Настоящее изобретение относится к соединениям, которые имеют новое использование в качестве источника моноксида углерода и, возможно, в качестве восстановителя при получении карбонильных комплексов переходных металлов.
Карбонильные комплексы являются соединениями, которые содержат моноксид углерода в качестве координированного лиганда. Моноксид углерода является распространенным лигандом в химии переходных металлов, отчасти из-за синергической природы его химической связи с переходными металлами. Связь СО с металлом состоит из двух компонент. Первая компонента связи основана на σ-донировании, то есть перекрывании неподеленной пары электронов у атома углерода с вакантной d-орбиталью металла. Вторая компонента включает обратное π-донирование из полностью заполненной d-орбитали металла в вакантную π* орбиталь атома углерода СО. Этот вторая компонента называется дативной пи-связью или обратным π-донированием.
Вышеописанное образование карбонильных комплексов переходных металлов является ключевым моментом для использования таких соединений при введении метки в молекулы белков, пептидов и широкого разнообразия других соединений. Во многих областях применения мечение этих молекул осуществляют с использованием так называемого набора для мечения веществ, который содержит необходимые реактивы. Современные наборы для введения метки основаны на использовании бороводорода в качестве восстановителя, которые также содержат тартрат, лактозу и боратный буфер, рН 11,5 и заполнены газообразным СО в качестве источника монооксида углерода. К недостаткам этих известных реакционных смесей относится медленное растворение СО в растворителе реакционной смеси, что приводит к снижению выхода карбонильных комплексов, невозможности промышленного производства большого количества флаконов, содержащих набор реактивов, заполненных СО, и медленной диффузии СО даже через герметично закрытые флаконы. Недостатком является также то, что показатель рН смесей довольно высок.
Задачей настоящего изобретения является создание модифицированной формы СО и борогидрида натрия, которая не имела бы вышеупомянутых недостатков.
Задача решена тем, что заявляются новые производные боранокарбоната следующей формулы:
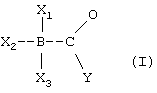
где X1 означает - Н;
Х3 и Х2 являются заместителями, которые могут быть одинаковьми или различными и которые выбирают из группы, состоящей из - Н, - NHxRy при х+у=3, или - R, где R означает заместитель, который связан через атом углерода с атомом азота или бора соответственно и представляет собой метил или этил;
Y означает группу - ОН, - OH2, - OR или - NHR, в которой R означает заместитель, который связан через атом углерода с атомом азота или бора соответственно и представляет собой метил или этил; или их соли для использования в качестве источника моноксида углерода (СО), а также в качестве восстановителя при получении карбонильных комплексов металлов в водном растворе.
Когда Y означает группу -ОН2, то соединения представляют собой кислоты, которые могут быть депротонированы (например с использованием NaOH). В этом случае соединения, выделяемые в чистом виде, являются солями (боранокарбонат-анион R3В-СОО2-+ катион соответствующего металла, например, Li+, Na+, Ca2 + Mg2+ и других). Соединение проявляет функцию восстановителя только в случае, когда по меньшей мере один из радикалов X1, Х2 и Х3 означает водород. Для обеспечения устойчивости предпочтительно, чтобы два радикала из X1, Х2 и Х3 представляли собой Н. Монооксид углерода выделяется при нагревании водного раствора, содержащего такое соединение.
Преимущества вышеуказанных соединений заключаются в следующем. Впервые СО получен в водной среде при регулируемых условиях (рН, температура). Карбонильные комплексы заявляемых металлов можно получить в воде в хорошо подобранных условиях, а не в органических растворителях, или при высоком давлении, либо при высокой температуре. Источник СО и восстановитель могут присутствовать в том же самом единственном соединении, что является удобным, поскольку для восстановления фактически всегда требуется получение карбонилов. В случае когда металлом, который служит комплексообразователем, является 99mТc, или 188/186Re, наборы для введения метки можно получить без необходимости заполнения флаконов токсичным и летучим СО. Главным преимуществом указанного варианта осуществления изобретения является то, что получают молекулу, которая объединяет различные функциональные группы в одном соединении. Такое соединение может действовать одновременно в качестве восстановителя и в качестве источника СО in situ, в котором получают только СО, если присутствует протонный растворитель (аналогично воде) или кислота Льюиса. Путем модификации заместителей в различных положениях можно получить различные типы соединений. Эти соединения могут быть подразделены на следующие группы:
1. Соединение борана с карбонатом, в котором X1, X2 и Х3 означают - Н, a Y означает -ОН2, и/или соответствующие соли моно- или дидепротонированного боран-карбоната [Н3ВСО2]2-.
2. Боранаминокислоту (аммонийкарбоксиборан) в котором Х2 означает NН3, X1 и Х3 означают - Н, a Y означает - ОН и/или соответствующие соли монодепротонированного боранкарбоната аммония [(NН3)Н2ВСO2]-.
3. Алкилированные боранаминокислоты (триалкиламмонийкарбоксибораны), в которых Х2 означает - NHxRy при х+y=3, где R означает заместитель, который связан через атом углерода с атомом азота и представляет собой предпочтительно алкил или арил, Х1 и Х3 означают - Н, а Y означает - ОН.
4. Соединения формулы I, в которых X1 и Х3 означают -Н; Х2 органический заместитель, связанный через атом углерода с атомом бора, а Y означает - ОН2.
5. Соединения формулы I, в которых X1, Х2 и Х3 имеют вышеуказанные в пунктах 1-4 значения, а Y означает OR', где R' означает заместитель, связанный через атом углерода с атомом кислорода, предпочтительно метил или этил.
6. Соединения формулы I, в которых X1, X2 и Х3 имеют вышеуказанные в пунктах 1-4 значения, а Y означает NH2, NHR" или NR"2, где R" означает заместитель, связанный через атом углерода с атомом азота, предпочтительно метил или этил.
В качестве конкретных примеров указанных соединений можно привести:
Производные боранкарбоната: [Н3В-СООН2], [Н3В-СООН]М, [Н3В-СОО]М2, Na [Н3В-СООСН3], в которых М означает катион щелочного металла;
боранокарбонатов формулы Na [Н3ВСОМНСН3], M[H3B-CONH2], в которых М означает катион щелочного металла;
аммоний-боранокарбонатов формулы [Н3N-ВН2-СООН], [Н3N-ВН2-СОО]Li, [(СН3)3N-ВН2-СООН], [(СН3)Н2N-ВН2-СООН], [(СН3)Н2Н-ВН2-СОО]Li, [(СН3) H2N-BH2-СООСН3]; и аммоний-боранокарбонатов формулы [Н3N-ВН2-СОNН2], [(CH3)2HN-BH2-CONHC2H5].
Соединения настоящего изобретения могут быть получены с помощью или аналогично методам, которые описаны Burg и др. в журнале J. Am. Chem. Soc., вып.59, стр.780 (1936) для ВН3СО; Malone и др., Inorg. Chem., вып.6, стр.817 (1967) для М2 [Н3В-СОО] и М [Н3В-СООС2Н5]; Howe и др., Inorg. Chem., вып.10, стр.930(1971) для М[Н3В-CONH2]; Spielvogel и др., J. Am. Chem.Soc., вып.102, стр.6343 (1980) для [Н3N-ВН2-СООН] и [(СН3)3N-ВН2-СONНС2Н5]; Spielvogel и др., Inorg. Chem., вып.23, стр.4322 (1984) для [(СН3)Н2N-ВН2-СООСН3]; Spielvogel и др., Inorg. Chem., вып.23, стр.1776 (1984) и J. Am. Chem. Soc., вып.98, стр.5702 (1976) для [H3N-BH2-CONH2], [(CH3)2HN-BH2-CONHC2H5].
Изобретение далее относится к способу получения карбонильных комплексов переходных металлов, в котором одно или несколько соединений, определенных выше, используется в качестве источника СО и, возможно, в качестве восстановителя. Вкратце этот способ заключается в высвобождении СО из любого соединения настоящего изобретения, в частности из одного или нескольких соединений, приведенных в п.1-6, в воде или буферном растворе за счет протолиза и последующих гидролитических реакций. В свою очередь, металл, с которым карбонил должен образовывать комплекс, восстанавливается гидридным заместителем, присоединенным к бору. Соединения настоящего изобретения, в частности указанные в п.1-6, растворяют в воде или буфере, и затем добавляют металл либо в виде твердого тела, либо в виде раствора. Протонирование и гидролиз соединений настоящего изобретения, в частности соединений 1-6, приводит к высвобождению СО. В то же самое время гидриды, присоединенные к бору (-Н), восстанавливают центр металла до валентности, где металл способен координировать выделенный СО. В этот момент образуются карбонильные комплексы. В соответствии с настоящим изобретением способ получения карбонильных комплексов, таким образом, включает смешивание соединений бороводорода изобретения с водным раствором металла в виде иона металла или (пер) металлата. Под используемом в данной заявке термином "Металл" следует понимать все формы металла, то есть также ионы металла и (пер) металлаты.
Соединения и способ настоящего изобретения пригодны для образования любого карбонильного комплекса, в частности в которых переходной металл в карбонильном комплексе переходного металла выбирают из групп металлов V-B-VIII-B. Более конкретно, заявляемый способ пригоден для получения карбонильных комплексов следующих переходных металлов: ванадия (V), хрома (Сr), молибдена (Мо), вольфрама (W), марганца (Мп), технеция (Тc), рения (Re), железа (Fe), рутения (Ru), осмия (Os), кобальта (Со), родия (Rh), иридия (Ir) и никеля (Ni) и их радиоактивных изотопов.
В объеме данного изобретения предлагается, кроме того, набор для получения карбонильных комплексов переходных металлов, включающий в свой состав любое соединение, полученное в соответствии с настоящим изобретением, в водном растворе, стабилизатор, например как тартрат, глюкогептонат, лактат, цитрат, и буферную систему, например как боратный или фосфатный буфер. В предпочтительном варианте набор данного изобретения содержит по меньшей мере 2 мг боран-карбоната, предпочтительно в боратном буфере (рН 9,1) в бескислородной среде в атмосфере азота. Предпочтительно, чтобы суммарный объем раствора после прибавления раствора радиоактивного металла не превышал 1 мл. Однако в некоторых обстоятельствах можно использовать большие объемы раствора, например 2 или 3 мл. Подходящие условия для инкубации включают нагревание раствора в течение примерно 20 минут до 75°С.
Соединения данного изобретения можно использовать также в воде для восстановления органических соединений с избирательностью и реакционной способностью, сопоставимой с бороводородом или цианбораном.
Кроме того, было установлено, что Н3ВСО может быть получен непрерывно из Н3В-THF (тетрагидрофуран) и последующего взаимодействия in situ со спиртовым раствором гидроксида калия с выходом К2[Н3ВСО2]. Ключевым моментом процесса получения является регулирование равновесного состояния между Н3ВСО и Н3В-THF. THF селективно конденсируется из потока газа при -50°С, в то время как проходит Н3ВСО (т.кип.-64°С), уносимый потоком моноксида углерода. После этого полученную газовую смесь непосредственно пропускают через этанольный раствор КОН при температуре -78°С. Нуклеофильная атака [ОН-] у высокоэлектрофильного атома углерода в Н3ВСО приводит к образованию К2[Н3ВСO2] с высоким его выходом. При необходимости, сам Н3ВСО может быть выделен в холодной ловушке при температуре -78°С. Этот способ получения Н3ВСО проще и удобнее, чем катализируемые простым эфиром методы или те, которые проводят при высоком давлении, и позволяет получить выход продукта до нескольких грамм и более.
Таким образом, данное изобретение относится к способу получения боранкарбоната, включающего стадии:
a) взаимодействия комплекса ВН3-THF или аналогичного аддукта в THF или смеси THF и другого органического апротонного растворителя с СО, что дает в результате Н3ВСО;
b) пропускания образованного таким образом Н3ВСО через холодный раствор гидроксида с моно- или дикатионным противоионом и алифатическим спиртом; и
c) через соответствующее время, необходимое для реакции, нагревания полученного спиртового раствора для осаждения боранкарбоната.
Аналогичным продуктом присоединения является, например, Н3В (Et2O). Гидроксид выбирают, например, из группы, состоящей из гидроксида калия, гидроокиси натрия или гидроокиси тетраалкиламмония. Алифатический спирт может быть выбран из группы, состоящей из метанола, этанола и изопропанола.
Соединение Н3ВСО входит также в объем этого изобретения. Это соединение обладает восстанавливающими свойствами и может использоваться для той цели, например, при получении карбонильных комплексов без СО при высоком давлении, как описано выше, но тогда в апротонном или только слабо протонных растворителях. Представляется также возможность использовать Н3ВСО in situ, в то время как соединение образуется, когда СО пропускают через растворы "металлов" в THF, например, используемые для синтеза макроскопического [ТсС3(СO)3]2 или аналога Re.
Соединения согласно изобретению находят более широкое использование, чем только для получения карбонильных комплексов, но их можно также использовать в других обстоятельствах, где необходимо получение источника СО в водном растворе. Изобретение также относится к использованию боранокарбоната или его производных в качестве восстановителя органических субстратов, например как сложные эфиры, имины или альдегиды, в воде. Восстановительная способность этих соединений сопоставима с ВН4- или цианобораном, и они могут, таким образом, быть заменителями, например, для цианоборана в крупномасштабных объемных промышленных процессах.
Настоящее изобретение продемонстрировано нижеследующими примерами, которые служат исключительно для иллюстрации изобретения.
Пример 1
Получение К2Н3ВСO2
1.Синтез ВН3СО
4 г NaBH4 осторожно прибавляли к 15 мл концентрированной НзРO4 (высушенной в течение ночи в высоком вакууме при комнатной температуре) при пониженном давлении (1 мбар) при интенсивном перемешивании в течение 2 часов. Выделившийся ВН3 сушили путем пропускания через охлаждаемую ловушку при -78°С и конденсировали во второй охлаждаемой ловушке при -200°С, содержащей 70 мл сухого диметилэтаноламина (DME). Вторую ловушку отсоединяли от первой ловушки и вакуумной линии. Температуру доводили до -40°С. Затем в этой ловушке создавали повышенное давление с использованием сухого СО 1,3 бар. Реакционную смесь затем перемешивали в охлаждающей ванне при -40°С (сухой лед с ацетонитрилом) при давлении 1,3 бар СО в течение ночи.
2. Синтез К2Н3ВСО3
Выходное отверстие для газа ловушки подсоединяли к 100 мл двухгорлой круглодонной колбе (оснащенной входным отверстием для газа и парциальным конденсатором горячего орошения), содержащей 50 мл сухого этанола и 3 г КОН. Охлаждающую ванну ловушки затем удаляли и выделяющийся ВН3СО медленно пропускали через раствор КОН в этаноле при 0°С. DME раствор медленно нагревали до 80°С и затем ловушку трехкратно очищали струей СО. После прекращения выделения ВН3СО, этанольный раствор кипятили с обратным холодильником в течение 30 мин. После охлаждения раствора до комнатной температуры К2Н3ВСO2 осаждался в виде белого порошка, который затем фильтровали через фильтр из огнеупорного стекла, промывали охлажденным льдом этанолом и сушили при пониженном давлении.
Пример 2
Эксперимент по введению метки с использованием лиофилизированного набора
Набор для введения метки получали путем лиофилизирования 1 мг К2[ВН3СОО] в 0,1 мл 0,1М PBS (забуференный фосфатом физиологический раствор), рН 7,5 во флаконе, который очищали струей N2. В качестве альтернативного варианта можно использовать 0,1М боратный буфер, рН 8,5.
Для мечения прибавляли 1 мл солевого раствора, содержащего [99mTcO4]. Установлено, что выход не зависит от абсолютного количества [99mTcO4]-. Полученный таким образом раствор нагревали затем до 75°С в течение 20 мин.
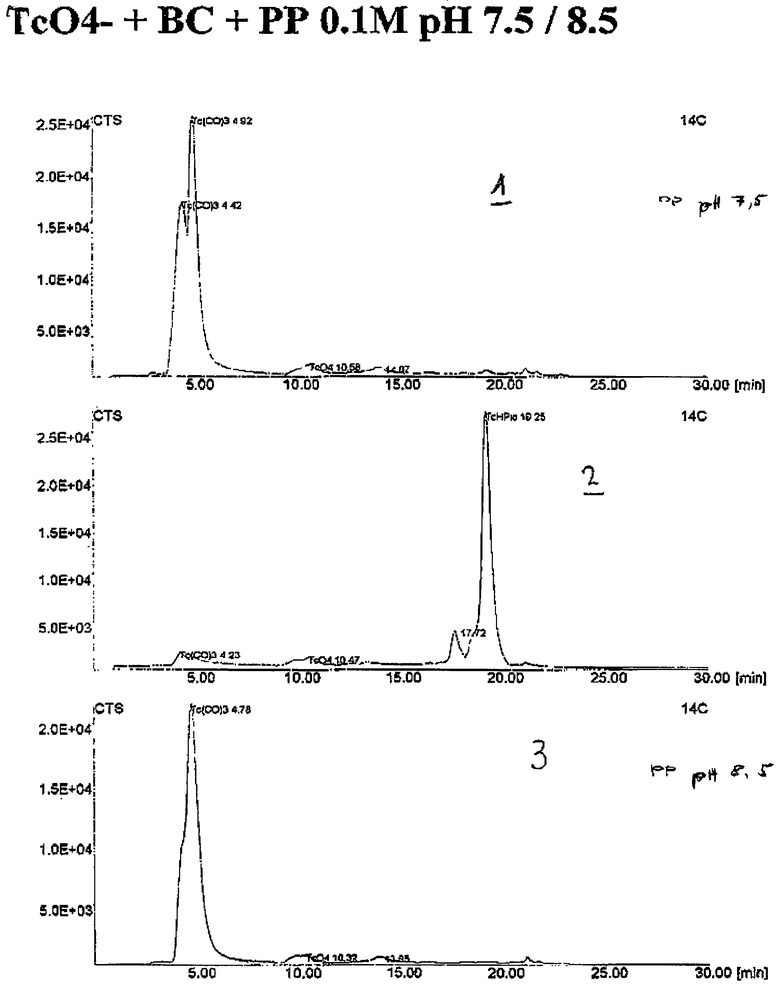
Выход варьирует от 80 до 100% (следы 1 на чертеже) при рН 7,5; следы 3 на чертеже при рН 8,5.
Для установления идентичности соединения, пиколиновую кислоту прибавляли непосредственно к реакционному раствору, в котором получали карбонильный комплекс. С помощью ВЭЖХ обнаружен комплекс [99mTc(ОН2) (пик) (СO)3] (чертеж, следы 2) при сравнении с "холодным" материалом, при этом в данном случае аналогичный комплекс был получен с "холодным" рением. "Горячий" материал установлен с помощью детектора-счетчика радиоактивности, в то время как "холодный материал обнаруживают с помощью УФ-детектора.
Пример 3
Эксперимент по введению метки с использованием так называемого "мокрого набора"
Флакон, содержащий 2 мг боран-карбоната и элюата пертехнетата, выходящего из генератора, в боратном буфере, рН 9,1, в суммарном объеме 1 мл нагревали в течение 20 мин до 75°С. Выход по мечению полученного таким образом продукта [99mТc(ОН2) (СO)3]-был выше 97%.
Пример 4
Получение дикалий(карбоксилато)тригидробората из Н3ВTHF
Используемый реакционный аппарат состоял из 250 мл трехгорлой круглодонной колбы, подсоединенной через стеклянную трубку к охлаждаемой ловушке. Другие два горлышка колбы герметизировали каучуковой перегородкой. В колбу помещали трубку из политетрафторэтилена (PTFE) для введения газа. Из выходного отверстия охлаждаемой ловушки PTFE-трубку помещали в 400 мл пробирку Schlenk (Шленка). С закраины пробирки Schlenk tube помещали политеновую трубку, выходящую в барботер с силиконовым маслом, который изолировал реакционный аппарат от окружающей атмосферы.
Охлаждаемую ловушку и пробирку Schlenk погружали в сосуды Дьюара, содержащие изопропанол. Аппарат прочищали струей сухого не содержащего кислорода азота в течение 30 минут, обеспечивая одновременно охлаждение охлаждаемой ловушки до -50°С, а пробирку Schlenk до -78°С путем введения в соответствующие сосуды Дьюара сухого льда.
Раствор, содержащий 5,0 гидроокиси калия в 200 мл абсолютного этанола, прибавляли к содержимому пробирки Schlenk и давали постепенно охладиться до -78°С. Через аппарат быстро пропускали струю монооксида углерода и 30 мл раствора 1 моль дм -3 боран-тетрагидрофуранового комплекса в тетрагидрофуране вводили в круглодонную колбу. СО пропускали в раствор так, чтобы приблизительно один пузырек в секунду выходил из аппарата через барботер с силиконовым маслом. Температуру средней части охлаждаемой ловушки поддерживали в интервале от -45 до -55°С, добавляя время от времени сухой лед.
После двухчасового пропускания монооксида углерода 20 мл диметоксиэтана вводили в круглодонную колбу и еще 20 мл порцию диметоксиэтана вводили в среднюю часть охлаждаемой ловушки. Монооксид углерода пропускали через реакционный аппарат по методике, описанной выше. Через 1 час пробирку Schlenk отсоединяли от остальной части аппарата и давали нагреться до комнатной температуры. Спиртовый раствор кипятили с обратным холодильником в течение 45 минут. Полученный белый преципитат отфильтровывали, промывали двумя 5 мл порциями абсолютного этанола и сушили при пониженном давлении с выходом 1,26 г продукта (43% в расчете на ВН3THF) в виде белого порошка. Найдено К, 38,85% (обнаружен гравиметрическим методом в виде K2Na[CO(NO2)6]); для СН4ВКО2 необходимо К, 39,9%. 6н, (200 МГц, D2O,25°C) 0,80 (1:1:1:1 квартет. 1J(H-11В)=80 Hz; 1:1:1:1:1:1:1 септет, 1J(Н-10В)=27 Гц).
Пример 5
Восстановление органического субстрата натрия бензальдегид-2-сульфоната боранокарбонатом в воде.
Калия боранокарбонат (100 мг) и натрия бензальдегид-2-сульфонат (40 мг) смешивали в воде (1 мл) и оставляли на 30 мин при комнатной температуре. Количественный выход натрия 2-(гидроксиметил)-бензолсульфоната подтверждался по исчезновению 1Н-ЯМР-сигнала в исходном соединении при δ=10,77 и появлению этого сигнала в продукте при δ=5,04. В конце эксперимента реакционная смесь не имела запаха, что свидетельствует о восстановлении сульфонатной группы.
Пример 6
Пример получения соединения I, где Y=-OC2H5, или -ОСН3, или O-(N-гидроксисукцинимид)(NНS).
i) Соединение, в котором Y=-ОСН3: Na[H3BCOOH], получают с соответствии с методикой, описанной в примере Примера 4. 100 мг (1,22 ммоль) растворяли в СН3ОН (3 мл) и прибавляли к полученному раствору 1,5 эквивалента дициклогексилкарбодиимида (DCC). Полученный раствор нагревали до температуры 50°С 3 часа, в течение которых образовался преципитат. Реакционный раствор охлаждали до комнатной температуры и затем отгоняли растворители при пониженном давлении. Остаток перемешивали с тетрагидрофураном для удаления избытка DCC. Затем остаток перемешивали с ацетонитрилом с получением раствора, в котором продукт находился в растворенном состоянии, в то время как образовавшаяся в результате дициклогексилмочевина и исходный материал находились в нерастворенном состоянии. Раствор фильтровали и полученный фильтрат помещали в холодильник и держали при температуре -20°С, в результате чего получали 65 мг (64%) аналитически чистого белого преципитата Na[H3BCOOCH3].
Аналитические данные: Вычислено для NаВС2Н6O2: С, 25,06% Н, 6,30%. Найдено: С 24,82%; Н, 6,03%. ИК 2265 см-1 (В-Н септет), 1703 см-1 (С=O септет).
ii) Y=-ОС2Н5: 100 мг (1,2 ммоль) Na[H3BCOOCH3], полученного по описанной ранее методике, растворяли в 10 мл С2Н5OН и кипятили с обратным холодильником в течение 2,5 часов, что привело к переэтерификации продукта, и чистый продукт Na[H3BCOOC2H5] можно было выделить путем отгонки растворителя при пониженном давлении. Выход продукта составляет 120 мг, 92%.
Аналитические данные: Вычислено для NаВС3Н8О2: С, 32,79% Н, 7,34%. Найдено С, 31,05% Н, 7,12%. ИК 2290 см-1 (В-Н септет), 1698 см-1 (СО септет).
iii) Y=-NHS: 200 мг (2,4 ммоль) Nа[Н3ВСООСН3] тщательно перемешивали с 1 г NHS. Затем смесь помещали в пробирку Schlenk в атмосфере азота N2 и поддерживали ее при температуре примерно 100°С, пока смесь не превратилась в гомогенный расплав. Через 20 минут при этой же температуре смесь отверждали путем охлажения и затем ее растворяли в диметоксиэтане. Эта операция обеспечила удаление избыточного количества NHS. Затем прибавляли ацетонитрил (5 мл), в котором выделенное вещество и продукт находились в растворенном состоянии, но не продукты разложения. Никакого присутствия исходного материала не обнаружено после этой операции. Последующее разведение смеси диэтиловьм эфиром 1:1 и охлаждение до -20°С дало аморфный белый преципитат Na[H3BCOO-NHS], 105 мг, 49%.
Аналитические данные: Вычислено для NaBC5H7O4N: С, 33,57%, Н, 3,94%, N 7,83%. Найдено: С, 31,11% Н, 3,56% N, 8,24%, ИК-спектр 2270 (широкий) см-1 (В-Н септет), 1680 см-1 (плечо при 1705) (С=O септет).
Пример 7
Пример получения соединения I, в котором Y=-N(C2H5)2.
120 мг (1,44 ммоль) Nа[Н3ВСООСН3]], полученного по описанной ранее методике, растворяли в этаноле и прибавляли к полученному раствору 2,5 эквивалента HN(C2H5)2. Раствор перемешивали при температуре 30-40°С в течение ночи. Растворитель отгоняли при пониженном давлении, остаток смешивали с THF и затем смесь фильтровали. Прибавление диэтилового эфира к раствору с THF до появления помутнения раствора с последующим выдерживанием реакционной смеси при температуре 0°С в течение 48 часов обеспечило получение 102 мг (52%) Na[H3BCON(C2H5)2].
Пример 8
Пример получения соединения I, в котором Y=-ОН и Х2=N(СН3)3.
25 г (250 ммоль) (СН3)3NНСl перемешивали в THF. Затем к полученной суспензии прибавляли 15,6 г (250 ммоль) Н3ВСN, и полученную смесь кипятили с обратным холодильником в течение 48 часов. Завершение реакционного взаимодействия определяли, если не выделялось больше никакого Н2. Растворитель отгоняли и после сублимирования остатка получали 18 г чистого (CH3)3NBH2CN, который непосредственно использовали в следующей стадии.
11,9 г (120 ммоль) (CH3)3NBH2CN растворяли в CH2Cl2 и прибавляли [(С2Н5)3О][ВF4]. Раствор затем кипятили с обратным холодильником в течение 24 часов. Растворитель отгоняли при пониженном давлении и остаток растворяли в 50 мл этанола.
К полученной смеси затем прибавляли 2 мл концентрированного раствора хлористоводородной соли и полученную смесь кипятили с обратным холодильником еще 48 часов. Растворитель отгоняли и остаток экстрагировали три раза с использованием СН2Сl2, что дало (СН3)3NВН2СООН в виде белого твердого продукта. Выход составляет 11,3 г (80%).
Аналитические данные: Вычислено для BC4H12O2N: С, 41,08% Н, 10,04%, N 11,98%. Найдено: С, 40,51% Н, 9,73% N, 11,25%. ИК-спектр 2365 (широкий) см-1 (В-Н септет), 1642 см-1 (С=O септет).
Пример 9
Пример получения соединения, в котором Y=-NHC2H5 и X2=N(СН3)3.
Это соединение получали в первых стадиях реакционного процесса по методике, описанной в предыдущем примере. После завершения реакции с [(С2Н5)3О][ВF4], растворитель не отгоняли, а прибавляли непосредственно 0,5 М NaOH, и полученную реакционную смесь интенсивно перемешивали в течение 2 часов при комнатной температуре. Органический слой отделяли, а водную фазу экстрагировали три раза с использованием СН2Сl2. После отгонки растворителя получали соединение [(СН3)3NВН2(СONН(С2Н5)] в виде вязкой жидкости.
Аналитические данные: Вычислено для ВС6Н17NО: С, 55,43% Н, 13,18%, N 10,77% Найдено: С, 56,20% Н, 12,98% N, 10,43%. ИК 2332 (широкий) см-1 (В-Н септет), 1587 см-1 (С=O септет).
Пример 10
Пример получения соединения, в котором Y=-ОСН3 и Х2=N(СН3)3.
Исходное соединение (СН3)3NBН2СООН получали по методике, описанной в примере 8. 2,5 г (21,6 ммоль) растворяли в СН3ОН и затем прибавляли 1,5 эквивалента DCC.
Полученную смесь нагревали до температуры 50°С в течение 20 часов, охлаждали затем до комнатной температуре и растворитель отгоняли. Промывание остатка диэтиловым эфиром обеспечило удаление избытка DCC. Остаток затем перемешивали с THF и фильтровали из раствора дициклогексилмочевины. После упаривания THF получали (СН3)3NВН2СОСН3 в виде бесцветной и вязкой жидкости.
Аналитические данные: Вычислено для BC5H14NO2: С, 45,85% Н, 10,77%, N, 10,69%. Найдено: С, 44,33% Н, 10,14% N, 11,23%. ИК-спектр 2297 (широкий) см-1 (В-Н септет), 1699 см-1 (С=O септет).
Пример 11
Пример получения соединения, в котором Y=-NR2 и Х2=N(СН3)3.
Соединение (СН3)3NВН2СОСН3, полученное по методике примера 10, подвергали взаимодействию в THF с любым вторичным амином HNR2 с выходом соединений общей формулы (СН3)3NВН2СОNR2 через 10 часов при температуре 50°С. Реакция протекала при этих условиях до полного завершения, и указанные соединения получают обычно в виде бесцветной жидкости.
Пример 12
Пример получения карбонильных комплексов элементов из группы VIB с боранокарбонатом в качестве восстановителя/ агента передачи СО.
К2Сr2O7 (20 мг, 0,07 ммоль) растворяли в 5 мл воды. К полученному раствору прибавляли 110 мг (3,4 ммоль) Na [Н3ВСO2Н] (1), растворенный в 5 мл воды. Цвет раствора постепенно менялся от оранжево-желтого до коричнево-зеленоватого в течение 12 часов при комнатной температуре. После этого времени к раствору прибавляли 300 мкл концентрированной HCl для деструкции избытка 1. Происходило сильное выделение газа. Раствор затем упаривали досуха и повторно растворяли в 2 мл этилового спирта. Затем прибавляли 300 мг [Net4]Cl и полученный раствор перемешивали в течение ночи. Образовавшийся мелкокристаллический желто-коричневый преципитат отфильтровывали (12 мг). ИК-спектр дает типичную картину fас-М(СО)3 при 2082 и 1986 см-1, что свидетельствует об образовании карбонильного комплекса. Хотя этот комплекс не охарактеризован структурно, мы предполагаем, что он представляет собой [NEt4]2[CrCl4(CO)3].
Пример 13
Пример получения карбонильных комплексов элементов из группы VIIIB с боранокарбонатом в качестве восстановителя /агента передачи СО.
Свежеприготовленный водный раствор Na[H3BCO2H] (130 мг,1,6 ммоль) перемешивался в атмосфере N2, и затем прибавляли по каплям раствор RuO4 (26 мг, 0,016 ммоль) в 2 мл тетрагидрофурана в течение 2 часов. Раствор приобретал коричнево-черный цвет. Через 4 часа при комнатной температуре выпадал в небольшом количестве черный осадок (RuO2), и раствор имел желтоватый цвет. Преципитат отфильтровывали. ИК-спектр раствора показал типичную картину fас-М(СО)3, что свидетельствует об образовании части [Ru(СО)3]2. Этот раствор подкисляли трифторуксусной кислотой и упаривали досуха. Анализ остатка (22 мг) свидетельствовал о наличии νCO при 2095 и 2003 см-1, что согласуется с образованием Ru(ОН2)3(СО)3][СF3СO2]2.
Пример 14
Примеры, иллюстрирующие использование Nа[Н3ВСO2Н] в качестве восстановителя органических субстратов
Имины:
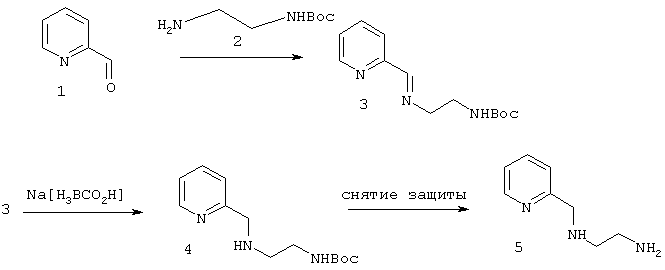
Амин 4 и 5 получали путем реакции восстановления имина 3 при использовании в качестве восстановителя Na[H3BCO2H]. 214 мг (2 ммоль) 1 прибавляли к раствору, содержащему 330 мг (2,2 моль) 2 в 20 мл метанола через молекулярное сито. Раствор постепенно стал желтого цвета, и ход протекания реакции контролировали с использованием высокоэффективной жидкостной хроматографии. Через 12 часов взаимодействие компонентов полностью завершилось. После этого растворитель удаляли в вакууме и остаток промывали водой для удаления избыток 2. Без последующей очистки остаток повторно растворяли в метаноле и прибавляли 240 мг Nа[Н3ВСO2Н]. Полученную смесь перемешивали при комнатной температуре в течение 3 часов и затем кипятили с обратным холодильником в течение еще 2 часов. Желтый цвет постепенно тускнел. Реакцию гасили подкисленной водой до полного прекращения газовыделения. Полученное соединение 4 очищали методом колоночной хроматографии. Выход продукта составил 420 мг в виде бесцветного масла. 1Н ЯМР(СDСl3): 7,27, 8,66, 7,44, 7,79 (пир7Н), 3,92 (СН2), 2,75, 3,40 (CH2), 1,22 (трет-бутил-Н), 13С ЯМР: 157,6, 149,2, 135,8, 122,5, 120,6, 53,9, 50,6, 44,8, 83,2, 40,4, 25,3.
Элементный анализ: Вычислено: С 62,13%, Н 8,42%, N 16,72%. Найдено С 6,93%, Н 8,52%, N 16,44%
Соединение затем обрабатывали для снятия защитной группы с использованием трифторуксусной кислоты с количественным выходом соединения 5.
Восстановление кетона до спирта
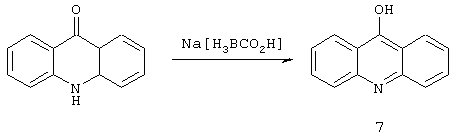
97 мг (0,5 ммоль) состава 6 растворяли в метаноле и прибавляли 81 мг Nа[Н3ВСO2Н]. Смесь затем кипятили с обратньм холодильником в течение приблизительно 3 часов и ход протекания реакции контролировали с использованием высокоэффективной жидкостной хроматографии. По завершении этого интервала времени исходный материал полностью испарился (исчез). Избыток Nа[Н3ВСO2Н] гасили несколькими каплями концентрированной НСl и смесь затем сушили досуха. Очистку осуществляли на силикагеле с последующей обработкой смесью МеОН / CH2Cl2 в виде подвижной фазы. Получено 72 мг соединения 7 в виде слегка желтого порошка.
1Н ЯМР(СDСl3): 8,12, 7,88, 7,55, 7,40, 13С ЯМР: 158,7, 150,0, 130,9, 126,5, 121,5 Элементный анализ: Вычислено: С 79,98%, Н 4,65%, N 7,17%. Найдено С 78,80%. Н 4,69%, N 7, 55%.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ЦИКЛИЧЕСКИХ ДИКЕТОНОВ | 2005 |
|
RU2384562C2 |
| ПРИМЕНЕНИЕ СОЕДИНЕНИЙ ДЛЯ ПОВЫШЕНИЯ АКТИВНОСТИ ПИРУВАТДЕГИДРОГЕНАЗЫ | 1999 |
|
RU2242224C2 |
| ПРОИЗВОДНЫЕ 4-АМИНОПИПЕРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2266282C2 |
| ДИФОСФИНЫ И МЕТАЛЛОКОМПЛЕКСЫ | 2006 |
|
RU2408600C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПИРАЗОЛОВ | 2015 |
|
RU2712192C2 |
| НАЦЕЛИВАЮЩИЕ АМИНОКИСЛОТНЫЕ ЛИПИДЫ | 2013 |
|
RU2654210C2 |
| СПОСОБ ПОЛУЧЕНИЯ 3-ГАЛОГЕН-4,5-ДИГИДРО-1Н-ПИРАЗОЛОВ | 2003 |
|
RU2326877C2 |
| НОВЫЕ 2-ГЕТЕРОАРИЛ-ЗАМЕЩЕННЫЕ БЕНЗОТИОФЕНЫ И БЕНЗОФУРАНЫ 709 | 2008 |
|
RU2472789C2 |
| ПРОИЗВОДНЫЕ НАФТИРИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2002 |
|
RU2296764C2 |
| АЗОЛОВЫЕ ПРОИЗВОДНЫЕ БЕНЗОЛА | 2014 |
|
RU2641891C2 |
Настоящее изобретение относится к новым производным боранокарбоната формулы I:
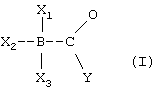
где X1 означает -Н; Х3 и Х2 являются заместителями, которые могут быть одинаковыми или различными и которые выбирают из группы, состоящей из -Н, -NHxRy при х+у=3, или -R, где R означает заместитель, который связан через атом углерода с атомом азота или бора соответственно и представляет собой метил или этил; Y означает группу -ОН, -OH2, -OR или -NHR, в которой R означает заместитель, который связан через атом углерода с атомом азота или бора соответственно и представляет собой метил или этил; или их соли. Технический результат изобретения состоит в использовании полученных соединений в качестве источника моноксида углерода (СО), а также в качестве восстановителя при получении карбонильных комплексов металлов в водном растворе. Изобретение также включает в свой объем способ получения боранокарбоната и способ восстановления с использованием Н3ВСО в качестве восстанавливающего агента. 6 н. и 14 з.п.ф-лы, 1 ил.
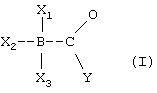
где X1 означает -Н;
X2 и Х3 являются заместителями, которые могут быть одинаковыми или различными и которые выбирают из группы, состоящей из -Н, -NHxRy при х+у=3, или -R, где R означает заместитель, который связан через атом углерода с атомом азота или бора соответственно и представляет собой метил или этил;
Y означает группу -ОН, -ОН2, -OR или -NHR, в которой R означает заместитель, который связан через атом углерода с атомом азота или кислорода соответственно, и представляет собой метил или этил;
или их соли для использования в качестве источника моноксида углерода (СО), и необязятельно в качестве восстановителя при получении карбонильных комплексов металлов в водном растворе.
аммоний-боранокарбаматов: [H3N-BH2-CONH2], [(CH3)2HN-BH2-CONHC2H5].
а) взаимодействия ВН3-THF или Н3В(Еt2О) в THF (тетрагидрофуран) или смеси THF и другого органического апротонного растворителя с СО, приводя к образованию Н3ВСО;
b) пропускания образованного таким образом Н3ВСО через холодный раствор гидроксида с моно- или дикатионным противоионом и алифатическим спиртом; и
c) через соответствующее время, необходимое для реакции, нагревания полученного спиртового раствора для осаждения боранокарбоната.
| WO 9848848 A 05.11.1998 | |||
| DE 19648313 A1 28.05.1997 | |||
| МИХАЙЛОВ Б.М | |||
| Химия бороводородов | |||
| - М.: Наука, 1967, с | |||
| Канатное устройство для подъема и перемещения сыпучих и раздробленных тел | 1923 |
|
SU155A1 |

