Изобретение относится к новым производным пиразоло/3,4-в/ пиридина формулы I
R1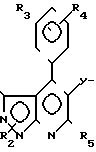 z (I) где R1-H, С1-8-алкил, С3-6-циклоалкил, или
z (I) где R1-H, С1-8-алкил, С3-6-циклоалкил, или 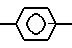 R6, где R6-Н, Сl, Br, Fr, R2 - связан с азотом в положении 1- или 2-пиразолопиридинового кольца и означает С1-8-алкил, фенил, бензил, 2, 3- или 4- пиридил или алкоксифенил, R3 - фтор, хлор или бром, R4-Н, R5-C1-8-алкил, или С3-7-циклоалкил. У- -СН=СН- , Z -
R6, где R6-Н, Сl, Br, Fr, R2 - связан с азотом в положении 1- или 2-пиразолопиридинового кольца и означает С1-8-алкил, фенил, бензил, 2, 3- или 4- пиридил или алкоксифенил, R3 - фтор, хлор или бром, R4-Н, R5-C1-8-алкил, или С3-7-циклоалкил. У- -СН=СН- , Z - 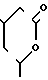 или группу -Q-CH2-W-CH2-CO2R12, где Q-CH/OH/, W-C(R′′)(OH), где R′′-H, R12-H или алкильная часть химически или физиологически гидролизуемого алкильного сложного эфира или натрий, калий или 1/2-кальция.
или группу -Q-CH2-W-CH2-CO2R12, где Q-CH/OH/, W-C(R′′)(OH), где R′′-H, R12-H или алкильная часть химически или физиологически гидролизуемого алкильного сложного эфира или натрий, калий или 1/2-кальция.
Известно, что некоторые метаболические продукты брожения такие, как компактин и др. являются ингибиторами НМ6-СоА редуктазы, которая является ферментом, ограничивающим скорость при биосинтезе холестерина.
Цель изобретения - синтез новых соединений, обладающих ценными свойствами.
Цель достигается соединениями формулы I.
П р и м е р 1. Этил/E/-7-/4-/4′′-фторфенил/-1′, 3′-диметил-6-1/1′′-метил-этил) -пиразоло(3,4-b)пиридин-5-ил/-3,5-дигидроксигепт-6-еноат (соединение 1-1-1).
Данное соединение получалось с помощью синтеза, включающего следующие стадии реакции, пример 1-а-пример 1-g.
П р и м е р 1-а. Этил 4-/4-фторфенил/-1,3-диметил-6-/1′-метилэтил/пиразоло/3, 4-b/-пиридин-5-илкарбоксилат (соединение VII-I).
Синтез дигидросоединения
2,22 г /0,02 моля/5-амино-1,3-диметилпиразола и 5,3 г /0,02 моля/этил 2-/4′-фторбензилиден /-4-метил-3-оксо-пентаноата смешивались и нагревались при температуре примерно 130оС в течение 1 ч. Вещества, имеющие низкую точку кипения, отгонялись при пониженном давлении с помощью роторного испарителя. Затем реакционная смесь растворялась в хлороформе, промывалась насыщенным водным раствором карбоната натрия и водой и сушилась над безводным сульфатом магния. Хлороформ выпаривался и остаточное масло очищалось с помощью хроматографии на силикагельной колонке и получался дигидропиразоло /3,4-b/пиридин/XI-I/.
ПЯМР/СДСI3/δ млн.дол.: 0,81 /д. I=7 Гц, 3Н/, 1,0-1,3/ м., 6Н/, 1,97/ с. , 3Н/, 2,64 /м., IH/, 3,44/д. I=3 Гц, IH/, 3,81 /с., 3H/, 4,06/ кв. I=7 Гц, 2Н/, 4,48 /д., I= =3 Гц, IH/, 6,84 /м., 4Н/
Метод окисления -1.
7,54 г дигидросоединения, полученного на стадии, описанной выше, растворялось в 15 мл ледяной уксусной кислоты и к смеси добавлялось 2,2 г хромового ангидрида. Смесь перемешивалась при комнатной температуре /15-20оC/. После подтверждения исчезновения исходных веществ с помощью тонкослойной хроматографии к смеси добавлялось 100 мл воды. Смесь экстрагировалась хлороформом. Хлороформный слой встряхивался вместе с насыщенным водным раствором карбоната натрия и водой и затем сушился над безводным сульфатом магния.
Хлороформ отгонялся. Остаточное масло очищалось с помощью хроматографии на силикагельной колонке /элюент: 1% метанол (хлороформ) и получалось желаемое соединение в виде белых кристаллов.
Точка плавления: 60-64оС, выход: 52% (в расчете на аминопиразол).
Метод окисления -2.
1 г дигидросоединения, полученного на стадии, описанной выше, растворялся в ацетоне, содержащем небольшое количество этанола, и к смеси добавлялся перманганат калия (1,5-кратное мол количество). Смесь перемешивалась при комнатной температуре в течение 1 дня. После подтверждения полного исчезновения непрореагировавшего дигидросоединения с помощью тонкослойной хроматографии диоксид марганца удалялся с помощью фильтрации. Фильтрат концентрировался и остаточное масло обрабатывалось таким же образом, как в методе окисления -1, и получалось желаемое соединение.
Выход: 60% /в расчете на аминопиразол/.
П р и м е р 1-b. 4-/4′-фторфенил/-5-гидроксиметил-1,3-диметил-6-/1′-метилэтил/ пиразоло /3,4-b/пиридин/соединение VI-I/.
5,0 г /0,014 мол/ соединения VII-I растворялось в сухом толуоле в атмосфере азота и охлаждалось до 0оС в ледяной ванне. К этому раствору, по каплям, добавлялось 35 мл 16 мас.% раствора диизобутилалюминийгидридтолуол и затем смесь перемешивалась при 0оС в течение 2 ч. После подтверждения полного исчезновения соединения VII-I с помощью тонкослойной хроматографии к смеси при 0оС добавлялся насыщенный водный раствор хлорида аммония для завершения реакции. К реакционной смеси добавлялся диэтиловый эфир и органический слой отделялся. Желированное вещество растворялось путем добавления водного раствора гидроокиси натрия и вновь экстрагировалось этиловым эфиром. Экстракт этилового эфира собирался и высушивался над безводным сульфатом магния. Экстракт отделялся с помощью фильтрации и растворитель отгонялся, при этом получалось 3,9 г слегка желтого желаемого соединения.
Выход: 88%. точка плавления: 174-175оС.
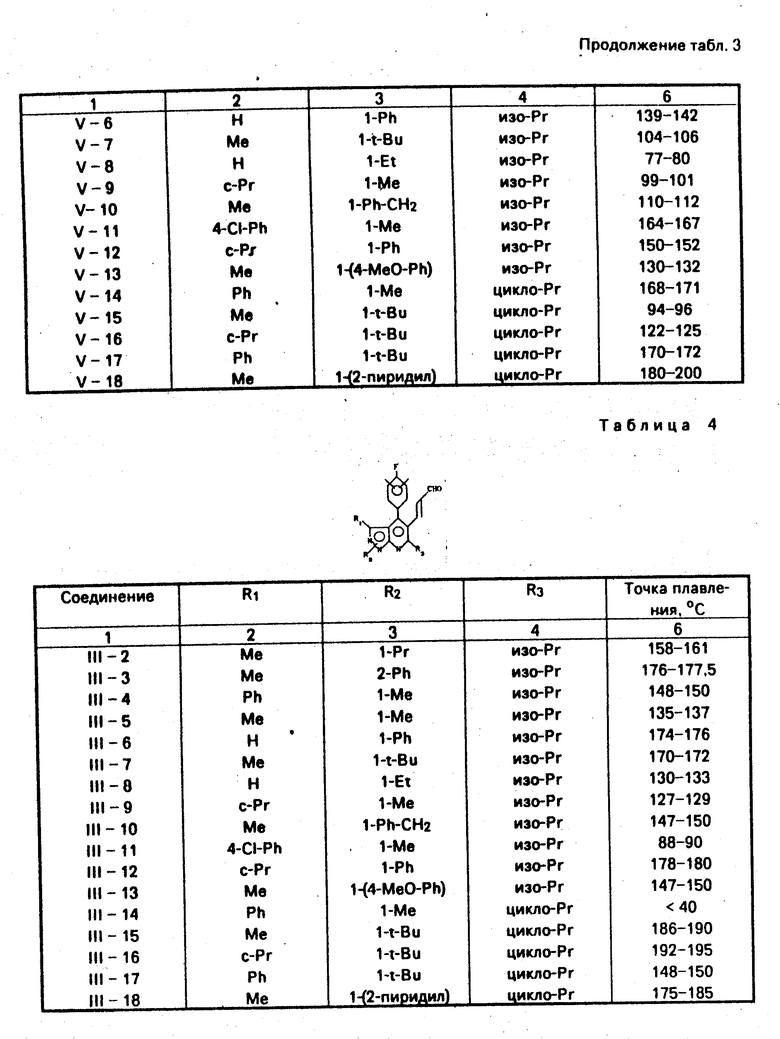
П р и м е р 1-с. /4-(4′-фторфенил/-1,3-диметил-6-/1′- метилэтил/пиралозо/3,4-b/пиридин-5-ил/карбоксиальдегид /соединение V-I/
4,2 г /19 ммол/ хлорхромата пиридиния, 0,69 г безводного ацетата натрия и 3,8 г /12 ммол/ соединения VI-I суспендировались в 50 мл сухого дихлорметана при комнатной температуре. Реакционный раствор перемешивался в течение 1 ч и затем к нему добавлялось 100 мл диэтилового эфира. Смесь тщательно перемешивалась. Реакционная смесь подвергалась фильтрации через слой целита, и экстракт выпаривался при пониженном давлении досуха. Остаток подвергался хроматографии на силикагельной колонке (элюент: хлороформ) для получения 2,9 г (выход: 78%) желаемого соединения в виде слегка желтого вещества.
Точка плавления: 144-146оС.
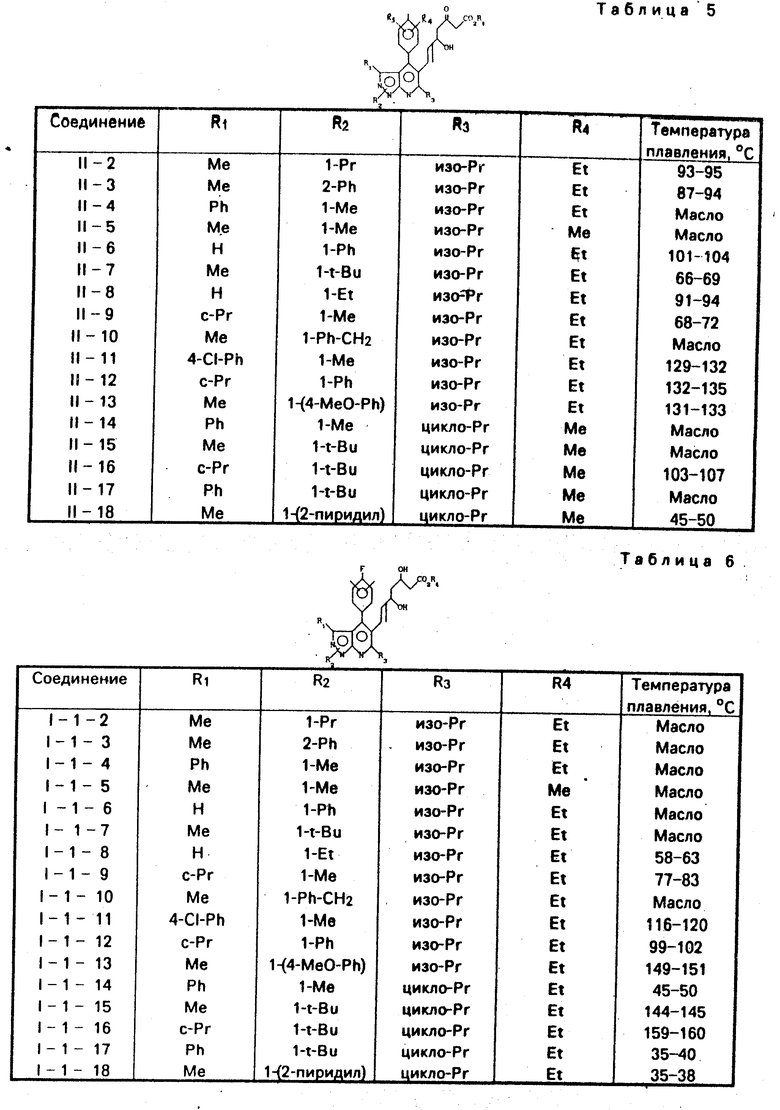
П р и м е р ы 1-d и 1-е. /E/-3-/4′-/4′′-фторфенил/-1′,3′-диметил-6′- 1′′-метил-этил/пиразоло/3,4-b/пиридин-5′-ил/про- пенальдегид /cоединение III-I/
П р и м е р 1-d. 14,5 г /40 ммол/циc-1-этокси-2/-три-н-бутилстаннил/этилена растворялось в 50 мл сухого тетрагидрофурана и раствор охлаждался до -78оС в атмосфере азота. К этому раствору, по каплям, добавлялось 26 мл /40 ммол/ 15 мас.% раствора н-бутиллитий-н-гексан. Смесь перемешивалась в течение 20 мин и затем к ней по каплям добавлялся раствор 2,5 г /8 ммол/ соединения V-I, растворенного в 20 мл сухого тетрагидрофурана. Реакционная смесь перемешивалась при -78оС в течение 1 ч, затем для завершения реакции к ней добавлялось 26 мл насыщенного раствора хлористого аммония. Органический слой экстрагировался диэтиловым эфиром. Эфирный экстракт промывался насыщенным водным раствором хлорида натрия и сушился над безводным сульфатом магния. Растворитель отгонялся при пониженном давлении, остаток подвергался жидкостному разделению между н-гексаном и ацетонитрилом. Ацетонитрильный слой подвергался перегонке при пониженном давлении и получалось по существу чистое соединение IV-I.
П р и м е р 1-е. Соединение IV-I, получаемое в примере 1-d, растворялось в 70 мл тетрагидрофурана, и к смеси добавлялось 20 мл воды и 3 г п-толуолсульфокислоты. Смесь перемешивалась при комнатной температуре в течение 2 ч. Реакционный раствор осторожно нейтрализовался водным раствором гидроокисинатрия. Затем к нему добавлялся диэтиловый эфир, и несколько раз проводилось экстрагирование. Экстракт промывался насыщенным водным раствором хлористого натрия и сушился над безводным сульфатом магния. Затем растворитель отгонялся при пониженном давлении. Остаток подвергался хроматографии на силикагельной колонке /элюент: этилацетат /н-гексан=1/9/объем/объем/, давая желаемое соединение в виде желтого вещества.
Количество: 2,2 г /выход:79%/.
Точка плавления:133-134оС
П р и м е р 1-f. Этил-/E/-7-/4′-/4′′ фторфенил/-1′,3′ диметил-6′-/1′′-метилэтил /пиразоло/3,4-b/пиридин-5′-ил/-5- гидрокси-3-оксогепто-6-еноат /соединение II-I/. 1,25 г 60% гидрида натрия промывалось сухим петролейным эфиром, сушилось в токе азота и затем суспендировалось в 200 мл сухого гидрофурана. Суспензия охлаждалась до -15оС в атмосфере азота, к ней по каплям добавлялось 3,9 мл /30 ммол/ этилацетоацетата. Смесь перемешивалась в течение 15 мин. Затем к ней, по каплям, добавлялось 20 мл /30 ммол/ 15 мас. %. раствора н-бутиллития-н-гексан, и смесь перемешивалась в течение 30 мин. Далее к смеси добавлялся раствор 2,1 г /6,1 ммоля/ соединения III-I, растворенного в сухом тетрагидрофуране, и смесь перемешивалась в течение 1 ч. К реакционной смеси, при -15оС, добавлялось 10 мл насыщенного водного раствора хлористого аммония, смесь три раза экстрагировалась диэтиловым эфиром. Эфирный раствор промывался насыщенным водным раствором хлористого натрия, сушился над безводным сульфатом магния и затем выпаривался при пониженном давлении досуха. Остаток подвергался хроматографии на силикагельной колонке/элюент: этилацетат/хлороформ =1/9/объем/объем//, давая 2,5 г/ выход:89%/ желаемого соединения в виде белого вещества.
Точка плавления: 95-98оС.
П р и м е р I-g. Метод восстановления 1. Этил/E/-7-/4′-/4′′-фторфенил/-1′, 3′-ди- метил-6′-/1′′-метилэтил/пиразоло /3,4-b/пиридин-5′-ил/-3,5-дигидроксигепт-6- еноат (соединение 1-1-1).
2,32 г (4,96 ммоля) соединения II-I растворялось в 20 мл этанола в атмосфере азота, смесь охлаждалась до 0оС Затем к ней добавлялось 740 мг (20 ммол) боргидрида натрия, смесь перемешивалась в течение 1 ч. Смесь осторожно нейтрализовалась путем добавления 10% водного раствора соляной кислоты и затем 3 раза экстрагировалась диэтиловым эфиром. Диэтилово-эфирный раствор промывался насыщенным водным раствором хлористого натрия, сушился над безводным сульфатом магния и затем выпаривался при пониженном давлении досуха. Остаточное масло очищалось с помощью хроматографии на силикагельной колонке, давая чистый желаемый продукт в виде бесцветного вязкого маслянистого вещества.
Количество: 1,81 г (выход:78%)
ЯМР /δ млн. дол. в СДСl3/ 1,28 /т., I=8 Гц, 3Н/, 1,32/д, I=8 Гц, 6Н/, 1,4-1,8/ м. , IH/, 1,92 / c., 3H/, 2,2-2,6/ м., 3Н/, 2,9-3,8/м., 2Н/, 3,42/ гепталет. , I= 8 Гц, IH/, 4,06/с., 3Н/, 4,1-4,6 /м., 4Н/, 5,1-5,5/ м., IH/, 6,6-6,7/м., IH/, 6,9-7,3/ м., 4Н/
Метод восстановления 2.
Этил/E/-7-/6′-циклопропил-4′-/4′′- фторфенил/-1′-метил-31- фенилпиразоло/3,4-b/пиридин-5′-ил/-3,5-дигидрокси- гепт-6-еноат (соединение 1-1-14).
200 мл диэтилового эфирного раствора примерно 0,15 мол/л боргидрида цинка перемешивалось в атмосфере азота при охлаждении раствора при -70оС. К смеси, постепенно, по стенке реактора добавлялся раствор 3,75 г /7,12х10-3 мол/ соединения 11-14, растворенного в 40 мл сухого диэтилового эфира. Далее смесь перемешивалась при -70оС в течение 6 ч. После подтверждения значительного исчезновения исходных веществ с помощью тонкослойной хроматографии к смеси при -70оС добавлялось 40 мл метанола и затем 100 мл воды для завершения реакции. К реакционному раствору для доведения рН до 4 добавлялся диэтиловый эфир и разбавленные уксусная кислота и продукт экстрагировался диэтиловым эфиром.
Диэтилово-эфирный слой промывался водой до тех пор, пока диэтилово-эфирный слой не становился нейтральным, далее он промывался насыщенным водным раствором хлористого натрия. Диэтилово-эфирный слой сушился над безводным сульфатом магния и затем растворитель выпаривался с помощью испарителя. Остаток подвергался хроматографии на силикагельной колонке /элюент: бензол/этилацетат/давая 3,09 г (82,0%) желаемого соединения в виде слегка желтого порошка.
П р и м е р 2. Натрий /E/-7-/4′-/4′′-фторфенил/-1′,3′-диметил -6′-/1′′-метилэтил/пиразоло/3,4-b/пиридин-5′-ил/-3,5-дигидрок- сигент -6-еноат (соединение 1-5-1).
200 мг (0,43 ммол) соединения 1-1-1 растворялось в 2 мл этанола, и к смеси по каплям добавлялось 0,85 мл 0,5 н. водного раствора гидроокиси натрия. Смесь перемешивалась при комнатной температуре в течение 1 ч. Затем этанол отгонялся при пониженном давлении, к смеси добавлялось 2 мл воды и смесь экстрагировалась диэтиловым эфиром. Водный слой сушился вымораживанием, давая 180 мг (выход:91%) гигроскопичного слегка желтого порошка.
Точка плавления: 258-264оС (с разложением).
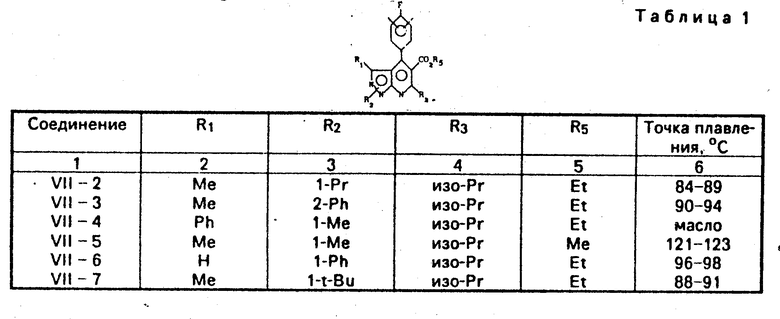
Таким же способом, как в примере 1-а были получены соединения VII-2-VII-18. Физические свойства соединений, полученных при этом, показаны в табл. 1. ПЯМР соединения VII-4 /СДСl3/δ млн. дол.: 0,96 /т., I=8 Гц, 3Н/, 1,42 /д. , I=7 Гц, 6Н/, 3,27 /гепталет, I=7 Гц, IH/, 4,02/ кв., I=8 Гц, 2Н/, 4,18 /м. , 3Н/, 6,6-7,3/ м., 9Н/ ПЯМР соединения VII-8 /СДСl3/δ млн. дол.: 1,07 /т., I= 8 Гц, 3Н/, 1,42/д., I=7 Гц, 6Н/, 1,59/т., I=8 Гц, 3Н 3,41 /гепталет, I=7 Гц, IH/, 4,20 /кв., I=8 Гц, 2Н/, 4,70 /кв., I=8 Гц, 2Н/, 7,1-8,0/м., 5Н/
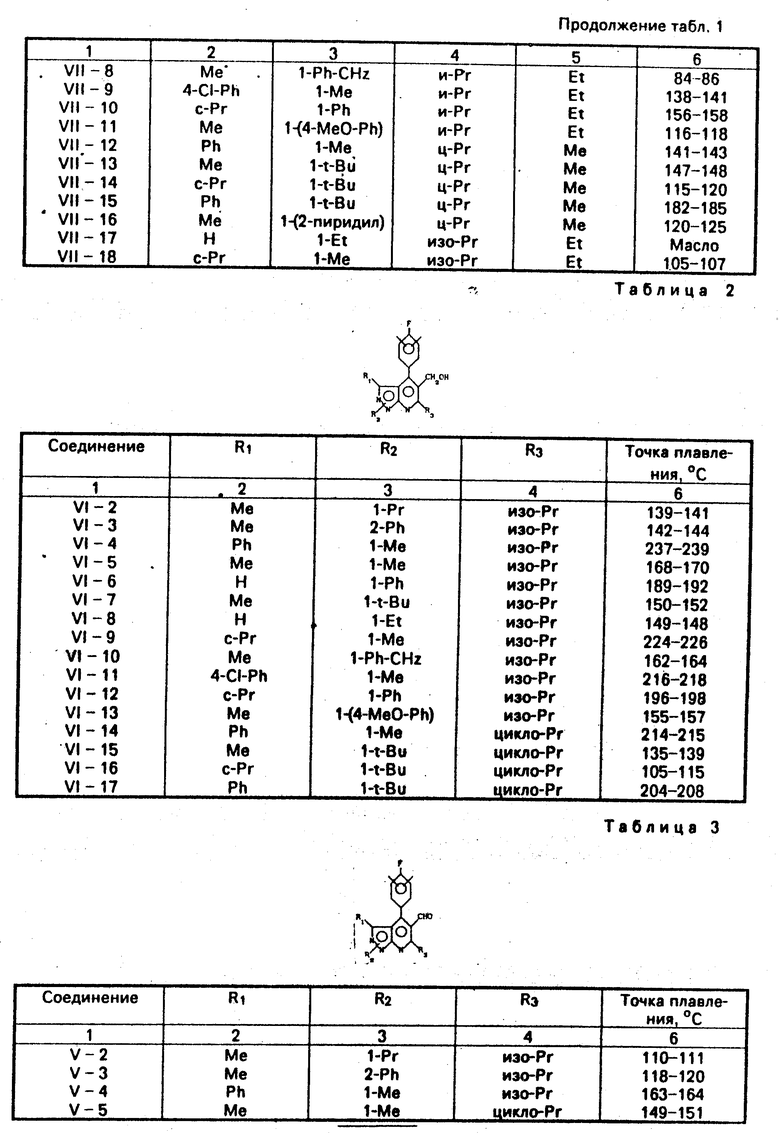
Таким же способом, как в примере 1-b, были получены соединения VI-2-VI-17.
Физические свойства полученных соединений показаны в табл. 2.
Таким же способом, как в примере 1-с, были получены соединения V-2-V-18. Физические свойства полученных соединений, показаны в табл. 3.
Таким же способом, как в примерах 1-d и 1-е, были получены соединения III-2-III-18. Физические свойства полученных соединений показаны в табл. 4.
Таким же способом, как в примере 1-f, получены соединения II-2-II-18. Физические свойства полученных соединений показаны в табл. 5.
ПЯМР соединения II-4 /CДСl3/δ млн. дол.:
1,24 /т., I=9 Гц, 3Н/, 1,32 /д., I=8 Гц, 6Н/, 2,1-2,5 /м., 2Н/, 2,6-2,9 м. , 1Н/, 3,2-3,7/ м.,, 3Н/, 3,9-4,7/ м., 3Н/. 4,11 /c., 3H/, 4,9-5,4/ м., 1Н/,6,3-7,2/м., 1 ОН/,
ПЯМР соединения II-5-/CДCl3/δ млн. дол.:
0,8-1,5/ м. , 4Н/, 1,27/т., I=7 Гц, 3Н/, 1,89/ м., 3Н/, 2,1-2,4 /м., 1Н/, 2,51/д. , I= 6 Гц, 2Н/, 2,6-3,1/м., IH/, 3,40/C., 2H/, 3,94/c., 3H/, 4,17/кв. I= 7 Гц, 2Н/, 4,4-4,8/м., 1Н/, 5,3-5,7/ м., 1Н/, 6,4-6,8/м., 1Н/, 7,0-7,4/ м., 4H/, ПЯМР соединения II-10 /CДСl3/δ млн. дол.:
1,26 /т., I=7 Гц, 3Н/, 1,33/д., I=7 Гц, 6Н/, 1,92/c., 3H/, 2,2-2,6/м., 1Н/, 2,45/д.,I=6 Гц, 2Н/, 3,41/с., 2Н/, 3,43/гепталет I=7 Гц, 1Н/, 4,22/кв. , I=7 Гц, 2Н/, 4,4-4,8/м., 1Н/, 4,8-5,6/м., 1Н/, 5,65/ c., 2H/, 6,4-6,8/м., 1Н/, 7,0-7,7/м., 9Н/. ПЯМР соединения II-14 /СДСl3/δ млн. дол.:
0,9-1,5/м., 4Н/, 1,24/т., I=8 Гц, 3Н/, 2,1-2,5/м.,1Н/,
2,5-2,7/м., 2H/, 3,31/с., 2Н/, 4,05/c., 3H/, 4,12/кв., I=8 Гц, 2Н/
4,4-4,8 /м. , 1Н/, 5,35/д., д., I=17 Гц, I=6 Гц, 1Н/, 6,4-7,2/м., 10Н/ ПЯМР соединения II-15 /CДСl3/δ млн. дол.:
0,9-1,4 (м., 4Н/, 1,26-/т., I=8 Гц, 3Н/, 1,74/c., 9H/,
1,88/c., 3H/, 2,2-2,4-/м., 1Н/, 2,4-2,6/м., 2Н/, 3,35/c., 2H/, 4,14/кв. , I=8 Гц, 2Н/, 4,3-4,7/м., 1Н/, 5,40/д., д., I=17 Гц, I=6 Гц, 1Н/, 6,50/д., I= 17 Гц, 1Н/, 6,9-7,4/м. , 4Н/ ПЯМР соединения II-16 /СДСl3/δ млн. дол.: 0,4-1,4/м. , 8Н/, 1,28/т. , I= 8 Гц, 3Н/, 1,75/c., 9H/, 2,1-2,5/м., 2H/, 2,4-2,6/м. , 2H/, 3,43/c/. , 2H/, 4,23/кв., I=8 Гц, 2H/, 4,4-4,7/м., 1Н/, 6,52/ д. , д. I=17 Гц, I=6 Гц, 1Н/,6,64/ д., 1Н, I=6 Гц/, 6,9-7,6/м., 4Н/, ПЯМР соединения II-17 /СДСl3/δ млн. дол.:
0,8-1,5/м. , 4Н/, 1,20/т., I=8 Гц, 3Н/, 1,80/c., 9H/,2,0-2,7/м., 1Н/, 2,3-2,5/м. , 2Н/, 3,30/c. , 2H/, 4,10/кв., I=8 Гц, 2Н/, 4,3-4,7/м., 1Н/, 5,35/д.,д., I=17 Гц, I=6 Гц, 1Н/, 6,3-7,3/м., 10Н/,
Таким способом, как в примере 1-0 получены соединения 1-1-2-1-1-18. Физические свойства соединений, полученных при этом показаны в табл. 6.
ПЯМР соединения 1-1-2 /СДСl3/δ млн. дол.:
1,29/т. , I=8 Гц, 3Н/, 1,36/д., I=8 Гц, 6Н/, 1,5-1,8/м., 1H/, 1,98/c., 3H/, 2,2-2,7/м. , 3Н/, 2,9-3,7/м., 2Н/, 3,47/гепталет, I=8 Гц, 1Н/, 3,8-4,6/м. , 2Н/, 4,20/кв. , I= 8 Гц, 2Н/, 6,1-5,6/м., 1Н/, 6,3-6,7 /м., 1Н/, 6,9-7,7/м., 7Н/, 8,3-8,6/м., 2Н/- ПЯМР соединения 1-1-3 /СДСl3/δ млн.дол.:
1,1-1,5/м. , 9Н/, 1,6-1,7/м. , 1Н/, 1,9-2,0/м., 3Н/,2,3-2,5/м., 2Н/, 2,8-3,2/м. , 1Н/, 3,3-3,7/м., 3Н/, 3,8-4,5/м., 4Н/, 5,2-5,6/м., 1H/, 6,4-6,7/м. , 1H/, 7,0-7,6/м. , 7H/, 8,3-8,5/м. , 2H/, ПЯМР соединения 1-2-4 /СДСl3/δ млн. дол.:
1,25/т., I=8 Гц, 3H/, 1,33/д., I=8 Гц, 6H/, 1,7-2,0/м., 1H/,
2,2-2,6/м. , 3Н/, 2,9-3,8/м., 3Н/, 3,8-4,6/м., 2H/, 4,10/кв., I=8 Гц, 2H/, 4,12/c., 3H/, 4,9-5,4/м., 1H/, 6,3-7,2/м., 10H/, ПЯМР соединения 1-1-5 /СДСl3/δ млн.дол.:
0,8-11, /м. , 4H/, 1,28/т., I=7 Гц, 3H/, 1,4-1,8/м., 2Н/, 1,89/c., 3H/, 2,1-2,6/м., 4Н/, 3,0-3,8/с., 2Н/, 3,98/c., 3H/,
4,18/кв. , I= 7 Гц, 2Н/ 4,3-4,6/м., 1Н/, 5,3-5,7/м., 1H/, 6,4-6,8/м., 1H/, 6,9-7,3/м., 4H/, ПЯМР соединения 1-1-6 /СДСl3/δ млн.дол.:
1,29 /т., I=7 Гц, 3H/, 1,37/д., I=7 Гц, 6H/, 1,4-1,9/м., 2H/,
2,2-4,7/м. , 2H/, 3,0-3,9/м., 3H/, 3,9-4,7/м., 2H/, 4,19/кв., I=7 Гц, 2H/, 5,1-5,5/м. , 1H/, 6,6-6,9/м., 1H/, 7,0-8,6/м., 10H/, ПЯМР соединения 1-1-7 /СДСl3/δ млн.дол.:
1,28/т. , I=7 Гц, 3H/, 1,32.д., I=7 Гц, 6H/, 1,4-1,8/м., 2H/, 1,84/c., 9H/, 1,89/c. , 3H/2,3-2,6/м. , 3Н/3,1-3,7/м., 1Н/, 3,41/гепталет, I=7 Гц, 1H/, 3,9-4,7/м. , 2H/, 4,17/кв., I=7 Гц, 2Н/, 5,1-5,5/м., 1Н/, 6,4-6,7/м., 1H/, 6,9-7,3/м., 4Н/, ПЯМР соединения 1-1-10 /СДСl3/δ млн.дол.:
1,29/т., I=7 Гц, 3H/, 1,33/д., I=7 Гц, 6H/, 1,4-1,9/м., 2H/,
1,90/c., 3H/, 2,2-2,6/м., 3H/, 3,1-3,7-/м., 1H/, 3,43/гепталет, I=7 Гц, 1H/, 3,8-4,5/м., 2Н/, 4,19/кв, I=7 Гц, 2H/, 5,1-5,5/м., 1H/, 5,62/c., 2H/, 7,3-7,7/м., 1H/, 6,9-7,6/м., 9Н/, ПЯМР соединения 1-1-14 /СДСl3/δ млн. дол.:
0,9-1,3/м. , 4H/, 1,4-1,8/м. , 2H/, 1,28/т., I=8 Гц, 3H/, 2,3-2,5/м., 3H/, 4,1-4,2/м. , 1Н/, 4,3-4,5/м., 1H/,4,18/кв. I=8 Гц, 2H/, 4,13/c., 3H/, 5,45/д. , д., I=17 Гц, I=6 Гц, 1H/, 6,6-7,3/м., 10Н/, ПЯМР соединения 1-1-15 /СДСl3/δ млн. дол.:
0,9-1,4/м. , 4H/, 1,29/т., I=8 Гц, 3H/, 1,4-1,8/м., 2H/,1,78/c., 9H/, 1,87/c. , 3H/, 2,3-2,5/м., 1H/, 2,4-2,5/м., 2H/,4,0-4,1/м., 1H/, 4,18/кв., I= 8 Гц, 2H/, 4,3-4,4/м., 1H/, 5,50/д.,д.,I=17 Гц, I=6 Гц, 1H/, 6,55/д., I= 17 Гц, 1H/, 7,0-7,3/м., 4Н/, ПЯМР соединение 1-1-16 (СДСl3) δ млн. дол.:
0,4-0,9/м. , 4H/, 0,9-1,5/м., 6H/, 1,29/т., I=8 Гц, 3Н/, 1,74/c., 9H/, 2,2-2,5/м. , 2H/, 2,4-2,5/м., 2H/, 3,9-4,5/м., 2H/, 4,19/кв., I=8 Гц, 2H/, 5,50/д. , д. , I=17 Гц, I=6 Гц, 1H/, 6,58/д., I=17 Гц, 1H/, 6,9-7,4/м., 4H/ ПЯМР соединения 1-1-17 (СДСl3) δ млн.дол.:
0,9-1,5/м. , 6H/, 1,28/т., I=8 Гц, 3H/, 1,86/c., 9H/,2,2-2,6/м., 1H/, 2,3-2,5/м. , 2H/, 3,9-4,5/м., 2H/, 4,18/кв., I=8 Гц, 2H/, 5,45/д.,д., I=17 Гц, I=6 Гц, 1H/,
6,63/д., I=17 Гц, 1Н/, 6,8-7,3/м., 9H/,
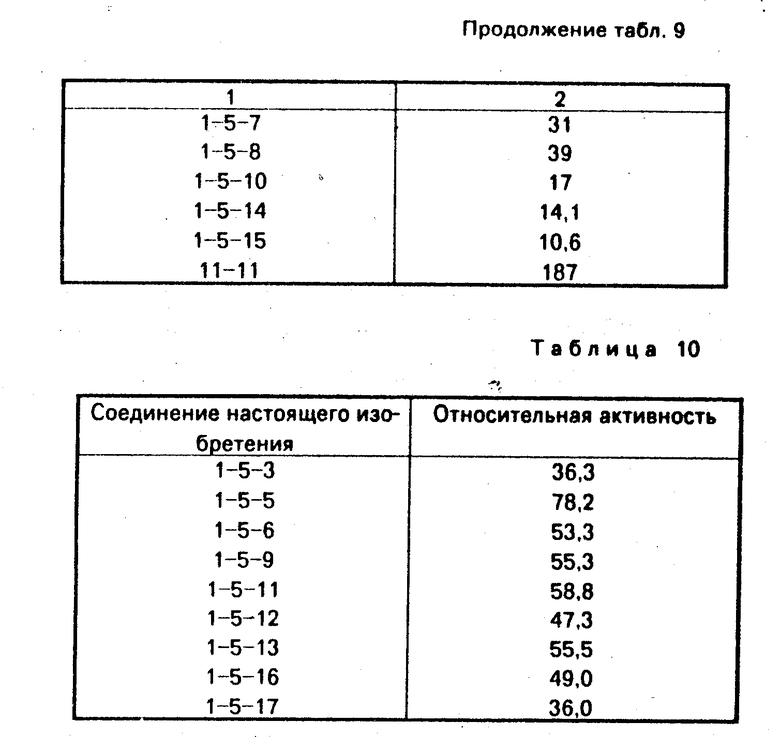
Таким же способом, как в примере 2, были получены соединения 1-5-2-1-5-18. Физические свойства соединений, полученных при этом, показаны в табл. 7. ПЯМР соединения 1-5-8 (СДСl3) δ млн. дол.:
1,0-1,7/м., 2H/, 1,29/д., I=7 Гц, 6H/, 1,47/т., I=7 Гц, 3H/,
1,7-2,3/м. , 3H/, 3,1-3,3/м., 1H/, 3,3-3,9/м., 1H/, 3,53/гепталет, I=7 Гц, 1H/, 4,0-4,3/м. , 1H/, 4,49/кв., I=7 Гц, 2H/, 5,2-5,5/м., 1H/, 6,4-6,7/м., 1H/, 7,0-7,7/м., 5H/, ПЯМР соединения 1-5-14 (СДСl3) δ млн.дол.:
0,9-1,3/м. , 4H/, 1,3-1,8/м. , 2H/, 1,9-2,0/м., 1H/, 2,4-2,6/м., 2H/, 3,5-3,7/м. , 1H/, 4,04/c., 3H/, 4,1-4,2/м., 1H/, 5,51/д.,д., I=17 Гц, I=6 Гц, 1H/, 6,44/д. , I=17 Гц, 1H/, 6,8-7,2/м., 9Н/, ПЯМР соединения 1-5-15 (СДСl3) δ млн.дол.:
1,0-1,2/м., 4H/, 1,3-1,8/м., 2H/, 1,71/c., 9H/, 1,77/c., 3H/,1,9-2,1/м. , 1H/, 2,4-2,5/м., 2H/, 3,5-3,6/м., 1H/,4,1-4,2/м., 1H/, 5,52/д.,д., I=17 Гц, I= 6 Гц, 1Н/, 6,38/д., I=17 Гц, 1H/, 7,1-7,3/м., 4H/, ПЯМР соединения 1-5-16 (СДСl3) δ млн. дол.:
0,4-0,7/м. , 4H/, 0,9-1,2/м., 4H/, 1,3-1,7/м., 2Н/, 1,68/c., 9H/, 1,7-1,8/м., 1H/, 1,9-2,0/м., 1H/, 2,4-2,5/м., 2H/, 3,5-3,6/м., 1H/., 4,1-4,2/м. , 1H/, 5,52/д.,д., I=17 Гц, I=6 Гц, 1H/, 6,40/д., I=17 Гц, 1H/, 7,2-7,4/м., 4Н/. ПЯМР соединения 1-5-17 (СДСl3) δ млн. дол.;
1,0-1,2/м. , 4H/, 1,3-1,8/м., 2H/, 1,80/c., 9H/, 1,9-2,1/м., 1H/, 2,5-2,6/м., 2H/, 3,5-3,6/м., 1H/,4,1-4,2/м., 1H/, 5,51/д., д., I=17 Гц, I=6 Гц, 1H/, 6,42/д., I=17 Гц, 1H/, 6,8-7,2/м., 9H/.
П р и м е р 3. /E/-7-/4′-/4′′-фторфенил/-1′,3′-диметил-6′-/1′-метилэтил/пиразоло /3,4-b/пиридин-5′-ил/-3,5-дигидроксигепт- 6-еновая кислота (соединение 1-2-1).
0,25 г (0,53 ммоля) соединение 1-1-1 растворялось в 3 мл этанола, в раствор по каплям добавлялось 1,06 мл 0,5 н. водного раствора гидроокиси натрия. Этанол отгонялся при пониженном давлении, а затем в смесь добавлялось 3 мл дистиллированной воды. Раствор промывался диэтиловым эфиром. Водный слой осторожно нейтрализовался 1%-ной соляной кислотой и экстрагировался диэтиловым эфиром. Эфирный слой сушился над безводным сульфатом магния и упаривался при пониженном давлении, давая требуемое соединение.
Количество: 0,21 г/выход:90%/,
ПЯМР (ДМСО-d6/ δ млн. дол.: 1,29/д., I=7 Гц, 6Н/, 1,83/c., 3H/, 2,1-2,3/м. , 2H/, 2,4-2,6/м., 1H/, 3,0-3,6/м., 4H/, 3,96/c., 3H/, 4,3-4,8/м., 2Н/, 5,2-5,6/м., 1H/, 6,3-6,6/м., 1H/, 7,2-7,4/м., 4H/, 11,5-12,0/шир. с., 1Н/.
Соединения данного изобретения проявляют высокую ингибиторную активность против биосинтеза холестерина, при котором НМG-СоА редуктаза действует как фермент, ограничивающий скорость, и таким образом способны подавлять или понижать количество холестерина в крови как липопротеин. Таким образом, соединения настоящего изобретения являются полезными в качестве лечебных агентов против гиперлипидемии, гиперлипопротеинами и атеросклероза.
Они могут преобразовываться в разнообразные подходящие выпускные или готовые формы препаратов в зависимости от способа назначения. Соединения настоящего изобретения могут назначаться в форме свободных кислот или в виде физиологически гидролизуемых и приемлемых сложных эфиров или лактонов, или фармацевтически приемлемых солей.
Фармацевтическая композиция изобретения предпочтительно назначается орально в форме соединения настоящего изобретения самого по себе, или в виде порошков, гранул, таблеток или капсул, сформулированных с помощью смешения соединения настоящего изобретения с подходящим фармацевтически приемлемым носителем, включающим связующее, такое как гидроксипропилцеллюлоза, сироп, аравийская камедь, желатин, сорбит, камедь трагаканта, поливинилпирролидон или СМС-Са, эксципиент, такой как лактоза, сахар, кукурузный крахмал, фосфат кальция, сорбит, глицин или порошок кристаллической целлюлозы, смазочный агент, такой как стеарат магния, тальк, полиэтиленгликоль или двуокись кремния, и дезинтегратор, такой как картофельный крахмал.
Однако фармацевтическая композиция данного изобретения не ограничивается таким оральным назначением, она применима и для парентерального назначения. Например, она может назначаться в форме суппозитория, образованного с использованием масляного основного материала, такого как масло какао, полиэтиленгликоль, ланолин или триглицерид жирной кислоты, трансдермального терапевтического основания, образованного с использованием жидкого парафина, белого вазелина, высших спиртов, мази Макроголоя, гидрофильной мази или гидрогельного основного материала, инъекционной препаративной формы, приготавливаемой с помощью использования одного или более материалов, выбираемых из группы, состоящей из полиэтиленгликоля, гидрогельного основного материала, дистиллированной воды, дистиллированной воды для инъекций и эксципиента, такого как лактоза, или кукурузный крахмал, или препаративной формы для назначения через слизистую оболочку, такую как слизистая оболочка глаза, носовая слизистая оболочка и слизистая оболочка рта.
Кроме того, соединения настоящего изобретения могут комбинироваться с основными ионообменными смолами, которые способны связывать желчные кислоты и все же не абсорбируются желудочно-кишечным трактом.
Суточная доза соединения формулы I составляет 0,05-500 мг, предпочтительно 0,5-50 мг для взрослого человека. Оно назначается для приема от одного до трех раз в день. Доза может конечно варьироваться в зависимости от возврата, веса и состояния болезни пациента.
Соединения формул II-VII являются новыми, они являются важными промежуточными соединениями для получения соединений формулы I. Соответственно настоящее изобретение относится также к соединениям формул II-VII и к процессам их получения.
Примеры фармакологических испытаний.
Испытание А: Ингибирование биосинтеза холестерина из ацетата ин витро.
Раствор фермента приготавливался из печени самцов крыс Уистар, которым была вставлена канюля и выводилась желчь в течение 24 ч. Печень вырезалась в сумерки и микросомы и фракция, плавающая сверху, способная осаждаться 40-80% раствором сульфата аммония (суп фракция), приготавливались из гомогената печени в соответствии с модифицированным методом Knauss et al, Kuroda, M., et. al., Biochim. Biophys. Acta,: 489, 119/1977/. Для анализа биосинтеза холестерина, микросомы (0,1 мг белка) и суп фракция (1,0 мг белка) инкубировались в течение 2 ч при 37оС в 200 мкл реакционной смеси, содержащей АТФ, 1 мМ, Глютатион, 6 мМ, Глюкоза-1-фосфат, 10 мМ, NAD; 0,25 мМ, NADP 0,25 мМ, СоА, 0,04 мМ и 0,2 мМ (2-14С) ацетат натрия (0; 2 μ Сi) с 4 мкл раствора испытуемого соединения, растворенного в воде или диметилсульфоксиде. Для остановки реакции и опыления к реакционной смеси добавлялся 1 мл 15% EtoN-КОН и смесь нагревалась при 75оС в течение 1 ч. Не способные к омылению липиды экстрагировались петролейным эфиром и подсчитывалась введенная радиоактивность 14С. Ингибирующая активность соединений выражалась показателем IC50.
Испытание В: Ингибирование биосинтеза холестерина в клетках культуры.
Клетки Нер G2 высевались на 12 ячеистых пластин и инкубировались с модифицированной bagle средой Duibecco (DME), содержащей 10% бычьей зародышевой сыворотки (IBS) при 37оС, 5% СО2, до тех пор, пока клетки не становились сливающимися в течение примерно 7 дней. Клетки подвергались воздействию DME среды, содержащей 5% сыворотки с недостаточным содержанием липопротеина (LpDS), полученной с помощью метода ультрацентрифугирования в течение почти 24 ч. Среда заменялась 0,5 мл свежей 5% LpDS, содержащей DME перед анализом, и добавлялось 10 мкл раствора испытуемого соединения, растворенного в воде или ДМСО. Через 0 часов (В-1) или 4 ч (В-2) после добавления соединений добавлялось 0,2 μ Сi (214С) ацетата натрия (20 мкл). После дополнительного инкубирования в течение 4 ч с (214С) ацетатом натрия среда удалялась, клетки промывались солевым раствором, буферированным фосфатом (РВS), охлажденным при 4оС. Клетки соскабливались резиновой палочкой и собирались в трубки с РВS и дигерировались с 0,2 мл 0,5 н. КОН при 37оС. Некоторая доза жидкости от стадии дигерирования использовалась для анализа белка, а остальное количество омылялось 1 мл 15% EtОН-КОН при 75оС в течение 1 ч. Неомыляемые липиды экстрагировались петролейным эфиром, подсчитывалась 14С радиоактивность. Результаты подсчета проверялись по клеточному белку и обозначались величиной ДРМ/мг (распадов в минуту/мг) белка. Ингибиторная активность соединения выражалась величиной IС50.
Испытание С: Ингибирование биосинтеза холестерина ин виво. Самцам крыс Spragne-Dawley массой около 150 г давали корм в виде обычной еды Ourina и воду по потребности, и перед использованием для испытания на ингибирование ин виво биосинтеза холестерина крыс подвергали воздействию освещения по схеме 12 ч свет /12 ч темнота (2:00 после полудня - 2:00 до полудня темное время). Животных разделяли на группы, состоящие из пяти крыс, так, чтобы средняя величина массы тела была в каждой группе. Испытуемые соединения в дозе 0,02-0,2 мг/кг массы тела (0,4 мл/100 г веса тела) растворялись в воде или суспендировались в 0,5% метилцеллюлозе и назначались орально за 2-3 ч до наступления сумерек (8:00 после полудня), когда биосинтез холестерина у крыс достигал максимума. В качестве контроля крысам назначали только воду или носитель. Через 90 мин после назначения пробы, крыс инъецировали интраперитонально 10 мк Сi (2-14С) ацетата натрия в объеме 0,2 мл на каждую спустя 2 ч брали пробы крови, сразу же отделяли сыворотку. Общие липиды экстрагировались в соответствии с методом Folch и др. и омылялись смесью ЕтОН-КОН. Неомыляемые липиды экстрагировались петролейным эфиром, подсчитывалась радиоактивность, введения в неомыляемые липиды.
Ингибиторная активность отмечалась как процентное уменьшении числа отсчета (показатель фоновой радиации) в испытуемых группах (распады) в минуту/2 мл сыворотки (2 ч) по сравнению с показателем в контрольной группе.
Что касается соединений настоящего изобретения показатели ингибиторной активности против биосинтеза холестерина, при котором НМG-СоА редуктаза служит в качестве ограничивающего скорость фермента, измерялись с помощью описанных выше испытаний А и В. Результаты показаны в табл. 2, 2-2, 3, 3-2, и 3-3. Дополнительно представлены также результаты измерений с помощью испытания С.
Показатели ингибиторной активности ссылочного соединения с помощью испытания А приведены ниже:
Ссылочное соединение IC50(молярная концентрация)
CS - 514 1,1 x 10-8
В табл. 8 представлены показатели относительной активности в расчете на активность CS - 514 по испытанию А, которая оценивалась равной 1.
Структура ссылочного соединения: /1/CS-514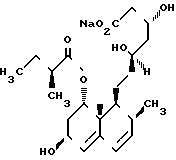
Ингибиторная активность по испытанию В-1 приведена ниже: Ссылочное IC50 (моляр- соединение ная концентрация) СS-514 1,1x106
В табл. 9 показаны данные относительной активности в расчете на активность соединения СS-514 по испытанию В-1, оцененную равной 1.
Дополнительно по результатам испытания В-1 в табл. 10 показаны данные ингибиторной активности соединения настоящего изобретения в концентрации 1.0х10-7 моль/л.
Результаты измерения ингибиторной активности по данным испытания С.
Процентное снижение показателей отсчета после орального назначения 0,2 мг/кг соединения 1-5-4, 1-5-5 и 1-5-7 было соответственно 53, 49 и 52% относительно измеренной величины контрольной группы. Процентное снижение подсчетов после орального назначения 0,2 мг/кг соединения CS-514 в тех же условиях было 39%.
Соединения настоящего изобретения проявляли активность, превосходящую показатели ссылочного соединения СS-514 в испытаниях А, В и С.
Испытание Д: Острая токсичность.
0,5% суспензия в СМС испытуемого соединения назначалась орально самцам мышей ICR (группа из трех мышей). Острая токсичность определялась на основании смертности спустя семь дней. В случае соединения 1-5-1, 1-5-2, 1-5-4. 1-5-7, 1-5-10, 1-5-11, 1-5-12, 1-5-14 и 1-5-15 настоящего изобретения смертность была 0 % даже, когда они назначались орально в количестве 1000 мг/кг соответственно.
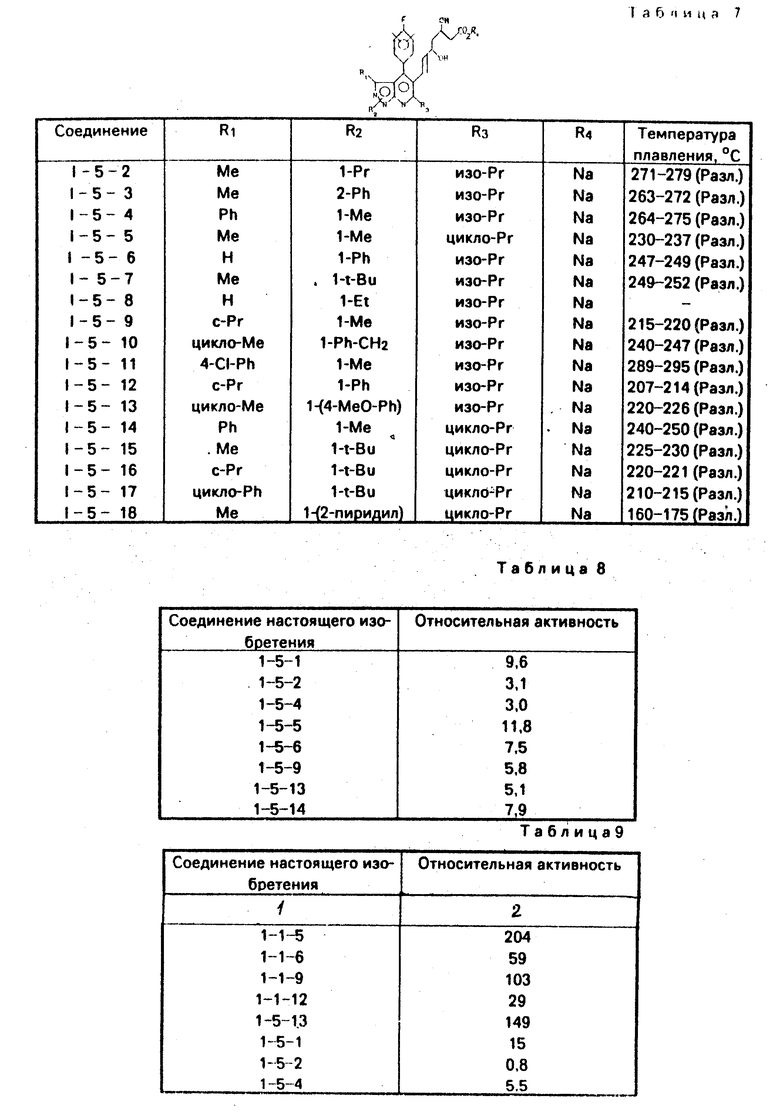
Сущность изобретения: 5-диоксиалкеновым производным пиразоло [3,4-в] пиридин ф-лы I, приведенной в описании, где R1-R5; Y и Z имеют значения, приведенные в тексте описания.
R1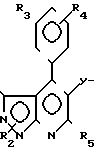 Z
Z
где R1 - водород, C1 - C8-алкил, C3 - C6-циклоалкил
или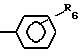
где R6 - водород, хлор, бром или фтор;
R2 связан с азотом азота в положении 1 или 2, пиразоло-пиридинового кольца и означает C1 - C8 - алкил, фенил, бензил, 2,3-или 4-пиридил или алкоксифенил;
R3 - фтор, хлор или бром;
R4 - водород;
R5 - C1 - C8-алкил или C3 - C7-циклоалкил;
Y - СН=СН-;
Z - группа формулы
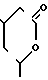
или группа формулы - Q-CH2-W-CH2-CO2R12,
где Q-CH(OH), W-C(R11)(OH), где R11 - водород, R12 - водород или алкильная часть химически или физиологически гидролизуемого алкильного сложного эфира или натрий, калий или 1 - 2 кальция.
| A.Endo, J.Med.Chem, 28(4, 401, 1985. |
