Изобретение относится к области молекулярной генетики и биотехнологии, конкретно к способам обнаружения ошибок спаривания оснований в нуклеиновых кислотах (точечных мутаций), что является основой для автоматизации молекулярно-эпидемиологических исследований различных заболеваний. Предлагаемое техническое решение касается использования фагового дисплея для поиска пептидов, экспонированных на поверхности фаговой частицы и проявляющих способность связываться с неспаренностями.
Точечные мутации лежат в основе многих серьезных многофакторных заболеваний, таких как рак, атеросклероз и т.д. Выявление полиморфизма генов и влияние точечных мутаций в медицине и генетике крайне актуально. С полиморфизмом генов связывают лекарственную устойчивость патогенных микроорганизмов, чувствительность организма-хозяина к инфекциям, к неблагоприятным факторам окружающей среды (радиации, химическим мутагенам), к эффективности вакцинации и т.п. Определение точечных мутаций в генах по многим локусам из большого количества образцов ДНК является основой молекулярно-эпидемиологических исследований различных заболеваний и требует создания быстрых и простых методов [1]. В связи с широким использованием однонуклеотидных полиморфных маркеров для обнаружения геномных локусов, влияющих на фенотипическую вариабельность различных признаков, таких как устойчивость к инфекциям, опухолям, неблагоприятному воздействию мутагенных факторов окружающей среды и т.п., особенно возрастает потребность в методах с высокой пропускной способностью, пригодных для автоматизации, а именно в связи с развитием технологии чипов.
Известны и широко используются способы, основанные на предварительной амплификации анализируемого фрагмента ДНК (или кДНК) в полимеразной цепной реакции (ПЦР) и конформационно обусловленных различиях в электрофоретической подвижности цепей ДНК с мутациями и без них. Одним из простых является способ использования одноцепочечного конформационного полиморфизма (SSCP, single-stranded conformational polymorphism) [2]. Этот способ заключается в том, что дикий и мутантный тип ДНК-мишени амплифицируют в ПЦР, денатурируют и после медленной ренатурации разделяют электрофорезом в ПААГ-геле в неденатурирующих условиях (обычно с добавлением глицирина). Одноцепочечная ДНК ренатурирует в трехмерную структуру в соответствии с первичной нуклеотидной последовательностью. Изменения в последовательности (вплоть до замены одного нуклеотида) влияют на третичную структуру и приводят к изменению подвижности образца в геле, что может быть визуализировано известными способами (окраской этидиум бромидом, серебром, введением радиоактивной или флюоресцентной метки). SSCP показывает высокую чувствительность при анализе небольших фрагментов ДНК (не более 200 п.о.); при увеличении длины фрагмента до 400 п.о. и более чувствительность способа резко падает, что является его основным недостатком.
Другим широко используемым способом является денатурирующий градиентный гель-электрофорез (DGGE-denaturing gradient gel electrophoresis), который также основан на различиях в миграции ДНК-фрагментов дикого и мутантного типов [3]. В отличие от SSCP, DGGE более трудоемок, так как после ПЦР и денатурации ДНК проводят перекрестное смешивание образцов для получения смеси гетеро- и гомодуплексов, которую анализируют по подвижности в геле с градиентом денатурирующего агента (мочевины). Оптимальным для анализа являются фрагменты длиной от 400-800 п.о. Кроме длины исследуемого фрагмента чувствительность способа также зависит от нуклеотидного окружения мутации, что требует дополнительных ухищрений, а именно - подбора определенных праймеров с повышенным содержанием G/C - пар нуклеотидов. Эти недостатки, а также необходимость применения дорогостоящей аппаратуры, ограничивают использование DGGE.
Иным подходом к определению ошибок спаривания оснований в нуклеиновых кислотах являются способы, основанные на химическом или ферментативном расщеплении двуспиральных фрагментов нуклеиновых кислот в месте локализации неспаренного основания в искусственно сформированном гетеродуплексе ДНК-ДНК (для химического) и РНК-ДНК (для ферментативного расщепления). Недостатком при использовании ферментативного расщепления является необходимость применения меченой РНК, которая является нестабильным субстратом, а сам способ по чувствительности не превышает 50%. Химическое расщепление не требует получения РНК, однако, для детекции расщепленной ДНК используется токсичный химический агент-пиридин [4]. Несмотря на высокую чувствительность (до 100%) и широкий диапазон анализируемых длин (до 1,7 тыс. п.о.), необходимость использования высокотоксических веществ значительно ограничивает использование данного способа в широких медико-генетических исследованиях, не говоря о широком внедрении в практику.
Известен способ обнаружения ошибочного спаривания одной или большего числа пар оснований путем расщепления резолвазой [5]. Способ предусматривает контакт эталонной и тестируемой нуклеиновых кислот в одноцепочечных формах для образования гетеродуплекса, который иммобилизуется на гелеподобном носителе, с резолвазой (например, Т4 эндонуклеазой VII), способной распознавать по меньшей мере одно ошибочное спаривание с последующим расщеплением до образования продуктов, анализируемых гель-электрофорезом. Недостатком данного способа является то, что на конечном этапе для получения результата требуется проведение электрофореза, что затрудняет полную автоматизацию процесса.
В настоящее время интенсивно изучается вопрос о возможности детекции неспаренных оснований в составе двуцепочечной ДНК с помощью ферментов системы репарации. В частности, показано, что белок MutS может специфически связываться с ДНК, содержащей неспаренные основания, а образовавшийся комплекс может детектироваться по изменению подвижности при электрофорезе [6, 7]. Основные проблемы, ограничивающие широкое использование Mut-белков, заключаются в низких значениях констант связывания (особенно к А/С паре), высокой чувствительности к конкретному нуклеотидному окружению и неспецифическом связывании с ДНК при различных вариантах первичной структуры.
Технической задачей изобретения является создание унифицированного (быстрого и простого) способа для определения однонуклеотидных замен (точечных мутаций) на дуплексе ДНК, использующего ДНК-белковые взаимодействия. При этом ключевую роль должна играть возможность быстрого подбора белкового лиганда, специфичного для нуклеотидных замен в конкретном геномном локусе, который будет подвергаться массовому анализу.
Перспективным направлением для решения этой проблемы может оказаться использование технологии "фагового дисплея", который становится наиболее распространенным подходом к исследованию механизма связывания белков с различными субстратами.
Возможность селекции фагов, связывающихся со специфическими фрагментами ДНК и РНК, из состава рекомбинантной библиотеки, содержащей случайные пептидные вставки в поверхностных фаговых белках, была продемонстрирована в ряде экспериментов по изучению взаимодействий белков с нуклеиновыми кислотами [8, 9].
Использование "фагового дисплея" для определения ошибочно спаренных оснований в нуклеиновых кислотах в литературе не описано.
Поставленная задача решается путем разработки стратегии отбора фагов, несущих пептидные аналоги функционально активных модулей белков репарационной системы, способных избирательно связываться с участками неспаренных оснований на двуцепочечных ДНК конкретного локуса, что является основой создания способа для обнаружения точечных мутаций в генах и изучения их полиморфизма. Стратегия основана на комбинации "фагового дисплея" с использованием биотинилированных праймеров для получения ПЦР-фрагментов и парамагнитных частиц со стрептавидином для физического разделения комплексов ДНК-фаг.
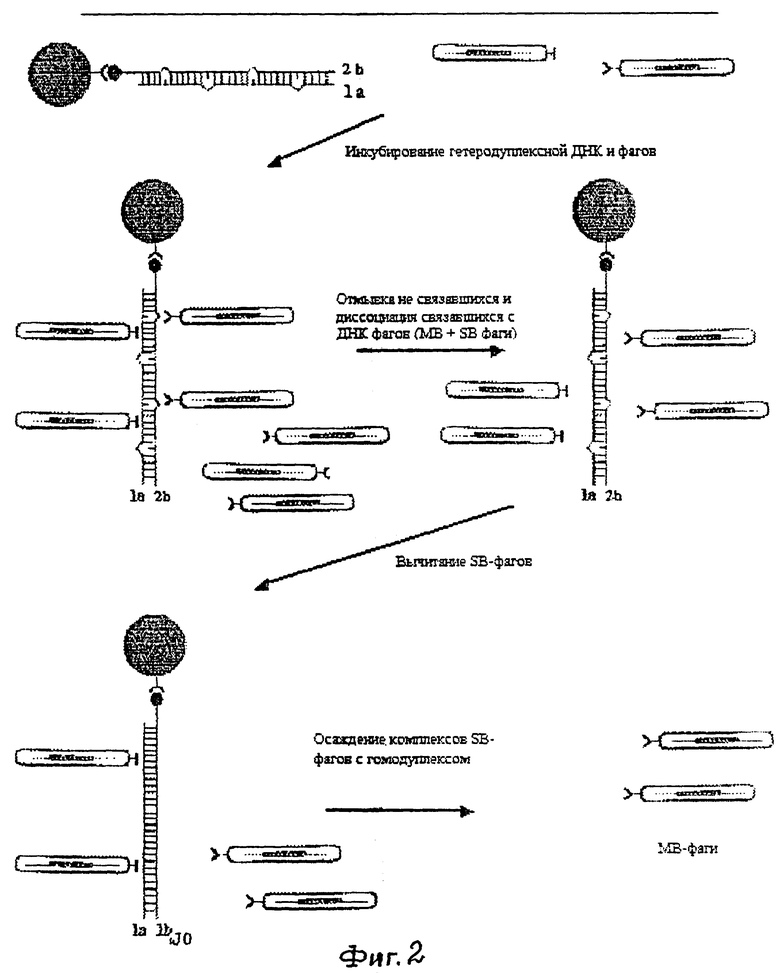
Сущность предлагаемого способа определения ошибочно спаренных оснований в ДНК (точечных мутаций) заключается в том, что на начальной стадии процесса получают одноцепочечную форму тестируемой ДНК (например, в ПЦР амплификации с последующим денатурированием) и эталонной нуклеиновой кислоты - гомологичный участок ДНК дикого типа (например, в ПЦР с последующим денатурированием или химически синтезированный олигонуклеотид), при этом реакцию амплификации проводят с праймерами, один из которых биотинилирован по 5'-концу; затем осуществляют контакт между цепочками и временную иммобилизацию полученного гетеродуплекса (например, с помощью магнитных микрочастиц Fе3O4, несущих на поверхности несколько молекул стрептавидина); иммобилизованный гетеродуплекс приводят в контакт с пептидами, экспонированными на поверхности фаговой частицы (например, со статистическими (рандомизированными) пептидами в составе фаговой библиотеки HPL); проводят полное удаление несвязавшихся с гетеродуплексами ("ненужных") фагов, реассоциацию связавшихся фагов и отделение их от гетеродуплекса (например, с помощью магнитного разделения с использованием системы "Dynal") и последующее повторное ("вычитающее") связывание реассоциированных фагов с гомодуплексом (например, неденатурированным гомологичным ПЦР-фрагментом ДНК дикого типа); в завершение проводят удаление связавшихся с гомодуплексом фаговых частиц вместе с гетеродуплексом (например, с использованием магнитной системы Dynal) от несвязавшихся, которые размножают на чувствительном штамме E.coli (например, Е. coli К 91).
Отселектированные подобным образом фаги избирательно (предпочтительно) связываются с ошибочно спаренными основаниями в составе любого гетеродуплекса. Для усиления чувствительности процедуру селекции (раунд) можно повторить. Полученные фаги могут быть иммобилизованы на различные носители и использоваться в полностью автоматизированных процессах (например, в чипах).
Изобретение иллюстрируется следующими графическими материалами.
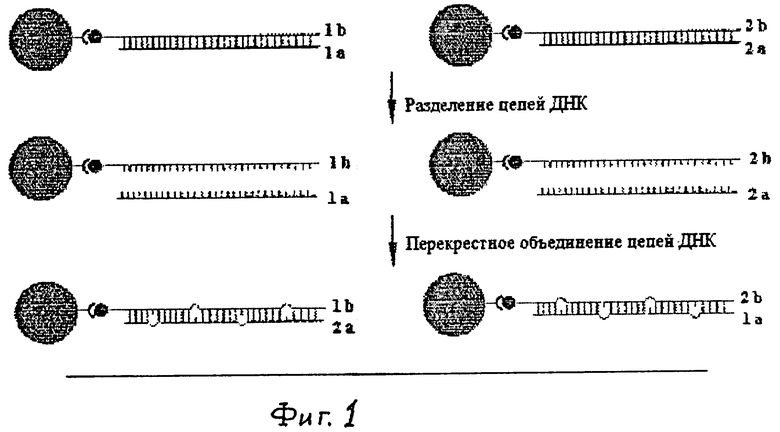
- Фиг.1. Схема получения гетеродуплексов.
- Фиг.2. Схема селекции фагов.
Для лучшего понимания сущности предлагаемого технического решения ниже следуют примеры его осуществления.
Пример 1. Получение тестируемой и эталонной ДНК в одноцепочечной форме с обеспечением последующего контакта между цепочками для получения временно иммобилизованного гетеродуплекса с неспаренными основаниями
Гетеродуплексы получают путем смешивания различных цепей фрагментов ДНК, отличающихся точечными несоответствиями (один как пример тестируемого, другой - эталонный), полученными в реакции амплификации с праймерами, один из которых биотинилирован с 5' конца. (Фиг.1). Праймеры имеют следующую структуру:
- Р5: 5'-*АСТ ССА ССА ТТА GCA ССС ААА G (биотинилированный)
- Н: 5'-TGA ТТТ САС GGA GGA TGG TG (небиотинилированный)
Состав реакционной смеси: буфер (75 мМ трис-HCl, 20 мМ (NH4)2SO4, 0,01% Twin-20, 4 мМ MgCl2); dATP, dGTP, dTTP, dCTP каждый 0,2 мМ; праймеры каждый 0,5 мкМ (5-10 рмоль); Taq - ДНК полимераза 2,5 е.а./100 мкл; геномная ДНК 0,3 мкг. ПЦР проводят в объеме 25 мкл. Температурный режим: 94°С - 1 мин, 58°С - 0,6 мин, 72°С - 0,4 мин, 38 циклов. Качество продукта контролируют в 4% полиакриламидном гель-электрофорезе (соотношение акриламид /бисакриламид 19:1).
Выделение биотинилированного ПЦР-фрагмента ДНК осуществляют с использованием системы магнитных микрочастиц Fе3O4, несущих на поверхности несколько молекул стрептавидина (фирма Dynal, Норвегия). Для этого микрочастицы, отмытые от буфера хранения, смешивают с раствором ДНК и инкубируют 15 мин при комнатной температуре для связывания биотина в составе ПЦР-фрагмента со стрептавидином на магнитной частице. Оптимальное соотношение составляет 5 мкг микрочастиц к 1 мкл реакционной смеси ПЦР. Суммарно, к 40 мкл суспензии микрочастиц в буфере 2×BW (WB:5 мМ трис НСl, pH 7,5; 0,5 мМ EDTA; 1 мМ NaCl) добавляют 40 мкл ПЦР-смеси. После инкубации микрочастицы со связавшимися через биотин-стрептавидиновое взаимодействие молекулами ДНК осаждают, поместив пробирку в магнитное поле. После этого супернатант отбрасывают, а осадок дважды промывают в буфере BW путем суспендирования с последующим повторным магнитным разделением.
Разделение цепей ДНК ПЦР-фрагментов проводят в соответствии с рекомендациями фирмы Dynal с небольшими модификациями. Для этого выделенную описанным выше способом ДНК денатурируют в 10 мкл 0,1 М NaOH в течение 10 мин при комнатной температуре. Затем пробирку помещают в магнитное поле и осаждают микрочастицы на стенке пробирки. При этом осаждается биотинилированная цепь. В растворе остается цепь ДНК, не имеющая биотиновой метки. Супернатант отбирают в отдельную пробирку и нейтрализуют добавлением 5 мкл 0,2 М НСl и 1 мкл 1 М трис-HCl, pH 7,5. Осадок микрочастиц, связанных с ДНК, промывают последовательно 50 мкл 0,1 М NaOH, 50 мкл буфера BW, 50 мкл буфера ТЕ. После этого комплиментарные цепи перекрестно объединяют между собой при нейтральном pH. Таким образом, биотинилированная цепь одного типа первичной структуры объединяется с небиотинилированной цепью другого (фиг.1). Для ренатурации смесь прогревают 10 мин при 95°С и затем медленно охлаждают до 30°С в течение 30-40 мин. Гомодуплексы, используемые в стадии “вычитания” и для оценки эффективности связывания, представляют собой 2 типа исходных ПЦР-фрагментов (1b-1а и 2b-2а) с полностью спаренными основаниями.
Пример 2. Приведение иммобилизованного гетеродуплекса в контакт с пептидами, экспонированными на фаговой частице, с последующей диссоциацией связавшихся фагов (после предварительного полного удаления несвязавшихся фагов) и отделением их от гетеродуплекса.
Гетеродуплексы, получение которых описано в Примере 1, используют для селекции фагов, избирательно связывающихся с неспаренными участками. Общая схема одного раунда селекции представлена на фиг.2.
Для селекции была использована пептидная фаговая библиотека рIII- HPL 15, любезно предоставленная G.P.Smith (University of Columbia, Missouri, U.S.A.). Библиотека представляет собой коллекцию 15-мерных пептидов, экспонированных на поверхности нитчатого фага f1 E.coli в составе минорного белка оболочки pIII, со статистически выбранной пептидной вставкой длиной 15 а.к. в минорный А белок [10]. Концентрация фаговых частиц в исходной библиотеке составляет 1014 вирионов/мл.
Все манипуляции с фагами, а именно размножение и титрование с использованием чувствительных клеток E.coli К.91 Kan (Kmr) проводят в соответствии с методами [11]. В селекционном раунде проводят инкубацию в буфере связывания (ВС, 10 mM NaCl; 20 mМ Tris-HCl, pH 7,8; 5 mМ MgCl) фаговых частиц с искусственными гетеродуплексами в соотношении 10:1. Лимитирующим в данном соотношении было количество биотинилированного гетеродуплекса, обеспечивающее компактность магнитного осадка и визуальный контроль за ним. Эти требования были соблюдены при внесении в инкубационную смесь не менее 2×109 виpиoнoв/мкл.
После 30 мин инкубации при 37°С получают магнитный осадок, содержащий взаимодействующие с гетеродуплексом фаги. Осадок после удаления магнитного супернатанта, содержащего несвязавшиеся фаги, многократно промывают до полного устранения фагового фона, что контролируют титрованием промывочных фракций. Осадок ресуспендируют в буфере диссоциации (BD, 1 M NaCl,) в течение 10 мин при температуре 50°С для разделения фагов и гетеродуплексных фрагментов. Освободившиеся от фагов гетеродуплексы удаляют путем магнитного осаждения, а к оставшимся фагам добавляют 5 мкл гомодуплексов при соотношении (10:1).
Селекция начинается со стадии инкубации фагов с биотинилированными гетеродуплексами. В первом раунде гетеродуплексы инкубируют с фагами из исходной фаговой библиотеки. В последующих раундах инкубацию проводят с фагами, полученными в предыдущем раунде.
Условия инкубации были подобраны, исходя из необходимости обеспечения максимального специфического связывания фагов к неспаренным участкам. Исходя из предположения, что фаговые пептиды функционально имитируют взаимодействие белка Mut S с неспаренными участками ДНК [7], при выборе условий руководствовались данными, что в отличие от Zn finger белков, связывающихся с ДНК в присутствии ионов Zn [9], белки Mut семейства не являются металлозависимыми и, как показали эксперименты, связывание их с ДНК происходит при физиологических значениях pH, концентрации соли и в диапазоне температур от 20 до 40°С [6, 7].
Объем инкубационной смеси был равен 10 мкл, что с одной стороны было вызвано необходимостью обеспечения более высокой концентрации взаимодействующих компонентов (фагов и дуплексов), а с другой стороны - это был минимальный объем, в котором проведение магнитного разделения поддается визуальному контролю.
После инкубации, как видно из фиг.2, все связавшиеся с ДНК фаги находятся в магнитном осадке, а не связавшиеся - в супернатанте, который в дальнейшем удаляется. Причем среди фагов, связавшихся с ДНК, есть как нужные, взаимодействующие только с неспаренными участками (mismached bind (МВ)-фаги), так и фаги, которые связываются со спаренными участками ДНК (sequence bind (8В)-фаги) на протяженности всего дуплекса. Спаренные области ДНК фрагмента, как показано выше, идентичны у гомо- и гетеродуплексов. Полное удаление всех фагов (кроме SB- и МВ-фагов), в том числе и связавшихся с другими субстратами (магнитными частицами, стенками пробирок и пр.), осуществляют путем серии "отмывок" при последовательной смене буфера с последующим его удалением после магнитного связывания комплексов гетеродуплекс - фаг (фиг.2). Необходимое число "отмывок" было определено экспериментальным путем. Для этого после каждой "отмывки" определяли количество фагов, оставшихся в магнитном супернатанте. Результаты определения необходимого количества "отмывок" для полного удаления несвязавшихся фагов представлены в таблице 1.
Для увеличения эффективности удаления неспецифических фагов объем отмывочного буфера многократно (в 20 раз) превышал объем инкубационной смеси. Однако, как видно из таблицы, после пятой "отмывки" остается достаточно много "неспецифических" фагов, и только после десятой фаговый фон исчезает.
После того как "неспецифические" фаги полностью удалены, проводят стадию диссоциации комплекса гетеродуплекс - фаг. Диссоциацию проводят в буфере, содержащем NaCl (до 1 М) и при повышенной температуре (до 50°С). Такие достаточно "жесткие" условия отмывки способствуют более эффективной диссоциации фаговых частиц и ДНК. При этих условиях жизнеспособность фаговых частиц не нарушается [12]. Титрование фагов после диссоциации показывает, что общее количество фагов, специфически связавшихся с гетеродуплексами, составляет 104-105 частиц, причем в этот общий пул входят как MB-, так и SB-фаги.
Пример 3. Проведение "вычитающего" связывания реассоциированных фагов с гомодуплексом для удаления вместе с дуплексом, с последующим размножением оставшихся (нужных фагов) на чувствительном штамме E.coli.
Для удаления "ненужных" SB-фагов, пользуясь тем, что спаренные участки у гомо- и гетеродуплексов идентичны, вводят дополнительную стадию "вычитания" - связывание с гомодуплексами (фиг.2). Количество оставшихся после "вычитания" MB- фагов составляет 101-102 частиц, т.е. доля "нужных" фагов составляет не более 1% от общего количества связывающихся с ДНК. Предполагая, что связывание фагов с дуплексом по всей длине является равновероятным событием, и учитывая, что протяженность неспаренных оснований составляет не более 1,5% (общее число 447 н.п.-100%, 7 неспаренных оснований - 1,5%), полученные результаты являются вполне ожидаемыми.
Полученные в первом раунде фаги размножают до необходимого титра (109 вирионов в мкл) на чувствительной к фагам культуре E.coli K 91. В общей сложности было проведено 5 раундов селекции.
Пример 4. Оценка эффективности селекции.
Эффективность селекции оценивали по разнице величин оптической плотности (при ОП600) после диссоциации связавшихся отселектированных фагов с гетеро- и гомодуплексами. Для этого после соответствующего размножения и концентрирования этих фагов (до 109 вирионов /мкл) проводили инкубацию с дуплексами (отдельно для гомо- и гетеродуплекса). После инкубации следовала 10-кратная отмывка, с последующей стадией диссоциации. Все процедуры проводили, как описано выше. После диссоциации проводили измерение оптической плотности.
В качестве дополнительного контроля брали фаги из исходной библиотеки.
Полученные MB-фаги были оценены по способности отличать гетеродуплексы с неспаренными основаниями от полностью спаренных дуплексов (гомодуплексов). Для сравнения проводили инкубацию фаговых частиц из исходной фаговой библиотеки (pIII НРL-15).После связывания диссоциацию фагов и ДНК дуплексов проводили в условиях, описанных выше. Количество диссоциированных фагов определяли по оптической плотности (ОП600).
Результаты оценки эффективности связывания представлены в таблице 2.
Как видно из таблицы, отселектированные на гетеродуплексах фаги более эффективно связываются именно с данными типом дуплекса. Разница по эффективности связывания с гетеро- и гомодуплексами составляет от 5-ти (в 3 раунде) до 3-х (в 5 раунде) раз. В то же время фаги из исходной фаговой библиотеки практически не связываются с гетеродуплексом. Изменения эффективности связывания зависят, вероятно, от доли МВ-фагов в общем пуле фагов, полученных после размножения в каждом раунде.
Таким образом, предложена система селекции фагов, избирательно (предпочтительно) связывающихся с ошибочно связанными основаниями в составе любого гетеродуплекса. Полученные фаги могут быть использованы для анализа полиморфизма генов, выявления точечных мутаций, а при иммобилизации на различные носители и в полностью автоматизированных процессах, например в чипах.
Литература
1. Grompe M. // Nat. genetics - 1993, v.5, p.111-116.
2. Orita M., Iwahana M., Kanazawa H. et al. // Proc. Natl. Acad. Sci. USA. - 1989, v.86, p.2766-2770.
3. Borresen A.-L., Hoving E., Smith-Sorensen В., Malkin D., Lystad S., Andersen Т., Nesland J.M., Isselbacher K.J., Friend S.H. // Proc. Natl. Acad. Sci. USA. - 1991, v.88, p.8405-8409.
4. Anon A. // Nat. Genetics - 1998, v.20, p.217-218.
5. Патент США №5876941, кл. С 12 Q 1/68 // Бюлл. ИСМ - 2000, вып.46, №3, стр.72.
6. Jiricny J., Su Sh.-S., Wood S.G., Modrich P. // Nucl. Acids Res. - 1988, v.16, №16, p.7843-7853.
7. Lishanski A., Ostrander E.A., Rine J. // Proc. Natl. Acad. Sci. USA - 1994, v.91, p.2674-2678.
8. Smith P.G., Petrenko V.A. // Chem. Rev. - 1997, v.97, p.391-410.
9. Choo Y, Klug A. // Proc. Natl. Acad. Sci. USA. - 1994, v.91, p.11163-11167.
10. Friesen W.J, Darby M.K. // J. Biol. Chem - 1997, v.272, №17, p.10994-10997.
11. Choo Y., Sanchez-Garcia I., Klug A. // Nature - 1994, v.372, p.642-645.
12. Smith G.P., Scott J.K. // Methods in Enzymology - 1993, v.2217, p.225-229.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ термической диссоциации для проведения селекции ДНК-аптамеров | 2019 |
|
RU2723373C1 |
| СПОСОБ СКРИНИНГА БИБЛИОТЕКИ ФАГОВОГО ДИСПЛЕЯ | 2005 |
|
RU2402777C2 |
| Способ направленного истощения олигонуклеотидных библиотек для снижения неспецифической адсорбции при твердофазной селекции аптамеров на основе нуклеиновых кислот | 2015 |
|
RU2618872C1 |
| СПОСОБ ИДЕНТИФИКАЦИИ НАТИВНЫХ ПАР ФРАГМЕНТОВ ДНК ИЛИ РНК, ПРИСУТСТВУЮЩИХ В ОДНИХ И ТЕХ ЖЕ ЖИВЫХ КЛЕТКАХ | 2015 |
|
RU2600873C1 |
| СПОСОБ ИДЕНТИФИКАЦИИ НАТИВНЫХ ПАР ФРАГМЕНТОВ ДНК ИЛИ РНК, ПРИСУТСТВУЮЩИХ В ОДНИХ И ТЕХ ЖЕ ЖИВЫХ КЛЕТКАХ | 2013 |
|
RU2578009C2 |
| БИБЛИОТЕКА ЗАВИСИМЫХ ОТ КОНЦЕНТРАЦИИ ИОНОВ СВЯЗЫВАЮЩИХ МОЛЕКУЛ | 2012 |
|
RU2812861C1 |
| СВЯЗЫВАЮЩИЕ ЭЛЕМЕНТЫ С ИЗМЕНЕННЫМИ ДИВЕРСИФИЦИРОВАННЫМИ ДОМЕНАМИ ОСТОВА | 2017 |
|
RU2778694C2 |
| БИБЛИОТЕКИ TCR | 2015 |
|
RU2722696C2 |
| КОМПОЗИЦИИ НА ОСНОВЕ СТАБИЛИЗИРОВАННЫХ ФИБРОНЕКТИНОВЫХ ДОМЕНОВ, СПОСОБЫ И ОБЛАСТИ ИХ ПРИМЕНЕНИЯ | 2011 |
|
RU2603272C2 |
| КОМПОЗИЦИИ И СПОСОБЫ С УЧАСТИЕМ НУКЛЕИНОВЫХ КИСЛОТ, НАЦЕЛЕННЫХ НА НУКЛЕИНОВЫЕ КИСЛОТЫ | 2014 |
|
RU2662932C2 |
Изобретение относится к области молекулярной генетики и биотехнологии, конкретно к способам обнаружения ошибок спаривания оснований в нуклеиновых кислотах (точечных мутаций). Предложена стратегия отбора фагов, несущих пептидные аналоги функционально активных модулей белков репарационной системы, способных избирательно связываться с участками неспаренных оснований на двуцепочечных ДНК конкретного локуса. Стратегия основана на комбинации “фагового дисплея” с использованием биотинилированных праймеров для получения ПЦР-фрагментов и парамагнитных частиц со стрептавидином для физического разделения комплексов ДНК-фаг. Отселектированные таким образом фаги избирательно (предпочтительно) связываются с ошибочно спаренными основаниями в составе любого гетеродуплекса. Изобретение позволяет автоматизировать молекулярно-эпидемиологические исследования различных заболеваний. 3 з.п. ф-лы, 2 ил., 2 табл.
| GROMPE M | |||
| Nat | |||
| genetics, 1993, vol.5, p.111-116 | |||
| ORITA M | |||
| et al., Proc | |||
| Natl | |||
| Acad | |||
| Sci | |||
| USA, 1989, vol.86 p.2766-2770 | |||
| BORRESEN A.-L | |||
| et al., Proc | |||
| Natl | |||
| Acad | |||
| Sci | |||
| USA, 1991, vol.88, p.8405-8409 | |||
| ANON A.Nat, genetics, 1998, vol.20, p.217-218 | |||
| US 5876941 A, 02.03.1999 | |||
| JIRICNY J | |||
| et al., Nucl.Acids Res., 1988, vol.16, No 16, p.7843-7853 | |||
| LISHANSKI A | |||
| et al | |||
| Proc | |||
| Natl | |||
| Acad | |||
| Sci | |||
| USA, 1994, vol.91, p.2674-2678 |

