Настоящее изобретение относится к новым производным 4-(трифторметансульфонилоксифенил)пропионовой кислоты и содержащим их фармацевтическим композициям, которые применяют в качестве ингибиторов хемотаксиса полиморфонуклеарных и мононуклеарных клеток, особенно при лечении нейтрофилзависимых патологий.
Определенные клетки крови (макрофаги, гранулоциты, нейтрофилы, полиморфонуклеатированные), восприимчивые к химическому стимулу (при стимуляции веществами, названными хемокинами), мигрированием вдоль градиента концентрации стимулирующего агента посредством процесса, названного хемотаксисом. Основные известные стимулирующие агенты или хемокины представлены продуктами разложения комплемента С5а, некоторыми N-формилпептидами, генерированными в результате лизиса бактериальной поверхности, или пептидами синтетического происхождения, такими как формил-метионил-лейцил-фенилаланин (f-MLP) и в основном различными цитокинами, в том числе интерлейкином-8 (IL-8, обозначенным также CXCL8). Интерлейкин является эндогенным хемотактическим фактором, продуцируемым большей частью содержащих ядро клетками, такими как фибробласты и макрофаги.
При некоторых патологических состояниях, отмеченных обостренным рекрутингом нейтрофилов, более серьезное повреждение участка связано с инфильтрацией нейтрофильных клеток. Недавно была широко показана роль нейтрофильной активации при определении повреждения, связанного с реперфузией после ишемии и легочной гипероксией.
Биологическая активность IL-8 опосредуется взаимодействием интерлейкина с мембранными рецепторами CXCR1 и CXCR2, которые принадлежат к семейству семи трансмембранных рецепторов, экспрессируемых поверхностью нейтрофилов человека и некоторых типов Т-клеток (L. Xu et al., J. Leukocyte Biol., 57, 335, 1995). Известны селективные лиганды, которые могут быть различными для CXCR1 и CXCR2: GRO-α является примером CXCR2-селективного хемотактического фактора.
Широко описана потенциальная патологическая роль IL-8 в легочных заболеваниях (повреждение легкого, острый респираторный дистресс-синдром, астма, хроническое легочное воспаление и муковисцидоз) и особенно при патогенезе COPD (хроническое закупоривающее легочное заболевание) посредством пути рецептора CXCR2 (D. WP Hay and H.M. Sarau., Current Opinion in Pharmacology 2001, 1:242-247).
Характерная аккумуляция нейтрофилов имеет место при острых и хронических патологических состояниях, например в очень воспаленных и не поддающихся лечению областях псориатических повреждений. Нейтрофилы хемотактически притягиваются и активируются синергическим действием хемокинов, IL-8 и Gro-a, высвобождаемых стимулированными кератиноцитами, а также фракции С5а/С5а-дезArg, продуцируемой посредством активации пути альтернативного комплемента (T. Terui et al., Exp. Dermatol., 9, 1, 2000).
Авторами настоящего изобретения недавно описан новый класс «омега-аминоалкиламидов R-2-арилпропионовых кислот» в качестве ингибиторов хемотаксиса полиморфонуклеарных и мононуклеарных клеток (WO 02/068377). Новый класс включает в себя соединения, являющиеся ингибиторами от селективных ингибиторов С5а до имеющих двойное действие ингибиторов С5а/IL-8.
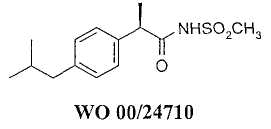
Кроме того, новые классы амидов R-2-арилпропионовых кислот и N-ацилсульфонамидов описаны в качестве эффективных ингибиторов IL-8-индуцируемого хемотаксиса и дегрануляции нейтрофилов (WO 01/58852; WO 00/24710).
Авторами настоящего изобретения обнаружен новый класс производных 2-(R)-фенилпропионовых кислот в качестве ингибиторов хемотаксиса полиморфонуклеарных и мононуклеарных клеток. В частности, соединения их изобретений являются сильнодействующими ингибиторами IL-8-индуцируемого хемотаксиса нейтрофилов и С5а-индуцируемого хемотаксиса нейтрофилов и моноцитов с улучшенными фармакокинетическими характеристиками и профилем фармакологической активности.
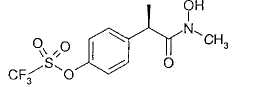
Настоящее изобретение, таким образом, предлагает производные 2-(R)-фенилпропионовых кислот формулы (I):
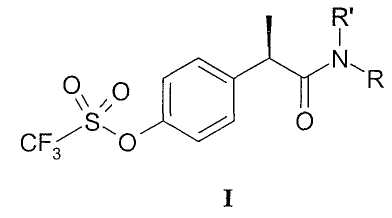
и их фармацевтически приемлемые соли,
где
R' выбран из
- Н, ОН и,
когда R' представляет собой Н, R выбран из
- Н, С1-С5-алкила, С3-С6-циклоалкила, С2-С5-алкенила, С1-С5-алкокси;
- гетероарильной группы, выбранной из замещенного и незамещенного пиридина, пиримидина, пиррола, тиофена, фурана, индола, тиазола, оксазола;
- аминокислотного остатка, состоящего из неразветвленного или разветвленного С1-С6-алкила, С3-С6-циклоалкила, С2-С6-алкенила, С1-С6-фенилалкила, замещенного одной дополнительной карбоксигруппой (СООН);
- остатка формулы -СН2-СН2-Z-(CH2-CH2O)nR', где R' представляет собой Н или С1-С5-алкил, n равно целому числу от 0 до 2 и Z представляет собой кислород или серу;
- остатка формулы -(СН2)n-NRaRb, где n равно целому числу от 0 до 5 и каждый из Ra и Rb, которые могут быть одинаковыми или разными, представляет собой С1-С6-алкил, С2-С6-алкенил или, в альтернативном случае, Ra и Rb вместе с атомом азота, с которым они связаны, образуют гетероцикл из 3-7 членов формулы (II)
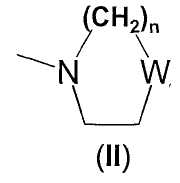
где W представляет собой простую связь, О, S, N-Rc, причем Rc представляет собой Н, С1-С6-алкил или С1-С6-алкилфенил, и n равно целому числу от 0 до 3;
- остатка формулы SO2Rd, где Rd представляет собой С1-С6-алкил, С3-С6-циклоалкил, С2-С6-алкенил, арил и гетероарил;
когда R' представляет собой ОН, то R выбран из Н, С1-С5-алкила, С3-С6-циклоалкила, С2-С5-алкенила.
Настоящее изобретение далее предлагает применение соединений формулы (I) в качестве лекарственных средств. В частности, такие лекарственные средства являются ингибиторами хемотаксиса полиморфонуклеарных и мононуклеарных клеток.
Соединения формулы (I), в общем, включены в общие формулы ингибиторов IL-8 и С5а, описанных ранее в WO 01/58852, WO 00/24710 и WO 02/068377. Обнаружено, что соединения формулы (I) имеют существенно преимущественные характеристики по сравнению с особенно предпочтительными соединениями цитированных выше изобретений.
Соединения изобретения принадлежат к химическому классу арилтрифлатов. В исследованиях по медицинской химии трифлатную группу считают обычной биоизостерической заменой фенольного гидроксила или метоксигруппы; несмотря на очень низкую эффективность соответствующих 4-гидрокси- и 4-метоксианалогов, соединения формулы (I) неожиданно оказались сильнодействующими ингибиторами IL-8-индуцированного хемотаксиса PMN человека. Кроме того, трифлатная группа на фенильном кольце специфически придает высокую аффинность у рецепторов CXCR1 и CXCR2 IL-8. Соединения формулы (I), по сравнению с известными ингибиторами IL-8- и/или С5а-индуцированного хемотаксиса PMN, как неожиданно было обнаружено, являются очень сильнодействующими ингибиторами при ингибировании GRO-α-индуцированного хемотаксиса PMN, что указывает на специфическое действие на СХСR2-опосредуемый путь.
Известно, что в противоположность реакционноспособному характеру алифатических трифлатов, ароматические трифлаты (арилтрифлаты) являются как химически, так и биологически стабильными. Вследствие электроноакцепторных свойств и липофильности трифлатная группа предотвращает окисление ароматического кольца посредством изоферментных систем цитохрома Р450. Трифлатная часть содействует повышению метаболической стабильности соединений формулы (I), замедляя метаболизм (гидроксилирование ароматического кольца/заместителя и последующее сопряжение), который обычно имеет место, когда аналоги, несущие электронодонорные группы, вводят in vivo.
В связи с этим свойством новый класс проявляет более высокую пероральную биологическую доступность, более высокое t1/2 и более низкое связывание белка по сравнению с классами цитированных выше изобретений.
Эти характеристики обеспечивают оптимальный общий фармакологический профиль этих лекарственных средств и обеспечивают возможность терапевтического применения их при различных хронических или острых патологических состояниях.
Когда R' представляет собой Н, предпочтительные группы R представляют собой
Н, С1-С5-алкил, С3-С6-циклолкил, С1-С5-алкокси, С1-С2-карбоксиалкил;
гетероарильную группу, выбранную из замещенного и незамещенного пиридина, тиазола, оксазола;
остаток формулы-(СН2)n-NRaRb, где n равно целому числу 2 или 3, более предпочтительно 3, и группа NRaRb представляет собой N,N-диметиламин, N,N-диэтиламин, 1-пиперидил, 1-пирролидинил, 4-морфолил, 1-пирролидил, 1-пиперазинил, 1-(4-метил)пиперазинил;
остаток формулы SO2Rd, где Rd представляет собой С1-С2-алкил, С3-С6-циклоалкил.
Когда R' представляет собой ОН, предпочтительные группы R представляют собой Н, С1-С5-алкил, С3-С6-циклоалкил.
Особенно предпочтительными соединениями изобретения являются:
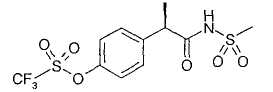
1. - R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-метансульфонилпропионамид;
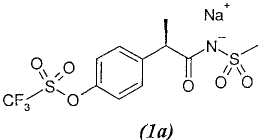
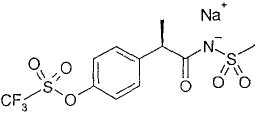
1а. - натриевая соль R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-метансульфонилпропионамида;
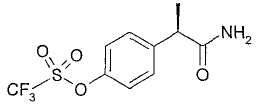
2. - R(-)-2-[(4'-трифторметансульфонилокси)фенил]пропионамид;
3. - R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-метилпропионамид;
4. - R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-изопропоксипропионамид;
5. - R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-циклопентилпропионамид;
6. - R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-[3-(N'-пиперидинил)пропил]пропионамид;
6а. - гидрохлорид R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-[3-(N'-пиперидинил)пропил]пропионамида;
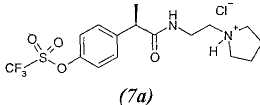
7. - R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-[2-(N'-пирролидинил)этил]пропионамид;
7а. - гидрохлорид R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-[2-(N'-пирролидинил)этил]пропионамида;
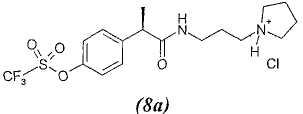
8. - R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-[3-(N'-пирролидинил)пропил]пропионамид;
8а. - гидрохлорид R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-[3-(N'-пирролидинил)пропил]пропионамида;
9. - R(+)-2-[(4'-трифторметансульфонилокси)фенил]-N-(2-гидроксиэтоксиэтил)пропионамид;
10. - R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-[2-(4'-трифторметил)тиазолил]пропионамид;
11. - R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-метил-N-гидроксипропионамид.
Наиболее предпочтительным соединением в списке является соединение 1 и относящаяся к нему соль натрия 1а.
Соединения изобретения являются сильнодействующими ингибиторами хемотаксиса PMN человека, индуцированного IL-8. Соединения изобретения, у которых R представляет собой остаток формулы -(СН2)n-NRaRb, являются двойными ингибиторами хемотаксиса PMNs, индуцированного С5а и IL-8.
Соединения изобретения формулы (I) обычно выделяют в форме их аддитивных солей как с органическими, так и неорганическими фармацевтически приемлемыми кислотами или основаниями. Примеры таких кислот выбраны из хлористоводородной кислоты, серной кислоты, фосфорной кислоты, метансульфоновой кислоты, фумаровой кислоты, лимонной кислоты.
Примерами таких оснований являются гидроксид натрия, гидроксид калия, гидроксид кальция, (D,L)-лизин, L-лизин, трометамин.
Соединения формулы (I) получают, исходя из R(-)-2-(4'-трифторметансульфонилоксифенил)пропионовой кислоты, согласно методологиям, ранее описанным в WO 01/58852, WO 00/24710 и WO 02/068377.
Например, соединения формулы (I), у которых R представляет собой SO2Rd и Rd представляет собой С1-С2-алкил или С3-С6-циклоалкил, можно получить обработкой эквимолярного количества R(-)-2-(4'-трифторметансульфонилоксифенил)пропионовой кислоты эквимолярным количеством подходящего сульфонамида RdSO2NH2 в инертном растворителе в присутствии эквимолярного количества или небольшого избытка конденсирующего агента, например карбодиимида (такого как дициклогексилкарбодиимид), растворимый карбодиимид (например, гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида) или 1,1'-карбонилдиимидазола, и противооснования, выбранного из группы, состоящей из триэтиламина, 4-(N,N-диметиламино)пиридина, 1,8-диазабицикло[5.4.0]ундец-7-ена и 1,5-диазабицикло[4.3.0]нон-5-ена.
Соединения изобретения формулы (I) оценивали in vitro на их способность ингибировать хемотаксис полиморфонуклеарных лейкоцитов (далее обозначаемые PMNs) и моноцитов, индуцированный фракциями IL-8 и GRO-α и С5а. Для этой цели, чтобы выделить PMNs из гепаринизированной крови человека, взятой у здоровых взрослых волонтеров, мононуклеарные клетки удаляли при помощи седиментации на декстране (согласно методике, описанной W.J. Ming et al., J. Immunol., 138, 1469, 1987) и эритроциты удаляли гипотоническим раствором. Жизнеспособность клеток вычисляли исключением трипаном синим, тогда как пропорцию циркулирующих полиморфонуклеарных клеток оценивали на центрифугированных клетках после окрашивания Diff Quick.
В анализе индуцированного IL-8 хемотаксиса рекомбинантный IL-8 человека (Pepro Tech) применяли в качестве стимулирующего агента в экспериментах хемотаксиса: лиофилизованный белок растворяли в объеме HBSS, содержащем 0,2% бычьего сывороточного альбумина (BSA), так чтобы получить исходный раствор, имеющий концентрацию 10-5 М, который разводят в HBSS до концентрации 10-9 М для анализов хемотаксиса.
Ингибирование GRO-α-индуцированного хемотаксиса оценивали аналогичным анализом.
В анализе C5а-индуцированного хемотаксиса фракции hr-C5a и HrC5a-дезArg (Sigma) применяли в качестве стимулирующих агентов в экспериментах хемотаксиса, получая при этом практически идентичные результаты. Лиофилизованный С5а растворяли в объеме HBSS, содержащем 0,2% BSA, так чтобы получить исходный раствор, имеющий концентрацию 10-5 М, который разводят в HBSS до концентрации 10-9 М для анализов хемотаксиса.
В экспериментах хемотаксиса PMN инкубировали с соединениями изобретения формулы (I) в течение 15' при 37°С в атмосфере, содержащей 5% СО2.
Хемотактическую активность С5а оценивали на циркулирующих полиморфонуклеарных клетках человека (PMN), ресуспендированных в HBSS при концентрации 1,5×106 PMN на мл.
Во время анализа хемотаксиса (согласно W. Falket et al., J. Immunol. Methods, 33, 239, 1980) применяли фильтры без PVP с пористостью 5 мкм и микрокамеры, подходящие для репликации.
Соединения изобретения формулы (I) оценивали при концентрации между 10-6 и 10-10 М; для этой цели их добавляли при одинаковой концентрации как к нижним порам, так и верхним порам микрокамеры. Оценку способности соединений изобретения формулы (I) ингибировать хемотаксис моноцитов человека проводили согласно способу, описанному Van Damme J. еt al. (Eur. J. Immunol., 19, 2367, 1989).
Связывание белка определяли следующим образом. Пробы плазмы крыс в двух повторностях для каждого соединения при концентрации 50 мкг/мл инкубировали при 37°С в течение 20 минут при осторожном встряхивании. Затем пробы ультрафильтровали через устройства микроразделения Centrifree® центрифугированием при 1500 g в течение 15 минут. Ультрафильтрат подвергали количественному анализу ВЭЖХ-МС/МС (колонка Luna C18, 150×2 мм внутр. диаметр, 5 мкм (Phenomenex), подвижная фаза: элюент А) 0,02М НСОО-NH4 + (рН 4,3 c НСООН); элюент В) СН3ОН).
Фармакокинетический профиль (t1/2, пероральная биологическая доступность и т.д.) заявленных соединений оценивали на самцах мышей после внутривенного и перорального введения. Фармакокинетический анализ проводили с применением концентраций соединений в плазме при различном времени. Данные оценивали Kinetica 2000TM, версия 3,0 программного обеспечения [InnaPhase Corporation, World headquarters, 1700 Race Street, Philadelphia, PA 19103 USA].
Обнаружено, что соединения формулы (I), оцененные ex vivo в крови в общем согласно методике, описанной Patrignani et al., in J. Pharmacol. Exper. Ther., 271, 1705, 1994, являются полностью неэффективными в качестве ингибиторов ферментов циклооксигеназ (СОХ).
В большинстве случаев соединения формулы (I) не препятствуют продуцированию ПГЕ2, индуцируемому в мышиных макрофагах стимуляцией липополисахаридами (LPS, 1 мкг/мл), при концентрации между 10-5 и 10-7 М. Ингибирование продуцирования ПГЕ2, которое может быть зарегистрировано, имеется, главным образом, при пределе статистического значения, и более часто оно ниже 15-20% основной величины. Пониженная эффективность при ингибировании СО создает преимущество для терапевтического применения соединений изобретения ввиду того, что ингибирование синтеза простагландинов создает стимул для клеток макрофагов для амплификации синтеза TNF-α (индуцированного LPS или пероксидомводорода), который является важным медиатором нейтрофической активации и стимулом для продуцирования цитокина интерлейкина-8.
Ингибиторы активации СXCR1 и CXCR2 находят полезные применения, как подробно описано выше, особенно при лечении хронических воспалительных патологий (например, псориаза), в которых, как предполагается, активация обоих рецепторов IL-8 играет критическую патофизиологическую роль в развитии заболевания.
Действительно, известно, что активация CXCR1 является важной в IL-опосредуемом хемотаксисе PMN (Hammond M et al., J. Immunol., 155, 142, 1995). С другой стороны, предполагается, что активация CXCR2 является важной при пролиферации и ангиогенезе IL-опосредуемых эпидермальных клеток псориатических пациентов (Kulke R et al., J. Invest Dermatol, 110, 90, 1998).
Кроме того, антагонисты CXCR2 находят особенно полезные терапевтические применения при лечении важных легочных заболеваний, подобных хроническому закупоривающему легочному заболеваний COPD (D. WP Hay and H.M. Sarau., Current Opinion in Pharmacology 2001, 1:242-247).
С точки зрения экспериментального доказательства, обсуждаемого выше, и роли, выполняемой интерлейкином-8 (IL-8) и агентами, имеющими общее с ним происхождение, в процессах, которые включают в себя активацию и инфильтрацию нейтрофилов, соединения изобретения являются особенно полезными при лечении таких заболеваний, как псориаз (R. J. Nicholoff et al., Am. J. Pathol., 138, 129, 1991), кишечные хронические воспалительные патологии, такие как неспецифический язвенный колит (Y. R. Machida et al., Clin. Sci., 82, 273, 1992), хроническое закупоривающее легочное заболевание (COPD), буллезная пузырчатка, ревматоидный артрит (M. Selz et al., J. Clin. Invest., 87, 463, 1981), идиопатический фиброз (E. J. Miller, ранее цитированная публикация и P. C. Carre et al., J. Clin. Invest., 88, 1882, 1991), гломерулонефрит (T. Wada et al., J. Exp. Med., 180, 1135, 1994).
Соединения настоящего изобретения являются также эффективными при профилактике и лечении повреждений, вызванных ишемией и реперфузией, в частности при защите от функционального повреждения при трансплантации органов, особенно трансплантации почек.
На экспериментальной модели трансплантации почек у крыс доказано, что соединения изобретения являются активными в сохранении почечной функции сразу после повреждения из-за ишемии/реперфузии, которое следует за изогенной трансплантацией почек, тем, что они предотвращают инфильтрацию лейкоцитов в трансплантате, которая имеет место после постишемической реперфузии.
Экспериментальная модель трансплантации почек у крыс
Изучение проводили на модели изогенного трансплантата почки с применением крыс в качестве доноров и реципиентов трансплантатов. Создавали анастомоз между почечной артерией реципиента и донора, а также анастомоз «бок в бок» почечной вены. Васкулярные зажимы освобождали через 30 минут (теплая ишемия). «Родную» правую почку затем удаляли. Животных помещали в отдельные метаболические клетки для измерения суточного диуреза как показателя восстановления почечной функции. Через 16 и 24 часа почечную функцию анализировали измерением концентрации креатинина в плазме. Через двадцать четыре часа после трансплантации почки животных умерщвляли. Почечный трансплантат удаляли, делали его срезы и помещали в раствор Dubosq-Brazil для анализа общепринятой гистологии световым микроскопом. Кроме того, дополнительные фрагменты почки замораживали в жидком азоте и применяли для иммуногистохимического анализа инфильтрата воспалительных клеток (полиморфонуклеарные клетки, клетки, положительные по МНС класса II).
Все соединения изобретения проявляли защиту от повреждения у трансплантированных крыс, обработанных внутривенно перед трансплантацией почки и подкожно через два часа после трансплантации, при концентрации, составляющей от 5 мг/кг до 30 мг/кг.
Кроме того, соединения изобретения являются особенно полезными при лечении меланомы и ангиогенеза.
Активность in vitro на клетках меланомы определяли следующим образом.
Пролиферация клеток меланомы
Планшеты с девяносто шестью лунками, содержащими 2-6×103 клеток меланомы/лунку, предварительно обрабатывали выбранными соединениями изобретения, стимулировали интерлейкином-8 (CXCL8) и культивировали в течение 3-4 дней. Затем к каждой лунке добавляли бромид 3-(4,5-диметилтиазолил-2)-2,5-дифенилтетразолия (МТТ, 400 мкг/мл) и планшеты инкубировали в течение 2 часов. Среду удаляли и для лизирования клеток добавляли 100 мкл диметилсульфоксида. Величина поглощения, определенная на аппарате для прочтения микропланшетов, измеряла изменения в пролиферации клеток.
Анализ инвазии с помощью планшета Matrigel
Клетки меланомы помещали в планшет (5×103 клеток на шестилуночные планшеты) и давали им возможность прикрепляться в течение 24 часов. После 5 дней обработки выбранными соединениями изобретения клетки высвобождали из планшетов непродолжительным воздействием смеси трипсин-этилендиаминтетрауксусная кислота, подсчитывали и центрифугировали. Камеры для инвазии Biocoat Matrigel примировали согласно инструкциям изготовителя. CXCL8, растворенный в бессывороточной среде, помещали в нижнюю лунку для действия в качестве хемоаттрактанта и 3×103 клеток в 500 мкл бессывороточной среды помещали в верхнюю камеру планшета Matrigel и планшет инкубировали при 37°С в течение 22 часов. Клетки на нижней поверхности фильтра окрашивали Diff-Quick и количественно определяли анализатором изображения, присоединенным к микроскопу.
Следовательно, следующей целью настоящего изобретения является предложение применения соединений формулы (I) при изготовлении лекарственного средства для лечения псориаза, неспецифического язвенного колита, меланомы, ангиогенеза, хронического закупоривающего легочного заболевания (COPD), буллезной пузырчатки, ревматоидного артрита, идиопатического фиброза, гломерулонефрита и при профилактике и лечении повреждений, вызванных ишемией и реперфузией.
В таблице I указана биологическая активность примерных соединений настоящего изобретения по сравнению с примерными соединениями цитированных выше патентных документов.
(10-9 М)
(10-8 М)
(10-8 М) WO 00/24710
WO 00/24710 12
12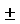 18
18 96
96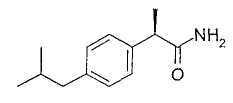
WO 01/58852127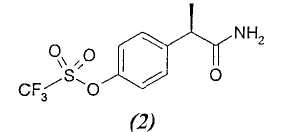 23
23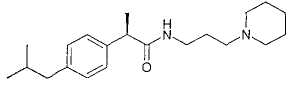
WO 02/0683772106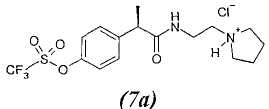 1218
1218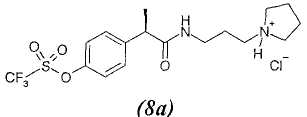 1310
1310
#Испытан при 10-6М
В таблице II указаны физико-химические, фармакологические и фармакокинетические характеристики примерных соединений формулы (I) по сравнению с примерными соединениями цитированных выше патентных документов. Соединения формулы (I) проявляют более высокую пероральную биологическую доступность, более высокое t1/2 и более низкое связывание белка по сравнению с примерными соединениями.
 (2)
(2)
Фармацевтические композиции, включающие в себя соединение изобретения и его подходящий носитель, также находятся в пределах объема настоящего изобретения.
Соединения изобретения вместе с обычно применяемым вспомогательным средством, носителем, разбавителем или эксципиентом могут быть фактически изготовлены в форме фармацевтических композицией и их стандартных лекарственных форм и в такой форме их можно применять в твердом виде, таком как таблетки или наполненные капсулы, или жидкостей, таких как растворы, суспензии, эмульсии, эликсиры или капсулы, наполненные ими, причем все они предназначены для перорального применения, или в форме стерильных инъецируемых растворов для парентерального (в том числе подкожного) применения. Такие фармацевтические композиции и их стандартные лекарственные формы могут включать в себя ингредиенты в общепринятых пропорциях с дополнительными активными соединениями или составными агентами или без них, и такие стандартные лекарственные формы могут содержать любое подходящее эффективное количество активного ингредиента, соизмеримое с предназначенным диапазоном суточных доз для применения.
При применении в качестве фармацевтических средств соединения данного изобретения обычно вводят в форме фармацевтической композиции. Такие композиции можно получать способом, хорошо известным в фармацевтической области, они включают в себя, по меньшей мере, одно активное соединение. Соединения данного изобретения обычно вводят в фармацевтически эффективном количестве. Количество соединения, вводимое в действительности, обычно определяют на основе приемлемых обстоятельств, включающих в себя подвергаемое лечению состояние, выбранный путь введения, реальное вводимое соединение, возраст, масса и восприимчивость отдельного пациента, тяжесть симптомов пациента и тому подобное.
Фармацевтические композиции изобретения можно вводить различными путями, включающими пероральный, ректальный, трансдермальный, подкожный, внутривенный, внутримышечный и интраназальный пути. В зависимости от предназначенного пути доставки, соединения предпочтительно изготовляют в виде либо инъецируемых, либо пероральных композиций. Композиции для перорального введения могут быть в форме нерасфасованных жидких растворов или суспензий или нерасфасованных порошков. Однако более обычно композиции представляют в виде стандартных лекарственных форм для облегчения достижения точной дозировки. Термин «стандартные лекарственные формы» относится к физически дискретным единицам, подходящим в качестве стандартных доз для субъекта-человека и других млекопитающих, причем каждая единица содержит предварительно определенное количество активного вещества, вычисленное для достижения требуемого терапевтического действия, в сочетании с подходящим фармацевтическим эксципиентом. Обычные стандартные лекарственные формы включают в себя предварительно наполненные, содержащие предварительно измеренное количество жидких композиций ампулы или шприцы, или пилюли, таблетки, капсулы или тому подобное в случае твердых композиций. В таких композициях кислотное соединение обычно является меньшим компонентом (от приблизительно 0,1 до приблизительно 50 мас.% или предпочтительно от приблизительно 1 до приблизительно 40 мас.%), причем остальное количество составляют различные наполнители или носители и вспомогательные средства для обработки, помогающие образованию требуемой лекарственной формы.
Жидкие формы, подходящие для перорального введения, могут включать в себя подходящий водный или неводный наполнитель с буферами, суспендирующими и дисперсирующими агентами, красящими веществами, корригентами и тому подобное. Жидкие формы, в том числе инъецируемые композиции, описанные ниже, всегда хранят в отсутствие света, чтобы избежать любого каталитического действия света, такое как образование гидропероксида или пероксида. Твердые формы могут включать в себя, например, любой из следующих ингредиентов или соединений подобной природы: связывающее вещество, такое как микрокристаллическая целлюлоза, трагакантная камедь или желатин; эксципиент, такой как крахмал или лактоза, дезинтегрирующий агент, такой как альгиновая кислота, примогель или кукурузный крахмал; смазывающее вещество, такое как стеарат магния; вещество, придающее скольжение, такое как коллоидальный диоксид кремния; подслащивающий агент, такой как сахароза или сахарин; или корригент, такой как мята перечная, метилсалицилат или апельсиновый корригент.
Инъецируемые композиции обычно основаны на инъецируемом стерильном солевом растворе, или забуференном фосфатом солевом растворе или других инъецируемых носителях, известных в данной области. Как указано выше, кислотное производное формулы (I) в таких композициях является обычно меньшим компонентом, часто составляющим от 0,05 до 10 мас.%, причем остальное количество составляет инъецируемый носитель и тому подобное. Средняя суточная доза будет зависеть от различных факторов, таких как серьезность заболевания и состояния пациента (возраст, пол и масса). Доза обычно варьирует от 1 мг или нескольких мг до 1500 мг соединений формулы (I) в день, такую дозу необязательно делят для нескольких введений. Благодаря низкой токсичности соединений изобретения можно также вводить более высокие дозы на протяжении длительных периодов времени. Описанные выше компоненты для перорально вводимых или инъецируемых композиций являются лишь репрезентативными. Дополнительные вещества, а также способы обработки и тому подобное описаны в части 8 публикации “Remington's Pharmaceutical Sciences Handbook”, 18th Edition, 1990, Mack Publishing Company, Easton, Pennsylvania, которая включена здесь в качестве ссылки.
Соединения изобретения можно также вводить в формах для продолжительного высвобождения или из систем доставки лекарственного средства с длительным высвобождением. Описание репрезентативных материалов для длительного высвобождения можно также найти во включенных материалах в указанном выше справочнике Ремингтона.
Настоящее изобретение будет иллюстрировано при помощи следующих примеров, которые не истолковываются как ограничение объема изобретения.
ПРИМЕРЫ
Примерами аббревиатур являются: ТГФ для тетрагидрофурана, DBU для 1,8-диазабицикло[5.4.0]ундец-7-ена.
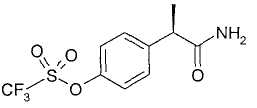
Все соединения, описанные в примерах, получают, исходя из R-(-)-2-(4'-трифторметансульфонилоксифенил)пропионовой кислоты, полученной как описано ранее в WO 03/043625.
Пример 1
R-(-)-2-[(4'-Трифторметансульфонилокси)фенил]-N-метансульфонилпропионамид
1,1'-Карбонилдиимидазол (7,21 г, 44,5 ммоль) добавляют при комнатной температуре к раствору R-(-)-2-(4'-трифторметансульфонилоксифенил)пропионовой кислоты (13,28 г, 44,5 ммоль) в безводном CH2Cl2 (130 мл). Образовавшийся раствор оставляют при перемешивании в течение 1 час 30'. Затем добавляют метансульфонамид (4,23 г, 44,5 ммоль), спустя 1 час при комнатной температуре добавляют DBU (6,65 мл, 44,5 ммоль). Образовавшуюся смесь оставляют для перемешивания при комнатной температуре на протяжении ночи. Органическую фазу промывают 0,5М HCl (2×50 мл), 5% раствором NaH2PO4 (3×50 мл) и водой до достижения нейтральности. После сушки над Na2SO4 и выпаривания растворителя полученный остаток обрабатывают изопропиловым простым эфиром. Образованный осадок отделяют фильтрованием и маточные растворы упаривают при пониженном давлении, получая при этом неочищенное твердое вещество, которое после суспендирования в н-гексане (50 мл) при комнатной температуре в течение 2 ч дает чистый R-(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-метансульфонилпропионамид (13,2 г, 35,15 ммоль) в виде белого порошка (выход 79%).
Т.пл. 98-100°С. [α]D=-49,4 (с=0,5; СН3ОН). 1Н ЯМР (CDCl3): δ 7,40 (д, 2Н, J=7 Гц); 7,23 (д, 2Н, J=7 Гц); 3,68 (кв., 1Н, J=7 Гц); 3,15 (с, 3Н); 1,42 (д, 3Н, J=7 Гц).
Пример 2а
Натриевая соль R-(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-метансульфонилпропионамида
R-(-)-2-[(4'-Трифторметансульфонилокси)фенил]-N-метансульфонилпропионамид (6,89 г, 18,35 ммоль) растворяют в этаноле (35 мл) и к раствору по каплям добавляют 1М NaOH (объемный стандарт) (18,35 мл). Раствор оставляют для перемешивания в течение 30' при комнатной температуре. Спирт выпаривают при пониженном давлении и водный раствор замораживают и лиофилизуют на протяжении ночи. Чистую натриевую соль получают в виде белого порошка (7,29 г, 18,35 ммоль).
[α]D=-27,2 (с=0,5; СН3ОН).
Пример 2
R-(-)-2-[(4'-Трифторметансульфонилокси)фенил)]пропионамид
Тионилхлорид (4,8 мл, 67 ммоль) добавляют при комнатной температуре к раствору R-(-)-2-(4'-трифторметансульфонилоксифенил)пропионовой кислоты (10 г, 33,5 ммоль) в безводном толуоле (10 мл). Раствор кипятят с обратным холодильником в течение 2 ч. После охлаждения до комнатной температуры толуол выпаривают при пониженном давлении и неочищенный масляный остаток растворяют в CH2Cl2 (25 мл) и в раствор в течение 1 час барботируют аммиак. Органический раствор промывают водой (3×15 мл), сушат над Na2SO4 и упаривают при пониженном давлении, получая при этом светло-коричневое неочищенное твердое вещество, которое очищают суспендированием в н-гексане (100 мл) в течение 2 часов. Чистый R-(-)-2-[(4'-трифторметансульфонилокси)фенил]пропионамид (8,1 г, 27,2 ммоль) выделяют фильтрованием в вакууме в виде белого твердого вещества (выход 81%).
Т.пл. 67-69°С. [α]D=-12 (с=1; СН3ОН). 1Н ЯМР (CDCl3): δ 7,69 (д, 2Н, J=7 Гц); 7,22 (д, 2Н, J=7 Гц), 5,37 (ушир. с, 2Н, СОNH 2); 3,63 (кв., 1Н, J=7 Гц); 1,53 (д, 3Н, J=7 Гц).
Пример 3
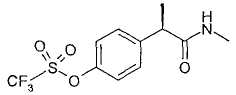
R-(-)-2-[(4'-Трифторметансульфонилокси)фенил)]-N-метилпропионамид
Тионилхлорид (4 мл) добавляют при комнатной температуре к раствору R-(-)-2-(4'-трифторметансульфонилоксифенил)пропионовой кислоты (1 г, 3,35 ммоль) в безводном толуоле (2,5 мл). Раствор кипятят с обратным холодильником в течение 2 ч. После охлаждения до комнатной температуры толуол выпаривают при пониженном давлении и неочищенный масляный остаток растворяют в CH2Cl2 (10 мл). Органический раствор добавляют по каплям к раствору метиламина (0,414 мл, 10,08 ммоль) в CH2Cl2 (5 мл). Смесь оставляют для перемешивания при комнатной температуре в течение 3 час. Растворитель выпаривают при пониженном давлении для отгонки избыточного амина и неочищенный продукт снова разбавляют CH2Cl2 (10 мл), промывают насыщенным раствором NaHCO3 (2×5 мл) и водой (3×15 мл), сушат над Na2SO4 и упаривают при пониженном давлении, получая при этом неочищенный остаток в виде оранжевого масла. Неочищенное масло очищают флэш-хроматографией (элюент: CH2Cl2/СН3ОН, 98:2), получая при этом чистый R-(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-метилпропионамид в виде прозрачного масла (0,78 г, 2,51 ммоль) (выход 75%).
[α]D=-19 (с=0,5; СН3ОН). 1Н ЯМР (CDCl3): δ 7,48 (д, 2Н, J=7 Гц); 7,24 (д, 2Н, J=7 Гц); 5,35 (ушир.с, 1Н, СОNH); 3,55 (кв., 1Н, J=7 Гц); 2,72 (д, 3Н, J=3 Гц); 1,55 (д, 3Н, J=7 Гц).
Пример 4
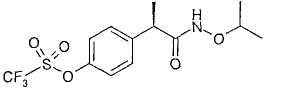
R-(-)-2-[(4'-Трифторметансульфонилокси)фенил]-N-изопропоксипропионамид
К суспензии гидрохлорида N-изопропилгидроксиламина (0,14 г, 1,67 ммоль) и NaHCO3 (0,19 г, 3,34 ммоль) в безводном ТГФ (5 мл) добавляют R-(-)-2-(4'-трифторметансульфонилоксифенил)пропионилхлорид, полученный, исходя из соответствующей кислоты (0,5 г, 1,67 ммоль), как описано в примере 3, и раствор оставляют для перемешивания при комнатной температуре в течение 3 ч. После выпаривания растворителя неочищенный продукт разбавляют CH2Cl2 (10 мл), промывают водой (2×10 мл), сушат над Na2SO4 и упаривают при пониженном давлении, получая при этом масляный остаток. Неочищенное масло очищают обработкой н-гексаном и образовавшийся осадок после фильтрования дает чистый R-(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-изопропоксипропионамид в виде белого порошка (0,45 г, 1,28 ммоль) (выход 77%).
[α]D=-24 (с=0,5; СН3ОН). 1Н-ЯМР (CDCl3): δ 8,15 (ушир.с, 1Н, CONH); 7,45 (д, 2Н, J=7 Гц); 7,20 (д, 2Н, J=7 Гц); 3,65 (м, 1Н); 3,50 (кв., 1Н, J=7 Гц); 1,55 (д, 3Н, J=7 Гц); 1,2 (д, 6Н, J=7 Гц).
Согласно такой же методике, как описана для примера 3, синтезировали следующие амины, исходя из коммерчески доступных аминов или из аминов, полученных согласно методике, описанной в WO 01/58852; WO 00/24710 и WO 02/068377.
Пример 5
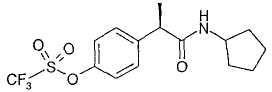
R-(-)-2-[(4'-Трифторметансульфонилокси)фенил]-N-циклопентилпропионамид
[α]D=-35 (с=1; СН3ОН). 1Н-ЯМР (CDCl3): δ 7,52 (д, 2Н, J=7 Гц); 7,28 (д, 2Н, J=7 Гц); 5,55 (ушир.с, 1Н, CONH); 3,58 (кв., 1Н, J=7 Гц); 3,48 (м, 1Н); 2,85 (м, 4Н); 2,36 (м, 4Н); 1,58 (д, 3Н, J=7 Гц).
Пример 6
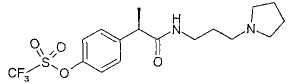
R-(-)-2-[(4'-Трифторметансульфонилокси)фенил]-N-[3-(N'-пиперидинил)пропил]пропионамид
[α]D=-26 (с=2; СН3ОН). 1Н-ЯМР (CDCl3): δ 8,11 (ушир.с, 1Н, CONH); 7,72 (д, 2Н, J=7 Гц); 7,25 (д, 2Н, J=7 Гц); 3,88 (кв., 1Н, J=7 Гц); 3,55 (м, 2Н); 3,30-2,95 (м, 3Н); 2,70 (м, 2Н); 2,48 (м, 2Н); 2,25 (м, 2Н); 2,05 (м, 2Н); 2,00-1,74 (м, 2Н); 1,54 (д, 3Н, J=7 Гц).
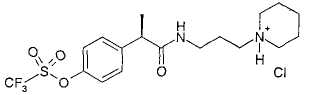
Пример 6а
Гидрохлорид R-(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-[3-(N'-пиперидинил)пропил]пропионамида
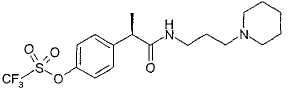
R-(-)-2-[(4'-Трифторметансульфонилокси)фенил]-N-[3-(N'-пиперидинил)пропил]пропионамид (0,15 г, 0,35 ммоль) растворяют в CH2Cl2 (3 мл). Добавляют 3 н. HCl (0,5 мл) и после перемешивания при комнатной температуре в течение 1 ч растворители выпаривают при пониженном давлении, и неочищенный продукт разбавляют безводным этиловым эфиром (5 мл). Образованный осадок выделяют фильтрованием в вакууме, получая при этом чистый гидрохлорид R-(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-[3-(N'-пиперидинил)пропил]пропионамида в виде белого порошка (0,128 г, 0,28 ммоль).
[α]D=-12 (с=2; СН3ОН).
Пример 7
R-(-)-2-[(4'-Трифторметансульфонилокси)фенил]-N-[2-(N'-пирролидинил)этил]пропионамид
[α]D=-34 (с=1; СН3ОН). 1Н-ЯМР (CDCl3): δ 8,65 (ушир.с, 1Н, CONH); 7,75 (д, 2Н, J=7 Гц); 7,22 (д, 2Н, J=7 Гц); 4,02 (м, 2Н); 3,85-3,74 (м, 3Н); 3,31 (м, 2Н); 3,0-2,80 (м, 2Н); 2,41-2,12 (м, 4Н); 1,65 (д, 3Н, J=7 Гц).
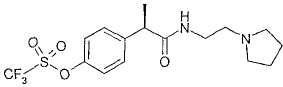
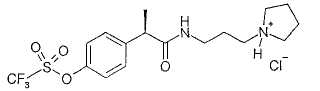
Пример 7а
Гидрохлорид R-(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-[2-(N'-пирролидинил)этил]пропионамида
Соединение получают по методике, описанной в примере 6а.
[α]D=-22 (с=1; СН3ОН).
Пример 8
R-(-)-2-[(4'-Трифторметансульфонилокси)фенил]-N-[3-(N'-пирролидинил)пропил]пропионамид
[α]D=-41 (с=1; СН3ОН). 1Н-ЯМР (CDCl3): δ 8,01 (ушир.с, 1Н, CONH); 7,62 (д, 2Н, J=7 Гц); 7,15 (д, 2Н, J=7 Гц); 3,80 (кв., 1Н, J=7 Гц); 3,52 (м, 2Н); 3,31 (м, 2Н); 2,95 (м, 2Н); 2,78 (м, 2Н); 2,15-1,90 (м, 6Н); 1,55 (д, 3Н, J=7 Гц);
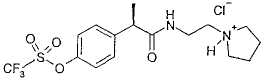
Пример 8а
Гидрохлорид R-(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-[3-(N'-пирролидинил)пропил]пропионамида
Соединение получают по методике, описанной в примере 6а.
[α]D=-17 (с=1; СН3ОН).
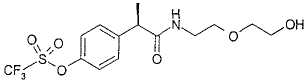
Пример 9
R-(+)-2-[(4'-Трифторметансульфонилокси)фенил]-N-(2-гидроксиэтоксиэтил)пропионамид
Раствор R-(-)-2-(4'-трифторметансульфонилоксифенил)пропионовой кислоты (0,53 г, 1,79 ммоль) в тионилхлориде (1 мл) кипятят с обратным холодильником в течение 2 ч. После охлаждения при комнатной температуре и упаривания при пониженном давлении неочищенный масляный остаток растворяют в CH2Cl2 (2 мл) и добавляют по каплям к раствору 2-гидроксиэтоксиэтиламина (0,36 мл, 3,58 ммоль) в CH2Cl2 (4 мл). Смесь оставляют для перемешивания при комнатной температуре на протяжении ночи. Раствор промывают водой (3×10 мл), сушат над Na2SO4 и упаривают при пониженном давлении, получая при этом неочищенный масляный остаток. Неочищенное вещество очищают обработкой в изопропиловом простом эфире (на протяжении ночи при комнатной температуре), получая при этом, после фильтрования, R-(+)-2-[(4'-трифторметансульфонилокси)фенил]-N-(2-гидроксиэтоксиэтил)пропионамид в виде воскообразного твердого вещества (0,48 г, 1,25 ммоль) (выход 70%).
[α]D=+6 (с=1; СН3ОН). 1Н ЯМР (CDCl3): δ 7,78 (д, 2Н, J=7 Гц); 6,95 (д, 2Н, J=7 Гц); 5,92 (ушир. с, 1Н, СОNH); 3,68 (м, 2Н); 3,55-3,44 (м, 7Н); 1,52 (д, 3Н, J=7 Гц).
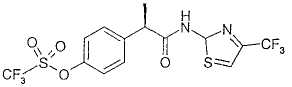
Пример 10
R-(-)-2-[(4'-Трифторметансульфонилокси)фенил]-N-[2-(4'-трифторметил)тиазолил]пропионамид
Раствор R-(-)-2-(4'-трифторметансульфонилоксифенил)пропионовой кислоты (1,012 г, 3,39 ммоль) в тионилхлориде (2 мл) кипятят с обратным холодильником в течение 2 ч. После охлаждения при комнатной температуре и упаривания при пониженном давлении неочищенный масляный остаток растворяют в CH2Cl2 (2 мл) и добавляют по каплям к раствору 2-амино-4-трифторметилтиазола (1,14 г, 6,78 ммоль) в CH2Cl2 (4 мл). 2-Амино-4-трифторметилтиазол получают, как описано в Moazzam M. et al., Indian J. Chem., 27B (11), pages 1051-1053 (1988). Образовавшуюся смесь оставляют для перемешивания при комнатной температуре на протяжении ночи. Раствор промывают насыщенным раствором NaHCO3 (2×5 мл), водой (3×10 мл), сушат над Na2SO4 и упаривают при пониженном давлении, получая при этом неочищенный масляный остаток. После обработки неочищенного вещества изопропиловым эфиром на протяжении ночи при комнатной температуре и фильтрования образованного осадка, выделяют чистый R-(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-[2-(4'-трифторметил)тиазолил]пропионамид в виде светло-коричневого твердого вещества (0,94 г, 2,10 ммоль) (выход 62%).
Т.пл. 138-141°С. [α]D=-50 (с=0,5; СН3ОН). 1Н ЯМР (CDCl3): δ 10,68 (ушир. с, 1Н, CONH); 7,45 (д, 2Н, J=7 Гц); 7,28 (д, 2Н, J=7 Гц), 7,06 (с, 1Н); 3,88 (кв, 1Н, J=7 Гц); 1,67 (д, 3Н, J=7 Гц).
Пример 11
R-(-)-2-[(4'-Трифторметансульфонилокси)фенил]-N-метил-N-гидроксипропионамид
Раствор N,N-диметилформамида (0,42 мл, 5,42 ммоль) в CH2Cl2 (4 мл) охлаждают при Т=-20°С и по каплям добавляют раствор оксалилхлорида (0,16 мл; 1,83 ммоль) в CH2Cl2 (5 мл). В конце добавления температуру повышают до 0°С и после перемешивания 30' добавляют R-(-)-2-(4'-трифторметансульфонилоксифенил)пропионовую кислоту (0,5 г, 1,67 ммоль) и 4-метилморфолин (0,185 мл, 1,67 ммоль). После перемешивания при Т=0°С в течение 30' добавляют гидрохлорид N-метилгидроксиламина (0,27 г, 3,3 ммоль) и 4-метилморфолин (0,73 мл, 6,6 ммоль). Температуре дают возможность повыситься до комнатной температуры и смесь оставляют для перемешивания на протяжении ночи. Образованный осадок отделяют фильтрованием и маточные растворы упаривают при пониженном давлении. Неочищенный масляный остаток растворяют в CH2Cl2 (5 мл) и раствор промывают 1 н. HCl (2×5 мл), водой (2×10 мл), насыщенным раствором NaHCO3 (2×10 мл), сушат над Na2SO4 и упаривают при пониженном давлении, получая при этом неочищенный масляный остаток. Очистка неочищенного продукта флэш-хроматографией (CH2Cl2/СН3ОН, 99:1) дает чистый R-(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-метил-N-гидроксипропионамид в виде светло-желтого масла (0,355 г, 1,08 ммоль) (выход 65%).
[α]D=-23 (с=1,5; СН3ОН). 1Н ЯМР (ДМСО-d6): δ 10,05 (ушир. с, 1Н, OH); 7,48 (с, 4Н); 4,40 (кв, 1Н, J=7 Гц); 3,10 (с, 3Н); 1,40 (д, 3Н, J=7 Гц).
В таблице III приводятся химические названия и структурные формулы для соединений примеров 1-11.
| название | год | авторы | номер документа |
|---|---|---|---|
| N-(2-АРИЛПРОПИОНИЛ)СУЛЬФОНАМИДЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ | 1999 |
|
RU2255084C2 |
| ОМЕГА-АМИНОАЛКИЛАМИДЫ R-2-АРИЛПРОПИОНОВЫХ КИСЛОТ В КАЧЕСТВЕ ИНГИБИТОРОВ ХЕМОТАКСИСА ПОЛИМОРФНОЯДЕРНЫХ И ОДНОЯДЕРНЫХ КЛЕТОК, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2272024C2 |
| (R)-2-АРИЛПРОПИОНАМИДЫ, ПОЛЕЗНЫЕ ПРИ ИНГИБИРОВАНИИ ИЛ-8-ИНДУЦИРОВАННОГО ХЕМОТАКСИСА НЕЙТРОФИЛОВ, СПОСОБ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИНГИБИРУЮЩАЯ ХЕМОТАКСИС НЕЙТРОФИЛОВ, ИНДУЦИРОВАННЫЙ ИНТЕРЛЕЙКИНОМ-8 | 2001 |
|
RU2273630C2 |
| (R)-АРИЛАЛКИЛАМИНОПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2006 |
|
RU2458051C2 |
| 2-АРИЛУКСУСНЫЕ КИСЛОТЫ, ИХ ПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2004 |
|
RU2356887C2 |
| (R)-4-(ГЕТЕРОАРИЛ)ФЕНИЛЭТИЛЬНЫЕ ПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2008 |
|
RU2475486C2 |
| ПРОИЗВОДНЫЕ 2-АРИЛПРОПИОНОВОЙ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, ИХ ВКЛЮЧАЮЩИЕ | 2005 |
|
RU2410372C2 |
| АМИДИНЫ И ИХ ПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2004 |
|
RU2375346C2 |
| ПРИМЕНЕНИЕ ХИРАЛЬНЫХ АРИЛКЕТОНОВ В ЛЕЧЕНИИ НЕЙТРОФИЛ-ЗАВИСИМЫХ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ | 2003 |
|
RU2345759C2 |
| СУЛЬФОНОВЫЕ КИСЛОТЫ, ПРОИЗВОДНЫЕ УКАЗАННЫХ КИСЛОТ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2004 |
|
RU2345063C2 |
Описываются производные 2-(R)-фенилпропионовой кислоты формулы (I) и их фармацевтически приемлемые соли, где R' выбран из Н, ОН и, когда R' представляет собой Н, R выбран из Н, С1-С5-алкила, С3-С6-циклоалкила, С1-С5-алкокси, тиазолила, замещенного CF3, остатка формулы -CH2-CH2-Z-(CH2-CH2O)nR', где n равно 2 и Z представляет собой кислород, остатка формулы -(CH2)n-NRaRb, остатка формулы SO2Rd, когда R' представляет собой ОН, то R выбран из С1-С5-алкила. Соединения применимы при ингибировании хемотаксической активации нейтрофилов (лейкоцитов PMN), индуцированной взаимодействием интерлейкина-8 (IL-8) с мембранными рецепторами CXCR1 и CXCR2. Соединения являются применимыми для профилактики и лечения патологий, порожденных указанной активацией. Описываются также применение соединений для изготовления лекарственных средств для лечения псориаза, неспецифического язвенного колита, меланомы, ангиогенеза, хронического закупоривающеего легочного заболевания (COPD), буллезной пузырчатки, ревматоидного артрита, идиопатического фиброза, гломерулонефрита и при профилактике и лечении повреждений, вызванных ишемией и реперфузией, фармацевтическая композиция и способ получения соединений формулы (I), где R' представляет собой Н и R группу SO2Rd. 4 н. и 4 з.п. ф-лы, 3 табл.
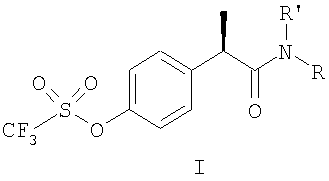
1. Производные 2-(R)-фенилпропионовой кислоты формулы (I):
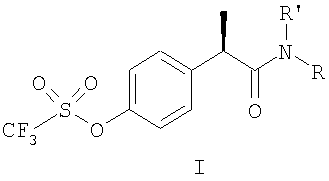
и их фармацевтически приемлемые соли, где
группа R' выбрана из
Н, ОН и,
когда R' представляет собой Н, R выбран из
Н, С1-С5-алкила, С3-С6-циклоалкила, С1-С5-алкокси;
тиазолила, замещенного CF3,
остатка формулы -CH2-CH2-Z-(CH2-CH2O)nR', где n равно 2 и Z представляет собой кислород;
остатка формулы -(CH2)n-NRaRb, где n равно целому числу 2 или 3, и Ra и Rb вместе с атомом азота, с которым они связаны, образуют гетероцикл из 5-6 членов формулы (II)
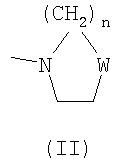
где W представляет собой простую связь, и n равно целому числу 1 или 2;
остатка формулы SO2Rd, где Rd представляет собой C1-С6-алкил;
когда R' представляет собой ОН, то R выбран из
С1-С5-алкила.
2. Соединение по п.1, где,
когда R' представляет собой Н, R выбран из
Н, С1-С5-алкила, С3-С6-циклолкила, С1-С6-алкокси, тиазолила, замещенного CF3;
остатка формулы -(CH2)n-NRaRb, где n равно целому числу 2 или 3, более предпочтительно 3, и группа NRaRb представляет собой 1-пиперидил, 1-пирролидил;
остатка формулы SO2Rd, где Rd представляет собой С1-С2-алкил;
когда R' представляет собой ОН, то R представляет собой С1-С5-алкил.
3. Соединения по п.1 или 2, выбранные из:
R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-метансульфонилпропионамида;
натриевой соли R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-метансульфонилпропионамида;
R(-)-2-[(4'-трифторметансульфонилокси)фенил]пропионамида;
R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-метилпропионамида;
R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-изопропоксипропионамида;
R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-циклопентилпропионамида;
R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-[3-(N'-пиперидинил)пропил]пропионамида;
гидрохлорида R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-[3-(N'-пиперидинил)пропил]пропионамида;
R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-[2-(N'-пирролидинил)этил]пропионамида;
гидрохлорида R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-[2-(N'-пирролидинил)этил]пропионамида;
R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-[3-(N'-пирролидинил)пропил]пропионамида;
гидрохлорида R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-[3-(N'-пирролидинил)пропил]пропионамида;
R(+)-2-[(4'-трифторметансульфонилокси)фенил]-N-(2-гидроксиэтоксиэтил)пропионамида;
R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-[2-(4'-трифторметил)тиазолил]пропионамида;
R(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-метил-N-гидроксипропионамида.
4. Соединения по п.1, которые являются R-(-)-2-[(4'-трифторметансульфонилокси)фенил]-N-метансульфонилпропионамидом и его натриевой солью.
5. Соединения по п.1 для применения в качестве лекарственных средств, предназначенных для лечения псориаза, неспецифического язвенного колита, меланомы, ангиогенеза, хронического закупоривающего легочного заболевания (COPD), буллезной пузырчатки, ревматоидного артрита, идиопатического фиброза, гломерулонефрита и при профилактике и лечении повреждений, вызванных ишемией и реперфузией.
6. Применение соединений по любому из пп.1-4 для изготовления лекарственных средств для лечения псориаза, неспецифического язвенного колита, меланомы, ангиогенеза, хронического закупоривающего легочного заболевания (COPD), буллезной пузырчатки, ревматоидного артрита, идиопатического фиброза, гломерулонефрита и при профилактике и лечении повреждений, вызванных ишемией и реперфузией.
7. Фармацевтическая композиция, для лечения псориаза, неспецифического язвенного колита, меланомы, ангиогенеза, хронического закупоривающего легочного заболевания (COPD), буллезной пузырчатки, ревматоидного артрита, идиопатического фиброза, гломерулонефрита и при профилактике и лечении повреждений, вызванных ишемией и реперфузией, включающая в себя соединение по любому из пп.1-4 в смеси с подходящим носителем.
8. Способ получения соединений формулы (I) по п.1, у которых R' представляет собой Н, и R представляет собой SO2Rd, где Rd представляет собой С1-С2алкил, включающий в себя обработку R-(-)-2-(4'-трифторметансульфонилоксифенил)пропионовой кислоты подходящим сульфонамидом RdSO2NHz, где Rd представляет собой С1-С2-алкил, в присутствии конденсирующего агента.
| WO 00/24710 A1, 04.05.2000 | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| RU 2001113733 A, 27.12.2003. | |||

