Настоящее изобретение относится к ацилированным нуклеозидам, которые представляют собой пролекарства-предшественники ингибитора РНК-зависимой полимеразы вирусной РНК вируса гепатита С (HCV). При введении этих соединений оральным путем они легко абсорбируются из желудочно-кишечного тракта и эффективно превращаются в крови в исходную форму нуклеозида. Эти лекарства являются ингибиторами репликации РНК-зависимой вирусной РНК и используются в качестве ингибиторов HCV NS5B полимеразы, в качестве ингибиторов HCV репликации и для лечения гепатита С у млекопитающих.
Изобретение относится к нуклеозидным пролекарствам, которые являются ингибиторами HCV репликации. В частности, изобретение касается применения ацилированных производных пиримидиновых нуклеозидов, которые обеспечивают улучшенную абсорбацию лекарства при оральном введении нуклеозида.
Вирус гепатита С во всем мире представляет собой значительную проблему для здоровья и является основной причиной хронических заболеваний печени (Воуеr N. et al., J. Hepatol. 2000, 32:98-112). Для пациентов, инфицированных HCV, существует угроза развития цирроза печени и затем гепатоцеллюлярной карциномы, поэтому HCV является основным фактором для пересадки печени.
Согласно данным Всемирной Организации Здравоохранения во всем мире инфицировано 200 миллионов человек и каждый год инфицируется, по крайней мере, 3-4 миллиона человек. Около 20% инфицированных лиц побеждают инфекцию и освобождаются от вируса, а остальные могут оставаться носителями вируса HCV всю оставшуюся жизнь. У 10-20% инфицированных людей в конце концов развиваются цирроз или рак, разрушающие печень. Заражение вирусом происходит парентерально при использовании зараженной крови или ее составляющих (продуктов), инфицированных матерей или матерей-носителей к их потомству. Современное лечение HCV инфекций, которое сводится к иммунотерапии с помощью рекомбинантного интерферона-α самого по себе или в сочетании с нуклеозидным аналогом рибавирином, приносит ограниченную лечебную пользу, так как быстро развивается невосприимчивость. Таким образом, существует неотложная потребность в улучшенных терапевтических средствах, которые эффективно побороли бы хроническое HCV заболевание.
Согласно классификации HCV являются членом семейства вирусов Flaviviridae, которое включает роды flaviviruses, pestiviruses и hepaciviruses, которые включают вирусы гепатита С (Rice, RAU/02.10.2006 C.M.Flaviviridae: The viruses and their replication, b: Fields Virology, Editors: Fields B.N., Knipe D.M., Howley P.M., Lippincott-Raven Publishers, Philadelphia, Pa., Chapter 30, 931-959, 1996). HCV представляет собой оболочечный вирус, содержащий геном из позитивно направленной одноцепочечной РНК в приблизительно 9,4 кб. Геном вируса состоит из 5′-нетранслируемого региона (UTR), протяженной открытой рамки считывания (ORF), кодирующей предшественник полипротеина приблизительно из 3011 аминокислот, и короткий 3′ UTR. 5′ UTR является самой высококонсервативной частью генома HCV и важна для инициации и контроля трансляции полипротеина.
Генетическим анализом HCV было определено шесть основных генотипов, показывающих>30% отличий в последовательностях ДНК. Каждый генотип включает серию нескольких близкородственных подтипов, которые показывают 20-25% отличий в нуклеотидных последовательностях (Simmonds P., 2004, J. Gen. Vilor., 85:3173-88). Более 80 подтипов уже охарактеризованы. В США примерно 70% инфицированных заражено типом 1а и 1b. Тип 1b - наиболее распространенный подтип в Азии. (X. Forns, J. Bukh, Clinics in Liver Disease, 1999, 3:6930716; J. Bukh et al., Semin. Liv. Dis. 1995, 15:41-63). К сожалению, заражения Типом 1 более устойчивы к терапии, чем генотипы Типа 2 или 3 (N.N.Zein, Clin. Microbiol. Rev., 2000, 13:223-235).
Генетическая организация и полипротеиновый процессинг части ORF, относящейся к неструктурному белку, у пестивирусов и гепатовирусов очень похожи. Эти вирусы с положительно-направленными цепочками РНК имеют одну обширную ORF, кодирующую все вирусные белки, необходимые для вирусной репликации. Эти белки экспрессируются как полипротеин, который ко- и посттрансляционно создается как клеточными, так и кодируемыми вирусом протеиназами с получением в результате зрелых вирусных белков. Эти вирусные белки, которые отвечают за репликацию вирусной геномной РНК, локализованы ближе к С-концу. Две третьих ORF называются неструктурными (NS) белками. И у пестивирусов, и у гепатовирусов в зрелых неструктурных (NS) белках на участке от аминоконца региона, кодирующего неструктурные белки, до карбоксиконца ORF, последовательно содержатся р7, NS2, NS3, NS4A, NS4B, NS5A и NS5B.
NS белки пестивирусов и гепатовирусов распределяются в последовательностях доменов, которые характерны для определенных функциональных белков. Например, NS3 белки вирусов обеих групп включают основные аминокислотные последовательности (мотивы), характерные для сериновых протеиназ и геликаз (Gorbalenya et al. Nature 1988, 333:22, Bazan and Fletterick, Virology, 1989, 171:637-639; Gorbalenya et al., Nucleic Acid Res. 1989, 17. 3889-3897). Аналогично NS5B белки пестивирусов и гепатовирусов несут последовательности, характерные для РНК-направленных РНК-полимераз (Koonin E.V., Dolja V.V., Grit. Rev. Biochem. Molec. Biol. 1993, 28:375-430).
Фактическое предназначение и функции NS белков пестивирусов и гепатовирусов в жизненном цикле вирусов, в основном, аналогичны. В обоих случаях NS3 сериновая протеиназа ответственна за протеолитический процессинг предшественников полипротеина ниже его положения в ORF (Wiskerchen, Collet, Virology, 1991, 184:341-350; Bartenschlager et al. J. Virol. 1993 67:3835-3844; Eckart et al. Biochem. Biophys. Res. Comm. 1993 192:399-406; Grakoui et al. J.Virol. 1993 67:2832-2843; Grakoui et al. Proc. Natl. Acad. Sci. USA 1993 90:10583-10587; Ilijikata et al. J.Virol. 1993 67:4665-4675; Tome et al. J. Virol. 1993 67:4017-4026). NS4A белок в обоих случаях действует как кофактор NS3 сериновой протеиназы (Bartenschlager et al. J. Virol. 1994 68:5045-5055; Failla et al. J. Virol. 1994 68:3753-3760; Xu et al. J. Virol. 1997 71:53 12-5322). NS3 белок обоих типов вирусов также функционирует как геликаза (Kim et al. Biochem. Biophys. Res. Comm. 1995 215:160-166; Jin, Peterson Arch. Biochem. Biophys. 1995, 323:47-53; Warrener, Collett J. Virol. 1995 69:1720-1726). Наконец, NS5B белки пестивирусов и гепатовирусов обладают предсказанной заранее активностью РНК-направленных РНК-полимераз (Behrens et al. ЕМВО 1996 15:12-22; Lechmann et al. J. Virol. 1997 71:8416-8428; Yuan et al. Biochem. Biophys. Res. Comm. 1997 232:231-235; Hagedom, PCTWO 97/12033; Zhong et al. J. Virol. 1998 72:9365-9369).
В настоящее время существует ограниченное количество одобренных терапевтических методов, которые повсеместно были бы признаны пригодными для лечения HCV инфекции. Новые и уже существующие терапевтические подходы для лечения HCV и ингибирования HCV NS5B полимеразы описаны в следующих работах: R.G.Gish, Sem. Liver. Dis., 1999 19:5; Di Besceglie, A.M. And Bacon, B. R., Scientific American, October: 1999 80-85; G. Lake-Bakaarm Current and Future Therapy for Chronic Hepatitis С Virus Liver Disease, Curr. Drug Targ Infect Dis. 2003 3(3):247-253; P. Hoffmann et al. Recent patents on experimental therapy for hepatitis С vims infection (1999-2002), Exp.Opin. Ther. Patents 2003 13(11):1707-1723; F. F. Poordad et al. Developments in Hepatitis С therapy drug during 2000-2002, Exp.Opin. Emerging Drugs 2003 8(1):9-25; M.P. Walker et al.. Promising Candidates for the treatment of chronic hepatitis C, Exp.Opin. Investig. Drugs 2003 12(8): 1269-1280; S. - L. Tan et al. Hepatitis С Therapeutics: Current Status and Emerging Strategies, Natire Rev. Drug Discov. 2002 1:867-881; R. De Francesco et al. Approaching a new era for hepatitis С virus therapy: inhibitors of the NS3-4A serine protease and the NS5B RNA-dependent RNA polymerase. Antiviral Res. 2003 58:1-16; Q.M. Wang et al. Hepatitis С virus encoded proteins: targets for antiviral therapy. Drugs of the Future 2000 25(9):933-8-944; J. A. Wu and Z. Hong, Targeting NS5B-Dependent RNA Polymerase forAnti-HCV Chemotherapy Cur. Dmg Targ.-Inf. Dis. 2003 3:207-219.
В этих работах упоминаются соединения сразу же по мере их создания. Сочетанная (комбинационная) терапия на основе двух или трех средств, направленных на одни и те же или различные цели, стала стандартной терапией для подавления или замедления распространения устойчивых штаммов вируса, а соединения, раскрытые в вышеупомянутых работах, могут использоваться при сочетанной терапии вместе с соединениями настоящего изобретения.
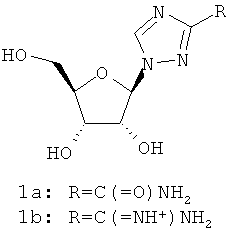
Рибавирин (1а; амид 1-((2R, 3R, 4S, 5R)-3,4-Дигидрокси-5-гидроксиметил-тетрагидро-фуран-2-ил)-1Н-[1,2,4]триазол-3-карбоновой кислоты; VIRAZOLE®) является синтетическим неиндуцирующим интерферон аналогом нуклеозида с антивирусной активностью широкого спектра действия. Рибавирин обладает in vitro активностью в отношении ряда ДНК- и РНК-вирусов, включая Flaviviridae (Gary L. Davis, Gastroenterology 2000 118:S104-S114). При монотерапии рибавирин снижает уровни сывороточной амино-трансферазы до нормального у 40% пациентов, но он не снижает в сыворотке уровни HCV-RNA. Рибавирин показывает также значительную токсичность, и известно, что он вызывает анемию. Рибавирин не утвержден к применению против HCV как средство монотерапии, но применение этого соединения одобрено в сочетанной терапии вместе с интерфероном α-2а и интерфероном α-2b. Вирамидин 1b является пролекарством, превращающимся в 1а в гепатоцитах.
Интерфероны (IFNs) уже применяются для лечения хронического гепатита в течение примерно десяти лет. IFNs представляют собой гликопротеины, вырабатываемые иммунными клетками в ответ на вирусное заражение. Распознают два различных типа интерферонов: Тип 1 включает несколько интерферонов альфа и один интерферон β, Тип 2 включает интерферон γ. Интерферон Типа 1 продуцируется, главным образом, инфицированными клетками и предохраняет соседние клетки от de novo заражения. IFNs ингибируют репликацию многих вирусов, включая HCV, и при использовании в качестве монотерапевтического средства для лечения гепатита С IFN подавляет сывороточную HCV-RNA до недетектируемых уровней.
Кроме этого, IFN нормализует уровни аминотрансферазы в сыворотке. К сожалению, действие IFN временное. Прекращение (перерыв) лечения приводит к 70% случаев рецидивов и только в 10-15% случаях наблюдается устойчивая длительная вирологическая реакция с нормальными уровнями сывороточной аланинтрансферазы (L. - B. Davis, см. выше).
Неким ограничением ранней IFN терапии является быстрое выведение (клиренс) белка из крови. Химическая дериватизация IFN с помощью полиэтиленгликоля (PEG) привела к существенно улучшенным фармакокинетическим свойствам белков. PEGASIS® представляет собой конъюгат интерферона α-2b и 12 кДа моно-метокси PEG. (В.A. Luxon et al., Clin. Therap.2002 24(9): 13631383; A. Kozlowski and J.M.Harris, J. Control. Release, 2001, 72:217-224).
Интерферон α-2а и интерферон α-2b недавно были одобрены как средства для монотерапии при лечении HCV, ROFERON-A® является рекомбинантной формой интерферона α-2а. PEGASIS® (Roche) является пэгилированной (т.е. модифицированной полиэтиленгликолем) формой интерферона α-2а. INTRON-A® (Sobering Corporation) является рекомбинантной формой интерферона α-2b, a PEG-INTRON® (Schering Corporation) является пэгилированной формой интерферона α-2b.
Другие формы интерферона α, так же, как и интерферона β, γ, τ и ω, в настоящее время находятся на стадии клинических испытаний в целях лечения HCV. Например, INFERGEN® (интерферон альфакон-1) от biterMune, OMNIFERON® (природный интерферон) от Viragen, ALBUFERON® от Human Genome Sciences, REBIF® (интерферон (3-1 а) от Ares-Serono, Omega Interferon от BioMedicine, Oral Interferon Alpha от Amarillo Bosciences, а также интерферон γ-1b от InterMune проходят испытания,
Сочетанная терапия HCV рибавирином и интерфероном-α в настоящее время показала себя как оптимальная терапия. Сочетание рибавирина и PEG-IFN (см. ниже) приводит к продолжительной реакции на вирус у 54-56% пациентов. SVR достигает 80% для Типа 2 и 3 HCV. (Walker, см. выше). К сожалению, это сочетание дает также и побочные эффекты, которые вызывают клинические проблемы. С подкожным введением IFN-a связывают такие симптомы, как депрессия, гриппоподобные симптомы и кожные реакции, а гемолитическую анемию связывают с рибавирином, вводимым непрерывно.
Другими макромолекулярными соединениями, которые проходят в настоящее время предклинические и клинические испытания, в плане лечения вирусного гепатита С, являются: Interleukin-10 от Schering-Plough, IPSO 1 от bitermenron, Merimebodlib (VX-497) от Vertex, HEPTAZYME® от RPI, IDN-6556 от Idun Pharma, XTL-002 от XTL, HCV/MFS9 от Chiron, CIVACIR® (иммуноглобулин к гепатиту С) от NABI, ZADAXIN® (тимозин α-1) от SciClone, тимозин плюс пегилированный интерферон от SciClone, CEPLENE®; терапевтическая вакцина от Epimmune/Genencor, терапевтическая вакцина от Merix, терапевтическая вакцина, ChronVacC, от Tripep.
Другие микромолекулярные методы включают рибозимы, нацеленные на HCV RNA. Рибозимы - это небольшие молекулы природного происхождения с эндонуклеазной активностью, которые катализируют секвенс-специфичное расщепление РНК. Альтернативный подход заключается в применении антисмысловых олигонуклеотидов, связывающих РНК и стимулирующих опосредованное РНК-азойН расщепление.
В настоящее время уже идентифицирован ряд потенциальных молекулярных мишеней для разработки и испытания их в качестве анти-HCV терапевтических средств, например, NS2-NS3 аутопротеаза, N3 протеаза, N3 геликаза и NS5B полимераза. РНК-зависимая РНК-полимераза абсолютно необходима для репликации одноцепочечной смысловой РНК генома, и поэтому этот энзим вызывает значительный интерес у медиков и фамацевтов.
Нуклеозидные ингибиторы NS5B полимеразы могут действовать либо как субстрат неприродного происхождения, что приводит в результате к терминации цепи, либо как конкурентный ингибитор, который конкурирует со связыванием нуклеотида с полимеразой. Для того чтобы функционировать как терминатор цепи, нуклеозидный аналог должен быть поглощенным клеткой и превращенным in vivo в трифосфат, чтобы конкурировать за сайт нуклеотидного связывания полимеразы. Эта конверсия в трифосфат обычно происходит при участии клеточных киназ, что обуславливает дополнительные структурные требования к потенциальному нуклеозидному ингибитору полимеразы. К сожалению, это ограничивает прямую оценку нуклеозидов как ингибиторов HCV репликации в исследованиях, проводимых на клетках, поддающихся in situ фосфорилированию.
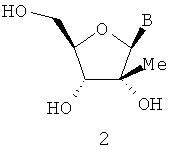

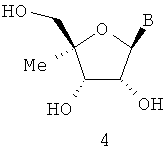
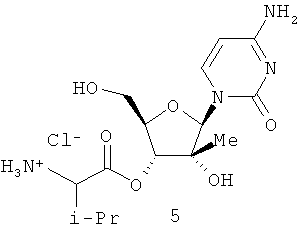
В=аденин, тимидин, урацил, цитидин, гуанин и гипоксантин.
В WO 0190121, опубликованной 29 ноября 2001 г., J.-P.Sommadossi и Р.Lacolla описали и дали примеры активности анти-HCV полимеразы в части 1′-алкил- и 2′-алкилнуклеозидов формул 2 и 3. В WO 01/92282, опубликованной 6 декабря 2001, J.-P.Sommadossi и Р.Lacolla раскрыли и описали примеры лечения Flaviviruses и Pestiviruses 1′-алкил- и 2′-алкилнуклеозидами формул 2 и 3. В WO 03/026675, опбликованной 3 апреля 2003 г., G. Gosselin описал применение 4′-алкилнуклеозидов формулы 4 для лечения Flaviviruses и Pestiviruses.
В WO 2004003000, опубликованной 8 января 2004 г., J.-P.Sommadossi et al. описали 2′- и 3′-пролекарства на основе 1′-, 2′-, 3′- и 4′-замещенных β-D и β-L-нуклеозидов. В WO 2004/002422, опубликованной 8 января 2004 г., описан 2′-С-метил-3′-O-валиновый эфир рибофуранозилцитидина для лечения заражения Flaviviridae. Idenix сообщил о клинических испытаниях родственного соединения NM283, которое, как полагают, является эфиром валина 5 и аналога 2 цитидина (В=цитозин). В WO 2004/002999, опубликованной 8 января 2004 г., J.-P.Sommadossi et al. описали серию 2′ или 3′ пролекарств из 1′, 2′, 3′ или 4′-разветвленных нуклеозидов для лечения заражений флавивирусами, в том числе HCV инфекций.
В WO 2004/046331, опубликованной 3 июня 2004 г., J.-P.Sommadossi et al. описали 2′-разветвленные нуклеозиды и мутацию Flaviviridae. В WO 03/026589, опубликованной 3 апреля 2003 г., G.Gosselin et al. описали способы лечения вирусного гепатита C с помощью 4′-модифицированных нуклеозидов. В WO 2005009418, опубликованной 3 февраля 2005 г., R.Storer et al. описали пуриновые нуклеозидные аналоги для лечения заболеваний, вызываемых, в том числе, Flaviviridae.
Другие патентные заявки раскрывают применение определенных нуклеозидных аналогов для лечения вирусного гепатита С. В WO 01/32153, опубликованной 10 мая 2001 г., R.Storer описал производные нуклеозидов для лечения вирусных заболеваний. В WO 01/60315, опубликованной 23 августа 2001 г., Н.Ismaili et al. описали способы лечения или профилактики заражений, вызванных Flaviviruses, с помощью производных нуклеозидов. В WO 02/18404, опубликованной 7 марта 2002 г., R.Devos et al. описали 4′-замещенные нуклеозиды для лечения вирусного HCV. В WO 01/79246, опубликованной 25 октября 2001 г., К.A.Watanabe описал производные 2′ или 3′-гидроксиметил-нуклеозидов для лечения вирусных заболеваний. В WO 02/32920, опубликованной 25 апреля 2002 г., и в WO 02/48165, опубликованной 20 июня 2002 г., L.Stuyver et al. описали производные нуклеозидов для лечения вирусных заболеваний.

В WO 03/105770, опубликованной 24 декабря 2003 г., В. Bhar et al. описали ряд карбоциклических нуклеозидных производных, которые ингибируют РНК-зависимую РНК-полимеразу вируса. Нуклеозиды, раскрытые в этой публикации, являются, в основном, 2′-метил-2′-гидроксизамещенными нуклеозидами. В WO 2002/057425, опубликованной 25 июля 2002 г., S.S.Carroll et al. описали производные нуклеозидов, которые ингибируют РНК-зависимую вирусную полимеразу, и способы лечения HCV заболеваний.
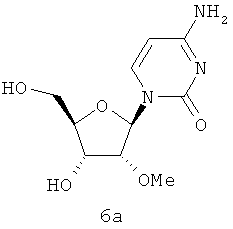
В WO 02/057287, опубликованной 25 июля 2002 г., S.S.Carroll et al. описали родственные 2α-метил и 2β-метилрибозные производные, в которых основания необязательно замещены 7Н-пирроло[2,3-(1]пиримидиновым радикалом 6. В той же самой заявке приведен один пример на 3β-метил-нуклеозид. S.S.Carroll et al. (J.Biol.Chem. 2003 278(14):11979-11984), описали ингибирование полимеразы HCV 2′-O-метилцитидином (6а). В WO 2004/009020, опубликованной 29 января 2004 г., D.В.Olsen et al. описали ряд тионуклеозидных производных как ингибиторов РНК-зависимой РНК-полимеразы вируса.
РСТ публикация №WO 99/43691, на имя Emory University, озаглавленная «2′-Фторнуклеозиды», описывает применение определенных 2′-фторнуклеозидов для лечения HCV. US патент №6348587, выданный Emory University, озаглавленный «2′-Фторнуклеозиды», раскрывает ряд семейств 2′-фторнуклеозидов, полезных при лечении гепатита В, HCV, HIV и анормальной клеточной пролиферации. Обе конфигурации 2′-фторзаместителя раскрыты.
Eldrup et al. (Oral Session V, Hepatitis С Virus, Flaviviridae; 16th International Conference on Antiviral Research (Apr. 27, 2003, Savannah, Ga.)) охарактеризовали взаимосвязь между структурой и активностью для 2′-модифицированных нуклеозидов в плане ингибирования HCV.
Bhat et al. (Oral Session V, Hepatitis С Virus, Flaviviridae; 16th International Conference on Antiviral Research (Apr. 27, 2003, Savannah, Ga.) p A75) описали синтез и фармакокинетические свойства нуклеозидных аналогов как возможных ингибиторов репликации РНК HCV. Авторы сообщили, что 2′-модифицированные нуклеозиды показывают высокую ингибирующую активность в исследованиях на клеточных репликонах.
Olsen et al. (Oral Session V, Hepatitis C Virus, Flaviviridae; 16th International Conference on Antiviral Research (Apr. 27, 2003, Savannah, Ga.) p A76) также показали действие 2'-модифицированных нуклеозидов на репликацию РНК HCV.
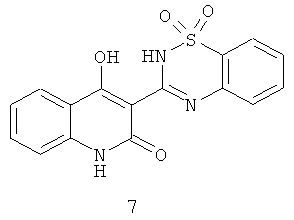
Ненуклеозидные аллостерические ингибиторы обратной транскриптазы HIV обладают доказанным эффективным терапевтическим действием как сами по себе, так и в сочетании с нуклеозидными ингибиторами и с ингибиторами протеазы. Несколько классов ненуклеозидных HCV NS5B ингибиторов уже описаны и находятся в настоящее время на стадии испытаний, включая бензимидазолы (Н.Hashimoto et al. WO 01/47833, Н.Hashimoto et al. WO 03/000254, P.L.Beaulien et al. WO 03/020240:A2; P.L.Beaulien et al. US 6448281 Bl P.L.Beaulien et al. WO 03/007945 A1); индолы (P.L.Beaulien et al. WO 03/0010141 A2); бензотиадиазины, например 7 (D.Dhanak et al. WO 01/85172 A1; D. Dhanak et al. WO 03/037262 A2; К.J.Duffy et al. WO 03/099801 A1; D.Chai et al. WO 2004052312, D.Chai et al. WO 2004052313, D.Chai et al. WO 02/098424, J.K.Pratt et al. WO 2004/041818 A1; J.K.Pratt et al. WO 2004/087577 A1), тиофены, например 8 (С.К.Chan et al. WO 02/100851)

Бензотиофены (D.С.Young and T.R.Bailey WO 00/18231); β-кетопируваты (S.Attamura et al. US 6492423 B1, A.Attamura et al. WO 00/06529); пиримидины (С.Gardelli et al. WO 02/06246 A1); пиримидиндионы (T.R.Bailey and D.C.Young WO 00/13708); триазины (K.-H. Chung et al. WO 02/079187 A1); производные роданина (T.R.Bailey and D.C.Young WO 00/10573, J.C.Jean et al. WO 01/77091 A2); 2,4-диоксопираны (R.A.Love et al. EP 256628 A2); производные фенилаланина (M.Wang et al. J.Biol. Chem. 2003 278:2489-2495).
Для разработки новой стратегии анти-HCV терапии целенаправленно была выбрана NS3 протеаза. В WO 98/22496, опубликованной 28 мая 1998 г., M.R.Attwood et al. описали механизм, в основе которого ингибирование активных центров протеазы (M.R.Attwood et al. Antiviral Chemistry and Chemotherapy 1999 10:259-273; M.R.Attwood et al. Preparation and use of amino acid derivatives as anti-viral agents, German Patent Pub. DE 19914474). В WO 98/17679, опубликованной 30 апреля 1998 г., R.D.Tung et al. раскрыли механизм действия пептидных ингибиторов на NS3 протеазу.
В WO 99/07734, опубликованной 18 февраля 1999 г., и в WO 00/09543, опубликованной 9 августа 1999 г., M. Llinas-Brunet et al. описали пептидные ингибиторы протеазы. В WO 00/59929, опубликованной 12 октября 2000 г., Y.S.Tsantrizos et al. описали макроциклические трипептиды, являющиеся мощными ингибиторами HCV NS3 протеазы. Серия родственных патентов Boehringer-Ingleheim раскрывает сходные между собой ингибиторы протеазы и уже привела к идентификации трипептидного производного BILN 2061 (M. Llinas-Brunet et al. Bioorg. Med. Chem. Lett. 2000 10(20):2267-70; J.Med.Chem. 2004 47(26): 6584-94; J.Med.Chem. 2004 47(7): 1605-1608; Angew. Chem. Int. Ed. Eng. 2003 42(12): 1356-60).
Другие трипептидные ингибиторы, обнаруженные Bristol-Myers Squibb, были описаны, между прочим, в WO 03/099274, опубликованной 4 декабря 2003 г., в WO 2004/032827, опубликованной 22 апреля 2004 г., в WO 03/053349, опубликованной 3 июля 2003 г., в WO 2005/046712, опубликованной 26 мая 2005 г. и в WO 2005/051410, опубликованной 9 июня 2005 г. В WO 2004/072234, опубликованной 26 августа 2004 г. и в WO 2004/093798, опубликованной 4 ноября 2004 г., Enanta Pharmaceuticals были описаны и другие трипептидные ингибиторы протеазы. В WO 2005/037214, опубликованной 28 апреля 2005 г., L.M.Blatt et al. описали другие трипептидные производные, ингибирующие HCV NS3 протеазу В WO 2005/030796, опубликованной 7 апреля 2005 г., S.Venkatraman et al. описали макроциклические ингибиторы NS3 сериновой протеазы HCV. В WO 2005/058821, опубликованной 30 июня 2005 г., F.Velazquer et al. описали ингибиторы HCV NS3/NS4a сериновой протеазы. В WO 02/48172, опубликованной 20 июня 2007 г., Z. Zhu описал диарилпептиды как ингибиторы NS3 протеазы. В WO 02/08187 и в WO 02/08256, опубликованных 31 января 2002 г., A.Saksena et al. описали пептидные ингибиторы HCV NS3 протеазы. В WO 02/08251, опубликованной 31 января 2002 г., M.Lim-Wilby et al. описали пептидные ингибиторы NS3 протеазы. В US 6004933, опубликованном 21 декабря 1999 г., L.W.Spruce et al. описали гетероциклические производные пептидов, которые ингибируют цистеиновые протеазы, включая эндопептидазу HCV.
Исследуются также и несубстратспецифичные ингибиторы NS3 протеазы, такие как производные 2,4,6-тригидрокси-3-нитробензамида (Sudo К. et al., BBRC 1997 238:643-647; Sudo К. et al. Antiviral Chemistry and Chemotherapy 1998 9:186), включая RD3-4082 и RD3-4078, причем первый из названных замещен по амиду 14С-цепочкой, а последний подвергнут обработке пара-феноксифенильной группы.
SCH 68631, фенантренхинон, является ингибитором HCV протеазы (Chu M. et al., Tetrahedron Lett. 1996 37:7229-7232). В другом примере теми же самыми авторами SCH 351633, выделенный из гриба Penicillium griseofulvum, был охарактеризован как ингибитор протеазы (Chu M. et al. Bioorg. Med. Chem. Lett. 1999 9:1949-1952). Наномолекулярная эффективность действия против HCV NS3 протеазного энзима уже достигнута конструированием селективных ингибиторов на основе макромолекулы eglin с.Eglin с, выделенной из пиявки, является сильным ингибитором нескольких сериновых протеаз, таких как протеазы А и В из S. griseus, а-химотрипсин, химаза и субтилизин (Qasim M. A. et al., Biochemistry 1997 36:1598-1607).
Тиазолидиновые производные, которые показывают релевантное ингибирование в исследовании HPLC с обращенной фазой в отношении слитого белка NS3/4A и субстрата NS5A/5B (Sudo К. et al. Antiviral Research 1996 32:9-18), особенно соединение RD-1-6250, несущее слитый циннамоильный остаток, замещенный длинноцепочечным алкилом, представляют собой RD4 6205 и RD4 6193. Тиазолидины и бензанилиды идентифицированы N. Kakiuchi et al. в FEBS Let. 1998 421:217-220 и N. Takeshita et al. Anal. Biochem. 1997 247:242-246.
Имидазолидиноны как ингибиторы NS3 сериновой протеазы HCV раскрыты в WO 02/08198 от Sobering Corporation, опубликованной 31 января 2002 г., и в WO 02/48157 от Bristol Myers Squibb, опубликованной 20 июня 2002. В WO 02/48116, опубликованной 20 июня 2002 г., P. Glunz et al. описали пиримидиноновые ингибиторы NS3 протеазы.
Другие энзимные мишени для противо-НСУ-терапии включают HCVIRES сайт (Internal Entry Site) и HCV геликазу. Об IRES ингибиторах уже были сообщения от Immusol, Rigel Pharmaceuticals (R803) и Anadys (ANA 245 и ANA 246). Vertex описал ингибитор геликазы HCV.
Сочетанная терапия, при которой могут подавляться устойчивые мутантные штаммы, стала общепринятьм подходом в антивирусной химиотерапии. Описанные здесь нуклеозидные ингибиторы могут быть скомбинированы с другими нуклеозидными ингибиторами HCV полимеразы, ненуклеозидными ингибиторами HCV полимеразы и ингибиторами HCV протеазы. Так как возникли и разрабатываются другие классы лекарственных против HCV, например, ингибиторы внедрения вируса, ингибиторы геликазы, IRES ингибиторы, рибозимы и антисмысловые олигонуклеотиды, они также могут быть отличными кандидатами для использования в сочетанной терапии. Производные интерферона уже успешно скомбинированы с рибавирином, и интерфероны и химически модифицированные интерфероны окажутся полезными в сочетании с описанными здесь нуклеозидами.
Производные нуклеозидов часто представляют собой эффективные антивирусные (например, HIV, HCV, Herpes simplex, CMV) и антираковые химиотерапевтические средства. К сожалению, их практическое использование часто ограничено двумя факторами. Во-первых, скудные фармакокинетические свойства часто ограничивают абсорбцию нуклеозида из пищеварительного тракта и внутриклеточную концентрацию нуклеозидных производных, а во-вторых, субоптимальные физические свойства ограничивают выбор лекарственного состава, который мог бы быть использован для увеличения степени выделения активного ингредиента.
Albert ввел термин «пролекарство», чтобы охарактеризовать соединение, которое само по себе не обладает биологической активностью, но способно метаболически трансформироваться и превращаться в активную лекарственную субстанцию (A.Albert, Selective Toxicity, Chapman and Hall, London, 1951). За последнее время появилось много обзоров о пролекарствах (Р.Ettmayer et al. J.Med.Chem. 2004 47(10):2393-2404; К.Beaumont et al. Curr. Dmg Metab. 2003 4:461-485; H.Bundgaard, Design of Prodrugs: Bioreversible derivatives for various functional groups and chemical entities in Design of Prodrugs, H.Bundgaard (ed) Elsiver Science Publishers, Amsterdam 1985; G.M.Paulette et al. Adv. Drug Deliv. Rev. 1997 27:235-256; R.J.Jones and N.Bischofberger, Antiviral Res. 1995 27:1-15, и С.R.Wagner et al., Med. Res. Rev. 2000 20:417-45).
Несмотря на то, что метаболическая трансформация может катализироваться специальными ферментами, часто - гидролазами, активное вещество может также быть регенерировано в результате неспецифических химических процессов.
К фармацевтически приемлемым пролекарствам относят соединения, которое метаболизируется, например, гидролизуется или окисляется, в хозяине с образованием соединения настоящего изобретения. При биоконверсии не должно быть образования токсикологически обремененных фрагментов. Типичные примеры пролекарств включают соединения, которые имеют биологические лабильные защитные группы, связанные с функциональным остатком активного соединения. Алкилирование, ацилирование и другая липофильная модификация гидроксильной группы (групп) в остатке сахара применяются при создании пронуклеотидов. Эти пронуклеотиды можно гидролизовать или деалкилировать in vivo для получения активного соединения.
Факторами, ограничивающими оральную биодоступность, зачастую являются абсорбция из желудочно-кишечного тракта и выделение при первом прохождении кишечными стенками и печенью. Оптимизация трансцеллюлярной абсорбции при прохождении через ЖКТ требует D(7,4) больше нуля. Оптимизация коэффициента распределения, однако, не обеспечивает успеха. Может быть, пролекарство должно избегать в энтероцитах активных факторов переноса при просачивании. Внутриклеточный метаболизм в энтероците может приводить к пассивному транспорту или активному транспорту метаболита насосами просачивания обратно внутрь кишечной полости. Пролекарство должно также быть устойчивым по отношению к нежелательным биотрансформациям в крови перед тем, как достигнуть мишени - клеток или рецепторов.
Хотя предполагаемые пролекарства иногда могут быть созданы с помощью рационального мышления, основываясь на химической функциональности данной молекулы, химическая модификация активного компонента приводит к совершенно иной, новой молекулярной структуре, которая может проявить нежелательные физические, химические и биологические свойства, отсутствующие в соединении-предшественнике. Установленные требования для идентификации метаболитов могут ставить сложные задачи, если многочисленные пути метаболизма приводят к множеству разных метаболитов. Так что идентификация пролекарств остается сомнительным и порождающим проблемы занятием. Более того, оценка фармакологических свойств потенциальных пролекарств является сложным и дорогостоящим делом. Фармакокинетические результаты, полученные на модельных животных, могут иметь отличия, если экстрагировать их для человека.
Объектом настоящего изобретения является обеспечение новых соединений, способов и композиций для лечения субъекта-хозяина, зараженного вирусом гепатита С.
Настоящее изобретение касается новых диацильных производных 4-амино-1-((2R, 3R, 4R, 5R)-3-фтор-4-гидрокси-5-гидроксиметил-3-метил-тетрагидро-фуран-2-ил)-1Н-пиримидин-2-она (называемого также (2R)2′-дезокси-2-метил-2-фтор-цитидин).
Соединения данного изобретения обладают структурой согласно формуле I.
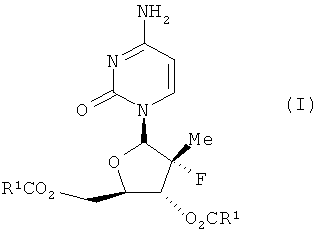
где R1 выбран из группы, состоящей из С2-5 неразветвленного или разветвленного амина, C2-5 неразветвленного или разветвленного алкенила, C2-5 неразветвленного или разветвленного алкинила, C2-5 низшего галоалкила, С3-6 циклоалкила и C2-4 алкокси; или представляют собой соответствующие ей гидраты, сольваты или соли в результате присоединения кислот.
Соединения настоящего изобретения полезны при лечении расстройства, вызванного HCV.
Изобретение, кроме того, включает способы лечения HCV, описываемые в данном изобретении соединениями и фармацевтическими композициями, включающими эти соединения.
В одном из вариантов данного изобретения предлагается соединение согласно формуле I, где R1 имеет указанные выше значения.
В другом варианте настоящего изобретения предлагается соединение согласно формуле I, где R1 представляет собой этил, н-пропил, изо-пропил, н-бутил или изобутил.
Еще в одном варианте настоящего изобретения предлагается соединение согласно формуле I, где R1 представляет собой этил или изопропил.
Еще в одном варианте настоящего изобретения предлагается соединение согласно формуле I, где R1 представляет собой изопропил, а соединение является гидрохлоридной или сульфатной солью.
Еще в одном варианте настоящего изобретения предлагается соединение согласно формуле I, где R1 представляет собой изопропил, а соединение является гидрохлоридной солью.
Еще в одном варианте настоящего изобретения предлагается соединение согласно формуле I, где R1 представляет собой этокси, н-пропокси или изопропокси.
Еще в одном варианте настоящего изобретения предлагается соединение согласно формуле I для применения в терапии, особенно для применения в лечении заболевания, вызванного вирусом HCV.
Еще один вариант изобретения касается применения соединения формулы I для изготовления лекарственного средства для лечения заболевания, вызванного вирусом HCV.
Соединение формулы I может, в частности, использоваться для приготовления лекарственного средства для введения нуждающемуся в этом пациенту в терапевтически эффективной зоне, предпочтительно в дозе от 0,1 до 10 г в день, более предпочтительно в дозе от 0,5 до 7 г в день и наиболее предпочтительно в дозе от 1,0 г до 6,0 г в день.
Соединение формулы I может также использоваться для приготовления лекарственного средства, которое может дополнительно включать терапевтически эффективное количество, по меньшей мере, одного модулятора иммунной системы, например, интерферона, химически дериватизированного интерферона, интерлейкина, фактора некроза опухоли или колониестимулирующего фактора и/или, по меньшей мере, одного антивирусного агента, который ингибирует репликацию HCV, такого как ингибитор протеазы HCV, другой нуклеозидный ингибитор полимеразы HCV, ненуклеозидный ингибитор полимеразы HCV, ингибитор геликазы HCV, ингибитор примазы HCV или слитый ингибитор HCV.
Еще один вариант настоящего изобретения предусматривает способ лечения заболевания, опосредованного вирусом HCV, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективной дозы соединения согласно формуле I, которое охарактеризовано выше.
Еще один вариант настоящего изобретения предусматривает способ лечения заболевания, опосредованного вирусом HCV, включающий введение пациенту, нуждающемуся в этом, дозы от 0,1 г до 10 г в день соединения согласно формуле I, которое охарактеризовано выше.
Еще в одном варианте изобретения доза составляет 0,5-7 г в день, а еще в другом варианте доза составляет 1,0-6,0 г в день.
В другом варианте настоящего изобретения предусматривается способ лечения заболевания, опосредованного вирусом HCV, включающий совместное введение пациенту, нуждающемуся в этом, терапевтически эффективной дозы соединения согласно формуле I, которое охарактеризовано выше, и терапевтически эффективного количества по меньшей мере, одного модулятора иммунной системы и/или, по меньшей мере, одного антивирусного агента, который ингибирует репликацию HCV.
В другом варианте настоящего изобретения предусматривается способ лечения заболевания, опосредованного вирусом HCV, включающий совместное введение пациенту, нуждающемуся в этом, терапевтически эффективной дозы соединения согласно формуле I, которое охарактеризовано выше, и терапевтически эффективного количества, по меньшей мере, одного модулятора иммунной системы, причем этот модулятор иммунной системы представляет собой интерферон, интерлейкин, фактор некроза опухоли или колониестимулирующий фактор.
Другой вариант настоящего изобретения предусматривает способ лечения заболевания, опосредованного вирусом HCV, включающий совместное введение пациенту, нуждающемуся в этом, терапевтически эффективной дозы соединения согласно формуле I, которое охарактеризовано выше, и терапевтически эффективного количества, по меньшей мере, одного модулятора иммунной системы, причем этот модулятор иммунной системы представляет собой интерферон или химически дериватизированный интерферон.
Другой вариант настоящего изобретения предусматривает способ лечения заболевания, опосредованного вирусом HCV, включающий совместное введение пациенту, нуждающемуся в этом, терапевтически эффективной дозы соединения согласно формуле I, которое охарактеризовано выше, и терапевтически эффективного количества, по меньшей мере, одного иного антивирусного соединения.
Другой вариант настоящего изобретения предусматривает способ лечения заболевания, опосредованного вирусом HCV, включающий совместное введение пациенту, нуждающемуся в этом, терапевтически эффективной дозы соединения согласно формуле I, которое охарактеризовано выше, и терапевтически эффективного количества по меньшей мере, одного иного антивирусного соединения, которое является ингибитором протеазы HCV, другим нуклеозидным ингибитором полимеразы HCV, ненуклеозидным ингибитором полимеразы HCV, ингибитором геликазы HCV, ингибитором примазы HCV или слитым ингибитором HCV.
Другой вариант настоящего изобретения предусматривает фармацевтическую композицию, включающую соединение согласно формуле I, которое охарактеризовано выше, смешанное с по меньшей мере одним фармацевтически приемлемым носителем, разбавителем или наполнителем.
Другой вариант настоящего изобретения предусматривает способ получения соединения согласно формуле I, которое охарактеризовано выше, причем способ включает стадии (i)-(v), перечисленные в пункте 15 формулы изобретения и проиллюстрированные в примерах. Способ включает обработку I в основной водной органической среде, которая может быть гомогенной или двухфазной с ацилирующим агентом, как определено здесь, в присутствии DMAP и достаточного количества основания для поддержания рН раствора при рН, по крайней мере, около 7,5.
Данный способ позволяет проводить ацилирование без сопутствующей реакции гетероциклического основания.
Выражение «а» или «an» величина, в используемом здесь смысле, означает одну или несколько таких единиц, например, соединение имеет отношение к одному или нескольким соединениям или, по меньшей мере, одному соединению. Так что термины «а» (или «an»), «один или несколько» и «по меньшей мере, один» могут использоваться здесь взаимозаменяющим образом.
Термины «необязательный» или «необязательно» в том смысле, в котором они здесь используются, означают, что впоследствии описываемое событие или обстоятельство может происходить, но необязательно, потому описание включает примеры, когда это событие или обстоятельство имеет место, и примеры, когда оно не происходит. Например, «необязательная связь» означает, что связь может присутствовать и может не быть, и описание тогда включает одинарные, двойные или тройные связи.
Термин «независимо» используется здесь, чтобы показать, что переменная (величина) используется в любом примере без связи с присутствием или отсутствием переменной, имеющей такое же или иное определение в рамках одного и того же соединения. Так что в соединении, в котором R появляется дважды и определен как «независимо углерод или азот», оба R могут быть углеродами, оба могут быть азотами, или один R может быть углеродом, а другой - азотом.
Термин «алкенил» в используемом здесь значении означает незамещенный углероводородный цепочечный радикал из 2-10 атомов углерода, имеющий одну или две олефиновых двойных связи [предпочтительно одну олефиновую двойную связь], «С2-10 алкенил» при использовании здесь означает алкенил, состоящий из 2-10 атомов углерода. Примерами являются винил; 1-пропенил, 2-пропенил (аллил) или 2-бутенил (кротил).
Термин «алкил» означает здесь неразветвленную или разветвленную цепочку насыщенного моновалентного углеводородного остатка, состоящего из 1-10 атомов углерода. Термин «низший алкил» означает прямой или разветвленный остаток углеводородной цепочки, состоящий из 1-6 атомов углерода. «C2-10 алкил» означает здесь алкил, состоящий из 1-10 атомов углерода. Примеры алкильных групп включают (но без ограничения ими) низшие алкильные группы, которые включают метил, этил, пропил, i-пропил, n-бутил, i-бутил, t-бутил или пентил, изопентил, неопентил, гексил, гептил и октил. Термин (ар) алкил или (гетероарил) алкил указывает на то, что алкильная группа необязательно замещена арилом или гетероарилом соответственно.
Термин «алкинил» означает здесь радикал, представляющий собой неразветвленную или разветвленную углеводородную цепочку из 2-10 атомов углерода, предпочтительно из 2-5 атомов углерода, и имеющий одну тройную связь. «С2-10 алкинил» означает здесь алкинил, состоящий из 2-10 атомов углерода. Примерами являются этинил, 1-пропинил, 2-пропинил, 1-бутинил, 2-бутинил или 3-бутинил.
Термин «циклоалкил» означает здесь насыщенное карбоциклическое кольцо, содержащее от 3 до 8 атомов углерода, то есть циклопропил, циклобутил, циклопентил, циклогексил, циклогептил или циклооктил. «С3-7 циклоалкил» означает здесь циклоалкил, содержащий 3-7 атомов углерода в карбоциклическом кольце.
Термин «галоалкил» означает здесь неразветвленную или разветвленную алкильную цепочку, как определено выше, в которой 1, 2, 3 или более атомов водорода замещены галогеном. «C1-3 галоалкил» означает здесь галоалкил с 1-3 атомами углерода и 1-8 галогенными заместителями. Примерами являются 1-фторметил, 1-хлорметил, 1-бромметил, 1-йодометил, трифторметил, трихлорметил, трибромметил, трийодметил, 1-фторэтил, 1-хлорэтил, 1-бромэтил, 1-йодэтил, 2-фторэтил, 2-хлорэтил, 2-бромэтил, 2-йодэтил, 2,2-дихлорэтил, 3-бромпропил или 2,2,2-трифторэтил.
Термин «алкокси», используемый здесь, означает -O-алкильную группу, в которой алкил определен выше. Примерами являются метокси, этокси, n-пропилокси, i-пропилокси, n-бутилокси, i-бутилокси, t-бутилокси. «Низший алкокси» означает здесь алкоксигруппу с «низшей алкильной» группой, как определено выше. «C1-10 алкокси» означает-O-алкил, где алкил представляет собой C1-10.
Термин «диацильное» производное, используемое здесь, означает дериватизированное нуклеозидное производное, в соответствии с описанным здесь, в котором 3′- и 5′-гидрокси относятся к эфиру -OC(=O)R1 и OC(=O)R2, где R1 и R2 - такие, как определено в пункте 1.
Термин «ацилирующий агент» используемый здесь, относится к ангидриду, галоиду кислоты, хлоркарбонилалкоксиду (например, этилхлорформиату) или активированному производному N-замещенной альфа аминокислоты. Термин «ангидрид» в используемом здесь смысле относится к соединениям общей структуры R1C(O)-O-C(O)R1, где R имеет значение, определенное в пункте 1. Термин «галоидкислоты» относится здесь к соединениям общей формулы R1C(O)X, где Х - галоген. Термин «ацилимидазол» относится здесь к соединению общей формулы R1C(O)X, где Х - является N-имидазолилом. Термин «активированное производное» соединения в используемом здесь смысле относится к транзиентной реактивной форме оригинального соединения, которое делает соединение активным в нужной химической реакции, в которой исходное (оригинальное) соединение только умеренно реакционно-способно или вовсе нереакционно-способно. Активация достигается образованием производного или химической группировки в молекуле с более высоким содержанием свободной энергии, чем у исходного соединения, что придает активированной форме большую восприимчивость к реакции с другим реагентом. В контексте настоящего изобретения активация карбоксигруппы чрезвычайно важна и соответствующие активирующие агенты или группировки, которые активируют карбоксигруппу, описываются детально ниже.
Особый интерес с точки зрения настоящего изобретения вызывают ангидриды карбоновых кислот и хлориды карбоновых кислот.
Выражение «гетерогенная водная смесь растворителей» означает здесь смесь воды и органического сорастворителя, что в результате дает двухфазную или гетерогенную смесь. Эта гетерогенная водная смесь растворителей может образовываться из сорастворителя с ограниченной водной растворимостью или ионная сила водного компонента может быть отрегулирована так, чтобы ограничивать растворимость сорастворителя в водной фазе.
Термин «гидроксид щелочного металла» относится к соединению МОН, где М представляет собой литий, натрий, калий или цезий, «бикарбонат щелочного металла» относится к группе М2СО3, где М является натрием или калием, а «карбонат щелочного металла» относится к группе М2СО3, где М - натрий или калий. Специалисту в данной области понятно, что и другие основания могут использоваться в объеме данного изобретения для поддержания рН на желаемом уровне.
Аббревиатуры, используемые в данном описании, включают: ацетил (Ас), уксусная кислота (НОАс), 1-N-гидроксибензотриазол (HOBt), атмосферы (Atm), жидкостная хроматография над высоким давлением (HPLC), метил (Me), трет-бутоксикарбонил (Воc), ацетонитрил (MeCN), ди-трет-бутил-пирокарбонат или bос ангидрид (ВОС2О), 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид (EDCI), бензил (Вn), бутил (Вu), метанол (МеОН), бензилоксикарбонил (cbz или Z), точка плавления (mр), карбонилдиимидазол (CDI), MeSO2 - (мезил или Ms), 1,4-диазабицикло [2.2.2]октан (DABCO), масс-спектр (ms), метил-т-бутиловый эфир (МТВЕ), 1,5-диазабицикло [4.3.0]-нон-5-ен (DBN), 1,8-диазабицикло [5.4.0] ундек-7-ен (DBU), N-метилморфолин (NMM), N-метилпирролидон (NMP), 1,2-дихлорэтан (DCE), N,N′-дициклогексилкарбодиимид (DCC), дихромат пиридинил (PDC), дихлорметан (DCM), пропил (Рг), фунтов на квадратный инч (psi), диизопропилэтиламин (DIPEA, Hunigs′ Base), пиридин (руг), комнатная температура, rt или RT, N,N-диметилацетамид (DMA), трет-бутилдиметилсилил или t-BuMe2Si, (TBDMS), 4-N,N-диметиламинопиридин (DMAP), триэтиламин (EtsN или TEA), N,N-диметилформамид (DMF), диметилсульфоксид (DMSO), трифторуксусная кислота (TFA), тонкослойная хроматография (TLC), этилацетат (ЕtOАс), тетрагидрофуран (THF), диэтиловый эфир (Et2O), триметилсилил или Me3Si (TMS), этил (Et), моногидрат п-толуолсульфокислоты (TsOH или pTsOH), H-Me-C6H4SO2- или тозил (Ts), изопропил (i-Pr), N-уретан-N-карбоксиангидрид (UNCA), этанол (ЕtOН). Применяемые обычно номенклатурные обозначения, в том числе префиксы «нормальный» (n), «изо» (i-), «вторичный» (sec-), «третичный» (tert-) и «новый» (neo), имеют свои обычные значения при использовании вместе с алкильными остатками. (J.Rigaudi and D.R.Klesney, Nomenclature in Organic Chemistry, IUPAC 1979 Pergamon Press, Oxford).
Примеры репрезентативных соединений, охватываемых данным изобретением и подпадающих под объем настоящего изобретения, представлены в Таблице I. Эти примеры и их выполнение предназначены для того, чтобы специалисты в данной области могли бы с большей легкостью понять и воспроизвести настоящее изобретение. Это не должно расцениваться как ограничение объема изобретения, а только лишь с целью иллюстративности и репрезентативности представляемых данных.
В общем номенклатура, используемая в описании этой заявки, основывается на AUTONOM™ в 4.0 компьютеризированной системе Beilstein Institute для воссоздания IUРАС систематической номенклатуры. Существует расхождение между изображенной структурой и наименованием, присвоенным этой структуре, считается, что изображенная структура имеет больший вес и влияние. Кроме того, если стереометрия структуры или части структуры не показана, с помощью, например, рельефных или пунктирных линий, строение части этой структуры следует интерпретировать как охватывающую все изомеры этой структуры. Система нумерования для этих кольцевых систем является такой, как показано ниже.
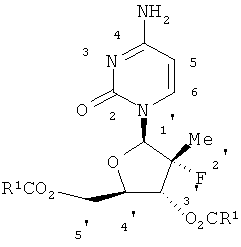
Соединения и их получение
Ингибирующая активность (2R)-2′-дезокси-2′-фтор-2′-С-метилцитидина в отношении HCV полимеразы уже описана (J.L.Clark et al. J. Med. Chem. 2005 48(17):5504-8; J.Clark, опубликованная заявка US №2005/0009737, обе публикации включены здесь во всей своей полноте в качестве ссылки). В клинической практике желательно сначала вводить высокие дозы I-6, чтобы быстро ингибировать HCV полимеразу, а затем более низшие виральные уровни в условиях, при которых минимизируется способность вируса мутировать, и отбор устойчивых штаммов. Значительно высокие уровни могут затруднять успешное достижение цели исходным нуклеозидом.
Пролекарства предусматривают стратегию улучшения фармакокинетических и физических свойств соединения и тем самым оптимизировать биодоступность. В US публикации №2005/0009737 предлагаются общие подходы к нуклеотидным пролекарствам на базе 2′-дезокси-2′-фтор-2′-метилнуклеозидам. Несмотря на то, что кандидаты в пролекарства с виду обманчиво просты, идентификация соединений с соответствующими физико-химическими и фармакодинамическими свойствами, in vivo трансформации и профиль безопасности - есть комплекс разнообразных задач, что требует ответственных экспериментов. Затруднения в идентификации пролекарств для орального применения включают поддерживание значительной водной растворимости, липофильности и химической стабильности в то же самое время, что дает возможность быстрого и эффективного выделения активного остатка после введения. Дополнительно должен быть минимизирован важный неэстеразный метаболизм и опосредованный транспортером клиренс Пролекарства (К.Beaumont et al. Curr. Dmg Metab. 2003 4(6):461-485). Кроме того, ингибирование или индукция цитохром Р450-энзимов может вызывать неблагоприятные взаимодействия лекарства с лекарством, что нежелательно. Неожиданно было обнаружено, что диэфиры низших алкилов I-6, обозначенных в Таблице I, имеют значительно улучшенную биодоступность I-6.
Фармакокинетические характеристики потенциальных пролекарств оценивали на крысах и обезьянах, чтобы попробовать минимизировать внутривидовую вариабельность и генетические полиморфизмы, которые могут приводить к внутривидовым артефактам. В том смысле, что энзиматические трансформации ответственны за гидролиз эфирных связей, специфическая аффинность пролекарства может зависеть от специфической структуры эстеразы и/или пептидазы, которая может катализировать трансформацию. Уже часто показывали, что активность эстеразы крыс существенно выше, чем у человека (J.A.Fix et al. Pharm. Res, 1990 7(4):384-387; W.Li et al. Antimicrob. Agents Chemother 1998 42(3):647-653). Другой потенциально важный параметр - это биоконверсия цитидинового основания в уридин с помощью дезаминазы. Хотя трифосфаты цитодина и уридина являются ингибиторами полимеразы, in vivo фосфорилирование уридина менее эффективно, чем цитидина. Так что делают вывод о том, что повышенные уровни уридина нежелательны. О недостаточной эффективности, проявляемой производным уридина в НСl репликоне (J.Clark et al. J.Med. Chem., см. выше), уже сообщалось.
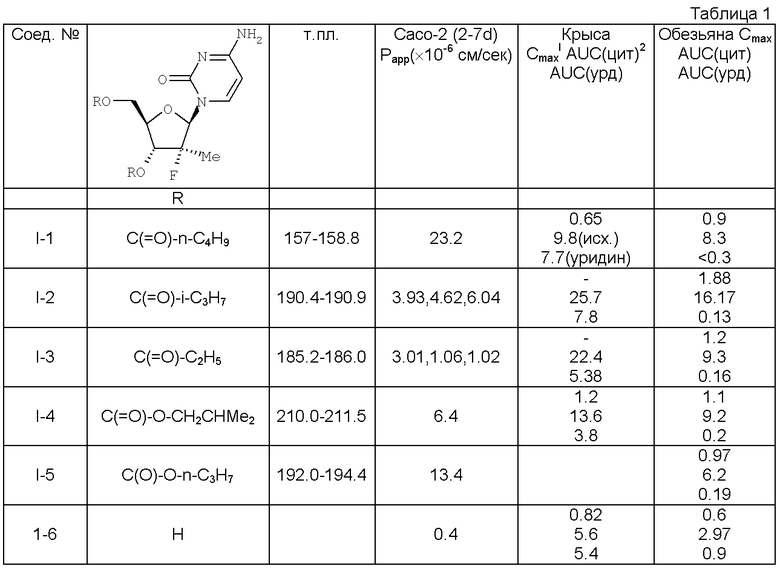
1. Cmax представляет собой пик концентрации 1-7 в крови.
2. AUC(cyt) - площадь над кривой для цитидинового нуклеозида.
3. AUC(urd) - площадь над кривой уридинового нуклеозида.
4. ЕС90 в репликоне равно 5,40±2,6 µМ (J.Clark et al. J.Med.Chem., см. выше).
Неожиданно было установлено, что C2-5 алкильные диэфиры I-6 проявляют отличные пролекарственные свойства. В крови и крыс, и обезьян наблюдаются существенно более высокие уровни фторированных нуклеотидов. Более того, отношение цитидина к уридину во фторированном исходном основании выше в обоих образцах, когда нуклеозид вводят в виде диэфира.
Кроме того, биэфиры способны образовывать два различных моноэфира и неэтерифицированный нуклеозид. Фармакокинетический анализ в этой ситуации может быть сложным (комплексным), если все образцы содержатся в крови в значительных количествах. Присутствие множества метаболитов в крови добавляет дополнительное время при установлении, является ли пролекарство безопасным. Неожиданно гидролиз обоих эфиров оказался абсолютно легким, и единственным важным метаболизмом, обнаруживаемом в крови помимо исходного нуклеозида, был 3'-моноэфир, который быстро превращается в I-6.
Для того, чтобы далее оценить потенциальное функционирование пролекарств в организме человека, оценивали транспорт мнимых пролекарств через Сасо-2 клетки. Сасо-2 клетки обычно используют, чтобы оценить потенциальную абсорбцию/проницаемость молекул (G.Gaviraghi et al. in Pharmacokinetic optimization in drug research. Biological, Pharmacokinelic and Computatinal Strategies. B.Testa et al. eds. Wiley Interscience VCH, Zurich 2001 pp.3-14). Было обнаружено, что проницаемость Сасо-2 является приемлемой для C2-5 алкильных диэфиров I-6.
Помимо эффективной in vivo биотрансформации пролекарство для орального введения должно также показывать подходящие физико-химические свойства для образования лекарства и обеспечить абсорбцию из кишечника в тот отдел, где нужная и желаемая биотрансформация может происходить. Особенно соответствующими являются растворимость в воде, коэффициент разделения и стабильность в жидкой среде желудочной и кишечной области. Величины всех этих параметров показаны в Таблице 2.
1. Вычисленный коэффициент разделения октанол/вода.
2. Экспериментально определенный коэффициент распределения при рН 7,4.
3. Растворимость в водной среде, воде или буфере рН 6,5 (мг/мл).
4. Стабильность в симулированном желудочном соке (SGF; рН 1,2).
5. Стабильность в симулированной жидкости кишечника (SIP; рН 7,4).
6. Растворимость в SGF составляет 13,3 мг/мл.
7. не определяли
8. Средой является SIF, растворимость в SGF составляет 13,6 мг/мл.
9. Средой является SIF, растворимость в SGF составляет 0,16 мг/мл.
10. Измерено в воде и буфере при рН 6.
11. Незначительная деградация отмечалась в течение этого периода времени.
12. Определяли при рН 5,0.
Коэффициент распределения «масло/вода» Po/w (смешанный Р вычисляется как Po/w) является важной характеристикой для орально применяемых лекарств. Лекарственная субстанция должна обладать существенной водной растворимостью, чтобы раствориться в композиции и жидкостях желудочно-кишечного тракта (ЖКТ) для того, чтобы взаимодействовать с клетками эндотелия в желудке и кишечнике, и значительную растворимость в жирах, чтобы преодолеть липидные бислойные мембраны в этих клетках и мгновенно попасть в кровоток. Оптимальный диапазон log P соединения для оральной биодоступности находится между 1 и 3, который характерен для соединений настоящего изобретения.
Выражение Po/w, используемое здесь, называется коэффициентом разделения октанол/вода. Выражение смешанный Po/w в используемом здесь смысле относится к вычисленному Po/w. Компьютерные программы, с помощью которых вычисляется Po/w, доступны химикам-фармацевтам и медикам. Термин «коэффициент распределения» относится к экспериментально определяемому коэффициенту распределения между октанолом и водным буферным раствором. Коэффициент разделения и коэффициент распределения в целом схожи, но последний из них является функцией от рН водного раствора, тогда как Po/w независим от рН.
Растворимость в виде орально принимаемого пролекарства должна быть выше, чем, по крайней мере, приблизительно 0,1 мг/мл для оптимального состава, а время полужизни в искусственно вызванном желудочном соке и искусственно смоделированной кишечной жидкости должны быть 1-2 часа и 2-4 часа соответственно для того, чтобы обеспечить достаточное время для того, чтобы пройти через желудок и абсорбироваться в кишечнике.
Соединения настоящего изобретения стандартным образом получают в одну стадию ацилированием 1-6 в водном органическом растворителе. Растворитель может быть либо гомогенным водным раствором, либо двухфазным раствором. рН водного органического поддерживают на уровне 7,5 добавлением основания, чтобы нейтрализовать кислоту, получающуюся при ацилировании. Основание может быть гидроксидом щелочного или щелочноземельного металла или четвертичным амином. Реакцию проводят в присутствии DMAP, который, как известно специалистам в данной области, является катализатором ацилирования. Преимуществом заявленного настоящего способа является то, что целевой продукт может быть получен без ацилирования гетероциклическим основанием. Не требуется никаких защитных групп, что позволяет избежать стадии защиты и снятия защиты. Далее процесс описан в нижеследующих примерах.
Дозировки и введение
Соединения настоящего изобретения могут входить в состав множества используемых орально дозировочных форм в сочетании с носителями. Для орального применения могут использоваться таблетки, таблетки с покрытием, твердые и мягкие желатиновые капсулы, растворы, эмульсии, сиропы или суспензии. Соединения настоящего изобретения эффективны при введении в виде суппозиториев в числе других типов введения. Самым привычным и стандартным методом введения обычно является оральный с использованием стандартных дневных дозировочных режимов, которые могут быть отрегулированы в соответствии с тяжестью заболевания и реакцией пациента на антивирусное лечение.
Соединение или соединения настоящего изобретения, а также их фармацевтически приемлемые соли, вместе с одним или несколькими носителями, наполнителями и разбавителями могут быть в форме фармацевтических композиций или единичных дозировочных форм. Эти фармацевтические композиции и единичные дозировочные формы могут включать стандартные ингредиенты в принятых обычно пропорциях, с добавленными активными соединениями или без них, и единичные дозировочные формы могут содержать некое эффективное количество соответственного активного ингредиента, при использовании в установленных суточных диапазонах дозировок. Фармацевтические композиции могут применяться в твердом виде, например, как таблетки или заполненные капсулы, в полутвердом виде, в виде порошков, составов с непрерывным выделением активного начала, или в жидком виде, например, в виде суспензий, эмульсий или заполненных капсул для орального приема; или в форме суппозиториев для ректального или вагинального применения. Типичный препарат должен содержать от около 5% до около 95% активного соединения или соединений (вес/вес). Считается, что термин «препарат» или «дозировочная форма» включает и жидкий, и твердый состав на основе активного соединения, и специалисту в данной области будет понятно, что активный ингредиент может входить в состав различных препаратов в зависимости от предпочитаемой дозы и фармацевтических препаратов.
Термин «наполнитель» в используемом здесь смысле означает соединение, которое используется для приготовления фармацевтической композиции и в целом является безопасным, нетоксичным, не является неприемлемым ни в биологическом, ни в каком-то ином смысле, и включает наполнители, которые приемлемы для использования в ветеринарии, а также в фармацевтике для людей. Соединения настоящего изобретения можно вводить сами по себе, но вообще следует вводить в сочетании с одним или несколькими подходящими фармацевтическими наполнителями, разбавителями или носителями, подобранными с учетом типа введения и стандартной фармацевтической практики.
«Фармацевтически приемлемая соль» как форма активного ингредиента может также вначале придать желаемое фармакокинетическое свойство активному ингредиенту, которое отсутствует у его несолевой формы, и может даже положительно воздействовать на фармакодинамику активного ингредиента в плане его терапевтической активности в организме. Термин «фармацевтически приемлемая соль» соединения в используемом здесь смысле означает соль, которая является фармацевтически приемлемой и которая обладает желаемой фармакологической активностью исходного соединения. Такие соли включают: (1) соли присоединения кислот, образованные неорганическими кислотами, например, соляная кислота, водороднобромистая кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., или образованные органическими кислотами, такими как гликолевая, пировиноградная кислота, молочная кислота, малоновая кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, виннокаменная кислота, лимонная кислота, 3-(4-гидроксибензоил) бензойная кислота, коричная кислота, янтарная кислота, метансульфокислота, этансульфокислота, 1,2-этандисульфокислота, 2-гидроксиэтансульфокислота, бензолсульфокислота, 4-хлорбензолсульфокислота, 2-нафталинсульфокислота, 4-толуолсульфокислота, камфорсульфокислота, лаурилсерная кислота, глюконовая кислота, глутаминовая кислота, салициловая кислота, муконовая кислота и т.п.Должно быть понятно, что все отсылки на фармацевтически приемлемые соли включают формы присоединения растворителя (сольваты) или кристаллические формы (полиморфы), согласно данному здесь определению, или соли присоединения тех же самых кислот.
Препараты в твердой фазе включают порошки, таблетки, пилюли, капсулы, суппозитории и диспергируемые гранулы. Твердым носителем может быть одно или несколько веществ, которые могут также действовать как разбавители, отдушки, солюбилизаторы, любриканты, суспендирующие агенты, связующие, консерванты, дезинтегрирующие таблетки агенты или инкапсулирующий материал. В порошках носитель обычно представляет собой тонкоразмолотое твердое вещество, которое является смесью с тонкоразмолотым активным компонентом. В таблетках активный компонент, в основном, как правило, смешан с носителем, имеющим необходимую связывающую способность, в подходящих пропорциях, и уплотнен до нужного размера и формы. Подходящие носители включают (но без ограничения перечисленными далее): карбонат магния, стеарат магния, тальк, сахар, лактозу, пектин, декстрин, крахмал, желатин, трагакант, метилцеллюлозу, карбоксиметилцеллюлозу натрия, низкоплавящийся воск, масло какао и т.п. Препараты твердых форм могут содержать, в дополнение к активному компоненту, красители, ароматизаторы, стабилизаторы, буферы, искусственные или природные подсластители, диспергаторы, загущающие средства, солюбилизирующие агенты и т.п.
Жидкие составы тоже пригодны для орального введения и включают жидкие составы, в том числе эмульсии, сиропы, эликсиры и водные суспензии. Они включают препараты в твердой форме, которые, как подразумевается, превращаются в препараты в жидкой форме почти сразу же после их употребления. Эмульсии могут готовиться в растворах, например, в водных растворах пропиленгликоля, или они могут содержать эмульгирующие агенты, такие как лецитин, сорбитан моноолеат или (камедь) акации. Водные суспензии можно готовить, диспергируя тонкомолотый активный компонент в воде с вязким материалом, таким как природные или синтетические смолы, полимеры, метилцеллюлоза, натрий карбоксиметилцеллюлоза или др. хорошо известные суспендирующие агенты.
Соединения настоящего изобретения могут быть приготовлены как составы для введения в виде суппозиториев. Низкоплавящийся воск, такой как смесь глицеридов жирных кислот или масло какао, сначала расплавляется, а активный компонент диспергируется до гомогенного состояния, например, путем перемешивания. Расславленная гомогенная смесь затем выливается в заготовки-формы определенного размера, ей дают остыть и затвердеть.
Соединения настоящего изобретения могут быть приготовлены как средства для вагинального применения. Пессарии, тампоны, кремы, гели, пасты, пены или спреи, содержащие в дополнение к активному ингредиенту такие носители, которые, как известно в данной области, являются подходящими и уместными.
Подходящие составы наряду с фармацевтическими носителями, разбавителями и наполнителями описаны в: Remington: The Science and Practice of Pharmacy 1995, издано E.W.Martin, Mack Publishing Company, 19th edition, Taston, Pennsylvania. Опытный химик-фармацевт может модифицировать составы в рамках инструкций к описанию и создать в результате множество составов для конкретного типа введения, обеспечив стабильность композиций настоящего изобретения и не принеся ущерба их терапевтической активности.
Модификация соединений настоящего изобретения для того, чтобы сделать их более растворимыми в воде или другом средстве, например, может быть легко выполнена за счет минимальных модификаций (например, за счет подбора состава солей), что хорошо известно специалистам в данной области. Также в компетенции специалистов в данной области находится и возможность модифицировать тип введения и дозированный режим в отношении конкретного соединения с тем, чтобы управлять фармакокинетикой заявленных соединений для максимально благоприятного воздействия на пациентов.
Термин «терапевтически эффективное количество» в используемом здесь смысле означает количество, требующееся для уменьшения симптомов заболевания у больных. Дозу следует подобрать в индивидуальных условиях в каждом конкретном случае. Эти дозировки могут колебаться в широких пределах в зависимости от многочисленных факторов, таких как тяжесть заболевания, которое надо лечить, возраста и общего состояния здоровья пациента, других лекарств, которые получает пациент, тип и форма введения, а также предпочтения и опыт лечащего врача. При оральном введении суточная дозировка между около 0.1 и около 10 г/день будет приемлемой при монотерапии и/или при сочетанной терапии. Предпочтительная суточная дозировка составляет от около 0,5 и до около 7,5 г/день, более предпочтительно 1,5 и около 6,0 г/день. Вообще лечение начинается с большой первоначальной «Загружающей дозы», чтобы быстро ослабить или элиминировать вирус с последующим снижением дозы до уровня, достаточного для предотвращения возобновления заражения. Любой специалист в данной области, подготовленный для лечения заболеваний, описываемых здесь, сможет без чрезмерного экспериментирования и на основе собственных знаний, опыта и описания данной заявки установить терапевтически эффективное количество соединений настоящего изобретения для данного заболевания и данного пациента.
Терапевтическая эффективность может быть установлена исходя из опытов по функционированию печени, но без ограничения уровнями протеинов, которые являются сывороточными белками (например, альбумин, факторы свертывания, щелочная фосфатаза, аминотрансферазы (например, аланинтрансаминаза, аспартат-трансаминаза), 5′-нуклеозидаза, γ-глутаминилтранспептидаза и др.), синтез билирубина, синтез холестерола и синтез билевых кислот; метаболическая функция печени, включая (но не только) метаболизм углеводов, метаболизм аминокислот и аммиака.
Альтернативно терапевтическая эффективность может регистрироваться путем измерения HCV-RNA. Результаты этих тестов позволят оптимизировать дозу.
В вариантах изобретения активное соединение или соль могут вводиться в сочетании с другим антивирусным агентом, таким как рибавирин, другой нуклеозидный ингибитор HCV полимеразы, ненуклеозидный ингибитор HCV полимеразы, ингибитор HCV протеазы, ингибитор HCV геликазы или слитый ингибитор HCV. При введении активного соединения или его производного или соли в сочетании с другим антивирусным агентом активность исходного соединения может увеличиваться. Когда лечение представляет собой сочетанную терапию, такое введение может быть совместным или поочередным по отношению к нуклеозидным производным. «Совместное введение» в используемом здесь смысле включает введение агентов в одно и то же время или в разное время. Введение двух или более агентов в одно и то же время может достигаться при использовании одного состава, содержащего два или более активных ингредиентов или практически одновременным введением двух или нескольких дозировочных форм с одним активным агентом.
Следует понимать, что данные здесь ссылки на лечение распространяются и на профилактику, а также и на лечение уже имеющихся состояний. Кроме того, термин «лечение» HCV инфекции, в используемом здесь смысле, включает также лечение и профилактику заболевания или состояния, связанного с HCV инфекцией или опосредованного 4CV инфекцией, или их клинических симптомов.
Пример 1
(2R, 3R, 4R, 5R)-5-(4-амино-2-оксо-2Н-пиримидин-1-ил)-4-фтор-4-метил-3-пропионилокси-тетрагидрофуран-2-илметиловый эфир пропионовой кислоты (I-3)
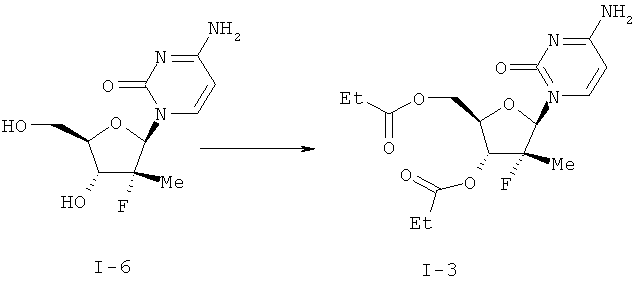
К суспензии I-6 (30 г, 0,116 ммоль), DMAP (1,4 г, 11,57 ммоль) в THF (300 мл) и воде (150 мл) добавляют TEA (35,1 г, 0,347 моль) с получением прозрачного раствора (рН около 11). Реакционную смесь охладили до 5-10°С и по каплям стали добавлять пропионовый альдегид (30,1 г, 0,23/моль), перемешивая двухфазную реакционную смесь. Значения рН регистрируют и поддерживают на уровне около 11-12, одновременно подкапывая раствор КОН. Течение отслеживают с помощью HPLC диализа и после того, как был добавлен пропионилхлорид, в реакционной смеси содержалось около 52% диэфира, 30% моноэфиров и 15% исходного вещества. При условиях, определенных выше, снова по каплям добавляют дополнительно пропионовый ангидрид (45,2 г, 0,347 моль). Реакционную смесь выдерживают в течение ночи без перемешивания. HPLC органическая фаза показала …96% диэфира и 2% моноэфира. Эти фазы собирают и водные фазы экстрагируют THF (100 мл). Объединенные органические фазы промывают брином. Органическую фазу отфильтровывают, выпаривают, а остаток растворяют в воде (са. 500 мл) и затем разбавляют БРА (са. 100 мл). Полученную смесь медленно охлаждают до комнатной температуры при перемешивании. Получившийся осадок отфильтровывают, промывают водой и гептаном, высушивают при около 60°С под вакуумом в течение са. 60 часов до получения 3,45 г (80,3%) 1-3 с чистотой 90,7% согласно HPLC.
Пример 2
(2R, 3R, 4R, 5R)-5-(4-амино-2-оксо-2Н-пиримидин-1-ил)-4-фтор-3-изобутирилокси-4-метил-тетрагидрофуран-2-илметиловый эфир изомасляной кислоты (I-2)

К охлаждаемой на льду суспензии I-6 (970 г, 3,74 моль) и DMAP (50 г, 0,412 моль) в THF (10 л) добавляют TEA (2,3 кг, 16,5 моль) и воду (7 л), в результате получают прозрачный раствор. К смеси при перемешивании медленно добавляют изобутирилхлорид (3 эквивалента), поддерживая температуру при са. 0°С. Еще 1,2 и затем 0,7 эквивалента изобутирилхлорида добавляют до тех пор, пока HPLC не покажет, что реакция практически движется к завершению (всего са. 1,95 кг). Реакционную смесь подкисляют конц. НСl до рН са. 6,4 и органическую фазу промывают EtOAc (2×10 л). Объединенные экстракты промывают водой (1×15 л). Органическую фазу отфильтровывают и концентрируют в вакууме. Остаток растворяют в IРА (са. 20 кг) и добавляют гептан (14,2 кг). Раствор нагревают до са. 74-75°C с получением чистого раствора, затем са. 5 л удаляют дистилляцией. Получившийся раствор медленно охлаждают до комнатной температуры. Осадок формируется при са. 42-43°С. Охлаждение до 5°С продолжается медленно, затем в течение ночи осуществляют перемешивание. Полученный твердый осадок отфильтровывают и фильтрат промывают смесью IPA/гептан (1:8) (13,4 кг), высушивают под вакуумом при около 60-70°C с получением 1,295 кг (86,65%) 1-2 с чистотой 99,45% согласно HPLC.
Пример 3
Изобутиловый эфир (2R, 3R, 4R, 5R)-5-(4-амино-2-оксо-2Н-пиримидин-1-ил)-4-фтор-2-изобутоксикарбонилоксиметил-4-метил-тетрагидрофуран-3-ил эфира карбоновой кислоты (I-4).

Суспензию 1-6 (700 мг), DMAP (33 мг) в THF (7 мл) разбавляют брином (7 мл). Добавляют разбавленный NaOH, пока рН не станет са. 11. Реакционную смесь охлаждают в ледяной бане и по каплям добавляют изобутил хлорформиат (1,11 г) в перемешиваемую двухфазную реакционную смесь, поддерживая рН до около 11 добавлением NaOH, если нужно. HPLC анализ показывает главным образом дикарбонат, загрязненный са. 15% монокарбонатов. К охлаждаемому на льду раствору добавляют еще дополнительный 1 экв. изобутилхлорформиата. HPLC показывает практически завершение реакции. Получившуюся смесь оставляют на ночь при комнатной температуре. Добавляют ЕtOАс (са. 50 мл) и рН водной фазы регулируют до 7,5 конц. НСl. Фазы разделяют, органическую фазу промывают водой (3х) и выпаривают досуха с получением бесцветного осадка (са. 1,22 г). Осадок растворяют в горячем ацетоне, что приводит к прозрачному раствору, который медленно охлаждают до комнатной температуры, что приводит к выпадению осадка. Осадок растворяют в IРА (са. 20 мл), что затем приводит к образованию осадка, который фильтруют и последовательно промывают IPA гептаном, затем сушат с получением 0,85 г (68,5%) 1-4 с чистотой 97,5% согласно HPLC.
Пример 4
Пропиловый эфир (2R, 3R, 4R, 5R)-5-(4-амино-2-оксо-2Н-пиримидин-1-ил)-4-фтор-4-метил-2-пропоксикарбонилоксиметил-тетрагидро-фуран-3-ил эфира карбоновой кислоты; солянокислая соль (I-5)
У суспензии I-6 (700 мг, 2,70 ммоль), DMAP (33 мг, 0,27 ммоль) в THF (7 мл) и разбавленной брином (7 мл) добавляют разбавленный КОН для поддержания рН са. 11. Реакционную смесь охлаждают до са 5°С и к двухфазной реакционной смеси при перемешивании по каплям добавляют n-пропилхлорформиат (1,0 г). HPLC показал образование целевого продукта наряду с са. 20% монокарбонатов. Две дополнительные порции пропилхлорформиата добавляют (2×1 эквивалент) к холодному раствору до тех пор, пока HPLC анализ не покажет, что реакция пришла к завершению. Реакционную смесь разбавляют ЕtOАс (50 мл) и рН водного раствора доводят до СА. 6,5 конц. НСl. Фазы разделяют, и органическую фазу промывают водой (3х), выпаривают досуха с получением бесцветного осадка (са. 1,1 г). Горячий IPA раствор подкисляют 4N НСl (са. 1 мл) и выпаривают досуха. Получившийся осадок перерастворяют в горячем ЕtOН (са. 115 мл) и перемешивают в течение ночи при комнатной температуре. Образовавшуюся твердую массу отфильтровывают и остаток промывают смесью МеОН/гептан (1:1). Оставшийся осадок высушивают в вакууме при около 60°C с получением 0,325 г (25,8%) 1-5 с чистотой 97,5% согласно HPLC.
Пример 5
Определение фармакокинетических параметров на крысах
Использовали интактных крыс-самцов мыши IGS Wistar Han Rats Crl:WI (GLxx/BRL/Han) IGS BR (Hanover Wistar), весом 200-250 г. Группы из трех животных использовали для каждого уровня доз исследуемого соединения. Животным был организован нормальный доступ к корму и питью во время эксперимента. Исследуемая субстанция была приготовлена как водная суспензия, содержащая Captex355EP, Capmul MCM, EtOH и пропиленгликоль (30:20:20:30) при дозе, эквивалентной 10 мг/кг I-6 и вводилась орально через желудочный зонд. Образцы крови (0,3 мл) отбирали у обработанных таким образом крыс через 0,25, 0,5, 1, 3, 5 и 8 часов из шейной канюли и через 24 часа кардиальной пункцией. К пробам добавляли оксалат калия/NaF и хранили их на льду в течение всей процедуры отбора проб. Пробы подвергали вращению в рефрижераторной центрифуге при -4°C с такой скоростью, с какой было возможно, а плазму образцов хранили при -80°С во фризере до начала анализа. Аликвоты плазмы (0,05 мл) смешивали с 0,1 мл ацетонитрила. Добавляли внутренний стандарт (0,05 мл в воде) и 0,02 мл чистого растворителя. Серию калибровочных стандартов готовили, смешивая 0,05 мл аликвоты плазмы от небработанных крыс с 0,1 мл ацетонитрила, 0,02 мл аликвотами стандартного раствора в смеси метанол:вода (1:1) и 0,05 мл аликвотами внутреннего стандарта в воде. Каждый образец плазмы и калибровочного стандарта тщательно взбалтывали на вортексе и затем центрифугировали при 3000 об/мин, в течение 5 минут для осаждения белка. Супернатант (100 µл каждый) после центрифугирования переносили в 96-луночный планшет, содержащий 200 мл водной подвижной фазы для LC/MS/MS анализа.
Анализ проб:
Пролекарства анализировали, используя высокоэффективную жидкостную хроматографию в тандеме масс-спектрометрией (HPLC/MS/MS). Для разделения использовали Thermo Aquasil С18 4,6×50 мл колонку (5 µМ). Для ионизации процесса использовали Electrospray lonization (ESI). Подвижная доза А содержала 5 мМ ацетата аммония в воде с 0,1% муравьиной кислоты, а мобильная фаза В содержала МеОН с 0,1% муравьиной кислоты. Элюцию выполняли при следующем градиенте со скоростью протекания 1 мл/мин:
Пример 6
Определение фармакокинетических параметров на обезьянах
Использовались три самца Cynomolgus обезьян весом 8-10 кг. В течение эксперимента животным дозволялся открытый доступ к корму и воде. Вес животных в течение всего периода введения лекарства, а также неблагоприятные реакции у животных регистрировали. Исследуемая субстанция была приготовлена как водная суспензия, содержащая в своем составе гипромеллозу (2910, 50 спз), USP, полисорбат 80, NF, бензиловый спирт, NF (5,0, 4,0 и 9,0 мг/мл) и стерильную воду (в количестве, достаточном для получения 1,0 мл), при дозе эквивалентной 10 мг/кг 1-6 и 0,5 мг/кг вводили орально через желудочный зонд. Образцы крови (0,5-1,0 мл) собирали через 0,083, 0,25, 0,5, 1, 3, 5, 8 и 24 часа. Обработки проб и анализ выполняли так же, как описано в эксперименте на крысах. Пробу 5 мл мочи брали от каждой обезьяны перед введением дозы за 0-8 часов. Образцы мочи хранили при -80°С и анализировали с помощью LC/MS/MS. Стандартные кривые были составлены для чистой плазмы животных, содержащей NaF и оксалат калия.
Пример 7
Сасо протокол
Материалы:
Сухие Krebs-Henseleit буфер, дигидрат хлорида кальция и бикарбонат натрия были получены от Sigma (st. Louis, МО). Сасо-2 клетки (Пассаж ≈ 100) были получены от Roche Basel. DMEM (обогащенная среда, 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота (HEPES) и сыворотка быка были получены от JRH Bioscience (Lenexa, KS). MEM ненезаменимые аминокислоты, L-глутамин, пенициллин и стрептомицин были получены от GIBCO, habs, Life Tech. LLC (Grand Island, NY). Вкладыши в лунки для клеточных культур (6,5 мм диаметром, 1,12 см2, размером пор 0,4 µм), были получены от Costar (Cambridge, МА).
Клеточные культуры:
Клетки выращивали в 75 см2 колбах при поддержании температуры 37°С в атмосфере 5% СO2 и 95% воздуха. Культуральная среда содержала DMEM/обогащенную среду с добавлением 5% сыворотки быка, 25 мМ HEPES, 1% MEM ненезаменимых аминокислот, 1% L-глутамина, 100 ЕД/мл пенициллина и 100 рг/мл стрептомицина. Культуры пассажировали каждую неделю в соотношении разведения 1-3. Для исследования проницаемости Сасо-2 клетки в пассажах номер 110-120 засевали при плотности 400000 клеток/см2 на переносимые поликарбонатные фильтры в вставляющиеся вкладыши и давали расти в течение 7 дней перед тем, как использовать.
Krebs-Henseleit Буфер:
Krebs-Henseleit бикарбонатный буфер, содержащий 10 мМ глюкозы и 25 м СаСl2, с установленными рН 6,5 и 7,4 готовят по инструкции на упаковке. Сухие соли количественно растворяют в примерно 90% требуемого объема Millipore воды. Дигидрат хлорида кальция и бикарбонат натрия добавляют прежде, чем установить рН с помощью 1н. НСl или 1н. NaOH. Дополнительная порция Millipore воды добавлялась, когда раствор нужно было довести до конечного объема. Раствор стерилизовали фильтрацией, используя мембраны с пористостью 0,22 микрон и хранили в холодильнике (~ 20°С) перед использованием.
Приготовление клеточных культур:
Дифференцированные клетки были получены от Cell Culture Core Facility в уравновешенном при 37°С состоянии в атмосфере 5% СO2 и 95% воздуха. Вставки, содержащие сасо-2 монослои, были орошены при 37°С уравновешенным Krebs-Henseleit буфером, рН 7,4.
Метод:
Клеточные вкладыши (вставки) использовали как диффузионные камеры. рН Krebs-Henseleit буфера в апикальной и базолатеральной камерах был 6,5 и 7,4, соответственно, а начальная концентрация субстрата на апикальной стороне была 100 µM. Клетки были преинкубированы с исследуемым соединением с апикальной камере в течение примерно 30 минут при 37°С в атмосфере 5% СО2 и 95% воздуха. Эксперименты начинали, когда клеточные вставки с 100 µМ соединений в Krebs-Henseleit буфере рН 665 были перенесены в новый планшет с предварительно уравновешенным буфером в базолатеральной камере. Образцы с донорной стороны на 0 минуте, а также как в донорной, так и ресиверной сторон через 30 минут собирали для анализа.
Контроль после эксперимента:
Для оценки превращения в диффузионной системе использовали Lucifer Yellow. Вслед за последним отбором проб для определения исследуемых соединений Lucifer Yellow добавлялся в апикальную камеру, чтобы обнаружить первоначальную концентрацию 100 µМ. Спустя 60 минут инкубации 250 µл были удалены из базальной камеры и исследованы.
Вычисление коэффициента проницаемости (Рарр):
Рарр вычисляли по следующей формуле:
Papp=(V×dC)/(А×Со×dt)=(см/сек)
где V - объем (см3) принимаемого раствора, А - площадь поверхности (см2) вставки, Со - начальная концентрация (нМ), a dC/dt - представляет собой изменение концентрации в принимающей (ресиверной) камере с течением времени, то есть уклон (нМ/мин) (тангенс угла наклона) концентрации в принимающей камере по отношению ко времени. Концентрации в каждой точке отбора проб корректировали, чтобы рассчитать аликвоты, удаленные или перемещенные донорскими вставками в новые планшеты в зависимости от эксперимента.
Пример 8
Фармацевтические композиции заявленных соединений для введения разными способами были приготовлены, как описано в примере 8.
Композиция для содержащего влагу гранулированного состава для орального введения (А):
Ингредиенты смешивают, гранулируют и распределяют в твердые желатиновые капсулы, содержащие около 500 мг активного соединения. Композиция для орального применения (В):
Ингредиенты собирают, гранулируют, используя в качестве растворителя воду. Состав высушивают затем и формируют таблетки, содержащие около 500 мг активного соединения, в соответствующем таблетирующем устройстве.
Композиция для орального применения (С):
Ингредиенты смешивают с получением суспензии для орального применения Состав для суппозиториев (D):
Ингредиенты расплавляют вместе и перемешивают на паровой бане. Затем выливают в формы на 2,5 г общего веса.
Особенности (признаки) изобретения, раскрытые в предшествующем описании или приведенной далее формуле изобретения, выраженные в виде их специфических форм или в специальных терминах для того, чтобы дать представление об их функциях или о способе или процессе для достижения провозглашенного результата, соответственно могут либо сами по себе, либо в сочетании друг с другом, использоваться для воспроизведения изобретения в самых его разнообразных формах.
Вышеприведенное изобретение детально описано с помощью иллюстративных примеров, чтобы достичь ясности и понимания. Специалисту в данной области будет очевидно, что все изменения и модификации, которые могут быть осуществлены, подпадают под объем формулы изобретения. Поэтому понятно, что вышеприведенное описание следует расценивать как иллюстративное и не ограничивающее. Объем данного изобретения должен поэтому определяться не ссылками на вышеприведенное описание; а должен вместо этого соответствовать следующей далее формуле изобретения наряду с полным объемом эквивалентов, которые охватываются пунктами формулы изобретения.
Все патенты, патентные заявки и публикации, цитированные здесь, включены в качестве ссылок во всей их полноте во всех отношениях в той же самой степени, как если бы на каждый отдельный патент, заявку на патент или публикацию было бы представлено аналогичное отдельное обозначение.
Настоящее изобретение относится к соединениям формулы (I), где R1 выбран из этила, н-пропила, изо-пропила или изобутила, и к его фармацевтически приемлемым солям. Кроме того, изобретение относится к фармацевтической композиции на основе указанных соединений, предназначенной для лечения заболевания, опосредованного вирусом гепатита С (HCV), а также к способу лечения заболевания, опосредованного вирусом гепатита С (HCV), и к способу избирательного O-ацилирования нуклеозида II для получения O-ацил нуклеозида I в щелочных реакционных условиях, включающему стадии: (i) растворение II и DMAP в гетерогенной смеси воды и растворителя и добавление водного основания для регулирования рН от около 7,5 до около 12; (ii) необязательное добавление достаточного количества насыщенного водного NaCl для получения двухфазной реакционной смеси; (iii) добавление ацилирующего агента и дополнительного основания, достаточного для сохранения рН от около 7,5 до около 12; (iv) мониторинг реакции и прерывание добавления указанного ацилирующего агента и указанного основания при достижении конверсии достаточного уровня; (v) необязательное контактирование O-ацилнуклеозида с фармацевтически приемлемой кислотой для образования фармацевтически приемлемой соли. 4 н. и 5 з.п. ф-лы, 2 табл.
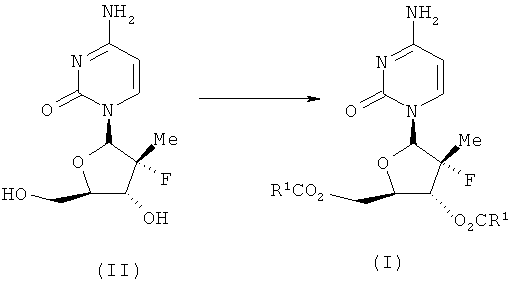
1. Соединение формулы I
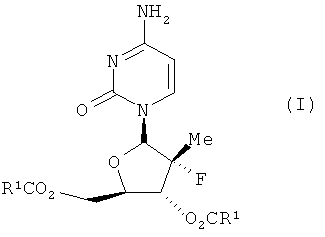
где R1 выбран из группы, состоящей из этила, н-пропила, изопропила или изобутила, и
его фармацевтически приемлемые соли.
2. Соединение по п.1, в котором R1 является этилом или изопропилом.
3. Соединение по п.1, в котором R1 является изопропилом, а соединение представляет собой солянокислую или сульфатную соль.
4. Соединение по пп.1-3, предназначенное для лечения заболевания, опосредованного вирусом гепатита С (HCV).
5. Фармацевтическая композиция, предназначенная для лечения заболевания, опосредованного вирусом гепатита С (HCV), включающая терапевтически эффективное количество соединения по пп.1-4 в сочетании с одним фармацевтически приемлемым носителем, разбавителем или наполнителем.
6. Способ лечения заболевания, опосредованного вирусом гепатита С (HCV), включающий введение пациенту, нуждающемуся в этом, терапевтически эффективной дозы соединения по пп.1-4.
7. Способ по п.6, отличающийся тем, что пациенту вводят дозу от 0,1 до 10 г в сутки.
8. Способ по п.6 или 7, дополнительно включающий введение, по крайней мере, одного модулятора иммунной системы и/или, по крайней мере, одного антивирусного агента, который ингибирует репликацию HCV.
9. Способ избирательного O-ацилирования нуклеозида II для получения O-ацил нуклеозида I в щелочных реакционных условиях
где R1 выбран из группы, состоящей из этила, н-пропила, изопропила или изобутила, включающий стадии:
(i) растворение II и DMAP в гетерогенной смеси воды и растворителя и добавление водного основания для регулирования рН от около 7,5 до около 12;
(ii) необязательное добавление достаточного количества насыщенного водного NaCl для получения двухфазной реакционной смеси;
(iii) добавление ацилирующего агента и дополнительного основания, достаточного для сохранения рН от около 7,5 до около 12;
(iv) мониторинг реакции и прерывание добавления указанного ацилирующего агента и указанного основания при достижении конверсии достаточного уровня;
(v) необязательное контактирование O-ацилнуклеозида с фармацевтически приемлемой кислотой для образования фармацевтически приемлемой соли.
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| J.L.CLARK ET AL // J.MED.CHEM., V.48, 2005, P.5504-5508 | |||
| Метил-2-О-бензоил-3-фтор-3-дезокси-Д-рибофуранозиды в качестве промежуточных продуктов в синтезе биологически активных 3 @ -фтор-3 @ -дезоксирибонуклеозидов | 1987 |
|
SU1521738A1 |

