Уровень техники
Некоторые С-7-замещенные стероиды, например эплеренон, являются хорошо известными в отношении их антагонистической активности в отношении альдостерона и таким образом они пригодны для лечения и профилактики заболеваний кровеносной системы. Эплеренон является предметом нескольких патентов и заявок, например патентов США № 4559332 и 5981744 и публикаций международных заявок WO98/25948 и WO97/21720. Однако в результате появления новых и расширенных клинических применений эплеренона возрастает необходимость в усовершенствованных способах производства его и других близких стероидов. Основным затруднением для эффективного синтеза эплеренона и близких стероидных соединений является введение в положение С-7 карбоксильной группы или функциональной группы, которая может быть трансформирована в карбоксильную группу.
Известно, что аллильные производные и, в частности, аллильные ацетаты, бензоаты, пивалаты и тому подобное, взаимодействуют с нуклеофильными реагентами в присутствии кислоты Льюиса в способе, называемом "аллилированием", как уже было описано ранее. Реакцию аллилирования использовали для целого ряда субстратов. Например, было показано, что при аллилировании гликали дают аллилгликозиды, гликозилцианиды и гликозалазиды (Yadav J.S. et al., Tetrahedron Lett., 2001, 42, 4057). Аллильные ацетаты и карбонаты дают соответствующие цианиды (Yasushi T. et al., J. Org. Chem., 1993, 58, 16). Богатые электронами ароматические и гетероароматические соединения дают соответствующие аллилированные продукты (Malkov A.V. et al., J. Org. Chem., 1999, 64, 2751). Однако реакция аллилирования до настоящего времени не применялась к стероидам для получения 7-замещенных стероидов, пригодных для превращения в 7-карбоксизамещенные стероиды, такие как эплеренон. 3,17-Диацетокси-7-гидроксиандрост-5-ен или соответствующие 7-метансульфонаты подвергали взаимодействию с фенолом и анизолом с использованием жесткого катализатора хлорида алюминия (Negi A.S. et al., Steroids, 1995, 60, 470). Образовавшиеся в результате 7-арильные производные получают с низким выходом в виде смеси С-7-эпимеров. Кроме того, 7-арильные производные, в лучшем случае, будет трудно применять для получения 7-карбоксизамещенных стероидов.
Сущность изобретения
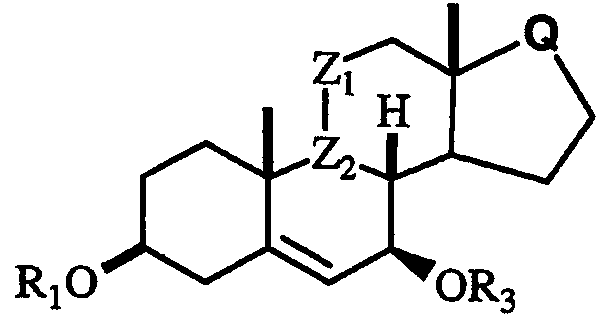
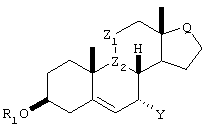
Данное изобретение относится к способам получения новых 7-карбоксизамещенных стероидных соединений формулы I,
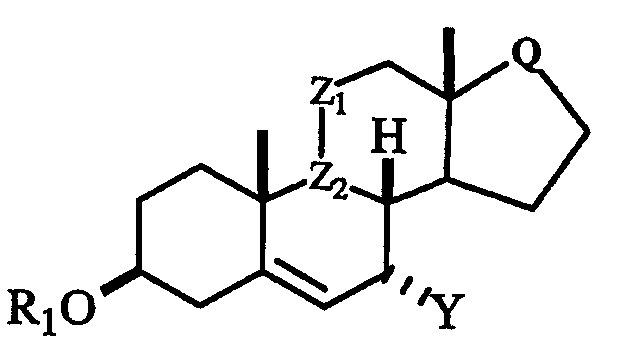
формула I
в которой R1 является -COR2;
R2 представляет собой С1-6алкил или С1-6алкокси;
Z1 является СН2 или
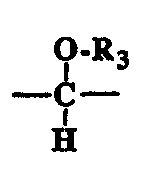
, где OR3 находится в α-конфигурации;
R3 представляет собой Н или -COR2;
Z2 является -СН-; или
Z1 и Z2, взятые вместе, образуют углерод-углеродную двойную связь;
Q представляет собой
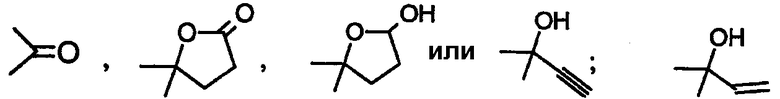 ;
;
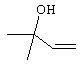
Y является -CN, -CH2-CH=CH2,
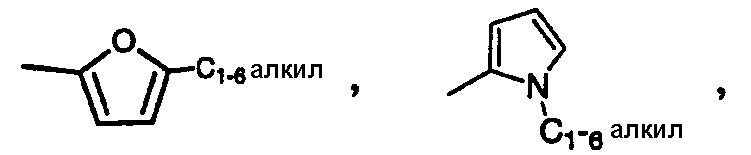
CHR4C(O)Ar, CHR4C(O)C1-6алкил, CHR4C(O)XAr или CHR4C(O)XC1-6алкил;
где R4 = ОС1-6алкил или арил,
Х = О или S.
Данные новые промежуточные соединения пригодны для получения 7-карбоксизамещенных стероидных соединений и, в частности, изобретение относится к новым и преимущественным способам получения метилового эфира 9,11-α-эпокси-17-α-гидрокси-3-оксопрегн-4-ен-α-21-дикарбоновой кислоты γ-лактона (эплеренона; эпоксимексренона).
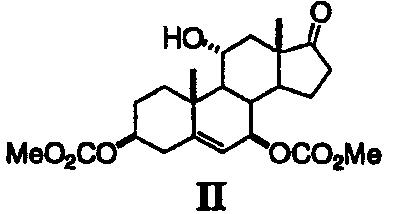
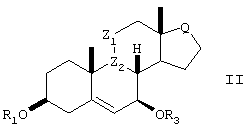
Ключевой стадией в способах по настоящему изобретению является взаимодействие нового стероидного промежуточного соединения формулы II,
формула II
в которой R1 и R3 независимо выбраны из Н, С(O)OR2 или COR2, и, по меньшей мере, один из R1 или R2 представляет собой C(O)OR2 или COR2;
Z1, Z2, R2 и Q имеют значения, определенные для формулы I;
с нуклеофильным реагентом, выбранным из группы С1-4триалкилсилилцианидов, С1-4триалкилсилиленоэфиров, С1-4триалкилсилилоксикетентиоацеталей (т.е. RCH=C(OSiRC1-C6алкил)SRC1-6алкил), аллилтриС1-4алкилсиланов, аллилтри-С1-4алкилстаннанов, 2-С1-4алкилфуранов и 2-С1-4алкилпирролов в присутствии катализатора - кислоты Льюиса.
В соединении формулы II имеется структурный элемент производного аллилового спирта в положениях -С-5, -С6, -С7-OR3. Новые схемы синтеза, в которых используется преимущество реакции "аллилирования", подробно описаны в разделе "Описание предпочтительных вариантов осуществления".
Описание предпочтительных вариантов осуществления
Определения
При подробном описании используются следующие определения. Термин "алкил", сам или как часть другого заместителя, означает, если не указано иначе, нормальный либо разветвленный, или циклический углеводородный радикал или их сочетание. Примеры насыщенных углеводородных радикалов включают, но не ограничиваются такими группами, как метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, циклогексил, (циклогексил)этил, циклопропилметил, их гомологи и изомеры, например, н-пентил, н-гексил, н-гептил, н-октил и тому подобное.
Термин "арил", (Ar), используется ли он самостоятельно или в сочетании с другими терминами (например, арилокси, арилтиокси, аралкил), означает, если не указано иначе, ароматический заместитель, который может представлять собой единичное кольцо или множественные кольца (до трех колец), которые сконденсированы вместе или связаны ковалентно.
Термин "нуклеофильный реагент" означает богатые электронами реагенты, которые обладают тенденцией к атаке ядра углерода, как описано Morrison R.T. et al., Organic Chemistry, sixth edition, Prentice Hall pub., 1992, p. 172.
Термин "кислота Льюиса" означает акцептор электронной пары, как определено McQuarrie D.A. et al., General Chemistry, third edition, W.H.Freeman and Company pub., 1991, p. 665.
Описание предпочтительных вариантов осуществления
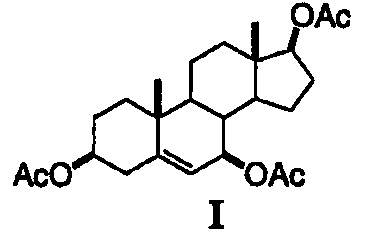
Заявители, с удивлением, установили, что карбоксипроизводные С-7-гидрокси-С-5Δ6-еновых стероидов формулы II вступают в реакцию аллилирования с различными нуклеофильными реагентами с образованием соответствующих С-7-замещенных стероидных производных формулы I, как представлено в таблице 1. Подходящие нуклеофильные реагенты включают, но не ограничиваются ими, С1-4триалкилсилилцианиды, С1-4триалкилсилиленоэфиры, триалкилсилилоксикетентиоацетали (RCH=C(OSiR3)SR), аллилтри-С1-4алкилсиланы, аллилтри-С1-4алкилстаннаны, 2-С1-4аллилфураны и 2-С1-4алкилпирролы в присутствии катализатора - кислоты Льюиса. Подходящие кислоты Льюиса включают, но не ограничиваются ими, трифлаты переходных элементов (OTf=OSO2CF3), такие как Sc(OTf)3, Ce(OTf)3, Fe(ClO4)2, Cu(ClO4)2 и Yb(OTf)3, и комплексы молибдена(II), такие как Mo(CO)5(OTf2) и [Mo(CO)4Br2]2.
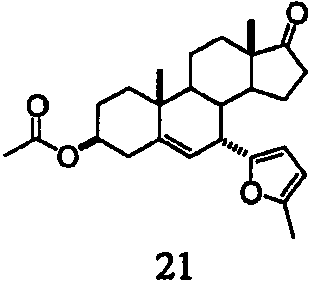
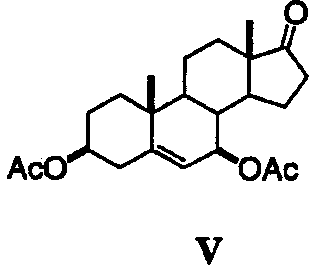
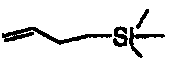
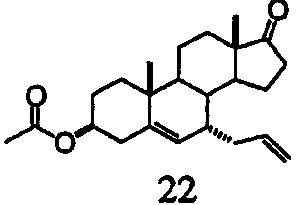
В тех же условиях, однако, индол и орцинол дают выход < 10% и сложные смеси. Винилтриметилсилан и триметилсилилацетилен не подвергаются взаимодействию.
Схемы, представляющие сущность изобретения
Как уже отмечалось выше, соединения формулы I можно использовать в качестве исходных соединений при синтезе эплеренона. На схемах I и II приводится схематичная диаграмма примеров применения соединения формулы I, полученного способом по данному изобретению, для получения эплеренона.
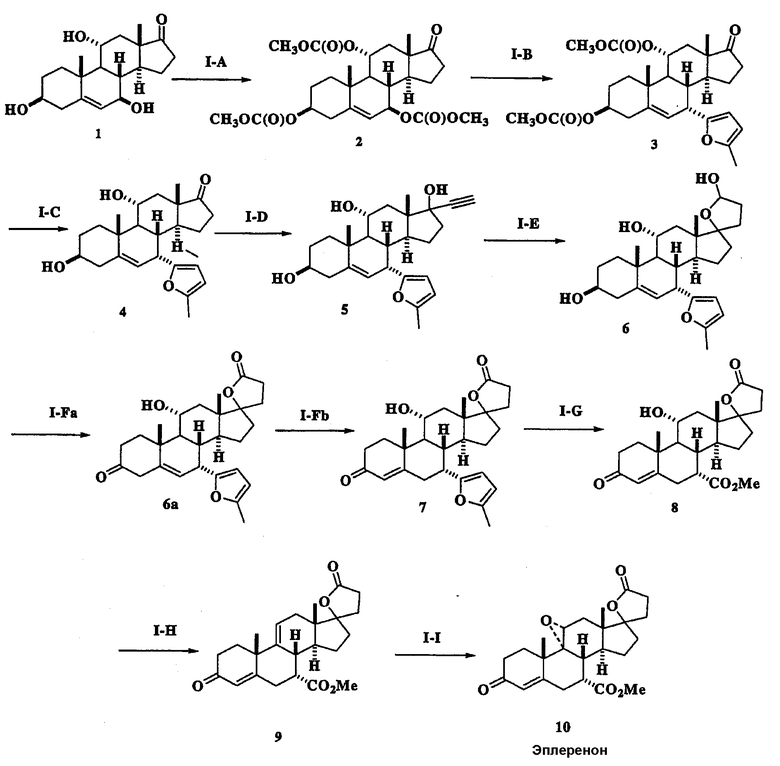
Схема I
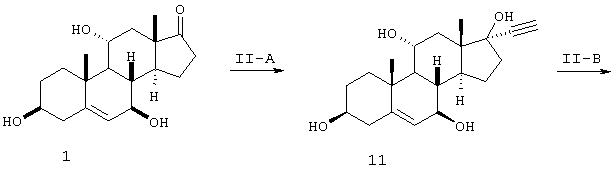
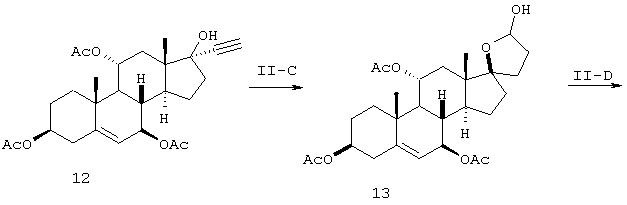
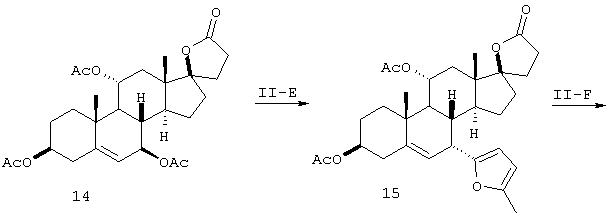
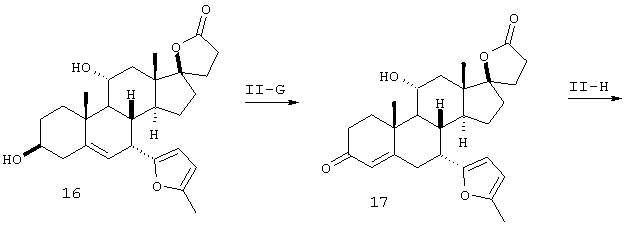
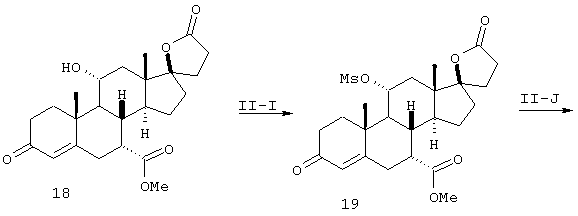
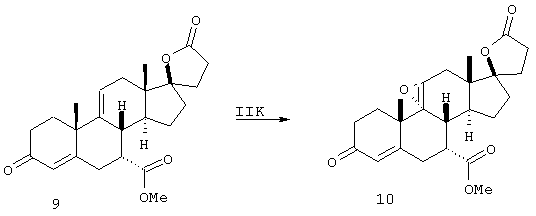
Схема II
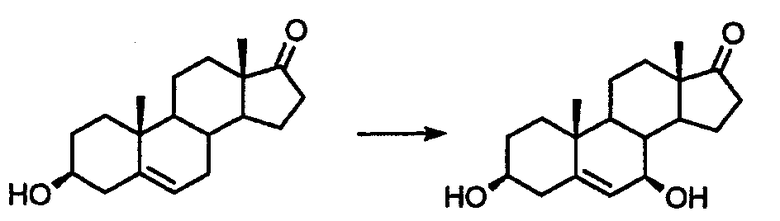
Исходное соединение 1 (схема I), (3β, 7β, 11α-тригидрокси-5-андростен-17-он) для схем I-II получают одним из двух путей. Первый путь представляет собой вначале контактирование 5-андростен-3β-ол-17-она с глубинной культурой Diplodia qossypina ATCC 20517 (синоним Botryodiplodia theobromae IFO 6469) с получением 5-андростен-3β,7β-диол-17-она (смотри пример 10), затем контактирование 5-андростен-3β,7β-диол-17-она с глубинной культурой Aspergillus ochraceus ATCC 18500 с получением 5-андростен-3β,7β,11α-триол-17-она (схема I). Альтернативно можно подвергнуть 5-андростен-3β-ол-17-он контактированию с глубинной культурой Absidia coerulea ATCC 6647 с получением 5-андростен-3β,7β,11α-триол-17-она (схема I).
Стадии IA и IIB
Гидроксильные промежуточные соединения 1 и 11 (схема II) ацилируют с использованием ацилирующего реагента в присутствии третичного органического основания способами, хорошо известными в данной области, с получением соединений 2 и 12. Ацилирующие реагенты включают ангидриды низших алкановых кислот, хлорангидриды низших алкановых кислот, низшие алкилкарбонилхлориды, ангидриды низших алкилкарбоновых кислот и тому подобное. Подходящие третичные органические основания включают пиридин, 4-диметиламинопиридин, триэтиламин, диизопропилэтиламин и тому подобное. Альтернативно можно получить смешанные карбонаты (RO-CO-O-) взаимодействием с алкоксикарбонилоксибензотриазолом в присутствии третичного органического основания при модификации опубликованных способов (Harada T. et al., J. Carbohydrate Chem., (1995), 14, 165).
Стадии I-B и II-E
Взаимодействие триацилированных соединений 2 (схема I) и 14 (схема II) с нуклеофильным реагентом в присутствии кислоты Льюиса, обычно в инертном растворителе, таком как ацетонитрил или метиленхлорид, дает, соответственно, соединения 3 (схема I) и 15 (схема II). Подходящие нуклеофильные реагенты включают, но не ограничиваются ими, триалкилсилилцианиды, 3-силил-замещенные алкены, енолацетаты, простые силиленолэфиры, аллилстаннаны, N-алкилпирролы, N,N-диалкиланилины, силиленолтиоэфиры, сложные силиленолэфиры и богатые электронами гетероароматические соединения, такие как 2-алкилзамещенный фуран. Подходящие кислоты Льюиса включают, но не ограничиваются ими, трифлаты переходных элементов (OTf=OSO2CF3), такие как Sc(OTf)3, Ce(OTf)3 и Yb(OTf)3, и комплексы молибдена(II), такие как Mo(CO)5(OTf2) и [Mo(CO)4Br2]2.
Стадии I-С и II-F
Гидролиз ацильных групп соединения 3 (схема I) и 15 (схема II) с получением соединения 4 (схема I) или 16 (схема II) проводят с использованием гидроокиси щелочного металла, бикарбоната или карбоната, таких как гидроокись натрия, карбонат калия, бикарбонат натрия, гидроокись цезия, бикарбонат лития и тому подобное, с использованием метанола в качестве растворителя, необязательно вместе с сорастворителем.
Стадии I-D и II-A
17-Оксо промежуточные соединения 4 (схема I) и 1 (схема II) подвергают взаимодействию с ацетиленом с получением соответствующего аддукта 5 (схема I) и 11 (схема II) согласно способам, описанным в литературе (например, смотри Schwede W. et al., Steroids, (1998), 63, 166; Corey E.J. et al., J. Amer. Chem. Soc. (1999), 121, 710-714; Schwede W. et al., Steroids (1998), 63(3), 166-177; Ali H. et al., J. Med. Chem. (1993), 36(21), 3061; Turuta A.M. et al., Mendeleev Commun. (1992), 47-8; Kumar V. et al., Tetrahedron (1991), 47(28), 5099; Page P.C., Tetrahedron (1991), 47, 2871-8; Curts S.W. et al., Steroids (1991), 56, 8; Kataoka H. et al., Chem. Lett. (1990), 1705-8; Christiansen R.G. et al., J. Med. Chem. (1990), 33(8), 2094-100). Необязательно тригидроксипроизводное 1 на стадии II-A можно триметилсилилировать без его выделения до добавления ацетилена. Силилирование проводят с помощью гексаметилдисилазана и мягкого кислотного катализатора, такого как триметилсилилхлорид или сахарин. После добавления ацетилена триметилсилильные группы удаляют во время обработки реакционной смеси слабой минеральной кислотой, уксусной кислотой, фосфорной кислотой, фторидом тетраалкиламмония и тому подобное.
Стадия Е
Лактольные промежуточные соединения 6 (схема I) и 13 (схема II) получают гидроформилированием соединения 5 и 12 с использованием окиси углерода и водорода в присутствии каталитического количества катализатора родия и координационного лиганда родия согласно способам, описанным в литературе (Wuts P.G.M. et al., J. Org. Chem. 1989, 54, 5180; Botteghi C. et al., Tetrahedron, 2001, 57, 1631). Реакцию проводят под давлением 14-500 фунтов/кв. дюйм, предпочтительно в пределах 100-200 фунтов/кв. дюйм. Соотношение водорода и окиси углерода находится в пределах от 1/5 до 5/1, обычно составляет 1/1. Подходящие катализаторы на основе родия включают ацетат родия, хлорид родия и дикарбонилацетилацетонатородий II. Подходящие лиганды включают триарилфосфины, триалкилфосфиты, бидентатные фосфины, такие как ксантфос, бидентатные фосфиты и тому подобное.
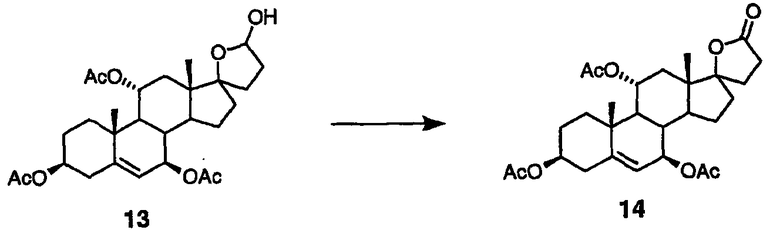
Стадии I-Fa и II-D
Окисление соединения 6 (схема I) в соединение 6а (схема I) и соединения 13 (схема II) в соединение 14 (схема II) можно проводить с помощью различных обычных окислителей. Примеры подходящих окислителей включают: йодосукцинимид/йодид тетрабутиламмония (Kraus, George A. Bioorganic & Medicinal Chemistry Letters (2000), 10(9), 895-897; Barrett, A.G.M. et al., J. Org. Chem. (1989), 54(14), 3321); реагент Джонса (хромовая кислота в ацетоне) (Panda J. et al., Tetrahedron Letters (1999), 40, 6693; Tomioka K. et al., J. Org. Chem. (1988), 53(17), 4094); карбонат серебра (Chow T.J. et al., J. Chem. Soc., Perkin Transactions 1, (1999), 1847); хлорхромат пиридиния (Uchiyama M. et al., Tetrahedron Letters (2000), 41(51), 10013; Vanderiei J.M. de L., Synthetic Communications (1998), 28(16), 3047; Kassou M. et al., Journal of Organic Chemistry (1997), 62, 3696; Rehnberg N. et al., J. Org. Chem. (1990), 55(14), 4340-9; RuO4/соли тетраалкиламмония/N-оксид трет-амина (Jeewoo K. et al., Chem. Lett. (1995), (4), 299); дихромат пиридиния (Paquette L.A. et al., J. Am. Chem. Soc. (1995), 117(4), 1455-6); гипохлорит натрия/ N-оксид трет-амина (Waldemar A. et al., Chem. Rev., (2001), 101, 3499); алкоголяты алюминия/ацетон (Ooi T. et al., Synthesis (2002), 279); Djerassi C. et al., Org. React. (1951), 6, 207); триацетоксипериодоиндан (Martin J.C. et al., J. Amer. Chem. Soc., (1991), 113, 7277).
Стадия Fb
В тех случаях, когда окисление на стадии Fa приводит к образованию несопряженной двойной связи в положении 5-6, миграцию двойной связи в соединении 6а (схема I) из положения С5-6 в положение С4-5 проводят взаимодействием соединения 6а (схема I) с органической или неорганической кислотой в инертном органическом растворителе или водной смеси растворителей при температуре 0°С-80°С. Подходящие органические кислоты включают, но не ограничиваются ими, толуолсульфоновую кислоту, метансульфоновую кислоту, бензолсульфоновую кислоту, трифторуксусную кислоту, щавелевую кислоту, трихлоруксусную кислоту и тому подобное. Подходящие неорганические кислоты включают, но не ограничиваются ими, соляную кислоту, бромистоводородную кислоту, фосфорную кислоту, перхлорную кислоту и тому подобное. Альтернативно, катализатор может представлять собой третичное органическое основание, такое как триэтиламин, диазабициклоундекан (DBU) и тому подобное, или неорганическое основание, такое как гидроокись натрия, гидроокись калия, гидроокись кальция и тому подобное. Миграция двойной связи была уже описана ранее (Bakshi et al., патент США № 5237064; Pollack et al., J. Amer. Chem. Soc., 1987, 109, 5048; Tsubuki et al., J. Org. Chem., 1992, 57, 2930; Zenge et al., J. Amer. Chem. Soc., 1991, 113, 3838).
Стадии I-H и II-I,J
Дегидратацию 11-гидрокси промежуточных соединений 7 (схема I) и 18 (схема II) проводят с использованием пентахлорида фосфора, как было ранее описано (патент США № 4559332). Альтернативно, 11-гидрокси промежуточные соединения можно превратить в сульфониловый эфир, например, в метансульфонат или п-толуолсульфонат, с последующей обработкой основанием для проведения дегидратации, как описано в заявках WO97/21720 и WO98/25948.
Стадии I-H и II-H
Превращение фуранового кольца в соединении 7 (схема I) в метиловый эфир соединения 8 (схема II) проводят озонолизом, окислением и эстерификацией, как описано в примерах.
Стадии I-J и II-K
Превращение известного промежуточного соединения 9 (схема I) в соединение 10 (схема I) (эплеренон) описано в патентах США № 4559332 и 5981744.
Примеры
Получение исходного соединения 1, способ 1
Стадия 1: Биоконверсия 5-андростен-3β-ол-17-она в 5-андростен-3β,7β-диол-17-он
Биоконверсию 5-андростен-3β-ол-17-она в 5-андростен-3β,7β-диол-17-он проводят с использованием глубинной культуры Diplodia gossypina ATCC 20571 (синоним Botryodiplodia theobromae IFO 6469) в ферментере емкостью 10 л.
(А) Стадия первичного посева
Замороженные вегетативные клетки Diplodia gossypina ATCC 20571 размораживают, переносят на чашки с картофельным-декстрозным агаром (PDA) и инкубируют при 28°С в течение 72 ч. Единичные колонии мицелия (диаметром 6-7 мм) используют для посева в покрытые силиконом качалочные колбы с метками емкостью 500 мл, содержащие 100 мл среды для первичного посева. Среда для первичного посева состоит из (на литр воды RO, т.е. воды подвергнутой обратноосмотическому обессоливанию): декстрина, 50 г; соевой муки, 35 г; церелозы, 5 г; хлорида кобальта гексагидрата, 2 мг; силиконового противовспенивателя (SAG 471), 0,5 мл; перед стерилизацией значение рН доводят до 7,0-7,2 гидроокисью натрия (2 н. раствором). Diplodia gossypina ATCC 20571 инкубируют в течение 48 ч при 28°С с использованием термостата-встряхивателя с контролируемой окружающей средой при 280 об/мин (орбитальный ход 1").
(В) Стадия вторичного посева
В ферментеры для вторичного посева емкостью 10 л вносят 1,2 мл вегетативной культуры первичного посева (количество внесенного посевного материала составляет 0,012% [об./об.]). Среда для вторичного посева состоит из (на литр воды RO): церелозы, 60 г; соевой муки, 25 г; соевого масла, 30 мл; сульфата магния гептагидрата, 1 г; дигидрофосфата калия, 0,74 г; полиоксиэтиленсорбитана моноолеата, 2 мл; силиконового противовспенивателя (SAG 471), 0,5 мл; перед стерилизацией значение рН доводят до 3,95-4,00 концентрированной серной кислотой. Ферментеры, содержащие среду для вторичного посева, стерилизуют в течение 20 мин при 121°С с использованием как согревающего пара (в паровой рубашке), так и введения пара. Скорость перемешивания во время стерилизации равняется 200 об/мин. После стерилизации рН среды доводят до 4,0 с использованием стерильной серной кислоты (5% раствора). Diplodia gossypina ATCC 20571 инкубируют при 28°С с использованием следующих первоначальных параметров: перемешивание, 100 об/мин; противодавление = 5 фунтов/кв. дюйм избыточного давления; воздушный поток = 2,5 SLM (0,25 об./об/мин); нижнее значение DO (растворенный кислород), 30%; без контроля значения рН. Когда значение DO вначале уменьшается до 30%, то воздушный поток увеличивают до 5 SLM (0,5 об./об/мин). Когда культура вновь достигает нижнего значения DO, то значение DO поддерживают на 30% регуляцией воздушного потока. Культуры вторичного посева собирают примерно через 60 ч после посева, когда значение скорости потребления кислорода (СПК) находится в пределах примерно от 10 до примерно 15 мМ/л/час.
(С) Биоконверсия стероидов
В ферментеры для биоконверсии стероидов емкостью 10 л вносят 500 мл вегетативной культуры второго посева (количество внесенного посевного материала составляет 5% [об./об.]). Среда для биоконверсии стероидов является такой же, как среда для вторичного посева. Условия стерилизации и доведение рН являются такими же, как описано для среды вторичного посева. Diplodia gossypina ATCC 20571 инкубируют при 28°С с использованием в основном таких же первоначальных параметров, которые использовали для культивирования вторичного посева, за исключением того, что нижнее значение DO повышают с 30% до 50%. Когда значение DO вначале уменьшается до 50%, то воздушный поток увеличивают с 2,5 SLM (0,25 об./об/мин) до 5 SLM (0,5 об./об/мин). Когда культура вновь достигает нижнего значения DO, то значение DO поддерживают равным 50%, регулируя перемешивание. Через 24 ч после посева в ферментер вносят микронизированный 5-андростен-3β-ол-17-он, разведенный в минимальном объеме 0,2% полиоксиэтиленсорбитана моноолеата, с интервалом 1 ч, пока не вносят в целом 400 г. Примерно через 3 суток после посева в ферментер емкостью 10 л вносят еще 100 г церелозы.
Культуры для биоконверсии анализируют ежедневно на выработку 5-андростен-3β,7β-диол-17-она с использованием ТСХ. Один миллилитр цельной культуральной жидкости экстрагируют 10 мл метанола. Клетки отделяют от водно-метанольной смеси центрифугированием (3000×g в течение 10 мин) и несколько микролитров наносят на пластинку для ТСХ. Пластинку для ТСХ обрабатывают в системе циклогексан:этилацетат:метанол (90:60:15) и продукт проявляют опрыскиванием пластинки для ТСХ 50% серной кислотой с последующим прокаливанием в сушильном шкафу. Продукт сравнивают с соответствующим стандартом, который становится синим при опрыскивании 50% серной кислотой. Биоконверсия 5-андростен-3β-ол-17-она в 5-андростен-3β,7β-диол-11-он заканчивается примерно через 4 суток после посева.
(D) Выделение
Цельную культуральную жидкость, полученную при сборе, центрифугируют и обогащенные твердые частицы отделяют центрифугированием. Обогащенные твердые частицы экстрагируют 10 л метиленхлорида и обогащенный экстракт выделяют центрифугированием. Экстракт обрабатывают и концентрируют примерно до 1 л упариванием, и взвесь кристаллов охлаждают до -10°С. Кристаллы отделяют фильтрованием, промывают холодным метиленхлоридом для удаления окраски и высушивают с получением 227 г очищенного кристаллического 5-андростен-3β,7β-диол-17-она.
Стадия 2: биоконверсия 5-андростен-3β,7β-диол-17-она в 5-андростен-3β,7β,11α-триол-17-он
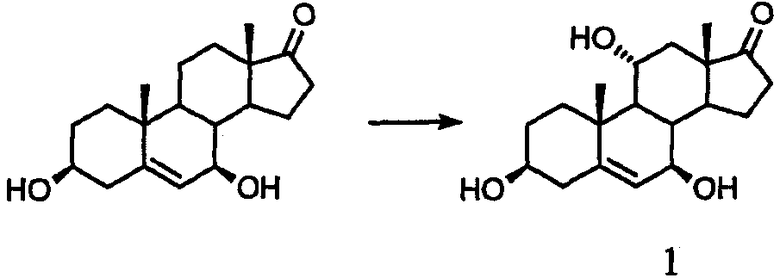
Биоконверсию 5-андростен-3β,7β-диол-17-она в 5-андростен-3β,7β,11α-триол-17-он проводят с использованием глубинной культуры Aspergillus ochraceus ATCC 18500 (синоним NRRL 405) в ферментере емкостью 10 л.
(А) Стадия первичного посева
Культуры Aspergillus ochraceus ATCC 18500 первичного посева готовят, как описано для Diplodia gossypina ATCC 20571 на стр. 14-15.
(В) Стадия вторичного посева
В ферментеры для вторичного посева емкостью 10 л вносят 1,2 мл вегетативной культуры первичного посева (количество внесенного посевного материала составляет 0,012% [об./об.]). Среда для вторичного посева состоит из (на литр воды RO): церелозы, 40 г; соевой муки, 25 г; соевого масла, 30 мл; сульфата магния гептагидрата, 1 г; дигидрофосфата калия, 0,74 г; нонилфеноксиполиэтоксиэтанола, 0,25 мл; силиконового противовспенивателя (SAG 471), 0,5 мл; перед стерилизацией значение рН доводят до 3,95-4,00 концентрированной серной кислотой. Ферментеры, содержащие среду для вторичного посева, стерилизуют в течение 20 мин при 121°С с использованием как согревающего пара, так и введения пара. Скорость перемешивания во время стерилизации равняется 200 об/мин. После стерилизации рН среды доводят до 4,0 с использованием стерильной серной кислоты (5% раствора). Aspergillus ochraceus ATCC 18500 инкубируют при 28°С с использованием следующих первоначальных параметров: перемешивание, 100 об/мин; противодавление = 5 фунтов/кв. дюйм избыточного давления; воздушный поток = 2,5 SLM (0,25 об./об/мин); нижнее значение DO, 50%; без контроля значения рН. Когда значение DO вначале уменьшается до 50%, то воздушный поток увеличивают до 5 SLM (0,5 об./об/мин). Когда культура вновь достигает нижнего значения DO, то значение DO поддерживают равным 50% с использованием регуляции перемешивания. Культуры вторичного посева собирают примерно через 50-54 ч после посева, когда значение СПК находится в пределах примерно от 20 до примерно 26 мМ/л/час.
(С) Биоконверсия стероидов
В ферментеры для биоконверсии стероидов емкостью 10 л вносят 500 мл вегетативной культуры второго посева (количество внесенного посевного материала составляет 5% [об./об.]). Среда для биоконверсии стероидов в основном является такой же, как среда для вторичного посева, за исключением того, что концентрацию нонилфеноксиполиэтоксиэтанола повышают с 0,25 мл/л до 2 мл/л и значение рН до стерилизации доводят до 2,95-3,00 концентрированной серной кислотой. Условия стерилизации являются такими же, как описаны для среды вторичного посева. После стерилизации рН среды доводят до 3,0 с использованием стерильной серной кислоты (5% раствор). Aspergillus ochraceus ATCC 18500 инкубируют при 28°С с использованием в основном таких же первоначальных параметров, которые использовали для культивирования вторичного посева, за исключением того, что вначале перемешивание проводят при 200 об./мин. Примерно через 18 ч после посева 200 г микронизированного 5-андростен-3β,7β-диол-17-она, разведенного в минимальном объеме 0,2% нонилфеноксиполиэтоксиэтанола, вносят в ферментер емкостью 10 л.
Культуры для биоконверсии анализируют ежедневно на выработку 5-андростен-3β,7β,11α-триол-17-она с использованием ТСХ, как описано в примере 10. Биоконверсия 5-андростен-3β,7β-диол-17-она в 5-андростен-3β,7β,11α-триол-17-он заканчивается примерно через 4 суток после посева.
(D) Выделение
Твердые частицы в цельной культуральной жидкости отделяют центрифугированием. Жидкость отбрасывают. Обогащенные твердые частицы экстрагируют 10 л смеси 80% ацетон/20% вода при температуре 45°С-50°С, и теплый экстракт осветляют фильтрованием. Обогащенный фильтрат концентрируют упариванием для удаления ацетона с получением водной суспензии сырых кристаллов. Сырые кристаллы отделяют фильтрованием и маточный раствор отбрасывают. Содержащие воду кристаллы растирают в 600 мл метиленхлорида для удаления примесей, растворяют в 700 мл метанола (при нагревании до 55°С) и затем обесцвечивают с помощью 5 г угля Darco G-60. После фильтрования для удаления угля фильтрат концентрируют для кристаллизации продукта. Затем метанол удаляют добавлением 300 мл н-бутилацетата и концентрированием до густой взвеси кристаллов. Кристаллы отфильтровывают, промывают н-бутилацетатом и высушивают с получением 158 г очищенного кристаллического 5-андростен-3β,7β,11α-триол-17-она.
Получение соединения 1, способ 2: биоконверсия 5-андростен-3β-ол-17-она в 5-андростен-3β,7β,11α-триол-17-он
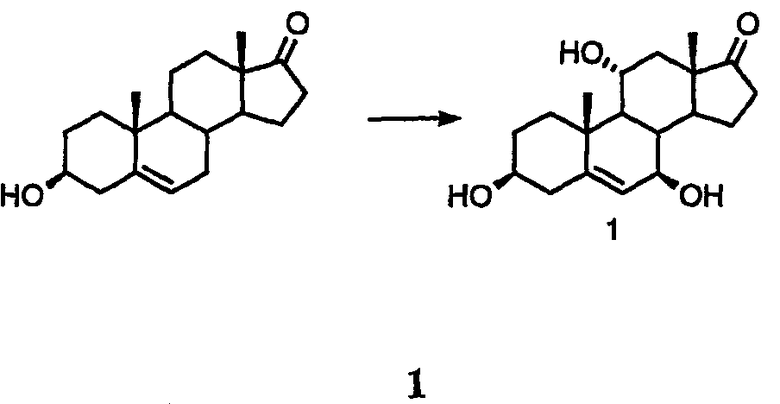
Биоконверсию 5-андростен-3β-ол-17-она в 5-андростен-3β,7β,11α-триол-17-он проводят с использованием глубинной культуры Absidia coerulea ATCC 6647 в ферментере емкостью 10 л.
(А) Стадии первичного посева
Культуры Absidia coerulea ATCC 6647 первичного посева готовят, как описано для Diplodia gossypina ATCC 20571 на стр. 14-15.
(В) Стадия вторичного посева
В ферментеры для вторичного посева емкостью 10 л вносят 1,2 мл вегетативной культуры первичного посева (количество внесенного посевного материала составляет 0,012% [об./об.]). Среда для вторичного посева состоит из (на литр воды RO): декстрина, 50 г; соевой муки, 35 г; церелозы, 5 г; хлорида кобальта гексагидрата, 2 мг; силиконового противовспенивателя (SAG 471), 0,5 мл; перед стерилизацией значение рН доводят до 4,95-5,00 концентрированной серной кислотой. Ферментеры, содержащие среду для вторичного посева, стерилизуют в течение 20 мин при 121°С с использованием как согревающего пара, так и введения пара. Скорость перемешивания во время стерилизации равняется 200 об/мин. После стерилизации рН среды доводят до 5,0 с использованием стерильной серной кислоты (5% раствора). Absidia coerulea ATCC 6647 инкубируют при 28°С с использованием следующих первоначальных параметров: перемешивание, 100 об/мин; противодавление = 5 фунтов/кв. дюйм избыточного давления; воздушный поток = 2,5 SLM (0,25 об./об/мин); нижнее значение DO, 30%; без контроля значения рН. Когда значение DO вначале уменьшается до 30%, то воздушный поток увеличивают до 5 SLM (0,5 об./об/мин). Когда культура вновь достигает нижнего значения DO, то значение DO поддерживают на 30%, регулируя перемешивание. Культуры вторичного посева собирают примерно через 76 ч после посева, когда значение СПК находится в пределах примерно от 4 до примерно 7 мМ/л/час.
(С) Биоконверсия стероидов
В ферментеры для биоконверсии стероидов емкостью 10 л вносят 500 мл вегетативной культуры второго посева (количество внесенного посевного материала составляет 5% [об./об.]). Среда для биоконверсии стероидов состоит из (на литр воды RO): декстрина, 50 г; соевой муки, 35 г; церелозы, 20 г; силиконового противовспенивателя (SAG 471), 0,5 мл; перед стерилизацией значение рН доводят до 2,95-3,00 концентрированной серной кислотой. Условия стерилизации являются такими же, как описаны для среды вторичного посева. После стерилизации рН среды доводят до 3,0 с использованием стерильной серной кислоты (5% раствор). Absidia coerulea ATCC 6647 инкубируют при 28°С с использованием таких же первоначальных параметров, которые использовали для культивирования вторичного посева. Примерно через 17 ч после посева в ферментер емкостью 10 л вносят 200 г микронизированного 5-андростен-3β-ол-17-она, разведенного в минимальном объеме 0,2% октилфеноксиполиэтоксиэтанола.
Культуры для биоконверсии анализируют ежедневно на выработку 5-андростен-3β,7β,11α-триол-17-она с использованием ТСХ, как описано в примере 1. Биоконверсия 5-андростен-3β-ол-17-она в 5-андростен-3β,7β,11α-триол-17-он заканчивается примерно через 6-7 суток после посева.
(D) Выделение
Твердые частицы в цельной культуральной жидкости отделяют центрифугированием. Жидкость отбрасывают. Обогащенные твердые частицы экстрагируют 10 л смеси 80% ацетон/15% вода при температуре 45°С-50°С и теплый экстракт осветляют фильтрованием. Обогащенный фильтрат концентрируют упариванием для удаления ацетона с получением водной суспензии сырых кристаллов. Сырые кристаллы отделяют фильтрованием и маточный раствор отбрасывают. Содержащие воду кристаллы растирают в 600 мл метиленхлорида для удаления примесей, растворяют в 700 мл метанола (при нагревании до 55°С) и затем обесцвечивают с помощью 5 г угля Darco G-60. После фильтрования для удаления угля фильтрат концентрируют для кристаллизации продукта. Затем метанол удаляют добавлением 300 мл н-бутилацетата и концентрируют до густой взвеси кристаллов. Кристаллы отфильтровывают, промывают н-бутилацетатом и высушивают с получением 75,5 г неочищенного кристаллического 5-андростен-3β,7β,11α-триол-17-она.
Сырые кристаллы растирают в 600 мл метиленхлорида для удаления дополнительных примесей, растворяют в 700 мл метанола (при нагревании до 55°С) и затем обесцвечивают 5 г угля Darco G-60. После фильтрования для удаления угля фильтрат концентрируют для кристаллизации продукта. Затем метанол удаляют добавлением 300 мл н-бутилацетата и концентрированием до густой взвеси кристаллов. Кристаллы отфильтровывают, промывают н-бутилацетатом и высушивают с получением 42,1 г очищенного кристаллического 5-андростен-3β,7β,11α-триол-17-она.
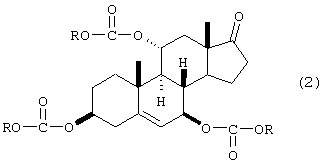
Пример 1: получение трикарбоната 2
К 250 мл 3 н. раствора в круглодонной колбе добавляют триол 1 (схема I) (10,00 г, 31 ммоль), растворенный в пиридине (100 мл). К данному раствору добавляют триэтиламин (31 мл, 218 ммоль), карбометоксибензотриазол (24,2 г, 125 ммоль) и 4-N,N-диметиламинопиридин (1,2 г, 9,4 ммоль). Суспензию перемешивают в течение 2 ч, в течение этого периода времени все вещества растворяются. Добавляют дополнительное количество карбометоксибензотриазола (12 г, 62 ммоль) и триэтиламина (10 мл, 73 ммоль). По растворении твердых частиц реакция заканчивается. Медленно добавляют воду (300 мл) и смесь охлаждают на ледяной бане. Осадок отфильтровывают и промывают 10% HCl (2×35 мл) и гексаном (3×50 мл) и высушивают в вакуумном сушильном шкафу в течение 24 ч с получением указанного в заголовке соединения 2 (схема I). С13 ЯМР (CDCl3) δ 217,78, 155,60, 155,23, 154,88, 144,48, 122,35, 78,58, 76,81, 75,39, 55,29, 54,93, 51,09, 49,47, 47,79, 38,48, 37,89, 36,19, 36,08, 27,96, 23,58, 19,07, 14,40.
Пример 2: получение фурана 3 (схема I)
Раствор трикарбоната 2 (1,0 г, 2,02 ммоль) в 7 мл ацетонитрила при комнатной температуре обрабатывают 2-метилфураном (0,2 мл, 2,22 ммоль) и 0,298 г Sc(OTf)3 в течение 1 ч. Данные ТСХ (смесь 30% EtOAc/Hex) показывают, что реакция закончилась. Хроматография на силикагеле со смесью 25% EtOAc/Hex дает 0,92 г (выход 96%) фурана 3. С13 ЯМР (CDCl3) δ 217,88, 171,08, 155,34, 154,93, 152,38, 151,49, 140,72, 123,98, 110,56, 106,45, 77,50, 75,89, 60,51, 54,98, 54,71, 47,45, 46,57, 38,73, 37,66, 36,21, 35,91, 27,96, 22,22, 19,14, 13,98, 13,77.
Пример 3: получение диола 4 (схема I)
Раствор дикарбоната 3 (1,0 г) в 10 мл МеОН обрабатывают 500 мг К2СО3 и подогревают до 40°С. Затем смесь перемешивают, пока данные ТСХ показывают, что реакция закончилась. По окончании суспензию выливают в воду и продукт выделяют EtOAc. Концентрирование органической фракции дает диол 4 в виде вязкого масла. 1Н ЯМР (CDCl3) δ 5,7(с, Н), 5,45(д, J=5,7 Гц, 1Н), 3,45(м, 1Н), 3,29(т, J=5,1 Гц, 1Н), 2,09(с, 3Н), 1,1(с, 3Н), 0,75(с, 3Н).
Пример 4: получение алкина 5 (схема I)
Раствор 2,8 г (25,0 ммоль) трет-BuOK в 50 мл ТГФ при -10°С барботируют ацетиленом в течение 30 мин. Затем медленно добавляют раствор кетона 11 в 10 мл ТГФ, продолжая барботирование ацетиленом. Смесь перемешивают при -10°С в течение 1 ч и затем добавляют 2,0 мл уксусной кислоты в 10 мл воды. Продукт выделяют экстракцией EtOAc после того, как смесь выливают в воду. Толуол используют для удаления уксусной кислоты азеотропной перегонкой. Данные ЯМР свидетельствуют о наличии небольших количеств уксусной кислоты и толуола; выход 2,25 г. С13 ЯМР (CDCl3) δ 153,77, 151,21, 142,66, 122,83, 110,12, 106,1, 87,3, 79,42, 74,03, 72,29, 69,48, 50,61, 47,49, 45,70, 43,64, 42,87, 39,38, 39,06, 38,29, 37,68, 31,84, 23,7, 21,26, 19,27, 14,19, 13,9.
Пример 5: получение лактола 6 (схема I)
Раствор 1,55 г алкина 5 (схема I), 27 мг Rh2(OAc)2 и 92 мг Ph3Р в 20 мл EtOAc находится под давлением 100 фунтов/кв. дюйм СО и 100 фунтов/кв. дюйм Н2 и его нагревают до 80°С в течение ночи. Смесь концентрируют и хроматографируют на силикагеле с использованием смеси 90% EtOAc/Hex с получением 2 фракций. Данные ЯМР показывают, что фракция 1 представляет собой выделенное исходное соединение. Фракция 2 является желаемым лактолом. Данные 13С ЯМР показывают сигналы смеси лактола при 94,8 и 94,5 м.д.
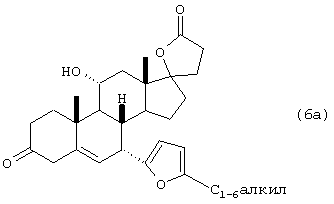
Пример 6: получение 5,6-енона 6а (схема I)
Смесь 2,0 г лактола, 50 мг KBr, 12 мг ТЕМРО, 800 мг NaHCO3 в 20 мл СН2Cl2 и 5 мл воды охлаждают до 5°С. Затем данную смесь медленно обрабатывают 8 мл 1,1М раствора NaOCl, поддерживая температуру ниже 6°С. После добавления смесь перемешивают еще в течение 30 мин и затем продукт выделяют с использованием СН2Cl2 с получением соединения 6а (схема I). С13 ЯМР (CDCl3) δ 209,82, 176,37, 153,19, 151,78, 143,34, 128,15, 121,41, 110,62, 106,53, 94,35, 72,0, 55,39, 50,44, 47,99, 44,41, 42,26, 39,03, 38,63, 37,1, 35,67, 31,9, 29,19, 23,39, 18,33, 15,69, 14,07.
Пример 7: получение 4,5-енона 7 (схема I) с использованием кислоты
Смесь 5,6-енона 6а (схема I) (500 мг) и щавелевой кислоты (200 мг) в этаноле (10 мл) нагревают при 40°С в течение 3 ч. Этанол удаляют при пониженном давлении и остаток растворяют в этилацетате (50 мл), органический раствор промывают водой (2×50 мл), высушивают над сульфатом натрия и концентрируют. Остаток очищают колоночной хроматографией на силикагеле с получением 4,5-енона 7 (схема I).
Пример 8: получение 4,5-енона 7 с использованием основания
Смесь 5,6-енона 6а (500 мг) и DBU (200 мг) в тетрагидрофуране (5 мл) кипятят с обратным холодильником в течение 3 ч, затем охлаждают, разбавляют раствором хлорида аммония и экстрагируют этилацетатом. Экстракт высушивают над сульфатом натрия и концентрируют. Остаток очищают колоночной хроматографией на силикагеле с получением 4,5-енона 7.
Пример 9: получение диенона 9 (схема I)
Раствор 1,2 г спирта 7 растворяют в 10 мл ТГФ и охлаждают до -33°С. Затем одной порцией добавляют PCl5 (950 мг). Раствор перемешивают в течение 3 ч при -33°С и затем быстро гасят добавлением воды. Продукт выделяют EtOAc с получением диена 9 (схема I). Его очищают хроматографией на силикагеле с использованием смеси EtOAc/гексан.
Пример 10: получение метиловых эфиров из фурановых заместителей
Способ А

Раствор производного фурана 8 (схема I) (1,0 г, 2,280 ммоль) в 100 мл метиленхлорида охлаждают до -79°С. Смесь О3/О2 пропускают через раствор в течение 10 мин, затем смесь подогревают до комнатной температуры и концентрируют до получения твердого остатка, который переводят в 50 мл смеси метанол/метиленхлорид 1/1, обрабатывают 1,0 мл пиридина и перемешивают при комнатной температуре в течение 18 ч. Затем раствор охлаждают до -80°С. Затем через раствор пропускают смесь О3/О2 в течение 4 мин. Затем смесь разбавляют 100 мл этилацетата и экстрагируют 70 мл водного раствора бикарбоната натрия. Водную фазу подкисляют водным раствором соляной кислоты до рН 0,5, затем экстрагируют метиленхлоридом и концентрируют до получения пены (масса: 250 мг). Пену растворяют в смеси толуол/метанол, обрабатывают триметилсилилдиазометаном (0,5 мл 2,0М раствора в гексане, 1,0 ммоль) при комнатной температуре, затем раствор концентрируют с получением эфира 9 в виде масла.
Способ В
Стадия 1) 5α,17β-Дигидроксипрегн-9-(11)-ен-3-он-7α,21-дикарбоновой кислоты бис-γ-лактон 8а (схема I)
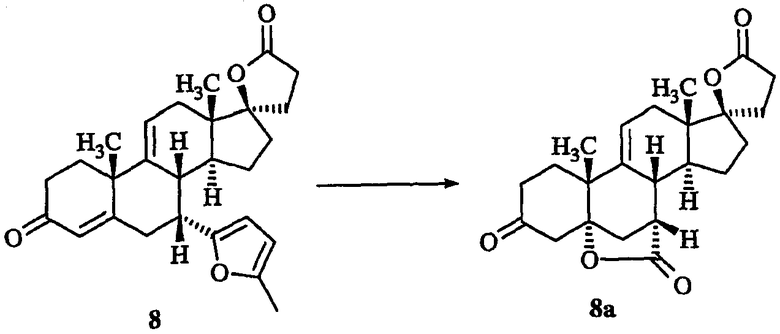
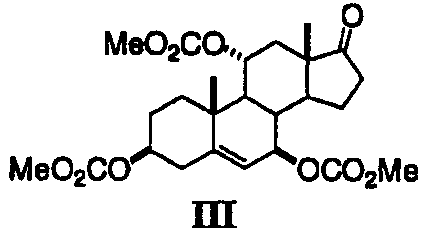
Смесь 17β-гидрокси-7α-(5'-метил-2'-фурил)-прегна-4,9(11)-диен-3-он-21-карбоновой кислоты γ-лактона (100 г, 0,23778 моль) и ацетата калия (50,0 г, 0,5094 моль, 2,14 экв) в ацетоне (500 мл) и воде (150 мл) охлаждают до -10°С и обрабатывают суспензией дибромантина (34,0 г, 0,1189 моль, 0,50 молярных экв) в воде (100 мл), пока не происходит повышение окислительно-восстановительного потенциала. В этой точке данные ЖХ указывают на полное превращение в ендион (III-цис). Затем реакционную смесь, содержащую ендион (III-цис), гасят изобутилвиниловым эфиром (1,0 мл, 0,768 г, 7,668 ммоль, 0,032 экв), концентрируют до получения густой суспензии, разбавляют метиленхлоридом (200 мл) и обрабатывают при 20°С концентрированной соляной кислотой (50,0 мл, 0,50 моль, 2,10 экв). Смесь перемешивают при 20-25°С в течение 2 ч, в это время данные ЖХ указывают на полное превращение в ендион (III-транс). Органическую фазу, содержащую ендион (III-транс), отделяют, разбавляют метиленхлоридом (80 мл) и метанолом (300 мл) и охлаждают до -48°С. Через данную смесь барботируют О3/О2, пока данные ЖХ не указывают на полное исчезновение ендиона (III-транс), затем смесь гасят диметилсульфидом (30,0 мл, 25,38 г, 0,4085 моль, 1,72 экв), перемешивают при -20°С в течение 16 ч, концентрируют до объема ≈300 мл, разбавляют метанолом (350 мл), концентрируют до объема примерно 300 мл, разбавляют изопропанолом (40 мл) и метанолом (80 мл), затем обрабатывают теплым (55-60°С) раствором бикарбоната калия (120 г, 1,1986 моль, 5,04 экв) в воде (240 мл). Данную суспензию охлаждают до 5-10°С, затем добавляют в течение 3 ч перекись водорода (50% раствор, 66,0 г, содержащий 33,0 г (0,9703 моль, 4,08 экв) перекиси водорода). Смесь перемешивают в течение 4 ч и гасят диметилсульфидом (40 мл, 33,84 г, 0,5447 моль, 2,29 экв). После перемешивания при 20-25°С в течение 23 ч смесь разбавляют метиленхлоридом (100 мл) и водой (80 мл) и подкисляют до рН 3,0 концентрированной соляной кислотой. Двухфазную смесь нагревают до 36°С, затем фазы разделяют и водную фазу экстрагируют метиленхлоридом (100 мл). Органические фазы объединяют, промывают водой (75 мл) и водную фазу экстрагируют обратно в метиленхлорид (25 мл). Органические фазы объединяют, концентрируют до объема 150 мл, затем обрабатывают бензолсульфоновой кислотой (1,0 г 90% чистого соединения, содержащего 0,90 г, (5,690 ммоль, 0,0239 экв) бензолсульфоновой кислоты) и ацетоном (50 мл). Затем смесь концентрируют при нормальном давлении до объема 160 мл, затем разбавляют ацетоном (250 мл), концентрируют до объема 200 мл, охлаждают до 12°С и фильтруют. Осадок на фильтре промывают холодным ацетоном (2×25 мл) и сушат в атмосфере азота с получением указанного в заголовке соединения. 13С ЯМР (100 МГц, CDCl3) δ 206,08, 176,47, 175,41, 139,63, 124,00, 94,89, 90,97, 47,08, 43,90, 42,36, 41,58, 41,07, 38,93, 36,97, 35,16, 33,01, 32,42, 32,42, 31,35, 29,10, 23,08, 22,98 и 14,23; ЯМР (400 МГц, CDCl3) 0,94, 1,40, 1,4-2,8 и 5,70; МС (CI, NH3) m/е = 385 (Р + Н, 100%).
Стадия 2) 17β-гидрокси-7α-карбометоксипрегна-4,9(11)-диен-3-он-21-карбоновой кислоты γ-лактон 9 (схема I)
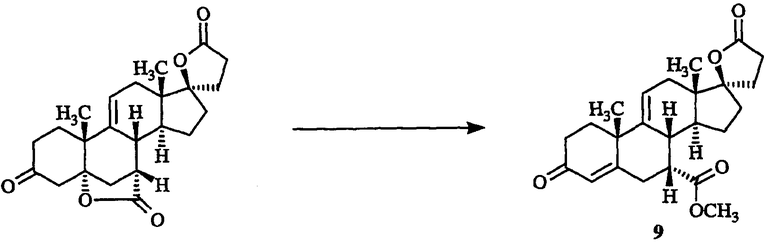
Смесь 5α,17β-дигидроксипрегн-9(11)-ен-3-он-7α,21-дикарбоновой кислоты бис-γ-лактон (50,0 г, 0,13005 моль) и бикарбоната калия (16,92 г, 0,1690 моль, 1,3 экв) в ацетоне (200 мл) и воде (100 мл) перемешивают при 45°С в течение 2 ч, по данным ЖХ в это время заканчивается превращение 5,7-лактона (VII) в карбоновую кислоту (VI). Затем полученную смесь обрабатывают диметилсульфатом (22,92 г, 0,1817 моль, 1,40 экв), перемешивают при 45°С в течение 3 ч, затем обрабатывают раствором бикарбоната калия (1,3 г, 0,0130 моль, 0,100 экв) в воде (10 мл) с последующей обработкой неразбавленным триэтиламином (1,81 мл, 1,314 г, 0,0130 моль, 0,100 экв). Смесь перемешивают при 45°С в течение 1 ч, гасят концентрированной соляной кислотой (1,92 мл, 2,304 г, содержащие 0,852 г (0,0234 моль, 0,180 экв) соляной кислоты), охлаждают до 0°С, концентрируют при пониженном давлении до объема 150 мл (температура резервуара 13°С), затем фильтруют и остаток на фильтре промывают водой (2×25 мл) и высушивают с получением указанного в заголовке соединения 9 (схема I).
Пример 11: получение эплеренона
Диенон 9 (схема I) окисляют, как описано в патентах США № 4559332 и 5981744 и заявках WO97/21720 и WO98/25948 с получением эплеренона.
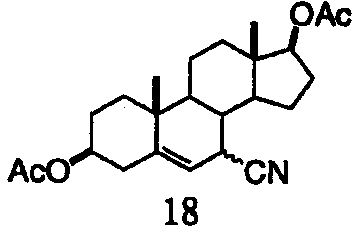
Пример 12: аллилирование триметилсилилцианида с использованием соединения I
Раствор триацетата I (таблица 1) (1,0 г, 2,24 ммоль) в 10 мл СН2Cl2 охлаждают до 14°С и обрабатывают 0,6 мл триметилсилилцианида (TMSCN) и 100 мг Sc(OTf)3. Смесь перемешивают в течение 5 ч и экстрагируют этилацетатом. Во время концентрирования экстракта из раствора выпадают кристаллы. Их отфильтровывают и высушивают с получением нитрила 18 в виде смеси изомеров. 13С ЯМР (CDCl3, в виде смеси) δ 147,31, 146,0, 131,68, 129,39, 128,12, 119,23, 115,47, 115,04, 62,74, 82,50, 51,13, 49,0, 47,72, 44,38, 43,67, 43,05, 37,32, 37,04, 36,32, 33,58, 32,09, 32,0, 27,92, 27,79, 26,75, 23,68, 23,32, 20,45, 19,13, 18,26, 12,30.
Пример 13: аллилирование аллилтриметилсилана с использованием соединения V
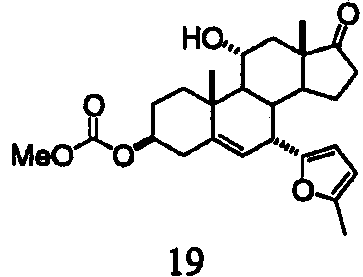
Раствор триацетата V (таблица 1) и аллилтриметилсилана в ацетонитриле обрабатывают при комнатной температуре Sc(OTf)3. Через 1 ч медленно добавляют воду для осаждения продукта. Фильтрование и высушивание дают аллильное производное 19. 13С ЯМР (CDCl3) δ 221,05, 170,89, 193,87, 137,62, 127,15, 116,36, 74,26, 47,81, 46,29, 38,61, 37,49, 36,23, 35,61, 35,31, 31,57, 28,04, 22,61, 20,56, 19,64, 13,48.
Пример 14: добавление ацетилена к 17-оксо промежуточным соединениям
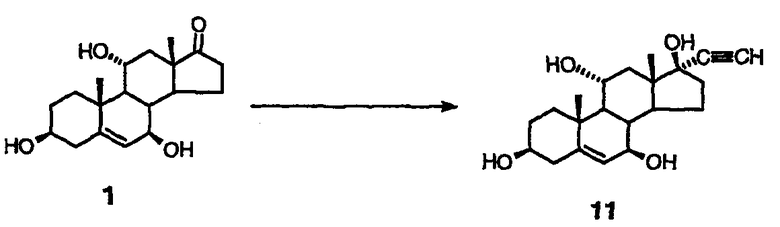
Стадия 1:
Гексаметилдисилазан (HMDS) (100 мл) добавляют к перемешиваемой суспензии 50,0 г триола 1 в 400 мл метиленхлорида. Добавляют сахарин (0,57 г) и смесь кипятят с обратным холодильником в течение 3 ч, в продолжение этого периода времени суспензия постепенно превращается в прозрачный янтарный раствор. Добавляют воду (5 мл) для гашения любого избытка HMDS. После кипячения с обратным холодильником в течение 5 мин смесь фильтруют через пропитанный СН2Cl2 слой 32,6 г магнезоля на воронке с фильтром-фриттой для грубой очистки емкостью 350 мл. Фильтрат должен быть прозрачным и почти бесцветным. Осадок на фильтре промывают 2×100 мл СН2Cl2. Объединенные фильтраты концентрируют при пониженном давлении и остаточное количество метиленхлорида удаляют выпариванием порциями 2×500 мл тетрагидрофурана (ТГФ), концентрируют досуха после каждого добавления с получением белого твердого вещества.
Стадия 2:
Суспензию трет-бутилата калия (42,0 г) в 500 мл ТГФ охлаждают до -9°С±5°С на бане со льдом/метанолом. В смесь барботируют ацетилен непосредственно под поверхностью при умеренном перемешивании в течение, по меньшей мере, 1 ч. Добавляют вышеуказанное силилированное стероидное промежуточное соединение в ТГФ (400 мл) в течение 30 мин, поддерживая температуру реакционной смеси при 0°С±5°С. После добавления смесь перемешивают еще в течение часа при 5°С±5°С. Медленно добавляют воду (100 мл), давая смеси нагреться до 15°С±5°С. Медленно добавляют 125 мл 10% HCl для снижения значения рН до 2,5-3. Смесь перемешивают при рН 2,5-3, добавляя при необходимости небольшие количества 5% HCl для поддержания значения рН в пределах от 2,5 до 3, в течение 1-2 ч при 20°С±5°С. Когда гидролиз завершается, добавляют полунасыщенный раствор NaHCO3 для повышения рН до 5,5-6. Смесь разбавляют этилацетатом (500 мл) и фазы разделяют. Водную фазу экстрагируют этилацетатом и объединенные этилацетатные фазы промывают водой, насыщенным раствором соли, высушивают над сульфатом магния и концентрируют с получением аддукта 2. 13С ЯМР (ДМСО-d6) δ 141,99, 127,38, 89,37, 77,73, 75,24, 72,13, 70,54, 67,68, 54,13, 49,57, 47,43, 43,94, 42,58, 40,52, 40,22, 39,01, 38,09, 31,95, 25,8, 18,58, 14,09.
Пример 15: гидроксиацетилирование
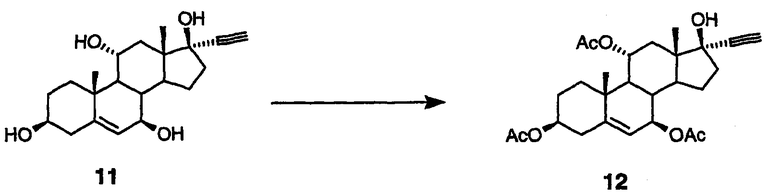
Смесь тетраола 11 (схема II) (50,00 г, 144 ммоль), растворенную в пиридине (150 мл), охлаждают до < 10°С на ледяной бане. Добавляют диметиламинопиридин (DMAP) (1,7 г, 14 ммоль) с последующим медленным добавлением уксусного ангидрида (41,4 мл, 439 ммоль) со скоростью, достаточной для поддержания температуры раствора ниже 10°С. После добавления реакционную смесь подогревают до комнатной температуры. Смесь разводят этилацетатом (75 мл) и водой (50 мл), перемешивают в течение 5 мин и слои разделяют. Органический слой промывают 10% HCl (4×25 мл) с последующим промыванием Н2О (2×50 мл), высушивают над MgSO4 и концентрируют. Продукт перекристаллизовывают из толуола (100 мл). 13С ЯМР (CDCl3) δ 170,68, 170,10, 143,48, 128,90, 128,10, 125,17, 122,59, 86,63, 78,21, 75,07, 74,40, 72,79, 71,47, 50,16, 48,07, 47,02, 38,76, 37,83, 37,67, 36,92, 27,66, 24,18, 21,74, 21,44, 18,65, 13,06.
Пример 16: гидроформилирование аддуктов ацетилена
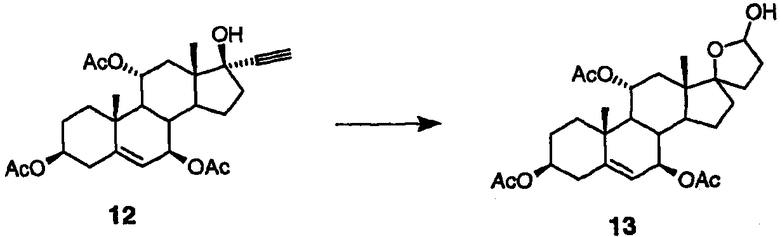
Раствор триацетата 12 (схема II) (25,4 г, 54 ммоль), PPh3 (2,13 г, 8,1 ммоль) и Rh2(ОАс)4 (716 мг, 1,62 ммоль) в этилацетате (200 мл) нагревают при 80°С в смеси водород/окись углерода 1/1 под давлением 170 фунтов/кв. дюйм в течение 12 ч. Смесь концентрируют при пониженном давлении и продукт 13 (схема II) очищают колоночной хроматографией (смесь EtOAc/Hex 70/30 и 500 г силикагеля). ЯМР-спектр данного соединения был сложным в результате наличия изомеров с открытым кольцом и закрытым кольцом и не был полностью охарактеризован.
Пример 17: окисление лактола в лактон
Смесь лактола 4 (схема I) (25 г, 50 ммоль), метиленхлорида (250 мл), воды (38 мл), 2,2,6,6-тетраметилпиперидин-1-оксил (ТЕМРО) (156 мг, 1 ммоль), KBr (595 мг, 5 ммоль) и NaHCO3 (5,5 г, 65 ммоль) охлаждают до ≤ 10°С на ледяной бане. Медленно добавляют 1,1М раствор гипохлорита натрия (NaOCl) (50 мл, 55 ммоль). Смеси дают нагреться до комнатной температуры и разводят водой (50 мл). Слои разделяют и органический слой промывают насыщенным раствором соли (2×50 мл). Органический слой высушивают над MgSO4, фильтруют и концентрируют с получением соединения 5 в виде не совсем белой пены. 13С ЯМР (CDCl3) δ 177,94, 172,60, 172,15, 171,58, 145,49, 124,36, 96,18, (79,22, 78,90, 78,59, CDCl3), 76,59, 74,57, 72,63, 52,14, 49,55, 47,75, 40,00, 39,75, 39,61, 38,65, 37,47, 32,74, 30,85, 29,56, 26,01, 23,61, 23,37, 23,17, 23,11, 20,52, 16,19.
Пример 18: фуранилирование
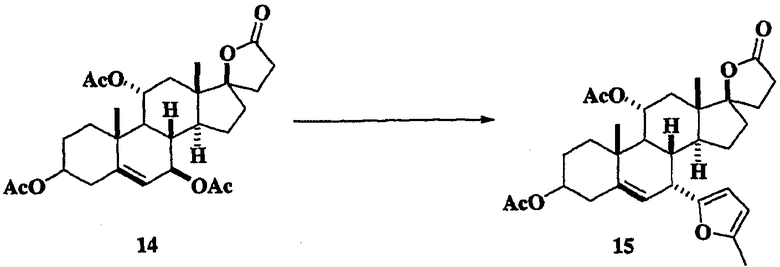
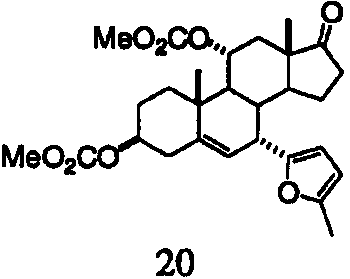
Раствор триацетата 14 (схема I) (1,30 г, 2,58 ммоль), 2-метилфурана (0,8 мл) в 25 мл ацетонитрила при 20°С обрабатывают 250 мг Sc(OTf)3 и перемешивают в течение 1 ч. Продукт выделяют экстракцией EtOAc и очищают хроматографией на силикагеле смесью 40% EtOAc/Hex с получением 1,0 г (выход 74%) фурана 15. 13С ЯМР (CDCl3) δ 176,27, 170,45, 169,85, 152,53, 150,96, 140,60, 123,45, 110,05, 106,01, 94,95, 73,39, 71,37, 46,50, 45,40, 44,60, 38,55, 38,37, 38,06, 37,78, 37,74, 36,89, 35,41, 31,81, 30,72, 28,96, 28,93, 27,69, 23,07, 22,63, 21,74, 20,98, 18,80, 14,83, 14,13, 14,06, 13,62.
Пример 19: гидролиз ацетата
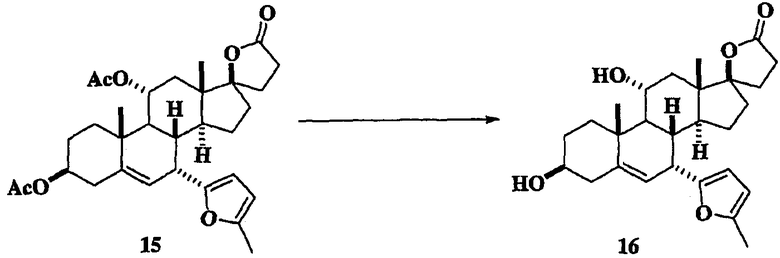
Смесь 810 мг диацетата 15 (схема II), 112 мг К2СО3 в 20 мл метанола перемешивают при комнатной температуре. Данные ТСХ указывают на то, что реакция не закончилась и, таким образом, добавляют еще 100 мг К2СО3 и перемешивание продолжают, пока данные ТСХ не покажут, что реакция закончилась. Смесь подкисляют 1М раствором HCl и продукт экстрагируют EtOAc. Хроматография на силикагеле с 100% EtOAc дает 610 мг (выход 89,5%) диола 16 (схема II). 13С ЯМР (CDCl3) δ 176,68, 153,20, 150,79, 142,05, 122,32, 109,80, 105,94, 95,3, 71,83, 68,64, 50,13, 45,81, 44,88, 42,73, 42,62, 39,01, 38,56, 37,73, 36,81, 35,44, 31,57, 30,84, 29,06, 23,13, 18,81, 15,27, 13,64.
Пример 20: окисление соединения 16 с получением соединения 17
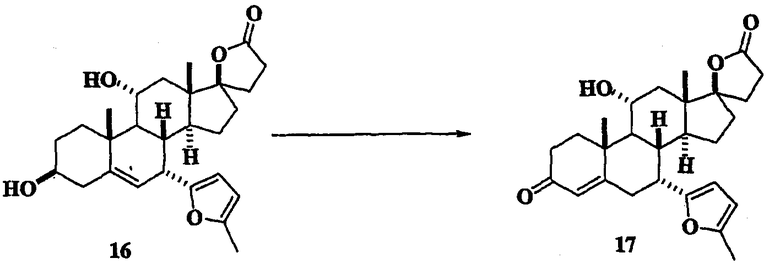
Диол 16 (схема II) растворяют в 2 мл толуола и 0,1 мл ацетона и обрабатывают 50 мг изопропилата алюминия и нагревают до 100°С. Через несколько часов оказывается, что превращения не произошло, так что добавляют 0,1 мл циклогексанона и смесь нагревают в течение ночи. Продукт 17 (схема II) выделяют хроматографией на силикагеле с использованием этилацетата в качестве элюента. 13С ЯМР (CDCl3) δ 199,96, 177,05, 170,32, 153,34, 150,74, 126,7, 109,14, 106,33, 95,54, 69,08, 52,50, 46,26, 46,0, 43,58, 40,13, 39,05, 38,8, 37,93, 36,92, 35,66, 34,59, 31,33, 29,47, 23,06, 18,83, 16,01, 13,90.
| название | год | авторы | номер документа |
|---|---|---|---|
| 7-КАРБОКСИЗАМЕЩЕННЫЕ СТЕРОИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ ЭПЛЕРЕНОНА | 2003 |
|
RU2304144C2 |
| ПРОИЗВОДНЫЕ АНДРОСТЕН(АН)ОНА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЙ СОЛЬВАТ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ, СПОСОБ ЛЕЧЕНИЯ | 1994 |
|
RU2144037C1 |
| СПОСОБ ПОЛУЧЕНИЯ 17-(3-ГИДРОКСИПРОПИЛ)-17-ГИДРОКСИСТЕРОИДОВ | 2008 |
|
RU2466137C2 |
| СПОСОБ ПОЛУЧЕНИЯ 3-КЕТО-7α-АЛКОКСИКАРБОНИЛЗАМЕЩЕННОГО Δ4,5-СТЕРОИДА, СПОСОБЫ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1997 |
|
RU2261865C2 |
| СПОСОБ ПОЛУЧЕНИЯ 7β-ЗАМЕЩЕННЫХ 4-АЗАαАНДРОСТАН-3-ОНОВ И СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ 7β-АЛКИЛ-АНДРОСТ-5-ЕН-3-ОНОВ | 1993 |
|
RU2114117C1 |
| ПОЛУГИДРАТ 16 АЛЬФА-БРОМО-3 БЕТА-ГИДРОКСИ-5 АЛЬФА-АНДРОСТАН-17-ОН, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, ПРИМЕНЕНИЕ СОЕДИНЕНИЙ | 2000 |
|
RU2295534C2 |
| КОНДЕНСИРОВАННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СУКЦИНИМИДНЫЕ СОЕДИНЕНИЯ | 2001 |
|
RU2298554C2 |
| СПОСОБ ПОЛУЧЕНИЯ 7α-МЕТИЛСТЕРОИДОВ, СОЕДИНЕНИЕ | 2003 |
|
RU2305105C2 |
| СТЕРОИДНОЕ СОЕДИНЕНИЕ | 1995 |
|
RU2160279C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ЭПЛЕРЕНОНА | 2003 |
|
RU2339642C9 |
Описан способ получения новых 7-замещенных стероидных соединений общей формулы I, где
R1-Н или -COR2; где R2-С1-6алкил, С1-6алкокси, Z1--СН2- или 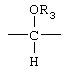 , где R3 находится в α-конфигурации; R3-Н или -COR2, Z2-CH- или Z1 и Z2 вместе
, где R3 находится в α-конфигурации; R3-Н или -COR2, Z2-CH- или Z1 и Z2 вместе
- двойная связь, Q - 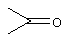
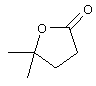
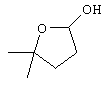
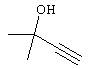
Y--CN, -СН2-СН=СН2 или 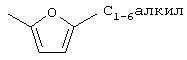
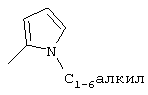 ,
,
CHR4C(O)Ar, CHR4С(O)С1-6алкил, CHR4C(O)XAr или CHR4C(O)XC1-6алкил, где R4-OC1-6алкил или арил, Х=O или S, являющихся промежуточными соединениями в синтезе эплеренона.
6 н. и 1 з.п. ф-лы, 1 табл.
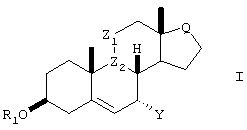
в которой R1 представляет собой Н или -COR2;
Z1 является СН2 или 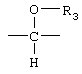 , где OR3 находится в α-конфигурации;
, где OR3 находится в α-конфигурации;
R3 представляет собой Н или -COR2;
R2 представляет собой C1-6алкил или С1-6алкокси;
Z2 является -СН-; или
Z1 и Z2, взятые вместе, образуют углерод-углеродную двойную связь;
Q представляет собой
 ,
,  ,
, 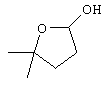 или
или 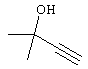 ;
; 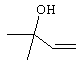 ;
;
Y является -CN, -СН2-СН=СН2 или
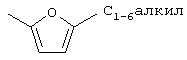 ,
, 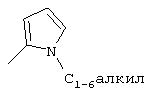 ,
,
CHR4C(O)Ar, CHR4С(O)С1-6алкил, CHR4C(O)XAr или CHR4C(O)XC1-6алкил;
где R4 - ОС1-6алкил или арил,
арил Ar - ароматический заместитель, состоящий из 1-3 колец, сконденсированных или ковалентно связанных между собой,
Х - О или S,
включающий взаимодействие стероидного промежуточного соединения формулы II
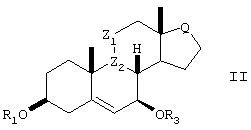
в которой R1 и R3, Z1, Z2, R2 и Q имеют значения, определенные для формулы I,
с нуклеофильным реагентом, выбранным из группы, состоящей из C1-4триалкилсилилцианида, С1-4триалкилсилиленолэфира, триалкилсилилоксикетентиоацеталя RCH=C(OSiR3)SR, аллилтри-C1-4алкилсилана, аллилтри-С1-4алкилстаннана, 2-С1-4аллилфурана и 2-C1-4алкилпиррола, в присутствии катализатора - кислоты Льюиса.
R1 представляет собой Н или -COR2;
Z1 является СН2 или , где OR3 находится в α-конфигурации;
R3 представляет собой Н или -COR2;
R2 представляет собой С1-6алкил или С1-6алкокси;
Z2 является -CH-; или Z1 и Z2, взятые вместе, образуют углерод-углеродную двойную связь;
Q представляет собой
, , или ; ;
Y является -CN, -CH2-CH=СН2,
,
а) взаимодействия кетостероида формулы 1
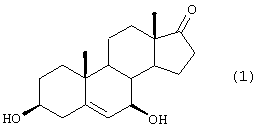
с C1-С6алкилхлорформиатом, или бензилхлорформиатом, или алкоксикарбонилбензотриазолом в присутствии третичного органического основания с получением трикарбоната формулы 2
в которой R представляет собой C1-С6алкил или бензил;
b) взаимодействия триацилсоединения формулы 2 с 2-C1-6алкилфураном в присутствии катализатора - кислоты Льюиса с получением диацилэфира формулы 3
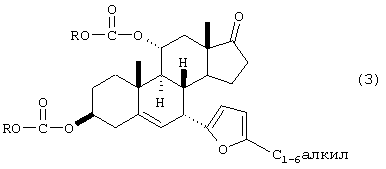
с) гидролиза диацилэфира формулы 3 в присутствии основания с получением дигидроксиэфира формулы 4
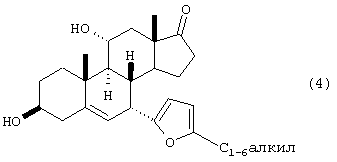
d) взаимодействия соединения формулы 4 с ацетиленом в присутствии сильного основания с получением ацетиленового соединения формулы 5
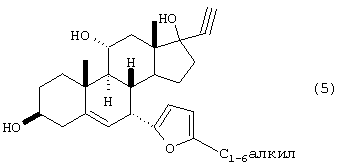
е) взаимодействия ацетиленового соединения формулы 5 с окисью углерода в присутствии лиганда родиевого катализатора с получением лактола формулы 6
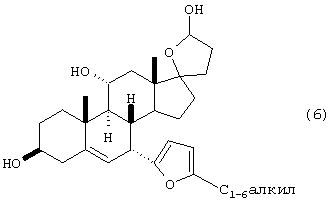
f) окисления лактола формулы 6 с получением лактона формулы 6а
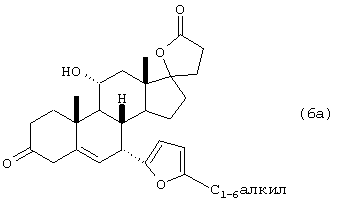
g) изомеризации 4,5-двойной связи в соединении 6а с получением лактона формулы 7
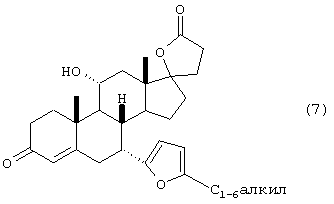
h) бромирования, озонирования, окисления и эстерификации соединения формулы 7 с получением сложного эфира формулы 8
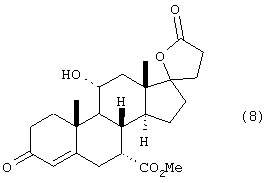
i) дегидратации соединения формулы 8 с получением промежуточного соединения формулы 9
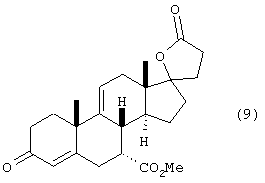
j) окисления диенона формулы 9, посредством чего получают эплеренон формулы 10
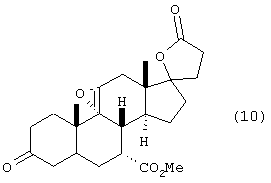
в которой R1 представляет собой Н или -COR2;
Z1 представляет собой СН2 или , где OR3 находится в α-конфигурации;
R3 представляет собой Н или -COR2;
R2 представляет собой C1-6алкил или C1-6алкокси;
Z2 является -СН-; или
Z1 и Z2, взятые вместе, образуют углерод-углеродную двойную связь;
Q представляет собой
, , или ; ;
с нуклеофильным реагентом, выбранным из группы, состоящей из C1-4триалкилсилилцианида, С1-4триалкилсилиленоэфира, триалкилсилилоксикетентиоацеталя RCH=С(OSiR3)SR, аллилтри-C1-4алкилсилана, аллилтри-С1-4алкилстаннана, 2-С1-4аллилфурана и 2-C1-4алкилпиррола, в присутствии катализатора - кислоты Льюиса.
а) взаимодействия соединения формулы 1
с ацетиленом с получением соединения формулы 11
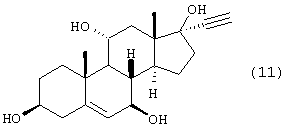
b) ацилирования соединения формулы 11 с получением соединения формулы 12
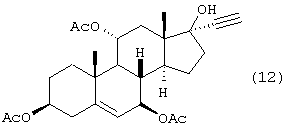
с) гидроформилирования соединения формулы 12 с получением соединения формулы 13

d) окисления соединения формулы 13 с получением соединения формулы 14
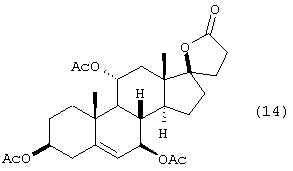
е) взаимодействия соединения формулы 14 с 2-алкилфураном в присутствии кислоты Льюиса с получением соединения формулы 15
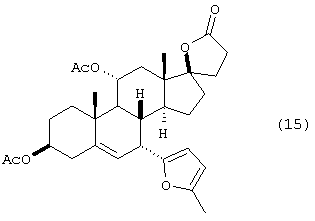
f) гидролиза соединения формулы 15 с получением соединения формулы 16
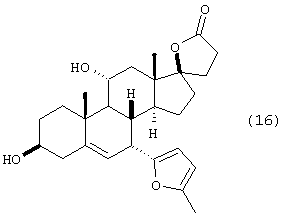
g) окисления соединения формулы 16 с получением соединения формулы 17
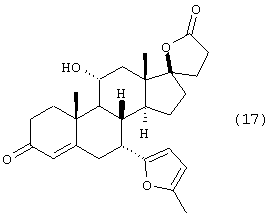
h) превращения фуранового кольца соединения формулы 17 в метоксикарбонильное соединение формулы 18
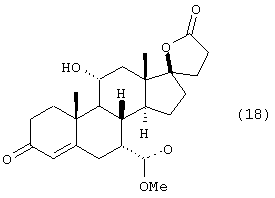
i) превращения соединения формулы 18 в сульфонатный сложный эфир формулы 19
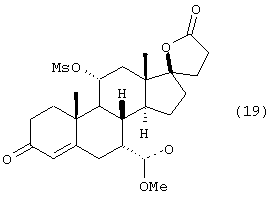
j) удаления сульфонатного сложного эфира формулы 19 с получением соединения формулы 9
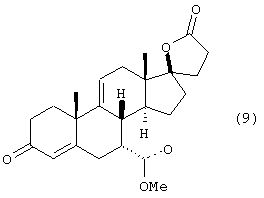
k) окисления соединения формулы 9 с получением эплеренона формулы 10
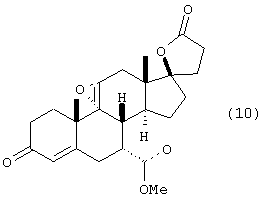
| US 5227375 A, 13.07.1993 | |||
| WO 9721720, A, 19.06.1997 | |||
| US 4559332, A, 17.12.1985. |

