Перекрестная ссылка на родственную заявку
По данной заявке испрашивается приоритет в соответствии с предварительной заявкой США, серийный 60/128596, поданной 9 апреля 1999, описание которой включено здесь в качестве ссылки.
Область изобретения
Настоящее изобретение относится к водным фармацевтическим композициям эритропоэтина, свободным от продуктов сыворотки крови человека, стабилизированным аминокислотой и сорбитан моно-9-октадеценоат поли(окси-1,2-этандиил) производным. Настоящее изобретение также относится к водным стабильным, содержащим консервирующие добавки фармацевтическим композициям эритропоэтина, которые содержат антибактериальное количество крезола и аминокислоту.
Предпосылки изобретения
Эритропоэтин (ЕРО) представляет собой гликопротеиновый гормон, секретируемый в почках в ответ на тканевую гипоксию, который стимулирует выработку красных клеток крови в костном мозге (1). Ген ЕРО был клонирован и экспрессирован в клетках яичников китайских хомячков (2, 3). Этот рекомбинантный человеческий эритропоэтин (эпоэтин альфа, rhEPO) имеет аминокислотную последовательность, идентичную последовательности человеческого эритропоэтина мочи и, хотя существуют различия в гликозилировании белка, что влияет на эффективность in vivo, оба являются неотличимыми в функциональных и иммунологических анализах (4, 5).
В клинических испытаниях на сегодня rhEPO оценивали на здоровых субъектах, а также на пациентах с различными анемическими состояниями (6, 7). ЕРО стимулирует быстрый гематологический ответ у здоровых добровольцев, при условии адекватного поступления железа для поддержания усиленного синтеза гемоглобина (8). Большая часть испытаний изучала надежность и эффективность rhEPO при лечении пациентов с хронической почечной недостаточностью, находящихся на диализе, и больных, которым диализ еще не назначен. Другие показания, одобренные в США, включают вторичную анемию при химеотерапии рака и анемию, вызванную лечением зидовудином при инфекции вируса иммунодефицита человека. Во всем мире ЕРО используется для лечения анемии, связанной с ревматоидным артритом, преждевременными родами, миелофиброзом, с трансплантацией костного мозга, ожогами, серповидно-клеточной анемией, β-таласемией, для увеличения объема крови перед забором аутологичной донорской крови и используется в качестве вспомогательного средства перед хирургическим вмешательством (6, 7).
Хотя rhEPO обычно хорошо переносится, наблюдали редкие случаи кожных высыпаний и крапивницы, что предполагает наличие аллергической гиперчувствительности к некоторым компонентам композиции эпоэтина альфа, вероятно к альбумину сыворотки человека. Более того, несмотря на скрининг крови, если фармацевтический агент получают с использованием продуктов человеческой крови, существует риск заражения инфекционным агентом. Таким образом, необходимы фармацевтические композиции rhEPO, которые являлись бы стабильными и свободными от продуктов человеческой крови, таких как альбумин.
В патенте США 4992419 описаны лиофилизированные композиции эритропоэтина, содержащие 5-50 г/л мочевины, 1-50 г/л аминокислот, 0,05-5 г/л поверхностно-активных веществ и не содержащие альбумина сыворотки человека. Описываются водные композиции, но они показывают ограниченную стабильность по сравнению с композициями, содержащими альбумин сыворотки человека, и, таким образом, не являются коммерчески приемлемыми. Поэтому потребитель, либо врач, либо пациент, должен приготовить композицию непосредственно перед введением. Лиофилизированные композиции не являются предпочтительными среди клинических композиций эритропоэтина, так как процесс приготовления является длительным, создает риск плохой очистки белковой композиции, или приготовление может быть неправильным и для сохранения достаточной активности лекарственного препарата обычно требуются определенные добавки, такие как стабилизаторы. Следовательно, обычно предпочтительными являются водные композиции эритропоэтина.
В патенте США 5376632 описаны водные, лиофилизированные, или высушенные распылением композиции или β- или γ-циклодекстрины, содержащие эритропоэтин и не содержащие других дополнительных стабилизаторов, таких как альбумин сыворотки человека, альбумин бычьей сыворотки, лецитин, метилцеллюлоза, полиэтиленгликоль, серосодержащие восстанавливающие агенты, мочевина, аминокислоты и поверхностно-активные вещества (колонка 4, строки 51-54).
В патенте США 5661125 описаны водные композиции эритропоэтина, содержащие антибактериальные консерванты, такие как бензиловый спирт, парабены, фенолы и их смеси. Композиции, свободные от альбумина сыворотки крови человека, получали и исследовали на стабильность относительно соответствующих композиций, содержащих альбумин сыворотки крови человека. Результаты показали существенное осаждение эритропоэтина в HSA-свободных крезольных (0,5%) и хлоркрезольных (0,3%) композициях, даже при 0oС (столбец 8, строки 1-29), делая этих композиции, таким образом, непригодными для клинического применения. Более того, композиции ЕРО, содержащие крезол в области от 0,2-0,5%, описываются как имеющие слабое увеличение стабильности и более быстрое разрушение ЕРО (стоблец 6, строки 21-25). Коммерчески доступные мультидозированные композиции на настоящее время содержат 1% бензилового спирта.
Коммерчески доступные композиции rhEPO получают в цитратном буфере с активностью 1000 Ед/0,5 мл, 2000 Ед/мл, 5000 Ед/мл и 10000 Ед/мл (10К) как для внутривенной (вв), так и для подкожной (пк) инъекции. Эти композиции клинически подтверждены как непосредственная причина дискомфорта у пациентов, связанного с п.к. введением композиций с цитратным буфером (9, 10). Поскольку предпочтительное использование rhEPO переключилось с в.в. на п.к., скоро стало очевидно, что местное введение настоящего состава не является наилучшим. Ни нагревание лекарственного средства перед инъекцией до комнатной температуры, ни снижение объемов до не более 1 мл не было достаточно для предотвращения болезненных ощущений на месте инъекции. Ни человеческий альбумин, который используется в качестве стабилизатора в композиции, ни снижение осмотического давления раствора не оказалось причиной боли, предполагаемым кандидатом стал цитратный буфер rhEPO. Композиция без консервантов с единичной дозой 40000 Ед/мл является коммерчески полезной, если содержит фосфат натрия, а не цитратный буфер. Для стабильности необходима более низкая доза композиции, содержащая фосфатный буфер вместо менее предпочтительного цитратного буфера.
Краткое описание изобретения
Настоящее изобретение относится к фармацевтическим композициям эритропоэтина, содержащим: (а) рН буферирующий агент; (b) стабилизирующее количество сорбитан моно-9-октадеценоат поли(окси-1,2-этандиил) производного; (с) стабилизирующее количество аминокислоты и (d) фармацевтическое количество эритропоэтина, где композиция не содержит мочевину или продукта крови человека. Предпочтительные композиции в качестве стабилизирующих агентов включают комбинации полисорбата 80 и глицина. Особенно предпочтительные композиции в качестве стабилизирующих агентов включают комбинации полисорбата 80 и глицина в системе фосфатного буфера.
Настоящее изобретение также относится к фармацевтическим композициям эритропоэтина, содержащим: (а) рН буферирующий агент; (b) стабилизирующее количество альбумина сыворотки человека; (с) стабилизирующее количество аминокислоты; (d) антибактериальное количество крезола и (е) фармацевтическое количество эритропоэтина. Предпочтительные композиции в качестве стабилизирующих агентов включают комбинации альбумина человека и глицина. Особенно предпочтительные композиции в качестве стабилизирующих агентов включают комбинации альбумина сыворотки человека и глицина в системе фосфатного буфера с м-крезолом, используемым в концентрации около 0,3%.
Композиции по настоящему изобретению проявляют фармакокинетические свойства, такие как абсорбция, биодоступность, подобные современным фармацевтическим продуктам, и уровни концентрации сыворотки подобны коммерчески доступным в настоящее время фармацевтическим композициям рекомбинантного человеческого эритропоэтина. Более того, композиции по настоящему изобретению причиняют при введении меньший дискомфорт у пациента и обладают значительно более короткой длительностью дискомфорта в месте инъекции. Таким образом, настоящее изобретение относится к свободным от альбумина сыворотки крови человека или с консервирующими добавками фармацевтическим композициям эритропоэтина, которые могут использоваться вместо эритропоэтина и могут быть составлены для снижения дискомфорта у пациента.
Краткое описание чертежей
Фиг. 1 - графики: средняя концентрация эритропоэтина сыворотки - время (не скорректированы с основными уровнями эритропоэтина) у субъектов, получающих единичную 150 Ед/кг п.к. дозу rhEPO с консервантом, 0,3% м-крезолом, или без него. Концентрации сывороточного эритропоэтина определяли радиоиммунным анализом (РИА).
Фиг. 2 - графики: средняя концентрация эритропоэтина сыворотки - время (не скорректированы с основными уровнями эритропоэтина) у субъектов, получающих единичную 150 Ед/кг п.к. дозу rhEPO (2K) с новым стабилизатором или без него. Концентрации сывороточного эритропоэтина определяли радиоиммунным анализом (РИА).
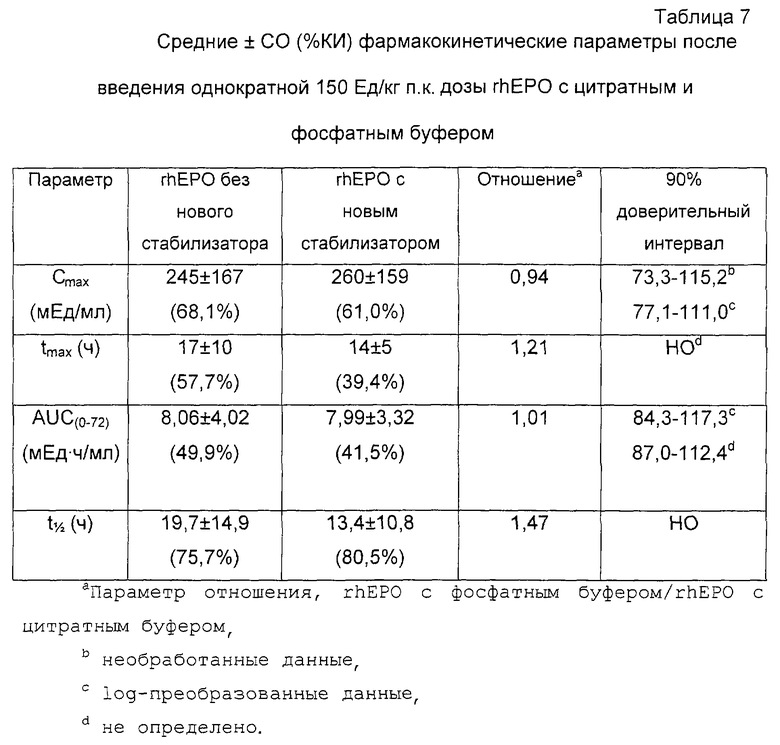
Фиг. 3 - графики: средняя концентрация эритропоэтина сыворотки - время (не скорректированы с основными уровнями эритропоэтина) у субъектов, получающих единичную 750 Ед/кг п.к. дозу rhEPO (40K) с новым стабилизатором или без него. Концентрации сывороточного эритропоэтина определяли с помощью (РИА).
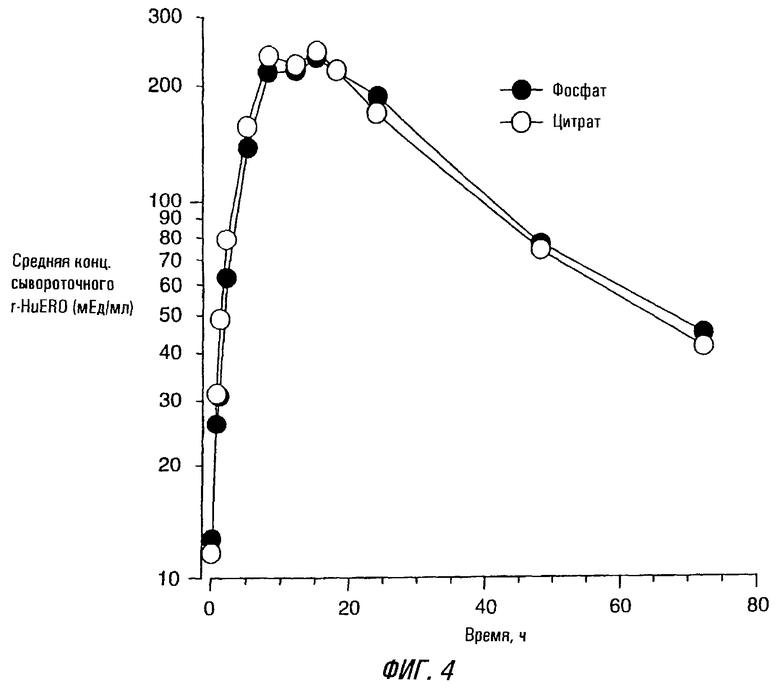
Фиг. 4 - графики: средняя концентрация эритропоэтина сыворотки - время (не скорректированы с основными уровнями эритропоэтина) у субъектов, получающих единичную 150 Ед/кг п.к. дозу rhEPO с цитратным и фосфатным буфером. Концентрации сывороточного эритропоэтина определяли радиоиммунным анализом (РИА).
Подробное описание
Настоящее изобретение относится к устойчивым водным фармацевтическим композициям эритропоэтина, которые содержат аминокислоты и сорбитан моно-9-октадеценоат поли(окси-1,2-этандиил) производные в качестве стабилизирующих агентов в буфере, свободном от продуктов крови человека или мочевины. Таким образом, настоящее изобретение относится к фармацевтическим композициям эритропоэтина, содержащим:
(a) рН буферирующий агент;
(b) стабилизирующее количество сорбитан моно-9-октадеценоат поли(окси-1,2-этандиил) производного;
(c) стабилизирующее количество аминокислоты;
(d) фармацевтическое количество эритропоэтина,
и где композиция не содержит мочевину или продукт крови человека.
Настоящее изобретение также охватывает водные композиции эритропоэтина с консервантом крезолом, стабилизированные комбинацией альбумина сыворотки крови человека и аминокислотой. Следовательно, настоящее изобретение относится к фармацевтическим композициям эритропоэтина, содержащим:
(a) рН буферирующий агент;
(b) стабилизирующее количество альбумина сыворотки крови человека;
(c) стабилизирующее количество аминокислоты;
(d) антибактериальное количество крезола и
(e) фармацевтическое количество эритропоэтина.
Одной из задач для данных композиций было минимизировать описанные в клинической практике случаи дискомфорта у пациента, связанные с подкожным введением композиций с цитратным буфером. Следовательно, во всех композициях по настоящему изобретению особенно предпочтительными являются системы фосфатного буфера.
Количество буферирующего агента, используемого в фармацевтических композициях по настоящему изобретению, в значительной степени зависит от конкретного используемого буфера и желаемого рН композиции. Предпочтительные области рН растворов находятся между около 5-8, более предпочтительные между около 6-7,5, а наиболее предпочтительным является значение рН, равное приблизительно 6,9. В соответствии с этими значениями рН количество буфера обычно находится в области от около 10 до около 30 мМ. Использование буферной системы из двухосновного фосфата натрия и одноосновного фосфата натрия является предпочтительным. Другие подходящие буферные системы для поддержания желаемой области рН от около 5 до около 8 включают, но ими не ограничиваются, цитрат натрия/лимонная кислота, ацетат натрия/уксусная кислота и любой другой фармацевтически приемлемый рН буферирующий агент, известный в данной области.
Может быть добавлен агент, устанавливающий рН, такой как, но ими не ограничивающийся, хлористоводородная кислота, лимонная кислота, гидроксид натрия или соль любого из них, в частности для этих целей может быть использован цитрат натрия.
Осмотический агент, используемый в композициях по настоящему изобретению, представляет собой любой агент, способный придать композициям по настоящему изобретению изоосмотичность с кровью человека. Обычные осмотические агенты широко известны в данной области и включают, но ими не ограничиваются, хлорид натрия, маннит, глицин, глюкозу и сорбит. В композициях по настоящему изобретению использование хлорида натрия в качестве осмотического агента является предпочтительным.
Сорбитан моно-9-октадеценоат поли(окси-1,2-этандиил) производные, включающие, но ими не ограничивающиеся, полисорбат 80 или полисорбат 20, являются примерами производных, используемых в качестве стабилизирующих агентов для предотвращения адсорбции эритропоэтина на поверхностях контейнеров, в которых находятся содержащие эритропоэтин композиции. В настоящем изобретении используется широкое разнообразие сорбитан моно-9-октадеценоат поли(окси-1,2-этандиил) производных, известных специалистам в данной области. Количество полисорбата 20 или 80, используемое в композициях по настоящему изобретению, находится в интервале от около 0,01 до около 1,0 мг на 1 мл для композиции, содержащей 1000-100000 Ед эритропоэтина на флакон. В композициях по настоящему изобретению использование полисорбата 80 является предпочтительным, при этом использование 0,3 мг/мл полисорбата 80 является более предпочтительным.
Аминокислоты, включающие, но ими не ограничивающиеся, глицин, L-изолейцин, L-лейцин, L-2-фенилаланин, L-глутамовую кислоту и L-треонин, используются в количествах от 0,1 до 50 г/л и предпочтительно в количествах от 0,25 до 20 г/л. L-Аланин и L-аргинин не являются предпочтительными в композициях по настоящему изобретению, так как их включение вызывает конформационные изменения в структуре ЕРО. Глицин является предпочтительной аминокислотой и используется предпочтительно в количестве от 0,25 до 20 г/л, в частности в количестве 0,5 г/л, в композициях, содержащих полисорбат 80 и не содержащих альбумина сыворотки крови человека. Глицин является предпочтительной аминокислотой, и в композициях, содержащих альбумин сыворотки крови человека и консервант крезол, он используется предпочтительно в количестве от 0,5 до 50 г/л, в частности 2,0 г/л. В композициях по настоящему изобретению, подходящей для использования является любая аминокислота, либо в L-, либо в D-изомерной форме, за исключением L-аргинина и L-аланина.
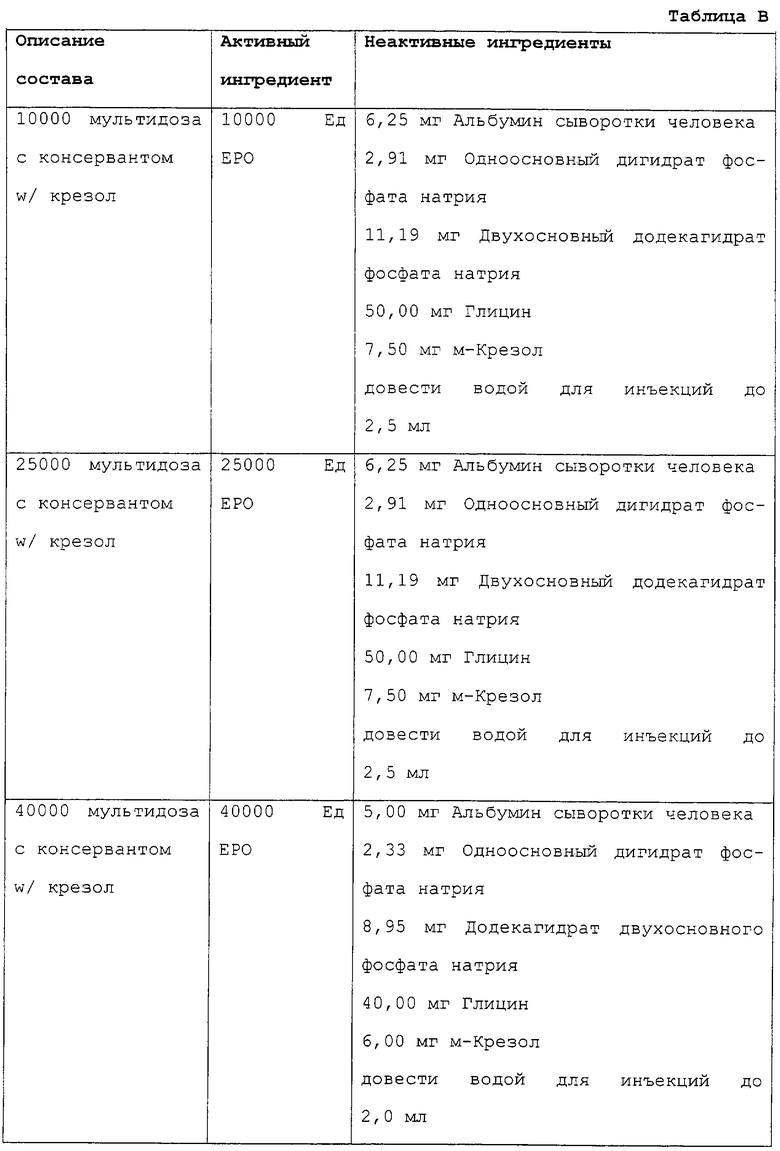
Наиболее предпочтительные композиции выбраны из группы, входящей в перечисленные в таблице А или таблице В.
Неожиданно было обнаружено, что водные композиции эритропоэтина, использующие систему фосфатного буфера, содержащие аминокислоту и сорбитан моно-9-октадеценоат поли(окси-1,2-этандиил) производное и не содержащие мочевину или продукт человеческой крови, представляют собой стабильные композиции. В частности, композиции, содержащие глицин и полисорбат 80, обеспечивают прекрасную и неожиданную стабильность по сравнению с предшествующими композициями, описанными в данной области.
Эритропоэтин находится в композициях в терапевтически эффективных количествах. Эритропоэтин может включать такие белки, которые имеют биологическую активность человеческого эритропоэтина, а также аналоги эритропоэтина, изоформы эритропоэтина, миметики эритропоэтина, фрагменты эритропоэтина, гибридные белки эритропоэтина, слитые белковые олигомеры и мультимеры вышеперечисленных белков, гомологи вышеперечисленных белков, гликозилированные примеры вышеуказанных белков и мутеины вышеперечисленных белков, независимо от их биологической активности и, более того, независимо от метода синтеза или промышленного получения, включая, но ими не ограничиваясь, рекомбинант, полученный из кДНК, либо из геномной ДНК, синтетическими, трансгенными и методами генной активации. Характерные примеры эритропоэтина включают эпоэтин альфа (EPREX®, ERYPO®), новый белок, стимулирующий эритропоэз (NESP) (гипергликозилированный аналог рекомбинантного человеческого эритропоэтина (Epoetin), описанный в европейской патентной публикации ЕР 640619), слитые белки аналога человеческого эритропоэтина - альбумина сыворотки человека, описанные в международной патентной публикации WO 9966054, мутанты эритропоэтина, описанные в международной патентной публикации WO 9938890, эритропоэтин омега, который может быть получен из ApaI рестрикционного фрагмента гена эритропоэтина человека, описанного в патенте США 5688679, измененный гликоэилированием человеческий эритропоэтин, описанный в международной патентной публикации WO 9911781, PEG конъюгированные аналоги эритропоэтина, описанные в WO 9805363 или патенте США 5643575. Характерные примеры клеточных линий, модифицированных для экспрессии эндогенного человеческого эритропоэтина, описаны в международных патентных публикациях WO 9905268 и WO 9412650.
Действие эритропоэтина может быть прослежено путем измерения гематокрита по отношению к целевому гематокриту, находящемуся в области 30-33%. Регулирование дозы может проводиться с помощью мониторинга гематокрита. Флаконы для однократного использования эритропоэтина содержат 2000, 3000, 4000, 10000 или 40000 единиц эритропоэтина (1 Ед соответствует приблизительно 8,4 нг рекомбинантного эритропоэтина). В виду того, что композиции в первом воплощении по настоящему изобретению содержат консерванты и представляют пользу, находясь в мультидозированном виде, композиции предпочтительно содержат многократно повторенное число единиц эритропоэтина, представленного в одноразовом флаконе. В настоящее изобретение включены композиции, содержащие 1000-100000 единиц или более эритропоэтина на флакон. Обычно предполагается, что эффективное количество находится от около 1 до 500 Ед/кг массы тела и более предпочтительно от 50 до 300 Ед/кг массы тела, особенно эритропоэтин вводимый п.к. Эффективное количество, более того, зависит от видов и размера субъекта, подвергающегося лечению, индивидуального состояния или заболевания, которое лечат, его серьезности и пути введения. В любом случае используемая доза должна быть нетоксична для пациента.
Неожиданно было обнаружено, что эритропоэтин может быть представлен в виде стабильной законсервированной водной композиции, содержащей <0,5% крезола и содержащей альбумин сыворотки крови человека и аминокислоты в качестве стабилизирующих агентов. Предпочтительные уровни крезола составляют от 0,2 до 0,5%, наиболее предпочтительный составляет 0,3%. Глицин является предпочтительным аминокислотным стабилизатором. Настоящее изобретение относится к фосфатбуферированным композициям, содержащим крезол в качестве консерванта, действующим с активностью 10000 Ед/2,5 мл, 25000 Ед/2,5 мл и 40000 Ед/2,0 мл (40К). Более высокая единичная доза композиции снижает число подкожных инъекций и улучшает переносимость пациентом.
Данные примера 1 по настоящему изобретению показывают, что использование цитрата или фосфата в качестве буфера и включение в композицию м-крезола не вызывают снижения характеристик абсорбции и биодоступности, а также параметров безопасности эритропоэтина. Таким образом, можно сделать вывод, что новые мультидозированные фосфатбуферированные композиции эритропоэтина с консервантами сопоставимы с выпускаемыми в настоящее время композициями для однократного использования, и мультидозированный эритропоэтин может быть использован при всех показаниях, принятых в настоящее время для эритропоэтина.
Будучи фармакокинетически сравнимой с однократно используемыми EPREX композициями, новая композиция, следовательно, предполагает также клиническое использование. Более того, мультидозированный фосфатбуферированный эритропоэтин обладает возможным дополнительным преимуществом по сравнению с однократно используемым эритропоэтином во флаконах; экономическим преимуществом. Использование эритропоэтина для лечения анемии или предепонирование аутологичной крови требует повторных введений гормона один раз каждые 4 недели, один раз каждые 2 недели, один раз в неделю, дважды в неделю или три раза в неделю. Предназначенная для одноразового использования любая неиспользованная часть однократно используемого эритропоэтина должна быть выброшена, тогда как, используя подходящее количество мультидозированного законсервированного эритропоэтина, количество выброшенного эритропоэтина будет минимизировано. Кроме того, бактерицидное свойство законсервированных композиций по настоящему изобретению делает их более подходящими для возможности самостоятельного введения и для лечения большого числа пациентов в стационарных условиях, что приносит существенные сбережения в затратах здравоохранения. Мультидозированные флаконы эритропоэтина также обеспечивают дополнительное удобство при введении лекарственного средства путем минимизации числа одноразовых флаконов и ампул, которые пациент/здравохранение использует при высоких объемах доз. Более высокая активность (25000 Ед/2,5 мл и 40000 Ед/2,0 мл) мультидозированного эритропоэтина может также минимизировать объемы инъекций, особенно в тех случаях, когда требуются высокие дозы. Мультидозированный эритропоэтин может также быть в виде одноразового флакона, используемого в качестве вводимой дозы. Например, рекомендуемая доза для программы донорской аутологичной крови составляет 600 Ед/кг дважды в неделю (7). Для пациента весом 70 кг рекомендуемый режим дозировки будет приблизительно 40К дважды в неделю. В этом случае одноразовый 40К сосуд может использоваться для разового введения лекарства.
Следующие примеры иллюстрируют настоящее изобретение, однако его не ограничивают.
Пример 1.
Для исследования примера 1, была выбрана композиция с активностью 10000 Ед, так как данная активность представляет собой приемлемую дозу, а также максимально рекомендуемый объем для п.к. инъекции, равный приблизительно 1 мл. Путем снижения объема инъекции высокая активность мультидозированного эритропоэтина потенциально дает возможность снижения точек подкожных инъекций, что несомненно дает преимущества для улучшения переносимости инъекций пациентом.
Цель: сравнение безопасности и фармакокинетических свойств подкожно вводимого рекомбинантного эритропоэтина человека (эпоэтин альфа, rhEPO), полученного в фосфатном буфере и содержащего 0,3% м-креазола в качестве консерванта (10000 Ед/мл) с коммерчески доступной цитратно-буферной композицией rhEPO (без консерванта).
Пациенты и методы
Пациенты
Восемнадцать здоровых добровольцев в возрасте от 20 до 38 лет (средний возраст 26,9 лет) и массой 60,8-87,4 кг (средняя масса 72,3 кг) были включены в испытание и полностью прошли его. Средний возраст субъектов (пациентов) группы I (28,1 лет) был чуть больше, чем таковой у субъектов группы II (25,8 лет), хотя не ожидалось, что это различие повлияет на результаты испытания. Включенные в испытания пациенты не имели клинически существенных патологических изменений в лабораторных исследованиях крови или биохимии сыворотки. У них были отрицательные показатели в отношении токсичности мочи, ВИЧ и поверхностных антигенов гепатита В. У них не было любого из ниже перечисленных случаев: гипертензии (например, диастолическое кровяное давление ≥95 мм рт. ст.); случаев любых первичных гематологических заболеваний; значимых случаев заболевания печени, почек, сердечно-сосудистой системы, желудочно-кишечной системы, мочеполовой системы, метаболического, неврологического заболевания; случаев анемии или припадков; известной чувствительности на продукты млекопитающих или к человеческому альбумину сыворотки; пристрастия и злоупотребления напитками, содержащими кофеин; участия в любом другом клиническом испытании, или в переливании крови, или в донорском заборе крови в течение 30 дней до начала испытания; действия rhEPO в течение трех месяцев до начала испытания; болезни в течение семи дней до начала испытания и значимых случаев ненормальных физических данных до испытания или клинических лабораторных данных в течение 14 дней до начала испытания. Все субъекты оценивались на безопасность и все заборы крови для фармакокинетических анализов проводились в соответствии с графиком испытания. Все испытания выполнялись при согласии комитета института по этике и самих пациентов.
Ход испытания
Испытание включало фазу I исследования одного центра, открытое, рандомизированное, перекрестное исследование с двумя периодами здоровых мужчин-добровольцев. Восемнадцать субъектов были случайным образом распределены в одну из двух групп последовательной обработки (девять субъектов/группа). rhEPO вводили в два независимых периода введения дозы в виде болюса п.к. инъекцией в верхнюю часть бедра. Каждый период введения дозы был разделен 14-дневным периодом вымывания препарата. Субъекты помещались в клинику, по крайней мере, за 12 часов до и находились до 72 часов после введения дозы в каждом из двух периодов введения дозы, но не между периодами. Режим дозирования приведен в таблице 1.
В данном испытании использовали две композиции ЕРО. Эпоэтин альфа (коммерчески доступный как EPREX®) содержит 10000 Ед/мл rhEPO в цитратном буфере. Композиция rhEPO с консервантом содержит 25000 Ед rhEPO в 2,5 мл фосфатного буфера с консервантом м-крезолом.
Забор образцов крови
Последовательные образцы крови получали с помощью непосредственного пунктирования вены перед и после введения ЕРО. Образцы венозной крови (5 мл) для определения концентраций эритропоэтина сыворотки получали за ~30, 20 и 10 минут до введения дозы (3 основных образца) и после введения дозы через приблизительно следующее время: через 30 минут и через 1, 2, 5, 8, 12, 15, 18, 24, 30, 36, 48, 60 и 72 часа. Каждый образец сыворотки разделяли на две аликвоты. Все образцы сыворотки хранили при -20oС. Образцы сыворотки транспортировали в сухом льду. Лабораторно-клинические исследования натощак (анализ крови, биохимия сыворотки и анализ мочи) проводили сразу до введения первой дозы в 1 день, на утро 4-го дня, сразу перед введением дозы на 16-й день и утром 19-го дня.
Биоаналитические методы
Для определения концентраций эритропоэтина сыворотки использовали методику набора для радиоиммунного анализа (РИА) (Diagnostic Systems Laboratory [DSL] , Webster TX). Коммерчески доступный РИА представляет собой метод двойных антител, конкурентный метод, который в качестве первичного антитела использует поликлональную иммунную сыворотку кролика к эритропоэтину мочи человека и в качестве метки использует 125I-меченный человеческий эритропоэтин мочи. В наборе калибраторов и образцах качественного контроля, представленных в DSL наборе эритропоэтин мочи заменяли эпоэтином альфа. Стандартные концентрации, используемые в методе, составляли 7,8, 15,6, 31,3, 50, 62,5, 100 и 125 мЕд/мл. Чувствительность, определенная как среднее из определений нижней границы стандарта, дающего приемлемую точность, составила 8,6 мЕд/мл и диапазон метода был расширен до 2000 мЕд/мл путем разведений образцов качественного контроля.
Определения безопасности
Основные показатели состояния организма регистрировались непосредственно перед введением каждой дозы (1 и 16 день) и через 6, 24, 48 и 72 часа после введения дозы. Определения безопасности основаны на изучении отклонений и изменений в клинических лабораторных испытаниях от основных и характере неблагоприятных событий. Кроме того, перед испытанием оценивались изменения основных показателей состояния организма, включая кровяное давление и результаты физического осмотра.
Анализ данных
Концентрации сыворотки после введения дозы были скоррегированы с основными концентрациями эритропоэтина до введения дозы путем вычитания из каждого значения концентрации сыворотки после введения средней основной концентрации эритропоэтина, определенной по усредненным уровням эритропоэтина из трех образцов, собранных за 30, 20 и 10 минут до введения дозы. Концентрации сыворотки эритропоэтина до введения дозы не брались в расчет средней величины, если они были ниже количественного уровня чувствительности анализа. Фармакокинетические параметры определяли по данным концентрации сыворотки, скоррегированными с основной концентрацией эритропоэтина.
Фармакокинетические параметры рассчитывали с помощью независимых методов на цифровом оборудовании операционной компьютерной системы VAX 8600, используя программу BIOAVL, версия 8,0, (Scientific Computer Systems, RWJPRI). Были определены следующие фармакокинетические параметры: пик концентрации сыворотки (Сmax); время для достижения пика концентрации сыворотки (tmax); площадь под кривой концентрация-время (AUC) от начала отсчета времени (ноль) до времени забора последнего образца крови (AUC0-72) подсчитывали с использованием формулы линейных трапеций и заключительный период полураспада (t1/2) определяли как 0,693/константа скорости распада. Константа скорости распада определялась линейной регрессией следующих друг за другом точек в заключительной линейной области логарифмического графика концентрации-времени. Для каждого случая обработки рассчитывали среднее квадратичное отклонение (СO) и средний коэффициент изменчивости (КИ) фармакокинетических параметров. Рассчитывали отношение средних параметров (композиция с консервантом/композиция без консерванта).
Результаты
Результаты безопасности
Доля неблагоприятных событий (явлений) была одинаково распределена среди обработанных групп (подвергнутых лечению) (39%, rhEPO без консерванта; 33%, rhEPO с консервантом). Не было обнаружено клинически существенных изменений показателей основных или лабораторно-клинических анализов до введения дозы или кровяного давления и никаких примечательных изменений в результатах осмотра физического состояния и в основных показателях состояния организма относительно измеренных до введения дозы. Интересно, что средние показатели общего билирубина уменьшились почти до половины основного значения к 4-му дню после терапии. Однако значение этого снижения было одинаковым в обеих обрабатываемых группах и средние уровни общего билирубина оставались в пределах нормы. Подобные изменения в других исследованиях функции печени (щелочная фосфатаза, ACT, АЛТ, ЛДГ) не отмечались. Таким образом, параметры безопасности для двух обрабатываемых групп оказались одинаковыми.
Фармакокинетические результаты
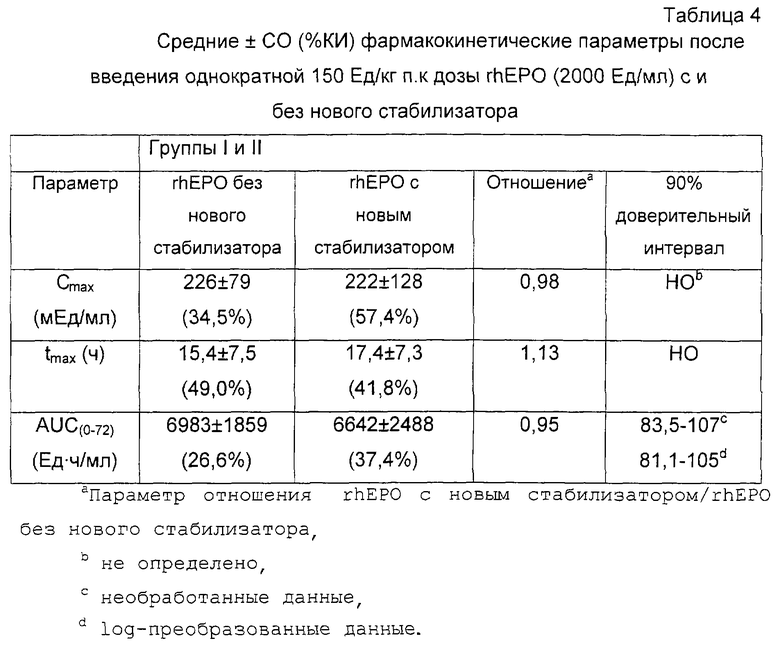
Графики средняя концентрация эритропоэтина сыворотки - время (не скоррегированы с основными уровнями эритропоэтина) у всех 18 субъектов после приема единичной дозы 150 Ед/кг п.к. rhEPO с и без консерванта м-крезола были соизмеримы в каждой эталонной точке времени (фиг. 1). Все субъекты имели основные концентрации эритропоэтина в пределах нормальных физиологических значений (<8,6-22 мЕд/мл).
Фармакокинетические параметры были определены из сывороточных данных, скоррегированных со средними основными концентрациями эритропоэтина до введения дозы (таблица 2). Значения Сmах находятся в области от низкого 85 мЕд/мл до высокого 558 мЕд/мл (среднее значение 198±123 мЕд/мл) для rhEPO с консервантом и от 61 мЕд/мл до высокого 601 мЕд/мл (среднее значение 226±138 мЕд/мл) для rhEPO без консерванта. Необходимо отметить, что два субъекта имели необычно высокие значения Сmах (то есть субъект 111 для rhEPO с консервантом [558 мЕд/мл] и субъект 118 для rhEPO без консерванта [601 мЕд/мл]) по сравнению с другими 16 субъектами (то есть <400 мЕд/мл). Когда значение Сmах было рассчитано без этих двух значений, среднее значение Сmах для rhEPO композиций с и без консерванта были более соизмеримы (177±89 мЕд/мл и 196±102 мЕд/мл соответственно).
Для композиции rhEPO с консервантом среднее значение эритропоэтина AUC(0-72ч) колеблющееся в области от 2973 до 11185 мЕд•ч/мл, было 5754±2284 мЕд•ч/мл. Точно так же композиция rhEPO без консерванта имела среднее значение эритропоэтина АUС(0-72ч) 6521±2769 мЕд•ч/мл, колеблющееся в области от 3096 до 14235 мЕд•ч/мл (таблица 2). Необходимо отметить, что два субъекта имели необычно высокие значения АUС(0-72ч) (то есть субъект 111 для rhEPO с консервантом [11185 мЕд•ч/мл] и субъект 118 для rhEPO без консерванта [14235 мЕд•ч/мл] ) по сравнению с другими 16 субъектами (то есть <10000 мЕд/мл). Когда значение АUС(0-72ч) рассчитывали без этих двух значений, среднее значение АUС(0-72ч) Для rhEPO композиций с и без консерванта были более соизмеримы (5460±1960 и 6000±2100 мЕд/мл соответственно).
Среднее tмax было одинаковым для rhEPO с и без консерванта (12±5 и 13±5 ч соответственно). Диапазон для tмах был одинаковым для обеих rhEPO композиций (8-24 ч). Заключительные значения периода полураспада были одинаковы для rhEPO с и без консерванта (20,2±8,1 и 19,8±6,6 ч соответственно).
Заключительные значения периода полураспада, наблюдаемые в обеих частях этого испытания (~20 ч), были сходны со значениями, полученными после п.к. инъекций, описанными в литературе (11, 12). Эти значения периода полураспада, однако, были выше чем, те которые наблюдали ранее после в.в. введения (~5 ч) (11-14). Эти более высокие значения периода полураспада не были неожиданными, в случае, если скорость абсорбции на месте инъекции, а не скорость элиминирования (выведения) являлась лимитирующим скорость фактором.
Хотя настоящее испытание проводилось на здоровых субъектах мужского пола, сходные характеристики абсорбции и графики безопасности можно ожидать в других популяциях больных, таких как мужчины и женщины, больные раком или страдающие хронической почечной недостойностью, дети с почечной недостаточностью, пациенты в программе предепонирования аутологичной крови или пациенты плановой хирургии с избирательной тактикой.
В итоге, подкожное введение единичных доз rhEPO (150 Ед/мл) с или без 0,3% м-крезола в качестве консерванта было безопасно и хорошо переносилось здоровыми субъектами мужского пола. Основываясь на сопоставимых случаях неблагоприятных событий, лабораторно-клинических показателях, основных показателях состояния организма и данных физического осмотра графики безопасности rhEPO композиций с или без консервантов были эквивалентны. Предложенная дробная концентрация мультидозированных EPREX потенциально обеспечивает большую клиническую пользу пациентам и поставщикам лекарственных средств.
Пример 2.
Сравнительная безопасность, переносимость и фармакокинетические свойства двух композиций рекомбинаного человеческого эритропоэтина с или без нового стабилизатора после единичного подкожного введения здоровому субъекту.
Цель: сравнить безопасность, переносимость и фармакокинетические свойства двух концентраций (2000 [самая низкая на данный момент] и 40000 Ед/мл [самая высокая на данный момент]) рекомбинантного человеческого эритропоэтина в фосфатном буфере (EPREX®, эпоэтин альфа, rhEPO), приготовленных с новым стабилизатором (глицин и полисорбат 80), с коммерчески доступными композициями EPREX®, которые в качестве стабилизатора используют альбумин сыворотки человека (HSA).
Пациенты и методы
Пациенты
Двадцать четыре добровольца в возрасте от 18 до 35 лет (средний возраст 21,9 лет) и массой между 57,2-92,3 кг (средняя масса 74,3 кг) принимали участие в первом испытании. Средний возраст двух групп последовательной обработки в первом испытании был практически одинаковым (21,8 и 22,0 лет). Двадцать три из двадцати четырех субъектов полностью прошли испытание. Один субъект прошел все оценки в первый период введения дозы, но решил не проходить второй период введения дозы. Данные о безопасности и переносимости этого субъекта, который получил только rhEPO без нового стабилизатора, были включены в данные о безопасности. Данные концентрации ЕРО сыворотки этого субъекта, однако, не были включены в фармакокинетический анализ.
Другие двадцать четыре добровольца в возрасте от 18 до 40 лет (средний возраст 23,8 лет) и массой между 62,2-93,4 кг (средняя масса 73,1 кг) были зарегистрированы и полностью прошли второе испытание. Средний возраст двух групп последовательной обработки был практически одинаковым (24,5 и 23,1 лет).
Все зарегистрированные субъекты в этих двух испытаниях имели показатель гемоглобина, не превышающий 16,0 г/дл; не имели клинически существенных патологических значений в лабораторных испытаниях крови или биохимии сыворотки; имели отрицательные показатели токсичности мочи, ВИЧ и поверхностных антигенов гепатита В и антител гепатита С. У субъектов не отмечалось случаев гипертензии (например, измеренное в сидячем положении диастолическое кровяное давление ≥95 мм рт. ст.); не было случаев любых первичных гематологических заболеваний; не было значимых случаев заболевания печени, почек, сердечно-сосудистой системы, желудочно-кишечной системы, мочеполовой системы, метаболического, неврологического заболевания; не было случаев анемии или припадков. Субъекты не имели аллергии на продукты млекопитающих или на HSA; не отмечали пристрастия и злоупотребления напитками, содержащими кофеин; не были злостными курильщиками (>10 сигарет/день); не принимали участия в любом другом клиническом испытании, или в переливании крови, или в донорском заборе крови в течение 90 дней до начала испытания; не подвергались действию rhEPO в течение трех месяцев до начала испытания. Все субъекты были оценены на безопасность и все сборы крови для фармакокинетических анализов проводились в соответствии с графиком испытания. Все испытания выполнялись при согласии комитета по этике и самих пациентов.
Ход испытания
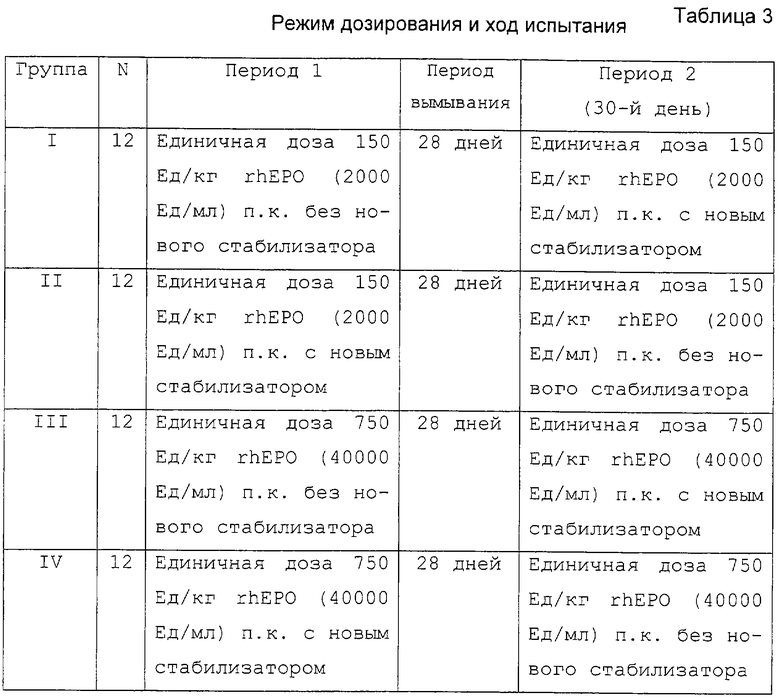
Оба испытания включали фазу 1, проводили испытания одного центра, двойное-слепое исследование, рандомизированное, перекрестное исследование с двумя периодами здоровых мужчин добровольцев. В каждом испытании 24 субъекта были случайным образом распределены в одну из двух групп последовательной обработки (12 субъектов/группа). ЕРО вводили в два независимых периода введения дозы в виде болюса п.к. инъекцией в верхнюю часть бедра. Субъектов помещали в клинику (исследовательский центр), по крайней мере, за 12 часов до и находились, по крайней мере, 36 часов после введения дозы в каждом из двух периодов введения дозы. Вне периода нахождения в исследовательском центре субъекты приходили в клинику для необходимых оценок и забора проб крови. Каждый период введения дозы был разделен 28-дневным периодом вымывания. Субъектов не помещали в клинику в период между введениями дозы (3-29 дней). Режим дозирования приведен в таблице 3.
В данном испытании использовали две композиции ЕРО. В первом испытании использовали композицию ЕРО с активностью 2000 Ед/мл (2К) с HSA в качестве стабилизатора и новую композицию rhEPO с активностью 2К в фосфатном буфере, содержащую глицин и полисорбат 80 в качестве нового стабилизатора. Во втором испытании использовали композицию ЕРО с активностью 40000 Ед/мл (40К) с HSA в качестве стабилизатора и новую композицию rhEPO с активностью 40К в фосфатном буфере, содержащую глицин и полисорбат 80 в качестве нового стабилизатора.
Забор образцов крови
Последовательные образцы крови получали с помощью непосредственного пунктирования вены перед и после введения ЕРО; гепариновая пробка не использовалась. Образцы венозной крови (5 мл) для определения концентраций эритропоэтина сыворотки получали за ~30, 20 и 10 минут до введения дозы (3 основных образца) и после введения дозы через приблизительно следующее время: 0,5, 1, 2, 5, 8, 12, 15, 18, 24, 30, 36, 48, 60 и 72 часа (все группы). У субъектов во втором испытании дополнительно брали образцы крови на 96, 120, 144 и 168 часы. Каждый образец сыворотки разделяли на две аликвоты. Все образцы сыворотки хранили при -20oС. Образцы сыворотки транспортировали в сухом льду. Лабораторно-клинические методы натощак (анализ крови, биохимия сыворотки и анализ мочи) проводили сразу до введения первой дозы в 1 день, на утро 4-го дня, сразу перед введением дозы на 30-й день и утром 33-го дня.
Биоаналитические методы
Анализы образцов на уровни сывороточного эритропоэтина выполняли в LabCorp. Для определения концентраций эритропоэтина сыворотки использовали набор методик для радиоиммунного анализа (РИА), производимый Diagnostic Systems Laboratory (DSL), Webster, TX. Коммерчески доступный РИА представляет собой метод двойных антител, конкурентный метод, который в качестве первичного антитела использует поликлональную иммунную сыворотку кролика к эритропоэтину мочи человека и в качестве метки использует 125I-меченный человеческий эритропоэтин мочи. DSL набор был изменен путем замещения эритропоэтина мочи на эпоэтин альфа в наборе калибраторов и образцах качественного контроля. Стандартные концентрации, используемые в методе, равнялись 7,8, 15,6, 31,3, 50, 62,5, 100 и 125 мЕд/мл. Чувствительность, определенная как среднее из определений нижней границы стандарта, дающего приемлемую точность, составила 7,8 мЕд/мл и диапазон метода был расширен до 5000 мЕд/мл путем разведений образцов качественного контроля.
Определение безопасности
Основные показатели состояния организма регистрировались непосредственно перед введением каждой дозы (1 и 30 дней) и через 6, 24, 30, 36, 48, 60 и 72 часа после введения каждой дозы. Определения безопасности основаны на изучении отклонений и изменений в клинических лабораторных испытаниях от основных (до 72 часов после введения дозы) и характере неблагоприятных событий. Кроме того, перед испытанием оценивались изменения основных показателей состояния организма, включая кровяное давление и результаты физического осмотра.
Определение переносимости
Испытания для оценки переносимости в месте инъекции поводили в течение 45 минут после каждой инъекции (1 и 30 дней). Поскольку каждая доза была разделена на 3, 4 или 5 инъекций, оценивалось наиболее болезненное место инъекций. Имелись две шкалы оценки боли: визуальная аналоговая шкала (VAS), состоящая из 10 см горизонтальной линии без градаций, и устная описательная шкала (VDS), состоящая из пяти вертикально помещенных ячеек со сходными описаниями. Конечные точки на VAS шкале находились в области от "нет болезненных ощущений", до "худшее из того, что можно представить" и эти же точки на шкале VDS находились в области от "нет боли" до "очень сильная боль". Субъекта затем спрашивали относительно продолжительности боли. Если боль продолжалась дольше 45 минут, испытания переносимости проводили через часовые интервалы до возвращения к норме.
Анализ данных
Концентрации сыворотки после введения дозы были скоррегированы с основными концентрациями эритропоэтина до введения дозы путем вычитания из каждого значения концентрации сыворотки после введения средней основной концентрации эритропоэтина, определенной по усредненным уровням эритропоэтина из трех образцов, собранных приблизительно за 30, 20 и 10 минут до введения дозы. Концентрации сыворотки эритропоэтина до введения дозы не брались в расчет средней величины, если они были ниже количественного уровня чувствительности анализа. Фармакокинетические параметры определяли по данным концентрации сыворотки, скоррегированным с основной концентрацией эритропоэтина.
Фармакокинетические параметры рассчитывали с помощью образцов независимых методов, используя программу WinNonlin, версия 1,1 (Scientific Consulting, Inc). Были определены следующие фармакокинетические параметры: пик концентрации сыворотки (Сmах); время для достижения пика концентрации сыворотки (tmax); площадь под кривой концентрация-время (AUC) от начала отсчета времени (ноль) до времени забора последнего образца крови AUC(0-72) [группы I и II] и AUC(0-168) [группы III и IV] подсчитывали с использованием формулы линейных трапеций; заключительный период полураспада (t1/2 определяли как 0,693/константа скорости распада. Константа скорости распада определялась линейной регрессией следующих друг за другом опорных точек в заключительной линейной области логарифмического графика концентрации-времени. В линейной регрессии использовали минимальное из трех опорных точек. Для тех, у кого коэффициент корреляции был меньше чем 0,975, соответствующие значения t1/2 не были эритропоэтизированы. Для каждого случая обработки рассчитывали среднее квадратичное отклонение (СО) и средний коэффициент изменчивости (КИ) фармакокинетических параметров. Рассчитывали отношение средних параметров (rhEPO с новым стабилизатором/rhEPO без нового стабилизатора).
Статистические исследования проводили и на необработанных, и на log-преобразованных параметрах биодоступности, AUC. В исследование различных моделей входили необработанные данные AUC и log-преобразованные данные AUC в качестве зависимой переменной, и эффекты в результате последовательной обработки группы, субъектов, находящихся в группах последовательной обработки, и промежуток времени в качестве независимой переменной. Исследование эффекта последовательной обработки группы проводили при уровне 10%, используя среднюю площадь находящихся в группах последовательной обработки субъектов в качестве вектора ошибок. Период времени эффекта был исследован на уровне 5%, используя остаточный вектор ошибок. Средняя площадь ошибок в вышеуказанной модели использовалась для оценки вариабельности субъектов в группе. Определенные наименьшие площади и вариабельность субъектов в группе в вышеуказанной модели использовались для создания 90% доверительных интервалов для отношения среднего EPREX® с новым стабилизатором/среднего HSA-содержащего EPREX®. Основываясь на 40% КИ, полученного в предыдущем исследовании, объем выборки из 24 субъектов должен быть достаточным для того, чтобы оценить отношение в пределах ±20% истинной величины с доверительностью 90%. Сmах, tmax и t1/2 как необработанных, так и преобразованных параметров были сообщены описательно.
В качестве незапланированных статистических исследований были проанализированы три параметра местной переносимости с помощью перекрестных методов (11). Перед проведением статистического исследования, множества VAS и значения продолжительности болезненных ощущений преобразовывали в нормированные. Перекрестные исследования для преобразования VAS и значения продолжительности болезненных ощущений проводили с помощью Proc glm в SAS®. Ход исследования предусматривал 28-дневный период вымывания для устранения возможности разброса между периодами введения дозы. Таким образом, вычисления были основаны на стандартной перекрестной модели без эффекта разброса. Результаты боли, оцененной VDS, анализировали с помощью критерия суммы рангов Уилкоксона-Манна-Уитни, согласно методам перекрестных проб, описанных Кохом и другими (12). Вкратце, данная методика включает в себя ранжирование периодов, различающихся у всех пациентов в эксперименте, и затем использование теста Уилкоксона-Манна-Уитни на различие между двумя группами последовательностей.
Результаты
Результаты безопасности
Доля всех неблагоприятных событий была одинаково распределена среди всех обработанных групп. Доля событий для композиций с активностью 2К составила 63% (rhEPO без нового стабилизатора) и 61% (rhEPO с новым стабилизатором), и для композиций с активностью 40К составила 54% (rhEPO без нового стабилизатора) и 46% (rhEPO с новым стабилизатором). Все неблагоприятные события были кратковременны и проходили без вмешательства, за исключением парацетамола, который давали пяти субъектам при умеренной головной боли и умеренных симптомах, подобных гриппу (три получающие 2К rhEPO и два получающие 40КБ rhEPO). Наиболее частым неблагоприятным явлением во всех группах, получавших ЕРО, была головная боль.
Как возможно связанные с приемом исследуемого лекарственного препарата неблагоприятные события были классифицированы у субъектов, принимающих композицию rhEPO с активностью 40К, на: шелушение кожи (n=4), головная боль (n= 2), миалгия (n=1) и усталость (n=l). У субъектов, принимающих композицию rhEPO с активностью 2К, неблагоприятные события, возможно связанные с приемом исследуемого лекарственного препарата, включают головную боль (n=2) и усталость (n= 2). Все четыре случая высыпания на коже, вызванные ЕРО, произошли между третьим и восьмым днями после введения rhEPO с новым стабилизатором; все случаи были умеренными и проходили за два или три дня. За исключением четырех сообщений о шелушении кожи не было клинически существенных различий в характере, и доля неблагоприятных событий отмечалась в любой из двух композиций при обеих концентрациях rhEPO. Не было обнаружено клинически существенных изменений в лабораторно-клинических анализах, кровяном давлении, результатах осмотра физического состояния и в основных показателях состояния организма, измеренных в течение первых четырех дней после введения любой из двух композиций исследуемого лекарства при обеих концентрациях rhEPO. Во всех обрабатываемых группах на 4-й день наблюдалось незначительное увеличение ретикулоцитов по сравнению с основным. Наряду с этим во всех группах на 4-й день имелось существенное уменьшение среднего количества общего билирубина по сравнению с основным. Минорные изменения показателей биохимии сыворотки наблюдались в течение четырех дней исследования; однако ни один не рассматривался как клинически значимый. Таким образом, параметры безопасности для всех обрабатываемых групп оказались одинаковыми.
Результаты переносимости
Средние значения VAS после единичной 150 Ед/кг п.к. дозы rhEPO (2K) с и без нового стабилизатора существенно не различались (p=0,608). Аналогично средние значения VAS после единичной 750 Ед/кг п.к. дозы rhEPO (40K) с и без нового стабилизатора существенно не различались (p=0,402). При оценке VDS тяжести боли посредством критерия суммы рангов Уилкоксона разность между этими двумя композициями существенно не различались (p=0,610) для субъектов групп I и II и для субъектов групп III и IV (p=0,581).
Общая средняя продолжительность боли при всех введениях лекарственного препарата составляла 1,01 минуты для субъектов групп I и II и 3,03 минуты для субъектов групп III и IV. Продолжительность боли, как сообщалось, длилась до трех минут или меньше для субъектов в группах I или II и до двух минут или меньше для субъектов в группах III и IV. Результаты продолжительности боли были согласованы с результатами, полученными для других параметров переносимости. Не наблюдалось статистически значимого различия в продолжительности боли после инъекции у субъектов, получавших EPREX® с и без нового стабилизатора, в группах I и II (р=0,159) и группах III и IV (р= 0,951).
Сравнение боли при двух различных концентрациях эпоэтина альфа показало, что боль наблюдалась после всех введений исследуемого лекарственного препарата (2К rhEPO) в группах I и II. Напротив, в группах III и IV субъектов (40К rhEPO) вообще не наблюдалось боли через 24-48 часов после п.к. введения (50%). Кроме того, средние значения боли (VAS, VDS) были большее после единичной 150 Ед/кг дозы rhEPO (2K), чем после единичной 750 Ед/кг дозы rhEPO (40K). Однако наблюдалась более высокая доля случаев боли, также как и высокое среднее значение боли, после единичной 150 Ед/кг дозы rhEPO (2K), чем после единичной 750 Ед/кг дозы rhEPO (40K). Это может быть связано с тем, что для достижения требуемой дозы в группах I и II было необходимо большее количество инъекций при введении исследуемого лекарственного средства из-за более слабой активности композиции (2К) по сравнению с используемой в группах III и IV (40К). Также необходимо отметить, что иголка, используемая при обоих введениях субъектам группы I и II, была более тонкая и более гибкая, чем используемая у субъектов группы III и IV, что делало более трудным проведение п. к. инъекций. Наконец, средняя продолжительность боли была дольше в группах III и IV (40К), чем в группах I и II (2К), но это было вероятно из-за двух субъектов, получавших 40К rhEPO, у которых наблюдалась самая длительная боль после обеих обработок, по сравнению со всеми субъектами во всех обрабатываемых группах.
Фармакокинетические результаты
Графики средняя концентрация эритропоэтина сыворотки - время у всех субъектов, получавших либо единичную 150 Ед/кг п.к. дозу rhEPO (группы I и II) или единичную 750 Ед/кг п.к дозу rhEPO (группы III и IV) с или без нового стабилизатора представлены на фиг.2 и 3 соответственно (не скоррегированы с основными уровнями эритропоэтина). Все субъекты (n=48) до введения дозы имели основные концентрации эритропоэтина в пределах нормальных физиологических значений (<7,8-32 мЕд/мл). Через 4 дня после введения единичной 150 Ед/кг п. к. дозы rhEPO (2K) с или без нового стабилизатора средние уровни эритропоэтина сыворотки (36±11 и 33±11 мЕд/мл соответственно) достигали средних уровней эндогенного эритропоэтина до введения дозы (16±6 и 16±7 мЕд/мл соответственно). Аналогично через 7 дней после введения единичной 750 Ед/кг п.к. дозы rhEPO (40K) с или без нового стабилизатора средние уровни эритропоэтина сыворотки (26±9 и 23±7 соответственно) также достигали средних уровней эндогенного эритропоэтина до введения дозы.
Фармакокинетические параметры были определены из сывороточных данных, скоррегированных со средними основными концентрациями эритропоэтина до введения дозы. В группах I и II значения Сmах находились в области от низкого 86,6 мЕд/мл до высокого 681 мЕд/мл (среднее значение 222±128 мЕд/мл) после введения единичной 150 Ед/кг п.к. дозы rhEPO (2K) с новым стабилизатором и от 119 мЕд/мл до высокого 377 мЕд/мл (среднее значение 226±79 мЕд/мл) для rhEPO без нового стабилизатора (таблица 4). Один субъект, получавший единичную 150 Ед/кг дозу rhEPO (2K) с новым стабилизатором, имел необычно высокое значение Сmах и один субъект той же группы имел необычно низкое значение Сmах. Причины таких необычных значений Сmах не известны. В группах III и IV значения Сmах находились в области от низкого 1194 мЕд/мл до высокого 4334 мЕд/мл (среднее значение 2978±808 мЕд/мл) после введения единичной 750 Ед/кг п. к. дозы rhEPO (40K) с новым стабилизатором и от 1892 мЕд/мл до высокого 3997 мЕд/мл (среднее значение 3065±705 мЕд/мл) для rhEPO без нового стабилизатора (таблица 5). Не было обнаружено статистически значимых различий значений Сmах (р<0,05) в композициях rhEPO обеих активностей 2К и 40К с или без нового стабилизатора.
В группах I и II значения AUC(0-72) находились в области от низкого 3238 до высокого 11318 (среднее значение 6647±2488 мЕд•ч/мл) после введения единичной 150 Ед/кг п. к. дозы rhEPO (2К) с новым стабилизатором. Аналогично значения AUC(0-72) находились в области от низкого 4234 до высокого 10968 (среднее значение 6983±1855 мЕд•ч/мл) после введения единичной 150 Ед/кг п. к. дозы rhEPO (2K) без нового стабилизатора (таблица 4). В группах III и IV AUC(0-168) находилось в от низкого 48347 до высокого 136290 (среднее значение 102768±21500 мЕд•ч/мл) после введения единичной 750 Ед/кг п.к. дозы rhEPO (40K) с новым стабилизатором. Аналогично значения AUC(0-168) находились в области от низкого 69537 до высокого 136689 (среднее значение 104897±15781 мЕд•ч/мл) после введения единичной 750 Ед/кг п.к. дозы rhEPO (40K) без нового стабилизатора (таблица 5). Необходимо отметить, что эффект последовательной обработки был статистически значимым между rhEPO (40K) с новым стабилизатором и rhEPO (40K) без нового стабилизатора.
Среднее значение tмax было одинаковым после введения единичной 150 Ед/кг п. к. дозы rhEPO (2K) с и без нового стабилизатора (17,4±7,3 и 15,4±7,5 ч соответственно) (таблица 4). Подобно среднее значение tмax после введения единичной 750 Ед/кг п.к. дозы rhEPO (40K) с и без нового стабилизатора было 16,7±6,8 и 16,6±4,8 ч соответственно (таблица 5). Значения tмax находились в области от 8-30 часов для обеих 2К композиций rhEPO. Для концентраций 40000 Ед/мл rhEPO tмax находился в области 8-36 часов для композиций с новым стабилизатором и 12-30 часов без нового стабилизатора.
Хотя сообщалось только о небольшом проценте субъектов групп I и II, имеющих заключительные значения периода полураспада, наблюдаемые значения были сходными после введения единичной 150 Ед/кг п.к. дозы rhEPO (2K) с и без нового стабилизатора (19,7±12,8 и 20,1±10,4 ч соответственно). После введения единичной 750 Ед/кг п.к. дозы rhEPO (40K) с или без нового стабилизатора у субъектов групп III и IV значения периода полураспада были сходны (25,7±14,9 и 23,8±11,8 ч соответственно).
EPREX® композиция с глицином и полисорбатом 80, в качестве нового белкового стабилизатора, представляет собой альтернативный состав для пациентов и производителей лекарственных препаратов, и включает в себя все признаки, одобренные в настоящее время для EPREX®. Различные активности ЕРО (то есть 2К, 4К, 10К и 40К) с новым стабилизатором могут взаимозаменяемо использоваться со всеми различными активностями HSA-содержащими ЕРО (то есть 2К, 4К, 10К и 40К).
Пример 3.
Исследование фармакокинетических свойств, безопасности и переносимости двух композиций рекомбинаного человеческого эритропоэтина после единичного подкожного введения здоровому субъекту.
Цель: сравнить фармакокинетические свойства, переносимость и безопасность подкожного введения единичной дозы двух композиций рекомбинантного человеческого эритропоэтина (эпоэтин альфа, rhEPO) в фосфатном или цитратном буфере и оценить значение буферного компонента в индуцировании боли.
Пациенты и методы
Пациенты
Восемнадцать здоровых добровольцев, находящихся в возрасте от 18 до 40 лет (средний возраст 27 лет), и массой между 62,6-82,2 кг (средняя масса 70,1 кг) были включены в испытание и полностью прошли это испытание. Включенные в испытание субъекты не имели клинически существенных патологических изменений в лабораторных исследованиях крови или биохимии сыворотки; имели отрицательные показатели в отношении токсичности мочи, ВИЧ и поверхностных антигенов гепатита В. У субъектов не отмечалось случаев гипертензии (то есть диастолическое кровяное давление >95 мм рт.ст.); не было случаев любого первичного гематологического заболевания; не было случаев существенного заболевания печени, почек, сердечно-сосудистой системы, желудочно-кишечной системы, мочеполовой системы, метаболического, неврологического заболевания; не было случаев анемии или припадков; известной чувствительности на продукты млекопитающих или к человеческому альбумину сыворотки; случаев злоупотребления алкоголем или наркотиками на протяжении прошедших двух лет; субъекты не принимали участия в любом другом клиническом испытании, или в переливании крови, или в донорском заборе крови в течение 30 дней до начала испытания; субъекты не подвергались действию rhEPO в течение трех месяцев до начала испытания; субъекты не имели существенных отклонений физических данных до испытания или клинических лабораторных данных в течение 14 дней до начала испытания. Все субъекты оценивались на безопасность и все заборы крови для фармакокинетических анализов проводились в соответствии с графиком испытания. Все испытания выполнялись при согласии комитета по этике и самих пациентов.
Ход испытания
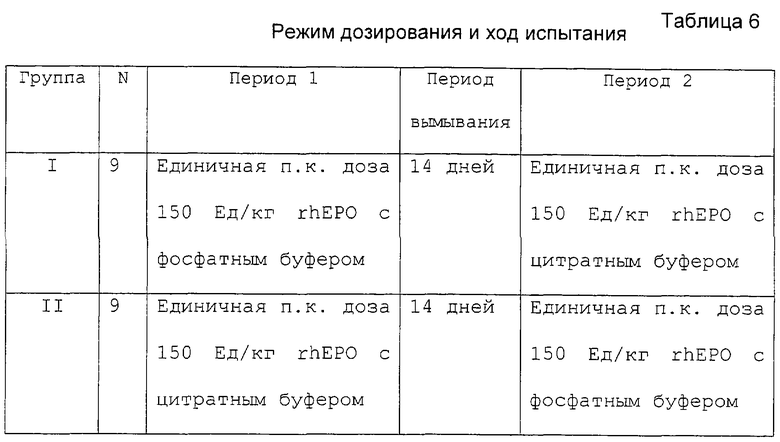
Испытание включало фазу I, исследования одного центра, открытое, рандомизированное, перекрестное исследование с двумя периодами здоровых мужчин добровольцев. Восемнадцать субъектов были случайным образом распределены в одну из двух групп последовательной обработки (девять субъектов/группа). rhEPO вводили в два независимых периода введения дозы в виде болюса п.к. инъекцией в верхнюю часть бедра. Каждый период введения дозы был разделен 14-дневным периодом вымывания. Субъекты помещались в клинику, по крайней мере, за 12 часов до и находились до 72 часов после введения дозы в каждом из двух периодов введения дозы, но не между периодами. Режим дозирования приведен в таблице 6.
В данном испытании использовали две композиции ЕРО. Эпоэтин альфа (коммерчески доступен как EPREX® 10000 Ед/мл) готовили в цитратном буфере и новую композицию rhEPO (10000 Ед/мл) готовили в фосфатном буфере. Обе композиции были приготовлены в Ortho Biologies Division of Ortho McNeil Janssen Pharmaceuticals, Inc. (Manati, Puerto Rico).
Забор образцов крови
Последовательные образцы крови получали с помощью непосредственного пунктирования вены перед и после введения ЕРО; гепариновая пробка не использовалась. Образцы венозной крови (5 мл) для определения концентраций эритропоэтина сыворотки получали за ~30, 20 и 10 минут до введения дозы (3 основных образца) и после введения дозы через приблизительно следующее время: 30 минут и 1, 2, 5, 8, 12, 15, 18, 24, 48 и 72 часа. Каждый образец сыворотки разделяли на две аликвоты. Все образцы сыворотки хранили при -20oС.
Образцы сыворотки транспортировали в сухом льду. Лабораторно-клинические методы натощак (анализ крови, биохимия сыворотки и анализ мочи) проводили сразу до введения первой дозы в 1 день, на утро 2-го дня, сразу перед введением дозы на 16-й день и утром 17-й день.
Биоаналитические методы
Анализы образцов на уровни сывороточного эритропоэтина выполняли в RWJPRI. Для определения концентраций эритропоэтина сыворотки использовали методику набора для радиоиммунного анализа (РИА) (Diagnostic Systems Laboratory [DSL], Webster, TX). Коммерчески доступный РИА представляет собой метод двойных антител, конкурентный метод, который в качестве первичного антитела использует поликлональную иммунную сыворотку кролика к эритропоэтину мочи человека и в качестве метки использует 125I-меченный человеческий эритропоэтин мочи. DSL набор был изменен путем замещения эритропоэтина мочи на эпоэтин альфа в наборе калибраторов и образцах качественного контроля. Стандартные концентрации, используемые в методе, равнялись 7,8, 15,6, 31,25, 50, 62,5, 100 и 125 мЕд/мл. Чувствительность, определенная как среднее из определений нижней границы стандарта, дающего приемлемую точность, составлила 8,6 мЕд/мл и диапазон метода был расширен до 2000 мЕд/мл путем разведений образцов качественного контроля.
Определение безопасности
Основные показатели состояния организма регистрировались непосредственно перед введением каждой дозы (1 и 16 день) и через 6, 24, 48 и 72 часа после введения дозы. Определения безопасности основаны на изучении отклонений и изменений в клинических лабораторных испытаниях от основных и характере неблагоприятных событий. Кроме того, перед испытанием оценивались изменения основных показателей состояния организма, включая кровяное давление и результаты физического осмотра.
Определение переносимости
Испытания для оценки переносимости в месте инъекции поводили в течение 45 минут после каждой инъекции в день введения. Имелись две шкалы оценки боли: визуальная аналоговая шкала (VAS), состоящая из 10 см горизонтальной линии без градаций и устная описательная шкала (VDS), состоящая из пяти вертикально помещенных ячеек со сходными описаниями. Конечные точки на VAS шкале находились в области от "нет болезненных ощущений", до "худшее, что можно представить" и эти же точки на школе VDS находились в области от "нет боли" до "очень сильная боль". Кроме того, субъекта спрашивали о продолжительности боли.
Анализ данных
Концентрации сыворотки после введения дозы были скоррегированы с основными концентрациями эритропоэтина до введения дозы путем вычитания из каждого значения концентрации сыворотки после введения средней основной концентрации эритропоэтина, определенной по усредненным уровням эритропоэтина из трех образцов, собранных за 30, 20 и 10 минут до введения дозы. Концентрации сыворотки эритропоэтина до введения дозы не брались в расчет средней величины, если они были ниже количественного уровня чувствительности анализа. Фармакокинетические параметры определяли по данным концентрации сыворотки, скоррегированным с основной концентрацией эритропоэтина.
Фармакокинетические параметры рассчитывали с помощью независимых методов на цифровом оборудовании операционной компьютерной системы VAX 8600, используя программу BIOAVL, версия 8,0 (Scientific Computer Systems, RWJPRI). Были определены следующие фармакокинетические параметры: пик концентрации сыворотки (Сmах); время для достижения пика концентрации сыворотки (tmax); площадь под кривой концентрация-время (AUC) от начала отсчета времени (ноль) до времени забора последнего образца крови (AUC0-72) подсчитывали с использованием формулы линейных трапеций; заключительный период полураспада (t1/2) определяли как 0,693/константа скорости распада. Константа скорости распада определялась линейной регрессией следующих друг за другом опорных точек в заключительной линейной области логарифмического графика концентрации-времени. Минимальное из трех точек значений использовали в регрессии. Для таких регрессий с коэффициентами с формулой (r2)<0,95, соответствующие значения t1/2 не сообщались. Для каждого случая обработки рассчитывали среднее квадратичное отклонение (СO) и средний коэффициент изменчивости (КИ) фармакокинетических параметров. Рассчитывали отношение средних фармакокинетических параметров (фосфат/цитрат).
Статистические исследования проводили и на необработанных и на log-преобразованных параметрах биодоступности. В исследование различных моделей входили данные с одним из интересующих параметров биодоступности (AUC, Cmax - и необработанные и log-преобразованные) в качестве зависимой переменной и эффекты в результате последовательной обработки группы, субъектов, находящихся в группах последовательной обработки, и промежуток времени в качестве независимой переменной. Исследование эффекта последовательной обработки группы проводили на уровне 10%, используя среднюю площадь находящихся в группах последовательной обработки субъектов в качестве вектора ошибок. Период времени эффекта был исследован на уровне 5%, используя остаточный вектор ошибок. Средняя площадь ошибок в вышеуказанной модели использовалась для оценки вариабельности субъектов в группе. Определенные наименьшие площади и вариабельность субъектов в группе в вышеуказанной модели использовались для создания 90% доверительных интервалов для отношения средних параметров биодоступности rhEPO с фосфатным буфером к rhEPO с цитратным буфером.
Статистические исследования проводились на трех параметрах устойчивости. Перед проведением статистического исследования множества VAS и значения продолжительности болезненных ощущений преобразовывали в нормированные. Все три параметра анализировались с помощью средних значений перекрестных исследований. Перекрестные исследования для преобразования VAS и значения продолжительности болезненных ощущений проводили с помощью Proc glm в SAS®. Ход исследования предусматривал 14-дневный период вымывания для устранения возможности разброса между периодами введения дозы. Таким образом, вычисления были основаны на стандартной перекрестной модели без эффекта разброса. Результаты боли, оцененной VDS, анализировали с помощью критерия суммы рангов Уилкоксона-Манна-Уитни, согласно методам перекрестных проб, описанных Кохом и другими. Вкратце, данная методика включает в себя ранжирование периодов, различающихся у всех пациентов в эксперименте, и затем использование теста Уилкоксона-Манна-Уитни на различие между двумя группами последовательностей.
Результаты
Результаты безопасности
В течение обоих периодов введения дозы наблюдались шесть клинических неблагоприятных случаев (AEs): два AEs наблюдались у двух субъектов (11%) после введения композиции с цитратным буфером и четыре AEs наблюдались у четырех субъектов (22%) после введения композиции лекарственного препарата с фосфатным буфером. Два сообщенных AEs случая после инъекции композиции с цитратным буфером затрагивали систему органов дыхания (то есть фарингит и синусит). Четыре сообщенных AEs случая после инъекции композиции с фосфатным буфером включали: судороги мышцы, головную боль, боль в ногах и дерматит. Все группы AEs случаев были классифицированы как умеренные и кратковременного характера.
Снижение концентраций общего билирубина наблюдалось у всех субъектов после обеих инъекций обоих составов. Средний общий билирубин уменьшился почти в половину от основного значения до введения дозы в течение двух дней наблюдения. Однако величина этого снижения была подобна в обеих обрабатываемых группах и средние уровни общего билирубина остались в пределах нормального диапазона. Это снижение казалось кратковременным, в виду того что два основных уровня до введения дозы (промежуток 16 дней) были весьма сопоставимы. Подобные изменения в других исследованиях функции печени (щелочная фосфатаза, ACT, АЛТ, ЛДГ) не отмечались.
За исключением неожиданного уменьшения в общем билирубине сыворотки, при общем физическом осмотре, в клинических лабораторных исследованиях, кровяном давлении и основных показателях состояния организма не имелось клинически существенных случаев в течение первых двух дней после введения лекарственного препарата. Таким образом, параметры безопасности для двух обрабатываемых групп оказались одинаковыми.
Результаты переносимости
У двух пациентов, получающих композицию с цитратным буфером, были пропущены записи боли в течение каждого периода введения. Среднее значение VAS для всех 18 фосфатных инъекций было 21,5±27,5 мм, по сравнению с 34,3±28,6 мм для всех 16 цитратных инъекций (p=0,0692). Принимая во внимание, что болевые ощущения после фосфатного раствора не зависели от периода инъекции, боль после цитратного состава была более интенсивна в течение периода I, чем в течение периода II (39,3±31,7 мм против 29,4±26,3 мм).
Различие между этими двумя составами в VDS переносимости было статистически существенным (p= 0,0339). Тринадцать субъектов (72%) либо не ощущали боли, либо ощущали умеренную боль и пять субъектов (28%) имели среднюю/интенсивную/очень сильную боль после введения состава с фосфатным буфером. Соответствующие наблюдения после введения состава с цитратным буфером были сделаны у 8 (44%) и 10 (56%) субъектов соответственно. Кроме того, VDS ощущение боли воспринималось как слегка более интенсивное, когда последовательность обработки была цитратом/фосфатом.
Продолжительность боли была существенно дольше (p=0,0057) после составов с цитратным буфером (1,5±2,0 минуты), чем после состава с фосфатным буфером (0,4±0,5 минут). Принимая во внимание, что различия были очевидны уже в течение периода I (среднее значение 0,8 против 0,3 минут), оно становится выраженным в течение периода II (среднее значение 2,2 против 0,5 минут), то есть когда цитратную композицию вводят субъекту, которому уже вводили композицию с фосфатным раствором. Необходимо отметить, что обе композиции лекарственного средства не регистрировались в течение периода I. В настоящем испытании было отмечено, что стандартная EPREX® композиция (с цитратом) вызывает более существенное усиление боли на месте инъекции, чем когда композиция использует в качестве буфера фосфат. Болевое ощущение, вызванное цитратом, было более интенсивно, когда испытания включали последовательность цитрат/фосфат в противоположность фосфат/цитратной последовательности, демонстрируя, что основные события опыта могут изменить ощущение боли. К тому же, касаясь продолжительности боли, различие между этими двумя композициями было очень существенно (p= 0,0057), однако, в этой последовательности вызванная цитратным буфером боль воспринималась гораздо дольше, чем последовательность фосфат/цитрат.
Фармакокинетические результаты
Графики средняя концентрация эритропоэтина сыворотки - время (не скоррегированы с основными уровнями эритропоэтина) у всех 18 субъектов после приема единичной дозы 150 Ед/кг п.к. rhEPO с цитратным или фосфатным буфером сходны (фиг. 4). Все субъекты имели основные концентрации эритропоэтина до введения дозы в пределах нормальных физиологических значений (<8,6-18 мЕд/мл).
Фармакокинетические параметры были определены из сывороточных данных, скоррегированных со средними основными концентрациями эритропоэтина до введения дозы (таблица 7). Значения Сmах находятся в области от низкого 74 мЕд/мл до высокого 583 мЕд/мл (среднее значение 260±169 мЕд/мл) для rhEPO с цитратным буфером и от 64 мЕд/мл до высокого 642 мЕд/мл (среднее значение 245±167 мЕд/мл) для rhEPO с фосфатным буфером. Несколько субъектов в каждой группе имели необычно высокие значения Сmах. Не наблюдалось статистически значимых различий (p>0,05) в значениях Сmах у двух композиций. Для композиции ЕРО с цитратным буфером среднее значение эритропоэтина AUC(0-72) было 7,992±13,319 мЕд•ч/мл, в пределах от 3,215 до 13,077 мЕд•ч/мл. Аналогично композиция rhEPO с фосфатным буфером имела среднее значение эритропоэтина AUC(0-72) 8,059±4,021 мЕд•ч/мл, в пределах от 3,445 до 17,996 мЕдoч/мл (таблица 7). Не имелось статистически существенного различия (р>0,05) в AUC(0-72) у этих двух составов. Среднее tmax было одинаковым у rhEPO с цитратным и фосфатным буфером (14±5 и 17±10 ч соответственно). Значения tmax были подобны для обеих композиций rhEPO (8-24 часа).
Средние заключительные значения периода полураспада были одинаковы для rhEPO с цитратным и фосфатным буфером (13,4±10,8 и 19,7±14,9 ч соответственно).
Ссылки
1. Koury ST, Bondurat MC. Koury MJ. (1988). Localization of erythropoietin synthesizing cells in murine kidneys by in situ hybridization. Blood 71:524-527.
2. Jacobs К, Shoemaker С, Rudersdorf R, Neill SD, Kaufman RJ, Mufson A. et al. (1985). Isolation and characterization of genomic and cDNA clones of human erythropoietin. Nature 313:806-810.
3. Lin FK, Suggs S, Lin Ck, Browne JK, Smalling R, Egrie JC et al. (1985). Cloning and expression of the human erythropoietin gene. Proc Nati Acad Sci USA 82:7580-7584.
4. Jelkmann W. (1992). Erythropoietin: structure, control of production, and function. Physiol Rev 72:449-489.
5. Egric JC, Browne JK, Lai P, Lin FK (1986). Characterization and biological effects of recombinant human erythropoietin. Immunobiology 172: 213-224.
6. Faulds D, Sorkin EM (1989). Epoetin (Recombinant Human Erythropoietin): A review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in anemia and the stimulation of erythropoiesis Drugs 38:863-899.
7. Markham A, Bryson HM (1995). Epoetin alfa: A review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in nonrenal applications Drugs 49:232-254.
8. Breymann С, Bauer С, Major A et al. (1996). Optimal timing of repeated rh-erythropoietin administration improves its effectiveness in stimulating erythropoiesis in healthy volunteers. Brit J Heamatol 92:295-301.
9. Granolleras С, Leskopf W, Shaldon S, Fourcade J (1991). Experience of pain after subcutaneous administration of different preparations of recombinant human erythropoietin: a randomized, double-blind crossover study. Clin Nephrol 36:294-298.
10. Frenken LA, Van Lier HJ, Jordans JG et al. (1993). Identification of the component part in an epoetin alfa preparation that causes pain after subcutaneous inject. Am J Kidney Dis 22:553-556.
11. Halstenson CE, Macres M, Katz SA et al. (1991). Comparative pharmacokinetics and pharmacodynamics of epoetin alfa and epoetin beta. Clin Pharmacol Ther 50:702-712.
12. Ateshkadi A, Johnson CA, Oxton LL, Hammond TG, Bohenek WS, Zimmerman SW (1993). Pharmacokinetics of intraperitoneal, intravenous, and subcutaneous recombinant human erythropoietin in patients on continuous ambulatory peritoneal dialysis. Am J Kidney Dis 21:635-642.
13. McMahon FG, Vargas R, Ryan M et al. (1990). Pharmacokinetics and effects of recombinant human erythropoietin after intravenous and subcutaneous injections in healthy volunteers. Blood 76:1718-1722.
14. Salmonson Т, Danielson BG, Wikstrom B. (1990). The pharmacokinetics of recombinant human erythropoietin after intravenous and subcutaneous administration to healthy subjects. Brit J clin Pharmacol 29:709-713.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАКОКИНЕТИЧЕСКОЕ И ФАРМАКОДИНАМИЧЕСКОЕ МОДЕЛИРОВАНИЕ ВВЕДЕНИЯ ЭРИТРОПОЭТИНА | 2000 |
|
RU2248215C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ hGLP-1, ЭКСЕНДИНА-4 И ИХ АНАЛОГОВ | 2007 |
|
RU2419452C2 |
| ЛЕЧЕНИЕ НЕВРОЛОГИЧЕСКОЙ ДИСФУНКЦИИ С ПРИМЕНЕНИЕМ СУЛЬФАМАТОВ ФРУКТОПИРАНОЗЫ И ЭРИТРОПОЭТИНА | 2002 |
|
RU2317086C2 |
| КОМПОЗИЦИИ ИНСУЛИНОВ ДЛИТЕЛЬНОГО ДЕЙСТВИЯ | 2011 |
|
RU2564104C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ПЛАЗМИНОГЕН, И ЕЕ ПРИМЕНЕНИЕ | 2015 |
|
RU2711989C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ВАЛСАРТАНА | 2008 |
|
RU2487710C2 |
| КОМПОЗИЦИИ ИНСУЛИНОВ ДЛИТЕЛЬНОГО ДЕЙСТВИЯ | 2015 |
|
RU2642662C2 |
| СПОСОБЫ И КОМПОЗИЦИИ, КОТОРЫЕ ОБЕСПЕЧИВАЮТ МОДУЛЯЦИЮ ИММУННЫХ ОТВЕТОВ, СВЯЗАННЫХ С ВВЕДЕНИЕМ БИОФАРМАЦЕВТИЧЕСКОГО ЛЕКАРСТВЕННОГО СРЕДСТВА | 2014 |
|
RU2662558C2 |
| СПОСОБ ЛЕЧЕНИЯ ОСТЕОПОРОЗА И ИСПОЛЬЗУЕМАЯ В НЕМ КОМПОЗИЦИЯ | 2007 |
|
RU2506070C2 |
| КОМБИНИРОВАННЫЕ ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ, СОДЕРЖАЩИЕ ЭРИТРОПОЭТИН И ПРЕПАРАТЫ ЖЕЛЕЗА | 1997 |
|
RU2188033C2 |





Изобретение относится к водным фармацевтическим композициям эритропоэтина, которые являются свободными от продуктов человеческой сыворотки, стабилизированным аминокислотой и сорбитан моно-9-октадеценоат поли(окси-1,2-этандиил) производным. Настоящее изобретение также относится к водным стабильным, содержащим консерванты фармацевтическим композициям эритропоэтина, которые содержат антибактериальные количества крезола и аминокислоту. Фармацевтические композиции стабильны и свободны от продуктов человеческой крови, таких как альбумин. 2 с. и 50 з.п. ф-лы, 4 ил., 9 табл.
| СПОСОБ ПОЛУЧЕНИЯ ИНЪЕКЦИОННОЙ ФОРМЫ ЭРИТРОПОЭТИНА | 1988 |
|
RU2043118C1 |
| ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ УВЕЛИЧЕНИЯ БИОЛОГИЧЕСКОЙ ДОСТУПНОСТИ АКТИВНОГО ВЕЩЕСТВА С ИСПОЛЬЗОВАНИЕМ ДАННОГО СОСТАВА | 1991 |
|
RU2104715C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРЕДСТАВЛЯЮЩАЯ МИКРОЭМУЛЬСИЮ С БИОЛОГИЧЕСКИ АКТИВНЫМ МАКРОМОЛЕКУЛЯРНЫМ МАТЕРИАЛОМ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1989 |
|
RU2122403C1 |
| US 4992419 А, 12.02.1991. | |||

