Область изобретения
Настоящее изобретение относится к оксазоло-, тиазоло- и селеназоло[4,5-с]хинолин-, тетрагидрохинолин-4-аминам и их аналогам, а также к промежуточным соединениям, используемым для синтеза этих продуктов. Изобретение относится также к фармацевтическим композициям, состоящим из указанных выше соединений, а также к применению этих соединений в качестве иммуномодуляторов и для стимулирования цитокинетического биосинтеза, включая биосинтез интерферона-α и(или) биосинтез фактора-α некроза опухоли.
Предпосылки к созданию изобретения и аналоги
В первом отчете по синтезу 1Н-имидазо[4,5-с]хинолиновой циклической системы Васkman и др., I. Org. Сhеm. 15, 1278-1284 (1950), сообщили о синтезе 1-(6-метокси-8-хинолинил)-2-метил-1Н-имидазо[4,5-с]хинолина и о возможном использовании этого соединения в качестве антималярийного препарата. В дальнейшем сообщалось о синтезе различных замещенных 1Н-имидазо[4,5-с]хинолинов. Например, Jаin и др., J. Меd. Сhеm. 11, 87-92 (1968), синтезировали соединение 1-[2-(4-пиперидил)этил]-1Н-имидазо[4,5-с]хинолин в качестве возможного противосудорожного и сердечно-сосудистого средства. Кроме того, Баранов и др., Сhет.Аbs. 85, 94362 (1976), и Веrеnуi и др., J. Неtеrосусlic Сhеm. 18, 1537-1540 (1981), также сообщали о синтезе некоторых 2-оксоимидазо[4,5-с]хинолинов.
На основании выше перечисленных работ было установлено, что 1Н-имидазо[4,5-с]хинолин-4-амины и 1- и 2-замещенные производные этих соединений являются эффективными антивирусными средствами, бронхолитическими средствами и иммуномодуляторами. Такие соединения описаны в патентах США №№ 4689338, 4698348, 4929624, 5037986, 5266675, 5268376, 5346905, 5389640, 5605899, 5352784, 5446153 и 5482936. Shen и др. в патентах США №№ 4038396 и 4131677 описывают некоторые оксазоло- и тиазолопиридины, обладающие антивоспалительными, обезболивающими и жаропонижающими свойствами.
Краткое изложение существа изобретения
Оксазоло- и тиазоло[4,5-с]хинолин-4-амины формулы I:

отличающиеся тем, что
заместитель R1 выбран из группы, состоящей из атомов кислорода и серы;
заместитель R2 выбран из группы, состоящей из
- атома водорода;
- алкила;
- алкил-ОН (гидроксиалкила);
- алкил-Х-алкила;
- алкил-O-С(O)-N(R5)2;
- морфолинила, пирролидинила;
- алкил-Х-арильного радикала;
- алкенил-Х-арильного радикала;
каждый из заместителей R3 и R4 независимо представляет собой атом водорода или взятые вместе заместители R3 и R4 образуют конденсированную ароматическую или [1,5]нафтиридиновую систему;
Х представляет собой -O- или простую связь;
R5 представляет собой атом водорода,
а также фармацевтически приемлемые соли на основе этих соединений.
Вторым предметом настоящего изобретения являются фармацевтические композиции, содержащие терапевтически эффективные количества соединения общей формулы I(а) и фармацевтически приемлемый наполнитель.
Фармацевтические композиции, для стимулирования образования цитокина в человеческих клетках, содержащие терапевтически эффективное количество соединения общей формулы I(а):

где заместитель R1 выбран из группы, состоящей из атомов кислорода и серы;
заместитель R2 выбран из группы, состоящей из
- атома водорода;
- алкила;
- алкил-ОН (гидроксиалкила);
- алкил-Х-алкила;
- алкил-O-С(O)-N(R5)2;
- морфолинила, пирролидинила;
- алкил-Х-арильного радикала;
- алкенил-Х-арильного радикала;
каждый из заместителей R3 и R4 независимо представляет собой атом водорода или взятые вместе заместители R3 и R4 образуют конденсированную ароматическую или [1,5]нафтиридиновую систему;
Х представляет собой -O- или простую связь;
R5 представляет собой атом водорода,
а также фармацевтически приемлемые соли на основе этих соединений.
В изобретении описаны также фармацевтически приемлемые соли на основе соединений I(а).
Соединения общей формулы I(а) способны стимулировать цитокинетический биосинтез в организме животных и, в частности, в организме человека. Цитокинез, который может быть стимулирован указанными в изобретении соединениями, включает в себя, но не ограничен только этими примерами, биосинтез интерферонов, в частности интерферона-α и фактора-α некроза опухоли. В связи с этим в изобретении описан также способ стимулирования цитокинетического биосинтеза в животном путем введения в организм животных эффективного количества композиции, содержащей в своем составе соединение формулы I(а). Благодаря своей способности стимулировать цитокинетический биосинтез приведенные в изобретении соединения являются эффективными препаратами для лечения различных заболеваний, включая вирусные заболевания и различные опухоли. Кроме того, изобретение предоставляет способ лечения таких заболеваний путем введения в организм терапевтически эффективных количеств композиции, в состав которой входит соединение формулы I(а).
Еще одним предметом настоящего изобретения являются промежуточные соединения общей формулы II
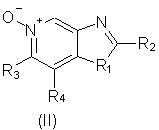
отличающиеся тем, что
заместитель R1 выбран из группы, состоящей из атомов кислорода и серы;
заместитель R2 выбран из группы, состоящей из
- алкила;
- алкила-ОН;
- алкил-Х-алкила;
- алкил-Х-арила;
- морфолинила;
каждый из заместителей R3 и R4 независимо представляет собой атом водорода или взятые вместе заместители R3 и R4 образуют конденсированную ароматическую или [1,5]нафтиридиновую систему;
Х представляет собой -O- или простую связь.
Подробное описание изобретения
Настоящее изобретение включает в себя соединения общей формулы I, фармацевтические композиции, содержащие соединения общей формулы I(а), и терапевтические способы использования соединений формулы I(а), а также промежуточные соединения общей формулы II, которые используются для синтеза соединений формул I и Iа.
Применяемые в данном описании термины “алкил” и “алкенил” относятся к линейной или разветвленной углеводородной группе или к циклической группе (например, циклоалкил и циклоалкенил), которая содержит от 1 до 20, предпочтительно от 1 до 10 и наиболее предпочтительно от 1 до 8, углеродных атомов, если не указано иначе. Типичными алкильными группами являются, например, метильная, этильная, н-пропильная, изопропильная, н-бутильная, изобутильная, втор-бутильная, трет-бутильная, н-пентильная, н-гексильная, н-гептильная, н-октильная и другие подобные группы. Примерами циклических групп являются циклопропильная, циклопентильная, циклогексильная, циклогексенильная и адамантильная группы. В тех случаях, когда используется суффикс “алк”, например для “алкокси” и для других подобных случаев, он имеет такое же значение.
Термин “арил” (“арильный радикал”) относится к карбоциклическому ароматическому циклу или циклической системе. Арильная группа предпочтительно представляет собой шестичленное кольцо, такое как фенильная группа, или ароматическую полициклическую систему, такую как нафтильная группа. Наиболее предпочтительной арильной группой является фенильная группа, которая может быть незамещенной или замещенной одним или большим количеством заместителей, как это указано ниже. Другими примерами арильных групп являются бифенильная, флуоренильная и инденильная группировки.
Подходящие гетероарильные группы включают в себя фурильную, тиенильную, пиридильную, хинолинильную, тетразолильную, имидазольную группировки и т.п. В тех случаях, когда заместители R3 и R4 используются вместе и образуют пяти- или шестичленные гетероароматические циклы, в качестве гетероатома используют азот, кислород или серу, а цикл может содержать один или большее количество таких атомов. Предпочтительно в качестве гетероатома используют азот или серу. Предпочтительные гетероароматические циклы, образованные с участием R3 и R4, проиллюстрированы следующими формулами, в которых две линии указывают место, в котором происходит их конденсация.

Термины “гетероциклический” и “гетероциклический радикал” относятся к неароматическим циклам или циклическим системам, содержащим один или большее количество гетероатомов (например, O, S, N). К примерам гетероциклических групп относятся пирролидинильная, тетрагидрофуранильная, морфолинильная, пиперидиновая, пиперазиновая, тиазолидинильная, имидазолидильная и другие подобные группы.
Все указанные выше циклы или циклические системы могут быть незамещенными или содержать один или большее количество заместителей, выбранных из группы, состоящей из таких радикалов, как алкильный, алкоксильный, алкилтиорадикал, гидроксильный, галоидный атом, галоидоалкильный, полигалоидоалкильный, пергалоидоалкильный (например, трифторметильный), трифторалкоксильный (например, трифторметоксильный), нитрильный, аминорадикал, алкиламинный, диалкиламинный, алкилкарбонильный, алкенилкарбонильный, арилкарбонильный, гетероарилкарбонильный, арильный, арилалкильный, гетероарильный, гетероарилалкильный, гетероциклильный, гетероциклоалкильный, нитрильный и алкоксикарбонильный. Предпочтительными заместителями являются С1-4 алкильный, С1-4 алкоксильный, галоидный атом, аминный, алкиламинный, диалкиламинный, гидроксильный, С1-4алкоксиметильный и трифторметильный радикалы.
Термин “галоидо” относится к галоидному атому, такому, например, как фтор, хлор, бром или иод.
Настоящее изобретение включает в себя описанные здесь соединения в любой их фармацевтически приемлемой форме, включая изомеры, такие как диастереомеры и энантиомеры, соли, сольваты, различные полиморфные состояния и т.п.
Как отмечалось выше, соединения, имеющие формулу I и I(а), способны образовывать “фармацевтически приемлемые соли”. Фармацевтически приемлемые соли соединений I и I(а) включают соли, полученные из таких нетоксичных неорганических кислот как соляная, азотная, фосфорная, серная, бромистоводородная, иодистоводородная, фтористоводородная, фосфористая кислоты, а также соли, полученные из таких нетоксичных органических кислот, как алифатические моно- и дикарбоновые кислоты, фенилзамещенные жирные кислоты, гидроксиалкановые кислоты, диалкановые кислоты, ароматические кислоты, алифатические и ароматические сульфоновые кислоты и т.д. Таким образом такие соли включают сульфаты, пиросульфаты, бисульфаты, сульфиты, бисульфиты, нитраты, фосфаты, первичные и вторичные кислые фосфаты, метафосфаты, пирофосфаты, хлориды, бромиды, иодиды, ацетаты, трифторацетаты, пропионаты, каприлаты, изобутираты, оксалаты, малонаты, сукцинаты, соли пробковой кислоты, себацинаты, фумараты, малеаты, соли миндальной кислоты, бензоаты, хлорбензоаты, метилбензоаты, гидроксинафтоаты, ксинофоаты, динитробензоаты, фталаты, бензолсульфонаты, толуолсульфонаты, фенилацетаты, соли лимонной кислоты, лактаты, малеаты, тартраты, метансульфонаты и т.д. Также предлагаются соли аминокислот, такие как аргинаты и глюконаты, галактуронаты (см., например, Веrgе и др., “Фармацевтические соли”, J. Рhаrm. Sci. 1977; 66:1).
Соли соединений получали с помощью обычных способов при взаимодействии свободного основания с необходимым количеством выбранной кислоты. Соединения в виде свободного основания могут быть получены обработкой солевой формы этих соединений основанием и в результате последующего выделения свободного основания с помощью обычных способов.
Предпочтительными соединениями I и I(а) являются такие соединения, в которых заместитель R1 представляет собой атом кислорода или серы. Предпочтительными заместителями R2 являются алкильный и алкоксиалкильный заместитель, причем особенно предпочтительными являются заместители с С1-4 алкильной группой.
Предпочтительно, чтобы заместители R3 и R4 использовались вместе, образуя конденсированный бензольный или пиридиновый цикл, который может быть замещенным или незамещенным.
Наиболее предпочтительными соединениями являются соединения формулы III или IV


в которой заместитель R2 выбирается, как определено выше, а заместитель R представляет собой атом водорода, алкильную, алкоксильную, алкилтиольную, гидроксильную, галоидную, галоидоалкильную, полигалоидоалкильную, пергалоидоалкильную (например, трифторметильную), трифторалкоксильную (например, трифторметоксильную), нитрильную, аминную, алкиламинную, диалкиламинную, алкилкарбонильную, алкенилкарбонильную, арилкарбонильную, гетероарилкарбонильную, арильную, арилалкильную, гетероарильную, гетероарилалкильную, гетероциклильную, гетероциклоалкильную, нитрильную и алкоксикарбонильную группу.
Примерами этих соединений являются:
2-метилтиазоло[4,5-с]хинолин-4-амин;
тиазоло[4,5-с]хинолин-4-амин;
2-этилтиазоло[4,5-с]хинолин-4-амин;
2-пропилтиазоло[4,5-с]хинолин-4-амин;
2-пентилтиазоло[4,5-с]хинолин-4-амин;
2-бутилтиазоло[4,5-с]хинолин-4-амин;
2-(1-метилэтил)тиазоло[4,5-с]хинолин-4-амин;
2-(2-фенил-1-этенил)тиазоло[4,5-с]хинолин-4-амин;
2-(2-фенил-1-этил)тиазоло[4,5-с]хинолин-4-амин;
2-(4-аминотиазоло[4,5-с]хинолин-2-ил)-1,1-диметилэтилкарбамат;
2-(этоксиметил)тиазоло[4,5-с]хинолин-4-амин;
2-(метоксиметил)тиазоло[4,5-с]хинолин-4-амин;
2-(2-метилпропил)тиазоло[4,5-с]хинолин-4-амин;
2-бензилтиазоло[4,5-с]хинолин-4-амин;
8-метил-2-пропилтиазоло[4,5-с]хинолин-4-амин;
(4-аминотиазоло[4,5-с]хинолин-2-ил)метанол;
2-метилоксазоло[4,5-с]хинолин-4-амин;
2-этилоксазоло[4,5-с]хинолин-4-амин;
2-бутилоксазоло[4,5-с]хинолин-4-амин;
2-пропилтиазоло[4,5-с]хинолин-4,8-диамин;
2-пропилоксазоло[4,5-с]хинолин-4-амин;
8-бром-2-пропилтиазоло[4,5-с]хинолин-4-амин;
7-метил-2-пропилтиазоло[4,5-с]хинолин-4-амин;
2-бутил-7-метилоксазоло[4,5-с]хинолин-4-амин;
7-метил-2-пропилоксазоло[4,5-с]хинолин-4-амин;
7-фтор-2-пропилоксазоло[4,5-с]хинолин-4-амин;
7-фтор-2-пропилтиазоло[4,5-с]хинолин-4-амин;
2-пропил-7-(трифторметил)тиазоло[4,5-с]хинолин-4-амин;
2-(4-морфолино)тиазоло[4,5-с]хинолин-4-амин;
2-(1-пирролидино)тиазоло[4,5-с]хинолин-4-амин;
2-бутилтиазоло[4,5-с][1,5]нафтиридин-4-амин;
2-пропилтиазоло[4,5-с][1,5]нафтиридин-4-амин;
7-хлор-2-пропилтиазоло[4,5-с]хинолин-4-амин;
7-метокси-2-пропилтиазоло[4,5-с]хинолин-4-амин
и фармацевтически приемлемые соли на основе этих соединений, особенно солянокислые соли на основе этих соединений.
Получение соединений
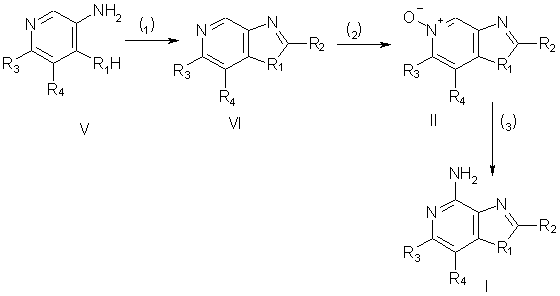
Соединения согласно настоящему изобретению могут быть получены в соответствии со схемой 1, в которой заместители R1, R2, R3 и R4 выбираются, как определено выше.
На стадии (1) процесса по схеме Iа соединение V обрабатывают карбоновой кислотой или эквивалентным ей соединением, в результате чего получают соединение формулы VI. Подходящими заменителями карбоновой кислоты являются ангидриды кислот, хлорангидриды, ортоэфиры и 1,1-диалкоксиалконоаты. Карбоновую кислоту или ее заменитель выбирают таким образом, чтобы получить требуемый заместитель R2 в соединении VI. Например, при использовании триэтилортоформиата будет получаться соединение VI, в котором заместителем R2 является атом водорода, а при использовании уксусного ангидрида будет получаться соединение VI, в котором заместитель R2 представляет собой метильную группу. Реакция может протекать в отсутствие растворителя в присутствии кислоты, такой, например, как полифосфорная кислота, или предпочтительно в присутствии карбоновой кислоты общей формулы R2С(O)OН. Реакцию проводят при достаточном нагревании, которое необходимо для отгонки спирта или воды, образующихся в качестве побочного продукта. Соединение V является доступным соединением или может быть получено обычными способами (см., например, Васhmаn и др., Journal of the Аmеriсаn Сhеmiсаl Sосiеty, 69, стр.365-371 (1947); Аmbrogi и др., Synthesis, стр.656-658 (1992); Аdler и др., Journal of the Chemical Societly, стр.1794-1797 (1960); Sus и др., Justus Liebis Аnnаlеn dеr Сhеmiе, 583, стр.150-160 (1953), и Sus и др., Justus Liebigs Аnnаlеn dеr Сhеmiе, 593, стр.91-126 (1955).
На стадии (2) реакции по схеме Iа проводят окисление соединения VI, в результате чего получают N-оксид, имеющий формулу II. Окисление проводят под действием обычных окислителей, способных образовывать N-оксиды. Предпочтительные условия проведения реакции заключаются в обработке раствора соединения VI в хлороформе 3-хлорпербензойной кислотой при комнатной температуре. Окисление можно также проводить, используя надуксусную кислоту в подходящем растворителе, таком, например, как этил- или метилацетат.
На стадии (3) процесса по схеме I проводят аминирование N-оксида (II), в результате чего получают соединение формулы I. Стадия (3) включает в себя (i) реакцию соединения (II) с ацилирующим агентом и (ii) последующую обработку полученного продукта аминирующим агентом. Фаза (i) стадии (3) заключается в обработке N-оксида формулы II ацилирующим агентом. Подходящими ацилирующими агентами являются хлорангидриды алкил- или арилсульфоновой кислот (например, бензолсульфонилхлорид, метансульфонилхлорид, п-толуолсульфонилхлорид). Арилсульфонилхлориды являются предпочтительными ацилирующими агентами. Наиболее предпочтительным из них является пара-толуолсульфонилхлорид. Фаза (ii) стадии (3) заключается во взаимодействии продукта, образовавшегося на фазе (i), с избытком аминирующего агента. Подходящими аминирующими агентами являются аммиак (например, в форме гидроксида аммония) и аммонийные соли (например, карбонат аммония, бикарбонат аммония, фосфат аммония). Предпочтительным является использование гидроксида аммония. Реакцию предпочтительно проводить, растворяя или диспергируя N-оксид формулы II в инертном растворителе, таком как дихлорметан или хлороформ, добавляя к раствору или суспензии аминирующий агент и после этого медленно добавляя ацилирующий агент. Образовавшийся продукт или фармацевтически приемлемая соль на его основе могут быть выделены из реакционной смеси обычными способами.
Согласно другому способу стадию (3) можно проводить, (i) обрабатывая N-оксид формулы II изоцианатом, а затем (ii) проводя гидролиз полученного продукта. Фаза (i) включает в себя реакцию N-оксида с изоцианатом, в котором изоцианатная группа связана с карбонильной группой. Предпочтительными изоцианатами являются трихлорацетилизоцианат и ароилизоцианаты, такие, например, как бензоилизоцианат. Реакцию изоцианата с N-оксидом проводят в практически безводных условиях, добавляя изоцианат к раствору N-оксида в инертном растворителе, таком как дихлорметан. Фаза (ii) заключается в гидролизе продукта, образующегося на фазе (i). Гидролиз осуществляют с помощью обычных способов, таких как нагревание в присутствии воды или низшего спирта в присутствии катализатора, такого как низший алкоксид щелочного металла или аммония.
Схема I реакции
Приведенные в изобретении соединения, в которых заместителем R1 является атом кислорода или серы, а заместители R3 и R4 образуют ароматическое кольцо, можно получить в соответствии со схемой II, в которой природа заместителей R и R2 выбирается, как определено выше.
На стадии (1) реакции по схеме II, проводят реакцию 3-аминохинолин-4-ола или 3-аминохинолин-4-тиола (соединение VII) с карбоновой кислотой или эквивалентным ей соединением, в результате чего получают оксазоло- или тиазоло[4,5-с]хинолин, имеющий формулу VIII. Подходящими заменителями карбоновой кислоты являются ангидриды кислот, хлорангидриды кислот, ортоэфиры и 1,1-диалкоксиалконоаты. Карбоновую кислоту или ее заменитель выбирают таким образом, чтобы получить требуемый заместитель R2 в соединении VIII. Например, при использовании триэтилортоформиата будет получаться соединение VIII, в котором заместителем R2 является атом водорода, а при использовании уксусного ангидрида будет получаться соединение VIII, в котором заместитель R2 представляет собой метильную группу. Реакция может протекать в отсутствие растворителя, в присутствии кислоты, такой, например, как полифосфорная кислота, или предпочтительно в присутствии карбоновой кислоты общей формулы R2С(O)OН. Реакцию проводят при достаточном нагревании, которое необходимо для отгонки спирта или воды, образующихся в качестве побочного продукта. 3-Аминохинолин-4-ол или 3-аминохинолин-4-тиол (соединение VII) являются доступными продуктами или могут быть получены известными способами.
На стадии (2) реакции по схеме II происходит окисление оксазоло- или тиазоло[4,5-с]хинолина (VIII), приводящее к образованию оксазоло- или тиазоло[4,5-с]хинолин-5N-оксида (соединение IX), являющегося предшественником соединения II. Окисление проводят под действием обычных окислителей, способных образовывать N-оксиды. Предпочтительные условия проведения реакции заключаются в обработке раствора соединения VIII в хлороформе 3-хлорпербензойной кислотой при комнатной температуре. Окисление можно проводить также, используя надуксусную кислоту в подходящем растворителе, таком, например, как этил- или метилацетат.
На стадии (3) реакции по схеме II проводят аминирование N-оксида (IX) до оксазоло[4,5-с]хинолин-4-амина (соединение III) или тиазоло[4,5-с]хинолин-4-амина (соединение IV); оба эти соединения являются производными соединения I. Стадия (3) включает в себя (i) реакцию соединения (IX) с ацилирующим агентом и (ii) последующую обработку полученного продукта аминирующим агентом. Фаза (i) стадии (3) заключается в обработке N-оксида формулы IX ацилирующим агентом. Подходящими ацилирующими агентами являются хлорангидриды алкил- или арилсульфоновой кислоты (например, бензолсульфонилхлорид, метансульфонилхлорид, п-толуолсульфонилхлорид). Арилсульфонилхлориды являются предпочтительными ацилирующими агентами. Наиболее предпочтительным из них является пара-толуолсульфонилхлорид. Фаза (ii) стадии (3) заключается во взаимодействии продукта, образовавшегося на фазе (i), с избытком аминирующего агента. Подходящими аминирующими агентами являются аммиак (например, в форме гидроксида аммония) и аммонийные соли (например, карбонат аммония, бикарбонат аммония, фосфат аммония). Предпочтительно использование гидроксида аммония. Реакцию предпочтительно проводить, растворяя или диспергируя N-оксид (соединение IX) в инертном растворителе, таком как дихлорметан или хлороформ, добавляя к раствору или суспензии аминирующий агент и после этого медленно добавляя ацилирующий агент. Образовавшийся продукт или фармацевтически приемлемая соль на его основе может быть выделена из реакционной смеси обычными способами.
Согласно другому способу стадию (3) можно проводить, (i) обрабатывая N-оксид формулы IX изоцианатом и затем (ii) проводя гидролиз полученного продукта. Фаза (i) включает в себя реакцию N-оксида с изоцианатом, в котором изоцианатная группа связана с карбонильной группой. Предпочтительными изоцианатами являются трихлорацетилизоцианат и ароилизоцианаты, такие, например, как бензоилизоцианат. Реакцию изоцианата с N-оксидом проводят в отсутствие воды, добавляя изоцианат к раствору N-оксида в инертном растворителе, таком как дихлорметан. Фаза (ii) заключается в гидролизе продукта, образующегося на фазе (i). Гидролиз осуществляют с помощью обычных способов, таких как нагревание в присутствии воды или низшего спирта в присутствии катализатора, такого как низший алкоксид щелочного металла или аммония.
Схема II реакции

Приведенные в изобретении соединения, в которых заместителем R1 является атом серы, можно получить в соответствии со схемой III, в которой заместители R2, R3 и R4 выбираются, как определено выше.
На стадии (1) реакции по схеме III соединение формулы Х обрабатывают галоидоацильным соединением общей формулы R2С(O)Z, в котором заместитель R2 выбирается, как определено выше, а Z - атом хлора или брома. В результате такой обработки получают амид формулы XI. Реакцию можно проводить в присутствии третичного амина, добавляя с контролируемой скоростью (например, по каплям) галоидацильное соединение к раствору или суспензии соединения Х в подходящем растворителе, таком как пиридин или дихлорметан.
На стадии (2) реакции по схеме III амид формулы XI реагирует с пятисернистым фосфором с образованием соединения XII. Для протекания реакции пятисернистый фосфор добавляют к раствору или суспензии соединения XI в соответствующем растворителе, например в пиридине, и нагревают полученную смесь.
Стадии (3) и (4) реакции по схеме III можно проводить таким же образом, как и стадии (2) и (3) реакции, протекающие по схеме I. В результате получают соответственно N-оксид формулы XIII, являющийся предшественником соединения II, и соединение XIV, являющееся предшественником соединения I.
Схема III реакции

Приведенные в изобретении соединения, в которых заместителем R1 является атом серы, а заместители R3 и R4 образуют замещенное ароматическое кольцо, можно получить в соответствии со схемой IV реакции, в которой заместители R и R2 выбираются, как определено выше.
На стадии (1) реакции по схеме IV соединение формулы XV обрабатывают галоидоацильным соединением общей формулы R2С(O)Z, в котором заместитель R2 выбирается, как определено выше, а Z - атом хлора или брома. В результате такой обработки получают N-(4-гидроксихинолин-3-ил)амид формулы XVI. Реакцию можно проводить, добавляя с контролируемой скоростью (например, по каплям) галоидацильное соединение к раствору или суспензии соединения XV в подходящем растворителе, таком как дихлорметан, причем реакцию проводят в присутствии третичного амина.
На стадии (2) реакции по схеме IV N-(4-гидроксихинолин-3-ил)амид формулы XVI реагирует с пятисернистым фосфором с образованием тиазоло[4,5-с]хинолина (соединение XVII). Для протекания реакции пятисернистый фосфор добавляют к раствору или суспензии соединения XVI в соответствующем растворителе, например в пиридине, и нагревают полученную смесь.
Стадии (3) и (4) реакции по схеме IV можно проводить таким же образом, как и стадии (2) и (3) реакции по схеме II. В результате получают соответственно тиазоло[4,5-с]хинолин-5N-оксид формулы XVIII, являющийся предшественником соединения II, и тиазоло[4,5-с]хинолин-4-амин (соединение IV), являющееся предшественником соединения I.
Схема IV реакции

Заместители в положение 2 могут быть введены за счет взаимодействия соединения XIX

в котором заместитель R1 представляет собой атом кислорода или серы, а заместитель R выбирается, как определено выше, с соответствующим литирующим агентом, таким как диизопропиламид лития и н-бутиллитий, в полярном апротонном растворителе. В результате этого взаимодействия происходит образование соединения, имеющего литированную 2-метильную группу. После этого полученное литийсодержащее соединение может реагировать с соответствующим реагентом, содержащим функциональную группу, способную замещать литированную 2-метильную группу. Примерами подходящих реагентов являются галогениды, такие как йодистый метил или хлорметилметиловый эфир, альдегиды, такие как бензальдегид, и кетоны, такие как ацетон. Полученные соединения в дальнейшем могут быть подвергнуты окислению и аминированию с помощью способов, описанных выше при синтезе соединений III и IV.
Некоторые соединения общей формулы I могут быть получены непосредственно из других соединений I. Например, нитрирование 2-пропилтиазоло[4,5-с]хинолин-4-амина приводит к образованию 8-нитро-2-пропилтиазоло[4,5-с]хинолин-4-амина, а восстановление этого нитросоединения дает 2-пропилтиазоло[4,5-с]хинолин-4,8-диамин.
Фармацевтические композиции и биологическая активность
Фармацевтические композиции согласно настоящему изобретению содержат терапевтически эффективные количества соединения I(а) вместе с фармацевтически приемлемым наполнителем.
Используемый в данном описании термин “терапевтически эффективное количество” означает количество соединения, достаточное для стимулирования желаемого терапевтического эффекта, такого как цитокинетический биосинтез, противоопухолевая активность и(или) противовирусная активность. Точное количество активного соединения, используемого в фармацевтической композиции согласно настоящему изобретению, будет изменяться в широких пределах в зависимости от различных факторов, таких как физическая и химическая природа соединения, а также природа наполнителя композиции, предписанная дозировка и, кроме того, от тяжести заболевания. Тем не менее полагают, что предлагаемая в изобретении композиция содержит достаточное количество активного ингредиента, чтобы обеспечить дозу от 100 нг на 1 кг веса пациента до приблизительно 50 мг/кг, предпочтительно от 10 мкг/кг до 5 мг/кг веса пациента. Фармацевтические композиции можно использовать в любом виде, а именно в виде таблеток, лепешек, составов для парентерального введения, сиропов, кремов, мазей, аэрозолей, различного рода пластырей и т.п. Используемая форма дозировки будет также зависеть от характеристик состава, который нужно вводить в организм. Например, некоторые соединения I(а), особенно такие соединения, в которых в качестве заместителя R1 используют атом серы, обладают относительно низкой оральной биодоступностью и быстро подвергаются метаболизму при попадании в кровь. Эти свойства делают такие соединения особенно эффективными для лечения таких болезней, при которых желательно локальное введение соединения, модифицирующего иммунную реакцию. К числу таких болезней относятся астма, базалиома, внутриэпителиальные цервикальные новообразования и т.д.
Показано, что приведенные в изобретении соединения способствуют образованию некоторых цитокинов в опытах, выполненных в соответствии с приведенными ниже Способами испытания. Эти результаты свидетельствуют о том, что такие соединения могут быть эффективными в качестве модификаторов иммунной реакции, которые способны модулировать иммунную реакцию различными способами, и тем самым могут быть эффективными препаратами для излечения различных расстройств.
Цитокины, которые образуются при введении в организм описанных в изобретении соединений, обычно включают интерферон-α (IFN-α ) и(или) фактор-α некроза опухоли (ТNF-α ), а также некоторые интерлейкины (IL). Цитокинины, чей биосинтез может быть стимулирован описанными в изобретении соединениями, включают IFN-α , ТNF-α , IL-1,6, 10 и 12, а также различные другие цитокины. Цитокины ингибируют образование вирусов и рост опухолевых клеток, что делает описанные в изобретении соединения эффективными веществами для лечения опухолей и вирусных заболеваний.
Помимо способности стимулировать образование цитокинов описанные в изобретении соединения воздействуют на другие аспекты врожденной иммунной реакции. Например, при введении цитокинов может быть стимулирована активность природных “клеток-убийц”. Эти соединения могут также активировать макрофаги, которые, в свою очередь, стимулируют выделение оксида азота и образование дополнительных цитокинов. Кроме того, эти соединения способны вызвать разрастание и дифференциацию В-лимфоцитов и тем самым могут быть полезны для созревания in vitro дендритных клеток.
Соединения, описываемые в настоящем изобретении, оказывают также влияние на приобретенную иммунную реакцию. Например, хотя полагают, что они не оказывают непосредственного влияния на Т-клетки или продуцирование Т-лимфоцитных цитокинов, эти соединения опосредованно через цитокин Т-фага-помощника типа 1 (Тh1) влияют на образование IFN-γ , в то время как за счет цитокина Т-фага-помощника типа 2 (Тh2) происходит ингибирование образования IL-4, IL-5 и IL-13. Этот факт указывает на то, что эти соединения эффективны при лечении болезней, при которых желательно повышение роли Тh1 и уменьшение роли Тh2. Учитывая способность соединений Iа ингибировать иммунную реакцию Тh2, можно ожидать, что эти соединения будут эффективны при лечении атопии, например аллергических дерматитов, астмы, аллергии, аллергических ринитов и красной волчанки; для усиления клеточного иммунитета, а возможно также для лечения рецидивирующих грибковых заболеваний и хламидоза.
Модифицирующее влияние соединений на иммунную реакцию делает их полезными препаратами для лечения различных заболеваний. Обладая способностью стимулировать цитокины, такие как INF-α и(или) ТNF-α , эти соединения особенно полезны для лечения вирусных заболеваний и опухолей. Такая иммуномодулирующая активность позволяет считать, что описанные в данном изобретении соединения полезны для лечения, помимо прочего, таких болезней, как вирусные заболевания, например остроконечные кондиломы, простые бородавки, подошвенные бородавки, гепатит В, гепатит С, герпесы простого типа I и II, контагиозный моллюск, ВИЧ, вирусы цитомегалии и ветряной оспы, цервикальная внутриэпителиальная неоплазия, вирусная папиллома человека и связанные с ней новообразования; грибковые заболевания, например аспергиллез, криптококковый менингит; раковые заболевания, например, базально-клеточный рак, лейкемия волосяных клеток, саркома Капоши, рак почки, плоскоклеточный рак, миелогенная лейкемия, множественная миеломная болезнь, меланома, лимфома не по Ходжкину, кожная лимфома Т-клеток и другие раковые образования; паразитарные болезни, например, пневмония, криптоспоридоз, гистоплазмоз, токсоплазмоз, трипаносомная инфекция, лейшманиоз и бактериальные заразные болезни, например туберкулез, микобактериальный авиум. Помимо лечения приведенных выше заболеваний описанные в настоящем изобретении соединения могут использоваться также для лечения таких заболеваний, как экзема, эозинофилия, основной тромбоцитоз, лепра, множественный склероз, синдром Оммена, ревматоидный артрит, красная волчанка, дисковидная волчанка, болезнь Боуена, папулез Боуена; эти соединения могут ускорять или стимулировать заживление ран, включая хронические раны.
Таким образом, настоящее изобретение предлагает способ стимулирования цитокинетического биосинтеза в живом организме, заключающийся во введении в организм животного эффективного количества соединения Iа. Количество соединения, эффективное для стимулирования цитокинетического биосинтеза, - это количество, достаточное для того, чтобы один или более типов клеток, таких как моноциты, макрофаги, дендритные клетки и В-клетки, могли образовать один или большее количество цитокинов, таких, например, как IFN-α , TNF-α , IL-1, 6, 10 и 12, в количестве, превышающем фоновое количество этих цитокинов. Точное количество будет меняться в зависимости от различных факторов, но ожидается, что доза соединения будет находиться в пределах от 100 нг на 1 кг веса пациента до 50 мг/кг, предпочтительно от приблизительно 10 мкг/кг до 5 мг/кг. Изобретение предлагает также способ лечения вирусной инфекции в организме животного, заключающийся во введении эффективного количества соединения Iа в организм животного. Эффективное количество для лечения или предотвращения вирусной инфекции - это такое количество, которое приводит к уменьшению одного или большего количества признаков вирусной инфекции, таких как вирусное повреждение, вирусная нагрузка, скорость образования вируса и гибели вирусов, по сравнению с ситуацией, наблюдаемой у контрольных животных. Точное количество будет меняться в зависимости от различных факторов, но ожидается, что доза соединения будет находиться в пределах от 100 нг/кг до 50 мг/кг, предпочтительно от приблизительно 10 мкг/кг до 5 мг/кг.
Приведенные в настоящем изобретении соединения могут быть введены в организм в виде единственного терапевтического агента или могут составлять часть терапевтического препарата в комбинации с одним или несколькими другими агентами. Примеры подходящих агентов, которые могут быть использованы в комбинации с приведенными в настоящем изобретении соединениями, модифицирующими иммунную реакцию, являются, помимо прочих веществ, анальгетики, антибактериальные вещества, противогрибковые, противовоспалительные, противоопухолевые агенты, антивирусные, бронхолитические средства, наркотические и стероидные препараты.
Следующие примеры приведены для иллюстрации изобретения, которое, однако, никоим образом не ограничивается только этими примерами.
ПРИМЕРЫ
Пример 1
2-Метилтиазоло[4,5-с]хинолин-5N-оксид

Часть А
Суспензию около 12 г З-аминохинолин-4-тиола в смеси уксусного ангидрида (150 мл) и уксусной кислоты (300 мл) кипятили с обратным холодильником в течение ночи. Реакционную смесь фильтровали для удаления мелкого твердого осадка. Фильтрат упаривали в вакууме. Остаток разбавляли этанолом и кипятили с обратным холодильником в течение 30 минут. Полученный раствор концентрировали в вакууме, и остаток разбавляли водой. Водный слой подщелачивали гидроксидом натрия и затем экстрагировали диэтиловым эфиром. Эфирные экстракты соединяли вместе, сушили над сульфатом магния и фильтровали. Фильтрат упаривали и в результате получили 12,8 г неочищенного продукта. 800 мг этого продукта перекристаллизовывали из гексана; получили 2-метилтиазоло[4,5-с]хинолин в виде желтых игольчатых кристаллов, температура плавления которых составляла 95,5-97,5° С. Анализ: рассчитано для С11H8N2S: %С, 65,97; %Н, 4,03; %N, 13,99. Найдено: %С, 65,96; %Н, 4,16; %N, 14,08.
Часть В
5,0 г (25 ммоль) 2-метилтиазоло[4,5-с]хинолина, 9,5 г 3-хлорпербензойной кислоты (в виде 50-60%-ного раствора) и 150 мл дихлорметана перемешивали при комнатной температуре в течение 3 часов. Реакционный раствор разбавляли 300 мл дихлорметана и затем экстрагировали водным раствором карбоната натрия для удаления кислот. Органический слой промывали водой, разбавляли этилацетатом для удаления мути, сушили над сульфатом магния и затем концентрировали в вакууме. В результате получили 4,5 г сырого продукта. Малую часть этого продукта перекристаллизовывали из метанола. Получили желтые иглы гидрата 2-метилтиазоло[4,5-с]хинолин-5N-оксида; температура плавления очищенного продукта 150-160° С. Анализ: рассчитано для С11Н8N2OS+0,75 Н2O: %С, 57,50; %Н, 4,17; %N, 12,19. Найдено: %С, 57,58; %Н, 4,10; %N, 11,93.
Пример 2
2-Метилтиазоло[4,5-с]хинолин-4-амин
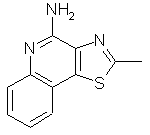
1,5 г 2-метилтиазоло[4,5-с]хинолин-5N-оксида (6,9 ммоль) добавляли к смеси 10 мл дихлорметана и 25 мл гидроксида аммония. При интенсивном перемешивании к реакционной смеси добавляли раствор 2,0 г тозилхлорида (10,4 ммоль) в 10 мл дихлорметана. Реакционную смесь кипятили с обратным холодильником, добавляя дополнительные количества дихлорметана и гидроксида аммония для завершения реакции, о котором судили по данным тонкослойной хроматографии. Из полученной смеси отгоняли дихлорметан и из оставшейся водной фракции отфильтровывали желтый твердый продукт. Его промывали водой и сушили. В результате было получено 1,2 г неочищенного вещества. Этот продукт растворяли в разбавленной соляной кислоте, раствор выдерживали над активированным углем и затем фильтровали. Фильтрат подщелачивали разбавленным раствором гидроксида натрия. Полученный осадок отфильтровывали, промывали водой и перекристаллизовывали из смеси метанола с дихлорметаном. В результате получили 0,46 г 2-метилтиазоло[4,5-с]хинолин-4-амина в виде белого порошка, температура плавления которого составляла 184-187° С. Анализ: рассчитано для С11Н9N3S: %С, 61,37; %Н, 4,21; %N, 19,52. Найдено: %С, 61,32; %Н, 4,52; %N, 19,68.
Пример 3
Альтернативный способ синтеза 2-метилтиазоло[4,5-с]хинолин-4-амина
Трихлорацетилизоцианат (2,0 мл, 16,8 ммоль) добавляли к суспензии 2-метилтиазоло[4,5-с]хинолин-5N-оксида (3,03 г, 14,0 ммоль) в дихлорметане (150 мл). Реакционную смесь перемешивали при комнатной температуре в течение 50 минут. Дихлорметан концентрировали в вакууме, в результате чего получали неочищенный N-(2-метилтиазоло[4,5-с]хинолин-4-ил)трихлорацетамид. Этот амид растворяли в метаноле, к раствору добавляли метилат натрия (1 мл 25%-ного раствора метилата натрия в метаноле) и кипятили его с обратным холодильником в течение 40 минут. После этого метанол упаривали в вакууме. Оставшийся коричневый твердый продукт промывали водой и сушили; в результате получили 2,85 г неочищенного продукта. Этот продукт обрабатывали активированным углем и затем перекристаллизовывали из этилацетата. Полученный твердый 2-метилтиазоло[4,5-с]хинолин-4-амин имел температуру плавления 184-186° С. Анализ: рассчитано для С11H9N3S: %С, 61,37; %Н, 4,21; %N, 19,52. Найдено: %С, 61,48; %Н, 4,17; %N, 19,60.
Пример 4
Солянокислый 2-метилтиазоло[4,5-с]хинолин-4-амин
К раствору 0,5 г 2-метилтиазоло[4,5-с]хинолин-4-амина в 15 мл метанола добавляли 0,2 мл концентрированной (12,1 М) соляной кислоты, а затем 15 мл изопропанола. Смесь нагревали до кипения для удаления основной части метанола. Полученный осадок отфильтровывали, промывали изопропанолом и сушили. В результате получили солянокислый 2-метилтиазоло[4,5-с]хинолин-4-амин в виде твердого продукта, плавящегося при 323-325° С. Анализ: рассчитано для С11H9N3S· НСl: %С, 52,48; %Н, 4,00; %N, 16,69. Найдено: %С, 52,46; %Н, 4,08; %N, 16,52.
Пример 5
Гидрат солянокислого тиазоло[4,5-с]хинолин-4-амина
Часть А
Приблизительно 18,5 г 3-аминохинолин-2-тиола добавляли к 26,0 мл триэтилортоформиата. Смесь нагревали на паровой бане в течение 20 минут. Добавляли к ней 400 мл муравьиной кислоты и затем кипятили с обратным холодильником в течение ночи. Муравьиную кислоту упаривали в вакууме. Остаток соединяли с этанолом и кипятили с обратным холодильником в течение 30 минут, после чего этанол упаривали в вакууме. Остаток суспендировали в воде и суспензию подщелачивали, добавляя гидроксид натрия, при этом образовывался осадок, который экстрагировали несколькими порциями дихлорметана. Экстракты соединяли, сушили над сульфатом магния и концентрировали. В результате получили желтое твердое вещество, после перекристаллизации которого из смеси гексанов получали 13,1 г желтого кристаллического тиазоло[4,5-с]хинолина, плавящегося при 104-106° С.
Часть В
К суспензии тиазоло[4,5-с]хинолина (12,5 г, 67 ммоль) в метилацетате (300 мл) добавляли 21 мл надуксусной кислоты (32%-ный раствор в уксусной кислоте; 100 ммоль). Смесь кипятили с обратным холодильником в течение ночи и затем охлаждали до комнатной температуры. Осадок отфильтровывали и суспендировали в воде (100 мл). К суспензии добавляли 100 мл водного раствора бикарбоната натрия и смесь перемешивали в течение 1 часа. Твердый тиазоло[4,5-с]хинолин-5N-оксид отфильтровывали, промывали водой и сушили.
Часть С
0,72 мл трихлорацетилизоцианата (6,0 ммоль) добавляли к суспензии 1,10 г тиазоло[4,5-с]хинолин-5N-оксида (5,4 ммоль) в 100 мл дихлорметана. Реакционную смесь перемешивали при комнатной температуре в течение 45 минут, после чего дихлорметан упаривали в вакууме и получали неочищенный N-(тиазоло[4,5-с]хинолин-4-ил)трихлорацетамид. Этот амид перемешивали при комнатной температуре в течение 2 часов в 2M растворе аммиака в метаноле; метанол упаривали в вакууме. Полученный остаток суспендировали в воде, добавляли карбонат натрия и перемешивали 10 минут. Образовавшийся твердый коричневый продукт промывали водой и сушили. Затем суспендировали его в воде, добавляли 100 мл 6 N соляной кислоты и смесь нагревали на паровой бане. Смесь фильтровали и фильтрат медленно охлаждали до комнатной температуры. Полученный осадок отфильтровывали и сушили, в результате получили 0,75 г продукта в виде коричневых игольчатых кристаллов. Этот продукт растворяли при нагревании в 100 мл воды, к раствору добавляли активированный уголь и смесь перемешивали в течение 5 минут. Затем смесь фильтровали через слой Сеlite®. Фильтрат нагревали на паровой бане для удаления большей части воды и затем охлаждали до комнатной температуры. Образовавшийся при охлаждении осадок отфильтровывали и сушили, в результате чего получали 0,30 г белого кристаллического гидрата солянокислого тиазоло[4,5-с]хинолин-4-амина; температура плавления этого вещества 284-285° С. Анализ: рассчитано для С10H3S· НСl· Н2O: %С, 46,97; %Н, 3,94; %N, 16,43. Найдено: %С, 46,96; %Н, 3,99; %N, 16,34.
Пример 6
Тиазоло[4,5-с]хинолин-4-амин

Часть А
8,7 г тиазоло[4,5-с]хинолин-2-тиола (0,04 моля) суспендировали в водном растворе, содержащем 1,4 г гидроксида натрия (0,04 моля). К суспензии добавляли несколько капель 50%-ного раствора гидроксида натрия до полного растворения твердого продукта. Колбу с реакционной смесью опускали в баню с холодной водой и, выдерживая температуру смеси в пределах 25-35° С, по каплям в течение 30 минут добавляли 13,5 мл 30%-ного пероксида водорода (0,08 моля). Баню убирали и реакционную смесь перемешивали в течение 15 минут. После этого добавляли 2,5 г 95,98%-ной серной кислоты. Через 30 минут реакционную смесь подщелачивали 50%-ным раствором гидроксида натрия до рН 9-9,5. Затем реакционную смесь подкисляли соляной кислотой до рН 2,5, при этом наблюдалось осаждение коричневого твердого продукта. После нагревания смеси на паровой бане в течение 15 минут осадок растворялся. После охлаждения раствора до комнатной температуры опять образовывался осадок. Смесь подщелачивали 50%-ным гидроксидом натрия до рН 9. Образовавшийся маслообразный продукт экстрагировали этилацетатом. Экстракты соединяли, промывали водой, сушили над сульфатом магния и концентрировали в вакууме до получения 3,3 г тиазоло[4,5-с]хинолина в виде коричневого твердого вещества, температура плавления которого составляла 104,4-105° С. Анализ: рассчитано для С10Н6N2S: %С, 64,19; %Н, 3,25; %N, 15,04. Найдено: %С, 64,15; %Н, 3,26; %N, 14,9.
Часть В
К раствору тиазоло[4,5-с]хинолина (2,8 г) в метилацетате добавляли 4,7 мл 32%-ной надуксусной кислоты. В течение нескольких минут наблюдалось образование осадка. Реакционную смесь нагревали до кипения и затем разбавляли, добавляя еще 10 мл метилацетата. Большая часть осадка при этом растворялась. Через 1 час к смеси добавляли еще 3,1 мл надуксусной кислоты. После этого реакционную смесь нагревали в течение ночи и затем охлаждали до комнатной температуры. Метилацетат и уксусную кислоту удаляли в результате азеотропной перегонки с гептаном. Оставшийся маслообразный продукт суспендировали в воде. Смесь подщелачивали насыщенным раствором бакарбоната натрия и затем экстрагировали этилацетатом. Экстракты соединяли, промывали водой, сушили над сульфатом магния и затем концентрировали в вакууме до получения 0,6 г тиазоло[4,5-с]хинолин-5N-оксида в виде оранжевого твердого продукта, плавящегося при 178,4° С (с разложением).
Часть С
К охлажденной до 5° С суспензии тиазоло[4,5-с]хинолин-5]N-оксида (0,3 г) в смеси гидроксида аммония (5 мл) и дихлорметана (50 мл) по каплям добавляли раствор тозилхлорида (0,3 г) в воде, поддерживая при этом температуру в пределах 4-6° С. По завершении добавления тозилхлорида реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Анализ с помощью способа тонкослойной хроматографии свидетельствует о наличии в смеси исходного материала. Поэтому реакционную смесь охлаждали и добавляли еще 1 эквивалент тозилхлорида. Реакционную смесь перемешивали при комнатной температуре в течение 20 часов. После этого дихлорметан упаривали в вакууме. Остаток суспендировали в малом количестве воды. Смесь отфильтровывали. Выделенный твердый продукт промывали водой, сушили и затем перекристаллизовывали из изопропанола. Получено 0,2 г тиазоло[4,5-с]хинолин-4-амина в виде оранжевого порошка. Температура плавления 172,4° С (с разложением). Анализ: рассчитано для С10H7N3S: %С, 59,68; %Н, 3,50; %N 20,88. Найдено: %С, 59,82; %Н, 3,20; %N, 19,50.
Пример 7
2-Этилтиазоло[4,5-с]хинолин-5N-оксид

Часть А
1,0 г 2-метилтиазоло[4,5-с]хинолина (10,0 ммоль; из примеров 2 или 3) в атмосфере азота помещали в высушенную колбу. Добавляли 50 мл безводного тетрагидрофурана, и реакционную смесь охлаждали до -78° С на бане с сухим льдом. К смеси по каплям добавляли 6,7 мл 1,5 М раствора диизопропиламида лития (10,0 ммоль) в гексане. Через 30 минут добавляли 0,95 мл иодистого метила (15,0 ммоль). Через 40 минут смесь нагревали до комнатной температуры и выливали в воду, а затем экстрагировали диэтиловым эфиром (250 мл). Экстракт промывали тремя порциями воды по 100 мл каждая, сушили над сульфатом магния и концентрировали в вакууме до получения 2,8 г коричневого масла. Этот продукт очищали способом высокоэффективной жидкофазной хроматографии, используя в качестве элюента 3:1 смесь гексана и этилацетата. В результате получили 1,47 г 2-этилтиазоло[4,5-с]хинолина в виде желтого масла.
Часть В
К раствору 2-этилтиазоло[4,5-с]хинолина (0,53 г) в 20 мл хлороформа добавляли 0,44 г 3-хлорпербензойной кислоты. Смесь перемешивали в течение 2 часов при комнатной температуре и затем разбавляли 20 мл дихлорметана, промывали водным раствором бикарбоната натрия, тремя порциями воды по 100 мл каждая, сушили над сульфатом магния и затем концентрировали в вакууме. После перекристаллизации образовавшегося желтого твердого продукта из этилацетата получили 0,32 г твердого 2-этилтиазоло[4,5-с]хинолин-5N-оксида, температура плавления которого 128° С. Анализ: рассчитано для С12Н10N2OS: % C, 62,59; % Н, 4,38; % N, 12,16. Найдено: % С, 62,59; % Н, 4,27; % N, 12,12.
Пример 8
2-Этилтиазоло[4,5-с]хинолин-4-амин

Часть А
К суспензии 0,90 г 2-этилтиазоло[4,5-с]хинолин-5N-оксида (3,9 ммоль) в 60 мл дихлорметана добавляли 0,51 мл трихлорацетилизоцианата (4,3 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут и затем концентрировали в вакууме до получения 1,80 г N-(2-этилтиазоло[4,5-с]хинолин-4-ил)трихлорацетамида в виде желтого твердого вещества.
Часть В
N-(2-Этилтиазоло[4,5-с]хинолин-4-ил)трихлорацетамид (0,40 г) суспендировали в 20 мл 2 М раствора аммиака в метаноле и затем перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь концентрировали в вакууме. Остаток промывали водой и сушили, в результате чего получили 0,19 г неочищенного продукта, который перекристаллизовывали из смеси этилацетата и гексана. Полученный 2-этилтиазоло[4,5-с]хинолин-4-амин имеет вид коричневых игольчатых кристаллов, температура плавления которых составляет 170-172° С. Анализ: рассчитано для С12Н11N3S: % С, 62,85; % Н, 4,83; % N, 18,32. Найдено: % С, 62,58; % Н, 4,78; % N, 18,08.
Пример 9
Альтернативный способ синтеза 2-этилтиазоло[4,5-с]хинолин-4-амина
Часть А
К суспензии 3-аминохинолин-4-тиола (15 г) в пропионовой кислоте (100 мл) добавляли 20 мл ангидрида пропионовой кислоты. Реакционную смесь кипятили с обратным холодильником в течение ночи и затем фильтровали для удаления осадка. Фильтрат концентрировали в вакууме. Остаток смывали 200 мл дихлорметана, промывали сначала водным раствором бикарбоната натрия, а затем водой и сушили над сульфатом магния. Полученный раствор пропускали через слой силикагеля, используя в качестве элюента сначала 1:1 смесь этилацетата с гексаном, а затем чистый этилацетат. Фильтрат упаривали и получали 2,6 г 2-этилтиазоло[4,5-с]хинолина в виде желтого масла.
Часть В
К раствору 5 г 2-этилтиазоло[4,5-с]хинолина в 100 мл этилацетата добавляли 7,4 мл 32% -ной надуксусной кислоты. Смесь перемешивали при комнатной температуре в течение двух суток. Полученный осадок отфильтровывали, промывали гексаном и сушили. В результате получали 3,4 г 2-этилтиазоло[4,5-с]хинолин-5N-оксида.
Часть С
К суспензии 2-этилтиазоло[4,5-с]хинолин-5N-оксида (9,0 г, 39,1 ммоль) в 500 мл дихлорметана добавляли 6,5 мл трихлорацетилизоцианата (54 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов и затем концентрировали ее в вакууме до получения неочищенного N-(2-этилтиазоло[4,5-с]хинолин-4-ил)трихлорацетамида. Этот продукт добавляли к раствору аммиака в метаноле (500 мл 2 М раствора) и перемешивали при комнатной температуре в течение 2 часов, после чего реакционную смесь концентрировали в вакууме. Остаток смывали дихлорметаном. Промывали двумя порциями по 150 мл каждая водного раствора бикарбоната натрия, а затем тремя порциями по 150 мл воды, сушили над сульфатом магния и концентрировали в вакууме. После перекристаллизации полученного остатка из 1,2-дихлррэтана получали вещество в виде коричневых игольчатых кристаллов. Этот материал суспендировали в воде, добавляли к суспензии один эквивалент концентрированной соляной кислоты и смесь нагревали для растворения твердого продукта. Раствор обрабатывали активированным углем, а затем фильтровали. Фильтрат охлаждали и подщелачивали карбонатом натрия. Полученный осадок отфильтровывали и промывали водой. После перекристаллизации из 1,2-дихлорэтана получили 2-этилтиазоло[4,5-с]хинолин-4-амин в виде желтых игольчатых кристаллов; температура плавления 169-171 ° С. Анализ: рассчитано для С12Н11N3S: % С, 62,85; % Н, 4,83; % N, 18,32. Найдено:% С, 62,79; % Н, 4,86; % N, 18,22.
Пример 10
Солянокислый 2-этилтиазоло[4,5-с]хинолин-4-амин
К раствору 2-этилтиазоло[4,5-с]хинолин-4-амина (4,25 г) в теплом изопропаноле добавляли концентрированную соляную кислоту (18,5 ммоль). Смесь кипятили с обратным холодильником для уменьшения общего объема и удаления воды и затем охлаждали до комнатной температуры. Полученный осадок отфильтровывали и сушили. Полученный твердый солянокислый 2-этилтиазоло[4,5-с]хинолин-4-амин имел температуру плавления 268-270 ° С. Анализ: рассчитано для С12Н11N3S • НСl: % С, 54,23; % Н, 4,55; % N, 15,81. Найдено:% С, 54,25; % Н, 4,63; % N, 15,71.
Пример 11
2-Пропилтиазоло[4,5-с]хинолин-5"N-оксид

Часть А
Используя методику, приведенную в части А примера 7, сначала проводили реакцию между 2-метилтиазоло[4,5-с]хинолином (2,50 г, 12,5 ммоль) и диизопропиламидом лития. Последующая обработка полученного продукта йодистым этилом приводила к получению 0,28 г 2-пропилтиазоло[4,5-с]хинолина в виде желтого кристаллического вещества с температурой плавления 54° С. Анализ: рассчитано для С13Н12N3S: % С, 68,39; % Н, 5,30; % N, 12,27. Найдено: % С, 68,41; % Н, 5,19; % N, 12,31.
Часть В
Используя методику, приведенную в части В примера 7, 2-пропилтиазоло[4,5-с]хинолин (1,05 г, 4,6 ммоль) окисляли 3-хлорпербензойной кислотой до 2-пропилтиазоло[4,5-с]хинолин-5М-оксида. Получено 0,65 г целевого продукта в виде желтого твердого вещества с температурой плавления 123° С. Анализ: рассчитано для С13Н12N2OS: % С, 63,91; % Н, 4,95; % N, 11,47. Найдено: % С, 63,53; % Н, 4,88; % N, 11,44.
Пример 12
2-Пропилтиазоло[4,5-с]хинолин-4-амин

Используя методику, приведенную в примере 8, проводили реакцию 2-пропилтиазоло[4,5-с]хинолин-5N-оксида (0,63 г) с трихлорацетилизоцианатом и полученный промежуточный амид подвергали гидролизу, используя в качестве гидролизующего агента раствор аммиака в метаноле. В результате получили 0,22 г белого кристаллического 2-пропилтиазоло[4,5-с]хинолин-4-амина, температура плавления которого составляла 140-142 ° С. Анализ: рассчитано для С13Н13N3S: % С, 64,17; % Н, 5,38; % N, 17,27. Найдено: % С, 64,31; % H, 5,39; % Н, 17,13.
Пример 13
Альтернативный способ синтеза 2-пропилтиазоло[4,5-с]хинолин-4-амина
Часть А
Используя методику, приведенную в части А примера 9, суспензию 15 г 3-аминохинолин-4-тиола в масляной кислоте обрабатывали ангидридом масляной кислоты, в результате чего получали желтый маслообразный 2-пропилтиазоло[4,5-с]хинолин.
Часть В
Используя методику, приведенную в части В примера 9, 2-пропилтиазоло[4,5-с]хинолин (46 г) окисляли надуксусной кислотой до 2-пропилтиазоло[4,5-с]хинолин-5N-оксида, представляющего собой желтое кристаллическое твердое вещество.
Часть С
К раствору 20 г 2-пропилтиазоло[4,5-с]хинолин-5N-оксида в 500 мл хлороформа добавляли 50 мл гидроксида аммония. Смесь охлаждали на ледяной бане и затем по каплям добавляли раствор 16 г тозилхлорида в хлороформе. По окончании добавления тозилхлорида реакционную смесь кипятили с обратным холодильником в течение 2 часов и затем разбавляли дополнительным количеством хлороформа и воды. Разделяли водный и органический слои. Органический слой промывали водным раствором бикарбоната натрия, сушили над сульфатом магния и после этого концентрировали в вакууме. После перекристаллизации остатка из 1,2-дихлорэтана получали 2-пропилтиазоло[4,5-с]хинолин-4-амин в виде твердого коричневого вещества с температурой плавления 140-142 ° С. Анализ: рассчитано для С13Н13N3S: % С, 64,17; % Н, 5,38; % N, 17,27. Найдено:% С, 64,10; % Н,5,47; % N, 17,29.
Пример 14
Солянокислый 2-пропилтиазоло[4,5-с]хинолин-4-амин
Используя методику, приведенную в примере 10, 1,75 г 2-пропилтиазоло[4,5-с]хинолин-4-амина обрабатывали 1 эквивалентом концентрированной соляной кислоты. В результате получали желтовато-белый кристаллический продукт, представляющий собой солянокислый 2-пропилтиазоло[4,5-с]хинолин-4-амин; температура плавления 234-237 ° С. Анализ: рассчитано для С13Н13N3S • НСl: % С, 55,81; % Н, 5,04; % N, 15,02. Найдено: % С, 55,86; % Н, 5,02; % N, 14,99.
Пример 15
2-Пентилтиазоо[4,5-с]хинолин-5N-оксид

Часть А
Используя методику, приведенную в части А примера 7, сначала проводили реакцию между 2-метилтиазоло[4,5-с]хинолином (2,0 г, 10 ммоль) и диизопропиламидом лития (5,5 мл 2 М раствора в бензоле), а затем обрабатывали полученный продукт 1-иодбутаном (1,8 мл). В результате получили 1,1 г желтого твердого продукта, представляющего собой 2-пентилтиазоло[4,5-с]хинолин; температура плавления 62-64 ° С.
Часть В
К суспензии 1,25 г 2-пентилтиазоло[4,5-с]хинолина в 50 мл метилацетата добавляли 1,50 мл 32%-ного раствора надуксусной кислоты в уксусной кислоте. Реакционную смесь кипятили с обратным холодильником в течение 6 часов, после чего охлаждали до комнатной температуры, разбавляли дихлорметаном и промывали сначала водным раствором бикарбоната натрия, а затем водой. Органический слой сушили над сульфатом магния и концентрировали в вакууме. В результате получили 1,20 г бледно-желтого твердого продукта. После перекристаллизации этого вещества из этилацетата получили 0,90 г белого кристаллического 2-пентилтиазоло[4,5-с]хинолин-5N-оксида, температура плавления которого составляла 142-144° С. Анализ: рассчитано для С15Н16N2OS: %С, 66,14; %Н, 5,92; %N, 10,19. Найдено: %С, 65,63; %Н, 5,83; %N, 10,28.
Пример 16
2-Пентилтиазоло[4,5-с]хинолин-4-амин

К раствору 0,78 г 2-пентилтиазоло[4,5-с]хинолин-5N-оксида в 50 мл дихлорметана добавляли 0,51 мл трихлорацетилизоцианата. Реакционную смесь перемешивали при комнатной температуре в течение около 75 минут и затем концентрировали в вакууме. В результате чего получали неочищенный N-(2-пентилтиазоло[4,5-с]хинолин-4-ил)трихлорацетамид. К амиду добавляли 40 мл 2 М раствора аммиака в метаноле и затем дихлорметан, чтобы перевести весь продукт в раствор. По завершении реакции, о чем судили по данным анализа способом тонкослойной хроматографии, реакционную смесь концентрировали в вакууме. Остаток смешивали с дихлорметаном и водным раствором бикарбоната натрия. Органический слой отделяли, промывали сначала водным раствором бикарбоната натрия, а затем водой, сушили над сульфатом магния и затем концентрировали в вакууме до получения твердого белого продукта. После перекристаллизации этого вещества из этилацетата получали желтовато-белое кристаллическое вещество, представляющее собой 2-пентилтиазоло[4,5-с]хинолин-4-амин, плавящийся при 119-121° С. Анализ: рассчитано для С15Н17N3S: %С, 66,39; %Н, 6,31; %N, 15,48. Найдено: %С, 66,21; %Н, 6,35; %N, 15,39.
Пример 17
2-Бутилтиазоло[4,5-с]хинолин-5N-оксид

Часть А
Используя методику, приведенную в части А примера 7, сначала проводили реакцию между 2-метилтиазоло[4,5-с]хинолином (2,50 г, 12,5 ммоль) и диизопропиламидом лития (7,0 мл 2 М раствора в бензоле), а затем обрабатывали полученный продукт 1-иодпропаном (3,0 г). В результате получали 1,19 г желтого маслообразного продукта, представляющего собой 2-бутилтиазоло[4,5-с]хинолин.
Часть В
Используя методику, приведенную в части В примера 15, 2-бутилтиазоло[4,5-с]хинолин (1,33 г) окисляли надуксусной кислотой. В результате получали 0,5 г твердого 2-бутилтиазоло[4,5-с]хинолин-5N-оксида, температура плавления которого составляла 133-135 °С.
Пример 18
2-Бутилтиазоло[4,5-с]хинолин-4-амин

Используя методику, приведенную в примере 16, 2-бутилтиазоло[4,5-с]хинолин-5N-оксид (0,50 г) превращали в соответствующий амид, который затем подвергали гидролизу, в результате чего получили 0,25 г желтого кристаллического 2-бутилтиазоло[4,5-с]хинолин-4-амина, температура плавления которого составляла 149-151 ° С. Анализ: рассчитано для С14Н15N3S: %С, 65,34; %Н, 5,87; %N, 16,33. Найдено: %С, 64,88; %Н, 5,84; %N, 16,03.
Пример 19
2-( 1 -Метилэтил)тиазоло[4,5-с]хинолин-5N-оксид
Часть А
Используя методику, приведенную в части А примера 7, 2-метилтиазоло[4,5-с]хинолин (1,50 г, 7,5 ммоль) сначала обрабатывали диизопропиламидом лития (15,0 мл 2М раствора в бензоле), а затем иодистым метилом (2,4 мл). В результате получали 0,97 г желтого маслообразного продукта, представляющего собой 2-(1-метилэтил)тиазоло[4,5-с]хинолин.
Часть В
Используя методику, приведенную в части В примера 15, 2-(1-метилэтил)тиазоло[4,5-с]хинолин (0,95 г) окисляли надуксусной кислотой. В результате получили 0,84 г твердого продукта желтого цвета, представляющего собой 2-(1-метилэтил)тиазоло[4,5-с]хинолин-5N-оксид; температура плавления 161-162° С.
Пример 20
2-( 1 -Метилэтил)тиазоло[4,5-с]хинолин-4-амин

Используя методику, приведенную в примере 16, 2-(1-метилэтил)тиазоло[4,5-с]хинолин-5N-оксид (0,84 г) превращали в соответствующий амид и затем подвергали его гидролизу, в результате чего получали 0,16 г желтого продукта, имеющего форму игольчатых кристаллов и представляющего собой 2-(1-метилэтил)тиазоло[4,5-с]хинолин-4-амин, температура плавления которого составляла 163-165° С. Анализ: рассчитано для С13Н13N3S:%С, 64,17; %Н, 5,38; %N, 17,27. Найдено: %С, 63,49; %Н, 5,36; %N, 17,09.
Пример 21
2-(2-Фенил-1-этенил)тиазоло [4,5-с]хинолин-5N-оксид

Часть А
Используя методику, приведенную в части А примера 7, 2-метилтиазоло[4,5-с]хинолин (5,0 г, 25 ммоль) сначала обрабатывали диизопропиламидом лития (15,0 мл 2 М раствора в бензоле), а затем бензальдегидом (3,8 мл). В результате получали 5,3 г желтого твердого 1-фенил-2-тиазоло[4,5-с]хинолин-2-ил-1-этанола; температура плавления 147-148° С.
Часть В
К суспензии 2,16 г 1-фенил-2-тиазоло[4,5-с]хинолин-2-ил-1-этанола в 40 мл воды при нагревании на паровой бане добавляли по каплям концентрированную соляную кислоту до полного растворения твердого продукта. Нагревание реакционной смеси продолжали до полного завершения реакции, о чем судили по результатам анализа реакционной системы способом тонкослойной хроматографии. После этого реакционную смесь охлаждали до комнатной температуры, при этом наблюдалось образование осадка. Смесь нейтрализовали, добавляя к ней карбонат натрия, после чего при перемешивании добавляли дихлорметан до полного растворения осадка. Разделяли органическую и водную фазы. Водный слой экстрагировали дихлорметаном. Органический слой соединяли вместе с дихлорметановыми экстрактами, промывали водой, сушили над сульфатом магния и концентрировали в вакууме до получения 22 г твердого продукта зеленого цвета. После перекристаллизации этого продукта из этилацетата получили 1,55 г зеленого кристаллического 2-(2-фенил-1-этенил)тиазоло[4,5-с]хинолина. Анализ: рассчитано для С18Н12N3S: %С, 74,97; %Н, 4,19; %N, 9,71. Найдено: %С, 74,89; %Н, 4,17; %N, 9,72.
Часть С
К суспензии 1,20 г 2-(2-фенил-1-этенил)тиазоло[4,5-с]хинолина в 50 мл метилацетата добавляли 1,32 мл 32%-ного раствора надуксусной кислоты в уксусной кислоте; в результате появлялся осадок. К реакционной смеси добавляли этанол в количестве, необходимом до полного растворения осадка, после чего смесь кипятили с обратным холодильником в течение ночи, а затем охлаждали до комнатной температуры. Образовавшийся осадок отфильтровывали, сушили и затем перекристаллизовывали из смеси метанол/дихлорметан. В результате получили твердый 2-(2-фенил-1-этенил)тиазоло[4,5-с]хинолин-5N-оксид желтого цвета; температура плавления этого оксида 268-270° С. Анализ: рассчитано для С18Н12N2OS: %С, 71,03; %Н, 3,97;%N, 9,20. Найдено: %С, 69,94; %Н, 3,87; %N, 9,05.
Пример 22
2-(2-Фенил-1-этенил)тиазоло[4,5-с]хинолин-4-амин

Используя методику, приведенную в примере 16, 2-(2-фенил-1-этенил)тиазоло[4,5-с]хинолин-5N-оксид (0,67 г) превращали в соответствующий трихлорацетамид и затем подвергали его гидролизу, в результате чего получили 0,43 г желтого кристаллического 2-(2-фенил-1-этенил)тиазоло[4,5-с]хинолин-4-амина, температура плавления которого составляла 239-241 °С. Анализ: рассчитано для С18Н13N3S: %С, 71,26; %Н, 4,32; %N, 13,85. Найдено: %С, 70,73; %Н, 4,15; %N, 13,68.
Пример 23
2-(2-Фенил-1-этил)тиазоло[4,5-с]хинолин-5N-оксид

Часть А
К суспензии 1,16 г 2-(2-фенил-1-этенил)тиазоло[4,5-с]хинолина (пример 21, часть В) в 200 мл уксусной кислоты добавляли небольшое количество катализатора (5% палладия, нанесенного на активированный уголь). Смесь в течение суток подвергали восстановлению в аппарате Парра при давлении водорода 3,5 кг/см2. После этого катализатор отфильтровывали, фильтрат концентрировали в вакууме и остаток растворяли в дихлорметане, промывали сначала водным раствором бикарбоната натрия и затем водой, сушили над сульфатом магния и концентрировали в вакууме. В результате получили 0,88 г маслянистого твердого 2-(2-фенил-1-этил)тиазоло[4,5-с]хинолина.
Часть В
Используя методику, приведенную в части В примера 15, 2-фенилэтилтиазоло[4,5-с]хинолин (0,90 г) окисляли надуксусной кислотой. В результате получили 0,63 г оранжевого твердого 2-(2-фенил-1-этил)тиазоло[4,5-с]хинолин-5N-оксида; температура плавления 165-169° С. Анализ: рассчитано для С18Н14N2OS: %С, 70,56; %Н, 4,60; %N, 9,14. Найдено: %С, 69,59; %Н, 4,50; %N, 9,04.
Пример 24
2-(2-Фенил-1-этил)тиазоло[4,5-с]хинолин-4-амин

Используя методику, приведенную в примере 16, 2-(2-фенил-1-этил)тиазоло[4,5-с]хинолин-5N-оксид (0,63 г) превращали в соответствующий трихлорацетамид, который затем подвергали гидролизу, в результате чего получили 0,21 г 2-(2-фенил-1-этил)тиазоло[4,5-с]хинолин-4-амина в виде желтого кристаллического вещества, температура плавления которого составляла 158-159° С. Анализ: рассчитано для С18Н15N3S: %С, 70,79; %Н, 4,95; %N 13,75. Найдено: %С, 70,29; %Н, 4,90; %N, 13,66.
Пример 25
2-Метил-1 -(тиазоло[4,5-с]хинолин-2-ил)-2-пропанол-5N-оксид

Часть А
В сухую колбу, в которой находилось 8,40 г 2-метилтиазоло[4,5-с]хинолина, в атмосфере азота вводили 150 мл безводного тетрагидрофурана. Реакционную смесь охлаждали сухим льдом до -78° С и по каплям добавляли к ней 23 мл 2,0 М раствора диизопропиламида лития в бензоле. Приблизительно через 50 мин к смеси добавляли 5 мл ацетона и нагревали ее до 10° С. Через несколько часов смесь обрабатывали водой, разбавляли хлороформом и затем промывали водой. Органический слой сушили над сульфатом магния и концентрировали в вакууме. Остаток диспергировали в 200 мл воды, смесь нагревали и медленно добавляли концентрированную соляную кислоту (6 N) до полного растворения твердого продукта. К раствору добавляли активированный уголь и при перемешивании нагревали раствор в течение 5 минут. Активированный уголь отфильтровывали, фильтрат нейтрализовали бикарбонатом натрия и затем экстрагировали хлороформом. Хлороформный экстракт несколько раз промывали водой, сушили над сульфатом магния и затем концентрировали в вакууме до получения 8,0 г светло-коричневого твердого продукта. После перекристаллизации этого продукта из смеси дихлорметана и гексанов получили 5,0 г желтого кристаллического 2-метил-1-(тиазоло[4,5-с]хинолин-2-ил)-2-пропанола, температура плавления которого составляла 155-157° С. Анализ: рассчитано для С14Н14N2OS: %С, 65,08; %Н, 5,46; %N, 10,84. Найдено: %С, 64,97; %Н, 5,33; %N 10,90.
Часть В
К суспензии 3,0 г 2-метил-1-(тиазоло[4,5-с]хинолин-2-ил)-2-пропанола в 200 мл метилацетата добавляли 4,8 мл 32%-ного раствора надуксусной кислоты в уксусной кислоте. Реакционную смесь кипятили с обратным холодильником в течение ночи, после чего охлаждали до комнатной температуры и концентрировали в вакууме. Остаток растворяли в дихлорметане, к раствору добавляли бикарбонат натрия и тщательно перемешивали. Образовавшийся осадок отфильтровывали и растворяли в смеси метанола и дихлорметана. Этот раствор концентрировали в вакууме, остаток после отгонки растворителя добавляли к дихлорметану и затем полученную смесь фильтровали для удаления нерастворившегося продукта. Фильтрат концентрировали в вакууме и в результате получали 2,6 г целевого N-оксида. После перекристаллизации небольшой части этого продукта (0,2 г) из смеси метанол/вода получили твердый 2-метил-1 -(тиазоло[4,5-с]хинолин-2-ил)-2-пропанол-5N-оксид, температура плавления которого составляла 187-189° С. Анализ: рассчитано для С14Н14N2О2S • 1/3 Н2О: %С, 59,98; %Н, 5,27; %N, 9,99. Найдено: %С, 60,09; %Н, 5,03; %N, 10,00.
Пример 26
2-(4-Аминотиазоло[4,5-с]хинолин-2-ил)-1,1-диметилэтилкарбамат

К раствору 2,4 г 2-метил-1-(тиазоло[4,5-с]хинолин-2-ил)-2-пропанол-5N-оксида в 250 мл дихлорметана добавляли 3,2 мл трихлорацетилизоцианата. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, а затем концентрировали в вакууме. Остаток перемешивали в течение 2 часов с 2 М раствором аммиака в метаноле (150 мл), после чего метанол отгоняли в вакууме. Оставшийся после отгонки продукт диспергировали в смеси дихлорметана и этилацетата и затем промывали водным раствором бикарбоната натрия. Нерастворившийся продукт отфильтровывали, промывали сначала водой и затем дихлорметаном, после чего перекристаллизовывали из смеси метанола и дихлорметана. В результате получили 1,6 г твердого 2-(4-аминотиазоло[4,5-с]хинолин-2-ил)-1,1-диметилэтилкарбамата, температура плавления которого составляла 222-223° С. Анализ: рассчитано для С15Н16N4O2S: %С, 56,94; %Н, 5,09; %N, 17,70. Найдено: %С, 56,71; %H, 5,08; %N, 17,52.
Пример 27
2-(Этоксиметил)тиазоло[4,5-с]хинолин -5N-оксид

Часть А
К суспензии 4,6 г З-аминохинолин-4-тиола (26,1 ммоль) в 50 мл этоксиуксусной кислоты добавляли 6 мл этоксиацетилхлорида (53,8 ммоль). Смесь нагревали при 60° С в течение ночи, после чего концентрировали в вакууме для удаления части этоксиуксусной кислоты. При добавлении к остатку 100 мл воды образовывался осадок. Смесь подщелачивали 50%-ным раствором гидроксида натрия. Осадок отфильтровывали, промывали водой и сушили. Получили твердый пушистый 2-(этоксиметил)тиазоло[4,5-с]хинолин зеленого цвета.
Часть В
К раствору 1,0 г 2-(этоксиметил)тиазоло[4,5-с]хинолина в этаноле добавляли 1,0 мл 32%-ного раствора надуксусной кислоты в уксусной кислоте. Реакционную смесь перемешивали в течение недели при комнатной температуре, после чего концентрировали в вакууме и подвергали азеотропной перегонке с гептаном для удаления уксусной кислоты. Остаток растворяли в дихлорметане, промывали сначала водным раствором бикарбоната натрия, а затем водой, сушили над сульфатом магния и концентрировали в вакууме. После перекристаллизации оставшегося продукта из изопропанола получили желтый кристаллический 2-(этоксиметил)тиазоло[4,5-с]хинолин-5N-оксид; температура плавления 138-140° С. Анализ: рассчитано для С13Н12N2O2S: %С, 59,98; %Н, 4,65; %N 10,76. Найдено: %С, 59,85; %Н, 4,66; %N, 10,71.
Пример 28
2-(Этоксиметил)тиазоло[4,5-с]хинолин-4-амин
 К раствору 1,0 г 2-(этоксиметил)тиазоло[4,5-с]хинолин-5N-оксида в 50 мл дихлорметана добавляли 0,7 мл трихлорацетилизоцианата и перемешивали смесь при комнатной температуре в течение 2 часов, после чего концентрировали в вакууме до получения N-(2-(этоксиметил)тиазоло[4,5-с]хинолин-4-ил)трихлорацетамида. Амид обрабатывали метанолом и добавляли 1 эквивалент метилата натрия. Реакционную смесь перемешивали 30 мин при комнатной температуре и затем концентрировали в вакууме. Реакцию проводили еще один раз, используя 2 г N-оксида. Полученные в обоих опытах продукты соединили вместе и перекристаллизовали из изопропанола. В результате получили 2,25 г слабо-желтого 2-(этоксиметил)тиазоло[4,5-с]хинолин-4-амина, имеющего форму игольчатых кристаллов. Температура плавления этого продукта 149-151° С. Анализ: рассчитано для С13Н13N3OS: %С, 60,21; %Н, 5,05; %N, 16,20. Найдено: %С, 59,86; %Н, 4,97; %N, 16,16.
К раствору 1,0 г 2-(этоксиметил)тиазоло[4,5-с]хинолин-5N-оксида в 50 мл дихлорметана добавляли 0,7 мл трихлорацетилизоцианата и перемешивали смесь при комнатной температуре в течение 2 часов, после чего концентрировали в вакууме до получения N-(2-(этоксиметил)тиазоло[4,5-с]хинолин-4-ил)трихлорацетамида. Амид обрабатывали метанолом и добавляли 1 эквивалент метилата натрия. Реакционную смесь перемешивали 30 мин при комнатной температуре и затем концентрировали в вакууме. Реакцию проводили еще один раз, используя 2 г N-оксида. Полученные в обоих опытах продукты соединили вместе и перекристаллизовали из изопропанола. В результате получили 2,25 г слабо-желтого 2-(этоксиметил)тиазоло[4,5-с]хинолин-4-амина, имеющего форму игольчатых кристаллов. Температура плавления этого продукта 149-151° С. Анализ: рассчитано для С13Н13N3OS: %С, 60,21; %Н, 5,05; %N, 16,20. Найдено: %С, 59,86; %Н, 4,97; %N, 16,16.
Пример 29
2-(Метоксиметил)тиазоло[4,5-с]хинолин-5N-оксид

Часть А
К смеси 2,8 г З-аминохинолин-4-тиола и 15 мл метоксиуксусной кислоты добавляли 1,8 мл метоксиацетилхлорида, реакционную смесь выдерживали в течение 1 часа при 140° С и затем охлаждали до комнатной температуры. После этого добавляли небольшое количество воды, подщелачивали смесь 10%-ным раствором гидроксида натрия и затем экстрагировали 300 мл дихлорметана. Экстракт промывали сначала водным раствором бикарбоната натрия, затем водой, сушили над сульфатом магния и, наконец, концентрировали в вакууме до получения неочищенного темного маслообразного продукта. Масло растворяли в дихлорметане и очищали на хроматографической колонке с силикагелем, используя в качестве элюента 1:1 смесь гексана и этилацетата. Элюат концентрировали в вакууме и в результате получали 2,3 г твердого оранжевого 2-(метоксиметил)тиазоло[4,5-с]хинолина.
Часть В
Используя методику, приведенную в части В примера 27, проводили окисление 2-(метоксиметил)тиазоло[4,5-с]хинолина (1,7 г). В результате получили 1,8 г желтого кристаллического 2-(метоксиметил)тиазоло[4,5-с]хинолин-5N-оксида, температура плавления которого составляла 151-153° С. Анализ: рассчитано для С12H10N2ОS: %С, 58,52; %Н, 4,09; %N, 11,37. Найдено: %С, 57,95; %Н, 3,98; %N, 11,3.
Пример 30
2-(Метоксиметил)тиазоло[4,5-с]хинолин-4-амин

Используя методику, приведенную в примере 28, при обработке 2-(метоксиметил)тиазоло[4,5-с]хинолин-5N-оксида (1,3 г) получали соответствующий трихлорацетамид, который в дальнейшем подвергали гидролизу, в результате которого получали светло-желтые игольчатые кристаллы 2-(метоксиметил)тиазоло[4,5-с]хинолин-4-амина. Температура плавления полученного продукта составляла 183-185° С. Анализ: рассчитано для С12Н11N30S: %С, 58,76; %Н, 4,52; %N, 17,13. Найдено: %С, 58,69; %Н, 4,34; %N, 17,14.
Пример 31
2-(2-Метилпропил)тиазоло[4,5-с]хинолин-5N-оксид

Часть А
4,6 г З-аминохинолин-4-тиола добавляли к 80 г полифосфорной кислоты. Добавляли еще 3,5 мл изовалериановой кислоты и нагревали смесь при 140° С в течение 2 часов, после чего выливали ее в 300 мл ледяной воды. Смесь фильтровали через слой Сеlite® для удаления нерастворимого материала. Фильтрат охлаждали ледяной водой и подщелачивали 50%-ным раствором гидроксида натрия, после чего экстрагировали хлороформом. Экстракт промывали водой, сушили над сульфатом магния и затем концентрировали в вакууме до получения маслообразного продукта. Масло растворяли в дихлорметане и подвергали хроматографической очистке на силикагеле, используя в качестве элюента 1:1 смесь этилацетата и гексанов. Элюат концентрировали в вакууме и получали 2-(2-метилпропил)тиазоло[4,5-с]хинолин.
Часть В
Используя методику, приведенную в части В примера 27, проводили окисление 2-(2-метилпропил)тиазоло[4,5-с]хинолина (5,2 г). В результате получили 2,5 г твердого желтого 2-(2-метилпропил)тиазоло[4,5-с]хинолин-5N-оксида.
Пример 32
2-(2-Метилпропил)тиазоло[4,5-с]хинолин-4-амин

Используя методику, приведенную в примере 28, в результате обработки 2-(2-метилпропил)тиазоло[4,5-с]хинолин-5N-оксида (2,5 г) получали соответствующий трихлорацетамид, который в дальнейшем подвергали гидролизу, в результате которого получали светло-желтые пластинчатые кристаллы 2-(2-метилпропил)тиазоло[4,5-с]хинолин-4-амина. Температура плавления полученного продукта составляла 123-125° С. Анализ: рассчитано для С14Н15N3S: %С, 65,34; %Н, 5,87; %N, 16,33. Найдено: %С, 64,87; %Н, 5,79; %N, 16,18.
Пример 33
2-Бензилтиазоло[4,5-с]хинолин-5N-оксид

Часть А
К охлажденному раствору 2 г фенилуксусной кислоты в 10 мл дихлорметана добавляли по каплям 1,5 г тионилхлорида. Смесь перемешивали в течение часа при комнатной температуре и получали при этом раствор, содержащий фенилацетилхлорид. К суспензии 3-аминохинолин-4-ола в 10 мл дихлорметана добавляли 4,3 мл триэтиламина и полученную смесь охлаждали на ледяной бане. К этой охлажденной смеси добавляли по каплям приготовленный предварительно раствор фенилацетилхлорида. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Полученный липкий маслянистый осадок разбавляли водой (10 мл) и затем быстро перемешивали в течение 1 часа. Смесь фильтровали. Согласно данным тонкослойной хроматографии целевой продукт содержался как в выделенном при фильтровании твердом продукте, так и в фильтрате. Фильтрат разбавляли дихлорметаном и водой. Органический слой отделяли, сушили над сульфатом магния и концентрировали в вакууме. Остаток соединяли с предварительно полученным твердым продуктом и перекристаллизовывали из смеси 80:20 изопропанол:вода. В результате получили 1,3 г N-(гидроксихинолин-3-ил)фенилацетамида в виде игольчатых кристаллов. Температура плавления 253-255° С. Анализ: рассчитано для С17Н14N2О2: %С, 73,37; %Н, 5,07; %N, 10,07. Найдено: %С, 73,16; %Н, 5,03; %N, 10,07.
Часть В
К суспензии 1,0 г N-(4-гидроксихинолин-3-ил)фенилацетамида в пиридине добавляли 1,6 г пятисернистого фосфора. Смесь кипятили с обратным холодильником до полного завершения реакции, после чего реакционную смесь концентрировали в вакууме и подвергали азеотропной перегонке с водой для удаления пиридина. Остаток соединяли с водой, нейтрализовали карбонатом натрия и затем экстрагировали дихлорметаном. Экстракт промывали водой, сушили над сульфатом магния и концентрировали в вакууме до получения твердого 2-бензилтиазоло[4,5-с]хинолина.
Часть С
Используя методику, приведенную в части В примера 27, 2-бензилтиазоло[4,5-с]хинолина (3,3 г) окисляли до получения 2,1 г 2-бензилтиазоло[4,5-с]хинолин-5N-оксида. Целевой продукт получали в виде желтого твердого вещества, температура плавления которого составляла 185-186° С. Анализ: рассчитано для С17Н12N2OS: %С, 69,84; %Н, 4,14; %N 9,58. Найдено: %С, 69,51; %Н, 4,06; %N, 9,55.
Пример 34
Солянокислый 2-бензилтиазоло[4,5-с]хинолин-4-амин

1,2 мл трихлорацетилизоцианата (10,3 ммоль) добавляли к раствору 2,0 г 2-бензилтиазоло[4,5-с]хинолин-5N-оксида (6,8 ммоль) в 100 мл дихлорметана. Смесь перемешивали 2 часа при комнатной температуре и затем концентрировали. В результате получали неочищенный N-(2-бензилтиазоло[4,5-с]хинолин-4-ил)трихлорацетамид. Этот амид растворяли в метаноле и к раствору добавляли 1 эквивалент метилата натрия. Реакционную смесь нагревали в течение 30 минут на паровой бане и затем охлаждали до комнатной температуры. Образовавшийся осадок отфильтровывали и диспергировали в смеси метанола и изопропанола. К суспензии добавляли 1 эквивалент соляной кислоты, при этом происходило растворение твердого продукта. При стоянии выкристаллизовывалось белое твердое вещество. Его отфильтровывали, промывали изопропанолом и сушили. В результате получили 1,5 г солянокислого 2-бензилтиазоло[4,5-с]хинолин-4-амина, температура плавления которого составляла 152-155° С. Анализ: рассчитано для С17Н13N3S • НСl: %С, 62,28; %Н, 4,30; %N, 12,82. Найдено: %С, 62,05; %Н, 4,23; %N, 12,82.
Пример 35
8-Метил-2-пропилтиазоло[4,5-с]хинолин-5N-оксид

Часть А
К раствору 1 г 6-метил-3-нитрохинолин-4-ола в 25 мл этанола и 0,5 мл гидроксида аммония добавляли 0,10 г катализатора, представляющего собой активированный уголь с нанесенной на него платиной (10%). Восстановление 6-метил-3-нитрохинолин-4-ола проводили в атмосфере водорода в аппарате Парра при комнатной температуре. По окончании восстановления катализатор отфильтровывали и реакционную смесь концентрировали в вакууме. Остаток соединяли с водой и нагревали. Для полного растворения твердого продукта к смеси добавляли по каплям соляную кислоту и затем к раствору добавляли активированный уголь. Через некоторое время смесь фильтровали и к фильтрату добавляли 2 мл концентрированной (12 N) соляной кислоты. Такую перекристаллизацию проводили три раза, в результате чего получили 0,50 г солянокислого 3-амино-6-метилхинолин-4-ола. Температура плавления этого продукта превышала 310° С. Анализ: рассчитано для C10H10N2O· HCl: %С, 57,02; %Н, 5,26; %N, 13,30. Найдено: %С, 56,92; %Н, 5,16; %N, 13,24.
Часть В
К суспензии солянокислого 3-амино-6-метилхинолин-4-ола в 400 мл дихлорметана добавляли сначала 11,46 мл триэтиламина, а затем 4,46 мл хлорангидрида масляной кислоты. Смесь нагревали на паровой бане в течение 30 мин, после чего разбавляли бикарбонатом натрия и фильтровали. Фильтрат промывали водным раствором бикарбоната натрия и затем концентрировали в вакууме. После перекристаллизации остатка из изопропанола получали полугидрат 3-бутироамидо-5-метилхинолин-4-ола; температура плавления 274-277° С. Анализ: рассчитано для С14Н16N2O2·1/2 H2O: %С, 66,39; %Н, 6,76; %N, 11,06. Найдено: %С, 66,56; %Н, 6,46; %N, 11,03.
Часть С
К смеси 7,12 г полугидрата 3-бутироамидо-6-метилхинолин-4-ола в пиридине добавляли 12,9 г пятисернистого фосфора. Реакционную смесь кипятили с обратным холодильником в течение 90 мин, после чего добавляли в нее смесь льда и карбоната натрия и экстрагировали дихлорметаном. Экстракт концентрировали в вакууме. Остаток разбавляли толуолом и снова концентрировали в вакууме для получения неочищенного твердого вещества. Полученный продукт очищали на хроматографической колонке, используя в качестве элюента 20%-ный раствор дихлорметана в этилацетате. В результате получили твердый 8-метил-2-пропилтиазоло[4,5-с]хинолин желтого цвета.
Часть D
Используя методику, приведенную в части В примера 27, 8-метил-2-пропилтиазоло[4,5-с]хинолин (4,0 г) окисляли с помощью 3-хлорпербензойной кислоты. Полученный неочищенный продукт (4,19 г) перекристаллизовывали из изопропанола и в результате получили 2,0 г твердого 8-метил-2-пропилтиазоло[4,5-с]хинолин-5N-оксида; температура плавления 143-145° С. Анализ: рассчитано для С14Н14N2ОS: %С, 65,09; %Н, 5,46; %N, 10,84. Найдено: %С, 64,86; %Н, 5,40; %N 10,88.
Пример 36
8-Метил-2-пропилтиазоло[4,5-с]хинолин-4-амин
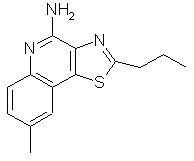
Используя методику, приведенную в примере 28, превращали 8-метил-2-пропилтиазоло[4,5-с]хинолин-5N-оксид в N-(8-метил-2-пропилтиазоло[4,5-с]хинолин-4-ил)трихлорацетамид, который в дальнейшем подвергали гидролизу, в результате которого получили 1,32 г твердого кристаллического 8-метил-2-пропилтиазоло[4,5-с]хинолин-4-амина; температура плавления полученного продукта составляла 147-149° С. Анализ: рассчитано для С14Н15N3S: %С, 65,34; %Н, 5,87; %N, 16,33. Найдено: %С, 64,97; %Н, 5,76; %N, 16,25.
Пример 37
(4-Аминотиазоло[4,5-с]хинолин-2-ил)метанол

Часть А
К суспензии 5 г 3-аминохинолин-4-ола в 50 мл дихлорметана добавляли 7,3 мл триэтиламина. Смесь охлаждали на ледяной бане и по каплям добавляли к ней 3 мл ацетоксиацетилхлорида, после чего перемешивали при комнатной температуре в течение 2 часов. Полученный осадок разбавляли водой (10 мл), быстро перемешивали в течение 10 мин и отфильтровывали. Согласно данным тонкослойной хроматографии целевое вещество содержалось как в твердом продукте, так и в фильтрате. Фильтрат концентрировали в вакууме, полученный остаток смешивали с водой и отфильтровывали. Твердый продукт, полученный на обеих стадиях, соединяли вместе и перекристаллизовывали из смеси 80:20 изопропанол : вода. В результате получили N-(4-гидроксихинолин-3-ил)ацетоксиацетамид; температура плавления 224-225° С.
Часть В
Используя методику, приведенную в части В примера 33, проводили реакцию между N-(4-гидроксихинолин-3-ил)ацетоксиацетамидом (5,3 г) и пятисернистым фосфором. В результате получили 2,9 г твердого тиазоло[4,5-с]хинолин-2-ил-метилацетата.
Часть С
Используя методику, приведенную в части В примера 27, 2,8 г тиазоло[4,5-с]хинолин-2-ил-метилацетата окисляли с помощью надуксусной кислоты в твердый кристаллический продукт коричневого цвета, представляющий собой тиазоло[4,5-с]хинолин-2-ил-метилацетат-5N-оксид.
Часть D
0,65 мл трихлорацетилизоцианата добавляли к раствору 1,0 г тиазоло[4,5-с]хинолин-2-ил-метилацетат-5N-оксида в 50 мл дихлорметана. Смесь перемешивали 2 часа при комнатной температуре и затем концентрировали в вакууме. Остаток растворяли в метаноле. К раствору добавляли 1 эквивалент метилата натрия и смесь перемешивали в течение ночи при комнатной температуре. Полученный осадок отфильтровывали и сушили. В результате получили 0,68 г белого твердого (4-аминотиазоло[4,5-с]хинолин-2-ил)метанола; температура плавления 247-249° С. Анализ: рассчитано для С11Н9N3ОS: %С, 57,13; %Н, 3,92; %N, 18,17. Найдено: %С, 56,85; %Н, 3,96; %N, 17,83.
Пример 38
2-Метилоксазоло[4,5-с]хинолин-4-амин

Часть А
6 г 3-аминохинолин-4-ола кипятили с 8 эквивалентами уксусного ангидрида до полного завершения реакции, которое определяли способом тонкослойной хроматографии. Реакционную смесь охлаждали, разбавляли ледяной водой и подщелачивали 10%-ным раствором гидроксида натрия, после чего экстрагировали дихлорметаном. Экстракт промывали водой и солевым раствором, сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали на хроматографической колонке (силикагель; элюент смесь метанола с этилацетатом). В результате получили 5,1 г 2-метилоксазоло[4,5-с]хинолина.
Часть В
Смесь 5,0 г 2-метилоксазоло[4,5-с]хинолина, 5 эквивалентов надуксусной кислоты и этанола перемешивали при комнатной температуре. Через 2 часа добавляли дополнительное количество надуксусной кислоты (2 эквивалента) и перемешивание продолжали еще 3 часа. Реакционную смесь концентрировали в вакууме. Остаток подвергали азеотропной перегонке с гептаном, после чего получили 4,2 г 2-метилоксазоло[4,5-с]хинолин-5N-оксида.
Часть С
3,6 мл трихлорацетилизоцианата медленно добавляли к охлажденной смеси 4,0 г 2-метилоксазоло[4,5-с]хинолин-5N-оксида и дихлорметана. Реакционную смесь перемешивали в течение нескольких часов и затем концентрировали в вакууме. К полученному неочищенному N-(2-метилоксазоло[4,5-с]хинолин-4-ил)трихлорацетамиду добавляли 2 М раствор аммиака в метаноле и смесь перемешивали 1 час. Реакционную смесь концентрировали в вакууме, разбавляли водой и затем экстрагировали дихлорметаном. Экстракт промывали водой и рассолом, сушили над сульфатом магния и вновь концентрировали в вакууме. Остаток очищали на хроматографической колонке (силикагель; элюент смесь этилацетата с гексаном). В результате получили 1,2 г твердого 2-метилоксазоло[4,5-с]хинолин-4-амина; температура плавления 195-197° С. Анализ: рассчитано для С11Н9N3О: %С, 66,32; %Н, 4,55; %N, 21,09. Найдено: %С, 65,96; %Н, 4,44; %N, 20,68.
Пример 39
2-Этилоксазоло[4,5-с]хинолин-4-амин
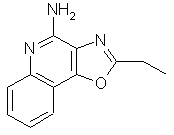
Часть А
6 г солянокислого 3-аминохинолин-4-ола кипятили с обратным холодильником с 8 эквивалентами пропионового ангидрида до полного завершения реакции, которое определяли способом тонкослойной хроматографии. Реакционную смесь охлаждали, разбавляли ледяной водой и подщелачивали 10%-ным раствором гидроксида натрия, после чего экстрагировали дихлорметаном. Экстракт промывали водой и солевым раствором, сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали на хроматографической колонке (силикагель; элюент смесь метанола с этилацетатом). В результате получили 4,0 г 2-этилоксазоло[4,5-с]хинолина.
Часть В
Смесь 3,5 г 2-этилоксазоло[4,5-с]хинолина, 4,5 мл 32%-ного раствора надуксусной кислоты в уксусной кислоте и 40 мл метилацетата нагревали в течение нескольких часов при 50° С, после чего концентрировали в вакууме. Остаток смешивали с гексаном и после фильтрования получали 2,5 г твердого 2-этилоксазоло[4,5-с]хинолин-5N-оксида.
Часть С
2 мл трихлорацетилизоцианата медленно добавляли к охлажденной смеси 2-этилоксазоло[4,5-с]хинолин-5N-оксида (2,5 г) и дихлорметана. Реакционную смесь перемешивали в течение нескольких часов и затем концентрировали в вакууме. К полученному неочищенному N-(2-этилоксазоло[4,5-с]хинолин-4-ил)трихлорацетамиду добавляли 2 М раствор аммиака в метаноле и смесь перемешивали 1 час. Реакционную смесь концентрировали в вакууме, разбавляли водой и затем экстрагировали дихлорметаном. Экстракт промывали водой и рассолом, сушили над сульфатом магния и вновь концентрировали в вакууме. Остаток очищали на хроматографической колонке (силикагель; элюент смесь этилацетата с гексаном). В результате получили твердый 2-этилоксазоло[4,5-с]хинолин-4-амин; температура плавления 175-178° С. Анализ: рассчитано для С12Н11N3О: %С, 67,59; %Н, 5,20; %N, 19,71. Найдено: %С, 67,19; %Н, 4,86; %N, 20,43.
Пример 40
2-Бутилоксазоло[4,5-с]хинолин-5N-оксид
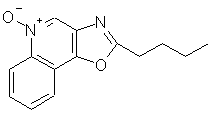
Часть А
Смесь 1,97 г солянокислого З-аминохинолин-4-ола (10,0 ммоль), 1,01 г триэтиламина (10,1 ммоль) и 9,3 г ангидрида валерьяновой кислоты (50,0 ммоль) кипятили с обратным холодильником в течение 18 часов, после чего охлаждали до комнатной температуры и выливали в лед. С помощью 10%-ного раствора гидроксида натрия рН смеси доводили до 12. Смесь перемешивали до тех пор, пока не таял весь лед, и затем экстрагировали диэтиловым эфиром. Эфирные экстракты сушили над сульфатом магния и концентрировали в вакууме до получения коричневого твердого продукта. Этот продукт очищали способом флеш-хроматографии, используя в качестве элюента смесь 3:2 этилацетата и дихлорметана. В результате получили 1,45 г 2-бутилоксазоло[4,5-с]хинолина.
Часть В
При перемешивании к раствору 1,4 г 2-бутилоксазоло[4,5-с]хинолина (6,2 ммоль) в 50 мл этанола добавляли 1,6 г 32%-ного раствора надуксусной кислоты в уксусной кислоте (6,8 ммоль). Смесь перемешивали при комнатной температуре в течение 3 суток и затем дезактивировали, добавляя насыщенный раствор карбоната калия. Разделяли органический и водный слои. Органический слой концентрировали в вакууме до получения коричневого твердого продукта. Этот продукт смешивали с диэтиловым эфиром и затем отфильтровывали. В результате получили 0,6 г 2-бутилоксазоло[4,5-с]хинолин-5N-оксида, температура плавления которого составляла 120-121° С. Анализ: рассчитано для С14Н14N2O2: %С, 69,41; %Н, 5,82; %N, 11,56. Найдено: %С, 69,22; %Н, 5,76; %N,11,59.
Пример 41
2-Бутилоксазоло[4,5-с]хинолин-4-амин

0,6 г трихлорацетилизоцианата (3,40 ммоль) в атмосфере азота добавляли при перемешивании к раствору 0,55 г 2-бутилоксазоло[4,5-с]хинолин-5N-оксида (2,27 ммоль) в 20 мл безводного дихлорметана. Реакционную смесь выдерживали в течение 2 часов при комнатной температуре и затем концентрировали в вакууме до получения неочищенного маслообразного N-(2-бутилоксазоло[4,5-с]хинолин-4-ил)трихлорацетамида. Этот продукт переносили в 25 мл метанола и к раствору добавляли 0,49 мл 25%-ного раствора метилата натрия (2,27 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 2 часов и затем концентрировали в вакууме. Остаток смывали этилацетатом и промывали водой. Органический слой концентрировали в вакууме до получения твердого продукта оранжевого цвета. Этот продукт дважды очищали способом флеш-хроматографии, используя в качестве элюента при первой очистке этилацетат, а при второй 30%-ный раствор дихлорметана в этилацетате. В результате получили 0,15 г 2-бутилоксазоло[4,5-с]хинолин-4-амина; температура плавления 96-98° С. Анализ: рассчитано для С14Н15N3О: %С, 69,69; %Н, 6,27; %N, 17,41. Найдено: %С, 69,23; %Н, 6,06; %N, 17,07.
Пример 42
2-Пропилтиазоло[4,5-с]хинолин-4,8-диамин

Часть А
К раствору 2-пропилтиазоло[4,5-с]хинолин-4-амина (1 г, 4,11 ммоль; из примера 12) в серной кислоте (10 мл) добавляли 0,46 г азотнокислого натрия (4,52 ммоль). Смесь перемешивали 30 мин при комнатной температуре, после чего выливали в лед, нейтрализовали (до рН 7) гидроксидом аммония (150 мл) и экстрагировали дихлорметаном. Экстракт промывали водным раствором бикарбоната натрия, сушили над сульфатом магния и затем концентрировали в вакууме до получения 1 г желтого твердого продукта. После перекристаллизации этого продукта из смеси изопропанол/вода получили 0,84 г твердого желтого 8-нитро-2-пропилтиазоло[4,5-с]хинолина, имеющего температуру плавления 228-230° С. Анализ: рассчитано для С13Н12N4О2S: %С, 54,15; %Н, 4,20; %N, 19,43. Найдено: %С, 54,22; %Н, 4,05; %N, 19,04.
Часть В
К раствору 1,31 г 8-нитро-2-пропилтиазоло[4,5-с]хинолина в этаноле добавляли 0,13 г катализатора (палладий, нанесенный на активированный уголь). Смесь восстанавливали водородом в аппарате Парра. После этого катализатор отфильтровывали и промывали его на фильтре этанолом. Фильтрат концентрировали в вакууме при 50° С и затем сушили в атмосфере азота. В результате получили желтые кристаллы 2-пропилтиазоло[4,5-с]хинолин-4,8-диамина; температура плавления 190-192° С. Анализ: рассчитано для С13H14N4S: %С, 60,44; %Н, 5,46; %N, 21,69. Найдено: %С, 60,11; %Н, 5,45; %N 21,96.
Пример 43
2-Пропилоксазоло[4,5-с]хинолин-4-амин

Часть А
Смесь 1,97 г солянокислого З-аминохинолин-4-ола (10,0 ммоль), 3,15 г ангидрида масляной кислоты (20 ммоль) и 25 мл пиридина кипятили с обратным холодильником в течение ночи, после чего охлаждали и выливали в лед. Смесь подщелачивали 1 N раствором гидроксида натрия (до рН 11) и затем экстрагировали тремя порциями диэтилового эфира по 100 мл каждая. Осадок отфильтровывали. Эфирные экстракты соединяли вместе, сушили над сульфатом магния и затем концентрировали до получения 1,1 г твердого 2-пропилоксазоло[4,5-с]хинолина желтовато-белого цвета.
Часть В
К раствору 1,0 г 2-пропилоксазоло[4,5-с]хинолина (4,7 ммоль) в 30 мл хлороформа при перемешивании добавляли 3-хлорпербензойную кислоту (1 эквивалент; в виде 60%-ного раствора). Смесь перемешивали при комнатной температуре в течение ночи и затем дезактивировали, добавляя к ней насыщенный раствор карбоната калия. Разделяли органический и водный слои. Водный слой экстрагировали дихлорметаном. Органические слои соединяли вместе и концентрировали. Полученный продукт очищали способом флеш-хроматографии, используя в качестве элюента 8:2 смесь этилацетата и дихлорметана. В результате получили 1,0 г твердого 2-пропилоксазоло[4,5-с]хинолин-5N-оксида коричневого цвета.
Часть С
0,9 г трихлорацетилизоцианата (5,25 ммоль) при перемешивании добавляли к раствору 0,8 г 2-пропилоксазоло[4,5-с]хинолин-5N-оксида (3,5 ммоль) в 30 мл хлороформа. Реакционную смесь перемешивали в течение 2,5 часов при комнатной температуре и затем концентрировали в вакууме до получения неочищенного N-(2-пропилоксазоло[4,5-с]хинолин-4-ил)трихлорацетамида. Этот амид растворяли в 50 мл метанола, добавляли к раствору 1 эквивалент метилата натрия (в виде 25%-ного раствора) и смесь кипятили с обратным холодильником в течение 2 часов. Реакционную смесь концентрировали в вакууме, остаток смывали диэтиловым эфиром и водой. Эфирный слой отделяли и концентрировали в вакууме до получения твердого коричневого продукта. Этот продукт очищали способом флеш-хроматографии, используя две хроматографические колонки (в первой колонке в качестве элюента использовали 8:2 смесь этилацетата с дихлорметаном, во второй 1:1 смесь этилацетата и дихлорметана). В результате получили 0,1 г 2-пропилоксазоло[4,5-с]хинолин-4-амина в виде желтого порошка; температура плавления 159,0-160,0° С. Анализ: рассчитано для С13Н13N3О: %С, 68,71; %Н, 5,77; %N, 18,49. Найдено: %С, 68,03; %Н, 5,77; %N, 18,14.
Пример 44
8-Бром-2-пропилтиазоло[4,5-с]хинолин-4-амин

1,0 г 2-пропилтиазоло[4,5-с]хинолин-4-амина (0,41 ммоль) добавляли к 15 мл уксусной кислоты и смесь нагревали до 60° С. Затем по каплям добавляли 0,10 мл брома (1,94 ммоль), смесь перемешивали при 60° С в течение 18 часов, после чего разбавляли ее водой и образовавшийся осадок отфильтровывали. В результате получили 0,25 г желтого твердого 8-бром-2-пропилтиазоло[4,5-с]хинолин-4-амина; температура плавления 177-180° С. Анализ: рассчитано для С13Н12ВrN3S: %С, 48,46; %Н, 3,75; %N, 13,04. Найдено: %С, 47,98; %Н, 3,95; %N, 12,70.
Пример 45
7-Метил-2-пропилтиазоло[4,5-с]хинолин-4-амин

Часть А
Смесь 37,8 мл диэтилэтоксиметилмалоната (187 ммоль) и 20 мл м-толуидина (187 ммоль) нагревали при 100° С в течение около 3 часов. При охлаждении смеси до комнатной температуры она затвердевала. К этой затвердевшей смеси добавляли 350 мл растворителя Dowtherm А и полученный раствор кипятили с обратным холодильником в течение около 30 минут, после чего смесь охлаждали до комнатной температуры. Образовавшийся осадок отфильтровывали, споласкивали ацетоном и сушили. В результате получили 33 г порошка этил-4-гидрокси-7-метил-3-хинолинкарбоксилата коричневого цвета.
Часть В
32 г этил-4-гидрокси-7-метил-3-хинолинкарбоксилата (138 ммоль) диспергировали в 500 мл 10%-ного водного раствора гидроксида натрия и затем кипятили с обратным холодильником в течение около 30 минут. Полученную смесь охлаждали до комнатной температуры и подкисляли ее концентрированной соляной кислотой. Образовавшийся при этом осадок отфильтровывали, хорошо споласкивали водой и затем сушили в печи. В результате получили 28 г 4-гидрокси-7-метил-3-хинолинкарбоновой кислоты. 2 г этого продукта дважды перекристаллизовывали из Н,N-диметилформамида, при этом получали пушистое белое твердое вещество, температура плавления которого составляла 264-265° С. Анализ: рассчитано для С11Н9NO3: %С, 65,02; %Н, 4,46;%N, 6,89. Найдено: %С, 65,22; %Н, 4,42; %N, 6,88.
Часть С
32 г 4-гидрокси-7-метил-3-хинолинкарбоновой кислоты загружали в круглодонную колбу. Затем колбу нагревали в бане, заполненной расплавленным сплавом Вуда, при 310° С в течение нескольких минут до тех пор, пока продукт полностью не расплавится с образованием светло-коричневой вязкой жидкости и не прекратится выделение пузырей. После этого реакционную смесь охлаждали до комнатной температуры и продукт перекристаллизовывали из смеси этилацетата и этанола. В результате получали 9,8 г 7-метил-4-хинолинола. В процессе перекристаллизации часть твердого продукта не растворялась; этот материал отфильтровывали и затем вновь перекристаллизовывали, при этом получили 1,1 г 7-метил-4-хинолинола в виде желто-коричневых пластинчатых кристаллов; температура плавления 233-235° С. Анализ: рассчитано для С10H9NО: %С, 75,45; %Н, 5,70; %N, 8,80. Найдено: %С, 75,23; %Н, 5,54; %N, 8,76.
Часть D
К горячему раствору (125° С) 10,5 г 7-метил-4-хинолинола в 125 мл пропионовой кислоты медленно добавляли 6 мл 70%-ной азотной кислоты. Смесь перемешивали в течение около 1,5 часа и затем охлаждали до комнатной температуры. Полученный осадок отфильтровывали, хорошо споласкивали этанолом и водой и сушили. В результате получили 6,9 г слабо-желтого твердого 7-метил-З-нитро-4-хинолинола.
Часть Е
Смесь, содержащую 11,8 г 7-метил-3-нитрохинолинола (58 ммоль), около 300 мл метанола, 50 мл гидроксида аммония и 1 г катализатора (10% палладия, нанесенных на активированный уголь), загружали в аппарат Парра и проводили реакцию восстановления в атмосфере водорода (при давлении 2,4 - 2,8 кг/см2) в течение около 1 часа. Реакционную смесь пропускали через слой фильтрующего материала Сеlite® и твердый осадок на фильтре тщательно промывали метанолом. Фильтрат обрабатывали активированным углем и затем концентрировали в вакууме до получения пушистого твердого продукта бледно-зеленого цвета. После обработки этого продукта ацетонитрилом получили 8,5 г 3-амино-7-метил-4-хинолинола.
Часть F
К суспензии 800 мг 3-амино-7-метил-4-хинолинола (4,6 ммоль) в 30 мл дихлорметана в атмосфере азота добавляли сначала 0,71 мл триэтиламина (5,1 ммоль), а затем 0,53 мл хлорангидрида масляной кислоты (5,1 ммоль). Смесь перемешивали при комнатной температуре в течение около 2 часов. После этого с помощью способа тонкослойной хроматографии (силикагель и элюент - смесь 9:1 дихлорметан-метанол) было установлено, что в реакционной смеси еще содержался исходный продукт. Поэтому реакционную смесь нагревали до кипения и затем позволяли ей сохнуть в течение около 30 минут. После этого вводили дополнительное количество растворителя и смесь кипятили с обратным холодильником еще в течение 1 часа. После этого анализ с помощью тонкослойной хроматографии не показывал наличия исходного материала в реакционной смеси. Полученный осадок отфильтровывали, споласкивали дихлорметаном и водой и получали 650 мг бледно-коричневых кристаллических пластинок N-(4-гидрокси-7-метилхинолин-4-ил)бутирамида.
Часть G
К смеси 630 мг N-(4-гидрокси-7-метилхинолин-4-ил)бутирамида (2,6 ммоль) в 20 мл пиридина в атмосфере азота добавляли 1,15 г пятисернистого фосфора (2,6 ммоль) и смесь нагревали до кипения. Реакционная смесь становилась ярко-желтой, и весь твердый продукт переходил в раствор. Смесь кипятили с обратным холодильником около 2 часов,и затем охлаждали до комнатной температуры и экстрагировали водой, водным раствором бикарбоната натрия и дихлорметаном. Органический слой обрабатывали насыщенным раствором сульфата меди, сушили над сульфатом магния и концентрировали в вакууме до получения маслообразного продукта. Этот продукт сушили в высоком вакууме и в результате получили 410 мг твердого оранжевого 7-метил-2-пропилтиазоло[4,5-с]хинолина.
Часть Н
К смеси 2 г 7-метил-2-пропилтиазоло[4,5-с]хинолина и 100 мл хлороформа добавляли 2,4 г 3-хлорпербензойной кислоты (в виде 57-86%-ного раствора). Смесь перемешивали 2 часа при комнатной температуре. Анализ с помощью тонкослойной хроматографии не показал наличия никакого исходного материала в реакционной смеси, но обнаружил два новых продукта. Смесь перемешивали при комнатной температуре еще в течение 1 часа и затем экстрагировали дихлорметаном и водным раствором бикарбоната натрия. Органический слой сушили над сульфатом магния и концентрировали в вакууме до получения желто-оранжевого масла. Это масло сушили в высоком вакууме и в результате получили 2,1 г твердого 7-метил-2-пропилтиазоло[4,5-с]хинолин-5N-оксида.
Часть I
К смеси 2,1 г 7-метил-2-пропилтиазоло[4,5-с]хинолин-5N-оксида (8,1 ммоль) и 100 мл дихлорметана в атмосфере азоте добавляли 1,4 мл трихлорацетилизоцианата (12,1 ммоль). Полученный темно-коричневый раствор перемешивали при комнатной температуре в течение около 2 часов, после чего реакционную смесь концентрировали в вакууме до получения маслообразного N-(7-метил-2-пропилтиазоло[4,5-с]хинолин-4-ил)трихлорацетамида. К этому маслу добавляли метанол и 1,9 мл 25%-ного раствора метилата натрия в метаноле (8,1 ммоль) и перемешивали при комнатной температуре 1 час. Полученный осадок отфильтровывали и дважды перекристаллизовывали из изопропанола. В результате получили 500 мг 7-метил-2-пропилтиазоло[4,5-с]хинолин-4-амина в виде желто-коричневого порошка; температура плавления 186-187° С. Анализ: рассчитано для С14Н15N3S: %С, 65,34; %Н, 5,87; %N, 16,33. Найдено: %С, 64,95; %Н, 5,77; %N, 16,08.
Пример 46
2-Бутил-7-метилоксазоло[4,5-с]хинолин-4-амин

Часть А
Смесь 5 г 3-амино-7-метил-4-хинолинола (28,7 ммоль) и 28 мл валерьянового ангидрида (143,5 ммоль) кипятили с обратным холодильником в атмосфере азота в течение около 20 часов. После этого смесь охлаждали до комнатной температуры, подщелачивали 10%-ным раствором гидроксида натрия и перемешивали еще 1 час, а затем экстрагировали дихлорметаном. Экстракт сушили над сульфатом магния и концентрировали в вакууме до получения темно-коричневой жидкости. Эту жидкость подвергали хроматографической очистке (колонки с силикагелем, в качестве элюента использовали смесь 3:2 этилацетат: дихлорметан) и в результате получили 4,7 г темно-коричневого полутвердого соединения. Часть этого соединения (около 700 мг) очищали на хроматографической колонке, заполненной силикагелем (в качестве элюента использовали смесь 95:5 дихлорметан: метанол). Очищенный таким образом продукт представлял собой 2-бутил-7-метилоксазоло[4,5-с]хинолин, температура плавления которого составляла 52-55° С. Анализ: рассчитано для С15Н16N2О: %С, 74,97; %Н, 6,71; %N, 11,66. Найдено: %С, 74,80; %Н, 6,73; %N 11,53.
Часть В
К раствору 3,9 г 2-бутил-7-метилоксазоло[4,5-с]хинолина (16,2 ммоль) в 100 мл хлороформа добавляли 4,6 г 3-хлорпербензойной кислоты (в виде 57-86%-ного раствора). Смесь перемешивали 4 часа при комнатной температуре, после чего промывали водным раствором бикарбоната натрия, сушили над сульфатом магния и концентрировали в вакууме до получения 4,2 г темно-коричневого-оранжевого масла, представляющего собой 2-бутил-7-метилоксазоло [4,5-с]хинолин-5N-оксид.
Часть С
К смеси 4,2 г 2-бутил-7-метилоксазоло[4,5-с]хинолин-5N-оксида (16 ммоль) и 100 мл дихлорметана в атмосфере азота добавляли 2,9 мл трихлорацетилизоцианата (24 ммоль). Смесь перемешивали при комнатной температуре в течение около 3 часов, после чего концентрировали в вакууме. Образовавшийся остаток смывали метанолом и соединяли с 3,7 мл 25%-ного раствора метилата натрия в метаноле (16 ммоль), а затем перемешивали при комнатной температуре в течение ночи. Метанол испаряли и оставшийся остаток очищали на хроматографической колонке (силикагель; элюент - смесь 95:5 дихлорметан:метанол). В результате получали твердое коричневое вещество. Перекристаллизация этого вещества из ацетонитрила давала 550 мг мелких игольчатых кристаллов коричневого цвета, представляющих собой 2-бутил-7-метилоксазоло[4,5-с]хинолин-4-амин; температура плавления 187-188° С. Анализ: рассчитано для С15Н17N3О + 0,1 Н2O: %С, 70,07; %Н, 6,74; %N, 16,34. Найдено: %С, 70,07; %Н, 6,49; %N, 16,58.
Пример 47
7-Метил-2-пропилоксазоло[4,5-с]хинолин-4-амин

Часть А
Смесь 3,4 г 3-амино-7-метил-4-хинолинола (20 ммоль) и 16 мл масляного ангидрида кипятили с обратным холодильником в атмосфере азота в течение ночи. После этого смесь охлаждали до комнатной температуры и выливали в лед, а затем подщелачивали до рН 12 10%-ным раствором гидроксида натрия и экстрагировали дихлорметаном. Экстракт сушили над сульфатом магния и концентрировали в вакууме. Остаток после экстракции еще содержал некоторое количество исходного ангидрида, поэтому к нему добавляли 10%-ный раствор гидроксида натрия и перемешивали 1 час при комнатной температуре. Смесь вновь экстрагировали дихлорметаном; экстракт сушили над сульфатом магния и концентрировали в вакууме до получения коричневого масла. После очистки этого масла на хроматографической колонке (силикагель; элюент - смесь 3:2 этилацетат: дихлорметан) получили 3,1 г 7-метил-2-пропилоксазоло[4,5-с]хинолина в виде светло-коричневого масла, которое затвердевало при стоянии; температура плавления 65-68° С. Анализ: рассчитано для С14Н14N2О: %С, 74,31; %Н, 6,24; %N, 12,38. Найдено: %С, 73.69; %Н, 6,07; %N, 12,15.
Часть В
К раствору 3 г 7-метил-2-пропилоксазоло[4,5-с]хинолина в 100 мл хлороформа добавляли 3,8 г 3-хлорпербензойной кислоты (в виде 57-86%-ного раствора). Смесь перемешивали при комнатной температуре в течение ночи, после чего промывали дважды водным раствором бикарбоната натрия, сушили над сульфатом магния и концентрировали в вакууме до получения 3,1 г бледно-оранжевого твердого 7-метил-2-пропилоксазоло [4,5-с]хинолин-5N-оксида.
Часть С
К раствору 3,1 г 7-метил-2-пропилоксазоло[4,5-с]хинолин-5N-оксида (12,8 ммоль) в 100 мл дихлорметана в атмосфере азота добавляли 2,3 мл трихлорацетилизоцианата (19,2 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов и затем концентрировали в вакууме. К образовавшемуся остатку оранжевого цвета добавляли последовательно 100 мл метанола и 2,9 мл 25%-ного раствора метилата натрия в метаноле (12,8 ммоль), после чего перемешивали при комнатной температуре в течение ночи. Полученный осадок отфильтровывали и перекристаллизовывали из изопропанола. В результате получили 450 мг белого кристаллического 7-метил-2-пропилоксазоло[4,5-с]хинолин-4-амина; температура плавления 188-189° С. Анализ: рассчитано для С14Н15N3О + 0,2 Н2О: %С, 68,66; %Н, 6,34; %N, 17,16. Найдено: %С, 68,44; %Н, 6,11; %N, 17,42.
Пример 48
7-Фтор-2-пропилоксазоло[4,5-с]хинолин-4-амин

Часть А
Смесь 50,0 г 3-фторанилина (0,45 моль) и 91 мл диэтилэтоксиметилмалоната (0,45 моль) нагревали при 100° С в атмосфере азота в течение 3 часов. После охлаждения смеси до комнатной температуры она затвердевала. К смеси добавляли 200 мл растворителя Dowtherm А, нагревали ее при 240° С в течение 4 часов и затем охлаждали до комнатной температуры. Полученный осадок отфильтровывали, промывали гексаном и сушили в вакуумном сушильном шкафу. В результате получили 71,5 г этил-7-фтор-4-гидрокси-З-хинолинкарбоксилата.
Часть В
Суспензию 65 г этил-7-фтор-4-гидрокси-3-хинолинкарбоксилата (0,28 моль) в 250 мл 10%-ного раствора гидроксида натрия кипятили с обратным холодильником в течение 3 часов, при этом суспензия превращалась в раствор. Полученную смесь охлаждали до комнатной температуры и фильтровали через фильтровальную бумагу в вакууме. Фильтрат подкисляли концентрированной соляной кислотой. Полученный осадок собирали, промывали водой и сушили. В результате получили 53,5 г твердой белой 7-фтор-4-гидрокси-3-хинолинкарбоновой кислоты.
Часть С
25 г 7-фтор-4-гидрокси-3-хинолинкарбоновой кислоты помещали в круглодонную колбу и нагревали до 330-350° С. В процессе нагревания наблюдалось выделение углекислого газа, и вещество становилось жидким. Через приблизительно 2 минуты добавляли еще 25 г 7-фтор-4-гидрокси-3-хинолинкарбоновой кислоты. Нагревание продолжали еще 4-6 минут, в ходе этого повторного нагревания уже не наблюдалось выделения углекислого газа. Раствор охлаждали до комнатной температуры, образовавшийся твердый продукт отфильтровывали и в результате получили 36,6 г твердого розового 7-фтор-4-хинолинола.
Часть D
К горячему раствору (125° С) 35 г 7-фтор-4-хинолинола (214 ммоль) в 200 мл пропионовой кислоты добавляли 20 мл 70%-ной азотной кислоты. Смесь перемешивали при 125° С около 1,5 часов и затем охлаждали до комнатной температуры. Полученный желтый осадок отфильтровывали, споласкивали последовательно водой и этанолом и затем перекристаллизовывали из смеси N,N-диметилформамид/вода. В результате получили 18 г 7-фтор-3-нитро-4-хинолинола.
Часть Е
Смесь, содержащую 17 г 7-фтор-3-нитро-4-хинолинола (81,7 ммоль), 80 мл гидроксида аммония, 200 мл метанола и 1 г катализатора (10%-ный палладий, нанесенный на активированный уголь), загружали в аппарат Парра и в течение 1 часа проводили реакцию восстановления в атмосфере водорода (при давлении 2,1 кг/см2). После этого катализатор отфильтровывали. Фильтрат обрабатывали активированным углем и затем концентрировали в вакууме до получения твердого продукта темно-коричневого цвета, который становился еще более темным в процессе сушки. Этот продукт растворяли в метаноле и добавляли к нему раствор соляной кислоты в диэтиловом эфире. Почти сразу же образовывался серый осадок. Суспензию перемешивали несколько часов при комнатной температуре. Затем осадок отфильтровывали и тщательно споласкивали эфиром. В результате получили 6,6 г солянокислого З-амино-7-фтор-4-хинолинола.
Часть F
Смесь 3,4 г солянокислого 3-амино-7-фтор-4-хинолинола (19,1 ммоль), 2,9 мл триэтиламина (21,0 ммоль) и 15,6 мл ангидрида масляной кислоты (95,5 ммоль) в атмосфере азота кипятили с обратным холодильником в течение 18 часов. После этого смесь выливали в лед и доводили величину рН до 12, добавляя 10%-ный раствор гидроксида натрия. Полученную суспензию перемешивали до полного плавления льда и затем смесь экстрагировали дихлорметаном. Экстракт сушили над сульфатом магния и затем концентрировали в вакууме до получения маслообразного продукта. Это масло очищали на хроматографической колонке, заполненной силикагелем, используя в качестве элюента сначала дихлорметан, а затем смесь 9:1 дихлорметан : метанол. В результате получили 2,6 г светло-коричневого твердого 7-фтор-2-пропилоксазоло[4,5-с]хинолина.
Часть G
Смесь 2,6 г 7-фтор-2-пропилоксазоло[4,5-с]хинолина (11,3 ммоль), 3,3 г 3-хлорпербензойной кислоты (в виде 57-86%-ного раствора) и 90 мл хлороформа перемешивали в течение около 3 часов при комнатной температуре. Анализ с помощью способа тонкослойной хроматографии показал наличие исходного материала в реакционной смеси. Поэтому добавляли еще 0,5 эквивалента 3-хлорпербензойной кислоты и смесь дополнительно перемешивали при комнатной температуре еще в течение 2 часов. После этого тонкослойная хроматография не показывала наличия в реакционной смеси никакого исходного материала. Реакционную смесь разбавляли дихлорметаном и дважды промывали водным раствором бикарбоната натрия. Органический слой сушили над сульфатом магния и концентрировали в вакууме. В результате получили 2,8 г твердого 7-фтор-2-пропилоксазоло[4,5-с]хинолин-5N-оксида в виде оранжево-желтого твердого маслянистого вещества.
Часть Н
К раствору 2,8 г 7-фтор-2-пропилоксазоло[4,5-с]хинолин-5N-оксида (11,3 ммоль) в 50 мл дихлорметана в атмосфере азота добавляли 2,0 мл трихлорацетилизоцианата (17 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, после чего дихлорметан отгоняли в вакууме. Остаток растворяли в метаноле, добавляли к раствору 2,4 мл 25%-ного раствора метилата натрия в метаноле (11,3 ммоль) и перемешивали при комнатной температуре в течение ночи, после чего отфильтровывали небольшое количество твердого продукта. Фильтрат концентрировали в вакууме. Остаток растворяли в дихлорметане, сушили над сульфатом магния и затем концентрировали в вакууме до получения коричневого масла. Этот продукт очищали на хроматографической колонке с силикагелем, используя в качестве элюента смесь 95:5 дихлорметан : метанол. Полученный бледно-коричневый твердый липкий материал перекристаллизовывали из ацетонитрила и в результате получили 200 мг 7-фтор-2-пропилоксазоло[4,5-с]хинолин-4-амина в виде порошка цвета ржавчины; температура плавления 184-187° С. Анализ: рассчитано для С13Н12FN3О: %С, 63,67; %Н, 4,93; %N, 17,13. Найдено: %С, 63,43; %Н, 4,57; %N, 16,74.
Пример 49
7-Фтор-2-пропилтиазоло[4,5-с]хинолин-4-амин
Часть А
К суспензии 3 г солянокислого 3-амино-7-фтор-4-хинолинола (14,0 ммоль) в 50 мл тетрагидрофурана в атмосфере азота при комнатной температуре добавляли сначала 6,4 мл триэтиламина (46,2 ммоль), а затем по каплям 1,6 мл хлорангидрида масляной кислоты (15,4 ммоль). Реакционную смесь перемешивали в течение 4 часов, добавляли к ней водный раствор бикарбоната натрия и перемешивание продолжали еще 1 час. Образовавшуюся двухфазную смесь фильтровали для удаления твердых продуктов. Эти твердые продукты промывали диэтиловым эфиром и выделяли в виде бледно-розового порошка. После удаления растворителя в вакууме из тетрагидрофуранового слоя был получен темно-розовый твердый продукт. Все твердые продукты соединяли вместе и получали из них 3,0 г N-(7-фтор-4-гидроксихинолин-3-ил)бутанамида. После перекристаллизации из смеси этилацетат/этанол порции этого продукта (300 мг) получали пушистое бледно-серое твердое вещество, плавящееся при 306-30° C. Анализ: Рассчитано для C13H13FN2O2: %С, 62,90; %Н, 5,28; %N, 11,28. Найдено: %С, 62,95; %Н, 5,43; %N, 11,14.
Часть В
К смеси 2,6 г N-(7-фтор-4-гидроксихинолин-3-ил)бутанамида (10,5 ммоль) и 80 мл пиридина в атмосфере азота добавляли 4,7 г пятисернистого фосфора (10,5 ммоль). Смесь кипятили с обратным холодильником в течение 2 часов и затем охлаждали до комнатной температуры и экстрагировали водным раствором бикарбоната натрия и дихлорметаном. Органический слой отделяли, дважды промывали водой, сушили над сульфатом магния и концентрировали в вакууме. В результате получали твердый продукт цвета ржавчины, который после перекристаллизации из метанола давал 1,8 г пластинчатых кристаллов 7-фтор-2-пропилтиазоло[4,5-с]хинолина, окрашенных в цвет ржавчины.
Часть С
К раствору 1,8 г 7-фтор-2-пропилтиазоло[4,5-с]хинолина (7,3 ммоль) в 50 мл хлороформа добавляли 2,1 г 3-хлорпербензойной кислоты (в виде 57-86%-ного раствора) и смесь перемешивали при комнатной температуре. Анализ с помощью способа тонкослойной хроматографии (элюирование на силикагеле смесью дихлорметан/метанол в соотношении 95:5) показал наличие исходного материала в реакционной смеси, поэтому добавляли еще 0,5 эквивалента 3-хлорпербензойной кислоты и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь дважды промывали водным раствором бикарбоната натрия, сушили над сульфатом магния и концентрировали в вакууме. В результате получили 1,8 г бледно-оранжевого твердого 7-фтор-2-пропилтиазоло[4,5-с]хинолин-5N-оксида.
Часть D
К смеси 1,8 г 7-фтор-2-пропилтиазоло[4,5-с]хинолин-5N-оксида (6,9 ммоль) и 50 мл дихлорметана в атмосфере азота добавляли 1,2 мл трихлорацетилизоцианата (10,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, после чего концентрировали в вакууме до получения N-(7-фтор-2-пропилтиазоло[4,5-с]хинолин-4-ил)трихлорацетамида в виде масла оранжевого цвета. Это масло растворяли в метаноле и добавляли к раствору 1,5 мл 25%-ного раствора метилата натрия в метаноле. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Образовавшийся осадок отфильтровывали и перекристаллизовывали сначала из ацетонитрила, а затем из метанола. В результате получили 1,1 г 7-фтор-2-пропилтиазоло[4,5-с]хинолин-4-амина в виде коричневатого порошка; температура плавления 192,5-193,5° С. Анализ: рассчитано для С13Н12FN3S: %С, 59,75; %Н, 4,63; %N, 16,08. Найдено: %С, 59,55; %Н, 4,69; %N, 16,12.
Пример 50
2-Пропил-7-(трифторметил)тиазоло[4,5-с]хинолин-4-амин

Часть А
Смесь 40 мл 3-(трифторметил)анилина (0,32 ммоль) и диэтилэтоксиметилмалоната нагревали в атмосфере азота при 100° С в течение 3 часов. После охлаждения до комнатной температуры раствор становился твердым и было получено 102 г твердого диэтил-2-{[3-(трифторметил)анилино]метилен}малоната, окрашенного в кремовый цвет.
Часть В
Смесь 80 г 2-{[3-(трифторметил)анилино]метилен}малоната (0,24 моль) и растворителя Dowtherm А нагревали в атмосфере азота до 240° С и перемешивали в течение 3 часов. После этого реакционную смесь охлаждали до комнатной температуры и перемешивание продолжали еще в течение 16 часов. Твердые продукты отделяли от раствора фильтрованием и промывали гексаном. В результате было получено 47,5 г твердого этил-4-гидрокси-7-(трифторметил)-3-хинолинкарбоксилата желтовато-белого цвета.
Часть С
Смесь 43,4 г этил-4-гидрокси-7-(трифторметил)-3-хинолинкарбоксилата (0,521 моль) и 150 мл 10%-ного раствора гидроксида натрия нагревали до кипения с обратным холодильником. Для облегчения растворения эфира к смеси в течение одного часа добавляли 150 мл метанола. После кипячения в течение 2 часов получали гомогенный раствор. Этот раствор кипятили еще 2 часа и затем оставляли на ночь остывать до комнатной температуры. Метанол отгоняли при пониженном давлении, а оставшийся водный раствор подкисляли концентрированной соляной кислотой. Образовавшийся осадок отфильтровывали, промывали водой и затем сушили в вакуумном сушильном шкафу при 120° С в течение 24 часов. В результате было получено 38,5 г твердой 4-гидрокси-7-(трифторметил)-3-хинолинкарбоновой кислоты белого цвета.
Часть D
34,1 г 4-гидрокси-7-(трифторметил)-3-хинолинкарбоновой кислоты (0,132 моль) помещали в круглодонную колбу. Затем колбу нагревали в бане из расплавленного сплава Вуда в течение 5 минут; в процессе нагревания наблюдалось выделение углекислого газа и плавление твердого продукта. Через 5 минут выделение углекислого газа прекращалось, поэтому колбу вынимали из нагревательной бани и охлаждали до комнатной температуры. Отфильтровывали образовавшийся твердый продукт, представляющий собой 27,75 г 7-(трифторметил)-4-хинолинола.
Часть Е
Смесь 22,7 г 7-(трифтометил)-4-хинолинола (0,106 моль) и 106 мл пропионовой кислоты нагревали до 120° С, добавляли по каплям 10 мл 70%-ной азотной кислоты и нагревание продолжали еще в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, образовавшийся осадок отфильтровывали и промывали водой и диэтиловым эфиром. В результате было получено 13,3 г твердого 3-нитро-7-(трифторметил)-4-хинолинола желтовато-белого цвета.
Часть F
В аппарат Парра загружали 40 мл метанола, 10 мл гидроксида аммония, 12,8 г 3-нитро-7-(трифторметил)-4-хинолинола (49,6 ммоль), 1 г палладиевого катализатора (10% палладия, нанесенного на активированный уголь) и в течение 4 часов проводили реакцию восстановления в аппарате Парра в атмосфере водорода при давлении водорода 2,8 кг/см2. По окончании реакции восстановления смесь фильтровали и катализатор промывали метанолом и дихлорметаном. Объединенные вместе органические растворы концентрировали в вакууме и в результате получали твердое вещество зеленого цвета. Этот продукт растворяли в метаноле и добавляли к нему 150 мл 1 N соляной кислоты в безводном диэтиловом эфире. Сразу же после добавления кислоты наблюдалось образование осадка. Реакционную смесь перемешивали в течение 16 часов, осадок отфильтровывали, промывали диэтиловым эфиром и затем сушили в вакуумном сушильном шкафу при 80° С. В результате было получено 9,3 г твердого солянокислого 3-амино-7-(трифторметил)-4-хинолинола желтовато-белого цвета.
Часть G
К смеси 3,5 г солянокислого 3-нитро-7-(трифторметил)-4-хинолинола (13,2 ммоль), 6,1 мл триэтиламина (43,6 ммоль) и 30 мл безводного тетрагидрофурана по каплям добавляли 1,5 мл хлорангидрида масляной кислоты (14,5 ммоль) и смесь перемешивали в течение 16 часов. После этого добавляли небольшое количество водного раствора бикарбоната натрия и перемешивание продолжали еще 0,5 часа. Тетрагидрофуран отгоняли в вакууме, оставшийся твердый продукт перемешивали с диэтиловым эфиром, затем отфильтровывали, промывали водой и диэтиловым эфиром и в течение ночи сушили в вакуумном сушильном шкафу при 80° С. В результате получили 3,3 г твердого N-[4-гидрокси-7-(трифторметил)хинолин-3-ил]бутанамида, окрашенного в кремовый цвет.
Часть Н
Смесь 3,0 г N-[4-гидрокси-7-(трифторметил)хинолин-3-ил]бутанамида (10,05 ммоль), 4,5 г пятисернистого фосфора (10,05 ммоль) и 30 мл пиридина кипятили с обратным холодильником в течение 6 часов, после чего раствор охлаждали до комнатной температуры и разбавляли его дихлорметаном и водным раствором бикарбоната натрия. Органический слой отделяли, промывали водой и рассолом, сушили над сульфатом магния и концентрировали в вакууме до получения желтого твердого продукта. Этот продукт обрабатывали гексаном и после фильтрования было получено 1,7 г твердого 2-пропил-7-(трифторметил)тиазоло[4,5-с]хинолина, окрашенного в коричневый цвет. Из гексанового фильтрата после упаривания растворителя получили еще 0,6 г твердого желтого продукта.
Часть I
К смеси 2,0 г 2-пропил-7-(трифторметил)тиазоло[4,5-с]хинолина (6,75 ммоль) и 30 мл хлороформа добавляли 1,93 г 3-хлорпербензойной кислоты (6,88 ммоль) и полученный раствор перемешивали в течение 24 часов. Затем реакционную смесь разбавляли водным раствором бикарбоната натрия и экстрагировали дихлорметаном. Органический слой отделяли, сушили над сульфатом магния и концентрировали в вакууме. В результате получили 1,98 г твердого желтого 2-пропил-7-(трифторметил)тиазоло[4,5-с]хинолин-5N-оксида.
Часть J
К смеси 1,3 г 2-пропил-7-(трифторметил)тиазоло[4,5-с]хинолин-5N-оксида (4,16 ммоль) и 20 мл безводного дихлорметана в атмосфере азоте добавляли 0,75 мл трихлорацетилизоцианата (6,24 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 16 часов, после чего растворитель удаляли при пониженном давлении. Остаток растворяли в 40 мл метанола и добавляли к раствору 1,43 мл 25%-ного раствора метилата натрия (6,24 ммоль) в метаноле. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Образовавшийся осадок отфильтровывали, промывали небольшим количеством метанола и сушили в течение 16 часов в вакуумном сушильном шкафу при 80° С. В результате получили 0,96 г белого твердого 2-пропил-7-(трифторметил)тиазоло[4,5-с]хинолин-4-амина; температура плавления 215-216° С. Анализ: рассчитано для С14Н12F3N3S: %С, 54,01; %Н, 3,89; %N, 13,50. Найдено: %С, 53,82; %Н, 3,66; %N, 13,37.
Пример 51
2-(Метилсульфонил)тиазоло[4,5-с]хинолин-5N-оксид

Часть А
К 5,4 г N4-(2-метилпропил)хинолин-3,4-диамина (25 ммоль) добавляли 9 мл дисульфида углерода (150 ммоль) и 55 мл этанола и смесь на паровой бане кипятили с обратным холодильником в течение 2 часов. Полученный осадок отфильтровывали, промывали этанолом и сушили на воздухе. В результате получили 4,4 г неочищенного продукта. 1 г этого продукта растворяли в горячем растворе гидроксида натрия и затем осаждали уксусной кислотой. Осадок отфильтровывали в горячем состоянии, промывали гексаном и после сушки на воздухе получили твердый тиазоло[4,5-с]хинолин-2-тиол; температура плавления 282-284° С. Анализ: рассчитано для С10H6N2S2: %С, 55,02; %Н, 2,77; %N 12,83. Найдено: %С, 54,96; %Н, 2,69; %N, 12,74.
Часть В
К раствору 13,65 г тиазоло[4,5-с]хинолин-2-тиола (63 ммоль) в 160 мл метанола добавляли 15,8 мл 25%-ного раствора метилата натрия (69 ммоль) в метаноле и 3,9 мл иодистого метила (63 ммоль). Смесь нагревали на паровой бане в течение 1 часа, после чего растворитель отгоняли в вакууме. Оставшееся светлое зеленовато-желтое твердое вещество смешивали с водой, отфильтровывали и промывали водой. В результате было получено 9,8 г неочищенного продукта. После перекристаллизации 1 г этого продукта из метанола получили твердый 2-(метилтио)тиазоло[4,5-с]хинолин, имеющий температуру плавления 116-119° С. Анализ: рассчитано для С11H8N2S2: %С, 56,87; %Н, 3,47; %N, 12,06. Найдено: %С, 57,09; %Н, 3,57; %N, 12,04.
Часть С
К смеси 7,7 г 2-(метилтио)тиазоло[4,5-с]хинолина (33 ммоль) и 100 мл уксусной кислоты добавляли 27,8 мл 32%-ной надуксусной кислоты (132 ммоль), смесь нагревали в течение 4 часов при 60° С, а затем в течение ночи выдерживали при комнатной температуре. Образовавшийся осадок отфильтровывали и получали 5,6 г неочищенного продукта. Фильтрат концентрировали в вакууме и остаток разбавляли 100 мл толуола. После удаления толуола в вакууме получали дополнительно 4 г неочищенного продукта. 1 г этого продукта перекристаллизовывали из N,N-диметилформамида и в результате получали твердый 2-(метилсульфонил)тиазоло[4,5-с]хинолин-5N-оксид, окрашенный в желтый цвет; температура плавления 245-247° С. Анализ: рассчитано для С11Н8N2О3S2: %С, 47,13; %Н, 2,88; %N, 9,99. Найдено: %С, 47,08; %Н, 3,08; %N, 10,14.
Пример 52
2-(4-Морфолино)тиазоло[4,5-с]хинолин-5N-оксид

Смесь 2,5 г 2-(метилсульфонил)тиазоло[4,5-с]хинолин-5N-оксида (8,9 ммоль) и приблизительно 50 мл морфолина нагревали на паровой бане в течение 9 часов. Образовавшийся осадок отфильтровывали и получали 0,9 г твердого желтого неочищенного продукта. Фильтрат охлаждали в ледяной бане. В результате было получено еще 0,8 г твердого желтого неочищенного продукта. Эти твердые продукты объединяли вместе и небольшую часть этого вещества (0,5 г) перекристаллизовывали из метанола. В результате был получен твердый 2-(4-морфолино)тиазоло[4,5-с]хинолин-5N-оксид, имеющий температуру плавления 241-242° С. Анализ: рассчитано для С14H13N4О2S: %С, 58,52; %Н, 4,56; %N, 14,62. Найдено: %С, 58,24; %Н, 4,38; %N, 14,43.
Пример 53
2-(4-Морфолино)тиазоло[4,5-с]хинолин-4-амин

К смеси 1,2 г 2-(4-морфолино)тиазоло[4,5-с]хинолин-5N-оксида (4,2 ммоль) и 24 мл дихлорметана добавляли 18 мл гидроксида аммония. Смесь охлаждали и медленно добавляли к ней раствор 0,88 г тозилхлорида (4,6 ммоль) в 10 мл дихлорметана. После этого смесь нагревали до комнатной температуры и перемешивали в течение ночи. Органическую фазу отделяли, промывали водным раствором бикарбоната натрия, сушили над сульфатом магния и концентрировали в вакууме до получения желтого твердого продукта. Этот продукт очищали на хроматографической колонке, затем растворяли в соляной кислоте и переосаждали гидроксидом натрия. Осадок отфильтровывали и переосаждали из метанола. В результате было получено 0,26 г твердого 2-(4-морфолино)тиазоло[4,5-с]хинолин-4-амина, температура плавления которого составляла 225-227° С. Анализ: рассчитано для С14Н14N4ОS: %С, 58,72; %Н, 4,93; %N, 19,57. Найдено: %С, 58,47; %Н, 4,63; %N, 19,23.
Пример 54
2-(1-Пирролидино)тиазоло[4,5-с]хинолин-4-амин

Часть А
Смесь 2,5 г 2-(метилсульфонил)тиазоло[4,5-с]хинолин-5N-оксида (8,9 ммоль) и приблизительно 70 мл пирролидина кипятили с обратным холодильником на паровой бане в течение 3 суток. Образовавшийся желтый осадок отфильтровывали и получали 0,4 г 2-(4-пирролидино)тиазоло[4,5-с]хинолин-5N-оксида. Фильтрат охлаждали в ледяной бане. Получившийся осадок отфильтровывали. В результате было получено еще 0,7 г твердого желтого 2-(1-пирролидино)тиазоло[4,5-с]хинолин-5N-оксида. Эти продукты объединяли вместе.
Часть В
К смеси 0,8 г 2-(1-пирролидино)тиазоло[4,5-с]хинолин-5N-оксида (2,95 ммоль) и 50 мл дихлорметана добавляли 12 мл гидроксида аммония. Смесь охлаждали и медленно добавляли к ней раствор 0,6 г тозилхлорида (3,2 ммоль) в 10 мл дихлорметана. После этого смесь нагревали до комнатной температуры и перемешивали в течение ночи. Органическую фазу отделяли, промывали водным раствором бикарбоната натрия, сушили над сульфатом магния и концентрировали в вакууме до получения желтого твердого неочищенного продукта. Этот продукт очищали способом флеш-хроматографии, после чего смешивали с горячим метанолом, охлаждали и отфильтровывали. В результате было получено 0,14 г твердого 2-(1-пирролидино)тиазоло[4,5-с]хинолин-4-амина; температура плавления 259-261° С. Анализ: рассчитано для С14Н14N4S: %С, 62,20; %Н, 5,22; %N, 20,49. Найдено: %С, 61,76; %Н, 5,25; %N, 20,72.
Пример 55
Ксинофоат 2-пропилтиазоло[4,5-с]хинолин-4-амина

3,0 г 2-пропилтиазоло[4,5-с]хинолин-4-амина (12,3 ммоль) и 2,3 г 1-гидрокси-2-нафтойной кислоты (12,3 ммоль) по отдельности растворяли в метаноле, при необходимости используя добавки дихлорметана. Эти два раствора объединяли вместе, и полученный раствор упаривали для уменьшения объема. Образовавшийся осадок отфильтровывали. В результате было получено 3,6 г бесцветного кристаллического ксинофоата 2-пропилтиазоло[4,5-с]хинолин-4-амина; температура плавления этого продукта 185-189° С (с разложением). Анализ: рассчитано для С24Н21N3О3S: %С, 66,80; %Н, 4,91; %N, 9,74. Найдено: %С, 66,18; %Н, 5,07; %N, 9,78.
Пример 56
3-Гидрокси-2-нафтоат 2-пропилтиазоло[4,5-с]хинолин-4-амина

Раствор 1,9 г 3-гидрокси-2-нафтойной кислоты (10 ммоль) в 30 мл метанола добавляли к раствору 2,4 г 2-пропилтиазоло[4,5-с]хинолин-4-амина (10 ммоль) в 70 мл горячего метанола. Сразу же при добавлении образовывался осадок. Смесь нагревали еще 5 минут и затем охлаждали до комнатной температуры. Осадок отфильтровывали, промывали метанолом и сушили. В результате было получено 4,0 г коричневого порошка. Этот продукт перекристаллизовывали из смеси метанол/дихлорметан и получили 3,2 г белого порошкообразного З-гидрокси-2-нафтоата 2-пропилтиазоло[4,5-с]хинолин-4-амина. Анализ: рассчитано для С24H21N3О3S: %С, 66,80; %Н, 4,91; %N, 9,74. Найдено: %С, 66,28; %Н, 4,92; %N, 9,59.
Пример 57
2-Бутилтиазоло[4,5-с][1,5]нафтиридин-5N-оксид

Часть А
Смесь, содержащую 7,5 г 3-нитро[1,5]нафтиридин-4-ола, 200 мл метанола, 50 мл гидроксида аммония и 0,75 г платинового катализатора (5% платины, нанесенной на активированный уголь), выдерживали в аппарате Парра в течение 6 часов. После этого реакционную смесь фильтровали для удаления катализатора, а затем вторично фильтровали через фильтр Сеlite® . Фильтрат концентрировали в вакууме и в результате получили 6,1 г твердого коричневого 3-амино[1,5]нафтиридин-4-ола.
Часть В
К суспензии 5,2 г 3-амино[1,5]нафтиридин-4-ола (32 ммоль) в 100 мл пиридина добавляли по каплям 4,3 г хлорангидрида валериановой кислоты (35 ммоль) и смесь кипятили с обратным холодильником в течение 2 часов. Пиридин удаляли. Остаток смывали горячей водой и затем охлаждали. Образовавшийся серый осадок отфильтровывали, тщательно промывали горячей водой и сушили в сушильном шкафу. В результате получили 2,3 г твердого N-(4-гидрокси[1,5]нафтиридин-3-ил)пентамида серого цвета.
Часть С
К суспензии 2,3 г N-(4-гидрокси[1,5]нафтиридин-3-ил)пентамида (9,4 ммоль) в 150 мл пиридина добавляли 4,2 г пятисернистого фосфора (9,4 ммоль). Смесь кипятили с обратным холодильником в течение 2 часов. Пиридин удаляли. Остаток смывали смесью воды, 10%-ного раствора карбоната натрия и 10%-ного раствора гидроксида натрия (в количестве, достаточном для того, чтобы обеспечить рН смеси более 8) и затем дважды экстрагировали дихлорметаном. Дихлорметановые экстракты объединяли вместе, промывали рассолом, сушили и концентрировали в вакууме. Остаток разбавляли толуолом и затем концентрировали в вакууме. В результате было получено 2 г черного сиропообразного вещества. Этот материал очищали на хроматографической колонке, заполненной силикагелем, и в результате получили 1,4 г 2-бутилтиазоло[4,5-с][1,5]нафтиридина в виде янтарной жидкости. Анализ способом масс-спектрометрии высокого разрешения (ЕI): рассчитано для С13Н13N3S (М+) 243,0830. Найдено 243,0825.
Часть D
К раствору 1,4 г 2-бутилтиазоло[4,5-с][1,5]нафтиридина (5,8 ммоль) в 100 мл хлороформа добавляли 1,1 г 3-хлорпербензойной кислоты (в виде 57-86%-ного раствора) в 50 мл хлороформа. Смесь перемешивали при комнатной температуре в течение 2,5 часов и затем разбавляли ее дихлорметаном, дважды промывали 10%-ным раствором гидроксида натрия, промывали рассолом, сушили и концентрировали в вакууме до получения светло-желтого сиропообразного вещества, которое затвердевало при стоянии. После очистки этого вещества на хроматографической колонке с силикагелем получили 1,2 г бледно-желтого твердого продукта. Перекристаллизация этого материала из смеси петролейного эфира (15 мл) и гексанов (100 мл) позволила получить 2-бутилтиазоло[4,5-с][1,5]нафтиридин-5N-оксид, температура плавления которого составляла 65-69° С. Анализ: рассчитано для С13Н13N3OS: %С, 60,21; %Н, 5,05; %N, 16,20. Найдено: %С, 60,43; %Н, 5,17; %N, 16,18. Анализ способом масс-спектрометрии высокого разрешения (ЕI): рассчитано для С13Н13N3OS (М+) 259,0779. Найдено 259,0789.
Пример 58
2-Бутилтиазоло[4,5-с][ 1,5]нафтиридин-4-амин

Раствор 0,5 г 2-бутилтиазоло[4,5-с][1,5]нафтиридин-5N-оксида (1,9 ммоль) в 100 мл дихлорметана охлаждали на ледяной бане и по каплям добавляли к нему раствор 0,4 г трихлорацетилизоцианата (2,1 ммоль) в 25 мл дихлорметана. Смесь перемешивали 8 часов при комнатной температуре и затем добавляли раствор аммиака в метаноле в таком количестве, чтобы смесь имела щелочную реакцию, после чего выдерживали ее в течение ночи. После этого смесь разбавляли еще дихлорметаном, дважды промывали 10%ным раствором гидроксида натрия, один раз рассолом, сушили и концентрировали в вакууме до получения 0,6 г бледно-желтого твердого вещества. Этот продукт очищали на хроматографической колонке с силикагелем и затем перекристаллизовывали из ацетонитрила (8 мл). В результате было получено 0,15 г белого кристаллического 2-бутилтиазоло[4,5-с] [1,5]нафтиридин-4-амина, температура плавления которого составляла 136-138° С. Анализ: рассчитано для С13Н14N4S: %С, 60,44; %Н, 5,46; %N, 21,69. Найдено: %С, 60,12; %Н, 5,42; %N, 21,51. Масс-спектрометрия высокого разрешения (ЕI): рассчитано для С13Н13N4S (М+) 258,0941. Найдено 258,0939. Сигналы в ЯМР спектре в СDСl3 (м.д.): 8,637 (дд) (1Н, J=3,6; 1,2 Гц), 8,048 дд (1Н, J=8,5; 1,2 Гц), 7,486 дд (1Н, J=8,5; 3,6 Гц), 5,691 уш.синглет (2Н), 3,196 т (2Н, J=7 Гц), 1,918 квинтет (2Н, J=7 Гц), 1,509 секстет (2Н, J=7 Гц), 1,003 т (3Н, J=7 Гц).
Пример 59
2-Пропилтиазоло[4,5-с][1,5]нафтиридин-5N-оксид

Часть А
Используя методику, приведенную в части В примера 57, 1,8 г 3-амино[1,5]нафтиридин-4-ола (11,2 ммоль) обрабатывали 1,3 г хлорангидрида масляной кислоты (12,3 ммоль), в результате чего получали 1,2 г N-(4-гидрокси[1,5]нафтиридин-3-ил)бутанамида в виде серого угольного порошка, температура плавления которого превышала 360° С.
Часть В
Реакцию между 1,2 г N-(4-гидрокси[1,5]нафтиридин-3-ил)бутанамида (5,2 ммоль) и 2,3 г пятисернистого фосфора (5,2 ммоль) проводили по методике, приведенной в части С примера 57. В результате было получено 0,9 г 2-пропилтиазоло[4,5-с][1,5]нафтиридина в виде сиропа янтарного цвета.
Часть С
Окисление 0,9 г 2-пропилтиазоло[4,5-с][1,5]нафтиридина проводили по методике, приведенной в части D примера 57; в результате было получено 0,7 г твердого бледно-желтого 2-пропилтиазоло[4,5-с][1,5]нафтиридин-5N-оксида, температура плавления которого составляла 139-142° С. Анализ: рассчитано для С12Н11N3ОS: %С, 58,76; %Н, 4,52; %N, 17,13. Найдено: %С, 58,66; %Н, 4,59; %N, 17,16. Масс-спектрометрия высокого разрешения (ЕI): Рассчитано для С12Н11N3ОS (М+) 245,0623. Найдено 245,0612.
Пример 60
2-Пропилтиазоло[4,5-с][1,5]нафтиридин-4-амин

Используя общую методику, приведенную в примере 58, 0,5 г 2-пропилтиазоло[4,5-с][1,5]нафтиридин-5N-оксида (2 ммоль) аминировали до 2-пропилтиазоло[4,5-с][1,5]нафтиридин-4-амина. Выход целевого продукта, представляющего собой игольчатые кристаллы цвета слоновой кости, составлял 0,2 г, температура плавления 135-136° С. Анализ: рассчитано для С12Н12N4S: %С, 58,99; %Н, 4,95; %N, 22,93. Найдено: %С, 59,06; %Н, 4,96; %N, 22,97. Масс-спектрометрия высокого разрешения (ЕI): рассчитано для С12Н12N4S (М+) 244,0783. Найдено 244,0785.
Пример 61
2-Пропилпиридо[3,4-d][1,3]тиазол-5N-оксид

Часть A
Суспензию 1,0 г 3-нитропиридин-4-ола (7,1 ммоль) в 110 мл метанола и небольшое количество используемого в качестве катализатора никеля Ренея загружали в аппарат Парра и подвергали гидрированию в течение 4 часов. Реакционную смесь подкисляли раствором соляной кислоты в этаноле и затем фильтровали для удаления катализатора. Фильтрат вновь пропускали через фильтр Сеlite® , а затем концентрировали в вакууме до получения 1,2 г коричневого порошка 3-аминопиридин-4-ола. Температура плавления полученного спирта 199-200° С.
Часть В
К суспензии 8,5 г 3-аминопиридин-4-ола (46 ммоль) в 100 мл дихлорметана добавляли сначала 33 мл N,N-диизопропилэтиламина (180 ммоль), а затем по каплям раствор 5,4 г хлорангидрида масляной кислоты (51 ммоль) в 100 мл дихлорметана. Реакционную смесь перемешивали 3 часа при комнатной температуре, после чего кипятили с обратным холодильником еще в течение 3 часов. После этого смесь фильтровали для удаления черного осадка, и фильтрат концентрировали в вакууме. Светло-коричневый остаток обрабатывали 250 мл горячего этилацетата и затем оставляли остывать в течение ночи. Смесь фильтровали для удаления образовавшихся твердых продуктов (9,1 г) и эти продукты промывали свежим этилацетатом. Фильтрат концентрировали в вакууме, в результате чего получили 13 г светло-янтарного сиропа. Этот сироп растворяли в воде и дважды экстрагировали этилацетатом. Экстракты объединяли вместе, промывали рассолом, сушили и концентрировали в вакууме до получения 2,5 г янтарного сиропообразного продукта. После очистки на хроматографической колонке было получено 1,2 г светло-янтарного сиропообразного N-(4-гидроксипирид-3-ил)бутанамида, который становился твердым при стоянии.
Часть С
Используя методику, приведенную в части С примера 57, 1,1 г N-(4-гидроксипирид-3-ил)бутанамида (6,1 ммоль) обрабатывали 2,7 г пятисернистого фосфора (6,1 ммоль) и получали 0,4 г 2-пропилпиридо[3,4-d1][1,3]тиазол в виде янтарного сиропа, который становился твердым при стоянии. Температура плавления 44-47° С.
Часть D
Используя методику, приведенную в части D примера 57, 0,4 г 2-пропилпиридо[3,4-d][1,3]тиазола (2,2 ммоль) окисляли до 2-пропилпиридо[3,4-d1][1,3]тиазол-5N-оксида (0,2 г), который после перекристаллизации из этилацетата (7 мл) имел вид коротких игольчатых кристаллов цвета слоновой кости, температура плавления которых составляла 137-139° С. Анализ: рассчитано для С9Н10N2OS: %С, 55,65; %Н, 5,19; %N, 14,42. Найдено: %С, 55,47; %Н, 5,25; %N, 14,34.
Пример 62
Трифторацетат 2-пропилпиридо[3,4-d][1,3]тиазол-4-амин

Раствор 0,11 г трихлорацетилизоцианата (0,6 ммоль) в 5 мл дихлорметана добавляли по каплям к охлажденному на ледяной бане раствору 0,1 г 2-пропилпиридо[3,4-d][1,3]тиазол-5N-оксида (0,5 ммоль) в 20 мл дихлорметана. Реакционную смесь перемешивали при комнатной температуре в течение 5 часов, после чего добавляли еще 0,2 г трихлорацетилизоцианата и перемешивание при комнатной температуре продолжали в течение ночи. Затем реакционную смесь нагревали до кипения с обратным холодильником, выдерживали короткое время при этой температуре и потом перемешивали при комнатной температуре еще 3 часа. Через смесь в течение 1 часа при комнатной температуре барботировали аммиак, затем смесь разбавляли дихлорметаном, дважды промывали 10%-ным раствором гидроксида натрия и один раз рассолом, сушили и концентрировали в вакууме до получения сиропообразного продукта янтарного цвета. Реакцию повторяли еще один раз. Продукты реакции объединяли вместе, всего было получено 0,1 г янтарного сиропообразного вещества. Этот материал очищали с помощью способа полупрепаративной жидкофазной хроматографии на приборе системы Gilson (колонка С18 Rainin Microsobr, 21,4× 250 мм, частицы размером 8 мкм, размер пор 60А, скорость подачи раствора 10 мл/мин, градиент элюирования от 2 до 95% В в течение 5 мин, где А=0,1%-ный раствор трифторуксусной кислоты в воде и В=0,1%-ный раствор трифторуксусной кислоты в ацетонитриле, детектирование фракций по поглощению при 254 нм). Полученные в результате такого полупрепаративного способа разделения фракции были проанализированы способом LС-АРСI/МS, и соответствующие фракции были лиофилизированы для получения целевого продукта в виде соли трифторуксусной кислоты; температура плавления 160-162° С. Анализ: рассчитано для С9Н11N4S + СF3СООН: %С, 42,99; %Н, 3,94; %N, 13,67. Найдено: %С, 42,84; %Н, 3,98; %N, 13,52. Масс-спектрометрия высокого разрешения (ЕI): рассчитано для С9Н11N4S (М+) 193,0674. Найдено 193, 0681.
Пример 63
7-Хлор-2-пропилтиазоло[4,5-с]хинолин-4-амин

Часть А
Смесь 35 г 7-хлор-4-гидроксихинолина (0,195 моль; продукт компании Аldrich, Милуоки, штат Висконсин) и 350 мл 70%-ной азотной кислоты кипятили с обратным холодильником в течение 75 мин, после чего горячую реакционную смесь выливали в лед. Образовавшийся ярко-желтый осадок отфильтровывали и три раза промывали кипящим этилацетатом. В результате было получено 17,3 г твердого бледно-желтого 7-хлор-3-нитро-4-гидроксихинолина.
Часть В
Смесь 4,48 г 7-хлор-3-нитро-4-гидроксихинолина (20 ммоль), 22,6 г дигидрата хлористого олова (II) (100 ммоль) и 200 мл этанола кипятили с обратным холодильником в течение 4 часов, после чего охлаждали до комнатной температуры и выливали в 250 мл воды. С помощью насыщенного раствора бикарбоната натрия рН смеси доводили до нейтрального значения и затем смесь фильтровали для удаления соли олова. Фильтрат экстрагировали этилацетатом; объединенные органические фракции сушили над сульфатом магния, фильтровали и затем концентрировали в вакууме до получения 1,8 г зеленого порошка 3-амино-7-хлор-4-гидроксихинолина.
Часть С
0,76 мл хлорангидрида масляной кислоты (7,3 ммоль) в атмосфере азота добавляли по каплям к смеси 1,3 г 3-амино-7-хлор-4-гидроксихинолина (6,7 ммоль), 3,0 мл триэтиламина (21,5 ммоль) и 20 мл безводного тетрагидрофурана. Реакционную смесь выдерживали при комнатной температуре в течение ночи, образовавшийся осадок отфильтровывали, промывали сначала водой и затем тетрагидрофураном, после чего сушили в вакууме. В результате такой обработки было получено 1,05 г коричневого порошка N-(7-хлор-4-гидроксихинолин-3 -ил)бутанамида.
Часть D
Смесь 0,9 г N-(7-хлор-4-гидроксихинолин-3-ил)бутанамида (3,4 ммоль), 1,51 г пятисернистого фосфора (3,4 ммоль) и 25 мл пиридина кипятили с обратным холодильником в течение 2,5 часов в атмосфере азота, после чего охлаждали до комнатной температуры. Реакционную смесь перемешивали со 100 мл дихлорметана и 100 мл насыщенного раствора бикарбоната натрия, при этом происходило распределение реакционной смеси между органической и водной фазами. Водный слой экстрагировали двумя порциями дихлорметана по 100 мл каждая. Органические фракции объединяли вместе, промывали водой, сушили над сульфатом магния, фильтровали и затем концентрировали в вакууме до получения неочищенного продукта реакции. Этот материал очищали на хроматографической колонке с силикагелем (в качестве элюента использовали смесь 97:3 дихлорметан:метанол; количество силикагеля 10 г) и в результате получили 0,62 г 7-хлор-2-пропилтиазоло[4,5-с]хинолина в виде золотисто-желтого твердого вещества.
Часть Е
В атмосфере азота 0,7 г 3-хлорпероксибензойной кислоты (в виде 57-86%-ного раствора) добавляли к смеси 0,5 г 7-хлор-2-пропилтиазоло[4,5-с]хинолина (1,9 ммоль) и 20 мл хлороформа. Через 2 часа выдерживания этой смеси при комнатной температуре к ней добавляли еще 0,2 г 3-хлорпербензойной кислоты и смесь выдерживали при комнатной температуре еще 14 часов. После этого реакционную смесь разбавляли дихлорметаном и дважды промывали насыщенным раствором бикарбоната натрия. Органическую фракцию сушили над сульфатом магния, фильтровали и концентрировали в вакууме до получения 0,52 г твердого оранжевого 7-хлор-2-пропилтиазоло[4,5-с]хинолин-5N-оксида.
Часть F
К смеси 0,50 г 7-хлор-2-пропилтиазоло[4,5-с]хинолин-5N-оксида (1,8 ммоль) и 20 мл дихлорметана в атмосфере азота добавляли 0,32 мл трихлорацетилизоцианата (2,7 ммоль), смесь выдерживали при комнатной температуре в течение 2 часов, после чего концентрировали в вакууме. Полученный маслообразный остаток растворяли в 10 мл метанола, добавляли 1 мл 25%-ного раствора метилата натрия (4,4 ммоль) и смесь выдерживали при комнатной температуре в течение 2,5 суток. Полученный осадок отфильтровывали и промывали гексаном. В результате было получено 0,28 г целевого продукта в виде золотисто-желтого порошка. 50 мг этого продукта перекристаллизовывали из метанола и получали твердый 7-хлор-2-пропилтиазоло[4,5-с]хинолин-4-амин золотисто-желтого цвета; температура плавления 159-160° С. Данные 1Н ЯМР-спектроскопии (300 МГц, ДМСО-d6): δ 7,82 (д,J=8,5; Гц, 1Н), 7,60 (д,J=2,0 Гц, 1H), 7,27 (дд,J=8,5; 2,1 Гц, 1H), 7,10 (с, 2Н), 3,16 (т, J=7,4 Гц, 2Н), 1,87 (секстет, J=7,4 Гц, 2Н), 1,02 (т, J=7,4 Гц, 3Н). МS (ЕI) m/е 277,0441 (277,0440 рассчитано для С13Н12СlN3S).
Пример 64
7-Метокси-2-пропилтиазоло[4,5-с]хинолин-4-амин

Часть А
Смесь 12,3 г 3-метоксианилина (0,1 моль) и 21,6 г диэтилэтоксиметиленмалоната (0,1 моль) нагревали при 120° С в течение 3 часов, после чего охлаждали до комнатной температуры и выдерживали в вакууме -в течение ночи. В результате было получено 28,5 г диэтил-2-[3-(метоксианилино)метилен]малоната в виде оранжевого масла.
Часть В
В колбу, снабженную магнитной мешалкой, патрубком для ввода азота, ловушкой Дина-Старка и обратным холодильником, заливали приблизительно 200 мл растворителя Dowtherm А. Растворитель нагревали до интенсивного кипения и добавляли к нему 20,0 г 2-[3-(метоксианилино)метилен]малоната (68 ммоль). Смесь нагревали в течение 0,5 часа, после чего полученный коричневый раствор охлаждали до комнатной температуры. Образовавшийся осадок отфильтровывали, промывали ацетоном и сушили на воздухе. В результате было получено 12,5 г этил-4-гидрокси-7-метоксихинолин-3-карбоксилата в виде желтого порошка.
Часть С
Суспензию 12,0 г этил-4-гидрокси-7-метоксихинолин-3-карбоксилата (48 ммоль) в 200 мл 10%-ного водного раствора гидроксида натрия кипятили с обратным холодильником в течение 1,5 часов. После этого реакционную смесь охлаждали до комнатной температуры и доводили величину рН смеси до 3, добавляя к ней по каплям концентрированную соляную кислоту. Поученный осадок отфильтровывали, дважды промывали водой и сушили в течение ночи в вакуумном сушильном шкафу при 80° С. Получено 10,4 г 4-гидрокси-7-метоксихинолин-3-карбоновой кислоты.
Часть D
Суспензию 4,0 г 4-гидрокси-7-метоксихинолин-3-карбоновой кислоты в 75 мл Dowtherm А кипятили с обратным холодильником в течение 2 часов. Полученный коричневый раствор медленно охлаждали до комнатной температуры. Осадок отфильтровывали и сушили в вакуумном сушильном шкафу при 80° С в течение 2,5 суток. В результате получили 3,1 г твердого светло-коричневого 7-метоксихинолин-4-ола.
Часть Е
Смесь 5,0 г 7-метоксихинолин-4-ола (28,5 ммоль) и 50 мл пропионовой кислоты нагревали до кипения с обратным холодильником, затем в течение 15 мин по каплям добавляли к ней 3,2 мл 70%-ной азотной кислоты (50 ммоль) и смесь кипятили с обратным холодильником в течение 2 часов, после чего охлаждали до комнатной температуры. Образовавшийся осадок отфильтровывали, промывали сначала холодным этанолом и затем гексанами и сушили. В результате было получено 3,9 г твердого серого 7-метокси-3-нитрохинолин-4-ола.
Часть F
Смесь 4,5 г 7-метокси-3-нитрохинолин-4-ола (20,4 ммоль), 250 мл метанола, 5 мл гидроксида аммония и 400 мг палладиевого катализатора (10% палладия, нанесенного на активированный уголь) загружали в аппарат Парра и проводили гидрирование в атмосфере водорода (при давлении водорода 2,8 кг/см2) в течение 2 часов. По окончании реакции смесь фильтровали. Фильтрат концентрировали в вакууме до получения твердого вещества зеленого цвета. Этот продукт растворяли в 20 мл метанола и к раствору добавляли 75 мл 1 N раствора соляной кислоты в диэтиловом эфире. Образовавшийся осадок отфильтровывали и сушили. В результате было получено 2,6 г твердого солянокислого 3-амино-7-метоксихинолин-4-ола розового цвета.
Часть G
К раствору, содержащему 1,0 г солянокислого 3-амино-7-метоксихинолин-4-ола (5,26 ммоль), 2,35 мл триэтиламина (16,8 ммоль), 30 мл дихлорметана и 10 мл N,N-диметилформамида, по каплям добавляли 0,63 мл хлорангидрида масляной кислоты (6,1 ммоль). Смесь выдерживали в течение ночи при комнатной температуре, после чего N,N-диметилформамид отгоняли в вакууме, а оставшееся твердое вещество распределяли между 100 мл дихлорметана и 100 мл воды. Органическую фракцию промывали водой, сушили над сульфатом магния, фильтровали и концентрировали в вакууме до получения 0,86 г N-(4-гидрокси-7-метоксихинолин-3-ил)бутанамида в виде твердого вещества коричневого цвета.
Часть Н
Смесь 0,66 г N-(4-гидрокси-7-метоксихинолин-3-ил)бутанамида (2,54 ммоль), 20 мл пиридина и 1,13 г пятисернистого фосфора (2,54 ммоль) кипятили с обратным холодильником в атмосфере азота и затем охлаждали до комнатной температуры. Смесь фильтровали и фильтрат распределяли между 100 мл дихлорметана и 100 мл насыщенного водного раствора бикарбоната натрия. Водную фракцию экстрагировали 100 мл дихлорметана. Органические фракции объединяли вместе, промывали водой, сушили над сульфатом магния, фильтровали и концентрировали в вакууме до получения твердого продукта. Этот продукт очищали на хроматографической колонке, заполненной силикагелем (15 г SiO2, в качестве элюента использовали смесь 95:5 дихлорметан:метанол) и получили 0,45 г 7-метокси-2-пропилтиазоло[4,5-с]хинолина в виде бледно-желтого порошка.
Часть I
Используя методику, приведенную в части Е примера 63, окисляли 0,40 г 7-метокси-2-пропилтиазоло[4,5-с]хинолина (1,55 ммоль) до 7-метокси-2-пропилтиазоло[4,5-с]хинолин-5N-оксида, представляющего собой твердый оранжевый продукт.
Часть J
Используя методику, приведенную в части F примера 63, N-оксид, полученный в части I этого примера, обрабатывали трихлорацетилизоцианатом и образовавшийся амид подвергали гидролизу. В результате было получено 190 мг целевого желтовато-белого твердого продукта. При перекристаллизации этого продукта из метанола получили 7-метокси-2-пропилтиазоло[4,5-с]хинолин-4-амин в виде желтовато-белых игольчатых кристаллов; температура плавления 152-154° С. Данные 1Н ЯМР спектроскопии (300 МГц, ДМСО-d6): δ 7,67 (д, J=8,7 Гц, 1Н), 7,06 (д, J=2,4 Гц, 1Н), 6,91 (дд, J=8,7; 2,5 Гц, 1Н), 6,82 (с, 2Н), 3,86 (с, 3Н), 3,11 (т, J=7,4 Гц, 2Н), 1,85 (секстет, J=7,4 Гц, 2Н), 1,01 (т, J=7,4 Гц, 3Н). МS (ЕI) m/е 273,0934 (273,0936 рассчитано для С14Н15N3OS).
СТИМУЛИРОВАНИЕ СИНТЕЗА ИНТЕРФЕРОНА (α ) В КЛЕТКАХ ЧЕЛОВЕКА
Для оценки стимулирования синтеза интерферона соединениями, описанными в данном изобретении, использовали систему клеток крови человека in vitro. Активность определяли, исходя из количества интерферона, выделяемого в среду культуры. Количество интерферона определяли, оценивая биоактивность образца.
Подготовка культуры на основе клеток крови
Цельную кровь собирали венепункцией в специальные ампулы с ЭДТА. Одноядерные клетки периферийной крови (РВМ) отделяли от цельной крови с помощью системы LeucoPREPTM (полученной от компании Becton Dickinson) или с помощью раствора Ficoll-Paque® (полученного из компании Pharmacia LKB Biotechnology Inc, г. Пискатавэй, штат Нью-Джерси). РВМ в количестве 1· 106 клеток/мл суспендировали в среде RPMI 1640 (полученной от компании GIBC(О), Гранд-Айлэнд, штат Нью-Йорк). Эта среда содержала 25 ммоль НЕРЕS (N-2-гидроксиэтилпиперазин-N'-2-этансульфоновой кислоты) и L-глутамин (к которому добавляли 1%-ный раствор пенициллина/стрептомицина). Помимо этого к среде RPMI добавляли также дезактивированную на 10% (в результате нагревания при 56° С в течение 30 минут) сыворотку бычьего эмбриона. По 200 мкл суспензии РВМ помещали в 96 стерилизованных ячеек с плоским дном (чашки Петри) (полученные от компании Becton Dickinson Labware, Линкольн-Парк, штат Нью-Джерси).
Подготовка соединения
Соединения солюбилизировали в этаноле, диметилсульфоксиде (ДМСО) или водном растворе культуры ткани с последующим разбавлением водным раствором культуры ткани, 0,01 N раствором гидроксида натрия или 0,01 N раствором соляной кислоты. (Выбор растворителя зависит от химических свойств исследуемого соединения). При добавлении к образцам культуры конечная концентрация этанола или ДМСО не должна превышать 1%. На начальной стадии исследования соединений их концентрация находилась в интервале от 0,1 до 5 мкг/мл. Соединения, оказавшиеся эффективными при концентрации 0,5 мкг/мл, в дальнейшем были исследованы в более широком диапазоне концентраций.
Инкубация
Раствор исследуемого соединения в объеме, не превышающем 50 мкл, помещали в чашки Петри, в которых находилось по 200 мкл разбавленной цельной крови или РВМ в среде культуры. В контрольные чашки Петри добавляли растворитель и(или) среду (но не добавляли исследуемое соединение) и при необходимости доводили общий объем образца в каждой чашке Петри до 250 мкл. Чашки Петри закрывали пластмассовыми крышками, аккуратно перемешивали образцы и выдерживали их в течение 48 часов при 37° С в атмосфере, содержащей 5% углекислого газа.
Разделение
После окончания инкубационного периода крышки на чашках Петри с образцами заливали парафином и образцы центрифугировали в течение 10-15 минут при скорости вращения 1000 об/мин и температуре 4° С. Использовали центрифугу Dsmon IЕС Моdеl СRU-5000. Около 200 мкл среды отбирали из 4-8 ячеек и переносили в стерильную пробирку объемом 2 мл, предназначенную для работы при криогенных температурах. До проведения анализа образцы хранили при температуре -70° С.
Анализ Интерферона/Расчеты
Для анализа интерферона применяли способ определения биологической активности, используя А549 клетки рака легкого человека с признаками энцефаломиокардита. Подробно способ определения биологической активности описан Дж.Л.Бреннаном (G.L.Вгennan) и Л.X.Кроненбергом (L.H.Kronenberg) в статье "Автоматизированное определение биологической активности интерферонов в микроиспытательных ячейках", Biotechniques, июнь/июль, 78, 1983, приведенной в списке литературы. В кратком изложении этот способ заключается в следующем. Клетки А549 ячейки культивировали с исследуемыми образцами и стандартными растворами интерферона при 37° С в течение 12-24 часов. Затем подвергшиеся инкубации клетки инфицировали посевом вируса энцефаломиокардита. Перед количественным определением цитопатологического эффекта вирусов инфицированные клетки культивировали дополнительно в течение некоторого времени при 37° С. Для количественного определения цитопаталогического эффекта вирусов исследуемую систему окрашивали и затем спектрофотометрически определяли поглощение света. Результаты выражены в единицах альфа, единиц/мл, количество которых оценивали, исходя из данных, полученных для стандартного образца NIH HU IF-L. При проведении анализа способом нейтрализации интерферон был идентифицирован практически полностью, как α -интерферон, в то же время при использовании А549 клетки с вирусом энцефаломиокардита был обнаружен античеловеческий интерферон кролика (β -антиферон) и античеловеческий интерферон козы (α -интерферон).
С помощью приведенных выше способов испытания была исследована способность описанных в настоящем изобретении химических соединений стимулировать образование интерферона в клетках человека. Результаты приведены таблице 1, в которой знак "+" указывает на то, что соединение стимулирует образование α -интерферона при приведенной концентрации, знак "-" показывает, что соединение не стимулирует образования α -интерферона, а знак "±" указывает, что при используемой концентрации исследуемое соединение не проявляет четко выраженного действия.


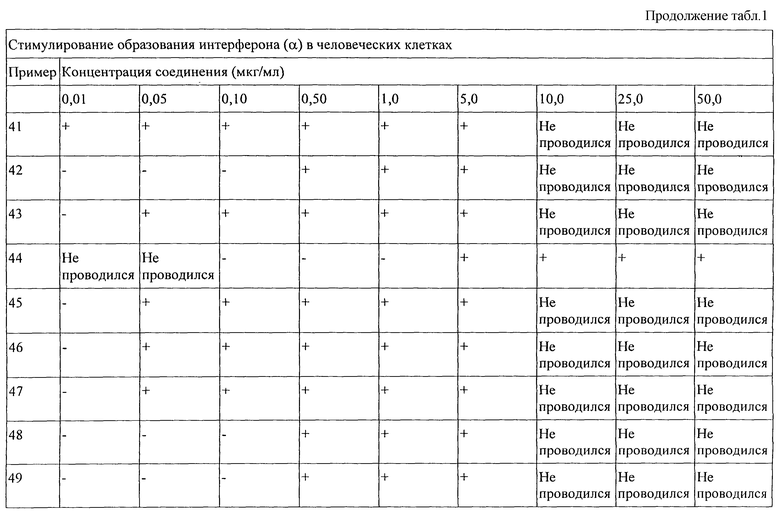

СТИМУЛИРОВАНИЕ СИНТЕЗА ЦИТОКИНА В КЛЕТКАХ ЧЕЛОВЕКА
Определение активности полученных в изобретении соединений для стимулирования образования цитокина проводили с использованием клеток человека in vitro. Активность определяли, исходя из количества интерферона α и фактора некроза опухоли (α )(IFN и TNF, соответственно), выделяемого в среду культуры. Для такой оценки использовали способ, описанный Testerman и др. в статье "Стимулирование цитокина под действием иммуномодуляторов Imiquimod и S-27609", Journal of Leukocyte Вiology, 58, стр.65-372 (сентябрь, 1995).
Приготовление клеток крови для культуры
Цельную кровь от здоровых доноров собирали венепункцией и помещали в специальные ампулы с ЭДТА. Одноядерные клетки периферийной крови (РВМС) отделяли от цельной крови с помощью центрифугирования с использованием аппарата Histopaque® -1077 (Sigma Chemicals, Сент-Луис, шт. Миссури). РВМС суспендировали в среде RРМI 1640, содержащей 10% сыворотки бычьего эмбриона, 2 ммоль/л L-глутамина и 1%-ный раствор пенициллина/стрептомицина (комплект RРМI). Концентрация клеток крови в такой суспензии составляла (3-4)× 106 клеток в 1 мл суспензии. Суспензию РВМС помещали в 48 стерилизованных ячеек с плоским дном (чашки Петри) (Соstаr, Кембридж, штат Миннесота, или Bector Dickinson Labware, Линколь-Парк, штат Нью-Джерси), содержащие равный объем комплекта RРМI с исследуемым соединением.
Подготовка соединения
Соединения солюбилизировали в диметилсульфоксиде (ДМСО). При добавлении к образцам культуры конечная концентрация ДМСО не должна превышать 1%. Концентрация испытываемых соединений обычно находится в интервале от 0,12 до 30 мкмоль/л.
Инкубация
Раствор применяемого для испытания соединения с концентрацией 60 мкмоль/л сначала добавляли в чашку Петри, в которой находился комплект RРМL, и затем несколько раз проводили последовательное разбавление образца в 3 раза. После этого добавляли равный объем суспензии РМВС; концентрация исследуемого соединения при этом сохранялась в интервале от 0,12 до 30 мкмоль/л. Конечная концентрация суспензии РВМС составляла (1,5-2)· 106 клеток/мл. Чашки Петри закрывали пластмассовыми крышками, аккуратно перемешивали образцы и выдерживали их в течение 18-24 часов при 37° С в атмосфере, содержащей 5% углекислого газа.
Разделение
После окончания инкубационного периода ячейки с образцами центрифугировали в течение 5-10 минут при скорости вращения 1000 об/мин (приблизительно 200 g) и температуре 4° С. Полученный верхний слой отбирали с помощью стерильной полипропиленовой пипетки и переносили в стерильную полипропиленовую пробирку. До проведения анализа образцы хранили при температуре от -30 до -70° С. Анализ образцов на содержание интерферона (α ) проводили либо способом ЕLISA, либо с помощью количественного определения биологической активности; анализ образцов на содержание фактора некроза опухоли (α ) осуществляли с помощью способа ЕLISA.
Количественное определение биологической активности интерферона
Для анализа интерферона применяли способ определения биологической активности, используя А549 клетки рака легкого человека с признаками энцефаломиокардита. Детали способа определения биологической активности описаны Дж.Л.Бреннаном (G.L.Вrennan) и Л.X.Кроненбергом (L.H.Кronenberg) в статье "Автоматизированное определение биологической активности интерферонов в микроиспытательных ячейках", Biotechniques, июнь/ июль, 78, 1983. В кратком изложении этот способ заключается в следующем. Клетки А549 ячейки культивировали с разбавленными исследуемыми образцами и стандартными растворами интерферона при 37° С в течение 24 часов. Затем подвергшиеся инкубации клетки инфицировали посевом вируса энцефаломиокардита. Перед количественным определением цитопатологического эффекта вирусов инфицированные клетки культивировали дополнительно в течение 24 часов при 37° С. Для количественного определения цитопатологического эффекта вирусов исследуемую систему окрашивали кристаллвиолетом и затем визуально наблюдали за видом культуры. Результаты выражены в единицах альфа, единиц/мл, количество которых оценивали, исходя из данных, полученных для стандартного образца NIH человеческого лейкоцита интерферона.
Анализ интерферона (α ) и фактора некроза опухоли (α ) способом ELISA
Концентрацию интерферона (α ) определяли способом ЕLISA, используя стандартный набор Human Мulti-Sресiеs (поставляемый компанией РВL Вiomedical Laboratories, Нью-Брунсвик, штат Нью-Джерси). Определение проводили в соответствии с прилагаемыми инструкциями производителя.
Концентрацию фактора некроза опухоли (α ) определили методом ЕLISA, используя наборы, поставляемые компаниями Genzime, Кэмбридж, штат Миннесота, R&αμπ; D Systems, Миннеаполис, штат Миннеаполис, или Рharmingen, Сан-Диего, штат Калифорния. Определение проводили в соответствии с прилагаемыми инструкциями производителя.
В нижеследующей таблице 2 знак "+" показывает, что соединение стимулировало образования указанного цитокина при приведенной концентрации; знак "-" указывает на то, что соединение не стимулировало образования указанного цитокина при приведенной концентрации, а знак "±" указывает на то, что при используемой концентрации исследуемое соединение не проявляет четко выраженного действия.

Выше приведено описание нескольких областей применения настоящего изобретения. Детальное описание изобретения и приведенные примеры даны здесь лишь для достижения полной ясности в понимании сути изобретения и областей его применения. Для специалистов в данной области будет очевидно, что описанные варианты осуществления изобретения могут быть различными способами модифицированы без нарушения сущности изобретения. Однако данное изобретение не ограничивается только точными деталями составов и структур, проиллюстрированных в различных примерах, но охватывает все стороны, приведенные в нижеследующей формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОЦИКЛИЛЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ | 2001 |
|
RU2351598C2 |
| АРИЛЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ НА ИХ ОСНОВЕ, СПОСОБЫ ЛЕЧЕНИЯ ВИРУСНОГО ЗАБОЛЕВАНИЯ НА ИХ ОСНОВЕ, СПОСОБЫ ЛЕЧЕНИЯ ОПУХОЛЕВОГО ЗАБОЛЕВАНИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2308456C2 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 1992 |
|
RU2193561C2 |
| ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ И СПОСОБ ИНДУКЦИИ БИОСИНТЕЗА ЦИТОКИНОВ НА ИХ ОСНОВЕ | 2000 |
|
RU2248975C2 |
| МОЧЕВИНОЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ И СПОСОБ ИНДУКЦИИ БИОСИНТЕЗА ЦИТОКИНОВ НА ИХ ОСНОВЕ | 2000 |
|
RU2265020C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ПРИМЕНЕНИЕМ ЭТИХ СОЕДИНЕНИЙ | 2007 |
|
RU2478635C2 |
| ПРОТИВОВИРУСНЫЕ СОЕДИНЕНИЯ | 2006 |
|
RU2441010C2 |
| ИНГИБИТОРЫ IAP | 2005 |
|
RU2401840C2 |
| ТИОЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2304143C2 |
| АМИДОЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ | 2000 |
|
RU2295343C2 |
Изобретение относится к оксазоло- и тиазоло[4,5-с]хинолин-4-аминам общей формулы (I), в которой R1 выбран из группы, состоящей из атомов кислорода и серы; R2 выбран из атома водорода; алкила; алкил-ОН (гидроксиалкила); алкил-Х-алкила; алкил-O-С(O)-N(R5)2; морфолинила, пирролидинила; алкил-Х-арильного радикала; алкенил-Х-арильного радикала; каждый из заместителей R3 и R4 представляет собой атом водорода или взятые вместе заместители R3 и R4 образуют конденсированную ароматическую или [1,5]нафтиридиновую систему; Х представляет собой -O- или простую связь; R5 представляет собой атом водорода. Также описаны промежуточные соединения, фармацевтическая композиция и способ стимулирования цитокинетического биосинтеза на основе этих соединений. Технический результат - получены новые соединения, обладающие ценными биологическими свойствами. 9 н. и 12 з.п. ф-лы, 2 табл.
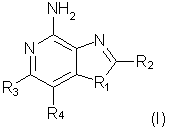
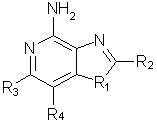
отличающееся тем, что
заместитель R1 выбран из группы, состоящей из атомов кислорода, и серы;
заместитель R2 выбран из группы, состоящей из
- атома водорода;
- алкила;
- алкил-ОН (гидроксиалкила);
- алкил-Х-алкила;
- алкил-O-С(O)-N(R5)2;
- морфолинила, пирролидинила;
- алкил-Х-арильного радикала;
- алкенил-Х-арильного радикала;
причем каждый из заместителей R3 и R4 независимо представляет собой атом водорода;
или взятые вместе заместители R3 и R4 образуют конденсированную ароматическую, или [1,5] нафтиридиновую систему;
X представляет собой -O- или простую связь; R5 представляет собой атом водорода; а также фармацевтически приемлемые соли на основе этих соединений.
2-метилтиазоло[4,5-с]хинолин-4-амин;
тиазоло[4,5-с]хинолин-4-амин;
2-этилтиазоло[4,5-с]хинолин-4-амин;
2-пропилтиазоло[4,5-с]хинолин-4-амин;
2-пентилтиазоло[4,5-с]хинолин-4-амин;
2-бутилтиазоло[4,5-с]хинолин-4-амин;
2-(1-метилэтил)тиазоло[4,5-с]хинолин-4-амин;
2-(2-фенил-1-этенил)тиазоло[4,5-с]хинолин-4-амин;
2-(2-фенил-1-этил)тиазоло[4,5-с]хинолин-4-амин;
2-(4-аминотиазоло[4,5-с]хинолин-2-ил)-1,1-диметилэтилкарбамат;
2-(этоксиметил)тиазоло[4,5-с]хинолин-4-амин;
2-(метоксиметил)тиазоло[4,5-с]хинолин-4-амин;
2-(2-метилпропил)тиазоло[4,5-с]хинолин-4-амин;
2-бензилтиазоло[4,5-с]хинолин-4-амин;
8-метил-2-пропилтиазоло[4,5-с]хинолин-4-амин;
(4-аминотиазоло[4,5-с]хинолин-2-ил)метанол;
2-метилоксазоло[4,5-с]хинолин-4-амин;
2-этилоксазоло[4,5-с]хинолин-4-амин;
2-бутилоксазоло[4,5-с]хинолин-4-амин;
2-пропилтиазоло[4,5-с]хинолин-4,8-диамин;
2-пропилоксазоло[4,5-с]хинолин-4-амин;
8-бром-2-пропилтиазоло[4,5-с]хинолин-4-амин;
7-метил-2-пропилтиазоло[4,5-с]хинолин-4-амин;
2-бутил-7-метилоксазоло[4,5-с]хинолин-4-амин;
7-метил-2-пропилоксазоло[4,5-с]хинолин-4-амин;
7-фтор-2-пропилоксазоло[4,5-с]хинолин-4-амин;
7-фтор-2-пропилтиазоло[4,5-с]хинолин-4-амин;
2-пропил-7-(трифторметил)тиазоло[4,5-с]хинолин-4-амин;
2-(4-морфолино)тиазоло[4,5-с]хинолин-4-амин;
2-(1-пирролидино)тиазоло[4,5-с]хинолин-4-амин;
2-бутилтиазоло[4,5-с] [1,5]нафтиридин-4-амин;
2-пропилтиазоло[4,5-с] [1,5]нафтиридин-4-амин;
7-хлор-2-пропилтиазоло[4,5-с]хинолин-4-амин;
7-метокси-2-пропилтиазоло[4,5-с]хинолин-4-амин;
и фармацевтически приемлемые соли на основе этих соединений.

где заместитель R1 выбран из группы, состоящей из атомов кислорода, и серы;
заместитель R2 выбран из группы, состоящей из
- атома водорода;
- алкила;
- алкил-ОН (гидроксиалкила);
- алкил-Х-алкила;
- алкил-O-С(O)-N(R5)2;
- морфолинила, пирролидинила;
- алкил-Х-арильного радикала;
- алкенил-Х-арильного радикала;
каждый из заместителей R3 и R4 независимо представляет собой атом водорода;
или взятые вместе заместители R3 и R4 образуют конденсированную ароматическую или [1,5] нафтиридиновую систему;
Х представляет собой -O- или простую связь, R5 представляет собой атом водорода; а также фармацевтически приемлемые соли на основе этих соединений с фармацевтически приемлемым носителем.

отличающееся тем, что
заместитель R1 выбран из группы, состоящей из атомов кислорода и серы; заместитель R2 выбран из группы, состоящей из
- алкила;
- алкила-ОН;
- алкил-Х-алкила;
- алкил-Х-арила;
- морфолинила;
- SO2CH3;
каждый из заместителей R3 и R4 независимо представляет собой атом водорода;
или взятые вместе заместители R3 и R4 образуют конденсированную ароматическую или [1,5] нафтиридиновую систему;
Х представляет собой -О- или простую связь.
2-метилтиазоло[4,5-с]хинолин-5N-оксида;
2-этилтиазоло[4,5-с]хинолин-5N-оксида;
2-пропилтиазоло[4,5-с]хинолин-5N-оксида;
2-пентилтиазоло[4,5-с]хинолин-5N-оксида;
2-бутилтиазоло[4,5-с]хинолин-5N-оксида;
2-(1-метилэтил)тиазоло[4,5-с]хинолин-5N-оксида;
2-(2-фенил-1-этенил)тиазоло[4,5-с]хинолин-5N-оксида;
2-фенилэтилтиазоло[4,5-с]хинолин-5N-оксида;
2-метил-1-тиазоло[4,5-с]хинолин-2-ил-2-пропанол-5N-оксида;
2-(этоксиметил)тиазоло[4,5-с]хинолин-5N-оксида;
2-(метоксиметил)тиазоло[4,5-с]хинолин-5N-оксида;
2-(2-метилпропил)тиазоло[4,5-с]хинолин-5N-оксида;
2-бензилтиазоло[4,5-с]хинолин-5N-оксида;
8-метил-2-пропилтиазоло[4,5-с]хинолин-5N-оксида; и
2-бутилоксазоло[4,5-с]хинолин-5N-оксида.
Приоритет по пунктам и признакам:
| Способ получения тиазоло [4,5-с] хинолина или его кислотно-аддитивных солей | 1986 |
|
SU1544190A3 |
| WO 9305042 А1,18.03.1993 | |||
| WO 9502597 А1, 26.01.1995 | |||
| WO 9502598 А1, 26.01.1995 | |||
| US 4038396 А, 26.07.1977 | |||
| СПОСОБ ПОЛУЧЕНИЯ ФРАКЦИИ ФЕНОЛЬНЫХ ВЕЩЕСТВ ИЗ ЧАГИ | 2013 |
|
RU2530637C1 |

