Предпосылки изобретения
Настоящее изобретение относится к циклическим пептидам, которые обладают агонистической активностью в отношении соматостатина, как видно из формулы (1), показанной и определенной ниже, или к их фармацевтически приемлемым солям, фармацевтическим композициям, содержащим указанные пептиды, и к их использованию в качестве агонистов подтипов рецептора соматостатина. Пептиды согласно настоящему изобретению связываются селективно с подтипом 5 рецептора соматостатина и проявляют агонистический эффект в отношении того подтипа рецепторов соматостатина, с которым указанные пептиды связываются.
Соматостатин (SRIF) представляет циклический тетрадекапептидный гормон, содержащий дисульфидный мостик между положением 3 и положением 14 (Heiman, et al., Neuroendocrinology, 45:429-436 (1987)) и обладающий способностью ингибировать высвобождение гормона роста (ГР, GH) и тиреотропного гормона (ТТГ, TSH), ингибировать высвобождение амилина, инсулина и глюкагона, снижать секрецию желудочного сока и высвобождение нейротрансмиттеров. Метаболизм соматостатина аминопептидазами и карбоксипептидазами сокращает период его действия. В связи с коротким сроком жизни нативного соматостатина разрабатываются различные аналоги соматостатина например, для лечения акромегалии (Raynor, et al., Molecular Pharmacol. 43:838 (1993)).
Были идентифицированы и охарактеризованы пять различных рецепторов соматостатина (Hoyer, et al., Naunyn-Schmiedeberg's Arch. Pharmacol., 350:441 (1994)). Соматостатин связывается с пятью различными подтипами рецептора (SSTR) с относительно высокой и равной аффинностью для каждого подтипа. Связывание с различными подтипами соматостатина имеет место при лечении различных указанных ниже состояний и/или заболеваний (SSTR-2) (Raynor, et al., Molecular Pharmacol. 43:838 (1993); Lloyd, et al., Am. J. Physiol., 268:G102 (1995)), тогда как за ингибирование инсулина ответственен тип 5 рецептора соматостатина (SSTR-5) (Coy, et al., 197:366-371 (1993)). Активация типов 2 и 5 связана с подавлением гормона роста и более конкретно с аденомами, секретирующими ГР (при акромегалии), а также с аденомами, секретирующими тиреотропный гормон (ТТГ). Активация типа 2, но не типа 5, имеет место при лечении аденом, секретирующих пролактин. Другие показания, связанные с активацией подтипов соматостатина, представляют ингибирование инсулина и/или глюкагона и более конкретно сахарного диабета, ангиопатии, пролиферирующей ретинопатии, феномена «утренней зари» и нефропатии; ингибирование секреции желудочного сока и более конкретно пептических язв, тонкокишечного свища и свища поджелудочной железы, синдрома раздраженной толстой кишки, демпинг - синдрома, водянистого стула, СПИД - ассоциированной диареи, диареи, вызванной химиотерапией, острого или хронического панкреатита и опухолей, секретирующих гастроинтестинальный гормон; лечение злокачественной опухоли, такой как гепатома; ингибирование ангиогенеза, лечение воспалительных заболеваний, таких как артрит; ретинопатия; хроническая реакция отторжения трансплантата; ангиопластика; предупреждение отторжения трансплантированных сосудов и желудочно-кишечное кровотечение. Предпочтительно иметь аналог, который является селективным для специфического подтипа рецептора соматостатина, отвечающего за требуемую биологическую реакцию, что таким образом позволит снизить взаимодействие с другими подтипами рецептора, которое могло бы привести к нежелательным побочным эффектам.
Пептиды формулы (I) представляют подгруппу, входящую в группу соединений, описанных и заявленных в совместно рассматриваемой заявке на выдачу патента США №08/855204, поданной 13 мая 1997 года, права на которую частично переданы владельцу прав на настоящее изобретение. Соединения формулы (I) согласно настоящей заявке конкретно не описаны в заявке на выдачу патента США №08/855204. Было обнаружено, что соединения формулы (I) согласно настоящему изобретению обладают агонистической активностью в отношении соматостатина. Указанное открытие явилось неожиданным, поскольку ранее в заявке на выдачу патента США №08/855204 было показано, что раскрытые в нем соединения обладают антагонистической активностью в отношении соматостатина.
Сущность изобретения
Одним объектом настоящего изобретения является пептид формулы (I)
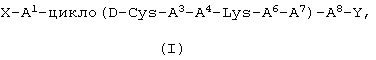
или его фармацевтически приемлемая соль,
где
Х представляет собой Н, 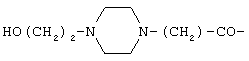 или
или
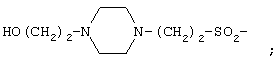
А1 и А3 каждый независимо представляет собой D- или L-изомер аминокислоты, выбираемой из группы, состоящей из Phe, Tyr, Tyr(I), Trp, 3-Pal, 4-Pal, Сра и Nal;
А4 представляет собой L-Trp, D-Trp, L-β-метил-Trp или D-β-метил-Trp;
А6 представляет собой -NH-(CHR1)n-CO-, где n равно 2, 3 или 4;
А7 представляет собой L-или D-Cys;
А8 представляет собой D-или L-изомер аминокислоты, выбираемой из группы, состоящей из Phe, Tyr, Tyr(I), Trp, Nal, Сра, Val, Leu, He, Ser и Thr;
Y представляет собой NR2R3, где R2 и R3 каждый представляет собой независимо Н или (C1-C5)алкил;
R1 выбран из группы, состоящей из Н, (C1-C4) алкила и -СН2-арила; причем указанный арил необязательно замещен группой, выбираемой из группы, состоящей из фенила, 1-нафтила и 2-нафтила, где указанная необязательно замещенная группа необязательно замещена одним или более заместителями, каждый из которых независимо выбран из группы, состоящей из (C1-6) алкила, (С2-6) алкенила, (C2-6) алкинила, арила, арил (C1-6) алкила, (С1-6)алкокси, -N(R4R5), -COOH, -CON(R4R5), галогена, -ОН, -CN и -NO2;
R4 и R5 каждый представляет собой независимо Н или (C1-3) алкил;
где Cys в А2 соединен с Cys в А7 дисульфидной связью, образуемой из тиоловых групп каждого Cys.
Предпочтительно группа пептидов указанного выше пептида формулы (I) включает такие пептиды, в которых
Х представляет собой Н;
А1 представляет собой L-Phe, D-Phe, L-Cpa или D-Cpa;
А3 представляет собой L-Tyr, L-Trp или L-3-Pal;
А4 представляет собой D-Trp;
А6 представляет собой β-Ala или Gaba;
А7 представляет собой L-Cys;
А8 представляет собой Thr, L-Trp, L-Leu или L-Nal; и
R2 и R3 каждый представляет собой Н; или их фармацевтически приемлемую соль.
Предпочтительные пептиды из указанной выше группы пептидов представляют:
Сра-цикло(D-Cys-3-Pal-D-Trp-Lys-Gaba-Cys)-Nal-NH2;
Сра-цикло (D-Cys-3-Pal-D-Trp-Lys-β-Ala-Cys)-Nal-NH2;
Phe-цикло(D-Cys-3-Pal-D-Trp-Lys-Gaba-Cys)-Nal-NH2;
Phe-цикло(D-Cys-Tyr-D-Trp-Lys-Gaba-Cys)-Nal-NH2;
Phe-цикло(D-Cys-Trp-D-Trp-Lys-Gaba-Cys)-Nal-NH2;
Phe-цикло(D-Cys-Tyr-D-Trp-Lys-Gaba-Cys)-Trp-NH2;
D-Phe-цикло(D-Cys-Tyr-D-Trp-Lys-Gaba-Cys)-Nal-NH2;
D-Phe-цикло(D-Cys-Tyr-D-Trp-Lys-Gaba-Cys)-Leu-NH2 и
Phe-цикло(D-Cys-Tyr-D-Trp-Lys-Gaba-Cys)-Thr-NH2; или их фармацевтически приемлемую соль.
Предпочтительные пептиды из указанной выше группы пептидов представляют:
Сра-цикло(D-Cys-3-Pal-D-Trp-Lys-Gaba-Cys)-Nal-NH2 и
Сра-цикло(D-Cys-3-Pal-D-Trp-Lys-β-Ala-Cys)-Nal-NH2; или их фармацевтически приемлемую соль.
Другим объектом настоящего изобретения является фармацевтическая композиция, применяемая для проявления агонистического ответа в отношении действия соматостатина у человека или другого животного, которая включает эффективное количество пептида формулы (I) или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
Другим объектом настоящего изобретения является способ проявления агонистического ответа в отношении действия соматостатина у человека или у другого млекопитающего в случае наличия такой потребности, который включает введение эффективного количества пептида формулы (I) или его фармацевтически приемлемой соли человеку или другому животному.
Еще одним объектом настоящего изобретения является способ селективного связывания подтипа рецептора типа 5 соматостатина человека или другого животного, который включает введение эффективного количества пептида формулы (I) или его фармацевтически приемлемой соли человеку или другому животному.
Еще одним объектом настоящего изобретения является способ лечения заболевания или состояния у человека или другого животного, при наличии потребности, который включает введение эффективного количества пептида формулы (I) или его фармацевтически приемлемой соли человеку или другому животному, причем указанное состояние выбирают из группы, включающей синдром Кушинга, гонадотропиному, гиперпаратиреоз, болезнь Педжета, випому, гиперплазию панкреатических островков, гиперинсулинизм, гастриному, синдром Золлингера - Эллисона, СПИД-ассоциированную гиперсекреторную диарею и вызванную другими состояниями, синдром раздраженной толстой кишки, панкреатит, болезнь Крона, системный склероз, рак щитовидной железы, псориаз, гипотензию, приступы паники, склеродому, непроходимость тонкой кишки, гастроэзофагеальный рефлюкс, дуоденогастральный рефлюкс, болезнь Грейвса, поликистоз яичников, кровотечение в верхней части желудочно-кишечного тракта, ложную кисту поджелудочной железы, асцит поджелудочной железы, лейкоз, менингиому, раковую кахексию, акромегалию, рестеноз, гепатому, рак легкого, меланому, подавление ускоренного роста солидной опухоли, снижение веса тела, лечение невосприимчивости к инсулину, синдром X, пролонгирование выживания панкреатических клеток, фиброз, гиперлипидемию, гиперамилинемию, гиперпролактинемию и пролактинемию.
Еще одним объектом настоящего изобретения является способ ингибирования при наличии потребности секреции гормона роста, инсулина, глюкагона или экзокринной секреции поджелудочной железы у человека или другого животного, который включает введение пептида формулы (I) или его фармацевтически приемлемой соли указанному человеку или другому животному.
Еще одним объектом настоящего изобретения является способ визуализации in vivo клеток, содержащих рецепторы соматостатина, у человека или другого животного, который включает введение пептида формулы (I) при условии, что по меньшей мере один из А1, А3 или А8 представляет Tyr(1), или его фармацевтически приемлемой соли указанному человеку или другому животному.
Еще одним объектом настоящего изобретения является способ визуализации in vitro клеток, содержащих рецепторы соматостатина, который включает введение пептида формулы (I) при условии, что по меньшей мере один из А1, А3 или А8 представляет Tyr(1), или его фармацевтически приемлемой соли указанному человеку или другому животному. Такие пептиды согласно настоящему изобретению могут применяться либо in vivo для обнаружения клеток, содержащих рецепторы соматостатина (например, раковых клеток), либо in vitro в качестве радиолиганда в тесте на связывание рецептора соматостатина.
Принятые трехбуквенные обозначения используются для обозначения аминокислот в пептиде согласно настоящему изобретению. В указанной далее в описании формуле дисульфидная связь между тиольной группой в боковой цепи остатка А2 (например, D-Cys) и тиольной группой в боковой цепи остатка А7 (например, L-Cys или D-Cys) не показана. Ниже даны расшифровки некоторых сокращений аминокислот: Сра=п-хлорфенилаланин; Nal=β-(2-нафтил)аланин; 3-Pal=β-(3-пиридил)аланин; 4-Pal=β-(4-пиридил)аланин и Gaba=4-аминомасляная кислота. Определение «-NH-(СН2)n-СО-, где n равно 2, 3 или 4» охватывает такие аминокислоты, как β-Ala и Gaba.
Если особо не оговорено иное, трехбуквенное сокращение аминокислот относится к L-изомеру.
Термин «алкил» включает алкильные группы указанной длины либо в линейной, либо в разветвленной конфигурации. Примерами таких алкильных групп являются метил, этил, пропил, изопропил, бутил, фтор-бутил, трет-бутил, пентил, изопентил и т.п. В том случае, когда термин С0-алкил включен в определение, он обозначает одинарную ковалентную связь.
Термин «алкенил» включает углеводородные группы, содержащие одну или более двойных связей, и указанное число атомов углерода находится либо в линейной, либо в разветвленной конфигурации. Примеры таких алкенильных групп включают этенил, пропенил, изопропенил, бутенил, фтор-бутенил, трет-бутенил, пентенил, изопентенил, гексенил, изогексенил и т.п.
Термин «алкинил» включает такие алкинильные группы, т.е. углеводородные группы, имеющие одну или более тройных связей, которые содержат указанное число атомов углерода либо в линейной, либо в разветвленной конфигурации. Примеры таких алкинильных групп включают этинил, пропинил, бутинил, пентинил, изопентинил, гексинил, изогексинил и т.п.
Термин «алкокси» охватывает те алкоксигруппы с указанным числом атомов углерода, которые находятся либо в линейной, либо в разветвленной конфигурации. Примеры таких алкоксигрупп включают метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, трет-бутокси, пентокси, изопентокси, гексокси, изогексокси и т.п.
Термин «арил» включает известные в технике ароматические кольца, которые могут быть моноциклическими или бициклическими, такие как фенил и нафтил.
Термин «галоген» включает хлор, бром, иод и фтор.
Подробное описание
Любой средний специалист в данной области на основании приведенного описания изобретения может использовать его с полной отдачей. Приведенные ниже конкретные варианты осуществления изобретения даны лишь для иллюстрации изобретения и никоим образом не ограничивают объем настоящего изобретения.
Пептиды согласно настоящему изобретению могут быть и были синтезированы на амидной МВНА-смоле Ринка (4-(2',4'-диметоксифенил-Fmoc-аминометил)феноксиацетамидонорлейцил-МВНА-смоле) с использованием стандартной процедуры твердофазного синтеза FMOC-химии с последующим отщеплением от смолы с помощью смеси ТФУ/фенол/Н2О/триизопропилсилан (83 мл/5 г/10 мл/ 2 мл). Пептиды циклизуют в смеси СН3CN/Н2О (5 мл/5 мл) с использованием смолы EKATHIOX™ (EKAGEN Corporation, San Carlos, CA) и очищают на C18 силикагеле (Rainin Instruments Co., Woburn, MA, в настоящее время Varian Analytical, Walnut Creek, CA) с использованием буфера на основе смеси ацетонитрил/0,1% трифторуксусная кислота. Гомогенность оценивают методом аналитической ВЭЖХ и устанавливают ее уровень >95% для каждого пептида. Пептиды характеризуют методом масс-спектрометриии.
Синтез иод-содержащих Tyr (Tyr(1)) пептидов формулы (I) согласно настоящему изобретению (например, хлорамин-Т методом) хорошо известен и описан в литературе, и средний специалист в данной области в состоянии его осуществить. (См., например, Czernick, et al., J. Biol. Chem. 258:5525 (1993) и Европейский патент №389180 Bl.)
Пептид формулы (I), где Х представляет собой
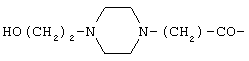 или
или 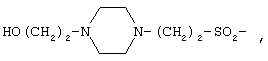 может быть синтезирован с помощью способов и методик, описанных в патенте США №5552520, содержание которого полностью включено в настоящее описание.
может быть синтезирован с помощью способов и методик, описанных в патенте США №5552520, содержание которого полностью включено в настоящее описание.
Ниже приведено подробное описание синтеза соединения в примерах 1 и 2. Другие пептиды из группы соединений формулы (I) могут быть получены при введении соответствующих модификаций, известных специалистам в области пептидного синтеза.
ПРИМЕР 1
Стадия 1 = Получение Fmoc-Cpa-S-тритил-D-Cys-Pal-N-in-t-Boc-D-Trp-N-ε-t-Boc-Lys-β-Ala-S-тритил-Cys-Nal-4-(2',4'-диметоксифениламинометил)феноксиацетамидонорлейцил-4-метил-бензгидриламиновой смолы.
Амидную МВНА - смолу Ринка (Novabiochem, Inc., San Diego, CA) (0,5 г; 0,265 ммоль) помещают в реакционный сосуд пептидного синтезатора 24-RV, включающего в собранном виде объединение вибратора (от Burrell Wrist-Action Laboratory Shaker), распределителя растворителя и вакуумного насоса. Пептидный синтезатор запрограммирован для проведения следующего реакционного цикла:
а. Диметилформамид;
b. 25% Пиперидин в диметилформамиде (добавляемый вручную) (2 раза в течение 15 минут с одноразовым промыванием ДМФ между добавлениями);
с. Промывание ДМФ (3×10 мл, по 1 минуте каждое).
Смолу перемешивают с FMOC-Nal (1,06 ммоль), гексафторфосфатом 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметил-урония (HBUT) (1,007 ммоль) и диизопропилэтиламином (2,12 ммоль) в диметилформамиде в течение примерно 1,5 часов и полученную аминокислотную смолу затем циклизуют в ходе осуществления стадий (а)-(с) в указанной выше программе промывания/разблокирования.
С применением той же процедуры связывают последовательно с Nal-смолой указанные ниже аминокислоты: Fmoc-S-тритил-Cys, Fmoc-β-Ala, N-ε-t-Boc-Lys, Fmoc-(N-in-t-Boc)-D-Trp, Fmoc-Pal, Fmoc-S-тритил-D-Суз и Fmoc-p-Cl-Phe.
После промывания ДМФ (3×10 мл, примерно по 1 минуте каждое) и сушки в вакууме получают готовую пептидную смолу весом 0,749 г.
Стадия 2: Получение Н-Сра-цикло(D-Cys-Pal-D-Trp-Lys-β-Ala-Cys)-Nal-NH2
Пептидную смолу, полученную на стадии 1 примера 1 (0,36 г; 0,087 ммоль), смешивают со свежеприготовленным раствором ТФУ (8,8 мл), фенола (0,5 г), Н2О (0,5 мл) и триизопропилсилана (0,2 мл) при комнатной температуре и перемешивают в течение примерно 2,5 часов. Избыток ТФУ выпаривают при пониженном давлении с получением маслянистого остатка. Затем к указанному маслянистому остатку добавляют эфир и свободный линейный пептид осаждают, фильтруют и промывают сухим эфиром. Далее неочищенный пептид растворяют в 10 мл смеси СН3CN/Н2О (5 мл/5 мл) и затем добавляют 200 мг смолы EKATHIOX™. Смесь перемешивают в течение ночи и фильтруют. Фильтрат выпаривают до малого объема и затем наносят на колонку (22-250 мм) с тонкоизмельченным сорбентом октадецилсилановым силикагелем (5 мкм). После элюирования линейным градиентом (от 20% до 40% в течение 60 минут) ацетонитрила в воде (оба растворителя содержат 0,1% трифторуксусной кислоты) получают фракции, которые исследуют с помощью аналитической высокоэффективной жидкостной хроматографии (ВЭЖХ) и объединяют соответствующие фракции с целью достижения максимальной чистоты. Лиофилизация раствора от воды дает 26 мг продукта в виде белого пушистого порошка. Указанный продукт является гомогенным, по данным ВЭЖХ на C18 силикагеле с использованием того же элюента, что и в описанной выше процедуре, и линейного градиента (от 30% до 70% в течение 15 минут). (Время удерживания - 6,313 минуты). Метод инфузионной масс-спектрометрии подтверждает состав циклического октапептида, М.м. 1133,8.
ПРИМЕР 2
Стадия 1: Получение Fmoc-Сра-S-тритил-D-Cys-Pal-in-t-Вос-D-Trp-N-ε-t-Boc-Lys-Gaba-S-тритил-Cys-Nal-4-(2',4'-диметоксифениламинометил)феноксиацетамидонорлейцил-4-метилбензгидриламиновой смолы
Амидную МВНА - смолу Ринка (Novabiochem, Inc., San Diego, CA) (0,2 г; 0,106 ммоль) помещают в реакционный сосуд №3 (RV-3) пептидного синтезатора 24-RV. Пептидный синтезатор запрограммирован на выполнение следующего реакционного цикла:
а. Диметилформамид;
b. 25% Пиперидин в диметилформамиде (добавляемый вручную) (2 раза в течение 15 минут с однократным промыванием ДМФ между добавлениями);
с. Промывание ДМФ (3×10 мл, каждый раз по 1 минуте).
Смолу перемешивают с FMOC-Nal (0,424 ммоль), гексафторфосфатом 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (HBUT) (0,403 ммоль) и диизопропилэтиламином (0,848 ммоль) в диметилформамиде в течение примерно 1,5 часов и затем полученную аминокислотную смолу циклизуют в ходе стадий (а)-(с) с включением указанной выше программы промывания.
С использованием той же процедуры проводят последовательное связывание с пептидной смолой указанных ниже аминокислот: Fmoc-S-тритил-Cys, Fmoc-Gaba, N-ε-t-Boc-Lys, Fmoc-(N-in-t-Boc)-D-Trp, Fmoc-Pal, Fmoc-S-тритил-D-Cys и Fmoc-Сра. После промывания ДМФ (3×10 мл, примерно по 1 минуте каждое) и сушки в вакууме получают готовую смолу весом 0,31 г.
Стадия 2: Получение Н-Сра-цикло(D-Cys-Pal-D-Trp-Lys-Gaba-Cys)-Nal-NH2
Пептидную смолу, полученную на стадии 1 примера 2, смешивают со свежеприготовленным раствором ТФУ (8,3 мл), фенола (0,5 г), Н2О (1 мл) и триизопропилсилана (0,2 мл) при комнатной температуре и перемешивают в течение примерно 2,5 часов. Избыток ТФУ выпаривают при пониженном давлении с получением маслянистого остатка. Затем к указанному маслянистому остатку добавляют эфир и свободный линейный пептид осаждают, фильтруют и промывают сухим эфиром. Далее неочищенный пептид растворяют в 10 мл смеси СН3CN/Н2О и затем добавляют 200 мг смолы EKATHIOX™. Смесь перемешивают в течение ночи и фильтруют. Фильтрат выпаривают до малого объема и затем наносят на колонку (22-250 мм) с тонкоизмельченным сорбентом октадецилсилановым силикагелем (5 мкм) и элюируют линейным градиентом (от 20% до 100% в течение 60 минут) ацетонитрила в воде, причем оба растворителя содержат 0,1% трифторуксусной кислоты. Фракции исследуют с помощью аналитической высокоэффективной жидкостной хроматографии (ВЭЖХ) и объединяют соответствующие фракции с целью достижения максимальной чистоты. Лиофилизация раствора от воды дает 13 мг продукта в виде белого пушистого порошка. Указанный продукт является гомогенным, по данным ВЭЖХ на C18 силикагеле с использованием того же элюента, что был указан выше (от 20% до 80% в течение 15 минут). (Время удерживания - 9,195 минуты). Анализ методом инфузионной масс-спектрометрии подтверждает состав циклического октапептида, М.м. 1147,83.
Пептиды согласно настоящему изобретению могут быть получены в виде фармацевтически приемлемых солей. Примеры таких солей включают, не ограничиваясь, соли органических кислот (например, уксусной, молочной, малеиновой, лимонной, яблочной, аскорбиновой, янтарной, бензойной, метансульфоновой, толуолсульфоновой или памоевой кислоты), неорганических кислот (например, соляной кислоты, серной кислоты или фосфорной кислоты) и полимерных кислот (например, танниновой кислоты, карбоксиметилцеллюлозы, полимолочной кислоты, полигликолевой кислоты или сополимеров полимолочной - гликолевой кислот). Типичный метод получения соли пептида согласно настоящему изобретению известен в данной области техники и может быть осуществлен с помощью стандартных методик солевого обмена. При этом ТФУ-соль пептида согласно настоящему изобретению (ТФУ-соль получают при очистке пептида методом препаративной ВЭЖХ, элюируя ТФУ-содержащими буферными растворами) может быть превращена в другую соль, такую как ацетат, при растворении пептида в небольшом количестве 0,25 н. водного раствора уксусной кислоты. Полученный раствор наносят на полупрепаративную ВЭЖХ колонку (Zorbax, 300 SB, C-8). Колонку элюируют (1) 0,1 н. водным раствором ацетата аммония в течение 0,5 часа, (2) 0,25 н. водным раствором уксусной кислоты в течение 0,5 часа и (3) линейным градиентом (от 20% до 100% раствора В в течение 30 минут) при скорости элюирования 4 мл/мин (раствор А представляет собой 0,25 н. водный раствор уксусной кислоты; раствор В представляет собой 0,25 н. уксусную кислоту в смеси ацетонитрил/вода, 80:20). Фракции, содержащие пептид, собирают и лиофилизуют досуха.
Аффинность пептида согласно настоящему изобретению к подтипам 1-5 рецептора соматостатина человека, (sst1, sst2, sst3, sst4 и sst5) определяют измерением уровня ингибирования связывания (125I-Tyr11) SRIF-14 с СНО-К1 трансфицированными клетками.
Ген рецептора человека sst1 клонируют в виде геномного фрагмента. Сегмент PstI-XmnI размером 1,5 т.п.н., содержащий 100 п.н. 5'-нетранслируемого участка, 1,17 т.п.н. полного кодирующего участка и 230 п.н. 3'-нетранслируемого участка, модифицируют добавлением линкера Bg1ll. Полученный фрагмент ДНК клонируют в сайте BamHI на pCMV-81 с получением плазмиды экспрессии млекопитающего (предоставлено Dr.Graeme Bell, Университет Чикаго). Клональная клеточная линия, стабильно экспрессирующая sst1-рецептор, была получена при трансфекции в СНО - К1 клетки (АТСС) с использованием метода соосаждения с фосфатом кальция (1). Плазмида p-RSV-neo (АТСС) была включена в качестве селективного маркера. Клональную клеточную линию выделяют на среде RPMI 1604, содержащей 0,5 мг/мл G418 (Gibco), циклически клонируют и размножают в культуре.
Ген sst2 рецептора соматостатина человека выделяют в виде BamHI фрагмента геномной ДНК размером 1,7 т.п.н. и субклонируют в плазмидном векторе pGEM3Z (Promega), любезно предоставленном д-ром Г. Беллом (Dr. G.Bell, Университет Чикаго). Вектор экспрессии в клетках млекопитающих конструируют при введении BamHI-HindII фрагмента размером 1,7 т.п.н. в совместимые сайты рестрикционных эндонуклеаз на плазмиде pCMV 5. Клональную клеточную линию получают при трансфекции в СНО-К1 клетки с использованием метода соосаждения с фосфатом кальция. Плазмиду pRSV-neo включают в качестве селективного маркера.
sst3 Человека выделяют в виде геномного фрагмента, причем полная кодирующая последовательность содержится в BamHI/HindIII фрагменте размером 2,4 т.п.н. Плазмиду экспрессии в клетках млекопитающих pCMV-h3 конструируют при введении NcoI-HindIII фрагмента размером 2,0 т.п.н. в сайт EcoR1 на векторе pCMV после модификации концов и добавления EcoR1 линкеров. Клональную клеточную линию, стабильно экспрессирующую sst3 рецептор, получают при трансфекции в СНО-К1 клетки (АТСС) с использованием метода соосаждения с фосфатом кальция. Плазмиду pRSV-neo (ATCC) включают в качестве селективного маркера. Клональные клеточные линии выделяют на среде RPMI 1640, содержащей 0,5 мг/мл G418 (Gibco), циклически клонируют и размножают в культуре.
Плазмида экспрессии sst4 рецептора человека pCMV-HX была предоставлена д-ром Г. Беллом (Dr. Graeme Bell, Университет Чикаго). Вектор, содержащий геномный фрагмент NheI-NheI размером 1,4 т.п.н., кодирующий sst4 человека, 456 п.н. на 5'-нетранслируемом участке и 200 п.н. на 3'-нетранслируемом участке, клонируют в сайтах XbaI/EcoR1 на pCMV-HX. Клональную клеточную линию, стабильно экспрессирующую sst4 рецептор, получают при трансфекции в СНО-К1 клетки (ATCC) с использованием метода соосаждения с фосфатом кальция. Плазмиду pRSV-neo (ATCC) включают в качестве селективного маркера. Клональные клеточные линии выделяют на среде RPMI 1640, содержащей 0,5 мг/мл G418 (Gibco), циклически клонируют и размножают в культуре.
Ген sst5 человека получают методом ПЦР с использованием в качестве матрицы геномного клона λ, который был любезно предоставлен д-ром Г. Беллом (Dr. Graeme Bell, Университет Чикаго). Полученный ПЦР фрагмент размером 1,2 т.п.н. содержит 21 пару оснований на 5'-нетранслируемом участке, полностью кодирующий участок и 55 п.н. на 3'-нетранслируемом участке. Клон вставляют в сайт EcoRl плазмиды pBSSK(+). Вставку восстанавливают в виде HindIII-XbaI фрагмента размером 1,2 т.п.н. для субклонирования в векторе экспрессии млекопитающих pCMV 5. Клональную клеточную линию, стабильно экспрессирующую sst5 рецептор, получают при трансфекции в СНО-К1 клетки (АТСС) с использованием метода соосаждения с фосфатом кальция. Плазмиду pRSV-neo (АТСС) включают в качестве селективного маркера. Клональные клеточные линии выделяют на среде RPMI 1640, содержащей 0,5 мг/мл G418 (Gibco), циклически клонируют и размножают в культуре.
СНО-К1 клетки, стабильно экспрессирующие один из sst-рецепторов человека, выращивают в среде RPMI 1640, содержащей 10% фетальной сыворотки теленка и 0,4 мг/мл генетицина. Клетки собирают с использованием 0,5 мМ ЭДТА и центрифугируют при 500g в течение примерно 5 минут при температуре примерно 4°С. Осадок ресуспендируют в 50 мМ Трис, рН 7,4, и центрифугируют дважды при 500g в течение примерно 5 минут при температуре примерно 4°С. Клетки лизируют озвучиванием и центрифугируют при 39000g в течение примерно 10 минут при температуре примерно 4°С. Осадок ресуспендируют в том же буфере и центрифугируют при 50000g в течение примерно 10 минут при температуре примерно 4°С и мембраны в полученном осадке хранят при -80°С.
Эксперименты по конкурентному ингибированию связывания 125I-Tyr11)SRIF-14 проводят дублирование в 96-луночных планшетах из пропилена. Клеточные мембраны (10 мкг белка/лунка) инкубируют с (125I-Tyr11) SRIF-14 (0,05 нМ) в течение примерно 60 минут при температуре примерно 37°С в 50 мМ HEPES (рН 7,4), 0,2% БСА, 5 мМ MgCl2, 200 КМЕ/мл трасилола, 0,02 мг/мл бацитрацина и 0,02 мг/мл фенилметилсульфонилфторида.
Связывание, определяемое свободным (125I-Tyr11) SRIF-14, выделяют немедленным фильтрованием через стекловолоконные фильтровальные пластинки GF/C (Unifilter, Packard), предварительно выдержанные в 0,1% полиэтиленимине (P.E.I.), с использованием устройства для сбора клеток Filtermate 196 (Packard). Фильтры промывают 50 мМ HEPES при температуре примерно 0-4°С в течение примерно 4 сек и подсчитывают радиоактивность с помощью Packard Top Count.
Специфическое связывание получают при вычитании неспецифического связывания (определяемого в присутствии 0,1 мкМ SRIF-14) из общего связывания. Данные по связыванию анализируют на компьютере методом нелинейной регрессии (MDL) и определяют значение константы ингибирования (Ки).
Определение, является ли соединение согласно настоящему изобретению агонистом или антагонистом, проводят следующим образом.
Функциональный тест: Ингибирование внутриклеточного образования цАМФ:
СНО-К1 клетки, экспрессирующие подтип рецепторов соматостатина человека (SRIF-14), вносят в 24-луночные планшеты с тканевой культурой в среде RPMI 1640, содержащей 10% ФСТ и 0,4 мг/мл генетицина. Среду меняют за день до эксперимента.
Клетки в количестве 105 клеток/лунка промывают 2 раза по 0,5 мл свежей RPMI 1640, содержащей 0,2% БСА с добавкой 0,5 мМ (1) 3-изобутил-1-метилксантина (ИБМК) и инкубируют в течение примерно 5 минут при температуре примерно 37°С.
- Образование циклического АМФ стимулируется при добавлении 1 мМ форсколина (ФСК) в течение примерно 15-30 минут при температуре примерно 37°С.
- Агонистический эффект соединения определяют при одновременном добавлении ФСК (1 мкМ), SRIF-14 (от 10-12 М до 10-6 М) и исследуемого соединения (от 10-10 M до 10-5 М).
- Антагонистический эффект соединения определяют при одновременном добавлении ФСК (1 мкМ), SRIF-14 (от 1 до 10 нМ) и исследуемого соединения (от 10-10 М до 10-5 М).
Реакционную среду удаляют и добавляют 200 мл 0,1 н. HCl. Уровень цАМФ определяют методом радиоиммуноанализа (набор FlashPlate SMP001A, New England Nuclear).
Как хорошо известно специалистам в данной области техники, возможные направления применения соматостатина разнообразны и многопрофильны. Так, введение пептида согласно настоящему изобретению с целью стимуляции рецепторов соматостатина может иметь тот же эффект или то же применение, что и сам соматостатин. В их числе можно назвать, например, применение для ингибирования секреции гормона роста, инсулина, глюкагона и экзокринной секреции поджелудочной железы (патент США №4853371); для лечения рестеноза (патент США №5147856); для лечения гепатомы (патент США №5411943); для лечения рака легкого (патент США №5073541); для лечения меланомы (заявка на выдачу патента США №08/089410, поданная 9 июля 1993 г.); для подавления ускоренного роста солидной опухоли (патент США №5504069); для снижения веса тела (заявка на выдачу патента США №08/854941, поданная 13 мая 1997 г.); для лечения невосприимчивости к инсулину и синдрома Х (заявка на выдачу патента США №08/854943, поданная 13 мая 1997 г.); для пролонгирования выживания клеток поджелудочной железы (патент США №5688418); для лечения фиброза (заявка № PCT/US97/14154); для лечения гиперлипидемии (заявка на выдачу патента США №08/855311, поданная 13 мая 1997 г.); для лечения гиперамилинемии (заявка на выдачу патента США №08/440061, поданная 12 мая 1995 г.); для лечения гиперпролактинемии и пролактиномы (заявка на выдачу патента США №08/852221, поданная 7 мая 1997 г.); синдрома Кушинга (см. Clark, R.V. et al., Clin. Res., 38, p.943A, 1990); гонадотропиномы (см. Ambrosi В. et al., Acta Endocr. (Copenh.) 122, 569-576, 1990); гиперпаратиреоза (см. Miller, D., et al., Canad. Med. Ass. J., Vol.145, pp.227-228, 1991); болезни Педжета (см. Palmieri, G.M.A., et al., J. of Bone and Mineral Research, 7, (Suppl. 1), p. S240 (Abs. 591), 1992); випомы (см. Koberstein, В., et al., Z. Gastroenterology, 28, 295-301, 1990 и Christensen, С., Acta Chir. Scand. 155, 541-543, 1989); гиперплазии панкреатических островков и гиперинсулинизма (см. Laron, Z., Israel J. Med. Sci., 26, №1, 1-2, 1990; Wilson, D.C., Irish J. Med. Sci., 158, №1, 31-32, 1989 и Micic, D., et al., Digestion, 16, Suppl. 1.70. Abs. 193, 1990); гастриномы (см. Bauer, F. E., et al., Europ. J. Pharmacol., 183, 55, 1990); синдрома Золлингера - Эллисона (см. Mozell, E., et al., Surg. Gynec. Obstet., 170, 476-484, 1990); СПИД-ассоциированной гиперсекреторной диареи и вызванной другими состояниями (обусловленной СПИДом, см. Cello, J.P., et al., Gastroenterology, 98, №5, Part 2, Suppl., A163, 1990; обусловленной повышенным содержанием гастрин-высвобождающего пептида, см. Alhindawi, R., et al., Can. J. Surg., 33, 139-142, 1990; вторичной при реакции «кишечный трансплантат против хозяина», см. Bianco, J.A., et al., Transplantation, 49, 1194-1195, 1990; диареи, связанной с химиотерапией, см. Petrelli, N., et al., Proc. Amer. Soc. Clin. Oncol., Vol.10, P 138, Abstr. №417, 1991); синдрома раздраженной толстой кишки (см. O'Donnell, L.J.D., et al., Aliment. Pharmacol. Therap., Vol.4, 177-181, 1990); панкреатита (см. Tulassay, Z., et al., Gastroenterology, 98, №5, Part 2, Suppl., A238, 1990; болезни Крона (см. Fedorak, R.N., et al., Can. J. Gastroenterology, 3, №2, 53-57, 1989); системного склероза (см. Soudah, H., et al., Gastroenterology, 98, №5, Part 2, Suppl., A129, 1990); рака щитовидной железы (см. Modigliani, E., et al., Ann. Endocr. (Paris), 50, 483-488, 1989); псориаза (см. Camisa, С., et al., Cleveland Clinic J. Med., 57, №1, 71-76, 1990); гипотензии (см. Hoeldtke, R.D., et al., Arch. Phys. Med. Rehabil., 69, 895-898, 1988 и Kooner, J.S., et al., Brit. J. Clin. Pharmacol., 28, 735P-736P, 1989); приступов паники (см. Abelson, J.L., et al., Clin. Psychopharmacol., 10, 128-132, 1990); склеродомы (см. Soudah, Н., et al., Clin. Res., Vol.39, р.303А, 1991); непроходимости тонкой кишки (см. Nott, D.M., et al., Brit. J. Surg., Vol.77, р.А691, 1990); гастроэзофагеального рефлюкса (см. Branch, M.S., et al., Gastroenterology, Vol.100, No.5, Part 2, Suppl., p.A425, 1991); дуоденогастрального рефлюкса (см. Hasler, W., et al., Gastroenterology, Vol.100, №5, Part 2, Suppl., p.A448, 1991); болезни Грейвса (см. Chang, Т.C., et al., Brit. Med. J., 304, p.158, 1992); поликистоза яичников (см. Prelevic, G. M., et al., Metabolism Clinical and Experimental, 41, Suppl. 2, pp. 76-79, 1992); кровотечения из верхней части желудочно-кишечного тракта (см. Jenkins, S. A., et al., Gut., 33, pp.404-407, 1992 и Arrigoni, A., et al., American Journal of Gastroenterology, 87, p.1311, (abs. 275), 1992); ложной кисты и асцита поджелудочной железы (см. Hartley, J.E., et al., J. Roy. Soc. Med., 85, pp.107-108, 1992); лейкоза (см. Santini, et al., 78, (Suppl. 1), p.429А (Abs. 1708), 1991); менингиомы (см. Koper, J. W., et al., J. Clin. Endocr. Metab., 74, pp.543-547, 1992) и раковой кахексии (см. Bartlett, D. L., et al., Surg. Forum., 42, pp.14-16, 1991).
Соответственно настоящее изобретение относится к фармацевтическим композициям, включающим в качестве активного ингредиента по меньшей мере один из пептидов формулы (I) в сочетании с фармацевтически приемлемым носителем.
В целом эффективная дозировка для проявления активности согласно настоящему изобретению, например, для лечения акромегалии находится в диапазоне от 0,01 до 200 мг/кг/сутки, предпочтительно от 0,5 до 100 мг/кг/сутки.
Пептид согласно настоящему изобретению может вводиться перорально, парентерально (например, внутримышечной, внутрибрюшинной, внутривенной или подкожной инъекцией или имплантатом), назальным, вагинальным, ректальным, сублингвальным или местным способами введения и может быть приготовлен с использованием фармацевтически приемлемых носителей с целью образования дозированных форм, подходящих для каждого из способов введения.
Твердые дозированные формы, предназначенные для перорального введения, включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых дозированных формах активное соединение смешивают по меньшей мере с одним инертным фармацевтически приемлемым носителем, таким как сахароза, лактоза или крахмал. Твердые дозированные формы могут также включать в соответствии с обычной практикой дополнительные вещества, отличные от указанных инертных разбавителей, например смазывающие вещества, такие как стеарат магния. В случае капсул, таблеток и пилюль дозированные формы могут также включать забуферивающие средства. Таблетки и пилюли могут быть приготовлены при дополнительном нанесении энтеросолюбильных оболочек.
Жидкие дозированные формы, предназначенные для перорального введения, включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы, эликсиры, содержащие инертные разбавители, обычно применяемые в фармацевтической технике, такие как вода. Кроме указанных инертных разбавителей композиции могут также включать адъюванты, такие как увлажнители, эмульгаторы и суспендирующие средства, а также подсластители, корригенты и ароматизаторы.
Препараты согласно настоящему изобретению, предназначенные для парентерального введения, включают стерильные водные или неводные растворы, суспензии или эмульсии. Примерами неводных растворителей или носителей являются пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло и кукурузное масло, желатин и инъецируемые органические сложные эфиры, такие как этилолеат. Указанные дозированные формы могут также содержать адъюванты, такие как консерванты, увлажнители, эмульгаторы и диспергирующие средства. Они могут быть простерилизованы, например, фильтрованием через фильтр, удерживающий бактерии, путем включения в композиции стерилизующих средств, облучением композиций или нагреванием композиций. Они могут быть также изготовлены в виде стерильных твердых композиций, которые растворяются в стерильной воде или в некоторых других стерильных инъецируемых средах непосредственно перед использованием.
Композиции, предназначенные для ректального или вагинального введения, представляют предпочтительно суппозитории, которые могут содержать, в дополнение к активному веществу, наполнители, такие как масло какао или воск для суппозитория.
Композиции, предназначенные для назального или сублингвального введения, также готовят с использованием стандартных наполнителей, известных в данной области техники.
Соединение согласно настоящему изобретению может также вводиться в составе композиции с замедленным высвобождением, такой как описано в приведенных ниже патентах и заявках на патент. Патент США №5672659 относится к композициям с замедленным высвобождением, содержащим биологически активный агент и полиэфир. Патент США №5595760 относится к композициям с замедленным высвобождением, содержащим биологически активный агент в гелеобразной форме. Патент США №5821221 относится к полимерным композициям с замедленным высвобождением, содержащим биологически активный агент и хитозан. Заявка на выдачу патента США №08/740778, поданная 1 ноября 1996 г., относится к композициям с замедленным высвобождением, содержащим биологически активный агент и циклодекстрин. Заявка на выдачу патента США №09/015394, поданная 29 января 1998 года, относится к абсорбируемым композициям с замедленным высвобождением, содержащим биологически активный агент. Заявка на выдачу патента США №09/121653, поданная 23 июля 1998 года, относится к способу получения микрочастиц, содержащих терапевтическое средство, такое как пептид в среде масло-в-воде. Заявка на выдачу патента США №09/131472, поданная 10 августа 1998 года, относится к комплексам, содержащим терапевтическое средство, такое как пептид, и фосфорилированный полимер. Заявка на выдачу патента США №09/184413, поданная 2 ноября 1998 года, относится к комплексам, содержащим терапевтическое средство, такое как пептид, и полимер, несущий неполимеризуемый лактон. Раскрытия указанных выше патентов и заявок включены в настоящее описание в качестве ссылок.
Дозировка активного ингредиента в композициях согласно настоящему изобретению может варьировать, однако необходимо, чтобы активный ингредиент применялся в таком количестве, которое обеспечивало бы достижение приемлемой дозы. Выбор дозировки зависит от требуемого терапевтического эффекта, от способа введения и от длительности курса лечения. В целом, для достижения эффективного высвобождения гормона роста вводятся ежедневные дозы от 0,0001 до 100 мг/кг массы тела человека и другого животного, например млекопитающего.
Предпочтительный диапазон дозировки составляет от 0,01 до 5,0 мг/кг массы тела в день, которая вводится либо однократно, либо дробно более мелкими дозами.
Если особо не оговорено иное, все используемые в настоящем описании технические и научные термины соответствуют общепринятым значениям, известным специалистам в данной области техники. Следует также отметить, что все публикации, заявки на получение патента, патенты и другие цитированные материалы включены в настоящее описание в качестве ссылок.
| название | год | авторы | номер документа |
|---|---|---|---|
| АГОНИСТЫ РЕЦЕПТОРОВ НЕЙРОМЕДИНА В И СОМАТОСТАТИНА | 2000 |
|
RU2263680C2 |
| АНТАГОНИСТЫ РЕЦЕПТОРОВ СОМАТОСТАТИНА | 1997 |
|
RU2179172C2 |
| АГОНИСТЫ И АНТАГОНИСТЫ УРОТЕНЗИНА-II | 2001 |
|
RU2263679C2 |
| АНТАГОНИСТЫ СОМАТОСТАТИНА | 2002 |
|
RU2328504C2 |
| АНТАГОНИСТЫ СОМАТОСТАТИНА | 2002 |
|
RU2263678C2 |
| СПОСОБ РЕГУЛИРОВАНИЯ ФОЛЛИКУЛЯРНОГО РЕЗЕРВА ЯИЧНИКА, СПОСОБ ЛЕЧЕНИЯ ОТКЛОНЕНИЙ В РОСТЕ ПОКОЯЩИХСЯ ФОЛЛИКУЛОВ У ЖЕНЩИН, СРЕДСТВО СТИМУЛЯЦИИ РАЗВИТИЯ ФОЛЛИКУЛОВ И СРЕДСТВО ОПРЕДЕЛЕНИЯ ВЛИЯНИЯ СОЕДИНЕНИЙ НА УСКОРЕНИЕ ИЛИ ЗАМЕДЛЕНИЕ РОСТА ФОЛЛИКУЛОВ ПРИ ПРОВЕДЕНИИ ТОКСИКОЛОГИЧЕСКИХ ИСПЫТАНИЙ (ВАРИАНТЫ) | 2004 |
|
RU2418604C9 |
| ЛИГАНДЫ РЕЦЕПТОРОВ МЕЛАНОКОРТИНОВ | 2006 |
|
RU2439079C2 |
| ЛИГАНДЫ РЕЦЕПТОРОВ МЕЛАНОКОРТИНОВ | 2006 |
|
RU2380372C2 |
| АГОНИСТЫ СОМАТОСТАТИНА | 2002 |
|
RU2259375C2 |
| КОНЪЮГАТЫ ЦИТОТОКСИЧЕСКИХ СРЕДСТВ С ПЕПТИДАМИ | 2007 |
|
RU2457218C2 |
Предложены агонисты соматостатина формулы (I): X-A1-цикло(D-Cys-A3-A4-Lys-A6-A7)-A8-Y, или его фармацевтически приемлемая соль, где Х представляет собой Н; А1 представляет собой L-Сра, L-Phe, L-Trp или L-Nal; А3 представляет собой L-3-Pal или L-4-Pal; А4 представляет собой D-Trp; А6 представляет собой -NH-(CHR1) n-СО-, где n равно 2, 3 или 4; А7 представляет собой L- или D-Cys; А8 представляет собой D- или L-изомер аминокислоты, выбранной из группы, состоящей из Nal, Phe, Сра и Trp; Y представляет собой NH2; R1 представляет собой Н; причем Cys в А2 соединен с Cys в А7 дисульфидной связью, образуемой тиольными группами каждого Cys. 6 н. и 2 з.п. ф-лы.
X-A1-цикло(D-Cys-A3-A4-Lys-A6-A7)-A8-Y, (I)
или его фармацевтически приемлемая соль,
где Х представляет собой Н;
А1 представляет собой L-Сра, L-Phe, L-Trp или L-Nal;
А3 представляет собой L-3-Pal или L-4-Pal;
А4 представляет собой D-Trp;
А6 представляет собой -NH-(CHR1) n-СО-, где n равно 2, 3 или 4;
А7 представляет собой L- или D-Cys;
А8 представляет собой D- или L-изомер аминокислоты, выбранной из группы, состоящей из Nal, Phe, Сра и Trp;
Y представляет собой NH2;
R1 представляет собой Н;
причем Cys в А2 соединен с Cys в А7 дисульфидной связью, образуемой тиольными группами каждого Cys.

