Изобретение относится к области выделения и очистки карбоната стронция, в том числе изотопнообогащенного, полученного методом электромагнитной сепарации.
Известен способ извлечения стронция из целестиновых руд (патент РФ №2050323, МПК6 С 01 F 11/36), включающий обработку целестина содовым раствором, отделение карбонатного продукта и его обработку азотной кислотой с концентрацией 3,0-3,5% маc., взятой при соотношении эквивалентов HNO3: SrСО3=(1,5-1,8):1, с получением раствора нитрата стронция.
Недостатком этого способа является невозможность получения чистого продукта, что потребует дальнейшей химической переработки раствора нитрата стронция.
Известен также способ получения стронциевого концентрата (патент РФ №2022940, МПК5 С 01 F 11/38), включающий разложение апатита азотной кислотой с получением азотно-кислой вытяжки, выдерживание ее при 40-60°C, отделение осадка стронциевого концентрата от жидкой фазы, охлаждение жидкой фазы до 25-30°С и выделение осадка стронциевого концентрата.
Недостатком этого способа также является необходимость проведения многостадийной переработки полученного концентрата для его очистки и выделения в требуемой химической форме.
Наиболее близким к заявленному техническому решению является способ переработки стронциевого концентрата в карбонат стронция (патент РФ №2024432, МПК5 С 01 F 11/18), включающий выщелачивание стронциевого концентрата азотной кислотой с получением стронцийсодержащего осадка, перевод солей стронция в раствор путем обработки осадка оборотным водным раствором, отделение нерастворимого осадка и его промывку, высаливание из полученного раствора кристаллов нитрата стронция подачей азотной кислоты, обезвоживание кристаллов, их растворение, осаждение из раствора примесей с последующим получением карбоната стронция. При этом отделяют и промывают крупнодисперсную часть нерастворимого остатка, а высаливание проводят из растворов, содержащих мелкодисперсную часть остатка, причем перед обезвоживанием кристаллы очищают от примесей путем противоточного контактирования с азотной кислотой в восходящем потоке.
Основным недостатком этого способа при использовании его для получения карбоната изотопнообогащенного стронция являются значительные потери дорогостоящего вещества на стадии получения кристаллов нитрата стронция высаливанием их добавлением азотной кислоты к водному раствору нитрата стронция. Экспериментально установлено, что при соотношении 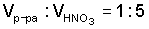 концентрация стронция в маточном растворе составляет 1,5-1,7 г/л, что недопустимо при переработке дорогостоящего изотопнообогащенного вещества.
концентрация стронция в маточном растворе составляет 1,5-1,7 г/л, что недопустимо при переработке дорогостоящего изотопнообогащенного вещества.
К тому же при значительном содержании меди, железа и свинца в исходном концентрате требуется увеличение расхода азотной кислоты при промывке кристаллов нитрата стронция, что также приводит к дополнительным потерям вещества. Экспериментально установлено, что содержание стронция в промывочной кислоте составляет 0,040-0,070 г/л.
Кроме того, этот способ не обеспечивает очистки стронция от примеси бария.
Техническим результатом изобретения является получение чистого карбоната стронция, в том числе изотопнообогащенного, при минимальных потерях на всех стадиях химической переработки.
Технический результат достигается способом получения карбоната стронция, включающим обработку концентрата стронция кислотой, получение стронцийсодержащего осадка, перевод стронция в раствор, отделение нерастворимого остатка и его промывку, получение кристаллов нитрата стронция, очистку кристаллов от примесей промывкой их азотной кислотой, обезвоживание кристаллов, их растворение, отделение примесей и последующее получение карбоната стронция, при этом согласно изобретению, раствор, полученный от обработки кислотой концентрата стронция, предварительно очищают путем осаждения и отделения примесей группы железа в виде гидроксидов, а бария и свинца в виде сульфатов, стронцийсодержащий осадок осаждают в виде карбоната стронция добавлением в раствор карбоната аммония и аммиака, с последующим прокаливанием осадка при 600-700°С, стронций переводят в раствор обработкой прокаленного осадка карбоната стронция азотной кислотой при соотношении (1:2)-(1:3). Также тем, что обезвоженные кристаллы нитрата стронция растворяют в воде при массовом соотношении воды к нитрату стронция, равном 1-2:1, и тем, что карбонат стронция осаждают карбонатом аммония при рН 9-10.
В заявленном техническом решении предложенные операции предварительного отделения примесей в виде гидроксидов, сульфатов и аммиачных комплексов, прокаливание стронцийсодержащего осадка с последующим его растворением и отделением примесей и осаждение карбоната стронция карбонатом аммония при рН 9-10 позволяют выделить чистый карбонат стронция с минимальными потерями на всех стадиях химической переработки.
Проведенный анализ общедоступных источников об уровне техники не позволил выявить техническое решение, тождественное заявленному, на основании чего делается вывод о неизвестности последнего, т.е. соответствии представленного в настоящей заявке изобретения критерию "новизна".
Сопоставительный анализ заявленного решения с известными техническими решениями позволил выявить, что представленная совокупность отличительных признаков не известна для специалиста в данной области и не следует явным образом из известного уровня техники, на основании чего делается вывод о соответствии представленного в настоящей заявке изобретения критерию "изобретательский уровень".
Предложенный способ получения металлического карбоната стронция реализовали следующим образом.
Пример 1
50 г концентрата, содержащего 28 г стронция, а также примеси меди, железа, хрома, свинца, бария, кремния, кальция, алюминия и т.д., обработали раствором НСl (1:5) при нагревании и разбавили водой до концентрации стронция 100 г/л. Провели отделение примеси бария и основного количества свинца в виде сульфатов, добавив к раствору 0,84 мл серной кислоты (из расчета 0,03 мл Н2SO4 на 1 г стронция). Раствор с осадком прогрели в течение 2 часов и выстаивали в течение суток. Осадок сульфатов отфильтровали, промыли дистиллированной водой. Возвратные потери стронция с сульфатным шламом составили 4,8%. К раствору стронция добавили аммиак до рН 8-9 и осадили гидроксиды примесей группы железа. Осадок отфильтровали и промыли. Шламы гидроксидов проанализировали на содержание стронция. Оно составило 0,02%. К раствору добавили карбонат аммония из расчета 1 г (NН4)2СО3 на 1 г стронция и избыток 15 г/л и аммиак до рН 9-10. После выстаивания в течение 8-10 часов осадок карбоната стронция отфильтровали, промыли до обесцвечивания промывных вод. Фильтрат проанализировали на содержание стронция. Оно составило 2,7 мг/л. Осадок карбоната стронция прокалили при 600°C.
Прокаленный карбонат стронция растворили в азотной кислоте (1:2), нерастворимый остаток оксида кремния отфильтровали и промыли водой. Содержание стронция в шламе оксида кремния составило 0,01%.
Раствор упарили до получения влажных кристаллов нитрата стронция, которые промыли азотной кислотой сначала методом репульпации (из расчета 40 мл HNO3 на 1 г стронция), а затем на фильтре. Азотнокислый фильтрат проанализировали на содержание стронция. Оно составило 48 мг/л. Кристаллы нитрата стронция просушили при 110-120°С и растворили в воде при соотношении масс и 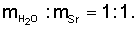 Нерастворимый остаток оксида кремния отфильтровали и промыли водой. Содержание в шламе оксида кремния составило 0,01%. В раствор нитрата стронция добавили аммиак до рН 9-10, карбонат аммония из расчета 1 г (NН4)2СО3 на 1 г Sr и избыток 10 г (NH4)2CO3 на 1 л раствора и осадили карбонат стронция. Раствор с осадком выстаивали 8-10 часов, осадок отфильтровали, промыли, просушили и прокалили при 600°С. Карбонатные фильтраты проанализировали на содержание стронция. Оно составило 3,2 мг/л. Выход карбоната стронция составил 99,5%. Полученный карбонат стронция проанализировали на содержание примесей по 70 элементам. Оно составило 0,007%, в том числе 0,0006% бария и 0,0009% свинца.
Нерастворимый остаток оксида кремния отфильтровали и промыли водой. Содержание в шламе оксида кремния составило 0,01%. В раствор нитрата стронция добавили аммиак до рН 9-10, карбонат аммония из расчета 1 г (NН4)2СО3 на 1 г Sr и избыток 10 г (NH4)2CO3 на 1 л раствора и осадили карбонат стронция. Раствор с осадком выстаивали 8-10 часов, осадок отфильтровали, промыли, просушили и прокалили при 600°С. Карбонатные фильтраты проанализировали на содержание стронция. Оно составило 3,2 мг/л. Выход карбоната стронция составил 99,5%. Полученный карбонат стронция проанализировали на содержание примесей по 70 элементам. Оно составило 0,007%, в том числе 0,0006% бария и 0,0009% свинца.
Пример 2
Получение карбоната стронция провели как в примере 1, но карбонат стронция прокаливали при 700°C, растворили его в азотной кислоте (1:3), а растворение обезвоженных кристаллов нитрата стронция проводили при соотношении 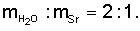 Возвратные потери стронция с осадком сульфатов составили 4,7%. Содержание стронция в шламах гидроксидов составило 0,03%. Содержание стронция в шламах оксида кремния составило 0,01%. Содержание стронция в азотнокислых фильтратах составило 51 мг/л. Содержание стронция в карбонатных фильтратах составило 2,9 мг/л. Выход карбоната стронция составил 99,4%. Содержание суммы примесей в карбонате стронция составило 0,005%.
Возвратные потери стронция с осадком сульфатов составили 4,7%. Содержание стронция в шламах гидроксидов составило 0,03%. Содержание стронция в шламах оксида кремния составило 0,01%. Содержание стронция в азотнокислых фильтратах составило 51 мг/л. Содержание стронция в карбонатных фильтратах составило 2,9 мг/л. Выход карбоната стронция составил 99,4%. Содержание суммы примесей в карбонате стронция составило 0,005%.
Пример 3
Получение карбоната стронция провели как в примере 1, но карбонат стронция прокалили при 500°C, растворили его в азотной кислоте (1:1), а растворение обезвоженных кристаллов нитрата стронция провели при соотношении 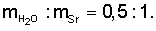
Возвратные потери стронция с осадком сульфатов составили 4,8%.
Содержание стронция в шламах гидроксидов составило 0,02%. Содержание стронция в шламах оксида кремния составило 0,17%. Содержание стронция в азотнокислых фильтратах составило 52 мг/л. Содержание стронция в карбонатных фильтратах составило 3,2 мг/л. Выход карбоната стронция составил 99,3%.
Содержание суммы примесей в карбонате стронция составило 0,046%, в том числе 0,031% кремния.
Время упаривания раствора при получении кристаллов нитрата стронция увеличилось.
Пример 4
Получение карбоната стронция провели по патенту РФ №2024482, МПК С 01 F 11/18, выбранному в качестве наиболее близкого технического решения. 50 г концентрата, содержащего 28 г стронция, а также примеси меди, железа, хрома, бария, свинца, кремния, алюминия, кальция и т.д., обработали азотной кислотой при нагревании. Суспензию упарили до получения осадка нитратов стронция и примесей. Осадок растворили в минимальном количестве воды (60 мл). К полученной сусупензии добавили азотной кислоты из расчета 40 мл HNO3 на 1 г стронция. Кристаллы нитрата стронция отделили от маточного раствора и промыли азотной кислотой сначала методом репульпации, а затем на фильтре, как в примере 1. В азотнокислых растворах определили содержание стронция. Оно составило 798 мг/л и 67 мг/л для маточного раствора и промывной кислоты соответственно.
Кристаллы нитрата стронция просушили при 110-120°С, растворили в воде, отфильтровали и промыли нерастворимый остаток. В полученном растворе осадили карбонат стронция, добавив аммиак до рН 9-10 и карбонат аммония из расчета 1 г (NН4)2СО3 на 1 г стронция и избыток 10 г/л. Раствор с осадком выстаивали в течение 8-10 часов, затем осадок отфильтровали, промыли, сушили при 120-150°С и прокалили при 600°С. Выход карбоната стронция составил 95,2%.
Полученный карбонат стронция проанализировали на содержание суммы примесей. Она составила 0,572%, в том числе 0,476% бария, 0,041% свинца и 0,037% кремния.
Полученные результаты показали, что при использовании известного технического решения по сравнению с предложенным, значительно увеличились потери стронция на стадии получения кристаллов нитрата стронция и их промывки и снизилась степень его химической очистки, что особенно недопустимо при получении карбоната изотопнообогащенного стронция.
Предложенный способ получения карбоната стронция был опробован на производстве стабильных изотопов. Он позволил выделить 99,2% карбоната стронция - 88 со степенью очистки 99,999%.
Разработанный способ позволяет применять стандартное оборудование, дешевые и доступные реактивы, не требует большого расхода электроэнергии.
Способ пригоден и с экологической точки зрения, т.к. образующиеся азотнокислые фильтраты нейтрализуются раствором щелочи или карбонатными фильтратами, полученными при получении карбоната стронция.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПЕРЕРАБОТКИ ТЕТРАГИДРАТА НИТРАТА КАЛЬЦИЯ ПРОИЗВОДСТВА АЗОФОСКИ | 1991 |
|
RU2031840C1 |
| СПОСОБ ОЧИСТКИ НИТРАТА СТРОНЦИЯ ОТ ПРИМЕСЕЙ БАРИЯ В ТЕХНОЛОГИИ ПОЛУЧЕНИЯ КАРБОНАТА СТРОНЦИЯ | 1993 |
|
RU2024433C1 |
| СПОСОБ ПОЛУЧЕНИЯ КАРБОНАТА СТРОНЦИЯ ВЫСОКОЙ ЧИСТОТЫ | 2009 |
|
RU2412116C1 |
| СПОСОБ ПОЛУЧЕНИЯ КОНЦЕНТРАТА СТРОНЦИЯ | 1991 |
|
RU2048439C1 |
| СПОСОБ ПОЛУЧЕНИЯ КАРБОНАТА БАРИЯ | 2001 |
|
RU2195428C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОКСИДА КАЛЬЦИЯ | 2002 |
|
RU2214966C1 |
| СПОСОБ ПОЛУЧЕНИЯ КАРБОНАТА СТРОНЦИЯ | 2002 |
|
RU2223223C1 |
| СПОСОБ ПЕРЕРАБОТКИ СТРОНЦИЕВОГО КОНЦЕНТРАТА В КАРБОНАТ СТРОНЦИЯ | 1993 |
|
RU2024432C1 |
| Способ получения карбоната кальция | 2017 |
|
RU2676292C1 |
| СПОСОБ ПОЛУЧЕНИЯ СТРОНЦИЙСОДЕРЖАЩЕГО КОНЦЕНТРАТА | 1991 |
|
RU2032622C1 |
Способ относится к выделению и очистке карбоната стронция, в том числе изотопнообогащенного, полученного методом электромагнитной сепарации. Концентрат стронция обрабатывают кислотой, получают стронцийсодержащий осадок, переводят стронций в раствор с отделением и промывкой нерастворимого остатка, получают кристаллы нитрата стронция, очищают кристаллы от примесей промывкой их азотной кислотой, кристаллы обезвоживают, растворяют и отделяют примеси с последующим получением карбоната стронция. При этом раствор, полученный от обработки концентрата стронция, предварительно очищают путем осаждения и отделения примесей группы железа в виде гидроксидов, а бария и свинца в виде сульфатов, стронцийсодержащий осадок осаждают в виде карбоната стронция добавлением в раствор карбоната аммония и аммиака с последующим прокаливанием осадка при 600-700°С. Стронций переводят в раствор обработкой прокаленного осадка карбоната стронция азотной кислотой при соотношении (1:2) -(1:3), обезвоженные кристаллы нитрата стронция растворяют в воде при массовом соотношении воды к нитрату стронция, равном (1-2):1, а карбонат стронция осаждают карбонатом аммония при рН 9-10. Предварительное отделение примесей в виде гидроксидов, сульфатов и аммиачных комплексов, прокаливание стронцийсодержащего осадка с последующим его растворением и отделением примесей и осаждение стронция карбонатом аммония при рН 9-10 позволяют выделить 99,2% карбоната стронция-88 со степенью очистки 99,999%. Техническим результатом является получение чистого карбоната стронция при минимальных потерях на стадиях переработки. 2 з.п. ф-лы.
| СПОСОБ ПЕРЕРАБОТКИ СТРОНЦИЕВОГО КОНЦЕНТРАТА В КАРБОНАТ СТРОНЦИЯ | 1993 |
|
RU2024432C1 |
| Способ получения карбоната стронция | 1977 |
|
SU645938A1 |
| СПОСОБ ПОЛУЧЕНИЯ КОНЦЕНТРАТА СТРОНЦИЯ | 1991 |
|
RU2048439C1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| US 4666688 А, 19.05.1987 | |||
| JP 59083934 А, 15.05.1984 | |||
| JP 55023055 А, 19.02.1980. | |||

