В данной заявке испрашиваются приоритеты от 15 февраля 2000 г. согласно предварительной заявке США №60/182676 и от 16 июня 2000 г. согласно заявке США, серийный №09/595365, каждая из которых включена здесь путем ссылки в полном объеме.
Область изобретения
Настоящее изобретение относится к области нуклеозидных аналогов.
Предшествующий уровень техники
Рибавирин (1-β-D-рибофуранозил-1,2,4-триазол-3-карбоксамид) представляет собой нуклеозидный аналог, который продемонстрировал эффективность в лечении вирусных заболеваний как в виде монотерапии (респираторно-синцитиальный вирус, Hall, С.В.; McBride, J.Т.; Walsh, Е.Е.; Bell, D.М.; Gala, С.L; Hildreth, S.; Ten Eyck, L.G.; W.J. Hall. Aerosolized ribavirin treatment of infants with respiratory syncytial viral infection. N. Engl. J. Med. 1983, 308, 1443-1447), так и в комбинационной терапии с интерфероном-альфа (вирус гепатита С, Reichard, О.; Norkrans, G.; Fryden, A.; Braconier, J.-H.; Sonnerborg, A.; Weiland, 0. Randomized, double blind, placebo controlled trial of interferon alpha 2B with and without ribavirin for chronic hepatitis C. Lancet 1998, 351, 83-87). В недавно описанных исследованиях показано, что эффективность рибавирина in vivo может являться результатом не только непосредственного ингибирования вирусной репликации, но также его способности усиливать опосредованный Т клетками иммунитет (Hultgren, С.; Milich, D.R.; Weiland, О.; Sallberg, M. The antiviral compound ribavirin modulates the Т helper Typel/Type 2 subset balance in hepatitis В and С virus-specific immune responses. J. Gen. Virol. 1998, 79, 2381-2391; Ning, Q.; Brown, D.; Parodo, J.; Cattral, M.; Fung, L.; Gorczynski, R.; Cole, E.; Fung, L; Ding, J. W.; Liu, M. F.; Rotstein, 0.; Phillips, M. J.; Levy, G. Ribavirin inhibits viral-induced macrophage production of tumor necrosis factor, interleukin-1, procoagulant activity fgl2 prothrombinase and preserves Th1 cytokine production but inhibits Th2 cytokine response. J. Immunol. 1998, 160, 3487-3493; Martin, M. J.; Navas, S.; Quiroga, J. A.; Pardo, M.; Carreno, V. Effects of the ribavirin-interferon alpha combination on cultured peripheral blood mononuclear cells from chronic hepatitis С patients. Cytokine 1998, 79, 2381-2391). Этот иммуномодуляторный эффект рибавирина можно продемонстрировать in vitro путем измерения уровней цитокинов типа 1, продуцируемых активированными Т клетками как от людей, так и от мышей (Tarn, R. С.; Pai, В.; Bard, J.; Lim, С.; Averett, D. R.; Phan, U. Т.; Milovanovic, Т. Ribavirin polarizes human Т cell responses towards a Type 1 cytokine profile. J. Hepatol. 1999, 30, 376-382), а также с помощью других измерений. Индукция изменения цитокинов типа 1 рибавирином является функционально значимой in vivo в мышиных системах (Tarn, R. С.; Lim, С.; Bard, J.; Pai, В. Contact hypersensitivity responses following ribavirin treatment in vivo are influenced by Type 1 cytokine polarization, regulation of IL-10 expression and costimulatory signaling. J. Immunol. 1999, 163, 3709-3717).
Иммунные системы млекопитающих содержат два основных класса лимфоцитов: В лимфоциты (В клетки), которые происходят из костного мозга; и Т лимфоциты (Т клетки), которые происходят из тимуса. В клетки в значительной степени ответственны за гуморальный иммунитет (то есть продуцирование антител), тогда как Т клетки являются в значительной степени ответственными за клеточно-опосредованный иммунитет.
Принято считать, что Т клетки подразделяются на два подкласса, хелперные Т клетки и цитотоксические Т клетки. Хелперные Т клетки активируют другие лимфоциты, включая В клетки и цитотоксические Т клетки, а также макрофаги, путем высвобождения растворимых белковых медиаторов, называемых цитокинами, которые вовлечены в клеточно-опосредованный иммунитет. Лимфокины, как этот термин используют здесь, представляют собой подгруппу цитокинов.
Принято считать, что хелперные Т клетки также подразделяются на два подкласса, клетки типа 1 и типа 2. Клетки типа 1 продуцируют интерлейкин 2 (ИЛ-2), фактор некроза опухоли (ФНОα) и интерферон гамма (ИФНγ) и являются ответственными прежде всего за клеточно-опосредованный иммунитет, такой как гиперчувствительность замедленного типа и противовирусный иммунитет. Напротив, клетки типа 2 продуцируют интерлейкины ИЛ-4, ИЛ-5, ИЛ-6, ИЛ-9, ИЛ-10 и ИЛ-13 и прежде всего вовлечены в содействие гуморальным иммунным ответам, таким как те, которые наблюдаются в ответ на аллергены, например переключение изотипа антител lgE и lgG4 (Mosmann, 1989, Annu Rev Immunol 7:145-173).
Подразумевается, что термины «ответы» типа 1 и типа 2, как их используют здесь, включают в себя полный диапазон эффектов, являющихся результатом индукции лимфоцитов типа 1 и типа 2 соответственно. Среди прочего такие ответы включают в себя изменение продуцирования соответствующих цитокинов посредством транскрипции, трансляции, секреции и, возможно, других механизмов, повышенную пролиферацию соответствующих лимфоцитов, а также другие эффекты, связанные с повышенным продуцированием цитокинов, включая эффекты подвижности.
Предшествующие заявки (09/462714, 09/291097, 09/291093, 09/471513, 60/164365, 60/164366, 60/172097, 60/175111), каждая из которых включена здесь путем ссылки, относятся к аспектам недавних открытий авторов изобретения, включающих в себя влияние различных нуклеозидов (которые определены здесь как включающие в себя производные и аналоги природных нуклеозидов) на селективное модулирование лимфоцитарных ответов относительно друг друга. Среди прочего авторы изобретения показали, что ответ либо типа 1, либо типа 2 может быть селективно супрессирован, в то время как другой из них либо индуцирован, либо остается относительно незатронутым, а также ответы либо типа 1, либо типа 2 можно селективно индуцировать, в то время как другой из них либо супрессирован, либо остается относительно незатронутым. Авторы изобретения также открыли удивительный факт, что некоторые нуклеозиды, эффективные в селективном модулировании ответов типа 1 и типа 2 относительно друг друга, склонны обладать бимодальным эффектом. Среди прочего некоторые нуклеозиды, которые склонны к общей супрессии или индукции обеих активностей типа 1 и типа 2 в относительно более высокой дозе, склонны к селективному модулированию типа 1 и типа 2 относительно друг друга в более низких дозах.
Показано, что Viramidine™ (1-β-D-рибофуранозил-1,2,4-триазол-3-карбоксамидина гидрохлорид, вирамидин) обладает активностью в десяти различных вирусах, которая сравнима с рибавирином (J.Т. Witkowski, R.К. Robins, G.P. Khare, R.W. Sidwell, J. Med. Chem., 16, 935-937, 1973; R.W. Sidwell, J.H. Huffman, D.L. Barnard, D.Y. Pifat, Antiviral Research, 10, 193-208, 1988; В. Gabrielsen, M.J. Phelan, L. Barthel-Rosa, C. See, J.W. Huggins, D.F. Kefauver, T.P. Monath, M.A. Ussery, G.N. Chmurny, E.M. Schubert, K. Upadhya, C. Kwong, D.A. Carter, J.A. Secrist III, J.J. Kirsi, W.M. Shannon, R.W. Sidwell, G.D. Kini, R.K. Robins, J. Med. Chem., 35, 3231-3238, 1992). Кроме того, Viramidine™ подобно рибавирину является ингибитором IMP дегидрогеназы (инозинмонофосфат-дегидрогеназы) (R.C. Willis, R.K. Robins, J.E. Seegmiller, Molecular Pharmacology, 18, 287-295, 1980). Кроме того, предварительные токсикологические исследования позволяют предположить, что Viramidine™ является менее токсичным, чем рибавирин (D.Y. Pifat, R.W. Sidwell, P.G. Canonico, Antiviral Research, 9, 136, 1988). Также недавние исследования в лаборатории авторов изобретения (R. Tarn, K. Ramasamy, ICN Pharmaceuticals, Inc., неопубликованные результаты, 1999) выявили, что Viramidine™ и рибавирин проявляют сходные иммуномодуляторные свойства. Эти результаты вместе с низкой биологической доступностью и токсичностью, присущими рибавирину, убедили авторов изобретения не только разрабатывать Viramidine™ для других вирусных заболеваний, но также получать другие производные Viramidine™, включая синтез пролекарств Viramidine™, и проводить их скрининг в качестве потенциальных противовирусных агентов.
Влияние других соединений, представляющих собой нуклеозидные аналоги, на селективное модулирование лимфоцитарных ответов относительно друг друга ранее не исследовано или не документировано. Авторы изобретения открыли, что бимодальный эффект или селективное модулирование ответов типа 1 и типа 2 относительно друг друга также имеют место после введения других соединений, представляющих собой нуклеозидные аналоги, таких как пролекарственные формы этих соединений.
Существует множество барьеров, которые предстоит преодолеть при разработке биологически активных соединений до клинически полезных агентов. Многие сильнодействующие биологически активные соединения никогда не станут клинически полезными агентами из-за их нежелательных биофармацевтических свойств, которые включают в себя низкую биологическую доступность вследствие низкой проникающей способности через биологические барьеры, такие как гематоэнцефалический барьер (ГЭБ) и кишечный барьер. Хотя на биологическую доступность лекарственного средства влияют многие факторы, нежелательные физико-химические свойства (например, заряд, липофильность, потенциал образования водородной связи, размер) многих лекарственных средств, вероятно, являются одним из наиболее часто встречающихся факторов, которые препятствуют проникновению лекарственных средств через биологические барьеры. Следовательно, оптимизация физико-химических характеристик (заряда, липофильности, потенциала образования водородной связи, размера) лекарственного средства, возможно, является наиболее вероятной общей стратегией, способствующей транспорту лекарственных средств через такие мембранные барьеры.
Одной из возможных стратегий оптимизации физико-химических свойств лекарственных средств является стратегия пролекарств (Н. Bundgaard, Design of Prodrugs, Elsevier, Amsterdam, 1985; N. Bodor, L. Prokai, W. M. Wu, H. Farag, S. Jonalagadda, M. Kawamura, J. Simpkins, Science, 257. 1698-1700, 1992; H.Е. Taylor, К.В. Sloan, J. Pharm. Sci., 87, 5-20, 1998). Термин «пролекарство» используют для описания агента, который должен претерпевать химическое или ферментативное превращение до активного или исходного лекарства после введения, так, чтобы метаболический продукт или исходное лекарство могли после этого проявлять желаемый фармацевтический ответ. Посредством получения временных и биологически обратимых производных некоторых полярных функциональных групп в малых органических молекулах нежелательные физико-химические характеристики (например, заряд, потенциал образования водородной связи) этих групп «замаскированы» без необратимого изменения фармакологических свойств этих молекул. Данную стратегию весьма успешно применили в тех случаях, когда в получение пролекарственного производного вовлечено превращение карбоксильной или гидроксильной функциональной группы в сложный эфир, который легко гидролизуется in vivo либо химическим, либо ферментативным путем. В качестве перспективной общей концепции пролекарства авторы изобретения ожидают, что введение других группировок в исходное лекарство повысит его биологическую доступность, всасывание и противовирусные эффекты.
Несмотря на существование еще не определенных механизмов, авторы изобретения открыли, что огромные потенциальные преимущества можно получить в результате селективного модулирования ответов типа 1 и типа 2 относительно друг друга. Авторы изобретения пришли к выводу, например, что специфическое модулирование типа 1 относительно типа 2 может быть полезным при лечении широкого разнообразия состояний и заболеваний в диапазоне от инфекций, инвазий, опухолей и гиперчувствительности до аутоиммунных заболеваний.
Эти открытия являются особенно важными, поскольку современные стратегии лечения для многих из перечисленных выше заболеваний обладают ограниченной эффективностью, значительными побочными эффектами, либо обоими недостатками. Лечение аутоиммунного заболевания, например, часто ограничено паллиативными мерами, удалением токсичных антител (как при тяжелой псевдопаралитической миастении) и введением вредных лекарственных средств, включая кортикостероиды, хлорохиновые производные и антиметаболические или противоопухолевые лекарственные средства, а также таких лекарственных средств, как циклоспорины, которые нацелены на клетки иммунной системы.
Краткое изложение сущности изобретения
Настоящее изобретение относится к соединениям, представляющим собой новые нуклеозидные аналоги, и родственным соединениям, таким как пролекарства, к их терапевтическому применению и синтезу.
В одном аспекте изобретения предложены соединения, представляющие собой нуклеозидные аналоги формулы 1:

Еще в одном аспекте изобретения предложена фармацевтическая композиция, содержащая терапевтически эффективное количество карбоксамидина формулы 1 либо его фармацевтически приемлемого сложного эфира или соли, в смеси по меньшей мере с одним фармацевтически приемлемым носителем.
Еще в одном аспекте изобретения предложена фармацевтическая композиция, содержащая пролекарственную форму карбоксамидина формулы 1 либо его фармацевтически приемлемого сложного эфира или соли, в смеси по меньшей мере с одним фармацевтически приемлемым носителем.
В следующем аспекте изобретения соединение согласно формуле 1 применяют в лечении любого состояния, при котором имеется положительный ответ на введение этого соединения, и в соответствии с любым препаратом и протоколом, при которых достигнут положительный ответ. Среди прочего полагают, что соединения формулы 1 можно применять для лечения инфекции, инвазии, рака, опухоли или другой неоплазмы, гигантоклеточного артериита или аутоиммунного заболевания.
Краткое описание графических материалов
Фиг.1 представляет собой примерную синтетическую схему для синтеза соединения согласно формуле 1.
Фиг.2 представляет собой графическое изображение влияния рассматриваемого соединения и других соединений на синтез цитокинов типа 1 в SEB-активированных Т клетках человека.
Фиг.3 представляет собой графическое изображение влияния 0,625-10 мкМ концентрации рассматриваемого соединения на синтез цитокинов типа 1 в SEB-активированных Т клетках человека.
Фиг.4 представляет собой графическое изображение влияния рассматриваемого соединения на КГЧ ответы у мышей BALB/c.
Фиг.5 представляет собой графическое изображение пикового ответа и пикового диапазона рассматриваемого соединения и других соединений по отношению к синтезу цитокинов типа 1 в SEB-активированных Т клетках человека.
Подробное описание изобретения
Там, где следующие термины используют в данном описании, их используют, как определено ниже.
Термины «нуклеозид» и «соединение, представляющее собой нуклеозидный аналог», являются взаимозаменяемыми и относятся к соединению, состоящему из любой пентозной или модифицированной пентозной группировки, присоединенной к конкретному положению гетероцикла, ароматического гетероцикла, либо к природному положению пурина (9-положение) или пиримидина (1-положение), либо к эквивалентному положению в аналоге.
Термин «нуклеотид» относится к фосфатному сложному эфиру, образованному по 5'-положению нуклеозида.
Термин «гетероцикл» относится к одновалентному насыщенному или ненасыщенному карбоциклическому радикалу, имеющему по меньшей мере один гетероатом, такой как N, О или S, в пределах кольца, каждое доступное положение которого может быть возможно независимо замещено, например, гидрокси, оксо, амино, имино, низшим алкилом, бромо, хлоро и/или циано. В данный класс заместителей включены пурины, пиримидины.
Термин «пурин» относится к азотистым бициклическим гетероциклам.
Термин «пиримидин» относится к азотистым моноциклическим гетероциклам.
Термин «D-нуклеозиды» относится к нуклеозидным соединениям, которые имеют D-рибозную сахарную группировку (например, аденозин).
Термин «L-нуклеозиды» относится к нуклеозидным соединениям, которые имеют L-рибозную сахарную группировку.
Термины «L-конфигурация» и «D-конфигурация» используют на протяжении всего настоящего изобретения для описания химической конфигурации рибофуранозильной группировки соединений, которая сшита с пирроло-пиримидиновым участком молекулы.
Термин «С-нуклеозиды» используют на протяжении всего описания для характеристики типа связи, которая образована между рибозной сахарной группировкой и гетероциклическим основанием. В С-нуклеозидах эта связь начинается от положения С-1 рибозной сахарной группировки и присоединяет атом углерода гетероциклического основания. Связь, которая образуется в С-нуклеозидах, принадлежит к типу углерод-углерод.
Термин «N-нуклеозиды» используют на протяжении всего описания для описания типа связи, которая образована между рибозной сахарной группировкой и гетероциклическим основанием. В N-нуклеозидах эта связь начинается от положения С-1 рибозной сахарной группировки и присоединяет атом азота гетероциклического основания. Связь, которая образуется в N-нуклеозидах, принадлежит к типу углерод-азот.
Термин «защитная группа» относится к химической группе, которую добавляют к атому кислорода или азота для предотвращения его дальнейшего взаимодействия в течение процесса получения производных по другим группировкам в той молекуле, в которой локализован этот атом кислорода или азота. Широкое разнообразие защитных групп для кислорода и азота известно специалистам в области органического синтеза.
Термин «низший алкил» относится к метилу, этилу, н-пропилу, изопропилу, н-бутилу, mpem-бутилу, изобутилу или н-гексилу. Дальнейшими примерами данного термина является циклическая, разветвленная или нормальная цепь из атомов углерода в количестве от одного до шести.
Термин «арил» относится к одновалентному ненасыщенному ароматическому карбоциклическому радикалу, имеющему единственное кольцо (например, фенил) или два конденсированных кольца (например нафтил), который может быть возможно замещен гидроксилом, низшим алкилом, хлоро и/или циано.
Термин «гетероцикл» относится к одновалентному насыщенному или ненасыщенному карбоциклическому радикалу, имеющему по меньшей мере один гетероатом, такой как N, О, S, Se или Р, в пределах кольца, каждое доступное положение которого может быть возможно незамещенным или независимо замещенным, например гидрокси, оксо, амино, имино, низшим алкилом, бромо, хлоро и/или циано.
Термин «моноциклический» относится к одновалентному насыщенному карбоциклическому радикалу, имеющему по меньшей мере один гетероатом, такой как О, N, S, Se или Р, в пределах кольца, каждое доступное положение которого может быть возможно независимо замещено сахарной группировкой или любыми другими группами, подобными бромо, хлоро и/или циано, так что эта моноциклическая кольцевая система в конечном итоге является ароматизированной [например тимидин].
Термины «иммуномодулятор» и «модулятор» используют здесь взаимозаменяемо, и они относятся к природным или синтетическим продуктам, способным модифицировать нормальную или аберрантную иммунную систему посредством стимуляции или супрессии.
Термин «эффективное количество» относится к количеству соединения формулы (1), которое будет восстанавливать иммунную функцию до нормальных уровней или повышать иммунную функцию выше нормальных уровней для того, чтобы элиминировать инфекцию.
Соединения формулы 1 могут иметь множественные асимметрические центры. Соответственно их можно получить либо в оптически активной форме, либо в виде рацемической смеси. В объем изобретения, как описано и заявлено, включены индивидуальные оптические изомеры и их нерацемические смеси, а также рацемические формы соединений формулы 1.
Термин «α» и «β» указывает на конкретную стереохимическую конфигурацию заместителя при асимметричном атоме углерода в химической структуре, как изображено.
Термин «энантиомеры» относится к паре стереоизомеров, которые представляют собой ненакладывающиеся зеркальные отражения друг друга. Смесь пары энантиомеров в соотношении 1:1 представляет собой «рацемическую» смесь.
Термин «изомеры» относится к различным соединениям, которые имеют одну и ту же формулу. «Стереоизомеры» представляют собой изомеры, которые различаются только типом ориентации атомов в пространстве.
«Фармацевтически приемлемые соли» могут представлять собой любые соли, полученные из неорганических и органических кислот или оснований.
Соединения
Соединения, представляющие собой нуклеозидные аналоги по настоящему изобретению, в целом описаны формулой 1:
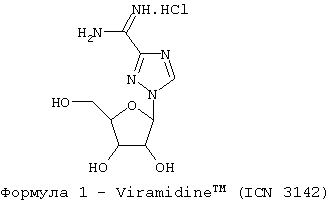
где химическая конфигурация этого соединения может представлять собой L-конфигурацию или D-конфигурацию. Примерный синтез рассматриваемых соединений (здесь: Viramidine™) может следовать методике, как описано ниже и показано на фиг.1.
3-Циано-1 -(2,3,5-три-O-ацетил-β-D-рибофуранозил)-1,2,4-триазол (7): Смесь 3-циано-1,2,4-триазола (18,8 г, 200 ммоль) (6), 1,2,3,5-тетра-O-ацетил-β-D-рибофуранозы (63,66 г, 200 ммоль) и бис(пара-нитрофенил)фосфата (1 г) помещали в колбу RB (500 мл). Эту колбу помещали в предварительно нагретую масляную баню при 165-175°С в вакуум, создаваемый водным аспиратором, при перемешивании в течение 25 минут. Вытесняемую уксусную кислоту собирали в охлаждаемый во льду уловитель, который помещался между аспиратором и колбой RB. Колбу извлекали из масляной бани и давали ей охладиться. Когда температура колбы достигала примерно 60-70°С, вводили EtOAc (300 мл) и насыщенный NaHCO3 (150 мл) и экстрагировали в EtOAc. Водный слой снова экстрагировали EtOAc (200 мл). Объединенный EtOAc экстракт промывали насыщенным NaHCO3 (300 мл), водой (200 мл) и рассолом (150 мл). Органический экстракт высушивали над безводным Na2SO4, фильтровали, а фильтрат выпаривали до сухости. Остаток растворяли в эфире (100 мл), который после охлаждении до 0°С в течение 12 ч давал бесцветные кристаллы. Это твердое вещество отфильтровывали, промывали минимальным количеством холодного EtOH (20 мл) и высушивали в высоком вакууме над твердым NaOH. Выход: 56,4 г (80%). Т. пл. 96-97°С. 1H ЯМР (CDCl3): δ 2.11 (s, 3Н, СОСН3), 2.13 (s, 3Н, СОСН3), 2.14 (s, 3Н, СОСН3), 4.22 (dd, 1H), 4.46 (m, 2H), 5.52 (t, 1H, J=6.0 Гц), 5.70 (m, 1H), 6.01 (d, 1H, CrH J=3.6 Гц) и 8.39 (s, 1H, C5H). Аналитически вычислено для C14H16N4O7 (352,30): С, 47,73; Н, 4,58; N, 15,90. Обнаружено: С, 47,70; Н, 4,63; N, 16,01.
1-β-D-Рибофуранозил-1,2,4-триазол-3-карбоксамидин (Viramidine™) гидрохлорид (8): Смесь (7) (14,08 г, 40,0 ммоль), NH4Cl (2,14 г, 40,0 ммоль) и безводного аммиака (150 мл) нагревали в стальном баллоне при 85°С в течение 18 ч. Этот стальной баллон охлаждали, открывали и содержимое выпаривали до сухости. Остаток кристаллизовали из MeCN-EtOH с получением 10,6 г (95%) 8. Т. пл. 177-179°С. 1H ЯМР (ДМСО-d6): δ 3.44-4.2 (m, 3Н), 4.40 (m, 2H), 5.04 (t, 1H), 5.29 (m, 1H), 5.74 (m, 1H), 5.87 (d, 1H, C1'H), 8.96 (bs, 3Н) и 9.17 (s, 1H, C5H). Аналитически вычислено для C8H14ClN5O4 (279,68): С, 34,35; Н, 5,05; N, 25,04; Cl, 12,69. Обнаружено: С, 34,39; Н, 5,10; N, 25,14; Cl, 12,71.
Альтернативно синтез можно проводить из коммерчески доступного Ribavirin™ следующим образом:
2',3',5'-Три-O-ацетил-1-β-D-рибофуранозил-1,2,4-триазол-3-карбоксамидин (9). Суспензию 1-β-D-рибофуранозил-1,2,4-триазол-3-карбоксамида (Ribavirin™) (28,4 г, 116,4 ммоль) (5) в уксусном ангидриде (200 мл) и пиридине (50 мл) перемешивали при комнатной температуре в течение ночи. Полученный в результате прозрачный раствор концентрировали в вакууме с получением прозрачной пены (43,1 г, количественно). Эта пена являлась гомогенной по результатам ТСХ, и ее использовали непосредственно для следующей стадии без очистки. Небольшое количество очищали флэш-хроматографией для получения аналитического образца. 1H ЯМР (300 МГц, ДМСО-d6): δ 2.01, 2.08, 2.09 (3s, 9H, СОСН3), 4.10 (m, 1H), 3.52 (m, 2H), 5.58 (t, 1H), 5.66 (m, 1H), 6.33 (d, 1H, J=3.0 Гц, C1H), 7.73, 7.92 (2s, 2H, CONH2), 8.86 (s, 1H, С5Н триазол). Аналитически (C10H18N4O8): С, Н, N.
3-Циано-2',3',5'-три-O-ацетил-1-β-D-рибофуранозил-1,2,4-триазол (10). К раствору 9 (43,1 г, 116,4 ммоль) в хлороформе (500 мл) добавляли триэтиламин (244 мл), и эту смесь охлаждали до 0°С в ледяной солевой бане. Добавляли по каплям оксихлорид фосфора (30,7 мл, 330 ммоль) при перемешивании, раствору давали нагреться до комнатной температуры. После перемешивания смеси при комнатной температуре в течение 1 ч ТСХ (гексан/ацетон 3:1) показала полное исчезновение исходного материала. Эту коричневую реакционную смесь концентрировали до сухости в вакууме и остаток растворяли в хлороформе (500 мл). Этот органический раствор промывали насыщенным водным бикарбонатом натрия (3×200 мл), высушивали над безводным сульфатом натрия и концентрировали в вакууме. Остаток подвергали хроматографии на силикагеле (флэш-хроматография) с 20%-ным ацетоном в гексане с получением 33,14 г (81% от рибавирина) чистого 10 в виде аморфного твердого вещества. Это твердое вещество было идентично во всех отношениях аутентичному образцу: т. пл. 101-103°С; ИК (бромид калия) ν 2250 (CN), 1750 (С=0), см-1. 1H ЯМР (300 МГц, CDCl3): δ 2.04, 2.06, 2.07 (3s, 9H, ацетилметилы), 4.15 (dd, 1H), 4.40 (m, 1Н), 5.47 (t, 1H), 5.63 (dd, 1Н), 5.95 (d, 1H, J=3.2 Гц, C1H), 8.34 (s, 1H, C5H триазол).
1-β-D-Рибофуранозил-1,2,4-триазол-3-карбоксамидин гидрохлорид (8). К суспензии 10 (4,0 г, 11,4 ммоль) в метаноле (100 мл) добавляли молярный раствор метанольного метоксида натрия (12 мл), и эту смесь перемешивали при комнатной температуре в течение ночи. Этот раствор подкисляли до рН 4 промытой метанолом смолой Dowex H+, смолу отфильтровывали, а фильтрат концентрировали до сухости в вакууме. Остаток растворяли в минимальном количестве метанола (15 мл) и переносили в сосуд под давлением. Добавляли хлорид аммония (0,61 г, 11,4 ммоль) и раствор метанола, насыщенный при 0°С сухим газообразным аммиаком (75 мл), сосуд запаивали, и этот раствор перемешивали при комнатной температуре в течение ночи. Этот раствор концентрировали до сухости в вакууме, и полученный в результате остаток кристаллизовали из ацетонитрила/этанола с получением 8 в виде кристаллического твердого вещества (2,95 г, 93%). Данный образец был идентичен во всех отношениях аутентичному образцу.
В некоторых фармацевтических лекарственных формах предпочтительной является пролекарственная форма соединений, особенно включая ацилированные (ацетилированные или другие) производные, пиридиновые сложные эфиры и различные солевые формы настоящих соединений, и эту форму можно вводить по способу лечения состояния пациента. Обычному специалисту в данной области техники будет понятно, как проще модифицировать настоящие соединения до пролекарственных форм, чтобы способствовать доставке активных соединений к сайту-мишени в пределах организма-хозяина или пациента. Обычному специалисту в данной области техники также будет понятно преимущество благоприятных фармакокинетических параметров пролекарственных форм, где они применимы, при доставке настоящих соединений к сайту-мишени в пределах организма-хозяина или пациента, чтобы максимизировать предполагаемый эффект этого соединения.
Рассматриваемый пример образования пролекарственной формы соединений, раскрытых здесь, является следующим. Одним из простейших пролекарств Viramidine™ является три-O-ацетильное производное Viramidine™. Это три-O-ацетильное производное получают, как изображено на схеме 1:
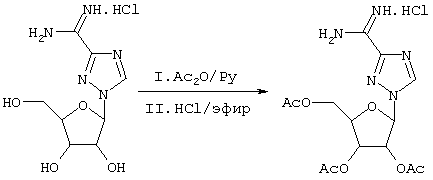
Схема 1
5'-Ретиноильное производное Viramidine™ является другой простой пролекарственной формой и его получают следующим образом:

Схема 2
Другие 5'-производные Viramidine™ включают в себя следующие производные, показанные на схеме 3:
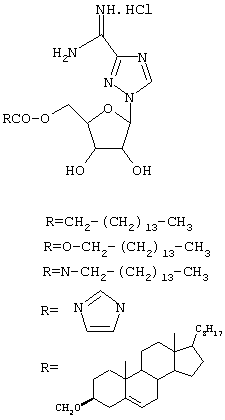
Схема 3
Большинство этих соединений можно получить, как описано (С. Sergheraert, С. Pierlot, A. Tartar, Y. Henin, M. Lemaitre, J. Med. Chem., 36, 826-830, 1993).
Синтез пролекарственной формы Viramidine™ на основе кумарина можно осуществить следующим образом:
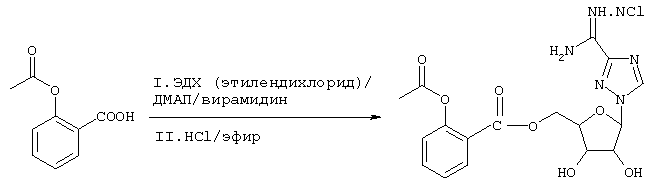
Схема 4
Эфиры аминокислот считаются лучшими пролекарственными формами в связи с возможным вовлечением стереоселективного переносчика. Аминокислотные производные Viramidine™ можно синтезировать, как показано ниже:
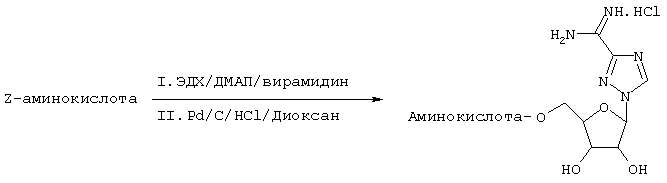
Схема 5
Для специфичной доставки лекарств в печень и билиарную систему привлекательным кандидатом является эндогенная транспортная система желчных кислот. Синтез конъюгатов Viramidine™ с желчными кислотами можно осуществить, как представлено ниже:

Схема 6
Другим классом пролекарств или пролекарственных форм являются нуклеотидные производные. Получение защищенных 5'-монофосфатных производных показано ниже. Посредством защиты отрицательных зарядов фосфатов нейтральными заместителями будут образовываться более липофильные производные, которые, как ожидают, будут превращаться обратно в соответствующие монофосфаты сразу при попадании внутрь клетки.

Схема 7
R1 представляет собой алкильные группы, такие как СН3С(O)S-СН2СН2-; (СН3)2СНС(O)S-СН2СН2-; (СН3)3СС(O)S-СН2СН2-; (СН3)3СС(O)ОСН2-; С6Н5С(O)S-СН2СН2- или HOCH2CH2SS-CH2CH2-.
Фосфорамидаты аминокислот представляют собой другой класс пролекарств, которые можно синтезировать, как описано ниже:
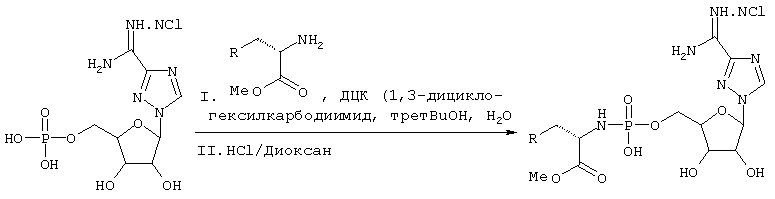
R=любой, за исключением водорода
Схема 8
Другие производные монофосфатных пролекарств представлены ниже:

R = алкил, липиды, витамины, желчные кислоты, производные холестерина
Схема 8А
Пролекарства Viramidine™ на основе салицилата можно получить по следующей схеме:
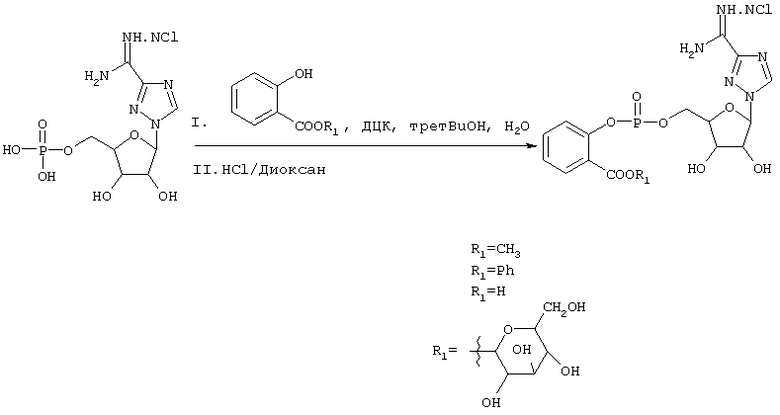
Схема 9
Пролекарства нуклеозид-5'-ди- или трифосфатов будут более интересны, поскольку они будут обходить больше метаболических стадий.
Следующие соединения являются потенциальными нуклеотидными липофильными пролекарствами и их получают, как изображено ниже:

Схема 10

Схема 11
Следующие соединения представляют собой другой класс потенциального фосфонатного пролекарства Viramidine™.

Схема 12
Другие возможные пролекарства включают в себя возможные комбинации групп, показанных в патентных заявках РСТ WO 98/39342, WO 98/39343, WO 98/39344 и WO 99/45016.
Пролекарства Viramidine™ можно получить не только посредством модификации сахарного участка исходной молекулы, но также посредством получения производных амидиновой функциональной группы. Следующие соединения представляют собой несколько классов пролекарств, которые можно получить модификацией амидиновой группы, как описано ниже:
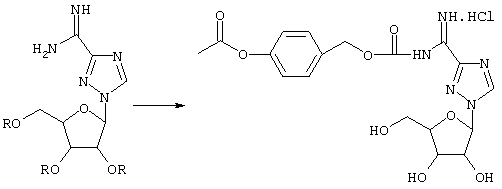
Схема 13

Схема 14

Схема 15
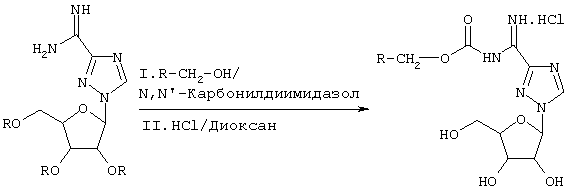
Схема 16
R=СН3-
R = фенил
R=R1-S-S-Ph- и
R1 = алкил, липиды, витамины, желчные кислоты, производные холестерина
Применение
Полагают, что соединения согласно формуле 1 будут применять для лечения широкого разнообразия состояний, и в действительности любого состояния, которое дает положительный ответ на введение одного или более чем одного из соединений. Среди прочего конкретно полагают, что соединения по изобретению можно применять для лечения инфекции, инвазии, рака или опухоли, либо аутоиммунного заболевания. Далее полагают, что соединения по изобретению можно применять для направленного лечения состояний или заболеваний в конкретных органах пациента, таких как печень или сердце.
Рассматриваемые инфекции, подлежащие лечению соединениями по настоящему изобретению, включают в себя респираторно-синцитиальный вирус (RSV), вирус гепатита В (HBV), вирус гепатита С (HCV), вирус простого герпеса типа 1 и типа 2, генитальный герпес, герпес роговицы, энцефалитный герпес, опоясывающий герпес, вирус иммунодефицита человека (ВИЧ), вирус гриппа А, вирус hantann (геморрагической лихорадки), вирус папилломы человека (HPV), корь и грибок.
Рассматриваемые инвазии, подлежащие лечению соединениями по настоящему изобретению, включают в себя протозойные инвазии, а также гельминтные и другие паразитарные инвазии.
Рассматриваемые раки и опухоли, подлежащие лечению, включают в себя те, которые вызваны вирусом, и в эффект может быть вовлечено ингибирование трансформации инфицированных вирусом клеток в неопластическое состояние, ингибирование распространения вирусов от трансформированных клеток к другим нормальным клеткам и/или остановка роста трансформированных вирусом клеток.
Рассматриваемые аутоиммунные и другие заболевания, подлежащие лечению, включают в себя артрит, псориаз, кишечное заболевание, ювенильный диабет, волчанку, рассеянный склероз, подагру и подагрический артрит, ревматоидный артрит, отторжение трансплантата, гигантоклеточный артериит, аллергию и астму.
Кроме того, другие рассматриваемые применения соединений согласно настоящему изобретению включают в себя применение в качестве промежуточных соединений при химическом синтезе других нуклеозидных или нуклеотидных аналогов, которые, в свою очередь, являются полезными в качестве терапевтических агентов или для других целей.
Еще в одном аспекте способ лечения млекопитающего включает в себя введение терапевтически и/или профилактически эффективного количества фармацевтического препарата, содержащего соединение по настоящему изобретению. В данном аспекте эффект может относится к модулированию некоторой части иммунной системы млекопитающего, особенно к модулированию профилей лимфокинов типа 1 и типа 2 относительно друг друга. Когда происходит модулирование лимфокинов типа 1 и типа 2, в частности полагают, что это модулирование может включать в себя либо супрессию лимфокинов как типа 1, так и типа 2, и более предпочтительно стимуляцию лимфокинов типа 1, либо относительное повышение ответа типа 1 по сравнению с ответом типа 2.
В частности, полагают, что Viramidine™ (1,39 мкг/мл) повышает экспрессию и синтез цитокинов типа 1 в (предпочтительно активированных) Т-лимфоцитах, и результаты различных экспериментов представлены на фиг.2-5. На фиг.2 представлено влияние 5 мкМ вирамидина (соединения согласно формуле 1), рибавирина и левовирина на синтез цитокинов типа 1 в SEB-активированных Т клетках человека (n=5 доноров), где вирамидин демонстрирует явное повышение ответа типа 1 по сравнению с контролем с триазолом. Фиг.3 представляет собой графическое представление эффекта вирамидина доза-ответ в диапазоне 0,625-10 мкМ в отношении синтеза цитокинов типа 1 в SEB-активированных (SEB, стафилококковый энтеротоксин В) Т клетках человека (данные представляют 4 индивидуальных доноров). Эффект in vivo, проявляющийся в повышенном ответе типа 1 в анализе контактной гиперчувствительности (КГЧ) вирамидина ясно продемонстрирован на фиг.4, а на фиг.5 показано сравнение между вирамидином и левовирином/рибавирином в отношении нуклеозидной концентрации пикового ответа и пикового диапазона ответов (на оси у изображено число респондентов в конкретном эксперименте).
Получение препарата Т клеток человека и активация in vitro
Мононуклеарные клетки периферической крови выделяли от здоровых доноров или пациентов с ревматоидным артритом с помощью центрифугирования в градиенте плотности с последующим Т-клеточным обогащением с использованием Lymphokwik (One Lambda, Canoga Park CA). Контаминирующие моноциты удаляли путем прилипания к пластику. Очищенные Т клетки представляли собой >99% CD2+, <1% HLA-DR+ и <5% CD25+, и их поддерживали в среде RPMI-AP5 (среда RPMI-1640, содержащая 20 мМ буфера HEPES, рН 7,4, 5% аутологичной плазмы, 1% L-глутамина, 1% пенициллина/стрептомицина и 0,05% 2-меркаптоэтанола).
Для определения уровней цитокинового белка Т клетки (1×106 клеток в объеме 1 мл) активировали добавлением 10 нг ФМА (форбол-12-миристат-13-ацетат) плюс 0,5 мкг иономицина (оба реагента от Calbiochem, La Jolla, CA) и инкубировали в 24-луночных планшетах в присутствии от 0 до 20 мкМ нуклеозида в течение промежутка времени до 48 ч при 37°С и 5% CO2 в увлажненном инкубаторе. После активации супернатанты анализировали на продуцирование цитокинов клеточного происхождения. Для исследований пролиферации и жизнеспособности вышеуказанный протокол модифицировали на 96-луночный формат планшета, используя 0,2×106 клеток в объеме 0,2 мл и активацию 2 нг ФМА и 0,1 мкг иономицина. В отдельных экспериментах 5×106 Т-клеток в 2 мл активировали 20 нг ФМА плюс 1 мкг иономицина. Альтернативно клетки можно активировать in vitro с помощью SEB, следуя опубликованным методикам. Здесь суммарную РНК выделяли из Т клеток после 6-24 ч инкубации и анализировали с помощью ОТ-ПЦР для определения уровней мРНК различных цитокинов и медиаторов воспаления. Также в отдельных экспериментах Т клетки человека дополнительно очищали (используя реагенты обогащения клеток от Stem Cell Technologies, Vancouver, ВС) с получением чистых популяций подгрупп Т клеток CD4+ (<1% CD8+ при использовании реагента для выделения Т клеток человека CD4+ RosetteSep) и CD8+ (<1% CD4+ при использовании реагента для выделения Т клеток человека CD4+ RosetteSep), после чего 1×106 клеток на мл активировали ФМА и иономицином, как в экспериментах с суммарными Т клетками.
Анализ внеклеточных цитокинов
Уровни цитокинов человека определяли в клеточных супернатантах после соответствующего разведения, используя наборы для ELISA (иммуноферментный твердофазный анализ), специфичные для ИЛ-2, ИФНγ, ФНОα, ИЛ-4 и ИЛ-5 (Biosource International, Camarillo, CA). Уровни цитокинов мышей определяли, используя наборы для ELISA, специфичные для мышиных ИФНγ и ИЛ-4 (R и D Systems, Minneapolis, MN). Все результаты ELISA выражали в пг/мл. Некоторые данные представлены в виде процента от активированного контроля, вычисленного как отношение уровня цитокинов активированных Т клеток в присутствии тестируемого нуклеозида к уровню цитокинов необработанных активированных Т клеток × 100%. Нулевой эффект тестируемых нуклеозидов по отношению к уровням цитокинов будет давать процент от значения активированного контроля, равный 100%. Альтернативно данные представляли в виде процентного изменения от активированного контроля ([(тест пг/мл - активированный контроль пг/мл)/активированный контроль пг/мл]×100%). Нулевой эффект тестируемых нуклеозидов по отношению к уровням цитокинов будет составлять 0%.
Контактная гиперчувствительность (КГЧ)
Реактивность на контактный аллерген, ДНФБ (динитрофторбензол), определяли у мышей BALB/c, как описано ранее (Ishii, N., К. Takahashi, Н. Nakajima, S. Tanaka, P. W. Askenase, 1994. DNFB contact sensitivity (CS) in BALB/c mice. J. Invest. Dermatol. 102: 321). Коротко, мышей подвергали сенсибилизации нанесением 20 мкл 0,3% ДНФБ в ацетоне: оливковом масле, 4:1, на бритое брюшко интактных мышей. Для оптимального проявления КГЧ мышам проводили стимуляции на обеих сторонах каждого уха 20 мкл 0,12%-ного ДНФБ через 5 суток после сенсибилизации. Несенсибилизированным мышам также проводили стимуляции и использовали их в качестве контролей в каждом эксперименте. Через 24 ч проводили измерения толщины уха, и ответ на ДНФБ оценивали путем вычитания значений после стимуляции из значений до стимуляции. Где указано, вводили 7-β-D-рибофуранозил-4-оксопирроло[2,3-d]пиримидин-5-карбоксамидин в дозе 6,2 мкг в 50 мкл ЗФР (забуференный фосфатом физиологический раствор) (0,3 мг/кг) или 12,4 мкг в 100 мкл ЗФР (0,6 мг/кг) путем внутрибрюшинной инъекции в момент стимуляции с ДНФБ. Эти дозы 7-β-D-рибофуранозил-4-оксопирроло[2,3-d]пиримидин-5-карбоксамидина давали максимальный эффект в предварительных исследованиях оптимизации. После конечных измерений толщины уха мышей умерщвляли цервикальной дислокацией и извлекали подмышечные/латеральные подмышечные лимфатические узлы. После выделения суммарной клеточной РНК из выделенных клеток лимфатических узлов проводили ОТ-ПЦР и Саузерн-блот анализы для мониторинга уровней ИФНγ, ИЛ-2 и ИЛ-10 мРНК у мыши.
Дополнительные эксперименты
В целом полагают, что сдвиг иммунного ответа в направлении ответа типа 1 является благоприятным. Следовательно, полагают, что соединения, являющиеся объектом изобретения, могут быть особенно полезны в лечении вирусных заболеваний (предпочтительно вирусных инфекций, при которых ответ типа 1 снижен или супрессирован). Чтобы подтвердить эффективность модулирования иммунного ответа, провели различные эксперименты, и нижеследующее представляет собой примерное краткое изложение некоторых экспериментов, проведенных с рассматриваемыми соединениями:
In vitro - вирамидин ингибировал вирусную инфекцию Punta Того LLC-МК2 (клетки почек обезьяны резуса) с EC50, равной 8 мг/мл (штамм Adames) и 12 мг/мл (штамм Balliet), - CC50 составляла 320 мг/мл (нормирование по вирусу 1,0-1,2).
In vivo - введение вирамидина подкожно или перорально приводило в результате к 100%-ной выживаемости (10 C57BL/6 мышей на группу) при подкожной инъекции PTV (штамм Adames).
В течение 24 ч после инфекции PTV in vivo минимально эффективная подкожная доза составляла 32 мг/кг для рибавирина, а для вирамидина она составляла 96 мг/кг, даваемых подкожно дважды в день в течение 5 суток. В течение 24 ч после инфекции PTV in vivo минимально эффективная пероральная доза составляла 20 мг/кг для рибавирина и 40 мг/кг для вирамидина, даваемых подкожно дважды в день в течение 5 суток.
В целом наиболее предпочтительными применениями согласно настоящему изобретению являются те, при которых активные соединения являются относительно менее цитотоксичными по отношению к клеткам-хозяевам, не являющимся мишенями, и относительно более активными против мишени. В этом отношении также может быть предпочтительным, что L-нуклеозиды могут обладать повышенной стабильностью по сравнению с D-нуклеозидами, что может привести к лучшей фармакокинетике. Этот результат может быть достигнут, поскольку L-нуклеозиды могут не распознаваться ферментами и, следовательно, они могут иметь более длительные периоды полураспада.
Полагают, что соединения по настоящему изобретению будут вводить in vivo, in vitro или ex vivo в любом подходящем фармацевтическом препарате и согласно любому подходящему протоколу. Таким образом, введение может иметь место перорально, парентерально (включая подкожные инъекции, внутривенные, внутримышечные, введение путем интрастернальной инъекции или инфузионных методик), посредством ингаляционного распыляемого раствора, либо ректальным, местным путем и так далее, и в препаратах стандартных форм, содержащих общепринятые нетоксичные фармацевтически приемлемые носители, адъюванты и растворители.
Например, полагают, что соединения согласно настоящему изобретению можно включать в препараты в смеси с фармацевтически приемлемым носителем. Например, соединения по настоящему изобретению можно вводить перорально в виде фармакологически приемлемых солей. Поскольку соединения по настоящему изобретению являются в основном растворимыми в воде, их можно вводить внутривенно в физиологическом солевом растворе (то есть забуференном до рН примерно от 7,2 до 7,5). Для этой цели можно использовать общепринятые буферы, такие как фосфаты, бикарбонаты или цитраты. Конечно, обычный специалист в данной области техники может модифицировать препараты в пределах указаний данного описания, чтобы обеспечить различные препараты для конкретного пути введения, не делая композиции по настоящему изобретению нестабильными или не подвергая риску их терапевтическую активность. В частности, модификацию настоящих соединений, например, для того, чтобы сделать их более растворимыми в воде или в другом носителе, в частности, можно легко осуществить с помощью незначительных модификаций (образования соли, этерификации и так далее), которые находятся в пределах компетентности обычного специалиста в данной области техники. Также в пределах компетентности обычного специалиста в данной области техники находятся модификации пути введения и режима дозировки конкретного соединения с целью управления фармакокинетикой настоящих соединений для достижения максимально полезного эффекта у пациентов.
Кроме того, соединения согласно по настоящему изобретению можно вводить отдельно или в комбинации с другими агентами для лечения вышеуказанных инфекций или состояний. При комбинационных терапиях согласно настоящему изобретению вводят по меньшей мере одно соединение по настоящему изобретению или его функциональное производное и по меньшей мере один другой фармацевтически активный ингредиент. Этот активный ингредиент (ингредиенты) и фармацевтически активные агенты можно вводить отдельно или вместе, и раздельное введение можно осуществлять одновременно или по отдельности в любом порядке. Количества активного ингредиента (ингредиентов) и фармацевтически активного агента (агентов) и относительные хронометражи введения следует выбирать таким образом, чтобы достичь желаемого комбинированного терапевтического эффекта. Предпочтительно комбинационная терапия включает в себя введение одного соединения по настоящему изобретению или его физиологически функционального производного и одного из агентов, упомянутых здесь ниже.
Примерами других лекарств или активных ингредиентов, предполагаемых как эффективные при комбинировании с модулятором согласно формуле 1, являются противовирусные агенты, такие как интерферон, включая, но не ограничиваясь ими, интерферон α и γ, рибавирин, ацикловир и AZT™; противогрибковые агенты, такие как толнафтат, фунгизон (Fungizone™), лотримин (Lotrimin™), мицелекс (Mycelex™), нистатин и амфотерацин; противопаразитарные агенты, такие как минтезол (Mintezol™), никлоцид (Niclocide™), вермокс (Vermox™) и флагил (Flagyl™), кишечные агенты, такие как иммодиум (Immodium™), ломотил (Lomotil™) и фазим (Phazyme™); противоопухолевые агенты, такие как интерферон α и γ, адриамицин (Adriamycin™), цитоксан (Cytoxan™), имуран (Imuran™), метотрексат, митрацин (Mithracin™), тиазофурин (Thiazofurin™), таксол (Taxol™); дерматологические агенты, такие как акловат (Aclovate™), циклокорт (Cyclocort™), денорекс (Denorex™), флорон (Floron™), оксорален (Oxsoralen™), каменноугольный деготь и салициловая кислота; противомигреневые препараты, такие как эрготаминовые соединения; стероиды и иммуносупрессанты, не перечисленные выше, включая циклоспорины, дипрозон (Diprosone™), гидрокортизон; флорон (Floron™), лидекс (Lidex™), топикорт и вализон; и метаболические агенты, такие как инсулин, а также другие лекарственные средства, которые точно не попадают в вышеуказанные категории, включая цитокины, такие как ИЛ2, ИЛ4, ИЛ6, ИЛ8, ИЛ10 и ИЛ12. Особенно предпочтительными первичными лекарствами являются AZT, 3ТС, 8-замещенные гуанозиновые аналоги, 2,3-дидезоксинуклеозиды, интерлейкин II, интерфероны, такие как IαB-интерфероны, тукарезол (tucaresol), левамизол, изопринозин и циклолигнаны.
Примеры таких дополнительных терапевтических агентов включают в себя агенты, которые являются эффективными для модулирования иммунной системы или связанных с ней состояний, такие как AZT, ЗТС, 8-замещенные гуанозиновые аналоги, 2',3'-дидезоксинуклеозиды, интерлейкин II, интерфероны, такие как α-интерферон, тукарезол, левамизол, изопринозин и циклолигнаны. Некоторые соединения согласно настоящему изобретению могут быть эффективными для усиления биологической активности некоторых агентов согласно настоящему изобретению посредством снижения метаболизма или инактивации других соединений, а также как таковые, и их вводят совместно для данного предназначенного эффекта.
В отношении дозировки обычному специалисту в данной области техники будет понятно, что терапевтически эффективное количество будет варьировать в зависимости от инфекции или состояния, которые нужно лечить, тяжести этого состояния, режима лечения, который следует применять, фармакокинетики применяемого агента, а также от пациента (животного или человека), которого лечат. Полагают, что различные альтернативные дозировки также являются подходящими, включая дозировки между 0,5 мг/кг и 0,1 мг/кг и менее, но также дозировки между 0,5 мг/кг и 1,0 мг/кг и более. Далее полагают, что, хотя при некоторых болезненных состояниях успеха лечения можно достичь при относительно низких концентрациях рассматриваемых соединений в плазме, при других вирусных инфекциях могут быть необходимы относительно высокие дозировки. Однако полагают, что подходящий режим следует разрабатывать посредством введения небольшого количества, а затем повышения этого количества до тех пор, пока побочные эффекты не станут чрезмерно вредными, либо пока предназначенный эффект не будет достигнут.
Введение активного соединения может варьировать в диапазоне от непрерывного введения (внутривенное капельное вливание) до нескольких пероральных введений в сутки (например, четыре раза в сутки) и может включать в себя среди прочих путей введения пероральное, местное, парентеральное, внутримышечное, внутривенное, подкожное, чрескожное (которое может включать в себя агент, усиливающий проникновение), трансбуккальное введение и введение суппозиториев.
Для изготовления фармацевтических композиций согласно настоящему изобретению терапевтически эффективное количество одного или более чем одного из соединений согласно настоящему изобретению предпочтительно тщательно смешивают с фармацевтически приемлемым носителем согласно общепринятым методикам фармацевтического компаундирования для получения дозы. Носитель может принимать широкое разнообразие форм в зависимости от формы препарата, желаемой для введения, например пероральной или парентеральной. При изготовлении фармацевтических композиций в пероральной лекарственной форме можно использовать любую из обычных фармацевтических сред. Так, для жидких пероральных препаратов, таких как суспензии, эликсиры и растворы, можно использовать подходящие носители и добавки, включая воду, гликоли, масла, спирты, корригенты, консерванты, красящие агенты и тому подобное. Для твердых пероральных препаратов, таких как порошки, таблетки, капсулы, и для таких твердых препаратов, как суппозитории, можно использовать подходящие носители и добавки, включая крахмалы, сахарный носитель, такой как декстроза, маннит, лактоза и родственные носители, разбавители, гранулирующие агенты, смазывающие агенты, связывающие агенты, разрыхляющие агенты и тому подобное. Если желательно, таблетки или капсулы с помощью стандартных методик могут быть покрыты энтеросолюбильной оболочкой или изготовлены в форме продолженного высвобождения.
Носитель для парентеральных препаратов обычно будет содержать стерильную воду или водный раствор хлорида натрия, хотя можно включать другие ингредиенты, включая те, которые способствуют диспергированию. Конечно, там, где нужно использовать стерильную воду и поддерживать ее в стерильном состоянии, композиции и носители также должны быть стерилизованными. Можно также готовить инъецируемые растворы, и в этом случае можно использовать подходящие жидкие носители, суспендирующие агенты и тому подобное.




Изобретение относится к области медицины и касается способа лечения инфекции вирусом гепатита С (HCV) или инфекции вирусом гепатита В (HBV), при котором вводят карбоксамидин формулы 1 или его фармацевтически приемлемую соль в дозе 0,1-40,0 мг/(кг массы тела), при этом также вводят α-интерферон. Соединение формулы 1 находится в D-конфигурации. Способ по данному изобретению позволяет эффективно лечить инфекцию вирусом гепатита С и гепатита В. 6 з.п. ф-лы, 5 ил.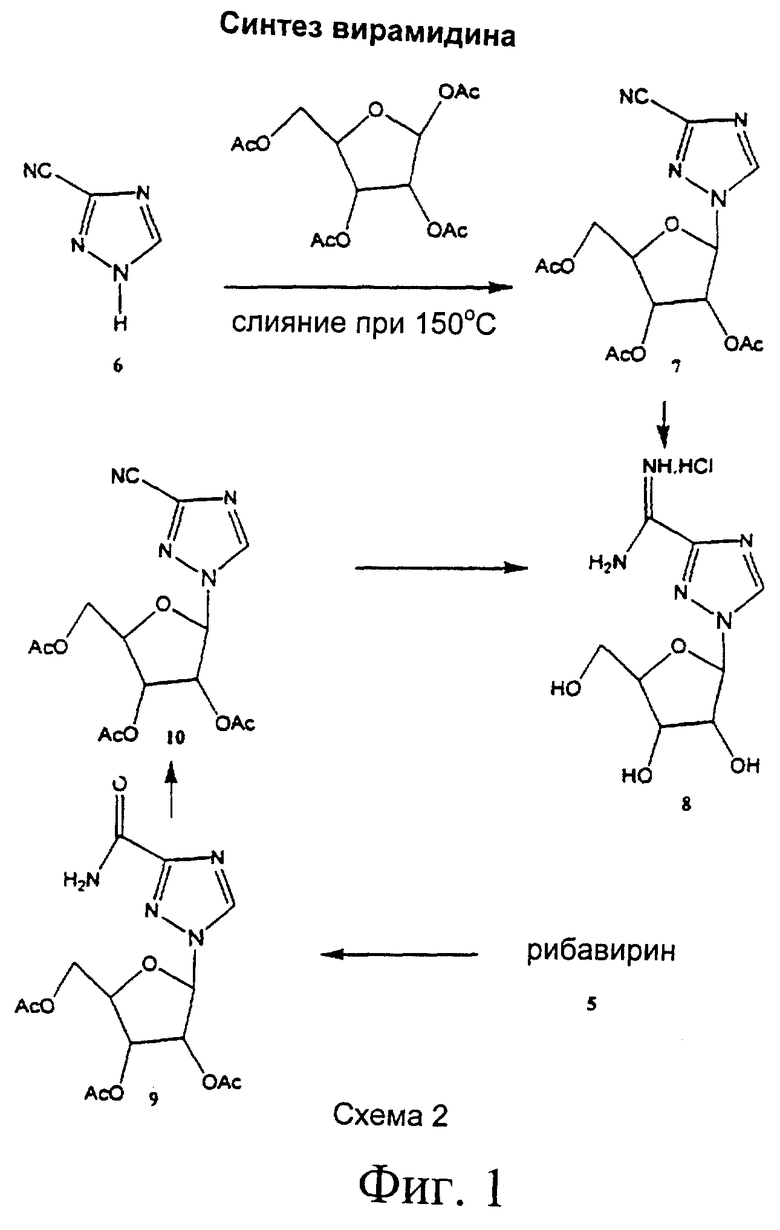

где это соединение находится в D-конфигурации.
| Автореферат АБД Medline | |||
| Sidwell R.W., Huffman J.H | |||
| Effects of ribamidine, a 3-carboxamidine derivative of ribavirine on experimentally induced Phlebovirus infections | |||
| Antiviral | |||
| Res | |||
| Механическая топочная решетка с наклонными частью подвижными, частью неподвижными колосниковыми элементами | 1917 |
|
SU1988A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Автореферат АБД Medline | |||
| Sidwell R.W., Huffman J.H | |||
| Effects of ribamidine, a 3-carboxamidine derivative of ribavirine | |||

