Область техники
Данное изобретение относится к новому диазепановому производному или его соли, которые пригодны в качестве фармацевтического препарата, в частности в качестве ингибитора активированного фактора Х коагуляции крови, а также к такому фармацевтическому средству.
Предпосылки создания изобретения
В связи с изменениями европейского и американского стиля жизни и повышением уровня пожилого населения за последние годы повысилось число пациентов с тромбоэмболическими заболеваниями, включая инфаркт миокарда, тромбоз сосудов головного мозга и периферический артериальный тромбоз, и социальная значимость их лечения все более и более повышалась. Так же, как и фибринолизная терапия и антитромбоцитная терапия, антикоагуляционная терапия является частью лекарственной терапии при лечении и профилактике тромбоза (Sogo Rinsho, 41: 2141-2145, 1989). В частности, безопасность, которая противостоит долговременному введению, и точная и правильная экспрессия антикоагулирующей активности являются существенными при профилактике тромбоза. Варфарин-калий часто используется во всем мире как единственный пероральный антикоагулянт, но использование в клинике этого лекарственного средства сопряжено с трудностями, так как чрезвычайно сложно контролировать антикоагулирующее действие благодаря свойствам, основанным на механизме его действия (J.Clinical Pharmacology, 32, 196-209, 1992 и N.Eng.J.Med., 324(26), 1865-1875, 1991), из-за чего большие усилия были направлены на разработку более пригодных и более легко применимых антикоагулянтов.
Тромбин регулирует превращение фибриногена в фибрин, что является конечной стадией коагуляции, и также в большой степени связан с активацией и агрегацией тромбоцитов ("T-PR and Pro-UK" edited by S.Matsuo, published by Gakusai Kikaku, pp.5-40, "Blood Coagulation", 1986), и его ингибитор был центром исследований по антикоагулянтам, как мишень для разрабатываемых фармацевтических препаратов. Однако ингибиторы тромбина, которые можно вводить перорально, не были предложены на рынок вплоть до настоящего времени из-за их низкой биодоступности при пероральном введении и проблем относительно безопасности (Biomed.Biochim. Acta, 44, 1201-1210, 1985).
Активированный фактор X коагуляции крови является ключевым ферментом, который расположен в точке соединения внешних и внутренних реакций каскада коагуляции и находится выше (слева) тромбина, в результате чего существует возможность того, что подавление этого фактора является более эффективным, чем подавление активности тромбина, и такой ингибитор может специфично подавлять эту систему коагуляции (Thrombosis Research, (19), 339-349, 1980).
Амидинонафтилалкилбензольные производные или их соли были известны как соединения, обладающие подавляющим действием на фактор Х коагуляции крови (Japanese Patent Laid-Open No. 208946/1993; Thrombosis Haemostasis, 71(3), 314-319, 1994; Thrombosis Haemostasis, 72(3), 393-396, 1994).
В WO 96/16940 указывается, что амидинонафтильное производное или его соль, представленное следующей ниже формулой, является соединением, обладающим действием, подавляющим активированный фактор Х коагуляции крови (прототип 1).
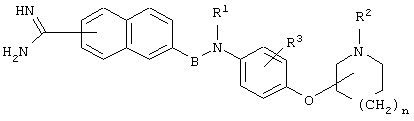
(Значения символов см. в ссылке)
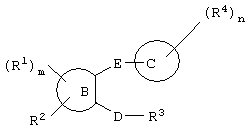
В WO 99/00121, WO 99/00126, WO 99/00127, WO 99/00128, WO 00/39111, WO 00/39117 и WO 00/39118 фенилендиамидные соединения и т.д., представленные следующей ниже формулой, упоминаются как ингибиторы фактора Ха (прототип 2).
(Значения символов см. в ссылке)
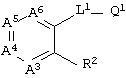
Кроме того, в WO 99/32477 широкий ряд соединений, представленных следующей далее формулой, упоминается в качестве антикоагулянтов (прототип 3).
(Значение символов см. в ссылке)
Описание изобретения
Настоящими изобретателями было получено диазепановое производное, представленное следующей далее формулой (I), или его соль и обнаружено, что оно обладает превосходным подавляющим действием на активированный фактор Х коагуляции крови и, в частности, обладает превосходной активностью при пероральном введении, следствием чего было создание данного изобретения.
Таким образом, данное изобретение относится к диазепановому производному, представленному следующей далее формулой (I), или его соли, а также к фармацевтической композиции, в частности к ингибитору активированного фактора Х коагуляции крови, содержащему производное диазепана или его соль в качестве активного ингредиента.
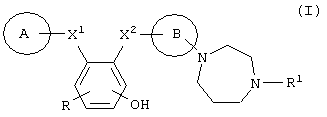
Символы в приведенной выше формуле имеют следующие значения.
Кольца А и В являются одинаковыми или различными, и каждое представляет собой арил или гетероарил, который может иметь 1-3 заместителя;
X1 представляет -C(=O)-NR2- или –NR2-C(=O)-;
X2 представляет -C(=O)-NR3- или –NR3-С(=O)-;
R представляет атом водорода, атом галогена, низший алкил или -О-низший алкил и
R1, R2 и R3 являются одинаковыми или различными, и каждый представляет собой атом водорода или низший алкил.
Соединение данного изобретения (I) имеет структуру, отличную от структуры соединений, упомянутых в прототипе 1, в отношении того, что оно имеет диазепановую составляющую и четыре циклических фрагмента и что атом азота диазепана непосредственно связан с кольцом В. Кроме того, соединение данного изобретения имеет структуру, отличную от прототипа 2 в том отношении, что оно имеет диазепановый фрагмент. К тому же в прототипе 3 конкретно не названы соединения, имеющие диазепановый фрагмент. Таким образом, отличительным признаком соединения (I) данного изобретения в плане химического строения является то, что диазепаниларил или диазепанилгетероарил связан с бензольным кольцом через амидную связь, что указанное бензольное кольцо дополнительно связано с арилом или гетероарилом через амидную связь и, кроме того, что указанное бензольное кольцо имеет ОН группу.
Далее соединение данного изобретения будет проиллюстрировано в деталях.
Термин "низший" в определении для формулы в описании означает прямую или разветвленную углеродную цепь, имеющую 1-6 атомов углерода, если не указано иначе. Поэтому примерами "низшего алкила" для R, R1 и R3 и примерами для заместителей, которые будут упоминаться позднее, являются метил, этил, пропил, изопропил, бутил, изобутил, вторичный бутил, третичный бутил, пентил, изопентил, неопентил, трет-пентил, 1-метилбутил, 2-метилбутил, 1,2-диметилпропил, гексил, изогексил, 1-метилпентил, 2-метилпентил, 3-метилпентил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2,3-диметилбутил, 3,3-диметилбутил, 1-этилбутил, 2-этилбутил, 1,1,2-триметилпропил, 1,2,2-триметилпропил, 1-этил-1-метилпропил и 1-этил-2-метилпропил. Среди них предпочтительны имеющие 1-3 атома углерода и наиболее предпочтительны метил и этил.
"Арил" означает ароматическое углеводородное кольцо, включая конденсированное кольцо, и оно, предпочтительно, является арилом, имеющим 6-14 атомов углерода, и, более предпочтительно, фенилом, нафтилом и т.д.
"Гетероарил" означает гетероциклический арил, имеющий 1-4 одинаковых или различных гетероатомов, выбранных из группы, состоящей из N, S и О, включая конденсированное кольцо, и его конкретными примерами являются фурил, тиенил, пирролил, имидазолил, пиразолил, изотиазолил, изоксазолил, триазолил, тетразолил, пиридил, пиримидинил, пиридазинил, пиразинил, индолил, индазолил, индолидинил, хинолил, изохинолил, хиназолинил, хинолидинил, хиноксалинил, циннолинил, бензимидазолил, имидазопиридил, бензофуранил, дигидробензофуранил, нафтилидинил, 1, 2-бензоизоксазолил, бензоксазолил, бензотиазолил, оксазолопиридил, изотиазолопиридил и бензотиенил, хотя данное изобретение не ограничивается ими.
Примерами "заместителя" для "арила или гетероарила, которые могут иметь 1-3 заместителя", являются необязательно замещенный низший алкил, низший алкенил, низший алкинил, С3-8-циклоалкил, -O-необязательно замещенный низший алкил, атом галогена, NH2, -NH-низший алкил, -N-(низший алкил)2, -С-(=NH)-NH2, -C(=N-OH)-NH2, -С(=NH)-NH-C(=O)-O-низший алкил, СООН, -С(=O)-O-необязательно замещенный низший алкил, -С(=O)- O-необязательно замещенный C6-14-арил, -С(=O)-O-необязательно замещенный гетероарил, CN, NO2, ОН, -O-СО-необязательно замещенный низший алкил, -O-CO-NH2, -O-CO-NH-низший алкил, -O-СО-N-(низший алкил)2, SH, -C(=O)-NH2, -С(=O)-NH(низший алкил) и -С(=O)-N-(низший алкил)2, хотя данное изобретение не ограничивается этим.
Примерами заместителя для "необязательно замещенного низшего алкила" и "необязательно замещенного гетероарила" являются атом галогена, -СООН, -С(=O)-O-ниэший алкил, ОН, NH2, -NH-низший алкил и -N-(низший алкил)2, хотя данное изобретение не ограничивается этим.
Примерами "атома галогена" являются атом фтора, атом хлора, атом йода и атом брома. Особенно предпочтительны атом хлора и атом брома.
В данном случае X2 представляет собой -C(=O)-NR3- или –NR3-C(=O)- и, более предпочтительно, он является -NR3-C(=O)-. R2 и R3 являются одинаковыми или различными и каждый представляет собой атом водорода или низший алкил, и атом водорода является более предпочтительным.
Соединение данного изобретения включает различные стереоизомеры, такие как геометрические изомеры, таутомеры и оптические изомеры, или в виде смесей, или в изолированных формах.
Соединение (I) данного изобретения может образовывать аддитивную соль кислоты. Кроме того, оно может образовывать соль с основанием в зависимости от типа заместителя. Конкретными примерами таких солей являются аддитивные соли кислоты, образованные минеральной кислотой, такой как соляная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, азотная кислота и фосфорная кислота, или органической кислотой, такой как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, метансульфоновая кислота и этансульфоновая кислота, или кислой аминокислотой, такой как аспарагиновая кислота и глутаминовая кислота, и соли неорганических оснований, таких как основания натрия, калия, магния, кальция и алюминия, или органических оснований, таких как метиламин, этиламин и этаноламин, основной аминокислоты, такой как лизин и орнитин и соль аммония.
К тому же, в данное изобретение также включены гидраты, фармацевтически приемлемые различные сольваты и полиморфизм данного соединения (I). Кстати, само собой разумеется, что данное изобретение не ограничивается соединениями, названными в последующих примерах, но включает все диазепановые производные, представляемые формулой (I), и их фармацевтически приемлемые соли.
Соединение данного изобретения включает все так называемые пролекарства, т.е. соединения, которые могут превращаться в соединение, представленное формулой (I), или его соль при метаболизме in vivo. Примерами групп, которые образуют пролекарства соединения данного изобретения, являются группы, названные в Prog.Med., 5: 2157-2161 (1985) и названные в "lyakuhin no Kaihatsu" (Development of Pharmaceuticals), опубликованной Hirokawa Shoten in 1990, Vol.7, "Molecular Design", pp.163-198.
Способы получения
Типичные способы получения соединения данного изобретения будут показаны здесь далее.
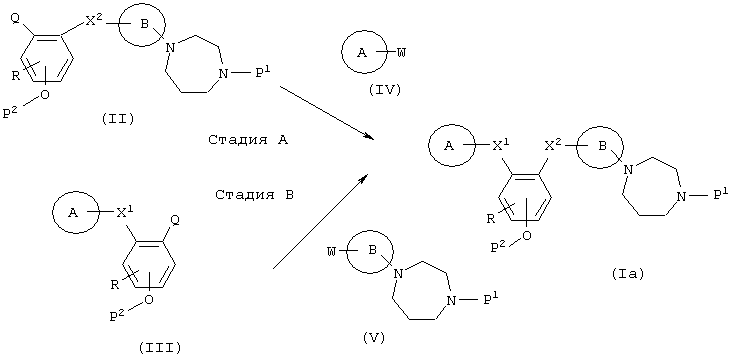
(в формулах А, В, R, Х1 и X2 имеют значения, указанные выше, и W являются такими, что когда Q представляет собой –NH2 или -NН-низший алкил, W является -СООН, в то время как, когда Q представляет собой -СООН, W является –NH2 или -NH-низшим алкилом; Р1 является атомом водорода, низшим алкилом или защитной группой для амина; и Р2 является атомом водорода или защитной группой для фенола.)
Стадия А
Эта стадия представляет собой реакцию синтеза (Iа), при которой амин и карбоновая кислота, включающие комбинацию соединения (II) и соединения (IV), взаимодействуют, предпочтительно, в присутствии конденсирующего агента. Эта реакция может осуществляться в соответствии с обычной реакцией ацилирования.
Примерами конденсирующего агента, который преимущественно используется, являются N,N-дициклогексилкарбодиимид (ДЦК), 1-этил-3-[3-(N,N-диметиламино)пропил]карбодиимид, карбонилдиимидазол, дифенилфосфорилазид (ДФФА) и диэтилфосфорилцианид.
Также можно превращать карбоновую кислоту в активные производные соответствующей карбоновой кислоты и затем конденсировать с амином.
Примерами используемого активного производного карбоновой кислоты являются активный сложный эфир, полученный реакцией с соединением фенольного типа, таким как п-нитрофенол, или N-гидроксиаминного типа, таким как 1-гидроксисукцинамид и 1-гидроксибензотриазол, моноалкиловый сложный эфир карбоновой кислоты, смешанный ангидрид кислоты, полученный реакцией с органической кислотой, и смешанный ангидрид кислоты типа фосфорной кислоты, полученный реакцией с фосфорилхлоридом и N-метилморфолином; азид кислоты, полученный реакцией сложного эфира с гидразином и алкилнитритом; галогенангидриды, такие как хлорангидрид или бромангидрид; и ангидрид кислоты симметричного типа. Обычно вышеуказанная реакция проводится в растворителе в диапазоне температур от охлаждения до комнатной температуры, хотя, в некоторых случаях, она должна проводиться в безводных условиях, в зависимости от типа реакции ацилирования.
Примерами применяемого растворителя являются инертные растворители, которые не участвуют в реакции, такие как диметилформамид, диоксан, тетрагидрофуран, эфир, дихлорэтан, дихлорметан, хлороформ, четыреххлористый углерод, диметоксиметан, диметоксиэтан, этилацетат, бензол, ацетонитрил и диметилсульфоксид, и их смесь, и предпочтительным является соответствующий выбор в зависимости от применяемого способа.
Кроме того, в зависимости от используемого метода, в некоторых случаях реакция гладко протекает в присутствии основания или при использовании такого основания в качестве растворителя, где основание представляет собой N-метилморфолин, триэтиламин, триметиламин, пиридин, гидрид натрия, трет-бутоксид калия, бутиллитий, натрийамид или тому подобное.
Стадия В
Эта стадия представляет собой реакцию синтеза (Iа), при которой реагируют амин и карбоновая кислота, включая комбинацию соединения (III) и соединения (V). Эта реакция проводится таким же образом, что и на стадии А.
Когда Р1 в соединении (Iа) данного изобретения является защитной группой для амина и защитная группа не отщепляется во время стадий А и В, удаление защитной группы с использованием способа, подходящего для отщепления защитной группы Р1, производится тогда, когда это возможно, с получением соединения данного изобретения (I), в котором R1 является атомом водорода. Кроме того, когда Р2 в соединении (Iа) данного изобретения является защитной группой для фенола и защитная группа не отщепляется во время стадий А и В, отщепление с использованием метода, подходящего для удаления защитной группы Р2, производится, когда это возможно, с получением соединения данного изобретения (I).
В отношении защитной группы для амина, примеры которой приведены для Р1, до настоящего времени не существует конкретного ограничения, так как она является группой, которая обычно используется для защиты амина, и примерами ее являются низший алкоксикарбонил, аралкилоксикарбонил, ацил, низший алкил, аралкил и сульфонил.
В отношении защитной группы для фенола, примеры которой даны для Р2, до настоящего времени не существует конкретного ограничения, так как она является группой, которая обычно используется для защиты фенола, и примерами ее являются необязательно замещенный низший алкил, аралкил, три(низший алкил)силил, низший алкилкарбонил, низший алкилоксикарбонил и сульфонил. "Аралкил" означает группу, где атом водорода вышеуказанного выше алкила замещается арилом, и его конкретными примерами являются бензил и фенилэтил. Конкретными примерами "ацила" являются формил, ацетил, пропионил и бутирил.
Также возможно проводить обычное N-алкилирование с использованием соединения данного изобретения (I), где R1 представляет собой атом водорода так, чтобы получить соединение данного изобретения (I), где R1 является низшим алкилом.
Соединение, представленное формулой (I), может быть также получено необязательной комбинацией стадий, которые обычно могут выбираться специалистом в данной области, таких как известные алкилирование, ацилирование, окисление, восстановление и гидролиз. Кроме того, способ, показанный на следующей схеме реакций, является особенно эффективным синтезом соединения, представленного формулой (I).
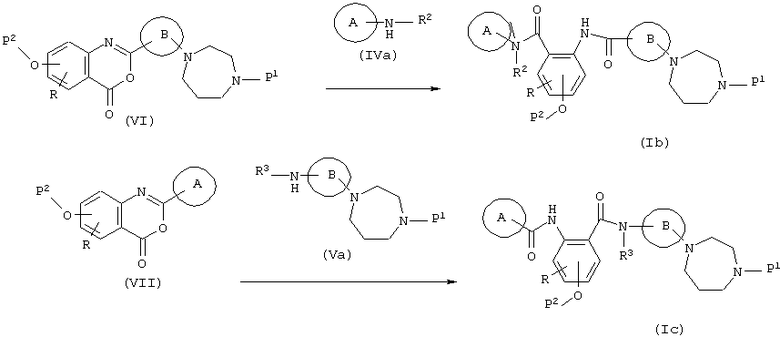
(в формулах А, В, Р1, Р2, R, R2 и R3 имеют значения, указанные выше. )
Этот способ представляет собой реакцию, при которой соединение (VI) и амин (IVa) или соединение (VII) и амин (Va) взаимодействуют с образованием амидной связи с получением соединения (Ib) или соединения (Iс) и которая осуществляется в названном выше инертном растворителе при комнатной температуре или при нагревании. Кроме того, в зависимости от применяемого способа, в некоторых случаях реакция протекает гладко в присутствии основания или использовании такого основания в качестве растворителя, когда основание является N-метилморфолином, триэтиламином, гриметиламином, пиридином, гидридом натрия, трет-бутоксидом калия, бутиллитием, амидом натрия или тому подобным.
Способы получения исходных соединений
Здесь далее будут проиллюстрированы типичные способы получения исходных соединений для соединения (I) данного изобретения.
Способ получения 1
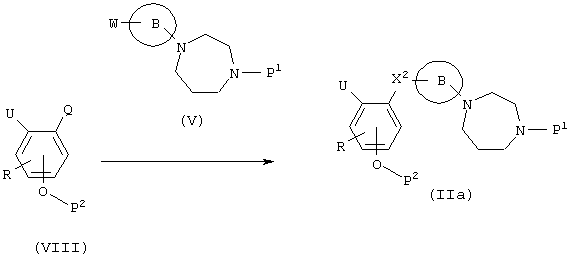
(в формулах В, X2, P1, P2, R, Q и W имеют значения, указанные выше, и U представляет -СООН, -COOP3, -NH2-, -NH-низший алкил, -NH-P4, -N(P4) -низший алкил или NO2, где P3 и P4 являются защитными группами для карбоксила и амина соответственно).
Способ получения 1
Этот способ получения представляет собой реакцию образования амидной связи конденсацией карбоновой кислоты с амином, включая комбинацию соединения (VIII) и соединения (V). Эта реакция осуществляется таким же образом, как и на вышеуказанной стадии А.
Когда U в соединении (IIа) представляет NO2, соединение, где U является NH2, может быть получено реакцией восстановления, в то время как, когда U представляет -COOP3, -NH-P4 или -(P4)-низший алкил, соединения, где U является -СООН, -NH2 или -NH-низшим алкилом, могут быть получены способом, который пригоден для отщепления каждой из защитных групп.
Способ получения 2
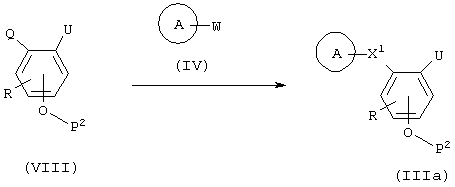
(в формулах А, Х1, Р2, R, Q, W и U имеют те же значения, что указаны выше)
Способ получения 2
Этот способ представляет собой реакцию, при которой амидная связь образуется конденсацией карбоновой кислоты и амина, включая комбинацию соединения (VIII) и соединения (IV). Эта реакция осуществляется таким же образом, как и на представленной выше стадии А. Когда U в соединении (IIIa) представляет NO2, соединение, где U является NH2, может быть получено реакцией восстановления, в то время как, когда U представляет собой -COOP3, -NH-P4 или –N(P4)-низший алкил, соединения, где U является -СООН, -NH2 или -NН-низшим алкилом, могут быть получены способом, который пригоден для отщепления каждой из защитных групп.
Соединения, представленные формулами (II) и (III), могут быть также получены с помощью необязательной комбинации стадий, которые обычно могут быть выбраны специалистом в данной области, таких как известные алкилирование, ацилирование, окисление, восстановление и гидролиз. Кроме того, способ, представляемый следующей схемой реакций, является особенно эффективным для синтеза соединений, представляемых формулами (II) и (III).
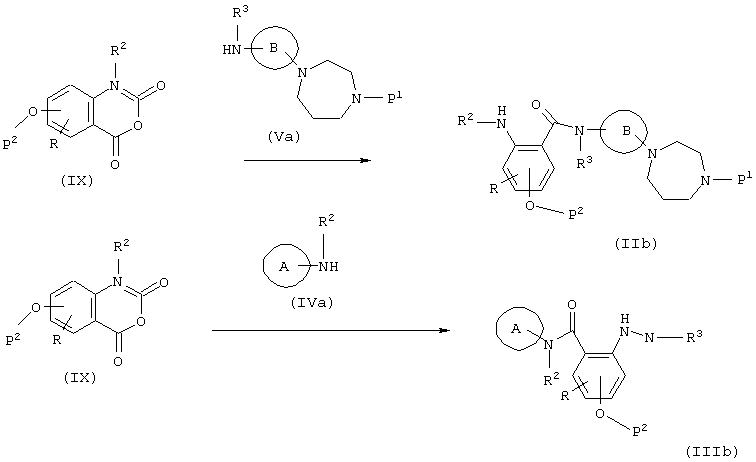
(в формулах А, В, R, R2, R3, Р1 и Р2 имеют значения, указанные выше).
Этот способ представляет собой реакцию, где амидная связь образуется при реакции соединения (IX) с амином (Va) или амином (IVa) с получением соединения (IIb) или соединения (IIIb), и она проводится в вышеуказанном, инертном растворителе при комнатной температуре или при нагревании. Кроме того, в зависимости от применяемого способа, в некоторых случаях реакция гладко протекает в присутствии основания или при использовании такого основания в качестве растворителя, причем основанием является N-метилморфолин, триэтиламин, триметиламин, пиридин, гидрид натрия, трет-бутоксид калия, бутиллитий, амид натрия или тому подобное.
Соединение данного изобретения, полученное таким путем, может быть выделено и очищено известными методами, такими как экстрагирование, осаждение, разделительная хроматография, дробная кристаллизация, перекристаллизация. Соединение данного изобретения может быть также превращено в желаемые соли с помощью обычных реакций образования солей.
Кроме того, соединение данного изобретения может существовать в форме оптических изомеров, когда оно имеет асимметричные атомы углерода. Эти оптические изомеры могут быть разделены обычным путем фракционированной кристаллизации, при которой изомер перекристаллизовывается вместе с соответствующей солью, или колоночной хроматографии.
Промышленное применение
Соединение данного изобретения проявляет сильное антикоагулирующее действие путем специфичного ингибирования активированного фактора Х коагуляции крови. Соответственно, соединение применимо в качестве ингибитора коагуляции крови или лекарственного средства для профилактики и лечения заболеваний, которые вызваны тромбозом или эмболией.
Примеры таких заболеваний включают цереброваскулярные заболевания, такие как инсульт, церебральный тромбоз, церебральная эмболия, преходящее нарушение мозгового кровообращения (ПНМК, TIA), субарахноидальное кровоизлияние (спазм сосудов) и тому подобное, ишемические заболевания сердца, такие как острый или хронический инфаркт миокарда, нестабильная стенокардия, тромболиз коронарных артерий и тому подобное, заболевания сосудов легких, такие как тромбоз легких, эмболия легких и тому подобное, и различные сосудистые заболевания, такие как закупорка периферических артерий, тромбоз глубоких вен, синдром диссеминированной внутрисосудистой коагуляции, тромбообразование после операции по замене кровеносного сосуда искусственным или после установки искусственного клапана, повторная окклюзия и повторный стеноз после операции шунтирования коронарных артерий, повторная окклюзия и повторный стеноз после операции ЧВКА (чрезкожной внутрипросветной коронарной ангиопластики) или ЧВКР (чрезкожной внутрипросветной коронарной реканализации) и тромбообразование во время экстракорпорального кровообращения.
Кроме того, было высказано предположение о возможности использования соединения, обладающего подавляющим активированный фактор Х коагуляции крови действием, в качестве лекарственного средства с использованием для профилактики и лечения инфекции, вызванной вирусом гриппа, на основе его активности по подавлению роста вируса гриппа (Japanese Patent Laid-Open No. 227971/1994), и поэтому, как предполагают, соединение данного изобретения также обладает таким же действием.
Высокая активность соединения данного изобретения по подавлению активированного фактора Х коагуляции крови была подтверждена следующими испытаниями.
1) Испытание по измерению времени коагуляции крови с помощью активированного фактора Х коагуляции крови человека.
К 90 мкл плазмы крови человека добавляли 10 мкл лекарственного препарата или физиологического раствора и 50 мкл фактора Ха человека (Enzyme Research Labs), инкубацию проводили при 37°С в течение 3 минут, добавляли 100 мкл 20 мМ CaCl2, предварительно нагретого до 37°С, и время до коагуляции измеряли с помощью коагулометра (КС10 от Amelung). Что касается плазмы крови человека, каждые 45 мл крови собирали из локтевой вены шести здоровых добровольцев, используя шприц, в котором содержалось 5 мл 3,8% цитрата натрия, и центрифугировали при 4°С в течение 15 минут при 3000 об/мин и отделенную плазму крови объединяли и замораживали до использования. Относительно фактора Ха человека, была выбрана концентрация, при которой время коагуляции, когда добавлялся физиологический раствор (контроль), составляло примерно 30-40 секунд. Значение CT2 (концентрация, при которой время коагуляции увеличивается двукратно) определяли, нанося на график концентрации лекарственного средства и относительные значения (кратность) времени коагуляции по отношению к контролю с последующим представлением линейной регрессии. Результат показан в следующей далее таблице 1.
2) Испытание по измерению времени коагуляции с помощью бычьего тромбина
К 50 мкл плазмы человека добавляли 50 мкл лекарственного препарата или физиологического раствора, инкубацию проводили при 37°С в течение 3 минут, добавляли 50 мкл раствора тромбина (500 единиц тромбина (полученного от быка; Mochida Pharmaceuticals)), предварительно нагретого до 37°С, и измеряли время до коагуляции с помощью коагулометра (КС10 от Amelung). Что касается плазмы крови человека, каждые 45 мл крови собирали иэ локтевой вены шести здоровых добровольцев, используя шприц, в котором содержалось 5 мл 3,8% цитрата натрия, и центрифугировали при 4°С в течение 15 минут при 3000 об/мин и отделенную плазму крови объединяли и замораживали до использования. В отношении тромбина, была выбрана концентрация, при которой время коагуляции, когда добавлялся физиологический раствор (контроль), составляло примерно 20 секунд. Значение СТ2 (концентрация, при которой время коагуляции увеличивается двукратно) определяли, нанося на график концентрации лекарственного средства и относительные значения (кратность) времени коагуляции по отношению к контролю с последующим представлением линейной регрессии. Результат показан в следующей далее таблице 1.
3) Испытание по измерению подавления активности фермента по методу с синтетическим субстратом
В 96-луночный планшет добавляли 80 мкл реакционного буфера (рН 8,4), 15 мкл лекарственного препарата и 30 мкл 2 мМ раствора синтетического субстрата S-2222 (Chromogenix), затем добавляли 25 мкл 0,025 Ед/мл активированного фактора Х коагуляции крови человека (фактор Ха; Enzyme Research Labs), реакция осуществлялась при 37°С в течение 10 минут, изменения в поглощении при 405 нм измеряли с помощью Bio-Rad, модель 3550, и рассчитывали IC50.
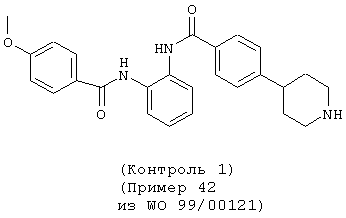
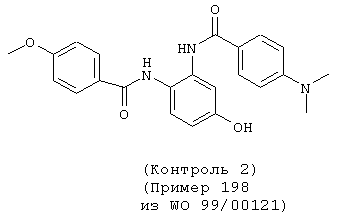
В результате измерений в вышеуказанных 1), 2) и 3) было подтверждено, что соединение данного изобретения специфически подавляет активированный фактор Х коагуляции крови и проявляет сильное антикоагулирующее действие в отношении крови. Например, соединения, показанные в примерах 1 и 3 данного изобретения, как подтверждено, явно продлевали время коагуляции при низкой концентрации, проявляя превосходное действие против коагуляции крови по сравнению с примером 42 (контроль 1) и примером 198 (контроль 2) из WO 99/00121, которые, как полагают, имеют структуру, наиболее сходную с соединениями данного изобретения.
4) Испытание по измерению ex vivo времени коагуляции у мышей (пероральное введение)
Лекарственное средство, которое растворено или суспендировано в 0,5% метилцеллюлозе, принудительно вводили per os (100 мг/кг) через пероральный зонд самцам мышей ICR (20-30 г; Nippon SLC), голодавших в течение 12 часов или более, и через 30 минут и 2 часа под анестезией диэтиловым эфиром отбирали 0,9 мл крови из нижней полой вены шприцом, содержащим 100 мкл 3,8% цитрата натрия, и плазму крови отделяли центрифугированием при 3000 об/мин в течение 10 минут. Используя полученную плазму крови, измеряли время наружного свертывания крови (ПВ; РТ) и время внутреннего свертывания крови (ЧАТВ; АРТТ) в соответствии со следующими методами а) и b).
а) Время наружного свертывания крови (ПВ)
Чистый тромбопластин Орто (54 мг/флакон; лиофилизованный препарат; Ortho Clinical Diagnostic) растворяли в 2,5 мл воды Milli-Q и предварительно прогревали при 37°С. Указанную выше отделенную плазму крови (50 мкл) прогревали при 30°С в течение 1 минуты, добавляли вышеуказанный раствор тромбопластина и измеряли время свертывания. Для измерения времени свертывания использовали Amelung КС 10А.
b) Время внутренней коагуляции (ЧАТВ)
К 50 мкл вышеуказанной плазмы крови добавляли 50 мкл Hemoliance Thrombosil I (Dia latron), смесь прогревали при 37°С в течение 3 минут, добавляли 50 мкл 20 мМ раствора CaCl2, предварительно нагретого до 37°С, и измеряли время свертывания. Для измерения времени свертывания использовали КС 10А, производимый Amelung.
Также изучали зависимость антикоагулирующего действия от дозы и временные изменения при изменении вводимой дозы или по времени свертывания крови.
5) Испытание по измерению ex vivo времени коагуляции у обезьян cynomolgus (пероральное введение)
Лекарственное средство (5 мг/мл), которое было растворено (суспендировано) в 0,5% метилцеллюлозе, принудительно вводили per оs в дозе 2 мл/кг (10 мг/кг) через пероральный зонд после отбора крови перед введением лекарственного средства самцу обезьяны cynomolgus (весом примерно 4 кг), голодавшему в течение 12 часов или более, и через 1, 2, 4, 6 и 8 часов 2 мл крови отбирали из бедренной вены с применением 1/10 объема 3,8% раствора цитрата натрия и плазму крови отделяли центрифугированием при 3000 об/мин в течение 10 минут. Используя полученную плазму крови, определяли время наружного свертывания крови (ПВ) и время внутреннего свертывания крови (ЧАТВ) по вышеуказанным методам а) и b). Кстати, эксперимент проводили без анестезии.
В результате испытаний 4) и 5) было подтверждено, что соединение данного изобретения обладает действием по увеличению времени свертывания также и при пероральном введении.
Фармацевтическую композицию, которая содержит в качестве активного ингредиента одно или более соединений данного изобретения, представленных формулой (I), или их фармацевтически приемлемые соли, изготавливают в виде таблеток, разбавленных порошков, тонких гранул, гранул, капсул, пилюль, растворов, инъекций, суппозиториев, мазей, пластырей и тому подобное с использованием обычно применяемых носителей, наполнителей и других добавок и вводимых или перорально, или парентерально.
Выбор клинической дозировки соединения данного изобретения у людей, по возможности, происходит с учетом симптомов, веса тела, возраста, пола и тому подобное каждого пациента, которому нужно лечение, и обычно составляет от 0,1 до 500 мг для перорального введения или от 0,01 до 100 мг для парентерального введения в сутки взрослому, причем суточная доза делится на один или несколько приемов в сутки. Так как доза изменяется при разных условиях, в некоторых случаях может быть достаточна меньшая доза, чем в указанном интервале.
Твердая композиция для использования при пероральном введении по данному изобретению применяется в виде таблеток, разбавленных порошков, гранул и тому подобное. В такой твердой композиции одно активное вещество или более смешивают с, по меньшей мере, одним инертным разбавителем, таким как лактоза, маннит, глюкоза, гидроксипропилцеллюлоза, микрокристаллическая целлюлоза, крахмал, поливинилпирролидон, метакремниевая кислота или алюминат магния. Обычно кроме инертного разбавителя композиция может содержать другие добавки, такие как увеличивающий скольжение агент (например, стеарат магния), дезинтегрирующий агент (например, целлюлозогликолят кальция), стабилизирующий агент (например, лактозу) и солюбилизирующее вспомогательное средство (например, глютаминовую кислоту и аспарагиновую кислоту). Если необходимо, таблетки или пилюли могут быть покрыты пленкой вещества, растворимого в желудке или кишечнике, такого как сахароза, желатин, гидроксипропилцеллюлоза, фталат гидроксипропилметилцеллюлозы и тому подобное.
Жидкие композиции для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы, эликсиры и тому подобное и содержат обычно используемый инертный разбавитель, такой как чистая вода или этиловый спирт. Кроме инертного разбавителя эти композиции могут также содержать вспомогательные агенты, такие как солюбилизирующий агент или солюбилизирующее вспомогательное средство, повышающий смачивание агент, суспендирующий агент и тому подобное, а также подсластители, улучшающие вкус и запах вещества, ароматизаторы и антисептики.
Инъекционные препараты для парентерального введения включают асептические водные или неводные растворы, суспензии и эмульсии. Примеры разбавителя для использования в водных растворах и суспензиях включают дистиллированную воду для инъекций и физиологический раствор. Примеры разбавителей для использования в неводных растворах и суспензиях включают пропиленгликоль, полиэтиленгликоль, растительное масло (например, оливковое масло), спирт (например, этиловый спирт), полисорбат 80 (торговая марка) и тому подобное.
Такая композиция может дополнительно содержать такие добавки, как изотоническое средство, антисептический агент, повышающий смачивание агент, эмульгатор, диспергирующее средство, стабилизатор (например, лактозу) и повышающий растворение агент или вспомогательное солюбилизирующее средство. Эти композиции стерилизуют фильтрованием через задерживающие бактерии фильтры, добавлением в смесь бактерицидного вещества или облучением. Альтернативно, для них может применяться сначала изготовление стерильных твердых композиций и затем растворение их в стерильной воде или стерильном растворителе для инъекций перед их употреблением.
Наилучший способ осуществления изобретения
Следующее далее описание конкретно иллюстрирует способ получения соединений данного изобретения со ссылкой на примеры получения соединений настоящего изобретения. В связи с тем, что исходные материалы для соединений настоящего изобретения включают новые соединения, способы их получения также описаны в виде справочных примеров.
Справочный пример 1
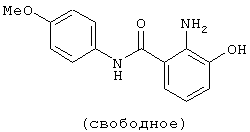
3-Гидрокси-2-нитробензойную кислоту (1,83 г) растворяли в 50 мл N,N-диметилформамида, затем добавляли 1,23 г 4-метоксианилина, 2,50 г 1-этил-3-диметиламинопропилкарбодиимид-гидрохлорида, 1,35 г 1-гидроксибензотриазола и 1,81 мл триэтиламина и смесь перемешивали при комнатной температуре в течение 66 часов. Реакционный раствор концентрировали в вакууме, добавляли воду и смесь экстрагировали этилацетатом. Органический слой промывали рассолом, сушили безводным сульфатом магния и концентрировали в вакууме. К полученному остатку добавляли хлороформ и полученный осадок отфильтровывали с получением 2,04 г 3-гидрокси-4'-метокси-2-нитробензанилида. Фильтрат очищали колоночной хроматографией на силикагеле, используя смесь хлороформ-метанол (98:2) в качестве элюирующего растворителя, к полученному неочищенному продукту добавляли хлороформ и полученный осадок отфильтровывали с получением дополнительно 0,24 г 3-гидрокси-4'-метокси-2-нитробензанилида.
Справочный пример 2
3-Гидрокси-4'-метокси-2-нитробензанилид (1/15 г) суспендировали в 50 мл метанола, добавляли 300 мг порошка 10% палладий на угле и смесь перемешивали в атмосфере водорода при комнатной температуре в течение 1 часа. Реакционный раствор фильтровали через целит и промывали метанолом и фильтрат концентрировали в вакууме с получением 966 мг 2-амино-3-гидрокси-4'-метоксибензанилида.
Справочный пример 3
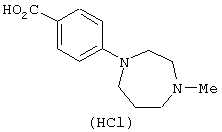
4-(4-Метил-1,4-диаэепан-1-ил)бензонитрил (18,86 г) растворяли в 185 мл 12 N водного раствора соляной кислоты, перемешивали в течение 12 часов и концентрировали в вакууме. Добавляли воду, смесь перемешивали при комнатной температуре и полученный осадок отфильтровывали и промывали водой. Полученное твердое вещество сушили в вакууме с получением 18,25 г гидрохлорида 4-(4-метил-1,4-диазепан-1-ил)бензойной кислоты.
Справочный пример 4
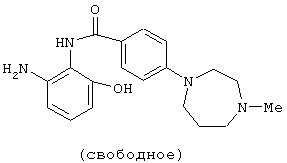
Гидрохлорид 4-(4-метил-1,4-диазепан-1-ил)бензойной кислоты (6,09 г) растворяли в 23 мл тионилхлорида и перемешивали при 60°С в течение 1 часа. Реакционный раствор концентрировали в вакууме, добавляли толуол и смесь снова концентрировали в вакууме. Полученный остаток суспендировали в 120 мл пиридина, добавляли 3,82 г 2-амино-3-нитрофенола при 0°С и смесь перемешивали при комнатной температуре в течение 3 дней. Добавляли этанол (10 мл) с последующим перемешиванием в течение 1 часа. Реакционный раствор концентрировали в вакууме, к полученному остатку добавляли хлороформ и смесь подщелачивали 200 мл 5% водного раствора бикарбоната натрия и экстрагировали хлороформом. Полученный органический слой сушили над безводным сульфатом натрия и концентрировали в вакууме. Полученный остаток очищали колоночной хроматографией на силикагеле, используя смесь хлороформ-метанол-насыщенный водный аммиак (100:10:1) в качестве элюирующего растворителя. Очищенный продукт (4,92 г) из 4,93 г, полученных выше, растворяли в 130 мл этанола, добавляли 500 мг порошка 10% палладий на угле и смесь перемешивали в атмосфере водорода при комнатной температуре в течение 2 часов. Реакционный раствор фильтровали через целит и концентрировали в вакууме. Полученный остаток растворяли в 130 мл метанола, добавляли 1,50 г порошка 10% палладий на угле и смесь перемешивали в атмосфере водорода при комнатной температуре в течение 12 часов и затем перемешивали в атмосфере водорода при давлении в 3 атмосферы при комнатной температуре в течение 20 часов. Реакционный раствор фильтровали через целит и концентрировали в вакууме с получением 3,42 г 2'-амино-6'-гидрокси-4-(4-метил-1,4-диазепан-1-ил)бензанилида.
Справочный пример 5
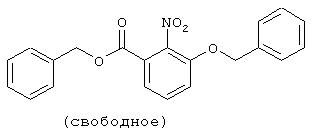
3-Гидрокси-2-нитробензойную кислоту (10,5 г) растворяли в 60 мл N,N-диметилформамида, затем добавляли 15 мл бензилбромида и 19,0 г карбоната калия при 0°С и смесь перемешивали в течение одной ночи при комнатной температуре. Реакционный раствор фильтровали через целит и концентрировали в вакууме. К полученному остатку добавляли воду и смесь экстрагировали эфиром, промывали рассолом и сушили над безводным сульфатом магния. Растворитель выпаривали в вакууме с получением 20,7 г бензил 3-бензилокси-2-нитробензоата.
Справочный пример 6
К 20,7 г бензил 3-бензилокси-2-нитробензоата добавляли 100 мл этанола и 120 мл 1 N водного раствора гидроксида натрия и смесь перемешивали при комнатной температуре в течение одной ночи, при 60°С в течение 3 часов и при 80°С в течение 5 часов. После того, как этанол испарится в вакууме, полученный водный раствор промывали эфиром и добавляли водный раствор соляной кислоты. Полученный осадок отфильтровывали и сушили в вакууме с получением 15,8 г 3-бензилокси-2-нитро-бензойной кислоты.
Справочный пример 7
К 5,47 г 3-бензилокси-2-нитробензойной кислоты добавляли 20 мл тионилхлорида и несколько капель N,N-диметилформамида и смесь перемешивали при 80°С в течение 30 минут. Реакционный раствор концентрировали в вакууме, к остатку добавляли 35 мл пиридина и 2,55 г 2-амино-5-хлорпиридина при 0°С и смесь перемешивали при комнатной температуре в течение одной ночи. Реакционный раствор концентрировали в вакууме, к полученному остатку добавляли насыщенный водный раствор бикарбоната натрия и смесь экстрагировали хлороформом. Органический слой сушили над безводным сульфатом магния, растворитель выпаривали в вакууме и остаток подвергали аэеотропной обработке толуолом с получением 7,44 г 3-бензилокси-N-(5-хлор-2-пиридил)-2-нитробензамида.
Справочный пример 8
К 7,44 г 3-бензилокси-N-(5-хлор-2-пиридил)-2-нитробензамида добавляли 40 мл трифторуксусной кислоты и 3,72 г пентаметилбензола и смесь перемешивали при 40°С в течение одной ночи. Реакционный раствор концентрировали в вакууме, к полученному остатку добавляли насыщенный водный раствор бикарбоната натрия до тех пор, пока остаток не становился щелочным, и смесь экстрагировали хлороформом. Органический слой экстрагировали 1 N водным раствором гидроксида натрия и водный слой подкисляли добавлением водного раствора соляной кислоты и экстрагировали хлороформом. Экстракт сушили безводным сульфатом магния, растворитель выпаривали в вакууме и к полученному остатку добавляли 200 мл этанольной суспензии никеля Ренея. Смесь перемешивали в атмосфере водорода в течение 6 часов, добавляли N,N-диметилформамид и нерастворимые вещества отфильтровывали. Растворитель выпаривали в вакууме и к полученному остатку добавляли воду. Полученный остаток отфильтровывали и сушили в вакууме с получением 4,58 г 2-амино-N-(5-хлор-2-пиридил)-3-гидроксибензамида.
Справочный пример 9
2-Амино-N-(5-хлор-2-пиридил)-3-гидроксибензамид (3,06 г) и 1,80 г N-хлорсукцинимида растворяли в 60 мл N,N-диметилформамида, раствор перемешивали при 50°С в течение 8 часов и при комнатной температуре в течение 4 часов и нерастворимые вещества отфильтровывали. После того, как растворитель выпаривали в вакууме, к полученному остатку добавляли 1 N водный раствор гидроксида натрия с последующим экстрагированием этилацетатом. Органический слой сушили над безводным сульфатом натрия, растворитель выпаривали в вакууме и полученный остаток очищали колоночной хроматографией на силикагеле. К полученному грубо очищенному продукту добавляли этанол и полученный осадок отфильтровывали и сушили в вакууме с получением 767 мг 2-амино-5-хлор-N-(5-хлор-2-пиридил)-3-гидроксибензамида.
Маточную жидкость концентрировали, добавляли смесь этилацетата и иэопропилового эфира и полученный осадок отфильтровывали и сушили в вакууме с получением дополнительно 942 мг вышеуказанного соединения.
Соединения справочных примеров 10 и 11 получали таким же образом, как в справочном примере 9.
Справочный пример 12
Этил 2-амино-5-хлор-3-гидроксибензоат (3,23 г) растворяли в 160 мл 3 N водного раствора соляной кислоты и перемешивали при 85°С в течение 3 часов и при 80°С в течение 5 дней. Реакционный раствор охлаждали до комнатной температуры, нерастворимые вещества отфильтровывали, к фильтрату добавляли 320 мл 1 N водного раствора гидроксида натрия и смесь перемешивали при комнатной температуре в течение 1 часа. Полученный остаток отфильтровывали, промывали чистой водой и сушили в вакууме с получением 1,55 г 2-амино-5-хлор-3-гидроксибензойной кислоты.
Справочный пример 13
2-Амино-5-хлор-3-гидроксибензойную кислоту (1,12 г) растворяли в 60 мл N,N-диметилформамида, затем добавляли 7,38 г 4-метоксианилина, 1,73 г 1-этил-3-диметиламинопропилкарбодиимидгидрохлорида, 1,21 г 1-гидроксибензотриазола и 1,26 мл триэтиламина и смесь перемешивали при комнатной температуре в течение 13 часов. Реакционный раствор концентрировали в вакууме, к полученному остатку добавляли этилацетат и смесь промывали чистой водой и рассолом, сушили над безводным сульфатом магния и концентрировали в вакууме. К полученному остатку добавляли хлороформ, смесь перемешивали в течение 30 минут и полученный осадок отфильтровывали, промывали хлороформом и сушили в вакууме с получением 0,96 г 2-амино-5-хлор-3-гидрокси-4'-метокси-2-бензанилида.
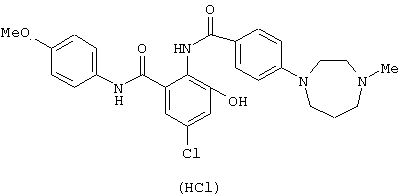
Пример 1
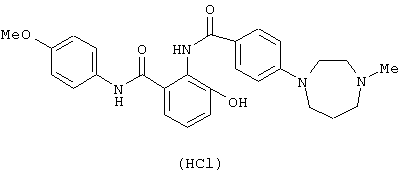
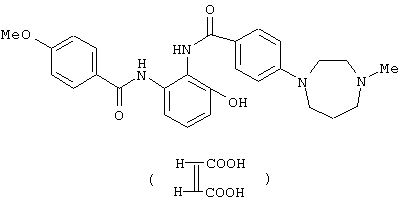
Гидрохлорид 4-(4-метил-1,4-диазепан-1-ил)бензойной кислоты (812 мг) растворяли в 8 мл тионилхлорида и перемешивали при 60°С в течение 30 минут. Реакционный раствор концентрировали и сушили в вакууме. Раствор, где 774 мг 2-aминo-4’-мeтoкcи-3-гидроксибензанилида растворено в 15 мл пиридина, добавляли к полученному остатку при 0°С и смесь перемешивали при комнатной температуре в течение 2 часов. Реакционный раствор концентрировали в вакууме, к полученному остатку добавляли толуол и смесь снова концентрировали в вакууме. К полученному остатку добавляли насыщенный водный раствор бикарбоната натрия и этилацетат и полученный осадок отфильтровывали. Этилацетатный слой маточной жидкости сушили над безводным сульфатом натрия и концентрировали в вакууме. Полученный остаток перемешивали с отфильтрованным осадком и очищали колоночной хроматографией на силикагеле, используя смесь хлороформ-метанол (98:2) в качестве элюирующего растворителя, с получением 873 мг 3-гидрокси-4’-метокси-2-{[4-(4-метил-1,4-диазепан-1-ил)бензоил]амино}бензанилида. Полученное соединение суспендировали в 10 мл этанола, добавляли 0,7 мл раствора 4 N соляной кислоты в этилацетате, смесь перемешивали и полученный осадок отфильтровывали, промывали этанолом и сушили в вакууме с получением 896 мг гидрохлорида 3-гидрокси-4'-метокси-2-{[4-(4-метил-1,4-диазапан-1-ил)бензоил]амино}бензанилида.
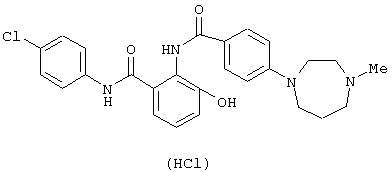
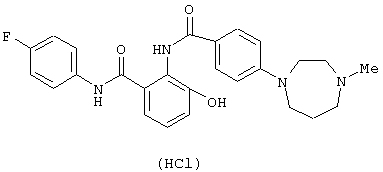
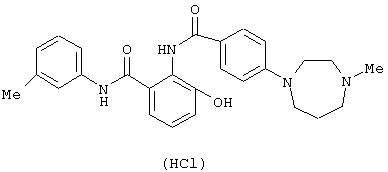
Соединения примеров 2-4, 8 и 9 были получены по методике примера 1.
Пример 5
2'-Амино-6'-гидрокси-4-(4-метил-1,4-диазепан-1-ил)бензанилид (2,03 г) растворяли в 60 мл пиридина, добавляли 1,12 г 4-метоксибензоилхлорида при 0°С и смесь перемешивали при комнатной температуре в течение 3 дней. Реакционный раствор концентрировали в вакууме, к полученному остатку добавляли 150 мл хлороформа и смесь подщелачивали 150 мл 5% водного раствора бикарбоната натрия и экстрагировали хлороформом. Полученный органический слой сушили над безводным сульфатом натрия и концентрировали в вакууме, добавляли толуол и смесь снова концентрировали в вакууме. Полученный остаток очищали колоночной хроматографией на силикагеле, используя смесь хлороформ-метанол-насыщенный водный аммиак (100:10:1) в качестве элюирукщего растворителя. Остаток перекристаллизовывали из этанола с получением 1,74 г 3-гидpoкcи-Nl-(4-мeтoкcибeнзoил)-N2-[4-(4-метил-1,4-диазепан-1-ил)бензоил]-1,2-фенилендиамина. 1,10 г 3-гидрокси-N1-(4-метоксибензоил)-N2-[4-(4-метил-1,4-диазепан-1-ил)бензоил]-1,2-фенилендиамина и 269 мг малеиновой кислоты растворяли в 11 мл 50% водно-этанольного раствора при нагревании, добавляли 11 мл воды и охлаждали. Полученные кристаллы отфильтровывали и сушили с получением 1,18 г малеата 3-гидрокси-N1-(4-метоксибензоил)-N2-[4-(4-метил-1,4-диазепан-1-ил)бензоил]-1,2-фенилендиамина.
Пример 6
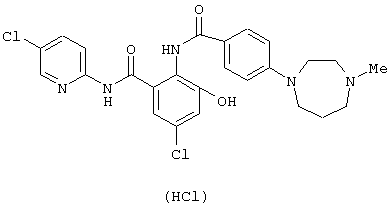
Гидрохлорид 4-(4-метил-1,4-диазепан-1-ил)бензойной кислоты (755 мг) растворяли в 2,2 мл тионилхлорида и перемешивали при 60°С в течение 30 минут. Реакционный раствор концентрировали и сушили в вакууме. К остатку добавляли раствор 891 мг 2-амино-5-хлор-N-(5-хлор-2-пиридил)-3-гидроксибензамида в 10 мл пиридина и смесь перемешивали при комнатной температуре в течение 13 часов. Реакционный раствор концентрировали в вакууме, к полученному остатку добавляли 20 мл уксусной кислоты и смесь перемешивали при комнатной температуре в течение 17 часов. Реакционный раствор концентрировали в вакууме, к полученному остатку добавляли насыщенный водный раствор бикарбоната натрия и смесь экстрагировали хлороформом, сушили безводным сульфатом натрия и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, используя смесь хлороформ-метанол-водный аммиак (97:3:0,3 до 95:5:0,5) в качестве элюирующего растворителя с получением грубо очищенного 5-хлор-N-(5-хлор-2-пиридил)-3-гидрокси-2-{[4-(4-метил-1,4-диазапан-1-ил)бензоил]амино}бензамида. Это вещество дополнительно очищали ODS колоночной хроматографией, используя смесь ацетонитрил-0,002 N водный раствор соляной кислоты (2:8 до 3:7) в качестве элюирующего растворителя, суспендировали в разбавленном водном растворе соляной кислоты и лиофилизовали с получением 492 мг гидрохлорида 5-хлор-N-(5-хлор-2-пиридил)-3-гидрокси-2-{[4-(4-метил-1,4-диазапан-1-ил)бензоил]амино}бензамида.
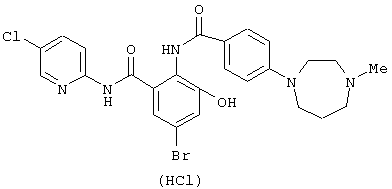
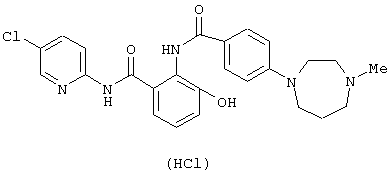
Соединение примера 7 синтезировали таким же образом, как и в примере 6.
Структурные формулы и физико-химические свойства соединений вышеприведенных справочных примеров и примеров представлены в таблицах 2-5. Символы в таблицах имеют следующие значения.
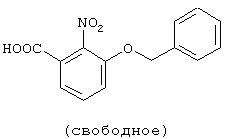
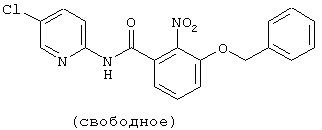
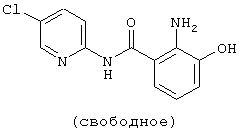
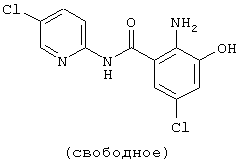
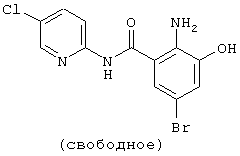
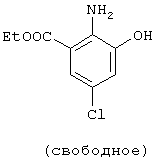
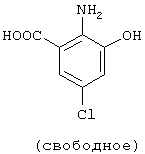
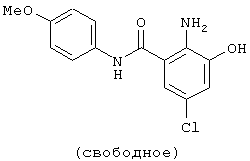
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ БЕНЗОЛА ИЛИ ИХ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2276137C2 |
| ПРОИЗВОДНОЕ БЕНЗОЛА ИЛИ ЕГО СОЛЬ | 2006 |
|
RU2395496C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ СУЛЬФОНАМИДНЫЕ ГРУППЫ, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ И АНТИАНГИОГЕННЫЙ АГЕНТ НА ИХ ОСНОВЕ | 2000 |
|
RU2239631C2 |
| ПРОИЗВОДНЫЕ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ И СПОСОБ ЛЕЧЕНИЯ РАССТРОЙСТВ, ХАРАКТЕРИЗУЮЩИХСЯ ПОРАЖЕНИЕМ НЕЙРОНОВ | 2000 |
|
RU2241709C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2658919C2 |
| ПРОИЗВОДНОЕ АМИНОБЕНЗОЙНОЙ КИСЛОТЫ ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ | 1993 |
|
RU2067979C1 |
| ИМИДАЗОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СПОСОБ ИНГИБИРОВАНИЯ АДЕНОЗИНДЕЗАМИНАЗЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1999 |
|
RU2243220C2 |
| АМИНОТРИАЗОЛОПИРИДИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2009 |
|
RU2552642C2 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ПОСРЕДНИКОМ КОТОРЫХ ЯВЛЯЕТСЯ ТАХИКИНИН | 1991 |
|
RU2073683C1 |
| 5-ЧЛЕННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЕ ДЛЯ ЛЕКАРСТВЕННЫХ ЦЕЛЕЙ | 2008 |
|
RU2515968C2 |
Описываются диазепановое производное общей формулы (I) или его фармацевтически приемлемая соль:
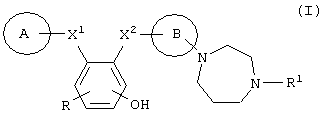
где кольцо В означает фенил; кольцо А означает пиридил, необязательно замещенный галогеном, или фенил, необязательно замещенный низшим алкилом, низшим алкокси или галогеном; X1 представляет -C(=O)-NR2- или –NR2-C(=O)-, где R2 - водород; X2 представляет -C(=O)-NR3- или –NR2-C (=O)-, где R3 - водород; R представляет собой атом водорода или атом галогена и R1 означает низший алкил, фармацевтическая композиция и ингибитор активированного фактора Х коагуляции крови, который может использоваться для профилактики и лечения заболеваний, вызванных тромбозом или эмболией. 3 н. и 2 з.п.ф-лы, 5 табл.
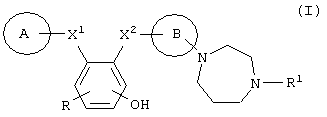
где кольцо В представляет собой фенил;
кольцо А представляет собой пиридил, необязательно замещенный галогеном, или фенил, необязательно замещенный низшим алкилом, низшим алкокси или галогеном;
Х1 представляет -C(=O)-NR2- или –NR2-C(=O)-, где R2 представляет собой атом водорода;
X2 представляет -C(=O)-NR3- или –NR3-C(=O)-, где R3 представляет собой атом водорода;
R представляет собой атом водорода или атом галогена;
R1 представляет собой низший алкил.
| (R)-5-БРОМ-N-(1-ЭТИЛ-4-МЕТИЛГЕКСАГИДРО-1H-1,4-ДИАЗЕПИН-6-ИЛ)-2-МЕТОКСИ-6-М ЕТИЛАМИНО-3-ПИРИДИНКАРБОКСАМИД, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРОТИВОРВОТНОЕ СРЕДСТВО, СОДЕРЖАЩЕЕ УКАЗАННОЕ СОЕДИНЕНИЕ, И ЕГО ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 1996 |
|
RU2156248C2 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| WO 00/39118 A1, 06.07.2000 | |||
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |

