Настоящее изобретение относится к омега-аминоалкиламидам (R)-2-арилпропионовых кислот в качестве ингибиторов хемотаксиса полиморфноядерных и одноядерных клеток. В частности, изобретение относится к C5a-индуцированному хемотаксису полиморфноядерных лейкоцитов и моноцитов, используемых при лечении патологических нарушений, включающих псориаз, ревматоидный артрит и нарушение, вызванное ишемией и реперфузией.
Исследования на животных показали, что некоторые аминоалкиловые сложноэфирные и амидные пролекарства рацемического ибупрофена и напроксена, в частности некоторые N-(3-диэтиламинопропил)амиды, обладают значительно большей обезболивающей и противовоспалительной активностью, чем исходные соединения, даже если установлено, что они являются слабыми ингибиторами синтеза простагландинов "in vitro". Найдено также, что все указанные пролекарства, за исключением глицинамида, значительно меньше раздражают слизистую оболочку желудка, чем их предшественники - свободные кислоты. (Shanbhag VR et al., J. Pharm. Sci., 81, 149, 1992 и приведенные ссылки 8-19).
Пикетопрофен [(±)-2-(3-бензоилфенил)-N-(4-метил-2-пиридинил)пропионамид] и амтолметингваяцил (называемый также гваяколовым эфиром толметинглицинамида, Eufans) являются дополнительными примерами нестероидных противовоспалительных (NSAI) пролекарств, находящих рассматриваемое терапевтическое применение. Умеренная противовоспалительная активность, незначительные побочные эффекты и хорошая желудочно-кишечная толерантность обнаружены для ряда N-[2-(1-пиперидинил)пропил]амидов некоторых NSAI-пролекарств, таких как рацемический ибупрофен, индометацин, п-хлорбензойная кислота, ацетилсалициловая кислота, диацетилгентизиновая кислота и адамантан-1-карбоновая кислота (Nawladonski F. and Reewuski, Pol. J. Chem., 52, 1805, 1978). Другие амиды рацемических 2-арилпропионовых кислот описаны в S. Biniecki et al., [PL 114050 (31. 01. 1981)], H. Akguen et al., [Arzneim-Forsch., 46, 891, 1986] и G. L. Levitt et al., [Russ. J. Org. Chem., 34, 346, 1998].
Противовоспалительная и обезболивающая активность "in vivo", сравнимая с аналогичной активностью предшественников - свободных кислот, и иногда большая, вместе с пониженным числом случаев поражения желудка, обнаружены для некоторых N-3-[(1-пиперидинил)пропил]амидов рацемического кетопрофена и флурбипрофена и для некоторых оснований Манниха, полученных взаимодействием их амидов с формальдегидом и вторичными аминами, такими как морфолин, пиперидин, дициклогексиламин, диметиламин, диэтиламин, дибензиламин и дибутиламин (N. Kawathekar et al, Indian J. Pharm. Sci., 60, 346, 1998).
В международной патентной заявке WO 00/40088 недавно сообщалось, что простой конверсии до амидного производного 2-арилуксусной и/или 2-арилпропионовой кислоты достаточно для превращения селективного COX-1-ингибитора в COX-2-селективный ингибитор, что объясняет пониженную разлагаемость указанных амидов в желудке, длительное время считавшихся только NSAI-пролекарствами.
Ранее было установлено, что ингибирование ферментов циклооксигеназы свойственно самому S-энантиомеру 2-арилпропионовой кислоты, соединенному с частью R-CoA-тиоэфира, претерпевающего биоконверсию "in vivo". Поэтому, слабая корреляция между ферментативным ингибированием "in vitro" и обезболивающими действиями "in vivo", обнаруженная для некоторых R,S-2-арилпропионовых кислот (Brune K. et al., Experientia, 47, 257, 1991), вызвала предположение, что могут быть задействованы альтернативные механизмы, такие как ингибирование транскрипции kB-нуклеарного транскрипционного фактора (NF-kB) и/или ингибирование хемотаксиса нейтрофилов, индуцируемого интерлейкином-8 (IL-8).
R-энантиомеры флурбипрофена, кетопрофена, напроксена, тиапрофена и фенопрофена, фактически, описаны в WO 00/40088 как ингибиторы активации транскрипционного фактора NF-kB и заявлены для применения в лечении NF-kB-зависимых заболеваний (астма, опухоль, шок, болезнь Крона и неспецифический язвенный колит, атеросклероз и так далее). IL-8 является важным медиатором воспаления, и показано, что он является эффективным хемотаксическим/клеточным активатором для полиморфноядерных нейтрофилов и базофилов (PMNs) и T-лимфоцитов. Клеточные источники IL-8 включают моноциты, PMNs, эндотелиальные клетки, эпителиальные клетки и кератиноциты, когда стимулированы факторами, такими как липополисахарид, IL-1 и TNF-a. С другой стороны, найдено, что C5a фрагмент комплемента, в дополнение к тому, что является прямым медиатором воспаления, индуцирует как синтез IL-8, так и высокий уровень высвобождения IL-8 из моноцитов. Количество IL-8, выделенное из C5a-активированных моноцитов в одноядерных клетках периферической крови, 1000-кратно превосходит количество IL-8, высвобождаемое из сопоставимого числа PMNs в подобных условиях. Поэтому IL-8, высвобожденный из C5a-активированных моноцитов может играть существенную роль в развитии и пролонгировании клеточной инфильтрации и активации на участках инфекции, воспаления или повреждения ткани (Ember J.A. et al., Am. J. Pathol., 144, 393, 1994).
В ответ на иммунологическое и инфекционное события активация системы комплемента опосредует амплификацию воспалительной реакции как через прямое действие на мембрану, так и путем высвобождения ряда пептидных фрагментов, обычно известных как анафилатоксины, генерируемых путем ферментативного расщепления C3-, C4- и C5- фракций комплемента. Такие пептиды включают C3a, C4a, оба построенные из 77 аминокислот; в свою очередь, C5-конвертаза расщепляет фракцию комплемента C5, давая гликопротеин C5a из 74 аминокислот. Анафилатоксины участвуют в распространении воспалительного процесса путем взаимодействия с индивидуальными клеточными компонентами; общие их характеристики включают клеточное высвобождение вазоактивных аминов и лизосомальных ферментов, сокращение гладкой мышцы и повышенную сосудистую проницаемость. Кроме того, C5a вызывает хемотаксис и агрегацию нейтрофилов, стимулирует высвобождение лейкотриенов и окисленных кислородом видов, индуцирует транскрипцию IL-1 в макрофагах и продуцирование антител.
C5a-пептидный фрагмент комплемента называют "полным" провоспалительным медиатором. Напротив, другие воспалительные медиаторы, такие как селективные цитокины (IL-8, MCP-1 и RANTES, например) являются высоко селективными по отношению к самопритягивающимся клеткам, тогда как гистамин и брадикинин являются лишь слабыми хемотаксическими агентами. Убедительные доказательства подтверждают вовлечение C5a, "in vivo", в некоторые патологические состояния, включающие ишемию/реперфузию, аутоиммунный дерматит, мембранозно-пролиферативный идиопатический гломерулонефрит, гиперчувствительность дыхательных путей, и хронические воспалительные заболевания, такие как ARDS и COPD, болезнь Альцгеймера, болезнь Стилла (N.P. Gerard, Ann. Rev. Immunol., 12, 755, 1994).
В связи с нейровоспалительным потенциалом C5a/C5a-desArg, создаваемым как локальным продуцированием комплемента, так и активацией амилоидов, связанной с хемотаксисом астроцитов и микроглий, и активацией, непосредственно вызываемой C5a, ингибиторы комплемента могут быть предложены для лечения неврологических заболеваний, таких как болезнь Альцгеймера (McGeer & McGeer P.L., Drugs, 55, 738, 1998).
Поэтому считается, что регулирование локального синтеза фракций комплемента создает высокий терапевтический потенциал для лечения шока и предупреждения отторжения (отторжение, связанное с множественным повреждением, и сверхострое отторжение трансплантата) (Issekutz A.C. et al., Int. J. Immunopharmacol, 12, 1, 1990; Inagi R. et at., Immunol. Lett., 27, 49, 1991). Позднее поступило сообщение о том, что с учетом вовлеченности комплемента в патогенез как хронического интерстициального, так и острого гломерулярного почечных нарушений, ингибирование фракций комплемента ведет к предупреждению повреждений естественных и трансплантированных почек (Sheerin N.S. & Sacks S.H., Curr. Opinion Nephrol. Hypert., 7, 395, 1998).
Исследования на основе генной инженерии и молекулярной биологии привели к клонированию комплементарных рецепторов (CRs) и продуцированию CRs-агонистов и антагонистов. Рекомбинантный растворимый рецептор CR1 (sCRl), который блокирует ферменты, активирующие C3 и C5, идентифицирован как потенциальный агент для подавления C-активации при ишемическом/реперфузионном нарушении (Weisman H.F. et al., Science, 239, 146, 1990; Pemberton M. et al., J. Immunol., 150, 5104, 1993).
Сообщается, что циклический пептид F-[OPdChWR] противодействует связыванию C5a с рецептором CD38 на PMNs и ингибирует C5a-зависимый хемотаксис и продуцирование цитокинов макрофагами, и нейтропению, вызванную у крыс стимуляцией C5a и LPS (Short A. et al., Br. J. Pharmacol., 126, 551, 1999; Haynes D.R. et al., Biochem. Pharmacol., 60, 729, 2000). Сообщается, что как CGS 27913, антагонист C5aR, так и его димер CGS 32359, ингибируют, "in vitro", C5a-связывание с мембранами нейтрофилов, внутриклеточную мобилизацию Ca2+, высвобождение лизоцима, хемотаксис нейтрофилов и отек кожи у кроликов (Pellas T.C. et al., J. Immunol., 160, 5616, 1998).Наконец, селекция из библиотеки бактериофагов с помощью метода "индикации фагов" привела к выделению специфического антагониста C5aR, способного снижать воспалительную реакцию при заболеваниях, опосредованных иммунокомплексами, и при ишемии и реперфузионных нарушениях (Heller T. et al., J. Immunol., 163, 985, 1999).
Несмотря на терапевтический потенциал только два из вышеуказанных антагонистов C5a проявляют активность "in vivo"; к тому же терапевтическое применение данных антагонистов ограничено из-за их пептидной природы (Pellas T.C., Wennogle P., Curr. Pharm. Des., 10, 737, 1999).
Характерная аккумуляция нейтрофилов может наблюдаться при некоторых патологических нарушениях, например на сильно воспаленных и не поддающихся терапевтическому лечению участках псориатических поражений. Нейтрофилы хемотаксически притягиваются и активируются за счет синергического действия хемокинов, IL-8 и Gro-a, высвобождаемых стимулированными кератиноцитами, и фракции C5a/C5a-desArg, продуцируемой путем альтернативной активации комплементарных путей (T. Terui et al., Exp. Dermatol., 9, 1, 2000). Кроме того, во многих случаях весьма желательно комбинировать ингибирование хемотаксиса, индуцируемого C5a, и ингибирование хемотаксиса, индуцируемого IL-8, с помощью одного единственного средства.
Получены также непептидные антагонисты фракций комплемента, например, замещенные-4,6-диаминохинолины. В частности, найдено, что [N,N"-бис-(4-амино-2-метил-6-хинолил)]карбамид и [6-N-2-хлорциннамоил)-4,6-диамино-2-метилхинолин] являются селективными антагонистами C5R, их IC50 изменяются в пределах от 3,3 до 12 мкг/мл (Lanza T.J. et al., J. Med. Chem., 35, 252, 1992).
Недавно поступило сообщение о том, что некоторые ингибиторы серинпротеазы [нафамостатмезилат (FUT 175) и некоторые аналоги] являются ингибиторами как активации комплемента, так и продуцирования C3a/C5a (Ueda N. et al., Inflammation Res. 49, 42, 2000).
В патенте США 6069172 сообщается о применении фармацевтических составов аммониевых солей R(-)-кетопрофена для ингибирования хемотаксиса нейтрофилов, индуцируемого IL-8.
WO 00/24710 описывает N-ацилсульфонамиды R(-)-2-арилпропионовой кислоты как ингибиторы IL-8-зависимого хемотаксиса полиморфноядерных лейкоцитов.
Две недавно поступившие патентные заявки [WO 01/58852 и WO 01/79189] описывают некоторые R-2-арилпропионамиды и R-2-(аминофенил)пропионамиды, полезные для предупреждения активации лейкоцитов, индуцируемой IL-8.
Настоящими заявителями недавно обнаружено, что чисто формальное восстановление гетероароматического цикла некоторых R-2-арил-N-(пиридинил)пропионамидов вызывает заметную потерю активности (1 или 2 логарифмический порядок) в способности ингибировать хемотаксис PMN-нейтрофилов, индуцируемый IL-8. Неожиданно было обнаружено, что родственные R-2-арил-N-(пиперидинил)пропионамиды являются эффективными ингибиторами хемотаксиса PMN-лейкоцитов и моноцитов человека, индуцируемого фракцией C5a комплемента.
Эти неожиданные открытия основаны на новом семействе омега-аминоалкиламидов R-2-арилпропионовой кислоты, способных ингибировать хемотаксическую активность, индуцируемую C5a и другими хемотаксическими протеинами, чья биологическая активность связана с активацией рецептора 7-членного домена (7-TD), гомологичного рецептору C5a (например, рецептора C3a и рецептора CXCR2; Neote K. et al., Cell, 72, 415, 1993; Tornetta M.A., J. Immunol., 158,5277, 1997).
Настоящее изобретение относится к новому классу омега-аминоалкиламидов R-2-арилпропионовых кислот и содержащим их фармацевтическим композициям. Положение "омега" в алкильной цепи означает наиболее удаленный атом углерода, считая от атома N амидной группы, к которой присоединен указанный алкил. Такие амиды полезны для ингибирования хемотаксической активации, индуцируемой C5a и другими хемотаксическими протеинами, чья биологическая активность связана с активацией рецепторов 7-членных доменов (7-TD) гомологичных рецептору C5a. В частности, такие амиды полезны для ингибирования хемотаксической активации полиморфноядерных лейкоцитов, моноцитов и Т-лимфоцитов, индуцируемой C5a фракцией комплемента, и для лечения патологических нарушений, связанных с указанной активацией.
В следующих абзацах приведены определения выделяемых химических структур, из которых построены соединения по изобретению, предложенные для применения в целях единообразия в описании и приложенных пунктах, если не приведено точное определение, то используют более широкое определение.
Термин "алкил" означает одновалентные алкильные группы, имеющие предпочтительно 1-6 атомов углерода. Примерами указанных терминов служат такие группы, как метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил и тому подобное.
"Арил" означает ненасыщенную ароматическую карбоциклическую группу из 6-14 атомов углерода, имеющую единственный цикл (например, фенил) или многократно конденсированные циклы (например, нафтил). Предпочтительный арил включает фенил, дифенил, нафтил, фенантренил и тому подобное.
"Алкенил" означает алкенильные группы, предпочтительно имеющие 2-5 атомов углерода и один или более участков алкенильной ненасыщенности. Предпочтительные алкенильные группы включают этенил (-CH=CH2), н-2-пропенил (аллил, -CH2CH=CH2) и тому подобное.
"Алкилен", "алкенилен", алкинилен" означают группы, дизамещенные с обоих концов. Предпочтительные группы включают метилен, этилен, пропилен и тому подобное.
"Замещенный или незамещенный": если не оговорен особо отдельный заместитель, вышеуказанные группы, такие как группы "алкил", "алкенил", "арил" и прочее, необязательно могут быть замещены 1-5 заместителями, выбираемыми из группы, включающей "С1-C6-алкил", "С1-C6-алкиларил", "С1-C6-алкилгетероарил", "C2-C6-алкенил", первичные, вторичные или третичные аминогруппы или четвертичные аммониевые группы, "ацил", "ацилокси", "ациламино", "аминокарбонил", "алкоксикарбонил", "арил", "гетероарил", карбоксил, циано, галоген, гидрокси, меркапто, нитро, сульфокси, сульфонил, алкокси, тиоалкокси, тригалогенметил и тому подобное. Подразумевается, что в объеме данного изобретения указанное "замещение" включает также случаи, когда соседние заместители подвергаются замыканию цикла, в частности, когда вовлекаются вицинальные (смежные) функциональные заместители, образуя таким образом, например, лактамы, лактоны, циклические ангидриды или циклоалканы, а также ацетали, тиоацетали, аминали, полученные путем замыкания цикла, например, с целью образования защитной группы.
Термин "фармацевтически приемлемые соли" относится к солям или комплексам нижеуказанных соединений формулы I, сохраняющим требуемую биологическую активность. Примеры таких солей включают, но не в порядке ограничения, кислотно-аддитивные соли, образованные с неорганическими кислотами (например, хлористоводородной кислотой, бромистоводородной кислотой, серной кислотой, фосфорной кислотой, азотной кислотой и тому подобным), и соли, образованные с органическими кислотами, такими как уксусная кислота, щавелевая кислота, винная кислота, янтарная кислота, яблочная кислота, фумаровая кислота, малеиновая кислота, аскорбиновая кислота, бензойная кислота, дубильная кислота, памовая кислота, альгиновая кислота, полиглутаминовая кислота, нафталинсульфоновая кислота, нафталиндисульфоновая кислота и полигалактуроновая кислота.
Примеры солей также включают соли присоединения, образованные с неорганическими основаниями, такими как гидроксид натрия, и с органическими основаниями, такими как трометакин, L-лизин, L-аргинин и тому подобное.
Настоящее изобретение представляет соединения (R)-2-арилпропионамида формулы (I),
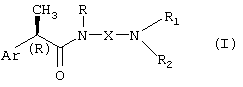
где
Ar означает замещенную или незамещенную арильную группу;
R означает водород, С1-C4-алкил, C2-C4-алкенил, C2-C4-алкинил, необязательно замещенный группой CO2R3, где R3 означает водород или линейную или разветвленную С1-C6-алкильную группу, или линейную или разветвленную C2-C6-алкенильную группу;
X означает:
линейный или разветвленный С1-C6-алкилен, C4-C6-алкенилен, C4-C6-алкинилен, необязательно замещенный группой CO2R3 или группой CONHR4, где R4 означает водород, линейный или разветвленный C2-C6-алкил, или группу OR3, где R3 принимает вышеуказанные значения;
(CH2)m-B-(CH2)n, группу, необязательно замещенную группой CO2R3 или CONHR4, принимающей вышеуказанные значения, где B означает атом кислорода или серы, m равно нулю или целому числу 2-3 и n означает целое число 2-3; или B означает группу CO, SO или CONH, m означает целое число 1-3 и n означает целое число 2-3;
либо X вместе с атомом азота омега-аминогруппы, с которым он связан, и с группой R1 образует неароматическое азотсодержащее 3-7-членное гетероциклическое, моноциклическое или полициклическое ядро, в котором атом азота имеет заместитель Rc, где Rc означает водород, С1-C4-алкил, С1-C4-гидроксилалкил, С1-C4-ацил, замещенный или незамещенный фенил, дифенилметил; R1 и R2 независимо выбирают из группы, включающей: водород, необязательно прерываемый атомом O или S линейный или разветвленный С1-C6-алкил, C3-C7-циклоалкил, C3-C6-алкенил, C3-C6-алкинил, арил-С1-C3-алкил, гидрокси-C2-C3-алкил;
либо R1 и R2 вместе с атомом N, с которым они связаны, образуют азотсодержащее 3-7-членное гетероциклическое ядро формулы (II)

где Y означает простую связь, CH2, O, S или группу N-Rc, принимающую вышеуказанные значения, и p означает целое число 0-3;
либо R1 принимает вышеуказанные значения, R2 означает группу формулы (III):
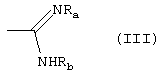
где Ra означает водород и Rb означает водород, гидрокси, С1-C4-алкил или группу NRdRe, где каждый из Rd и Re независимо означает водород, С1-C4-алкил или фенил;
или Ra и Rb, вместе с атомом азота, с которым они связаны, образуют 5-7-членное гетероциклическое ядро, моноциклическое или конденсированное с ядром бензола, пиридина или пиримидина;
при условии, что когда Ar означает 4-дифениловый остаток и X означает этиленовый или пропиленовый остаток, R1 и R2 не являются этилом;
при условии, что когда Ar означает 4-(2-фтор)дифениловый остаток и X означает замещенный CO2H группой бутилен, Ra и Rb не являются водородом;
и при дополнительном условии, что когда Ar означает фенил и X означает бутилен, R1 и R2 вместе не могут означать N-(2-метоксифенил)пиперазин.
Кроме того, настоящее изобретение также представляет соединения (R)-2-арилпропионамида формулы (I)
где
Ar означает замещенную или незамещенную арильную группу;
R означает водород, С1-C4-алкил, C2-C4-алкенил, C2-C4-алкинил, необязательно замещенный группой CO2R3, где R3 означает водород или линейную или разветвленную С1-C6-алкильную группу, или линейную или разветвленную C2-C6-алкенильную группу;
X означает:
линейный или разветвленный С1-C6-алкилен, C4-C6-алкенилен, C4-C6-алкинилен, необязательно замещенный группой CO2 R3 или группой CONHR4, где R4 означает водород, линейный или разветвленный C2-C6-алкил, или группу OR3, где R3 принимает вышеуказанные значения;
(CH2)m-B-(CH2)n, группу, необязательно замещенную группой CO2R3 или CONHR4, принимающей вышеуказанные значения, где B означает атом кислорода или серы, m равно нулю или целому числу 2-3 и n означает целое число 2-3; или B означает группу CO, SO или CONH, m означает целое число 1-3 и n означает целое число 2-3;
либо X вместе с атомом азота омега-аминогруппы, с которым он связан, и с группой R1 образует неароматическое азотсодержащее 3-7-членное гетероциклическое, моноциклическое или полициклическое ядро, в котором атом азота имеет заместитель Rc, где Rc означает водород, С1-C4-алкил, С1-C4-гидроксилалкил, С1-C4-ацил, замещенный или незамещенный фенил, дифенилметил;
R1 и R2 независимо выбирают из группы, включающей: водород, необязательно прерываемый атомом O или S линейный или разветвленный С1-C6-алкил, C3-C7-циклоалкил, C3-C6-алкенил, C3-C6-алкинил, арил-С1-C3-алкил, гидрокси-C2-C3-алкил;
либо R1 и R2 вместе с атомом N, с которым они связаны, образуют 3-7-членное азотсодержащее гетероциклическое ядро формулы (II)
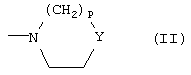
где Y означает простую связь, CH2, O, S или группу N-Rc, принимающую вышеуказанные значения, и p означает целое число 0-3;
либо R1 принимает вышеуказанные значения, R2 означает группу формулы (III):
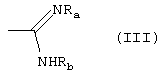
где Ra означает водород и Rb означает водород, гидрокси, С1-C4-алкил или группу NRdRe, где каждый из Rd и Re независимо означает водород, С1-C4-алкил или фенил; или Ra и Rb, вместе с атомом азота, с которым они связаны, образуют 5-7-членное гетероциклическое ядро, моноциклическое или конденсированное с ядром бензола, пиридина или пиримидина;
в качестве ингибиторов C5a-индуцируемого хемотаксиса полиморфноядерных лейкоцитов и моноцитов.
Фармацевтически приемлемые соли соединений формулы (I) также входят в объем настоящего изобретения.
Примеры арильных групп предпочтительно включают:
a) Ara - моно- или полизамещенную арильную группу или группы наиболее распространенных в настоящее время для терапевтического применения содержащих гетероциклические кольца 2-арилпропионовых кислот, выбираемых из группы, включающей: альминопрофен, беноксапрофен, карпрофен, фенбуфен, фенопрофен, флурбипрофен, ибупрофен, индопрофен, кетопрофен, локсопрофен, напроксен, пирпрофен и его дегидро- и дигидро- производные, пранопрофен, сурпрофен, тиапрофеновую кислоту, залтопрофен;
b) арилгидроксиметиларильную группу формулы (IVa), полученную восстановлением фенонкарбонила 2-арилпропионовых кислот, выбираемых из группы, включающей: кетопрофен, сурпрофен, тиапрофеновая кислота, как в виде отдельного (S',R) и/или (R',R) диастереомера, так и в виде диастереомерной смеси,
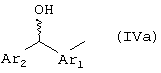
где в случае, когда Ar2 означает фенил, Ar1 выбирают из группы, включающей фенил и тиен-2-ил, и, когда Ar1 означает фенил, Ar2 выбирают из группы, включающей фенил, 4-тиенил, пиридил,
c) арил формулы (IVb):
φ-Arb (IVb),
где Arb означает фенил, моно- и полизамещенный необязательно замещенным гидрокси, меркапто, С1-C3-алкокси, С1-C3-алкилтио, хлором, фтором, трифторметилом, нитро, амино, необязательно замещенным С1-C7-ациламино; и φ означает водород; линейный или разветвленный С1-C5-алкильный, C2-C5-алкенильный или C2-C5-алкинильный остаток, (необязательно замещенный) С1-C3-алкоксикарбонилом, замещенным или незамещенным фенилом, 2-, 3- или 4-пиридилом, хинолин-2-илом; C3-C6-циклоалкильную группу; 2-фурил; 3-тетрагидрофурил; 2-тиофенил; 2-тетрагидротиофенил или остаток формулы (IVc)
A-(CH2)q- (IVc),
где A означает С1-C5-диалкиламиногруппу, С1-C8-(алканоил, циклоалканоил, арилалканоил)-С1-C5-алкиламиногруппу, например диметиламино, диэтиламино, метил-N-этиламино, ацетил-N-метиламино, пивалоил-N-этиламино; азотсодержащий 5-7-членный моноцикл, необязательно содержащий одну или две двойных связи и необязательно содержащий дополнительный гетероатом, отделенный, по меньшей мере, 2 атомами углерода от атома N, так, что образуется, например, группа: 1-пирролидино, 2,5-дигидропиррол-1-ил, 1-пиррол, 1-пиперидино, 1-пиперазино-4-незамещенный или 4-замещенный (метил, этил, 2-гидроксиэтил, бензил, бензгидрил или фенил), 4-морфолино, 4-3,5-диметилморфолино, 4-тиоморфолино; или альтернативно, остаток формулы (IVd)
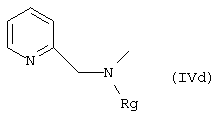
где Rg означает водород, С1-C3-алкил или остаток С1-C3-алкановой кислоты; q означает нуль или целое число 1,
d) 2-(фениламино)фенил формулы (IVe):
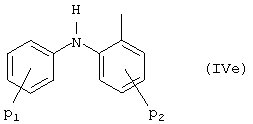
где P1 и P2 указывают, что две фенильных группы могут быть замещены независимо одной или более С1-C4-алкильными группами, С1-C3-алкоксигруппами, хлором, фтором и/или трифторметилом.
Предпочтительными соединениями в соответствие с изобретением являются соединения, в которых:
R означает водород,
X означает:
линейный алкилен, необязательно замещенный по С1 группой -CO2R3, принимающей вышеуказанные значения; линейным алкиленом, необязательно замещенным по С1 группой -CONHR4, где R4 означает OH; 2-бутинилен, цис-2-бутенилен, транс-2-бутенилен; 3-оксапентилен, 3-тиопентилен, 3-оксагексилен, 3-тиогексилен; (CH2)m-CO-NH-(CH2)n-, где каждый из m и n независимо означает целое число 2-3; (CHR')-CONH-(CH2)n, где n означает целое число 2-3 и R' означает метил, в абсолютной конфигурации R или S;
или X вместе с атомом N омега-аминогруппы образует азотсодержащее циклоалифатическое ядро, предпочтительно 1-метил-пиперидин-4-ил или 1,5-тропан-3-ил.
Предпочтительными соединениями являются также те, в которых NR1R2 означает группу NH2, диметиламино, диэтиламино, диизопропиламино, 1-пиперидинил, 4-морфолил, 4-тиоморфолил, или R1 и R2 вместе образуют остаток гуанидина, аминогуанидина, гидроксигуанидина, 2-амино-3,4,5,6-тетрагидропиримидила, 2-амино-3,5-дигидроимидазолила.
Примеры в особенности предпочтительных арильных групп включают:
4-изобутилфенил, 4-циклогексилметилфенил, 4-(2-метил)аллилфенил, 3-феноксифенил, 3-бензоилфенил, 3-ацетилфенил, отдельные диастереомеры (R) (S) и смесь диастереомеров (R,S), выбираемых из группы, включающей: 3-C6H5-CH(OH)-фенил, 3-CH3-CH(OH)-фенил, 5-C6H5-CH(OH)-тиенил, 4-тиенил-CH(OH)-фенил, 3-(пирид-3-ил)-CH(OH)-фенил, 5-бензоилтиен-2-ил, 4-тиеноилфенил, 3-никотиноилфенил, 2-фтор-4-фенил, 6-метокси-2-нафтил, 5-бензоил-2-ацетоксифенил и 5-бензоил-2-гидроксифенил.
В особенности предпочтительными арильными группами формулы (IVb) являются фенильные группы, 3-замещенные изопроп-1-ен-1-илом, изопропилом, пент-2-ен-3-илом; пент-3-илом; 1-фенилэтилен-1-илом; α-метилбензилом.
В особенности предпочтительными арилами формулы (IVc) являются 4-(пирролидин-1-ил)метилфенил, 3-хлор-4-(пирролидин-1-ил)метилфенил, 3-хлор-4-(2,5-дигидро-1-H-пиррол-1-ил)метилфенил, 3-хлор-4-(тиоморфолин-4-ил)фенил; 3-хлор-4-(пиперидин-1-ил)фенил, 4-((N-этил-N-хинолин-2-илметиламино)метил)фенил, 3-хлор-4-(морфолин-4-ил)фенил.
В особенности предпочтительными арилами формулы (IVe) являются 2-(2,6-дихлорфениламино)фенил; 2-(2,6-дихлорфениламино)-5-хлорфенил; 2-(2,6-дихлор-3-метилфениламино)фенил; 2-(3-трифторметилфениламино)фенил.
В особенности предпочтительными соединениями по изобретению являются:
(R)-2-[(4-изобутил)фенил]-N-(3-диметиламинопропил)пропионамид;
(R)-2-[(4-изобутил)фенил]-N-(4-диметиламинобутил)пропионамидгидрохлорид;
(R)-2-[(4-изобутил)фенил]-N-(3-N-морфолинилпропил)пропионамид;
(R)-2-[(4-изобутил)фенил]-N-(2-диметиламиноэтил)пропионамид;
(R)-2-[(4-изобутил)фенил)пропионил]-N-[2-(4-метилпиперазин-1-ил)этил]пропионамид;
(R)-N-(экзо-8-метил-8-аза-бицикло[3,2,1]окт-3-ил)-2-[(4-изобутилфенил)пропионамид;
(R)-2-[(4-изобутил)фенил]-N-(3-N-тиоморфолинилпропил)пропионамид;
(R)-2-[(4-изобутил)фенил]-N-[4-(N'-метил)пиперидинил]пропионамидгидрохлорид;
(R),(S')-2-[(4-изобутил)фенил]-N-(1-карбокси-2-диметиламиноэтил)пропионамид;
(R),(S')-2-[(4-изобутил)фенил]-N-[(1-карбокси-4-пиперидин-1-ил)бутил]пропионамид;
(R),(S')-2-[(4-изобутил)фенил]-N-(1-карбокси-4-аминобутил)пропионамид;
(R)-2-(4-изобутил)фенил-N-[2-(диметиламиноэтил)аминокарбонилметил]пропионамидгидрохлорид;
2-(2,6-дихлорфениламино)фенил-N-(3-диметиламинопропил)пропионамид;
(R),(R',S')-3-[3-(α-метил)бензил]фенил-N-(3-диметиламинопропил)пропионамид;
(R)-2-[(3-изопропил)фенил]-N-(3-диметиламинопропил)пропионамид;
(R)-2-[3-(пент-3-ил)фенил]-N-(3-диметиламинопропил)пропионамид;
(R)-2-[(4-изобутил)фенил]-N-(3-гуанидилпропил)пропионамид;
(R)-2-[(4-изобутил)фенил]-N-[(3-гидроксигуанидил)пропил]пропионамид;
(R)-2-[(4-изобутил)фенил]-N-[(3-аминогуанидил)пропил]пропионамид;
(R)-2-[(4-изобутил)фенил]-N-[3-(2-амино-2-имидазолин)пропил]пропионамид;
(R)-2-[(4-изобутил)фенил]-N-[N-метил-N-(2-гидроксиэтил)аминоэтокси]пропионамид;
(R),(S')-2-[(4-изобутил)фенил]-N-[1-карбокси-5-аминопентил]пропионамид.
Получение соединений формулы (I) может быть осуществлено посредством известных методик, таких как взаимодействие активированной формы R-2-арилпропионовой кислоты формулы (V) с амином формулы (VI) в условиях отсутствия рацемизации, предпочтительно в присутствии молярного избытка основания:
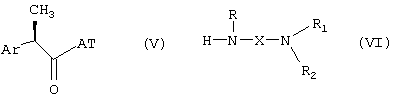
где
AT означает остаток, активирующий карбоксигруппу. Примерами активированных форм 2-арилпропионовой кислоты формулы (V, AT=OH) являются хлориды (AT=Cl), имидазолиды (AT=1-имидазол), сложные эфиры фенолов, таких как п-нитрофенол (AT=п-NO2-C6H4O-), или активированные формы, полученные взаимодействием в присутствии 1-гидроксибензотриазола (HOBZ) или карбодиимида, например дициклогексилкарбодиимида.
Ar, R, X, R1 и R2 принимают значения вышеуказанных групп, необязательно защищенных, где необходимо.
Взаимодействие активированной формы 2-арилпропионовой кислоты формулы (V) с защищенным амином формулы (VI) обычно выполняют при комнатной температуре, используя стандартные протонные или апротонные растворители и/или их смеси, предпочтительно безводные растворители, например, такие как метилацетат, этилацетат, этилформиат, нитрилы, такие как ацетонитрил, линейные или циклические простые эфиры, такие как диэтиловый эфир, сульфолан, диоксан, тетрагидрофуран, амиды, такие как диметилформамид, формамид, галогенированные растворители, такие как дихлорметан, ароматические углеводороды, такие как толуол, хлорбензол, или гетероароматические углеводороды, такие как пиридин и пиколин. Взаимодействия могут быть выполнены в присутствии основания; предпочтительными неорганическими основаниями являются карбонаты и бикарбонаты щелочных и щелочноземельных металлов, таких как, например, тонкоизмельченный карбонат калия, бикарбонат калия и карбонат магния и/или кальция. Полученные защищенные амиды могут быть превращены в амиды формулы (I) путем расщепления защитных групп и любых сложноэфирных групп, которые могут присутствовать. В особенности предпочтительным сложным эфиром такого типа является сложный аллиловый эфир, который может быть удален в строго выбранных условиях, например, через перенос аллильной группы к молекуле морфолина, которая в присутствии Pd(0) в качестве катализатора действует как переносчик H и как нуклеофильный акцептор, согласно методике, описанной в J. Org. Chem., 54, 751 1989.
Амиды формулы (I), где R2 означает группу формулы (III), могут быть получены взаимодействием первичных и вторичных аминов формулы (I) с изотиоуреидом или соответствующими солями изотиоурония формулы (IIIa)
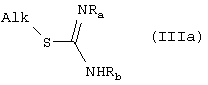
где Alk означает С1-C3-алкил и Ra и Rb принимают вышеуказанные значения.
Получение гидроксиизотиокарбамидов формулы (IIIa), где Rа означает OH и Rb означает H, описано в Bernd Clement, Arch. Pharm. (Wheineim) 319, 968 (1986); другие соединения формулы (IIIa) являются известными соединениями и могут быть получены общепринятыми способами алкилирования в основной среде соответствующих линейных и/или циклических тиокарбамидов и тиосемикарбазидов. Соединения формулы (IIIa) выделяют в виде солей изотиоурония и указанные соли могут быть подвергнуты взаимодействию с аминами формулы Ie по способу, описанному Bodansky M. et al., J. Am. Chem. Soc., 86, 4452, 1964. Альтернативно, избыток растворителя, такого как этилацетат (AcOEt), добавляют к водному раствору или суспензии соли изотиоурония формулы (IIIa) и при энергичном перемешивании соль нейтрализуют добавлением эквивалентного основного раствора (NaOH н, карбонат калия н), получая соответствующий изотиоуреид.
Амиды формулы (Ia)
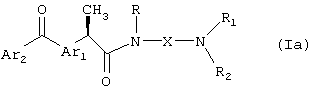
где Ar1, Ar2, X, R, R1 и R2 принимают вышеуказанные значения, можно подвергать взаимодействию по фенонкарбонильной группе, что приводит к диастереомерной паре R',S'-спиртов, которые необязательно разделяют фракционированной кристаллизацией и/или препаративной хроматографией, получая индивидуальные диастереомеры формулы (Ib):
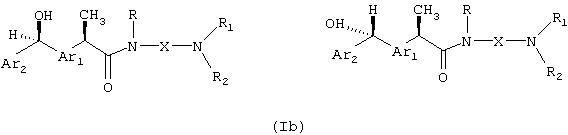
Принято обозначать абсолютной конфигурацией S' наиболее полярный диастереомер.
Соединения формулы (I) могут быть превращены в фармацевтически приемлемые соли за счет образования солей по присутствующим в структуре основным и кислотным группам путем использования, соответственно, фармацевтически приемлемых кислот или оснований. Примерами солей фармацевтически приемлемых оснований являются соли щелочных и щелочно-земельных металлов, предпочтительно лития, натрия и магния, или органических оснований, таких как трометанин, D-глюкозамин, лизин, аргинин.
Соединения формулы (I) обычно выделяют в форме их аддитивных солей как органических, так и неорганических фармацевтически приемлемых кислот. Примерами таких кислот являются: хлористоводородная, азотная, серная, фосфорная, муравьиная, уксусная, трифторуксусная, пропионовая, малеиновая и янтарная, малоновая и метансульфоновая, D и L-винные кислоты.
R-энантиомеры 2-арилпропионовых кислот формулы (Va):
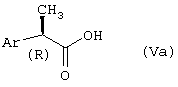
где Ar принимает вышеуказанные значения, являются слабыми ингибиторами циклооксигеназ и представляют собой обычно известные соединения.
Кислоты формулы (Vb):
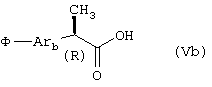
где φ и Arb принимают вышеуказанные значения, получают алкилированием станнанами полизамещенной 2-фенилпропионовой кислоты, несущей в орта-, мета- или параположении перфторбутансульфонатную группу, как описано ниже.
Соединения формулы (Vb) описаны в международной патентной заявке WO 01/58852.
В частности, 2-[3'-изопропил)фенил]пропионовая,
2-[3'-(α-метил)бензил)фенил]пропионовая и
2-[3'-(3-изопентил)фенил]пропионовая кислоты входят в число предпочтительных предшественников амидов формулы (I).
Каждая 2-арилпропионовая кислота может быть получена общим и стереоспецифическим синтезом или конверсией рацемата в один из индивидуальных энантиомеров после превращения в 2-арил-2-пропилкетены, как описано Larse R.D. et al., J. Am. Chem. Soc., 111, 7650, 1989 и Myers A.G., там же, 119, 6496, 1997. Стереоселективный синтез 2-арилпропионовых кислот обычно направлен на S-энантиомеры, но может быть легко изменен в целях получения R-энантиомеров путем выбора подходящего хирального вспомогательного агента. Применение арилалкилкетонов в качестве реагентов в синтезе α-арилалкановых кислот описано, например, B.M. Trost and J.H. Rigby, J. Org. Chem., 14, 2926, 1978; арилирование кислот Meldrum'a описано в J.T. Piney and R.A. Rowe, Tetrah. Lett., 21, 965, 1980; применение винной кислоты в качестве хирального вспомогательного агента в G. Castaldi et al., J. Org. Chem., 52, 3019, 1987; о применении сложных α-гидроксиэфиров в качестве хиральных реагентов сообщается в R.D. Larsen et al., J. Am. Chem. Soc., 111, 7650, 1989 и США 4.940.813, и приведенных там ссылках.
Способ получения 2-(2-OH-фенил)пропионовых кислот и их сложных эфиров описан в патенте Италии № 1283649. Испытанный и эффективный способ получения R-энантиомера (R,S)-2-(5-бензоил-2-ацетокси)пропионовой кислоты и кислот формулы (Vb), описанных выше, состоит в конверсии хлорангидридов указанных проп-1-кетеновых кислот путем взаимодействия с третичным амином, таким как диметилэтиламин, при последующем взаимодействии с R(-)-пантолактоном, что приводит к сложным эфирам R-энантиомеров указанных кислот с R-дигидро-3-гидрокси-4,4-диметил-2(3H)-фуран-2-оном. Последующее омыление сложного эфира с помощью LiOH дает соответствующую свободную кислоту.
Общий способ получения R(-)-2-арилпропионовых кислот формулы (Vb) включает взаимодействие гидроксиарилкетонов формулы (Vc), моно- или полизамещенных, с перфторбутансульфонилфторидом, приводящее к перфторбутансульфоновым эфирам формулы (Vd), где n означает целое число 1-9.
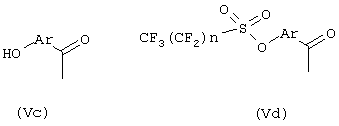
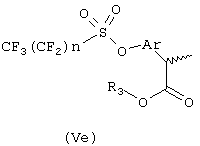
Соединения формулы (Vd) подвергают перегруппировке Willgerodt'a, получая после этерификации и метилирования по альфа углероду арилпропионовые производные формулы (Ve), где n означает целое число 1-9 и R3 означает С1-C4-алкил или C2-C4-алкенил.
Соединения формулы (Ve) подвергают взаимодействию с соответствующим трибутилстаннаном формулы Bu3SnR5, где R5 означает линейный или разветвленный С1-C6-алкил, линейный или разветвленный C2-C6-алкенил или линейный или разветвленный C2-C6-алкинил, незамещенный или замещенный арильной группой, получая соответствующие (R,S)-2-арилпропионаты формулы (Vf).
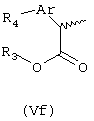
Алкенильная или алкинильная группы могут быть гидрированы в условиях каталитического гидрирования, что приводит к получению соответствующих насыщенных алкильных групп. Соединения формулы (Vf) подвергают процессу дерацемизации, как описано выше, состоящему в превращении соответствующих хлорангидридов кислот в кетены, которые путем взаимодействия с R(-)-пантонолактоном и последующего гидролиза превращают в чистые R-энантиомеры.
Амины формулы (VI) являются известными продуктами, чаще всего выпускаемыми промышленно, или могут быть получены известными способами. Синтез 4-диалкиламино-2-бутиниламина и из него цис- и транс-4-диалкиламино-2-бутениламина описан в R. Dalhome et al., J. Med. Chem., 9, 843, 1966 и T. Singh et al. там же, 12, 368, 1969, соответственно.
α-Аминокислоты с аминогруппой формулы -NR1'R2', связанной с концевым атомом углерода, получают известными способами, исходя из 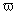 -гидрокси-α-аминокислот, карбокси- и аминогруппы которых соответственно защищены. Спиртовую группу превращают в бромид путем взаимодействия с трифенилфосфином и CBr4 (RG Weiss et al., J. Org. Chem. 36, 403, 1971 и M. Kang., там же, 64, 5528, 1966) с последующим взаимодействием полученного таким образом галогенида с, по меньшей мере, 2M избытком требуемого амина (т.е. диметиламина, пиперидина). Подходящими промышленными субстратами для данных целей являются серин и гомосерин: вышеуказанные гомологи получают исходя из α-аминокислот, защищенных по С1 и аминогруппе, свободную карбоксигруппу которых селективно восстанавливают до спирта путем восстановления в ТГФ при комнатной температуре избытком диборана.
-гидрокси-α-аминокислот, карбокси- и аминогруппы которых соответственно защищены. Спиртовую группу превращают в бромид путем взаимодействия с трифенилфосфином и CBr4 (RG Weiss et al., J. Org. Chem. 36, 403, 1971 и M. Kang., там же, 64, 5528, 1966) с последующим взаимодействием полученного таким образом галогенида с, по меньшей мере, 2M избытком требуемого амина (т.е. диметиламина, пиперидина). Подходящими промышленными субстратами для данных целей являются серин и гомосерин: вышеуказанные гомологи получают исходя из α-аминокислот, защищенных по С1 и аминогруппе, свободную карбоксигруппу которых селективно восстанавливают до спирта путем восстановления в ТГФ при комнатной температуре избытком диборана.
Настоящее изобретение относится к соединениям формулы (I), являющимся R-энантиомерами 2-арилпропионамидов, пригодным в качестве лекарственных средств.
Соединения по изобретению формулы (I) оценивают "in vitro" на их способность ингибировать хемотаксис полиморфноядерных лейкоцитов (здесь далее обозначенных PMNs) и моноцитов, индуцируемый фракциями C5a и C5a-desArg комплемента. С этой целью для выделения PMNs из гепаринизированной крови человека, отобранной у здоровых взрослых добровольцев, мононуклеары удаляют путем осаждения на декстране (согласно методике, описанной в W.J. Ming et al., J. Immunol., 138, 1469, 1987), а эритроциты с помощью гипотонического раствора. Жизнеспособность клеток оценивают путем исключения с помощью трипанового синего, тогда как долю PMNs оценивают по цитоцентрифугату после окрашивания с помощью Diff Quick.
Фракции hr-C5a и hrC5a-desArg (Sigma) используют как стимулирующие агенты в экспериментах на хемотаксис, получая практически идентичные результаты.
Лиофилизованный C5a растворяют в объеме HBSS, содержащем 0,2% BSA, так, чтобы получить основной раствор с концентрацией 10-5 M, подлежащий разбавлению HBSS до концентрации 10-9 M, для анализа на хемотаксис. В экспериментах на хемотаксис PMNs инкубируют с соединениями по изобретению формулы (I) в течение 15 минут при 37°C в атмосфере, содержащей 5% CO2.
Хемотаксическую активность C5a оценивают на циркулирующих полиморфонуклеарах (PMNs) человека, ресуспендированных в HBSS при концентрации 1,5×106 PMNs на мл.
В ходе анализа на хемотаксис (согласно W. Falket et al., J. Immunol. Methods, 33, 239, 1980) используют не содержащие PVP фильтры с пористостью 5 мкм и подходящие для проведения испытания микрокамеры.
Соединения по настоящему изобретению формулы (I) оценивают при интервале концентраций от 10-6 до 10-10 M; с этой целью их добавляют при одной и той же концентрации в нижнюю и верхнюю пористые части микрокамеры. Лунки в нижней части содержат раствор C5a или простой носитель, лунки в верхней части содержат суспензию в PMNs.
Ингибирование C5a-индуцируемой хемотаксической активности отдельными соединениями по настоящему изобретению формулы (I) оценивают, инкубируя микрокамеру для хемотаксиса в течение 60 мин при 37°C и в атмосфере, содержащей 5% CO2. Оценку способности соединений по изобретению формулы (I) ингибировать C5a-индуцируемый хемотаксис моноцитов человека проводят согласно вышеуказанной методике (Van Damme J. et al., Eur. J. Immunol., 19, 2367, 1989). Ингибирование C5a-индуцируемой хемотаксической активности отдельными соединениями по настоящему изобретению формулы (I) по отношению к моноцитам человека оценивают в интервале концентраций от 10-6 до 10-10 M, инкубируя микрокамеру для хемотаксиса в течение 120 мин при 37°C в атмосфере, содержащей 5% CO2.
Соединения по изобретению могут также быть оценены на их способность ингибировать IL-8-индуцируемый хемотаксис PMNs человека. С этой целью используют рекомбинантный интерлейкин-8 человека (rhIL-8, Pepro Tech): лиофилизованный протеин растворяют в HBSS (сбалансированный солевой раствор Ханка) при концентрации 100 мкг/мл и затем разбавляют до концентрации 10 нг/мл в экспериментах на хемотаксис. R(-)-2-[(4'-изобутил)фенил]пропионилметансульфонамид (ED50=10-9 M), описанный в WO 00/24710, используют в качестве эталона.
Результаты по ингибированию хемотаксиса, индуцируемого C5a и IL-8, приведены в таблице 1. Результаты показывают, что различные структуры амидной группы могут приводить к различной селективности соединений по настоящему изобретению.
Выбранный ряд соединений представляет двойные ингибиторы, ингибирующие хемотаксис, индуцируемый как C5a, так и IL-8, остальные являются селективными ингибиторами хемотаксиса индуцируемого C5a. Например, N-(1-метилпирид-4-ил)амиды, β-тропиламиды, N-(H2N-алкил)амиды формулы (I) - все являются селективными ингибиторами C5a-индуцируемого хемотаксиса PMN и моноцитов в интервале концентраций от 10-6 до 10-8 M. Все эти соединения проявляют слабую активность в качестве ингибиторов интерлейкин-8-индуцируемого хемотаксиса в том же интервале концентраций.
Выбранный ряд соединений по изобретению способен также ингибировать интерлейкин-8-индуцируемый хемотаксис PMN-лейкоцитов и T-лимфоцитов в дополнение к C5a-индуцируемому хемотаксису PMN-лейкоцитов и моноцитов в интервале концентраций от 10-6 до 10-8 M. В частности, соединения формулы (I), где R1 и R2 отличны от водорода, проявляют ингибирующую активность как в отношении C5a-индуцируемого хемотаксиса, так и IL-8-индуцируемого хемотаксиса. Обе активности присущи соединениям, у которых расстояние между концевым основным азотом и амидным азотом составляет 2-4 атома, при оптимальном значении n=3. Было установлено, что при таком структурном скелете соединения по изобретению играют двойную роль, ингибиторов C5a-индуцируемого хемотаксиса и IL-8-индуцируемого хемотаксиса.
Найдено, что соединения формулы (I), оцененные ex vivo в крови в целом по методике, описанной в Patrignani et al., J. Pharmacol. Exper. Ther., 271, 1705, 1994, совершенно не эффективны в качестве ингибиторов ферментов COX.
Почти во всех случаях соединения формулы (I) не препятствуют продуцированию PGE2, индуцируемому в макрофагах мышей путем стимуляции липополисахаридами (LPS, 1 мкг/мл) в интервале концентраций от 10-6 до 10-7 M. Ингибирование продуцирования PGE2, которое может быть зарегистрировано, по большей части находится в пределах статистической ошибки и чаще всего составляет ниже 15-20% от исходного уровня.
С учетом обсуждаемых выше экспериментальных данных и роли активации комплемента, через его C5a-фракцию, в патологических нарушениях, таких как псориаз (R.J. Nicholoff et al., Am. J. Pathol., 138, 129, 1991), пузырчатка и пемфигоид, ревматоидный артрит (M. Selz et al., J. Clin. Invest., 87, 463, 1981), хронические воспалительные патологии кишечника, такие как неспецифический язвенный колит (Y. R. Mahida et al., Clin. Sci., 82, 273, 1992), острый респираторный дистресс-синдром, муковисцидоз и идиопатический фиброз (E. J. Miller, предыдущая ссылка, и P. C. Carry et al., J. Clin. Invest., 88, 1882, 1991), хроническое обструктивное заболевание легких (COPD), гломерулонефрит (T. Wada et al., J. Exp. Med., 180, 1135, 1994), а также в предупреждении и лечении нарушения, вызванного ишемией или реперфузией, соединения по настоящему изобретению представляются в особенности полезными для достижения указанных терапевтических целей.
Таким образом, настоящее изобретение относится к соединениям формулы (I), пригодным для лечения патологий, включающих псориаз, пузырчатку и пемфигоид, ревматоидный артрит, хронические воспалительные патологии кишечника, включающие неспецифический язвенный колит, острый респираторный дистресс-синдром, системный и идиопатический фиброз легких, муковисцидоз, хроническое обструктивное легочное нарушение, гломерулонефрит, а также в предупреждении и лечении нарушения, вызываемого ишемией и реперфузией.
Изобретение также касается применения соединений формулы (I) в промышленном получении лекарственных препаратов для лечения и предупреждения указанных патологий. Соединения по изобретению вместе с обычно используемым вспомогательным средством, носителем, разбавителем или наполнителем могут быть представлены в форме фармацевтических композиций и их стандартных дозировок, и в такой форме могут быть использованы в виде твердых веществ, таких как таблетки или заполненные капсулы, либо жидкостей, таких как растворы, суспензии, эмульсии, эликсиры или заполненные ими капсулы, все для перорального применения, либо в форме стерильных растворов для инъекции для парентерального (включая подкожное) применения. Такие фармацевтические композиции и их дозированные лекарственные формы могут включать ингредиенты в общепринятых пропорциях, включая или не включая дополнительные активные соединения или действующие начала лекарственного вещества, и такие дозированные лекарственные формы могут содержать любое подходящее эффективное количество активного ингредиента, соизмеримое с рассчитанным на применение предполагаемым диапазоном суточных доз.
При использовании в качестве фармацевтических средств амиды по настоящему изобретению обычно вводят в форме фармацевтической композиции. Такие композиции могут быть получены хорошо известными в области фармации способами и включают, по меньшей мере, одно активное соединение. Обычно соединения по настоящему изобретению вводят в фармацевтически эффективном количестве. Реально вводимое количество соединения обычно определяется лечащим врачом с учетом соответствующих обстоятельств, включающих требующее лечения состояние, выбранный способ введения, конкретное вводимое соединение, возраст, массу и реакцию конкретного пациента, тяжесть наблюдаемых симптомов у пациента и тому подобное.
Фармацевтические композиции по изобретению могут быть введены различными способами, включающими пероральный, ректальный, трансдермальный, подкожный, внутривенный, внутримышечный и интраназальный. В зависимости от предполагаемого способа доставки соединения предпочтительно формулируют в виде композиций, предназначенных либо для инъекции, либо для перорального введения. Композиции для перорального введения могут быть выполнены в форме нерасфасованных жидких растворов или суспензий, либо нерасфасованных порошков. Однако чаще всего композиции представлены в дозированных лекарственных формах, что способствует точному дозированию. Термин "дозированные лекарственные формы" означает физически дискретные единицы, подходящие в качестве единичных доз для пациента, которым является человек или другое млекопитающее, каждая единица содержит заранее определенное количество активного ингредиента, рассчитанное в зависимости от требуемого терапевтического действия, вместе с подходящим фармацевтическим наполнителем. Типичные дозированные лекарственные формы включают заранее заполненные и измеренные ампулы или шприцы, содержащие жидкие композиции, или пилюли, таблетки, капсулы или тому подобное в случае твердых композиций. В таких композициях амидное соединение является обычно не основным по массе компонентом (приблизительно от 0,1 до 50 массовых % или предпочтительно от 1 до 40 массовых %), при этом остальное составляют различные растворители или наполнители и технологические добавки, полезные для формирования требуемой лекарственной формы.
Жидкие формы, пригодные для перорального введения, могут включать подходящий водный или неводный растворитель с буферными, суспендирующими и диспергирующими средствами, красителями, корригентами и тому подобное. Жидкие формы, включающие описанные ниже композиции для инъекций, обычно хранят в отсутствии света, чтобы избежать каталитического действия света, такого как образование гидропероксида или пероксида. Твердые формы могут включать, например, любые из следующих ингредиентов или близких им по природе соединений: связывающее вещество, такое как микрокристаллическая целлюлоза, камедь трагаканта или желатин; наполнитель, такой как крахмал или лактоза, диспергирующее средство, такое как альгиновая кислота, примогель или кукурузный крахмал; смазывающее вещество, такое как стеарат магния; средство для скольжения, такое как коллоидный диоксид кремния; подсластитель, такой как сахароза или сахарин; или коррегирующее средство, такое как мята перечная, метилсалицилат или апельсиновая вкусовая добавка.
Композиции для инъекции обычно основаны на пригодном для инъекции стерильном физиологическом растворе или содержащем фосфатный буфер солевом растворе, или других известных в данной области пригодных для инъекции носителях. Как упомянуто выше, амидное производное формулы (I) в таких композициях является обычно не основным по массе компонентом, часто содержащимся в пределах 0,05-10 массовых %, остальное представляет собой пригодный для инъекции носитель и тому подобное. Средняя суточная доза определяется рядом факторов, таких как тяжесть заболевания и характеристики пациента (возраст, пол и масса). Дозу обычно варьируют от 1 мг или нескольких мг до 1500 мг соединения формулы (I) в день, необязательно деля ее для многократных введений. Более высокие дозы можно также вводить благодаря низкой токсичности соединений по изобретению в течение длительного периода времени.
Описанные выше компоненты для перорально вводимых или предназначенных для инъекции композиций служат лишь характерными примерами. Дополнительные материалы, а также технологические приемы приведены в разделе 8 "Remington's Pharmaceutical Sciences Handbook", 18th Edition, 1990, Mack Publishing Company, Easton, Pennsylvania, включенного здесь в качестве ссылки.
Соединения по данному изобретению могут быть введены в формах замедленного высвобождения или из систем доставки лекарственных средств путем замедленного высвобождения. Описание характерных материалов для замедленного высвобождения могут также быть найдены в материалах, включенных в Remington's Handbook, смотри выше.
Настоящее изобретение далее иллюстрируется с помощью следующих примеров, которые не следует рассматривать как ограничивающие объем изобретения.
При описании соединений по изобретению формулы (I) условно принято указывать абсолютные конфигурации любых дополнительных хиральных заместителей, необязательно присутствующих в структуре указанных соединений, знаками штриха (например, R', S', S'' и прочее).
Примеры используемых сокращений: AcOH - уксусная кислота, AcOEt - этилацетат, BOC - N-трет-бутоксикарбонил-, DCC - дициклогексилкарбодиимид, DCU - дициклогексилкарбамид, DMF - диметилформамид (ДМФ), EtOH - этанол, Et2O - диэтиловый эфир, HOBZ - 1-гидроксибензотиазол, hr - час (ч), hrs - часы (ч), MeOH - метанол, r.t. - комнатная температура (комнатной температуре), THF - тетрагидрофуран (ТГФ), Z - N-бензилоксикарбонил.
Препаративные примеры:
Промежуточные соединения, используемые в приведенных ниже примерах, получены по следующим методикам.
1-Амино-4-диметиламинобутан
Гидрохлорид диметиламина (1,2 г; 12,5 ммоль) и, спустя 1 ч, 4-бромбутилфталимид (3,5 г; 12,4 ммоль) добавляют к суспензии K2CO3 (4,3 г; 31 ммоль) в ацетоне (5 мл) при 25°C; после чего суспензию нагревают до температуры кипения с обратным холодильником в течение ночи. После охлаждения до комнатной температуры смесь фильтруют и упаривают досуха; флэш-хроматография на силикагеле остаточного масла (элюент CHCL3/CH3OH 8:2) дает N-(4-диметиламинобутил)фталимид в виде белого твердого вещества (2,2 г; 8,94 ммоль).
Раствор указанного соединения в EtOH обрабатывают 35% водным гидразином (0,45 мл), нагревают до температуры кипения с обратным холодильником до исчезновения всех исходных реагентов (˜2 ч), фильтруют и упаривают досуха. Окончательная кристаллизация из смеси CH2Cl2/CH3OH (98:2) дает 0,85 г (7,32 ммоль; выход 82%) 1-амино-4-диметиламинобутана в виде белого твердого вещества.
1Н-ЯМР (CDCl3): δ 7,75 (м, 2Н); 7,65 (м, 2Н); 2,72 (м, 2Н); 2,35 (т, 2Н, J=7 Гц); 2,23 (с, 6Н); 1,75 (м, 2Н); 1,56 (уш.с, 2Н, NH2); 1,48 (м, 2Н).
1-Амино-4-метиламинобутан
Партию 1-амино-4-метиламинобутана получают по приведенной выше методике, используя метиламин вместо диметиламина.
1-(3-Аминопропил)тиаморфолин:
Раствор 3-BOC-аминопропилбромида (3,07 г; 12,9 ммоль) и тиаморфолина (2,6 мл; 25,8 ммоль) в CH2Cl2 (25 мл) нагревают до температуры кипения с обратным холодильником в течение 24 ч. Смесь охлаждают до комнатной температуры, фильтруют, промывают водой (2×50 мл), сушат над Na2SO4 и упаривают досуха в вакууме. Очистка флэш-хроматографией на силикагеле (элюент CHCl3/CH3OH 9:1) дает 1-(3-BOC-аминопропил)тиаморфолин (3,1 г; 11,96 ммоль) в виде прозрачного масла.
Расщепление защитной группы осуществляют растворением 1,4 г (5,4 ммоль) указанного соединения в 3н водном HCl (6 мл) при комнатной температуре; спустя 18 ч, раствор после подщелачивания добавлением водного 2н NaOH до достижения pH 8 экстрагируют CH2Cl2 (2×10 мл). Объединенные экстракты сушат над Na2SO4 и упаривают досуха, получая 1-(3-аминопропил)тиаморфолин в виде прозрачного масла (0,63 г; 3,96 ммоль).
1Н-ЯМР (CDCl3): δ 7,75 (м, 2Н); 7,65 (м, 2Н); 2,72 (м, 2Н); 2,35 (т, 2Н, J=7 Гц); 2,23 (с, 6Н); 1,75 (м, 2Н); 1,56 (уш.с, 2Н, NH2); 1,48 (м, 2Н).
1-(3-Аминопропил)-4-метилпиперазин (выделенный в виде хлористоводородной соли)
1Н-ЯМР (D2О): δ 3,75 (м, 7Н); 3,45 (м, 3Н); 3,15 (м, 2Н); 3,05 (м, 4Н); 2,20 (м, 2Н)
получают по той же методике, используя 4-метилпиперазин вместо тиаморфолина.
1-(3-Аминопропил)пиперидин
1Н-ЯМР (CDCl3): δ 2,85 (т, 2Н, J=8 Гц); 2,45 (м, 6Н); 1,90 (уш.с, 2Н, NH2); 1,8-1,62 (м, 6Н); 1,55 (м, 2Н)
получают по той же методике, используя пиперидин вместо тиаморфолина.
1-BOC-пропан-1,3-диамин
Водный раствор (5 мл) NaN3 (1,4 г; 21,5 ммоль) и 2-3 капли Aliquat 336 добавляют к перемешиваемому раствору 3-BOC-аминопропилбромида (5 г; 21,5 ммоль) в толуоле (10 мл); смесь нагревают до температуры кипения с обратным холодильником в течение 4 ч. После охлаждения до комнатной температуры органическую фазу отделяют, сушат над Na2SO4 и упаривают досуха в вакууме, получая 3-BOC-аминопропилазид (3,75 г; 18,3 ммоль) в виде прозрачного масла (выход 85%).
Раствор трифенилфосфина (4,8 г; 18,3 ммоль) в ТГФ (15 мл) добавляют по каплям к перемешиваемому раствору вышеуказанного азида в ТГФ (30 мл)/H2O (0,3 мл; 18,3 ммоль); перемешивание продолжают в течение 24 ч при комнатной температуре. После удаления растворителя досуха в вакууме остаток поглощают некоторым количеством EtOH, чтобы выделить белый осадок оксида трифенилфосфина, путем перемешивания в течение 6 ч при комнатной температуре. Наконец EtOH удаляют досуха при пониженном давлении, получая 3,22 г (18 ммоль) 1-BOC-пропан-1,3-диамина в виде светло-желтого масла.
1Н-ЯМР (CDCl3): δ 4,90 (уш.с, 1Н, CONH); 3,25 (м, 2Н); 2,85 (т, 2Н, J=7 Гц); 1,75 (т, 2Н, J=7 Гц); 1,60 (уш.с, 2Н, NH2); 1,55 (с, 9Н).
3-(BOC-метиламино)пропиламин
Получают по приведенной выше методике, используя 3-(BOC-метиламино)пропилбромид.
Метил-(S)-2-амино-3-диметиламинопропионат
2M раствор диметиламина в ТГФ (2,5 мл) добавляют по каплям к перемешиваемому раствору метил-(S)-2-BOC-амино-3-бромпропионата (0,45 г; 1,42 ммоль) (Weiss R.G. et al., J. Org. Chem, 36, 403, 1971; Kang M. et al., там же, 61, 5528, 1996) в безводном ТГФ (10 мл) при 25°C. Смесь перемешивают в течение ночи при комнатной температуре и упаривают досуха в вакууме. Остаток распределяют между Et2O (30 мл) и водным 0,5н NaOH (2×5 мл); эфирные экстракты объединяют, промывают насыщенным раствором соли, сушат над Na2SO4 и упаривают досуха, получая 0,34 г (1,22 ммоль) метил-(S)-2-амино-3-диметиламинопропионата в виде светло-желтого масла.
1Н-ЯМР (CDCl3): δ 7,45 (м, 5Н); 5,73 (уш.с, 1Н, CONH); 5,15 (с, 2Н); 4,32 (м, 1Н); 3,82 (с, 3Н); 2,75 (м, 2Н); 2,22 (с, 6Н).
Перемешиваемый раствор указанного сложного метилового эфира (0,34 г; 1,22 ммоль) в ацетонитриле (12 мл) обрабатывают триметилсилилиодидом (0,21 мл; 1,46 ммоль) при комнатной температуре; спустя 3 ч, смесь гасят, добавляя MeOH (0,24 мл; 5,9 ммоль), и упаривают в вакууме досуха. Остаток поглощают Et2O (2×10 мл); эфирные экстракты повторно экстрагируют, используя 30% водный AcOH (2×5 мл), собирают, подщелачивают до pH 8 и экстрагируют CH2Cl2 (2×10 мл). Экстракты в дихлорметане объединяют, сушат над Na2SO4, упаривают досуха, получая 0,16 г (1,1 ммоль) метил-(S)-2-амино-3-диметиламинопропионата.
1Н-ЯМР (CDCl3): δ 4,32 (м, 1Н); 3,82 (с, 3Н); 3,24 (уш.с, 2Н, NH2); 2,75 (м, 2Н); 2,22 (с, 6Н).
Метил-(S)-2-амино-5-(пиперидин-1-ил)пентаноат
При перемешивании и наружном охлаждении, поддерживая реакционную температуру в пределах 20-25°C, 0,03 молярных эквивалента 1н раствора B2H6 (диборана) в ТГФ добавляют к 0,01M раствору 1-гемиметилового эфира (S)-2-BOC-амино-1,5-пентадионовой кислоты в ТГФ (15 мл); спустя 2 ч, избыток диборана разрушают осторожным добавлением воды. После концентрации до небольшого объема в вакууме раствор разбавляют AcOEt (25 мл). Органическую фазу промывают 5% водным NaHCO3, насыщенным раствором соли и водой до нейтральной реакции, сушат над Na2SO4 и упаривают досуха.
Сырой остаток метил-(S)-2-BOC-амино-5-гидроксипентаноата обрабатывают трифенилфосфином и CBr4, получая сырой образец метил-(S)-2-BOC-амино-5-бромпентаноата.
Взаимодействие последнего соединения с пиперидином в ТГФ приводит к метил-(S)-2-BOC-амино-5-(пиперидин-1-ил)пентаноату, который при обработке трифторуксусной кислотой в дихлорметане дает бис-трифторацетатную соль метил-(S)-2-амино-5-(пиперидин-1-ил)пентаноата.
1Н-ЯМР (CDCl3): δ 4,32 (м, 1Н); 3,82 (с, 3Н); 3,54 (м, 1Н); 2,85 (т, 2Н, J=7 Гц); 2,45 (м, 6Н); δ 1,85 (уш.с, 2Н, NH2); δ 1,75-1,6 (м, 6Н); δ 1,5(м, 2Н).
Гидрохлорид 5-BOC-орнитинметилового эфира
Поддерживая реакционную температуру в пределах 0-5°C за счет наружного охлаждения, твердый 2-Z,5-BOC-орнитин (1 г, 2,7 ммоль; промышленно выпускаемый реагент) и, спустя 15 мин, метилиодид (0,34 мл, 5,4 ммоль) добавляют к перемешиваемой суспензии измельченного до тонкого порошка K2CO3 (0,38 г; 2,7 ммоль) в сухом ДМФ (20 мл). Смесь перемешивают дополнительно еще час при 0-5°C и при комнатной температуре в течение 1 ч, затем разбавляют EtOAc (40 мл) и фильтруют. Прозрачный раствор промывают водой (40 мл) и насыщенным раствором соли (3×30 мл); сушат над Na2SO4 и упаривают досуха. Последующая очистка флэш-хроматографией на силикагеле (элюент CHCl3/CH3OH 8:2) дает 2-Z,5-BOC-орнитинметиловый эфир (0,8 г; 2,1 ммоль).
Гидролитическое расщепление Z-защитной группы (выполнено по методике Meienhofer J. et. al, Tetrahedron. Lett., 3259, 1974) дает гидрохлорид 5-BOC-орнитинметилового эфира (0,73 г; 2,0 ммоль) в виде белого твердого вещества.
1Н-ЯМР (CDCl3): δ 9,25 (уш.с, 3Н, NH3 +); 5,40 (уш.с, 1Н, CONH); 4,40 (м, 1Н); 3,8 (с, 3Н); 3,0 (м, 2Н); 1,8 (м, 4Н); 1,4 (с, 9Н).
Экзо-8-метил-8-азабицикло[3,2,1]октан-3-амин (β-1H,5H-тропанамин)
Образец получают исходя из тропинона по методике Burks J.E. et al., Org. Proc. Res. Dev., 1, 198, 1997.
4-(N,N-Диметиламино)анилин
4-Нитроанилин (1,83 г; 13,24 ммоль) добавляют порциями к охлажденной (T=+4°C) муравьиной кислоте (3 мл; 66,2 ммоль). Добавляют формальдегид (37 мас.% раствор в воде; 2,72 мл; 29,13 ммоль) и полученную смесь нагревают до температуры кипения с обратным холодильником в течение 24 ч. После охлаждения до комнатной температуры добавляют 6н HCl (2,2 мл) и образовавшийся осадок фильтруют. Фильтрат разбавляют 1н NaOH (5 мл) и экстрагируют CH2Cl2 (3×20 мл); собранные органические экстракты сушат над Na2SO4 и упаривают в вакууме, получая твердый остаток, который после обработки смесью диизопропиловый эфир/ацетон 1:1 и фильтрации дает 4-нитро-N,N-диметиланилин в виде желтого порошка (1,65 г; 9,93 ммоль). Железо в порошке (2,145 г; 38,3 ммоль) и 37% HCl (28 мкл) суспендируют в 96% этиловом спирте (35 мл) и смесь нагревают до температуры кипения с обратным холодильником в течение 30 минут; наконец добавляют 4-нитро-N,N-диметиланилин (0,64 г; 3,84 ммоль) и смесь оставляют нагреваться до температуры кипения с обратным холодильником и при перемешивании в течение 2 ч. Горячую смесь фильтруют через рыхлый слой целита и после охлаждения до комнатной температуры фильтрат упаривают в вакууме. Маслянистый остаток разбавляют CH2Cl2 (25 мл) и промывают 1н NaOH (3×25 мл), сушат над Na2SO4 и упаривают в вакууме, получая 4-(N,N-диметиламино)анилин в виде светло-желтого масла (0,44 г; 3,26 ммоль).
1Н-ЯМР (CDCl3): δ 7,10 (д, 2Н, J=8 Гц); 6,60 (д, 2Н, J=8 Гц); 3,55 (уш.с, 2Н, NH2); 2,25 (с, 6Н).
По той же методике получают 4-(N,N-диметиламинометил)анилин в виде светло-желтого масла.
1Н-ЯМР (CDCl3): δ 7,12 (д, 2Н, J=8 Гц); 6,64 (д, 2Н, J=8 Гц); 3,50 (уш.с, 2Н, NH2); 3,28 (с, 2Н); 2,25 (с, 6Н).
N,N-Диметилбутин-2-илдиамин
Пропаргилбромид (1,3 мл, 17,4 ммоль) растворяют в ДМФ (30 мл) и добавляют калийфталимид (3,4 г; 18,4 ммоль). Смесь нагревают до температуры кипения с обратным холодильником в течение 5 ч. После охлаждения до комнатной температуры смесь разбавляют диэтиловым эфиром, промывают водой (3×50 мл), сушат над Na2SO4 и упаривают в вакууме, получая N-пропаргилфталимид в виде белого твердого вещества (3,15 г; 17 ммоль).
N-Пропаргилфталимид (0,64 г; 3,4 ммоль) растворяют в 1,4-диоксане (20 мл), затем добавляют диметиламин (8,5 мл; 17 ммоль), хлорид меди(I) (0,35 г) и параформальдегид (1 г). Раствор нагревают до температуры кипения с обратным холодильником в течение 3 ч. После охлаждения до комнатной температуры образовавшийся осадок фильтруют и фильтрат упаривают в вакууме, получая зеленый маслянистый остаток, который затем растворяют в CH2Cl2, промывают насыщенным раствором NaHCO3 (2×30 мл) и водой (2×30 мл). Органическую фазу сушат над Na2SO4 и упаривают в вакууме. Сырой продукт очищают обработкой диэтиловым эфиром, получая N-фталимидо-N',N'-диметилбутин-2-ил-1,4-диамин в виде светло-желтого твердого вещества (0,5 г; 2,05 ммоль).
Суспензию N-фталимидо-N',N'-диметилбутин-2-ил-1,4-диамина (0,5 г; 2,05 ммоль) в этиловом спирте (10 мл) обрабатывают гидразингидратом (98 мкл; 2 ммоль) и смесь нагревают до температуры кипения с обратным холодильником в течение ночи. После охлаждения до комнатной температуры осадок фильтруют и фильтрат упаривают в вакууме; сырой остаток обрабатывают ацетоном при комнатной температуре, получая после удаления образовавшегося осадка чистый продукт N,N-диметилбутин-2-ил-1,4-диамин в виде красного масла (0,2 г; 1,78 ммоль).
1Н-ЯМР (CDCl3): δ 3,52 (м, 2Н); 3,27 (м, 2Н); 2,35 (с, 6Н); 1,90-1,65 (уш.с, 2Н, NH2).
2-(Аминокси)-N-метил-N-(2-гидроксиэтил)]этиламин
a) (Z-Аминокси)уксусная кислота
Поддерживая реакционную температуру около 0-5°C за счет наружного охлаждения, бензилхлорформиат (1,41 мл, 10 ммоль) и водный 4н NaOH (2,23 мл) по каплям и поочередно добавляют к раствору в водном 2н NaOH (5 мл) 2,18 г (10 ммоль) гемигидрохлорида карбоксиметоксиламина [(промышленно выпускаемый реагент) называемый также гидрохлорид (аминокси)уксусной кислоты]. Перемешивание продолжают в течение 15 мин, после чего удаляют любые органические примеси с помощью Et2O (2×15 мл); затем добавлением колотого льда и подкисливанием до pH 2 с помощью 37% HCl получают твердый продукт, который фильтруют, промывают охлажденной водой и сушат в вакууме при T=40°C, получая 2,62 г (8,2 ммоль) (Z-аминокси)уксусной кислоты.
b) 2-(Z-аминокси)-N-метил-N-(2-гидроксиэтил)ацетамид
Тионилхлорид (0,78 мл, 9 ммоль) добавляют к перемешиваемому раствору (Z-аминокси)уксусной кислоты (2,62 г, 8,2 ммоль) в MeOH (10 мл). Смесь выдерживают в течение ночи при комнатной температуре, получая после обычного выпаривания растворителя в условиях глубокого вакуума сырой образец (Z-аминокси)ацетилхлорида. Без какой-либо дополнительной очистки раствор указанного соединения в СН2Cl2 (10 мл) добавляют по каплям при комнатной температуре в перемешиваемый раствор 2-метиламиноэтанола (1,44 мл, 18 ммоль) в СН2Cl2 (5 мл); спустя 18 ч, реакционную смесь разбавляют водным 1н HCl (15 мл). Органическую фазу отделяют; промывают водой (2×15 мл), сушат над Na2SO4 и упаривают, получая 2-(Z-аминокси)-N-метил-N-(2-гидроксиэтил)ацетамид (2,64 г, 7 ммоль) в виде прозрачного масла.
c) 2-(Z-Аминокси)-N-метил-N-(2-гидроксиэтил)этиламин
Селективное восстановление с помощью диборана 2-(Z-аминокси)-N-метил-N-(2-гидроксиэтил)ацетамида осуществляют по методике Brown'a (J. Am. Chem. Soc. 86, 3566, 1964 and J. Org. Chem., 38, 912, 1973), получая 2,1 г (5,8 ммоль) 2-(Z-аминокси)-N-метил-N-(2-гидроксиэтил)этиламина в виде масла.
d) 2-(Аминокси)-N-метил-N-(2-гидроксиэтил)этиламин
Расщепление бензилоксикарбонила путем гидрогенолиза проводят в присутствии формиата аммония по методике Makowski (Liebigs Ann. Chem., 1457, 1985), получают 2-(аминокси)-N-метил-N-(2-гидроксиэтил)этиламин (1,06 г, 4,64 ммоль) в виде прозрачного масла.1Н-ЯМР (CDCl3): δ 5,28 (уш.с, 2Н, ONH2); 4,67 (т, 2Н, J=7 Гц); 3,40 (м, 2Н); 2,75 (т, 2Н, J=7 Гц); 2,42 (т, 2Н, J=7 Гц); 2,21 (с, 3Н); 1,8 (уш.с, 1Н, OH).
2-Арилпропионилхлориды формулы V (общая методика).
Раствор 72,8 ммоль 2-арилпропионовой кислоты формулы V [например, (R)-2-(4-изобутилфенил)пропионовой кислоты, (R)(-)-ибупрофена, 72,8 ммоль] в тионилхлориде (37,5 мл) нагревают до температуры кипения с обратным холодильником в течение 3 ч. Смесь охлаждают до комнатной температуры; избыток реагента выпаривают досуха в вакууме; затем дважды подряд добавляют небольшие количества безводного диоксана и упаривают досуха в условиях глубокого вакуума до полного удаления любых следов остаточного тионилхлорида. Конечный маслянистый остаток используют в последующих взаимодействиях.
ИК (пленка) см-1: 1800 (ClC=0)
(S)-2-(4-Изобутилфенил)]-N-(3-диметиламинопропил)пропионамидгидрохлорид
Применяя предыдущую методику, (S)(+)-ибупрофен (реагент Fluka'a) превращают в соответствующий пропионилхлорид, обработка которого 3-диметиламинопропиламином по методике примера 1 позволяет получать образец (S)-2-(4-изобутилфенил)]-N-(3-диметиламинопропил)пропионамидгидрохлорида, т.пл. 97-98°C, [α]D=+27 (c=1; CH3OH).
1Н-ЯМР (D2О): δ 7,45-7,21 (м, 4Н); 3,75 (кв, 1Н, J1=7 Гц, J2=7 Гц); 3,45-3,15 (м, 2Н); 2,95 (т, 2Н, J=8 Гц); 2,85 (с, 6Н); 2,52 (д, 2Н, J=7 Гц); 1,98 (м, 1Н); 1,47 (д, 3Н, J=7 Гц); 0,90 (д, 6Н, J=7 Гц).
Пример 1
(R)-2-(4-изобутилфенил)-N-(3-диметиламинопропил)пропионамидгидрохлорид
При наружном охлаждении, поддерживая реакционную температуру ниже 40°C, раствор (R)-2-(4-изобутилфенил)пропионилхлорида (16,35 г; 72,8 ммоль) в CH2Cl2 (10 мл) медленно добавляют к перемешиваемому раствору 3-диметиламинопропиламина (19 мл; 152 ммоль). После выдерживания в течение ночи при комнатной температуре реакционную смесь разбавляют водой (100 мл), органическую фазу отделяют, промывают водой (50 мл) и сушат над Na2SO4. После удаления растворителя при пониженном давлении получают 20 г (68,8 ммоль) сырого (R)-2-(4-изобутилфенил)-N-(3-диметиламинопропил)пропионамида в виде светло-желтого масла.
Перемешиваемый раствор части указанного амида (58 ммоль) в изопропиловом спирте (200 мл) обрабатывают водным 37% HCl (6 мл), медленно добавляя при комнатной температуре; спустя 2 ч, реакционную смесь упаривают досуха при пониженном давлении. Следы остаточной воды удаляют азеотропной перегонкой в вакууме, добавляя небольшие количества безводного изопропилового спирта. Окончательная кристаллизация из AcOEt (300 мл) выделяет белый порошок, который фильтруют, промывают сухим AcOEt и сушат в течение 24 ч в условиях вакуума при T=40°C, получают 18 г (55 ммоль) (R)-2-(4-изобутилфенил)-N-(3-диметиламинопропил)пропионамидгидрохлорида.
Т.пл. 95-98°C,
[α]D=-26 (c=1,6; CH3OH).
1Н-ЯМР (D2О): δ 7,5-7,2 (м, 4Н); 3,75 (кв, 1Н, J1=7 Гц, J2=7 Гц); 3,45-3,15 (м, 2Н); 3,05 (т, 2Н, J=8 Гц); 2,80 (д, 6Н, J=4,5 Гц); 2,55 (д, 2Н, J=7 Гц); 1,95 (м, 1Н); 1,45 (д, 3Н, J=7 Гц); 0,93 (д, 6Н, J=7 Гц).
Пример 2
Используя 2-диметиламиноэтиламин и 4-диметиламинобутиламин вместо 3-диметилпропиламина в методике примера 1, получают следующие соединения:
(R)-2-(4-изобутилфенил)-N-(2-диметиламиноэтил)пропионамид·HCl
Т.пл. 90-93°C; [α]D=-16 (c=1; CH3OH).
1Н-ЯМР (CDCl3): δ 12,25 (уш.с, 1Н, NH+); 7,82 (уш.с, 1Н, CONH); 7,45 (д, 2Н, J=8 Гц); 7,05 (д, 2Н, J=8 Гц); 3,85 (м, 2Н); 3,70 (м, 1Н); 3,10 (м, 2Н); 2,80 (с, 3Н); 2,75 (с, 3Н); 2,55 (д, 2Н, J=7 Гц); 1,97 (м, 1Н); 1,65 (д, 3Н, J=7 Гц); 0,98 (д, 6Н, J=7 Гц).
(R)-2-(4-изобутилфенил)-N-(4-диметиламинобутил)пропионамид·HCl
Т.пл. 95-97°C; [α]D=-16 (c=0,52; CH3OH).
1Н-ЯМР (CDCl3): δ 7,25 (д, 2Н, J=8 Гц); 7,10 (д, 2Н, J=8 Гц); 6,18 (уш.с, 1Н, CONH); 3,60 (кв, 1Н, J1=7 Гц, J2=7 Гц); 3,25-3,15 (м, 2Н); 2,95 (м, 2Н); 2,75 (с, 6Н); 2,45 (д, 2Н, J=7 Гц); 1,85 (м, 1Н); 1,65 (м, 4Н); 1,48 (д, 3Н, J=7 Гц); 0,93 (д, 6Н, J=7 Гц).
Пример 3
(R)-2-(4-изобутилфенил)-N-2-(N-морфолинилэтил)пропионамид·HCl
Используя 1-аминоэтилморфолин в методике по примеру 1, получают сырой (R)-2-(4-изобутилфенил)-N-[2-(1-морфолинил)этил]пропионамид. Раствор 4,2н ацетилхлорида в абсолютном EtOH (3 мл) добавляют по каплям к перемешиваемому раствору указанного амида (0,416 г, 1,3 ммоль) в абсолютном EtOH (5 мл). Смесь перемешивают дополнительно 2 ч при комнатной температуре, после чего удаляют растворители при пониженном давлении. Остаток поглощают этиловым эфиром, выделяя 0,39 г (1,1 ммоль) (R)-2-(4-изобутилфенил)-N-[2-(1-морфолинил)этил]пропионамидгидрохлорида в виде белого твердого вещества, которое фильтруют и промывают тем же растворителем.
Т.пл. 123-125°C; [α]D=-36,3 (c=0,5; CH3OH).
1Н-ЯМР (CDCl3): δ 12,55 (уш.с, 1Н, NH+); 7,80 (уш.с, 1Н, CONH); 7,45 (д, 2Н, J=8 Гц), 7,05 (д, 2Н, J=8 Гц); 4,25 (м, 2Н); 3,95 (м, 1Н); 3,70 (м, 4Н); 3,41 (м, 1Н); 3,05 (м, 3Н); 2,75 (м, 2Н); 2,45 (д, 2Н, J=7 Гц); 1,97 (м, 1Н); 1,65 (д, 3Н, J=7 Гц); 0,95 (д, 6Н, J=7 Гц).
Пример 4
Использование в способе по примеру 3 следующих аминов, выбираемых из группы, включающей: 1-(3-аминопропил)морфолин, 1-(3-аминопропил)-4-тиоморфолин, 1-(2-аминоэтил)пиперазин-4-метил, 1-(3-аминопропил)пиперазин-4-метил, 1-(3-аминопропил)пиперидин и экзо-8-метил-8-аза-бицикло[3,2,1]-октан-3-амин, - вместо 1-(3-аминопропил)морфолина дает: (R)-2-(4-изобутилфенил)-N-3-(N-морфолинилпропил)пропионамид·HCl
Т.пл. 90-93°C
[α]D=-22,6 (c=0,5; CH3OH).
1Н-ЯМР (CDCl3): δ 12,55 (уш.с, 1Н, NH+); 7,80 (уш.с, 1Н, CONH); 7,45 (д, 2Н, J=8 Гц), 7,05 (д, 2Н, J=8 Гц); 4,25 (м, 2Н); 3,95 (м, 1Н); 3,70 (м, 4Н); 3,41 (м, 1Н); 3,05 (м, 3Н); 2,75 (м, 2Н); 2,45 (д, 2Н, J=7 Гц); 2,15 (м, 2Н); 1,97 (м, 1Н); 1,65 (д, 3Н, J=7 Гц); 0,95 (д, 6Н, J=7 Гц).
(R)-2-(4-изобутилфенил)-N-3-(N-тиоморфолинилпропил)пропионамид·HCl
Т.пл. 70-73°C; [α]D=-23 (c=0,5; CH3OH).
1Н-ЯМР (D2О): δ 8,15 (уш.с, 1Н, CONH); 7,40 (м, 4Н), 3,82 (кв, 1Н, J=7 Гц); 3,65 (м, 2Н); 3,41 (м, 1Н); 3,25 (м, 1Н); 3,15-2,80 (м, 8Н); 2,45 (д, 2Н, J=7 Гц); 1,95 (м, 3Н); 1,55 (д, 3Н, J=7 Гц); 0,95 (д, 6Н, J=7 Гц).
(R)-2-(4-изобутилфенил)-N-[2-(4-метилпиперазин-1-ил)этил]пропионамидгидрохлорид
Т.пл. выше 240°C; [α]D=-33,7 (c=0,5; CH3OH).
1Н-ЯМР (DMSO-d6): δ 7,15 (м, 4Н); 4,45 (м, 1Н); 4,13 (м, 2Н), 3,02 (м, 3Н); 2,75 (м, 4Н); 2,38 (д, 2Н, J=7 Гц); 1,85 (м, 1Н); 1,30 (д, 3Н, J=7 Гц); 0,81 (д, 6Н, J=7 Гц).
(R)-2-(4-изобутилфенил)-N-[3-(4-метилпиперазин-1-ил)пропил]пропионамид-бис-гидрохлорид
Т.пл. 216-220°C; [α]D=-20,5 (c=0,5; CH3OH).
1Н-ЯМР (D2O): δ 7,25 (м, 4Н); 3,75 (м, 1Н); 3,55 (м, 8Н), 3,25 (м, 2Н); 3,15 (м, 1Н); 3,00 (с, 3Н); 2,48 (д, 2Н, J=7 Гц); 1,95 (м, 3Н); 1,45 (д, 3Н, J=7 Гц); 0,90 (д, 6Н, J=7 Гц).
(R)-2-(4-изобутилфенил)-N-[3-(1-пиперидинил)пропил]пропионамидгидрохлорид
Т.пл. 76-80°C; [α]D=-29(c=0,5; CH3OH).
1Н-ЯМР (CDCl3): δ 11,4 (уш.с, 1Н, NH+); 7,45 (д, 2Н, J=8 Гц); 7,35 (уш.с, 1Н, CONH); 7,05 (д, 2Н, J=8 Гц), 3,85 (кв, 1Н, J=7 Гц); 3,45 (м, 4Н); 2,75 (м, 2Н); 2,52 (м, 4Н); 2,25 (м, 2Н); 2,05 (м, 2Н); 1,97 (м, 3Н); 1,60 (д, 3Н, J=7 Гц); 0,97 (д, 6Н, J=7 Гц).
(R)-2-(4-изобутилфенил)-N-[экзо-8-метил-8-аза-бицикло[3.2.1.]-окт-3-ил)пропионамидгидрохлорид
Т.пл. 72-75°C; [α]D=-3,3 (c=0,5; CH3OH).
1Н-ЯМР (CDCl3): δ 7,15 (д, 2Н, J=8 Гц), 7,05 (д, 2Н, J=8 Гц); 6,15 (уш.с, 1Н, CONH); 4,34 (м, 1Н); 3,75 (м, 2Н); 3,47 (кв, 1Н, J=7 Гц); 2,72 (с, 3Н); 2,60-2,38 (м, 4Н); 2,30-1,98 (м, 6Н); 1,98 (м, 2Н); 1,45 (д, 3Н, J=7 Гц); 0,9 (д, 6Н, J=7 Гц).
Пример 5
(R)-2-(4-изобутилфенил)-N-[3-аминопропил)пропионамидгидрохлорид
Раствор 3-BOC-аминопропиламина (3,22 г; 18 ммоль) в CH2Cl2 (10 мл) добавляют по каплям к перемешиваемой суспензии (R)(-)-ибупрофена (3 г; 17,5 ммоль), DCC (3,8 г; 18 ммоль) и HOBZ (2,8 г; 18 ммоль) в CH2Cl2 (50 мл) при 25°C. Перемешивание продолжают в течение 18 ч при комнатной температуре; после удаления DCU фильтрацией реакционную смесь упаривают досуха в вакууме. Оставшееся масло несколько раз поглощают ацетонитрилом; наконец собранные экстракты фильтруют, упаривают досуха, получая сырой образец (R)-2-(4-изобутилфенил)-N-3-(BOC-аминопропил)пропионамида, который кристаллизуют из горячего MeOH (50 мл), получая 3,4 г (9,25 ммоль, выход 53%) чистого (R)-2-(4-изобутилфенил)-N-(3-аминопропил)пропионамида при охлаждении до T=+4°C в течение 18 ч.
Суспензию указанного соединения в 10 мл водного 3н HCl перемешивают при комнатной температуре в течение 48 ч, получая (R)-2-(4-изобутилфенил)-N-3-(аминопропил)пропионамидгидрохлорид (1,9 г; 6,3 ммоль);
т.пл. 160-163°C;
[α]D=-31 (c=0,5; CH3OH).
1Н-ЯМР (CDCl3): δ 8,2 (уш.с, 1Н, NH3 +); 7,18 (д, 2Н, J=8 Гц); 7,05 (д, 2Н, J=8 Гц), 6,83 (уш.с, 1Н, CONH); 3,65 (кв, 1Н, J=7 Гц); 3,30 (м, 2Н); 3,00 (м, 2Н); 2,40 (д, 2Н, J=7 Гц); 1,95-1,74 (м, 3Н); 1,45 (д, 3Н, J=7 Гц); 0,92 (д, 6Н, J=7 Гц).
Пример 6
(R)-2-(4-изобутилфенил)-N-(1-метилпиперидин-4-ил)пропионамидгидрохлорид
Формиат аммония (15,4 г; 240 ммоль) и 10% Pd/C (3,14 г; 29 ммоль) добавляют к раствору 1-метил-4-пиперидона (3,26 мл; 26,5 ммоль) в водном метаноле (80 мл, CH3OH/H2O 9:1); смесь перемешивают 24 ч при комнатной температуре; катализатор удаляют фильтрованием через целит и выпаривают досуха при пониженном давлении, получая светло-желтый остаток 1-метил-4-аминопиперидина. Добавление по каплям 37% HCl (4,6 мл) к перемешиваемому раствору указанного амина в EtOH (50 мл) приводит к выделению белого осадка 1-метил-4-аминопиперидингидрохлорида, который отфильтровывают спустя 18 ч, после охлаждения в течение 18 ч до T=+4°C. Наконец, водный раствор гидрохлорида, обработанный избытком 0,1н NaOH (≈ 10 мл), экстрагируют CH2Cl2 (3×10 мл). После стандартной обработки растворитель выпаривают досуха, получая чистый 1-метил-4-аминопиперидин (1,4 г; 12,4 ммоль).
1Н-ЯМР (CDCl3): δ 2,85 (м, 2Н); 2,58 (м, 1Н); 2,25 (с, 3Н), 2,01 (м, 2Н); 1,85 (м, 2Н); 1,63 (уш.с, 2Н, NH2); 1,47 (м, 2Н).
При комнатной температуре раствор (R)-2-(4-изобутилфенил)пропионилхлорида (1,12 г; 5 ммоль) в CH2Cl2 (20 мл) медленно добавляют по каплям к раствору 1-метил-4-аминопиперидина (1,1 г; 10 ммоль) в CH2Cl2 (10 мл). Спустя 3 ч, реакционную смесь вновь разбавляют CH2Cl2 (10 мл), промывают 1н HCl (25 мл) и насыщенным раствором соли, сушат над Na2SO4, получая после выпаривания досуха растворителя (R)-2-(4-изобутилфенил)-N-(1-метилпиперидин-4-ил)пропионамидгидрохлорид в виде стеклообразного твердого вещества (1,2 г; 3,5 ммоль).
[α]D=-11 (c=0,5; CH3OH).
1Н-ЯМР (D2О): δ 7,28 (м, 5Н); 3,95 (м, 1Н); 3,75 (кв, 1Н, J=7 Гц), 3,54 (м, 2Н); 3,15 (м, 2Н); 2,90 (с, 3Н); 2,53 (д, 2Н, J=7 Гц); 2,28-2,05 (м, 2Н); 1,95-1,65 (м, 4Н); 1,45 (д, 3Н, J=7 Гц); 0,95 (д, 6Н, J=7 Гц).
Пример 7
Натриевая соль (R),(S)-2-(4-изобутилфенил)-N-(1-карбокси-2-диметиламиноэтил)пропионамида
Раствор (S)-метил-3-диметиламино-2-аминопропаноата (0,16 г; 1,1 ммоль) в CH2Cl2 (2 мл) добавляют по каплям к перемешиваемой суспензии (R)(-)-ибупрофена (0,23 г; 1,1 ммоль), DCC (0,23 г; 1,1 ммоль) и HOBZ (0,17 г; 1,1 ммоль) в CH2Cl2 (5 мл) при комнатной температуре. Перемешивание продолжают в течение 18 ч при комнатной температуре; после удаления DCU removal фильтрованием реакционную смесь упаривают досуха в вакууме. Остаток многократно поглощают ацетонитрилом; затем собранные экстракты фильтруют и упаривают досуха в вакууме. Последующая очистка флэш-хроматографией на силикагеле (элюент CH2Cl2/CH3OH 95:5) дает 0,3 г (0,88 ммоль) метил-(S),(R)-3-диметиламино-2-[2-(4-изобутилфенил)пропионил]аминопропаноата (выход 80%) в виде прозрачного масла.
Перемешиваемый раствор указанного сложного эфира (0,3 г; 0,88 ммоль) в диоксане (2 мл) обрабатывают стехиометрическим количеством водного н NaOH (0,88 мл) и выдерживают в течение 18 ч при комнатной температуре, после чего разбавляют охлажденной водой (20 мл). Замороженный раствор лиофилизуют, получая 0,307 г (0,88 ммоль) натриевой соли (R),(S)-2-(4-изобутилфенил)-N-(1-карбокси-2-диметиламиноэтил)пропионамида в виде белого твердого вещества.
Т.пл. выше 240°C;
[α]D=-25 (c=0,5; CH3OH)
1Н-ЯМР (CDCl3): δ 7,35 (м, 4Н); 6,25 (уш.с, 1Н, CONH); 4,72 (м, 1Н); 3,60 (м, 1Н); 2,51 (д, 2Н, J=7 Гц), 2,30 (д, 2Н, J=7 Гц); 2,22 (м, 6Н); 1,55 (д, 3Н, J=7 Гц); 0,95 (д, 6Н, J=7 Гц).
Пример 8
Натриевая соль (R),(S)-2-(4-изобутилфенил)-N-(1-карбокси-2-пиперидин-1-ил-бутил)пропионамида и (R),(S)-2-(4-изобутилфенил)-N-(1-этоксикарбонил-2-пиперидин-1-ил-бутил)пропионамид
Получают, используя (S)-метил-5-(пиперидин-1-ил)-2-аминопентаноат в методике по примеру 7 вместо (S)-метил-3-диметиламино-2-аминопропаноата.
Пример 9
R-2-[(4'-изобутилфенил]-N-[2-(диметиламиноэтил)аминокарбонилметил]пропионамидгидрохлорид
HOBZ (0,607 г; 4,49 ммоль) добавляют к перемешиваемому раствору (R)(-)-ибупрофена (1,01 г; 4,9 ммоль) в ДМФ (4 мл) при T=0°C и оставляют перемешиваться в течение 30 мин. Затем добавляют смесь N-(3-диметиламинопропил)глицинамидгидрохлорида (0,64 г; 4,47 ммоль) в ДМФ (8 мл) и триэтиламине (0,6 мл; 4,45 ммоль) и также добавляют, небольшими порциями N,N-дициклогексилкарбодиимид (1 г; 4,85 ммоль). Смесь перемешивают 2 ч при T=0°C и затем 18 ч при комнатной температуре. После фильтрации DCU большую часть ДМФ удаляют перегонкой при пониженном давлении. Остаток поглощают водой и экстрагируют Et2O (3×25 мл); органические экстракты объединяют, сушат над Na2SO4 и упаривают при пониженном давлении, получая прозрачное масло (1 г; 3,43 ммоль). Затем раствор полученного соединения в диоксане (3,5 мл) обрабатывают 1н NaOH (3,5 мл), перемешивают 24 ч при комнатной температуре, разбавляют водой (10 мл) и после этого подкисливают 2н HCl и экстрагируют CH2Cl2 (3×10 мл).
Затем органические экстракты объединяют, сушат над Na2SO4 и упаривают при пониженном давлении, получая R-2-[(4'-изобутил)фенил]-N-[2-(диметиламиноэтил)аминокарбонилметил]пропионамидгидрохлорид (0,68 г; 2,04 ммоль) в виде светло-желтого масла.
[α]D=-25 (c=0,5; CH3OH).
1Н-ЯМР (CDCl3): δ 7,24 (м, 2Н); 7,10 (м, 2Н); 6,10 (уш.с, 1Н, CONH), 3,55 (м, 1Н); 3,30 (м, 2Н); 2,45 (д, 2Н, J=7 Гц); 2,35 (м, 2Н); 2,18 (с, 6Н); 1,85 (м, 1Н); 1,52 (д, 3Н, J=7 Гц); 0,90 (д, 6Н, J=7 Гц).
Пример 10
(R)-2-[2-(2,6-дихлорфениламино)фенил]-N-3-(диметиламинопропил)пропионамид
Суспензию (R)-2-[2-(2,6-дихлорфениламино)]фенил]пропионовой кислоты (0,15 г; 0,48 ммоль), DCC (0,173 г; 0,84 ммоль) и HOBZ (0,075 г; 0,56 ммоль) в CH2Cl2 (6 мл) перемешивают 4 ч при комнатной температуре; затем добавляют по каплям раствор 3-(диметиламино)пропиламина (0,06 мл; 0,48 ммоль) в CH2Cl2 (5 мл). Перемешивание продолжают в течение 18 ч при комнатной температуре, затем выделившийся DCU фильтруют и растворитель удаляют при пониженном давлении. Остаток дважды поглощают ацетонитрилом, экстракты объединяют, фильтруют для полного удаления следов DCU и упаривают при пониженном давлении. Очистка флэш-хроматографией (элюент CH2Cl2/CH3OH 95:5) дает (R)-2-[2-(2,6-дихлорфениламино)фенил]-N-3-(диметиламинопропил)пропионамид (0,141 г; 0,36 ммоль; выход 75%) в виде прозрачного масла.
[α]D=-30 (c=1; CH3OH).
1Н-ЯМР (D2О): δ 7,38 (м, 4Н); 7,15 (м, 1Н); 7,05 (м, 1Н), 6,60 (м, 1Н+CONH); 4,25 (дд, 2Н, J1=7 Гц, J2=3 Гц); 3,30 (м, 2Н); 2,35 (м, 2Н); 2,10 (с, 6Н); 1,65 (м, 2Н); 1,65 (д, 3Н, J=7 Гц).
Пример 11
Следующие амиды получают, используя (R),(R',S')-2-[3-(α-гидроксибензил)фенил]пропионовую кислоту, 2-[3'-(α-гидроксиэтил)фенил]пропионовую кислоту и (R),(R',S')-2-[3'-(α-гидрокси,α-метилбензил)фенил]пропионовую кислоту в качестве исходного материала вместо (R)-2-[2-(2,6-дихлорфениламино)]фенил]пропионовой кислоты в методике по примеру 10.
(R),(R',S')-2-[3-(α-гидроксибензил)фенил]-N-3-(диметиламинопропил)пропионамид в виде бесцветного масла.
[α]D=-24 (c=1; CH3OH).
1Н-ЯМР (CDCl3): δ 7,41-7,3 (м, 3Н); 7,31-7,14 (м, 6Н); 5,75 (с, 1Н), 4,02 (уш.с, 1Н, ОН); 3,31 (м, 2Н); 2,38 (т, 2Н, J=8 Гц); 2,15 (с, 6Н); 1,75 (м, 2Н); 3,68 (кв, 1Н, J=7 Гц); 1,4 (д, 3Н, J=7 Гц).
(R),(R',S')-2-[3-(α-гидрокси,α-метилбензил)фенил]-N-3-(диметиламинопропил)пропионамид в виде бесцветного масла.
[α]D=-28 (c=1; CH3OH).
1Н-ЯМР (CDCl3): δ 7,41-7,3 (м, 3Н); 7,31-7,14 (м, 6Н); 4,02 (уш.с, 1Н, ОН), 3,31 (м, 2Н) 2,38 (т, 2Н, J=8 Гц); 2,15 (с, 6Н); 1,75 (м, 2Н); 3,68 (кв, 1Н, J=7 Гц); 1,4 (д, 3Н, J=7 Гц).
(R),(R',S')-2-[3-(α-гидроксиэтил)фенил]-(3-диметиламинопропил)пропионамид
1Н-ЯМР (DMSO-d6): δ 8,12 (уш.с, 1Н, CONH); 7,31 (с, 1Н), 7,25-7,10 (м, 3Н); 5,1 (уш.с, 1Н, ОН); 4,7 (м, 1Н); 3,62 (м, 1Н); 3,10 (м, 2Н); 2,91 (м, 2Н); 3,65 (с, 6Н); 1,73 (м, 2Н); 1,30 (м, 6Н).
Пример 12
(R),(R',S')-2-[3'-(α-метилбензил)фенил]-N-3-(диметиламинопропил)пропионамид в виде светло-желтого масла (1,2 г; 3,52 ммоль).
[α]D=-30(c=1; CH3OH)
1Н-ЯМР (CDCl3): δ 7,38-7,13 (м, 9Н); 6,60 (уш.с, 1Н, CONH); 4,20 (м, 1Н), 3,78 (м, 1Н); 3,27 (м, 2Н); 2,30 (м, 2Н); 2,12 (с, 6Н); 1,72 (д, 3Н, J=7 Гц); 1,65 (м, 2Н); 1,55 (д, 3Н, J=7 Гц) получают, используя в методике по примеру 1 (R),(R',S')2-[3-(α-метилбензил)фенил]пропионилхлорид вместо (R)-(2-(4-изобутилфенил)пропионилхлорида.
Альтернативное применение (R)-2-(3-изопропилфенил)пропионилхлорида, (R)-2-(3-изобутилфенил), (R)-2-[3-(стирен-1-ил)фенил]пропионилхлорида, (R)-2-[3'-(пент-3-ил)фенил]пропионилхлорида в методике по примеру 1 дает:
(R)-2-(3-изопропилфенил)-N-3-(диметиламинопропил)пропионамид
1Н-ЯМР (CDCl3): δ 7,21 (м, 4Н); 6,95 (уш.с, 1Н, CONH); 3,53 (м, 1Н), 3,30 (м, 2Н); 2,90 (м, 1Н); 2,37 (м, 2Н); 2,15 (с, 6Н); 1,65 (д, 3Н, J=7 Гц); 1,23 (д, 3Н, J=7 Гц).
(R)-2-(3-изобутилфенил)-N-3-(диметиламинопропил)пропионамид
[α]D=-30 (c=1; CH3OH).
1Н-ЯМР (CDCl3): δ 7,21-7,13 (м, 4Н); 6,85 (уш.с, 1Н, CONH); 3,53 (м, 1Н), 3,25 (м, 2Н); 2,48 (д, 2Н, J=7 Гц); 2,30 (т, 2Н, J=7 Гц); 2,09 (с, 6Н); 1,9 (м, 1Н); 1,55 (м, 2Н); 1,45 (д, 3Н, J=7 Гц); 0,95 (д, 3Н, J=7 Гц).
(R)-2-[3-(стирен-1-ил)фенил]-N-3-(диметиламинопропил)пропионамид
[α]D=-31 (c=1; CH3OH).
1Н-ЯМР (CDCl3): δ 7,8-7,13 (м, 9Н); 6,95 (уш.с, 1Н, CONH); 5,0 (с, 2Н), 3,53 (м, 1Н); 3,30 (м, 2Н); 2,37 (м, 2Н); 2,15 (с, 6Н).
(R)-2-[3'-(пент-3-ил)фенил]-N-3-(диметиламинопропил)пропионамид
[α]D=-28(c=1; CH3OH).
1Н-ЯМР (CDCl3): δ 7,25 (м, 3Н); 7,12 (м, 1Н); 7,08 (уш.с, 1Н, CONH); 3,65 (м, 1Н), 3,5-3,13 (м, 2Н); 2,75 (м, 2Н); 2,55 (с, 6Н); 2,35 (м, 1Н); 1,95 (м, 2Н); 1,70 (м, 2Н); 1,58 (м, 2Н); 1,50 (д, 3Н, J=7 Гц); 0,76 (т, 6Н, J=7 Гц).
(R)-2-[(3-бензоил)фенил]-N-(3-диэтиламинопропил)пропионамид:
[α]D=-11,5 (c=3; CH3OH)
1Н-ЯМР (CDCl3): δ 7,8 (м, 3Н); 7,70-7,55 (м, 3Н); 7,50-7,28 (м, 3Н), 7,25 (уш.с, 1Н, CONH); 3,75 (м, 1Н); 3,50-3,20 (м, 2Н); 3,3,15-2,80 (м, 6Н); 2,05 (м, 2Н); 1,65 (д, 3Н, J=7 Гц); 1,70-1,53 (м, 3Н); 1,50-1,45 (м, 3Н).
(R)-2-[(3-бензоил)фенил]-N-(3-диметиламинопропил)пропионамид
[α]D=-20(c=1; CH3OH)
1Н-ЯМР (CDCl3): δ 7,88-7,78 (м, 3Н); 7,75-7,58 (м, 3Н); 7,55-7,46 (м, 3Н), 7,25 (д, 1Н, CONH); 3,62 (м, 1Н); 3,28 (м, 2Н); 2,35 (м, 2Н); 2,12 (с, 6Н); 1,68-1,53 (м, 5Н).
Пример 13
(R)-2-(4-изобутилфенил)-N-3-(гуанидинилпропил)пропионамидгидрохлорид
(R)-2-[(4-изобутилфенил)-N-3-(аминопропил)пропионамидгидрохлорид по примеру 5 превращают в свободный амин и обрабатывают изотиоуронийхлоридом согласно методике Bodanszky M. et al, (J. Am. Chem. Soc., 86, 4452, 1964), получая (R)-2-(4-изобутилфенил)-N-3-(гуанидинилпропил)пропионамидгидрохлорид.
Т.пл. 142-146°C; [α]D=-24 (c=1 CH3OH).
1Н-ЯМР (D2О): δ 7,2 (д, 2Н, J=8 Гц), 7,1 (д, 2Н, J=8 Гц); 6,8 (уш.с, 1Н, CONH); 3,6 (кв, 1Н, J=7 Гц); 3,55 (м, 2Н); 2,95 (м, 2Н); 2,4 (д, 2Н, J=7 Гц); 2,0-1,8 (м, 3Н); 1,5 (д, 3Н, J=7 Гц); 0,9 (д, 6Н, J=7 Гц).
Альтернативное применение в той же методике хлористоводородной соли метилового эфира N-гидроксикарбамидотиокарбоновой кислоты и метилового эфира N-аминокарбамидотиокарбоновой кислоты дает:
(R)-2-(4-изобутилфенил)-N-[3-(гидроксигуанидинил)пропил]пропионамид·HCl
(R)-2-(4-изобутилфенил)-N-[3-(аминогуанидинил)пропил]пропионамид·HCl
Пример 14
(R)-2-(4-изобутилфенил)-N-[3-(имидазолин-2-ил)-аминопропил]пропионамид
(R)-2-[(4-Изобутилфенил)-N-3-(аминопропил)пропионамидгидрохлорид (смотри, пример 5) превращают в свободный амин и обрабатывают 2-метилтио-2-имидазолиниодгидратом (промышленный реагент) согласно указанной выше методике Bodanszky (J. Am. Chem. Soc., 86, 4452, 1964), получая (R)-2-(4'-изобутилфенил)-N-[3-(имидазолин-2-ил)аминопропил]пропионамид.
Т.пл. 155-168°C; [α]D=-15 (c=1 CH3OH).
1Н-ЯМР (D2О): δ 7,2 (д, 2Н,J=8 Гц); 7,1 (д, 2Н, J=8 Гц); 6,8 (уш.с, 1Н, CONH); 3,6 (кв, 1Н, J=7 Гц), 3,55 (м, 2Н); 3,40 (с, 4Н); 2,90 (м, 2Н); 2,35 (д, 2Н, J=7 Гц); 2,0-1,8 (м, 3Н); 1,55 (д, 3Н, J=7 Гц); 1,0 (д, 6Н, J=7 Гц).
Использование 2-метилтиотетрагидропиримидина в вышеуказанной методике дает: (R) 2-(4-изобутилфенил)-N-[3-(тетрагидропиримидин-2-ил)аминопропилпропионамид.
1Н-ЯМР (D2О): δ 7,2 (д, 2Н, J=8 Гц); 7,1 (д, 2Н, J=8 Гц); 6,8 (уш.с, 1Н, CONH); 3,6 (кв, 1Н, J=7 Гц), 3,55 (м, 2Н); 3,40 (с, 4Н); 2,90 (м, 2Н); 2,35 (д, 2Н, J=7 Гц); 2,0-1,8 (м, 5Н); 1,55 (д, 3Н, J=7 Гц); 1,0 (д, 6Н, J=7 Гц).
Пример 15
(R),(S')-2-(4-изобутилфенил)-N-[(1-карбокси-4-амино)бутил]пропионамид
Раствор (R)-2-(4-изобутилфенил)пропионилхлорида (0,54 г; 2,42 ммоль) в CH2Cl2 (10 мл) медленно добавляют по каплям к суспензии гидрохлорида 5-BOC-орнитинметилового эфира (0,69 г; 2,42 ммоль) и триэтиламина (0,68 мл; 4,84 ммоль) в CH2Cl2 при 25°C. Смесь оставляют перемешиваться в течение ночи при комнатной температуре, затем разбавляют водой (10 мл). Органическую фазу отделяют и промывают насыщенным раствором NaHCO3 (10 мл), сушат над Na2SO4 и упаривают, получая сырой продукт, который очищают флэш-хроматографией (элюент CHCl3/CH3OH 9:1), получая метиловый эфир (R),(S)-2-(4-изобутилфенил)пропионил-(5-BOC)-орнитина в виде прозрачного масла (0,6 г; 1,4 ммоль). Обработка указанного соединения 3н HCl (8 мл) в течение 18 ч при комнатной температуре с последующим выпариванием растворителя дает (R),(S')-2-(4-изобутилфенил)-N-[(1-метоксикарбонил-4-амино)бутил]пропионамидгидрохлорид (0,41 г, 1,25 ммоль).
К раствору указанного гидрохлорида в диоксане добавляют при комнатной температуре 4н NaOH (0,625 мл; 2,5 ммоль), смесь перемешивают в течение ночи и упаривают досуха при пониженном давлении. Остаток поглощают EtOAc (15 мл); органическую фазу промывают насыщенным раствором NaCl (2×15 мл) и сушат над Na2SO4. Выпаривание AcOEt дает (R),(S')-2-(4-изобутилфенил)-N-[(1-карбокси-4-амино)бутил]пропионамид в виде белого твердого вещества,
т.пл. выше 240°C;
[α]D=-29 (c=0,5; CH3OH).
1Н-ЯМР (DMSO-d6): δ 7,3 (д, 2Н); δ 7,1 (д, 2Н); 6,25 (уш.с, 1Н, CONH); 4,20 (м, 1Н); 3,70 (м, 1Н), 3,50 (м, 2Н); 2,5 (д, 2Н); 1,9 (м, 1Н); 1,8 (м, 4Н); 1,6 (д, 3Н); 0,95 (д, 6Н, J=7 Гц).
(R),(S')-2-(4'-изобутилфенил)-N-(1-карбокси-5-аминопентил)пропионамидгидрохлорид
Получают, используя соответствующее производное (L)-лизина вместо производного орнитина.
[α]D=-28,3 (c=1; CH3OH)
1Н-ЯМР (DMSO-d6): δ 12,62 (уш.с, 1Н, COOH); 8,25 (д, 1Н, CONH, J=8 Гц), 7,75 (д, 3Н, NH3 +); 7,25 (д, 2Н, J=8 Гц); 7,06 (д, 2Н, J=8 Гц); 4,15 (м, 1Н); 3,70 (м, 1Н); 2,63 (м, 2Н); 2,38 (д, 2Н, J=7 Гц); 1,92-2,78 (м, 1Н); 1,70-1,38 (м, 4Н); 1,35 (д, 3Н, J=7 Гц); 1,20 (м, 2Н); 0,92 (д, 6Н, J=7 Гц).
Пример 16
(R)-2-(4-изобутилфенил)-N-[(N'-метил,N'-2-гидроксиэтил)аминоэтокси]пропионамид
Раствор (R)-2-(4-изобутилфенил)пропионилхлорида (0,42 г; 1,875 ммоль) в CH2Cl2 (10 мл) медленно добавляют по каплям к раствору 0,85 г (3,75 ммоль) 2-(аминокси)-N-метил-N-(2-гидроксиэтил)этиламина в CH2Cl2 (10 мл) при 25°C. Смесь оставляют перемешиваться при комнатной температуре на 3 ч и затем разбавляют H2O (10 мл). Две фазы затем взбалтывают и органическую фазу отделяют, промывают водой (5 мл), сушат над Na2SO4 и упаривают, получая 0,59 г (1,43 ммоль) (R)-2-(4-изобутилфенил)-N-2-[(N'-метил,N'-2-гидроксиэтил)аминоэтокси]пропионамид в виде масла.
[α]D=-35 (c=1; CH3OH).
1Н-ЯМР (CDCl3): δ 7,25 (м, 4Н); 6,15 (уш.с, 1Н, CONH); 4,67 (т, 2Н, J=7 Гц), 3,40 (м, 2Н); 2,75 (т, 2Н, J=7 Гц); 2,55 (д, 2Н, J=7 Гц); 2,35 (уш.с, 1Н, ОН); 2,42 (т, 2Н, J=7 Гц); 2,21 (с, 3Н); 1,95 (м, 1Н); 1,53 (д, 3Н, J=7 Гц); 1,00 (д, 6Н, J=7 Гц).
Пример 17
R-2-[(4-изобутил)фенил]-N-[4-(диметиламино)-2-бутинил]пропионамид
R(-)-ибупрофен (0,34 г; 1,65 ммоль) растворяют в сухом CH2Cl2; добавляют DCC (0,37 г; 1,8 ммоль) и HOBZ (0,24 г; 1,78 ммоль) и раствор оставляют при комнатной температуре при перемешивании на 3 ч. К раствору добавляют N,N-диметилбутин-2-ил-1,4-диамин (0,2 г; 1,78 ммоль), растворенный в сухом CH2Cl2 (2 мл), и полученную смесь перемешивают в течение ночи. Спустя 18 ч DCU фильтруют и фильтрат разбавляют CH2Cl2, промывают насыщенным раствором NaHCO3 (2×10 мл), водой (2×10 мл) и насыщенным раствором соли, сушат над Na2SO4 и упаривают в вакууме, получая красный маслянистый сырой остаток. Последующая очистка флэш-хроматографией дает R(-)-2-[(4'-изобутил)фенил]-N-[4-(диметиламино)-2-бутинил]пропионамид в виде желтого масла (0,347; 1,155 ммоль).
[α]D=+4,4 (c=0,5; CH3OH)
1Н-ЯМР (CDCl3): δ 7,15-7,10 (м, 2Н); 7,09-7,05 (м, 2Н); 5,45 (уш.с, 1Н, CONH); 4,05 (м, 2Н), 3,55 (м, 1Н); 3,15 (с, 2Н); 2,47 (д, 2Н, J=7 Гц); 2,22 (с, 6Н); 1,85 (м, 1Н); 1,48 (д, 3Н, J=7 Гц); 0,91 (д, 6Н, J=7 Гц).
Пример 18
R-Z-2-[(4-изобутил)фенил]-N-[4-(диметиламино)-2-бутенил]пропионамид
R-2-[(4'-изобутил)фенил]-N-[4-диметиламино-2-бутинил]пропионамид по примеру 17 (0,08 г; 0,27 ммоль) растворяют в абсолютном EtOH (5 мл) и добавляют 5% палладий на карбонате кальция (катализатор Lindlar'a; 0,08 г). Смесь гидрируют при атмосферном давлении и комнатной температуре в течение 2 ч, затем фильтруют через рыхлый слой целита. Плотный осадок на фильтре сильно промывают EtOH, фильтрат упаривают в вакууме, получая чистый R-Z-2-[(4-изобутил)фенил]-N-[4-(диметиламино)-2-бутенил]пропионамид в виде светло-желтого масла (0,07 г; 0,23 ммоль).
[α]D=-26,5 (c=1,1; CH3OH)
1Н-ЯМР (CDCl3): δ 7,20-7,12 (д, 2Н, J=8 Гц), 7,10-7,05 (д, 2Н, J=8 Гц); 5,95 (уш.с, 1Н, CONH); 5,67-5,55 (м, 2Н); 3,93-3,85 (м, 2Н); 5,02 (м, 1Н); 3,05 (д, 2Н, J=8 Гц); 2,47 (д, 2Н, J=7 Гц); 2,25 (с, 6Н); 1,93 (м, 1Н); 1,55 (д, 3Н, J=7 Гц); 0,95 (д, 6Н, J=7 Гц).
Пример 19
R-2-[(4-изобутил)фенил]-N-[4-(диметиламинометил)фенил]пропионамид
R(-)-Ибупрофен (0,31 г; 1,5 ммоль) растворяют в тионилхлориде (5 мл) и раствор нагревают до температуры кипения с обратным холодильником в течение 90 минут. Полное исчезновение исходной карбоновой кислоты контролируют по ИК; после охлаждения до комнатной температуры и отделения растворителя добавлениями 1,4-диоксана маслянистый остаток разбавляют сухим ДМФ (5 мл) и добавляют по каплям к перемешиваемому раствору 4-(N,N-диметиламинометил)анилина (0,27 г; 1,8 ммоль) в сухом ДМФ (3 мл) при комнатной температуре. Раствор оставляют перемешиваться в течение ночи; растворитель упаривают в вакууме и остаток очищают флэш-хроматографией, получая R-2-[(4-изобутил)фенил]-N-[4-(диметиламинометил)фенил]пропионамид в виде светло-желтого масла (0,406 г; 1,2 ммоль).
[α]D=-98 (c=1; CH3OH)
1Н-ЯМР (CDCl3): δ 7,40-7,18 (м, 9Н); 3,75 (м, 1Н); 3,47 (с, 2Н); 2,50 (д, 2Н, J=7 Гц); 2,17 (с, 6Н); 1,95 (м, 1Н); 1,56 (д, 3Н, J=7 Гц); 0,94 (д, 6Н, J=7 Гц).
Следуя той же методике, получают R-2-[4-изобутил)фенил]-N-[4-(диметиламино)фенил]пропионамид.
[α]D=-131 (c=0,25; CH3OH)
1Н-ЯМР (CDCl3): δ 7,28-7,25 (м, 4Н); 7,22-7,15 (м, 2Н); 6,83-6,79 (уш.с, 1Н, CONH); 6,73-6,65 (м, 2Н), 3,72 (м, 1Н); 2,80 (с, 6Н); 2,48 (д, 2Н, J=7 Гц); 1,85 (м, 1Н); 1,52 (д, 3Н, J=7 Гц); 0,97 (д, 6Н, J=7 Гц).
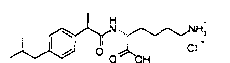
5±8
49±3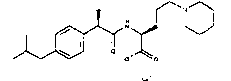
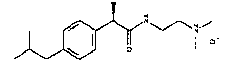

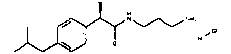
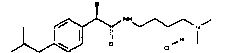
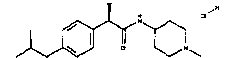

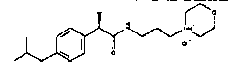
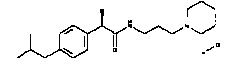
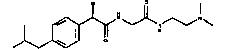
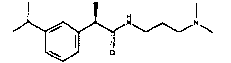
(с=10-6 М)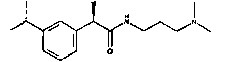

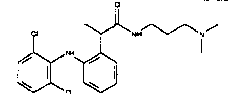
(с=10-6 М)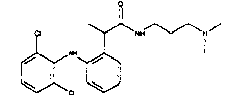
Экспериментальные данные in vivo на модели реакции гиперчувствительности замедленного типа (РГЗТ) и повреждения печени в результате ишемии/реперфузии (I/R)
(R)-2-(4-изобутилфенил)-N-(1-метилпиперидин-4-ил)пропионамид, (R)-2-(4-бромфенил)-N-(1-метилпиперидин-4-ил)пропионамид и (R)-2-(4-изобутилфенил)-N-(3-диметиламинопропил)-пропионамид исследовали на различных моделях животных в отношении повреждения РГЗТ и I/R. Эти модели являются соответственно представителями воспалительных заболеваний человека, таких как псориаз и повреждение печени в результате постишемической реперфузии.
Экспериментальный пример 1
(R)-2-(4-изобутилфенил)-N-(1-метилпиперидин-4-ил)пропионамид исследован на мышиной модели РГЗТ, используя два различных аптена (DNFB-фтор-2,4-динитробензол и оксазолон1,2) в дозе 30 мг/кг (обработка: подкожно за 8 ч до введения; за 30 мин до введения; после 8 ч; 16 ч после введения вещества, провоцирующего заболевание).
Эффективность на модели РГЗТ оценивали как 24 ч после введения провоцирующего вещества метилпероксидазным методом (МРО), оценкой веса уха мыши и эдемы (экстравазия жидкостью Эванс голубой). Более того, (R)-2-(4-изобутилфенил)-N-(1-метилпиперидин-4-ил)пропионамид тестировали на крысиной модели I/R3 печени при двух различных дозах, 13 мг/кг и 6,5 мг/кг (обработка: подкожно за 12 ч до ишемии; внутривенно 5 с перед реперфузией и подкожно 2 ч после реперфузии), как будет показано ниже.
Эффективность в отношении I/R повреждения печени оценивали путем измерения инфильтрата полиморфоядерных нейтрофилов (PMN) печени (МРО исследование и подсчет гистологических PMN) и оценкой гепатоцеллюлярного некроза (уровни ALT (аланин аминотрансферазы) и AST (аспартат аминотрансферазы) в сыворотке). Результаты эффективности (R)-2-(4-изобутилфенил)-N-(1-метилпиперидин-4-ил)пропионамида на вышеуказанных моделях показаны в Таблице 2.
Экспериментальный пример 2
(R)-2-(4-бромфенил)-N-(1-метилпиперидин-4-ил)пропионамид исследовали на описанной экспериментальной модели РГЗТ в дозе 30 мг/кг (обработка: подкожно за 8 ч до введения; за 30 мин до введения; после 8 ч; 16 ч после введения вещества, провоцирующего заболевание), используя в качестве аптена DNFB. Было найдено, что (R)-2-(4-бромфенил)-N-(1-метилпиперидин-4-ил)пропионамид эффективен в предотвращении PMN инфильтрации (-50% МРО уровней) и в уменьшении увеличения веса уха (-30%).
Экспериментальный пример 3
(R)-2-(4-изобутилфенил)-N-(3-диметиламинопропил)пропионамид тестировали на крысиной модели I/R3 печени в двух различных дозах, 13 мг/кг и 6.5 мг/кг (обработка: подкожно за 12 ч до ишемии, внутривенно 5 с перед реперфузией, подкожно 2 ч после реперфузии). Результаты опытов представлены в Таблице 3.
Вес уха: -40%
МРО: -70%
Вес уха: -50%
МРО: -80%
Ссылки:
1) Dilulio NA et al. Groα-mediated recruitment of neutrophils is required for elicitation of contact hypersensitivity. 1999. Eur J Immunol 29, 3485-3495.
2) Grabbe S et al. Beta2 integrins are required for skin homing of primed Т cells but not for priming naive Т cells. 2002. J Clin Invest 109, 183-192.
3) Bertini et. al 2004, PNAS, 101, 11791-11796.
Пример получения фармацевтической композиции.
Препарат для инъекции
В растворе, содержащем 100 г соединения, полученного в примере 8, 5 г динатрийгидрофосфата в 3 л дважды перегнанной воды устанавливают рН 6.5 с помощью 2н соляной кислоты, раствор стерильно фильтруют, разливают в стеклянные емкости для препаратов для инъекции, лиофилизируют в стерильных условиях и стерильно закупоривают. Каждая стеклянная емкость с препаратом для инъекции содержит 5 мг биологически активного вещества.
Таблетки
Смесь 1 кг биологически активного вещества (соединение примера 10), 4 кг лактозы, 1.2 кг картофельного крахмала, 0.2 кг талька и 0.1 кг стеарата магния тщательно перемешивают и прессуют в таблетки так, что каждая таблетка содержит 10 мг биологически активного вещества.
| название | год | авторы | номер документа |
|---|---|---|---|
| (R)-2-АРИЛПРОПИОНАМИДЫ, ПОЛЕЗНЫЕ ПРИ ИНГИБИРОВАНИИ ИЛ-8-ИНДУЦИРОВАННОГО ХЕМОТАКСИСА НЕЙТРОФИЛОВ, СПОСОБ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИНГИБИРУЮЩАЯ ХЕМОТАКСИС НЕЙТРОФИЛОВ, ИНДУЦИРОВАННЫЙ ИНТЕРЛЕЙКИНОМ-8 | 2001 |
|
RU2273630C2 |
| ЧЕТВЕРТИЧНЫЕ АММОНИЕВЫЕ СОЛИ ОМЕГА-АМИНОАЛКИЛАМИДОВ R-2-АРИЛПРОПИОНОВЫХ КИСЛОТ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ | 2002 |
|
RU2291857C2 |
| N-(2-АРИЛПРОПИОНИЛ)СУЛЬФОНАМИДЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ | 1999 |
|
RU2255084C2 |
| ПРИМЕНЕНИЕ ХИРАЛЬНЫХ АРИЛКЕТОНОВ В ЛЕЧЕНИИ НЕЙТРОФИЛ-ЗАВИСИМЫХ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ | 2003 |
|
RU2345759C2 |
| ПРОИЗВОДНЫЕ 2-АРИЛПРОПИОНОВОЙ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, ИХ ВКЛЮЧАЮЩИЕ | 2005 |
|
RU2410372C2 |
| ПРОИЗВОДНЫЕ 2-ФЕНИЛПРОПИОНОВОЙ КИСЛОТЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2005 |
|
RU2375347C2 |
| 2-АРИЛУКСУСНЫЕ КИСЛОТЫ, ИХ ПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2004 |
|
RU2356887C2 |
| АМИДИНЫ И ИХ ПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2004 |
|
RU2375346C2 |
| (R)-4-(ГЕТЕРОАРИЛ)ФЕНИЛЭТИЛЬНЫЕ ПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2008 |
|
RU2475486C2 |
| 2-АРИЛПРОПИОНОВЫЕ КИСЛОТЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2002 |
|
RU2317075C2 |
Изобретение относится к соединениям формулы (I)
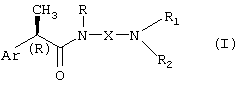
где Ar представляет фенил, замещенный группой, выбранный из изобутила, бензоила, изопропила, стирола, пентила, (2,6-дихлорфенил)амино, α-гидроксиэтила, α-гидроксибензила, α-метилбензила и α-гидрокси-α-метилбензила; R представляет водород; Х означает линейный C1-С6-алкилен, С4-С6-алкенилен, С4-С6алкинилен, необязательно замещенный группой CO2R3, где R3 означает водород, (CH2)m-B-(CH2)n группу, где В означает атом кислорода, m равно нулю и n означает целое число 2; или В означает группу CONH, m означает целое число 1 и n означает целое число 2 и т.д.; R1 и R2 независимо выбирают из группы, включающей: водород, линейный С1-С4-алкил, гидрокси-С2-С3-алкил и т.д. Предложен способ получения соединений формулы (I). Предложены ингибиторы С5-индуцируемого хемотаксиса полиморфноядерных лейкоцитов и моноцитов, представляющие омега-аминоалкиламиды (R) 2-арилпропионовой кислоты формулы (I). Фармацевтическая композиция, обладающая ингибирующей активностью в отношении хемотаксиса полиморфноядерных лейкоцитов и моноцитов, содержащая соединения формулы (I) в смеси с подходящим носителем. Технический результат - омега-алкиламиды (R)-2-арилпропионовых кислот, полезные для ингибирования хемотаксической активации, индуцируемой С5а и другими хемотаксическими протеинами. 4 н. и 14 з.п. ф-лы, 3 табл.
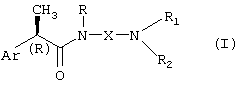
и их фармацевтически приемлемые соли,
где Ar означает фенил, замещенный группой, выбранной из изобутила, бензоила, изопропила, стирола, пентила, (2,6-дихлорфенил)амино, α-гидроксиэтила, α-гидроксибензила, α-метилбензила и α-гидрокси-α-метилбензила;
R означает водород;
Х означает:
линейный C1-С6-алкилен, С4-С6-алкенилен, С4-С6-алкинилен, необязательно замещенный группой СО2R3, где R3 обозначает водород;
(CH2)m-B-(CH2)n группу, где В означает атом кислорода, m равно нулю и n означает целое число 2; или В означает группу CONH, m означает целое число 1 и n означает целое число 2;
либо X вместе с атомом азота омега-аминогруппы, с которым он связан, и с группой R1 образует неароматическое азотсодержащее 6-членное гетероциклическое, моноциклическое ядро, в котором атом азота имеет заместитель Rc, где Rc означает С1-С4-алкил, или Х означает фенильную или фенилметильную группу;
R1 и R2 независимо выбирают из группы, включающей: водород, линейный С1-С4-алкил, гидрокси-С2-С3-алкил;
либо R1 и R2 вместе с атомом N, с которым они связаны, образуют азотсодержащее 6-членное гетероциклическое ядро формулы (II)
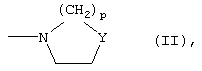
где Y означает СН2, О, S или группу N-Rc, принимающую вышеуказанные значения, и р означает целое число 2;
либо R1 принимает вышеуказанные значения, R2 означает группу формулы (III)
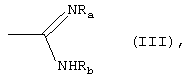
где Ra означает водород и Rb означает водород;
или Ra и Rb, вместе с атомом азота, с которым они связаны, образуют 5-6-членное гетероциклическое ядро, моноциклическое ядро.
(R)-2-[(4-изобутил)фенил]-N-(3-диметиламинопропил)пропионамид;
(R)-2-[(4-изобутил)фенил]-N-(4-диметиламинобутил)пропионамид гидрохлорид;
(R)-2-[(4-изобутил)фенил]-N-(3-N-морфолинилпропил)пропионамид;
(R)-2-[(4-изобутил)фенил]-N-(2-диметиламиноэтил)пропионамид;
(R)-2-[(4-изобутил)фенил)пропионил]-N-[2-(4-метилпиперазин-1-ил)этил]пропионамид;
(R)-N-(экзо-8-метил-8-аза-бицикло[3.2.1]окт-3-ил)-2-[(4-изобутилфенил)пропионамид;
(R)-2-[(4-изобутил)фенил]-N-(3-N-тиоморфолинилпропил)пропионамид;
(R)-2-[(4-изобутил)фенил]-N-[4-(N'-метил)пиперидинил]пропионамид гидрохлорид;
(R),(S')-2-[(4-изобутил)фенил]-N-(1-карбокси-2-диметиламиноэтил)пропионамид;
(R),(S')-2-[(4-изобутил)фенил]-N-[(1-карбокси-4-пиперидин-1-ил)бутил]пропионамид;
(R),(S')-2-[(4-изобутил)фенил]-N-(1-карбокси-4-аминобутил)пропионамид;
(R)-2-(4-изобутил)фенил-N-[2-(диметиламиноэтил)амино-карбонилметил]пропионамид гидрохлорид;
2-(2,6-дихлорфениламино)фенил-N-(3-диметиламинопропил)пропионамид;
(R),(R',S')-3-[3-(α-метил)бензил]фенил-N-(3-диметиламинопропил)пропионамид;
(R)-2-[(3-изопропил)фенил]-N-(3-диметиламинопропил)пропионамид;
(R)-2-[3-(пент-3-ил)фенил]-N-(3-диметиламинопропил)пропионамид;
(R)-2-[(4-изобутил)фенил]-N-(3-гуанидилпропил)пропионамид;
(R)-2-[(4-изобутил)фенил]-N-[(3-гидроксигуанидил)пропил]-пропионамид;
(R)-2-[(4-изобутил)фенил]-N-[(3-аминогуанидил)пропил]-пропионамид;
(R)-2-[(4-изобутил)фенил]-N-[3-(2-амино-2-имидазолин)пропил]-пропионамид;
(R)-2-[(4-изобутил)фенил]-N-[N-метил-N-(2-гидроксиэтил)-аминоэтокси]пропионамид;
(R),(S')-2-[(4-изобутил)фенил]-N-[1-карбокси-5-аминопентил]-пропионамид.
где Ar, X, R, R1 и R2, принимают значения, указанные по п.1, включающий взаимодействие активированной формы R-2-арилпропионовой кислоты формулы (V) с амином формулы (VI)
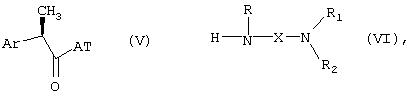
где AT означает остаток, активирующий карбоксигруппу R-2-арилпропионовой кислоты;
при условии, что Ar не замещен остатком дигидропиррола.
и их фармацевтически приемлемые соли,
где Ar означает фенил, замещенный группой, выбранной из изобутила, бензоила, изопропила, стирола, пентила, (2,6-дихлорфенил)амино, α-гидроксиэтила, α-гидроксибензила, α-метилбензила и α-гидрокси-α-метилбензила;
R означает водород;
Х означает линейный C1-С6-алкилен, С4-С6-алкенилен, С4-С6-алкинилен, необязательно замещенный группой CO2R3, где R3 обозначает водород; (СН2)m-В-(СН2)n группу, где В означает атом кислорода, m равно нулю и n означает целое число 2; или В означает группу CONH, m означает целое число 1 и n означает целое число 2;
либо Х вместе с атомом азота омега-аминогруппы, с которым он связан, и с группой R1 образует неароматическое азотсодержащее 6-членное гетероциклическое, моноциклическое ядро, в котором атом азота имеет заместитель Rc, где Rc означает С1-С4-алкил, или Х означает фенильную или фенилметильную группу;
R1 и R2 независимо выбирают из группы, включающей: водород, линейный С1-С4-алкил, гидрокси-С2-С3-алкил;
либо R1 и R2 вместе с атомом N, с которым они связаны, образуют азотсодержащее 6-членное гетероциклическое ядро формулы (II)
где Y означает СН2, О, S или группу N-Rc, принимающую вышеуказанные значения, и р означает целое число 2;
либо R1 принимает вышеуказанные значения, R2 означает группу формулы (III)
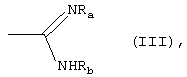
где Ra означает водород и Rb означает водород;
или Ra и Rb, вместе с атомом азота, с которым они связаны, образуют 5-6-членное гетероциклическое, моноциклическое ядро.
| TSUNEMATSU H | |||
| et al: "Synthesis and the stereoselective enzymatic hydrolysis of flurbiprofen-basic amino acid ethyl esters", JOURNAL OF DRUG TARGETING, 1995, vol.2, no.6, p.517-525 | |||
| SHANBHAG V.R: "Ester and amide prodrugs of ibuprofen and naproxen: synthesis, anti-inflammatory activity, and gastrointestinal toxicity", AMERICAN PHARMACEUTICAL |

