Предпосылки создания изобретения
Настоящее изобретение относится к новым способам получения 9,11-эпоксистероидных производных, в частности соединений ряда 20-спироксанов и их аналогов; к новым промежуточным соединениям, используемым для получения стероидных соединений; и к способам получения этих новых промежуточных соединений. Более конкретно, изобретение относится к новым и усовершенствованным способам получения метилгидро 9,11α-эпокси-17α-гидрокси-3-оксопрегн-4-ен-7α,21-дикарбоксилата, γ-лактона (называемого также эплереноном или эпоксимексреноном) (монометиловый эфир γ-лактона 9,11α-эпокси-17α-гидрокси-3-оксопрегн-4-ен-7α,21-дикарбоновой кислоты).
Способы получения соединений ряда 20-спироксанов описаны в патенте США №4559332. Соединения, продуцированные способом, описанным в патенте 4559332, имеют незамкнутое кислородсодержащее кольцо Е общей формулы:
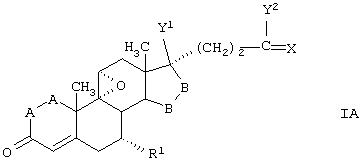
где
-А-А- представляет группу -СН2-СН2- или -СН=СН-;
R1 представляет α-ориентированный низший алкоксикарбонильный или гидроксикарбонильный радикал;
-В-В- представляет группу -СН2-СН2- или α- или β-ориентированную группу;
R6 и R7 представляют водород;
X представляет два атома водорода или оксо;
Y1 и Y2 вместе представляют кислородный мостик -О-, или
Y1 представляет гидрокси, и
Y2 представляет гидрокси, низший алкокси или, если Х представляет H2, также низший алканоилокси;
и соли таких соединений, в которых Х представляет оксо и Y2 представляет гидрокси, то есть соли соответствущих 17β-гидрокси-21-карбоновых кислот.
В патенте США №4559332 описан ряд методов получения эпоксимексренона и близких по структуре соединений формулы IA. Появление новых и более широких клинических возможностей применения эпоксимексренона приводит к необходимости усовершенствования способов получения его и других близких по структуре стероидов.
Краткое описание изобретения
Главной целью настоящего изобретения является разработка улучшенных способов получения эпоксимексренона, иных 20-спироксанов и других стероидов, имеющих общие структурные признаки. Некоторыми из конкретных целей настоящего изобретения являются: разработка улучшенного способа получения продуктов формулы IA и других близких по структуре соединений с высоким выходом; разработка такого способа, который предусматривает осуществление минимума стадий выделения; и разработка такого способа, который может быть реализован с разумными капитальными затратами и с приемлемыми затратами, связанными с конверсией.
В соответствии с этим, настоящее изобретение относится к ряду схем синтеза эпоксимексренона; промежуточных соединений, которые могут быть использованы для производства эпоксимексренона; и к синтезу таких новых промежуточных соединений.
Новые схемы синтеза подробно изложены в описании предпочтительных вариантов осуществления изобретения. Часть этих новых промежуточных соединений по настоящему изобретению представлены сразу далее.
Соединение формулы IV соответствует структуре
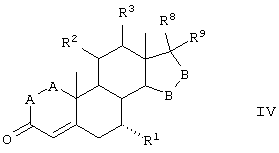
где:
-А-А- представляет группу -CHR4-CHR5- или -CR4=CR5-;
R3, R4 и R5 независимо выбраны из группы, включающей водород, галоген, гидрокси, низший алкил, низший алкокси, гидроксиалкил, алкоксиалкил, гидроксикарбонил, циано и арилокси;
R1 представляет альфа-ориентированный низший алкокси-карбонильный или гидроксикарбонильный радикал;
R2 представляет 11α-удаляемую группу, отщепление которой является эффективным для введения двойной связи между 9- и 11-атомами углерода;
-В-В представляет группу -CHR6-CHR7-, либо альфа- или бета-ориентированную группу:
где R6 и R7 независимо выбраны из группы, включающей водород, галоген, низший алкокси, ацил, гидроксиалкил, алкоксиалкил, гидроксикарбонил, алкил, алкоксикарбонил, ацилоксиалкил, циано и арилокси; и
R8 и R9 независимо выбраны из группы, включающей водород, гидрокси, галоген, низший алкокси, ацил, гидроксиалкил, алкоксиалкил, гидроксикарбонил, алкил, алкоксикарбонил, ацилоксиалкил, циано и арилокси; или R8 и R9 вместе образуют карбоциклическую или гетероциклическую кольцевую структуру, или R8 или R9 вместе с R6 или R7 образуют карбоциклическую или гетероциклическую кольцевую структуру, конденсированную с пентациклическим кольцом D.
Соединение формулы IVA соответствует формуле IV, где R8 и R9 вместе с кольцевым атомом углерода, к которому они присоединены, образуют структуру:
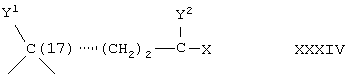
где X, Y1, Y2 и С(17) определены выше.
Соединение формулы IVB соответствует формуле IV, где R8 и R9 вместе образуют структуру формулы XXXIII:
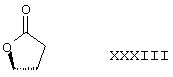
Соединения формул IVC, IVD и IVE, соответственно, соответствуют любой из формул IV, IVA или IVB, где каждый из -А-А- и -В-В представляют -СН2-СН2-, R3 представляет водород и R1 представляет алкоксикарбонил, предпочтительно, метоксикарбонил. Соединения, охватываемые формулой IV, могут быть получены путем взаимодействия низший-алкилсульфонилирующего или ацилирующего реагента или галогенид-образующего агента с соответствующим соединением формулы V.
Соединение формулы V соответствует структуре:
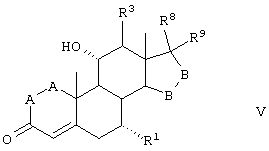
где: -А-А-, -В-В-, R1, R3, R8 и R9 определены в формуле IV.
Соединение формулы VA соответствует формуле V, где R8 и R9 вместе с кольцевым углеродом, к которому они присоединены, образуют структуру:
где X, Y1, Y2 и С(17) определены выше.
Соединение формулы VB соответствует формуле V, где R8 и R9 вместе образуют структуру формулы XXXIII:
Соединения формул VC, VD и VE, соответственно, соответствуют любой из формул V, VA или VB, где каждый из -А-А- и -В-В представляет -СН2-СН2-, R3 представляет водород и R1 представляет алкоксикарбонил, предпочтительно метоксикарбонил. Соединения, охватываемые формулой V, могут быть получены путем взаимодействия алкоксида щелочного металла с соответствующим соединением формулы VI.
Соединение формулы VI соответствует структуре:

где: -А-А-, -В-В-, R3, R8 и R9 определены в формуле IV.
Соединение формулы VIA соответствует формуле VI, где R8 и R9 вместе с кольцевым углеродом, к которому они присоединены, образуют структуру:
где X, Y1, Y2 и С(17) определены выше.
Соединение формулы VIB соответствует формуле VI, где R8 и R9 вместе образуют структуру формулы XXXIII:
Соединения формул VIC, VID и VIE, соответственно, соответствуют любой из формул VI, VIA или VIB, где каждый из -А-А- и -В-В- представляет -СН2-СН2-, и R3 представляет водород. Соединения формул VI, VIA, VIB и VIC получают путем гидролиза соединения, соответствующего формулам VII, VIIA, VIIB или VIIC, соответственно.
Соединение формулы VII соответствует структуре:
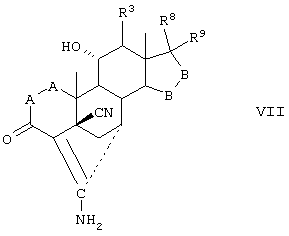
где: -А-А-, -В-В-, R3, R8 и R9 определены выше для формулы IV.
Соединение формулы VIIA соответствует формуле VII, где R8 и R9 вместе с кольцевым углеродом, к которому они присоединены, образуют структуру:
где X, Y1, Y2 и С(17) определены выше.
Соединение формулы VII В соответствует формуле VII, где R8 и R9 вместе образуют структуру формулы XXXIII:
Соединения формул VIIC, VIID и VIIE, соответственно, соответствуют любой из формул VII, VIIA или VIIB, где каждый из -А-А- и -В-В- представляет -СН2-СН2-, и R3 представляет водород. Соединение, охватываемое формулой VII, может быть получено путем цианидирования соединения, охватываемого формулой VIII.
Соединение формулы VIII соответствует структуре:
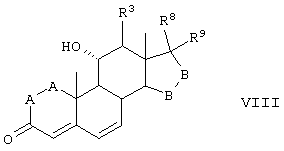
где: -А-А-, -В-В-, R3, R8 и R9 определены выше в формуле IV.
Соединение формулы VIIIA соответствует формуле VIII, где R8 и R9 вместе с кольцевым углеродом, к которому они присоединены, образуют структуру:
где X, Y1, Y2 и С(17) определены выше.
Соединение формулы VIII В соответствует формуле VIII, где R8 и R9 вместе образуют структуру формулы XXXIII:
Соединения формул VIIIC, VIIID и VIIIE, соответственно, соответствуют любой из формул VIII, VIIIA или VIIIB, где каждый из -А-А- и -В-В- представляет -СН2-СН2- и R3 представляет водород. Соединения, охватываемые формулой VIII, получают путем окисления субстрата, содержащего соединение формулы XXX, как описано ниже, путем ферментации, проводимой для введения 11-гидроксигруппы в субстрат в α-ориентации. Соединение формулы IX соответствует структуре:
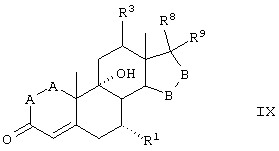
где: -А-А-, -В-В-, R3, R8 и R9 определены выше для формулы IV, а R1 определен для формулы V.
Соединение формулы IXA соответствует формуле IX, где R8 и R9 вместе с кольцевым углеродом, к которому они присоединены, образуют структуру:
где X, Y1, Y2 и С(17) определены выше.
Соединение формулы IXB соответствует формуле IX, где R8 и R9 вместе с кольцевым углеродом, к которому они присоединены, образуют структуру формулы XXXIII:
Соединения формул IXC, IXD и IXE, соответственно, соответствуют любой из формул IX, IXA или IXB, где каждый из -А-А- и -В-В- представляет -CH2-CH2- и R3 представляет водород. Соединения, охватываемые формулой IX, могут быть получены путем конверсии соответствующего соединения формулы X.
Соединение формулы XIV соответствует структуре;

где: -А-А-, -В-В-, R3, R8 и R9 определены выше в формуле IV.
Соединение формулы XIVA соответствует формуле XIV, где R8 и R9 вместе с кольцевым углеродом, к которому они присоединены, образуют структуру:
где X, Y1, Y2 и С(17) определены выше.
Соединение формулы XIVB соответствует формуле XIV, где R8 и R9 вместе с кольцевым углеродом, к которому они присоединены, образуют структуру формулы XXXIII:
Соединения формул XIVC, XIVD и XIVE, соответственно, соответствуют любой из формул XIV, XIVA или XIVB, где каждый из -А-А- и -В-В- представляет -CH2-CH2- и R3 представляет водород. Соединения, охватываемые формулой XIV, могут быть получены путем гидролиза соответствующего соединения формулы XV.
Соединение формулы XV соответствует структуре:
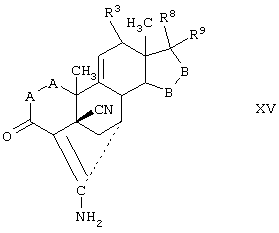
где: -А-А-, -В-В-, R3, R8 и R9 определены выше для формулы IV.
Соединение формулы XVA соответствует формуле XV, где R8 и R9 вместе с кольцевым углеродом, к которому они присоединены, образуют структуру:
где X, Y1, Y2 и С(17) определены выше.
Соединение формулы XVB соответствует формуле XV, где R8 и R9 вместе с кольцевым углеродом, к которому они присоединены, образуют структуру формулы XXXIII:
Соединения формул XVC, XVD и XVE, соответственно, соответствуют любой из формул XV, XVA или XVB, где каждый из -А-А- и -В-В- представляет -CH2-CH2- и R3 представляет водород. Соединения, охватываемые формулой XV, могут быть получены путем цианидирования соответствующего соединения, охватываемого формулой XVI.
Соединение формулы XXI соответствует структуре:
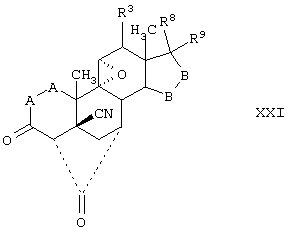
где: -А-А-, -В-В-, R3, R8 и R9 определены выше для формулы IV.
Соединение формулы XXIA соответствует формуле XXI, где R8 и R9 вместе с кольцевым углеродом, к которому они присоединены, образуют структуру:
где X, Y1, Y2 и С(17) определены выше.
Соединение формулы XXIB соответствует формуле XXI, где R8 и R9 вместе образуют структуру формулы XXXIII:
Соединения формул XXIC, XXID и XXIE, соответственно, соответствуют любой из формул XXI, XXIA или XXIB, где каждый из -А-А- и -В-В- представляют -СН2-СН2-, и R3 представляет водород. Соединения, охватываемые формулой XXI, могут быть получены путем гидролиза соответствующего соединения, охватываемого формулой XXII.
Соединение формулы XXII соответствует структуре:
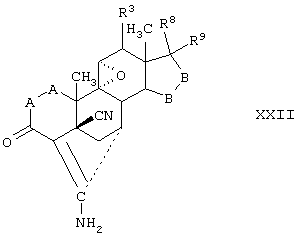
где: -А-А-, -В-В-, R3, R8 и R9 определены выше для формулы IV.
Соединение формулы XXIIA соответствует формуле XXII, где R8 и R9 вместе с кольцевым углеродом, к которому они присоединены, образуют структуру:
где X, Y1, Y2 и С(17) определены выше.
Соединение формулы XXIIB соответствует формуле XXII, где R8 и R9 вместе образуют структуру формулы XXXIII:
Соединения формул XXIIC, XXIID и XXIIE, соответственно, соответствуют любой из формул XXII, XXIIA или XXIIB, где каждый из -А-А- и -В-В- представляет -СН2-СН2-, и R3 представляет водород. Соединения, охватываемые формулой XXII, могут быть получены путем цианидирования соответствующего соединения формулы XXIII.
Соединение формулы XXIII соответствует структуре:
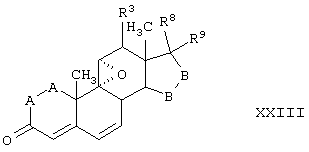
где: -А-А-, -В-В-, R3, R8 и R9 определены выше для формулы IV.
Соединение формулы XXIIIA соответствует формуле XXIII, где R8 и R9 вместе с кольцевым углеродом, к которому они присоединены, образуют структуру:
где X, Y1, Y2 и С(17) определены выше.
Соединение формулы XXIIIB соответствует формуле XXIII, где R8 и R9 вместе образуют структуру формулы XXXIII:
Соединения формул XXIIIC, XXIIID и XXIIIE, соответственно, соответствуют любой из формул XXIII, XXIIIA или XXIIIB, где каждый из -А-А- и -В-В- представляет -CH2-CH2-, а R3 представляет водород. Соединения, охватываемые формулой XXIII, могут быть получены путем окисления соединения формулы XXIV, как описано ниже.
Соединение формулы XXVI соответствует структуре:
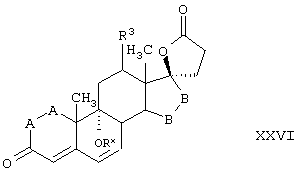
где: -А-А-, -В-В-, R3, R8 и R9 определены выше для формулы IV.
Соединение формулы XXVIA соответствует формуле XXVI, где каждый из -А-А- и -В-В- представляет -CH2-CH2-, и R3 представляет водород. Соединения, охватываемые формулой XXVI, могут быть получены путем окисления соединения формулы XXVII.
Соединение формулы XXV соответствует структуре:
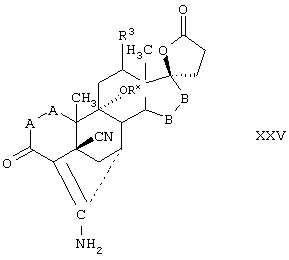
где; -А-А-, -В-В-, R3, R8 и R9 определены выше для формулы IV.
Соединение формулы XXVA соответствует формуле XXV, где каждый из -А-А- и -В-В- представляет группу -CH2-CH2-, и R3 представляет водород. Соединения, охватываемые формулой XXV, могут быть получены путем цианидирования соединения формулы XXVI.
Соединение формулы 104 соответствует структуре:
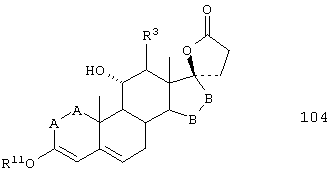
где: -А-А-, -В-В- и R3 определены выше для формулы IV, и R11 представляет С1-С4-алкил.
Соединение формулы 104А соответствует формуле 104, где каждый из -А-А- и -В-В- представляет -СН2-СН2-, и R3 представляет водород. Соединения, охватываемые формулой 104, могут быть получены путем термического разложения соединения формулы 103.
Соединение формулы 103 соответствует структуре:
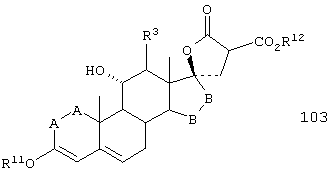
где: -А-А-, -В-В-, R3 и R11 определены выше для формулы 104, и R12 представляет С1-С4-алкил.
Соединение формулы 103А соответствует формуле 103, где каждый из -А-А- и -В-В- представляет -СН2-СН2-, и R3 представляет водород. Соединения, охватываемые формулой 103, могут быть получены путем взаимодействия соответствующего соединения формулы 102 с диалкилмалонатом в присутствии основания, такого как алкоксид щелочного металла.
Соединение формулы 102 соответствует структуре;
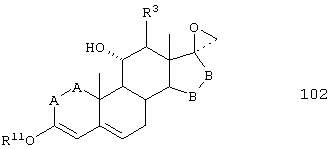
где: -А-А-, -В-В-, R3 и R11 определены выше для формулы 104.
Соединение формулы 102А соответствует формуле 102, где каждый из -А-А- и -В-В- представляет -СН2-СН2-, и R3 представляет водород. Соединения, охватываемые формулой 102, могут быть получены путем взаимодействия соответствующего соединения формулы 101 с триалкилсульфониевым соединением в присутствии основания.
Соединение формулы 101 соответствует структуре:
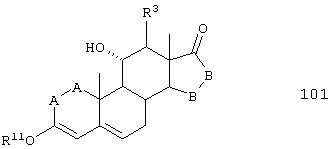
где: -А-А-, -В-В-, R3 и R11 определены выше для формулы 104.
Соединение формулы 101А соответствует формуле 101, где каждый из -А-А- и -В-В- представляет -СН2-СН2-, и R3 представляет водород. Соединения, охватываемые формулой 101, могут быть получены путем взаимодействия 11α-гидроксиандростен-3,17-диона или другого соединения формулы XXXVI с триалкил-орто-формиатом в присутствии кислоты.
Соединение формулы XL соответствует формуле;

где -Е-Е- выбрано из:

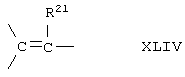
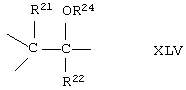
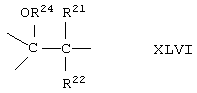
и
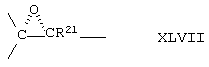
R21, R22 и R23 независимо выбраны из водорода, алкила, галогена, нитро и циано; R24 выбран из водорода и низшего алкила; R80 и R90 независимо выбраны из кето и заместителей, которыми могут быть R8 и R9 (как определено выше по отношению к формуле IV) ; и -А-А-, -В-В- и R3 определены для формулы IV.
Соединение формулы XLA соответствует формуле XL, где R21, R22 и R23 независимо выбраны из водорода, галогена и низшего алкила.
Соединение формулы XLB соответствует формуле XLA, где -Е-Е- соответствует формуле XLIII, XLIV, XLV или XLVII. Соединение формулы XLC соответствует формуле XLB, где -Е-Е- соответствует формуле XLV. Соединение XLD соответствует формуле XLB, где -Е-Е- соответствует формуле XLVII.
Соединение формулы XLE соответствует формуле XL, где R80 и R90 вместе с кольцевым углеродом, к которому они присоединены, представляют кето или:
где X, Y1, Y2 и С(17) определены выше или
Соединения формулы XLIE соответствуют формуле XL, где R80 и R90 вместе образуют кето.
Соединения формул XLF, XLG, XLH, XLJ, XLM и XLN соответствуют формулам XL, XLA, XLB, XLC, XLD и XLE, соответственно, в которых -А-А-, -В-В- и R3 определены выше.
Соединение формулы XLI соответствует формуле:
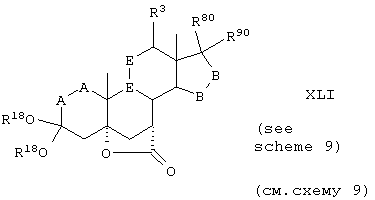
где -Е-Е- выбран из:
и
R18 представляет C1-C4-алкил, или группы R18O - вместе образуют O,O-оксиалкиленовый мостик; R21, R22 и R23 независимо выбраны из водорода, алкила, галогена, нитро и циано; R24 выбирают из водорода и низшего алкила; R80 и R90 независимо выбраны из кето и заместителей, которыми могут быть R8 и R9; и -А-А-, -В-В- и R3 определены в формуле IV.
Соединение формулы XLIA соответствует формуле XLI, где R21, R22 и R23 независимо выбраны из водорода, галогена и низшего алкила.
Соединение формулы XLIB соответствует формуле XLIA, где -Е-Е- соответствует формуле XLIII, XLIV, XLV или XLVII.
Соединение формулы XLIC соответствует формуле XLI, где R80 и R90 вместе с кольцевым углеродом, к которому они присоединены, представляют кето или:
где: X, Y1, Y2 и С(17) определены выше.
Соединения формулы XLID соответствуют формуле XLI, где заместитель XXXIV соответствует структуре XXXIII
Соединения формулы XLIE соответствуют формуле XL, где R80 и R90 вместе образуют кето.
Соединения формул XLIF, XLIG, XLIH, XLIJ, XLIM и XLIN соответствуют формулам XLI, XLIA, XLIB, XLIC, XLID и XLIE, соответственно, где -А-А-, -В-В- и R3 определены выше. Соединения, охватываемые формулой XLI, получают путем гидролиза соответствующих соединений формулы XL, показанной ниже.
Соединение формулы XLII соответствует формуле:
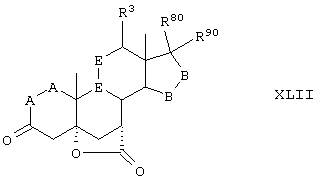
где -Е-Е- выбран из:
и
R21, R22 и R23 независимо выбраны из водорода, алкила, галогена, нитро и циано; R24 выбран из водорода и низшего алкила; R80 и R90 независимо выбраны из кето и заместителей, которыми могут быть R8 и R9, а -А-А-, -В-В- и R3 определены в формуле IV.
Соединение формулы XLIIA соответствуют формуле XLII, где R21, R22 и R23 независимо выбраны из водорода, галогена и низшего алкила.
Соединение формулы XLIIB соответствуют формуле XLIIA, где -Е-Е- соответствует формуле XLIII, XLIV, XLV или XLVII.
Соединение формулы XLIIC соответствует формуле XLII, где R80 и R90 вместе с кольцевым углеродом, к которому они присоединены, представляют кето или:
где X, Y1, Y2 и С(17) определены выше.
Соединения формул XLIID соответствуют формуле XLII, где заместитель XXXIV соответствует структуре XXXIII
Соединения формулы XLIIE соответствуют формуле XLII, где R80 и R90 вместе образуют кето. Соединения формул XLIIF, XLIIG, XLIIH, XLIIJ, XLIIM и XLIIN соответствуют формулам XLII, XLIIA, XLIIB, XLIIC, XLIID и XLIIE, соответственно, где -А-А- и -В-В- представляют -CH2-CH2-, и R3 представляет водород. Соединения, охватываемые формулой XLII, получают путем удаления защиты у соответствующего соединения формулы XLI.
Соединение формулы XLIX соответствуют структуре:
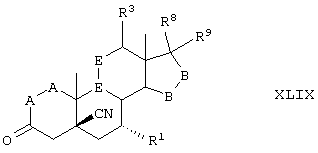
где -Е-Е- определен для формулы XL, а -А-А-, -В-В-, R1, R3, R8 и R9 определены в формуле IV.
Соединение формулы XLIXA соответствует формуле XLIX, где R8 и R9 вместе с кольцевым углеродом, к которому они присоединены, образуют структуру:
где X, Y1, Y2 и С(17) определены выше.
Соединение формулы XLIXB соответствует формуле XLIX, где R8 и R9 вместе образуют структуру формулы XXXIII:
Соединения формул XLIXC, XLIXD, XLIXE, соответственно, соответствуют любой из формул XLIX, XLIXA или XLIXB, где каждый из -А-А- и -В-В- представляет -CH2-CH2-, R3 представляет водород, и R1 представляет алкоксикарбонил, предпочтительно метоксикарбонил. Соединения, охватываемые формулой XLIX, могут быть получены путем взаимодействия спиртового или водного растворителя с соответствующим соединением формулы VI в присутствии подходящего основания.
Соединение формулы А203 соответствует структуре:

где -Е-Е- выбран из:
и
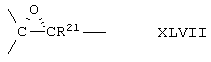
R18 выбран из С1-С4-алкила; R21, R22 и R23 независимо выбраны из водорода, алкила, галогена, нитро и циано; R24 выбран из водорода и низшего алкила; а -А-А-, -В-В- и R3 определены для формулы IV.
Соединение формулы А203А соответствует формуле А203, где R21, R22 и R23 независимо выбраны из водорода, галогена и низшего алкила.
Соединение формулы А203В соответствует формуле А203А, где -Е-Е- соответствует формуле XLIII, XLIV, XLV или XLVII.
Соединения формул А203С, A203D и А203Е, соответственно, соответствуют формулам А203, А203А и А203В, где каждый из -А-А- и -В-В- представляет -СН2-СН2-, и R3 представляет водород. Соединения, охватываемые формулой А203, получают путем восстановления соединения формулы А202, представленной ниже.
Соединение формулы А204 соответствует структуре:
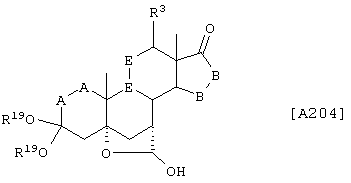
где R19 представляет С1-С4-алкил и -Е-Е-, -А-А-, -В-В- и R3 определены для формулы 203.
Соединение формулы А204А соответствует формуле А204, где R21, R22 и R23 независимо выбраны из водорода, галогена и низшего алкила.
Соединение формулы А204В соответствует формуле А204А, где -Е-Е- соответствует формуле XLIII, XLIV, XLV или XLVII.
Соединения формул А204С, A204D и А204Е, соответственно, соответствуют формулам А204, А204А и А204В, где каждый из -А-А- и -В-В- представляет -CH2-CH2-, и R3 представляет водород. Соединения, охватываемые формулой А204, получают путем гидролиза соответствующих соединений формулы А203.
Соединение формулы А205 соответствует структуре:
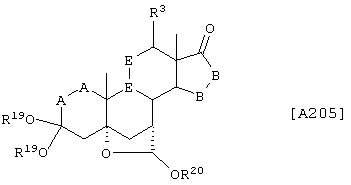
где R20 представляет С1-С4-алкил, а -Е-Е-, R19, -А-А-, -В-В- и R3 определены для формулы 204.
Соединение формулы А205А соответствует формуле А205, где R21, R22 и R23 независимо выбраны из водорода, галогена и низшего алкила.
Соединение формулы А205В соответствует формуле А205А, где -Е-Е- соответствует формулам XLIII, XLIV, XLV или XLVII.
Соединения формул А205С, A205D и А205Е, соответственно, соответствуют формулам А205, А205А и А205В, где каждый из -А-А- и -В-В- представляет -СН2-СН2-, и R3 представляет водород. Соединения, охватываемые формулой А205, могут быть получены путем взаимодействия соответствующего соединения формулы А204 со спиртом и кислотой.
Соединение формулы А206 соответствует структуре:
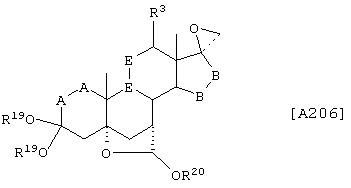
где R19, R20, -Е-Е-, -А-А-, -B-B- и R3 определены для формулы 205.
Соединение формулы А206А соответствует формуле А206, где R21, R22 и R23 независимо выбраны из водорода, галогена и низшего алкила.
Соединение формулы А206В соответствует формуле А206А, где -Е-Е- соответствует формулам XLIII, XLIV, XLV или XLVII.
Соединения формул А206С, A206D и А206Е, соответственно, соответствуют формулам А206, А206А и А206В, где каждый из -А-А- и -В-В- представляет -СН2-СН2-, и R3 представляет водород. Соединения, охватываемые формулой А206, могут быть получены путем взаимодействием соответствующего соединения формулы А205 с галогенидом триалкилсульфония.
Соединение формулы А207 соответствует структуре:
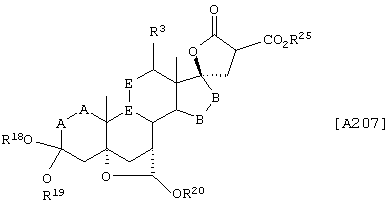
где R25 представляет С1-С4алкил и -Е-Е-, R19, R20, -А-А-, -В-В- и R3 определены для формулы А205.
Соединение формулы А207А соответствует формуле А207, где R21, R22 и R23 независимо выбраны из водорода, галогена и низшего алкила.
Соединение формулы А207В соответствует формуле А207А, где -Е-Е- соответствует формулам XLIII, XLIV, XLV или XLVII.
Соединения формул А207С, A207D и А207Е, соответственно, соответствуют формулам А207, А207А и А207В, где каждый из -А-А- и -В-В- представляет -СН2-СН2- и R3 представляет водород. Соединения, охватываемые формулой А207, могут быть получены путем взаимодействия соответствующего соединения формулы А206 с диалкилмалонатом.
Соединение формулы А208 соответствует структуре:
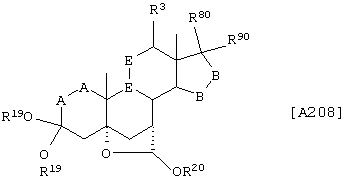
где -Е-Е-, R80 и R90 определены для формулы XLII; -А-А-, -В-В- и R3 определены в формуле 104; а R19, R20, -А-А-, -В-В- и R3 определены для формулы 205.
Соединение формулы А208А соответствует формуле А208, где R21 и R22 независимо выбраны из водорода, галогена и низшего алкила.
Соединение формулы А208В соответствует формуле А208А, где -Е-Е- соответствует формулам XLIII, XLIV, XLV или XLVII.
Соединение формулы А208С соответствует формуле А208, где R80 и R90 вместе с кольцевым углеродом, к которому они присоединены, представляют кето или:
где X, Y1, Y2 и С(17) определены выше.
Соединение формулы 208D соответствует формуле 208С, в которой заместитель XXXIV соответствует структуре XXXIII:
Соединения формул А208Е, A208F, A208G, А208Н и A208J, соответственно, соответствуют формулам А208, А208А, А208В, А208С и A208D, где каждый из -А-А- и -В-В- представляет -СН2-СН2, и R3 представляет водород. Соединения, охватываемые формулой А208, могут быть получены путем термического разложения соответствующих соединений формулы А207.
Соединение формулы А209 соответствует структуре:
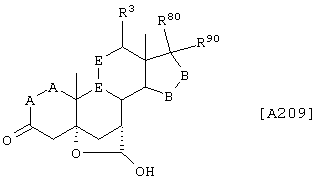
где R80 и R90 определены для формулы XLI, и -Е-Е-, -А-А-, -В-В- и R3 определены в формуле 205.
Соединение формулы А209А соответствует формуле А209, где R21 и R22 независимо выбраны из водорода, галогена и низшего алкила.
Соединение формулы А209В соответствует формуле А209А, где -Е-Е- соответствует формулам XLIII, XLIV, XLV или XLVII.
Соединение формулы А209С соответствует формуле А209В, где -Е-Е- соответствует формуле XLIV.
Соединение формулы A209D соответствует формуле А208, где R80 и R90 вместе с кольцевым углеродом, к которому они присоединены, представляют кето или:
где X, Y1, Y2 и С(17) определены выше.
Соединение формулы 209Е соответствует формуле 209D, где заместитель XXXIV соответствует структуре XXXIII:
Соединения формул A209F, A209G, А209Н, A209J, A209L и А209М, соответственно, соответствуют формулам А209, А209А, А209В, А209С, A208D и А209Е, где каждый из -А-А- и -В-В- представляет -CH2-CH2-, и R3 представляет водород. Соединения, охватываемые формулой А209, могут быть получены путем гидролиза соответствующего соединения формулы А208.
Соединение формулы А210 соответствует структуре;
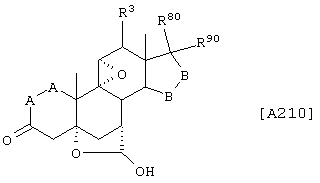
где R80 и R90 определены для формулы XLI, и заместители -А-А-, -В-В- и R3 определены для формулы IV.
Соединение формулы А210А соответствует формуле А210, где R80 и R90 вместе с кольцевым углеродом, к которому они присоединены, представляют кето или:
где X, Y1, Y2 и С(17) определены выше.
Соединение формулы А210В соответствует формуле А210А, где заместитель XXXIV соответствует структуре XXXIII:
Соединение формулы А210С соответствует формуле А210А, где R80 и R90 вместе образуют кето.
Соединения формул A210D, А210Е, A210F и A210G, соответственно, соответствуют формулам А210, А210А, А210В и А210С, где каждый из -А-А- и -В-В- представляет -CH2-CH2-, a R3 представляет водород. Соединения, охватываемые формулой 210, могут быть получены путем эпоксидирования соединения формулы 209, в котором -Е-Е- представляет  .
.
Соединение формулы А211 соответствует формуле:
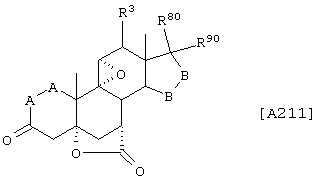
где -А-А-, -В-В- и R3 определены выше.
Соединение формулы А211А соответствует формуле А211, где R80 и R90 вместе образуют кето или:
где X, Y1, Y2 и С(17) определены выше.
Соединение формулы А211В соответствует формуле А211А, в которой заместитель XXXIV соответствует структуре XXXIII:
Соединение формулы А211С соответствует формуле А211А, где R80 и R90 вместе образуют кето.
Соединения формул A211D, А211Е, A211F и A211G, соответственно, соответствуют формулам А211, А211А, А211В и А211С, где каждый из -А-А- и -В-В- представляет -CH2-CH2-, и R3 представляет водород. Соединения, охватываемые формулой А211, могут быть получены путем оксиления соответствующего соединения формулы А210, либо путем эпоксидирования соответствующего соединения формулы А209, где -Е-Е- представляет . Соединения формулы А211 могут быть преобразованы в соединения формулы I способом, описанным ниже.
Соединение формулы L соответствует структуре:
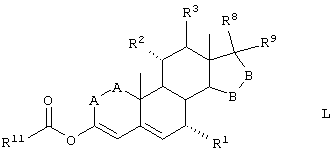
где R11 представляет С1-С4алкил, и -А-А-, -В-В-, R1, R2, R3, R8 и R9 определены выше.
Соединения формулы LA соответствуют формуле L, где R8 и R9 вместе с углеродом, к которому они присоединены, представляют:
где X, Y1 и Y2 определены выше.
Соединения формулы LB соответствуют формуле L, где R8 и R9 соответствуют структуре XXXIII
Соединения формул LC, LD, LE соответствуют формулам L, LA и LB, соответственно, где каждый из -А-А- и -В-В- представляет -CH2-CH2-, a R3 представляет водород.
Исходя из описания конкретных реакционных схем, представленных ниже, можно будет определить, которые из этих соединений являются наиболее подходящими для данной реакционной схемы. Соединения по настоящему изобретению могут быть использованы в качестве промежуточных соединений для получения эпоксимексренона и других стероидов.
Другие цели и признаки настоящего изобретения отчасти очевидны, а отчасти описаны ниже.
Краткое описание чертежей
На Фиг.1 представлена схема способа биологического превращения канренона или производного канренона в соответствующее 11α-гидроксисоединение;
на Фиг.2 представлена схема предпочтительного способа биологического превращения/11-α-гидроксилирования канренона и производных канренона;
на Фиг.3 представлена схема особенно предпочтительного способа биологического превращения /11-α-гидроксилирования канренона и производных канренона;
на Фиг.4 представлено распределение частиц канренона по размерам, полученное в соответствии со способом, проиллюстрированным на Фиг.2; и
на Фиг.5 представлено распределение по размерам частиц канренона, стерилизованного в ферментере для трансформации способом, проиллюстрированным на Фиг.3.
Соответствующие номера позиций на чертежах указывают на соответствующие части этих чертежей.
Описание предпочтительных вариантов осуществления настоящего изобретения
В соответствии с настоящим изобретением были разработаны различные новые схемы способов получения эпоксимексренона и других соединений, соответствующих формуле I:
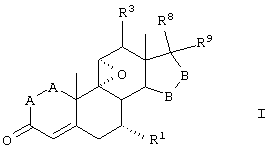
где:
-А-А- представляет группу -CHR4-CHR5- или -CR4=CR5-;
R3, R4 и R5 независимо выбраны из группы, включающей водород, галоген, гидрокси, низший алкил, низший алкокси, гидроксиалкил, алкоксиалкил, гидроксикарбонил, циано и арилокси;
R1 представляет α-ориентированный низший алкоксикарбонильный или гидроксиалкильный радикал; и
-В-В представляет группу -CHR6-CHR7- или альфа- или бета-ориентированную группу:
где R6 и R7 независимо выбраны из группы, включающей водород, галоген, низший алкокси, ацил, гидроксиалкил, алкоксиалкил, гидроксикарбонил, алкил, алкоксикарбонил, ацилоксиалкил, циано и арилокси; и
R8 и R9 независимо выбраны из группы, включающей водород, гидрокси, галоген, низший алкокси, ацил, гидроксиалкил, алкоксиалкил, гидроксикарбонил, алкил, алкоксикарбонил, ацилоксиалкил, циано и арилокси; либо R8 и R9, взятые вместе, представляют карбоциклическую или гетероциклическую кольцевую структуру, либо R8 или R9, взятые вместе с R6 или R7, представляют карбоциклическую или гетероциклическую кольцевую структуру, конденсированную с пентациклическим кольцом D.
Если это не оговорено особо, органические радикалы, называемые в настоящем описании "низшими", содержат максимум 7, а предпочтительно от 1 до 4 атомов углерода.
Низшим алкоксикарбонильным радикалом предпочтительно является радикал, происходящий от алкильного радикала, имеющего от 1 до 4 атомов углерода, такого как метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил и трет-бутил; особенно предпочтительными являются метоксикарбонил, этоксикарбонил и изопропоксикарбонил. Низшим алкокси-радикалом является, предпочтительно, радикал, происходящий от одного из вышеупомянутых С1-С4алкильных радикалов, особенно от первичного С1-С4-алкильного радикала; при этом особенно предпочтительным является метокси. Низшим алканоильным радикалом является, предпочтительно, радикал, происходящий от прямого алкила, имеющего от 1 до 7 атомов углерода; а особенно предпочтительными являются формил и ацетил.
Метиленовый мостик в 15,16-положении является, предпочтительно, β-ориентированным.
Предпочтительным классом соединений, которые могут быть получены способами настоящего изобретения, являются 20-спироксановые соединения, описанные в Патенте США №4559332, т.е. соединения, соответствующие формуле IA

где:
-А-А- представляет группу -СН2-СН2- или -СН=СН-;
-В-В- представляет группу -СН2-СН2- или альфа- или бета-ориентированную группу формулы IIIA:
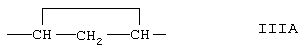
R1 представляет альфа-ориентированный низший алкоксикарбонильный или гидроксикарбонильный радикал;
Х представляет два атома водорода, оксо или =S;
Y1 и Y2, взятые вместе, представляют кислородный мостик -О- или
Y1 представляет гидрокси, и
Y2 представляет гидрокси, низший алкокси, или, если Х представляет Н2, то также низший алканоилокси.
Предпочтительно, 20-спироксановые соединения, полученные новыми способами настоящего изобретения, представляют соединения формулы I, где Y1 и Y2, взятые вместе, представляют кислородный мостик -O-.
Особенно предпочтительными соединениями формулы I являются соединения, в которых Х представляет оксо. Из 20-спироксановых соединений формулы 1А, где Х представляет оксо, наиболее предпочтительными являются те соединения, в которых Y1 вместе с Y2 представляют кислородный мостик -O-.
Как уже упоминалось, 17β-гидрокси-21-карбоновая кислота может быть также получена в форме ее солей. В частности, рассматриваются соли металлов и аммония, такие как соли щелочных металлов и щелочно-земельных металлов, например соли натрия, кальция, магния, а предпочтительно, соли калия; и соли аммония, происходящие от аммиака или подходящего, предпочтительно физиологически приемлемого органического азотсодержащего основания. В качестве оснований рассматриваются не только амины, например низшие алкиламины (такие как триэтиламин), гидрокси(низшие)алкиламины (такие как 2-гидроксиэтиламин, ди-(2-гидроксиэтил)амин или три-(2-гидроксиэтил)амин), циклоалкиламины (такие как дициклогексиламин), или бензиламины (такие как бензиламин и N,N'-дибензилэтилендиамин), но также и азотсодержащие гетероциклические соединения, например ароматические соединения (такие как пиридин или хинолин), или соединения, имеющие по крайней мере частично насыщенное гетероциклическое кольцо (такие как N-этилпиперидин, морфолин, пиперазин, или N,N'-диметилпиперазин).
Предпочтительными соединениями также являются соли щелочных металлов, особенно калиевые соли соединений формулы IA, где R1 представляет алкоксикарбонил, Х представляет оксо, а каждый из Y1 и Y2 представляет гидрокси.
Особенно предпочтительными соединениями формулы I и IA являются, например, следующие соединения:
9α,11α-эпокси-7α-метоксикарбонил-20-спирокс-4-ен-3,21-дион;
9α,11α-эпокси-7α-этоксикарбонил-20-спирокс-4-ен-3,21-дион;
9α,11α-эпокси-7α-изопропоксикарбонил-20-спирокс-4-ен-3,21-дион
и 1,2-дегидро-аналоги каждого из этих соединений;
9α,11α-эпокси-6α,7α-метилен-20-спирокс-4-ен-3,21-дион;
9α,11α-эпокси-6β,7β-метилен-20-спирокс-4-ен-3,21-дион;
9α,11α-эпокси-6β,7β,15β,16β-бисметилен-20-спирокс-4-ен-3,21-дион
и 1,2-дегидро-аналоги каждого из этих соединений;
9α,11α-эпокси-7α-метоксикарбонил-17β-гидрокси-3-оксо-прегн-4-ен-21-карбоновая кислота;
9α,11α-эпокси-7α-этоксикарбонил-17β-гидрокси-3-оксо-прегн-4-ен-21-карбоновая кислота;
9α,11α-эпокси-7α-изопропоксикарбонил-17β-гидрокси-3-оксо-прегн-4-ен-21-карбоновая кислота;
9α,11α-эпокси-17β-гидрокси-6α,7α-метилен-3-оксо-прегн-4-ен-21-карбоновая кислота;
9α,11α-эпокси-17β-гидрокси-6β,7β-метилен-3-оксо-прегн-4-ен-21-карбоновая кислота,
9α,11α-эпокси-17β-гидрокси-6β,7β,15β,16β-бисметилен-3-оксо-прегн-4-ен-21-карбоновая кислота,
и соли щелочных металлов, особенно калиевая или аммониевая соли каждой из этих кислот, а также соответствующие 1,2-дегидро-аналоги каждой из вышеупомянутых карбоновых кислот или их солей;
9α,11α-эпокси-15β,16β-метилен-3,21-диоксо-20-спирокс-4- ен-7α-карбоновой кислоты метиловый, этиловый и изопропиловый сложный эфир;
9α,11α-эпокси-15β,16β-метилен-3,21-диоксо-20-спирокса-1,4-диен-7α-карбоновой кислоты метиловый, этиловый и изопропиловый сложный эфир;
9α,11α-эпокси-3-оксо-20-спирокс-4-ен-7α-карбоновой кислоты метиловый, этиловый и изопропиловый сложный эфир;
9α,11α-эпокси-6β,6β-метилен-20-спирокс-4-ен-3-он;
9α,11α-эпокси-6β,7β,15β,16β-бисметилен-20-спирокс-4-ен-3-он;
9α,11α-эпокси-17β-гидрокси-17α(3-гидроксипропил)-3-оксо-адрост-4-ен-7α-карбоновой кислоты метиловый, этиловый и изопропиловый сложный эфир;
9α,11α-эпокси-17β-гидрокси-17α-(3-гидроксипропил)-6α,7α-метилен-андрост-4-ен-3-он,
9α,11α-эпокси-17β-гидрокси-17α-(3-гидроксипропил)-6β,7β-метилен-андрост-4-ен-3-он,
9α,11α-эпокси-17β-гидрокси-17α-(3-гидроксипропил)-6β,7β,-15β,16β-бисметилен-андрост-4-ен-3-он,
включая 17α-(3-ацетоксипропил)- и 17α-(3-формилокси-пропил)-аналоги вышеупомянутых андростановых соединений,
а также 1,2-дегидро-аналоги всех видов вышеупомянутых соединений андрост-4-ен-3-она и 20-спирокс-4-ен-3-она.
Химические названия соединений формул I и IA и соединений аналогов, имеющих те же самые характерные структурные особенности, приводятся в соответствии с общепринятой номенклатурой, а именно: названия для соединений, в которых Y1 вместе с Y2 представляет -O, происходят от 20-спироксана (например, соединение формулы IA, где Х представляет оксо, а Y1, взятый вместе с Y2, представляет -O, называется "20-спироксан-21-он"); название для соединений, в которых каждый из Y1 и Y2 представляет гидрокси, а Х представляет оксо, происходит от 17β-идрокси-17α-прегнен-21-карбоновой кислоты; а название для соединений, в которых каждый из Y1 и Y2 представляет гидрокси, а Х представляет два атома водорода, происходит от 17β-гидрокси-17α-(3-гидроксипропил)-андростана. Поскольку циклические формы и формы с незамкнутой цепью, то есть лактоны и 17β-гидрокси-21-карбоновые кислоты и их соли, соответственно, имеют настолько близкое сходство друг с другом, что последние могут считаться лишь гидратированной формой первых, то следует отметить, что в предшествующем и последующем описании, если это не оговорено особо, эти формы, как в конечных продуктах формулы I, так и в исходных и промежуточных соединениях аналогичной структуры, во всех случаях упоминаются вместе.
В соответствии с настоящим изобретением было разработано несколько отдельных схем для получения соединений формулы I с высоким выходом и при умеренных материальных затратах. Каждая из этих схем синтеза предусматривает получение серии промежуточных соединений. Ряд этих промежуточных соединений представляют собой новые соединения, а способы получения этих промежуточных соединений являются новыми способами.
Схема 1 (с использованием исходного канренона или родственного соединения)
В одной из предпочтительных схем получения соединений формулы I в качестве исходного соединения используют преимущественно канренон или родственное соединение, соответствующее формуле XIII (или, альтернативно, этот способ предусматривает использование в качестве исходного соединения андростендиона или родственного соединения)
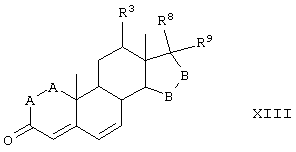
где:
-А-А- представляет группу -CHR4-CHR5- или -CR4=CR5-;
R3, R4 и R5 независимо выбраны из группы, включающей водород, галоген, гидрокси, низший алкил, низший алкокси, гидроксиалкил, алкоксиалкил, гидроксикарбонил, циано и арилокси;
-В-В- представляет группу -CHR6-CHR7- или альфа- или бета-ориентированную группу:
где R6 и R7 независимо выбраны из группы, включающей водород, галоген, низший алкокси, ацил, гидроксиалкил, алкоксиалкил, гидроксикарбонил, алкил, алкоксикарбонил, ацилоксиалкил, циано и арилокси; и
R8 и R9 независимо выбраны из группы, включающей водород, гидрокси, галоген, низший алкокси, ацил, гидроксиалкил, алкоксиалкил, гидроксикарбонил, алкил, алкоксикарбонил, ацилоксиалкил, циано и арилокси; либо R8 и R9, взятые вместе, представляют кето, карбоциклическую или гетероциклическую кольцевую структуру, либо R8 или R9, взятые вместе с R6 или R7, представляют карбоциклическую или гетероциклическую кольцевую структуру, конденсированную с пентациклическим кольцом D.
С использованием способа биологического превращения типа способа, проиллюстрированного на Фиг.1 и 2, 11-гидроксигруппу в α-ориентации вводят в соединение формулы XIII, в результате чего получают соединение формулы VIII:
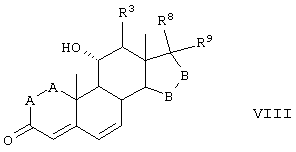
где -А-А-, -В-В-, R3, R8 и R9 определены в формуле XIII.
Соединение формулы XIIIA имеет, предпочтительно, структуру:
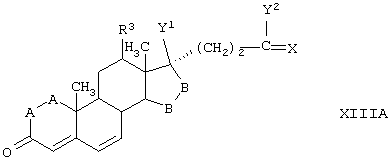
и 11α-гидрокси-продукт имеет структуру
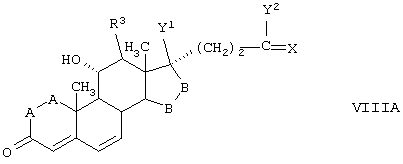
в каждой из которых:
-А-А- представляет группу -СН2-СН2- или -СН=СН-;
-В-В- представляет группу -CH2-CH2- или α- или β-ориентированную группу:
R3 представляет водород, низший алкил или низший алкокси;
Х представляет два атома водорода, оксо или =S;
Y1 и Y2, взятые вместе, представляют кислородный мостик -О- или
Y1 представляет гидрокси, и
Y2 представляет гидрокси, низший алкокси, или, если Х представляет H2, то также низший алканоилокси;
и соли соединений, в которых Х представляет оксо, a Y2 представляет гидрокси. Более предпочтительно, если соединение формулы VIIIA, полученное в этой реакции, соответствует формуле VIIIA, где каждый из -А-А-, -В-В- представляет -CH2-CH2-; R3 представляет водород; Y1 и Y2 и Х определены в формуле XIIIA; а R8 и R9, взятые вместе, образуют 20-спироксановую структуру:
.
Предпочтительными микроорганизмами, которые могут быть использованы в указанной стадии гидроксилирования, являются Aspergillus ochraceus NRRL 405, Aspergillus ochraceus ATCC 18500, Aspergillus niger ATCC 16888 и ATCC 26693, Aspergillus nidulans ATCC 11267, Rhizopus oryzae ATCC 11145, Rhizopus stolonifer ATCC 6227b, Streptomyces fradiae ATCC 10745, Bacillus megaterium ATCC 14945, Pseudomonas cruciviae ATCC 13262 и Trichothecium roseum ATCC 12543. Другими предпочтительными микроорганизмами являются Fusarium oxysporum f.sp.cepae ATCC 11171 и Rhizopus arrhizus ATCC 11145.
Другими микроорганизмами, которые обладают активностью для этой реакции, являются Absidia coerula ATCC 6647, Absidia glauca ATCC 22752, Actinomucor elegans ATCC 6476, Aspergillus flavipes ATCC 1030, Aspergillus fumigatus ATCC 26934, Beauveria bassiana ATCC 7159 и АТСС 13144, Botryosphaeria obtusa IMI 038560, Calonectria decora ATCC 14767, Chaetomium cochliodes ATCC 10195, Corynespora cassiicola ATCC 16718, Cunninghamella blakesleeana ATCC 8688a, Cunninqhainella echinuata ATCC 3655, Cunninghamella elegans ATCC 9245, Curvularia clavata ATCC 22921, Curvularia lunata ACTT 12017, Cylindrocarpon radicicola ATCC 1011, Epicoccum humicola ATCC 12722, Gongronella butleri ATCC 22822, Hypomyces chrysospermus ATCC IMI 109891, Mortierella isabellina ATCC 42613, Mucor mucedo ATCC 4605, Mucor griseo-cyanus ATCC 1207A, Myrothecium verrucaria ATCC 9095, Nocardia corallina ATCC 19070, Paecliomyces carneus ATCC 46579, Penicillum patulum ATCC 24550, Pithomyces atro-olivaceus IFO 6651, Pithomyces cynodontis ATCC 26150, Pycnosporium sp. ATCC 12231, Saccharopolyspora erythrae ATCC 11635, Sepedonium chrysospermum ATCC 13378, Stachylidium bicolor ATCC 12672, Streptomyces hygroscopicus ATCC 27438, Streptomyces purpurascens ATCC 25489, Syncephalastrum racemosum ATCC 18192, Thamnostylum piriforme ATCC 8992, Thielavia terricola ATCC 13807 и Verticillium theobromae ATCC 12474.
Другими организмами, которые, как ожидается, могут обладать активностью для 11α-гидроксилирования, являются Cephalosporium aphidicola (Phytochemistry (1996), 42(2), 411-415), Cochliobolus lunatas (J.Biotechnol. (1995), 42(2), 145-150), Tieghemella orchidis (Khim-Farm.Zh. (1986), 20(7), 871-876), Tieghemella hyalospora (Khim.-Farm.Zh. (1986), 20(7), 871-876), Monosporium olivaceum (Acta Microbiol.Pol., Ser.B. (1973), 5(2), 103-110), Aspergillus ustus (Acta Microbiol. Pol., Ser.B. (1973), 5(2), 103-110), Fusarium graminearum (Acta Microbiol.Pol., Ser.B. (1973), 5(2), 103-110), Verticillium glaucum (Acta Microbiol.Pol., Ser.B (1973), 5(2), 103-110) и Rhizopus nigricans (J.Steroid Biochem. (1987), 28(2), 197-201).
11β-Гидрокси-производные андростендиона и мексренона могут быть получены способами биологического превращения, описанными в Примерах 19А и 19В, соответственно. По аналогии, авторами настоящего изобретения было сделано предположение, что соответствующий β-гидрокси-изомер соединения формулы VIII, имеющий вместо C11-α-гидрокси-заместителя С11-β-гидрокси-заместитель, может быть получен аналогичным способом биологического превращения с использованием подходящих микроорганизмов, способных осуществлять 11β-гидроксилирование, таких как один или несколько микроорганизмов, описанных в настоящей заявке.
Перед осуществлением масштабной ферментации для гидроксилирования карненона или других субстратов формулы XIII, получают инокулят клеток в посевной системе для ферментации, содержащей посевной ферментер, либо два или более последовательно соединенных посевных ферментеров. Рабочую исходную суспензию спор вводят в первый посевной ферментер вместе с питательным раствором для культивирования клеток. Если желательно или необходимо, чтобы объем инокулята превышал объем, который продуцируется в первом посевном ферментере, то этот объем инокулята может быть увеличен в арифметической или геометрической прогрессии путем пропускания через остальные последовательно расположенные ферментеры в посевном агрегате для ферментации. Предпочтительно, чтобы инокулят, продуцируемый в посевной системе для ферментации, имел достаточный объем и содержал жизнеспособные клетки для достижения быстрой инициации реакции в производственном ферментере; и чтобы последовательные циклы продуцирования были относительно короткими, а также чтобы ферментер имел высокую производительность. Независимо от числа сосудов в системе посевных ферментеров, второй и последующий посевные ферментеры должны иметь, предпочтительно, такой размер, чтобы степень разбавления в каждой стадии этого ряда была, в основном, одной и той же. Первоначальное разбавление инокулята в каждом посевном ферментере может быть приблизительно таким же, как и разбавление в производственном ферментере. Канренон или другой субстрат формулы XIII загружают в производственный ферментер вместе с инокулятом и питательным раствором и проводят реакцию гидроксилирования.
Суспензию спор, загружаемую в посевную систему для ферментации, подают из сосуда с рабочим исходным раствором суспензии спор, взятой из множества сосудов, содержащих уже заготовленный рабочий исходный фонд клеток, который, до его использования, хранится в криогенных условиях. Этот рабочий фонд исходных клеток, в свою очередь, происходит от маточного фонда исходных клеток, который получают следующим образом. Образец спор, полученный из соответствующего источника, например из АТСС, сначала суспендируют в водной среде, такой как, например, физиологический раствор, питательный раствор или раствор поверхностно-активного вещества (например, неионогенного поверхностно-активного вещества, такого как Твин 20, при концентрации около 0,001% масс.) и эту суспензию распределяют по чашкам для культивирования, каждая из которых содержит твердую питательную смесь, обычно на основе негидролиэуемого полисахарида, такого как агар, где размножаются споры. Твердая питательная смесь, предпочтительно, содержит от около 0,5% до около 5% масс. глюкозы, от около 0,05% до около 5% масс. источника азота, например пептона; от около 0,05% до около 0,5% масс. источника фосфора, например фосфата аммония или щелочного металла, такого как дикалийбифосфат; от около 0,25 % до около 2,5% масс. дрожжевого лизата или экстракта (или другого источника аминокислоты, такого как мясной экстракт или бульон с сердечно-мозговым экстрактом); от около 1% до около 2% масс. агара или другого негидролизуемого полисахарида. Кроме того, но необязательно, твердая питательная смесь может содержать и/или содержит от около 0,1% до около 5% масс. экстракта солода. рН этой твердой питательной смеси составляет, предпочтительно, от около 5,0 до около 7,0, и если необходимо, он может быть скорректирован путем добавления гидроксида щелочного металла или ортофосфорной кислоты. Подходящими твердыми средами для культивирования являются следующие среды:
Число агаровых чашек, используемых для получения главного исходного клеточного фонда, может быть выбрано в зависимости от будущих требований для исходного маточного материала, но, в основном, это число составляет от около 15 до около 30 приготовленных таким образом чашек. После соответствующего периода культивирования, например в течение 7-10 дней, чашки отскребают для сбора спор в присутствии водного носителя, обычно физиологического раствора или буфера, и полученную маточную исходную суспензию распределяют по небольшим сосудам, например 1 мл суспензии помещают в каждый из множеств 1,5 мл сосудов. Для получения рабочей исходной суспензии спор для исследований или для промышленной ферментации содержимое одного или нескольких из этих сосудов с маточным исходным материалом второй генерации может быть распределено по чашкам с агаром и инкубировано способом, аналогичным способу, описанному выше для получения маточной исходной суспензии спор. Если предусматривается традиционное промышленное продуцирование, то для получения рабочего исходного материала второй генерации может быть использовано по крайней мере от 100 до 400 чашек. Содержимое каждой чашки отскребают в отдельный сосуд с рабочим исходным материалом, где каждый сосуд, обычно, содержит 1 мл продуцированного инокулята. Для постоянной консервации, как маточную исходную суспензию, так и инокулят второй генерации хранят преимущественно в паровой фазе в сосуде для криогенного хранения, содержащем жидкий N2 или другую криогенную жидкость.
В способе, проиллюстрированном на Фиг.1, получают водную среду для культивирования, которая содержит источник азота, такой как пептон, дрожжевое производное или его эквивалент, глюкозу, и источник фосфора, такой как как фосфатная соль. Споры микроорганизма культивируют в этой среде в посевной ферментационной системе. Предпочтительным микроорганизмом является Aspergillus ochraceus NRRL 405 (ATCC 18500). Затем полученную таким образом посевную среду вводят в ферментер для продуцирования вместе с субстратом формулы XIII. Бульон для ферментации размешивают и аэрируют в течение интервала времени, достаточного для прохождения реакции до нужной степени завершения.
Среда для посевного ферментера, предпочтительно, включает водную смесь, которая содержит от около 0,5% и около 5% масс. глюкозы, от около 0,05% до около 5% масс. источника азота, например пептона; от около 0,05% до около 0,5% масс. источника фосфора, например фосфата аммония или щелочного металла, такого как одноосновный фосфат аммония или дикалий-бифосфат; от около 0,25% до около 2,5% масс. дрожжевого лизата или экстракта (или другого источника аминокислоты, такого как экстракт барды) ; от около 1% до около 2% масс. агара или другого негидролизуемого полисахарида. Особенно предпочтительная среда для культивирования посевного материала содержит от около 0,05% до около 5% масс. источника азота, такого как пептон; от около 0,25% до около 2,5% масс. автолизированных дрожжей или дрожжевого экстракта; от около 0,5% до около 5% масс. глюкозы и от около 0,05% до около 0,5% масс. источника фосфора, такого как одноосновного фосфата аммония. Особо экономичные способы были разработаны с использованием другой предпочтительной посевной культуры, которая содержит от около 0,5% до около 5% масс. жидкого кукурузного экстракта, от около 0,25% до около 2,5% масс. автолизированных дрожжей или дрожжевого экстракта; от около 0,5% до около 5% масс. глюкозы и от около 0,05% до около 0,5% масс. одноосновного фосфата аммония. Жидкий кукурузный экстракт является особенно экономичным источником белков, пептидов, углеводов, органических кислот, витаминов, ионов металлов, микроэлементов и фосфатов. Вместо жидкого кукурузного экстракта или в дополнение к нему может быть использован раствор пульпы из других зерен. рН этой среды предпочтительно доводят до значений от около 5,0 до около 7,0, например путем добавления гидроксида щелочного металла или ортофосфорной кислоты. Если в качестве источника азота и углерода используется жидкий кукурузный экстракт, то рН, предпочтительно, доводят до значения от около 6,2 до около 6,8. Среду, содержащую пептон и глюкозу, предпочтительно доводят до рН от около 5,4 до около 6,2. Средами для культивирования, подходящими для использования в посевной ферментации, являются:
Споры микроорганизма вводят в эту среду из сосуда, обычно содержащего приблизительно 109 спор на мл суспензии. Оптимальная продуктивность посевной генерации достигается в том случае, когда при разбавлении культуральной средой в начале культивирования посевной культуры плотность популяции спор не снижается ниже примерно 107 на мл. Предпочтительно, споры культивируют в посевной системе ферментации до тех пор, пока объем осажденного мицелия (PMV) в посевном ферментере не будет составлять по крайней мере около 20%, а предпочтительно от около 35% до около 45%. Поскольку цикл в сосуде для ферментации посевного материала (или в любом сосуде из множества сосудов, которые составляют систему для посевной ферментации) зависит от первоначальной концентрации в этом сосуде, то может оказаться предпочтительным проводить две или три стадии посевной ферментации для ускорения всего процесса. Однако, желательно избегать использования значительно более чем три ферментера в системе, поскольку в том случае, когда ферментация посевного материала включает излишнее число стадий, то это может неблагоприятно повлиять на активность процесса. Ферментацию посевной культуры проводят при перемешивании при температуре в пределах от около 23 до около 37°С, а предпочтительно в пределах от около 24 до около 28°С.
Культуру из посевной системы ферментации вводят в производственный ферментер вместе с производственной средой для культивирования. В одном из вариантов осуществления изобретения в качестве субстрата для реакции служит нестерильный канренон или другой субстрат формулы XIII. Предпочтительно, если субстрат добавляют в производственный ферментер в виде 10%-30% масс. суспензии в среде для культивирования. Для увеличения площади поверхности, доступной для реакции 11α-гидроксилирования, перед введением субстрата формулы XIII в ферментер размер частиц этого субстрата уменьшают путем его пропускания через автономно работающий микронайзер (мельницу для тонкого помола). Также отдельно вводят стерильный исходный питательный раствор, содержащий глюкозу, и второй стерильный питательный раствор, содержащий дрожжевое производное, такое как автолизованные дрожжи (или эквивалентную аминокислотную композицию, составленную на основе альтернативных источников, таких как экстракт барды). Эта среда включает водную смесь, содержащую от около 0,5% и около 5% масс. глюкозы, от около 0,05% до около 5% масс. источника азота, например пептона; от около 0,05% до около 0,5% масс. источника фосфора, например фосфата аммония, или щелочного металла, такого как дикалийбифосфат; от около 0,25% до около 2,5% масс. дрожжевого лизата или экстракта (или другого источника аминокислоты, такого как экстракт барды); от около 1% до около 2% масс. агара или другого негидролизуемого полисахарида. Особенно предпочтительная производственная среда для культивирования содержит от около 0,05% до около 5% масс. источника азота, например пептона; от около 0,25% до около 2,5% масс. автолизированного дрожжевого экстракта; от около 0,5% до около 5% масс. глюкозы и от около 0,05% до около 0,5% масс. источника фосфора, такого как одноосновного фосфата аммония. Другая предпочтительная производственная среда содержит от около 0,5% до около 5% масс. жидкого кукурузного экстракта, от около 0,25% до около 2,5% масс. автолизированных дрожжей или дрожжевого экстракта; от около 0,5% до около 5% масс. глюкозы и от около 0,05% до около 0,5% масс. одноосновного фосфата аммония. рН среды для производственной ферментации предпочтительно корректируют способом, описанным выше для среды, предназначенной для посевной ферментации, при этом наиболее предпочтительными являются пределы значений рН, указанные для среды, содержащей пептон/глюкозу, и для среды, содержащей жидкий кукурузный экстракт, соответственно. Среды для культивирования, подходящие для реакций биологического превращения, указаны ниже:
Нестерильный раствор канренона и стерильный питательный раствор загружают путем цепной подачи в производственный ферментер от около пяти до около двадцати, предпочтительно от около десяти до около пятнадцати порций, и предпочтительно, в основном, равные порции каждого ингредиента в течение всего производственного цикла. При этом, перед инокуляцией бульоном для посевной ферментации, субстрат сначала вводят, предпочтительно, в количестве, достаточном для достижения концентрации от около 0,1% масс. до около 3% масс., а предпочтительно от около 0,5% масс. до около 2% масс., а затем периодически добавляют, в основном через каждые 8-24 часа, для достижения кумулятивного количества от около 1% до около 8% масс. Если дополнительное количество субстрата добавляют каждые 8 часов, то общее добавление будет немного меньше, например 0,25%-2,5% масс., чем в том случае, когда субстрат добавляют только через сутки. В последнем случае, может оказаться необходимым кумулятивное добавление канренона в пределах от 2% до около 8% масс. Дополнительной питательной смесью, добавляемой в процессе реакции ферментации, является предпочтительно концентрат, например смесь, содержащая от около 40% до около 60% масс. стерильной глюкозы, от около 16% до около 32% масс. стерильного дрожжевого экстракта или другого стерильного источника дрожжевого производного (или другого аминокислотного источника). Поскольку субстрат, подаваемый в производственный ферментер, показанный на Фиг.1, является нестерильным, то для подавления роста нежелательных микроорганизмов в бульон для ферментации периодически добавляют антибиотики. Антибиотики, такие как канамицин, тетрациклин и цефалексин, могут быть добавлены без какого-либо неблагоприятного воздействия на рост и биологическое превращение микроорганизмов. Эти антибиотики вводят в бульон для ферментации, предпочтительно, в концентрации, например от около 0,0004% до около 0,002% по полной массе бульона, содержащего, например, от около 0,0002% до около 0,0006% сульфата канамицина, от около 0,0002% до около 0,006% тетрациклина·HCl и/или от около 0,001% до около 0,003% цефалексина по полной массе бульона.
Обычно цикл производственной ферментации продолжается примерно 80-160 часов. Таким образом, порции каждого из субстратов формулы XIII и питательных растворов обычно добавляют примерно через каждые 2-10 часов, а предпочтительно через каждые 4-6 часов. В систему посевной ферментации и в производственный ферментер предпочтительно также вводить пеногаситель.
В способе, проиллюстрированном на Фиг.1, инокулят загружают в производственный ферментер, предпочтительно в количестве от около 0,5% до около 7%, более предпочтительно от около 1% до около 2% по объему полной смеси в ферментере, а концентрацию глюкозы поддерживают от около 0,01% до около 1,0%, предпочтительно от около 0,025% до около 0,5%, а более предпочтительно от около 0,05% до около 0,25% по массе, путем периодических добавлений, которые осуществляют предпочтительно порциями от около 0,05% до около 0,25% по массе полной партии загрузки. Температура ферментации предпочтительно регулируется в пределах от 20°С до 37°С, предпочтительно от 24°С до около 28°С, однако может оказаться желательным постепенно снижать температуру в процессе реакции, например на 2°С, но при этом поддерживать объем осажденного мицелия (PMV) ниже примерно 60%, а более предпочтительно ниже примерно 50%, что способствует предотвращению увеличения вязкости бульона при ферментации, которая может препятствовать достаточному размешиванию. Если рост биомассы выходит за пределы поверхности жидкости, то субстрат, присутствующий в биомассе, может оказаться за пределами реакционной зоны и быть недоступным для реакции гидроксилирования. Для поддерживания продуктивности желательно, чтобы PMV достигал 30-50%, а предпочтительно, 35-45% в первые 24 часа реакции ферментации, а после этого условия, предпочтительно, корректируют для регулирования последующего роста в пределах, установленных выше. В процессе реакции рН среды для ферментации корректируют в пределах от около 5,0 до около 6,5, предпочтительно от около 5,2 до около 5,8 и содержимое ферментера размешивают со скоростью от около 400 до около 800 об/мин. Уровень растворенного кислорода по крайней мере около 10% от насыщения достигается путем аэрации партии примерно при 0,2-1,0 об/об/мин, при этом давление в головной части ферментера составляет примерно в пределах от атмосферного давления до около 1,0 бар, а более предпочтительно порядка около 0,7 бар. Скорость размешивания может быть также увеличена, если это необходимо для поддержания минимальных уровней растворенного кислорода. Растворенный кислород поддерживают, преимущественно, на уровне, значительно превышающем около 10%, а фактически вплоть до около 50% для стимуляции превращения субстрата. Поддерживание рН в пределах 5,5±0,2 является также оптимальным условием для биологического превращения. Пенообразование регулируют, при необходимости, путем добавления обычно используемого пеногасителя. После добавления всего субстрата реакцию предпочтительно продолжают до тех пор, пока молярное отношение продукта формулы VIII к оставшемуся непрореагировавшему субстрату формулы XIII не будет составлять по крайней мере от около 9 до 1. Такое превращение может быть достигнуто в процессе 80-160-часового цикла, описанного выше.
Было установлено, что высокая степень превращения связана с истощением уровней исходных питательных веществ ниже исходного уровня загрузки, а поэтому путем регулирования скорости аэрации и скорости размешивания можно предотвратить выплескивание субстрата из жидкого бульона. В процессе, проиллюстрированном на Фиг.1, уровень питательных веществ истощается, а затем поддерживается на уровне, не превышающем около 60%, предпочтительно около 50%, от уровня первоначальной загрузки, а в процессах, проиллюстрированных на Фиг.2 и 3, уровень питательных веществ снижается до уровня и подерживается на уровне, не превышающем около 80%, предпочтительно около 70% от уровня первоначальной загрузки. Скорость аэрации, предпочтительно, не превышает 1 об/об/мин, а более предпочтительно составляет порядка около 0,5 об/об/мин; а скорость размешивания предпочтительно составляет не более 600 об/мин.
Особенно предпочтительный способ получения соединения формулы VIII проиллюстрирован на Фиг.2. Предпочтительным микроорганизмом для 11α-гидрокилирования соединения формулы XIII (например, канренона) является Aspergillus ochraceus NRRL 405 (ATCC 18500). В этом способе среда для культивирования предпочтительно содержит от около 0,5% до около 5% масс. жидкого кукурузного экстракта, от около 0,5% до около 5% масс. глюкозы, от около 0,1% до около 3% масс. дрожжевого экстракта и от около 0,05% до около 0,5% масс. фосфата аммония. Однако, могут быть также использованы и другие производственные среды для культивирования. Посевную культуру получают, в основном, способом, проиллюстрированным на Фиг. 1, с использованием любой среды для посевной ферментации, описанной в настоящей заявке. Суспензию не измельченного канренона или другого субстрата формулы XIII в среде для культивирования получают в асептических условиях в смесителе, предпочтительно в относительно высокой концентрации, составляющей от около 10% до около 30% по массе субстрата. Предпочтительно, асептические условия получения могут предусматривать стерилизацию или пастеризацию суспензии после смешивания. Полное количество стерильной суспензии субстрата, необходимое для получения партии продукта, вводят в производственный ферментер в начале цикла или путем периодической цепной подачи. Размер частиц субстрата уменьшают путем мокрого помола в насосе со сдвиговым действием, работающем в оперативном режиме, который подает суспензию в производственный ферментер, что позволяет избежать необходимости использовать автономно работающий микронайзер. Если асептические условия обеспечиваются путем пастеризации, а не стерилизации, то степень агломерации может быть незначительной, однако использование насоса со сдвиговым действием может оказаться желательным для осуществления позитивного регулирования размера частиц. Стерильную среду для культивирования и раствор глюкозы вводят в производственный ферментер, в основном, способом, аналогичным описанному выше. Все питательные компоненты перед их введением в производственный ферментер стерилизуют, а поэтому использования антибиотиков не требуется.
При предпочтительном осуществлении способа, проиллюстрированного на Фиг.2, инокулят вводят в производственный ферментер в количестве от около 0,5% до около 7%, при этом температура ферментации составляет от около 20°С до около 37°С, предпочтительно от около 24°С до около 28°С, а рН корректируют в пределах от около 4,4 до около 6,5, а предпочтительно от около 5,3 до около 5,5, например, путем введения газообразного аммиака, водного гидроксида аммония, водного гидроксида щелочного металла или ортофосфорной кислоты. Как и в способе, показанном на Фиг.1, температуру предпочтительно корректируют для регулирования роста биомассы так, чтобы PMV не превышал 55-60%. Первоначальная загрузка глюкозы, предпочтительно, составляет от около 1% до около 4% масс., а более предпочтительно 2,5%-3,5% масс., однако в процессе ферментации она может составлять предпочтительно ниже около 1,0% масс. Дополнительное количество глюкозы периодически подается порциями от около 0,2% до около 1,0% по полной массе загрузочной партии так, чтобы концентрация глюкозы в зоне ферментации поддерживалась в пределах от около 0,1% до около 1,5% масс., а предпочтительно от около 0,25% до около 0,5% масс. Источники азота и фосфора могут, но не обязательно, подаваться вместе с глюкозой. Однако, поскольку загрузка всего канренона производится в начале цикла для данной патрии, то необходимая подача азот- и фосфор-содержащих питательных веществ может быть также проведена одновременно, что позволит использовать для добавления в процессе реакции лишь раствор глюкозы. Скорость и тип размешивания значительно варьируются. Умеренное размешивание стимулирует массообмен между твердым субстратом и водной фазой. Однако для предупреждения разложения миелина микроорганизмов необходимо использовать мешалку с небольшими сдвиговыми усилиями. Оптимальная скорость размешивания варьируется в пределах от 200 до 800 об/мин в зависимости от вязкости культурального бульона, концентрации кислорода и условий размешивания, на которые оказывает влияние конфигурация сосуда, перегородки и мешалки. Обычно предпочтительная скорость размешивания составляет в пределах 350-600 об/мин. Предпочтительно, лопасти для размешивания осуществляют функцию аксиальной накачки сверху вниз, что обеспечивает хорошее размешивание сбраживаемой биомассы. Эту партию предпочтительно аэрируют при скорости, составляющей от около 0,3 до около 1,0 об/об/мин, а предпочтительно от около 0,4 до 0,8 около об/об/мин, а давление в головной части ферментера составляет, предпочтительно, от около 0,5 до около 1,0 бар на шкале отсчета. Температура размешивания, аэрация и противодавление предпочтительно регулируются для поддержания количества растворенного кислорода на уровне по крайней мере около 10% по объему в процессе биологического превращения. Продожительность всего цикла для данной партии обычно составляет от около 100 до около 140 часов.
Хотя принцип осуществления способа, проиллюстрированного на Фиг.2, основан на раннем введении, в основном, всей загрузки канренона, однако, при этом, следует отметить, что культивирование бульона для ферментации может быть осуществлено до загрузки всего объема канренона. Некоторая часть канренона может быть также добавлена в партию позже, но необязательно. Однако, в основном, через 48 часов после инициации ферментации в ферментер для трансформации должно быть введено по крайней мере около 75% стерильного канренона. Более того, желательно, чтобы по крайней мере около 25% масс. канренона было введено в начале ферментации или по крайней мере в первые 24 часа ферментации для стимуляции продуцирования фермента(ов) биологического превращения.
В более предпочтительном способе, проиллюстрированном на Фиг.3, полную загрузку партии и питательный раствор стерилизуют в производственном аппарате для ферментации, а затем вводят инокулят. Питательные растворы, которые могут быть использованы, а также предпочтительные из этих растворов, являются в основном, такими же, как и в способе, проиллюстрированном на Фиг.2. В этом варианте осуществления изобретения сдвиговое усилие лопастей мешалки разрушает агломераты субстрата, которые так или иначе образуются после стерилизации. Было установлено, что эта реакция протекает благоприятным образом, если размер частиц канренона составляет менее чем около 300 микрон и по крайней мере 75% масс. всех частиц имеет размер менее 240 микрон. Было установлено, что использование подходящей мешалки, например дисковой турбинной мешалки, при адекватной скорости порядка 200-800 об/мин, с максимальной скоростью, составляющей по крайней мере около 400 см/сек, обеспечивает скорость сдвига, достаточную для поддержания указанных значений размера частиц, несмотря на агломерацию частиц, которая обычно происходит после стерилизации в производственном ферментере. Остальные операции способа, проиллюстрированного на Фиг.3, являются, в основном, такими же, как в способе, проиллюстрированном на Фиг.2. Способы, проиллюстрированные на Фиг.2 и 3, имеют некоторые существенные преимущества по сравнению со способом, проиллюстрированным на Фиг.1. Основным преимуществом является возможность использования недорогостоящей питательной основы, такой как жидкий кукурузный экстракт. Другие преимущества реализуются благодаря исключению необходимости добавления антибиотиков, что упрощает процедуры подачи и позволяет осуществлять стерилизацию партии канренона или другого субстрата формулы XIII. Другим существенным преимуществом является возможность использования простого раствора глюкозы вместо сложного питательного раствора для добавления во время осуществления реакционного цикла.
В способах, проиллюстрированных на Фиг.1-3, продукт формулы VIII является кристаллическим твердым веществом, которое вместе с биомассой может быть выделено из реакционного бульона путем фильтрации или низкоскоростного центрифугирования. Альтернативно, этот продукт может быть экстрагирован из всего реакционного бульона с использованием органических растворителей. Продукт формулы VIII выделяют путем экстракции растворителем. Для максимального выделения жидкофазный фильтрат и фильтр с биомассой или осадок на центрифуге обрабатывают экстрагирующим растворителем, но обычно ≥95% продукта ассоциировано с биомассой. Обычно, для экстракции могут быть использованы углеводород, сложный эфир, хлорированный углеводород и кетоновые растворители. Предпочтительным растворителем является этилацетат. Другими, в основном подходящими растворителями являются толуол и метилизобутилкетон. Для экстракции из жидкой фазы может оказаться предпочтительным использовать объем растворителя, приблизительно равный объему реакционного раствора, с которым он контактирует. Для восстановления продукта из биомассы эту биомассу суспендируют в растворителе, предпочтительно в большом избытке относительно первоначальной загрузки субстрата, например 50-100 мл растворителя на грамм первоначальной загрузки канренона, и полученную суспензию, предпочтительно нагревают с обратным холодильником в течение периода времени примерно от 20 минут до нескольких часов для гарантии переноса продукта в фазу растворителя из углублений и пор биомассы. После этого биомассу удаляют путем фильтрации или центрифугирования и осадок на фильтре предпочтительно промывают свежим растворителем и деионизованной водой. Затем водную промывку и промывку растворителем объединяют и оставляют для разделения фаз. Продукт формулы VIII выделяют путем кристаллизации из раствора. Для максимизации выхода мицелий два раза подвергают контакту со свежим растворителем. После осаждения продукт выделяют из фазы растворителя для полного отделения водной фазы. Более предпочтительно растворитель выпаривают в условиях вакуума до тех пор, пока не начнется кристаллизация, а затем концентрированный экстракт охлаждают до температуры от около 0°С до около 20°С, а предпочтительно от около 10°С до около 15°С, в течение периода времени, достаточного для осаждения и роста кристаллов, обычно в течение от около 8 до около 12 часов.
Наиболее предпочтительными являются способы, проиллюстрированные на Фиг.2, а особенно способ, проиллюстрированный на Фиг.3. Эти способы осуществляют при низкой вязкости и они являются подходящими для точного контроля параметров процесса, таких как рН, температура и уровень растворенного кислорода. Кроме того, условия стерильности легко поддерживаются без введения антибиотиков.
Процессы биологического превращения являются экзотермическими, что требует отвода тепла с использованием ферментера с рубашкой или охлаждающего змеевика в производственном ферментере. Альтернативно, реакционный бульон может быть возвращен на повторный цикл через внешний теплообменник. Растворенный кислород, предпочтительно, поддерживают на уровне по крайней мере около 5%, а предпочтительно по крайней мере около 10% по объему, который является достаточным для обеспечения энергии для данной реакции и гарантии превращения глюкозы в СО2 и Н2О путем регулирования скорости потока воздуха, вводимого в реактор после измерения уровня кислорода в бульоне. рН предпочтительно поддерживают в пределах от около 4,5 до около 6,5.
В каждом из альтернативных способов 11-гидроксилирования субстрата формулы XIII продуктивность этого способа ограничена переносом массы от твердого субстрата в водную фазу или в область границы раздела фаз, где очевидно происходит реакция. Как указано выше, продуктивность, в основном, не ограничена скоростью массопереноса, при условии, что средний размер частиц субстрата уменьшен до менее чем около 300 микрон, и что по крайней мере 75% частиц имеет размеры меньше, чем 240 микрон. Однако, продуктивность этих процессов может быть еще больше увеличена в некоторых альтернативных вариантах осуществления изобретения, в которых обеспечивается основная загрузка канренона или другого субстрата формулы XIII в производственный ферментер в органическом растворителе. В соответствии с этим вариантом субстрат растворяют в не смешивающемся с водой растворителе и смешивают с исходной водной культуральной средой для пересева и с поверхностно-активным веществом. Подходящими несмешивающимися с водой растворителями являются, например, ДМФ, ДМСО, C6-C12-жирные кислоты, С6-С12-н-алканы, растительные масла, сорбитаны и водные растворы поверхностно-активного вещества. Размешивание этой загрузки способствует продуцированию эмульсионной реакционной системы, имеющей протяженную площадь межфазной поверхности для переноса массы субстрата из органической жидкой фазы в зоны реакции.
Во втором варианте осуществления изобретения сначала растворяют субстрат в смешивающемся с водой растворителе, таком как ацетон, метилэтилкетон, метанол, этанол или глицерин, в концентрации, в основном, превышающей его растворимость в воде. При получении исходного раствора субстрата при повышенной температуре растворимость возрастает, что способствует увеличению количества растворимой формы субстрата, вводимого в реактор, и, тем самым, увеличивает полезную нагрузку реактора. Теплый раствор субстрата загружают в производственный реактор для ферментации вместе с относительно холодной водной загрузкой, содержащей среду для культивирования и инокулят. При смешивании раствора субстрата с водной средой происходит осаждение субстрата. Однако, в условиях значительного сверхнасыщения и умеренно интенсивного размешивания вместо роста кристаллов происходит преимущественное образование центров кристаллизации, при этом образуются очень мелкие частицы с большой поверхностной площадью. Большая площадь поверхности стимулирует массоперенос между жидкой фазой и твердым субстратом. Кроме того, равновесная концентрация субстрата в водной жидкой фазе также увеличивается в присутствии смешивающегося с водой растворителя. В соответствии с этим увеличивается продуктивность.
Хотя данный микроорганизм не должен обязательно быть толерантным к высокой концентрации органического растворителя в водной фазе, однако желательно использовать концентрацию этанола, например, в пределах от около 3% до около 5% масс.
Третьим вариантом осуществления изобретения является солюбилизация субстрата в водном растворе циклодекстрина. Типичными циклодекстринами являются гидроксипропил-β-циклодекстрин и метил-β-циклодекстрин. Молярное отношение субстрат: циклодекстрин может составлять от около 1:0,5 до около 1:1,5, а более предпочтительно от около 1:0,8 до около 1:1. Затем эта смесь субстрат: циклодекстрин может быть добавлена, в условиях асептики, в реактор для биологического превращения.
11α-Гидроксиканренон и другие продукты реакции 11α-гидроксилирования (Формулы VIII и VIIIA) представляют собой новые соединения, которые могут быть выделены путем фильтрации реакционной среды и экстракции продукта из биомассы, собранной на среде после фильтрации. Для экстракции могут быть использованы стандартные органические растворители, например этилацетат, ацетон, толуол, хлорированные углеводороды и метилизобутилкетон. Затем продукт формулы VIII может быть перекристаллизован из органического растворителя аналогичного типа. Соединения формулы VIII используются, в основном, в качестве промежуточных соединений для получения соединений формулы I и, главным образом, соединений формулы IA.
Предпочтительно, соединения формулы VIII соответствуют формуле VIIIA, где -А-А- и -В-В- представляют -CH2-CH2-, R3 представляет водород, низший алкил или низший алкокси, а R8 и R9 вместе составляют 20-спироксановое кольцо:
Затем, в соответствии со способом, показанным на Схеме 1, соединение формулы VIII подвергают реакции с источником иона цианида в щелочных условиях с получением енаминового соединения формулы VII
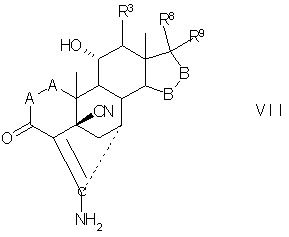
где -А-А-, -В-В-, R3, R8 и R9 определены выше.
Если субстрат соответствует формуле VIIIA, то продукт представляет собой соединение формулы VIIA:
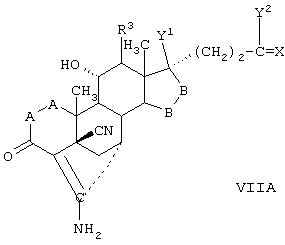
где -А-А-, -В-В-, R3, Y1, Y2 и Х определены в формуле XIIIA.
R3 представляет предпочтительно водород.
Цианидирование 11α-гидроксильного субстрата формулы VIII может быть осуществлено посредством его реакции с источником иона цианида, таким как цианогидрин кетона, наиболее предпочтительно цианогидрин ацетона, в присутствии основания и соли щелочного металла, наиболее предпочтительно LiCl.
Альтернативно, цианидирование может быть осуществлено без цианогидрина с использованием цианида щелочного металла в присутствии кислоты.
В способе с использованием цианогидрина кетона реакцию проводят в растворе, предпочтительно с использованием апротонного полярного растворителя, такого как диметилформамид или диметилсульфоксид. Образование енамина требует использования по крайней мере двух молей источника иона цианида на один моль субстрата, а предпочтительно небольшого избытка источника цианида. Основанием является предпочтительно азотистое основание, такое как диалкиламин, триалкиламин, алканоламин, пиридин или т.п. Однако, могут быть также использованы неорганические основания, такие как карбонаты щелочных металлов или гидроксиды щелочных металлов. Предпочтительно, субстрат формулы VIII первоначально присутствует в соотношении от около 20 до около 50% масс., а основание присутствует в соотношении от 0,5 до двух эквивалентов на эквивалент субстрата. Температура реакции не играет решающей роли, но при повышенной температуре выход продукта увеличивается. Так, например, если в качестве основания используют триэтиламин, то реакцию преимущественно проводят в пределах температуры от около 80°С до около 90°С. При этой температуре реакция протекает до ее полного завершения за период от около 5 до около 20 часов. Если в качестве основания используют диизопропилэтиламин, то реакцию проводят при 105°С, и эта реакция завершается через 8 часов. По окончании реакции растворитель удаляют в вакууме и остаточное масло растворяют в воде и нейтрализуют до рН 7 путем добавления разбавленной кислоты, предпочтительно хлористоводородной кислоты. Продукт осаждается из этого раствора, после чего его промывают дистиллированной водой и сушат воздухом. Выделение HCN может быть прекращено продувкой инертным газом и погашено в щелочном растворе. Осушенный осадок растворяют в хлороформе или другом подходящем растворителе, а затем экстрагируют концентрированной кислотой, например 6н НС1. Экстракт нейтрализуют до рН 7 путем добавления неорганического основания, предпочтительно гидроксида щелочного металла, и охлаждают до температуры в пределах 0°С. Полученный осадок промывают и сушат, а затем перекристаллизовывают из подходящего растворителя, например ацетона, с получением продукта формулы VII, который может быть использован в последующей стадии.
Альтернативно, реакция может быть проведена в водной системе растворителей, содержащей смешиваемый с водой органический растворитель, такой как метанол, или в двухфазной системе, содержащей воду и органический растворитель, такой как этилацетат. В этом альтернативном варианте, продукт может быть выделен путем разбавления реакционного раствора водой с последующей экстракцией продукта с использованием органического растворителя, такого как метиленхлорид или хлороформ, а затем обратной экстракцией из органического экстракта с использованием концентрированной минеральной кислоты, например 2н HCl. См. Патент США №3200113.
В соответствии с другим альтернативным способом реакция может быть проведена в смешиваемом с водой растворителе, таком как диметилформамид, диметилацетамид, N-метил, пирролидон или диметилсульфоксид, после чего раствор реакционного продукта разбавляют водой и делают его щелочным, например путем добавления карбоната щелочного металла, а затем охлаждают до 0-10°С, что приводит к осаждению продукта. Предпочтительно систему гасят гипогалитом щелочного металла или другим реагентом, эффективным для предотвращения выделения цианида. После фильтрации и промывки водой осажденный продукт может быть использован в последующей стадии данного процесса.
В соответствии с еще одним альтернативным способом, енаминовый продукт формулы VII может быть получен посредством реакции субстрата формулы VIII, в присутствии источника протонов, с избытком цианида щелочного металла, предпочтительно NaCN, в водном растворителе, содержащем апротонный смешиваемый с водой полярный растворитель, такой как диметилформамид или диметилацетамид. Источником протонов предпочтительно является минеральная кислота или C1-C5-карбоновая кислота, а наиболее предпочтительной является серная кислота. Интересно отметить, что если цианидирующим реагентом является коммерчески доступный реагент LiCN в ДМФ, то дискретный источник протонов добавлять не обязательно.
Источник ионов цианида, такой как соль щелочного металла, загружают в реактор, предпочтительно в соотношении от около 2,05 до около 5 молярных эквивалентов на эквивалент субстрата. Очевидно, что минеральная кислота, или другой источник протонов, стимулирует присоединение HCN по 4,5- и 6,7-двойным связям и предпочтительно присутствует в соотношении по крайней мере один молярный эквивалент на один молярный эквивалент субстрата; но при этом реакционная система должна оставаться щелочной, благодаря поддержанию избыточного количества цианида щелочного металла по отношению к количеству присутствующей кислоты. Реакцию предпочтительно проводят при температуре по крайней мере около 75°С, обычно при 60°С-100°С, за период времени от около 1 часа до около 8 часов, а предпочтительно от около 1,5 до около 3 часов. По окончании реакции реакционную смесь охлаждают, предпочтительно примерно до комнатной температуры; и енаминовый продукт осаждают путем подкисления реакционной смеси и смешивания ее с холодной водой, предпочтительно при температуре, близкой к температуре ледяной бани. Подкисление, очевидно, способствует замыканию 17-лактона, который имеет тенденцию к размыканию в щелочных условиях, преобладающих при реакции цианидирования. Реакционную смесь обычно подкисляют с использованием той же самой кислоты, которая присутствует в процессе реакции и которая является предпочтительно серной кислотой. Воду добавляют предпочтительно в соотношении от около 10 до около 50 молярных эквивалентов на один моль продукта.
Соединения формулы VII являются новыми соединениями и используются, в основном, в качестве промежуточных соединений для получения соединений формулы I, а особенно формулы IA. Предпочтительно, соединения формулы VII соответствуют формуле VIIA, где -А-А- и -В-В- представляют -СН2-СН2-, R3 представляет водород, низший алкил или низший алкокси, а R8 и R9 вместе составляют 20-спироксановое кольцо:
Наиболее предпочтительным соединением формулы VII является 5'R(5'α),7'β-20'-аминогексадекагидро-11'β-гидрокси-10'α,13'α-диметил-3',5'-диоксоспиро[фуран-2(3Н), 17'α(5'H)-[7,4]метено[4Н]-циклопента[а]фенантрен]-5'-карбонитрил.
При реакции превращения соединения формулы VIII в енамин Формулы VII в сыром продукте с помощью хроматографии наблюдали 7-циано-производное соединения формулы VIII. Было предположено, что в этом процессе превращения, 7-циано-сое-динение является промежуточным соединением. Кроме того, было сделано предположение, что реакция самого 7-циано-производного приводит к образованию второго промежуточного соединения, 5,7-дициано-производного соединения формулы VIII, которое, в свою очередь, реагирует с образованием сложного енэфира. См. например, работу R.Christiansen et al., The Reaction of Steroidal 4,6-Dien-3-Ones With Cyanide, Steroids, Vol.1, June 1963, которая вводится в настоящее изобретение посредством ссылки. Эти новые соединения также используются в качестве хроматографических маркеров, а также являются синтетическими промежуточными соединениями. В предпочтительном варианте осуществления этой стадии полного синтеза, показанного на Схеме 1, этими промежуточными соединениями являются 7α-циано-11α,17-дигидрокси-3-оксо-17α-прегн-4-ен-21-дикарбоновая кислота, γ-лактон и 5β,7α-дициано-11α,17-дигидрокси-3-оксо-17α-прегнан-21-дикарбоновая кислота, γ-лактон.
В следующей стадии синтеза, показанного на Схеме 1, енамин формулы VII гидролизуют с получением дикетонового соединения формулы VI:
где: -А-А-, -В-В-, R3, R8 и R9 определены в формуле XIII.
Для гидролиза может быть использована любая водная органическая или минеральная кислота. Предпочтительной является хлористоводородная кислота. Для увеличения выхода продукта в качестве сорастворителя предпочтительно используют смешиваемый с водой органический растворитель, такой как диметилацетамид или низший алканол. Более предпочтительным растворителем является диметилацетамид. Кислота должна присутствовать в соотношении по крайней мере один эквивалент на эквивалент субстрата формулы VII. В водной системе субстрат енамина VII может быть, в основном, превращен в дикетон формулы VI в течение около 5 часов при около 80°С. Проведение реакции при повышенной температуре увеличивает выход продукта, однако температура не играет решающей роли. Подходящую температуру выбирают, исходя из летучести системы растворителей и кислоты.
Предпочтительно, субстрат енамина формулы VII соответствует формуле VIIA:
и дикетоновый продукт соответствует формуле VIA:

где каждый из -А-А-, -В-В-, R3, Y1, Y2 и Х определены в формуле XIIIA. Предпочтительно, R3 представляет водород.
По окончании реакции раствор охлаждают до температуры от около 0°С до 25°С для кристаллизации продукта. Продукт в виде кристаллов может быть перекристаллизован из подходящего растворителя, такого как изопропанол или метанол, с получением продукта формулы VI, который может быть использован в последующей стадии этого способа; но обычно перекристаллизация не является необходимой стадией. Продукты формулы VI представляют собой новые соединения, которые, в основном, используются в качестве промежуточных соединений для получения соединений формулы I, а особенно соединений формулы IA. Предпочтительно, соединения формулы VI соответствуют соединениям формулы VIA, где -А-А- и -В-В- представляют -CH2-CH2-, R3 представляет водород, низший алкил или низший алкокси, а R8 и R9 вместе составляют 20-спироксановое кольцо:
Наиболее предпочтительным соединением формулы VI является 4'S(4'α),7'α-гексадекагидро-11'α-гидрокси-10'β,13'β-диметил-3',5,20'-триоксоспиро[фуран-2(3Н),17'β-[4,7]метано[17Н]циклопента[а]фенантрен]-5'β(2'Н)-карбонитрил.
В наиболее предпочтительном варианте осуществления настоящего изобретения, енаминовый продукт формулы VII получают из соединения формулы VIII способом, описанным выше, и превращают in situ в дикетон формулы VI. В этом варианте осуществления настоящего изобретения субстрат формулы VIII подвергают реакции с избытком цианида щелочного металла в водном растворителе, содержащем источник протонов, или, необязательно, с избытком цианогидрина кетона в присутствии основания и LiCI, как описано выше. Однако, вместо охлаждения реакционной смеси, проводят подкисление и добавление воды в соотношении, необходимом для осаждения енамина, при этом предпочтительно избегать значительного охлаждения реакционной смеси. Вместо этого, по окончании реакции цианидирования, к смеси добавляют воду и кислоту, предпочтительно минеральную кислоту, такую как серная кислота. Это количество добавляемой кислоты является достаточным для нейтрализации избытка цианида щелочного металла, которая обычно требует введения по крайней мере одного молярного эквивалента кислоты на один моль субстрата формулы VIII, а предпочтительно от около 2 до около 5 молярных эквивалентов кислоты на один эквивалент субстрата. Однако, во избежание значительного осаждения реакцию осуществляют при достаточно высокой температуре и при достаточно большом разбавлении, а реакцию гидролиза енамина в дикетон проводят в жидкой фазе. Таким образом, этот процесс реакции протекает с минимальными перерывами и дает высокий выход продукта. Гидролиз предпочтительно проводят при температуре по крайней мере 80°С, более предпочтительно в пределах от около 90°С до около 100°С, в течение периода времени обычно от около 1 часа до около 10 часов, а более предпочтительно от около 2 до около 5 часов. Затем реакционную смесь охлаждают предпочтительно до температуры от около 0°С до около 15°С, преимущественно в ледяной бане при температуре от около 5°С до около 10°С, для осаждения дикетонового продукта формулы VI. Этот твердый продукт может быть выделен путем фильтрации, а уменьшение количества примесей достигается путем промывки водой.
В следующей стадии синтеза Схемы 1 дикетоновое соединение формулы VI подвергают реакции с алкоксидом металла для размыкания кетонового мостика между 4- и 7-положениями посредством разрыва связи между карбонильной группой и 4-углеродом с образованием α-ориентированного алкоксикарбонильного заместителя в 7-положении и удалением цианида у 5-углерода. Продукт этой реакции представляет собой сложное гидроксиэфирное соединение, соответствующее формуле V:
где -А-А-, -В-В-, R3, R8 и R9 определены в формуле XIII, a R1 представляет низший алкоксикарбонил или гидроксикарбонил.
Алкоксид металла, используемый в реакции, соответствует формуле R10OM, где М представляет щелочной металл, a R10O- соответствует алкокси-заместителю R1. Выходы этой реакции являются наиболее удовлетворительными в том случае, если алкоксидом металла являются метоксид калия или метоксид натрия, но могут быть использованы и другие низшие алкоксиды. Наиболее предпочтительным является алкоксид калия. Могут быть также использованы феноксиды, другие арилоксиды, а также арилсульфиды. Реакцию обычно проводят в присутствии спирта, соответствующего формуле R10OH, где R10 определен выше. Могут быть использованы и другие подходящие растворители. Субстрат формулы VI присутствует предпочтительно в количестве от около 2% до около 12% масс., а более предпочтительно по крайней мере около 6% масс. R10OM присутствует предпочтительно в соотношении от около 0,5 до около 4 моль на один моль субстрата, более предпочтительно от около 1 до около 2 моль на один моль субстрата, и еще более предпочтительно около 1,6 моль на один моль субстрата. Температура не играет решающей роли, но реакция при повышенной температуре имеет более высокую продуктивность. Время реакции обычно составляет от около 4 до около 24 часов, а предпочтительно от около 4 до 16 часов. Обычно реакцию проводят при нагревании с обратным холодильником в зависимости от используемого растворителя.
Время, требуемое для достижения равновесия реакции, зависит от количества алкоксида, которое добавляют к реакционной смеси, и от способа его добавления. Алкоксид может быть добавлен одной порцией или несколькими порциями, либо его можно добавлять непрерывно. Если алкоксид добавляют несколькими порциями, то предпочтительно, чтобы примерно 1,6 эквивалентов метоксида калия были добавлены в две стадии. При этом двухстадийном добавлении к реакционной смеси сначала добавляют 1 эквивалент метоксида калия, а затем, через 90 минут, добавляют еще 0,6 эквивалента метоксида калия. Эта процедура двухстадийного добавления уменьшает время достижения равновесия по сравнению с добавлением 1,6 эквивалентов метоксида натрия одной порцией.
Поскольку равновесие является более благоприятным условием для продуцирования сложного гидроксиэфира при низких концентрациях дикетона, реакцию предпочтительно проводят при довольно высокой степени разбавления, например вплоть до 40:1 для реакции с использованием метоксида натрия. Было обнаружено, что значительно более высокая продуктивность может быть получена при использовании метоксида калия вместо метоксида натрия, поскольку для минимизации степени обратного цианидирования, где реагентом является метоксид калия, является, в основном, достаточно разбавление в пределах около 20:1.
В соответствии с настоящим изобретением, кроме того, было установлено, что реакция обратного цианидирования может быть ингибирована путем применения соответствующих химических или физических методов для удаления иона цианида в качестве побочного продукта из реакционной зоны. Таким образом, в другом варианте осуществления настоящего изобретения, реакция дикетона с алкоксидом щелочного металла может быть проведена в присутствии осаждающего агента для иона цианида, такого как, например, соль, содержащая катион, который образует нерастворимое цианидное соединение. Такими солями могут быть, например, иодид цинка, сульфат железа(3) или, в основном, любой галогенид, сульфат или другая соль щелочноземельного или переходного металла, которые являются более растворимыми, чем соответствующий цианид. Если иодид цинка присутствует в соотношении порядка около одного эквивалента на один эквивалент субстрата дикетона, то было обнаружено, что продуктивность реакции существенно увеличивается по сравнению с реакцией, осуществляемой в отсутствие галогенида щелочного металла.
Даже если для удаления иона цианида используют осаждающий агент, то это остается предпочтительным для проведения реакции при достаточно высокой степени разбавления, однако при использовании этого осаждающего агента молярное отношение растворитель:дикетоновый субстрат может быть значительно снижено по сравнению с реакциями, где такой агент отсутствует. Выделение сложного гидроксиэфира формулы V может быть проведено способами, либо предусматривающими, либо не предусматривающими экстракцию, как описано ниже.
Для стимуляции продуцирования сложного гидроксиэфира формулы V может также контролироваться равновесие реакции путем удаления этого сложного гидроксиэфира из реакционной смеси после его синтеза. Удаление сложного гидроксиэфира может быть осуществлено постадийно или непрерывно таким способом, как фильтрация. Удаление сложного гидроксиэфира может быть использовано для регулирования равновесия реакции, либо отдельно, либо в комбинации с химическим или физическим удалением цианида из реакционной смеси. Последующее нагревание полученного фильтрата приводит к смещению равновесия реакционной системы в сторону превращения оставшегося дикетона формулы VI в сложный гидроксиэфир формулы V.
При превращении дикетона формулы VI в сложный гидроксиэфир формулы V в неочищенном продукте наблюдалось присутствие 5-цианогидроксиэфира в небольших количествах, составляющих, обычно, менее чем около 5% масс. Было высказано предположение, что 5-цианогидроксиэфир представляет собой равновесное промежуточное соединение между дикетоном формулы VI и сложным гидроксиэфиром формулы V. Кроме того, было высказано предположение, что это равновесное промежуточное соединение образуется из дикетона в результате воздействия метоксида на 5,7-оксо-группу и протонирования энолята и из сложного гидроксиэфира путем присоединения иона побочного продукта цианида к функциональной 3-кето-Δ4,5-группе сложного гидрокси-эфира посредством реакции Михаэля.
Кроме того, в неочищенном продукте с помощью хроматографии наблюдалась 5-циано-7-кислота и 17-алкоксид сложного гидроксиэфира формулы V. Было высказано предположение, что 5-циано-гидроксиэфирное промежуточное соединение реагирует с ионом побочного продукта цианида (присутствующего в результате реакции децианирования, которая вводит двойную Δ4,5-связь) с продуцированием 5-циано-7-кислоты. Было высказано предположение, что воздействие иона цианида приводит к деалкилированию 7-сложноэфирной группы 5-циано-гидроксиэфира с образованием 5-циано-7-кислоты и соответствующего алкилнитрила.
Кроме того, было высказано предположение, что неустойчивое промежуточное соединение, 17-алкоксид, образуется в результате воздействия метоксида на 17-спиролактон сложного гидроксиэфира (или предшествующего промежуточного соединения, которое затем превращается в сложный гидроксиэфир). 17-Алкоксид легко превращается в сложный гидроксиэфир после обработки кислотой. Поэтому он обычно не обнаруживается в матричном продукте.
5-Циано-гидроксиэфир, 5-циано-7-кислота и 17-алкоксид являются новыми соединениями, которые могут быть использованы в качестве хроматографических маркеров и в качестве промежуточных соединений для получения сложного гидроксиэфира. Они могут быть выделены из сырого продукта в этой стадии Схемы 1 синтеза. Альтернативно, они могут быть синтезированы непосредственно для использования в качестве маркеров или промежуточных соединений. 5-Циано-гидроксиэфир может быть синтезирован посредством реакции раствора выделенного дикетона формулы VI с основанием, таким как алкоксид или амин, с последующим выделением полученного осадка. Предпочтительным соединением является 17-метилгидро-5β-циано-11α,17-дигидрокси-3-оксо-17α-прегнан-7α,21-дикарбоксилат, γ-лактон.
5-Циано-7-карбоновая кислота может быть синтезирована непосредственно посредством реакции дикетона формулы VI со слабым водным основанием, таким как ацетат натрия или бикарбонат натрия, с последующим выделением полученного остатка. Полученным соединением является, предпочтительно, 5-β-циано-11-α,17-дигидрокси-3-оксо-17α-прегнан-7α,21-дикарбоновая кислота, γ-лактон.
17-Алкоксид может быть синтезирован непосредственно путем реакции раствора сложного гидроксиэфира формулы V с алкоксидом с получением смеси 17-алкоксида и соответствующего сложного гидроксиэфира. Полученным соединением является предпочтительно диметил-11α,17-дигидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоксилат, γ-лактон.
Предпочтительно, дикетоновый субстрат формулы VI соответствует формуле VIA
и гидроксиэфирный продукт соответствует формуле VA:
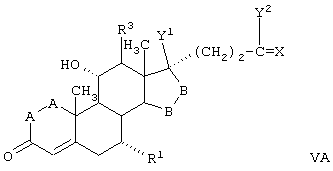
где каждый из -А-А-, -В-В-, R3, Y1, Y2 и Х определены в формуле XIIIA, a R1 определен в формуле V. Предпочтительно R3 представляет водород.
Продукты формулы V представляют собой новые соединения, которые, в основном, используются в качестве промежуточных соединений для получения соединений формулы I, а особенно формулы IA. Предпочтительно, соединения формулы V соответствуют формуле VA, где -А-А-, -В-В- представляют -CH2-CH2-, R3 представляет водород, низший алкил или низший алкокси, а R8 и R9, взятые вместе, представляют 20-спироксановое кольцо:
Наиболее предпочтительным соединением формулы V является метилгидро-11α,17α-дигидрокси-3-оксопрегн-4-ен-7α,21-дикарбоксилат, γ-лактон.
Соединение формулы V может быть выделено путем фильтрации или путем подкисления реакционного раствора, например минеральной кислотой, такой как водная HCl или серная кислота, с последующим охлаждением до комнатной температуры и экстракции продукта органическим растворителем, таким как метиленхлорид или этилацетат. Экстракт промывают водным раствором щелочной промывки, сушат и фильтруют, после чего растворитель удаляют. Альтернативно, реакционный раствор, содержащий продукт формулы V, может быть погашен концентрированной кислотой. Раствор продукта концентрируют, охлаждают до температуры от около 0°С до 25°С и твердый продукт выделяют путем фильтрации.
В предпочтительном варианте осуществления изобретения метанол и HCN удаляют после окончания периода прохождения реакции путем дистилляции, при этом минеральную кислоту (такую как соляная кислота или серная кислота) добавляют перед дистилляцией, а воду добавляют после дистилляции. Минеральная кислота может быть добавлена целиком одной порцией, постадийно или непрерывно. В предпочтительном варианте минеральную кислоту добавляют непрерывно в течение периода времени от около 10 до около 40 минут, а более предпочтительно от около 15 до около 30 минут. Аналогичным образом, вода может быть добавлена к кубовому остатку одной порцией, постадийно или непрерывно. В предпочтительном варианте осуществления изобретения концентрированную реакционную смесь после нагревания с обратным холодильником охлаждают, а затем добавляют воду. Перед добавлением воды эту смесь предпочтительно охлаждают до температуры в пределах от около 50°С до около 70°С, предпочтительно от около 60°С до около 70°С, а более предпочтительно до около 65°С. Затем добавляют воду, предпочтительно непрерывно в течение периода времени от около 15 минут до около 3 часов, а более предпочтительно от около 60 минут до около 90 минут, поддерживая примерно постоянную температуру. Продукт формулы V начинает кристаллизоваться из кубового остатка по мере добавления воды. После добавления воды к смеси разбавленную реакционную смесь поддерживают при той же самой температуре в течение около 1 часа, а затем охлаждают до около 15°С в течение дополнительного периода времени от около 4 до около 5 часов. Эту смесь поддерживают при температуре около 15°С в течение периода времени от около 1 до 2 часов. Выдерживание смеси при 15°С в течение более длительного периода времени приводит к увеличению выхода сложного цианоэфира в смеси. Этот способ выделения обеспечивает получение кристаллического продукта высокого качества без осуществления процедур экстракции.
В соответствии с другим предпочтительным способом выделения продукта формулы V метанол и HCN удаляют после окончания времени реакции путем дистилляции, причем воду и кислоту добавляют перед дистилляцией или в процессе дистилляции. Добавление воды перед дистилляцией упрощает операции, а постепенное ее добавление в процессе дистилляции позволяет поддерживать в кубе, в основном, постоянный объем. Продукт формулы V кристаллизуется из кубового остатка в процессе дистилляции. Этот способ выделения обеспечивает получение кристаллического продукта высокого качества без осуществления процедур экстракции.
В соответствии с еще одним альтернативным вариантом реакционный раствор, содержащий продукт формулы V, может быть погашен путем добавления минеральной кислоты, например 4н HCl, после чего растворитель удаляют путем дистилляции. Удаление растворителя является также эффективным для удаления остаточного HCN из реакционного продукта. Было установлено, что для очистки соединения формулы V, где соединение формулы V служит в качестве промежуточного соединения в процессе получения эпоксимексренона, описанного выше, не обязательно проводить множество экстракций растворителем. Фактически, часто такие экстракции могут быть совсем исключены. В том случае, когда для очистки используют экстракцию растворителем, то желательно дополнительно осуществлять промывки солевым насыщенным раствором и щелочные промывки. Но в том случае, когда экстракцию растворителем не проводят, то и промывки солевым насыщенным раствором и щелочные промывки также являются излишними. Исключение процедур экстракции и промывок, в основном, способствует увеличению продуктивности этого процесса, не оказывая, при этом, неблагоприятного влияния на выход и качество продукта, а также позволяет избежать необходимости сушки промытого раствора с использованием осушителя, такого как сульфат натрия.
Сырой 11α-гидрокси-7α-алкоксикарбонильный продукт снова растворяют в растворителе для проведения следующей реакционной стадии процесса, которая представляет собой превращение 11-гидрокси-группы в уходящую группу в 11-положении, в результате чего получают соединение формулы IV:

где: -А-А-, -В-В-, R3, R8 и R9 определены выше в формуле XIII; R1 определен в формуле V, а R2 представляет низший арилсульфонилокси, алкилсульфонилокси, ацилокси или галогенид. Предпочтительно, 11α-гидрокси этерифицируют посредством реакции с низшим алкилсульфонилгалогенидом, ацилгалогенидом или ангидридом кислоты, который добавляют в раствор, содержащий промежуточный продукт формулы V. Ангидриды низшей кислоты, такие как ангидриды уксусной кислоты и ангидриды тригалогенированной кислоты, такие как ангидрид трифторуксусной кислоты, могут быть использованы для получения соответствующих уходящих ацилокси-групп. Однако, предпочтительными являются галогениды низшей алкилсульфоновой кислоты, а особенно предпочтительным является метансульфонилхлорид. Альтернативно, 11α-гидрокси-группа может быть превращена в галогенид с помощью реакции подходящего реагента, такого как тионилбромид, тионилхлорид, сульфурилхлорид или оксалилхлорид. Другими реагентами для образования сложных эфиров 11α-сульфоновой кислоты являются тозилхлорид, бензолсульфонилхлорид и ангидрид трифторметансульфоновой кислоты. Эту реакцию проводят в растворителе, содержащем акцептор галоген-водорода, такой как триэтиламин или пиридин. Могут быть также использованы неорганические основания, такие как карбонат калия или карбонат натрия. Исходная концентрация сложного гидроксиэфира формулы V составляет предпочтительно от около 5% до около 50% масс. Этерифицирующий реагент предпочтительно присутствует в небольшом избытке. Особенно подходящим растворителем для данной реакции является метиленхлорид, но могут быть также использованы и другие растворители, такие как дихлорэтан, пиридин, хлороформ, метилэтилкетон, диметоксиэтан, метилизобутилкетон, ацетон, другие кетоны, простые эфиры, ацетонитрил, толуол и тетрагидрофуран. Температура реакции определяется, главным образом, летучестью растворителя. В случае метиленхлорида температура реакции составляет предпочтительно от около -10°С до около 10°С.
Гидроксиэфирный субстрат формулы V предпочтительно соответствует формуле VA:
и продукт соответствует формуле IVA:
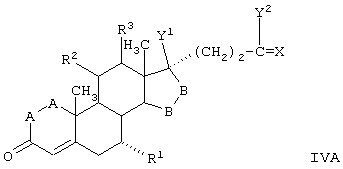
где каждый из -А-А-, -В-В-, R3, Y1, Y2 и Х определен в формуле XIIIA, R1 представляет низший алкоксикарбонил или гидроксикарбонил, а R2 определен в формуле IV. Предпочтительно, R3 представляет водород.
Продукты формулы IV представляют собой новые соединения, которые используются, в основном, в качестве промежуточных соединений для получения соединений формулы I, а особенно соединений формулы IA. Предпочтительно, соединения формулы V соответствуют формуле VA, где -А-А-, -В-В- представляют -CH2-CH2-, R3 представляет водород, низший алкил или низший алкокси, а R8 и R9 вместе представляют 20-спироксано-вое кольцо;
Более предпочтительным соединением формулы IV является метилгидро-17α-гидрокси-11α-(метилсульфонил)окси-3-оксо-прегн-4-ен-7α,21-дикарбоксилат, γ-лактон. Если желательно присутствие уходящей ацилокси-группы, то соединением формулы IV является, предпочтительно, 7-метилгидро-17-гидрокси-3-оксо-11α-(2,2,2-трифтор-1-оксоэтокси)-17α-прегн-4-ен-7α,21-дикарбоксилат, γ-лактон; или 7-метил-11α-(ацетилокси)-17-гидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоксилат, γ-лактон.
Если необходимо, то соединение формулы IV может быть выделено путем удаления растворителя. При этом предпочтительно реакционный раствор сначала промыть водным раствором щелочной промывки, например 0,5-2н NaOH, а затем промыть кислотой, например 0,5-2н HCl. После удаления реакционного растворителя продукт перекристаллизовывают, например путем растворения продукта в метиленхлориде, с последующим добавлением другого растворителя, такого как этиловый эфир, который снижает растворимость продукта формулы IV, что приводит к его осаждению в кристаллической форме.
При выделении продукта формулы IV или при получении реакционного раствора для превращения промежуточного соединения формулы IV в промежуточное соединение формулы II, как это будет описано ниже, все стадии экстракций и/или промывок могут быть исключены, а вместо этого раствор может быть обработан ионообменными смолами для удаления кислотных и основных примесей. Этот раствор сначала обрабатывают анионообменной смолой, а затем катионообменной смолой. Альтернативно, реакционный раствор может быть сначала обработан неорганическими адсорбентами, такими как основная окись алюминия или основная двуокись кремния, с последующей промывкой разбавленной кислотой. Основная двуокись кремния или основная окись алюминия могут быть, в основном, смешаны с реакционным раствором в соотношении, составляющем от около 5 до около 50 г на кг продукта, а предпочтительно от около 15 до около 20 г на кг продукта. В случае использования ионообменных смол или неорганических адсорбентов обработка может быть осуществлена просто путем суспендирования смолы или неорганического адсорбента реакционным раствором с перемешиванием при комнатной температуре и с последующим удалением смолы или неорганического адсорбента путем фильтрации.
В альтернативном и предпочтительном варианте осуществления настоящего изобретения полученное соединение формулы IV выделяют в неочищенной форме в виде концентрированного раствора путем удаления части растворителя. Этот концентрированный раствор непосредственно используют в следующей стадии процесса, в которой из соединения формулы IV удаляют уходящую 11α-группу, в результате чего получают сложный енэфир формулы II:
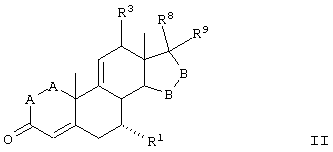
где: -А-А-, -В-В-, R3, R8 и R9 определены выше в формуле XIII, а R1 определен в формуле V. Для осуществления этой реакции заместитель R2 соединения формулы IV может быть любой уходящей группой, удаление которой является эффективным для образования двойной связи между 9- и 11-углеродами. Уходящей группой является предпочтительно низший алкилсульфонилокси- или ацилокси-заместитель, который удаляют посредством реакции с кислотой или солью щелочного металла. Для этого могут быть использованы минеральные кислоты, но предпочтительными являются низшие алкановые кислоты. Кроме того, предпочтительным реагентом для данной реакции является соль щелочного металла и используемой алкановой кислоты. Особенно предпочтительно, чтобы уходящая группа содержала мезилокси, а реагент для данной реакции содержал муравьиную кислоту или уксусную кислоту, или соль щелочного металла одной из этих кислот или другой низшей алкановой кислоты. В случае, если уходящей группой является мезилокси, а удаляемым реагентом является либо уксусная кислота и ацетат натрия, либо муравьиная кислота и формиат калия, то достигается относительно высокое отношение 9,11-олефина к 11,12-олефину. Если в процессе удаления уходящей группы присутствует свободная вода, то наблюдается тенденция к образованию примесей, в частности, 7,9-лактона
(где: -А-А-, R3, -В-В-, R8 и R9 определены выше в формуле XIII), которые трудно удалить из конечного продукта. Поэтому для удаления воды, присутствующей в муравьиной кислоте, используют уксусный ангидрид или другой осушитель. Содержание свободной воды в реакционной смеси перед осуществлением реакции должно поддерживаться на уровне ниже около 0,5%, предпочтительно ниже около 0,1% масс., как было определено путем анализа Карла Фишера для воды в расчете на массу всего реакционного раствора. Хотя предпочтительно, чтобы реакционная смесь выдерживалась практически в сухом состоянии, хорошие результаты могут быть получены и при содержании воды 0,3% масс. При этом предпочтительно, чтобы загрузочная реакционная смесь содержала от около 4% до около 50% масс. субстрата формулы IV в алкановой кислоте. Предпочтительно, чтобы эта смесь содержала от около 4% до около 20% масс. соли щелочного металла с кислотой. Если в качестве осушителя используется уксусный ангидрид, то предпочтительно, чтобы он присутствовал в соотношении от около 0,05 моль до около 0,2 моль на моль алкановой кислоты.
Было установлено, что количества побочного продукта 7,9-лактона и 11,12-олефина в реакционной смеси будут относительно малы, если реагент для элиминации содержит комбинацию трифторуксусной кислоты, трифторуксусного ангидрида и ацетата калия, который используется в качестве реагента для элиминации уходящей группы и образования сложного енэфира (9,11-олефина). Трифторуксусный ангидрид служит в качестве осушающего агента и должен присутствовать в количестве по крайней мере около 3% масс., более предпочтительно по крайней мере около 15% масс., а наиболее предпочтительно около 20% масс. в расчете на массу элиминирующего реагента, содержащего трифторуксусную кислоту.
В этой стадии Схемы синтеза 1, помимо 7,9-лактона, наблюдается присутствие и других примесей и побочных продуктов, которые могут быть использованы в качестве промежуточных соединений для синтеза и в качестве хроматографических маркеров. Новый 4,9,13-триен сложного енэфира формулы II (например, 7-метилгидро-17-метил-3-оксо-18-норпрегна-4,9(11),13-триен-7α,21-дикарбоксилат) был выделен из раствора продукта с помощью хроматографии. Очевидно, что продуцируемое количество этого соединения возрастает с увеличением времени реакции для этой стадии синтеза. Было высказано предположение, что это соединение образуется при протонировании лактона и полученный ион С17-карбония способствует миграции ангулярной метильной группы из С13-положения. Депротонирование этого промежуточного соединения приводит к образованию 4,9,13-триена.
Новое 5-циано-Δ11,12-соединение сложного енэфира формулы II (например, 7-метилгидро-5β-циано-17-гидрокси-3-оксо-17α-прегн-11-ен-7α,21-дикарбоксилата, γ-лактона) и новое 5-циано-соединение сложного енэфира формулы II (например, 7-метил-гидро-5-циано-17-гидрокси-3-оксо-17α-прегн-11-ен-7α,21-дикарбоксилата, γ-лактона) также были выделены из неочищенного продукта с помощью хроматографии. Было высказано предположение, что эти соединения образуются посредством дегидратации остаточной 5-циано-7-кислоты и 5-циано-гидроксиэфира, соответственно, которые присутствуют в растворе неочищенного продукта, полученного в третьей стадии Схемы синтеза 1.
Новый С17-эпимер сложного енэфира формулы II (например, 7-метилгидро-17-гидрокси-3-оксо-17α-прегна-4,9(11)-диен-7α,21-дикарбоксилата, γ-лактона) также был выделен из неочищенного продукта с помощью хроматографии. Было высказано предположение, что кислотные условия реакции элиминации могут приводить к рацемизации хирального С17-центра с получением 17-эпимера сложного енэфира. 17-эпимер может быть синтезирован непосредственно путем реакции соединения формулы IV с раствором формиата калия, муравьиной кислоты и уксусного ангидрида с последующим выделением 17-эпимера.
Хотя в растворе неочищенного продукта не наблюдалось примеси, 11-кетон сложного гидроксиэфира формулы V может быть получен путем окисления 11-гидрокси соответствующего сложного гидроксиэфира подходящим окислителем, таким как реактив Джонса. Полученным 11-кетоном является предпочтительно 7-метилгидро-17-гидрокси-3,11-диоксо-17α-прегна-4-ен-7α,21-дикарбоксилат, γ-лактон.
Альтернативно, уходящие 11α-группы соединения формулы IV могут быть удалены с получением сложного енэфира формулы II путем нагревания раствора формулы IV в органическом растворителе, таком как ДМСО, ДМФ или ДМА.
Кроме того, в соответствии с настоящим изобретением соединение формулы IV сначала подвергают реакции с алкенил-алканоатом, таким как изопропенилацетат, в присутствии кислоты, такой как толуолсульфоновая кислота или безводная минеральная кислота, такая как серная кислота, с образованием сложного эфира 3-енола
соединения формулы IV. Альтернативно, сложный эфир 3-енола может быть получен путем обработки соединения формулы IV ангидридом кислоты и основанием, таким как уксусная кислота и ацетат натрия. Другими альтернативными способами являются обработка соединения формулы IV кетеном в присутствии кислоты с получением соединения формулы IV(Z). Промежуточное соединение формулы IV(Z) затем подвергают реакции с формиатом щелочного металла или ацетатом в присутствии муравьиной или уксусной кислоты с получением Δ9,11-енолацетата формулы IV(Y):

который затем может быть превращен в сложный енэфир формулы II в органическом растворителе, предпочтительно спирте, таком как метанол, либо посредством термического разложения енолацетата, либо посредством его реакции с алкоксидом щелочного металла. Реакция элиминации является в высокой степени избирательной для сложного енэфира формулы II, при этом предпочтительность отдается 11,12-олефину и 7,9-лактону и эта избирательность сохраняется в процессе превращения енолацетата в енон.
Предпочтительно, субстрат формулы IV соответствует формуле IVA:
а продукт соответствует формуле IIA:
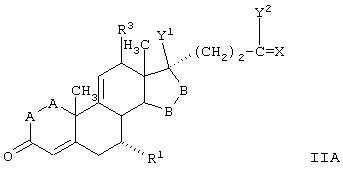
где каждый из -А-А-, -В-В-, R3, Y1, Y2 и Х определены в формуле XIIIA, R1 определен в формуле V, а R3 представляет предпочтительно водород.
Если необходимо, то соединение формулы может быть выделено путем удаления растворителя, растворения твердого продукта в холодной воде и экстрагирования органическим растворителем, таким как этилацетат. После соответствующих стадий промывки и сушки продукт выделяют путем удаления растворителя для экстракции. Затем сложный енэфир растворяют в растворителе, подходящем для превращения в продукт Формулы I. Альтернативно, сложный енэфир может быть выделен путем добавления воды к концентрированному раствору продукта и фильтрации твердого продукта с последующим предпочтительным удалением 7,9-лактона. Превращение субстрата формулы II в продукт формулы IA может быть проведено способом, описанным в патенте США №4559332, которое во всей своей полноте вводится в настоящее изобретение посредством ссылки, или более предпочтительно с помощью новой реакции с использованием галогенацетамидного стимулятора, как описано ниже.
В другом варианте осуществления настоящего изобретения сложный гидроксиэфир формулы V может быть превращен в сложный енэфир формулы II без выделения промежуточного соединения формулы IV. В этом способе сложный гидроксиэфир растворяют в органическом растворителе, таком как метиленхлорид; и к раствору добавляют либо ацилирующий агент, например метансульфонилхлорид, либо галогенирующий агент, например сульфурилхлорид. Полученную смесь размешивают и, если предусмотрено галогенирование, добавляют акцептор HCl, такой как имидазол. Эта реакция является в высокой степени экзотермичной и, следовательно, она должна проводиться при регулируемой скорости с полным охлаждением. После добавления основания полученную смесь нагревают до умеренной температуры, например от около 0°С до комнатной температуры или немного выше, и реакцию проводят обычно в течение периода времени от около 1 часа до около 4 часов. После завершения реакции растворитель удаляют, предпочтительно в условиях высокого вакуума (например, от около 24" до около 28" рт.ст.(дюйм рт.ст.)), от около -10°С до около +15°С, более предпочтительно от около 0 до около 5°С, для концентрирования раствора и удаления избытка основания. Затем, для превращения в сложный енэфир, субстрат снова растворяют в органическом растворителе, предпочтительно в галогенированном растворителе, таком как метиленхлорид.
Реагент для элиминации уходящей группы предпочтительно получают путем смешивания органической кислоты, соли органической кислоты и осушителя, предпочтительно муравьиной кислоты, формиата щелочного металла и уксусного ангидрида, соответственно в сухом реакторе. Добавление уксусного ангидрида сопровождается выделением тепла и приводит к выделению СО, а поэтому скорость добавления должна соответствующим образом регулироваться. Для стимуляции удаления воды температуру этой реакции поддерживают предпочтительно в пределах от около 60 до около 90°С, а наиболее предпочтительно от около 65 до около 75°С. Затем указанный реагент добавляют к раствору продукта соединения IV для осуществления реакции элиминации. По истечении периода времени от около 4 до около 8 часов реакционную смесь предпочтительно нагревают до температуры по крайней мере около 85°С, но предпочтительно не выше чем около 95°С, до полного удаления летучего дистиллята, а затем дополнительно нагревают до полного завершения реакции, обычно в течение от около 1 до около 4 часов. Реакционную смесь охлаждают и, после выделения стандартными методами экстракции, этот сложный енэфир может быть выделен, если это необходимо, путем выпаривания растворителя.
Кроме того, было обнаружено, что сложный енэфир формулы II может быть выделен из реакционного раствора альтернативным способом, который позволяет избежать необходимости осуществления стадий экстракции после реакции элиминации, что дает возможность уменьшить материальные затраты, увеличить выход и/или повысить продуктивность. В этом способе сложный енэфир осаждают путем разбавления реакционной смеси водой после удаления муравьиной кислоты. Затем продукт выделяют путем фильтрации. В этом случае, никакой экстракции не требуется.
В соответствии с другим альтернативным способом превращения соединения формулы V в сложный енэфир формулы II без выделения соединения формулы IV 11α-гидрокси-группу сложного гидроксиэфира формулы V заменяют галогеном, после чего сложный енэфир формулы II образуется in situ вследствие термического дегидрогалогенирования. Замену гидрокси-группы галогеном осуществляют посредством реакции с сульфурилгалогенидом, предпочтительно с сульфурхлоридом, при охлаждении в присутствии акцептора галогенводорода, такого как имидазол. Сложный гидроксиэфир растворяют в растворителе, таком как тетрагидрофуран, и охлаждают до температуры от около 0°С до около -70°С. Затем добавляют сульфурилхлорид и реакционную смесь нагревают до умеренной температуры, например до комнатной температуры, в течение периода времени, достаточного для завершения реакции элиминации, обычно в течение периода времени от около 1 до около 4 часов. Способ осуществления этого варианта изобретения не только позволяет объединить две стадии в одну, но также позволяет избежать использования: галогенированного реакционного растворителя; кислоты (такой как уксусная кислота) и осушителя (такого как уксусный ангидрид или сульфат натрия). Кроме того, эта реакция не требует условий нагревания с обратным холодильником и позволяет избежать продуцирования побочного продукта СО, образующегося при использовании уксусной кислоты в качестве осушающего агента.
В соответствии с особенно предпочтительным вариантом осуществления настоящего изобретения, дикетоновое соединение формулы VI может быть превращено в эпоксимексренон или другое соединение формулы I без выделения какого-либо промежуточного соединения в очищенной форме. В соответствии с этим предпочтительным способом реакционный раствор, содержащий сложный гидроксиэфир, гасят раствором сильной кислоты, охлаждают до комнатной температуры, а затем экстрагируют соответствующим растворителем для экстракции. Перед экстракцией к реакционной смеси предпочтительно добавляют водный раствор неорганической соли, например около 10% масс. насыщенного солевого раствора. Экстракт промывают и осушают путем азеотропной дистилляции для удаления метанолового растворителя, оставшегося после реакции расщепления кетона.
Затем, полученный концентрированный раствор, содержащий от около 5% до около 50% масс. соединения формулы V, подвергают контакту в холодном состоянии с ацилирующим или алкилсульфирующим реагентом с образованием сложного эфира сульфоновой кислоты или сложного эфира дикарбоновой кислоты. После завершения реакции алкилсульфирования или карбоксилирования реакционный раствор пропускают через колонку с кислотной, а затем с основной ионообменной смолой для удаления основных и кислотных примесей. После каждого пропускания колонку промывают соответствующим растворителем, например метиленхлоридом, для выделения из нее остаточного сложного эфира сульфоновой или дикарбоновой кислоты. Объединенный элюат и фракции промывки объединяют и концентрируют предпочтительно в вакууме, в результате чего получают концентрированный раствор, содержащий сложный эфир сульфоновой или дикарбоновой кислоты формулы IV. Затем этот концентрированный раствор подвергают контактировованию с сухим реагентом, содержащим агент, эффективный для удаления уходящей 11α-сложноэфирной группы и отщепления водорода с образованием 9,11-двойной связи. Предпочтительно, реагент для удаления уходящей группы содержит раствор сухого реагента, муравьиной кислоты/формиата щелочного металла/уксусного ангидрида, описанного выше. После завершения реакции реакционную смесь охлаждают, а муравьиную кислоту и/или другие летучие компоненты удаляют в вакууме. Остаток охлаждают до комнатной температуры, подвергают соответствующим стадиям промывки, а затем сушат, в результате чего получают концентрированный раствор, содержащий сложный енэфир формулы II. Этот сложный енэфир может быть затем превращен в эпоксимексренон или в другое соединение формулы I с использованием способа, описанного выше, или способа, описанного в патенте США №4559332.
В особенно предпочтительном варианте осуществления изобретения растворитель удаляют из реакционного раствора в условиях вакуума и продукт формулы IV распределяют между водой и соответствующим органическим растворителем, например этилацетатом. Затем водный слой подвергают обратной экстракции органическим растворителем и этот обратный экстракт промывают щелочным раствором, предпочтительно раствором гидроксида щелочного металла, содержащим галогенид щелочного металла. Органическую фазу концентрируют предпочтительно в вакууме и получают енэфирный продукт формулы II. Этот продукт формулы II может быть затем растворен в органическом растворителе, например в метиленхлориде, а после этого он может быть подвергнут реакции способом, описанным в патенте 322 с получением продукта формулы I.
В случае, если в реакции эпоксидирования используется тригалогенацетонитрил, то, как было установлено, очень важное значение имеет выбор растворителя, при этом в высокой степени предпочтительными являются галогенированные растворители, а особенно предпочтительным является метиленхлорид. Такие растворители, как дихлорэтан и хлорбензол, дают вполне достаточный выход, однако значительно лучший выход дает реакционная среда, содержащая метиленхлорид. Такие растворители, как ацетонитрил и этилацетат, в основном, дают недостаточный выход, а реакция в таких растворителях, как метанол или вода/тетрагидрофуран, дает небольшое количество нужного продукта.
Кроме того, в соответствии с настоящим изобретением было установлено, что значительное усовершенствование синтеза эпоксимексренона может быть реализовано путем использования для реакции эпоксидирования в качестве пероксидного активатора тригалогенацетамида вместо тригалогенацетонитрила. В особенно предпочтительном способе это эпоксидирование осуществляют посредством реакции субстрата формулы IIA с перекисью водорода в присутствии трихлорацетамида и соответствующего буфера. Эту реакцию, предпочтительно, проводят при рН в пределах от около 3 до около 7, а наиболее предпочтительно от около 5 до около 7. Однако, несмотря на это, эта реакция может быть успешно проведена при рН, имеющем значения вне указанных предпочтительных пределов.
Наиболее подходящие результаты получают с использованием буфера, содержащего дикалийбифосфат, и/или с использованием буфера, содержащего комбинацию дикалийбифосфата и бифосфата калия в соответствующих соотношениях от около 1:4 до около 2:1, а наиболее предпочтительно в пределах около 2:3. Могут быть также использованы боратные буферы, но они обычно дают более медленные реакции превращения, чем бифосфат калия или смеси K2HPO4/KH2PO4. Независимо от используемого буфера он должен обеспечивать рН в пределах, указанных выше. Независимо от полного состава буфера или точности рН, которую он может обеспечивать, было замечено, что реакция протекает намного более эффективно, если по крайней мере часть буфера содержит ион двухосновного бифосфата. Очевидно, что этот ион может, в основном, осаждаться в виде гомогенного катализатора при образовании аддукта или комплекса, содержащего стимулятор и гидроксипероксидный ион, продуцирование которого, в свою очередь, может играть важную роль в механизме реакции эпоксидирования. Таким образом, количественным требованием для двухосновного бифосфата (предпочтительно, исходя из K2HPO4) может быть только небольшая концентрация катализатора. В основном, предпочтительно, чтобы К2HPO4 присутствовал в количестве по крайней мере около 0,1 эквивалентов, например от около 0,1 до около 0,3 эквивалентов на эквивалент субстрата.
Эту реакцию осуществляют в подходящем растворителе, предпочтительно в метиленхлориде, но альтернативно могут быть также использованы и другие галогенированные растворители, такие как хлорбензол или дихлорэтан. Было установлено, что удовлетворительные результаты также дают толуол и смеси толуола и ацетонитрила. Не претендуя на какую-либо конкретную теорию, следует отметить, что реакция протекает более эффективно в двухфазной системе, в которой образующееся гидропероксидное промежуточное соединение распределяется по органической фазе с низким содержанием воды и реагирует с субстратом в органической фазе. Таким образом, предпочтительными растворителями являются такие растворители, у которых растворимость в воде является низкой. Эффективное выделение из толуола стимулируется путем включения другого растворителя, такого как ацетонитрил.
При превращении субстратов формулы II в продукты формулы I использование толуола дает преимущества, поскольку эти субстраты легко растворяются в толуоле в отличие от указанных продуктов. Таким образом, продукт осаждается в процессе реакции, когда превращение достигает 40-50%, что приводит к продуцированию трехфазной смеси, из которой этот продукт может быть легко выделен путем фильтрации. При осуществлении реакции превращения на этой стадии данного процесса, метанол, этил, ацетонитрил, взятые отдельно, и ТГФ и ТГФ/вода не являются такими эффективными, как галогенированные растворители или толуол.
Хотя трихлорацетамид является в высокой степени предпочтительным реагентом, однако могут быть также использованы и другие тригалогенацетамиды, такие как трифторацетамид и хлордифторацетамид. Может быть также использован тригалоген-бензамид и другие соединения, имеющие ариленовую, алкенильную или алкинильную группу (или другую группу, которая способствует переносу электрон-акцептирующего действия электрон-акцепторной группы на карбонил амида) между электрон-акцепторной тригалогенметильной группой и карбонилом амида. Могут быть также использованы гептафторбутирамиды, но с менее благоприятными результатами. Обычно пероксидный активатор может соответствовать формуле:
R°C(O)NH2
где R° представляет группу, имеющую по крайней мере такую же высокую электрон-акцепторную силу (измеряемую константой сигма), как и монохлорметильная группа. Для максимальной эффективности электрон-акцепторную группу предпочтительно присоединяют непосредственно к карбонилу амида. Более конкретно, пероксидный активатор может соответствовать формуле

где RP представляет группу, которая позволяет переносить электрон-акцептирующее действие электрон-акцепторной группы на карбонил амида и которую предпочтительно выбирают из арилена, алкенила, алкинила и -(СХ4X5)n-групп; X1, X2, X3, X4 и X5 независимо выбирают из галогена, водорода, алкила, галоген-алкила, циано и цианоалкила; а n равно 0, 1 или 2; при условии, что если n=0, то по крайней мере один из X1, X2 и X3 представляет галоген; а если RP представляет -(СХ4X5)n и n=1 или 2, то по крайней мере один из X4 и X5 представляет галоген. В случае, если любой из X1, X2, X3, X4 и X5 не является галогеном, то он предпочтительно представляет галогеналкил, а более предпочтительно пергалогеналкил. Особенно предпочтительными активаторами являются такие активаторы, в которых n=0, и по крайней мере два из X1, X2 и X3 представляют галоген; либо такие активаторы, в которых RP представляет -(СХ4X5)n; n=1 или 2; по крайней мере один из X4 и X5 представляет галоген, а другие из X4 и X5 представляют галоген или пергалогеналкил; и X1, X2 и X3 представляют галоген или пергалогеналкил. Каждый из X1, X2, X3, X4 и X5 предпочтительно представляет Cl или F, а наиболее предпочтительно Cl, хотя могут быть также использованы смешанные галогениды, то есть перхлоралкил или пербромалкил и их комбинации, при условии, что углерод, непосредственно связанный с карбонилом амида, замещен по крайней мере одной галогеновой группой.
Предпочтительно, чтобы пероксидный активатор присутствовал в соотношении по крайней мере около 1 эквивалента, более предпочтительно от около 1,5 до около 2 эквивалентов на эквивалент первоначально присутствующего субстрата. Перекись водорода должна быть загружена в реакцию по крайней мере в умеренном избытке, либо она должна постоянно добавляться по мере прохождения реакции эпоксидирования. Хотя реакция поглощает только от одного до двух эквивалентов перекиси водорода на один моль субстрата, однако предпочтительно, чтобы перекись водорода была загружена в значительном избытке по отношению к первоначально присуствующему субстрату и активатору. Не претендуя на какую-либо конкретную теорию, следует отметить, что механизм реакции предусматривает образование аддукта активатора и аниона пероксида и что эта реакция является обратимой со смещением равновесия в сторону обратной реакции и что поэтому необходимо использовать значительный первоначальный избыток перекиси водорода для прохождения реакции в прямом направлении. Точная температура реакции не имеет решающего значения и эта реакция может быть эффективно проведена при температуре в пределах от около 0°С до около 100°С. Оптимальная температура зависит от выбора растворителя. В основном, предпочтительная температура составляет от около 20°С до около 30°С, но при определенных растворителях, например в толуоле, эта реакция может быть преимущественно проведена при температуре от около 60°С до около 70°С. При температуре около 25°С для проведения реакции обычно требуется менее чем около 10 часов, в основном от около 3 до около 6 часов. Если необходимо, то в конце реакционного цикла, для достижения полного превращения субстрата, может быть добавлено дополнительное количество активатора и перекиси водорода.
В конце реакционного цикла водную фазу удаляют, органический реакционный раствор предпочтительно промывают для удаления водорастворимых примесей, после чего продукт может быть выделен путем удаления растворителя. Перед удалением растворителя реакционный раствор должен быть промыт по крайней мере от слабой до умеренно щелочной промывкой, например карбонатом натрия. Предпочтительно, реакционную смесь промывают последовательно: слабым восстанавливающим раствором, таким как слабый (например, около 3% масс.) раствор сульфита натрия в воде; щелочным раствором, например NaOH или КОН (предпочтительно около 0,5н); кислым раствором, таким как HCl (предпочтительно около 1н); и конечной нейтральной промывкой, содержащей воду или солевой раствор, предпочтительно насыщенным солевым раствором для минимизации потери продукта. Перед удалением реакционного растворителя может быть добавлен преимущественно другой растворитель, такой как органический растворитель, предпочтительно этанол, для того, чтобы этот продукт можно было выделить путем кристаллизации после дистилляции, проводимой для удаления более летучего реакционного растворителя.
При этом следует отметить, что новый способ эпоксидирования с использованием трихлорацетамида или другого нового пероксидного активатора имеет применение, выходящее далеко за рамки различных схем для получения эпоксимексренона, и фактически может быть использован для образования эпоксидов посредством олефиновых двойных связей в субстратах широкого ряда, подвергаемых реакции в жидкой фазе. Эта реакция является особенно эффективной в ненасыщенных соединениях, в которых олефины являются тетразамещеннымии и тризамещенными, то есть RaRbC=CRcRd и RaRbC=CRcH, где Ra-Rd представляют заместители, не являющиеся водородом. Эта реакция протекает наиболее быстро и полно, где субстрат представляет собой циклическое соединение с тризамещенной двойной связью, либо циклическое или ациклическое соединение с тетразамещенной двойной связью. Примерами субстратов для реакции эпоксидирования являются Δ 5,11-канренон и следующие субстраты:
Поскольку с тризамещенными и тетразамещенными двойными связями реакция протекает более быстро и более полно, то она является особенно эффективной для селективного эпоксидирования по таким двойным связям в соединениях, которые могут включать другие двойные связи, где атомы углерода олефина являются монозамещенными или даже дизамещенными.
Другими неограничивающими примерами, иллюстрирующими общую реакцию эпоксидирования, являются следующие реакции эпоксидирования:


Кроме того, следует отметить, что эта реакция может быть использована для преимущественного проведения реакции эпоксидирования монозамещенных или даже дизамещенных двойных связей, таких как 11,12-олефин в различных стероидных субстратах.
Однако, поскольку эпоксидируются преимущественно двойные связи с более высокой степенью замещения, например 9,11-олефин, с высокой степенью селективности, то способ настоящего изобретения является особенно эффективным для достижения высоких выходов и продуктивности в стадиях эпоксидирования в различных реакционных схемах, также описанных в настоящей заявке.
Было показано, что этот улучшенный способ является особенно преимущественным при его применении для получения соединения 
путем эпоксидирования соединения
 ; и
; и
с получением соединения
путем эпоксидирования соединения:
Было продемонстрировано множество преимуществ способа настоящего изобретения, в котором в качестве реагента для переноса кислорода в реакции эпоксидирования используют трихлорацетамид вместо трихлорацетонитрила. Система трихлорацетамидного реагента имеет низкое сродство для электронно-дефицитных олефинов, таких как α,β-ненасыщенные кетоны. Это позволяет проводить селективное эпоксидирование олефина с несопряженными двойными связями в субстрате, содержащем оба типа двойных связей. Кроме того, в комплексных субстратах, таких как стероиды, дизамещенные и тризамещенные олефины могут быть дифференцированы посредством реакции. Так, например, хорошая селективность наблюдается при эпоксидировании изомерных Δ-9,11 и Δ-11,12-соединений. В этом случае, 9,11-эпоксид образуется при минимальной реакции изомера, содержащего Δ-11,12-двойную связь. В соответствии с этим выход реакции, профиль продуктов и конечная чистота продукта являются, в основном, более высокими по сравнению с реакциями, в которых используется тригалогенацетонитрил. Кроме того, было обнаружено, что значительно избыточное продуцирование кислорода, наблюдаемое при использовании тригалогенацетонитрила, минимизируется при использовании трихлорацетамида, придавая большую надежность этому способу эпоксидирования. Кроме того, в противоположность реакции, стимулированной трихлорацетонитрилом, реакция с использованием трихлорацетамида дает минимальный экзотермический эффект, что облегчает регулирование температурного профиля реакции. По наблюдениям было установлено, что в этой реакции влияние перемешивания является минимальным, а производительность реактора является более высокой, и эта реакция имеет дополнительные преимущества по сравнению с реакцией, проводимой с использованием трихлорацетонитрила. Эта реакция является более пригодной для масштабного продуцирования, чем реакция, стимулированная трихлорацетонитрилом. Выделение продукта и его очистка являются простыми. Окисления Байера-Виллигера карбонильной функциональной группы (стимулированное пероксидом превращение кетона в сложный эфир) не наблюдалось, как было выявлено в эксперименте с использованием м-хлорпероксибензойной кислоты или других перкислот. Этот реагент является недорогостоящим, легко доступным и технологичным.
Кроме того, в неочищенном продукте, полученном в стадии Схемы синтеза 1, в которой сложный енэфир формулы II был превращен в соединение формулы I, с помощью хроматографии наблюдались следующие соединения:
(1) новый 11α,12α-эпоксид сложного енэфира формулы II, например 7-метилгидро-11α,12α-эпокси-17-гидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоксилат, γ-лактон;
(2) новый 4,5:9,11-диэпоксид сложного енэфира формулы II, например 7-метилгидро-4α,5α:9α,11α-диэпокси-17-гидрокси-3-оксо-17α-прегнан-7α,21-дикарбоксилат, γ-лактон;
(3) новый 12-кетон сложного енэфира формулы II, например 7-метилгидро-17-гидрокси-3,12-диоксо-17α-прегна-4,9(11)-диен-7α,21-дикарбоксилат, γ-лактон;
(4) новый 9,11-дигидрокси сложного енэфира формулы II, например 7-метилгидро-9α,11β,17-тригидрокси-3-оксо-17α-прегна-4-ен-7α,21-дикарбоксилат, γ-лактон;
(5) новый 12-гидрокси-аналог сложного енэфира формулы II, например 7-метилгидро-12α,17-дигидрокси-3-оксо-17α-прегна-4,9(11)-диен-7α,21-дикарбоксилат, γ-лактон; и
(6) новая 7-кислота соединения формулы I, например 9,11α-эпокси-17-гидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоновая кислота, γ-лактон.
Эти соединения используют в качестве синтетических промежуточных соединений и/или хроматографических маркеров для получения соединения формулы I, в частности эпоксимексренона.
Было высказано предположение, что 11α,12α-эпоксид сложного енэфира формулы II образуется через примесь, продуцированную в процессе предыдущей стадии, в которой соединение формулы IV превращается в сложный енэфир формулы II. Эта примесь была выделена путем хроматографии и представляет собой сложный Δ11,12-енэфир. Он обычно продуцируется вместе со сложным Δ9,11-енэфиром в отношении около 90:10 (Δ9,11-енэфир:Δ11,12-енэфир), хотя это отношение может варьироваться. В результате оксиления сложного Δ11,12-енэфира в процессе превращения сложного енэфира формулы II в соединение формулы I образуется 11α,12α-эпоксид.
4,5:9,11-Диэпоксид сложного енэфира формулы I выделяют с помощью хроматографии. Было высказано предположение, что он образуется в результате сверхэпоксидирования сложного енэфира. В сыром продукте он обычно присутствует в количестве около 5% масс. или менее, хотя это количество может варьироваться.
12-Кетон сложного енэфира формулы II выделяют с помощью хроматографии. Было высказано предположение, что он образуется в результате аллильного окисления сложного енэфира. В сыром продукте он обычно присутствует в количестве около 5% масс. или менее, хотя это количество может варьироваться. Уровень 12-кетона, обнаруженного в сыром продукте при использовании трихлорацетонитрила в качестве активатора перекиси водорода, был значительно выше, чем уровень, обнаруживаемый с использованием в качестве активатора трихлорацетамида.
9,11-Дигидрокси-продукт сложного енэфира формулы II выделяют с помощью хроматографии. В сыром продукте он обычно присутствует в количестве около 5% масс. или менее, хотя это количество может варьироваться. Было высказано предположение, что он образуется в результате гидролиза эпоксида формулы I.
12-Гидрокси-продукт сложного енэфира формулы II выделяют с помощью хроматографии. В сыром продукте он обычно присутствует в количестве около 5% масс. или менее, хотя это количество может варьироваться. Было высказано предположение, что он образуется в результате гидролиза 11,12-эпоксида с последующей элиминацией 11β-гидрокси.
Кроме того, соединения формулы I, полученные в соответствии с этим описанием, могут быть дополнительно модифицированы с получением метаболита, производного пролекарственного соединения или т.п., с улучшенными свойствами (такими как повышенная растворимость и абсорбция), что облегчает введение и/или эффективность эпоксимексренона. 6-Гидрокси соединения формулы I (например, 7-метилгидро-6β,17-дигидрокси-9,11α-эпокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоксилата, γ-лактона) являются новыми соединениями, которые были идентифицированы как возможный метаболит у крыс. 6-Гидрокси-метаболит может быть получен из соответствующего этилового эфира енола (например, 7-метилгидро-9α,11α-эпокси-3-этокси-17-гидрокси-17α-прегн-4-ен-7α,21-дикарбоксилата, γ-лактона). Этиловый эфир енола соединения формулы I может быть получен в соответствии с процедурой, описанной в работе R.M.Weier & L.M.Hofmann (J.Med.Chem. 1977, 1304), которая вводится в настоящее описание посредством ссылки. Затем этиловый эфир енола подвергают реакции с м-хлорпероксибензойной кислотой и получают соответствующее 6-гидрокси-соединение формулы I.
Было, кроме того, высказано предположение, что монокарбоновые соли эпоксимексренона, особенно соли калия и натрия, могут служить подходящей альтернативой для упрощения введения соединения формулы I индивидууму, для которого показано введение антагониста альдостерона. В мягких основных условиях можно осуществлять селективное размыкание спиролактона соединений формулы I без гидролиза С7-сложноэфирного заместителя с получением соответствующего 17β-гидрокси-17α-(3-пропионовая кислота)-аналога. Эти аналоги с разомкнутой цепью являются более полярными, чем их лактонные аналоги, и имеют более короткое время удерживания при анализе с помощью обращенно-фазовой ВЭЖХ. Кислотные условия обычно приводят к регенерации лактонного кольца.
В более жестких условиях спиролактон размыкается и сложный С7 гидролизуется с образованием соответствующих побочных продуктов, 7β-гидрокси-17α(3-пропионовая кислота)-7-кислотных аналогов соединений формулы I. Эти дикарбоновые кислоты имеют более короткое время удерживания при их анализе с помощью обращенно-фазовой ВЭЖХ. Кислотные условия (например, обработка разбавленной кислотой, такой как 0,1-4М соляная кислота), обычно приводят к регенерации лактонного кольца дикарбоновой кислоты.
Новый способ эпоксидирования настоящего изобретения является в высокой степени эффективным в качестве завершающей стадии Схемы синтеза 1. В особенно предпочтительном варианте осуществления изобретения весь процесс Схемы 1 протекает следующим образом:
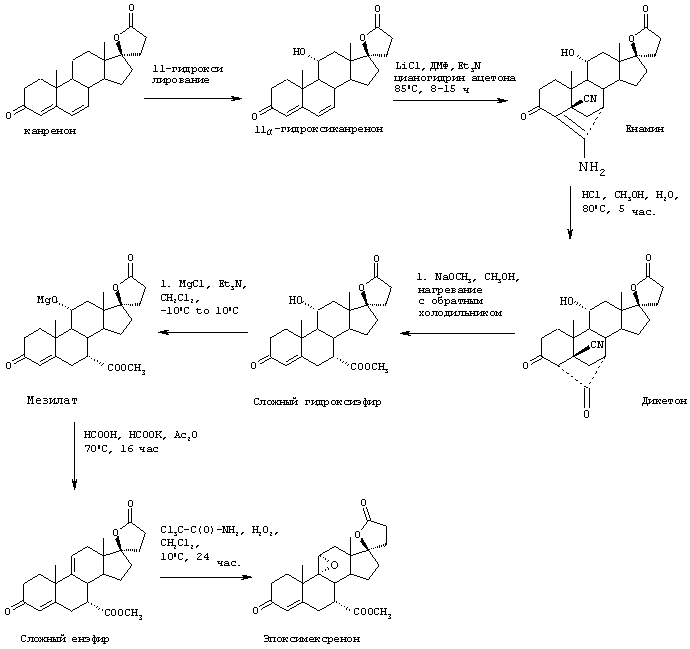
Схема 2.
Вторая схема новых реакций настоящего изобретения (Схема 2) начинается с использования канренона или другого субстрата соответствующей формулы XIII
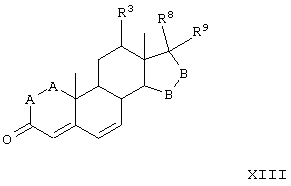
где: -А-А-, -В-В-, R3, R8 и R9 определены выше в формуле XIII.
В первой стадии этого способа субстрат формулы XIII превращают в продукт формулы XII
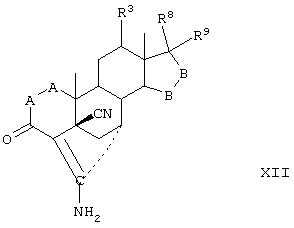
с использованием схемы реакции цианидирования, в основном аналогичной схеме, описанной выше для превращения субстрата формулы VIII в промежуточное соединение формулы VII. Предпочтительно, субстрат формулы XIII соответствует формуле XIIIA:
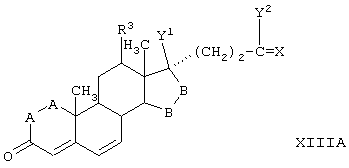
и енаминовый продукт соответствует формуле XIIA:
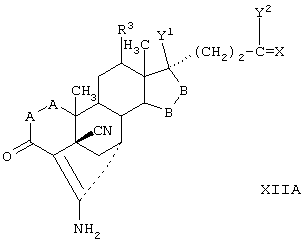
где каждый из -А-А-, -В-В-, R3, Y1, Y2 и Х определены в формуле XIIIA. Предпочтительно, R3 представляет водород.
Во второй стадии Схемы 2 енамин формулы XII гидролизуют с получением промежуточного дикетонового продукта формулы XI:
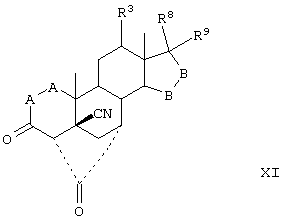
где: -А-А-, -В-В-, R3, R8 и R9 определены выше в формуле XIII, с использованием реакционной схемы, в основном аналогичной схеме, описанной выше для превращения субстрата формулы VIII в промежуточное соединение формулы VII. Предпочтительно, субстрат формулы XII соответствует формуле XIIA:
и дикетоновый продукт соответствует формуле XIA:
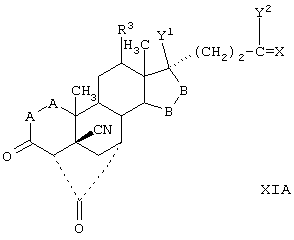
где каждый из -А-А-, -В-В-, R3, Y1, Y2 и Х определены в формуле XIIIA. Предпочтительно, R3 представляет водород.
Кроме того, в соответствии с реакционной схемой 2 дикетон формулы XI подвергают реакции с алкоксидом щелочного металла с образованием мексренона или другого продукта, соответствующего формуле X,
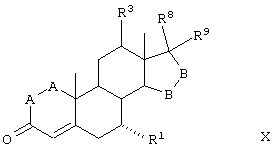
где каждый из -А-А-, -В-В-, R3, R8 и R9 определены в формуле XIII, а R1 определен в формуле V.
Этот способ осуществляют с использованием, в основном, реакционной схемы, описанной выше для превращения соединений формулы VI в соединения формулы V. Предпочтительно, субстрат формулы XI соответствует формуле XIA
а промежуточный продукт соответствует формуле ХА:
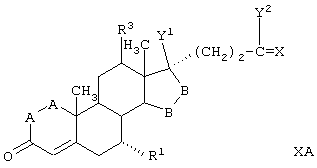
где каждый из -А-А-, -В-В-, R3, Y1, Y2 и Х определены в формуле XIIIA, a R1 определен в формуле V. Предпочтительно, R3 представляет водород.
Затем мексренон и другие соединения формулы Х подвергают 9α-гидроксилированию новым способом биологического превращения и получают продукты формулы IX:
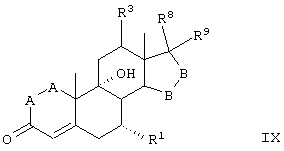
где: -А-А-, -В-В-, R3, R8 и R9 определены выше в формуле XIII, а R1 определен в формуле V.
Микроорганизмами, которые могут быть использованы в этой стадии гидроксилирования являются; Nocardia conicruria ATCC 31548, Nocardia aurentia ATCC 12674, Corynespora cassiicola ATCC 16718, Streptomyces hydroscopicus ATCC 27438, Mortierella isabellina ATCC 42613, Beauvria bassiana ATCC 7519, Penicillum purpurogenum ATCC 46581, Hypomyces chrysospermus IMI 109891, Thamnostylum piriforme ATCC 8992, Cunninghamella blakesleeana ATCC 8688a, Cunninghamella echinulata ATCC 3655, Cunninghamella elegans ATCC 9245, Trichothecium roseum ATCC 12543, Epicoccum Humicola ATCC 12722, Saccharopolyspora eythrae ATCC 11635, Beauvria bassiana ATCC 13144, Arthrobacter simplex, Bacterium cyclooxydans ATCC 12673, Cylindrocarpon radicicola ATCC 11011, Nocardia aurentia ATCC 12674, Norcardia restrictus ATCC 14887, Pseudomonas testosteroni ATCC 11996, Rhodococcus egui ATCC 21329, Mycobacterium fortuitum NRRL B8119 и Rhodococcus rhodochrous ATCC 19150. Эту реакцию осуществляют, в основном, способом, описанным выше со ссылками на Фиг.1 и 2. Особенно предпочтительным является способ, проиллюстрированный на Фиг.1.
Среда для культивирования, которая может быть использована в реакциях биологического превращения, предпочтительно содержит от около 0,05% до около 5% масс. доступного азота; от около 0,5% до около 5% масс. глюкозы; от около 0,25% до около 2,5% масс. дрожжевого производного; и от около 0,05% до около 5% масс. доступного фосфора. Особенно предпочтительными средами для культивирования являются следующие среды:
на основе соевой муки: от около 0,5% до около 3% масс. глюкозы; от около 0,1% до около 1% масс. соевой муки; от около 0,05% до около 5% масс. галогенида щелочного металла; и от около 0,05% до около 0,5% масс. дрожжевого производного, такого как автолизованные дрожжи или дрожжевой экстракт; от около 0,05% до около 5% масс. фосфатной соли, такой как К2HPO4; рН=7;
пептон-дрожжевой экстракт-глюкоза: от около 0,2% до около 2% масс. пептона; от около 0,05% до около 0,5% масс. дрожжевого экстракта; и от около 2% до около 5% масс. глюкозы;
среда Меллера-Хинтона: от около 10% до около 40% масс. говяжьего бульона; от около 0,35% до около 8,75% масс. казамино-кислот; и от около 0,15% до около 0,7% масс. крахмала.
Грибки могут быть культивированы в питательной среде на основе соевой муки или пептона, тогда как актиномицеты и эубактерии могут расти в среде на основе соевой муки (в которую добавлено 0,5%-1% масс. соли карбоновой кислоты, такой как формиат натрия для биологических трансформаций) или в бульоне Меллера-Хинтона.
Получение 11β-гидроксимексренона из мексренона путем ферментации обсуждается в Примере 19В. Аналогичные способы биологических трансформаций могут быть использованы для получения других исходных соединений и промежуточных соединений. В Примере 19А описано биологическое превращение андростендиона в 11β-гидроксиандростендион. В Примере 19С описано биологическое превращение мексренона в 11α-гидроксимексренон, Δ1,2-мексренон, 6β-гидроксимексренон, 12β-гидроксимексренон и 9α-гидроксимексренон. В Примере 19D описано биологическое превращение канренона в Δ9,11-канренон.
Продукты формулы IX являются новыми соединениями, которые могут быть выделены путем фильтрации, промыты подходящим органическим растворителем, например этилацетатом, и перекристаллизованы из того же самого или аналогичного растворителя. Они являются ценными, в основном как промежуточные соединения для получения соединения формулы I, а особенно соединений формулы IA. Предпочтительно, соединения формулы IX соответствуют формуле IXA, где -А-А-, -В-В- представляют -CH2-CH2-, R3 представляет водород, низший алкил или низший алкокси, а R8 и R9 вместе представляют 20-спироксановое кольцо:
В следующей стадии Схемы синтеза 2, продукт формулы IX подвергают реакции с дегидратирующим реагентом (подходящие дегидратирующие агенты, такие как PhSOCl или ClSO3Н, известны специалистам) с получением соединения формулы II
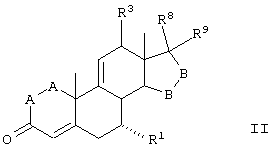
где: -А-А-, -В-В-, R3, R8 и R9 определены выше в формуле XIII, а R1 определен в формуле V. Предпочтительно, соединение формулы IX соответствует формуле IXA
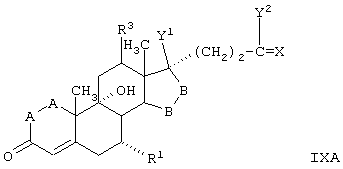
а промежуточный продукт соответствует формуле IIA
где каждый из -А-А-, -В-В-, R3, Y1, Y2 и Х определен в формуле XIIIA, а R1 определен в формуле V. Предпочтительно, R3 представляет водород.
В конечной стадии этой схемы синтеза продукт формулы II превращают в продукт формулы I путем эпоксидирования методом, описанным в патенте США 4559332; или предпочтительно новым способом эпоксидирования настоящего изобретения, описанным выше.
В особенно предпочтительном варианте осуществления изобретения весь процесс Схемы 2 осуществляют следующим образом:
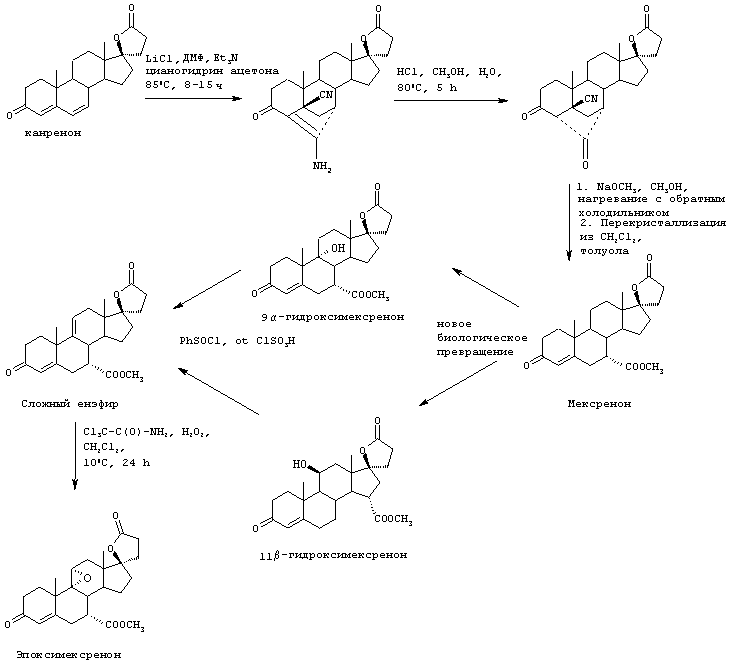
Схема 3.
В этом случае синтез начинают, исходя из субстрата, соответствующего формуле XX
где: -А-А- и R3 определены выше в формуле XIII, -B-В- определен в формуле XIII, за исключением того, что ни R6, ни R7 не являются частью кольца, конденсированного с кольцом D в 16,17-положениях, а R26 представляет низший алкил, предпочтительно метил. Предпочтительно, R3 представляет водород. В результате реакции субстрата формулы XX с илидом сульфония образуется эпоксидное промежуточное соединение, соответствующее формуле XIX:
где: -А-А-, -В-В-, R3 и R26 определены выше в формуле XX. R3 предпочтительно представляет водород.
В следующей стадии схемы синтеза 3 промежуточное соединение формулы XIX превращают в другое промежуточное соединение формулы XVIII:
где: -А-А-, -В-В-, R3 определены выше в формуле XX. R3 предпочтительно представляет водород. В этой стадии субстрат формулы XIX превращают в промежуточное соединение формулы XVIII с помощью реакции с NaCH(COOEt)2 в присутствии основания в растворителе.
После термообработки соединения формулы XVIII и его обработки водой и галогенидом щелочного металла получают декарбоксилированное промежуточное соединение, соответствующее формуле XVII:
где: -А-А-, -В-В-, R3 определены выше в формуле XX. R3 предпочтительно представляет водород.
Этот способ превращения соединения формулы XX в соединение формулы XVII соответствует в основном способу, описанному в патентах США 3897417, 3413288 и 3300489, которые во всей своей полноте вводятся в настоящее описание посредством ссылки. Хотя субстраты различаются, однако реагенты, механизмы и условия для введения 17-спиролактоновой части являются, в основном, теми же самыми.
В результате реакции промежуточного соединения формулы XVII с дегидрирующим реагентом получают другое промежуточное соединение формулы XVI:
где: -А-А-, -В-В-, R3 определены выше в формуле XX. R3 предпочтительно представляет водород.
Обычно используемыми дегидрирующими реагентами являются дихлордицианобензохинон (DDQ) и хлоранил (2,3,5,6-тетрахлор-п-бензохинон). Альтернативно, дегидрирование может быть проведено путем последующего галогенирования в 6-положении углерода, а затем посредством реакции дегидрогалогенирования.
Затем, промежуточное соединение формулы XVI превращают в енамин формулы XVB:
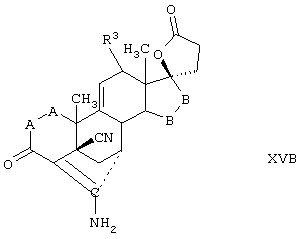
где -А-А-, -В-В- и R3 определены в формуле XX. Предпочтительно, R3 представляет водород.
Реакцию превращения осуществляют путем цианидирования, в основном, как описано выше для превращения 11α-гидроксисоединения формулы VIII в енамин формулы VII. Обычно, источником ионов цианида может быть цианид щелочного металла. Основанием является предпочтительно пирролидин и/или тетраметилгуанидин. Может быть использован и метаноловый растворитель.
Продукты формулы XVB являются новыми соединениями, которые могут быть выделены с помощью хроматографии. Эти и другие новые соединения формулы XV служат промежуточными соединениями для получения соединений формулы I, а особенно формулы IA. Соединения формулы XV соответствуют структуре:
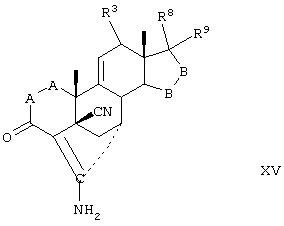
где -А-А-, -В-В-, R3, R8 и R9 определены в формуле XIII. В наиболее предпочтительных соединениях формулы XV и формулы XVB -А-А- и -В-В- представляют -CH2-CH2-, а R3 представляет водород.
В соответствии с гидролизом, описанным выше для получения дикетоновых соединений формулы VI, енамины формулы XVB могут быть превращены в дикетоны формулы XIVB;
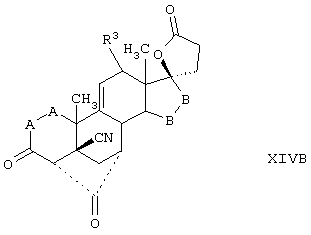
где -А-А-, -В-В- и R3 определены в формуле XX. Предпочтительно R3 представляет водород.
Особенно предпочтительными для синтеза эпоксимексренона являются те соединения формулы XIV, которые также охватываются формулой XIVB, определенной ниже.
Продукты формулы XIVB являются новыми соединениями, которые могут быть выделены путем осаждения. Эти и другие новые соединения формулы XIV служат промежуточными соединениями для получения соединений формулы I, а особенно формулы IA. Соединения формулы XIV соответствуют структуре:
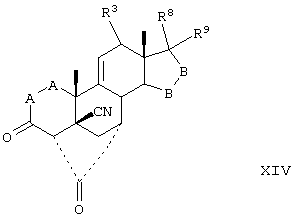
где -А-А-, -В-В-, R3, R8 и R9 определены в формуле XIII. В наиболее предпочтительных соединениях формулы XIV и XIVB -А-А- и -В-В- представляют -CH2-CH2-, a R3 представляет водород.
Затем соединения формулы XIVB превращают в соединения формулы XXXI, в основном способом, описанным выше для превращения дикетона формулы VI в сложный гидроксиэфир формулы V. В этом случае необходимо сначала выделить промежуточное соединение XXXI:
а затем провести реакцию его превращения в продукт формулы XXXII;
где -А-А-, -В-В- и R3 определены в формуле XX, а R1 определен в формуле V. Предпочтительно R3 представляет водород. Предпочтительными соединениями формулы XXXI являются соединения, охватываемые формулой IIA. Соединения формулы XXXI превращают в соединения формулы XXXII методом, описанным выше или описанным в Патенте США №4559332.
Предпочтительным соединением формулы XIV является
4'S(4'α),7'α-1',2',3',4,4',5,5',6',7',8',10',12',13',14',15',16'-гексадекагидро-10β-,13'β-диметил-3',5,20'-триоксоспиро[фуран-2(3Н),17'β-[4,7]метано[17Н]циклопента[а]фенантрен]5'-карбонитрил; а соединением формулы XV является 5'R(5'α),7'β-20'-амино-1',2',3',-4,5,6',7',8',10',12',13',14',15',16'-тетрадекагидро-10'α,13'α-диметил-3',5-диоксоспиро[фуран-2(3Н),17'α(5'Н)-[7,4]метено[4Н]-циклопента[а]фенантрен]-5'-карбонитрил. В наиболее предпочтительном варианте осуществления настоящего изобретения полная реакция в Схеме 3 протекает следующим образом:

Схема 4.
Первые три стадии Схемы 4 аналогичны стадиям Схемы 3, т.е. стадии получения промежуточного соединения формулы XVII, исходя из соединения, соответствующего формуле XX.
Затем промежуточное соединение формулы XVII эпоксидируют, например способом, описанным в Патенте США №4559332, с получением соединения формулы XXIV:
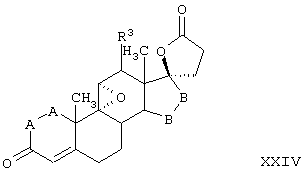
где -А-А-, -В-В- и R3 определены в формуле XX. Однако, в особенно предпочтительном варианте осуществления настоящего изобретения субстрат формулы XVII эпоксидируют по 9,11-двойной связи с использованием окислителя, содержащего пероксидный активатор типа амида, наиболее предпочтительно трихлорацетамид, в соответствии со способом, описанным выше в Схеме 1 для превращения сложного енэфира формулы II в продукт формулы I. Условия и соотношения реагентов для этой реакции являются, в основном, такими, как они были описаны для реакции превращения сложного енэфира формулы II в эпоксимексренон. Особенно предпочтительными соединениями формулы XXIV являются те соединения, в которых -А-А- и -В-В- являются такими, как они были определены для формулы XIII, а R3 представляет водород.
Было обнаружено, что реакция эпоксидирования субстрата формулы XVII может также давать очень хороший выход с использованием перкислоты, такой как, например, м-хлорпероксибензойная кислота. Однако трихлорацетамидный реагент дает наилучший результат в отношении минимизации образования побочного продукта в результате окисления Байера-Виллигера. Этот побочный продукт может быть удален, но для этого необходимо проводить растирание из растворителя, такого как этилацетат, с последующей кристаллизацией из другого растворителя, такого как метиленхлорид. Эпокси-соединение формулы XXIV подвергают дегидрированию с образованием двойной связи между 6- и 7-углеродами посредством реакции с дегидрирующим агентом (окислителем), таким как DDQ или хлоранил, или с использованием последовательной реакции бромирования/дегидробромирования (или другой реакции галогенирования/дегидрогалогенирования) с получением другого нового промежуточного соединения формулы XXIII.
где -А-А-, -В-В-, R3, R8 и R9 определены в формуле XX. Особенно предпочтительными соединениями формулы XXIII являются те соединения, где -А-А- и -В-В- являются такими, как они были определены для формулы XIII, a R3 представляет водород.
Хотя прямое окисление является эффективным для получения продукта формулы XXIII, однако эта реакция дает, в основном, низкие выходы. Поэтому реакцию окисления проводят предпочтительно в две стадии, сначала осуществляют галогенирование субстрата формулы XXIV в С-6-положении, а затем проводят дегидрогалогенирование с образованием 6,7-олефина. Реакцию галогенирования проводят предпочтительно с N-галогенорганическим реагентом, таким как, например, N-бромсукцинамид. Реакцию бромирования проводят в подходящем растворителе, таком как, например, ацетонитрил, в присутствии стимулятора галогенирования, такого как бензоилпероксид. Эта реакция эффективно протекает при температуре в пределах от около 50 до около 100°С, обычно при нагревании с обратным холодильником, в растворителе, таком как тетрахлорметан, ацетонитрил или их смеси. Однако, для завершения реакции обычно требуется от 4 до 10 часов. После этого реакционный растворитель выпаривают и остаток растворяют в несмешивающемся с водой растворителе, например в этилацетате. Полученный раствор последовательно промывают слабым щелочным раствором (таким как бикарбонат щелочного металла) и водой, или предпочтительно насыщенным солевым раствором для минимизации потерь продукта, после чего растворитель выпаривают и остаток растворяют в другом растворителе (таком как диметилформамид), пригодном для реакции дегидрогалогенирования.
Подходящий дегидрогалогенирующий реагент, например 1,4-диазабицикло[2.2.2]октан (DABCO), добавляют к раствору вместе с галогенидом щелочного металла, такого как LiBr, раствор нагревают до подходящей реакционной температуры, например 60-80°С, и реакцию проводят в течение нескольких часов, обычно от 4 до 15 часов, до полного завершения дегидробромирования. Если необходимо, то во время реакционного цикла может быть добавлено дополнительное количество дегидробромирующего реагента для доведения реакции до полного завершения. Затем продукт формулы XXIII может быть выделен, например путем добавления воды для осаждения продукта, который затем отделяют путем фильтрации и, предпочтительно, промывают еще некоторым количеством воды. Этот продукт предпочтительно перекристаллизовывают, например из диметилформамида.
Продукты формулы XXIII, такие как 9,11-эпоксиканренон, являются новыми соединениями, которые могут быть выделены путем экстракции/кристаллизации. Они имеют важное значение как промежуточные соединения для получения соединений формулы I, а особенно формулы IA. Например, они могут быть использованы в качестве субстратов для получения соединений формулы XXII.
С использованием, в основном, способа, описанного выше для получения соединений формулы VII, соединения формулы XXIII подвергают реакции с ионом цианида с получением новых эпоксиенаминовых соединений, соответствующих формуле XXII:
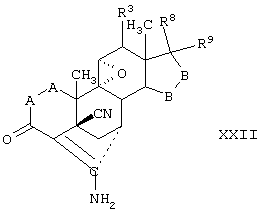
где -А-А-, -В-В-, R3, R8 и R9 определены в формуле XX. Особенно предпочтительными соединениями формулы XXII являются соединения, в которых -А-А- и -В-В- являются такими, как они были определены для формулы XIII, а R3 представляет водород.
Продукты формулы XXII являются новыми соединениями, которые могут быть выделены путем осаждения и фильтрации. Они имеют важное значение как промежуточные соединения для получения соединений формулы I, а особенно формулы IA. В наиболее предпочтительных соединениях формулы XXII, -А-А- и -В-В- представляют -СН2-СН2-, а R3 представляет водород.
С использованием, в основном, способа, описанного выше для получения соединений формулы VI, эпоксиенаминовые соединения формулы XXII превращают в новые эпоксидикетоновые соединения формулы XXI:
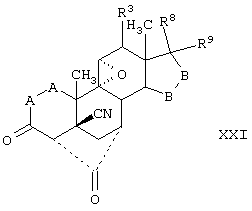
где -А-А-, -В-В-, R3, R8 и R9 определены в формуле XIII. В наиболее предпочтительных соединениях формулы XXI -А-А- и -В-В- представляют -СН2-СН2-, а R3 представляет водород.
Продукты формулы XXI являются новыми соединениями, которые могут быть выделены путем осаждения и фильтрации. Они имеют важное значение как промежуточные соединения для получения соединений формулы I, а особенно формулы IA. Особенно предпочтительными соединениями формулы XXI являются соединения, в которых -А-А- и -В-В- являются такими, как они были определены для формулы XIII. В наиболее предпочтительных соединениях формулы XXI -А-А- и -В-В- представляют -СН2-СН2-, а R3 представляет водород.
С использованием, в основном, способа, описанного выше для получения сложных гидроксиэфирных соединений формулы V из дикетоновых соединений формулы VI, эпоксидикетоновые соединения формулы XXI превращают в соединения формулы XXXII:
где -А-А-, -В-В- и R3 определены в формуле XX, а R1 определен в формуле V.
Так же, как и при реакции превращения дикетона формулы V в сложный гидроксиэфир формулы VI, 5-β-циано-7-эфирное промежуточное соединение также образуется при реакции превращения эпоксидикетона формулы XXI в соединения формулы XXXII. Оба вида этих 5-β-циано-7-эфирных промежуточных соединений могут быть выделены путем обработки соответствующего дикетона спиртом, таким как метанол, в присутствии основания, такого как триэтиламин. Предпочтительно, эти промежуточные соединения получают путем кипячения с обратным холодильником смеси дикетона в спирте, таком как метанол, содержащей от около 0,1 до около 2 эквивалентов триэтиламина на один моль дикетона, в течение от около 4 до около 16 часов. Продукты выделяют в чистом виде путем охлаждения смеси до около 25°С с последующей фильтрацией. Выделенные промежуточные соединения могут быть превращены в соединения формулы XXXII путем обработки основанием, таким как алкоксид щелочного металла, в растворителе, предпочтительно спирте, таком как метанол. Использование алкоксида в спирте дает равновесную смесь, аналогичную той смеси, которая образуется в случае обработки соответствующего дикетона формулы XXI при тех же условиях.
Помимо этого, в сыром продукте конечной стадии способа Схемы 4 с помощью хроматографии был обнаружен сложный 7β-эфир соединения формулы XXXII (например 7-метилгидро-9,11α-эпокси-17-гидрокси-3-оксо-17α-прегн-4-ен-7β,21-дикарбоксилат, γ-лактон). Алкоксид и/или цианид в растворе реагируют между собой с превращением сложного 7α-эфира в эпимерную смесь сложного 7α-эфира и его 7β-эфирного эпимера. Чистый сложный 7β-эфир может быть выделен из эпимерной смеси путем селективной кристаллизации.
Предпочтительно соединением формулы XXI является 4'S-(4'α),7'α-9',11α-эпоксигексадекагидро-10β-,13'β-диметил-3',5,20'-триоксоспиро[фуран-2(3Н),17'β-[4,7]метано[17Н]-циклопента[а]-фенантрен-5'-карбонитрил; соединением формулы XXII является 5'R(5'α),7'β-20'-амино-9,11β-эпоксигексадекагидро-10,13'-диметил-3',5-диоксоспиро[фуран-2(3Н),17'α-(5'Н)-[7,4]метен[4Н]-циклопента[а]фенантрен-5'-карбонитрил; а соединением формулы XXIII является 9,11α-эпокси-17α-гидрокси-3-оксопрегна-4,6-диен-21-карбоновая кислота, γ-лактон.
В особенно предпочтительном варианте осуществления настоящего изобретения полный реакционный процесс Схемы 4 заключается в следующем:
Схема 5.
Процесс Схемы 5 осуществляют, исходя из субстрата, соответствующего формуле XXIX:
где: -А-А-, -В-В-, R3 определены выше в формуле XX.
Нижеследующие микроорганизмы способны осуществлять 9α-гидроксилирование соединения формулы XXXV (такого как андростендион).
где: -А-А-, -В-В-, R3 определены выше в формуле XIII, с образованием соединения формулы XXIX в условиях, аналогичных условиям, описанным в Примере 19В:
Aspergillus niger ATCC 16888 и 26693, Corynespora cassiicola ATCC 16718, Curvularia clavata ATCC 22921, Mycobacterium fortuitum NRRL B8119, Nocardia canicruria ATCC 31548, Pycnosporium spp. ATCC 12231, Stysanus microsporus ATCC 2833, Syncephalastrum racemosum ATCC 18192 и Thamnostylum piriforme ATCC 8992.
Субстрат, соответствующий формуле XXIX, превращают в продукт формулы XXVIII:
посредством реакции с триметилортоформиатом, где: -А-А-, -В-В- и R3 определены выше в формуле XX.
После образования соединений формулы XXVIII, эти соединения превращают в соединения формулы XXVII способом, описанным выше для превращения субстрата формулы XX в соединение формулы XVII. Соединения формулы XXVII имеют следующую структуру:
где: -А-А-, -В-В-, R3 определены выше в формуле XX, a Rx представляет любую из стандартных гидрокси-защитных групп.
Альтернативно, С9-α-гидрокси-группа может быть защищена на более ранней стадии этой схемы синтеза, если желательна защита на этой стадии, то есть С9-гидрокси-группа соединения формулы XXVIII или С9-гидрокси-группа соединения формулы XXIX могут быть защищены любой из стандартных гидрокси-защитных групп.
Способом, описанным выше для получения соединений формулы XVI, соединения формулы XXVII окисляют с получением новых соединений, соответствующих формуле XXVI
где: -А-А-, -В-В- и R3 определены выше в формуле XX.
Особенно предпочтительными соединениями формул XXIX, XXVIII, XXVII и XXVI являются соединения, в которых -А-А- и -В-В- являются такими, как они были определены для формулы XIII, а R3 представляет водород.
Продукты формулы XXVI являются новыми соединениями, которые могут быть выделены путем осаждения/фильтрации. Они, в основном, могут быть использованы в качестве промежуточных соединений для получения соединений формулы I, а особенно соединений формулы IA. Особенно предпочтительными соединениями формулы XXVI являются соединения, в которых -А-А- и -В-В- являются такими, как они были определены для формулы XIII, а R3 представляет водород. В наиболее предпочтительных соединениях формулы XXVI, -А-А- и -В-В- представляют -СН2-СН2-, а R3 представляет водород.
Способом, описанным выше для цианидирования соединений формулы VIII, новые промежуточные соединения формулы XXVI превращают в новые 9-гидроксиенаминовые промежуточные соединения формулы XXV
где: -А-А-, -В-В-, R3 определены выше в формуле XX.
Продукты формулы XXV являются новыми соединениями, которые могут быть выделены путем осаждения/фильтрации. Они, в основном, используются в качестве промежуточных соединений для получения соединений формулы I, а особенно соединений формулы IA. Особенно предпочтительными соединениями формулы XXVI являются соединения, в которых -А-А- и -В-В- являются такими, как они были определены для формулы XIII, a R3 представляет водород. В наиболее предпочтительных соединениях формулы XXVI -А-А- и -В-В- представляют -CH2-CH2-, a R3 представляет водород.
Способом, описанным выше для получения дикетоновых соединений формулы VI, 9-гидроксиенаминовые промежуточные соединения формулы XXV превращают в дикетоновые соединения формулы XIVB. Следует отметить, что в данном случае эта реакция эффективна для одновременного гидролиза енаминовой структуры и дегидратации в 9,11-положениях для введения 9,11-двойной связи. Затем соединение формулы XIV превращают в соединение формулы XIII с использованием той же самой стадии, которая была описана выше в Схеме 3.
Предпочтительно, соединением формулы XIV является 4'S(4'α),7'α-1',2',3',4,4',5,5',6',7',8',10',12',13',14',15',16'-гексадекагидро-10β-13'β-диметил-3',5,20'-триоксоспиро[фуран-2(3Н),17'β-[4,7]метано[17Н]-циклопента[а]фенантрен]-5'-карбонитрил; соединением формулы XXV является 5'R(5'α),7'β-20'-аминогексадека-гидро-9'β-гидрокси-10'α, 13'α-диметил-3',5-диоксоспиро[фуран-2(3Н),17'α(5'H)-[7,4]метено[4Н]-циклопента[а]-фенантрен]-5'- карбонитрил; соединением формулы XXVI является 9α,17α-дигидрокси-3-оксопрегна-4,6-диен-21-карбоновая кислота, γ-лактон; а соединением формулы XXVII является 9α,17α-дигидрокси-3-оксопрегн-4-ен-21-карбоновая кислота, γ-лактон.
В особенно предпочтительном варианте осуществления изобретения весь способ Схемы 5 осуществляют следующим образом:
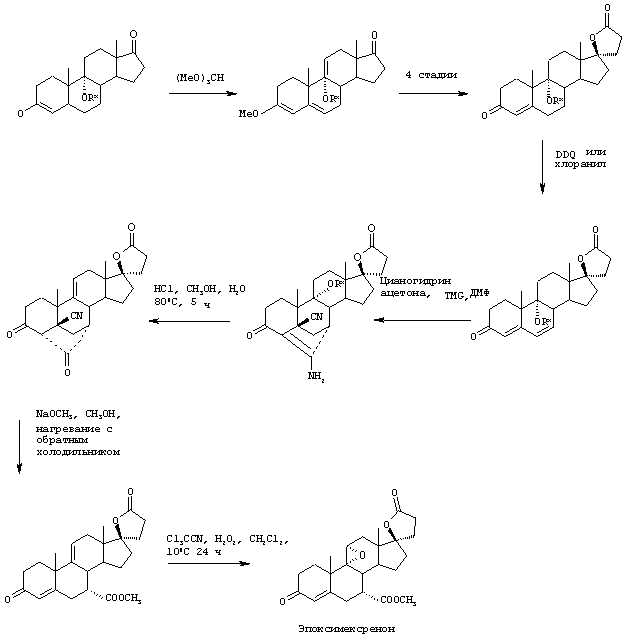
Схема 6.
Схема 6 представляет преимущественный способ получения эпоксимексренона и других соединений, соответствующих формуле I, исходя из 11α или 11β-гидроксилирования андростендиона или другого соединения формулы XXXV:
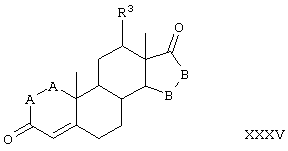
где: -А-А-, -В-В- и R3 определены выше в формуле XIII, с получением промежуточного соединения, соответствующего формуле XXXVI или его соответствующего 11β-гидрокси-изомера
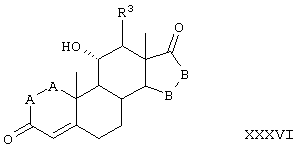
где: -А-А-, -В-В- и R3 определены выше в формуле XIII.
За исключением выбора субстрата, этот способ 11α-гидроксилирования осуществляют, в основном, как описано выше для Схемы 1. 11α-Гидроксилирование андростендиона или другого соединения формулы XXXV способны осуществлять нижеследующие микроорганизмы:
Absidia glauca ATCC 22752, Aspergillus flavipes ATCC 1030, Aspergillus foetidus ATCC 10254, Aspergillus fumigatus ATCC 26934, Aspergillus ochraceus NRRL 405 (ATCC 18500), Aspergillus niger ATCC 11394, Aspergillus nidulans ATCC 11267, Beauveria bassiana ATCC 7159, Fusarium oxysporum ATCC 7601, Fusarium oxysporum cepae ATCC 11171, Fusarium Lini ATCC IFO 7156, Gibberella fujikori ATCC 14842, Hypomyces chyrsospermus IMI 109891, Mycobacterium fortuitum NRRL B8119, Penicillum patulum ATCC 24550, Pycnosporium spp. ATCC 12231, Rhyzopus arrhizus ATCC 11145, Saccharopolyspora erythraea ATCC 11635, Thamnostylum piriforme ATCC 8992, Rhizopus oryzae ATCC 11145, Rhizopus stolonifer ATCC 6227b и Trichothecium roseum ATCC 12519 и ATCC 8685.
11β-Гидроксилирование андростендиона или другого соединения формулы XXXV способны осуществлять нижеследующие микроорганизмы:
Aspergillus fumigatus ATCC 26934, Aspergillus niger ATCC 16888 и ATCC 26693, Epicoccum oryzae ATCC 7156, Curvularia lunata ATCC 12017, Cunninghamella blakesleeana ATCC 8688a и Pithomyces atro-olivaceous IFO 6651.
Затем, 11α-гидроксиандрост-4-ен-3,17-дион или другое соединение формулы XXXVI превращают в простой эфир 11α-гидрокси-3,4-енола формулы (101):
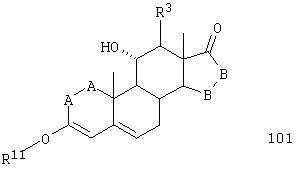
где: -А-А-, -В-В- и R3 определены выше в формуле XIII, а R11 обозначает метил или другой низший алкил (C1-C4) с помощью реакции с этерифицирующим реагентом, таким как триалкилортоформиат, в присутствии кислотного катализатора. Для осуществления этого превращения 11α-гидроксисубстрат подкисляют путем смешивания с кислотой, такой как, например, гидрат бензолсульфоновой кислоты или гидрат толуолсульфоновой кислоты в растворителе, таком как низший спирт, например этанол. Триалкилортоформиат, предпочтительно триэтилортоформиат вводят постепенно в течение 5-40 минут, поддерживая смесь в холодном состоянии, предпочтительно при температуре от около 0°С до около 15°С. Затем смесь нагревают и реакцию осуществляют при температуре от 20°С до около 60°С. Реакцию предпочтительно осуществляют при 30-50°С в течение 1-3 часов, а затем нагревают с обратным холодильником еще некоторое время, обычно в течение 2-6 часов до полного завершения реакции. Реакционную смесь охлаждают, предпочтительно до 0°С-15°С, а более предпочтительно при около 5°С, и растворитель удаляют в вакууме.
С использованием той же самой реакционной схемы, которая была описана выше в Схеме 3 для превращения соединения формулы XX в соединение формулы XVII, 17-спиролактонную часть формулы XXXIII вводят в соединение формулы 101. Так, например, субстрат формулы 101 может быть подвергнут реакции с илидом сульфония в присутствии основания, такого как гидроксид щелочного металла, в подходящем растворителе, таком как ДМСО, с получением промежуточного соединения формулы 102:
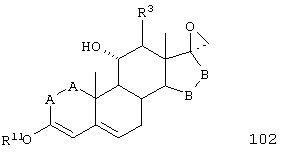
где: -А-А-, R3, R11 и -В-В- определены выше в формуле 101. Промежуточное соединение формулы 102 затем подвергают реакции со сложным диэфиром малоновой кислоты в присутствии алкоксида щелочного металла с получением пятичленного спиролактонового кольца и промежуточного соединения формулы 103:
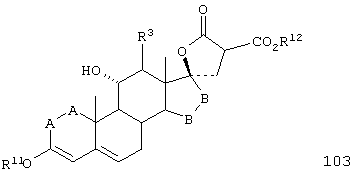
где: -А-А-, R3, R11 и -В-В- определены выше в формуле 102, а R12 представляет С1-С4алкил, предпочтительно этил. И наконец, соединение формулы 103 в подходящем растворителе, таком как диметилформамид, нагревают в присутствии галогенида щелочного металла, отщепляя, при этом, алкоксикарбонильную группу и получая промежуточное соединение формулы 104:
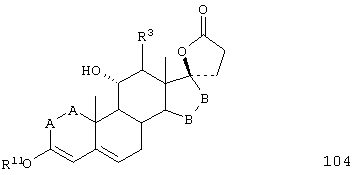
где снова -А-А-, R3, R11 и -В-В- определены выше в формуле 102.
Затем эфирное соединение 3,4-енола 104 превращают в соединение формулы XXIII, то есть соединение формулы VIII, в котором R8 и R9, взятые вместе, образуют часть формулы XXXIII. Эту стадию окисления осуществляют, в основном, таким же способом, которым осуществляли стадию окисления для превращения соединения формулы XXIV в промежуточное соединение формулы XXIII в Схеме синтеза 4. Прямое окисление может быть осуществлено с использованием такого реагента, как 2,3-дихлор-5,6-дициано-1,4-бензохинон (DDQ) или тетрахлорбензохинон (хлоранил); либо, предпочтительно, двухстадийное окисление осуществляют сначала путем бромирования, например с использованием N-галоген-бромирующего агента, такого как N-бромсукцинамида (NBS) или 1,3-дибром-5,5-диметилгидантоин (DBDMH), а затем путем дегидробромирования с использованием основания, например DABCO, в присутствии LiBr и при повышенной температуре. В случае, если для бромирования используют NBS, то для превращения эфира 3-енола в енон должна быть также использована кислота. DBDMH, ионный, а не свободнорадикальный бромирующий реагент, является сам по себе эффективным для бромирования и превращения эфира енола в енон.
Затем соединение формулы VIII превращают в эпоксимексренон или другое соединение формулы I в стадиях, описанных выше для Схемы 1.
Каждое из промежуточных соединений формул 101, 102, 103 и 104 является новым соединением, которое может быть, в основном, использовано в качестве промежуточного соединения для получения эпоксимекстренона или других соединений формул IA и I. В каждом из соединений формул 101, 102, 103 и 104 -А-А- и -В-В- представляют -CH2-CH2-, a R3 представляет водород, низший алкил или низший алкокси. R3 предпочтительно представляет водород. Наиболее предпочтительно соединением формулы 101 является 3-этокси-11α-гидроксиандрост-3,5-диен-17-он; соединением формулы 102 является 3-этоксиспиро[андрост-3,5-диен-17β,2'-оксиран]-11α-ол, соединением формулы 103 является этилгидро-3-этокси-11α-17α-дигидроксипрегна-3,5-диен-21,21-дикарбоксилат, гамма-лактон и соединением формулы 104 является 3-этокси-11α-17α-дигидроксипрегна-3,5-диен-21-карбоновая кислота, гамма-лактон.
В особенно предпочтительном варианте осуществления изобретения весь процесс Схемы 6 протекает следующим образом:
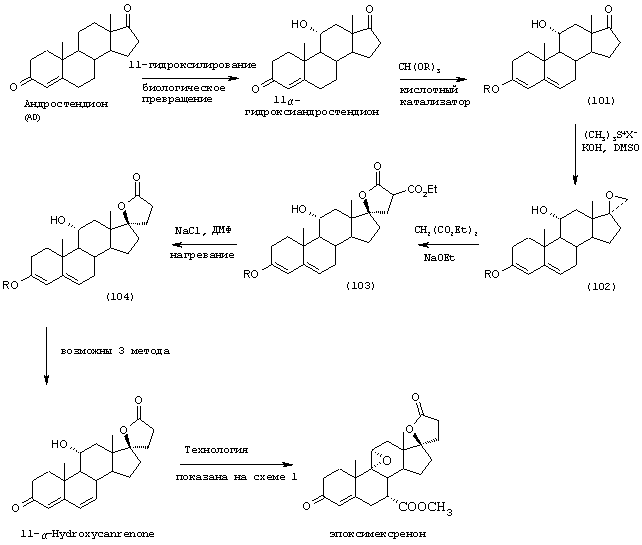
Было высказано предположение, что эпоксимексренон и другие соединения, соответствующие формуле I, могут быть также получены из 11β-гидроксиандростендиона или других соединений формулы XXXV, которые были 11β-гидроксилированы. Другими словами, эпоксимексренон и другие соединения, соответствующие формуле I, могут быть получены общим способом, представленным в Схеме 6 с использованием либо α-гидроксилированного субстрата формулы XXXV либо соответствующего β-гидроксилированного субстрата.
Схема 7.
Схема 7 представляет синтез эпоксимексренона и других соединений формулы I с использованием исходного субстрата, содержащего β-ситостерин, холестерин, стигмастерин или другое соединение формулы XXXVII:
где: -А-А-, R3 и -В-В- определены выше в формуле XIII; D-D представляет -CH2-CH2- или -СН=СН-; а каждый из R13, R14, R15 и R16 независимо выбирают из водорода или С1-С4алкила.
R3 предпочтительно представляет водород.
В первой стадии синтеза 11α-гидроксиандростендиона или другого соединения формулы XXXVI получают путем биологического превращения соединения формулы XXXVII. Способ такого биологического првращения осуществляют, в основном, способом, описанным выше для 11α-гидроксилирования канренона (или другого субстрата формулы XIII).
При синтезе 11α-гидроксиандростендиона 4-андростен-3,17-дион сначала получают путем биологического превращения соединения формулы XXXVII. Это первоначальное биологическое превращение может быть проведено способом, описанным в патенте США №3759791, который во всей своей полноте вводится в настоящее описание посредством ссылки. Затем 4-андростен-3,17-дион превращают в 11α-гидроксиандростендион, в основном, способом, описанным выше для 11α-гидроксилирования канренона (или другого субстрата формулы XIII).
Остальная часть синтеза Схемы 7 идентична Схеме 6. В особенно предпочтительном варианте осуществления изобретения весь процесс Схемы 7 протекает следующим образом:
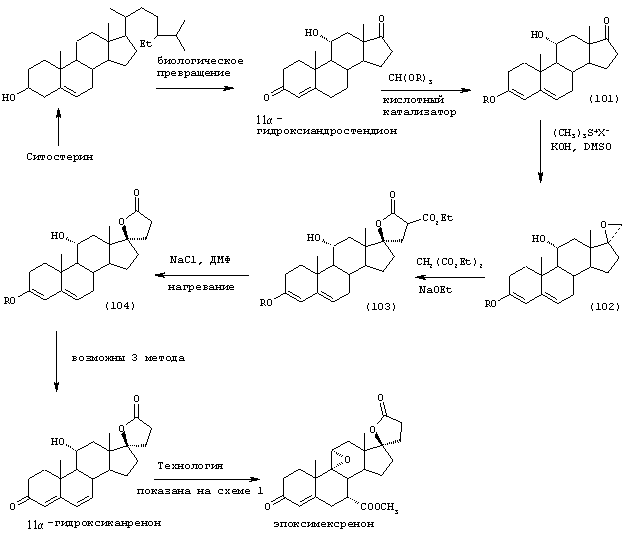
Было высказано предположение, что эпоксимексренон и другие соединения, соответствующие формуле I, могут быть также получены в соответствии с общим способом, представленным в Схеме 7, в том случае, если продуктом биологического превращения β-ситостерина или других соединений формулы XXXVIII является 11β-гидроксиандростендион или другие соединения формулы XXV, которые были 11β-гидроксилированы. Другими словами, эпоксимексренон и другие соединения, соответствующие формуле I, могут быть получены общим способом, представленным в Схеме 7, если биологическое превращение β-ситостерина или других соединений формулы XXXVIII приводит к получению либо α-гидроксилированного субстрата формулы XXXV либо соответствующего β-гидроксилированного субстрата.
Схема 8.
Значительная сложность в синтезе эпоксимексренона и родственных соединений заключается в необходимости стереоселективного введения α-алкоксикарбонильного заместителя у 7-углерода, не вызывая при этом нежелательных модификаций в других участках стероидной структуры. В соответствии с настоящим изобретением было установлено, что способ эффективного синтеза для введения 7α-алкоксикарбонильного заместителя включает следующие стадии: (i) первоначальное цианидирование в 7-углероде стероида, (ii) гидролиз 7-цианостероида с образованием смеси 7α-карбоновой кислоты и 7β-карбоновой кислоты стероидов, (iii) образование 5,7-лактонстероида из 7α-карбоновой кислоты стероида, и (iv) отделение 7β-карбоновой кислоты стероида от 5,7-лактонстероида. В результате опосредованной основанием реакции размыкания кольца, которая происходит между 5,7-лактонстероидом и алкилирующим реагентом, получают нужный 7α-алкоксикарбонильный стероид.
В соответствии с этим, процесс проведения Схемы 8 обычно направлен на получение 3-кето-7α-алкоксикарбонил-замещенного Δ4,5-стероида, предусматривающего проведение реакции алкилирующего реагента с 3-кето-4,5-дигидро-5,7-лактонстероидным субстратом в присутствии основания. Лактонный субстрат замещен кето-группой в 3-положении углерода и, кроме того, содержит часть:
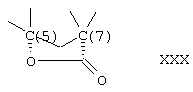
где С(5) представляет 5-углерод, а С(7) представляет 7-углерод стероидной структуры субстрата. Превращение 5,7-лактона в 7α-алкоксикарбонил предпочтительно осуществляют посредством реакции с алкилгалогенидом в присутствии основания. Алкилгалогенидным реагентом является предпочтительно иодид, а наиболее предпочтительно метилиодид.
Кроме того, в соответствии с настоящим изобретением был разработан преимущественный способ для получения 4,5-дигидро-5,7-лактонстероидного соединения, описанного выше. В этом способе 3-кето-Δ4,5-7α-циано-замещенный стероидный субстрат превращают в 7-карбоновую кислоту, а эту кислоту, в свою очередь, подвергают реакции с триалакилортоформиатом в подкисленном растворителе, низшем спирте, с получением 5,7-лактона. Реакция с ортоформиатными сложными эфирами способствует превращению 3-кето-группы в 3-ациклический или циклический кеталь-5,7-лактон (следует отметить, что сначала образуется лактон). Предпочтительным 3-кеталь-5,7-лактоном является 3-диалкилкеталь-5,7-лактон. Более предпочтительно алкильная часть спиртового растворителя является такой же, как и алкильная часть алкокси-групп ортоформиата (а наиболее предпочтительно все они являются метилом), поскольку: алкокси-части кеталя могут происходить либо от ортоформиата, либо от спирта; смешанные кетали не являются предпочтительными; а 3-диметокси является предпочтительным. Если кеталь является этиленкеталем, то алкильная часть спиртового растворителя не обязательно должна быть такой же, как алкильная часть алкокси-групп ортоформиата. 3-кеталь-5,7-лактон легко гидролизуется с образованием 3-кето-5,7-лактона, кристаллического соединения, которое может быть легко очищено. Поскольку, реакции лактонизации подвергается только 7α-карбоновая кислота, то обеспечивается полная стереоспецифичность. 7β-Кислота может быть затем удалена из реакционной смеси в виде соли, например путем обработки 7β-кислоты слабым основанием, таким как бикарбонат натрия.
7-Циано-субстрат для продуцирования 5,7-лактона может быть получен известным способом. Так например, субстрат, незамещенный в 7-положении углерода, может быть подвергнут реакции с небольшим избытком иона цианида, предпочтительно от около 1,05 до около 1,25 эквивалентов на эквивалент субстрата в слегка подкисленном растворе, содержащем растворяющую смесь вода/ДМСО. Предпочтительно, чтобы реакционная смесь включала карбоновую кислоту, например около одного эквивалента уксусной кислоты на эквивалент субстрата. При этом образуется как 7α-, так и 7β-CN-изомер, причем 7α-изомер является главным изомером. 7α-Циано-стероид может быть выделен стандартным способом. В этом дополнительном получении могут быть использованы и другие методы, известные специалистам.
В основном, в соответствии со Схемой 8, 5,7-лактон может быть образован из промежуточного 7-карбокси-соединения (которое само получают путем гидролиза промежуточного 7-циано-соединения), которое является замещенным в 17-положении либо кето-группой, либо R8, либо R9, где R8 и R9 определены выше, и которое имеет алифатическую, олефиновую, эпоксидную или гидрокси-замещенную конфигурацию в С-9 и С-11, то есть
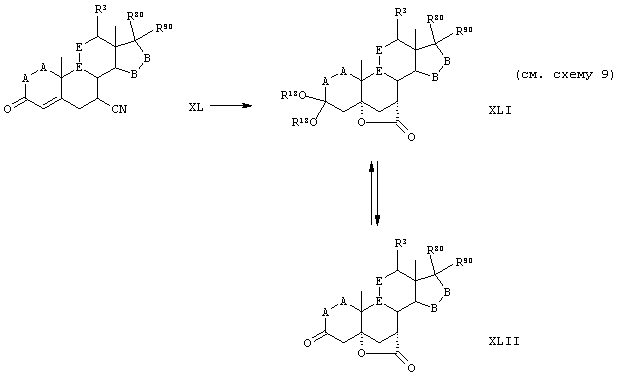
где -А-А-, -В-В- и R3 определены выше, R80 и R90 являются такими же, как R8 и R9, либо R80 и R90, взятые вместе, образуют кето, R18 определен ниже для Схемы 9, а -Е-Е- выбирают из
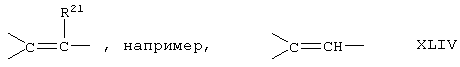
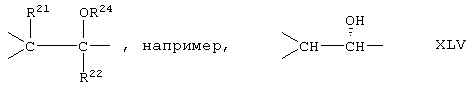
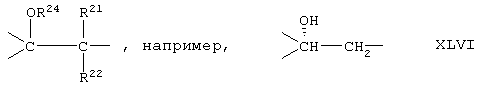
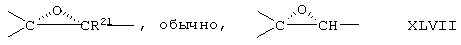
Соединение формулы XLII затем превращают в 7α-алкокси-карбонил;
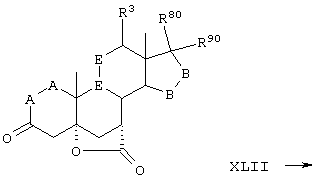
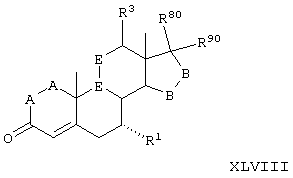
В каждом из соединений XL, XLI, XLII и XLVIII R80 и R90, взятые вместе, предпочтительно составляют кето или
где Y1, Y2, Х и С (17) определены выше, а наиболее предпочтительно, если R80 и R90, взятые вместе, составляют
R3 предпочтительно представляет Н, R1 предпочтительно представляет метоксикарбонил, а -А-А- и -В-В- предпочтительно представляют -СН2-СН2-. Следует отметить, что эти реакции могут быть также осуществлены с 3-кето-группой, защищенной путем ее превращения в каждую эфирную или кетальную форму и поддерживания ее в этой форме на протяжении всей последовательности реакций. Альтернативные способы Схемы 8 предусматривают использование различных промежуточных соединений, охватываемых формулами XLI и XLII, определенных выше.
Следует отметить, что реагентом, используемым для образования 5,7-лактона из 3-кето-Δ4,5-7-карбоновой кислоты в Схеме 8, является триалкилортоформиат, тот же самый реагент, который используется для превращения 11α-гидроксиандростендиона в промежуточное 3-енолэфир-3,5-диен-11α-гидрокси-соединение 101 Схемы 6, при этом, очевидно, что путь, по которому осуществляется Схема реакций 8, заивисит от замещения у С-7. В результате реакции с ортоформиатом в присутствии Н+ образуется промежуточный карбониевый ион, имеющий карбоксил в положении С-7, и положительный заряд при равновесии между С-3 и С-5. После потери протона С-3-ион карбония дает соединение формулы 101, а С-5-ион карбония дает лактон. В случае водорода у С-7 очевидно, что образуется преимущественно 3,5-диен-3-алкокси (эфир енола) из-за сопряжения двойных связей. В случае 7α-CO2-эаместителя у С-7, С-5-ион карбония захватывается карбокси и образуется 5,7-лактон. На этой стадии 3-кето-группа преимущественно превращается в кеталь, что способствует завершению реакции.
Предпочтительные варианты Схемы 8 описаны ниже в Схемах 9 и 10.
Схема 9.
Схема 9 начинается с использования того же самого субстрата, который был использован в Схеме 4, то есть соединения формулы XX. Этот субстрат сначала окисляют до соединения формулы В;
где: -А-А-, R3 и -В-В- определены выше в формуле XIII.
Реакцию окисления проводят в соответствии с любой из реакционных схем, описанных выше для превращения соединения формулы XXIV в промежуточное соединение формулы XXIII в синтезе Схемы 4. С использованием способов, описанных для схемы 8, соединение формулы В превращают в 7-циано-промежуточное соединение формулы С:
где -А-А-, R3 и -В-В- определены в формуле XIII. Затем соединение формулы С превращают в 5,7-лактон формулы D:
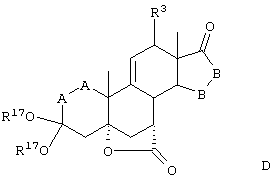
где -А-А-, R3 и -В-В- определены в формуле XIII, а R17 представляет С1-С4алкил, с использованием триалкилортоформиатного реагента, используемого ранее в Схеме 6. 5,7-Лактон формулы D легко выделяют из непрореагировавшего соединения 7-β-СООН, например путем удаления кислоты посредством промывки бикарбонатом, и, тем самым, создания нужной стереохимической С-7-структуры и предотвращения эпимеризации в последующих реакциях, которые осуществляют в основных условиях. Этерификация лактона путем реакции с алкилгалогенидом, как описано в Схеме 8, дает сложное енэфирное промежуточное соединение формулы II.
Продолжая синтез по Схеме 9, соединение формулы D превращают в соединение формулы II. С использованием 3-кето-группы, защищенной путем превращения в кеталь, 20-спироксановую группу формулы XXXIII избирательно вводят в 17-положение в соответствии с реакционной схемой, описанной выше для Схемы 3 и 6 (см. выше), и получают соединение формулы Е:
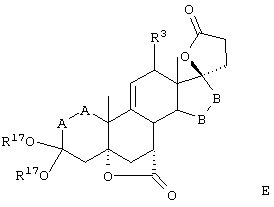
Поскольку 3-кетон является защищенным, то условия гидролиза могут быть выбраны так, чтобы они были оптимальны для воздействия на 17-кетон без образования побочных продуктов посредством реакции в 3-положении. После гидролиза 3-кеталевого соединения формулы Е в структуру с 3-кето-группой формулы F
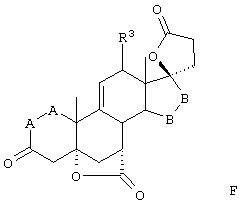
последнее промежуточное соединение подвергают реакции с алкилиодидом в присутствии основания по Схеме превращения 8 с получением промежуточного сложного енэфира формулы II. И, наконец, последнее промежуточное соединение превращают в эпоксимексренон или другое соединение формулы I с использованием любых способов, описанных выше для схемы 1.
Схема 9 имеет преимущества не только из-за возможности регулировать стереохимию, сообщаемую 5,7-лактонным промежуточным соединением, но она также дает дополнительные преимущества, которые заключаются в возможности использовать условия гидролиза в более широких пределах, не оказывая влияния на 17-спиролактон.
Подобно реакциям других схем синтеза настоящего изобретения реакции Схемы 9 могут быть использованы для превращения субстратов, отличающихся от субстратов, конкретно описанных выше. Так, например, превращение 3-кето- или 3-кеталь-7-циано-стероидов в 3-кето- или 3-кеталь-5,7-лактон, или превращение 3-кето- или 3-кеталь-5,7-лактона в 7α-алкоксикарбонил может быть осуществлено с использованием соединений, замещенных у 17-углерода группами R8 и R9, определенными выше, или, более предпочтительно, заместителем формулы:
где X, Y1 и Y2 определены выше, а С(17) означает 17-углерод. Однако эти важные преимущества реализуются, особенно с точки зрения экономичности способа, путем проведения реакций в определенной последовательности с использованием 17-кето-субстратов и в соответствии с конкретной реакционной схемой, описанной выше для введения 17-спиролактона и 7α-алкоксикарбонила в 3-кето-Δ9,11-стероид.
Лактоны формул D, Е и F являются новыми соединениями, которые могут быть использованы для получения эпоксимексренона и других соединений формул I и IA в соответствии со Схемой синтеза 9. В этих соединениях -А-А- и -В-В- предпочтительно представляют -СН2-СН2-, a R3 представляет водород, низший алкил или низший алкокси. Наиболее предпочтительным является соединение формулы D, в котором R17 представляет метокси.
В особенно предпочтительном варианте осуществления изобретения весь процесс Схемы 9 протекает следующим образом:

Схема 10.
Схема 10 является такой же, как Схема 9 вплоть до образования промежуточного 7-циано-соединения формулы С. В следующей стадии Схемы 10 7-циано-стероид подвергают реакции с триалкилортоформиатом в алканоловом растворителе, предпочтительно с триметилортоформиатом в метаноле, с одновременной защитой 3-кето- и 17-кето-групп путем превращения первого соединения в эфир енола, а последнего в кеталь. Затем 7-циано-группу восстанавливают до 7-формила, например путем реакции с гидридом диалкилалюминия, предпочтительно с гидридом диизобутилалюминия, в результате чего получают соединение формулы 203:
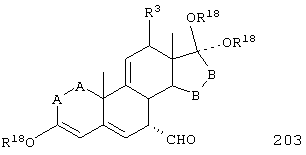
где -А-А-, R3 и -В-В- определены выше в формуле XIII, a R18 представляет С1-С4алкил.
Перед защитой кето-групп, как описано выше, их восстановление предупреждают с использованием гидрида диалкилалюминия. Затем промежуточное соединение формулы 203 подвергают реакции с разбавленной водной кислотой для селективного гидролиза 17-кеталя в присутствии избыточного количества спирта (R19OH), в результате чего получают промежуточное соединение формулы 204:
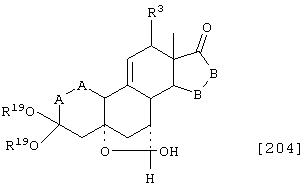
где R19 выбирают из низшего алкила (предпочтительно C1-C4), либо группы R19 в 3-положении образуют циклический O,O-оксиалкиленокси-заместитель у 3-углерода. Полуацеталь [204], кроме того, защищают путем обработки алканолом (R19OH) в присутствии безводной кислоты с получением промежуточного соединения формулы 205:
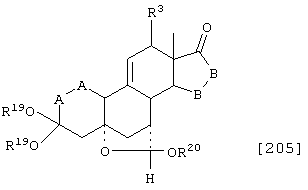
где: -А-А-, -В-В-, R3 и R19 определены выше, а R20 представляет С1-C4алкил.
17-Спиролактоновая часть может быть затем введена в соответствии с реакционными стадиями, описанными выше для Схем 3 и 6, которые осуществляют в нижеприведенной последовательности:
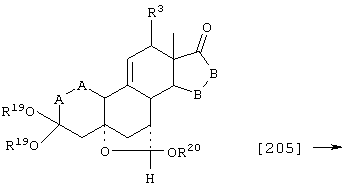

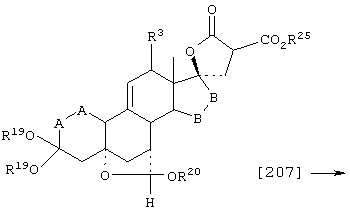
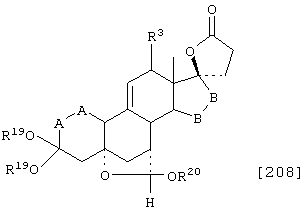
где -А-А-, -В-В-, R3, R19 и R20 определены выше, a R25 представляет С1-С4алкил.
После этого, у 3-положения снимают защиту путем стандартной реакции гидролиза с повторным введением 3-кето-группы и 5,7-полуацеталя и получают другое промежуточное соединение, соответствующее формуле 209:
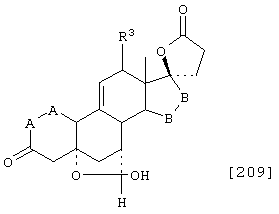
где -А-А-, -В-В- и R3 определены выше.
Затем 9,11-эпоксидную часть вводят любым из способов, описанных выше для превращения соединений формулы II в соединения формулы I. В окислительных условиях реакции эпоксидирования полуацеталь частично превращается в 5,7-лактон, в результате чего продуцируется другое промежуточное соединение, соответствующее формуле 211:
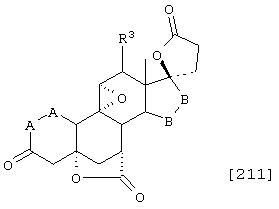
где: -А-А-, -В-В- и R3 определены выше.
Любой оставшийся продукт реакции промежуточного 9,11-эпокси-5,7-полуацеталя формулы 210:
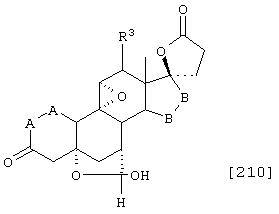
где: -А-А-, -В-В- и R3 определены выше, легко окисляется стандартными методами с получением соединения формулы 211. И, наконец, промежуточное соединение формулы 211 превращают в эпоксимексренон или другое соединение формулы I способом, описанным в Схеме 8 для превращения 5,7-лактона в 7α-алкокси-карбонильное соединение. Таким образом вся Схема 10 протекает как проиллюстрировано ниже; но, при этом, следует отметить, что по крайней мере следующие стадии могут быть осуществлены in situ без выделения промежуточного соединения. В целом синтез Схемы 10 осуществляют следующим образом:
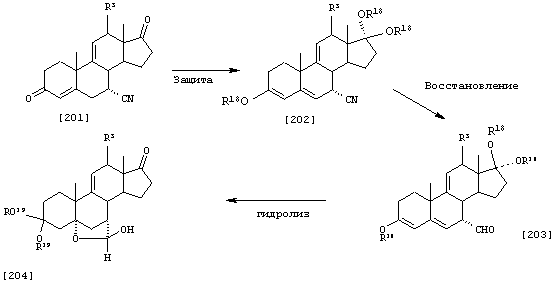
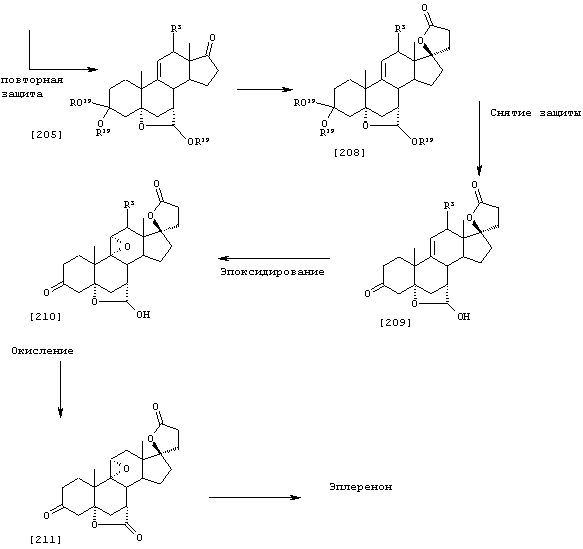
Как и в случае Схемы 9, реакции, описанные выше для Схемы 10, имеют важные преимущества, особенно с точки зрения экономичности способа; и, при этом, новые реакции Схемы 10 имеют также более общее применение по отношению к субстратам, не являющимся субстратами, конкретно описанными выше. Так, например, введение 7-формильной группы в эфир 3-енола стероида, защита полученного эфира 7-формил-Δ-5,6-3,4-енола, гидролиз с образованием 5,7-полуацетала и последующее деблокирование могут быть проведены на стероидах, замещенных в 17-положении группами R8 и R9, определенными выше или, более конкретно, заместителем формулы:
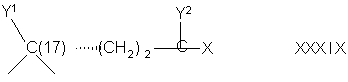
где каждый из X, Y1, Y2 и С(17) определены выше.
Альтернативные способы Схемы 10 предусматривают использование различных промежуточных соединений, охватываемых формулами A203-А210, соответственно описанных выше. Каждое из промежуточных соединений формулы А203-А211 является новым соединением, которое может быть использовано для получения эпоксимексренона и других соединений формулы I и IA в соответствии со Схемой синтеза 10.
В особенно предпочтительном варианте осуществления изобретения, весь процесс Схемы 10 протекает следующим образом:
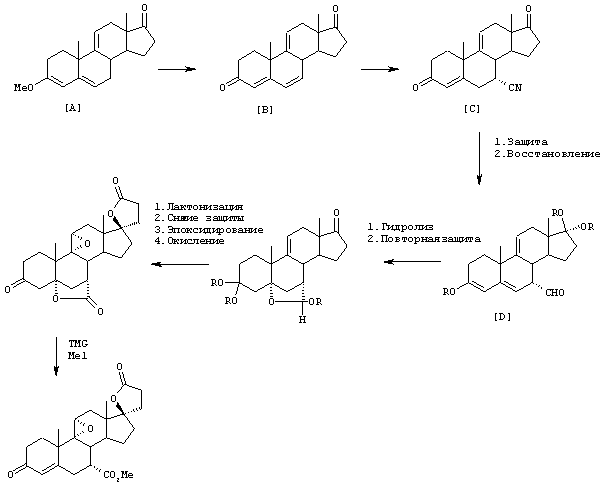
Исходя из нескольких схем, проиллюстрированных выше, следует отметить, что реакционные стадии, выбранные для использования в способах настоящего изобретения, обеспечивают значительную адаптивность в производстве эпоксимексренона и родственных соединений. Ключевыми отличительными признаками этих способов являются, inter alia: (а) биологическое превращение субстрата, такого как канренон, андростендион или β-ситостерин в 11α- или 9α-гидрокси-производное (с одновременным превращением β-ситостерина в 17-кето-структуру; (b) введение 9,11-двойной связи путем дегидратации соединения, содержащего либо 11α-, либо 9α-гидрокси-группу, с последующим введением эпокси-группы путем окисления 9,11-двойной связи; (с) присоединение 7α-алкоксикарбонила посредством образования енамина, гидролиза енамина с образованием дикетона и реакции этого дикетона с алкоксидом щелочного металла; (d) образование 20-спироксанового кольца в 17-положении; (е) образование 5,7-лактона и этерификация этого лактона с образованием 7-алкоксикарбонила; (f) защита 3-кетона путем превращения в эфир 3-енола или в 3-кеталь в процессе ряда превращений в других положениях (включая образование 20-спироксанового кольца в 17-положении). С некоторыми ограничениями, эти стадии четырехкомпонентного процесса (b)-(d) могут быть осуществлены почти в любой последовательности. Стадии процесса (е) и (f) обладают сравнительной адаптируемостью. Они обеспечивают путь получения эпоксимексренона и других соединений формулы I и были значительно упрощены по сравнению со способом, описанным в патенте США 4559332. Кроме того, они дают важные преимущества в отношении продуктивности и выхода.
В описаниях реакционных схем, представленных выше, получение, выделение и очистка реакционных продуктов могут быть, в основном, проведены методами, хорошо известными специалистам. Если это не указано особо, то условия, растворители и реагенты являются либо стандартными, либо они не имеют исключительно решающего значения, либо то и другое. Однако, некоторые из специфических процедур, конкретно описанных выше, имеют преимущества, которые обеспечивают увеличение общего выхода и/или продуктивности различных стадий процесса и схем данного процесса, и/или высокое качество промежуточных и конечных 9,11-эпокси-стероидных продуктов.
Эффективное использование 20-спироксановых соединений, продуцированных в соответствии с настоящим изобретением, описано в патенте США 4559332 (Grob), который во всей своей полноте вводится в настоящее описание посредством ссылки.
20-Спироксановые соединения, полученные в соответствии с настоящим изобретением, отличаются своими хорошими биологическими свойствами, а поэтому являются ценными фармацевтически активными ингредиентами. Так, например, они обладают сильным алдостерон-антагонистическим действием, которое заключается в том, что они снижают и нормализуют чрезмерно высокое накопление натрия и выведение калия, вызываемое альдостероном. Поэтому они действуют как калий-удерживающие диуретики и могут иметь важное терапевтическое применение, например для лечения гипертонии, сердечной недостаточности или цирроза печени.
Известны 20-спироксановые производные, обладающие альдостерон-антагонистическим действием, см., например, Fieser & Fieser: Steroids; p.708 (Reinhold Publ.Corp., New York, 1959) и описание патента Великобритании №1041534; также известны 17β-гидрокси-21-карбоновые кислоты и их соли с аналогичной активностью, см., например, патент США №3849404. Однако, соединения этого вида, используемые до настоящего времени в качестве лекарственных средств, имеют значительные недостатки, заключающиеся в том, что они всегда обладают некоторой секуально-специфической активностью, которая рано или поздно вызывает неблагоприятные последствия при обычной продолжительной терапии. Особенно нежелательными являются неблагоприятные эффекты, которые могут быть приписаны антиандрогенной активности известных антиальдостероновых препаратов.
Способы, процессы и композиции настоящего изобретения, а также условия и реагенты, используемые в настоящем изобретении, дополнительно описаны в нижеследующих примерах.
ПРИМЕР 1.
Культуры на скошенном агаре были получены с использованием среды для культивирования, описанной в Таблице 1
- рН 6,7
- доведение до рН 5 путем добавления 10% масс./об Н3РО4
- для скошенных сред:
7,5 мл в пробирках 180×18 мм
- для чашек (10 см ⊘)
25 мл в пробирках 200×20 мм
- стерилизация при 120°С в течение 20 минут
- рН после стерилизации: 5
Для продуцирования культур первой генерации колонии Aspergillus ochraceus суспендировали в дистиллированной воде (2 мл) в тест-пробирке; и 0,15 мл аликвоты этой суспензии наносили на каждую из скошенных сред, которые были получены, как описано выше. Эти скошенные среды инкубировали в течение семи дней при 25°С, после чего появлялась поверхностная культура, которая представляла собой белый пушистый мицелий. Обратная сторона была окрашена в оранжевый цвет в своей нижней части, а в верхней части - в желто-оранжевый цвет.
Скошенные культуры первой генерации суспендировали в стерильном растворе (4 мл), содержащем неионогенное поверхностно-активное вещество Твин 80 (3% масс.), и 0,15 мл аликвоты этой суспензии использовали для инокуляции скошенной культуры второй генерации, которая была получена с использованием среды для культивирования, описанной в Таблице 2.
- рН 5,3
- распределение в 1,5-мл-пробирках (180×18 мм)
- стерилизация при 120°С в течение 20 минут
Скошенные культуры второй генерации инкубировали в течение 10 дней при 25°С, в результате чего получали тяжелую массу спор золотистого цвета; обратная сторона была окрашена в коричнево-оранжевый цвет.
Была получена защитная среда, имеющая состав, указанный в Таблице 3.
Культуры от пяти скошенных культур второй генерации суспендировали в защитном растворе (15 мл) в 100 мл колбе. Суспензию разделяли на аликвоты (0,5 мл каждая) по 100×10 мм пробиркам для лиофилизации. Эти культуры предварительно замораживали при температуре от -70°С до -80°С в бане с ацетоном/сухим льдом в течение 20 минут, а затем сразу переносили в осушенную камеру, предварительно охлажденную до температуры от -40°С до -50°С. Предварительно охлажденные аликвоты лиофилизовали при остаточном давлении 50 мкм рт.ст. и при температуре ≤-30°С. По окончании лиофилизации в каждую пробирку с индикатором влажности и герметично запаянную пламенем добавляли две или три гранулы стерильного силикагеля.
Для получения маточной скошенной культуры, пригодной для крупномасштабной ферментации, одну аликвоту лиофилизованнной культуры, которая была получена способом, описанным выше, суспендировали в дистиллированной воде (1 мл) и 0,15 мл аликвоты этой суспензии использовали для инокуляции скошенных культур, которые были получены с использованием среды для культивирования, имеющей состав, указанный в Таблице 2. Эти маточные скошенные культуры инкубировали в течение семи дней при 25°С. По окончании инкубирования культуру, выращенную на скошенном агаре, хранили при 4°С.
Для получения стандартной скошенной культуры культуру, полученную от маточной культуры на скошенном агаре, суспендировали в стерильном растворе (4 мл), содержащем Твин 80 (3% масс.), и полученную суспензию распределяли в 0,15 мл аликвотах по скошенным культурам, которые были покрыты средой для культивирования, описанной в Таблице 2. Стандартные скошенные культуры могут быть использованы для инокуляции первичных посевных колб для лабораторной или промышленной ферментации.
Для приготовления первичной культуры в посевной колбе культуру от стандартной скошенной культуры, которую получали как описано выше, удаляли и суспендировали в растворе (10 мл), содержащем Твин 80 (3% масс.). 0,1-Аликвоту полученной суспензии вводили в 500 мл колбу с перегородкой, содержащую среду для культивирования, имеющую состав, указанный в Таблице 4.
- рН 5,2
- доведение до 5,8 путем добавления 20% NaOH
- распределение в 500 мл колбе с перегородкой, 100 мл
- распределение в круглодонных 2000 мл колбах с перегородками (3 перегородки), 500 мл
- стерилизация при 120°С в течение 20 минут
- рН после стерилизации около 5,7
Посевную колбу инкубировали на вращающемся шейкере (200 об/мин, смещение 5 см) в течение 24 часов при 28°С, в результате чего получали культуру в форме гранулоподобного мицелия с гранулами, имеющими диаметры 3-4 мм. После оценки под микроскопом было обнаружено, что посевная культура является чистой культурой, имеет синнематический рост и большие гифы, хорошо закрученные в спираль. рН суспензии составлял 5,4-5,6. PMV составлял 5-8%, как было определено путем центрифугирования (3000 об/мин × 5 мин).
Культуру для трансформации в колбе получали путем инокуляции среды для культивирования (100 мл), имеющей состав, указанный в Таблице 4, во второй 500 миллилитровой шейкерной колбе, содержащей биомассу (1 мл) из колбы с посевной культурой. Полученную смесь инкубировали на вращающемся шейкере (200 об/мин, смещение 5 см) в течение 18 часов при 28°С. После осмотра культуры было обнаружено, что она содержит гранулоподобный мицелий с гранулами диаметром 3-4 мм. После оценки под микроскопом было обнаружено, что посевная культура является чистой культурой, обнаруживает синнематический и филламентный рост, при котором апикальные (верхушечные) клетки состояли полностью из цитоплазмы, а более старые клетки были немного вакуолизированы. рН суспензии культуры составлял 5-5,2, a PMV определяли путем центрифугирования до 10%-15%. В соответствии с этим, культура оказалась подходящей для трансформации канренона в 11α-гидроксиканренон.
Канренон (1 г) тонко измельчали до образования частиц размером около 5 микрон и суспендировали в стерильной воде (20 мл). К этой суспензии добавляли: 40% (масс./об.) стерильного раствора глюкозы; 16% (масс./об.) стерильного раствора автолизованных дрожжей и стерильный раствор антибиотиков; содержание всех ингредиентов указано для 0 часов реакционного времени в Таблице 5. Раствор антибиотиков получали путем растворения сульфата канамицина (40 мг), тетрациклина·HCl(40 мг) и цефалексина (200 мг) в воде (100 мл). Суспензию стероида, раствор глюкозы и раствор автолизированных дрожжей постепенно добавляли к культуре, содержащейся в шейкерной колбе.
По мере прохождения реакции реакционную смесь периодически анализировали для определения содержания глюкозы и с помощью тонкослойной хроматографии определяли степень превращения в 11α-гидроксиканренон. Во время реакции к смеси для реакции ферментации добавляли дополнительное количество канренонового субстрата в регулируемых количествах для поддержания уровня глюкозы порядка около 0,1% масс. Схема добавления для стероидной суспензии раствора глюкозы, раствора автолизованных дрожжей и раствора антибиотиков представлена в Таблице 5. Реакцию трансформации осуществляли в течение 96 часов при 25°С на роторном шейкере (200 об/мин и смещение 5 см). Во время ферментации рН составлял в пределах 4,5-6. Когда PMV поднимался до 60% или выше, 10 мл часть бульонной культуры удаляли и заменяли 10 мл дистиллированной воды. Исчезновение канренона и появление 11α-гидроксиканренона непрерывно прослеживали в процессе прохождения реакции путем взятия образцов в интервалы времени через 4, 7, 23, 31, 47, 55, 71, 80 и 96 часов после начала цикла ферментации и последующего анализа этих образцов с помощью ТСХ. Прохождение реакции, определенное по этим образцам, показано в Таблице 6.
RF.=0,81
RF.=0,29
Пример 2.
Первичную культуру в посевной колбе получали способом, описанным в Примере 1. Получали питательную смесь, имеющую состав, указанный в Таблице 7
Первоначальную загрузку этой питательной смеси (4 л) вводили в ферментер для трансформации, имеющий геометрический объем 10 л. Этот ферментер имел цилиндрическую конфигурацию и отношение высоты к диаметру, которое составляло 2,58. Он был снабжен турбинным смесителем (400 об/мин), имеющим два дисковых колеса №2 с 6 лопастями каждый. Внешний диаметр этих рабочих колес смесителя составлял 80 мм, каждая лопасть имела радиальный размер 25 мм и высоту 300 мм, причем верхнее колесо располагалось на расстоянии 280 мм ниже от верха сосуда, нижнее колесо располагалось на 365 мм ниже от верха сосуда, а перегородки для сосуда имели высоту 210 мм и располагались радиально во внутренней части на расстоянии 25 мм от внутренней вертикальной стенки сосуда.
Посевную культуру (40 мл) смешивали с питательной загрузкой в ферментере и культуру для трансформации получали путем инкубирования в течение 22 часов при 28°С, при скорости аэрации 0,5 л/л/мин и при давлении 0,5 кг/см2. Через 22 часа PMV культуры составлял 20-25%, а рН=5-5,2.
Получали суспензию, содержащую канренон (80 г) в стерильной воде (400 мл), и 10 мл порцию добавляли в смесь, находящуюся в ферментере для трансформации. Одновременно добавляли 40% (масс./об.) стерильного раствора глюкозы, 16% (масс./об.) стерильного раствора автолизованных дрожжей и стерильный раствор антибиотиков в соотношениях, указанных в Таблице 8 на время реакции 0 часов. Раствор антибиотиков получали способом, описанным в Примере 1.
По мере прохождения реакции реакционную смесь периодически анализировали для определения содержания глюкозы и с помощью тонкослойной хроматографии определяли степень превращения в 11α-гидроксиканренон. Исходя из ТСХ-анализа образцов реакционного бульона, проводимого как описано ниже, к реакционной смеси добавляли дополнительное количество канренона по мере потребления канренонового субстрата. Проводили также мониторинг уровней глюкозы и, когда концентрация глюкозы падала примерно до 0,05% масс. или ниже, добавляли дополнительное количество раствора глюкозы для доведения концентрации примерно до 0,25% масс. Во время реакционного цикла в различные промежутки времени также добавляли питательные вещества и антибиотики. Схема добавления стероидной суспензии, раствора глюкозы, раствора автолизованных дрожжей и раствора антибиотиков представлена в Таблице 8. Реакция трансформации продолжалась 90 часов при скорости аэрации 0,5 объема воздуха на объем жидкости за 1 минуту (об/об/мин) при положительном давлении в головной части 0,3 кг/см2. Температуру поддерживали при 28°С до тех пор, пока PMV не достигнет 45%, после чего температуру снижали до 26°С и эту температуру поддерживали по мере возрастания PMV от 45% до 60%, а затем ее поддерживали при 24°С. Первоначальная скорость размешивания составляла 400 об/мин, а через 40 часов ее повышали до 700 об/мин. рН поддерживали в пределах 4,7-5,3 путем добавления 2М ортофосфорной кислоты или 2М NaOH, как указано. По мере образования пены ее гасили путем добавления нескольких капель пеногасителя SAG 471. В процессе реакции исчезновение канренона и появление 11α-гидроксиканренона прослеживали с интервалами времени в 4 часа с помощью ТСХ-анализа образцов бульона. Когда из бульона исчезало наибольшее количество канренона, то добавляли дополнительные количества канренона.
После добавления всего количества канренона реакцию завершали, когда ТСХ-анализ показывал, что концентрация канренонового субстрата по отношению к 11α-гидроксиканреноновому продукту снижалась примерно до 5%.
При завершении реакционного цикла бульон для ферментации фильтровали через марлевую ткань для отделения мицелия от жидкого бульона. Фракцию мицелия ресуспендировали в этилацетате с использованием около 65 объемов (5,2 литров) на один грамм канренона, загруженного в процессе реакции. Суспензию мицелия в этилацетате нагревали с обратным холодильником в течение одного часа с перемешиванием, а затем охлаждали примерно до 20°С и фильтровали на воронке Бюхнера. Мицелиальный осадок последовательно промывали этилацетатом (5 об. на один грамм канреноновой загрузки; 0,4 л) и деионизованной водой (500 мл) для удаления этилацетатного экстракта из осадка. Осадок на фильтре отбрасывали. Обогащенный экстракт, промывку растворителем и водную промывку собирали в сепараторе, а затем оставляли отстаиваться на 2 часа для разделения фаз.
Затем водную фазу отбрасывали, а органическую фазу концентрировали в вакууме до получения остаточного объема 350 мл. Кубовый остаток охлаждали до 15°С и эту температуру поддерживали при перемешивании в течение около одного часа. Полученную суспензию фильтровали для удаления кристаллического продукта, а фильтровальный осадок промывали этилацетатом (40 мл). После сушки определяли выход 11α-гидроксиканренона, который составлял 60 г.
Пример 3.
Суспензию спор получали из стандартной скошенной культуры способом, описанным в Примере 1. В 2000-миллилитровую круглодонную колбу с перегородками (3 перегородки, каждая размером 50 мм × 30 мм), содержащую питательный раствор (500 мл), имеющий состав, указанный в Таблице 4, вводили аликвоту (0,5 мл) суспензии спор. Полученную смесь инкубировали в колбе в течение 24 часов при 25°С на шейкере периодического действия (120 встряхиваний в мин; смещение 5 см), в результате чего получали культуру, которая, как было установлено после ее наблюдения под микроскопом, представляла собой чистую культуру с гифами, хорошо закрученными в спираль. рН культуры составлял около 5,3-5,5, a PMV (как было определено путем центрифугирования при 3000 об/мин в течение 5 мин) составлял 8-10%.
С использованием полученной таким образом культуры посевную культуру получали в ферментере из нержавеющей стали, имеющем вертикальную цилиндрическую конфигурацию, геометрический объем 160 л и относительный формат 2,31 (высота =985 мм, диаметр =425 мм). Этот ферментер был снабжен турбинным смесителем (400 об/мин), имеющим два дисковых рабочих колеса, каждое из которых имело 6 лопастей с внешним диаметром 240 мм, а каждая лопасть имела радиальный размер 80 мм и высоту 50 мм. Верхнее рабочее колесо располагалось на глубине 780 мм от верха ферментера, а второе рабочее колесо располагалось на глубине 995 мм от верха ферментера. Вертикальные перегородки имели высоту 890 мм и располагались радиально внутри на расстоянии 40 мм от внутренней вертикальной стенки ферментера. Смеситель работал на скорости 170 об/мин. Сначала в ферментер вводили питательную смесь (100 л), имеющую состав, указанный в Таблице 9, а затем вводили порцию пре-инокулята (1 л), полученного как описано выше и имеющего рН 5,7.
Инокулированную смесь инкубировали в течение 22 часов при скорости аэрации 0,5 л/л/мин и при давлении в головной части 0,5 кг/см2. Температуру поддерживали при 28°С до тех пор, пока PMV не достигал 25%, после чего температуру снижали до 25°С. рН поддерживали в пределах 5,1-5,3. Увеличение объема мицелия показано в Таблице 10, в которой также указаны рН и профили растворенного кислорода в реакции посевной культуры.
С использованием полученной таким образом посевной культуры цикл ферментации для трансформации осуществляли в вертикальном цилиндрическом ферментере из нержавеющей стали, имеющем диаметр 1,02 м, высоту 1,5 м и геометрический объем 1,4 м3. Этот ферментер был снабжен турбинным смесителем, имеющим два рабочих колеса, одно из которых располагалось на глубине 867 мм от верха реактора, а другое располагалось на глубине 1435 мм от верха реактора. Каждое рабочее колесо имело шесть лопастей, каждая из которых имела радиальный размер 95 см и высоту 75 см. Вертикальные перегородки имели высоту 1440 мм и располагались радиально внутри реактора на расстоянии 100 см от внутренней вертикальной стенки ферментера. Была получена питательная смесь (100 л), имеющая состав, указанный в Таблице 11:
Начальную загрузку (700 л) этой питательной смеси (рН=5,7) вводили в ферментер, а затем вводили посевной инокулят данного примера (7 л), полученный как описано выше.
Питательную смесь, содержащую инокулят, инкубировали в течение 24 часов при скорости аэрации 0,5 л/л/мин и при давлении в головной части 0,5 кг/см2. Температуру поддерживали при 28°С, а скорость перемешивания составляла 110 об/мин. Увеличение объема мицелия показано в Таблице 12, в которой также указаны рН и профили растворенного кислорода в реакции посевной культуры.
По окончании инкубирования наблюдалось осаждение мицелия, но этот осадок был, в основном, небольшим и относительно рыхлым. Диффузный мицелий суспендировали в бульоне. Конечный рН составлял 5,1-5,3.
К полученной таким образом культуре для трансформации добавляли суспензию канренона (1,250 кг; тонко измельченного до размера частиц в 5 микрон) в стерильной воде (5 л). Стерильный раствор добавок и раствор антибиотиков добавляли в соотношениях, указанных в Таблице 14 на время реакции 0. Состав раствора добавок представлен в Таблице 13.
Биологическую трансформацию осуществляли в течение около 96 часов при скорости аэрации 0,5 л/л/мин и при давлении в головной части 0,5 кг/см2, а рН составлял 4,7-5,3, который корректировали, если это это необходимо, путем добавления 7,5М NaOH или 4М Н3PO4. Первоначальная скорость аэрации составляла 100 об/мин, после чего через 40 часов ее повышали до 165 об/мин, а через 64 часа до 250 об/мин. Первоначальная температура составляла 28°С, а затем, когда PMV достигал 45%, температуру понижали до 26°С, а когда PMV увеличивался до 60%, то температуру понижали до 24°С. Для регулирования пенообразования, если это необходимо, добавляли несколько капель SAG 471. Уровни глюкозы в процессе реакции ферментации прослеживали с интервалами времени в 4 часа и, когда концентрация глюкозы падала ниже 1 г/л, в эту партию добавляли дополнительное количество стерильного раствора добавок (10 л). Исчезновение канренона и появление 11α-гидроксиканренона прослеживали в процессе реакции с помощью ВЭЖХ. Когда по крайней мере 90% исходного канренона превращалось в 11α-гидроксиканренон, добавляли еще 1,250 кг канренона. Когда в этом добавленном количестве обнаруживалось 90%-ное превращение канренона, то вводили еще 1,250 кг канренона. С использованием того же самого критерия добавляли дополнительные количества канренона (порции 1,250 кг) до тех пор, пока в реактор не будет введена вся загрузка (20 кг). После добавления всего количества канренона реакцию завершали, когда концентрация непрореагировавшего канренонового субстрата по отношению к количеству продуцированного 11α-гидроксиканренона составляла 5%. Схема добавления канренона, стерильного раствора добавок и раствора антибиотиков показана в Таблице 14.
Когда биологическое превращение было завершено, мицелий выделяли из бульона путем центрифугирования в корзиночной центрифуге. Как было определено с помощью ВЭЖХ, фильтрат содержал только 2% от полного количества 11α-гидроксиканренона в бульоне для сбора и, следовательно, был удален. Затем мицелий суспендировали в этилацетате (1000 л) в резервуаре для экстракции емкостью 2 м3. Эту суспензию нагревали в течение одного часа при перемешивании и в условиях нагревания с обратным холодильником с этилацетатом, а затем охлаждали и центрифугировали в корзиночной центрифуге. Осадок мицелия промывали этилацетатом (200 л), а затем сливали. Экстракт из растворителя, обогащенного стероидом, оставляли на один час для отделения водной фазы. Водную фазу экстрагировали дополнительным количеством этилацетатного растворителя (200 л), а затем отбрасывали. Объединенные фазы растворителя осветляли путем центрифугирования и помещали в концентратор (геометрический объем 500 л), а затем концентрировали в вакууме до остаточного объема 100 л. При выпаривании первоначальная загрузка в концентратор объединенного экстракта и промывочных растворов составляла 100 л и этот объем поддерживали постоянным путем непрерывного или периодического добавления объединенного раствора по мере улетучивания растворителя. После завершения стадии выпаривания кубовые остатки охлаждали до температуры 20°С и перемешивали в течение двух часов, а затем фильтровали на фильтре Бюхнера. Сосуд-концентратор промывали этилацетатом (20 л) и этот промывочный раствор затем использовали для промывания осадка на фильтре. Полученный продукт сушили в вакууме в течение 16 часов при 50°С. Выход 11α-гидроксиканренона составлял 14 кг.
Пример 4.
Лиофилизованные споры Aspergillus ochraceus NRRL 405 суспендировали в среде для культивирования с жидким кукурузным экстрактом (2 мл), имеющей состав, указанный в Таблице 15:
дистиллированная вода - достаточное количество до 1000 мл рН 4,6, доведенный до рН 6,5 путем добавления 20% NaOH;
распределение 50 мл по 250 мл колбам Эрленмейера;
стерилизация при 121°С в течение 20 минут.
Полученную суспензию использовали в инокуляте для размножения спор на агаровых чашках. Приготавливали десять чашек с агаром, каждая из которых содержала твердую среду для культивирования: глюкоза/дрожжевой экстракт/фосфат/агар, имеющую состав, указанный в Таблице 16:
дистиллированная вода, достаточное количество до 1000 мл, доведение рН до 6,5, стерилизация при 121°С, 30 минут
0,2 мл аликвоту суспензии переносили на поверхность каждой чашки. Эти чашки инкубировали в течение десяти дней при 25°С, после чего споры со всех чашек собирали в стерильную криогенную защитную среду, имеющую состав, указанный в Таблице 17:
дистиллированная вода, достаточное количество до 1000 мл, стерилизация при 121°С, 30 минут
Полученную суспензию распределяли по 20 сосудам, причем в каждый сосуд переносили 1 мл суспензии. Эти сосуды содержали основной фонд клеток, которые могут быть использованы для продуцирования рабочего фонда клеток в целях генерирования инокулята для биологического превращения канренона в 11α-гидроксиканренон. Эти сосуды, содержащие основной банк клеток, хранили в морозильной камере в паровой фазе жидкого азота при -130°С.
Для начала продуцирования рабочего фонда клеток споры из одного сосуда с исходным фондом клеток ресуспендировали в стерильной среде для культивирования (1 мл), имеющей состав, указанный в Таблице 15. Эту суспензию разделяли на десять 0,2 мл аликвот и каждую аликвоту использовали для инокуляции агаровой чашки с твердой средой для культивирования, имеющей состав, указанный в Таблице 16. Эти чашки инкубировали в течение десяти дней при 25°С. На третий день инкубирования нижняя часть среды для культивирования имела коричнево-оранжевую окраску. По окончании инкубирования наблюдалось интенсивное продуцирование спор, имеющих золотистую окраску. Споры из каждой чашки собирали способом, описанным выше для получения исходного клеточного фонда. Всего приготавливали одну сотню сосудов, каждый из которых содержал 1 мл суспензии. Эти сосуды содержали рабочий клеточный фонд. Сосуды с рабочим клеточным фондом также хранили в морозильной камере в паровой фазе жидкого азота при -130°С.
Среду для культивирования (50 мл), имеющую состав, указанный в Таблице 15, загружали в 250 мл колбу Эрленмейера. В колбу вводили аликвоту (0,5 мл) рабочей клеточной суспензии и смешивали со средой для культивирования. Инокулированную смесь инкубировали в течение 24 часов при 25°С для продуцирования первичной посевной культуры, имеющей объем осажденного мицелия приблизительно 45%. После визуального наблюдения за культурой было обнаружено, что она содержит гранулоподобный мицелий диаметром от 1 до 2 мм; а после наблюдения под микроскопом, она была идентифицирована как чистая культура.
Культивирование вторичной посевной культуры было инициировано путем введения среды для культивирования, имеющей состав, указанный в Таблице 15, в 2,8-литровую колбу Фернбаха с последующей инокуляцией среды частью (10 мл) первичной посевной культуры этого примера, получение которой было описано выше. Эту инокулированную смесь инкубировали при 25°С в течение 24 часов на роторном шейкере (200 об/мин, смещение - 5 см). По окончании инкубирования культура обладала теми же свойствами, которые были описаны выше для первичной посевной культуры, и эта культура может быть использована для реакции трансформации путем ферментации, при которой канренон биологически превращается в 11α-гидроксиканренон.
Трансформация была проведена в ферментере Biostat Braun Е, описание которого представлено ниже:
Канренон при концентрации 20 г/л суспендировали в деионизованной воде (4 л) и часть (2 л) среды для культивирования, имеющей состав, указанный в Таблице 18, добавляли во время размешивания смеси в ферментере при 300 об/мин.
деионизованная вода дост.кол. до 7,5 л
- стерилизация пустого ферментера в течение 30 мин при 121°С
Полученную суспензию размешивали в течение 15 минут, после чего объем доводили до 7,5 л путем добавления деионизованной воды. На этой стадии рН суспензии доводили до 5,2-6,5 путем добавления 20% масс. раствора NaOH, после чего суспензию стерилизовали путем нагревания при 121°С в течение 30 минут в ферментере Braun Е. рН после стерилизации составлял 6,3±0,2, а конечный объем составлял 7,0 л. Стерилизованную суспензию инокулировали частью (0,5 л) вторичной посевной культуры этого примера, которая была получена, как описано выше, и объем доводили до 8,0 л путем добавления 50% стерильного раствора глюкозы. Ферментацию проводили при температуре 28°С до тех пор, пока PMV не достигал 50%, после чего температуру понижали до 26°С, а затем, когда PMV превышал 50%, температуру снижали до 24°С для поддержания постоянного уровня PMV примерно ниже 60%. Воздух вводили через барботер при скорости 0,5 об/об/мин, исходя из первоначального объема жидкости, а давление в ферментере поддерживали на отметке 700 миллибар. Перемешивание начинали со скорости 600 об/мин, а затем, если это необходимо, постепенно повышали до 1000 об/мин для поддержания уровня растворенного кислорода свыше 30% об. Концентрацию глюкозы непрерывно прослеживали. После того, как первоначально высокая концентрация глюкозы падала ниже 1%, благодаря ее поглощению во время реакции ферментации, добавляли дополнительное количество глюкозы в виде 50%-ного стерильного раствора глюкозы для поддержания ее концентрации в пределах 0,05%-1% в течение всего оставшегося цикла для данной партии. Перед инокуляцией рН составлял 6,3±0,2. После снижения рН до около 5,3 в течение начального периода ферментации, рН поддерживали в пределах 5,5±0,2 в течение оставшегося цикла путем добавления гидроксида аммония. Пенообразование регулировали путем добавления полиэтиленгликолевого пеногасителя, имеющегося в продаже под торговым знаком SAG 471 OSI Specialties, Inc.
Рост культуры наблюдался в первые 24 часа цикла, в течение которых PMV составлял около 40%, рН составлял около 5,6, а содержание растворенного кислорода составляло около 50% об. Превращение канренона начиналось даже во время роста культуры. Концентрации канренона и 11α-гидроксиканренона прослеживали во время биологического превращения путем ежедневного анализа образцов. Образцы экстрагировали горячим этилацетатом и полученный раствор образца анализировали с помощью ТСХ и ВЭЖХ. Биологическое превращение считалось завершенным, если концентрация остаточного канренона составляла около 10% от первоначальной концентрации. Приблизительное время превращения составляло 110-130 часов.
После завершения биологического превращения мицелиальную биомассу выделяли из бульона путем центрифугирования. Супернатант экстрагировали равным объемом этилацетата, а водный слой отбрасывали. Мицелиальную фракцию ресуспендировали в этилацетате с использованием приблизительно 65 объемов на один грамм канренона, загруженного в реактор для ферментации. Затем мицелиальную суспензию нагревали с обратным холодильником в течение одного часа, помешивая при этом, а затем охлаждали до около 20°С и фильтровали на воронке Бюхнера. Мицелиальный осадок на фильтре дважды промывали 5 объемами этилацетата на один грамм канренона, загруженного в ферментер, а затем промывали деионизованной водой (1 л) для удаления остаточного этилацетата. Водный экстракт, обогащенный растворитель, промывку растворителем и водную промывку объединяли. Оставшийся истощенный мицелиальный осадок либо отбрасывали, либо снова экстрагировали в зависимости от результата анализа на присутствие в нем остаточных стероидов. Объединенные жидкие фазы оставляли на два часа для отстаивания. А органическую фазу концентрировали в вакууме до тех пор, пока остаточный объем не становился равным приблизительно 500 мл. Затем сосуд аппарата охлаждали до около 15°С путем медленного перемешивания примерно в течение около одного часа. Кристаллический продукт выделяли путем фильтрации и промывали охлажденным этилацетатом (100 мл). Растворитель удаляли из кристаллов путем выпаривания и кристаллический продукт сушили в вакууме при 50°С.
Пример 5.
Лиофилизованные споры Aspergillus ochraceus ATCC 18500 суспендировали в культуральной среде с жидким кукурузным экстрактом (2 мл), как описано в Примере 4. Десять агаровых чашек получали способом, также описанным в Примере 4. Эти чашки инкубировали и споры собирали, как описано в Примере 4 для получения основного клеточного фонда. Сосуды, содержащие основной клеточный фонд, хранили в морозильной камере в паровой фазе жидкого азота при -130°С.
Из сосуда с основным клеточным фондом получали рабочий клеточный фонд, как описано в Примере 4, и хранили в морозильной камере в жидком азоте при -130°С.
Среду для культивирования (300 мл), имеющую состав, указанный в Таблице 19, загружали в 2 л колбу с перегородками. В эту колбу вводили аликвоту (3 мл) рабочей клеточной суспензии. Инокулированную смесь инкубировали в течение 20-24 часов при 28°С на вращающемся шейкере (200 об/мин, смещение 5 см) с продуцированием первичной посевной культуры, которая имела объем осажденного мицелия приблизительно 45%. После визуального осмотра было обнаружено, что эта культура содержит гранулоподобный мицелий с диаметром гранул 1-2 мм; а после наблюдения под микроскопом было установлено, что эта культура является чистой.
Культивирование вторичной посевной культуры инициировали путем введения 8 л среды для культивирования, имеющей состав, указанный в Таблице 19, в 14-литровый стеклянный ферментер. Этот ферментер инокулировали 160-200 миллилитрами первичной посевной культуры этого примера. Получение этой культуры описано выше.
Инокулированную смесь культивировали при 28°С в течение 18-20 часов при размешивании со скоростью 200 об/мин и при скорости аэрации 0,5 об/об/мин. По окончании культивирования культура имела те же самые свойства, что и описанная выше первичная культура.
Трансформацию проводили в 60 л ферментере, в основном способом, описанным в Примере 4, за исключением того, что среда для культивирования имела состав, указанный в таблице 20, а первоначальная загрузка вторичной посевной культуры составляла 350-700 мл. Скорость перемешивания сначала составляла 200 об/мин, но затем ее увеличивали до 500 об/мин из-за необходимости поддерживать уровень растворенного кислорода выше 10% об. Приблизительное время биологического превращения для 20 г/л канренона составляло 80-160 часов.
Пример 6.
С использованием суспензии спор из рабочего клеточного фонда, продуцированного способом, описанным в Примере 4, были получены первичная и вторичная посевные культуры, также, в основном, способом, описанным в Примере 4. С использованием вторичной посевной культуры, продуцированной этим способом, осуществляли два цикла биологической трансформации в соответствии с модифицированным способом, проиллюстрированным на Фиг.1, и осуществляли два цикла биологической трансформации способом, проиллюстрированным на Фиг.2. Культуральная среда для трансформации, схема добавления канренона, время сбора и степень превращения для этих циклов указаны в Таблице 21. В цикле R2A использовали схему добавления канренона, в основном, описанную Примере 3, тогда как в цикле R2C использовали модифицированную схему Примера 3, только с двумя добавлениями канренона, одно в начале цикла, а другое через 24 часа. В циклах R2B и R2D всю загрузку канренона вводили в начале цикла и этот процесс осуществляли, в основном, способом, описанным в Примере 4, за исключением того, что перед введением в ферментер загрузку канренона стерилизовали в отдельном сосуде, а глюкозу добавляли по мере прохождения реакции данной партии. Для минимизации образования комков, образующихся после стерилизации, использовали гомогенизатор Уоринга. В циклах R2A и R2B канренон вводили в загрузочную партию в растворе метанола и, в этом отношении, эти циклы также отличались от циклов, описанных в Примерах 3 и 4, соответственно.

В циклах R2A и R2B концентрация метанола в забродившей среде аккумулировалась до около 6,0% и эта концентрация, как было обнаружено, ингибировала рост культуры и биологическое превращение. Однако, исходя из результатов проведения этих циклов, был сделан вывод, что метанол или другой смешивающийся с водой растворитель может быть с успехом использован при более низких концентрациях для увеличения загрузки канренона и обеспечения присутствия канренона в виде тонкоизмельченного осадка, что приводит к увеличению площади межфазной поверхности и способствует доставке канренона в зону реакции.
Канренон является стабильным при температуре стерилизации (121°С), но образует комки. Для дробления этих комков на мелкие частицы используют гомогенизатор Уоринга, что способствует успешному превращению канренона в нужный продукт.
Пример 7.
С использованием суспензии спор из рабочего клеточного фонда, продуцированного способом, описанным в Примере 4, были получены первичная и вторичная посевные культуры, также, в основном, способом, описанным в Примере 4. Описание и результаты Примера 7 представлены в Таблице 22. С использованием вторичной посевной культуры, продуцированнной этим способом, один цикл биологической трансформации (R3C) осуществляли, в основном, способом, описанным в Примере 3, а три цикла биологической трансформации осуществляли способом, описанным в общих чертах в Примере 5. В последних трех циклах (R3A, R3B и R3D) канренон стерилизовали в портативном резервуаре вместе со средой для культивирования, не содержащей глюкозы. Глюкозу подавали из другого резервуара в условиях асептики. Стерилизованную суспензию канренона вводили в ферментер либо до инокуляции, либо во время ранней стадии биологического превращения. В цикле R3B дополнительное количество стерильного канренона и среду для культивирования вводили через 46,5 часа. Комки канренона, образованные после стерилизации, дробили с помощью гомогенизатора Уоринга, что способствовало продуцированию тонкодисперсной суспензии, вводимой в ферментер. Трансформирующая среда для культивирования, схема добавления канренона, схема добавления питательных веществ, время сбора и степень превращения для этих циклов представлены в Таблицах 22 и 23.
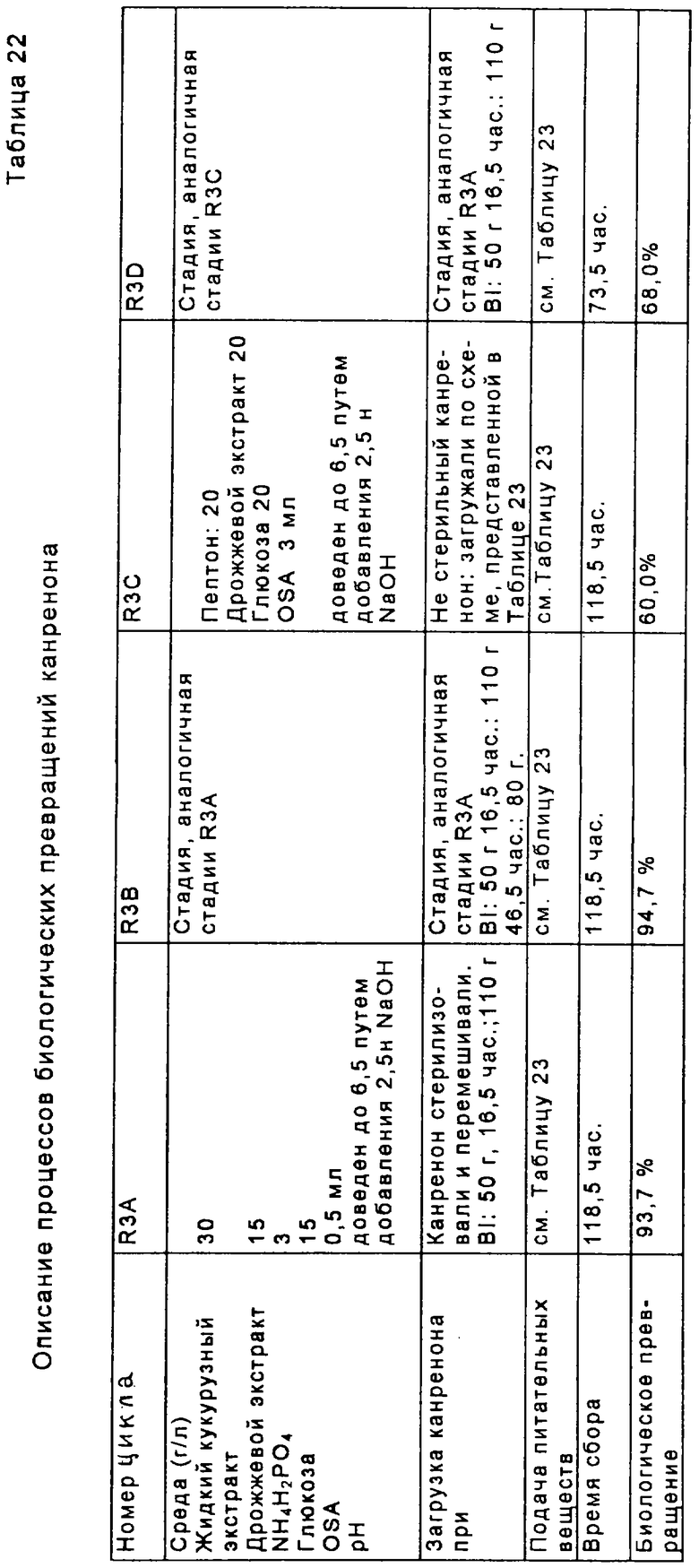
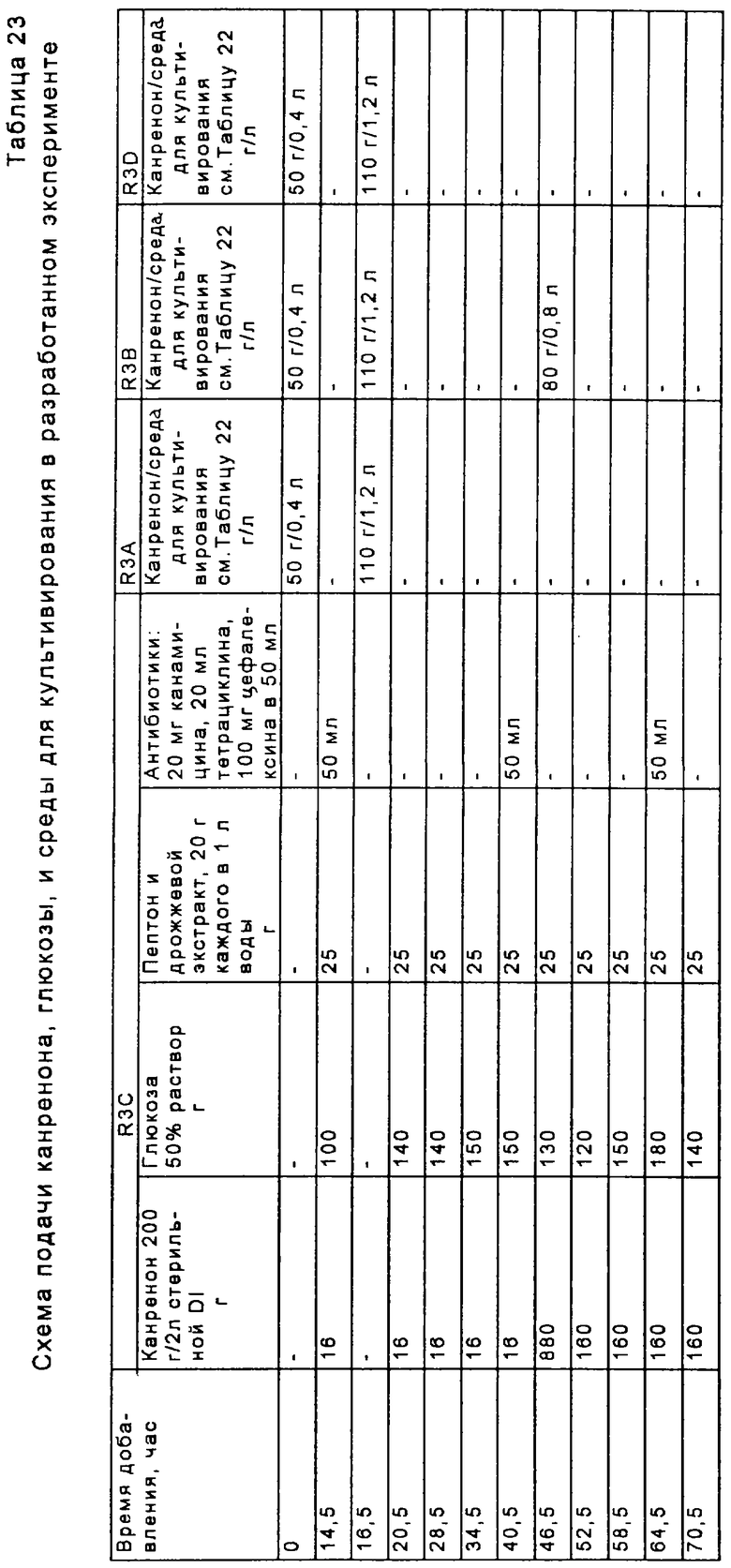
Благодаря росту филаментов, в ферментере наблюдался в высокой степени вязкий бульон во всех четырех циклах этого Примера. Для устранения препятствий, которые создает высокая вязкость в отношении аэрации, смешивания, регулирования рН и регулирования температуры, во время этих циклов увеличивали скорость аэрации и скорость размешивания. Реакции превращения проходили удовлетворительно даже в более жестких условиях, но на поверхности жидкого бульона образовывался плотный осадок. Благодаря этому осадку, некоторое количество непрореагировавшего канренона было выведено из бульона.
Пример 8.
Описание и результаты Примера 8 систематизированы в Таблице 24. Были осуществлены четыре цикла ферментации, в которых 11α-гидроксиканренон был продуцирован путем биологического превращения канренона. В двух из этих циклов (R4A и R4D) биологическое превращение осуществляли, в основном, тем же способом, который был описан для циклов R3A и R3D в Примере 6. В цикле R4C канренон был превращен в 11α-гидроксиканренон, в основном, тем же самым способом, который был описан в Примере 3. В цикле R4B процесс осуществляли, в основном, способом, описанным в Примере 4, то есть со стерилизацией канренона и среды для культивирования в ферментере непосредственно перед инокуляцией; все азотные и фосфорные питательные вещества вводили в начале цикла, а дополнительный раствор, содержащий только глюкозу, вводили в ферментер для поддержания уровня глюкозы по мере прохождения цикла. В последнем процессе (цикл R4B) концентрацию глюкозы прослеживали через каждые 6 часов и раствор глюкозы добавляли в указанном количестве для поддержания уровней глюкозы в пределах от 0,5 до 1%. Схемы добавления канренона для этих циклов представлены в Таблице 25.

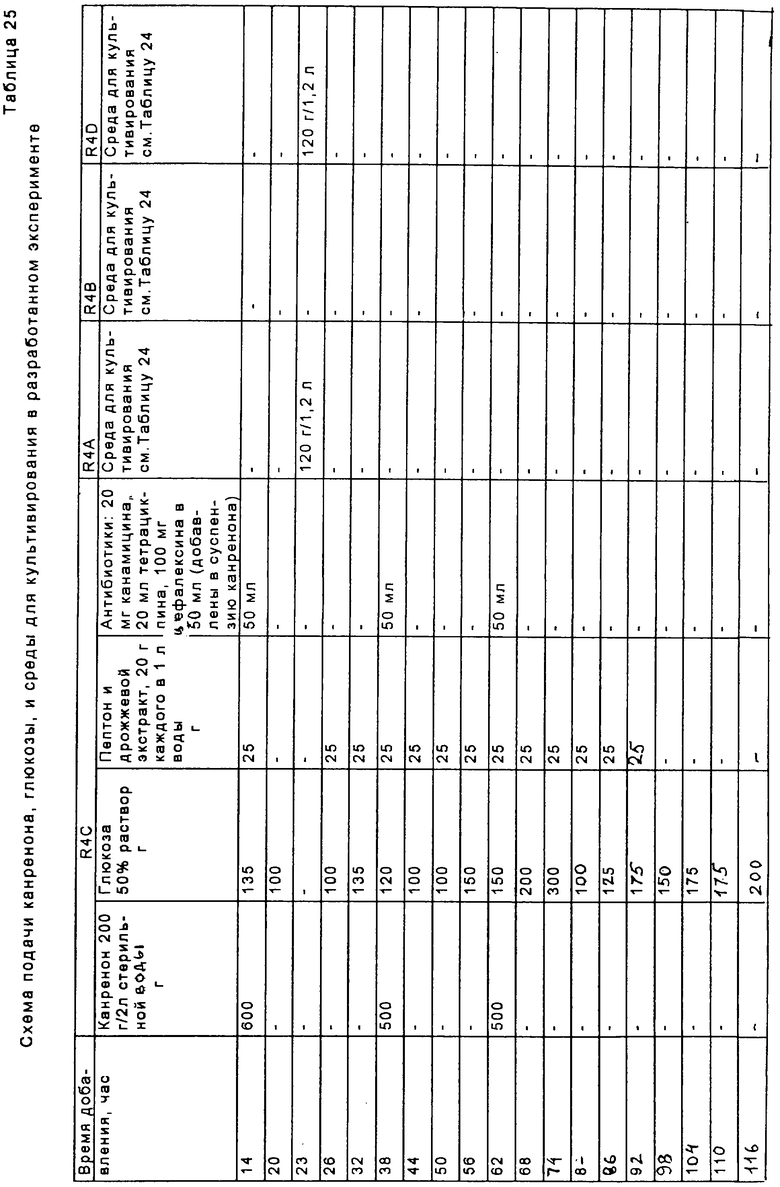
Во всех ферментерах проводили высокоскоростное размешивание и аэрацию в течение большей части цикла ферментации, поскольку забродившая среда становилась в высокой степени вязкой уже через один день и далее после инокуляции.
Пример 9.
Трансформирующие среды для культивирования, схемы добавления канренона, время сбора и степень превращения для циклов в этом Примере представлены в Таблице 26.
Четыре цикла биологического превращения проводили, в основном, способом, описанным для цикла R4B в Примере 8, за исключением процедур, описанных ниже. В цикле R5B верхнее рабочее колесо турбинного дискового смесителя, используемого для перемешивания в других циклах, заменяли морским ротором для накачки сверху вниз. Действие накачки сверху вниз обеспечивает аксиальную подачу бульона в центр ферментера и способствует снижению образования плотного осадка. Метанол (200 мл) добавляли сразу после инокуляции в цикле R5D. Поскольку канренон в ферментере был стерилизован, то все питательные вещества, за исключением глюкозы, подавали в начале цикла, что позволяло избежать необходимости цепной подачи источников азота, источников фосфора или антибиотиков.

Для обеспечения смачивания твердой фазы, выступающей над поверхностью жидкости, в каждый ферментер через 96 часов после начала цикла добавляли среду для культивирования (2 л). Проблемы, связанные с размешиванием, могут быть, в целом, преодолены либо путем добавления среды для культивирования, либо путем использования рабочего колеса насоса с накачкой сверху вниз (цикл R5B), но результаты этих циклов указывают на целесообразность и преимущества этого способа и свидетельствуют о том, что достаточное размешивание может быть осуществлено в соответствии со стандартной практикой.
Пример 10.
Три цикла биологического превращения проводили, в основном, способом, описанным в Примере 9. Трансформирующие среды для культивирования, схемы добавления канренона, время сбора и степень превращения для циклов в этом Примере представлены в Таблице 27.
Среду для культивировании (1,3 л) и стерильную воду (0,8 л) добавляли через 71 час в цикле R6A для пропитки плотного осадка мицелия, выступающего над поверхностью жидкого бульона. Для тех же самых целей среду для культивирования (0,5 л) и стерильную воду (0,5 л) добавляли через 95 часов в цикле R6B. Данные о равновесии материалов показали, что массовое равновесие лучше определять в том случае, когда уровень плотного осадка, выступающего над поверхностью жидкости, минимизирован.
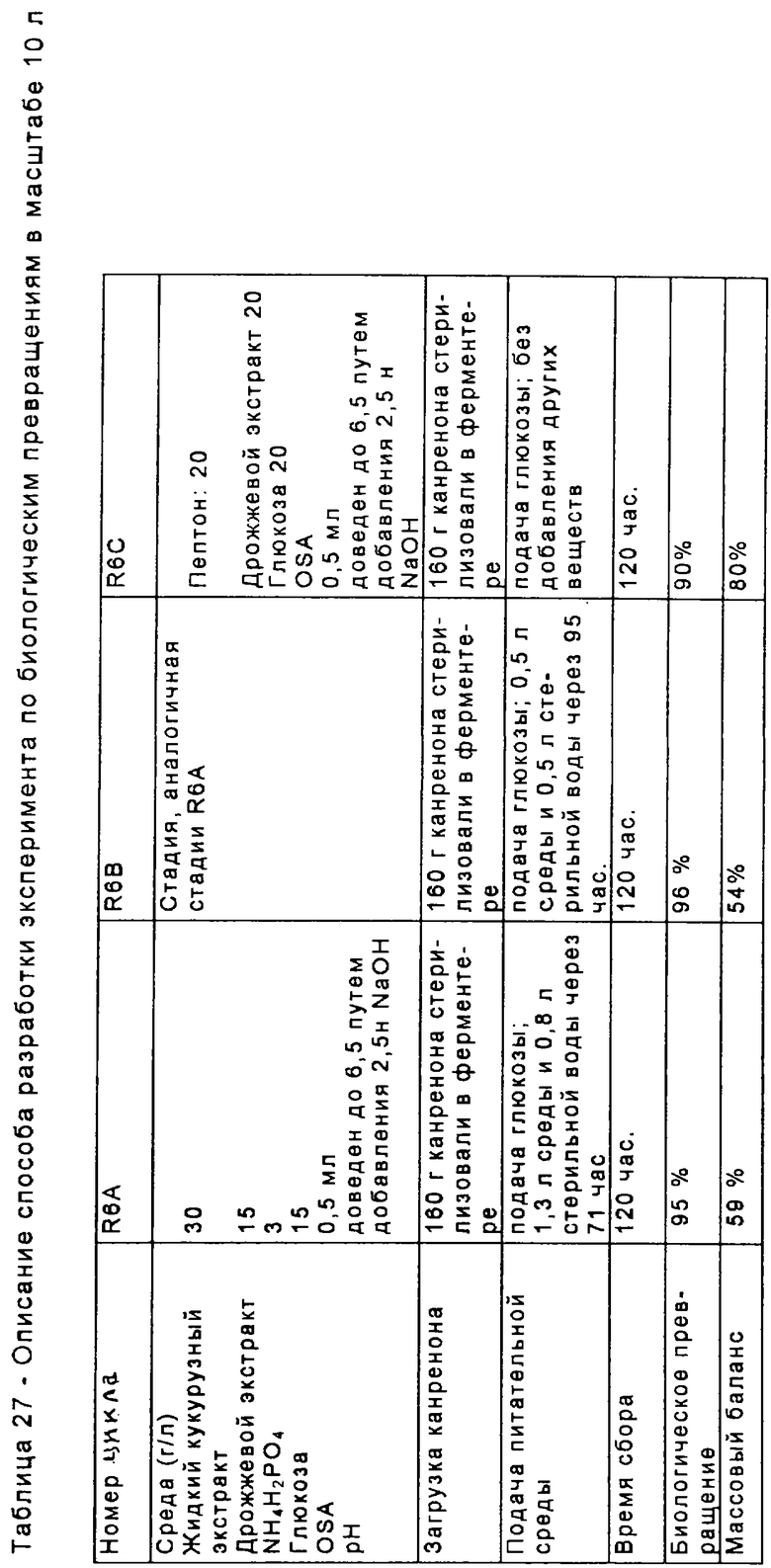
Пример 11.
Были проведены циклы ферментации для сравнения предварительной стерилизации канренона со стерилизацией канренона и среды для культивирования в ферментере для трансформации. Цикл R7A осуществляли способом, проиллюстрированным на Фиг.2, в условиях, сравнимых с условиями циклов R2C, R2D, R3A, R3B, R3D, R4A и R4D. Цикл R7B проиллюстрирован на Фиг.3 в условиях, сравнимых с условиями циклов, проводимых как описано в Примерах 4, 9 и 10 и цикла R4B. Трансформирующие среды для культивирования, схемы добавления канренона, время сбора и степени превращения для циклов в этом Примере представлены в Таблице 28.
Массовый баланс, определенный исходя из конечного образца, взятого из партии R7B, составлял 83%, что указывало на то, что значительной потери или разложения субстрата при биологическом превращении не наблюдалось. Было установлено, что размешивание для обоих циклов было адекватным.
Остаточная концентрация глюкозы превышала контрольный диапазон желательно на 5-10 г/литр в первые 80 часов реакции. Рыхлый осадок, который аккумулировался в головной части обоих ферментеров, не оказывал какого-либо заметного неблагоприятного влияния на производительность цикла.
Пример 12.
Эффективность экстракции определяли в серии циклов 1 л экстракции и систематизировали в Таблице 29. В каждом из этих циклов стероиды экстрагировали из мицелия с использованием этилацетата (объем ферментации 1 л/л). В каждом цикле проводили две последовательные экстракции. Исходя из ОФ-ВЭЖХ, при первой экстракции было выделено около 80% всего стероида; а при второй экстракции выход стероида увеличивался до 95%. При третьей экстракции должно было быть выделено еще 3% стероида. Остальные 2% стероида составляли потери в водной фазе надосадочной жидкости. Экстракт упаривали досуха в вакууме, но промывку каким-либо дополнительным растворителем не проводили. Обработка растворителем должна способствовать улучшению выхода начальной экстракции с точки зрения экономических затрат.
Метилизобутилкетон (MIBK) и толуол оценивали в качестве растворителей экстракции/кристаллизации для 11α-гидроксиканренона в масштабе 1 л бульона. С использованием вышеописанной схемы экстракции MIBK и толуол были сравнимы с этилацетатом как по эффективности экстракции, так и по продуктивности кристаллизации.
Пример 13.
В качестве частичной оценки способов, проиллюстрированных на Фиг.2 и 3, исследование размера частиц проводили с использованием канренонового субстрата, добавляемого в начале цикла ферментации в каждом из этих способов. Как описано выше, в способе, проиллюстрированном на Фиг.1, канренон перед его введением в ферментер тонко измельчали, но не стерилизовали, при этом рост нежелательных микроорганизмов подавляли путем добавления антибиотиков. В способах, проиллюстрированных на Фиг.2 и 3, канренон стерилизовали перед проведением реакции. В способе, проиллюстрированном на Фиг.2, канренон перед его введением в ферментер стерилизовали в смесителе. В способе, проиллюстрированном на Фиг.3, суспензию канренона в среде для культивирования стерилизовали в ферментере в начале цикла. Как обсуждалось выше, стерилизация может приводить к агломерации частиц канренона. Из-за ограниченной растворимости канренона в водной среде для культивирования продуктивность способа зависит от переноса массы из твердой фазы, а поэтому можно ожидать, что она зависит от площади межфазной поверхности, определяемой площадью поверхности макрочастиц твердого субстрата, которая, в свою очередь, зависит от гранулометрического состава этих частиц. Эти соображения служат, главным образом, в качестве сдерживающих факторов в способах, проиллюстрированных на Фиг.2 и 3.
Однако, было обнаружено, что размешивание в смесителе, Фиг.2, и в бродильном чане, Фиг.3, вместе с действием сдвигового насоса, используемого для подачи партии, Фиг.2, способствует разрушению этих агломератов на частицы с размером, достаточным для того, чтобы можно было осуществлять подачу нестерилизованного и тонко измельченного канренона в цикл, показанный на Фиг.1. Это было проиллюстрировано путем оценки гранулометрического состава для частиц канренона, присутствующих в начале реакционного цикла каждого из трех способов. См. Таблицу 30 и Фиг.4 и 5.
93,1% (120 ч)
R4C:
96,3% (120 ч)
94,6% (120 ч)
R4B:
95,2% (120 ч)
97,6% (120 ч)
R5B:
93,8% (120 ч)
Исходя из данных Таблицы 30, следует отметить, что смесители и насос со сдвиговым действием являются эффективными для снижения среднего размера частиц стерилизованного канренона до того же порядка величины, который характерен для нестерилизованного субстрата, но, при этом, все же остается значительное различие в размерах частиц, которое свидетельствует в пользу нестерилизованного субстрата. Несмотря на это различие, данные продуктивности реакции указывают на то, что способ с проведением предварительной стерилизации является, по крайней мере, таким же продуктивным, как и способ, проиллюстрированный на Фиг.1. Дополнительные преимущества могут быть реализованы в способе, проиллюстрированном на Фиг.2, с использованием нескольких стадий для дальнейшего снижения и регулирования размера частиц, например путем мокрого размола стерилизованного канренона и/или путем пастеризации вместо стерилизиции.
Пример 14.
Посевную культуру получали способом, описанным в Примере 5. Через 20 часов мицелий в ферментере с инокулятом имел мягкую консистенцию с 40% PMV. Его рН составлял 5,4, а 14,8 г/л глюкозы оставалось неутилизованным.
Была получена трансформирующая среда для культивирования (35 л), имеющая состав, указанный в Таблице 20. Для получения питательной среды глюкозу и дрожжевой экстракт стерилизовали отдельно и смешивали с получением однородной питательной массы с начальной концентрацией 30% масс. глюкозы и 10% масс. дрожжевого экстракта. рН питательной массы доводили до 5,7.
С использованием этой среды (Таблица 20) были проведены два цикла биологического превращения канренона в 11α-гидроксиканренон. Каждый из этих циклов проводили в 60-литровом ферментере, снабженном смесителем, содержащим одно турбинное рабочее колесо Rushton и два турбинных рабочих колеса Lightnin А315.
Первоначальная загрузка среды для культивирования в ферментер составляла 35 л. Тонко измельченный и нестерилизованный канренон добавляли до начальной концентрации 0,5%. Среду в ферментере инокулировали посевной культурой, полученной как описано в Примере 5 при начальной степени инокуляции 2,5%. Ферментацию осуществляли при температуре 28°С, при скорости перемешивания 200-500 об/мин, при скорости аэрации 0,5 об/об/мин и при противодавлении, достаточном для поддержания уровня растворенного кислорода по крайней мере 20% об. Культура для трансформации, развивающаяся во время производственного цикла, имела форму очень мелких овальных гранул (около 1-2 мм). Канренон и дополнительные питательные вещества вводили в ферментер путем цепной подачи, в основном способом, описанным в Примере 1. Питательные добавки вводили каждые четыре часа в соотношении 3,4 г глюкозы и 0,6 г дрожжевого экстракта на один литр бульона в ферментере.
В Таблице 31 приводятся скорость аэрации, скорость перемешивания, уровень растворенного кислорода, PMV и рН, определенные в соответствующие интервалы времени в течение каждого из циклов, описанных в данном примере, а также схема добавления глюкозы в течение данного цикла. В Таблице 32 показан профиль превращения канренона. Цикл R11A завершался через 46 часов; цикл R11B продолжался в течение 96 часов. В последнем цикле за 81 час достигалась 93%-ное превращение; через 84 часа проводили еще одну добавку питательной смеси, после чего подачу питательных веществ завершали. Следует отметить, что значительное изменение вязкости происходило в промежуток времени между прекращением подачи питательных веществ и завершением цикла.
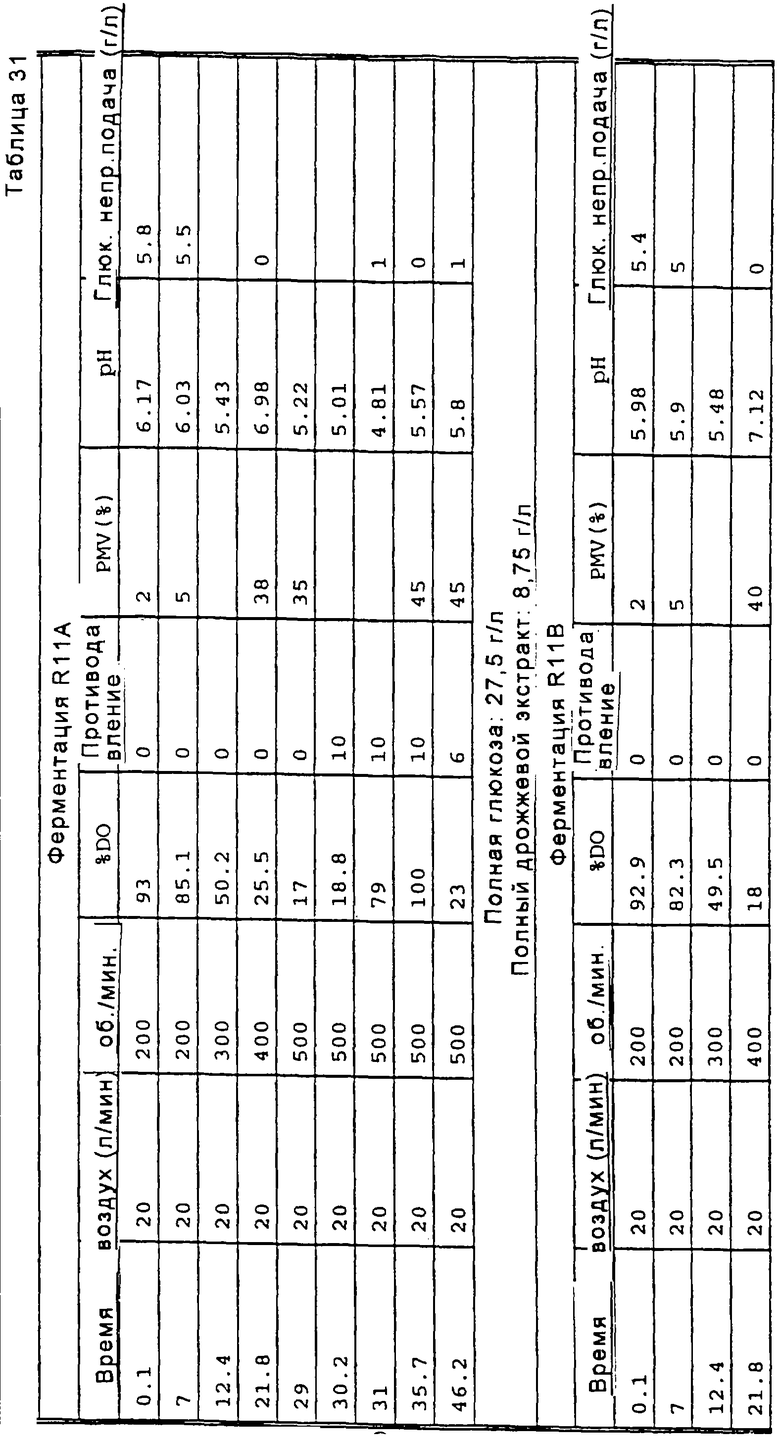
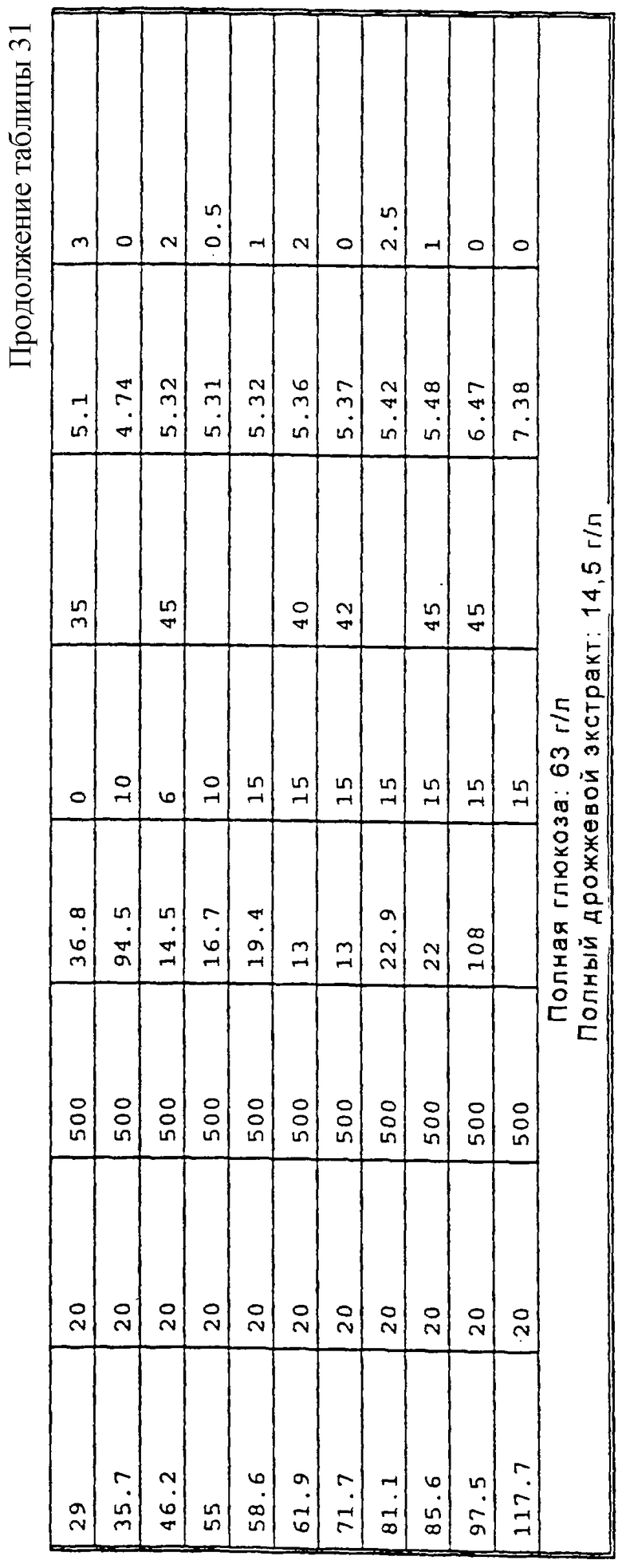
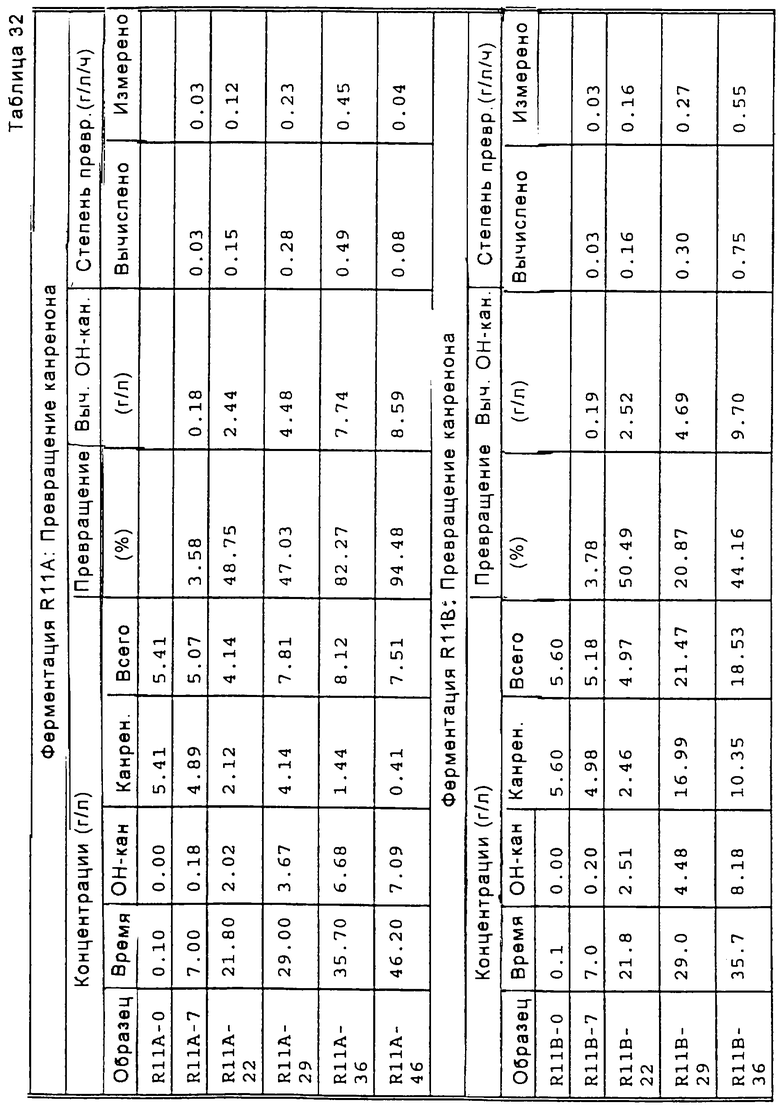

Пример 15.
Различные культуры тестировали на эффективность биологического превращения канренона в 11α-гидроксиканренон способом, в основном описанным выше.
Рабочий клеточный фонд для каждого из микроорганизмов Aspergillus niger ATCC 11394, Rhizopus arrhizus ATCC 11145 и Rhizopus stolonifer ATCC 6227b получали способом, описанным в Примере 5. Среду для культивирования (50 мл), имеющую состав, указанный в Таблице 18, инокулировали суспензией спор (1 мл) от рабочего клеточного фонда и помещали в инкубатор. Посевную культуру получали в инкубаторе путем ферментации в течение 20 часов при 26°С. Содержимое инкубатора перемешивали со скоростью 200 об/мин.
Аликвоты (2 мл) посевной культуры каждого микроорганизма использовали для инокуляции колб для трансформации, содержащих среду для культивирования (30 мл), указанную в Таблице 18. Каждую культуру использовали для инокуляции двух колб, а всего использовали шесть колб. Канренон (200 мг) растворяли в метаноле (4 мл) при 36°С и 0,5 мл аликвоты этого раствора вводили в каждую из этих колб. Биологическое превращение осуществляли, в основном, в условиях, описанных в Примере 5, при ежедневном добавлении 50% масс. раствора глюкозы (1 мл). Через первые 72 часа проводили наблюдения за ростом мицелия в соответствующих бродильных колбах для трансформации:
АТСС 11394 - хороший равномерный рост;
АТСС 11145 - хороший рост в первые 48 часов, однако наблюдалась агрегация мицелия в комки; рост не наблюдался в последние 24 часа;
АТСС 6227b - хороший рост; мицелиальная масса агрегируется в комки.
Образцы бульона были проанализированы на степень биологического превращения. Через три дня ферментация с использованием АТСС 11394 приводила к 80-90%-ному превращению в 11α-гидроксиканренон; АТСС 11145 давала превращение 50%; а АТСС 6227b давала превращение от 80 до 90%.
Пример 16.
С использованием метода, в основном описанного в Примере 15, на эффективность превращения канренона в 11α-гидроксиканренон были протестированы другие микроорганизмы. Тестированные микроорганизмы и результаты тестов представлены в Таблице 33.
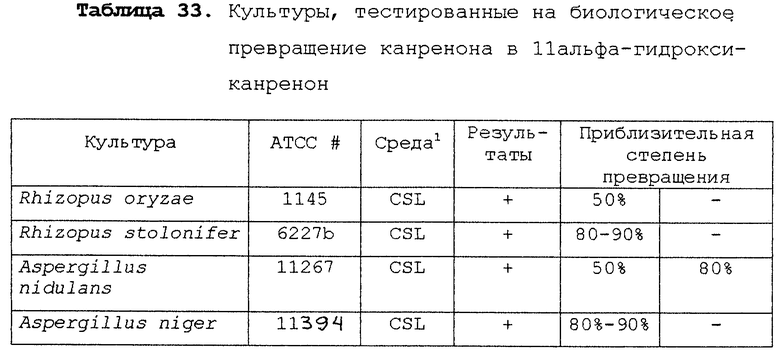
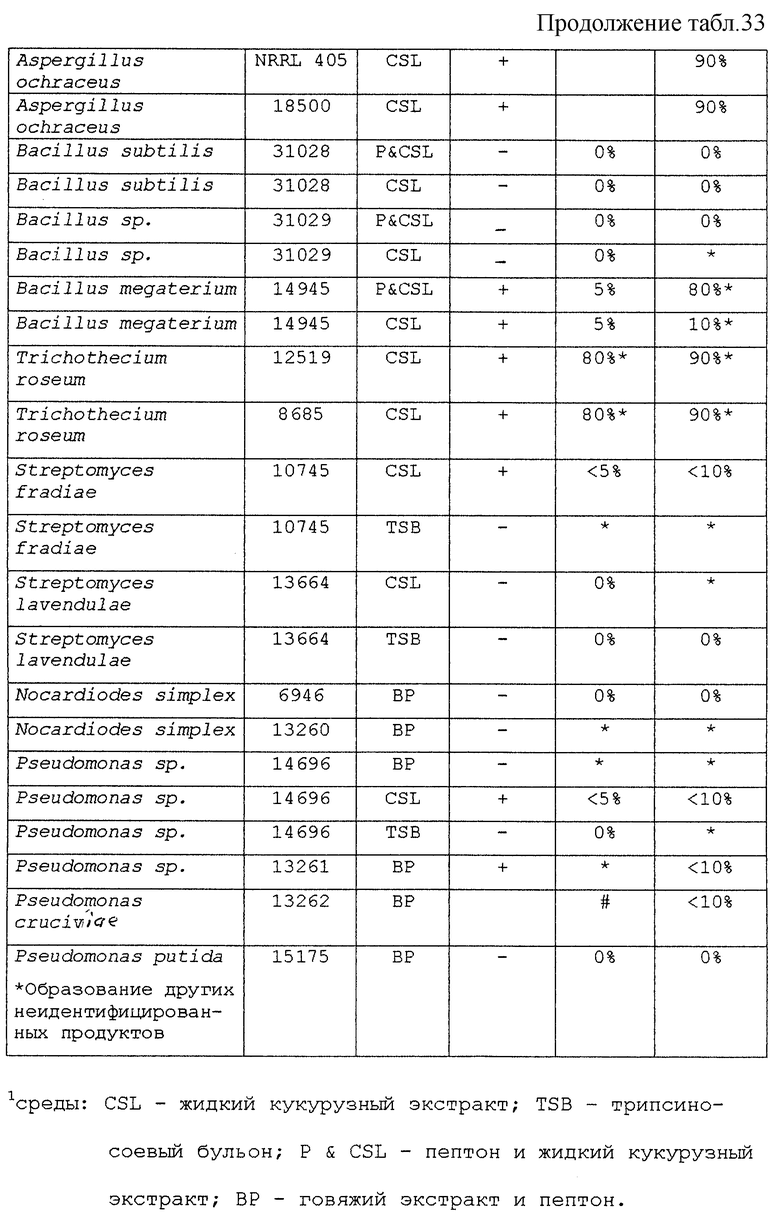
Пример 17.
Различные микроорганизмы тестировали на эффективность превращения канренона в 9α-гидроксиканренон. Сбраживающие среды, которые были получены для проведения циклов ферментации этого Примера, представлены в Таблице 34.
Грибы выращивали в среде с соевой мукой и в среде на основе пептона-дрожжевого экстракта-глюкозы; актиномицеты и эубактерии выращивали в среде с соевой мукой (+0,9% масс. формиата Na для биологических трансформаций) и в бульоне Миллера-Хинтона.
Исходные культуры инокулировали свежезамороженными исходными культурами спор (20 мл соевой муки в 250 мл колбе Эрленмейера). Колбы покрывали фильтром из матового стекла и биозащитным покрытием. Исходные культуры (24- или 48-часовые) использовали для инокуляции культур метаболизма (также 20 мл в 250 мл колбе Эрленмейера) с 10%-15% перекрещивающимся объемом, затем инкубировали в течение 24-48 часов и добавляли стероидный субстрат для реакции трансформации.
Канренон растворяли/суспенидировали в метаноле (20 мг/мл), фильтр стерилизовали и добавляли к культурам до конечной концентрации 0,1 мг/мл. Все колбы для осуществления реакции превращения путем ферментации встряхивали при 250 об/мин (амплитуда 2") при регулируемой комнатной температуре 26°С и влажности 60%.
Продукты биологической трансформации собирали через 5 и 48 часов или через 24 часа после добавления субстрата. Сбор начинали с добавления этилацетата (23 мл) или метиленхлорида в бродильную колбу. Затем колбы встряхивали в течение двух минут и содержимое каждой колбы выливали в коническую 50 мл пробирку. Для разделения фаз эти пробирки центрифугировали при 4000 об/мин в течение 20 минут при комнатной температуре. Органический слой из каждой пробирки переносили в 20 мл сосуд из боросиликатного стекла и выпаривали в скоростном вакуумном испарителе. Сосуды накрывали крышкой и хранили при -20°С.
Для получения материала в целях определения структуры, масштаб биологических трансформаций увеличивали до 500 мл путем увеличения числа шейкерных колб для ферментации до 25. На время сбора (через 24 или 48 часов после добавления субстрата) в каждую колбу отдельно добавляли этилацетат и колбы накрывали крышкой и помещали обратно в шейкер на 20 минут. Затем содержимое колб вышивали в полипропиленовые бутыли и центрифугировали для разделения фаз, либо выливали в разделительную воронку, в которой фазы разделяются под действием силы тяжести. Органическую фазу сушили и получали неочищенный экстракт стероидов, содержащийся в реакционной смеси.
Продукт реакции анализировали сначала с помощью тонкослойной хроматографии на флуоресцентных обращенных пластинках с силикагелем (254 мкм). К каждому сосуду, содержащему осушенный этилацетатный экстракт из реакционной смеси, добавляли этилацетат (500 мкл). Последующий анализ проводили с помощью высокоразрешающей жидкостной хроматографии и масс-спектрометрии. ТСХ-пластинки проявляли в смеси растворителей, хлороформе/метаноле, 95:5 об/об.
Еще один анализ проводили с помощью высокоразрешающей жидкостной хроматографии и масс-спектрометрии. Для этого использовали ВЭЖХ Waters, программное обеспечение Millennium, детектор на фотодиодной матрице и автосборник образцов. В обращенно-фазовой ВЭЖХ использовали колонку Waters NovaPak C-18 (размер частиц 4 мкм) с 4 мм патроном Radial-Pak. Хроматографию начинали с линейного градиента растворителя (25 минут) на колонке, инициализированной в воде:ацетонитриле (75:25), и заканчивали в воде:ацетонитриле (25:75). Затем использовали градиент до 100% ацетонитрила (3 минуты) и 4-минутную изократную промывку, после чего колонку регенерировали при первоначальных условиях.
Для ЖХ/МС ацетат аммония добавляли как в ацетонитриловую, так и в водную фазу при концентрации 2 нМ. На хроматографию это не оказывало существенного влияния. Элюат с колонки был разделен 22:1, при этом большая часть материала направлялась в детектор PDA. Остальные 4,5% материала направлялись в камеру масс-спектрометра Sciex API III для ионизации электронапылением. Масс-спектрометрию осуществляли в позитивном режиме. Аналогичную серию данных, полученных по ВЭЖХ-хроматограмме на одной длине волны с помощью детектора PDA, переносили в масс-спектрометр для совместного анализа УФ- и МС-данных.
Образцы масс-спектрометрической фрагментации могут быть использованы для отбора из гидроксилированных субстратов. Два предполагаемых гидроксилированных канренона, 11α-гидрокси- и 9α-гидрокси-, теряют воду при различных частотах и до соответствующей степени, что может быть использовано в качестве диагностики. Кроме того, 9α-гидроксиканренон образовывал аммониевый аддукт более легко, чем 11α-гидроксиканренон. В Таблице 35 систематизированы данные ТСХ, ВЭЖХ/УФ и ЖХ/МС для ферментации канренона и показано, какие из тестированных микроорганизмов являются эффективными для биологического превращения канренона в 9α-гидроксиканренон. Из них предпочтительным микроорганизмом является Corynespora cassiicola АТСС 16718.
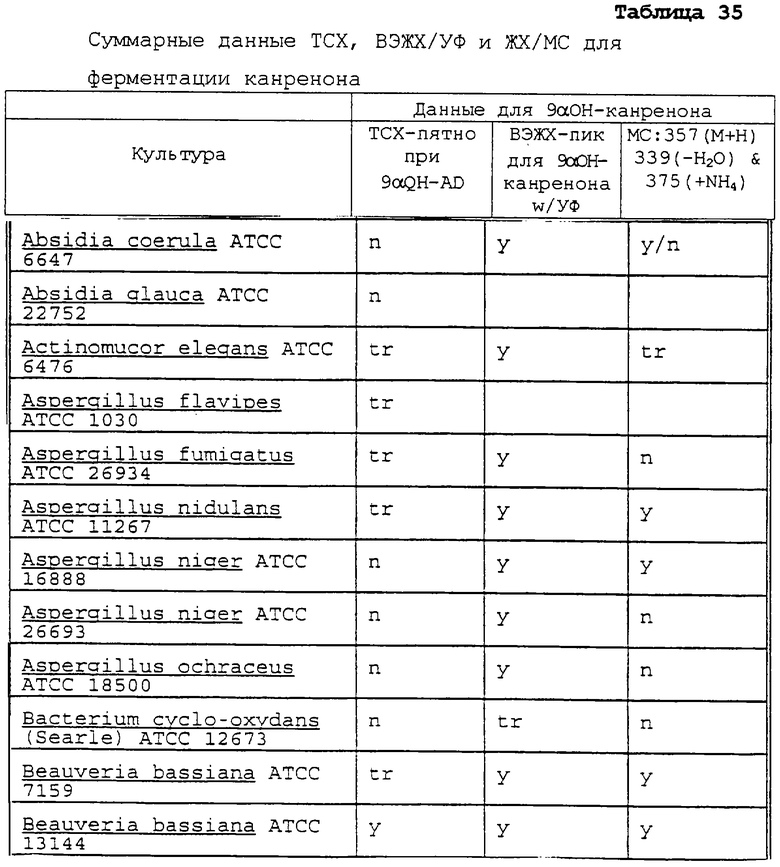




Пример 18.
Различные культуры тестировали на эффективность биологического превращения канренона в 11α-гидроксиканренон способом, описанным в общих чертах выше.
Рабочий клеточный фонд каждого из микроорганизмов Aspergillus ochraceus NRRL 405 (ATCC 18500), Aspergillus niger ATCC 11394, Aspergillus nidulans ATCC 11267, Rhizopus oryzae ATCC 11145, Rhizopus stolonifer ATCC 6227b, Trichothecium roseum ATCC 12519 и ATCC 8685 получали способом, в основном описанным в Примере 4. Среду для культивирования (50 мл), имеющую состав, указанный в Таблице 18, инокулировали суспензией спор (1 мл) от рабочего клеточного фонда и помещали в инкубатор. Посевную культуру получали в инкубаторе путем ферментации в течение около 20 часов при 26°С. Содержимое инкубатора перемешивали со скоростью 200 об/мин.
Аликвоты (2 мл) посевной культуры каждого микроорганизма использовали для инокуляции колб для трансформации, содержащих среду для культивирования (30 мл), указанную в Таблице 15. Каждую культуру использовали для инокуляции двух колб из всех 16 колб. Андростендион (300 мг) растворяли в метаноле (6 мл) при 36°С и 0,5 мл аликвоты этого раствора вводили в каждую из этих колб. Биологическое превращение осуществляли, в основном, в условиях, описанных в Примере 6, в течение 48 часов. Через 48 часов образцы бульона объединяли и экстрагировали этилацетатом, как описано в Примере 17. Этилацетат концентрировали путем выпаривания и образцы анализировали с помощью тонкослойной хроматографии для определения, имеет ли данный продукт хроматографическую подвижность, аналогичную подвижности стандартного 11α-гидроксиандростендиона (Sigma Chemical Co., St.Louis). Результаты представлены в Таблице 36. Положительные результаты обозначены "+".
Данные в Таблице 36 указывают на то, что каждая из перечисленных культур способна продуцировать соединение из андростендиона, имеющего то же самое значение Rf, что и стандартный 11α-гидроксиандростендион.
Aspergillus ochraceus NRRL 405 (ATCC 18500) снова тестировали способом, описанным выше, и культуральные продукты выделяли и очищали с помощью колоночной хроматографии на нормальной фазе силикагеля с использованием метанола в качестве растворителя. Фракции анализировали с помощью тонкослойной хроматографии. ТСХ-пластинки представляли собой пластинки Whatman с силикагелем K6F 60 , размером 10×20, толщиной 250 микрон. Использовали смесь растворителей хлороформ:метанол 95:5 об/об. Кристаллизованный продукт и стандартный 11α-гидроксиандростендион анализировали с помощью ЖХ-МС и ЯМР-спектроскопии. Оба соединения имели аналогичные профили и молекулярные массы.
, размером 10×20, толщиной 250 микрон. Использовали смесь растворителей хлороформ:метанол 95:5 об/об. Кристаллизованный продукт и стандартный 11α-гидроксиандростендион анализировали с помощью ЖХ-МС и ЯМР-спектроскопии. Оба соединения имели аналогичные профили и молекулярные массы.
Пример 19А.
Различные микроорганизмы тестировали на эффективность биологического превращения андростендиона в 11α-гидроксиандростендион способом, в основном описанным выше в Примерах 17 и 18.
Культуры каждого из микроорганизмов Aspergillus fumigatus ATCC 26934, Aspergillus niger ATCC 16888 и АТСС 26693, Epicoccum oryzae ATCC 7156, Curvularia lunata ATCC 12017, Cunninghamella blakesleeana ATCC 8688a и Pithomyces atroolivaceus IFO 6651 выращивали способом, в основном описанным в Примере 17. Среды для культивирования и ферментации (30 мл) имели состав, указанный в Таблице 34.
11α-Гидроксилирование андростендиона вышеперечисленными микроорганизмами анализировали с использованием, в основном, методов, аналогичных методам идентификации продуктов, описанным в Примерах 17 и 18. Результаты представлены в таблице 19А-1.
В Таблице 19А-1 "+" указывает на положительный результат, то есть Rf имеет ожидаемое значение при тонкослойной хроматографии и приблизительно соответствующую молекулярную массу при ЖХ/МС.
Эти результаты свидетельствуют о том, что перечисленные микроорганизмы способны осуществлять 11β-гидроксилирование андростендиона.
Пример 19В.
Различные микроорганизмы тестировали на эффективность биологического превращения мексренона в 11β-гидроксимексренон. Сбраживающие среды для этого примера получали, как описано в Таблице 34.
Условия ферментации и аналитические методы были аналогичны методам, описанным в Примере 17. ТСХ-пластинки и система растворителей описаны в Примере 18. Принцип хроматографического анализа заключался в следующем: 11α-гидроксимексренон и 11α-гидроксиканренон имели одинаковую хроматографическую подвижность, 11α-гидроксиканренон и 9α-гидроксиканренон имели такой же характер подвижности, как и 11α-гидроксиандростендион и 11β-гидроксиандростендион. Поэтому 11β-гидроксимексренон должен иметь подвижность, аналогичную подвижности 9α-гидроксиканренона. Следовательно, соединения, экстрагированные из культуральной среды, были протестированы по сравнению со стандартным 9α-гидроксиканреноном. Результаты представлены в Таблице 37.
P = PYG (пептон/дрожжевой экстракт/глюкоза)
S = соевая мука
SF = соевая мука + формиат
2 ? = спорное отличие от контроля без субстрата
Эти данные предположительно указывают на то, что основная часть микроорганизмов, перечисленных в этой таблице, продуцирует продукт, сходный или идентичный 11α-гидроксимексренону, образующемуся из мексренона.
Пример 19С.
Различные микроорганизмы были протестированы на эффективность в превращении мексренона в 11α-гидроксимексренон, Δ1,2-мексренон, 6β-гидроксимексренон, 12β-гидроксимексренон и 9α-гидроксимексренон. Мексренон может быть получен способом, описанным Weier в Патенте США №3787396 и R.M.Weier et al., J.Med.Chem., Vol.18, pp.817-821 (1975), которые вводятся в настоящее описание посредством ссылки. Среды для ферментации были получены, как описано в Примере 17, за исключением того, что в них был включен мексренон. Условия ферментации были, в основном, аналогичны условиям, описанным в Примере 17; аналитические методы также были аналогичны методам, описанным в Примерах 17 и 18. ТСХ-пластинки и система растворителей были аналогичны описанным в Примерах 17 и 18.
Тестированные микроорганизмы и результаты, полученные на их основе, представлены в Таблице 19С-1.
В Таблице 19С-1 "+" указывает на положительный результат, то есть Rf имеет ожидаемое значение при тонкослойной хроматографии и приблизительно ожидаемое время удерживания при ВЭЖХ, m/z 417:399 указывает на отношение высоты пиков молекулы 417 (гидроксимексренон) и молекулы 399 (мексренон). Стандарт имеет отношение высоты пиков 10:1 для m/z 417 - m/z 399.
Продукт, полученный от Beauveria bassiana АТСС 13144, выделяли из инкубационной смеси и анализировали с помощью ЯМР, который показал, что структурный профиль этого продукта соответствует 11α-гидроксимексренону. Аналогично, продукты, полученные от других микроорганизмов, перечисленных в Таблице 19С-1, также предположительно являются 11α-гидроксимексреноном.
В Таблице 19С-2 "+" указывает на положительный результат, то есть Rf имеет ожидаемое значение при тонкослойной хроматографии и приблизительно ожидаемое время удерживания при ВЭЖХ и т.п.
Продукт, полученный от Bacterium cyclooxydans ATCC 12673, выделяли из инкубационной смеси и анализировали с помощью ЯМР, который показал, что структурный профиль этого продукта соотвествует Δ1,2-мексренону. Аналогично, продукты, полученные от других микроорганизмов, перечисленных в Таблице 19С-2, также предположительно являются Δ1,2-мексреноном.
Продуцирование 6β и 12β-гидроксимексренона
Mortierella isabella ATCC 42613 выращивали, как описано в Примере 17, в присутствии мексренона. Продукты ферментации выделяли и очищали с помощью флэш-хроматографии. Очищенные продукты анализировали с помощью ЖХ/МС, как описано в Примерах 17 и 18, и с помощью протонного ЯМР и 13С-ЯМР. Данные этих анализов показали, что эти продукты включали 6β- и 12β-гидроксимексренон.
Микроорганизмы, перечисленные в Таблице 19С-3, выращивали в тех самых условиях, которые были описаны в Примере 17, в присутствии мексренона. Продукты ферментации анализировали с помощью ТСХ и ЖХ/МС, как описано в Примерах 17 и 18. В Таблице 19С-3 "+" указывает на положительный результат, то есть Rf имеет ожидаемое значение при тонкослойной хроматографии и приблизительно ожидаемое время удерживания при ВЭЖХ и т.п. Эти данные позволяют предположить, что полученные продукты включают 9α-гидроксимексренон.
Пример 19D.
Различные микроорганизмы были протестированы на эффективность в превращении канренона в Δ9,11-канренон. Среды для ферментации и условия культивирования были, в основном, такими же, как и в Примере 17, за исключением того, что в эту среду был включен канренон. Методы анализа были аналогичны методам, описанным в Примере 17 и 18. Микроорганизмы и результаты представлены ниже в Таблице 19D-4.
Продукты ферментации были проанализированы с помощью ТСХ и ЖХ/МС, как описано в Примерах 17 и 18. Символ "+" указывает на положительный результат, то есть Rf имеет ожидаемое значение при тонкослойной хроматографии и приблизительно ожидаемое время удерживания при ВЭЖХ и т.п.
Продукт, полученный от Comomonas testosteroni ATCC 11996 выделяли из культуральной среды и анализировали с помощью УФ-спектроскопии. Спектроскопический профиль подтверждал присутствие Δ9,11-канренона. Аналогично, продукты, полученные от микроорганизмов, перечисленных выше в Таблице 19D-1, предположительно также являлись Δ9,11-канреноном.
Пример 20А.
Схема 1: Стадия 1: Метод А: Получение 5'R(5'α),7'β-20'-аминогексадекагидро-11'β-гидрокси-10'а,13'α-диметил-3',5-ди-оксоспиро[фуран-2(3Н),17'α(5'Н)-[7,4]метено[4Н[циклопента-[а]фенантрен]-5'-карбонитрила
В эмалированный реактор емкостью 50 галлон (189,25 дм3) загружали при перемешивании 61,2 л (57,8 кг) ДМФ, а затем 23,5 кг 11-гидроксиканренона 1. К этой смеси добавляли 7,1 кг хлорида лития. Смесь перемешивали в течение 20 минут и загружали 16,9 кг цианогидрина ацетона, а затем 5,1 кг триэтиламина. Полученную смесь нагревали до 85°С и поддерживали при этой температуре в течение 13-18 часов. После завершения реакции добавляли 353 л воды, а затем 5,6 кг бикарбоната натрия. Смесь охлаждали до 0°С, переносили в эмалированный реактор емкостью 200 галлон (757 дм3) и медленно гасили с использованием 130 кг 6,7% раствора гипохлорита натрия. Продукт фильтровали и промывали 3×40 л порциями воды с получением 21,4 кг енаминового продукта.
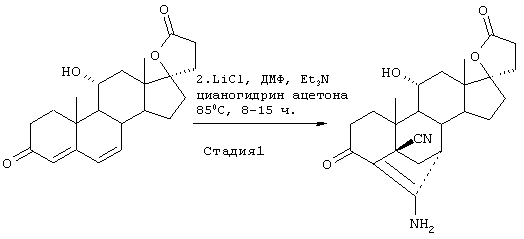
Н1 ЯМР (ДМСО-d6): 7,6(2Н, шир.д), 4,53(1Н, д, J=5,9), 3,71(1Н, м), 3,0-1,3(17Н, м), 1,20(5Н, м), 0,86(3Н, с), 0,51 (1Н, т, J=10).
Пример 20В. Получение 7α-циано-11α,17-дигидрокси-3-оксо-17α-прегн-4-ен-21-карбоновой кислоты, γ-лактона
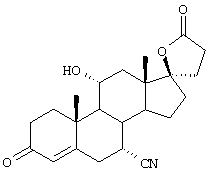
50,0 г 11-гидроксиканренона и 150,0 мл диметилацетамида добавляли в чистую сухую трехгорлую колбу, снабженную механической мешалкой, холодильником, термопарой и кожухом для нагревания. К этой смеси добавляли 16,0 мл раствора серной кислоты (полученного путем смешивания 50,0 мл серной кислоты (98,7% кислота сорта Baker) с 50,0 мл воды). Затем добавляли раствор цианида натрия, содержащий 15,6 г цианида натрия и 27,0 мл воды.
Полученную смесь нагревали в течение 7 часов при 80°С и степень превращения периодически отслеживали с помощью ТСХ или ВЭЖХ. Приблизительно через 7 часов ВЭЖХ указывала на присутствие 7-циано-соединения. Затем смесь перемешивали в течение ночи и оставляли охлаждаться до комнатной температуры (около 22°С). К этой смеси добавляли 200 мл воды, а затем 200 мл метиленхлорида и полученную двухфазную смесь перемешивали, после чего смесь оставляли для разделения фаз. Водный слой представлял собой гель. К этому водному слою, в безуспешной попытке разрушить этот гель, добавляли 100 мл раствора бикарбоната натрия. Затем водный слой отбрасывали.
Отделенный метиленхлоридный слой промывали 100 мл воды и полученную двухфазную смесь перемешивали. Затем смесь оставляли для разделения фаз и отделенный метиленхлоридный слой фильтровали через 200 г силикагеля (сито 200-400 меш, 60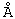 , Aldrich). Фильтрат концентрировали досуха при пониженном давлении и при 45°С, с использованием устройства для отсасывания воды, и получали около 53,9 г сырого твердого продукта. Затем этот сырой твердый продукт растворяли в 50 мл метиленхлорида и обрабатывали 40 мл 4н соляной кислоты в делительной воронке и двухфазную смесь оставляли для отстаивания. Метиленхлоридный слой промывали 50 мл воды. Объединенные водные слои экстрагировали 50 мл метиленхлорида. Затем объединенные метиленхлоридные слои сушили сульфатом натрия и получали 45 г твердого вещества, которое представляло собой смесь 11α-гидроксиканренона и продукта 7α-циано-11α,17-дигидрокси-3-оксо-17α-прегн-4-ен-21-карбоновой кислоты, γ-лактона.
, Aldrich). Фильтрат концентрировали досуха при пониженном давлении и при 45°С, с использованием устройства для отсасывания воды, и получали около 53,9 г сырого твердого продукта. Затем этот сырой твердый продукт растворяли в 50 мл метиленхлорида и обрабатывали 40 мл 4н соляной кислоты в делительной воронке и двухфазную смесь оставляли для отстаивания. Метиленхлоридный слой промывали 50 мл воды. Объединенные водные слои экстрагировали 50 мл метиленхлорида. Затем объединенные метиленхлоридные слои сушили сульфатом натрия и получали 45 г твердого вещества, которое представляло собой смесь 11α-гидроксиканренона и продукта 7α-циано-11α,17-дигидрокси-3-оксо-17α-прегн-4-ен-21-карбоновой кислоты, γ-лактона.
Образец этого продукта анализировали с помощью ВЭЖХ (колонка: 25 см × 4,6 мм, 5 мкм C18LL Altima); градиент растворителя: растворитель А=вода/трифторуксусная кислота=99,9/0,1, растворитель В=ацетонитрил/трифторуксусная кислота=99, 9/0,1, скорость потока=1,00 мл/мин, градиент=65:30 (об./об.) (А:В-сначала); 35:65 (об./об.) (А: В - через 20 минут); 10:90 (об./об.) (А: В - через 25 минут), детектор на диодной матрице, которая обнаруживала λмакс 238 нм.
Реакционную смесь анализировали с помощью ВЭЖХ-ЯМР с использованием следующих условий: ВЭЖХ-колонка: Zorbax RX-C8 (25 см × 4,6 мм, 5 микрон) с использованием градиента растворителей 75% D2O, 25% ацетонитрила - 25% D2O, 75% ацетонитрила в течение 25 минут со скоростью потока 1 мл/мин;
1Н ЯМР-спектр (полученный с использованием подавления растворителя WET) 5,84 (с, 1Н), 4,01(м, 1Н), 3,2(м, 1Н), 2,9-1,4 (м, полный сигнал не имеет значимого значения из-за подавления ацетонитрила), 0,93-0,86(с, перекрывающийся 3Н, и т,2Н).
Пример 20С. Получение 5β,7α-дициано-17-гидрокси-3-оксо-17α-прегнан-21-карбоновой кислоты, γ-лактона
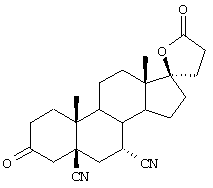
102 г (0,3 моль) 17-гидрокси-3-оксо-17α-прегна-4,6-диен-21-карбоновой кислоты, γ-лактона (канренона) суспендировали с 46,8 г (0,72 моль) цианида калия, 78,6 мл (1,356 моль) уксусной кислоты и 600 мл метанола в трехлитровой трехгорлой круглодонной колбе. К этой смеси добавляли 64,8 мл (0,78 моль) пирролидина, объединенную суспензию нагревали с обратным холодильником (64°С) и выдерживали в течение около 1,5 часов. Затем температуру суспензии снижали до 25°С-30°С в течение десяти минут с использованием охлаждающей бани. 120 мл концентрированной соляной кислоты медленно добавляли во время охлаждения, в течение которого наблюдалось осаждение твердого вещества рыжевато-коричневого цвета.
Эту смесь перемешивали при 25°С-30°С в течение 1,5 часов, а затем через 30 минут добавляли еще 500 мл воды. Эту смесь охлаждали до 5°С в бане со льдом и рН доводили от 3 до 5,5 (рН прослеживали с использованием полосок индикаторной бумаги) путем добавления 100 мл водного 9,5М гидроксида натрия (0,95 моль). Избыточный цианид разлагали путем добавления бытового отбеливателя. 25 мл (0,020 моль) добавляли для проведения негативного теста с иодидом крахмала. Холодную смесь (10°С) фильтровали и твердые вещества промывали водой до тех пор, пока промывка не приобретала нейтральный рН (рН-индикаторная бумага). Твердое вещество сушили при 60°С до получения постоянной массы 111,4 г.
Выделенное твердое вещество расплавляли при 244-246°С на блоке Фишера Джонса. Метаноловый раствор, содержащий твердое вещество, не обнаруживал абсорбции в УФ-области 210- 240 нм. ИК (CHCl3) см-1 2222 (цианид), 1775 (лактон), 1732 (3-кето). 1Н-ЯМР (пиридин-d5) м.д. 0,94(с, 3Н), 1,23(с, 3Н).
Пример 21А.
Схема 1: Стадия 2: Получение 4'S(4'α),7'α-гексадека-гидро-11'α-гидрокси-10'β,13'β-диметил-3',5,20'-триоксоспиро-[фуран-2(3Н),17'β-[4,7]метано[17Н]циклопента-[а]фенантрен]-5'β(2'Н)-карбонитрила
В эмалированный реактор емкостью 200 галлон (757 дм3) загружали 50 кг енамина 2, приблизительно 445 л 0,8н разбавленной соляной кислоты и 75 л метанола. Смесь нагревали в течение 5 часов до 80°С, а затем охлаждали в течение 2 часов до 0°С. Твердый продукт фильтровали с получением 36,5 кг сухого дикетонового продукта.
1Н ЯМР (ДМСО-d6): 4,53(1Н, д, J=6), 3,74(2H, м), 2,73 (1Н, дд, J=14,7), 2,65-2,14 (8Н, м), 2,05(1Н, т, J=11), 1,98-1,71(4Н, м), 1,64(1Н, м), 1,55(1Н, дд, J=13,5), 1,45-1,20 (7Н, м), 0,86(3Н, с).
Пример 21В.
Схема 1: Стадия 1 и 2: in situ-получение 4'S(4'α),7'α-гексадекагидро-11'α-гидрокси-10'β,13'β-диметил-3',5,20'-три-оксоспиро[фуран-2(3Н),17'β(4,7)-метано[17Н]циклопента[а]-фенантрен]-5'β(2'Н)-карбонитрила из 11α-гидроксиканренона
В реактор, снабженный холодильником, механической мешалкой, кожухом для нагревания, автоматическим регулятором и воронкой, загружали 100 г (280,54 моль) 11-гидроксиканренона, полученного способом, описанным в Примере 1, а затем добавляли 300 мл диметилацетамида (Aldrich). Смесь перемешивали до тех пор, пока 11-гидроксиканренона не был растворен. К этой смеси добавляли 31,5 мл 50% серной кислоты (Fisher), которая способствовала повышению температуры полученной смеси до около 10°С-15°С. Затем к смеси 11α-гидроксиканренона в течение 2-3 минут добавляли раствор цианида натрия, полученный путем растворения 31,18 г (617,20 моль)(Aldrich) цианида натрия в 54 мл деионизованной воды. После добавления раствора цианида натрия температура полученной смеси повышалась до около 20°С-25°С.
Эту смесь нагревали до 80°С и поддерживали при этой температуре в течение 2-3 часов. После того, как ВЭЖХ-анализ указывал на то, что реакция превращения 11α-гидроксиканренона в енамин была, в основном, завершена (превращение более чем на 98%), источник нагревания удаляли. Без выделения енамина, находящегося в смеси, к этой смеси в течение 3-5 минут добавляли еще 148 мл 50% серной кислоты. Затем к смеси в течение 10 минут добавляли 497 мл деионизованной воды.
Эту смесь нагревали до 102°С и поддерживали при этой температуре до тех пор, пока приблизительно 500 г дистиллята не было удалено из смеси. Во время реакции/дистилляции к смеси было добавлено 500 мл деионизованной воды четырьмя отдельными порциями по 125 мл. Каждую порцию добавляли к смеси после удаления эквивалентного количества дистиллята (приблизительно 125 мл). Реакция продолжалась в течение 2 часов. После того, как ВЭЖХ-анализ указывал на то, что реакция гидролиза енамина в дикетон была, в основном, завершена (превращение более чем на 98%), смесь охлаждали до около 80°С в течение 20 минут.
Эту смесь фильтровали через стеклянную воронку. Реактор промывали 1,2 л деионизованной воды для удаления остаточного продукта. Твердое вещество на фильтре три раза промывали приблизительно равными порциями (около 0,4 л) промывочной воды. В реакторе получали 1 л раствора метанола и деионизованной воды (1:1, об./об.) и фильтрат промывали 500 мл этого раствора. Затем фильтрат два раза промывали оставшимися 500 мл раствора метанол/вода. В воронке создавали вакуум для осушки фильтрата до степени, достаточной для его переноса. Фильтрат переносили в сушильную печь, где его сушили в вакууме в течение 16 часов с получением 84 г сухого дикетонового продукта, 4'S(4'α),7'α-гексадекагидро-11'α-гидрокси-10'β,13'β- диметил-3',5,20'-триоксоспиро[фуран-2(3Н),17'β(4,7)-метано-[17Н]циклопента[а]фенантрен]-5'β(2'Н)-карбонитрила. ВЭЖХ-анализ показал 94% нужного дикетона.
Пример 22.
Схема 1: Стадия 3А: Метод А: Получение метилгидро-11α,17α-дигидрокси-3-оксопрегн-4-ен-7α,21-дикарбоксилата, γ-лактона.
4-Горлая 5-литровая круглодонная колба была снабжена механической мешалкой, капельной воронкой для выравнивания давления с трубкой для впуска азота, термометром и холодильником с барботером. Этот барботер был подсоединен посредством системы тигоновых трубок к двум 2-литровым ловушкам, первая из которых была пустой и помещалась в реакционный сосуд для предупреждения обратного отсасывания материала во второй ловушке (1 л концентрированного раствора гидрохлорита натрия). В колбу, в 3 л метанола добавляли дикетон 3 (79,50 г; [масса не была скорректирована на чистоту, которая составляла 85%]). 25% Метаноловый раствор метоксида натрия (64,83 г) выливали в воронку и по каплям добавляли, размешивая в атмосфере азота, в течение 10 минут. После завершения добавления оранжево-желтую реакционную смесь нагревали с обратным холодильником в течение 20 часов. По истечении этого периода через капельную воронку к еще кипящей реакционной смеси по каплям добавляли 167 мл 4н HCl (Внимание: на этой стадии происходит выделение HCN!). Реакционная смесь, которая имела светлую окраску, становилась бледно-золотисто-оранжевого цвета. Затем холодильник заменяли головной насадкой для отвода и 1,5 л метанола удаляли путем дистилляции, при этом в колбу через воронку одновременно добавляли 1,5 л воды в соответствии со скоростью дистилляции. Реакционную смесь охлаждали до комнатной температуры и два раза экстрагировали 2,25 л аликвотами метиленхлорида. Объединенные экстракты последовательно промывали 750 мл аликвотами холодного насыщенного раствора NaCl, 1н NaOH и снова насыщенным раствором NaCl. Органический слой сушили сульфатом натрия в течение ночи, фильтровали и концентрировали в вакууме до объема ˜250 мл. Затем добавляли толуол (300 мл) и оставшийся метиленхлорид выпаривали при пониженном давлении, причем в течение этого времени на стенках колбы начинал образовываться продукт в виде белого твердого вещества. Содержимое колбы охлаждали в течение ночи и твердое вещество удаляли путем фильтрации. Это вещество промывали 250 мл толуола, а затем два раза 250 мл аликвотами простого эфира и сушили в ваккум-воронке, в результате чего получали 58,49 г белого твердого вещества, который, как показала ВЭЖХ, имел чистоту 97,3%. После концентрирования маточного раствора получали еще 6,76 г продукта с чистотой 77,1%. Полный выход, скорректированный на чистоту, составлял 78%.
1Н-ЯМР (CDCl3): 5,70(1Н, с), 4,08(1Н, с), 3,67(3Н, с), 2,9-1,6(19Н, м), 1,5-1,2(5Н,м), 1,03(3Н, с).
Пример 23.
Схема 1: Стадия 3В: Превращение метилгидро-11α,17α-дигидрокси-3-оксопрегн-4-ен-7α,21-дикарбоксилата, γ-лактона в метилгидро-17α-гидрокси-11α-(метилсульфонил)окси-3-оксопрегн-4-ен-7α,21-дикарбоксилат, γ-лактон
5-литровая четырехгорлая колба была снабжена так же, как описано выше, за исключением того, что к барботеру не была подсоединена система ловушек. В колбу при перемешивании в атмосфере азота добавляли сложный гидроксиэфир в количестве 138,70 г, а затем 1425 мл метиленхлорида. Реакционную смесь охлаждали до -5°С с использованием солевой/ледяной бани. Затем быстро добавляли метансульфонилхлорид (51,15 г, 0,447 моль), после чего медленно по каплям добавляли триэтиламин (54,37 г) в 225 мл метиленхлорида. Добавление, которое требует ˜30 минут, было сделано так, чтобы температура реакционной смеси никогда не поднималась выше около 5°С. После добавления перемешивание продолжали в течение 1 часа и содержимое реактора переносили в 12-литровую делительную воронку, в которую добавляли 2100 мл метиленхлорида. Этот раствор последовательно промывали 700 мл аликвотами холодной 1н HCl, 1н NaOH и насыщенным раствором NaCl (каждого). Водные промывки объединяли и подвергали обратной экстракции 3500 мл метиленхлорида. Все органические промывки объединяли в 9 л сосуде, в который добавляли 500 г нейтральной окиси алюминия с классом активности II и 500 г безводного сульфата натрия. Содержимое этого сосуда тщательно смешивали в течение 30 минут и фильтровали. Фильтрат упаривали досуха в вакууме с получением смолистой желтой пены. Эту пену растворяли в 350 мл метиленхлорида и по каплям при перемешивании добавляли 1800 мл простого эфира. Скорость добавления корректировали так, чтобы около половины эфира было добавлено в течение 30 минут. После добавления примерно 750 мл продукт начинал выделяться в виде кристаллического твердого вещества. Оставшееся количество эфира добавляли в течение 10 минут. Твердое вещество удаляли путем фильтрации и осадок на фильтре промывали 2 л эфира и сушили в вакуумной печи при 50°С в течение ночи, в результате чего получали 144,61 г (88%) почти белого твердого вещества, т.пл. 149-150°С. Продукт, полученный таким образом, имел обычно 98-99% чистоту, как показала ВЭЖХ (% площади). В одном цикле получали продукт, имеющий температуру плавления 153-153, 5°С, с чистотой 99,5%, как было определено по ВЭЖХ-площади.
1Н-ЯМр (CDCl3): 5,76(1Н, с), 5,18(1Н, дт), 3,68(3Н, с), 3,06(3Н, с), 2,85(1Н, м), 2,75-1,6(19Н, м), 1,43(3Н, с), 1,07(3Н, с).
Пример 24.
Схема 1: Стадия 3С: Метод А: Получение 7-метилгидро-17α-гидрокси-3-оксопрегна-4,9(11)-диен-7α,21-дикарбоксилата, γ-лактона
1-Литровая четырехгорлая колба была снабжена так же, как описано во втором примере. В эту колбу при размешивании в атмосфере азота добавляли муравьиную кислоту (250 мл) и уксусный ангидрид (62 мл). Затем добавляли формиат калия (6,17 г) и реакционную смесь нагревали в масляной бане до внутренней температуры 40°С (эту процедуру повторяли при 70°С с лучшими результатами) в течение 16 часов. По истечении 16 часов добавляли мезилат и внутреннюю температуру повышали до 100°С. Нагревание и перемешивание продолжали еще в течение 2 часов, после чего растворитель удаляли в вакууме на роторном испарителе. Остаток перемешивали с 500 мл ледяной воды в течение пятнадцати минут, а затем два раза экстрагировали 500 мл аликвотами этилацетата. Органические фазы объединяли и последовательно промывали 250 мл аликвотами холодного насыщенного раствора хлорида натрия (два раза), 1н раствором гидроксида натрия и снова насыщенным хлоридом натрия. Затем органическую фазу сушили сульфатом натрия, фильтровали и концентрировали досуха в ваккуме, в результате чего получали желтовато-белую пену, которую наносили путем распыления на стекло при помощи шпателя. Анализ 14,65 г полученного порошка (по % площади, ВЭЖХ) указывал, что этот порошок представлял собой смесь 82,1% 7-метилгидро-17α-гидрокси-3-оксопрегна-4,9(11)-диен-7α,21-дикарбоксилата, γ-лактона; 7,4% 7-метилгидро-17α-гидрокси-3-оксопрегна-4,11- диен-7α,21-дикарбоксилата, γ-лактона; и 5,7% 9α,17-дигидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоновой кислоты, бис(γ-лактона).
1Н-ЯМР (CDCl3): 5,74(1Н, с), 5,67(1Н, м), 3,61(3Н, с), 3,00(1Н, м), 2,84(1Н, ддд, J=2,6, 15), 2,65-2,42(6Н, м), 2,3-2,12(5Н, м), 2,05-1,72(4Н, м), 1,55-1,45(2Н, м), 1,42(3Н, с), 0,97(3Н, с).
Пример 25.
Схема 1: Стадия 3С: Метод В: Получение метилгидро-17α-гидрокси-3-оксопрегна-4,9(11)-диен-7α,21-дикарбоксилата, γ-лактона
В 5-литровую четырехгорлую колбу, которая была снабжена так же, как описано выше, добавляли при размешивании в атмосфере азота 228,26 г уксусной кислоты и 41,37 г ацетата натрия. С использованием масляной бани смесь нагревали до внутренней температуры 100°С. Затем добавляли мезилат (123,65 г) и нагревание продолжали еще тридцать минут. По окончании этого периода времени нагревание прекращали и добавляли 200 мл ледяной воды. Температуру снижали до 40°С и продолжали размешивать еще 1 час, после чего реакционную смесь медленно выливали в 1,5 л холодной воды в 5-литровой размешиваемой колбе. Продукт выделяли в виде смолистого масла. Это масло растворяли в 1 л этилацетата и промывали 1 л холодного насыщенного раствора хлорида натрия, 1н гидроксида натрия (каждого) и, наконец, снова насыщенным хлоридом натрия. Органическую фазу сушили сульфатом натрия и фильтровали. Фильтрат концентрировали досуха в вакууме с получением пены, которая оседала с образованием смолистого масла. Это масло растирали с простым эфиром в течение некоторого промежутка времени и оно, в конце концов, отверждалось. Полученное твердое вещество фильтровали и промывали еще эфиром с получением 79,59 г желто-белого твердого вещества. Это вещество состояло из 70,4% целевого сложного Δ9,11-енэфира 6; 12,3% сложного Δ11,12-енэфира 8; 10,8% 7-α,9-α-лактона 9 и 5,7% непрореагировавшего соединения 5.
Пример 26.
Схема 1: Стадия 3D: Метод А: Синтез метилгидро-9,11α-эпокси-17α-гидрокси-3-оксопрегн-4-ен-7α,21-дикарбоксилата, γ-лактона.
4-х горлый 500 мл-реактор с рубашкой был снабжен механической мешалкой, холодильником/барботером, термометром и капельной воронкой с трубкой для подачи азота. В этот реактор при размешивании в атмосфере азота загружали 8,32 г сырого сложного енэфира в 83 мл метиленхлорида. После этого добавляли 4,02 г двухосновного фосфата калия, а затем 12 мл трихлорацетонитрила. Через рубашку реактора пропускали воду для внешнего охлаждения и реакционную смесь охлаждали до 8°С. В капельную воронку в течение 10 минут добавляли 36 мл 30% перекиси водорода. После завершения добавления реакционная смесь, первоначально имеющая бледно-желтую окраску, становилась почти бесцветной. Во время добавления реакционную смесь поддерживали при температуре 9±1°С и перемешивали в течение ночи (всего 23 часа). К реакционной смеси добавляли метиленхлорид (150 мл) и все содержимое добавляли в ˜250 мл ледяной воды. Эту смесь экстрагировали три раза 150 мл аликвотами метиленхлорида. Объединенные метиленхлоридные экстракты промывали 400 мл холодного 3% раствора сульфита натрия для разложения какого-либо остаточного пероксида. После этого промывали 330 мл холодной промывки с 1н гидроксидом натрия, 400 мл холодной промывки с 1н соляной кислотой и, наконец, промывкой с 400 мл насыщенного солевого раствора. Органическую фазу сушили сульфатом магния, фильтровали, а осадок на фильтре промывали 80 мл метиленхлорида. Растворитель удаляли в вакууме с получением 9,10 г сырого продукта в виде бледно-желтого твердого вещества. Это вещество перекристаллизовывали из ˜25 мл 2-бутанона с получением 5,52 г почти белых кристаллов. После конечной перекристаллизации из ацетона (˜50 мл) получали 3,16 г длинных игольчатых кристаллов, т.пл. 241-243°С.
1Н-ЯМР (CDCl3): 5,92(1Н, с), 3,67(3Н, с), 3,13(1Н, д, J=5), 2,89(1H, м), 2,81-2,69(15Н, м), 1,72(1Н,дд, J=5,15), 1,52-1,22 (5Н, м), 1,04(3Н, с).
Пример 27.
Схема 1: Стадия 3: Вариант 1: Превращение 4'S(4'α),7'α-гексадекагидро-11'α-гидрокси-10'β,13'β-диметил-3',5,20'-три-оксоспиро[фуран-2(3Н),17'β(4,7)-метано[17Н]циклопента[а]-фенантрен]-5'β(2'Н)-карбонитрила в метилгидро-9,11α-эпокси-17α-гидрокси-3-оксопрегн-4-ен-7α,21-дикарбоксилат, γ-лактон.
В чистый и осушенный реактор загружали дикетон (20 г), а затем добавляли 820 мл МеОН и 17,6 мл 25% раствора NaOMe/MeOH. Реакционную смесь нагревали с обратным холодильником (˜67°С) в течение 16-20 часов. Продукт гасили 40 мл 4н HCl. Растворитель удаляли при атмосферном давлении путем отгонки. Затем добавляли 100 мл толуола, а остаточный метанол удаляли путем азеотропной перегонки с толуолом. После концентрирования сырой сложный гидроксиэфир 4 растворяли в 206 мл метиленхлорида и охлаждали до 0°С. После этого добавляли метансульфонилхлорид (5 мл), а затем медленно добавляли 10,8 мл триэтиламина. Этот продукт перемешивали в течение 45 минут. Растворитель удаляли путем вакуумной перегонки и получали сырой мезилат 5.
В отдельный осушенный реактор добавляли 5,93 г формиата калия, 240 мл муравьиной кислоты, в затем 118 мл уксусного ангидрида. Смесь нагревали до 70°С в течение 4 часов.
Смесь, содержащую муравьиную кислоту, добавляли в концентрированный раствор 5, полученный как описано выше. Эту смесь нагревали до 95-105°С в течение 2 часов. Смесь, содержащую продукт, охлаждали до 50°С и летучие компоненты удаляли путем вакуумной перегонки при 50°С. Продукт распределяли между 275 мл этилацетата и 275 мл воды. Водный слой подвергали обратной экстракции 137 мл этилацетата, промывали 240 мл холодного 1н раствора гидроксида натрия, а затем 120 мл насыщенного NaCl. После разделения фаз органический слой концентрировали путем вакуумной перегонки с получением сырого сложного енэфира.
Продукт растворяли в 180 мл метиленхлорида и охлаждали до 0-15°С. После этого добавляли 8,68 г дикалийбифосфата, а затем 2,9 мл трихлорацетонитрила. К полученной смеси в течение 3 минут добавляли 78 мл раствора 30% перекиси водорода. Реакционную смесь перемешивали в течение 6-24 час при 0-15°С. После прохождения реакции двухфазную смесь оставляли для разделения фаз. Органический слой промывали 126 мл 3% раствора сульфата натрия, 126 мл 0,5н раствора гидроксида натрия, 126 мл 1н соляной кислоты и 126 мл 10% насыщенного солевого раствора. Продукт сушили безводным сульфатом натрия или фильтровали на целите, после чего растворитель метиленхлорид удаляли путем перегонки при атмосферном давлении. Продукт два раза кристаллизовали из метилэтилкетона и получали 7,2 г эпоксимексренона.
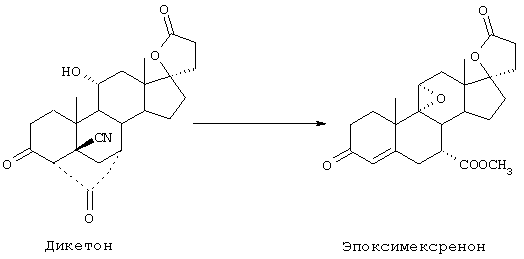
Пример 28.
Схема 1: Стадия 3: Вариант 2: Превращение 1'S(4'α),7'α-гексадекагидро-11'α-гидрокси-10'β,13'β-диметил-3',5,20'-три-оксоспиро[фуран-2(3Н), 17'β(4,7)-метано[17Н]циклопента[а]фенантрен]-5'β(2'Н)-карбонитрила в метилгидро-9,11α-эпокси-17α-гидрокси-3-оксопрегн-4-ен-7α,21-дикарбоксилат, γ-лактон, без выделения промежуточного соединения.
4-х горлая круглодонная 5 л-колба была снабжена механической мешалкой, капельной воронкой с трубкой для подачи азота, термометром и холодильником с барботером, присоединенным к скрубберу с гипохлоритом натрия. В колбу в 3,05 л метанола добавляли дикетон (83,20 г). В капельную воронку загружали 67,85 г 25% (масс./масс.) раствора метоксида натрия в метаноле. При перемешивании в атмосфере азота в колбу в течение 15 минут по каплям добавляли метоксид. В результате образовывалась темно-оранжевая/желтая суспензия. Реакционную смесь нагревали с обратным холодильником в течение 20 часов и по каплям добавляли 175 мл 4н хлористоводородной кислоты, продолжая при этом нагревание с обратным холодильником. (Осторожно, во время этой реакции выделяется HCN!). Обратный холодильник меняли на головную насадку с отводом и 1,6 л метанола удаляли путем дистилляции, добавляяя по каплям через воронку 1,6 л водного 10% раствора хлорида натрия в соответствии со скоростью дистилляции. Реакционную смесь охлаждали до комнатной температуры и дважды экстрагировали 2,25 л аликвотами метиленхлорида. Объединенные экстракты промывали холодными 750 мл аликвотами 1н гидроксида натрия и насыщенным раствором хлорида натрия. Органический слой сушили путем азеотропной перегонки метанола под давлением в одну атмосферу до конечного объема 1 л (0,5% от всего продукта было взято для анализа).
Концентрированный органический раствор (сложный гидроксиэфир) снова добавляли в первоначальную реакционную колбу, снабженную как описано ранее, но без ловушки для HCN. Эту колбу охлаждали до 0°С и добавляли 30,7 г метансульфонилхлорида, перемешивая в атмосфере азота. В капельную воронку в течение 15 минут по каплям вводили 32,65 г триэтиламина, поддерживая температуру при 5°С. Перемешивание продолжали в течение 2 часов, при этом реакционная смесь нагревалась до комнатной температуры. Затем приготавливали колонку, содержащую 250 г кислой ионообменной смолы Dowex 50 W × 8-100, и перед этим ее промывали 250 мл воды, 250 мл метанола и 500 мл метиленхлорида. Реакционную смесь удаляли из этой колонки и собирали. Затем готовили свежую колонку и вышеуказанную процедуру повторяли. После этого готовили третью колонку, содержащую 250 г основной ионообменной смолы Dowex 1 × 8-200 и предварительно обработанную как и колонка с кислой смолой, описанная выше. Реакционную смесь удаляли из этой колонки и собирали. Затем готовили четвертую колонку с ионообменной смолой и реакционную смесь снова удаляли и собирали. Затем через каждую колонку пропускали две 250 мл порции метиленхлоридных промывок и каждый прогон требовал ˜10 минут. Промывки растворителем объединяли с реакционной смесью и концентрировали в вакууме до объема ˜500 мл, а 2% от этого объема удаляли для количественной оценки. Затем остаток концентрировали до конечного объема 150 мл (неочищенный раствор мезилата).
В первоначальный 5-литровый реакционный аппарат добавляли 960 мл муравьиной кислоты, 472 мл уксусного ангидрида и 23,70 г формиата калия. Эту смесь нагревали при перемешивании в атмосфере азота до 70°С в течение 16 часов. Затем температуру повышали до 100°С и в течение 30 минут через капельную воронку добавляли неочищенный раствор мезилата. По мере отгонки метиленхлорида из реакционной смеси температуру понижали до 85°С. После того, как все вещество было удалено, температура снова повышалась до 100°С и ее поддерживали в течение 2,5 часов. Реакционную смесь охлаждали до 40°С, и муравьиную кислоту удаляли под давлением до достижения минимального объема перемешивания (˜150 мл). Остаток охлаждали до комнатной температуры и добавляли 375 мл метиленхлорида. Разбавленный остаток промывали холодными 1-литровыми порциями насыщенного раствора хлорида натрия, 1н карбоната натрия и снова раствора хлорида натрия. Органическую фазу сушили сульфатом магния (150 г) и фильтровали с получением темного рыжевато-коричневого раствора (неочищенного раствора сложного енэфира).
4-Горлый 1-литровый реактор с рубашкой снабжали механической мешалкой, холодильником/барботером, термометром и капельной воронкой с трубкой для подачи азота. В реактор загружали неочищенный раствор сложного енэфира (приблизительно 60 г) в 600 мл метиленхлорида с перемешиванием в атмосфере азота. После этого добавляли 24,0 г двухосновного фосфата калия, а затем 87 мл трихлорацетонитрила. Через рубашку реактора подавали воду для внешнего охлаждения и реакционную смесь охлаждали до 10°С. Эту смесь в течение 30 минут добавляли в капельную воронку с 147 мл 30% перекиси водорода. После завершения добавления первоначально окрашенная в темный рыжевато-коричневый цвет реакционная смесь приобретала светло-желтую окраску. В течение всего периода добавления реакционную смесь поддерживали при 10±1°С и перемешивание продолжали в течение ночи (всего 23 часа). Фазы отделяли и водную часть дважды экстрагировали 120 мл порциями метиленхлорида. Затем объединенные органические фазы промывали 210 мл 3% раствора сульфита натрия. Эту процедуру повторяли еще раз, после чего органические и водные части обнаруживали отрицательную реакцию на пероксид, о чем свидетельствовала индикаторная бумага с крахмалом/иодидом. Органическую фазу последовательно промывали 210 мл аликвотами холодного 1н гидроксида натрия, 1н хлористоводородной кислоты и, наконец, двумя промывками насыщенным солевым раствором. Органическую фазу сушили путем азеотропной перегонки до объема ˜100 мл, затем добавляли свежий растворитель (250 мл) и подвергали азеотропной перегонке аналогично до ˜100 мл и оставшийся растворитель удаляли в вакууме с получением 57,05 г сырого продукта в виде смолистой желтой пены. Затем часть (51,01 г) продукта сушили до постоянной массы 44,3 г и количественно оценивали с помощью ВЭЖХ. Анализ указывал на 27,1% эпоксимексренона.
Пример 29. Получение 3-этокси-11α-гидрокси-андроста-3,5-диен-17-она из 11α-гидроксиандростендиона
В реакционную колбу в атмосфере азота вводили 11α-гидроксиандростендион (429,5 г) и гидрат толуолсульфоновой кислоты (7,1). В реактор добавляли этанол (2,58 л) и полученный раствор охлаждали до 5°С. В этот раствор в течение 15 минут добавляли триэтилортоформиат (334,5 г) при температуре от 0°С до 15°С. После завершения добавления триэтилформиата реакционную смесь нагревали до 40°С и оставляли для реакции при этой температуре на 2 часа, после чего температуру повышали до температуры нагревания с обратным холодильником, и реакцию продолжали при нагревании с обратным холодильником в течение еще 3 часов. Реакционную смесь охлаждали в вакууме и растворитель удаляли в вакууме с получением 3-этокси-11α-гидроксиандроста-3,5-диен-17-она.
Пример 30. Получение енамина из 11α-гидроксиканренона
Схема 1: Стадия 1: Метод В: Получение 5'R(5'α),7'β-20'-аминогексадекагидро-11'β-гидрокси-10'α,13'α-диметил-3',5-диоксоспиро[фуран-2(3Н),17'α(5'Н)-[7,4]метено[4Н[циклопента[а]-фенантрен]-5'-карбонитрила.
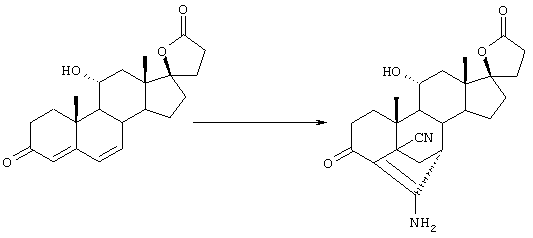
Цианид натрия (1,72 г) помещали в 25 мл 3-горлую колбу, снабженную механической мешалкой. Затем добавляли воду (2,1 мл) и смесь перемешивали при нагревании до тех пор, пока твердые вещества не растворялись. После этого добавляли диметилформамид (15 мл), а затем 11α-гидроксиканренон (5,0 г). К полученной смеси добавляли смесь воды (0,4 мл) и серной кислоты (1,49 г). Эту смесь нагревали до 85°С в течение 2,5 часа, за которые ВЭЖХ-анализ показал на полное превращение в продукт. Реакционную смесь охлаждали до комнатной температуры. Затем добавляли серную кислоту (0,83 г) и смесь перемешивали в течение получаса. Реакционную смесь добавляли к 60 мл воды, охлажденной в ледяной бане. Колбу промывали 3 мл ДМФ и 5 мл воды. Суспензию перемешивали в течение 40 минут и фильтровали. Осадок на фильтре дважды промывали 40 мл воды и сушили в вакуумной печи при температуре 60°С в течение ночи с получением 11α-гидроксиенамина, т.е. 5'R(5'α),-7'β-20'-аминогексадекагидро-11'β-гидрокси-10'α,13'α-диметил-3',5-диоксоспиро[фуран-2(3Н),-17'α(5'Н)-[7,4]метено[4Н]циклопента[а]фенантрен]-5'-карбонитрила (4,9 г).
Пример 31. Превращение 11α-гидроксиканренона в дикетон в одном резервуаре

В 50 мл 3-горлую колбу, снабженную механической мешалкой, добавляли цианид натрия (1,03 г). После добавления воды (1,26 мл) колбу слегка нагревали для растворения твердого вещества. Затем добавляли диметилацетамид [или диметилформамид] (9 мл), после чего 11α-гидроксиканренон (3,0 г). В реакционную колбу при перемешивании добавляли смесь серной кислоты (0,47 мл) и воды (0,25 мл). Полученную смесь нагревали до 95°С в течение 2 часов. ВЭЖХ-анализ указывал на завершение реакции. Затем добавляли серную кислоту (0,27 мл) и смесь перемешивали в течение 30 минут. После этого вводили дополнительное количество воды (25 мл) и серной кислоты (0,90 мл) и реакционную смесь перемешивали в течение 16 часов. Затем смесь охлаждали в ледяной бане до 5-10°С. Твердое вещество выделяли путем фильтрования через фильтр из спеченного стекла с последующей двойной промывкой водой (20 мл). Твердый дикетон, т.е. 4'S(4'α),7'α-гексадекагидро-11'α-гидрокси-10'β,13'β-диметил-3',5-20'-триоксоспиро[фуран-2(3Н),-17'β(4,7)метано[17Н]циклопента[а]фенантрен]-5'β(2'Н)-карбонитрил, сушили в вакуумной печи с получением 3,0 г твердого вещества.
Пример 32А-1.
Схема 1: Стадия 3А: Метод В: Получение метилгидро-11α,17α-дигидрокси-3-оксопрегн-4-ен-7α,21-дикарбоксилата, γ-лактона.
Суспензию 5,0 г дикетона, полученного способом, описанным в Примере 31, в метаноле (100 мл) нагревали с обратным холодильником и в течение 1 минуты добавляли 25% раствор метоксида калия в метаноле (5,8 мл). Смесь становилась гомогенной. Через 15 минут появлялся осадок. Смесь нагревали с обратным холодильником и примерно через 4 часа она снова становилась гомогенной. Нагревание с обратным холодильником продолжали полные 23,5 часа и добавляли 4,0 н HCl (10 мл). Все 60 мл раствора цианистого водорода в метаноле удаляли путем дистилляции. К остатку, полученному в результате дистилляции, в течение 15 минут добавляли воду (57 мл). Во время добавления воды температуру раствора повышали до 81,5°С и еще 4 мл раствора цианистого водорода/метанола удаляли путем дистилляции. После того как добавление воды было завершено, смесь становилась мутной и источник нагревания удаляли. Полученную смесь перемешивали в течение 3,5 часа и продукт медленно кристаллизовался. Суспензию фильтровали и собранное твердое вещество промывали водой, сушили в потоке воздуха на воронке, а затем сушили при 92°С (26 дюйм рт.ст.) в течение 16 часов с получением 2,98 г беловатого твердого вещества. Это твердое вещество представляло собой 91,4% сложного гидроксиэфира, т.е. метилгидро-11α,17α-дигидрокси-3-оксопрегн-4-ен-7α,21-карбоксилата, γ-лактона (по массе). Выход составлял 56,1%.
Пример 32А-2. Получение метилгидро-11α,17α-дигидрокси-3-оксопрегн-4-ен-7α,21-дикарбоксилата, γ-лактона.
Дикетон (40 г), полученный способом, описанным в Примере 31, загружали в очищенный, сухой 1-литровый реактор с рубашкой, снабженный нижним сливным отводом, холодильником, датчиком RTD и сборником для сбора фракций. В этот реактор загружали метанол (800 мл) и смесь перемешивали. Полученную суспензию нагревали до около 60-65°С и добавляли 25% раствор метоксида калия (27,8 мл). Смесь становилась гомогенной.
Полученную смесь нагревали с обратным холодильником. После примерно 1,5-часового нагревания с обратным холодильником к смеси при нагревании с обратным холодильником добавляли еще 16,7 мл 25% раствора метоксида калия. Смесь поддерживали при нагревании с обратным холодильником еще 6 часов. Превращение дикетона в сложный гидроксиэфир анализировали с помощью ВЭЖХ. После того, как ВЭЖХ-анализ показал отношение дикетона к сложному гидроксиэфиру менее около 10%, к смеси в течение около 15 минут при нагревании с обратным холодильником добавляли 77 мл 4М HCl (хлористоводородная кислота может быть заменена сравнимым количеством 1,5М-3М серной кислоты).
Затем смесь подвергали дистилляции и около 520 мл дистиллята метанола/HCN собирали и отбрасывали. Концентрированную смесь охлаждали до около 65°С. К смеси в течение 90 минут добавляли около 520 мл воды и во время добавления температуру поддерживали при около 65°С. Смесь постепенно охлаждали до около 15°С приблизительно в течение четырех часов, а затем перемешивали и поддерживали при температуре около 15°С еще в течение двух часов. Смесь фильтровали и отфильтрованный продукт два раза промывали около 200 мл воды (каждый раз). Отфильтрованный продукт сушили в вакууме (90°С, 25 мм рт.ст.). Получали приблизительно 25-27 г беловатого твердого вещества, содержащего, в основном, метилгидро-11α,17α-дигидрокси-3-оксопрегн-4-ен-7α,21-дикарбоксилат, γ-лактон.
Пример 32В-1. Получение 7-метилгидро-5β-циано-11α,17-дигидрокси-3-оксо-17α-прегнан-7α,21-дикарбоксилата, γ-лактона
В реакционную колбу загружали 4,1 г дикетона, полученного способом, описанным в Примере 31, 75 мл метанола и 1 мл 1н метанолового раствора гидроксида натрия. Суспензию перемешивали при комнатной температуре. Через несколько минут получали гомогенный раствор, а примерно через 20 минут наблюдался осадок. Перемешивание продолжали еще в течение 70 минут при комнатной температуре. По истечении этого времени твердый осадок фильтровали и промывали метанолом. Твердый осадок сушили в паровой камере, в результате чего получали 3,6 г 7-метилгидро-5β-циано-11α,17-дигидрокси-3-оксо-17α-прегнан-7α,21-дикарбоксилата, γ-лактона.
1Н-ЯМР (CDCl3) м.д. 0,95(с, 3Н), 1,4(с, 3Н), 3,03(д, 1Н, J=15), 3,69(с, 3Н), 4,1(м, 1Н).
13С-ЯМР (CDCl3) м.д. 14,6, 19,8, 22,6, 29,0, 31,0, 33,9, 35,17, 35,20, 36,3, 37,7, 38,0, 38,9, 40,8, 42,8, 43,1, 45,3, 45,7, 47,5, 52,0, 68,0, 95,0, 121,6, 174,5, 176,4, 207,0.
Пример 32В-2. Получение 7-метилгидро-5β-циано-9,11α-эпокси-17-гидрокси-3-оксо-17α-прегнан-7α,21-дикарбоксилата, γ-лактона
К 2,0 г (4,88 моль) 9,11-эпоксидикетона формулы 21, суспендированного в 30 мл безводного метанола, добавляли 0,34 мл (2,4 ммоль) триэтиламина. Суспензию нагревали с обратным холодильником и через 4,5 часа не наблюдалось присутствия исходного материала, на что указывала ВЭЖХ (Zorbax SB-C8, 150 × 4,6 мм, 2 мл/мин, линейный градиент 35:65 А:В-45:55 А:В в течение 15 минут, А=ацетонитрил/метанол 1:1, В=вода/0,1% трифторуксусная кислота, детекция при 210 нм). Смесь оставляли для охлаждения и выдерживали при около 26°С в течение около 16 часов. Полученную суспензию фильтровали и получали 1,3 г 7-метилгидро-5β-циано-9,11α-эпокси-17-гидрокси-3-оксо-17α-прегнан-7α,21-дикарбоксилата, γ-лактона, в виде белого твердого вещества. Фильтрат концентрировали досуха на роторном испарителе, а остаток растирали с 3-5 мл метанола. После фильтрации получали еще 260 мг 7-метилгидро-5β-циано-9,11α-эпокси-17-гидрокси-3-оксо-17α-прегнан-7α,21-дикарбоксилата, γ-лактона. Выход составлял 74,3%.
1Н ЯМР (400 МГц, дейтерохлороформ) δ: 1,00(с, 3Н), 1,45 (м, 1Н), 1,50(с, 3Н), 1,65(м, 2Н), 2,10(м, 2Н), 2,15-2,65 (м, 8Н), 2,80(м, 1Н), 2,96(м, 1Н), 3,12(д, J=13, 1Н), 3,35(д, J=7, 1Н), 3,67 (с, 3Н).
Пример 32С. Получение 5β-циано-11-α,17-дигидрокси-3-оксо-17α-прегнан-7α,21-дикарбоновой кислоты, γ-лактона
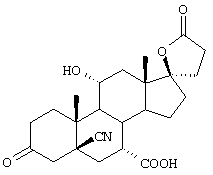
В реакционную колбу загружали 6,8 г дикетона (полученного способом, описанным в Примере 31), 68 мл ацетонитрила, 6,0 г ацетата натрия и 60 мл воды. Смесь нагревали и перемешивали с обратным холодильником. Через 1,5 часа смесь становилась почти гомогенной. Через 3 часа добавляли 100 мл воды, по мере дистилляции 50 мл ацетонитрила. Смесь охлаждали и осажденное твердое вещество (1,7 г) удаляли путем фильтрации. Фильтрат (рН=5,5) обрабатывали соляной кислотой для снижения рН примерно до 4,5 и твердое вещество осаждалось. Твердое вещество выделяли, промывали водой и сушили, в результате чего получали 4,5 г 5β-циано-11-α,17-дигидрокси-3-оксо-17α-прегнан-7α,21-дикарбоновой кислоты, γ-лактона.
1Н-ЯМР (ДМСО) м.д. 0,8(с, Н), 1,28(с, Н), 3,82(м, 1Н).
13С-ЯМР (ДМСО) м.д. 14,5, 19,5, 22,0, 28,6, 30,2, 33,0, 34,1, 34,4, 36,0, 37,5, 37,7, 38,5, 42,4, 42,6, 45,08, 45,14, 47,6, 94,6, 122,3, 176,08, 176,24, 207,5.
Пример 33.
Схема 1: Стадия 3А: Метод С: Получение метилгидро-11α,17α-дигидрокси-3-оксопрегн-4-ен-7α,21-дикарбоксилата, γ-лактона
Дикетон (30 г), полученный способом, описанным в Примере 31, загружали в очищенную и осушенную 3-горлую реакционную колбу, снабженную термометром, ловушкой Дина-Старка и механической мешалкой. Метанол (24 мл) загружали в реактор при комнатной температуре (22°С) и полученную суспензию перемешивали в течение 5 минут. В реактор загружали 25% масс. раствора метоксида натрия в метаноле (52,8 мл) и полученную смесь перемешивали при комнатной температуре 10 минут, в течение которых реакционная смесь превращалась в светло-коричневый прозрачный раствор и наблюдалось небольшое повышение температуры (на 2-3°С). Скорость добавления регулировали для предупреждения повышения температуры в резервуаре свыше 30°С. Затем смесь нагревали с обратным холодильником (около 67°С) и продолжали нагревать с обратным холодильником в течение 16 часов. Затем брали образец и анализировали с помощью ВЭЖХ на превращение. Реакционную смесь продолжали нагревать с обратным холодильником до тех пор, пока уровень остаточного дикетона не стал превышать 3% от загрузки дикетона. Во время нагревания с обратным холодильником в реакционный резервуар загружали 4н HCl (120 мл), в результате чего образовывался HCN, который гасили в скруббере.
После окончания реакции из реакционной смеси отгоняли 90-95% метанолового растворителя при атмосферном давлении. Температура в головной части в процессе дистилляции варьировалась от 67 до 75°С, а дистиллят, который содержал HCN, обрабатывали каустической содой и отбеливателем, а затем утилизовывали. После удаления метанола реакционную смесь охлаждали до комнатной температуры и по мере охлаждения смеси в пределах 40-45°С начинал осаждаться твердый продукт. Водный раствор, необязательно содержащий 5% масс. бикарбоната натрия (1200 мл) при 25°С, загружали в охлажденную суспензию, а затем полученную смесь охлаждали до 0°С примерно в течение 1 ч. Обработка бикарбонатом калия является эффективной для удаления остаточного непрореагировавшего дикетона из реакционной смеси. Суспензию перемешивали при 0°С в течение 2 часов для завершения осаждения и кристаллизации, после чего твердый продукт, метилгидро-11α,17α-дигидрокси-3-оксопрегн-4-ен-7α,21-дикарбоксилат, γ-лактон, выделяли путем фильтрации и осадок на фильтре промывали водой (100 мл). Продукт сушили при 80-90°С в вакууме под давлением 26" рт.ст. (дюйм мм.рт.) до постоянной массы. Содержание воды после сушки составляло менее 0,25% масс. Скорректированный молярный выход составлял в пределах 77-80% масс.
Пример 34.
Схема 1: Стадия 3А: Метод D: Получение метилгидро-11α,17α-дигидрокси-3-оксопрегн-4-ен-7α,21-дикарбоксилата, γ-лактона
Дикетон, полученный способом, описанным в Примере 31 (1 экв.), подвергали реакции с метоксидом натрия (4,8 экв.) в метаноловом растворителе в присутствии иодида цинка (1 экв.). Обработку продукта проводили либо способом, предусматривающим экстракцию и описанным в настоящей заявке, либо способом без экстракции, в котором отсутствовали стадии экстракции метиленхлоридом, промывки насыщенным солевым раствором и каустической содой и сушки сульфатом натрия. Кроме того, в способе, не предусматривающем экстракцию, толуол заменяли 5% масс. раствором бикарбоната натрия. В качестве продукта выделяли метилгидро-11α,17α-дигидрокси-3-оксопрегн-4-ен-7α,21-дикарбоксилат, γ-лактон.
Пример 35.
Схема 1: Стадия 3С: Метод С: Получение метилгидро-17α-гидрокси-3-оксопрегна-4,9(11)-диен-7α,21-дикарбоксилата, γ-лактона
Сложный гидроксиэфир, полученный как описано в Примере 34 (1,97 г), объединяли с тетрагидрофураном (20 мл) и полученную смесь охлаждали (-70°С). Затем добавляли сульфурилхлорид (0,8 мл) и смесь перемешивали в течение 30 минут, после чего добавляли имидазол (1,3 г). Реакционную смесь нагревали до комнатной температуры и перемешивали еще в течение 2 часов. Затем смесь разбавляли метиленхлоридом и экстрагировали водой. Органический слой концентрировали с получением неочищенного продукта метилгидро-17α-гидрокси-3-оксопрегна-4,9(11)-диен-7α,21-дикарбоксилата, γ-лактона (1,97 г). Небольшой образец неочищенного продукта анализировали с помощью ВЭЖХ. Анализ показал, что отношение 9,11-олефин:11,12-олефин:7,9-лактон составляло 75,5:7,2:17,3. Если реакцию осуществляли при 0°С или иначе, как описано выше, то эта реакция давала продукт, в котором соотношение 9,11-олефин:11,12-олефин:7,9-лактон составляло 77,6:6,7:15,7. Эту процедуру объединяли в одну стадию введения уходящей группы и ее удаления для последующего введения 9,11-олефиновой структуры сложного енэфира, т.е. реакция с сульфурилхлоридом приводит к замене 11α-гидроксигруппы сложного гидроксиэфира формулы V на галогенид с последующим дегидрогалогенированием с образованием Δ9,11-структуры. Таким образом, образование сложного енэфира эффективно протекает без использования сильной кислоты (такой как муравьиная кислота) или осушителя, такого как уксусный ангидрид. Также исключается стадия нагревания с обратным холодильником, осуществляемая в альтернативном способе, в котором образуется моноокись углерода.
Пример 36А.
Схема 1: Стадия 3С: Метод D: Получение метилгидро-17α-гидрокси-3-оксопрегна-4,9(11)-диен-7α,21-дикарбоксилата, γ-лактона.
Сложный гидроксиэфир (20 г), полученный как описано в Примере 34, и метиленхлорид (400 мл) добавляли в очищенную сухую трехгорлую круглодонную колбу, снабженную механической мешалкой, капельной воронкой и термопарой. Полученную смесь перемешивали при комнатной температуре до полного завершения образования раствора. Раствор охлаждали до 5°С с использованием ледяной бани. К раствору СН2Cl2, содержащему сложный гидроксиэфир, добавляли метансульфонилхлорид (5 мл), а затем сразу по каплям медленно добавляли триетиламин (10,8 мл). Скорость добавления корректировали так, чтобы температура реакции не превышала 5°С. Эта реакция была очень экзотермичной, а поэтому нуждалась в охлаждении. Реакционную смесь перемешивали в течение 1 ч при около 5°С. По завершении реакции (ВЭЖХ- и ТСХ-анализы) смесь концентрировали при около 0°С под давлением 26 дюйм рт.ст. до тех пор, пока не образовывалась густая суспензия. Полученную суспензию разбавляли CH2Cl2 (160 мл) и смесь концентрировали при около 0°С под давлением 26 дюйм рт.ст. с получением концентрата. Было установлено, что чистота концентрата (мезилатный продукт формулы IV, где R3=H и -А-А- и -В-В- представляют -CH2-CH2-, то есть превращение метилгидро-11α,17α-дигидрокси-3-оксо-прегн-4-ен-7α,21-дикарбоксилата, γ-лактона в метилгидро-17α-гидрокси-11α-(метилсульфонил)окси-3-оксопрегн-4-ен-7α,21-ди-карбоксилат, γ-лактон) составляет 82% (% площади, ВЭЖХ). Этот материал использовали в следующей реакции без выделения.
В чистый сухой реактор, снабженный механической мешалкой, холодильником, термопарой и кожухом для нагревания, добавляли формиат калия (4,7 г), муравьиную кислоту (16 мл) и уксусный ангидрид (8 мл, 0,084 моль). Полученный раствор нагревали до 70°С и перемешивали в течение примерно 4-8 часов. Добавление уксусного ангидрида приводило к экзотермической реакции и к образованию газа (СО), а поэтому для регулирования температуры и газообразования (давления) скорость добавления соответствующим образом корректировали. Время реакции для получения активного элиминирующего реагента зависело от количества воды, присутствующей в реакции (муравьиная кислота и формиат калия содержали около 3-5% воды, каждый). Реакция элиминирования является чувствительной к количеству присутствующей воды; то есть, если это количество >0,1% воды (KF), то уровень 7,9-лактонной примеси может быть увеличен. Этот побочный продукт трудно удалить из конечного продукта. Если KF составляет <0,1% воды, то в концентрат мезилата (0,070 моль), полученный в предшествующей стадии, вводили активный элиминирующий агент. Полученный раствор нагревали до 95°С, а летучий материал отгоняли и собирали в ловушке Дина-Старка. При прекращении выделения летучего материала ловушку Дина-Старка заменяли холодильником и реакционную смесь нагревали еще в течение 1 часа при 95°С. По истечении этого времени (ТСХ- и ВЭЖХ-анализ; <0,1% исходного материала) содержимое охлаждали до 50°С и начинали дистилляцию в вакууме (26 дюйм рт.ст./50°С). Смесь концентрировали до получения густой суспензии, а затем охлаждали до комнатной температуры. Полученную суспензию разбавляли этилацетатом (137 мл) и раствор перемешивали в течение 15 мин, а затем разбавляли водой (137 мл). Слои разделяли и водный слой снова экстрагировали этилацетатом (70 мл). Объединенный этилацетатный раствор один раз промывали насыщенным солевым раствором (120 мл) и два раза охлажденным на льду 1н раствором NaOH (120 мл, каждый). рН водного слоя измеряли, и если рН отработанной промывки составлял <8, то органический слой снова промывали. Если рН отработанной промывки составлял >8, то этилацетатный слой один раз промывали солевым раствором (120 мл) и концентрировали досуха на роторном испарителе с использованием водяной бани 50°С. В результате получали 92 г сложного енэфира, твердого продукта, то есть метилгидро-17α-гидрокси-3-оксопрегна-4,9(11)-диен-7α,21-дикарбоксилата, γ-лактона (выход 77% моль).
Пример 36В. Получение метилгидро-17α-гидрокси-3-оксо-прегна-4,9(11)-диен-7α,21-дикарбоксилата, γ-лактона.
В чистую сухую трехгорлую круглодонную 250 мл колбу, снабженную механической мешалкой, капельной воронкой и термопарой, добавляли 25 г (53,12 ммоль) сложного гидроксиэфира метилгидро-11α,17α-дигидрокси-3-оксопрегна-4-ен-7α,21-дикарбоксилата, γ-лактона, полученного как описано в Примере 34, а затем 150 мл метиленхлорида (Burdick & Johnson). Полученную смесь перемешивали при комнатной температуре до тех пор, пока не была получена светлая суспензия. Раствор охлаждали до -5°С в бане со льдом. К раствору метиленхлорида, содержащему сложный гидроксиэфир, добавляли метансульфонилхлорид (7,92 г, 69,06 ммоль) (Aldrich), а затем сразу по каплям медленно добавляли триэтиламин (7,53 г) (Aldrich). Скорость добавления корректировали так, чтобы температура реакции не превышала 0°С. Эта реакция была очень экзотермичной; а поэтому она нуждалась в охлаждении. Время добавления составляло 35 минут. Реакционную смесь перемешивали при около 0°С еще 45 минут. При завершении реакции (когда оставалось менее чем 1% гидроксиэфира, на что указывали ВЭЖХ- и ТСХ-анализы), смесь концентрировали путем упаривания приблизительно 110-125 мл метиленхлоридного растворителя при атмосферном давлении. Во время выпаривания температура реакции достигала приблизительно 40-45°С. Если еще через 45 минут реакция не завершалась, то в реактор может быть загружено еще 0,1 эквивалента метансульфонилхлорида и еще 0,1 эквивалента триэтиламина с последующей оценкой реакции на завершение. Полученная смесь содержала неочищенный продукт метилгидро-17α-гидрокси-11α-(метилсульфонил)окси-3-оксопрегн-4-ен-7α,21-дикарбоксилат, γ-лактон. Этот продукт был использован в последующей реакции без выделения.
Во второй 250-миллилитровый очищенный сухой реактор, снабженный механической мешалкой, холодильником, термопарой и кожухом для нагревания, добавляли безводный ацетат натрия (8,7 r)(Mallinkrodt), ледяную уксусную кислоту (42,5 мл)(Fisher) и уксусный ангидрид (0,5 мл)(Fisher). Полученный раствор нагревали до 90°С и перемешивали около 30 минут. Поскольку добавление уксусного ангидрида сопровождается повышением температуры, добавление проводили при такой скорости, чтобы она корректировала как температуру, так и давление. Уксусный ангидрид добавляли для снижения содержания воды в растворе до приемлемого уровня (менее чем около 0,1%). Если KF указывал на <0,1% воды, то раствор уксусной кислоты переносили в концентрат мезилата, полученного, как сообщалось в первом разделе этого примера. После переноса температура полученной смеси составляла от около 55°С до 60°С. Образование газа снижали с использованием уксусной кислоты и ацетата натрия вместо муравьиной кислоты и формиата калия, как описано в Примере 36А.
Полученную смесь нагревали до 135°С и поддерживали при той же температуре около 60-90 минут до тех пор, пока не прекращалось выделение летучих веществ. Летучие вещества, отгоняемые из смеси, собирали в ловушке Дина-Старка. После завершения реакции (ТСХ- и ВЭЖХ-анализ; <0,1% исходного вещества) источник нагревания удаляли. Когда температура смеси достигала 80°С, к этой смеси в течение 60-90 минут медленно добавляли 150 мл воды. По окончании добавления воды смесь охлаждалась до температуры от около 35°С до 45°С и начинала образовываться суспензия. Затем смесь охлаждали до 15°С и поддерживали при этой температуре от около 30 до 60 минут.
Полученную смесь фильтровали через стеклянную воронку.
Фильтрат промывали 100 мл воды. Затем фильтрат промывали второй раз еще 100 мл воды. Полученный фильтрат сушили при 70°С в вакууме с получением 25,0 г сухого енэфирного продукта, метилгидро-17α-гидрокси-3-оксопрегна-4,9(11)-диен-7α,21-дикарбоксилата, γ-лактона. ВЭЖХ-анализ указывал на присутствие 70% нужного 9,11-олефина, 15% 11,12-олефина и 5% 7,0-лактона.
Этот способ давал хороший результат (по сравнению с аналогичными способами), а именно: (i) снижает объемы растворителей, (ii) снижает количество отдельных рабочих стадий, необходимых для продуцирования сложного енэфира из сложного гидроксиэфира, (iii) позволяет избежать необходимости проведения промывок, (iv) при выделении конечного продукта позволяет заменить экстракцию осаждением водой; и (v) обеспечивает надежное элиминирование, которое ранее ассоциировалось с образованием смешанного ангидрида и выделением газа при использовании муравьиной кислоты вместо уксусной кислоты.
Пример 37А.
Схема 1: Стадия 3С: Метод Е: Получение метилгидро-17α-гидрокси-3-оксопрегна-4,9(11)-диен-7α,21-дикарбоксилата, γ-лактона.
В трехгорлую круглодонную 2 л колбу, снабженную механической мешалкой, капельной воронкой и термопарой, добавляли сложный гидроксиэфир (100 г, 0,22 моль). Баня с циркулирующим охлаждением была снабжена автоматическим регулятором температуры. Перед началом реакции колбу осушали из-за чувствительности метансульфонилхлорида к воде.
В колбу загружали метиленхлорид (1 л) и растворяли в нем сложный гидроксиэфир путем перемешивания. Раствор охлаждали до 0°С и в колбу через капельную воронку загружали метансульфонилхлорид (25 мл; 0,32 моль). В реакционную колбу через капельную воронку загружали триэтиламин (50 мл, 0,59 моль) и воронку промывали дополнительным количеством метиленхлорида (34 мл). Добавление триэтиламина было в высокой степени экзотермичным. Время добавления составляло около 10 мин при перемешивании и охлаждении. Загруженную смесь охлаждали до 0°С и выдерживали при этой температуре, размешивая при этом еще 45 минут, в течение которых головную область реакционной колбы продували азотом. Затем образец реакционной смеси анализировали с помощью тонкослойной хроматографии и высокоразрешающей жидкостной хроматографии для контроля за ходом реакции. После этого смесь перемешивали при 0°С еще 30 мин и снова оценивали на завершение реакции. Анализ показал, что на этой стадии реакция была, в основном, завершена; метиленхлоридный растворитель выпаривали при 0°С в вакууме при 26" рт.ст. Анализ дистиллята с помощью газовой хроматографии указывал на присутствие метансульфонилхлорида и триэтиламина. Затем в реакционную колбу загружали метиленхлорид (800 мл) и полученную смесь перемешивали в течение 5 минут при температуре в пределах 0-15°С. После этого растворитель снова выпаривали при 0-5°С под давлением 26" рт.ст. и получали мезилат формулы IV, где R3 представляет Н; -А-А- и -В-В-представляют -СН2-СН2-, a R1 представляет метоксикарбонил. Чистота продукта составляла около 90-95% (по площади).
Для получения элиминирующего реагента формиат калия (23,5 г; 0,28 моль), муравьиную кислоту (80 мл) и уксусный ангидрид (40 мл) смешивали в отдельном осушенном реакторе. Муравьиную кислоту и уксусный ангидрид подавали в реактор насосом и во время добавления уксусного ангидрида температуру поддерживали на уровне не более 40°С. Смесь элиминирующего реагента нагревали до 70°С для отвода воды из реакционной системы. Эту реакцию продолжали до тех пор, пока содержание воды не составляло ниже чем 0,3% масс., как было измерено с помощью анализа Карла Фишера. Затем раствор реагента для элиминации переносили в реактор, содержащий концентрированный сырой раствор мезилата, полученный как описано выше. Полученную смесь нагревали до максимальной температуры 95°С и летучий дистиллят собирали до тех пор, когда дистиллят уже не образовывался. Дистилляция прекращалась при около 90°С. После завершения дистилляции реакционную смесь перемешивали при 95°С еще 2 часа и завершение реакции контролировали с помощью тонкослойной хроматографии. Когда реакция была завершена, реактор охлаждали до 50°С, а муравьиную кислоту и растворитель удаляли из реакционной смеси в вакууме под давлением 26" рт.ст. при 50°С. Концентрат охлаждали до комнатной температуры, а затем вводили этилацетат (688 мл) и смесь этилацетата и концентрата перемешивали в течение 15 мин. На этой стадии вводили 12% солевой раствор (688 мл) для облегчения удаления из органической фазы водорастворимых примесей. Фазы оставляли на 20 минут для осаждения. Водный слой переносили в другой сосуд, в который было загружено дополнительное количество этилацетата (350 мл). Эту обратную экстракцию водного слоя осуществляли в течение 30 минут, после чего фазы оставляли для осаждения, а этилацетатные слои объединяли. К объединенным этилацетатным слоям добавляли насыщенный раствор хлорида натрия (600 мл) и перемешивание осуществляли в течение 30 минут. Затем фазы оставляли для осаждения. Водный слой был удален. Затем проводили дополнительную промывку хлоридом натрия (600 мл). Органическую фазу отделяли от второго отработанного промывочного раствора. После этого органическую фазу промывали 1н гидроксидом натрия (600 мл) при перемешивании в течение 30 минут. Фазы осаждали в течение 30 минут для удаления водного слоя. После оценки рН водного слоя было обнаружено, что он составлял >7. Дополнительную промывку насыщенным хлоридом натрия (600 мл) осуществляли в течение 15 минут. И, наконец, органическую фазу концентрировали в вакууме при 26 мм рт.ст. при 50°С и продукт выделяли путем фильтрации. После сушки конечный продукт представлял собой коричневое пенистое твердое вещество. Это вещество, кроме того, сушили при 45°С при пониженном давлении в течение 24 часов с получением 95,4 г енэфирного продукта, метилгидро-17α-гидрокси-3-оксопрегна-4,9(11)-диен-7α,21-дикарбоксилата, γ-лактона, который, как было проанализировано, составлял 68,8%. Молярный выход составлял 74,4%, который был скорректирован как на исходный сложный гидроксиэфир, так и на конечный сложный енэфир.
Пример 37В. Получение 7-метилгидро-17-метил-3-оксо-18-норпрегна-4,9(11),-13-триен-7α,21-дикарбоксилата
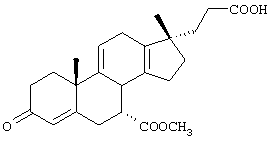
В реакционную колбу загружали 5,5 г мезилата, полученного способом, описанным в Примере 23, 55 мл 94,3% муравьиной кислоты и 1,38 г формиата калия. Смесь нагревали и перемешивали при нагревании с обратным холодильником (104°С) в течение двух часов. По истечении этих двух часов муравьиную кислоту отгоняли при пониженном давлении. Остаток растворяли в этилацетате и промывали 10%-ным карбонатом калия (50 мл). Выделенная водная часть имела желтую окраску. Этилацетат промывали 5%-ным гидроксидом натрия (50 мл). Водные части объединяли и подкисляли разбавленной хлористоводородной кислотой и нерастворившееся вещество экстрагировали этилацетатом. Этилацетат выпаривали досуха при пониженном давлении с получением 1,0 г остатка, 7-метилгидро-17-метил-3-оксо-18-норпрегна-4,9(11),13-триен-7α,21-дикарбоксилата.
1Н-ЯМР (CDCl3) м:.д.: 1,5(с, 3Н), 1,4(с, 3Н), 3,53(с, 3Н), 5,72(м, 1Н).
13С-ЯМР (CDCl3) м.д.: 25,1 и 25,4(18 СН3 и 19 СН3), 40,9 (10 С), 48,5(17 С), 51,4(ОСН3), 118,4(11 СН), 125,4(4 СН), 132,4 (9 С), 138,5 и 139,7(13 С и 14 С), 168,2(5 С), 172,4(7 СО), 179,6(22 СО), 198,9(3 СО).
Пример 37С. Получение 7-метилгидро-5β-циано-17-гидрокси-3-оксо-17α-прегн-11-ен-7α,21-дикарбоксилата, γ-лактона.
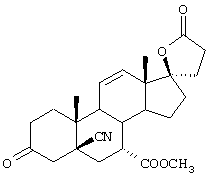
В реакционную колбу загружали 5,5 г мезилата, полученного как описано в Примере 23, 55 мл 94,3% муравьиной кислоты и 1,38 г формиата калия. Смесь нагревали и перемешивали с обратным холодильником (104°С) в течение двух часов. По истечении этих двух часов муравьиную кислоту отгоняли при пониженном давлении. Остаток растворяли в этилацетате и промывали 10% карбонатом калия (50 мл). Выделенная водная часть имела желтую окраску. Этилацетат промывали 5% гидроксидом натрия (50 мл). Этилацетат выпаривали досуха при пониженном давлении с получением 3,7 г остатка. Часть (3,4 г) остатка хроматографировали на 267 г силикагеля Merck (40-63 микрон). Продукт выделяли при схеме элюирования этилацетатом и толуолом (37:63)(об/об). После сушки этого продукта было получено 0,0698 г остатка, 7-метилгидро-5β-циано-17-гидрокси-3-оксо-17α-прегн-11-ен-7α,21-дикарбоксилата, γ-лактона.
1Н-ЯМР (CDCl3) м.д.: 1,03(с, 3Н), 1,22(с, 3Н), 3,70(с, 3Н), 5,60(д, 1Н, J=10), 5,98(д, 1Н, J=10).
МИК см-1: 2229 (CN), 1768 (лактон), 1710 (сложный эфир).
Пример 37D. Выделение 9α,17-дигидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоновой кислоты, бис(γ-лактона)
9α,17-Дигидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоновая кислота, бис(γ-лактон) является побочным продуктом элиминирования 11-мезилата. Чистый образец выделяли из реакционной смеси Примера 37А с помощью препаративной жидкостной хроматографии, а затем с помощью обращенно-фазовой препаративной ВЭЖХ. Таким образом, 73 г остатка хроматографировали на 2,41 кг силикагеля Merck (40-63 микрон) по схеме градиентного элюирования этилацетатом и толуолом (20:80, 30:70, 40:60, 60:40 об/об). Обогащенную смесь (10,5 г) енамина и 7,9-лактона получали во фракциях в соотношении 60:40. За ходом очистки следили с помощью ТСХ на пластинах с EMF, элюируя (60:40 об/об) этилацетатом и толуолом, и проводили визуализацию с помощью серной кислоты, SWUV. Затем часть (10,4 г) смеси очищали с помощью обращенно-фазовой ВЭЖХ на Kromasil C8 (7 мкм) с подвижной фазой: вода milliQ и ацетонитрил, 30:70 об/об. Из подвижной фазы был выделен 7,9-лактон (2,27 г) в виде кристаллов.
МИК см-1: 1762(7,9-лактон и 17-лактон), 1677, 1622(3-кето-Δ4,5).
1Н-ЯМР (CDCl3) м.д.: 1,00(с, 3Н), 1,4(с, 3Н), 2,05(д, 1Н), 2,78(д, 1Н), 5,87 (с, 1Н).
13С-ЯМР (CDCl3) м.д.: 13,2, 19,0, 22,2, 23,2, 26,8, 28,8, 29,5, 30,8, 33,1, 34,4, 35,1, 42,5, 43,6, 43,9, 45,0, 45,3, 89,9, 94,7, 129,1, 161,5, 176,0, 176,4, 196,9.
Вычислено: С 71,85 и Н 7,34; найдено: С 71,68 и Н 7,30.
Пример 37Е. Выделение 7-метилгидро-5-циано-17-гидрокси-3-оксо-17α-прегн-11-ен-7α,21-дикарбоксилата, γ-лактона
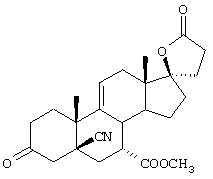
Соединение 7-метилгидро-5-циано-17-гидрокси-3-оксо-17α-прегн-11-ен-7α,21-дикарбоксилат, γ-лактон выделяли после проведения многократной препаративной жидкостной хроматографии реакционной смеси, полученной после реакции элиминирования 11-мезилокси-группы (Пример 24). Оно представляет собой часть скопления менее полярных примесей, как было оценено с помощью ТСХ на пластинках с EMF с использованием системы элюирования этилацетатом и метиленхлоридом (30:70 об/об) и визуализировано с помощью серной кислоты, SWUV. В основном, эти менее полярные примеси были выделены из сырого енамина посредством препаративной жидкостной хроматографии. В частности, 9,6 г сырого енаминового раствора хроматографировали на 534 г силикагеля Merck (40-63 микрон) с использованием схемы градиентного элюирования этилацетатом и толуолом (20:80, 30:70, 40:60, 60:40 об/об). Менее полярные примеси концентрировали во фракциях в соотношении 30:70. Этим же способом собирали 12,5 г пул из менее полярных примесей. Затем, этот продукт хроматографировали на 550 г силикагеля Merck (40-63 мкм) с использованием схемы градиентного элюирования этилацетатом и метиленхлоридом (5:95, 10:90, 20:80, 30:70 об/об). Обогащенная часть 20:80 фракций давала 1,2 г остатка. Дополнительная хроматография 1,2 г остатка на 53 г силикагеля Merck (40-63 мкм) с использованием градиента ацетона и метиленхлорида (3:97, 6:94, 10:90, 15:85 об./об.) давала 0,27 г 7-метилгидро-5-циано-17-гидрокси-3-оксо-17α-прегн-11-ен-7α,21-дикарбоксилата, γ-лактона из обогащенной части фракций 10:90.
МС М+425, вычислено для C25H31NO5 (425,52).
МИК 2222 см-1 (нитрил), 1767 см-1 (лактон), 1727 см-1 (сложный эфир и 3-кетон).
1Н-ЯМР (CDCl3) м.д. 0,92(с, 3Н), 1,47(с, 3Н), 2,95(м, 1Н), 3,65 (с, 3Н), 5,90(м, 1Н).
13С-ЯМР (CDCl3) м.д. 14,0(18 СН3), 23,5(15 CH2), 27,0 (19 СН3) 37,8, 38,5 и 40,9(7,8 и 14 СН), 52,0(ОСН3), 95,0(17 С), 121,5(23 CN), 123,5(11 СН), 135,3(9 С), 174,2 и 176,2 (22 и 24 СО), 206(3 СО).
Пример 37F. Получение 7-метилгидро-17-гидрокси-3-оксо-11α-(2,2,2-трифтор-1-оксоэтокси)-17α-прегн-4-ен-7α,21-дикарбоксилата
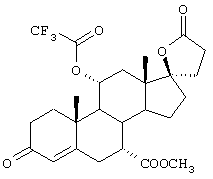
Сложный гидроксиэфир (2,0 г, 4,8 ммоль), полученный способом, описанным в Примере 34, добавляли в 40 мл метиленхлорида, находящегося в чистой сухой 3-горлой круглодонной колбе, снабженной механической мешалкой. Затем в раствор добавляли триэтиламин (0,61 г, 6,10 ммоль) и трифторуксусный ангидрид (1,47 г, 7,0 ммоль). Эту смесь перемешивали в течение ночи при комнатной температуре.
Затем смесь разбавляли еще 40 мл метиленхлорида. После этого смесь последовательно промывали 40 мл воды, 40 мл 1н HCl и 40 мл 1н раствора NaOH. Затем полученный раствор сушили сульфатом магния и концентрировали досуха, в результате чего получали 3,2 г светло-коричневого твердого вещества, 7-метилгидро-17-гидрокси-3-оксо-11α-(2,2,2-трифтор-1-оксоэтокси)-17α-прегн-4-ен-7α,21-дикарбоксилата.
Остаток анализировали и очищали с помощью хроматографии. Условия для ВЭЖХ: колонка - Waters Symmetry C18 (150 мм × 4,6 мм внут.диам.; размер частиц 5 микрон); темпрература колонки - комнатная; подвижная фаза - ацетонитрил/вода, 30/70 по объему; скрость потока - 1,0 мл/минут; объем инжекции - 20 микролитров; концентрация образца - 1,0 мг/мл; детекция УФ при 210 нм; давление - 1500 фунт/кв.дюйм (105,5 кг/см2); и время прогона - 45 минут. Условия для ТСХ: адсорбент - силикагель Merck 60 F254; система растворителей - этилацетат/толуол, 65/35 по объему; техника визуализации - коротковолновая; и количество нанесения - 100 микрограмм.
Пример 37G. Получение 7-метилгидро-11α-(ацетилокси)-17-гидрокси-3-оксо-17α-прегн-4-ен-7,21-дикарбоксилата, γ-лактона
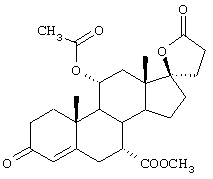
Сложный гидроксиэфир (2,86 г, 6,87 ммоль), полученный способом, описанным в Примере 34, добавляли в 40 мл метиленхлорида, находящегося в чистой сухой 3-горлой круглодонной колбе, снабженной механической мешалкой. Затем в раствор добавляли триэтиламин (1,39 г, 13,7 ммоль), диметиламинопиридин (0,08 г, 0,6 ммоль) и уксусный ангидрид (1,05 г, 10,3 ммоль). Эту смесь перемешивали в течение ночи при комнатной температуре.
Затем смесь разбавляли еще 150 мл этилацетата и 25 мл воды. После этого этилацетатный раствор промывали 25 мл раствором лимонной кислоты. Затем полученный раствор сушили сульфатом магния и концентрировали досуха, в результате чего получали 3,33 г светло-коричневого твердого вещества, 7-метилгидро-11α-(ацетилокси)-17-гидрокси-3-оксо-17α-прегн-4-ен-7,21-дикарбоксилата, γ-лактона.
Затем остаток анализировали и очищали с помощью хроматографии. Условия для ВЭЖХ: колонка - Waters Symmetry C18 (150 мм × 4,6 мм внут.диам.; размер частиц 5 микрон); температура колонки - комнатная; подвижная фаза - ацетонитрил/вода 30/70 по объему; скорость потока - 1,0 мл/минут; объем инжекции - 20 микролитров; концентрация образца - 1,0 мг/мл; детекция УФ при 210 нм; давление - 1500 фунт/кв.дюйм (105,5 кг/см2); и время прогона - 45 минут. Условия для ТСХ: адсорбент силикагель Merck 60 F254; система растворителей - метиленхлорид/метанол 95/5 по объему; техника визуализации - коротковолновая; и количество нанесения - 100 микрограмм.
Пример 37Н.
Схема 1: Стадия 3С: Метод F: Получение 7-метилгидро-17-гидрокси-3-оксо-17α-прегна-4,9(11)-диен-7,21-дикарбоксилата, γ-лактона
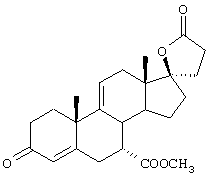
Формиат калия (1,5 г, 0,018 моль), муравьиную кислоту (60 мл, 1,6 мюль) и уксусный ангидрид (29,5 мл, 0,31 моль) добавляли в чистый, сухой 250 мл реактор, снабженный механической мешалкой, холодильником, термопарой и кожухом для нагревания. Затем раствор перемешивали 4 часа при 70°С и охлаждали до комнатной температуры, в результате чего получали реагент для элиминации, используемый для превращения мезилата формулы IV в продукт данного примера.
Предварительно полученный элиминирующий реагент ангидрид TFA/TFA добавляли к 70,0 г (0,142 моль) мезилата, полученного способом, описанным в Примере 23. Полученную смесь нагревали до 95-105°С в течение 2,5 часов, при этом степень превращения периодически контролировали с помощью ТСХ или ВЭЖХ. Полученный остаток охлаждали до 50°С, разбавляли ледяной водой (1,4 л) и перемешивали в течение 1 часа. Смесь оставляли для отстаивания в течение ночи при комнатной температуре. Слои отделяли и водную фазу снова эктрагировали этилацетатом (75 мл). Затем этилацетатный раствор последовательно промывали смесью воды/солевого раствора (70 мл), другой смесью воды/насыщенного солевого раствора (60 мл), 1н гидроксидом натрия (60 мл) и третьей смесью воды/насыщенного солевого раствора (60 мл). Крепость насыщенного солевого раствора составляла 12% масс. Затем этилацетатный раствор сушили сульфатом натрия, фильтровали и концентрировали досуха на роторном испарителе, в результате чего получали 4,5 г смеси нужного продукта и неизвестной примеси. Отношение примесь/продукт, определенное по ВЭЖХ-площади, составляло около 50/15, соответственно. Преобладающим продуктом этой реакции была примесь, которую идентифицировали как 7-метилгидро-17-гидрокси-3-оксо-17α-прегна-4,9(11)-диен-7,21-дикарбоксилат, γ-лактон.
Смесь очищали с помощью колоночной хроматографии и получали 1,9 г аналитически чистого 7-метилгидро-17-гидрокси-3-оксо-17α-прегна-4,9(11)-диен-7α,21-дикарбоксилата, γ-лактона.
Остаток снова анализировали и очищали с помощью хроматографии. Условия для ВЭЖХ: колонка - Waters Symmetry C18 (150 мм × 4,6 мм внут.диам.; размер частиц 5 микрон); темпрература колонки - комнатная; подвижная фаза - ацетонитрил/вода 30/70 по объему; скрость потока - 1,0 мл/минут; объем инжекции - 20 микролитров; концентрация образца - 1,0 мг/мл; УФ-детекция при 210 нм; давление - 1500 фунт/кв.дюйм (105,5 кг/см2); и время прогона - 45 минут. Условия для ТСХ: адсорбент силикагель Merck 60 F254; система растворителей - хлороформ/метил-трет-бутиловый эфир/изопропанол, 70/28/2 по объему; техника визуализации - 50% об. водной H2SO4/LWUV и 50% об. Н2SO4/фосфомолибденовая кислота; и количество нанесения - 100 микрограмм.
Пример 37I. Получение 7-метилгидро-17-гидрокси-3,11-диоксо-17α-прегн-4-ен-7α,21-дикарбоксилата, γ-лактона
Реактив Джонса получали путем растворения 6,7 г ангидрида хромовой кислоты (CrO3) в 6 мл концентрированной серной кислоты и эту смесь осторожно разбавляли дистиллированной водой до объема 50 мл. Одного мл этого реагента было достаточно для окисления 1 ммоль вторичного спирта с получением кетона.
Сложный гидроксиэфир (10,0 г, 24,0 ммоль), полученный способом, описанным в Примере 34, растворяли/суспендировали в 1200 мл ацетона. К этой смеси добавляли 8,992 мл реагента Джонса и объединенную смесь перемешивали в течение 10 минут. Аликвоту реакционной смеси после ее обработки водой и экстрагирования метиленхлоридом анализировали с помощью ВЭЖХ (колонка: Beckman Ultrasphere ODS С18, 4,6 мм × 250 мм, 5 микрон; градиент растворителя: ацетонитрил/вода=1/99-100/0 в течение 20 минут при скорости потока 1,5 мл/мин; детектор: УФ 210 нм). На завершение реакции указывало отсутствие какого-либо значимого количества исходного материала в реакционной смеси. Время удерживания для исходного материала (сложного гидроксиэфира) составляло 13,37 минут, а для продукта кетона - 14,56 минут.
Реакционную смесь обрабатывали путем добавления 200 мл воды и 300 мл метиленхлорида. Органический слой отделяли от водного слоя и снова промывали 200 мл воды. Органический слой отделяли от водного слоя и сушили сульфатом магния. Растворитель выпаривали с получением 9,52 г беловатого твердого вещества (выход сырого вещества 95,6%) 7-метилгидро-17-гидрокси-3,11-диоксо-17α-прегн-4-ен-7α,21-дикарбоксилата, γ-лактона.
Структуру подтверждали, исходя из масс-спектра (m/е 414). Н-ЯМР (ДМФ-d7) и С-ЯМР (ДМФ-d7). В Н-ЯМР характеристический пик 11-Н (4,51 м.д., дублет, J=5,8 Гц), обнаруженный в сложном гидроксиэфире, отсутствовал. В С-ЯМР был обнаружен пик при 208,97 м.д., который, предположительно, соответствует 11-кето-углероду.
С-ЯМР (400 МГц, ДМФ-d7) 208,97(11-кето), 197,70(3-кето), 176,00(22-лактон), 173,34(7-СООМе), 167,21(С5), 125,33 (С4), 93,63(С17) и другие пики в области от 15 до 57 м.д.
Пример 37J. Получение диметил-11α,17-дигидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоксилата, γ-лактона
Раствор 3,5 г (8,4 ммоль) сложного гидроксиэфира, полученного как описано в Примере 34, в 42 мл метанола смешивали с 4 мл метанолового 4н гидроксида калия (8 ммоль). Суспензию перемешивали в течение ночи при комнатной температуре и нагревали с обратным холодильником в течение одного часа. Метанол выпаривали в вакууме и остаток смешивали с 50 мл этилацетата. Этилацетат выпаривали в вакууме, а остаток гидролизовали 50 мл этилацетата. Осушенное твердое вещество объединяли с 50 мл ацетона и 2 мл метилиодида (32,1 ммоль). Смесь перемешивали при комнатной температуре в течение 18 часов. В течение этого времени большая часть твердых веществ растворялась. Смесь фильтровали и фильтрат выпаривали досуха в вакууме. Остаток гидролизовали этилацетатом, после чего твердые вещества удаляли путем фильтрации, а растворитель удаляли путем вакуумной дистилляции. Остаток определяли как смесь 78:22 (об./об.) диметил-11α,17-дигидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоксилата, γ-лактона и исходного материала сложного гидроксиэфира, как было определено с помощью 1Н-ЯМР. Эта смесь была пригодна для использования в качестве ВЭЖХ-маркера без дополнительной очистки.
1Н-ЯМР (CDCl3) показала следующий характерный спектр: м.д. 0,93(с, 3Н), 1,37(с, 3Н), 3,64(с, 3Н), 3,69(с, 3Н).
Пример 37К. Получение 11α,17-дигидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоксилата, γ-лактона
К 11,86 г (28,5 ммоль) сложного гидроксиэфира, полученного способом, описанным в Примере 34, добавляли 50 мл метанола и 20 мл 2,5М NaOH. Суспензию нагревали с обратным холодильником. Через 25 минут порцию исходного сложного эфира, который, как показала ВЭЖХ, осталась непрореагировавшей (Zorbax SB-C8 150 × 4,6 мм, 2 мл/мин, линейный градиент 35:65 А:В - 45:55 А:В в течение 15 минут, А=ацетонитрил/метанол 1:1, В=вода/0,1% трифторуксусная кислота, детекция при 210 нм) и добавляли 10 мл 10М NaOH. Через 1,5 часа, как показала ВЭЖХ, оставались лишь следовые количества непрореагировавшего сложного эфира. Реакционную смесь оставляли примерно на 64 часа при около 25°С.
Смесь разбавляли 100 мл воды, а затем сильно подкисляли путем добавления 20 мл концентрированной HCl. Полученный смолистый осадок перемешивали до тех пор, пока этот осадок не становился суспензией. Твердые вещества выделяли путем фильтрации, ресуспендировали в метаноле и фильтровали, в результате чего получали 3,75 г коричневого твердого вещества. Это вещество растворяли в 8 мл горячего ДМФ и смесь разбавляли 40 мл метанола. Кислоту кристаллизовали и выделяли путем фильтрации, в результате чего получали 1,7 г хлопьеобразного белого твердого вещества, 11α,17-дигидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоксилата, γ-лактона.
1Н-ЯМР (400 МГц, дейтеродиметилсульфоксид) δ 0,80(с, 3Н), 1,25 (с, 3Н), 1,2-2,7(м, 20Н), 3,8(шир.с, 1Н), 4,45(м, 1Н), 5,50(с, 1Н). Карбоксильный протон не наблюдался из-за присутствия пика HOD при 3,4 м.д.
Пример 38.
Схема 1: Стадия 3С: Метод G: Получение 7-метилгидро-17-гидрокси-3-оксо-17α-прегна-4,9(11)-диен-7,21-дикарбоксилата, γ-лактона
Повторяли процедуру Примера 37А за исключением того, что, благодаря обработке реакционного раствора ионнообменной смолой, основной окисью алюминия или основной двуокисью кремния, множества стадий промывки не проводили. Условия для обработки основной окисью алюминия или основной двуокисью кремния описаны в Таблице 38.Было установлено, что каждая из этих обработок была эффективной для удаления примесей без множественных стадий промывок Примера 44, в результате чего получали 7-метилгидро-17-гидрокси-3-оксо-17α-прегна-4,9(11)-диен-7,21-дикарбоксилат, γ-лактон.
Пример 39.
Схема 1: Стадия 3С: Метод Н: Получение 7-метилгидро-17-гидрокси-3-оксо-17α-прегна-4,9(11)-диен-7,21-дикарбоксилата, γ-лактона
Формиат калия (4 г) и трифторуксусную кислоту (42,5 мл) смешивали в 100 мл реакторе. Затем к смеси при регулируемой скорости добавляли трифторуксусный ангидрид (9,5 мл), поддерживая во время добавления температуру ниже 30°С. Затем раствор нагревали до 30°С в течение 30 минут и охлаждали до комнатной температуры, в результате чего получали элиминирующий реагент, используемый для превращения мезилата формулы IV в сложный енэфир формулы II.
Предварительно полученный элиминирующий реагент ангидрид TFA/TFA добавляли к раствору мезилата формулы IV, полученного ранее способом, описанным в Примере 37А. Полученную смесь нагревали до 40°С в течение 4,5 часов, при этом степень превращения периодически контролировали с помощью ТСХ или ВЭЖХ. При завершении реакции смесь переносили в 1-горлую колбу и концентрировали досуха при пониженном давлении и при комнатной температуре (22°С). К смеси добавляли этилацетат (137 мл), в результате чего твердофазный материал полностью растворялся, а затем добавляли смесь воды/насыщенного солевого раствора (137 мл) и полученную двухфазную смесь перемешивали в течение 10 минут. Затем смесь оставляли на 20 минут для разделения фаз. Крепость насыщенного солевого раствора составляла 24% масс. Водную фазу подвергали контакту с дополнительным количеством этилацетата (68 мл) и полученную таким образом двухфазную смесь перемешивали в течение 10 мин, после чего ее оставляли на 15 минут для разделения фаз. Этилацетатные слои после двух экстракций объединяли и промывали 24% масс. насыщенного солевого раствора (120 мл), еще одной аликвотой 24% масс. насыщенного солевого раствора (60 мл), 1н раствором гидроксида натрия (150 мл) и еще одной порцией насыщенного солевого раствора (60 мл). После каждого добавления водной фазы смесь перемешивали в течение 10 минут и оставляли на 15 минут для разделения фаз. Оставшийся раствор концентрировали досуха при пониженном давлении при 45°С с использованием устройства для отсасывания воды. Твердый продукт (8,09 г) анализировали с помощью ВЭЖХ, в результате чего было установлено, что этот продукт включает 83,4% (площади) сложного енэфира 7-метил-гидро-17-гидрокси-3-оксо-17α-прегна-4,9(11)-диен-7,21-дикарбоксилата, γ-лактона; 2,45% (площади) 11,12-олефина; 1,5% 7,9-лактона; и 1,1% непрореагировавшего мезилата.
Пример 40.
Схема 1: Стадия 3С: Метод I: Получение 7-метилгидро-17-гидрокси-3-оксо-17α-прегна-4,9(11)-диен-7,21-дикарбоксилата, γ-лактона
Мезилат, имеющий структуру, полученную в Примере 23 (1,0 г), изопропенилацетат(10 г) и п-толуолсульфоновую кислоту (5 мг) помещали в 50 мл колбу и нагревали до 90°С при размешивании. Через 5 часов смесь охлаждали до 25°С и концентрировали в вакууме при 10 мм рт.ст. Остаток растворяли в CH2Cl2 (20 мл) и промывали 5% водным NaHCO3. CH2Cl2-слой концентрировали в вакууме и получали 1,47 г желто-коричневого масла. Этот продукт перекристаллизовывали из CH2Cl2/Et2O и получали 0,50 г енолацетата формулы IV(Z).
Этот продукт добавляли к смеси ацетата натрия (0,12 г) и уксусной кислоты (2,0 мл), которую предварительно нагревали до 100°С при размешивании. Через 60 минут смесь охлаждали до 25°С и разбавляли CH2Cl2 (20 мл). Раствор промывали водой (20 мл) и сушили MgSO4. Осушающий агент удаляли путем фильтрации и фильтрат концентрировали в вакууме, в результате чего получали 0,4 г нужного 9,11-олефина, 7-метилгидро-17-гидрокси-3-оксо-17α-прегна-4,9(11)-диен-7,21-дикарбоксилата, γ-лактона. Неочищенный продукт содержал менее чем 2% примеси 7,9-лактона.
Пример 41. Термическое элиминирование мезилата в ДМСО
Схема 1: Стадия 3С: Метод J: Получение 7-метилгидро-17-гидрокси-3-оксо-17α-прегна-4,9(11)-диен-7,21-дикарбоксилата, γ-лактона
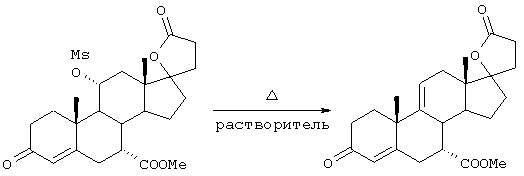
Смесь 2 г мезилата и 5 мл ДМСО в колбе нагревали при 80°С в течение 22,4 часа. ВЭЖХ-анализ реакционной смеси не обнаруживал исходного вещества. К реакционной смеси добавляли воду (10 мл) и осадок три раза экстрагировали метиленхлоридом. Объединенные метиленхлоридные слои промывали водой, сушили сульфатом магния и концентрировали с получением сложного енэфира, 7-метилгидро-17-гидрокси-3-оксо-17α-прегна-4,9(11)-диен-7,21-дикарбоксилата, γ-лактона.
Пример 42.
Схема 1: Стадия 3D: Метод В: Синтез метилгидро-9,11α-эпокси-17α-гидрокси-3-оксопрегн-4-ен-7α,21-дикарбоксилата, γ-лактона.
В 50-миллилитровой колбе грушевидной формы сложный енэфир формулы IIA (1,07 г анализируемого 74,4% сложного енэфира), трихлорацетамид (0,32 г) и дикалийбифосфат (0,70 г) в виде твердого вещества смешивали, перемешивая при этом с метиленхлоридом (15,0 мл). Затем пипеткой в течение 1 минуты добавляли перекись водорода (30% масс; 5,0 мл). Полученную смесь перемешивали 6 часов при комнатной температуре, в течение которых ВЭЖХ-анализ показал, что отношение эпоксимексренона к сложному енэфиру в реакционной смеси составляло приблизительно 1:1. В реакционную смесь вводили дополнительное количество трихлорацетамида (0,32 г) и реакцию продолжали, помешивая, еще 8 часов, после чего было установлено, что остаточное количество сложного енэфира снижалось до 10%. Затем добавляли дополнительное количество трихлорацетамида (0,08 г) и реакционную смесь оставляли на ночь, после чего в смеси оставалось только 5% непрореагировавшего сложного енэфира по отношению к эпоксимексренону.
Пример 43.
Схема 1: Стадия 3D: Метод С: Синтез метилгидро-9,11α-эпокси-17α-гидрокси-3-оксопрегн-4-ен-7α,21-дикарбоксилата, γ-лактона.
В 100 мл реактор добавляли сложный енэфир формулы IIA (5,4 г, в соответствии с количественным анализом 74,4% сложного енэфира). К этому сложному енэфиру добавляли трихлорацетамид (4,9 г) и дикалийбифосфат (3,9 г), оба в твердой форме, а затем добавляли метиленхлорид (50 мл). Смесь охлаждали до 15°С и в течение 10 минут добавляли 30% перекись водорода (25 г). Реакционную смесь оставляли до тех пор, пока она не нагревалась до 20°С, и при этой температуре перемешивали в течение 6 часов, по истечении которых проводили ВЭЖХ-анализ на степень превращения. Было установлено, что количество оставшегося енэфира составляло менее чем 1% масс.
Эту реакционную смесь добавляли в воду (100 мл) и смесь оставляли для разделения фаз, после чего метиленхлоридный слой удаляли. К метиленхлоридному слою добавляли гидроксид натрия (0,5н, 50 мл). Через 20 минут смесь оставляли для разделения фаз, а затем к метиленхлоридному слою добавляли HCl (0,5н; 50 мл), после чего смесь оставляли для разделения фаз и органическую фазу промывали насыщенным солевым раствором (50 мл). Метиленхлоридный слой сушили сульфатом магния и растворитель удаляли. В результате получали белое твердое вещество (5,7 г). Водный слой гидроксида натрия подкисляли и экстрагировали, а экстракт обрабатывали с получением еще 0,2 г продукта. Выход эпоксимексренона составлял 90,2%.
Пример 44.
Схема 1: Стадия 3D: Метод D: Синтез метилгидро-9,11α-эпокси-17α-гидрокси-3-оксопрегн-4-ен-7α,21-дикарбоксилата, γ-лактона
Сложный енэфир формулы IIA превращали в эпоксимексренон способом, описанным в Примере 43, за исключением следующих изменений: первоначальная загрузка содержала сложный енэфир (5,4 г, количественный анализ: 74,4% сложного енэфира), трихлорацетамид (3,3 г) и дикалийбифосфат (3,5 г). Затем добавляли перекись водорода (12,5 мл). Реакцию проводили в течение ночи при 20°С, после чего, как показала ВЭЖХ, наблюдалось 90%-ное превращение сложного енэфира в эпоксимексренон. Затем добавляли еще 3,3 г трихлорацетамида и 30% перекись водорода (5,0 мл) и реакцию проводили еще в течение 6 часов, по истечении которых количество остаточного сложного енэфира составляло только 2% из расчета загрузки сложного енэфира. После обработки, как описано в Примере 43, получали 5,71 г эпоксимексренона.
Пример 45.
Схема 1: Стадия 3D: Метод Е: Синтез метилгидро-9,11α-эпокси-17α-гидрокси-3-оксопрегн-4-ен-7α,21-дикарбоксилата, γ-лактона
Сложный енэфир формулы IIA превращали в эпоксимексренон способом, описанным в Примере 43. В реакции этого примера загрузка сложного енэфира составляла 5,4 г (количественный анализ: 74,4% сложного енэфира), загрузка трихлорацетамида составляла 4,9 г, загрузка перекиси водорода составляла 25 г, а загрузка дикалийбифосфата составляла 3,5 г. Реакцию проводили в течение 18 часов при 20°С. Количество остаточного сложного енэфира составляло менее чем 2%. После обработки получали 5,71 г эпоксимексренона.
Пример 45.
Схема 1: Стадия 3D: Метод F: Синтез метилгидро-9,11α-эпокси-17α-гидрокси-3-оксопрегн-4-ен-7α,21-дикарбоксилата, γ-лактона
Сложный енэфир формулы IIA превращали в эпоксимексренон способом, описанным в Примере 43, за исключением того, что температура реакции этого примера составляла 28°С. Загрузка в реактор содержала сложный енэфир (2,7 г), трихлорацетамид (2,5 г) и дикалийбифосфат (1,7 г), перекись водорада (17,0 г) и метиленхлорид (50 мл). Через 4 часа количество остаточного сложного енэфира составляло только 2% из расчета загрузки сложного енэфира. После обработки, как описано в Примере 43, получали 3,0 г эпоксимексренона.
Пример 47-1.
Схема 1: Стадия 3D: Метод G: Синтез метилгидро-9,11α-эпокси-17α-гидрокси-3-оксопрегн-4-ен-7α,21-дикарбоксилата, γ-лактона
Сложный енэфир формулы IIA (40 г, количественный анализ: 68,4% сложного енэфира) загружали в 1000 мл реактор с рубашкой и растворяли в 175 мл метиленхлорида. Раствор перемешивали по мере добавления трихлорацетамида (22,3 г) и дикалийбифосфата (6,0 г) в виде твердых веществ. Эту смесь перемешивали при 400 об/мин и температуру доводили до 27°С с помощью бани с постоянной температурой для регуляции циркулирования жидкости через рубашку реактора. Затем в течение 3-5 минут добавляли перекись водорода (72,8 мл, анализ: 30%). После добавления перекиси водорода смесь перемешивали при 400 об/мин и при 27°С. ВЭЖХ-анализ указывал на то, что за 5 часов реакция была завершена (99%). Через шесть часов добавляли 72,8 мл воды. Водную перекись водорода отделяли и один раз подвергали обратной экстракции 50 мл метиленхлорида. Объединенный метиленхлорид промывали 6% сульфитом натрия (62,3 мл) для разложения какой-либо оставшейся перекиси. Удаление метиленхлорида инициировали путем дистилляции при атмосферном давлении и помещения в вакуум. Таким образом был получен желтоватый остаток (48,7 г, анализ 55,4%). Это соответствовало выходу 94,8%, полученному по анализу, проведенному в расчете на молярный выход.
Часть (47,8 г) остатка объединяли с 498 мл этанола 3А (95% этанол, денатурированный 5% метанолом). Полученную смесь нагревали с обратным холодильником и 249 мл дистиллята удаляли при атмосферном давлении. Смесь охлаждали до 25°С и фильтровали. Промывку этанолом 3А (53 мл) использовали для облегчения переноса. Осушенное твердое вещество составляло 27,6 г (анализ: 87,0%), что соответствовало выходу 91%. Часть твердого вещества (27,0 г) растворяли в 292 мл метилэтилкетона при нагревании с обратным холодильником. Горячий раствор фильтровали через слой Solkafloc (измельченной в порошок целлюлозы) с другими 48,6 мл метилэтилкетона, использованного для облегчения переноса. 146 мл часть метилэтилкетона удаляли путем дистилляции при атмосферном давлении. Раствор охлаждали до 50°С и перемешивали в течение одного часа по мере кристаллизации продукта. Через один час смесь охлаждали до 25°С. Перемешивание продолжали в течение одного часа и твердое вещество фильтровали с 48,6 мл метилэтилкетона, использованного в качестве промывки. Твердое вещество сушили до постоянной массы 20,5 г, которая составляла выход 87,2% после перекристаллизации. Выход реакции и выделенные этанол, метилэтилкетон объединяли с получением полного выхода 75%.
Метилэтилкетоновый маточный раствор был подходящим для проведения повторного цикла с введением метиленхлоридного раствора из последующей реакции. Смесь объединенных метиленхлорида и метилэтилкетона упаривали досуха путем дистилляции при атмосферном давлении и в вакууме. Остаток объединяли с 19 объемами этанола 3А, исходя из содержания эпоксимексренона. Одну половину растворителя удаляли путем дистилляции при атмосферном давлении. После охлаждения до 25°С твердое вещество фильтровали и сушили. Твердое сухое вещество растворяли в 12 объемах метилэтилкетона при нагревании с обратным холодильником. Горячий раствор фильтровали через слой Solka Floc с 2 объемами метилэтилкетона, добавленного в качестве промывки. Фильтрат концентрировали путем дистилляции 6 объемов метилэтилкетона при атмосферном давлении. Этот раствор охлаждали до 50°С и перемешивали в течение одного часа по мере кристаллизации продукта. Через один час смесь охлаждали до 25°С. Перемешивание продолжали еще один час и твердый продукт фильтровали с 2 объемами метилэтилкетона, используемого в качестве промывки. Твердый продукт сушили до получения постоянной массы. Введение маточного раствора метилэтилкетона увеличивало общий выход до 80-85%.
Этот способ оказался особенно подходящим для масштабного продуцирования, поскольку он способствует максимизации производительности и минимизации объемов промывки и отходов.
Пример 47А.
Схема 1: Стадия 3D: Метод Н: Синтез метилгидро-9,11α-эпокси-17α-гидрокси-3-оксопрегн-4-ен-7α,21-дикарбоксилата, γ-лактона
Сложный енэфир формулы IIA (17 г, количественный анализ: 72% сложного енэфира) растворяли в метиленхлориде (150 мл), после чего добавляли трихлорацетамид (14,9 г) при медленном помешивании. Температуру смеси доводили до 25°С и в раствор енэфирного субстрата при перемешивании со скоростью 400 об/мин добавляли раствор дикалийбифосфата (10,6 г) в воде (10,6 мл). Затем к смеси субстрата/фосфата/трихлорацетамида в течение 3-5 минут добавляли перекись водорода (30% масс. раствора; 69,4 мл). Экзотермической реакции или выделения кислорода не наблюдалось. Полученную таким образом реакционную смесь перемешивали при 400 об/мин в течение 18,5 часов при 25°С. В процессе реакции какого-либо выделения кислорода не наблюдалось, но анализ на расход перекиси водорода показал, что во время реакции образовывалось некоторое количество кислорода. Реакционную смесь разбавляли водой (69,4 мл) и смесь перемешивали при около 250 об/мин в течение 15 минут. Во время этой реакции в температурном контроле не было необходимости и реакцию осуществляли, в основном, при комнатной температуре (подходящей является любая температура в пределах 5-25°С). Водный и органический слои оставляли для разделения и нижний метиленхлоридный слой удаляли.
Водный слой подвергали обратной экстракции метиленхлоридом (69,4 мл) в течение 15 мин, размешивая при 250 об/мин. Эти слои оставляли для разделения и нижний метиленхлоридный слой удаляли. Водный слой (177 г; рН=7) подвергали анализу на перекись водорода. Результат (12,2%) анализа указывал, что в реакции 0,0307 моль олефина поглощалось 0,0434 моль перекиси водорода. Избыточное поглощение перекиси водорода является показателем образования кислорода в данной реакции. Обратная экстракция небольшого объема метиленхлорида была достаточной гарантией отсутствия потери эпоксимексренона в водном слое. Этот результат совпадает с результатом, полученным с использованием второй экстракции большим количеством метиленхлорида, при которой был выделен только трихлорацетамид.
Объединенные метиленхлоридные растворы, полученные из вышеописанных экстракций, объединяли и промывали 3% масс. раствора сульфата натрия (122 мл) в течение по крайней мере 15 минут при перемешивании со скоростью около 250 об/мин. По окончании периода перемешивания был получен отрицательный тест с использованием иодида крахмала (KI-бумага; окраски не наблюдалось; в положительном тесте пурпурная окраска указывала на присутствие пероксида).
Водные и органические слои оставляли для их разделения, а нижний метиленхлоридный слой удаляли. Водный слой (рН=6) отбрасывали. Следует отметить, что добавление раствора сульфита натрия может вызвать небольшое повышение температуры, а поэтому такое добавление должно проводиться под температурным контролем.
Метиленхлоридную фазу промывали 0,5 н гидроксидом натрия (61 мл) в течение 45 минут при скорости перемешивания около 250 об/мин и при температуре в пределах 15-25°С (рН=12-13). Во время этой операции удаляли примеси, происходящие от трихлорацетамида. Подкисление щелочных водных фракций с последующей экстракцией метиленхлоридом подтвердило, что при этой операции теряется очень незначительное количество эпоксимексренона.
Метиленхлоридную фазу один раз промывали 0,1н соляной кислотой (61 мл) в течение 15 минут при скорости размешивания 250 об/мин и при температуре в пределах 15-25°С. Затем эти слои оставляли для разделения и нижний метиленхлоридный слой удаляли и снова промывали 10% масс. водного хлорида натрия (61 мл) в течение 15 минут при скорости перемешивания 250 об/мин и при температуре в пределах 15-25°С. И снова слои оставляли для разделения и органический слой удаляли. Органический слой фильтровали через слой Solkafloc, а затем упаривали досуха при пониженном давлении. Сушку проводили в водяной бане при температуре 65°С. В результате получали беловатое твердое вещество (17,95 г), которое подвергали ВЭЖХ-анализу. Анализ показал, что выход эпоксимексренона составил 66,05%. Скорректированный молярный выход для данной реакции составлял 93,1%.
Продукт растворяли в горячем метилэтилкетоне (189 мл) и полученный раствор подвергали дистилляции при атмосферном давлении до тех пор, пока не было удалено 95 мл кетонового растворителя. Температуру понижали до 50°С по мере кристаллизации продукта. Перемешивание продолжали в течение 1 ч при 50°С. Затем температуру понижали до 20-25°С и перемешивание продолжали еще 2 часа. Твердое вещество фильтровали, промывали МЕК (24 мл) и твердое вещество сушили до получения постоянной массы 9,98 г, ВЭЖХ-анализ которой указывал на содержание в ней 93,63% эпоксимексренона. Этот продукт снова растворяли в горячем МЕК (106 мл) и горячий раствор фильтровали через 10 микронный линейный фильтр под давлением. Затем наносили еще 18 мл МЕК в качестве промывки и отфильтрованный МЕК-раствор подвергали дистилляции при атмосферном давлении до тех пор, пока не было удалено 53 мл растворителя. Температуру понижали до 50°С по мере кристаллизации продукта; и перемешивание продолжали при 50°С в течение 1 часа. Затем температуру понижали до 20-25°С и продолжали перемешивать при этой температуре еще 2 часа. Твердый продукт фильтровали и промывали МЕК (18 мл). Затем твердый продукт сушили до получения постоянной массы 8,32 г, которая содержала 99,6% эпоксимексренона, на что указывал количественный ВЭЖХ-анализ. Конечная потеря после сушки составляла менее чем 1,0%. Общий выход эпоксимексренона, полученный при осуществлении реакции и обработки, описанных в этом Примере, составлял 65,8%. Этот общий выход представлял: выход реакции 93%, выделение после первой кристализации 78,9% и выделение после перекристаллизации 89,5%.
Пример 47В. Получение 7-метилгидро-11α,12α-эпокси-17-гидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоксилата, γ-лактона
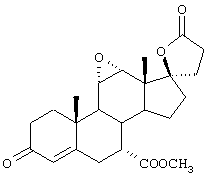
Δ11,12-Олефин сложного енэфира представляет собой побочный продукт элиминации 11-мезилата. Из реакционной смеси, полученной способом, описанным в Примере 37А, выделяли чистый образец с помощью повторной жидкостной хроматографии. Таким образом, 73 г остатка (полученного как описано в Примере 37А) хроматографировали на 2,41 кг силикагеля Merck (40-63 мкм) по схеме элюирования с использованием градиента этилацетата, толуола (20:80, 30:70, 40:60, 60:40 об./об.). Обогащенные Δ11,12-олефином части объединяли из выбранных 30:70-фракций. Отбор соответствующих фракций проводили, исходя из ТСХ на EMF-пластинках с использованием этилацетата/толуола (60:40 об./об.) и визуализации серной кислотой SWUV. 7,9 г сырого Δ11,12-олефина (80% по ВЭЖХ-площади), полученного после удаления растворителя, хроматографировали на 531 г силикагеля Merck (40-63 мкм) по схеме элюирования с использованием градиента этилацетата/метиленхлорида (10:90, 20:80, 35:65 об./об.). Чистый 7-метилгидро-17-гидрокси-3-оксо-17α-прегна-4,11-диен-7α,21-дикарбоксилат, γ-лактон (3,72 г) был получен из выбранных 20:80-фракций. Выбор фракций проводили исходя из ТСХ-оценки, как описано выше.
МИК см-1 1767 (лактон), 1727 (сложный эфир), 1668 и 1616 (3-кето-Δ4,5).
1Н-ЯМР (CDCl3) м.д. 1,05(с, 3Н), 1,15(с, 3Н), 3,66(с, 3Н), 5,58(дд,1Н), 5,80 (с, 1Н), 5,88(дд, 1Н).
13С-ЯМР (CDCl3) м.д. 17,41; 18,58; 21,73; 28,61; 32,28; 33,63; 34,91; 35,64; 35,90; 38,79; 42,07; 44,12; 48,99; 49,18; 51,52; 93,81; 126,43; 126,69; 133,76; 166,24; 172,91; 176,64; 198,56.
Раствор 1,6 г (3,9 ммоль) 7-метилгидро-17-гидрокси-3-оксо-17α-прегна-4,11-диен-7α,21-дикарбоксилата, γ-лактона в 16 мл метиленхлорида смешивали с 2,2 мл трихлорацетонитрила (22,4 ммоль) и 0,75 г дикалийфосфата (4,3 ммоль). Эту смесь перемешивали и объединяли с 6,7 мл 30% перекиси водорода (66 ммоль). Перемешивание продолжали при 25°С в течение 45 часов. По истечении этого времени добавляли 28 мл метиленхлорида и 39 мл воды. Органическую часть выделяли и промывали последовательно: а) 74 мл 3% сульфата натрия, b) 62 мл 1н гидроксида натрия, с) 74 мл 1н соляной кислоты и d) 31 мл 10% насыщенного солевого раствора. Органическую часть снова отделяли, сушили сульфатом магния и упаривали досуха в вакууме. 1,25 г остатка хроматографировали на 138,2 г силикагеля Merck (40-63 мкм) с использованием системы градиента метил-трет-бутилового эфира и толуола (40:60, 60:40, 75:25 об./об.). Соответствующие порции 60:40, 75:25-фракций объединяли, исходя из ТСХ-оценки, и получали 0,66 г чистого 7-метил-гидро-11α,12α-эпокси-17-гидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоксилата, γ-лактона. В ТСХ-системе использовали EMF-пластинки со схемой элюирования метил-трет-бутиловым эфиром и толуолом 75:25 (об./об.) и с использованием серной кислоты и SWUV для визуализации.
1Н-ЯМР (CDCl3) м.д. 1,09(с, 3Н), 1,30(с, 3Н), 3,05(АВ11,12 для 2Н), 3,67(с, 3Н), 5,80(с, 1Н).
13С-ЯМР (CDCl3) м.д. 14,2; 18,0; 21,2; 28,8; 31,9; 33,5; 34,6; 34,7; 35,1; 35,5; 37,4; 38,3; 41,8; 46,0; 47,2; 50,4; 51,7; 56,7; 94,0; 126,7; 165,2; 172,5; 176,7; 198,1
Вычислено: С 69,54 и Н 7,30; Найдено С 69,29 и Н 7,17.
Пример 47С. Выделение 7-метилгидро-4α,5α:9α,11α-диэпокси-17-гидрокси-3-оксо-17α-прегнан-7α,21-дикарбоксилата, γ-лактона
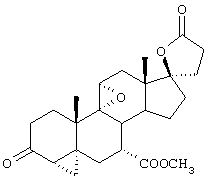
Неочищенный эпоксимексренон (157 г), полученный из 200 г сложного енэфира способом, описанным в Примере 26, подвергали хроматографии на 4,5 кг силикагеля Merck (40-63 мкм). 88,1 г порцию выделяли с использованием схемы элюирования ацетонитрилом и толуолом 10:90 (об./об.). Выделенное твердое вещество растворяли в 880 мл горячего метилэтилкетона и фильтровали через слой Solka Floc. Затем наносили еще 88 мл метилэтилкетона в качестве промывки. Фильтрат концентрировали путем удаления 643 мл растворителя и смесь охлаждали до комнатной температуры. Твердые вещества фильтровали и промывали метилэтилкетоном. После сушки получали 60,2 г эпоксимексренона, что составляло 96,8% в соответствии с ВЭЖХ-анализом. Фильтрат концентрировали досуха при пониженном давлении. 9,3 г остатка перекристаллизовывали из 99 мл метилэтилкетона и получали 2,4 г сухого твердого вещества. Порцию в 400 мг этого твердого вещества подвергали обращенно-фазовой препаративной ВЭЖХ на колонке YMC ODS AQ. Чистый 7-метил- гидро-4α,5α:9α,11α-диэпокси-17-гидрокси-3-оксо-17α-прегнан-7α,21-дикарбоксилат, γ-лактон (103 мг) выделяли с использованием схемы элюирования ацетонитрилом (24%), метанолом (4%) и водой (72%).
1Н-ЯМР (CDCl3) м.д. 0,98(с, 3Н), 1,32(с, 3Н), 2,89(м, 1Н), 3,07(с,д, 2Н), 3,73(с, 3Н).
МС, М+430, вычислено для С24Н30O7 (430,50).
Пример 47D. Выделение 7-метилгидро-17-гидрокси-3,12-диоксо-17α-прегна-4,9(11)-диен-7α,21-дикарбоксилата, γ-лактона
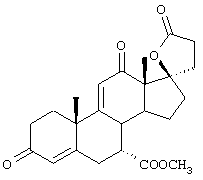
Маточный метилэтилкетоновый раствор, полученный способом, описанным в Примере 26, упаривали досуха при пониженном давлении. 4,4 г часть остатка подвергали хроматографии на 58,4 г BTR Zorbax LP (40 мкм). После элюирования градиентом метилэтилкетона и метиленхлорида (25:75-50:50 об./об.) получали 1,38 г продукта. 1,3 г часть этого продукта дополнительно очищали с помощью обращенно-фазовой препаративной ВЭЖХ с использованием в качестве подвижной фазы ацетонитрила (30%), метанола (5%) и воды (65%) и колонки YMC ODS AQ (10 мкм). Этот продукт получали из обогащенных фракций путем экстракции метиленхлоридом. Метиленхлорид выпаривали досуха и 175 мг остатка снова очищали посредством обращенно-фазовой препаративной ВЭЖХ с использованием в качестве подвижной фазы ацетонитрила (24%), метанола (4%) и воды (72%) и колонки YMC ODS AQ. Экстракция обогащенных фракций метиленхлоридом давала 30,6 мг чистого 7-метилгидро-17-гидрокси-3,12-диоксо-17α-прегна-4,9(11)-диен-7α,21-дикарбоксилата, γ-лактона.
1Н-ЯМР (CDCl3) м.д. 1,17(с, 3Н), 1,49(с, 3Н), 3,13(м, 1Н), 3,62(с, 3Н), 5,77(с, 1Н), 5,96 (с, 1Н).
13С-ЯМР (CDCl3) м.д. 13,1; 21,0; 28,0; 29,4; 33,1; 33,4; 33,9; 35,5; 36,7; 40,3; 41,5; 43,0; 43,4; 52,0; 55,0; 91,0; 123,7; 126,7; 163,2; 167,9; 171,8; 176,8; 197,4; 201,0.
Пример 47Е. Получение 9,11α-эпокси-17-гидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоновой кислоты, дигидрата, дикалиевой соли
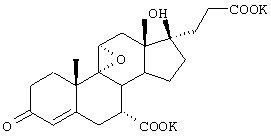
Получали суспензию, содержащую 2,0 г (4,8 ммоль) эпоксимексренона, полученного как описано в Примере 43, 10 мл воды, 3 мл диоксана и 9,3 мл 1,04н водного гидроксида калия (9,7 ммоль). Смесь перемешивали в течение 3 часов при 25°С. В течение первых двух часов образовывался желтый гомогенный раствор. Температуру повышали до 70°С и перемешивание продолжали еще 3 часа. Растворитель удаляли путем вакуумной дистилляции и остаток очищали с помощью обращенно-фазовой ВЭЖХ на 90 г силикагеля С18 с использованием воды в качестве элюента. Нужные фракции объединяли в соответствии с оценкой, проведенной с помощью ТСХ на EMF-пластинках с использованием метиленхлорида и метанола (7:3) в качестве элюента и SWUV для визуализации. Объединенные фракции концентрировали досуха в вакууме и остаток подвергали очистке с помощью обращенно-фазовой ВЭЖХ, проведенной как описано выше. Нужные фракции концентрировали досуха при пониженном давлении, а остаток растворяли в этаноле. Для определения температуры помутнения добавляли этилацетат, а затем добавляли гептан для завершения осаждения. Было выделено 0,55 г продукта 9,11α-эпокси-17-гидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоновой кислоты, дигидрата, дикалиевой соли, в виде желтого твердого вещества. Углеродный анализ соответствовал гидратированной структуре С23Н28O7K2 .1,75 Н2О: Вычислено С, 52,50 по ср. с 55,85 для гидратированной формы; найдено С 52,49. После проведения ТСХ на EMF-пластинках с использованием метиленхлорида, метанола и воды (6:3:0,5 об./об.) в качестве элюента и визуализации с помощью SWUV наблюдали Rf=0,29.
Пример 47F. Получение 9,11α-эпокси-17-гидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоновой кислоты, динатриевой соли
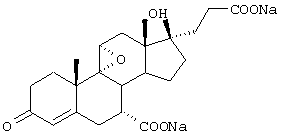
Около 5 мг (0,01 ммоль) эпоксимексренона, полученного способом, описанным в Примере 43, суспендировали в около 200 мкл метанола в 4 мл сосуде и разбавляли примерно 200 мкл 2,5 NaOH. Полученная смесь имела желтый цвет и была гомогенной. Затем смесь нагревали в масляной бане при 70°С. Через 10 минут 1 мкм образец, взятый из этой смеси, анализировали с помощью ВЭЖХ (Zorbax SB-C8, 150 × 4,6 мм, 2 мл/минут, градиент = 35:65 (об./об.) А: В, А = ацетонитрил/метанол (1:1), В=вода/0,1% трифторуксусная кислота, детекция при 210 нм) и этот анализ выявил два продукта при временах удерживания 4,86 и 2,93 минуты, которые соответствовали оксикислоте (незамкнутый лактон) и 7-карбоновой кислоте с незамкнутым лактоном, соответственно. Через 30 минут брали второй образец (0,05 мл) и подкисляли 0,05 мл 3н HCl, а затем нейтрализовали с использованием около 0,5 мл бикарбоната натрия. ВЭЖХ-анализ, как и выше, обнаруживал ожидаемые стероиды с замнутыми кольцами при временах удерживания 6,59 и 10,71 минут. Отношение 7-метилгидро-9,11α-эпокси-17-гидрокси-3- оксо-17α-прегн-4-ен-7α,21-дикарбоксилата, γ-лактона (10,71 мин) к соответствующей 7-карбоновой кислоте составляло 7:89.
Селективный гидролиз лактона можно проводить в мягких условиях. Второй 4 мл сосуд был приготовлен как описано выше, но без нагревания. Смесь обрабатывали ультразвуком в течение 5 минут. 0,05 мл образец разбавляли в 0,5 мл смеси метанола/ацетонитрила, 1:1 (об./сб.), и анализировали с помощью ВЭЖХ без предварительного подкисления. Полученный сложный 7-эфир карбоновой кислоты с незамкнутым лактоном имел время удерживания 4,85 минут, как наблюдалось выше, и не имел примесей в виде 7-карбоновой кислоты.
Пример 47G. Выделение 7-метилгидро-9α,11β,17-тригидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоксилата, γ-лактона
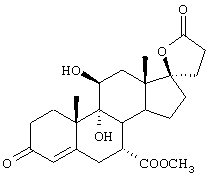
и 7-метилгидро-12α,17-дигидрокси-3-оксо-17α-прегна-4,9(11)-диен-7α,21-дикарбоксилата, γ-лактона
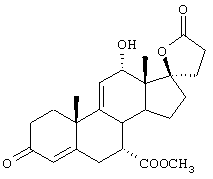
7-Метилгидро-9α,11β,17-тригидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоксилат, γ-лактон и 7-метилгидро-12α,17-дигидрокси-3-оксо-17α-прегна-4,9(11)-диен-7α,21-дикарбоксилат, γ-лактон выделяли после проведения препаративной жидкостной хроматографии 2-бутанонового маточного раствора, полученного в результате эпоксидирования сложного енэфира, как описано в Примере 26 (схема с использованием трихлорацетонитрила). Для этого первую кристаллизацию проводили с использованием указанного 2-бутанона. Однако, для перекристаллизации вместо ацетона использовали 2-бутанон (10 объмов на 1 г). Таким способом получали 2,8 г остатка, который очищали с помощью обращенно-фазовой препаративной ВЭЖХ. В качестве стационарной фазы использовали Cromasil C8 (10 мкм), а подвижная фаза состояла из воды milliQ и ацетонитрила в отношении 70:30 (об./об.). Кристаллизация наблюдалась в одной из обогащенных фракций. Твердое вещество (46,7 мг) выделяли и идентифицировали как 7-метилгидро-9α,11β,17-тригидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоксилат, γ-лактон. Маточный раствор упаривали досуха при пониженном давлении и остаток (123 мг) идентифицировали как 7-метилгидро-12α,17-дигидрокси-3-оксо-17α-прегна-4,9(11)-диен-7α,21-дикарбоксилат, γ-лактон. 7-метилгидро-9α,11β,17-тригидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоксилат, γ-лактон:
МС М+432, вычислено для C24H32O5 (432,51).
1Н-ЯМР (CDCl3) м.д. 1,23(с, 3Н), 1,54(с, 3Н), 3,00(м, 1Н), 3,14(м, 1Н), 3,74(с, 3Н), 5,14(с, 1Н, медленно обмениваемый), 5,79 (с, 1Н).
13С-ЯМР (CDCl3) м.д. 16,8; 22,7; 24,8; 29,0; 29,3; 32,1; 34,1; 34,7; 35,2; 35,7; 36,8; 40,7; 43,0; 45,0; 45,9; 52,9; 72,8; 77,4; 95,9; 127,4; 163,7; 176,7; 177,3; 199,4.
7-метилгидро-12α,17-дигидрокси-3-оксо-17α-прегна-4,9(11)-диен-7α,21-дикарбоксилат, γ-лактон:
МС М+441, вычислено для С24Н30О5 (414,50).
1Н-ЯМР (CDCl3) м.д. 0,87(с, 1Н), 1,40(с, 1H), 3,05(м, 1Н), 3,63 (с, 3Н), 3,99(м, 1Н); 5,72 (с, 1Н), 5,96(м, 1Н).
13С-ЯМР (CDCl3) м.д. 14,8; 24,0; 26,1; 29,7; 33,6; 33,8; 34,0; 36,3; 37,0; 37,4; 40,7; 40,9; 43,8; 48,1; 51,9; 69,1; 95,5; 122,7; 126,3; 145,9; 164,5; 173,2; 177,6; 198,2.
Пример 47Н. Получение 7-метилгидро-9,11α-эпокси-3-этокси-17-гидрокси-17α-прегн-4-ен-7α,21-дикарбоксилата, γ-лактона
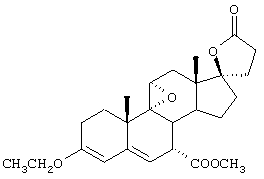
и 7-метилгидро-6β,17-дигидрокси-9,11α-эпокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоксилата, γ-лактона
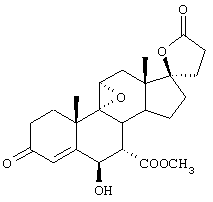
7-Метилгидро-9,11α-эпокси-3-этокси-17-гидрокси-17α-прегн-4-ен-7α,21-дикарбоксилат, γ-лактон получали методом, описанным в работе R.M.Weier & L.M.Hofmann (J.Med.Chem. 1977, 1304), которая вводится в настоящее описание посредством ссылки. 148 г (357 ммоль) 7-метилгидро-9,11α-эпокси-17-гидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоксилата, γ-лактона, полученного способом, описанным в Примере 43, объединяли с 311 мл абсолютного этанола и 155 мл (932 ммоль) триэтилортоформиата. Суспензию перемешивали при комнатной температуре и в качестве катализатора добавляли 10,4 г (54,7 ммоль) толуолсульфоновой кислоты (моногидрата). Перемешивание продолжали в течение 30 минут и реакцию гасили путем добавления 41,4 г (505 ммоль) измельченного в порошок ацетата натрия и 20,7 мл (256 ммоль) пиридина. Твердые вещества (70,2 г) удаляли путем фильтрации и фильтрат концентрировали досуха в вакууме. Остаток гидролизовали 300 мл этилацетата и 9,8 г твердых веществ удаляли путем фильтрации.
Фильтрат концентрировали досуха и остаток гидролизовали 100 мл метанола, содержащего 2 мл пиридина. 29,7 г твердых веществ удаляли путем фильтрации. В фильтрате наблюдалось дополнительное осаждение. Следовательно, фильтрат повторно фильтровали для удаления еще 21,9 г твердых веществ. Полученный фильтрат концентрировали досуха и остаток гидролизовали 50 мл метанола, содержащего 1 мл пиридина. 33,8 г твердых веществ выделяли путем фильтрации. Качественный ВЭЖХ-анализ указывал на то, что эта последняя порция твердых веществ была достаточно чистой (90% по площади 7-метилгидро-9,11α-эпокси-3-этокси-17-гидрокси-17α-прегн-4-ен-7α,21-дикарбоксилата, γ-лактона) для ее использования в последующей стадии реакции.
7-Метилгидро-9,11α-эпокси-3-этокси-17-гидрокси-17α-прегн-4-ен-7α,21-дикарбоксилат, γ-лактон:
1Н-ЯМР (CDCl3) м.д.: 1,02(с, 3Н), 1,27(с, 3Н), 1,30(т, 3Н), 3,12(м, 1Н), 3,28(м, 1Н), 3,66 (с, 3Н), 3,78(м, 2Н), 5,20 (с, 1Н), 5,29(д, 1Н).
8 г порцию эфира енола (7-метилгидро-9,11α-эпокси-3-этокси-17-гидрокси-17α-прегн-4-ен-7α,21-дикарбоксилата, γ-лактона) (18 ммоль), полученного в предыдущей стадии, растворяли в 120 мл 1,4-диоксана. Этот раствор объединяли со смесью 6,8 г 53% м-хлорпероксибензойной кислоты (20,9 ммоль), 18,5 мл 1,0н гидроксида натрия (18,5 ммоль) и 46 мл диоксана/воды (9:1). Температуру поддерживали при -3°С и смесь перемешивали в течение двух часов. Затем температуру повышали до 25°С и перемешивание продолжали еще 20 часов. Смесь объединяли с 400 мл холодной воды (10°С) и 23,5 мл 1,0н гидроксида натрия (23,5 ммоль). Смесь экстрагировали четыре раза 100 мл порциями метиленхлорида (каждый раз). Объединенные метиленхлоридные порции сушили сульфатом магния и растворитель надосадочной жидкости удаляли путем отгонки в вакууме. 13,9 г остатка растирали с 50 мл этилового эфира с получением 2,9 г твердого белого вещества. 2,4 г часть твердого вещества хроматографировали на 100 г силикагеля Merck (60 микрон). После первоначальной промывки 1 л этилацетата/гептана (1:1), продукт элюировали этилацетатом/гептаном в отношении 7:3. Обогащенные фракции объединяли, исходя из ТСХ-оценки (пластинки с EMF; элюент: этилацетат/гептан (7:3 об/об); визуализация с помощью SWUV). Таким образом получали 0,85 г обогащенного вещества и перекристаллизовывали его из 10 мл изопропанола с получением 0,7 г 7-метилгидро-6β,17-дигидрокси-9,11α-эпокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоксилата, γ-лактона. Фракции с большим количеством примесей объединяли и получали 0,87 г неочищенного 7-метилгидро-6β,17-дигидрокси-9,11α-эпокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоксилата, γ-лактона. Этот продукт хроматографировали на 67,8 г силикагеля Merck (40-63 мкм). Еще 0,69 г продукта выделяли с использованием толуола, содержащего 0,5-2,5% метанола. 7-Метилгидро-6β,17-дигидрокси-9,11α-эпокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоксилат, γ-лактон:
Вычислено: С 66,96 и Н 7,02: Найдено: С 66,68 и Н 7,16.
1Н-ЯМР (CDCl3): м.д.: 1,06(с, 3Н), 1,36(дм, 1Н), 1,63(с, 3Н), 2,92(м, 1Н), 3,02(дд, 1Н), 3,12(д, 1Н), 3,64(с, 3Н), 4,61(д, 1Н), 5,96 (с, 1Н).
13С-ЯМР (CDCl3) м.д.: 16,17; 21,32; 21,79; 24,36; 27,99; 28,94; 30,86; 31,09; 32,75; 33,19; 34,92; 36,77; 39,16; 43,98; 47,74; 51,56; 51,66; 65,36; 72,23; 94,79; 165,10; 171,36; 176,41, 199,59.
Пример 47 I. Получение 7-метилгидро-9,11α-эпокси-17-гидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоксилата, γ-лактона
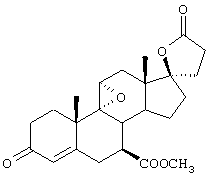
К 2 г (4,8 ммоль) 7-метилгидро-9,11α-эпокси-17-гидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоксилата, γ-лактона, полученного способом, описанным в Примере 43, добавляли 3,3 мл (14,4 ммоль) 25% метоксида натрия в метаноле. Полученную желтую суспензию нагревали до 50°С. Твердое вещество при этом не растворялось. К смеси добавляли 3,3 мл метанола (безводного метанола Aldrich). Затем смесь нагревали с обратным холодильником (65°С) и эта смесь становилась гомогенной. Через 30 минут твердый осадок препятствовал перемешиванию.
Затем добавляли около 25 мл безводного метанола и смесь переносили в 100 мл колбу. Эту смесь нагревали с обратным холодильником 16 часов, в течение которых она становилась темной и гомогенной. Смесь охлаждали до 25°С и добавляли 70 мл 3н HCl (экзотермическая реакция). Для охлаждения смеси добавляли несколько граммов льда и раствор экстрагировали двумя последовательными 25 мл порциями метиленхлорида. Темный раствор сушили сульфатом натрия и фильтровали через 2,5 см слой силикагеля (E.Merck, 70-230 меш, размер пор 60 ). Двуокись кремния элюировали 100 мл метиленхлорида. Затем элюированный метиленхлорид концентрировали в вакууме и получали 1 г коричневой пены, которая кристаллизовалась после добавления этилацетата. Слой двуокиси кремния элюировали второй раз 100 мл 10% этилацетатом/метиленхлоридом и элюированный раствор концентрировали, в результате чего получали 650 мг коричневой пены.
). Двуокись кремния элюировали 100 мл метиленхлорида. Затем элюированный метиленхлорид концентрировали в вакууме и получали 1 г коричневой пены, которая кристаллизовалась после добавления этилацетата. Слой двуокиси кремния элюировали второй раз 100 мл 10% этилацетатом/метиленхлоридом и элюированный раствор концентрировали, в результате чего получали 650 мг коричневой пены.
Тонкослойная хроматография (E.Merck, 60 F-254, силикагель 0,25 мм, толуол/этилацетат (1:1 об./об.)) выявила присутствие 7-метилгидро-9,11α-эпокси-17-гидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоксилата, γ-лактона и 7-метилгидро-9,11α-эпокси-17-гидрокси-3-оксо-17α-прегн-4-ен-7β,21-дикарбоксилата, γ-лактона в обоих образцах, хотя в первом образце присутствовало очень небольшое количество 7α-карбокси-эпимера. Первый образец растирали с горячим этилацетатом (77°С) и оставляли охлаждаться до 25°С. Затем смесь фильтровали с получением 400 мг беловатого твердого вещества, т.пл. 254-258°С. Н, 13С и 13С-АРТ подтверждал установленную структуру. Небольшое количество этилацетата оставалось в образце, но присутствия исходного соединения не было выявлено с помощью ВЭЖХ (Zorbax SB-C8, 150 × 4,6 мм, 2 мл/минут, изократная смесь =40:60 (об./об.) А:В, А = ацетонитрил/метанол (1:1), В = вода/0,1% трифторуксусная кислота, детекция при 210 нм) (ВЭЖХ показала 98,6% площади) и ТСХ (толуол-этилацетат, 1:1, об./об.).
FAB-MC подтвердила молекулярную массу 414 с М+Н при 415,2.
1Н-ЯМР (400 МГц, дейтерохлороформ) δ 0,95(с, 3Н), 1,50(с, 3Н), 1,45(м, 3Н), 1,55-2,7(м, 15Н), 2,85(т, J=13, 1H), 3,25(д, J=6, 1H), 3,65(с, 3Н), 5,78(с, 1H).
Пример 47J. Получение 9,11α-эпокси-17-гидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоновой кислоты γ-лактона
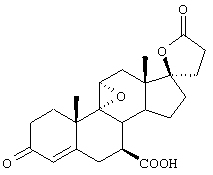
К 774 мг (1,82 ммоль) 7-метилгидро-9,11α-эпокси-17-гидрокси-3-оксо-17α-прегн-4-ен-7α,21-дикарбоксилата, γ-лактона, полученного способом, описанным в Примере 43, и суспендированного в 3 мл ацетонитрила, добавляли 3 мл (7,5 ммоль, 2,0 эквивалента) 2,5М гидроксида натрия. Смесь становилась желтой, а через 10 минут она становилась гомогенной.
Для слежения за ходом реакции аликвоты (0,1 мл) смеси гасили в 0,01 мл 3М серной кислоты и экстрагировали в стеклянном 4 мл сосуде с этилацетатом (0,2 мл). Разделение фаз осуществляли путем удаления нижней водной фазы пипеткой. Органическую фазу упаривали, а остаток анализировали с помощью ВЭЖХ методом, описанным в Примере 47Н. Через 50 минут при 25°С происходило небольшое изменение в составе смеси.
Смесь нагревали с обратным холодильником (около 90°С) в течение 50 минут. ВЭЖХ-анализ смеси указывал на присутствие 6% площади оставшегося исходного материала. Смесь перемешивали в течение 65 часов при 25°С. Подкисление, экстракция и ВЭЖХ-анализ аликвоты, проведенный как описано выше, подтвердили отсутствие оставшегося исходного материала.
Смесь сильно подкисляли путем добавления около 4 мл 3М серной кислоты и экстрагировали двумя частями (около 10 мл) метиленхлорида. Органические фазы объединяли и сушили сульфатом натрия. После концентрирования на роторном испарителе получали 780 г твердого вещества. Это твердое вещество перекристаллизовывали из диметилформамида/метанола с получением 503 мг (67%) желтовато-коричневого кристаллического твердого вещества. При быстром нагревании образец расплавлялся с выделением газа при температуре, близкой к 260°С. При медленном нагревании до 285°С образец медленно темнел, но оставался твердым.
1Н-ЯМР (диметилсульфоксид d-6, 400 МГц) δ 0,85(с, 3Н), 1,4(с, 3Н), 1,3-2,9(м, 19Н), 3,15(м, 1Н), 5,55(с, 1Н), 11,8(шир., 1Н).
Пример 47К.
Схема 1: Стадия 3D: Метод I: Синтез метилгидро-9,11α-эпокси-17α-гидрокси-3-оксопрегн-4-ен-7α,21-дикарбоксилата, γ-лактона.
0,2 М Раствор сложного енэфира формулы IIA в метиленхлориде объединяли с 2 эквивалентами бифосфата калия, растворенного в равной массе воды (50% масс./масс. водного раствора), 3 эквивалентами хлордифторацетамида и 22 эквивалентами перекиси водорода (добавленного в виде 30% водного раствора). Затем смесь перемешивали при 25°С в течение 23 часов. Реакционную смесь разбавляли количеством воды, равным загрузке перекиси водорода, а метиленхлорид отделяли. Метиленхлоридную часть один раз промывали 3% раствором сульфита натрия (с объемом, равным 1,75-кратной загрузке перекиси водорода). Метиленхлоридную часть отделяли и сушили сульфатом натрия. Раствор концентрировали путем дистилляции при атмосферном давлении до тех пор, пока температура в головной части не достигала 70°С. Остаток оценивали с помощью ВЭЖХ, 1Н- и 1С-ЯМР (CDCl3). Было определено, что выход эпоксимексренона составлял 54,2%, по площади ВЭЖХ.
Пример 47L.
Схема 1: Стадия 3D: Метод J: Синтез метилгидро-9,11α-эпокси-17α-гидрокси-3-оксопрегн-4-ен-7α,21-дикарбоксилата, γ-лактона.
Метод, описанный в Примере 47К, повторяли, но вместо хлордифторацетамида использовали гептафторбутирамид (CF3CF2CF2CONH2). Выход эпоксимексренона составил 58,4% по площади ВЭЖХ.
Пример 48. Эпоксидирование сложного енэфира формулы IIA с использованием толуола
Схема 1: Стадия 3D: Метод К: Синтез метилгидро-9,11α-эпокси-17α-гидрокси-3-оксопрегн-4-ен-7α,21-дикарбоксилата, γ-лактона.
Сложный енэфир формулы IIA превращали в эпоксимексренон способом, в основном, описанным в Примере 46, за исключением того, что в качестве растворителя был использован толуол. В реактор загружали такие вещества, как сложный енэфир (2,7 г), трихлорацетамид (2,5 г), дикалийбифосфат (1,7 г), перекись водорода (17,0 г) и толуол (50 мл). Реакцию оставляли для повышения температуры до 28°С и эта реакция завершалась через 4 часа. Полученную трехфазную смесь охлаждали до 15°С, фильтровали, промывали водой и сушили в вакууме с выходом 2,5 г продукта.
Пример 49.
Схема 4: Метод А: Эпоксидирование 9,11-диенона
Соединение, обозначенное XVIIA (соединение XVII, где оба -А-А- и -В-В- представляют -СН2-СН2-) (40,67 г), растворяли в метиленхлориде (250 мл) в однолитровой 3-горлой колбе, а затем охлаждали снаружи смесью из льда и соли. Затем добавляли бифосфат калия (22,5 г) и трихлорацетонитрил (83,5 г) и смесь охлаждали до 2°С, после чего в течение одного часа медленно добавляли 30% перекись водорода (200 г). Реакционную смесь перемешивали при 12°С в течение 8 часов и при комнатной температуре в течение 14 часов. Затем брали каплю органического слоя и оценивали на присутствие какого-либо исходного енона и было обнаружено, что оно составляет <0,5%. После этого добавляли воду (400 мл), перемешивали в течение 15 минут и слои отделяли. Органический слой последовательно промывали 200 мл иодида калия (10%), 200 мл тиосульфата натрия (10%) и 100 мл насыщенного раствора бикарбоната натрия, отделяя каждый раз слои. Органический слой сушили безводным сульфатом магния и концентрировали с получением сырого эпоксида (41 г). Этот продукт кристаллизовали из этилацетата: метиленхлорида с получением 14,9 г чистого продукта.
Пример 50.
Схема 4: Метод В: Эпоксидирование соединения XVIIA с использованием м-хлорпероксибензойной кислоты
Соединение XVIIA (18,0 г) растворяли в 250 мл метиленхлорида и охлаждали до 10°С. Затем в течение 15 минут, перемешивая, добавляли твердую м-хлорпербензойную кислоту (чистота 50-60%; 21,86 г). Повышения температуры при этом не наблюдалось. Реакционную смесь перемешивали в течение 3 часов и оценивали на присутствие диенона. Эту реакционную смесь последовательно обрабатывали раствором сульфита натрия (10%), раствором гидроксида натрия (0,5 н), раствором хлористоводородной кислоты (5%) и, наконец, 50 мл насыщенного солевого раствора. После сушки безводным сульфатом магния и выпаривания получали 17,64 г эпоксида и использовали непосредственно в следующей стадии. Было оценено, что этот продукт содержал продукт окисления Байера-Виллигера, который удаляли путем растирания с этилацетатом, с последующей кристаллизацией из метиленхлорида. Исходя из масштаба 500 г, осажденную м-хлорпербензойную кислоту фильтровали с последующей стандартной обработкой.
Пример 51.
Схема 4: Метод С: Эпоксидирование соединения XVIIA с использованием трихлорацетамида
Соединение XVIIA (2 г) растворяли в 25 мл метиленхлорида. Затем добавляли трихлорацетамид (2 г) и дикалийфосфат (2 г). Перемешивая при комнатной температуре, добавляли 30% перексись водорода (10 мл) и перемешивание продолжали в течение 18 часов с получением эпоксида (1,63 г). Продукт Байера-Виллигера не образовывался.
Пример 52.
В 2000 мл колбу загружали гидроксид калия (56,39 г; 1005,03 ммоль; 3,00 экв.) и суспендировали с диметилсульфоксидом (750,0 мл) при комнатной температуре. Затем в колбу вместе с ТГФ (956,0 мл) загружали триенон, соответствующий формуле XX (где R3 представляет Н, а каждый из -А-А- и -В-В- представляет -CH2-СН2-) (100,00 г; 335,01 ммоль; 1,00 экв.). После этого в колбу загружали метилсульфат триметилсульфония (126,14 г; 670,02 ммоль; 2,00 экв.) и полученную смесь нагревали с обратным холодильником в течение 1 ч при 80-85°С. За превращением в 17-спирооксиметилен следили с помощью ВЭЖХ. Приблизительно 1 л ТГФ выпаривали из реакционной смеси в условиях вакуума, после чего в течение 30 минут загружали воду (460 мл) и реакционную смесь охлаждали до 15°С. Полученную смесь фильтровали и твердый оксирановый продукт два раза промывали 200 мл аликвотами воды. Как было видно, продукт становился в высокой степени кристаллическим и проводить его фильтрацию было легко. Затем этот продукт сушили в вакууме при 40°С. Было выделено 104,6 г простого 3-метилового эфира енольной формы Δ-5,6,9,11-17-оксиранстероидного продукта.
Пример 53.
В сухой 500 мл реактор в потоке азота загружали этоксид натрия (41,94 г; 616,25 ммоль; 1,90 экв.). Затем в реактор загружали этанол (270,9 мл) и метоксид натрия суспендировали в этаноле. В эту суспензию загружали диэтилмалонат (103,90 г; 648,68 ммоль; 2,00 экв.), после чего добавляли оксиран-стероид, полученный как описано в Примере 52 (104,60 г; 324,34 ммоль; 1,00 экв.), и полученную смесь нагревали с обратным холодильником, то есть при 80-85°С. Нагревание продолжали в течение 4 часов, после чего завершение реакции контролировали с помощью ВЭЖХ. В реакционную смесь в течение 30 минут загружали воду (337,86 мл) и смесь охлаждалась до 15°С. Перемешивание продолжали 30 минут, а затем реакционную суспензию фильтровали с получением осадка на фильтре, содержащего тонкодисперсный аморфный порошок. Этот осадок на фильтре промывали два раза водой (200 мл каждый), а затем сушили при комнатной температуре в условиях вакуума. Выделяли 133,8 г 3-метил-енолэфир-Δ-5,6,9,11-17-спиролактон-21-этоксикарбонильного промежуточного продукта.
Пример 54.
3-Метил-енолэфир-Δ-5,6,9,11-17-спиролактон-21-этоксикарбонильное промежуточное соединение (формулы XVIII, где R3 представляет Н, а каждый из -А-А- и -В-В- представляет -CH2-CH2-; 133,80 г; 313,68 ммоль; 1,00 экв., полученное как описано в Примере 53) вместе с хлоридом натрия (27,50 г; 470,52 ммоль; 1,50 экв.), диметилформамидом (709 мл) и водой (5 мл) загружали при перемешивании в 2000 мл реактор. Полученную смесь нагревали с обратным холодильником при 138-142°С в течение 3 часов, после чего реакционную смесь анализировали с помощью ВЭЖХ на завершение реакции. Затем к смеси в течение 30 минут добавляли воду и вода охлаждалась до 15°С. Перемешивание продолжали в течение 30 минут, после чего реакционную суспензию фильтровали, в результате чего выделяли аморфный твердый реакционный продукт в виде осадка на фильтре. Этот осадок на фильтре два раза промывали (200 мл аликвотами воды), а затем сушили. После сушки получали 91,6 г продукта, 3-метиленолэфир-17-спиролактона (выход 82,3%; 96%, анализ по площади).
Пример 55.
Эфир енола, полученный как описано в Примере 54 (91,60 г; 258,36 ммоль; 1,00 экв.), этанол (250 мл), уксусную кислоту (250 мл) и воду (250 мл) загружали в 2000 мл реактор и полученную суспензию нагревали с обратным холодильником в течение 2 часов. Затем загружали воду (600 мл) в течение 30 минут и за это время реакционная смесь охлаждалась до 15°С. После этого реакционную суспензию фильтровали и остаток на фильтре два раза промывали водой (200 мл аликвотами). Затем осадок на фильтре сушили; и получали 84,4 г продукта, 3-кето Δ-4,5,9,11-17-спиролактона (соединение формулы XVII, где R3 представляет Н, а -А-А- и -В-В- представляют -CH2-CH2-; выход 95,9%).
Пример 56.
Соединение XVIIA (1 кг; 2,81 моль) загружали вместе с тетрахлорметаном (3,2 л) в 4-горлую 22-литровую колбу. В смесь добавляли N-бромсукцинамид (538 г), а затем ацетонитрил (3,2 л). Полученную смесь нагревали с обратным холодильником и поддерживали при этой температуре 68°С в течение приблизительно 3 часов, в результате чего образовывался прозрачный оранжевый раствор. Через 5 минут после нагрвания раствор становился темным. Через 6 часов источник нагревания удаляли и брали образцы смеси. Растворитель выпаривали в вакууме и к остатку на дне сосуда добавляли этилацетат (6 л). Полученную смесь перемешивали, после чего добавляли 5%-ный раствор бикарбоната натрия (4 л) и смесь перемешивали в течение 15 мин, после чего ее оставляли для разделения фаз. Водный слой удаляли и в смесь вводили насыщенный солевой раствор (4 л), после чего смесь перемешивали в течение 15 мин. Затем смесь снова оставляли для разделения фаз и органический слой упаривали в вакууме с получением вязкой суспензии. Затем добавляли диметилформамид (4 л) и выпаривание продолжали при температуре сосуда 55°С. Остаток на дне сосуда оставляли на ночь для остаивания, а затем добавляли DABCO (330 г) и бромид лития (243 г). Затем смесь нагревали до 70°С. После полуторачасового нагревания брали образцы жидкостной хроматографии, а после нагревания в течение 3,50 часов добавляли еще DABCO (40 г). После нагревания в течение 4,5 часов вводили воду (4 л) и полученную смесь охлаждали до 15°С. Эту суспензию фильтровали и осадок на фильтре промывали водой (3 л) и сушили на фильтре в течение ночи. Мокрый осадок (978 г) загружали снова в 22 л колбу и добавляли диметилформамид (7 л). Полученную таким образом смесь нагревали до 105°С, после чего осадок полностью растворялся в растворе. Источник нагревания удаляли и смесь в колбе перемешивали и охлаждали. В рубашку реактора выливали воду со льдом и смесь внутри реактора охлаждали до 14°С и выдерживали в течение 2 часов. Полученную суспензию фильтровали и дважды промывали 2,5 л аликвотами воды. Осадок на фильтре сушили в вакууме в течение ночи. Было получено 510 г светло-коричневого твердого продукта.
Пример 57.
В 2-литровую 4-горлую колбу загружали: 9,11-эпоксиканренон, полученный как описано в Примере 56 (100,00 г; 282,1 ммоль; 1,00 экв.), диметилформамид (650,0 мл), хлорид лития (30,00 г; 707,7 ммоль; 2,51 экв.) и цианогидрин ацетона (72,04 г; 77,3 мл; 846,4 ммоль; 3,00 экв.). Полученную суспензию подвергали механическому перемешиванию и обрабатывали тетраметилгуанидином (45,49 г; 49,6 мл; 395,0 ммоль; 1,40 экв.). Затем систему фильтровали с использованием охлаждаемого водой холодильника и холодильника с сухим льдом (заполненного сухим льдом в ацетоне) для предотвращения утечки HCN. Вытяжная труба от холодильника с сухим льдом была присоединена к скрубберу, заполненному значительным избытком хлорного отбеливателя. Эту смесь нагревали до 80°С.
Через 18 часов получали темный красновато-коричневый раствор, который охлаждали до комнатной температуры при перемешивании. В процессе охлаждения раствор барботировали азотом для удаления остаточного HCN при помощи вытяжной трубы, идущей к скрубберу с отбеливателем. Через 2 часа раствор обрабатывали уксусной кислотой (72 г) и перемешивали в течение 30 минут. Затем сырую смесь выливали, перемешивая, в воду со льдом (2 л). После этого перемешанную суспензию обрабатывали 10% водным HCl (400 мл) и перемешивали в течение 1 часа. Затем смесь фильтровали с получением темного кирпично-красного твердого вещества (73 г). Фильтрат помещали в 4-литровую делительную воронку и экстрагировали метиленхлоридом (3×800 мл); а органические слои объединяли и подвергали обратной экстракции водой (2×2 л). Метиленхлоридный раствор концентрировали в вакууме с получением 61 г темно-красного масла.
После того, как водные промывочные фракции были оставлены на ночь, образовывалось значительное количество осадка. Этот осадок собирали путем фильтрации и было определено, что он является чистым енаминовым продуктом (14,8 г).
После сушки, исходное красное твердое вещество (73 г) анализировали с помощью ВЭЖХ и было определено, что основным компонентом является 9,11-эпоксиенамин. Кроме того, ВЭЖХ показала, что енамин был главным компонентом красного масла, полученного в результате обработки метиленхлоридом. Вычисленный молярный выход енамина составлял 46%.
Пример 58.
9,11-эпоксиенамин (4,600 г; 0,011261 моль; 1,00 экв.), полученный как описано в Примере 57, вводили в круглодонную 1000 мл колбу. К смеси добавляли метанол (300 мл) и 0,5% масс. водной HCl (192 мл), после чего эту смесь нагревали с обратным холодильником в течение 17 часов. Затем метанол был удален в вакууме, что приводило к снижению количества вещества в перегонном кубе до 50 мл и к образованию белого осадка. К суспензии добавляли воду (100 мл), а затем ее фильтровали с получением белого твердого осадка, который три раза промывали водой. Выход твердого 9,11-эпоксидикетонового продукта составлял 3,747 г (81,3%).
Пример 59.
Эпоксидикетон, полученный способом, описанным в Примере 58 (200 мг; 0,49 ммоль), суспендировали в метаноле (3 мл) и к смеси добавляли 1,8-диазабицикло[5,4,0]ундек-7-ен (DBU). После нагревания с обратным холодильником в течение 24 часов смесь становилась гомогенной. Затем ее концентрировали досуха при 30°С на роторном испарителе и остаток распределяли между метиленхлоридом и 3,0н HCl. После концентрирования органической фазы получали желтое твердое вещество (193 мг), которое, как было определено, представляло собой 22% масс. эпоксимексренона. Выход составлял 20%.
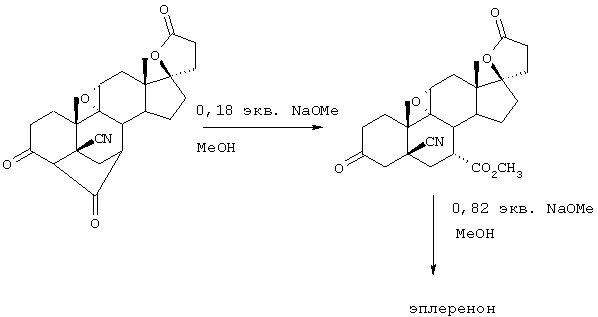
Пример 60.
К 100 мг дикетона (полученного в соответствии с Примером 58), суспендированного в 1,5 мл метанола, добавляли 10 микролитров (0,18 экв.) 25% (масс./масс.) раствора метоксида натрия в метаноле. Раствор нагревали с обратным холодильником. Через 30 минут дикетон не обнаруживался, но присутствовал сложный 5-цианоэфир. К смеси добавляли 46 микролитров 25% (масс./масс.) метанолового раствора натрия в метаноле. Смесь нагревали с обратным холодильником в течение 23 часов, при которых с помощью ВЭЖХ было выявлено, что основным продуктом является эпоксимексренон.
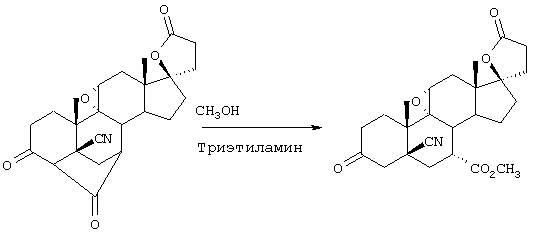
Пример 61.
К 2 г дикетона (полученного в соответствии с Примером 58), суспендированного в 30 мл сухого метанола, добавляли 0,34 мл триэтиламина. Суспензию нагревали с обратным холодильником в течение 4,5 часов. Смесь перемешивали при 25°С в течение 16 часов. Полученную суспензию фильтровали с получением 1,3 г сложного 5-цианоэфира в виде белого твердого вещества.
К 6,6 г дикетона, суспендированного в 80 мл метанола, добавляли 2,8 мл триэтиламина. Смесь нагревали с обратным холодильником в течение 4 часов и перемешивали при 25 об/мин в течение 88 часов, за которые продукт кристаллизовался из раствора. В результате фильтрации, а затем промывки метанолом, было получено 5,8 г сложного цианоэфира в виде белого порошка. Этот порошок кристаллизовали из хлороформа/метанола с получением 3,1 г кристаллического продукта, который был гомогенным, о чем свидетельствовала ВЭЖХ.
Исходя из вышеописанного можно сказать, что были достигнуты некоторые цели настоящего изобретения и получены другие преимущественные результаты.
Следует отметить, что хотя в вышеописанных композициях и способах могут быть внесены различные изменения, не выходящие за рамки объема настоящего изобретения, однако все варианты, описанные в настоящей заявке и проиллюстрированные в прилагаемых чертежах, имеют иллюстративный характер и не должны рассматриваться как некое ограничение существа изобретения.
Новые соединения
Кроме того, настоящее изобретение относится к дополнительным полициклическим органическим молекулам, которые могут быть использованы в качестве хроматографических маркеров при получении стероидных соединений, обладающих преимущественной биологической активностью, таких как спиронолактон или эпоксимексренон.
Короче говоря, было установлено, что некоторые соединения, содержащие замещенные или незамещенные стероидные ядра и замещенное или незамещенное карбоциклическое кольцо, конденсированное со стероидным ядром в 13,17-положении, могут быть использованы в качестве внутренних или хроматографических маркеров при получении стероидов, таких как спиронолактон и эпоксимексренон. В частности, соединение метил-2,3,3а,4,6,7,9,10,11,11а,12,13-додекагидро-3аβ,11аβ-диметил-1,9-диоксо-1Н-пенталено[1,6а-а]фенантрен-6α-карбоксилат:

может быть использовано в качестве хроматографического маркера. Одним из новых отличительных признаков этого соединения и родственных соединений настоящего изобретения является конденсированное карбоциклическое кольцо, присоединенное к кольцу D стероидного ядра. Спиронолактон и эпоксимексренон не обладают этим признаком, а вместо этого они содержат 20-спиролактонное кольцо.
Упоминаемое в настоящем описании стероидное ядро данного соединения соответствует следующей структуре:
Эта структура имеет стандартную нумерацию и обозначения колец, принятые для стероидных соединений. Стероидное ядро может быть насыщенным, ненасыщенным, замещенным или незамещенным. Оно содержит, предпочтительно, по крайней мере от одной до четырех ненасыщенных связей. Более предпочтительно, когда каждое из колец А, С и D содержит по крайней мере одну ненасыщенную связь. Стероидное ядро может быть также замещено, как более конкретно обсуждается ниже. Это ядро замещено, предпочтительно, по крайней мере С7-сложноэфирной группой.
Упоминаемое в настоящем описании карбоциклическое кольцо, конденсированное со стероидным ядром, соответствует четырех-, пяти- или шести-углеродному циклическому скелету. Оно может быть насыщенным, ненасыщенным, замещенным или незамещенным. Предпочтительно оно является насыщенным, либо имеет одну двойную связь и замещено гидрокси- или кето-группой. Кроме того, карбоциклическое кольцо предпочтительно имеет α-ориентацию по отношению к стероидному ядру.
В предпочтительном варианте осуществления изобретения карбоциклическое кольцо содержит пятиуглеродный циклический скелет и имеет α-ориентацию и указанное соединение выбирают из группы, состоящей из соединений, соответствующих следущим формулам:
 ;
;
где:
-А-А- представляет группу -CR102R102a-CR103R103a- или -CR102=CR103, где -CR102R102a- и -CR102= соответствует углероду С2, a CR103R103a- и =CR103- соответствуют углероду С3;
-D-D- представляет группу -CHR104-CH- или -CR104=C-;
-Е-Е- представляет группу -CH2-CR110- или -СН=С;
-А-Е- представляет группу -CR102R102a-CH2- или -CR102=CH, где -CR102R102a- и -CR102= соответствуют углероду С2, a -CH2- и=СН- соответствуют углероду С1;
-G-G- представляет группу -CR106R106a-CHR107- или -CR106=CR107-, где -CR106R106a- и -CR106= соответствуют углероду С6, a -CHR107- и =CR107- соответствуют углероду С7;
-J-J- представляет группу -CR108-CR109- или -С=С-, где -CR108- соответствует углероду С8, a -CR109- соответствует углероду С9;
-L-L- представляет группу -CR111R111a-CH2- или -CR111-CH-, где -CR111R111a- и -CR111= соответствуют углероду С11, a -CHR2- и =СН- соответствуют углероду С12;
-J-L- представляет группу -CR109-CR111R111a- или -C=CR111-, где -CR109- и -С= соответствуют углероду С9, a CR111R111а- и -CR111- соответствуют углероду С11;
-М-М- представляет группу -CR114-CH2- или -С=СН-, где -CR114- и -С= соответствуют углероду С14, a -CH2- и -С=Н- соответствуют углероду С15;
J-M- представляет группу -CR108-CR114- или -С=С-, где -CR108- соответствует углероду С8, a -CR114- соответствует углероду С14;
-Q-Q- представляет группу -CR120R120a-CR119R119a- или -CR120=CR119-, где -CR120R120a- и -CR120=CR119 соответствует углероду С20, a CR119R119a- и =CR119- соответствуют углероду С19;
-Q-T- представляет группу -CR119R119a-CHR118- или -CR119=CR118-, где -CR119R119a- и -CR119= соответствуют углероду С19, a -CHR118- и =CR118- соответствуют углероду С18;
R102 представляет водород, алкил, алкенил или алкинил;
R102a представляет водород; либо R102a представляет связь между атомом углерода С2 и атомом углерода С3 в том случае, если -А-А представляет группу -CR102=CR103-, а -А-Е- представляет группу -CR102R102a-CH2-, либо связь между атомом углерода С1 и атомом углерода С2 в том случае, если -А-Е- представляет группу -CR102=CH-, а -А-А- представляет группу -CR102R102a-CR103R103a-;
R103 представляет водород, гидрокси, защищенный гидрокси, R130O-, R130C(O)O-, R130OC(O)O- или R103, взятый вместе с R103a, образует оксо при условии, что -А-А- представляет -CR102R102a-CR103R103a-, когда R103 вместе с R103a образуют оксо;
R103a представляет водород, либо R103a вместе с R103 образуют оксо при условии, что -А-А- представляет -CR102R102a-CR103R103a-, когда R103a вместе с R103 образуют оксо;
R104 представляет водород, алкил, алкенил или алкинил;
R106 представляет водород, гидрокси или защищенный гидрокси, либо вместе с R106a образует оксо, либо вместе с R106 и атомом углерода, с которым они связаны, образуют циклопропильное, циклобутильное или циклопентильное кольцо при условии, что -G-G- представляет группу -CR106R106a-CR107R107a-, если R106 вместе с R106a образуют оксо;
R106a представляет водород, гидрокси или защищенный гидрокси, либо вместе с R106 образует оксо, либо вместе с R106 и атомом углерода, с которым они связаны, образует циклопропильное, циклобутильное или циклопентильное кольцо; или R106а вместе с R107а и атомами углерода, с которыми они связаны, образуют циклопропильное, циклобутильное или циклопентильное кольцо;
R107 представляет водород; гидроксикарбонил; низший алкил, алкенил, алкинил, арил, гетероарил или аралкил; галогеналкил; гидроксиалкил; алкоксиалкил; низший алканоил, алкеноил, алкиноил, арилоил, гетероарилоил или аралканоил; низший алкоксикарбонил, алкеноксикарбонил, алкиноксикарбонил, арилоксикарбонил, гетероарилоксикарбонил или аралкоксикарбонил; низший алканоилтио, алкеноилтио, алкиноилтио, арилоилтио, гетерароилтио или аралканоилтио; низший алкилтио, алкенилтио, алкинилтио, арилтио, гетероарилтио или аралкилтио; карбамил; алкоксикарбониламино; или циано; или
R107 вместе с R106a и атомами углерода, с которыми они связаны, образуют циклопропильное, циклобутильное или циклопентильное кольцо; или
R107 и R114, взятые вместе с атомами углерода С7, С8 и С14, образуют γ-лактон;
R108 представляет водород, гидрокси, защищенный гидрокси, алкил, алкенил, алкинил, R140O-, R140C(O)O-, R140OC(O)O-; или представляет связь между атомом углерода С8 и атомом углерода С9 в случае, если -J-J- представляет группу -С=С-, a -J-M- представляет группу -CR108-CR114-; либо представляет связь между атомом углерода С8 и атомом углерода С14 в случае, если -J-M- представляет группу -С=С-, а -J-J- представляет группу -CR108-CR114-;
R109 представляет водород, гидрокси, защищенный гидрокси, алкил, алкенил, алкинил, R150O-, R150C(O)O-, R150OC(O)O-; или представляет связь между атомом углерода С9 и атомом углерода С11 в случае, если -J-L- представляет группу -C=CR111-, а -J-J- представляет группу -CR108-CR109-; либо представляет связь между атомом углерода С9 и атомом углерода С8 в случае, если -J-J- представляет группу -С=С-, a -J-L- представляет группу -CR109-CR111R111a-;
R110 представляет водород или метил;
R111 представляет водород, гидрокси или защищенный гидрокси, либо R111 вместе с R111a образуют оксо при условии, что -J-L- представляет группу -CR109-CR111R111a-, a -L-L- представляет группу -CR111R111a-CH2-, если R111 вместе с R111a образует оксо;
R111a представляет водород, либо вместе с R111 образует оксо при условии, что -J-L- представляет группу -CR109-CR111R111a-, а -L-L- представляет группу -CR111R111a-CH2-, если R111 вместе с R111a образует оксо; либо R111a представляет связь между атомом углерода С11 и атомом углерода С9 в случае, если -J-L- представляет группу -C=CR111, a -L-L- представляет группу -CR111R111a-CH2-, либо R111a представляет связь между атомом углерода С11 и атомом углерода С12 в случае, если L-L-представляет группу CR111=CH, a -J-L- представляет группу CR109R109а-CR111R111а-;
R114 представляет водород, гидрокси, защищенный гидрокси, алкил, алкенил, алкинил, R160O-, R160C(O)O-, R160OC(O)O-; либо R114 и R107 вместе с атомами углерода С7, С8 и С14 образуют γ-лактон; либо R114 представляет связь между атомом углерода С14 и атомом углерода С8 в случае, если -J-M- представляет группу -С=C-, а -М-М- представляет группу -CR114-CH2-; либо R114 представляет связь между атомом углерода С14 и атомом углерода С15 в случае, если -М-М- представляет группу -С=СН-, а -J-M- представляет группу -CR108-CR114-;
R118 представляет водород, алкил, алкенил, алкинил, арил, гетероарил, алкилтио, алкенилтио или циано;
R118a представляет водород или связь между атомом углерода С18 и атомом углерода С19 в случае, если -Q-T- представляет группу -CR118=CR119-, a Q-Q- представляет группу CR119R119a-CR120R120a-;
R119 представляет водород, алкил или алкенил;
R119a представляет водород или связь между атомом углерода С19 и атомом углерода С20 в случае, если -Q-Q- представляет группу -CR120=CR119-, a -Q-T- представляет группу CR119R119a-CR118R118a-; либо связь между атомом углерода С19 и атомом углерода С18 в случае, если -Q-T- представляет группу -CR119=CR118-, a -Q-Q- представляет группу CR119R119a-CR120R120a-;
R120 представляет водород, гидрокси, защищенный гидрокси, либо вместе с R120a образует оксо; при условии, что -Q-Q- представляет группу CR119R119a-CR120R120a-, если R120 вместе с R120a образуют оксо;
R120a представляет водород, либо вместе с R120 образует оксо; при условии, что -Q-Q- представляет группу CR119R119a-CR120R120a-, если R120a вместе с R120 образуют оксо; и
R130, R140, R150 и R160 независимо представляют алкил, алкенил, алкинил, арил или гетероарил.
Более предпочтительно, это соединение соответствует соединению формулы С-3, где R107 представляет водород, гидроксикарбонил, низший алкил, низший алканоил, низший алкоксикарбонил, низший алканоилтио, низший алкилтио, карбамил, либо R107, взятый вместе с R106a и атомами углерода, с которыми они связаны, образуют циклопропильное кольцо; R106 представляет водород, гидрокси, либо взятый вместе с R106a и атомом углерода, с которым они связаны, образуют циклопропильное кольцо; R106a представляет водород, гидрокси или вместе с R106 и атомом углерода, с которым они связаны, образуют циклопропильное кольцо; либо R106a, взятый вместе с R107 и атомами углерода, с которыми они связаны, образуют циклопропильное кольцо; a R120 представляет кето.
Для обсуждения 13,17-конденсированных кольцевых соединений, описанных в настоящей заявке, используются нижеследующие определения.
Термин "низший алкил" означает алкильный радикал, имеющий от 1 до 6 атомов углерода, такой как метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил и трет-бутил, пентил и гексил. Этот радикал может быть прямым, разветвленным или циклическим, а также замещенным (в частности, арилом), незамещенным или гетерозамещенным.
Термин "низший алканоил" означает радикал, предпочтительно происходящий от прямого алкила, имеющего от 1 до 7 атомов углерода, и связанный с исходной молекулярной частью посредством карбонильной группы. Особенно предпочтительными являются формил и ацетил.
Термин "низший алкоксикарбонил" означает радикал, предпочтительно происходящий от прямого алкила, имеющего от 1 до 7 атомов углерода, и связанный с атомом кислорода, причем указанный атом кислорода присоединен к исходной молекулярной части посредством карбонильной группы. Особенно предпочтительными являются метоксикарбонил, этоксикарбонил, изопропоксикарбонил и н-гексилоксикарбонил.
Термин "низший алкенил" означает алкенильный радикал, имеющий от 2 до 6 атомов углерода, такой как этенил, пропенил, изопропенил, бутенил, изобутенил, втор-бутенил и трет-бутенил, пентенил и гексенил. Этот радикал может быть прямым или разветвленным, а также замещенным, незамещенным или гетерозамещенным. Термины "низший алкеноил" и "низший алкеноксикарбонил" являются такими, как они были определены для "низшего алканоила" и "низшего алкоксикарбонила", соответственно, за исключением того, что они происходят не от прямого алкила, а от прямого алкенила. Предпочтительно, кислород, связанный с алкенильным радикалом любой алкеноксикарбонильной группы, отделен от какого-либо ненасыщенного углерода по крайней мере одной метиленовой группой.
Термин "низший алкинил" означает алкинильный радикал, имеющий от 2 до 6 атомов углерода, такой как этинил, пропинил, изопропинил, бутинил, изобутинил, втор-бутинил и трет-бутинил, пентинил и гексинил. Этот радикал может быть прямым или разветвленным, а также замещенным, незамещенным или гетерозамещенным. Термины "низший алканоил" и "низший алкиноксикарбонил" являются такими, как они были определены для "низшего алканоила" и "низшего алкоксикарбонила", соответственно, за исключением того, что они происходят не от прямого алкила, а от прямого алкинила. Предпочтительно, кислород, связанный с алкинильным радикалом любой алкиноксикарбонильной группы, отделен от любого ненасыщенного углерода по крайней мере одной метиленовой группой.
"Арильная" часть предпочтительно содержит, либо отдельно, либо с различными заместителями, от 5 до 15 атомов и представляет собой фенил.
Термин "низший алкилтио" означает радикал, предпочтительно происходящий от прямого алкила, имеющего от 1 до 7 атомов углерода, и связанный с исходной молекулярной частью посредством атома серы. Особенно предпочтительным является метилтио.
Термин "низший алканоилтио" означает радикал, предпочтительно происходящий от прямого алкила, имеющего от 1 до 7 атомов углерода, и связанный с карбонильной группой, причем указанная карбонильная группа присоединена к исходной молекулярной части посредством атома серы. Особенно предпочтительным является ацетилтио.
Термины "низший алкенилтио" и "низший алкеноилтио" являются такими, как они были определены для "низшего алкилтио" и "низшего алканоилтио", соответственно, за исключением того, что они происходят не от прямого алкила, а от прямого алкенила. Предпочтительно, атом серы любой алкенилтио-группы отделен от любого ненасыщенного углерода по крайней мере одной метиленовой группой.
Термины "низший алкинилтио" и "низший алкиноилтио" являются такими, как они были определены для "низшего алкилтио" и "низшего алканоилтио", соответственно, за исключением того, что они происходят не от прямого алкила, а от прямого алкинила. Предпочтительно, атом серы любой алкинилтио-группы отделен от какого-либо ненасыщенного углерода по крайней мере одной метиленовой группой.
Термин "карбамил" означает радикал -NH2, связанный с исходной молекулярной частью посредством карбонильной группы. Карбамильная группа может быть моно-замещенной или ди-замещенной, а заместителями могут быть алкильный, алкенильный, алкинильный и арильный радикалы.
Группы, определенные выше, могут быть незамещенными или дополнительно замещенными. Такими дополнительными заместителями могут быть алкил, алкенил, алкинил, арил, гетероарил, карбокси, такой как алкокси, карбоксиалкил, ацил, ацилокси, галоген, такой как хлор или фтор, галогеналкокси, нитро, амино, амидо и кето. Группы, определенные выше, а также дополнительные заместители могут также содержать кислород, серу, фосфор и/или азот.
В настоящем описании "Me" означает метил; "Et" означает этил; а "Ас" означает ацетил.
В еще более предпочтительном варианте настоящего изобретения соединение выбирают из группы, состоящей из соединений, имеющих следующие формулы:
 ;
;
 ;
;
 ;
;
 ;
;
 ;
;
 ;
;
 ;
;
 ;
;
 ;
;
 ;
;
 ;
;
 ; и
; и
где R107, R106, R106a и R120 определены выше. Предпочтительно, R107 представляет водород, гидроксикарбонил, низший алкил, низший алканоил, низший алкоксикарбонил, низший алканоилтио, низший алкилтио, карбамил, либо вместе с R106a и атомами углерода, с которыми они связаны, образует циклопропильное кольцо; R106 представляет водород, гидрокси, либо вместе с R106a и атомом углерода, с которым они связаны, образует циклопропильное кольцо; R106a представляет водород, гидрокси, либо вместе с R106 и атомом углерода, с которым они связаны, образуют циклопропильное кольцо, либо вместе с R107 и атомами углерода, с которыми они связаны, образуют циклопропильное кольцо; a R120 представляет кето. Еще более предпочтительно, если это соединение, кроме того, соответствует соединению формулы С-5.
В еще более предпочтительном варианте осуществления изобретения соединение формулы С-3 выбирают из группы, состоящей из следующих соединений:

В наиболее предпочтительном варианте осуществления изобретения этим соединением является метил-2,2,3а,4,6,7,9,10,-11,11а,12,13-додекагидро-3αβ,11αβ-диметил-1,9-диоксо-1Н-пенталено[1,6а-а]фенантрен-6α-карбоксилат:
Это соединение формулы С-1 является особенно предпочтительным хроматографическим маркером при получении эпоксимексренона.
Новые соединения, обсуждаемые в настоящем описании, могут быть получены также в форме их солей.
Получение новых соединений
В основном, новые соединения, описанные непосредственно выше, могут быть получены с помощью реакции стероида, имеющего 20-спироксановое кольцо и стероидное ядро, описанное выше, с тригалогенированной алкановой кислотой. Предпочтительным реагентом для данной реакции является соль щелочного металла и используемой алкановой кислоты. Особенно предпочтительно, чтобы реагент для реакции включал 3-фторуксусную кислоту и соль щелочного металла и этой кислоты, такую как трифторацетат калия. Кроме того, осушитель, такой как трифторуксусный ангидрид, предпочтительно, используют в данной реакции для снижения уровня свободной воды, присутствующей в кислоте.
Стероидные соединения, используемые в качестве исходных соединений, предпочтительно имеют следующую формулу:
 ;
;
где -А-А-, -D-D-, -Е-Е-, -А-Е-, -G-G-, -J-J-, -L-L-, -J-L-, -М-М-, -J-M-, -Q-Q-и -Q-T- определены выше. Такие исходные соединения могут быть получены и/или выделены способами, аналогичными способам, описанным в Схеме 1 для рассмотренного ранее способа синтеза эпоксимексренона. Альтернативно, исходные соединения являются коммерчески доступными.
Исходная концентрация стероидного соединения формулы С-3 предпочтительно составляет по крайней мере около 0,1% по массе всей реакционной смеси, более предпочтительно от около 2% до около 20% масс., и даже более предпочтительно от около 5% до около 15% масс. Предпочтительно, чтобы тригалогеналкановая кислота присутствовала в избытке. Если используется трифторуксусный ангидрид, то его количество должно составлять по крайней мере около 3% по массе всей реакционной смеси, более предпочтительно от около 5% до около 25% масс., а наиболее предпочтительно от около 10% до около 15% масс.
Кроме того, реакционная температура должна превышать комнатную температуру (22°С). Предпочтительно, реакционная температура составляет от около 40°С до 100°С, более предпочтительно от около 50°С до 80°С, еще более предпочтительно от около 60°С до 70°С, а наиболее предпочтительно от около 60°С до 65°С. В данной реакции при повышении реакционной температуры до около 70°С или выше, продуцируется более высокое количество побочного продукта С14-лактона. Время реакции должно составлять по крайней мере около 30 минут, более предпочтительно от около 30 до около 6 часов, еще более предпочтительно от около 45 минут до около 4 часов, а наиболее предпочтительно, от около одного часа до двух часов. В предпочтительном варианте осуществления изобретения время реакции составляет от около одного часа до двух часов, а температура реакции поддерживается при около 60°С.
Нижепривиденная схема S-1 иллюстрирует особенно предпочтительный вариант этого способа:
Схема S-1.

Стероиды с конденсированными кольцами, имеющие различные заместители в различных положениях стероида, могут быть получены в соответствии с нижепривиденными реакционными схемами. Дополнительные процедуры и методы, которые конкретно не описаны в настоящей заявке и которые могут быть использованы для введения различных заместителей в различные положения стероида, известны каждому специалисту. Исходными соединениями могут быть либо стероид с конденсированным кольцом, либо стероид с 20-спироксановым кольцом. Для простоты описания в случае, когда исходным соединением является стероид с конденсированными кольцами, в нижеследующих реакционных схемах используются конкретные стероиды или группы стероидов в качестве иллюстративных исходных соединений. Однако, при этом следует отметить, что в той же самой серии реакций с использованием в качестве исходного соединения другого стероида с конденсированными кольцами, могут быть продуцированы другие стероидные производные с конденсированными кольцами или их аналоги. Аналогичным образом, для простоты описания в случае, когда исходным соединением является стероид с 20-спироксановым кольцом, в качестве исходных соединений используются некоторые конкретные стероиды с 20-спироксановым кольцом. При этом следует отметить, что в той же самой серии реакций с использованием в качестве исходного соединения другого стероида с 20-спироксановым кольцом, могут быть продуцированы другие стероидные производные с 20-спироксановым кольцом или их аналоги.
Стероиды, имеющие карбоновокислотный заместитель у С7, могут быть получены путем реакции омыления стероида, имеющего алкоксикарбонильный заместитель у С7, такого как соединение формулы С-1. Реакция омыления может быть проведена путем обработки исходного стероида щелочным реактивом, таким как гидроксид калия или натрия в подходящем растворителе, таком как метанол, этанол, изопропанол или т.п., при температурах вплоть до температуры кипения растворителя в присутствии или в отсутствие воды. Как проиллюстрировано на Схеме S-2, реакция омыления соединения формулы С-1 дает карбоновую кислоту формулы С-101.
Стероиды, имеющие карбоновоэфирные заместители у С7, не являющиеся метаноатом, могут быть получены с использованием карбоновых кислот, таких как С-101, в качестве исходных соединений. Обработка таких карбоновых кислот алкилирующим агентом, таким как алкилгалогенид, в присутствии основания (такого как бикарбонат натрия, карбонат натрия, бикарбонат калия или триэтиламин) в растворителе, таком как диметилформамид, приводит к получению нужных сложных эфиров. Примерами подходящих алкилирующих агентов являются этилиодид, этилбромид, изопропилиодид, гексилиодид, бензилбромид, аллилиодид и т.п.
Для синтеза карбамилов подходящим исходным соединением является также карбоновая кислота С-101. Обработка кислоты хлорформиатом, таким как изобутилхлорформиат или этилхлорформиат, в присутствии основания приводит к получению смешанного ангидрида. Обработка смешанного ангидрида амином (таким как диметиламин, метиламин или бенэиламин) приводит к образованию карбамила, где R1 и R2 являются заместителями на различных аминах.
Схема S-2.
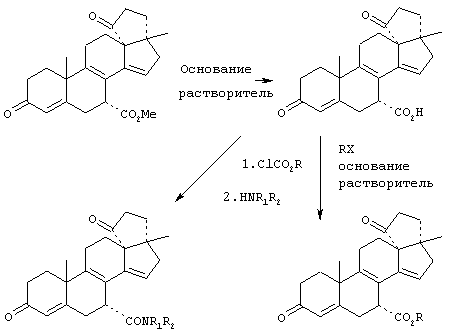
Некоторые модификации в положении С7 сделаны с использованием ненасыщенных кетонов, таких как соединения формулы С-105 (показанной ниже в Схеме S-3) в качестве исходных соединений. Сульфиды синтезируют путем присоединения подходящих тиолов в основных условиях. Примерами подходящих тиолов являются метилмеркаптан, этилмеркаптан и т.п. Подходящими основаниями являются пиперидин, триэтиламин и т.п.
Обработка ненасыщенных кетонов (таких как соединение формулы С-105) тиоалкановыми кислотами, такими как тиоуксусная кислота, приводит к образованию С7-тиоацильных соединений, таких как ацетилтио.
Конденсированный С6,С7-циклопропильный заместитель может быть введен путем обработки ненасыщенных кетонов (таких как соединение формулы С-105) метилидом диметилсульфоксония, который образуется путем обработки галогенида триметилсульфоксония подходящим основанием (таким как гидрид натрия) в подходящем растворителе.
Эти различные схемы синтеза проиллюстрированы в Схеме S-3, показанной ниже:
Схема S-3.
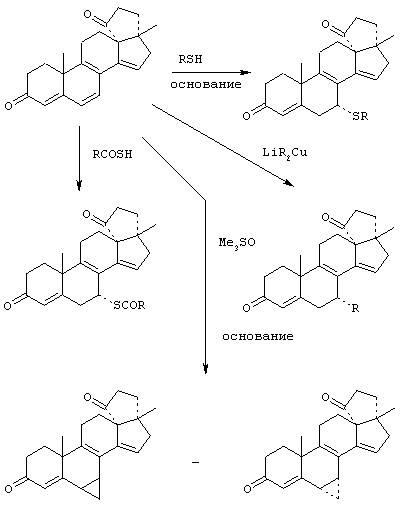
Стероиды, содержащие С6-спироциклопропильное кольцо, синтезируют способами, описанными ниже в Схеме S-4. Сначала еноны, такие как соединение формулы С-110, защищают в виде простого эфира енола С3 путем обработки сложным орто-эфиром, таким как триэтилортоформиат или триметилортоформиат, в присутствии кислоты, такой как п-толуолсульфоновая кислота. Полученный простой эфир енола обрабатывают реактивом Вильсмайера, образованным in situ путем добавления оксихлорида фосфора к диметилформамиду с образованием формильного соединения, такого как соединение формулы С-112. Восстановление формильной группы осуществляют с использованием гидридного восстановителя, такого как литий три-трет-бутоксиалюминий гидрид в растворителе, таком как тетрагидрофуран. Это приводит к получению промежуточного спирта, который после обработки кислотой элиминирует воду с образованием 6-метиленового соединения, такого как соединение формулы С-113. Подходящей кислотой является хлористоводородная кислота в водной среде. Обработка 6-метиленового соединения диазометаном приводит к образованию промежуточного пиразолина, который разлагается после нагревания с получением продукта, такого как спироциклопропиловое соединение формулы С-114. Защищенный простой эфир енола (такой как соединение формулы С-111) является универсальным промежуточным соединением и обработка гидридным восстановителем, таким как борогидрид натрия, с последующим кислотным гидролизом приводит к получению гидрокси-соединений, таких как соединения формулы С-115 и С-116. Эти различные стадии синтеза проиллюстрированы ниже на Схеме S-4:
Схема S-4.
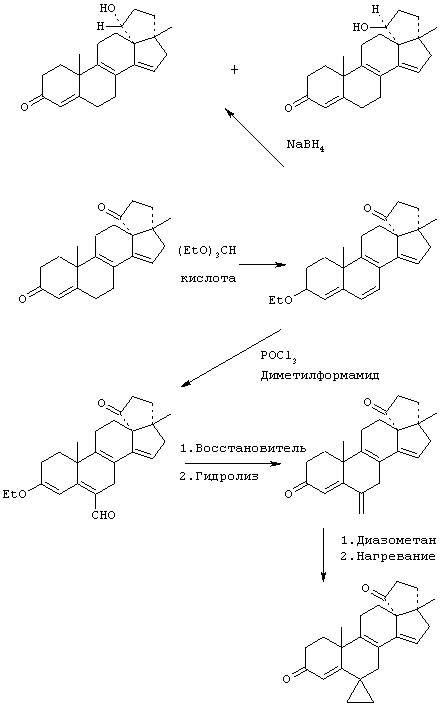
Стероиды, имеющие гидрокси-заместитель у С6 и сложно-эфирный заместитель у С7, могут быть синтезированы методами, проиллюстрированными ниже в Схеме S-5. Сложный эфир (такой как соединение формулы С-1) защищают у карбонила СЗ путем образования простого эфира 3,5-диенола (такого как соединение формулы С-117) с использованием сложного орто-эфира, такого как триэтилортоформиат или триметилортоформиат, в присутствии кислоты. Подходящей кислотой является п-толуолсульфоновая кислота. Обработка простого эфира енола окислителем, таким как мета-хлорпероксибензойная кислота, приводит к образованию гидрокси-соединения, такого как соединение формулы С-118.
Схема S-5
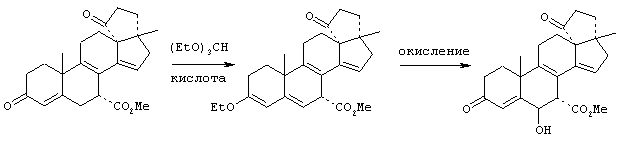
На Схеме S-6 проиллюстрировано введение двойной связи в положение С1-С2 стероида. Это может быть достигнуто путем обработки нужного стероида (такого как соединения формул С-1, С-108 и С-114) подходящим окислителем, таким как дихлордицианобензохинон, в подходящем растворителе (таком как диоксан) при температурах, доходящих вплоть до температуры кипения. Этим способом могут быть получены ненасыщенные соединения С1-С2, такие как соединения формул С-127, С-128 и С-129.
Схема S-6.
На Схеме S-7 проиллюстрировано введение двойной связи в конденсированное кольцо. Стероид (такой как соединение формулы С-114) обрабатывают сложным ортоэфиром (таким как триэтилортоформиат или триметилортоформиат) в присутствии кислотного катализатора (такого как п-толуолсульфоновая кислота) с получением простого эфира енола, где карбонил С3 является защищенным. В случае соединения формулы С-114, поскольку положение С6 является полностью замещенным, образованным простым эфиром енола, является эфир енола С2-С3 (такой как соединение формулы С-131). Обработка эфира енола сильным основанием, таким как диизопропиламид лития, при низкой температуре (от -78°С до -30°С), с последующей обработкой селенирующим агентом, таким как фенилселенилхлорид, приводит к образованию селенового производного, такого как соединение формулы С-132. Окисление селенового производного окислителем, таким как перекись водорода, например, при комнатной температуре, в присутствии основания, такого как пиридин, в растворителе, таком как метиленхлорид, приводит к элиминированию селено-группы и введению двойной связи. Гидролиз простого эфира енола приводит к получению кетона, такого как соединение формулы С-134.
Схема S-7.
На Схеме S-8 проиллюстрирован синтез изомеров с двойной связью соединения формулы С-1. Обработка различных спироксановых соединений, показанных на Схеме S-8, ацетатом калия, трифторуксусным ангидридом и трифторуксусной кислотой в условиях, аналогичных условиям синтеза соединения формулы С-1, приводит к образованию соединений формул С-121 и С-123.
Схема S-8.
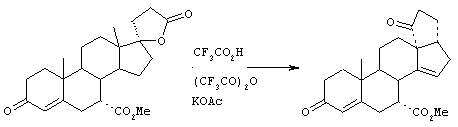
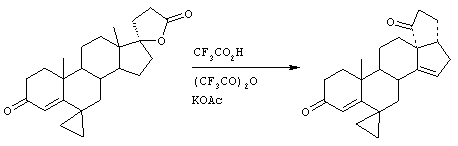
На Схеме S-9 проиллюстрирован альтернативный способ синтеза изомеров с двойной связью для этого семейства стероидов. В предварительно полученный енон (такой как соединение формулы С-24, синтез которого описан выше), уже содержащий конденсированное кольцо, вводят конденсированный С6-С7-циклопропан с использованием химического метода, описанного выше в Схеме S-3 для синтеза соединений, таких как соединения формул С-108 и С-109.
Схема S-9.
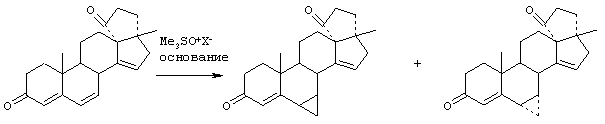
где X представляет галоген.
Конденсированные кольцевые стероиды, имеющие ароматическое кольцо А, могут быть получены путем обработки стероидов, таких как стероиды, описанные в работе P.Compain, et al., Tetrahedron, 52(31), 10405-10416 (1996) (которая вводится в настоящее описание посредством ссылки), трифторуксусной кислотой, ацетатом калия и трифторуксусным ангидридом, в основном, способом, аналогичным рассмотренному ранее для Схемы S-1.
Ожидается, что эти новые конденсированные кольцевые стероиды, содержащие ароматическое кольцо А и 3-гидрокси-заместитель, подвергаются всем химическим реакциям, которые являются типичными для фенолов. На Схеме S-10 проиллюстрирован синтез простого эфира 3-фенола из такого стероида с конденсированными кольцами. В частности предполагается, что обработка этих феноловых соединений основанием и алкилгалогенидом или алкилсульфонатом приводит к продуцированию соответствующего сложного эфира фенола. См. работы Feuer & Hooz, In The Chemisty of the Ether Linkage, Patai (Ed.), Interscience: New York, pp.446-450, 460-468 (1967); и Olson, W.T., J.Am.Chem.Soc., 69, 2451 (1947), которые вводятся в настоящее описание посредством ссылки.
Схема S-10.

Аналогично, Схема S-11 иллюстрирует синтез сложного эфира 3-фенола из стероида с конденсированными кольцами, имеющего ароматическое кольцо А и 3-гидрокси-заместитель. В частности предполагается, что обработка этих феноловых соединений ангидридом карбоновой кислоты или галогенидом карбоновой кислоты приводит к продуцированию соответствующего сложного эфира фенола. См. работу March, J., Advanced Organic Chemistry, Wiley: New York, pp.346-347 (1985), которая вводится в настоящее описание посредством ссылки.
Схема S-11.

Схема S-12 иллюстрирует синтез карбоната 3-фенола из стероида с конденсированными кольцами, имеющего ароматическое кольцо А и 3-гидрокси-заместитель. В частности, предполагается, что обработка этих феноловых соединений алкилгалогенформиатом приводит к продуцированию соответствующего карбоната фенола. См. работу March, J., Advanced Organic Chemistry, Wiley: New York, pp.346-347 (1985), которая вводится в настоящее описание посредством ссылки.
Схема S-12.
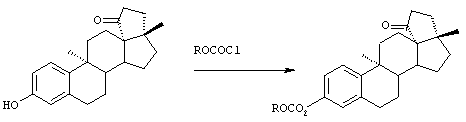
Схема S-13 иллюстрирует синтез орто-аллил-замещенных фениловых производных из стероида с конденсированными кольцами, имеющего ароматическое кольцо А и 3-гидрокси-заместитель. В частности ожидается, что обработка этих феноловых соединений основанием и аллилгалогенидом приводит к продуцированию соответствующего аллилфенилового эфира. Этот аллилфениловый эфир должен давать смесь орто-аллил-замещенных фениловых производных после термической перегруппировки. См. монографию Shine, H., J.Aromantic Rearrangements; Reaction Mechanisms in Organic Chemistry, Monograph 6, American Elsevier; New York, pp.89-120 (1967), которая вводится в настоящее описание посредством ссылки.
Схема S-13.
Схема S-14 иллюстрирует синтез орто-диалкилированных замещенных фениловых производных из стероида с конденсированными кольцами, имеющего ароматическое кольцо А и 3-гидрокси-заместитель. В частности ожидается, что обработка этих соединений спиртом и кислотой приводит к продуцированию соответствующих орто-диалкилированных фениловых производных. См., например, работу Calcott, W.S., J.Am.Chem.Soc., 61, 1010 (1939), которая вводится в настоящее описание посредством ссылки.
Схема S-14.
Схема S-15 иллюстрирует аллильное окисление стероида с конденсированными кольцами, имеющего ароматическое кольцо А и 3-гидрокси-заместитель. В частности, эти соединения могут быть окислены в аллильном положении посредством реакции с диоксидом селена и трет-бутилгидропероксидом с образованием соответствующего спирта. В результате дегидратации этого спирта получают соответствующий олефин. См., например, работу Schmuff, N.R., J.Org.Chem., 48, 1404 (1983), которая вводится в настоящее описание посредством ссылки.
Схема S-15.
Схема S-16 иллюстрирует защиту 3-карбонильной группы и восстановление 20-карбонильной группы новых стероидов с конденсированными кольцами настоящего изобретения. В частности, эти соединения могут быть подвергнуты реакции с триалкилортоформиатом и кислотой с образованием простого эфира 3-енола. Этот простой эфир 3-енола может быть затем подвергнут реакции с борогидридом натрия с восстановлением С-20 карбонильной группы в соответствующий С-20-спирт. Обработка С-20-спирта кислотой и водой обеспечивает деблокирование эфира 3-енола с образованием 3-кето-производного.
Схема S-16.
Схема S-17 иллюстрирует гидрирование олефиновых связей в новых стероидах с конденсированными кольцами настоящего изобретения. В частности предполагается, что гидрирование протекает постадийно. Сначала насыщается С-6,С-7-двойная связь, а затем насыщается С-8,С-14-двойная связь.
Схема S-17.
Схема S-18 иллюстрирует перегруппировку защищенных 11α-гидрокси-стероидов с конденсированными кольцами настоящего изобретения. В частности, сначала 11α-гидрокси-группу защищают подходящей защитной группой, такой как 2-метоксиэток-симетиловый эфир (МЕМ-эфир). Ожидается, что обработка защищенного 11α-гидрокси-стероида солью щелочного металла и тригалогеналкановой кислотой в присутствии ангидрида кислоты приводит к перегруппровке лактонной части в этих молекулах, как проиллюстрировано ниже. Удаление МЕМ-эфира бромидом цинка приводит к образованию перегруппированных спиртов, показанных ниже.
Схема S-18.
Схема S-19 иллюстрирует защиту 3-карбонильной группы и алкилирование 19-положения новых стероидов с конденсированными кольцами настоящего изобретения. В частности, сначала это соединение превращают в простой эфир 3-алкиленола, как показано на Схеме S-16. Обработка простого эфира 3-алкиленола диизопропиламидом лития (LDA) с последующей обработкой алкилгалогенидом приводит к образованию 19-алкилового производного. В результате гидролиза защитной группы простого эфира 3-алкиленола получают 3-кето-производное.
Схема S-19.
Схема S-20 иллюстрирует превращение простого метилового эфира эстрона в соответствующий спиролактон. Обработка лактона тригалогенированной алкановой кислотой, предпочтительно в присутствии соли щелочного металла используемой алкановой кислоты, в реакционных условиях, описанных ранее, приводит к перегруппировке лактона в соответствующий стероид с конденсированными кольцами. См., например, Otsubo, К., Tetrahedron Letters, 27(47), 5763 (1986).
Схема S-20.
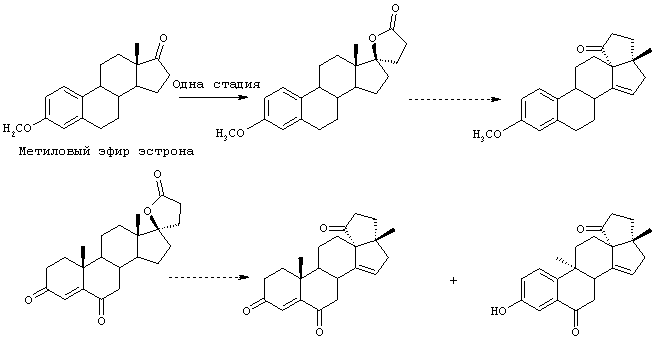
Новые соединения, описанные в настоящей заявке, могут быть, кроме того, подвергнуты способам биологического превращения, аналогичным способам, описанным ранее, с получением других новых стероидов с конденсированными кольцами, таких как стероиды, имеющие 9α-, 9β-, 11α- или 11β-гидрокси-заместитель, а также другие гидроксилированные стероиды с конденсированными кольцами. Если необходимо, то такие гидроксилированные стероиды могут быть затем окислены путем элиминации гидрокси-заместителя для введения олефиновой двойной связи, такой как Δ9,11-олефиновая двойная связь.
Исходя из вышеописанного, можно сказать, что были достигнуты некоторые цели настоящего изобретения и получены другие преимущественные результаты.
Следует отметить, что, хотя в вышеописанные композиции и способы могут быть внесены различные изменения, не выходящие за рамки объема настоящего изобретения, однако весь материал, описанный в настоящей заявке, имеет иллюстративный характер и не должен рассматриваться как некое ограничение сущности изобретения.
Нижеследующие неограничивающие примеры служат иллюстрацией различных аспектов настоящего изобретения.
Пример Х-1А. Получение метил-2,3,3а,4,6,7,9,10,11,11а,12,13-додекагидро-3аβ,11аβ-диметил-1,9-диоксо-1Н-пенталено[1,6а-а]фенантрен-6α-карбоксилата (Соединения С-1)
В очищенный сухой реактор, снабженный механической мешалкой, холодильником, термопарой и кожухом для нагревания, добавляли ацетат калия (6,7 г, 7,1 ммоль; Sigma-Aldrich 5128LG). Затем в реактор последовательно добавляли трифторуксусную кислоту (25,0 мл, 8,1 моль; Sigma-Aldrich 7125MG) и трифторуксусный ангидрид (4,5 мл, 31,0 ммоль; Sigma-Aldrich 11828PN). Затем раствор выдерживали в течение 30 минут при температуре от 25 до 30°С.
Предварительно полученный реагент, TFA/ангидрид TFA, добавляли к 5,0 г (9,6 ммоль) 7-метилгидро-17-гидрокси-11α-(метилсульфонил)окси-3-оксо-17α-прегна-4-ен-7α,21-дикарбоксилата, γ-лактона:
который получали способом, описанным в Примере 36. Полученную смесь нагревали в течение 60 минут при 60°С и степень превращения периодически оценивали с помощью ТСХ и/или ВЭЖХ. При завершении реакции (приблизительно 60 минут) смесь переносили в 1-горлую колбу и концентрировали при пониженном давлении при 50°С до тех пор, пока не образовывалась густая суспензия.
Полученную суспензию разбавляли 150 мл этилацетата и 80 мл смесью воды/насыщенного солевого раствора. Затем смесь оставляли для разделения фаз и водный слой повторно экстрагировали 80 мл этилацетата. Крепость насыщенного солевого раствора составляла 12% масс. Объединенный этилацетатный раствор один раз промывали 12% масс. насыщенного солевого раствора (80 мл), а затем один раз 1н раствором NaOH (80 мл) и, наконец, 2% масс. насыщенного солевого раствора (80 мл). Смесь оставляли для разделения фаз и отделенный этилацетатный слой концентрировали досуха при пониженном давлении при 45°С с использованием устройства для отсасывания воды, в результате чего получали около 3,8 г сырого твердого продукта. ВЭЖХ-анализ этого сырого продукта показал, что этот продукт содержал около 40% (по площади) соединения С-1.
Затем этот твердый продукт подвергали хроматографической очистке. После этой хроматографической очистки получали 210 мг метил-2,3,3а,4,6,7,9,10,11,11а,12,13-додекагидро-3аβ,11аβ-диметил-1,9-диоксо-1Н-пенталено[1,6а-а]фенантрен-6α-карбоксилата (Соединения С-1).
Данные масс-спектрометрии показали, что молекулярная масса составляет 380, а с помощью данных, полученных с высоким разрешением, была установлена формула C14H28O4. Масс-спектр, полученный методом ЭИ, имел пик М+ при m/z 380. Масс-спектр, полученный методом АПХИ, имел пики при m/z 381 (МН)+ и m/z 398 (MNH4)+. Углеродный и водородный анализы соответствовали предполагаемой молекулярной формуле.
ИК-спектр имеет два пика в области абсорбции карбонила: 1722 см-1 и 1667 см-1. Пик 1722 см-1 приписывается двум карбонилам, поскольку 13С-ЯМР-спектр имел сигналы при δ 217,7, обусловенные насыщенным кетоном, и при δ 172,7, обусловленные карбометоксикарбонилом. Отсутствие пика 1773 см-1 в ИК-спектре указывает на потерю лактонной части.
Данные 13С-АРТ и HETCOR-ЯМР указывают на присутствие углеродов следующих типов: 3 карбонила (δ 217,7, 198,4, 172,2); 4 полностью защищенных олефиновых углерода (δ 166,3, 141,8, 139,3, 121,8); 2 метинолефиновых углерода (δ 124,9, 122,0); 3 четвертичных алифатических углерода (δ 61,1, 50,7, 39,7); 1 метиновый алифатический углерод (δ 43,3); 8 метиленовых углеродов (δ 46,0, 37,5, 34,1, 33,3, 32,9, 31,9, 23,7, 22,2) и 3 метиловых углерода (δ 51,9, 23,6, 23,1).
Пример Х-1В. Получение (7α,13R,17β)-3',4',5',17-тетрагидро-14-гидрокси-17-метил-3,5'-диоксо-γ-лактона, циклопента[13,17]-18-норандроста-4,9(11)-диен-7-карбоновой кислоты (Соединения С-201)
В круглодонный 250 мл реактор, снабженный механической мешалкой, холодильником и кожухом для нагревания, добавляли ацетат калия (8,9 г, 90 ммоль), трифторуксусную кислоту (150 мл, 1,480 г/мл) и трифторуксусный ангидрид (33 мл, 1,487 г/мл). Полученный раствор перемешивали в течение около 10 минут при температуре от около 25°С до 30°С.
Предварительно полученный реагент, TFA/ангидрид TFA, добавляли к 15 г (30,0 ммоль) 7-метилгидро-17-гидрокси-11α-(метилсульфонил)окси-3-оксо-17α-прегна-4-ен-7α,21-дикарбоксилата, γ-лактона:
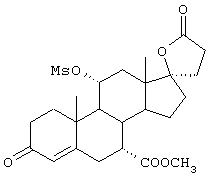
который получали способом, описанным в Примере 36. Полученную смесь нагревали в течение примерно 1-1,5 часа при около 60°С-70°С. Эту смесь концентрировали при пониженном давлении при 50°С до тех пор, пока не образовывалась густая суспензия. Полученную суспензию разбавляли 100 мл этилацетата и 2 раза промывали примерно 20% воды/насыщенного солевого раствора (каждый раз 80 мл), 1 раз 1н раствором гидроксида натрия (80 мл), а затем 1 раз смесью примерно 20% воды/насыщенного солевого раствора (80 мл). Сырой продукт сушили сульфатом магния, фильтровали и концентрировали, в результате чего получали около 18 г сырого материала.
Этот продукт два раза очищали с помощью колоночной хроматографии и получали около 3 г чистого (7α,13R,17β)-3',4',5',17-тетрагидро-14-гидрокси-17-метил-3,5'-диоксо-γ-лактона, циклопента[13,17]-18-норандроста-4,9(11)-диен-7-карбоновой кислоты (Соединения С-201).
Пример Х-1С. Получение [13S,17β]-3',4'-дигидро-3-гидрокси-9,17-диметилциклопента[13,17]гона-1,3,5(10)-триен-5'-[2'Н]-она (Соединения С-202)
В круглодонный 250 мл реактор, снабженный механической мешалкой, холодильником и кожухом для нагревания, добавляли ацетат калия (6 г, 61,1 ммоль), трифторуксусную кислоту (150 мл, 1,480 г/мл) и трифторуксусный ангидрид (26 мл, 1,487 г/мл). Полученный раствор перемешивали в течение около 10 минут при температуре от около 25°С до 30°С.
Предварительно полученный реагент, TFA/ангидрид TFA, добавляли к 15 г (43,7 ммоль) 17-гидрокси-3-оксо-17α-прегн-4-ен-21-карбоновой кислоты, γ-лактона (известного также как альдона; G.D.Searle & Co.):
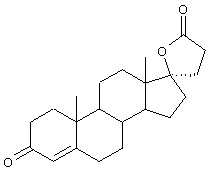
Полученную смесь нагревали в течение примерно 1-1,5 часа при 60°С-70°С. Эту смесь концентрировали при пониженном давлении при 50°С до тех пор, пока не образовывалась густая суспензия. Полученную суспензию растворяли в 100 мл этилацетата и 2 раза промывали примерно 20% воды/насыщенного солевого раствора (каждый раз 80 мл), 1 раз 1н раствором гидроксида натрия (80 мл), а затем 1 раз смесью примерно 20% воды/насыщенного солевого раствора (80 мл). Сырой продукт сушили сульфатом магния, фильтровали и концентрировали досуха при пониженном давлении при 50°С, в результате чего получали около 20 г неочищенного мокрого продукта.
Этот продукт два раза очищали с помощью колоночной хроматографии и получали около 125 г чистого [13S,17β]-3',4'-дигидро-3-гидрокси-9,17-диметилциклопента[13,17]гона-1,3,5(10)-триен-5'-[2'Н]-она (Соединения С-202).
Пример X-1D. Получение [13S,17β]-3',4'-дигидро-3-гидрокси-9,17-диметилциклопента[13,17]гона-1,3,5(10),6-тетраен-5'-[2'Н]-она (Соединения С-203)
В круглодонный 250 мл реактор, снабженный механической мешалкой, холодильником и кожухом для нагревания, добавляли ацетат калия (6 г, 61,1 ммоль), трифторуксусную кислоту (150 мл, 1,480 г/мл) и трифторуксусный ангидрид (26 мл, 1,487 г/мл). Полученный раствор перемешивали в течение около 10 минут при температуре от около 25°С до около 30°С.
Предварительно полученный реагент, TFA/ангидрид TFA, добавляли к 15 г (45,9 ммоль) 17-гидрокси-3-оксо-17α-прегн-4,9(11)-диен-21-карбоновой кислоты, γ-лактона (известного также как Δ-9,11-альдона):
который был получен из 3-метокси-3,5,9(11)-андростатриен-17-она (Upjohn). Полученную смесь нагревали при около 60°С-70°С в течение примерно 1-1,5 часа. Эту смесь концентрировали при пониженном давлении при 50°С до тех пор, пока не образовывалась густая суспензия. Полученную суспензию разбавляли 100 мл этилацетата и 2 раза промывали смесью примерно 20% воды/насыщенного солевого раствора (каждый раз 80 мл), 1 раз 1н раствором гидроксида натрия (80 мл), а затем 1 раз примерно 20% воды/насыщенного солевого раствора (80 мл). Сырой продукт сушили сульфатом магния, фильтровали и концентрировали досуха при пониженном давлении при 50°С, в результате чего получали около 18 г неочищенного мокрого продукта.
Этот продукт два раза очищали с помощью колоночной хроматографии и получали около 340 г чистого [13S,17β]-3',4'- дигидро-3-гидрокси-9,17-диметилциклопента[13,17]гона-1,3,5(10),6-тетраен-5'[2'Н]-она (Соединения С-203).
Пример Х-1Е. Получение [13S,17β]-3',4'-дигидро-17-метилциклопента-[13,17]-18-норандроста-4,6,8(14)-триен-3,5'[2'Н]-диона (Соединения С-204)
В круглодонный 250 мл реактор, снабженный механической мешалкой, холодильником и кожухом для нагревания, добавляли ацетат калия (8 г, 81,5 ммоль), трифторуксусную кислоту (150 мл, 1,480 г/мл) и трифторуксусный ангидрид (33 мл, 1,487 г/мл). Полученный раствор перемешивали в течение около 10 минут при температуре от около 25°С до около 30°С.
Предварительно полученный реагент, TFA/ангидрид TFA, добавляли к 15 г (44,0 ммоль) 17-гидрокси-3-оксо-17α-прегн-4,6-диен-21-карбоновой кислоты, γ-лактона (известного также как канренон; G.D.Searle & Co.):
Полученную смесь нагревали в течение примерно 1-1,5 часа при около 60°С-70°С. Эту смесь концентрировали при пониженном давлении при 50°С до тех пор, пока не образовывалась густая суспензия. Полученную суспензию растворяли в 100 мл этилацетата и 2 раза промывали смесью примерно 20% воды/насыщенного солевого раствора (каждый раз 80 мл), 1 раз 1н раствором гидроксида натрия (80 мл), а затем 1 раз смесью примерно 20% воды/насыщенного солевого раствора (80 мл). Сырой продукт сушили сульфатом магния, фильтровали и концентрировали досуха при пониженном давлении при 50°С, в результате чего получали около 18 г неочищенного мокрого продукта.
Этот продукт два раза очищали с помощью колоночной хроматографии и получали около 2,2 г чистого [13S,17β]-3',4'-дигидро-17-метилциклопента-[13,17]-18-норандроста-4,6,8(14)-триен-3,5'[2'Н]-диона (Соединения С-204).
Пример Х-2. Получение:
К перемешанному холодному (0°С) раствору 11α-гидроксиканренона (3,6 г, 10 ммоль) и триэтиламина (1,2 г, 12 ммоль) в метиленхлориде (20 мл) добавляли метансульфонилхлорид (1,1 г, 10 ммоль). Смесь перемешивали при охлаждении в течение 3 часов и оставляли для нагревания до комнатной температуры. Перемешивание продолжали до тех пор, пока тонкослойная хроматография не указывала на завершение реакции. Затем смесь разбавляли этилацетатом и экстрагировали водой, водным 5% раствором бикарбоната натрия и водой, а затем сушили сульфатом натрия. Осушитель фильтровали и фильтрат концентрировали в вакууме с получением неочищенного мезилата С-136, который может быть использован в последующей стадии:
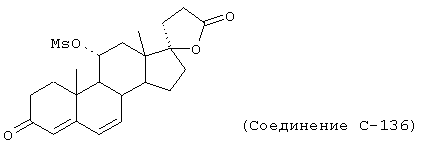
Раствор мезилата С-136 (4,3 г, 10 ммоль) подвергали реакции с трифторуксусной кислотой (25 мл), трифторуксусным ангидридом (4,5 мл) и ацетатом калия (6,7 г, 7,1 ммоль) способом, описанным для синтеза Соединения С-1 в Примере Х-1. Сырой продукт выделяли способом, аналогичным описанному для Соединения С-1 в Примере Х-1 и очищали с помощью хроматографии на силикагеле с использованием смесей этилацетата и толуола или этилацетата и гексана в качестве элюентов. Затем полученный продукт очищали путем перекристаллизации из спирта, спирта и воды или этилацетата и гексана с получением тетраена С-105.
Пример Х-3. Получение соединения:
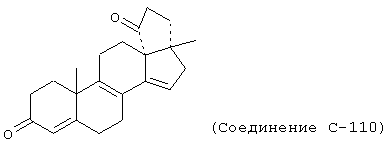
Мезилат С-138:
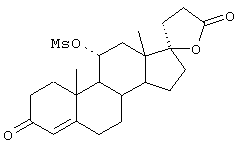
синтезировали и выделяли (методом, описанным в Примере Х-2 для синтеза мезилата С-136) с использованием 11α,17-дигидрокси-3-оксо-17-оксо-прегн-4-ен-21-карбоновой кислоты, γ-лактона (3,6 г, 10 ммоль), триэтиламина (1,2 г, 12 моль) и метансульфонилхлорида (1,1 г, 10 ммоль) в метиленхлориде (20 мл). Выделенный таким образом мезилат С-138 может быть использован в следующей стадии.
Раствор мезилата С-138 (4,4 г, 10 ммоль) подвергали реакции с трифторуксусной кислотой (25 мл), трифторуксусным ангидридом (4,5 мл) и ацетатом калия (6,7 г, 7,1 ммоль) способом, описанным для синтеза Соединения С-1 в Примере Х-1. Неочищенный продукт С-110 выделяли способом, аналогичным описанному для Соединения С-1 в Примере Х-1, и очищали с помощью хроматографии на силикагеле с использованием смесей этилацетата и толуола или этилацетата и гексана в качестве элюентов. Затем полученный продукт очищали путем перекристаллизации из спирта, спирта и воды или этилацетата и гексана.
Пример Х-4. Получение 3',4',5',17-тетрагидро-17β-метил-3,5'-диоксоциклопента[13R,17]-18-норандроста-4,8,14-триен-7α-карбоновой кислоты (Соединения С-101)
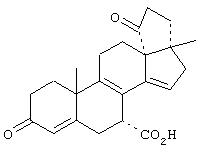
Раствор Соединения С-1 (3,8 г, 10 ммоль) и 1н водного раствора гидроксида натрия (35 мл) в этаноле (60 мл) нагревали с обратным холодильником в течение 8 часов. Реакционную смесь охлаждали до комнатной температуры, концентрировали на роторном испарителе в вакууме и остаточный водный слой три раза экстрагировали этилацетатом. Затем водный слой подкисляли 1н раствором хлористоводородной кислоты и три раза экстрагировали этилацетатом. Объединенные органические слои промывали водой и сушили сульфатом натрия. Осушитель фильтровали и фильтрат концентрировали на роторном испарителе. Остаточную неочищенную карбоновую кислоту С-101 кристаллизовали путем обработки этилацетатом и перекристаллизовывали из этилацетата и гексана или метанола или этанола и воды.
Пример Х-5. Получение 1-метилэтил-3',4',5',17-тетрагидро-17β-метил-3,5'-диоксоциклопента[13R,17]-18-норандроста-4,8,14-триен-7α-карбоксилата (Соединения С-102)
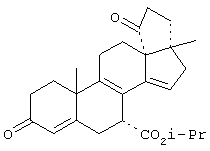
Смесь бикарбоната натрия (3,5 г) и раствор карбоновой кислоты С-101 (3,7 г, 10 ммоль) и изопропилиодида (3 мл) в диметилформамиде (35 мл) перемешивали в течение ночи при комнатной температуре. Реакционную смесь выливали в воду и водный раствор экстрагировали три раза этилацетатом. Объединенные органические слои промывали водой и сушили сульфатом натрия. Осушающий агент фильтровали и фильтрат концентрировали в вакууме. Полученный сырой изопропиловый сложный эфир С-102 кристаллизовали путем обработки этилацетатом или спиртом и очищали с помощью хроматографии на силикагеле, а затем перекристаллизовывали из этилацетата и гексана, либо спирта, либо спирта и воды.
Пример Х-6. Получение этил-3',4',5',17-тетрагидро-17β-метил-3,5'-диоксоциклопента[13R,17]-18-норандроста-4,8,14-триен-7α-карбоксилата
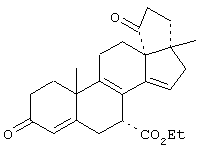
Смесь бикарбоната натрия (3,5 г) и раствор карбоновой кислоты С-101 (3,7 г, 10 ммоль) и этилиодида (3 мл) в диметилформамиде (35 мл) перемешивали в течение ночи при комнатной температуре. Реакционную смесь выливали в воду и водный раствор экстрагировали три раза этилацетатом. Объединенные органические слои промывали водой и сушили сульфатом натрия. Осушающий агент фильтровали и фильтрат концентрировали в вакууме. Остаточный сырой этиловый сложный эфир С-103 кристаллизовали путем обработки этилацетатом или спиртом и очищали с помощью хроматографии на силикагеле, а затем перекристаллизовывали из этилацетата и гексана, либо спирта, либо спирта и воды.
Пример Х-7. Получение гексил-3',4',5',17-тетрагидро-17β-метил-3,5'-диоксоциклопента[13R,17]-18-норандроста-4,8,14-триен-7α-карбоксилата (Соединения С-104)

Смесь бикарбоната натрия (3,5 г) и раствор карбоновой кислоты С-101 (3,7 г, 10 ммоль) и н-гексилиодида (3 мл) в диметилформамиде (35 мл) перемешивали в течение ночи при комнатной температуре. Реакционную смесь выливали в воду и водный раствор экстрагировали три раза этилацетатом. Объединенные органические слои промывали водой и сушили сульфатом натрия. Осушающий агент фильтровали и фильтрат концентрировали в вакууме. Остаточный сырой н-гексиловый сложный эфир С-104 кристаллизовали путем обработки этилацетатом или спиртом и очищали с помощью хроматографии на силикагеле, а затем перекристаллизовывали из этилацетата и гексана, либо из спирта, либо из спирта и воды.
Пример Х-8. Получение 3',4'-дигидро-17-метил-7α(метилтио)-циклопента[13,17]-18-норандроста-4,8,14-триен-3,5'(2'Н)-диона (Соединения С-106)
Раствор тетраена С-105 (3,2 г, 10 ммоль) в метаноле (40 мл) и пиперидине (4 мл) охлаждали до 5°С. Затем пропускали газообразный метилмеркаптан до тех пор, пока не наблюдался прирост в массе 7 г. Сосуд под давлением герметично закрывали и выдерживали при комнатной температуре в течение 20 часов. Затем раствор выливали в ледяную воду и осадок фильтровали, промывали водой и сушили на воздухе. Метилтиопродукт С-106 очищали путем перекристаллизации из метанола или этилацетата и гексана. См., например, процедуру, описанную в работе A.Karim & Е.А.Brown, Steroids, 20, 41 (1972), которая вводится в настоящее описание посредством ссылки.
Пример Х-9. Получение 7α-(ацетилтио)-3,4'-дигидро-17-метилциклопента[13,17]-18-норандроста-4,8,14-триен-3,5'(2'Н)-диона (Соединения С-107)
Раствор тетраена С-105 (3,2 г, 10 ммоль) в тиоуксусной кислоте (10 мл) нагревали в течение 1 часа при 85-95°С. Затем избыток тиоуксусной кислоты удаляли в вакууме и полученный сырой 7α-тиоацетат С-107 очищали путем перекристаллизации из подходящего растворителя, такого как метанол или этилацетат или этилацетат и гексан. См., например, процедуру, описанную в патенте США 3013012, J.A.Cella & R.C.Tweit, дек.12, 1961, который вводится в настоящее описание посредством ссылки.
Пример Х-10. Получение 1,2,4bR(4bR*),5,5aS*,7,7aR*,8,9,11,-12bS*-додекагидро-7а,12b-диметил-10aR*-циклопропал[1]пенталено[1,6а-а]фенантрен-3,10-диона
и
1,2,4bS(4bR*),5,5аS*,8,9,11,12,12bR*-додекагидро-7а,12b-диметил-10аS*-циклопропал[1]пенталено[1,6а-а]фенантрен-3,10-диона (Соединения С-109):
К раствору иодида триметилсульфоксония (1 г, 4,6 ммоль) в сухом диметилсульфоксиде (20 мл) добавляли гидрид натрия (220 мг 50%-ной дисперсии в минеральном масле, 4,6 ммоль). Смесь перемешивали при комнатной температуре в атмосфере азота до тех пор, пока не прекращалось выделение водорода. Затем добавляли раствор тетраена С-105 (1,12 г, 3,5 ммоль) в диметилсульфоксиде (4 мл) и перемешивание продолжали в течение 4 часов в атмосфере азота. Реакционную смесь разбавляли водой и полученный осадок фильтровали и сушили на воздухе. Полученный продукт представлял собой смесь 6β,7β-(Соединения С-108) и 6α,7α-(Соединения С-109) изомеров. Эти изомеры выделяли с помощью хроматографии на силикагеле, а затем отдельные изомеры очищали путем перекристаллизации из растворителей, таких как этилацетат и гексан, спирт или спирт и вода.
Пример Х-11. Получение соединения формулы:
К суспензии енона С-110 (3,2 г, 10 ммоль) в этилортоформиате (10 мл) и безводном этаноле (10 мл) добавляли моногидрат п-толуолсульфоновой кислоты (0,05 г). Реакционную смесь перемешивали в течение 30 минут при комнатной температуре и гасили путем добавления нескольких капель пиридина. После перемешивания еще в течение 5 минут при 0°С полученный осадок фильтровали, промывали небольшим количеством метанола и перекристаллизовывали из спирта или этилацетата и гексана, содержащих следовые количества пиридина, в результате чего получали чистый простой эфир енола С-111. Альтернативно, после добавления пиридина эта реакционная смесь может быть обработана путем удаления всех растворителей в вакууме и кристаллизации остатка путем добавления растворителей, таких как простой эфир, этилацетат или гексан. Сырой С-111 перекристаллизовывали как описано выше. См., например, процедуру, описанную в работе R.M.Weier & L.M.Hofmann, J.Med. Chem., 20, 1304-1303 (1977), которая вводится в настоящее описание посредством ссылки.
Пример Х-12. Получение соединения:
Осуществляли процедуру, описанную в работе R.M.Weier & L.M.Hofmann, J. Med. Chem., 20, 1304-1308 (1977), которая вводится в настоящее описание посредством ссылки.
Реактив Вильсмайера получали путем добавления оксихлорида фосфора (4,59 г, 30 ммоль) к диметилформамиду (30 мл) при 0°С. Через 5 минут добавляли раствор простого эфира енола С-111 (3,5 г, 10 ммоль) в диметилформамиде (5 мл) и реакционную смесь перемешивали в течение 2 часов при 0°С и в течение ночи при комнатной температуре. Реакционную смесь выливали в раствор водного ацетата натрия и перемешивали в течение 2 часов. Осадок фильтровали и сушили, в результате чего получали неочищенный альдегид С-112. Очистку осуществляли путем перекристаллизации из растворителей, таких как спирт, спирт и вода или этилацетат и гексан. Альтернативно, неочищенный альдегид С-112 выделяли из водного раствора ацетата натрия путем экстракции растворителем, таким как этилацетат. После сушки сульфатом натрия и удаления растворителя остаток очищали путем перекристаллизации или путем хроматографии на силикагеле с последующей перекристаллизацией.
Пример Х-13. Получение соединения:
К размешанному охлажденному раствору три-трет-бутокси-алюмогидрида лития (3,05 г, 12 ммоль) в тетрагидрофуране добавляли альдегид С-112 (3,78 г, 10 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 5 часов и гасили путем добавления воды и уксусной кислоты, при этом следили за тем, чтобы смесь оставалась слегка основной. Эту смесь концентрировали в вакууме и полученное твердое вещество суспендировали в этилацетате. Твердое вещество фильтровали и фильтрат концентрировали в вакууме. Остаток растворяли в минимальном объеме раствора ацетона и воды (3:1) и добавляли водный ацетон для подкисления (рН 1,5-2,0). Кислотную реакционную смесь перемешивали в течение 1 часа при комнатной температуре и концентрировали в вакууме. Полученный сырой диенон С-113 очищали путем перекристаллизации из растворителей, таких как спирт, спирт и вода или этилацетат и гексан. Альтернативно, сырой диенон С-113 очищали с помощью хроматографии на силикагеле, а затем перекристаллизовывали. См., например, процедуру, описанную в работе P.M.Weier & L.M.Hofmann, J.Med.Chem., 20, 1304-1308 (1977), которая вводится в настоящее описание посредством ссылки.
Пример Х-14. Получение 2',3',3'аα,4',6',10',11',11'аα,12',13'-декагидро-3'aR,3'а,11'а-диметил-13'aR*-спиро-[циклопропан-1,7'(9'Н)-[1Н]пенталено[1,6а-а]-фенантрен-1'9'-диона:
К раствору диенона С-113 (1,17 г, 0,05 ммоль) в тетрагидрофуране (30 мл) добавляли раствор диазометана (0,2 г, 0,07 ммоль) в простом эфире (7 мл). Полученный реакционный раствор хранили при комнатной температуре в течение нескольких дней. Затем добавляли уксусную кислоту для разложения остаточного диазометана и реакционную смесь концентрировали в вакууме. Остаток, который представлял собой промежуточный пиразолин, кристаллизовали из растворителя, такого как ацетон, гексан, этилацетат или этанол, а затем перекристаллизовывали. Это соединение превращали в спироциклопропан С-114. Полученное таким образом твердое вещество С-114 нагревали при 190°С в вакууме и полученное твердое вещество перекристаллизовывали из спирта, спирта и воды, ацетона и воды, или этилацетата и гексана. Альтернативно, раствор пиразолина в ацетоне обрабатывали эфиратом трифторида бора в течение около 1 часа при комнатной температуре. Затем добавляли воду и полученный осадок фильтровали и сушили на воздухе. Очистку проводили путем перекристаллизации. См., например, процедуру, описанную в патенте США 3499891, выданном 10 марта F.В.Colton & R.T.Nicholson, который вводится в настоящее описание посредством ссылки.
Пример Х-15. Получение соединения:
и

К раствору простого эфира енола С-111 (3,2 г, 10 ммоль) в метаноле (20 мл) добавляли борогидрид натрия (38 мг). Реакционную смесь перемешивали в течение 3 часов при комнатной температуре и обрабатывали 1н соляной кислотой в течение 30 минут. Затем реакционную смесь разбавляли водой и осадок фильтровали, промывали водой и сушили. Продукт, состоящий из смеси двух эпимерных спиртов (Соединения С-115 и Соединения С-116), хроматографировали на силикагеле для их разделения. Каждый из очищенных спиртов С-115 и С-116 кристаллизовали из растворителей, таких как спирт, спирт и вода, этилацетат и гексан, и ацетон и гексан. Альтернативно, разбавленную реакционную смесь экстрагировали этилацетатом, объединенные органические слои сушили сульфатом натрия и выделенный таким образом сырой продукт хроматографировали как описано выше с получением отдельных спиртов С-115 и С-116.
Пример Х-16. Получение соединения:
Соединение С-1 (3,8 г, 10 ммоль) превращали в простой эфир енола С-117 с использованием этилортоформиата (3,2 г, 10 ммоль), этанола (10 мл) и моногидрата п-толуолсульфоновой кислоты (0,05 г) в соответствии с процедурой, описанной для синтеза Соединения С-111 в Примере Х-11.
Пример Х-17. Получение метил-3',4',5',17-тетрагидро-6α-гидрокси-17β-метил-3,5'-диоксоциклопента-[13R,17]-18-норандроста-4,8,14-триен-7α-карбоксилата (Соединения С-118):
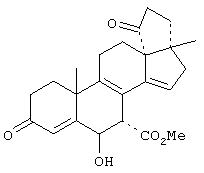
Раствор 57% м-хлорпероксибензойной кислоты (3,64 г) в 10% водном диоксане (20 мл) наполовину нейтрализовали 1н раствором гидроксида натрия. Этот раствор охлаждали до 0°С и порциями добавляли при размешивании холодный (0°С) раствор простого эфира енола С-117 (4,1 г, 10 ммоль) в 10% водном диоксане (20 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре, выливали в ледяную воду и экстрагировали метиленхлоридом или этилацетатом. Объединенные органические слои сушили сульфатом натрия. После выпаривания растворителя получали сырой сложный гидрокси-эфир С-118, который очищали с помощью хроматографии на силикагеле. См., например, процедуру, описанную в работе R.M.Weier & L.M.Hofmann, J.Med.Chem., 20, 1304-1308 (1977), которая вводится в настоящее описание посредством ссылки.
Пример Х-18. Получение соединения:
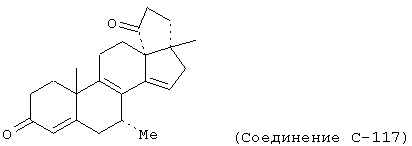
Диметилкупрат лития получали путем добавления метил-лития (19 мл 1,6М раствора в простом эфире, 30 ммоль) к перемешанной суспензии иодида меди (2,86 г, 15 ммоль) в простом эфире (30 мл) при 0°С в инертной атмосфере аргона. После перемешивания в течение 15 минут на холоде по каплям в течение 25 минут добавляли раствор тетраена С-105 (1,6 г, 15 ммоль) в тетрагидрофуране. Реакцию продолжали в течение еще 30 минут и реакционную смесь выливали в насыщенный раствор хлорида аммония при интенсивном размешивании. Водную смесь экстрагировали этилацетатом и метиленхлоридом. Объединенные органические слои промывали водным раствором хлорида аммония, водой и сушили сульфатом натрия. Растворитель удаляли в вакууме и остаток растворяли в этилацетате или метиленхлориде и обрабатывали п-толуолсульфоновой кислотой (100 мг) на паровой бане в течение периода времени от 30 минут до 1 часа. Органический раствор промывали водой и сушили сульфатом натрия. Растворитель удаляли в вакууме с получением сырого енона С-119. Очистку осуществляли путем перекристаллизации из спирта, спирта и воды или этилацетата и гексана. Альтернативно, сырой енон С-119 хроматографировали на силикагеле, после чего продукт перекристаллизовывали. См., например, процедуру, описанную в работе J.K.Grunwell et al., Steroids, 27, 759-771 (1976), которая вводится в настоящее описание посредством ссылки.
Пример Х-19. Получение:
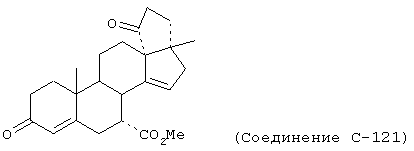
Сложный эфир С-120 (4,00 г, 10 ммоль):
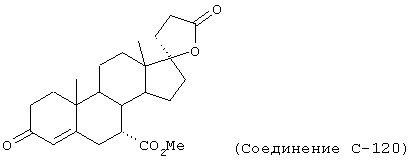
(полученный в соответствии с процедурой, описанной в работе R.M.Weier & L.M.Hofmann, J.Med.Chem., 18, 817 (1975), которая вводится в настоящее описание посредством ссылки) подвергали реакции с трифторуксусной кислотой (25 мл), ангидридом трифторуксусной кислоты (4,5 мл) и ацетатом калия (6,7 г, 7,1 ммоль) в соответствии с процедурой, описанной для синтеза Соединения С-1 в Примере Х-1. Сырой продукт С-121 выделяли в соответствии с процедурой, описанной для синтеза Соединения С-1 в Примере Х-1, и очищали с помощью хроматографии на силикагеле с использованием смесей этилацетата и толуола или этилацетата и гексана в качестве элюентов и полученный таким образом продукт дополнительно очищали путем перекристаллизации из спирта, спирта и воды или этилацетата и гексана.
Пример Х-20. Получение:
Енон С-122 (3,68 г, 10 ммоль):
(полученный в соответствии с процедурой, описанной в патенте США 3499891,выданном 10.03.1970 г. F.B.Colton & R.T.Nicholson, который вводится в настоящее описание посредством ссылки) подвергали реакции с ацетатом калия (6,7 г, 7,1 ммоль), трифторуксусной кислотой (25 мл) и ангидридом трифторуксусной кислоты (4,5 мл) в соответствии с процедурой, описанной для синтеза Соединения С-1 в Примере Х-1. Сырой продукт выделяли в соответствии с вышеуказанной процедурой и очищали с помощью хроматографии на силикагеле с использованием смесей этилацетата и толуола или этилацетата и гексана в качестве элюентов, и полученный таким образом продукт дополнительно очищали путем перекристаллизации из спирта, спирта и воды или этилацетата и гексана.
Пример Х-21. Получение:
и
Соединения С-125 и С-126 синтезировали в соответствии с процедурой, используемой выше для синтеза Соединений С-108 и С-1. К раствору иодида триметилсульфоксония (1 г, 4,6 ммоль) в сухом диметилсульфоксиде (20 мл) добавляли гидрид натрия (220 мг 50% дисперсии в минеральном масле, 4,6 ммоль). Смесь перемешивали при комнатной температуре в атмосфере азота до тех пор, пока не прекращалось выделение водорода.
Затем добавляли раствор триенона С-124 (1,14 г, 3,5 ммоль):
в диметилсульфоксиде (4 мл) и перемешивание продолжали в течение 4 часов в атмосфере азота. Триенон С-124 получали в соответствии с процедурой, описанной в Примере Х-2 для получения тетраена С-105, с использованием в качестве исходного соединения канренона вместо 11α-гидрокси-канренона. Реакционную смесь разбавляли водой и полученный осадок фильтровали и сушили на воздухе. Продукт представлял собой смесь 6β,7β-(Соединение С-125) и 6α,7α-(Соединение С-126) изомеров. Разделение этих изомеров осуществляли с помощью хроматографии на силикагеле и отдельные изомеры дополнительно очищали путем перекристаллизации из растворителей, таких как этил-ацетат и гексан, спирт или спирт и вода.
Пример Х-22. Получение метил-3',4',5',17-тетрагидро-17β-метил-3,5'-диоксоциклопента[13R,17]-18-норандроста-1,4,8,14-тетраен-7α-карбоксилата (Соединения С-127)
Диенон С-127 синтезировали в соответствии с процедурой, описанной в работе R.M.Weier & L.M.Hofmann, J.Med.Chem., 18, 817 (1975), которая вводится в настоящее описание посредством ссылки. Раствор Соединения С-1 (3,8 г, 10 ммоль) и дихлордицианобензохинона (2,72 г, 12 ммоль) в диоксане (80 мл) нагревали при перемешивании с обратным холодильником в течение 24 часов. Реакционную смесь концентрировали в вакууме, остаток гидролизовали метиленхлоридом, фильтровали и фильтрат промывали 2% сульфитом натрия, 5% гидроксидом натрия и насыщенным раствором хлорида натрия, а затем сушили сульфатом натрия. Осушающий агент фильтровали и фильтрат концентрировали в вакууме. Сырой продукт диенон С-127 очищали с помощью хроматографии на силикагеле с использованием смесей этилацетата и толуола в качестве элюентов и полученный таким образом продукт 27 дополнительно очищали путем перекристаллизации из спирта.
Пример Х-23. Получение 2',3',3'аα,4',6',11'аα,12',13'-октагидро-3'aR,3'а,11'а-диметил-13'aR*-спиро-[циклопропан-1,7'(9'Н)-[1Н]пенталено-[1,6а-а]-фенантрен-1'9'-диона (Соединения С-128)
С использованием процедуры и обработки, описанной выше в Примере Х-22 для синтеза диенона С-127, енон С-114 (3,48 г, 10 ммоль) превращали в диенон С-128 с использованием дихлордицианобензохиона (2,72 г, 12 ммоль) в диоксане (80 мл).
Пример Х-24. Получение 4bR(4bR*),5,5aS*,7,7aR*,8,9,11,-12,12bS*-декагидро-7А,12b-диметил-10R*-циклопропа(1)пенталено[1,6а-а]фенантрен-3,10-диона (Соединения С-129)
С использованием процедуры и обработки, описанной выше в Примере Х-22 для синтеза диенона С-127, енон С-108 (3,34 г, 10 ммоль) превращали в диенон С-129 с использованием дихлордицианобензохинона (2,72 г, 12 ммоль) в диоксане (80 мл).
Пример Х-25. Получение:
К холодному (0°С) размешанному раствору карбоновой кислоты С-101 (3,66 г, 10 ммоль) и N-метилморфолину (1,01 г, 10 ммоль) в тетрагидрофуране (35 мл) добавляли изобутилхлороформиат (1,36 г, 10 ммоль). Реакционную смесь перемешивали в течение 20 минут при 0°С, фильтровали и фильтрат концентрировали в вакууме. Остаток представлял собой смешанный ангидрид и был пригодным для использования в следующей стадии.
Газообразный диметиламин барботировали в холодный (0°С) раствор смешанного ангидрида (4,6 г, 10 ммоль) в тетрагидрофуране (40 мл) в сосуде под давлением. Через 15 минут этот сосуд под давлением герметично закрывали и реакционную смесь оставляли на 24 часа при комнатной температуре. Реакционную смесь нагревали до 40°С в течение 30 минут, а затем снова охлаждали до 0°С. После нагревания до комнатной температуры давление в сосуде снижали до атмосферного и оставляли для испарения избытка диметиламина. Реакционную смесь концентрировали в вакууме, остаток растворяли в этилацетате и экстрагировали 1н раствором гидроксида натрия и воды. После сушки сульфатом натрия органический слой упаривали и остаток очищали с помощью хроматографии на силикагеле с использованием смесей этилацетата и толуола в качестве элюентов. Полученный таким образом амид С-130 выделяли и дополнительно очищали путем перекристаллизации из спирта, спирта и воды или этилацетата и гексана.
Пример Х-26. Получение соединения формулы:
К суспензии енона С-114 (3,5 г, 10 ммоль) в этилортоформиате (10 мл) и безводном этаноле (10 мл) добавляли моногидрат п-толуолсульфоновой кислоты (0,05 г). Реакционную смесь перемешивали в течение 30 минут при комнатной температуре и гасили путем добавления нескольких капель пиридина. После перемешивания еще в течение 5 минут при 0°С полученный осадок фильтровали, промывали небольшим количеством метанола и перекристаллизовывали из спирта или этилацетата и гексана, содержащих следовые количества пиридина, в результате чего получали чистый простой эфир енола С-131. Альтернативно, после добавления пиридина эта реакционная смесь может быть обработана путем удаления всех растворителей в вакууме и кристаллизации остатка путем добавления растворителей, таких как гексан, простой эфир, этилацетат или гексан. Сырой простой эфир енола С-131 перекристаллизовывали, как описано выше.
Пример Х-27. Получение:
К охлажденному (-78°С) раствору бис(триметилсилил)амида лития (10 мл 1,0 М раствора в тетрагидрофуране) по каплям в течение 20 минут добавляли раствор простого эфира енола С-131 (3,8 г, 10 ммоль) в тетрагидрофуране (20 мл). Реакционную смесь перемешивали при -78°С в течение 10 минут и добавляли раствор фенилселенилхлорида (1,9 г, 10 ммоль) в тетрагидрофуране (5 мл). Реакционную смесь перемешивали в течение 5 минут и гасили путем добавления 1% раствора бисульфата натрия. Затем реакционную смесь разбавляли водой и экстрагировали этилацетатом. Объединенные органические слои промывали 5% водным раствором бикарбоната натрия и водой и сушили сульфатом натрия. Осушающий агент фильтровали и фильтрат концентрировали в вакууме. Остаток очищали с помощью хроматографии на силикагеле с использованием смесей этилацетата и толуола или этилацетата и гексана в качестве элюентов и получали чистый селенид С-132.
Пример Х-28. Получение:
К охлажденному (0°С) раствору селенида С-132 (г, 10 ммоль) и пиридина (I,61 мл, 20 ммоль) в метиленхлориде (40 мл) постепенно добавляли раствор перекиси водорода (3,1 г 30% раствора, 27 ммоль) в воде (3 мл). Температуру поддерживали при менее чем 30-35°С. После снижения экзотермичности водяную баню удаляли и реакционную смесь интенсивно перемешивали в течение 15 минут при комнатной температуре. Реационную смесь разбавляли метиленхлоридом и промывали 5% раствором бикарбоната натрия и водой и сушили сульфатом натрия. Осушающий агент фильтровали и фильтрат концентрировали в вакууме. Остаток очищали с помощью хроматографии на силикагеле с использованием смесей этилацетата и толуола или этилацетата и гексана в качестве элюентов и получали чистый ненасыщенный кетон С-133, который затем очищали путем перекристаллизации из этилацетата и гексана или спирта.
Пример Х-29. Получение:
Раствор кетона С-133 (2 г) в ацетоне (15 мл) обрабатывали 1н соляной кислотой (4 мл) в течение 1 часа при комнатной температуре. Реакционную смесь концентрировали в вакууме. Остаток разбавляли водой и экстрагировали этилацетатом. Объединенные органические слои промывали 5% раствором бикарбоната натрия и водой и сушили сульфатом натрия. Осушающий агент фильтровали и фильтрат концентрировали в вакууме с получением сырого кетона С-134. Этот сырой продукт очищали с помощью хроматографии на силикагеле с использованием смесей этилацетата и толуола в качестве элюентов и получали чистый кетон С-134, который дополнительно очищали путем перекристаллизации из этилацетата и гексана или спирта.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ПОЛУЧЕНИЯ ЭПЛЕРЕНОНА | 2003 |
|
RU2339642C9 |
| КОМБИНАЦИЯ, СОДЕРЖАЩАЯ АНТАГОНИСТ РЕЦЕПТОРА АНГИОТЕНЗИНА II И ЭПОКСИСТЕРОИДНЫЙ АНТАГОНИСТ РЕЦЕПТОРА АЛЬДОСТЕРОНА | 1996 |
|
RU2166330C2 |
| СПОСОБ ПОЛУЧЕНИЯ 7-ЗАМЕЩЕННЫХ СТЕРОИДНЫХ СОЕДИНЕНИЙ, СОЕДИНЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ ЭПЛЕРЕНОНА, ПРОДУКТ | 2003 |
|
RU2289586C2 |
| 7-КАРБОКСИЗАМЕЩЕННЫЕ СТЕРОИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ ЭПЛЕРЕНОНА | 2003 |
|
RU2304144C2 |
| СПОСОБ ПОЛУЧЕНИЯ 17-(3-ГИДРОКСИПРОПИЛ)-17-ГИДРОКСИСТЕРОИДОВ | 2008 |
|
RU2466137C2 |
| ОКСАЗОЛО- И ТИАЗОЛО-[4,5-С] -ХИНОЛИН-4-АМИНЫ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ СТИМУЛИРОВАНИЯ ЦИТОКИНЕТИЧЕСКОГО БИОСИНТЕЗА | 1999 |
|
RU2244717C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ | 2003 |
|
RU2360903C2 |
| СТЕРОИД С 17-СПИРОМЕТИЛЕНЛАКТОНОВОЙ ГРУППОЙ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1995 |
|
RU2168516C2 |
| СПОСОБЫ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2010 |
|
RU2531588C2 |
| ДИКЕТОН В КАЧЕСТВЕ ПРОМЕЖУТОЧНОГО ПРОДУКТА ПРИ ПОЛУЧЕНИИ 3-ГАЛОГЕН-1Н-ПИРАЗОЛОВ | 1996 |
|
RU2251543C2 |
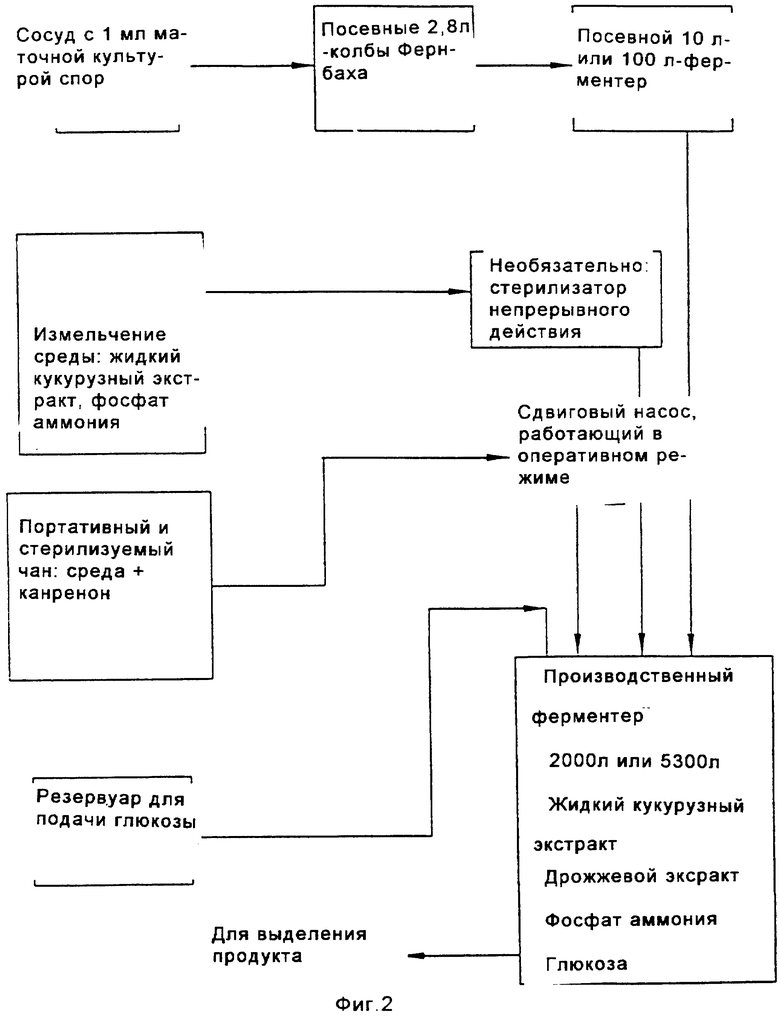
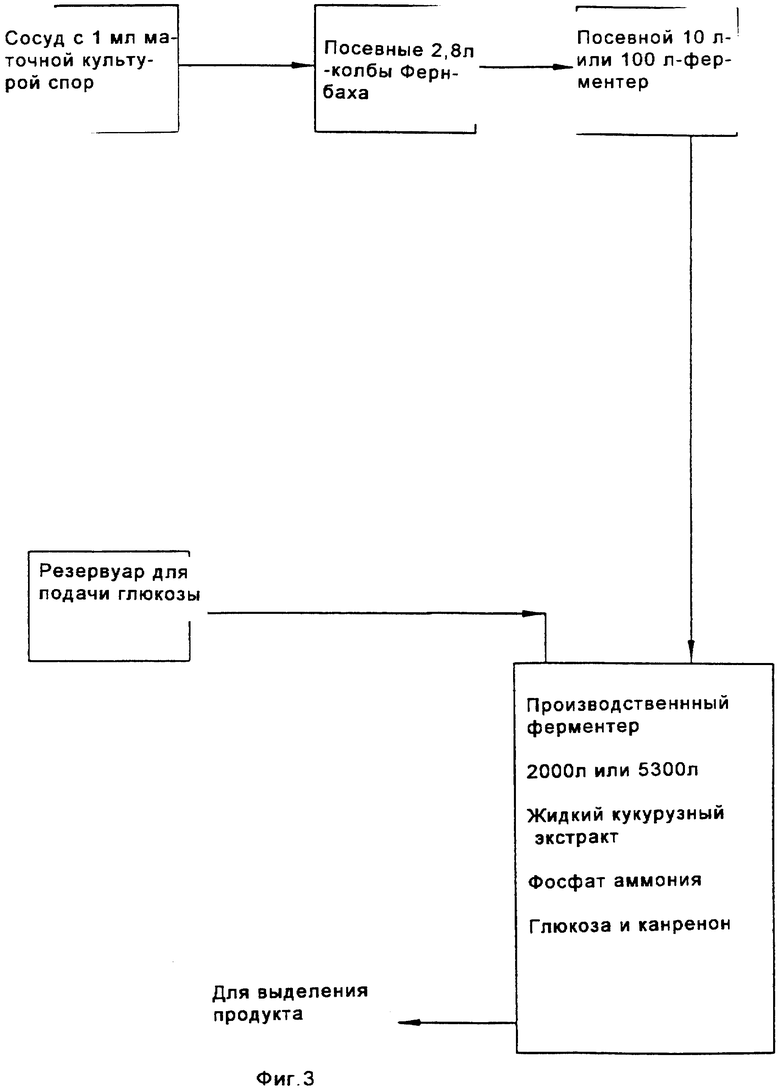
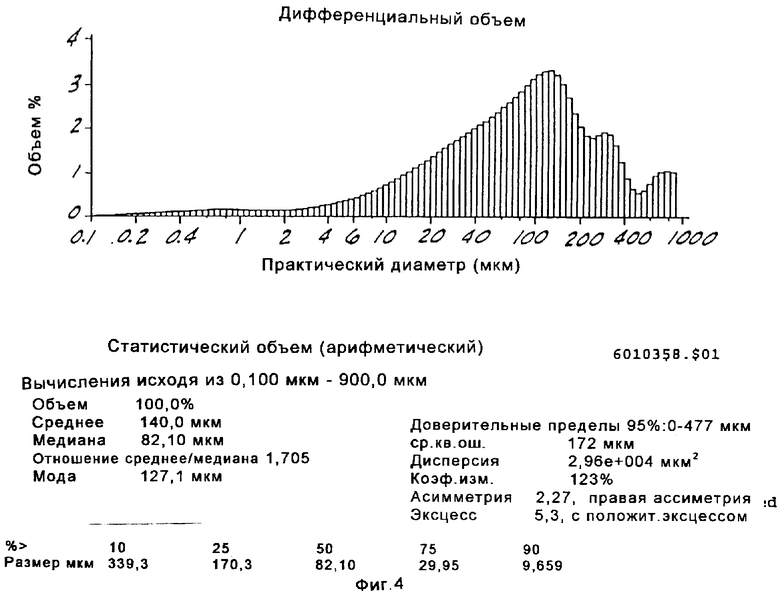
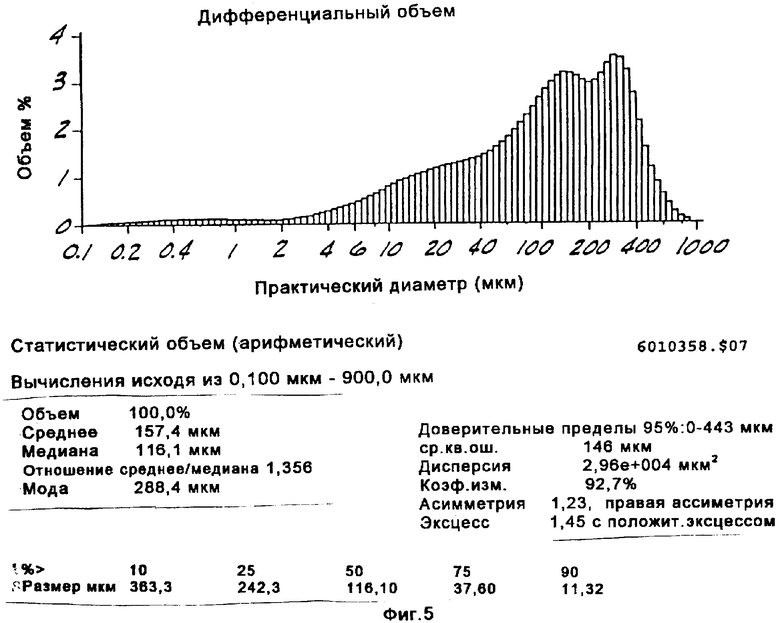
Описывается способ получения 3-кето-7α-алкоксикарбонил замещенного, Δ4,5-стероида формулы I
где 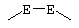 выбран из
выбран из 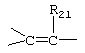 ,
,  или
или 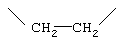 , R3 - Н, галоген, низший алкил, низший алкокси или CN; R21 - Н или алкил, R26 - С1-С4алкил, и R8 и R9 вместе образуют гетероциклическую кольцевую систему; действием алкилирующего агента с 4,5-дигидро-5,7-лактонстероидо формулы II, где R18 - С1-С4алкил или R18O - группы, взятые вместе, образуют O,O-оксиалкиленовый мостик или кетогруппу, а
, R3 - Н, галоген, низший алкил, низший алкокси или CN; R21 - Н или алкил, R26 - С1-С4алкил, и R8 и R9 вместе образуют гетероциклическую кольцевую систему; действием алкилирующего агента с 4,5-дигидро-5,7-лактонстероидо формулы II, где R18 - С1-С4алкил или R18O - группы, взятые вместе, образуют O,O-оксиалкиленовый мостик или кетогруппу, а  , R3, R8 и R9 имеют вышеуказанные значения, в присутствии основания. Соединения I применяют в качестве промежуточных соединений в улучшенных способах синтеза эпоксимексренона. 33 н. и 23 з.п.ф-лы, 43 табл., 5 ил.
, R3, R8 и R9 имеют вышеуказанные значения, в присутствии основания. Соединения I применяют в качестве промежуточных соединений в улучшенных способах синтеза эпоксимексренона. 33 н. и 23 з.п.ф-лы, 43 табл., 5 ил.
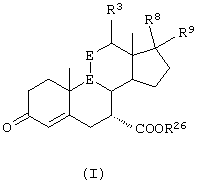
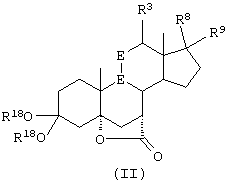
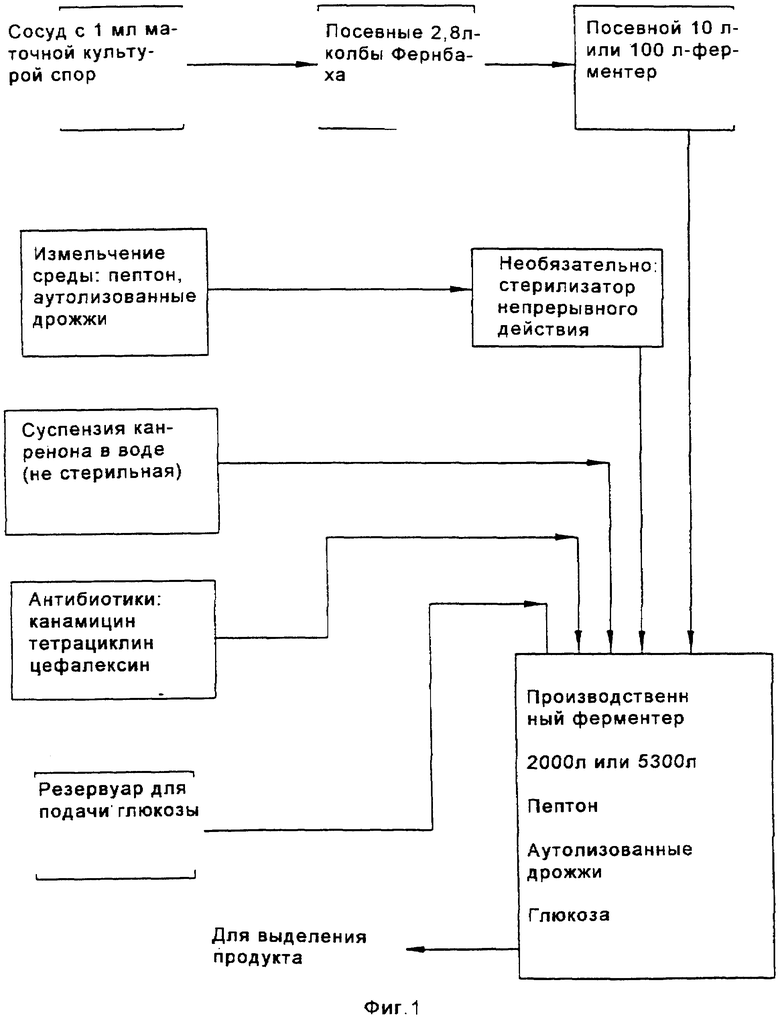

где 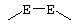 выбран из
выбран из 
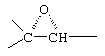 и
и 
R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R21 представляет собой водород или алкил;
R26 представляет собой С1-С4алкил;
R8 и R9, взятые вместе, образуют гетероциклическую кольцевую систему,
включающий взаимодействие алкилирующего агента с 4,5-дигидро-5,7-лактонстероидным субстратом общей формулы:
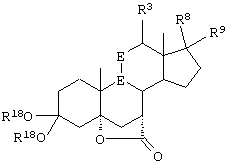
где R18 представляет собой С1-С4алкил, или R18О-группы, взятые вместе, образуют O,O-оксиалкиленовый мостик или кетогруппу, и 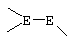 , R3, R8 и R9 имеют значения, указанные выше, в присутствии основания.
, R3, R8 и R9 имеют значения, указанные выше, в присутствии основания.

где , R3, R8, R9 и R18 имеют значения, где указанные в п.1, заключающийся в преобразовании соответствующего 7-цианзамещенного стероида в 7-карбоксизамещенный стероид и последующее преобразование 7-карбоксизамещенного стероида в сответствующий 5,7-лактонзамещенный стероид.
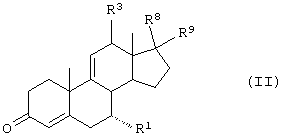 ,
,
где R1 представляет собой альфа-ориентированный C1-С4алкоксикарбонильный радикал;
R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R8 и R9, взятые вместе, образуют гетероциклическую кольцевую систему;
который включает алкилирование соединения формулы EI путем гидролиза с последующим взаимодействием с алкилирующим реагентом в присутствии основания, где соединение формулы EI имеет структуру:
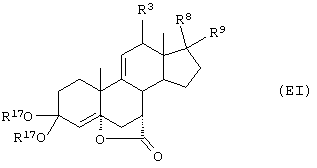 ,
,
где R3, R8 и R9 имеют значения, указанные выше;
R17 представляет собой С1-С4алкил.
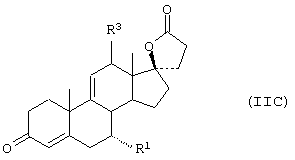 ,
,
где R1 представляет собой альфа-ориентированный C1-С4алкоксикарбонильный или гидроксикарбонильный радикал;
R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
который включает алкилирование соединения общей формулы Е путем гидролиза с последующим взаимодействием с алкилирующим реагентом в присутствии основания, где соединение формулы Е имеет структуру
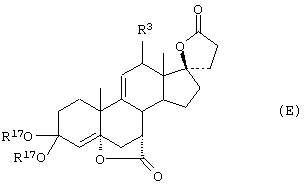 ,
,
где R3 имеет значения, указанные выше;
R17 представляет собой С1-С4алкил.
,
где R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R17 представляет собой С1-С4алкил;
который включает термическое разложение соединения, соответствующего формуле DE2, в присутствии галогенида щелочного металла, где указанное соединение формулы DE2 имеет структуру
 ,
,
где R12 представляет собой С1-С4алкил;
R3 и R17 имеют значения, указанные выше.
,
где R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R12 и R17 представляют собой С1-С4алкил;
который включает конденсацию соединения формулы DE1 с диалкилмалонатом в присутствии основания, где указанное соединение формулы DE1 имеет структуру:
 ,
,
где R3 и R17 имеют значения, указанные выше.
,
где R3 выбран из группы, включающей водород, галоген, низший алкил, низший алкокси или циано;
R17 представляет собой С1-С4алкил;
который включает взаимодействие соединения формулы D с илидом сульфония в присутствии основания, где указанное соединение формулы D имеет структуру:
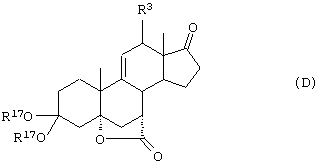 ,
,
где R3 и R17 имеют значения, указанные выше.
,
где R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R17 представляет собой С1-С4алкил;
который включает гидролиз соединения формулы С с образованием 7α-карбоновой кислоты и проведение реакции в кислотных условиях с триалкилортоформиатом, где соединение формулы С имеет структуру
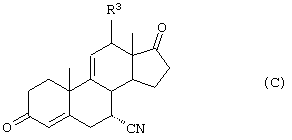 ,
,
где R3 имеет значения, указанные выше.
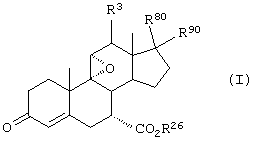 ,
,
где R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R26 представляет собой С1-С4алкил;
R80 и R90 независимо выбраны из R8 и R9 соответственно, или R80 и R90 вместе образуют кетогруппу;
который включает алкилирование соединения формулы А211 путем взаимодействия с алкилирующим реагентом в присутствии основания, где соединение формулы А211 имеет структуру
 ,
,
где R3, R80 и R90 имеют значения, указанные выше.
 ,
,
где R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R26 представляет собой С1-С4алкил;
который включает алкилирование соединения общей формулы 211 путем взаимодействия с алкилирующим реагентом в присутствии основания, где соединение формулы 211 имеет структуру
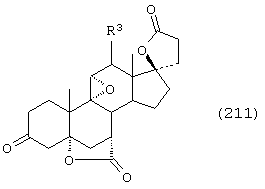 ,
,
где R3 имеет значения, указанные выше.
,
где R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R80 и R90 независимо выбраны из R8 и R9 соответственно, или R80 и R90 вместе образуют кетогруппу;
R8 и R9 независимо представляют собой гидроксикарбонилалкил, или R8 и R9 вместе представляют собой гетероциклическую кольцевую структуру;
который включает окисление соединения формулы А210, где соединение формулы А210 имеет структуру
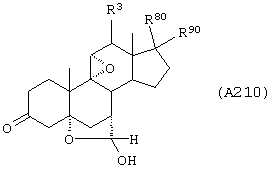 ,
,
где R3, R80 и R90 имеют значения, указанные выше.
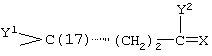
где Х представляет собой оксо;
Y1 представляет собой гидрокси;
Y2 представляет собой гидрокси или низший алкокси.
,
где R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R8 и R9 независимо представляют собой гидроксикарбонилалкил, или R8 и R9, взятые вместе, представляют собой гетероциклическую кольцевую структуру;
который включает взаимодействие промежуточного 3-кето-5,7-полуацеталя формулы А209С с пероксидным окислителем, где указанное соединение формулы А209С соответствует формуле
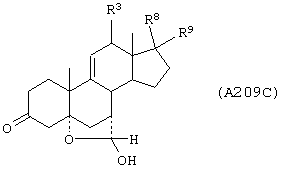
где R3, R8 и R9 имеют значения, указанные выше.
где Х представляет собой оксо;
Y1 представляет собой гидрокси;
Y2 представляет собой гидрокси или низший алкокси.
,
где R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R80 и R90 независимо выбраны из R8 и R9 соответственно, или R80 и R90 вместе образуют кетогруппу;
R8 и R9 независимо представляют собой гидроксикарбонилалкил, или R8 и R9 вместе представляют собой гетероциклическую кольцевую структуру;
который включает взаимодействие промежуточного 3-кето-5,7-полуацеталя формулы А209С с пероксидным окислителем, где указанное соединение формулы А209С соответствует формуле
где R3, R8 и R9 имеют значения, указанные выше.
где Х представляет собой оксо;
Y1 представляет собой гидрокси;
Y2 представляет собой гидрокси или низший алкокси.
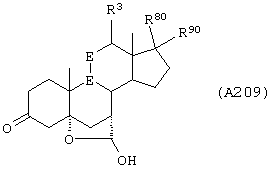
где R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R80 и R90 независимо выбраны из R8 и R9 соответственно, или R80 и R90 вместе образуют кетогруппу;
R8 и R9 независимо представляют собой гидроксикарбонилалкил или R8 и R9 вместе представляют собой гетероциклическую кольцевую структуру;
-Е-Е- выбран из группы, включающей
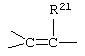 и
и
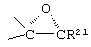
где R21 представляет собой водород или алкил;
который включает гидролиз соединения общей формулы А208
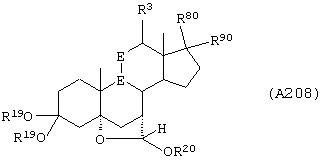 ,
,
где -Е-Е-, R3, R80 и R90 имеют значения, указанные выше;
R19 представляет собой С1-С4алкил, или R19O-группы, взятые вместе, образуют O,O-оксиалкиленовый мостик; R20 представляет собой C1-С4алкил.
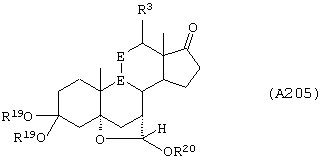 ,
,
где R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R19 представляет собой С1-С4алкил, или R19O-группы, взятые вместе, образуют O,O-оксиалкиленовый мостик;
R20 представляет собой С1-С4алкил;
-Е-Е- выбран из группы, включающей
и
где R21 независимо выбран из водорода и алкила;
который включает взаимодействие соединения общей формулы А204 с низшим спиртом и кислотой, где указанное соединение формулы А204 имеет структуру
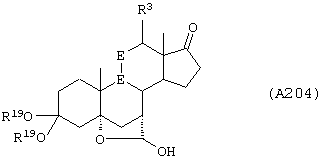 ,
,
где -Е-Е-, R3 и R19 имеют значения, указанные выше.
,
где R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R19 представляет собой C1-С4алкил, или R19O-группы, взятые вместе, образуют O,O-оксиалкиленовый мостик;
-Е-Е- выбран из группы, включающей
,
и
где R21 независимо выбран из водорода и алкила;
который включает гидролиз соединения общей формулы А203, где указанное соединение формулы А203 имеет формулу
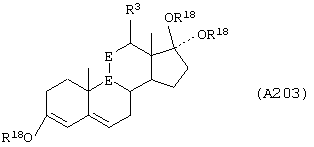
где -Е-Е- и R3 имеют значения, указанные выше;
R18 представляет собой С1-С4алкил, или R18О - группы, взятые вместе, образуют O,O-оксиалкиленовый мостик.
,
где R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R19 представляет собой С1-С4алкил, или R19O-группы, взятые вместе, образуют O,O-оксиалкиленовый мостик;
-Е-Е- выбран из группы, включающей
 и
и
где R21 независимо выбран из водорода и алкила;
который включает защиту кетозаместителей соединения формулы А201 путем взаимодействия с алканолом в кислых условиях в присутствии ортоформиата, где указанное соединение формулы А201 имеет структуру
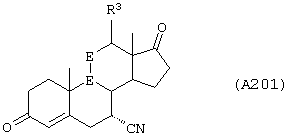 ,
,
где -Е-Е- и R3 имеют значения, указанные выше;
с получением, таким образом, промежуточного простого эфира 3-енола формулы А202
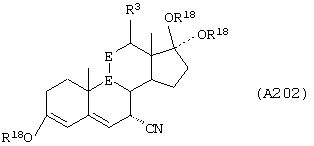 ,
,
где -Е-Е- и R3 имеют значения, указанные выше;
R18 представляет собой С1-С4алкил, или R18О-группы, взятые вместе, образуют O,O-оксиалкиленовый мостик;
восстановление указанного соединения формулы А202 с получением соединения формулы А203:
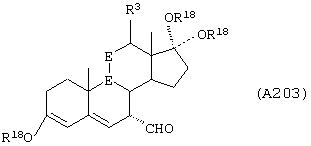 ,
,
где -Е-Е-, R3 и R18 имеют значения, указанные выше;
гидролиз указанного соединения формулы А203 с получением таким образом соединения формулы А204.
,
где R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R18 представляет собой С1-С4алкил, или R18О-группы при С-17, взятые вместе, образуют 0,0-оксиалкиленовый мостик;
-Е-Е- выбран из группы, включающей
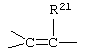 и
и
где R21 независимо выбран из водорода и алкила;
который включает восстановление соединения формулы А202:
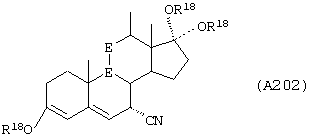
где -Е-Е-, R3 и R18 имеют значения, указанные выше.
и
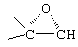
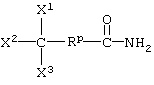 ,
,
где
Rp выбран из группы, включающей, алкенил, алкинил и -(СХ4X5)n-;
X1, X2, Х3, X4 и X5 независимо выбраны из галогена, водорода, алкила, галогеналкила, циано и цианоалкила;
n равно 0, 1 или 2;
при условии, что, когда n равно 0, тогда по крайней мере один из X1, X2 и X3 представляет собой галоген,
когда Rp представляет собой -(СХ4X5)n- и n равно 1 или 2, тогда по крайней мере один из X4 и X5 представляет собой галоген.
,
где R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R1 представляет собой альфа-ориентированный C1-4 алкоксикарбонильный радикал;
R8 и R9 независимо выбраны из гидроксикарбонилалкила; или R8 и R9, взятые вместе, образуют гетероциклическую кольцевую структуру.
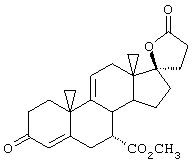

и продукт реакции эпоксидирования выбран из группы, включающей


и продукт реакции эпоксидирования выбран из группы, включающей
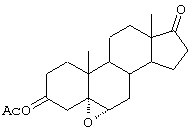
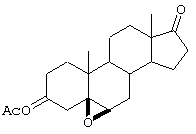



,
где R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R17 представляет собой С1-С4алкил.
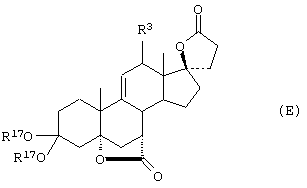 ,
,
где R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R17 представляет собой С1-С4алкил.
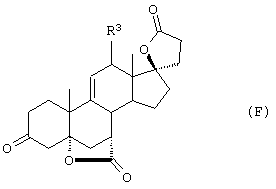 ,
,
где R3 представляет собой водород, галоген, низший алкил, низший алкокси и циано.
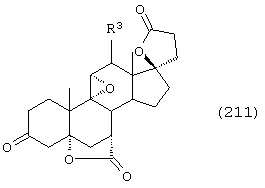 ,
,
где R3 представляет собой водород, галоген, низший алкил, низший алкокси и циано.

где R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R80 и R90 независимо выбраны из R8 и R9 соответственно, или R80 и R90 вместе образуют кетогруппу;
R8 и R9 независимо выбраны из гидроксикарбонилалкила; или R8 и R9, взятые вместе, образуют гетероциклическую кольцевую структуру.

где R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R80 и R90 независимо выбраны из R8 и R9 соответственно, или R80 и R90 вместе образуют кетогруппу;
R8 и R9 независимо выбраны из гидроксикарбонилалкила, или R8 и R9, взятые вместе, образуют гетероциклическую кольцевую структуру;
-Е-Е- выбран из
и
где R21 представляет собой водород или алкил.
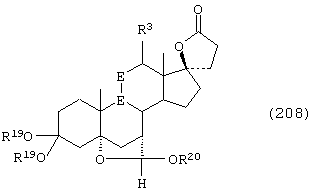
где R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R19 представляет собой С1-С4алкил, или R19O-группы, взятые вместе, образуют O,O-оксиалкиленовый мостик;
R20 представляет собой С1-С4алкил;
-Е-Е- выбран из
 и
и
где R21 представляет собой водород или алкил.
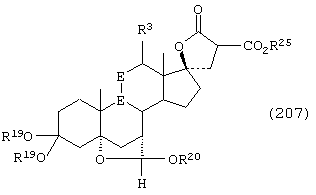
где R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R19 представляет собой С1-С4алкил, или R19O-группы, взятые вместе, образуют O,O-оксиалкиленовый мостик;
R20 представляет собой С1-С4алкил;
R25 представляет собой С1-С4алкил;
-Е-Е- выбран из
и
где R21 представляет собой водород или алкил.
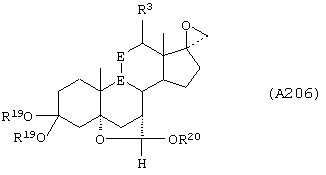
где R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R19 представляет собой С1-С4алкил, или R19O-группы, взятые вместе, образуют O,O-оксиалкиленовый мостик;
R20 представляет собой С1-С4алкил;
-Е-Е- выбран из
и
где R21 представляет собой водород или алкил.
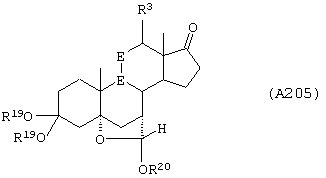 ,
,
где R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R19 представляет собой С1-С4алкил, или R19O-группы, взятые вместе, образуют O,O-оксиалкиленовый мостик;
R20 представляет собой C1-С4алкил;
-Е-Е- выбран из
и
где R21 представляет собой водород или алкил.
,
где R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R19 представляет собой С1-С4алкил, или R19O-группы, взятые вместе, образуют O,O-оксиалкиленовый мостик;
-Е-Е- выбран из
и
где R21 представляет собой водород или алкил.
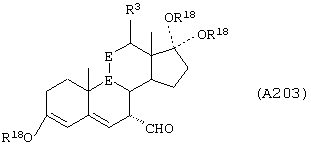
где R3 представляет собой водород, галоген, низший алкил, низший алкокси или циано;
R18 представляет собой С1-С4алкил, или R18O-группы у С-17, взятые вместе, образуют O,O-оксиалкиленовый мостик;
-Е-Е- выбран из
и
где R21 представляет собой водород или алкил.
-Е-Е- выбран из
и
где R21 представляет собой водород.
-Е-Е- представляет собой
где R21 представляет собой водород.
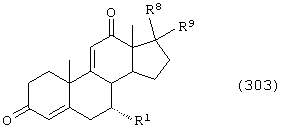
где R1 представляет собой альфа-ориентированный C1-С4алкоксикарбонильный или гидроксикарбонильный радикал;
R8 и R9 независимо представляют собой гидроксикарбонилалкил, или R8 и R9 вместе представляют собой гетероциклическую кольцевую структуру.
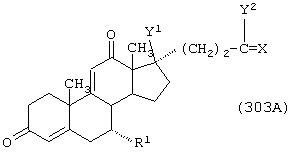
где R1 представляет собой альфа-ориентированный C1-С4алкоксикарбонильный или гидроксикарбонильный радикал;
Х представляет собой оксо;
Y1 представляет собой гидрокси;
Y2 представляет собой гидрокси или низший алкокси.
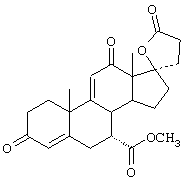
Приоритет по пунктам:
| US 4559332 А, 17.12.1985 | |||
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ФТОРЗАМЕЩЕННЫЕ ОЛЕФИНЫ, И ИХ ПРИМЕНЕНИЕ | 2006 |
|
RU2443746C2 |
| ЭЛЕКТРОЛИЗЕР С БИПОЛЯРНЫМИ АМАЛЬГАМНЫМИ | 0 |
|
SU165902A1 |
| Л.Физер, М.Физер | |||
| Стероиды | |||
| - М.: Мир, 1964 с.685. | |||

