Область техники, к которой относится изобретение
Настоящее изобретение относится к новым кристаллам фумаратных солей производных эритромицина и к способу их получения.
Предпосылки создания изобретения
Соединение, представленное формулой (I)

описано, например, в JP 6-56873 A (WO 93/24509) и в JP 9-100291 A (WO 97/06177). Известно, что указанное соединение обладает способностью улучшать деятельность пищеварительного тракта.
Получение указанного соединения описано, например, в JP 9-100291, Bioorg. and Med. Chem. Lett., vol. 4 (11), 1347 (1994) и JP 9-100291 A.
Традиционно имеется три типа кристаллов фумаратной соли соединения (I): кристаллическая форма А, кристаллическая форма С и кристаллическая форма D. Каждая из кристаллических форм А, С и D рассмотрена в JP 9-100291 и может быть получена, как описано в данном документе.
Кристаллическая форма А может быть получена из фумаратной соли соединения (I) перекристаллизацией из смешанного растворителя метанола и изопропанола. Мольное соотношение между соединением (I) и фумаратом составляет 2:1. Кристаллическая форма А дает дифракционную картину, как показано на фиг. 1, при измерении рентгеновской дифрактометрией с Cu-Kα-излучением.
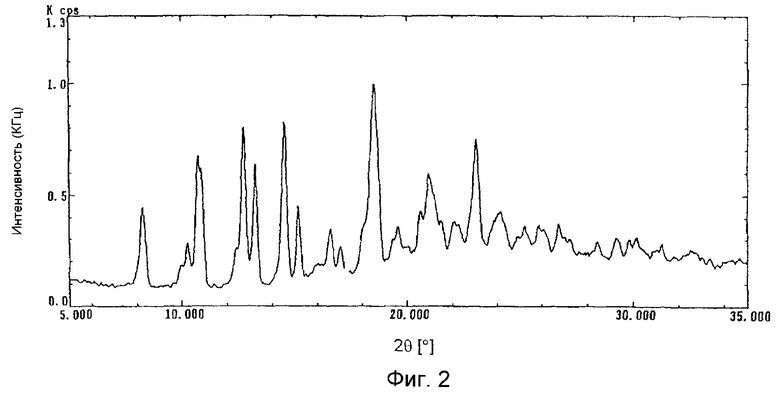
Кристаллическая форма С может быть получена из фумаратной соли соединения (I) обработкой этилацетатом. Мольное соотношение между соединением (I) и фумаратом составляет 1:1. Кристаллическая форма С дает дифракционную картину, как показано на фиг. 2, при измерении рентгеновской дифрактометрией с Cu-Kα-излучением.
Кристаллическая форма D может быть получена из фумаратной соли соединения (I) обработкой смешанным растворителем этилацетата и воды. Мольное соотношение между соединением (I) и фумаратом составляет 2:1. Кристаллическая форма D дает дифракционную картину, как показано на фиг. 3, при измерении рентгеновской дифрактометрией с Cu-Kα-излучением.
Указывается, что среди кристаллических форм А, С и D кристаллическая форма D имеет высокое качество как фармацевтический препарат и его исходный материал, поскольку она является лучшей по стабильности или другим свойствам по сравнению с другими кристаллическими формами (JP 9-100291 A).
Однако известная кристаллическая форма D, полученная обычными известными способами, как указано выше, имеет следующие недостатки: большой объем растворителя кристаллизации остается в кристалле в виде остаточного растворителя; остаточный растворитель труден для удаления в процессе сушки, и содержание в сухом продукте остаточного растворителя (степень сухости) не может быть меньше 1500 ч./млн. В данном случае остаточным растворителем, остающимся в кристаллической форме D, является этилацетат, который является менее токсичным и менее опасным для здоровья людей (смотри "Нормативы для остаточных растворителей в фармацевтических соединениях", приложенные к Извещению № 307 от 30 марта 1998 г, выпущенному директором Отдела оценки и лицензирования Бюро фармацевтической и медицинской безопасности Министерства здоровья и благосостояния, Япония). Однако естественно более желательно снизить содержание такого менее токсичного растворителя, остающегося в кристалле, в случае, когда кристалл предназначен для использования в качестве исходного продукта для фармацевтических препаратов. Предпочтительно содержание остаточного растворителя должно быть уменьшено до 1500 ч./млн или ниже, более предпочтительно 1000 ч./млн или ниже. Известная кристаллическая форма D также имеет другой недостаток, связанный с трудностью достижения малого размера частиц, что часто приводит к трудностям в таблетировании в процессе получения таблеток, содержащих указанный кристалл.
Изложение сущности изобретения.
В результате больших и интенсивных усилий по преодолению вышеуказанных проблем, имеющихся в уровне техники, авторы настоящего изобретения обнаружили структурно-новые кристаллы фумаратной соли соединения (I), которые отличаются от известных кристаллов, и предлагаемое изобретение основано на указанном открытии.
А именно, настоящее изобретение относится к кристаллической форме Е фумаратной соли соединения, представленного формулой (I):
где указанная кристаллическая форма Е характеризуется сильными рентгеновскими дифракционными пиками при углах дифракции (2θ) 5,6° и 10,4°, как установлено с помощью рентгеновской дифрактометрии с Cu-Kα-излучением.
Настоящее изобретение также относится к способу получения кристаллической формы Е фумаратной соли соединения, представленного формулой (I)
который включает обработку кристаллической формы С фумаратной соли соединения (I) в смешанном растворителе этилацетата и воды при 20-40°С, где указанная кристаллическая форма Е имеет сильные рентгеновские дифракционные пики при углах дифракции (2θ) 5,6° и 10,4°, как установлено с помощью рентгеновской дифрактометрии с Cu-Kα-излучением.
Кроме того, настоящее изобретение относится к кристаллической форме D фумаратной соли соединения, представленного формулой (I)
которую получают через кристаллическую форму Е фумаратной соли соединения (I), имеющей сильные рентгеновские дифракционные пики при углах дифракции (2θ) 5,6о и 10,4о, как установлено с помощью рентгеновской дифрактометрии с Cu-Kα-излучением.
Кроме того, настоящее изобретение также относится к способу получения кристаллической формы D фумаратной соли соединения, представленного формулой (I)
который включает получение кристаллической формы D через кристаллическую форму Е фумаратной соли соединения (I), которая имеет сильные рентгеновские дифракционные пики при углах дифракции (2θ) 5,6° и 10,4°, как установлено с помощью рентгеновской дифрактометрии с Cu-Kα-излучением.
Краткое описание чертежей.
На фиг. 1 представлена порошковая рентгенограмма дифракционных полос кристаллической формы А.
На фиг. 2 представлена порошковая рентгенограмма дифракционных полос кристаллической формы С.
На фиг. 3 представлена порошковая рентгенограмма дифракционных полос кристаллической формы D.
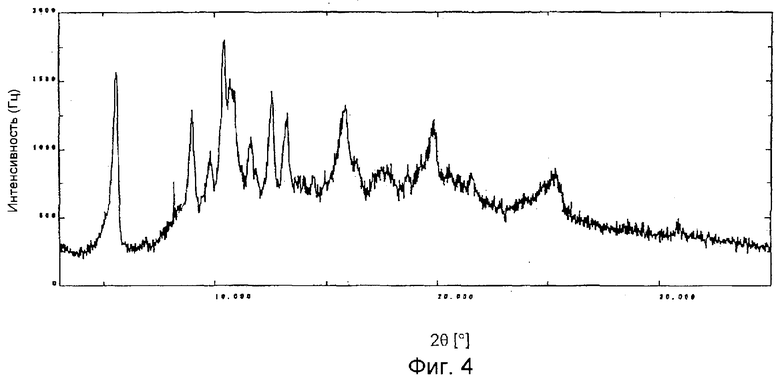
На фиг. 4 представлена порошковая рентгенограмма дифракционных полос кристаллической формы Е.
На фиг. 5 представлен DSK-спектр кристаллической формы Е.
Наилучший вариант осуществления изобретения.
Структурно-новый кристалл фумаратной соли производного эритромицина, представленного формулой (I), согласно настоящему изобретению (далее обозначаемый просто как "кристаллическая форма Е") дает дифракционную картину, как показано на фиг. 4, при измерении рентгеновской дифрактометрией с Cu-Kα-излучением. На фиг. 4 показаны сильные пики при углах дифракции (2θ) 5,6° и 10,4°.
Указанные углы рентгеновской дифракции могут быть измерены с использованием различных устройств, которые являются коммерчески доступными, такими как порошковый рентгеновский дифрактометр. Подробное описание принципов измерения порошковой рентгеновской дифрактометрии может быть найдено в Japanese Pharmacopoeia, 13th Edition, опубликованной Hirokawa Publishing Co. (1996), pp. B471-B475, Japanese Pharmacopoeia, 14th Edition, опубликованной Hirokawa Publishing Co. (2001), pp. B614-B619, и в других публикациях. Обычно, дифракционный угол имеет допустимую ошибку +0,2°.
Как использовано здесь, термин "степень сухости" относится к содержанию остаточного растворителя, которое достигает почти неизменного уровня в процессе сушки, более конкретно относится к содержанию в тот момент, когда сушка дает менее 100 ч./млн/час снижение содержания остаточного растворителя.
Настоящее изобретение описано далее ниже более подробно.
Способ получения кристаллической формы Е согласно настоящему изобретению может начинаться с кристаллической формы С.
Кристаллическая форма С может быть получена обработкой кристаллической формы А этилацетатом, как описано в JP 9-100241 А.
Далее, кристаллическая форма А может быть получена обработкой фумаратной соли соединения (I) смешанным растворителем метанола и изопропанола, как описано в JP 6-56873 А и JP 9-100241А.
Кристаллическая форма Е по настоящему изобретению может быть получена суспендированием кристаллической формы С в смешанном растворителе из этилацетата и воды. Кристаллическая форма С может быть использована либо в виде изолированного кристалла, либо в виде суспензии в растворителе, но предпочтительно используется как суспензия в растворителе. В предпочтительном варианте кристаллическую форму А обрабатывают этилацетатом с получением кристаллической формы С в виде суспензии в этилацетате, который дополнительно суспендируют добавлением воды.
В смешанном растворителе, используемом в указанной операции суспендирования, соотношение смешения между этилацетатом и водой обычно составляет 99:1-95:5, предпочтительно 97:3-95:5. Операцию суспендирования обычно осуществляют при температуре 20-40°С, предпочтительно 20-30°С. Температура ниже 20°С стремится стимулировать превращение в кристаллическую форму D. Операция суспендирования обычно продолжается в течение 30-300 мин, предпочтительно 60-240 мин.
Полученная кристаллическая форма Е может быть отделена от растворителя фильтрацией, центрифугированием или тому подобное. Выделенная кристаллическая форма Е может быть высушена при пониженном давлении или в других условиях, но предпочтительно сушится при пониженном давлении. Температура сушки составляет обычно 20-60°С, предпочтительно 30-50°С.
Кристаллическая форма Е может быть суспендирована в смешанном растворителе из этилацетата и воды при температуре ниже 20°С с получением кристаллической формы D. В смешанном растворителе, используемом здесь, соотношение смешения между этилацетатом и водой предпочтительно составляет 99:1-97:3. Суспендирование, предпочтительно осуществляют при температуре от -20 до 20°С и обычно продолжают в течение от 1 ч до 12 ч, предпочтительно 3-11 ч, более предпочтительно 5-10 ч.
Для того чтобы получить кристаллическую форму D со средним размером частиц, достаточным для того, чтобы избежать проблем с таблетированием (предпочтительно 90 мкм или более, более предпочтительно 100 мкм или более) из кристаллической формы Е, соотношение этилацетата и воды предпочтительно составляет 98,1:1,9-97:3 в смешанном растворителе, используемом для кристаллизации. Суспендирование осуществляют при температуре 10-20°С, предпочтительно 11-19°С, более предпочтительно 13-18°С. Для того чтобы стимулировать преобразование в кристаллическую форму D или улучшить выход кристаллической формы D, суспензия может быть дополнительно охлаждена до -20-10°С, предпочтительно до -15-10°С. Суспендирование обычно продолжают от нескольких минут до 20 часов, предпочтительно от 5 мин до 4 ч, более предпочтительно от 10 мин до 2 ч. В том случае, когда суспензию дополнительно охлаждают, операцию суспендирования обычно еще продолжают от нескольких минут до 20 часов, предпочтительно около 1 часа.
Необходимо учесть, что период времени, требуемый для отдельных процессов суспендирования, указанных выше, относится к минимальному периоду времени, необходимому для получения кристаллической формы Е, необходимому для получения кристаллической формы D из кристаллической формы Е и необходимому для получения кристаллической формы D с большим средним размером частиц из кристаллической формы Е. Отдельные операции суспендирования могут продолжаться дольше минимального периода времени, в зависимости от степени роста кристалла или готовности стадий получения.
При получении кристаллической формы D через кристаллическую форму Е кристаллическая форма D может быть также получена непрерывно из кристаллической формы С через кристаллическую форму Е простым регулированием температуры без выделения кристаллической формы Е в процессе получения.
Полученная кристаллическая форма D может быть отделена от растворителя фильтрацией, центрифугированием или тому подобное и затем высушена при пониженном давлении. Температура сушки составляет предпочтительно 20-70оС. Кристаллическая форма D по настоящему изобретению может полностью (100%) или частично состоять из молекул соединения, полученных через кристаллическую форму Е. В последнем случае кристаллическая форма D, полученная через кристаллическую форму Е, может быть представлена в любом процентном соотношении, пока содержание остаточного растворителя не превышает 1500 ч./млн, предпочтительно не превышает 1000 ч./млн, и/или отсутствуют проблемы с таблетированием.
Кристаллическая форма D, полученная через кристаллическую форму Е таким образом, обеспечивает содержание остаточного растворителя 1500 ч./млн или ниже, что не может быть достигнуто для известных из уровня техники кристаллических форм D. Кроме того, полученная таким образом кристаллическая форма D обуславливает содержание остаточного растворителя 1000 ч./млн или менее, а также легче сушится, чем известная кристаллическая форма D; поэтому она является более предпочтительной в качестве активного фармацевтического ингредиента. Кристаллическая форма D, частично полученная через кристаллическую форму Е, также обуславливает содержание остаточного растворителя 1500 ч./млн или менее, что не может быть достигнуто для известных кристаллических форм D. Указанная кристаллическая форма D дополнительно обуславливает содержание остаточного растворителя 1000 ч./млн или менее, а также легче сушится, чем известная кристаллическая форма D; она также поэтому является более предпочтительной в качестве активного фармацевтического ингредиента.
Кроме того, кристаллическая форма D с большим средним размером частиц, полученная в условиях, указанных выше, не может быть получена известными способами и позволяет избежать трудностей таблетирования; она поэтому является особенно предпочтительной в получении фармацевтических препаратов.
Содержание остаточного растворителя может быть определено известным образом, например, с помощью газовой хроматографии. Подробное описание методов газовой хроматографии можно найти в Japanese Pharmacopoeia, 13th Edition, опубликованной Hirokawa Publishing Co. (1996), pp. B83-B94, Japanese Pharmacopoeia, 14th Edition, опубликованной Hirokawa Publishing Co. (2001), pp. B98-B114, и в других публикациях. Обычно, газовая хроматография имеет ошибку измерения в пределах около +1%. Соединение по настоящему изобретению также может быть охарактеризовано средним размером его частиц известным образом или с использованием различных устройств, которые являются коммерчески доступными, таких как анализатор распределения сухих частиц по размеру. Обычно, такой анализатор распределения частиц по размеру имеет ошибку измерения в пределах около +5%.
Примеры
Настоящее изобретение дополнительно описано в следующих примерах, которые приводятся только в иллюстративных целях и не предназначены для ограничения объема изобретения.
В указанных примерах и сравнительных примерах рентгеновскую дифрактометрию осуществляют с использованием порошкового рентгеновского дифрактометра RINT-1100 (Rigaku), содержание остаточного растворителя определяют с использованием газового хроматографа GC-17А (Shimadzu Corp.) и средний размер частиц определяют с использованием анализатора распределения частиц по размеру RPS-95 (Seishin Enterprise Co., Ltd.). Содержание остаточного растворителя имеет ошибку измерения около +1%, тогда как средний размер частиц имеет ошибку измерения около +4%.
Пример 1
Фумаратную соль соединения (I) (50,0 г) растворяют в этилацетате (400 мл) и метаноле (40 мл) при комнатной температуре. Раствор затем концентрируют досуха при пониженном давлении при комнатной температуре. Полученный высушенный продукт перемешивают в этилацетате (415 мл) при 25°С в течение 1 ч с получением суспензии кристаллической формы С. К указанной суспензии добавляют воду (4,15 мл) с последующим перемешиванием при 25°С в течение 0,5 ч. Дополнительно добавляют воду (4,15 мл) и перемешивают при 25°С в течение 0,5 ч. Дополнительно добавляют воду (4,15 мл) и перемешивают при 25°С в течение 0,5 ч. Дополнительно добавляют воду (4,15 мл) и перемешивают при 25°С в течение 0,5 ч. Суспензию затем охлаждают до 20°С, перемешивают в течение 1 ч и фильтруют с получением сырого кристалла (43,7 г). Указанный сырой кристалл сушат при пониженном давлении при 40°С в течение 3 ч с получением кристалла фумаратной соли соединения (I) (34,1 г). Было подтверждено, что указанный кристалл является кристаллической формой Е, имеющей сильные пики при углах дифракции (2θ) 5,6° и 10,4° при измерении рентгеновской дифрактометрией. На фиг. 5 показан ДСК-спектр полученной кристаллической формы Е.
Пример 2
Фумаратную соль соединения (I) (20 г) перемешивают в этилацетате (166 мл) при 25°С в течение 2 ч с получением кристаллической формы С. После добавления воды (2,4%, 4,0 мл) указанную кристаллическую форму С постепенно охлаждают с обеспечением ее полного превращения в кристаллическую форму Е. Суспензию дополнительно охлаждают до 15°С и перемешивают в течение 3 ч с последующим охлаждением до -10°С. Полученный кристалл затем отделяют с получением сырой кристаллической формы D (20,4 г, средний размер частиц 302 мкм). Данную сырую кристаллическую форму D сушат при пониженном давлении при 25°С в течение 1 ч и дополнительно сушат при 60оС с получением кристаллической формы D фумаратной соли соединения (I). Установлено, что полученная кристаллическая форма D имеет содержание остаточного растворителя 78 ч./млн.
Пример 3
Повторяют такие же операции, как указано в примере 2, исходя из кристаллической формы С фумаратной соли соединения (I), за исключением добавления 2,6% воды, с получением кристаллической формы D с размером частиц 197 мкм через кристаллическую форму Е.
Пример 4
Фумаратную соль соединения (I) (11,6 кг) растворяют при 25°С в смешанном растворителе из этилацетата (104,6 кг) и метанола (9,2 кг). После концентрирования раствора к концентрированному остатку добавляют этилацетат (86,8 кг) при 25°С с последующим перемешиванием при 24°С в течение 1 ч с получением кристаллической формы С. После добавления воды (2,0%, 1,9 кг) указанную кристаллическую форму С постепенно охлаждают для преобразования его в кристаллическую форму Е. Суспензию дополнительно охлаждают до 15°С и перемешивают в течение 1 ч с последующим охлаждением до -10°С. Полученный кристалл затем центрифугируют с получением сырой кристаллической формы D (13,4 кг). Эту сырую кристаллическую форму D сушат при пониженном давлении при 60°С в течение 28 ч с получением кристаллической формы D фумаратной соли соединения (I) (10,5 кг, выход 90,5%, средний размер частиц 141 мкм). Было установлено, что полученная кристаллическая форма D имеет содержание остаточного растворителя 988 ч./млн. Кроме того, отсутствуют проблемы с таблетированием данной кристаллической формы D при использовании ее в качестве главного компонента для получения таблеток.
Пример 5
Фумаратную соль соединения (I) (11,6 кг) растворяют при 30°С в смешанном растворителе из этилацетата (94,2 кг) и метанола (9,1 кг). После концентрирования раствора к концентрированному остатку добавляют этилацетат (86,8 кг) при 22оС с последующим перемешиванием при 24°С в течение 1 ч с получением кристаллической формы С. После добавления воды (2,0%, 1,9 кг) указанную кристаллическую форму С постепенно охлаждают для преобразования в кристаллическую форму Е. Суспензию дополнительно охлаждают до 15°С и перемешивают в течение 1 ч с последующим охлаждением до -10°С. Полученный кристалл затем центрифугируют с получением сырой кристаллической формы D (13,2 кг). Данную сырую кристаллическую форму D сушат при пониженном давлении при 60оС в течение 10 ч с получением кристаллической формы D фумаратной соли соединения (I) (10,5 кг, выход 90,5%, средний размер частиц 197 мкм). Было установлено, что полученная кристаллическая форма D имеет содержание остаточного растворителя 845 ч./млн. Кроме того, отсутствуют проблемы с таблетированием данной кристаллической формы D при использовании в качестве главного компонента для получения таблеток.
Кристаллические формы С фумаратной соли соединения (I) обрабатывают аналогично в соответствии с примером 4 или 5 с получением кристаллических форм D через кристаллические формы Е (примеры 6-8).
Сравнительный пример 1
Фумаратную соль соединения (I) (10,8 кг) растворяют при 25°С в смешанном растворителе из этилацетата (87,6 кг) и метанола (8,5 кг). После концентрирования раствора к концентрированному остатку добавляют этилацетат (80,8 кг) при 25оС с последующим перемешиванием при 25°С в течение 1 ч с получением кристаллической формы С. После добавления воды (1,5%, 1,3 кг) указанную кристаллическую форму С охлаждают до 15°С и перемешивают в течение 1 ч с обеспечением его превращения в кристаллическую форму D. Суспензию дополнительно охлаждают до -10°С и перемешивают в течение 1 ч. Полученный кристалл затем центрифугируют с получением сырой кристаллической формы D (12,7 кг). Данную сырую кристаллическую форму D сушат при пониженном давлении при 60°С в течение 16 ч с получением кристаллической формы D фумаратной соли соединения (I) (10,8 кг, выход 87,4%, средний размер частиц 82 мкм). Имеются проблемы с таблетированием данной кристаллической формы D при использовании в качестве главного компонента для получения таблеток.
Кристаллическая форма С фумаратной соли соединения (I) обрабатывают аналогично в соответствии со сравнительным примером 1 с получением кристаллической формы D без стадии через кристаллическую форму Е (сравнительный пример 2).
В таблице представлены характеристики кристаллических форм D, полученных через кристаллические формы Е (примеры 2-8) и известных кристаллических форм D (сравнительные примеры 1 и 2).
60°С, 8 часов
60°С, 8 часов
60°С, 28 часов
60°С, 10 часов
60°С, 9 часов
60°С, 6 часов
60°С, 10 часов
60°С, 16 часов
45°С, 20 часов
Промышленная применимость
Кристаллическая форма Е фумаратной соли соединения (I) согласно настоящему изобретению позволяет получить кристаллическую форму D с наилучшими свойствами, включая сниженное содержание остаточного растворителя и высокую стабильность при приготовлении фармацевтических препаратов. В частности, кристаллическая форма Е характеризуется (1) возможностью получения фармацевтических препаратов высшего качества и (2) возможностью эффективного получения фармацевтических препаратов; поэтому она в высшей степени полезна при использовании при получении фармацевтических препаратов.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ КРИСТАЛЛИЧЕСКИЕ ФОРМЫ 1-(4-{ [6-АМИНО-5-(4-ФЕНОКСИ-ФЕНИЛ)-ПИРИМИДИН-4-ИЛАМИНО]-МЕТИЛ} -4-ФТОР-ПИПЕРИДИН-1-ИЛ)-ПРОПЕНОНА, ЕГО СОЛЕВЫЕ ФОРМЫ И СПОСОБ ПОЛУЧЕНИЯ | 2019 |
|
RU2836584C2 |
| СОЛИ ПРОИЗВОДНОГО ИНДАЗОЛА И ИХ КРИСТАЛЛЫ | 2017 |
|
RU2747399C2 |
| ПОЛИМОРФНЫЕ И ПСЕВДОПОЛИМОРФНЫЕ ФОРМЫ ФАРМАЦЕВТИЧЕСКОГО СОЕДИНЕНИЯ | 2010 |
|
RU2575173C2 |
| Полиморфные и псевдополиморфные формы фармацевтического соединения | 2010 |
|
RU2727509C2 |
| ПОЛИМОРФ РИФАКСИМИНА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2012 |
|
RU2593750C2 |
| СОЛЬ ПРОИЗВОДНОГО ПИРРОЛИДИН-3-ИЛ-УКСУСНОЙ КИСЛОТЫ И ЕЕ КРИСТАЛЛЫ | 2014 |
|
RU2640047C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА НАТРИЕВОЙ СОЛИ ПРАВАСТАТИНА | 2000 |
|
RU2247711C2 |
| КРИСТАЛЛИЧЕСКАЯ ФУМАРАТНАЯ СОЛЬ (S)-[3,4-ДИФТОР-2-(2-ФТОР-4-ЙОДФЕНИЛАМИНО)ФЕНИЛ][3-ГИДРОКСИ-3-(ПИПЕРИДИН-2-ИЛ)АЗЕТИДИН-1-ИЛ]МЕТАНОНА | 2016 |
|
RU2762181C2 |
| СОЛИ И ПОЛИМОРФЫ ЗАМЕЩЕННОГО ИМИДАЗОПИРИДИНИЛ-АМИНОПИРИДИНА | 2015 |
|
RU2732125C2 |
| СПОСОБ ПОЛУЧЕНИЯ И ОЧИСТКИ ПРОИЗВОДНЫХ ЭРИТРОМИЦИНА | 2001 |
|
RU2248981C2 |


Изобретение относится к кристаллической форме Е фумаратной соли производного эритромицина, представленного формулой (I), и способу ее получения. Указанная кристаллическая форма Е имеет сильные рентгеновские дифракционные пики при углах дифракции (2θ) 5,6° и 10,4°, как установлено с помощью рентгеновской дифрактометрии с Cu-Кα-излучением. Также предлагается кристаллическая форма D фумаратной соли производного эритромицина, представленного формулой (I), имеющая средний размер частиц 90 мкм или более и/или содержание остаточного растворителя 1500 ч./млн или менее. Способы получения указанной кристаллической формы D включают суспендирование указанной кристаллической формы Е в смеси этилацетата и воды в соотношении 99:1-97:3 при температуре от -20 до 20°C. Технический результат - снижение содержания остаточного растворителя и устранение трудностей при таблетировании. 7 н. и 7 з.п. ф-лы, 1 табл., 5 ил.



где указанная кристаллическая форма Е имеет сильные рентгеновские дифракционные пики при углах дифракции (2θ) 5,6° и 10,4°, как установлено с помощью рентгеновской дифрактометрии с Cu-Кα-излучением.
который включает обработку кристаллической формы С фумаратной соли производного эритромицина (I) в смешанном растворителе из этилацетата и воды при 20-40°С, где указанная кристаллическая форма Е имеет сильные рентгеновские дифракционные пики при углах дифракции (2θ) 5,6° и 10,4°, как установлено с помощью рентгеновской дифрактометрии с Cu-Кα-излучением.
имеющая средний размер частиц 90 мкм или более, полученная через кристаллическую форму Е фумаратной соли производного эритромицина (I), имеющую сильные рентгеновские дифракционные пики при углах дифракции (2θ) 5,6° и 10,4°, как установлено с помощью рентгеновской дифрактометрии с Cu-Кα-излучением.
включающий суспендирование кристаллической формы Е фумаратной соли производного эритромицина формулы (I) по п.1 в смеси этилацетата и воды в соотношении 99:1-97:3 при температуре от -20°С до 20°С.
включающий суспендирование кристаллической формы Е фумаратной соли производного эритромицина формулы (I) по п.1 в смеси этилацетата и воды в соотношении 98,1:1,9-97:3 при температуре 10-20°С и дополнительное охлаждение до температуры от -20°С до 10°С.
где содержание остаточного растворителя составляет 1500 ч./млн или менее.
где содержание остаточного растворителя составляет 1000 ч./млн или менее.
| Консольная опалубка | 1979 |
|
SU846697A1 |
| Способ борьбы с насекомыми и клещами | 1975 |
|
SU643068A3 |
| RU 98103451 C1, 10.12.1999. | |||

