Объектом данного изобретения является применение (R)-арилпропионовых кислот для получения лекарственных средств для лечения заболеваний людей и животных, на которые можно оказывать терапевтическое воздействие путем ингибирования активации NF-κВ (ядерный фактор каппа В).
Арилпропионовые кислоты и их производные используют уже давно в качестве нестероидных эффективных противовоспалительных и анальгетических лекарственных средств. Известными представителями этой группы биологически активных веществ являются ибупрофен, флурбипрофен, кетопрофен, напроксен, тиапрофеновая кислота и фенопрофен [Производные пропионовой кислоты; Goodman & Gilman's, Фармакологическая основа терапии, глава 27, стр.637 (девятое издание, 1996)].
На основании молекулярного строения с одним асимметрическим атомом углерода арилпропионовые кислоты и их производные являются хиральными, существующими также в виде (R)- и (S)-энантиомерных форм. Обычно при химическом синтезе эти биологически активные вещества получают в виде рацемата. Эти биологически активные вещества вплоть до (S)-напроксена [Williams: Энантиомеры при артритных нарушениях; Pharmac. Ther., том 46, стр.273-295 (1990); Evans: Энантиоселективные фармакодинамика и фармакокинетика хиральных нестероидных противовоспалительных лекарств. Eur J Clin Pharmacol 42: 237-256 (1992)] и последних дексибупрофена [Симпозиум: Новейшая информация о (S)(+)-ибупрофене; Going/Kitzbuhl 2-4 февраля 1996] и декскетопрофена [сертификат № 1831 от 22 июня 1993, стр.7; сертификат № 2144 от 9 июля 1996, стр.16] используются в лекарственных средствах до сих пор в виде рацематов.
Терапевтически желаемое тормозящее воспаление и болеутоляющее действие арилпропионовых кислот и их производных описывается, в основном, при ингибировании биосинтеза простагландинов [Vane и Botting: Обзор - механизм действия противовоспалительных лекарств. В сборнике: Улучшенные нестероидные противовоспалительные лекарства - ингибиторы фермента СОХ-2, стр.1-27, изд. Lancester: Kluwer Academic Publishers (1996)]. Это осуществляется за счет ингибирования участвующих в образовании простагландинов ферментов циклооксигеназы 1 и 2 (СОХ-1 и СОХ-2 или PGHS-1 и PGHS-2). Благодаря уменьшенному образованию простагландинов ослабляются находящиеся в зависимости от этих медиаторов воспаления воспалительные симптомы, как, например, боль, вздутие, эритема, образование отеков, согревание и ограничение функций. Ингибирование синтеза простагландинов считают общей отличительной чертой механизма противовоспалительного и анальгетического действия. Терапевтически желательное ингибирование продуцирования простагландинов в подвергнувшейся заболеванию ткани-мишени приводит в других органосистемах, которые имеют в наличии определенные концентрации простагландинов, к нежелательным последствиям действия лекарств. Особенно поражаются в результате нежелательного воздействия желудочно-кишечный тракт, почки, легкие и тромбоциты.
Известно, что в отношении ингибирования синтеза простагландинов имеются существенные различия между энантиомерными формами арилпропионовых кислот [Williams (см. выше); Evans (см. выше); Brooks и Day: Новые нестероидные противовоспалительные лекарства, изд-во Birkhauser, Базель, стр.119-126 (1985)]. В то время как все (S)-энантиомеры этих соединений обнаруживают четко выраженное ингибирование синтеза простагландинов, это действие не определяют в случае (R)-энантиомеров в терапевтически важной области концентраций. Таким образом, (R)-арилпропионовым кислотам и их производным не приписывают в терапевтических концентрациях ни желательных, ни нежелательных лекарственных воздействий, которые имеют какую-либо связь с ингибированием продуцирования простагландинов. Независимо от отсутствия таких нежелательных воздействий со специфическим механизмом действия (R)-энантиомеры этого класса биологически активных веществ могут обнаруживать нежелательное специфичное для вещества действие.
Вследствие давно известного терапевтического и экономического значения используемых в виде рацемата арилпропионовых кислот предпринимаются попытки обосновать смысл применения рацемических биологически активных веществ. В случае ибупрофена применение рацемата обосновывается, в основном, тем, что в организме человека или животного имеет место более или менее четкая инверсия (R)-ибупрофена в (S)-ибупрофен [Caldwell и др.: Метаболическая хиральная инверсия и имеющаяся в наличии энантиоселективность 2-арилпропионовых кислот и их биологические последствия; Biochemical Pharmacology, том 37, № 1, стр.105-114(1988)], так что также часть (R)-формы после инверсии в (S)-форму может быть эффективна в качестве ингибитора синтеза простагландинов. Кроме того, для (R)-ибупрофена описывается ингибирование полиморфно-ядерных лейкоцитов in vitro, которые при воспалительных заболеваниях могли бы оказаться полезными [Villanueva и др.: Эквипотенциальное ингибирование R(-)-, S(+)- и рацемическим ибупрофеном функции человеческих полиморфно-ядерных клеток in vitro; Br. J. clin. Pharmac., 35, 235-242 (1993)]. Терапевтическая важность этого механизма при применении рацемического ибупрофена не могла, однако, быть показана. В случае (R)-флурбипрофена инверсией пренебрегают.
Тот факт, что терапевтическое действие арилпропионовых кислот, по существу, приписывается ингибированию синтеза простагландина, привел к тому выводу, что применение чистых (S)-энантиомеров, при необходимости, рацемических соединений, но не чистых (R)-энантиомеров, является рациональным. Первоначально неожиданное открытие, что (R)-флурбипрофен обладает антиноцицептивным действием, которое не связано с ингибированием периферического биосинтеза простагландинов, вызвало развитие лекарственных средств на основе (R)-флурбипрофена [патент ФРГ 4028906 С2; европейский патент ЕР 0607128 В1; патенты США 5206029 и 5200198] в качестве анальгетиков без тормозящих воспаление эффективных компонентов. Позже было также описано болеутоляющее действие для (R)-кетопрофена [патент ФРГ 4319438 C1; международная заявка на патент WO 93/176671.
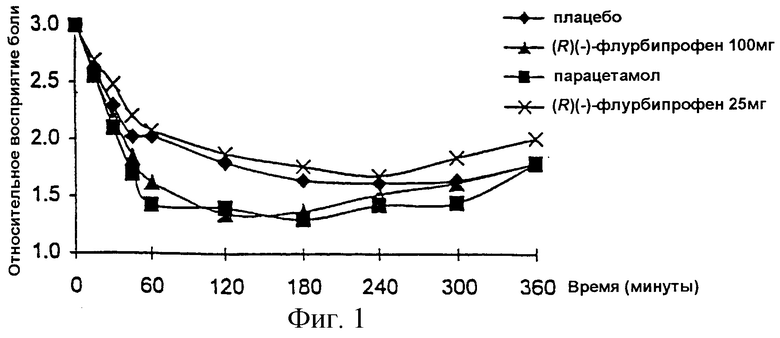
Более новые публикации подтверждают антиноцицептивное действие (R)-флурбипрофена [Geisslinger, Schaible: Новые взгляды на место приложения и способ антиноцицептивного действия энантиомеров флурбипрофена, J. Clin Pharmacol, 36, 513-520 (1996); Buritova, Besson: Периферические и/или центральные эффекты рацемического, S(+)- и R(-)-флурбипрофена на воспалительные ноцицептивные процессы: изучение белка c-Fos в спинном мозге крысы; British J. Pharmacology, 125, 87-101 (1998)]. В клинических исследованиях на больных смогли подтвердить болеутоляющее действие (R)-флурбипрофена [фиг.1] и (R)-кетопрофена [Cooper и др.: Аналгезирующая эффективность и безопасность (R)-кетопрофена при послеоперационной зубной боли; J. Clin Pharmacol, 38, 11S-18S (1998)].
Фиг.1: Контролируемое плацебо двойное слепое исследование на 180 женщинах с острой болью после эпизиотомии (кривые средних значений)
Госпитализированные больные были произвольно разделены на три группы, по 50 больных каждая, которым назначались лекарства, и группу, получающую плацебо (30 больных). Каждая больная получала в течение 48 часов после, в остальном, нормально протекающих родов разовую дозу подвергающегося исследованию назначенного лекарства (25 мг (R)(-)-флурбипрофена или 100 мг (R)(-)-флурбипрофена, или 1000 мг парацетамола) или перорально вводили плацебо. Незадолго до перорального введения изучаемых препаратов или плацебо и в точно установленные временные моменты исследования (15, 30, 45, 60, 120, 180, 240, 300 и 360 минут) больных опрашивали относительно их болевых ощущений. Эффективность отдельных препаратов оценивалась с помощью шкалы восприятия боли (0 = никакого восприятия боли, 1 = слабое, 2 = умеренное, 3 = сильное). Результаты протекания процесса во времени обобщены в приведенных на фиг.1 кривых средних значений для отдельных групп больных.
Экспериментальные исследования на животных подтверждают, что действие (R)-флурбипрофена может быть объяснено тормозящим воспаление и антиноцицептивным действием на центральную нервную систему [Birutova (см. выше); Neugebauer и др.: Антиноцицептивные эффекты R(-)- и S(+)-флурбипрофена на нейроны позвоночного дорсального роговидного отростка крысы, оказавшиеся гипервозбудимыми при остром воспалении коленного сустава; J. Pharmacol Exp Ther, 275, 618-628 (1995)]. Известное периферическое ингибирующее воспаление и антиноцицептивное действие флурбипрофена могло бы быть обнаружено, напротив, исключительно в случае (S)-энантиомеров [Birutova (см. выше) и Neugebauer (см. выше)]. Согласно современному уровню знаний, из этого проистекают существенные выводы, что для оптимального лечения периферических воспалительных заболеваний должны бы были использоваться в качестве выбранного средства (S)-арилпропионовые кислоты. Для уменьшения тесно связанного с ингибированием синтеза простагландинов нежелательного воздействия на желудочно-кишечный тракт и т.д. не следует, например, принимать (S)-флурбипрофен перорально, а следует наносить местно на воспаленные или болевые участки. (R)-Флурбипрофен следует, однако, вследствие центрального действия применять систематически [Birutova (см. выше)], например, перорально, внутримышечно или внутривенно.
Вопреки этим новейшим выводам о практически исключительно центральном действии (R)-флурбипрофена было неожиданно обнаружено, что (R)-флурбипрофен в определенных концентрациях является сильным и специфическим ингибитором активации ядерного фактора транскрипции NF-κВ. NF-κВ является повсеместным фактором транскрипции, который играет центральную роль в клетках при иммунных и воспалительных реакциях, а также при экспрессии цитокинов, хемокинов, клеточных адгезионных молекул, факторов роста, иммунорецепторов, белков острой фазы, различных ферментов и других факторов транскрипции [Lee, Burckart: Ядерный фактор каппа В: Важный фактор транскрипции и терапевтическая мишень, J. Clin. Pharm., 38, 981-993 (1998)].
Активация NF-κВ может быть ингибирована на различных стадиях каскада активации с помощью различных биологически активных веществ. Так, глюкокортикоиды ингибируют NF-κВ путем прямой ассоциации или путем усиления экспрессии. Циклоспорин и такролимус предотвращают активацию NF-κВ путем ингибирования фосфатазы, оказывающей воздействие на уровень кальция в моче, которая непрямо индуцирует расщепление I-κВ. Дезоксиспаргуалин ингибирует NF-κВ путем блокады смещения его ядра. Аспирин и салицилат ингибируют ранее упомянутые явления, которые индуцируют фосфорилирование I-κВ. Тепоксалин и антиоксиданты ингибируют активацию NF-κВ путем изменения окислительно-восстановительного состояния клетки. Необходимы дальнейшие исследования для разработки специфических ингибиторов для лечения заболеваний, на которые влияет NF-κВ [Lee, Burckart: Ядерный фактор каппа В: важный фактор транскрипции и терапевтическая мишень, J. Clin. Pharm., 38, 981-993 (1998)].
Известно, что (R)-ибупрофен и (S)-ибупрофен ингибируют активацию фактора транскрипции NF-κВ посредством сложного эфира форбола (ТРА), что приводит к регуляции активированной сложными эфирами форбола протеинкиназы С и вызванному, благодаря этому, фосфорилированию и инактивации I-κВ, однако, не в состоянии повлиять на активацию NF-κВ посредством 11а-15(S)-дигидрокси-9-оксо-5-цис, 13-транс-простадиеновой кислоты (PGE2) или липополисахарида (ЛПС). Применимость ибупрофена поэтому ограничена. [N.Scheuren и др. "Модуляция факторов транскрипции нестероидными противовоспалительными лекарствами", Naunyn-Schmiedeberg’s Arch. Pharmacol., том 354, № 4, приложение 1, 1996; N. Scheuren и др., "Энантиомеры нестероидного противовоспалительного лекарства ибупрофена являются сильными и специфическими ингибиторами транскрипции фактора NF-каппа, бета", Naunyn-Schmiedeberg’s Arch. Pharmacol., том 357, № 4, приложение, 1998; N. Scheuren и др. "Модуляция фактора транскрипции NF-каппа, бета энантиомерами нестероидного лекарства ибупрофена", Вr. J. Pharmacol., том 123, № 4, 1998; N. Scheuren и др., "Слабые ингибиторы циклооксигеназ могут усилить их антиноцицептивные эффекты модуляции факторов транскрипции", Adv. Exp. Med. Biol., том 433, 1997].
Изобретение поставило как раз задачу найти другие биологически активные вещества, которые ингибируют активацию NF-κВ.
Неожиданно было как раз обнаружено, что другие не рацемизирующиеся (R)-арилпропионовые кислоты могут вмешиваться в заболевание посредством специфического ингибирования шагов внутри каскада активации NF-κВ. Вследствие повсеместной функции фактора транскрипции NF-κВ при генной регуляции лекарственные средства с такими (R)-арилпропионовыми кислотами или их производными годятся не только для известного смягчения боли через антиноцицептивное действие на центральную нервную систему [патент ФРГ 4028906 С2], но при подходящих применении и дозе также используются при всех заболеваниях, при которых может быть терапевтически полезным ингибирование активации NF-κВ. Согласно изобретению эти лекарственные средства могут использоваться не только при болях и ревматических заболеваниях, но также при опухолях, иммунных заболеваниях, астме, шоке, воспалительных заболеваниях кишечника (болезнь Крона, язвенный колит), лучевых поражениях, артериосклерозе, болезни Альцгеймера, при лечении реакций отторжения после трансплантации тканей и органов и т.д. в соответственно подобранной дозе и фармацевтической готовой форме.
Сообщаемое здесь наблюдение, касающееся ингибирования образования NF-κВ, неожиданно, так как согласно уровню техники фармакологическим эффектам арилпропионовых кислот приписывались другие механизмы. Это приводило раньше к применению рацематов или (S)-энантиомеров при болях или воспалениях в более малых дозах.
В международной заявке на патент WO 98/09603 далее описывается применимость (R)-энантиомеров нестероидных противовоспалительных лекарств при опухолевых заболеваниях, в частности при раке толстой кишки и груди, кистозном фиброзе и болезни Альцгеймера.
Неожиданно было обнаружено, что (R)-флурбипрофен и другие не метаболизирующиеся в СоА-сложные тиоэфиры и тем самым рацемизующиеся (R)-арилпропионовые кислоты ингибируют активацию NF-κВ примерно в 100 раз сильнее, чем соответствующие (S)-энантиомеры. Для достижения удовлетворительного действия они, однако, должны использоваться в более высоких дозировках, чем обычно при известном терапевтическом применении рацемических арилпропионовых кислот. Вследствие хорошей переносимости по причине практически отсутствия воздействия этих дозировок (R)-арилпропионовых кислот на периферический биосинтез простагландинов и не происходящей рацемизации в (S)-энантиомеры возможно, однако, при применении (R)-энантиомеров установить такие высокие дозы, что может быть достигнуто желательное ингибирующее действие на активацию NF-κВ без того, чтобы нужно было опасаться происходящих от (S)-формы нежелательных воздействий. Биологически активные вещества поэтому используются предпочтительно в большинстве случаев свободными от (S)-энантиомеров, т.е. имеют оптическую чистоту выше 90%, в частности выше 99%, если не является желательным в качестве "побочного действия" также известное уменьшающее боль и воспаление действие (S)-энантиомеров. В противоположность (R)-ибупрофену не следует ожидать относящихся к этому нежелательных воздействий из-за отсутствующей инверсии R⇒S в случае не метаболизирующихся в СоА-сложный тиоэфир (R)-арилпропионовых кислот. Лекарственные средства по изобретению позволяют ожидать, таким образом, улучшенный спектр лечебного действия по сравнению с применением рацемических арилпропионовых кислот или их (S)-энантиомеров. Проведенные исследования на людях доказывают хорошую желудочно-кишечную переносимость (R)-флурбипрофена и других (R)-арилпропионовых кислот [Jerussi и др.: Клиническая эндоскопическая оценка гастродуоденальной толерантности к кетопрофену, флурбипрофену, рацемическому кетопрофену и парацетамолу: рандомизированное одноразовое-слепое, контролируемое плацебо исследование; J. Clin. Pharmacol, 38, 19S-24S (1998)], которая уже наметилась в ранее проведенных экспериментах на животных [патент ФРГ 4028906 С2].
С момента открытия ядерного фактора транскрипции NF-κВ примерно в течение десяти лет проводятся широкие исследовательские работы с помощью эндогенных и экзогенных веществ по изучению биологической функции и влияния на образование NF-κВ. Из известных фармакологических веществ ранее описывались, среди прочего, глюкокортикоиды, как, например, дексаметазон и преднизон, иммуносупрессоры, как, например, циклоспорин, такролимус и дезоксиспергуалин, в терапевтических концентрациях в качестве действующих на активацию NF-κВ. Из образующихся при биохимической инверсии (R)-ибупрофена в (S)-ибупрофен промежуточных метаболитов (R)-ибупрофен - коэнзим А - сложный тиоэфир также обнаруживал ингибирование активации NF-κВ, и поэтому возникло умозрительное предположение, что также (R)-ибупрофен через известную метаболическую активацию в человеческом организме в (R)-ибупрофен-СоА-сложный тиоэфир обнаруживал бы действие, которым сам (R)-ибупрофен не обладает. [Brune и др.: Лекарственные средства, содержащие ибупрофен-сложный тиоэфир, в качестве ингибиторов зависящего от Nf-κВ образования медиаторов воспалений и боли, заявка на патент ФРГ 19716713 A1, международная заявка на патент WO 98/47502].
Неожиданно было обнаружено, что другие терапевтически полезные производные арилпропионовых кислот, такие как флурбипрофен, кетопрофен, напроксен, тиапрофеновая кислота и фенопрофен, которые не обнаруживают заметного образования СоА - сложных тиоэфиров у людей и поэтому не рацемизуются, вызывают выраженное ингибирование активации NF-κВ и, таким образом, обладают потенциалом для связанных с влиянием этого механизма терапевтических эффектов. Эта группа в дальнейшем обозначается "не рацемизующиеся (сокращенно н.р.) (R)-арилпропионовые кислоты".
Лекарственные средства по изобретению на основе н.р. (R)-арилпропионовых кислот и их производных в качестве ингибиторов активации NF-κВ для лечения заболеваний, которые подвергаются воздействию посредством модификации активации NF-κВ, основываются на следующих экспериментальных исследованиях:
Фиг.2: Зависящее от концентрации влияние (R)- и (S)-флурбипрофена ((R)-ФЛУ и (S)-ФЛУ) на активацию фактора транскрипции NF-κВ в RAW-клетках. Анализ гельудерживания (анализ по сдвигу пятна при электрофорезе; комплект для определения сдвига в геле, фирма Берингер Маннхайм) показывает, что липополисахарид (ЛПС) (1 мкг/мл) приводит к активации NF-κВ (комплекс р50/р65 NF-κВ) (следы №№ 2 и 10). Микромолярные концентрации (R)-флурбипрофена (следы №№ 3, 4, 5, 6, 7 против следа № 2 в качестве контроля) были в состоянии ингибировать эту индуцированную ЛПС активацию NF-κВ. Денситометрическое определение показало, что (S)-флурбипрофен в отношении этих свойств был примерно в 100 раз менее активен (следы №№ 11, 12, 13, 14 против следа № 10 в качестве контроля).
Следы № 1 и 8 представляют соответственно нестимулированные контрольные клетки.
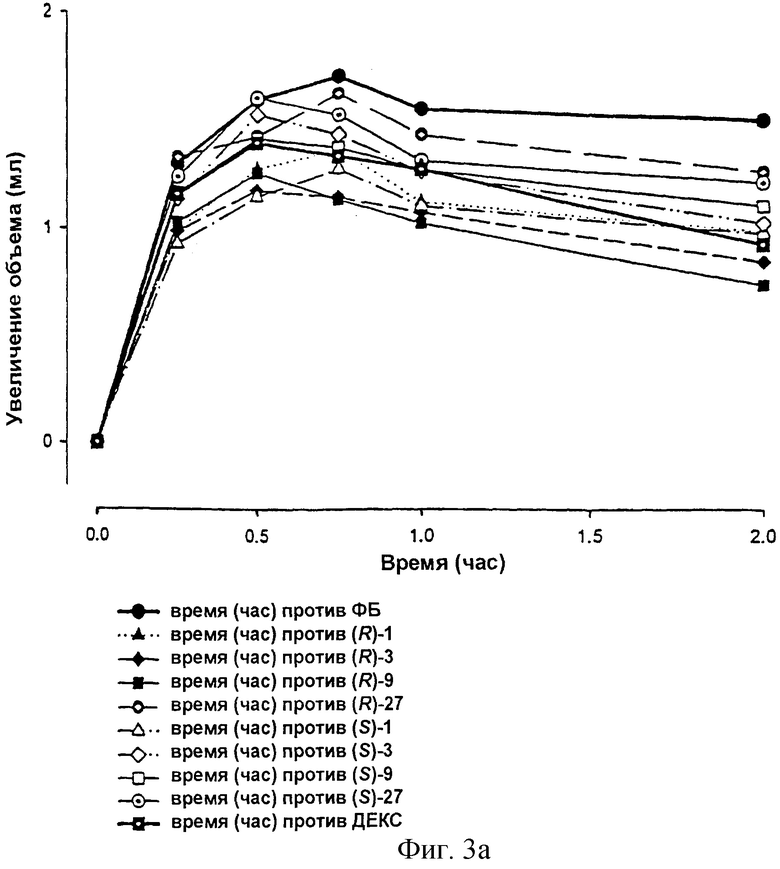
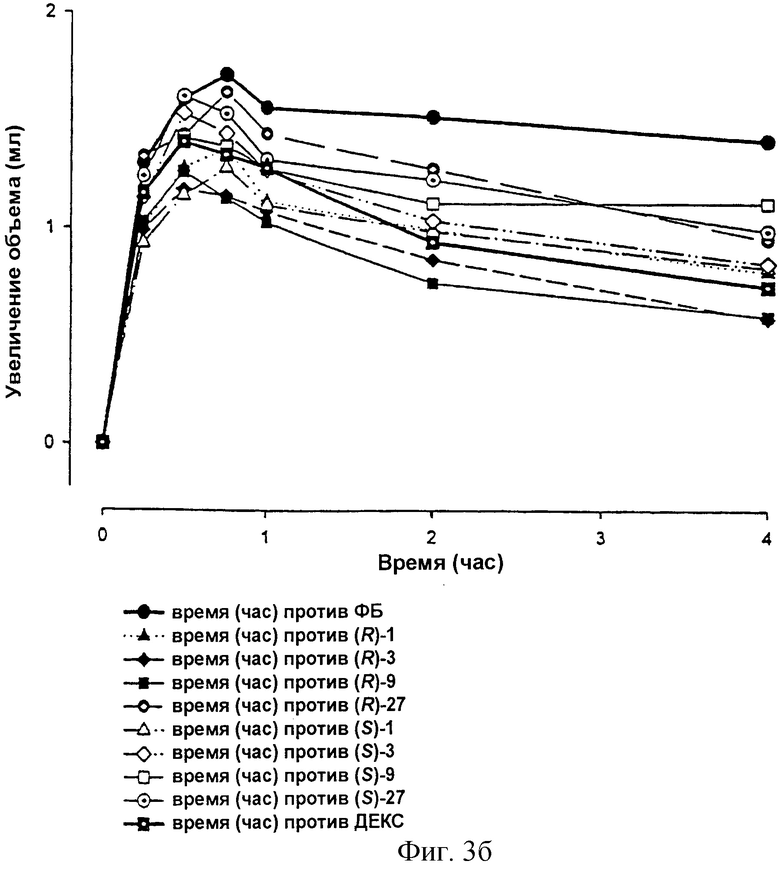
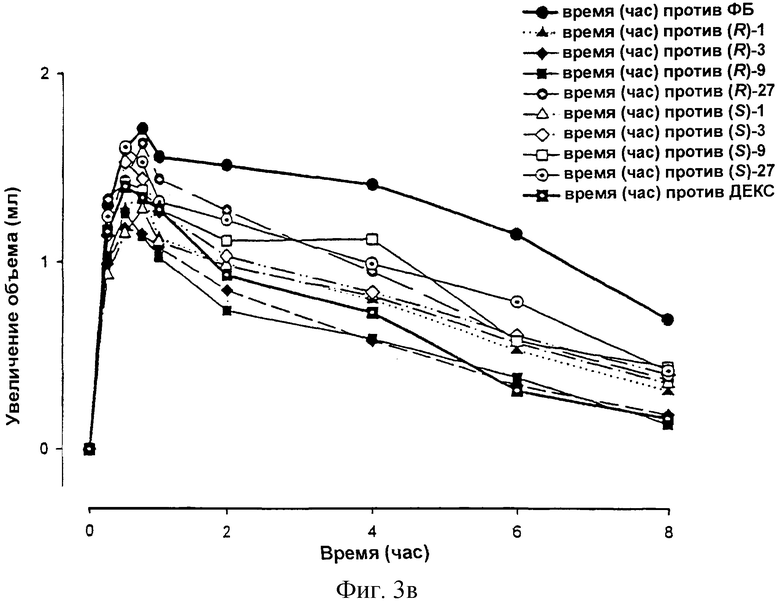
Так как ядерный фактор транскрипции NF-κВ является, среди прочего, ответственным за образование ряда ферментов с провоспалительными и отекообразующими свойствами, определяли влияние (R)-флурбипрофена на индуцированный зимозаном отек лапы крысы (методы, описанные: Meller ST и Gebhart GF: Внутриплантарный зимозан в качестве надежной, способной быть количественно оцененной модели тепловой и механической гипералгезии у крысы; European Journal of Pain, 1, 43-52 (1997). Фиг.3а-в объединяют полученные результаты.
Фиг.3а-в: Зависящее от времени увеличение объема лапы крысы (измеренное с помощью плетизмографа) после внутриподошвенного введения зимозана. После применения зимозана [Meller и Gebhart (см. выше)] в задней лапе крысы проявляется в качестве симптома воспаления увеличение объема лапы (группа, использующая плацебо, применение наполнителя = фосфатный буфер (ФБ)). На основании ингибирующего действия (R)-флурбипрофена на активацию NF-κВ при дозировках в области между 1 и 27 мг/кг массы тела (применение внутрибрюшинное) становится очевидным неожиданное уменьшение объема лапы. Этот эффект особенно был выражен между 2 и 6 часами после применения зимозана. Дексаметазон (ДЕКС) (0,5 мг/кг массы тела), известный ингибитор активации NF-κВ, использовали в качестве положительного контроля. (S)-Флурбипрофен также продемонстрировал в соответствии с ожиданием уменьшение объема лапы, причем этот эффект, однако, объясняется не ингибированием активации NF-κВ, а ингибированием синтеза провоспалительных простагландинов. (S)-Флурбипрофен является известным ингибитором циклооксигеназ.
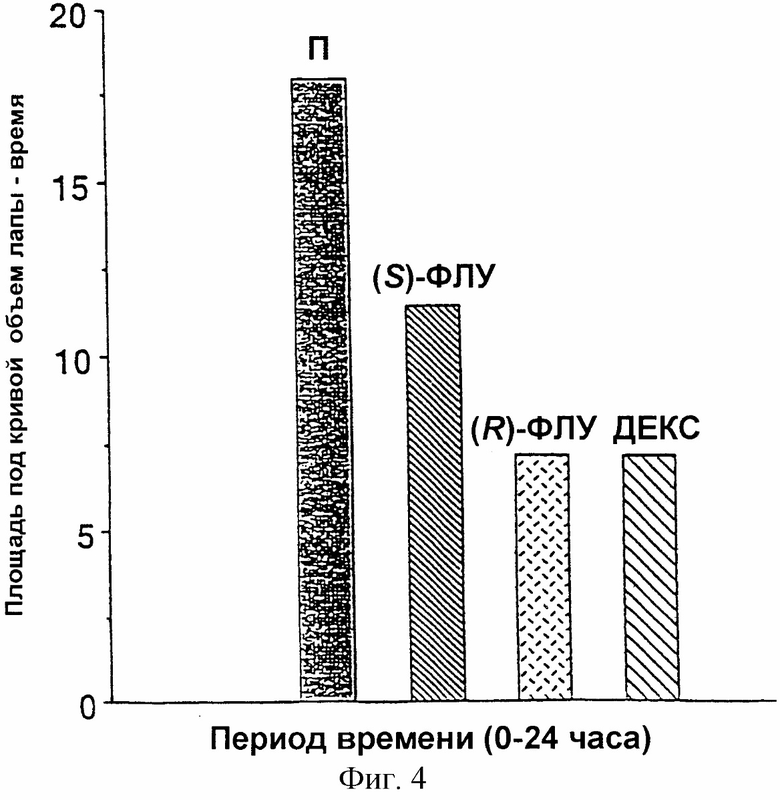
Фиг.4: Обобщение результатов воздействия 9 мг/кг (R)-флурбипрофена ((R)-ФЛУ), 9 мг/кг (S)-флурбипрофена ((S)-ФЛУ) и 0,5 мг/кг дексаметазона (ДЕКС) против плацебо (П) через 24 часа. Эффекты от 9 мг/кг (R)-флурбипрофена был сравнимы с таковыми от 0,5 мг/кг дексаметазона.
Известно получение и хиральное разделение арилпропионовых кислот и их производных. Например, следует сослаться на международную заявку на патент WO 93-17677 и на указанную там литературу.
Под производными арилпропионовых кислот имеют в виду согласно изобретению производные, расщепляющиеся в желудочно-кишечном тракте (при пероральном применении) или в крови в арилпропионовые кислоты, как, например, сложные алкиловые эфиры с 1-6 атомами углерода в алкильном остатке, которые при известных условиях могут содержать аминогруппы или гидроксильные группы, амиды или алкиламиды с 1-6 атомами углерода в алкильном остатке, а также фармацевтически переносимые соли, в особенности, соль аминокислоты с щелочным, щелочноземельным металлом, аммонийная соль аминокислоты, предпочтительно лизинат, меглюминат, трометамин, аргинат, или соль с алюминием. Такие соединения также известны.
Величина профилактической или лечебной дозы н.р. (R)-арилпропионовой кислоты при лечении в острый период и при хроническом лечении заболеваний варьируется соответственно силе жалоб подвергающегося лечению. Доза и частота дозировок различаются также в соответствии с возрастом, массой тела и реакцией отдельного больного. Обычно ежедневная доза н.р. (R)-арилпропионовой кислоты для представленных описанных жалоб должна быть примерно между 50 мг и 2000 мг, принимаемыми в один или несколько приемов. Предпочтительно дневная доза находится примерно между 100 мг и 500 мг, принимаемыми в один или несколько приемов. При уходе за больным лечение должно начинаться с низких дозировок, возможно от 20 мг до 200 мг, и дозировки увеличивают до примерно 1000 мг или выше, в каждом случае в зависимости от общей реакции больного. Далее рекомендуется, чтобы грудные дети, дети, больные старше 65 лет и больные с нарушенной функцией почек или печени сначала получали невысокую дозу, основываясь при этом на индивидуальной реакции и определении уровня в крови. В некоторых случаях может быть необходимо применение дозировки за пределами этой области, что является очевидным для специалиста. Далее отмечают, что лечащий домашний врач или врач-специалист в клинике в зависимости от общей реакции больного знает, как и когда прервать лечение, изменить или прекратить. Выражение "количество, которое достаточно для ингибирования NF-κВ, но недостаточно, чтобы вызвать отрицательные реакции (ингибирование синтеза простагландинов)" охватывается вышеприведенными дозированными количествами и инструкцией по дозированию. Может быть применен любой вид введения для обеспечения больного эффективной дозировкой н.р. (R)-арилпропионовой кислоты. Например, могут применяться пероральная, ректальная, чрескожная, парентеральная (подкожная, внутримышечная, внутривенная), внутриоболочечная, эпи- или перидуральная и им подобные формы введения. Возможными формами для применения являются, например, таблетки, дисперсии, суспензии, растворы, капсулы, пластыри и им подобные формы.
Фармацевтические готовые лекарственные формы представленного изобретения охватывают н.р. (R)-арилпропионовую кислоту в качестве биологически активного вещества или ее фармацевтически переносимое производное и фармацевтически переносимый носитель, и, на выбор, другие лекарственные добавки.
Выражения "фармацевтически переносимые производные" или "их фармацевтически переносимое производное" относятся к производным, полученным из фармацевтически переносимых нетоксичных кислот или оснований, включая неорганические кислоты и основания и органические кислоты и основания. Поскольку компоненты по данному изобретению являются кислотными, могут быть получены производные с фармацевтически переносимыми, нетоксичными основаниями, включая неорганические и органические основания. Подходящие фармацевтически переносимые основные добавляемые производные в качестве компонентов представленного изобретения охватывают соли с металлами, полученные из алюминия, кальция, лития, магния, калия, натрия и цинка, или органические соли, полученные из лизина, N,N’-дибензилэтилендиамина, холина, диэтаноламина, этилендиамина, меглюмина (N-метилглюкамин), трометамина, аргинина и алкиламинов с 1-6 атомами углерода в алкильном остатке.
Готовые лекарственные формы данного изобретения охватывают такие лекарственные формы, как суспензии, растворы, эликсиры и аэрозоли. В случае твердых используемых форм для перорального введения могут быть применены такие носители, как крахмал, сахар, микрокристаллическая целлюлоза, разбавители, вспомогательные средства для гранулирования, вещества, придающие скользкость, связывающие вещества, растворители и им подобные соединения. Твердым формам для перорального применения (как, например, порошки, капсулы и таблетки) отдается предпочтение перед жидкими формами для перорального применения. Предпочтительными твердыми формами для перорального применения являются таблетки. Таблетки могут по желанию быть покрыты стандартизированными водными или безводными средствами для покрытия.
В дополнение к обычным, приведенным выше формам применения компоненты по изобретению могут быть введены с помощью известных для этого средств в форме с замедленным высвобождением и/или в форме с быстрым высвобождением. Например, придающие гидрофобность добавки к пероральным формам применения действуют замедляющим образом, средства, способствующие распадаемости, и поверхностно-активные вещества способствуют растворимости и тем самым действуют ускоряюще, и, как известно, обе формы могут быть смешаны и представлены в виде гранул, чтобы часть биологически активного вещества высвобождалась быстро, а остаток с замедлением.
Фармацевтические готовые лекарственные формы по настоящему изобретению, которые годятся для перорального применения, могут в виде отдельных единиц, как, например, капсулы, драже или таблетки, или аэрозоли, содержать в каждом случае заданное количество биологически активного вещества в виде порошка или гранул, или в виде раствора или суспензии в водной жидкости, неводной жидкости, эмульсии масла в воде или жидкой эмульсии воды в масле. Такие готовые лекарственные формы могут быть получены любым фармацевтическим способом, но все способы предусматривают смешивание биологически активного вещества с носителем, который состоит из одной или нескольких необходимых составных частей. Обычно готовые лекарственные формы получают путем равномерного и тщательного смешивания биологически активного вещества с жидкими носителями или с мелкоизмельченными твердыми носителями, или с обоими и затем, если необходимо, путем формования продукта в желаемую форму применения.
Например, таблетку можно получить путем прессования или формования, на выбор, с одной или несколькими дополнительными составными частями. Прессованные таблетки могут быть получены путем прессования в соответствующем приспособлении, когда биологически активное вещество, представленное в сыпучей форме, как, например, порошкообразной или в виде гранул, смешивают, на выбор, со связывающим веществом, придающим скользкость веществом, с инертным разбавителем, диспергирующим или поверхностно-активным веществом. Формованные таблетки могут также быть получены путем формования в подходящем устройстве смеси измельченных до порошкообразного состояния компонентов, смоченных инертным жидким разбавителем, и последующего высушивания. Предпочтительно каждая таблетка содержит между 50 мг и 1000 мг биологически активного вещества и каждое драже или каждая капсула содержат примерно между 50 мг и 600 мг биологически активного вещества. Особенно предпочтительно таблетка, драже или капсула содержит одну из четырех дозировок, а именно 50 мг, 100 мг, 200 мг или 500 мг биологически активного вещества.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИБУПРОФЕНОВЫЙ ТИОЭФИР В КАЧЕСТВЕ ИНГИБИТОРА, ЗАВИСЯЩЕГО ОТ Nf-kB ОБРАЗОВАНИЯ МЕДИАТОРОВ ВОСПАЛЕНИЯ И БОЛИ | 1998 |
|
RU2204388C2 |
| НОВОЕ ЛЕЧЕНИЕ РАССЕЯННОГО СКЛЕРОЗА ( РС ) | 2011 |
|
RU2595861C2 |
| Способ ингибирования нуклеарного фактора каппа В с использованием 2-этил-6-метил-3-гидроксипиридиния L-2,6-диаминогексаноата в культуре клеток | 2017 |
|
RU2669348C1 |
| Способ ингибирования нуклеарного фактора каппа В с использованием 5-гидрокисиникотинат 3-(2,2,2-триметилгидразиний) пропионата калия в культуре клеток | 2017 |
|
RU2674443C1 |
| Способ ингибирования нуклеарного фактора каппа В с использованием 5-гидроксиникотината калия в культуре клеток | 2017 |
|
RU2675693C1 |
| БОЛЕУТОЛЯЮЩАЯ КОМПОЗИЦИЯ И СПОСОБ ДОСТИЖЕНИЯ БОЛЕУТОЛЯЮЩЕГО ЭФФЕКТА У ЧЕЛОВЕКА | 1993 |
|
RU2125873C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМБИНАЦИЯ И ЕЕ ПРИМЕНЕНИЕ | 2011 |
|
RU2575775C2 |
| ОМЕГА-АМИНОАЛКИЛАМИДЫ R-2-АРИЛПРОПИОНОВЫХ КИСЛОТ В КАЧЕСТВЕ ИНГИБИТОРОВ ХЕМОТАКСИСА ПОЛИМОРФНОЯДЕРНЫХ И ОДНОЯДЕРНЫХ КЛЕТОК, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2272024C2 |
| 2-АРИЛПРОПИОНОВЫЕ КИСЛОТЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2002 |
|
RU2317075C2 |
| АМИДИНЫ И ИХ ПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2004 |
|
RU2375346C2 |

Изобретение относится к области фармации, а именно к применению (R)-энантиомеров арилпропионовых кислот. Сущность изобретения - применение названных кислот для получения лекарственных средств, которые ингибируют каскад активации NF-κB. На основе их создана композиция. Технический результат - использование для лечения заболеваний, на которые может терапевтически положительно воздействовать ингибирование образования NF-κB. 5 з.п. ф-лы, 4 ил.
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |

