокинетической/фармакодинамической модели для определения режима дозирования ЕРО, который обеспечивает указанный желаемый один или несколько фармакодинамических ответов;
(c) отбор указанных одного или нескольких режимов дозирования ЕРО, которые обеспечивают указанные желаемые фармакодинамические ответы, причем указанные один или несколько режимов дозирования ЕРО позволяют поддерживать, по меньшей мере, концентрацию ЕРО в сыворотке, превышающую содержание до введения в течение приблизительно 5-30 суток между введениями ЕРО; и
(d) назначение указанного выбранного режима дозирования ЕРО пациенту.
(a) выбор одного или нескольких режимов дозирования ЕРО;
(b) применение фармакокинетической/фармакодинамической модели для определения фармакокинетического профиля указанных одного или нескольких режимов дозирования ЕРО; и
(c) отбор одного или нескольких режимов дозирования ЕРО, которые обеспечивают указанный желаемый фармакокинетический ответ, на основании указанного фармакокинетического профиля, причем указанные один или несколько режимов дозирования ЕРО позволяют поддерживать, по меньшей мере, концентрацию ЕРО в сыворотке, превышающую содержание до введения в течение приблизительно 5-30 суток между введениями EPО.
(а) выбор одного или нескольких желаемых фармакокинетических ответов;
(b) применение фармакокинетической/фармакодинамической модели для определения режимов дозирования ЕРО, которые обеспечивают указанный желаемый один или несколько фармакокинетических ответов; и
(c) отбор одного или нескольких режимов дозирования ЕРО, которые обеспечивают указанные желаемые фармакокинетические ответы, причем указанные один или несколько режимов дозирования ЕРО позволяют поддерживать, по меньшей мере, концентрацию ЕРО в сыворотке, превышающую содержание до введения в течение приблизительно 5-30 суток между введениями ЕРО.
(a) выбор одного или нескольких режимов дозирования;
(b) применение фармакокинетической/фармакодинамической модели для определения фармакодинамического профиля указанных одного или нескольких режимов ЕРО;
(c) отбор указанных режимов дозирования ЕРО, которые обеспечивают желаемый фармакодинамический ответ, на основании указанного фармакодинамического профиля, причем указанные один или несколько режимов дозирования ЕРО позволяют поддерживать, по меньшей мере, концентрацию ЕРО в сыворотке, превышающую содержание до введения в течение приблизительно 5-30 суток между введениями ЕРО; и
(d) назначение указанного режима дозирования ЕРО пациенту.
(a) получение фармакокинетических данных у пациентов;
(b) выбор уравнения на основании указанных данных; и (с) подгонка указанных фармакокинетических данных под указанное уравнение.
(a) нормализацию значений концентрации ЕРО в сыворотке на основе указанных фармакокинетических данных; и
(b) создание профилей ЕРО в сыворотке по времени на основе указанных нормализованных данных.
(a) получение значений исходной концентрации ЕРО в сыворотке из указанных фармакокинетических данных посредством усреднения значений концентрации ЕРО в сыворотке перед введением дозы во множестве временных точек;
(b) получение значения концентрации ЕРО в сыворотке после подкожного введения ЕРО;
(c) получение нормализованных значений концентрации ЕРО в сыворотке посредством вычитания значений концентрации ЕРО до введения дозы ЕРО из значений концентрации ЕРО в сыворотке; и
(d) расчет средних нормализованных значений концентрации ЕРО в сыворотке в каждой временной точке.
(a) нормализацию концентраций ЕРО в сыворотке;
(b) получение фармакодинамических данных;
(c) получение уравнения на основании указанной модели; и
(d) подгонку указанных фармакодинамических данных под указанное уравнение для создания указанной фармакодинамической модели.
(a) получение исходной концентрации ЕРО в сыворотке (Cbs) для каждой группы дозирования усреднением значений концентрации ЕРО в сыворотке до введения дозы во множестве временных точек для каждой группы дозирования; и
(b) корректировку Cbs добавлением Сbs к концентрации ЕРО в сыворотке, предсказанной на фармакокинетической модели, где указанное скорректированное значение Cbs можно использовать в качестве вынуждающей функции для фармакодинамического анализа.
(a) определение среднего количества клеток-предшественников до введения дозы;
(b) определение среднего количества ретикулоцитов до введения дозы;
(c) определение среднего количества эритроцитов до введения дозы;
(d) определение средней концентрации гемоглобина до введения дозы;
(e) получение профилей среднего количества ретикулоцитов по времени в соответствии с дозой ЕРО;
(f) получение профилей среднего количества эритроцитов по времени в соответствии с дозой ЕРО; и
(g) получение профилей средней концентрации гемоглобина по времени в соответствии с дозой ЕРО.
(a) получение фармакокинетических данных;
(b) расчет площади под кривой зависимости концентрации от времени;
(c) нормализация площади под кривой зависимости концентрации от времени по дозе; и
(d) получение уравнения для того, чтобы представить указанную биодоступность ЕРО посредством выполнения линейной регрессии указанных фармакокинетических данных.
| RU 97108814 А, 10.05.1999 | |||
| US 5541158 A, 30.07.1996 | |||
| US 5674534 A, 07.10.1997 | |||
| СОЛОВЬЕВ В.Н | |||
| и др | |||
| Фармакокинетика, М., Медицина, 1980, с.184-188 | |||
| ХОЛОДОВ Л.Е | |||
| и др | |||
| Клиническая фармакокинетика, М., Медицина, 1985, с.69-82 | |||
| SALMONSON Т.Scand | |||
| J.Urol | |||
| Nephrol | |||
| Способ приготовления консистентных мазей | 1919 |
|
SU1990A1 |
| MCMAHON et al | |||
| Blood, 01 November, 1990, vol | |||
| Аппарат, предназначенный для летания | 0 |
|
SU76A1 |
| Приспособление к секрету ровничной кардной машины для грубой шерсти | 1919 |
|
SU1718A1 |
| GOBBURU et al | |||
| Adv | |||
| Drug | |||
| Delivery Reviews, 1998, vol | |||
| Способ сопряжения брусьев в срубах | 1921 |
|
SU33A1 |
| Способ изготовления замочных ключей с отверстием для замочного шпенька из одной болванки с помощью штамповки и протяжки | 1922 |
|
SU221A1 |
| PORT et al | |||
| Brit J | |||
| Clin | |||
| Pharmacology, November, 1998, vol | |||
| Способ изготовления звездочек для французской бороны-катка | 1922 |
|
SU46A1 |

