Краткое описание изобретения
Настоящее изобретение относится к новым соединениям, полезным для ингибирования хемотаксической активации, индуцируемой компонентой C5a комплемента. Указанные соединения полезны в лечении патологий, связанных с хемотаксической активацией нейтрофилов и моноцитов, индуцированной компонентой C5a комплемента. В частности, соединения полезны в лечении сепсиса, псориаза, ревматоидного артрита, неспецифического язвенного колита, синдрома острой дыхательной недостаточности, идиопатического фиброза, гломерулонефрита и в предупреждении и лечении поражений, вызванных ишемией и реперфузией.
Уровень техники, к которой относится изобретение
В отношении иммунологических и инфекционных явлений активация системы комплемента является посредником в усилении воспалительной реакции как и посредством прямого мембранного действия, так и посредством выделения серии пептидных фрагментов, в общем, известных как анафилатоксины, получающихся при ферментативном расщеплении C3, C4 и C5 компонент комплемента. Эти пептиды включают в себя C3a и C4a, оба содержащие 77 аминокислотных остатка; в свою очередь, C5 конвертаза расщепляет C5 компонент комплемента, давая гликопротеин C5a, включающий 74 аминокислотных остатка.
C5a пептидный фрагмент комплемента был определен как "конечный" про-воспалительный медиатор, благодаря его хемотаксическому и воспалительному действию. В действительности, другие воспалительные медиаторы, такие как определенные цитокины (например, IL-8, MCP-I и RANTES), высокоселективны по отношению к "self-attracted" клеткам (клетки хемотаксически привлекающие клетки того же вида), в то время как другие, такие как гистамин и брадикинин, являются слабыми хемотаксическими агентами.
Убедительные подтверждения in vivo вовлечения C5a в отдельных патологических состояниях, которые включают ишемию/реперфузию, аутоиммунные дерматиты, мембранно-пролиферативный идиопатический гломерулонефрит, ирреспонзивность дыхательных путей и хронические воспалительные заболевания, респираторный дистресс-синдром и ХОЗЛ, болезнь Альцгеймера, форму ревматоидного артрита у детей (N.P. Gerard, Ann. Rev. Immunol., 12, 755, 1994).
В виду невро-воспалительного потенциала C5a/C5a-desArg, образующегося посредством как и локальной активации комплемента, так и амилоидной активации, связанной с астроцитарным и микроглиальным хемотаксисом и активацией, непосредственно индуцированной посредством C5a, были предложены ингибиторы комплимента для лечения неврологических заболеваний, таких как болезнь Альцгеймера (McGeer & McGeer P.L., Drugs, 55, 738, 1998).
Кроме того, контроль синтеза компонент комплемента рассматривается как многообещающая терапевтическая цель для лечения шокового состояния и предотвращения отторжения при трансплантации органов (полиорганная недостаточность и гиперострое отторжение трансплантанта) (Issekutz A.C. et al., Int. J. Immunopharmacol, 12, 1, 1990; Inagi R. et at., Immunol. Lett., 27, 49, 1991). В последнее время сообщалось, что способ ингибирования компонент комплемента был включен для предотвращения поражения родной и трансплантированной почек, принимая во внимание роль комплемента в патогенезе как хронических интерстициальных, так и острых гломерулярных поражений почек. (Sheerin N.S. & Sacks S.H., Curr. Opinion Nephrol. Hypert., 7, 395, 1998).
Характеристическая аккумуляция нейтрофилов имеет место при острых и хронических патологических состояниях, например, в сильно воспаленных и устойчивых к терапии областях псориатических поражений. Нейтрофилы хемотаксически привлекаются и активируются посредством синергетического действия хемокинов, IL-8 и Gro-α, выделяемыми стимулированными кератиноцитами, и C5a/C5a-desArg компонент, продуцируемых при альтернативном пути активации комплемента (T. Terui et al., Exp. Dermatol., 9, 1, 2000).Недавно нами был описан новый класс веществ “омега-аминоалкиламиды R-2-арилпропионовых кислот” как ингибиторы хемотаксиса полиморфноядерных и одноядерных клеток” (WO 02/068377). Указанный новый класс включает соединения с диапазоном действия от селективных C5a ингибиторов до дуальных C5a/IL-8 ингибиторов.
Кроме того, о четвертичных аммонийных солях омега-аминоалкиламидов R-2-арилпропионовых кислот сообщалось как о селективных ингибиторах C5a индуцированного хемотаксиса нейтрофилов и моноцитов (WO 03/029187).
Недавно нами были описаны новые классы “2-арилпропионилсульфонамиды” (WO 00/24710) и “2R-арилпропиониламиды” (WO 02/58858), “2-арилпропионовые кислоты” (WO 03/043625) и “2-арилуксусные кислоты” (WO 04/069782) как сильные и селективные ингибиторы CXCL8 индуцированного PMN хемотаксиса человека. Было найдено, что соединения, описанные в вышеуказанных заявках на патент, ингибируют CXCL8 индуцированный хемотаксис полиморфонуклеаров (PMN) при концентрациях в диапазоне от 10-7 M до 10-9 M; для сравнения, соединения по настоящему изобретению не ингибируют C5a и f-MLP индуцированный PMN хемотаксис в том же диапазоне концентраций.
Затем авторы описали новый класс “омега-аминоалкиламиды R-2-арилпропионовых кислот в качестве ингибиторов хемотаксиса полиморфно-ядерных и одноядерных клеток, индицированного анафилатоксином C5a” (WO 02/068377). В этой заявке на патент авторы сообщили, что омега аминогруппа в заместителе, связанном с N, является ключевой функцией (фармакофорной точкой) для наличия C5a ингибирующей активности. Было найдено, что некоторые соединения, соответствующие изобретению, способны ингибировать как C5a, так и CXCL8 индуцированный хемотаксис PMN благодаря гибкому (от 2 до 4 атомов) спейсеру между аминогруппой и основным остатком. Ключевая роль основной, положительно заряженной части для C5a ингибирования подтверждается активностью соответствующих четвертичных аммонийных солей, как описано в (WO 03/029187).
Подробное описание изобретения
В настоящее время авторы с удивлением обнаружили определенный класс 2-R-арилпропионамидов и 2-R-арилпропионилсульфонамидов, которые даже при отсутствии омега-аминоалкильной группы демонстрируют сильный и селективный ингибирующий эффект, направленный на C5a индуцированный хемотаксис PMN человека.
В настоящее время авторы обнаружили, что определенные 2-R-арилпропионамиды и 2-R-арилпропионилсульфонамиды с АКЦЕПТОРОМ ВОДОРОДНОЙ СВЯЗИ (атомом/группой), расположенным в хорошо известном положении химического пространства, демонстрируют на удивление мощный ингибирующий эффект, направленный на C5a индуцированный хемотаксис PMN человека. Любопытно, что эти соединения абсолютно лишены CXCL8 ингибирующего действия.
Понятие фармакофор определяется, как совокупность стерических и электронных функций в группе биологически активных соединений, необходимых для обеспечения соответствующей биологической активности. В общем, фармакофор может рассматриваться, как совокупность стерических и электронных функций, необходимых для обеспечения определенных взаимодействий между биологически активным соединением и его биологической цели.
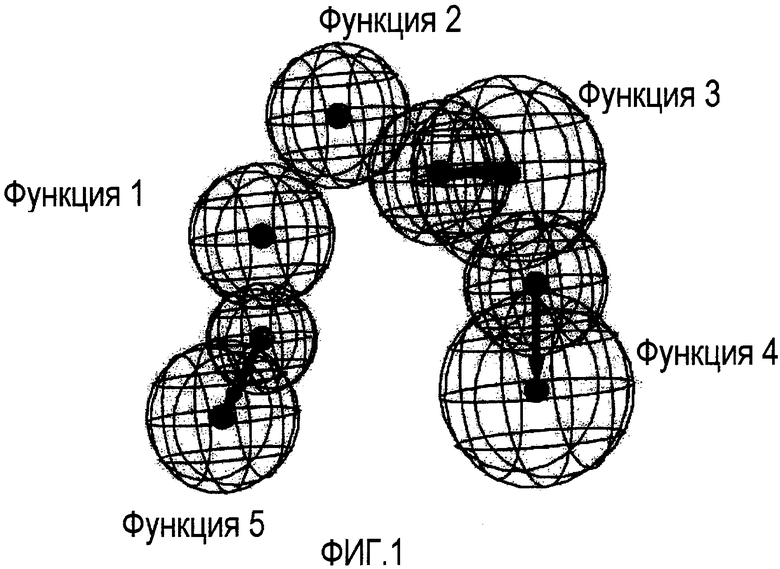
Рассчитываемая фармакофор-модель для ингибирования C5a изображена на Фиг. 1. Новая фармакофор-модель разделяет четыре из пяти функций с предыдущим описанным фармакофором, соответствующим CXCL8 ингибиторам (WO 04/069782); четыре общие функции (Функции 1-4) совершенно совместимы в 3D химическом пространстве.
Функция 5, соответствующая дополнительному Акцептору Водородной Связи, характерна для фармакофора, соответствующего C5a ингибиторам. Наличие на схеме Функции 5 является причиной высокой эффективности, а также наблюдаемой C5a/CXCL8 селективности соответствующих соединений. В сущности, все соединения, вполне соответствующие фармакофор-модели, приведенной на Фиг.1, теряют ингибирующий эффект по отношению к CXCL8.
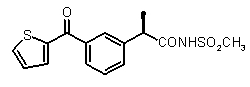
В Таблице 1 показан набор выбранных примеров эффективных и селективных CXCL8 ингибиторов. Соединения, лишенные дополнительной группы - акцептора водородной связи, не проявляют какой-либо ингибирующей активности по отношению к C5a (пункты таблицы 1, 2 и 3).
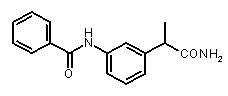
Предварительно сообщалось, что производные амида и сульфамида Кетопрофена (пункты таблицы 4 и 5) являются селективными CXCL8 ингибиторами с незначительной ингибирующей активностью по отношению к C5a, индуцируемому хемотаксису PMN.
Заслуживает внимания тот факт, что амидные производные Кетопрофена (пункты таблицы 4 и 5) могут хорошо подходить под C5a-фармакофор гипотезу с геометрической точки зрения; в соответствии с данным наблюдением, наблюдалась умеренная ингибирующая активность при высокой концентрации (c=10-6 M) лекарственного препарата (Таблица 1).
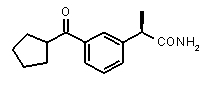
Хорошо известно, что карбонильная группа бензофенона является чрезвычайно слабым АКЦЕПТОРОМ ВОДОРОДНОЙ СВЯЗИ благодаря сильному электроноакцепторному эффекту двух фенильных групп; таким образом, производные Кетопрофена не удовлетворяют фармакофор-гипотезе из-за электронных свойств этой группы. Соответственно, усиление водородная-связь-акцепторных характеристик групп в области функции 5 - АКЦЕПТОРА ВОДОРОДНОЙ СВЯЗИ хорошо согласуется с возрастанием ингибирующего потенциала по отношению к C5a (в качестве примеров смотри Примеры 1-4, приведенные в Таблице 2) и в то же время с потерей ингибирующей активности по отношению к CXCL8.
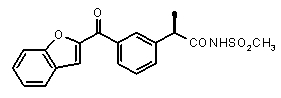
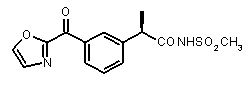
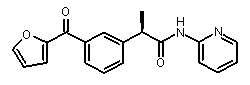
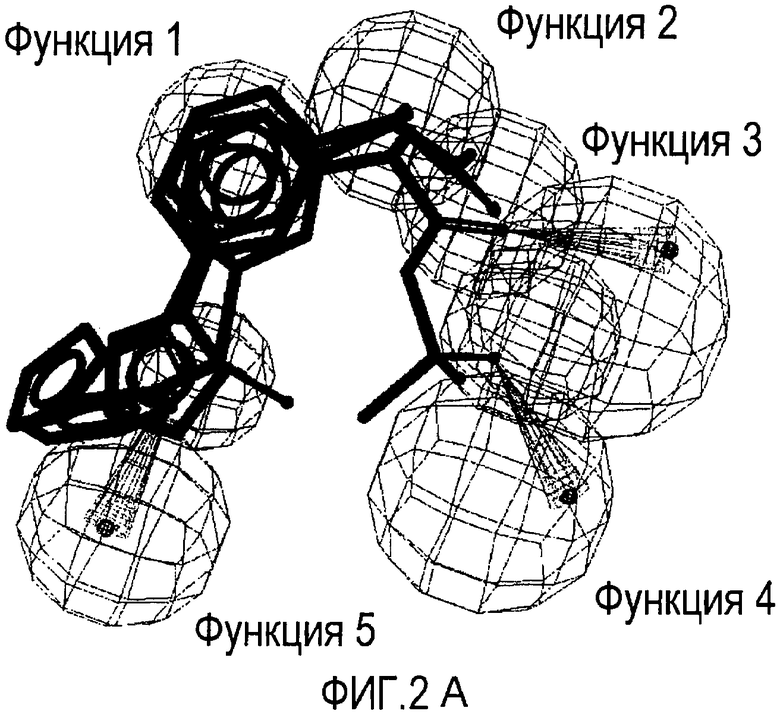
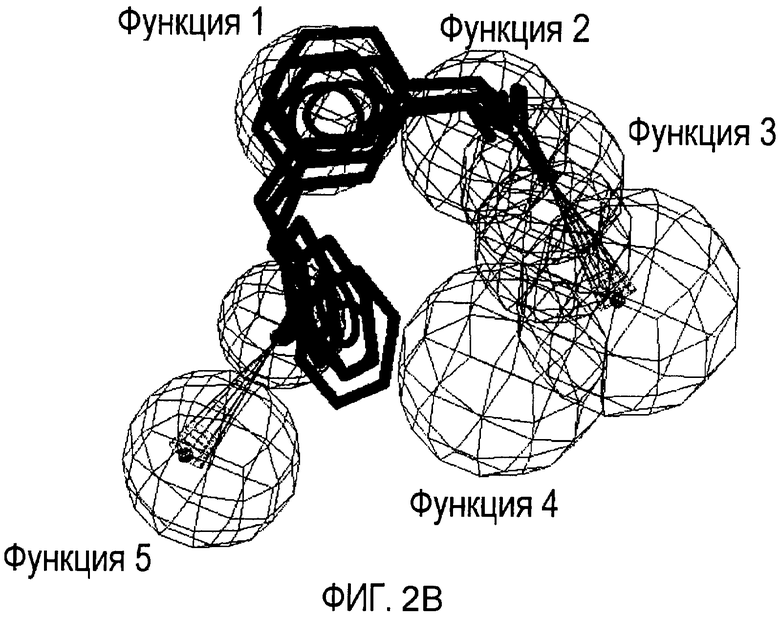
Совместимость с моделью для выбранных соединений из этого нового класса C5a ингибиторов показана на Фиг. 2a и Фиг. 2b.
ОБРАЗОВАНИЕ ФАРМАКОФОРА
Образование фармакофора осуществлялось с использованием программного обеспечения CatalystTM, версия 4.7 (Molecular Simulations, Inc., San Diego, CA), которое предназначено для идентификации основных конфигураций активных молекул посредством их химических функций. Конфигурация представляет собой совокупность относительных местоположений в 3D пространстве, где каждое ассоциировано с типом функции. Все соединения в тренинговом ряду были охарактеризованы в терминах их химических функций, ассоциированных в 3D пространстве. Кроме того, каждая химическая часть может рассматриваться программой, как более чем одна функция на основе выявленного подобия. Например, ароматическое кольцо может “устанавливать” как гидрофобные взаимодействия, так и π-π взаимодействия на целевом сайте, и это различное действие относится к разным функциям (ГИДРОФОБНОЕ, ГИДРОФОБНОЕ АРОМАТИЧЕСКОЕ).
Функциональная группа в молекуле может ассоциироваться с более чем одной функцией в зависимости от ее химических и физических свойств, и различные функциональные группы могут демонстрировать подобное поведение при взаимодействии с целью, таким образом, отмечаясь как одинаковые функции.
Анализ определения функций и выбор функций является ключевым шагом в создании фармакофор-гипотезы. Хорошо известно, что наиболее важные силы, участвующие в опознавании молекул, представляются электростатическими взаимодействиями, водородными связями и гидрофобными взаимодействиями. Авторы приняли определения индивидуальных функций, которые относят химическую природу группы к возможности наличия ценных специфических взаимодействий, ответственных за биологическую активность.
ОПРЕДЕЛЕНИЕ ФУНКЦИЙ
АКЦЕПТОР ВОДОРОДНОЙ СВЯЗИ (HBA) (липид)
Функция акцептора водородной связи липида выбирается из следующих типов атомов и групп атомов с доступной поверхностью: азот, кислород или сера (за исключением гипервалентной), которые имеют неподеленную пару и заряд, меньший или равный нулю.
После рассмотрения окружения липидов все основные амины (первичные, вторичные и третичные) включены в это определение. Водородная связь характеризуется строго “прямым” взаимодействием; эта функция является, таким образом, опосредованно связанной с теоретическим местоположением соответствующего донора водорода. Три позиции водородной связи, к примеру, рассмотренные в случае карбонильной группы (акцептор): первые две по направлению (идеальному) к неподеленным парам и третья вдоль направления C=O связи.
ДОНОР ВОДОРОДНОЙ СВЯЗИ (HBD)
Донор водородной связи выбирается из следующих типов атомов или групп атомов с доступной поверхностью:
некислые гидроксилы, тиолы, ацетиленовый водород и водород при азоте (за исключением тетразольного и трифторметил сульфонамидного водорода).
В качестве донора водородной связи не выбирается азот, который мог бы протонироваться из-за своей высокой основности.
ГИДРОФОБНАЯ (алифатика, ароматика)
Гидрофобная функция определяется как непрерывно связанная совокупность атомов, которые не расположены рядом с заряженными или электроотрицательными атомами, в конформере, так, что эти атомы имеют доступную поверхность. Гидрофобные группы включают в себя: фенил, циклоалкил, изопропил и метил.
Тем не менее, было необходимо различать ароматическую гидрофобную функцию и алифатическую для того, чтобы иметь хорошее соответствие с биологическими характеристиками.
Модель включала только ароматические атомы, далее только алифатические атомы.
Молекула подразумевает подбор конфигурации, если только она обладает набором соответствующих функций и специфической конформацией, такой, что ее функции могут быть совмещены с соответствующими “идеальными” позициями. Набор функций может считаться совместимым, если каждая функция располагается внутри границы допустимых отклонений, расположенной на определенном расстоянии от соответствующей идеальной точки-позиции.
Описание фигур
На Фигуре 1 графически показаны пять фармакофорных функции ингибиторов C5a. Следующие типы функций принимают участие в фармакофорной части: три Акцептора Водородной Связи, одна Гидрофобная Ароматическая и одна Гидрофобная Алифатическая функция. Соответствующие (ароматическая и алифатическая) гидрофобные функции представлены в виде сфер с радиусом 1,7 Е. Акцептор водородной связи представлен векторной функцией, включающей две сферы, чьи центры масс лежат на расстоянии 3,0 Е. Меньшая (1,7 Е радиус) сфера определяет место расположения атома акцептора водородной связи на лиганде, и большая сфера (2,3 Е) определяет проецируемую точку акцептора водородной связи на сайте рецептора.
Фигуры 2a и 2b иллюстрируют совместимость выбранных арилпропионовых производных, соответствующих Формуле I с фармакофор-моделью, приведенной на Фиг. 1.
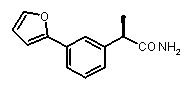
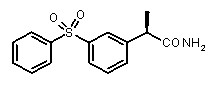
2a) Представленные соединения, соответствующие Формуле I: (R)-2-[(2-оксаксол-2-ил)фенил]пропионамид (пример 14), (R)-2-(3-бензолсульфонилфенил)пропионамид (пример 19) и N-{(R)-2-[3-фуран-2-карбонил)]пропионил}метансульфонамид (пример 23)
2b) Представленные соединения, соответствующие Формуле I: (R)-2-[3-(2-метоксифенокси)фенил]пропионамид (пример 10); (R)-2-[3-(2-метоксифениламино)фенил]пропионамид (пример 12); (R)-2-[3-(пиридин-2-иламино)фенил]пропионамид (пример 13).
КООРДИНАТЫ
Абсолютные координаты центров масс сфер каждой функции в Фиг. 1 приведены ниже:
Общие функции
Функция 1
ГИДРОФОБНАЯ АРОМАТИЧЕСКАЯ имеет декартовы координаты +2,588, +0,613, -1,940 соответственно вдоль XYZ осей.
Функция 2
ГИДРОФОБНАЯ АЛИФАТИЧЕСКАЯ имеет декартовы координаты +1,788, +2,693, +1,260 соответственно вдоль XYZ осей.
Функция 3
ПРОЕКЦИОННАЯ ТОЧКА АКЦЕПТОРА ВОДОРОДНОЙ СВЯЗИ имеет декартовы координаты -2,713, +2,333, +2,840 соответственно вдоль XYZ осей.
НАЧАЛЬНАЯ ТОЧКА АКЦЕПТОРА ВОДОРОДНОЙ СВЯЗИ 1 имеет декартовы координаты -0,233, +0,936, +1,877 соответственно вдоль XYZ осей.
Функция 4
ПРОЕКЦИОННАЯ ТОЧКА АКЦЕПТОРА ВОДОРОДНОЙ СВЯЗИ 2 (необязательного) имеет декартовы координаты -5,013, -1,188, -0,400 соответственно вдоль XYZ осей.
НАЧАЛЬНАЯ ТОЧКА АКЦЕПТОРА ВОДОРОДНОЙ СВЯЗИ 2 (необязательного) имеет декартовы координаты -2,688, -1,514, +1,472 соответственно вдоль XYZ осей.
Функция 5
ПРОЕКЦИОННАЯ ТОЧКА АКЦЕПТОРА ВОДОРОДНОЙ СВЯЗИ 3 имеет декартовы координаты -2,093, +3,893, +3,452 соответственно вдоль XYZ осей.
НАЧАЛЬНАЯ ТОЧКА АКЦЕПТОРА ВОДОРОДНОЙ СВЯЗИ 3 имеет декартовы координаты -1,815, +1,640, +1,497 соответственно вдоль XYZ осей.
Наличие функций 1, 2, 3, 5 (ГИДРОФОБНОЙ АЛИФАТИЧЕСКОЙ, ГИДРОФОБНОЙ АРОМАТИЧЕСКОЙ, АКЦЕПТОРА ВОДОРОДНОЙ СВЯЗИ 1, АКЦЕПТОРА ВОДОРОДНОЙ СВЯЗИ 3) необходимо для биологической C5a ингибирующей активности соответствующего класса (соединений).
Функция 4 (АКЦЕПТОР ВОДОРОДНОЙ СВЯЗИ 2) может быть, при необходимости, представлена посредством молекул из указанного класса, но наличие второго группы акцептора водородной связи не существенно.
Допустимое отклонение для всех расстояний между химическими функциями было установлено равным +0,5 Е, и допустимое отклонение для соответствующих геометрических углов ±20°.
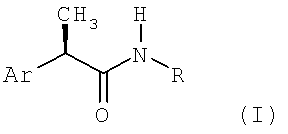
Настоящее изобретение относится к (R)-2-арилпропионамидам, соответствующим формуле (I):
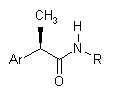
(I)
в которой
Ar - это фенильная группа, замещенная в 3 (мета) положении группой R1, выбираемой из:
линейного или разветвленного C1-C8-алканоила, C1-C6-циклоалканоила, гетероарилкарбонила, C1-C6-алкиламинокарбонила, ариламинокарбонила, C1-C6-алкиламино, C1-C6-ациламино, ариламино, бензоиламино, арилокси, гетероарила, C1-C6-алкоксикарбонила, C1-C6-арилоксикарбонила, C1-C8-алкансульфонила, арилсульфонила, или если R1 представлен аминогруппой, как определено выше, то R1 формирует 5-7 членный цикл с другим заместителем в 4 позиции;
R выбирается из:
- H, OH, C1-C5-алкила, C3-C6-циклоалкила, C2-C5-алкенила, C1-C5-алкокси;
- гетероарильная группа выбирается из: пиридина, пиримидина, пиррола, тиофена, фурана, индола, тиазола, оксазола;
- α или β карбоксиалкильный остаток может состоять из прямого или разветвленного C1-C6-алкила, C3-C6-циклоалкила, C2-C6-алкенила, C1-C6-фенилалкила, по желанию, замещенных другой карбоксильной (COOH) группой;
- остаток с формулой SO2Rd, в которой Rd - это C1-C6-алкил, C3-C6-циклоалкил, C2-C6-алкенил, арил, гетероарил.
Предпочтительны те соединения, соответствующие изобретению, в которых:
Ar - это фенильная группа, замещенная в положении 3 (мета) группой R1, выбираемой из:
линейного или разветвленного C1-C8-алканоила; 2-фурила, 2-оксазолила, 3-изоксазолила, 2-бензоксазолила, 3-бензоизоксазолила, 2-тиазолила, 2-пиридила; фуранкарбонила; бензофуранкарбонила; тиофенкарбонила; пиридинкарбонила; бензоиламинокарбонила; C1-C6-ациламино; бензоиламино; арилокси; ариламино, или R1 формирует спаренную бициклическую систему, выбираемую из 3-4-дигидро-1H-хинолил-2-она, 1,3-дигидроиндол-2-она, 1,3,4,5-тетрагидробензо[b]азепин-2-она;
R выбирается из:
- H, OH, C1-C5-алкила;
- 2-пиридила, 2-тиазолила; карбоксиалкильная группа может состоять из прямого или разветвленного C1-C6-алкила, C1-C6-фенилалкила;
остаток с формулой SO2Rd, в которой Rd - это C1-C6-алкил.
Примеры наиболее предпочтительных соединений формулы (I):
(R)-2-(3-изобутирилфенил)пропионамид,
(R)-2-(3-циклопентанкарбонилфенил)пропионамид,
(R)-2-[(3-(фуран-2-карбонил)фенил]пропионамид,
(R)-2-[(3-(бензофуран-2-карбонил)фенил]пропионамид,
(R)-2-[(3-(тиазол-2-карбонил)фенил]пропионамид,
(R)-2-[(3-(оксазол-2-карбонил)фенил]пропионамид,
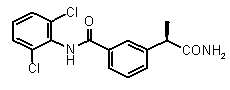
3-((R)-1-карамоилэтил)-N-(2,6-дихлорофенил)бензамид,
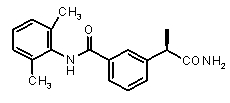
3-((R)-1-карбамоилэтил)-N-(2,6-диметилфенил)бензамид,
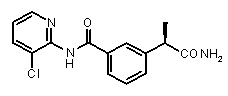
3-((R)-1-карбамоилэтил)-N-(3-хлоропиридин-2-ил)бензамид,
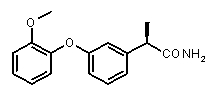
(R)-2-[3-(2-метоксифенокси)фенил)пропионамид,
(R)-2-[3-(2-хлорофениламино)фенил]пропионамид,
(R)-2-[3-(2-метоксифениламино)фенил]пропионамид,
(R)-2-[3-(пиридин-2-иламино)фенил]пропионамид,
(R)-2-(3-оксазол-2-ил)фенил]пропионамид,
(R)-2-(3-фуран-2-ил)фенил]пропионамид,
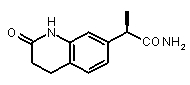
(R)-2-(оксо-1,2,3,4-тетрагидрохинолин-7-ил)пропионамид,
(R)-2-(3-бензолсульфонилфенил)пропионамид,
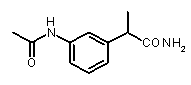
2-(3-ацетиламинофенил)пропионамид,
2-(3-бензоиламинофенил)пропионамид,
N-[(R)-2-(3-циклопентанкарбонилфенил)пропионил]метансульфонамид,
N-{(R)-2-[3-(фуран-2-карбонил)фенил]пропионил}метансульфонамид,
N-{(R)-2-[3-(5-метилфуран-2-карбонил)фенил]пропионил}метансульфонамид,
N-{(R)-2-[(3-(тиофен-2-карбонил)фенил]пропионил}метансульфонамид,
N-{(R)-2-[(3-(бензофуран-2-карбонил)фенил]пропионил}метансульфонамид,
N-{(R)-2-[(3-(оксазол-2-карбонил)фенил]пропионил}метансульфонамид,
(R)-2-[3-(фуран-2-карбонил)фенил]-N-пирид-2-илпропионамид,
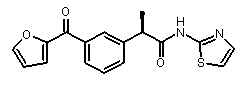
(R)-2-[3-(фуран-2-карбонил)фенил]-N-(2H-тиазол-2-ил)пропионамид,
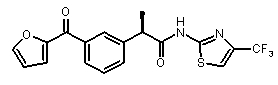
(R)-2-[3-(фуран-2-карбонил)фенил]-N-(4-трифторметил-2H-тиазол-2-ил)пропионамид,
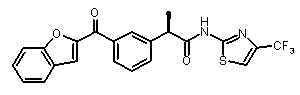
(R)-2-[(3-(бензофуран-2-карбонил)фенил]-N-(4-трифторметил-2H-тиазол-2-ил)пропионамид,
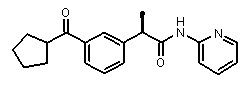
(R)-2-(3-циклопентанкарбонилфенил)-N-пирид-3-илпропионамид,
(R)-2-[3-(фуран-2-карбонил)фенил]-N-гидроксипропионамид,
(R)-2-[3-(тиазол-2-карбонил)фенил]-N-гидроксипропионамид,
2-{(R)-2-[3-(фуран-2-карбонил)фенил]пропиониламино}пропионовая кислота,
2-{(R)-2-[3-(фуран-2-карбонил)фенил]пропиониламино}уксусная кислота.
Соединения, соответствующие изобретению, - эффективные ингибиторы хемотаксиса PMNs человека, индуцированного C5a.
Таким образом, другой целью настоящего изобретения является использование соединений, соответствующих формуле (I), в приготовлении медикамента для лечения заболеваний, включающих в себя C5a индуцированный PMNs хемотаксис человека.
Для получения соединений, соответствующих формуле (I), были использованы известные методы получения амидов и ацилсульфонамидов (реакция Меншуткина); соответствующие карбоновые кислоты, в которых Ar определен выше, реагируют с аминами или сульфонамидами формулы RNH2, где R определен выше, в присутствии обычных активирующих карбоксильную группу реагентов в соответствии с методиками, ранее описанными в WO 01/58852; WO 00/24710 и WO 02/068377.
Соединения формулы (I), соответствующие изобретению, были протестированы in vitro по их способности ингибировать хемотаксис полиморфно-ядерных лейкоцитов (ниже используемые в сокращении PMNs) и моноцитов, индуцированный C5a и C5a-desArg компонентами комплемента. Для того чтобы выделить PMNs из гепаринизированной крови человека, взятой у здоровых взрослых добровольцев, мононуклеары (одноядерные клетки) были отделены посредством седиментации на декстране (в соответствии с процедурой, описанной в W.J. Ming et al., J. Immunol., 138, 1469, 1987), и красные кровяные клетки при помощи гипотонического раствора. Жизнеспособность клеток была подсчитана методом исключения с использованием трипанового синего, в то время как отношение циркулирующих полиморфонуклеаров (полиморфно-ядерных клеток) оценивалось на цитоцентрифугате после окрашивания с использованием Diff Quick (набор для окрашивания).
Рекомбинантные фракции C5a и C5a-desArg человека (Sigma) были использованы как стимулирующие агенты в экспериментах с хемотаксисом, давая практически идентичные результаты.
Лиофилизированный C5a растворяли в объеме HBSS, содержащем 0,2% альбумина бычий сыворотки BSA, чтобы, таким образом, получить основной раствор с концентрацией 10-5 M для дальнейшего разбавления в HBSS до концентрации 10-9 M для экспериментов по хемотаксису.
В экспериментах по хемотаксису PMNs инкубировались вместе с соединениями формулы (I), соответствующими изобретению, в течение 15' при 37°C в атмосфере, содержащей 5% CO2. Хемотаксическая активность C5a была оценена на циркулирующих полиморфонуклеарах человека, (PMNs) ресуспендированных в HBSS с концентрацией 1,5×106 PMNs на мл.
В ходе хемотаксических исследований (в соответствии с W. Falket et al., J. Immunol. Methods, 33, 239, 1980) использовались “PVP-free” (не содержащие поливинилпиридина) фильтры с пористостью 5 мкм и микрокамеры подходящие для данного теста.
Соединения формулы (I), соответствующие изобретению, тестировались с концентрацией, лежащей между 10-7 и 10-10 M; для этого они добавляли при одинаковой концентрации как в нижние лунки, так и в верхние лунки микрокамеры. Лунки в нижней части содержали раствор C5a или простой носитель, лунки в верхней части содержали суспензию PMNs.
Ингибирующая активность C5a-индуцированного хемотаксиса посредством индивидуальных соединений, соответствующих изобретению, формулы (I) была оценена инкубированием микрокамеры для хемотаксиса в течение 60 мин при 37°C в атмосфере, содержащей 5% CO2.
Оценка способности соединений, соответствующих изобретению, формулы (I) ингибировать C5a-индуцированный хемотаксис моноцитов человека проводилась в соответствии с методом, описанным Van Damme J. et al. (Eur. J. Immunol., 19, 2367, 1989). Ингибирующая активность C5a-индуцированного хемотаксиса индивидуальными соединениями, соответствующими изобретению, формулы (I) по отношению к моноцитам человека оценивалась при концентрации, лежащей между 10-7 и 10-10 M, путем инкубирования микрокамеры для хемотаксиса в течение 120 мин при 37°C в атмосфере, содержащей 5% CO2.
В качестве примера, данные ингибирования хемотаксиса PMN (концентрация между 10-7 и 10-8 M) некоторых типичных примеров соединений, соответствующих изобретению, приводятся в Таблице 2.
Соединения формулы (I) были оценены ex vivo в крови in toto в соответствии с процедурой, описанной Patrignani et al., in J. Pharmacol. Exper. Ther., 271, 1705, 1994. Почти во всех случаях соединения формулы (I) не затрагивают образование PGE2, индуцированного в макрофагах мыши посредством стимуляции липополисахаридами (LPS, 1 мкг/мл) при концентрации, лежащей между 10-5 и 10-7 M. Ингибирование образования PGE2 - главным образом, в пределе статистической значимости и, в общем, ниже 15-20% основного значения.
Таким образом, дальнейшей целью настоящего изобретения является использование соединений, соответствующих изобретению, в качестве лекарственных препаратов.
В виду экспериментальных данных, обсуждавшихся выше, и роли, которую играет каскад комплемента, а именно, его компонента C5a, в процессе, который включает активацию и инфильтрацию нейтрофилов, соединения, соответствующие изобретению, практически полезны в лечении заболеваний, таких как псориаз (R. J. Nicholoff et al., Am. J. Pathol., 138, 129, 1991), буллезный пемфигоид, ревматоидный артрит (M. Selz et al., J. Clin. Invest., 87, 463, 1981), кишечные хронические воспалительные патологии, такие как язвенный колит (Y. R. Mahida et al., Clin. Sci., 82, 273, 1992), синдром острой дыхательной недостаточности и идиопатический фиброз (E. J. Miller, previously cited, and P. C. Carre et al., J. Clin. Invest., 88, 1882, 1991), муковисцидоз, ХОЗЛ, гломерулонефрит (T. Wada et al., J. Exp. Med., 180, 1135, 1994) и предупреждение и лечение поражений, вызванных ишемией и реперфузией.
Кроме того, соединения, соответствующие изобретению, особенно полезны в лечении сепсиса.
In vivo активность в лечении сепсиса была продемонстрирована, как изложено ниже:
Лигирование слепой кишки и пункция (CLP)
Использовался образец мышиного полимикробного сепсиса и пораженной ткани (в соответствии с процедурой, описанной P. Villa et al., Journal of Endotoxin Research, 1997, 43 (3), 197-204), на основе хирургическим путем создаваемого дивертикулума слепой кишки, который затем пунктировался для создания обширного перитонита.
Полимикробный сепсис, вызванный лигированием слепой кишки и пункцией (CLP), у мышей приводит к воспалению и патологическим последствиям инфильтрации нейтрофилами легких, синдрому расстройства дыхания у взрослой особи (ARDS) и смерти.
После анестезии мыши были подвергнуты 1 см лапаротомии, и слепая кишка была изолирована. Слепая кишка лигировалась ниже илеоцекального клапана (без обструкции), пунктирована на антимезентериальной стороне иглой 18 размера, немного сдавлена, чтобы удостоверится, что отверстие открыто, и далее помещена обратно в брюшную полость. Надрез был закрыт, и мыши были приведены в сознание посредством 1 мл солевого раствора подкожно.
«Плацебо»-операция (симулирующее оперативное вмешательство) проводилась подобным образом, за исключением того, что кишка не пунктировалась. Антибиотики (гентамицин сульфат 3,2 мг/кг и клиндамицин фосфат 40 мг/кг) вводились подкожно, один раз ежедневно в течение 3 дней, сразу после оперативного вмешательства. Выживаемость контролировалась два раза в день в течение 10 дней. Животные были отобраны произвольно в группы, к которым применялся только носитель, или в группы, к которым применялось лечение, по 8-15 особей в группе.
Типичные примеры соединений, соответствующих настоящему изобретению, демонстрировали активность в лечении сепсиса в области концентраций от 1 до 50 мг/кг.
Для этого соединения, соответствующие изобретению, формулы (I) были легко составлены в фармацевтическую композицию с использованием обычных методик и наполнителей, таких как описанные в "Remington's Pharmaceutical Sciences Handbook" MACK Publishing, New York, 18th ed., 1990.
Соединения, соответствующие изобретению, могут применяться путем внутривенной инъекции, как болюс, в виде препаратов наносимых на кожу (кремы, лосьоны, спреи и мази), путем ингаляции, а также перорально в виде капсул, таблеток, сиропа, в составах с контролируемым высвобождением и подобного.
Средняя суточная доза зависит от нескольких факторов, таких как тяжесть заболевания, состояние, возраст, пол и вес пациента. Доза, в общем, будет варьироваться от 1 до 1500 мг в сутки соединения формулы (I), при желании, разделенная для многократного приема.
Следующие примеры иллюстрируют изобретения.
Материалы и методики
Амины формулы RNH2, использованные как реагенты для получения соединений формулы (I), являются известными продуктами, в общем, коммерчески доступными или они могут быть получены в соответствии с методиками, описанными в литературе.
Синтез 2-арилпропионовых кислот формулы ϕ-Ar3-C(CH3)H-CO2H и их R-энантиомеров описывается в Интернациональной заявке на патент WO 01/58852.
Перечень сокращений: THF: тетрагидрофуран; EtOAc: Этил ацетат; MeOH: метанол; EtOH: этанол; DCC: 1,3-Дициклогексилкарбодиимид; DCU: 1,3-Дициклогексилмочевина; DBU: 1,8-Диазобицикло[5.4.0]ундец-7-ен.
Приготовление интермедиатов 2-арилпропионовых кислот
А. 2-[(3-хлорокарбонил)фенил]пропионитрил
Коммерчески доступный 2-[(3-карбокси)фенил]пропионитрил (1,0 г, 5,70 ммоль) растворяли в SOCl2 (5 мл) и полученный раствор перемешивали при кипячении в течение 3 ч. После охлаждения до комнатной температуры смесь упаривали при пониженном давлении, что давало 2-[(3-хлорокарбонил)фенил]пропионитрил в виде масла желтого цвета с почти количественным выходом.
В. 2-(3-аминофенил)пропионитрил
К раствору 2-[(3-хлорокарбонил)фенил]пропионитрила (2,5 г, 14,25 ммоль) в CH2Cl2 (15 мл) добавляли тетрабутиламмоний бромид (0,07 ммоль), и смесь охлаждали до 0°C. При интенсивном перемешивании добавляли раствор азида натрия (1,275 г, 19,5 ммоль) в H2O (5 мл) и полученную смесь перемешивали при 0°C в течение 2 ч. Образовавшийся преципитат отфильтровывали, органическую фазу, содержащую соответствующий ацилазид, промывали H2O (3×25 мл), сушили над Na2SO4 и использовали как есть на следующей стадии. Органический раствор обрабатывали трифторуксусной кислотой (21,38 ммоль) и кипятили в течение 48 ч. По окончании реакции трифторуксусную кислоту упаривали при пониженном давлении, остаток разбавляли CH2Cl2 (50 мл), промывали последовательно насыщенным раствором NaHCO3 (2×25 мл) и H2O (50 мл). После сушки над Na2SO4 и упаривания растворителя при пониженном давлении получали 2-[(3-трифторацетиламино)фенил]пропионитрил.
Смесь 2-[(3-трифторацетиламино)фенил]пропионитрила (2,5 г, 9.25 ммол) и K2CO3 (2,55 г, 17,6 ммол) в H2O/CH3OH (3:1) (50 мл) нагревали при 60°C в течение 16 ч. После охлаждения до комнатной температуры и упаривания метанола, оставшуюся водную фазу экстрагировали CH2Cl2 (3×25 мл). Собранные органические экстракты сушили над Na2SO4 и упаривали при пониженном давлении, что давало 2-(3-аминофенил)пропионитрил в виде масла желтоватого цвета (1,2 г, 8,32 ммоль). Выход 58%.
1H-ЯМР (CDCl3): δ 7,08 (м, 1H); 6,64 (м, 2H); 6,57 (м, 1H); 3,72 (кв, 1H, J=7 Гц); 3,65 (уш.с, 2H, NH 2); 1,54 (д, 3H, J=7 Гц).
С. 2-(3-гидроксифенил)пропионитрил
2-(3-аминофенил)пропионитрил (1,0 г, 6,75 ммоль) суспендировали в воде (12 мл), затем при интенсивном перемешивании по каплям добавляли H2SO4 (1,5 мл, 27 ммоль). После перемешивания в течение 20 мин смесь охлаждали до 4°C, добавляли по каплям раствор NaNO2 (0,466 г, 6,75 ммоль) в воде (5 мл) и полученный раствор перемешивали при кипячении в течение 1 ч. После охлаждения до комнатной температуры к смеси добавляли этилацетат (10 мл), сырой продукт экстрагировали и органическую фазу промывали водой (3×10 мл) и рассолом (насыщенный раствор NaCl в воде) (3×10 мл). После сушки над Na2SO4 и упаривания растворителя при пониженном давлении получали 2-(3-гидроксифенил)пропионитрил в виде масла желтого цвета с почти количественным выходом.
1H-ЯМР (CDCl3): δ 7,20 (д, 1H); 6,88 (д, 1H, J=7 Гц) 6,80-6,72 (м, 2H); 4,90-4,60 (уш.с, 1H, OH); 3,75 (кв, 1H, J=7 Гц); 1,55 (д, 3H, J=7 Гц).
D. 2-(3-иодофенил)пропионитрил
2-(3-аминофенил)пропионитрил (1,0 г, 6,75 ммоль), полученный, как описано ранее, суспендировали в воде (12 мл) и при перемешивании по каплям добавляли 37% HCl (1,6 мл, 20,2 ммоль). Через 5 мин смесь охладили до 4°C, добавляли по каплям NaNO2 (0,466 г, 6,75 ммоль), растворенный в воде (5 мл), и полученный раствор перемешивали в течение 20 мин. К раствору производного бензолдиазоний хлорида добавляли по каплям водный раствор (5 мл) KI (1,13 г, 6,76 ммоль) при 4°C и полученную смесь перемешивали в течение 3 ч. К смеси добавляли EtOAc (15 мл), сырой продукт экстрагировали и промывали водой [экстракт] (3×10 мл) и рассолом (3×10 мл). После сушки над Na2SO4 и упаривания растворителя при пониженном давлении получали 2-(3-йодофенил)пропионитрил в виде масла желтого цвета (1,4 г, 5,4 ммоль). Выход 80%.
1H-ЯМР (CDCl3): δ 7,65 (д, 1H, J=7 Гц); 7,30-7,02 (м, 3H); 3,80 (кв, 1H, J=7 Гц); 1,55 (д, 3H, J=7 Гц).
Общая процедура для оптической очистки (R) энантиомеров
Оптическая очистка всех рацемических кислот, полученных по ниже описанным процедурам, осуществлялась по методике, описанной в Akgün, H.; et al., Arzneim.-Forsch./Drug Res. 1996, 46(II), 891-894, и используя наиболее подходящие хиральные амины.
(R)-2-[3-(изобутирил)фенил]пропионовая кислота (I)
Реакция была проведена в соответствии со следующей процедурой, описанной в Grey R. A., J. Org. Chem. 1984, 49, 2288-2289.
К суспензии ZnCl2 (0,390 г, 2,85 ммоль) в 5 мл сухого THF при 0°C в атмосфере азота добавляли коммерчески доступный изопропилмагний хлорид (2M в Et2O, 2,85 мл, 5,70 ммоль). После перемешивания в течение 20 мин добавляли катализатор (dppf)PdCl2 (1%, 0,057 ммоль) и, затем, по каплям добавляли раствор 2-(хлорокарбонил)фенилпропионитрила (5,72 ммоль), приготовленного, как описано выше, в сухом THF (5 мл). Смесь перемешивали в течение 1 ч при 0°C, далее 3 ч при комнатной температуре. После охлаждения до 0°C добавляли 3N HCl (10 мл) и Et2O (30 мл). Водный слой отделяли и органический слой промывали последовательно насыщенным раствором NaHCO3 (2×30 мл) и рассолом (30 мл). После осушки Na2SO4 и упаривания растворителя при пониженном давлении получали продукт, который после флэш-хроматографии (элюент n-гексан/EtOAc 95:5) давал 2-[3-(изобутирил)фенил]пропионитрил в виде масла желтого цвета (0,804 г, 4,64 ммоль). Выход 81%.
1H-ЯМР (CDCl3): δ 7,86 (с, 1H); 7,76 (д, 1H, J=7 Гц); 7,45-7,35 (м, 2H); 3,84 (кв, 1H, J=7 Гц); 3,45 (м, 1H); 1,68 (д, 3H, J=7 Гц); 1,1 (кв, 6H, J=7 Гц).
К раствору 2-[3-(изобутирил)фенил]пропионитрила (0,93 г, 4,62 ммоль) в 10 мл диоксана добавляли 37% HCl (10 мл). Смесь перемешивали при 70°C в течение 4 ч. После охлаждения до комнатной температуры диоксан упаривали, добавляли к остатку холодную воду (10 мл) и EtOAc (15 мл). Две фазы встряхивали и отделяли, органическую фазу экстрагировали 1N NaOH (2×5 мл). К собранным основным водным экстрактам добавляли 37% HCl для осаждения кислоты. По окончании осаждения 2-[3-(изобутирил)фенил]пропионовой кислоты фильтрованием выделяли чистый продукт в виде твердого вещества белого цвета (0,86 г, 3,95 ммоль). Выход 85%.
[α]D 25 (c=1, EtOH): -38°; 1H-ЯМР (CDCl3): δ 10,6 (уш.с, 1H, COOH); 7,86 (с, 1H); 7,76 (д, 1H, J=7 Гц); 7,45-7,35 (м, 2H); 3,79 (кв, 1H, J=7 Гц); 3,45 (м, 1H); 1,45 (д, 3H, J=7 Гц); 1,1 (д, 6H, J=7 Гц).
В соответствии с такими же экспериментальными процедурами, и используя соответствующие коммерческие реагенты Гриньяра в качестве исходных, были синтезированы следующие соединения:
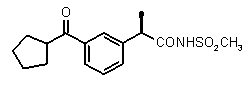
(R)-2-[3-(циклопентанкарбонил)фенил]пропионовая кислота (II)
[α]D 25 (c=1, EtOH): -43°; 1H-ЯМР (CDCl3): δ 7,86 (м, 1H); 7,79 (д, 1H, J=7 Гц); 7,52 (д, 1H, J=7 Гц); 7,37 (м, 1H); 3,82 (кв, 1H, J=7 Гц); 3,71 (м, 1H); 2,22 (м, 2H); 2,01 (м, 3H); 1,82 (м, 3H); 1,58 (д, 3H, J=7 Гц).
(R)-2-[3-(оксазол-2-карбонил)фенил]пропионовая кислота (III)
Из коммерческого реагента 2-(3-карбокси)фенилпропионитрила по следующим процедурам, описанным в Harn N. K. et al., Tetrahedron Letters, 1995, 36(52), 9453-9456, получали 2-[3-(1,3-оксазол-2-илкарбонил)фенил]пропионовую кислоту.
К раствору оксазола (0,5 мл, 7,6 ммоль) в 50 мл THF при -78°C в атмосфере азота добавляли н-BuLi (1,6 M раствор в гексане, 4,7 мл, 7,60 ммоль). После перемешивания в течение 20 мин добавляли ZnCl2 (2,071 г, 15,2 ммоль), смесь доводили до 0°C и перемешивали 45 мин. Затем добавляли CuI (1,45 г, 7,6 ммоль) и через 20 мин добавляли по каплям раствор 2-(хлорокарбонил)фенилпропионитрила (15,2 ммоль), полученного, как описано ранее, в 10 мл THF. Смесь перемешивали в течение 2 ч. Органическую фазу разбавляли EtOAc и промывали последовательно насыщенным раствором NaHCO3 (2×50 мл) и рассолом (50 мл). После осушки над Na2SO4 и упаривания растворителя при пониженном давлении получали остаток, который после флэш-хроматографии давал 2-[3-(оксазол-2-карбонил)фенил]пропионитрил в виде масла желтого цвета (1,27 г, 5,63 ммоль). Выход 74%.
1H-ЯМР (CDCl3): δ 8,48 (м, 2H); 7,70 (с, 1H); 7,61 (д, 1H, J=7 Гц); 7,46 (т, 1H, J=7 Гц); 7,28 (с, 1H); 4,03 (кв, 1H, J=7 Гц); 1,73 (д, 3H, J=7 Гц).
К раствору 2-[3-(оксазол-2-карбонил)фенил]пропионитрила (1 г, 4,43 ммоль) в 10 мл диоксана добавляли 37% HCl (10 мл). Смесь перемешивали при 70°C на 4 ч. После охлаждения до комнатной температуры диоксан упаривали, и добавляли к остатку холодную воду (10 мл) и EtOAc (15 мл). Две фазы встряхивали и отделяли, органическую фазу экстрагировали 1N NaOH (2×5 мл). К собранным основным водным экстрактам добавляли 37% HCl для осаждения требуемой кислоты. По окончании осаждения, после фильтрования получали чистую 2-[3-(оксазол-2-карбонил)фенил]пропионовую кислоту в виде твердого вещества белого цвета (0,87 г, 3,54 ммоль). Выход 80%.
[α]D 25 (c=1, EtOH): -43° (38%), 1H-ЯМР (CDCl3): δ 8,45 (м, 2H); 7,90 (с, 1H); 7,68 (д, 1H, J=7 Гц); 7,50 (т, 1H, J=7 Гц); 7,38 (с, 1H); 3,90 (кв, 1H, J=7 Гц); 1,56 (д, 3H, J=7 Гц).
В соответствии с такими же экспериментальными процедурами, и используя тиазол в качестве исходного реагента, было синтезировано следующее соединение:
(R)-2-[3-(тиазол-2-карбонил)фенил]пропионовая кислота (IV)
[α]D 25 (c=1, MeOH): -36°,1H-ЯМР (CDCl3): δ 8,44 (м, 2H); 8,10 (д, 1H, J=3 Гц); 7,73 (д, 1H, J=3 Гц); 7,63 (д, 1H, J=7 Гц); 7,51 (т, 1H, J=7 Гц); 3,90 (кв, 1H, J=7 Гц); 1,60 (д, 3H, J=7 Гц).
В соответствии с такими же экспериментальными процедурами, и используя фуран в качестве исходного реагента, было синтезировано следующее соединение:
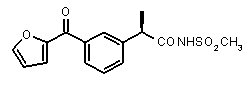
(R)-2-[3-(фуран-2-карбонил)фенил]пропионовая кислота (V)
[α]D 25 (c=1, MeOH): -41°,1H-ЯМР (CDCl3): δ 7,86 (м, 1H); 7,82 (м, 1H, J=7 Гц); 7,64 (с, 1H); 7,49 (м, 1H); 7,41 (м, 1H); 7,16 (д, 1H, J=7 Гц); 6,53 (м, 1H); 3,79 (кв, 1H, J=7 Гц); 1,51 (д, 3H, J=7 Гц).
(R)-2-[3-(бензофуран-2-карбонил)фенил]пропионовая кислота (VI)
Из коммерчески доступного реагента 2-(3-карбокси)фенилпропионитрила по следующим процедурам, описанным в Galli C., Synthesis, 1979, 303-304, была синтезирована 2-[3-(бензофуран-2-карбонил)фенил]пропионовая кислота.
К раствору 2-(3-карбокси)фенилпропионитрила (1,03 г, 5,88 ммоль) в 50 мл сухого ацетонитрила в атмосфере азота добавили 2,3-бензофуран (1,65 мл, 14,7 ммоль) и трифторуксусный ангидрид (3,3 мл, 23,52 ммоль). Смесь перемешивали 5 ч. Растворитель упаривали при пониженном давлении, остаток разбавляли CHCl3 и промывали последовательно насыщенным раствором NaHCO3 (2×50 мл) и рассолом (50 мл). После осушки над Na2SO4 и упаривания растворителя при пониженном давлении получали остаток, который после флэш-хроматографии давал 2-[3-(бензофуран-2-карбонил)фенил]пропионитрил в виде масла желтого цвета (1,05 г, 3,82 ммоль). Выход 65%.
1H-ЯМР (CDCl3): δ 8,04 (м, 2H); 7,76 (д, 1H, J=8 Гц); 7,68-7,54 (м, 5H); 7,36 (м, 1H); 4,03 (кв, 1H, J=7 Гц); 1,74 (д, 3H, J=7 Гц).
К раствору 2-[3-(бензофуран-2-карбонил)фенил]пропионитрила (1 г, 3,63 ммоль) в 10 мл диоксана добавляли 37% HCl (10 мл). Смесь перемешивали при 70°C на 4 ч. После охлаждения до комнатной температуры диоксан упаривали и добавляли к осадку холодную воду (10 мл) и CHCl3 (15 мл). Две фазы встряхивали и разделяли, органическую фазу экстрагировали 1N NaOH (2×5 мл). К собранным основным водным экстрактам добавляли 37% HCl до pH=2 и кислую фазу экстрагировали CHCl3 (3×10 мл). После осушки над Na2SO4 и упаривания растворителя при пониженном давлении, после фильтрации получали чистую 2-[3-(бензофуран-2-карбонил)фенил]пропионовую кислоту в виде порошка белого цвета (1,06 г, 3,60 ммоль). Количественный выход.
[α]D 25 (c=1, EtOH): -58° (35%), 1H-ЯМР (CDCl3): δ 7,82 (с, 1H); 7,72 (д, 1H, J=8 Гц); 7,51 (д, 1H, J=8 Гц); 7,42 (д, 2H, J=8 Гц); 7,28 (т, 2H, J=8 Гц); 7,11 (т, 1H, J=8 Гц); 6,38 (м, 1H); 4,23 (уш.с, 1H, COOH); 3,65 (кв, 1H, J=7 Гц); 1,36 (д, 3H, J=7 Гц).
В соответствии с такими же экспериментальными процедурами, и используя 2-метилфуран в качестве исходного реагента, были синтезированы следующие соединения:
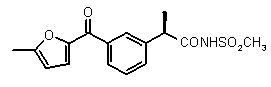
(R)-2-[3-(5-метилфуран-2-карбонил)фенил]пропионовая кислота (VII)
[α]D 25 (c=1, MeOH): -72°,1H-ЯМР (CDCl3): δ 7,94 (м, 1H); 7,56 (м, 3H); 7,10 (д, 1H, J=4 Гц); 6,25 (д, 1H, J=4 Гц); 3,85 (кв, 1H, J=7 Гц); 2,52 (с, 3H); 1,64 (д, 3H, J=7 Гц).
(R)-2-[3-(2,6-дихлорофенилкарбомоил)фенил]пропионовая кислота (VIII)
К раствору коммерческого 2,6-дихлороанилина (1,4 г, 8,64 ммоль) и пиридина (0,69 мл, 8,64 ммоль) в 10 мл сухого CH2Cl2 при комнатной температуре по каплям добавляли 2-[(3-хлорокарбонил)фенил]пропионитрил (1,67 г, 8,64 ммоль), приготовленный, как описывалось ранее. Смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь охладили до 0°C, добавили 1N HCl и органическую фазу промыли 1N HCl (2×10 мл). Органический слой промыли последовательно насыщенным раствором NaHCO3 (2×30 мл) и рассолом (30 мл). После осушки над Na2SO4 и упаривания растворителя при пониженном давлении получали чистый 2-[3-(2,6-дихлорофенилкарбамоил)фенил]пропионитрил в виде масла желтого цвета (1,929 г, 6,05 ммоль), выход (70%).
1H-ЯМР (DMSO-d6): δ 10,4 (уш.с, 1H, CONH); 8,10-8,25 (м, 2H); 7,80-7,55 (м, 5H); 4,02 (кв, 1H, J=7 Гц); 1,55 (д, 3H, J=7 Гц).
К раствору 2-[3-(2,6-дихлорофенилкарбомоил)фенил]пропионитрила (1,929 г, 6,05 ммоль) в 15 мл диоксана добавили 37% HCl (8 мл). Смесь перемешивали при 40°C на ночь. После охлаждения до комнатной температуры диоксан упаривали, добавляли к остатку холодную воду (10 мл) и EtOAc (15 мл). Две фазы встряхивали и разделяли, органический слой экстрагировали 1N NaOH (2×5 мл). К собранным основным водным экстрактам добавляли 37% HCl для осаждения требуемой кислоты. По окончании осаждения, после фильтрации получали чистую 2-[3-(2,6-дихлорофенилкарбомоил)фенил]пропионовую кислоту в виде твердого белого вещества (1,32 г, 3,93 ммоль). Выход 40%.
[α]D 25 (c=1, EtOH): -32° (30%), 1H-ЯМР (DMSO-d6): δ 10,4 (уш.с, 1H, CONH); 8,12-8,22 (м, 2H); 7,75-7,60 (м, 5H); 3,95 (кв, 1H, J=7 Гц); 1,50 (д, 3H, J=7 Гц).
В соответствии с такими же экспериментальными процедурами, и используя соответствующие коммерчески доступные производные анилина, были синтезированы следующие соединения:
(R)-2-[3-(2,6-диметилфенилкарбамоил)фенил]пропионовая кислота (IX)
[α]D 25 (c=1, EtOH): -32°, 1H-ЯМР (DMSO-d6): δ 9,75 (уш.с, 1H, CONH); 8,00-7,90 (м, 2H); 7,60-7,40 (м, 3H); 7,10 (с, 2H); 3,70 (кв, 1H, J=7 Гц); 2,15 (с, 6H, J=7 Гц); 1,35 (д, 3H, J=7 Гц).
(R)-2-[3-(3-хлоропиридин-2-илкарбамоил)фенил]пропионовая кислота (X)
[α]D 25 (c=1, EtOH): -28°, 1H-ЯМР (CDCl3): δ 8,70 (уш.с, 1H, CONH); 8,20 (д, 1H, J=9 Гц); 7,80-7,68 (м, 3H); 7,40-7,18 (м, 3H); 3,80 (кв, 1H, J=7 Гц); 1,58 (д, 3H, J=7 Гц).
(R)-2-{3-[(2-метокси)фенокси]фенил}пропионовая кислота (XI)
Реакция была осуществлена по следующим процедурам, описанным в Evans D. A. et al., Tetrahedron Letters, 1998, 39, 2937-2940.
К раствору 2-(3-гидроксифенил)пропионитрила (0,118 г, 0,80 ммоль), приготовленного, как описано ранее, в сухом CH2Cl2 (6 мл) последовательно добавляли молекулярные сита (4Е), CuOAc (0,145 мг, 0,80 ммоль) и пиридин (0,33 мл, 4,0 ммоль). После перемешивания в течение 20 мин добавляли коммерчески доступную 2-метоксифенилбороновую кислоту (0,243 г, 1,60 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь охлаждали до 0°C, добавляли 0,5N HCl и органическую фазу промывали (3×10 мл) 0,5N HCl. После осушки над Na2SO4 и упаривания растворителя при пониженном давлении получали остаток, который после флэш-хроматографии (элюирующая смесь n-гексан/EtOAc 9:1) давал 2-{3-[(2-матокси)фенокси)фенил]пропионитрил в виде масла желтоватого цвета (0,172 г, 0,68 ммоль). Выход 85%.
1H-ЯМР (CDCl3): δ 7,20-7,10 (м, 2H); 6,98-6,80 (м, 5H); 6,70 (д, 1H, J=7 Гц); 3,75 (с, 3H); 3,48 (кв, 1H, J=7 Гц); 1,45 (д, 3H, J=7 Гц).
К раствору 2-{3-[(2-метокси)фенокси)фенил]пропионитрила (0,17 г, 0,68 ммоль) в 5 мл диоксана добавляли 37% HCl (5 мл). Смесь перемешивали при 70°C на 4 ч. После охлаждения до комнатной температуры диоксан упаривали, добавляли к остатку холодную воду (10 мл) и этилацетат (10 мл). Две фазы встряхивали и разделяли, органическую фазу экстрагировали 1N NaOH (2×5 мл). К собранным основным водным экстрактам добавляли 37% HCl для осаждения требуемой кислоты. По окончании осаждения, после фильтрования получали чистую 2-{3-[(2-метокси)фенокси)фенил]пропионовую кислоту в виде воскообразного вещества белого цвета (0,166 г, 0,61 ммоль). Выход 90%.
[α]D 25 (c=1, EtOH): -41° (38%), 1H-ЯМР (CDCl3): δ 7,22-7,12 (м, 2H); 7,00-6,85 (м, 5H); 6,72 (д, 1H, J=7 Гц); 3,75 (с, 3H); 3,55 (кв, 1H, J=7 Гц); 1,50 (д, 3H, J=7 Гц).
(R)-2-[3-(2-хлорофениламино]фенил]пропионовая кислота (XII)
Из 2-(3-амино)фенилпропионитрила и по следующим описанным процедурам (Wolfe J. P. et al., J. Am. Chem. Soc., 1996, 118, 7215-7216, Wolfe J. P. et al., Tet. Lett., 1997, 38, 6359-6362, Wolfe J. P. et al., J. Org. Chem., 2000, 65, 1144-1157, Ferreira I. C. F. R. et al., Tetrahedron, 2003, 59, 975-981), была синтезирована 2-[3-(2-хлорофениламино)фенил]пропионовая кислота.
Смесь 2-бромохлоробензола (0,58 мл, 5,5 ммоль), 2-(3-амино)фенилпропионитрила (0,72 г, 5 ммоль), Pd(OAc)2 (3 мол.%), rac-BINAP (4 мол.%) и Cs2CO3 (2.28 г, 7 ммоль) в сухом толуоле (15 мл) заполняли в Ar атмосфере в пробирковидную колбу Шленка, и смесь нагревали при 100°C в течение 20 ч. После охлаждения до комнатной температуры добавляли воду (25 мл) и Et2O (25 мл). Фазы отделяли, и водную фазу экстрагировали Et2O (2×10 мл). После осушки над Na2SO4 и упаривания растворителя при пониженном давлении получали остаток, который после флэш-хроматографии давал 2-[3-(2-хлорофениламино)фенил]пропионитрил в виде бесцветного масла (0,64 г, 2,5 ммоль). Выход 50%.
1H-ЯМР (CDCl3): δ 7,22 (д, 1H, J=3 Гц); 7,09 (м, 1H); 7,00 (м, 1H); 6,72 (м, 2H); 6,64 (м, 2H); 6,57 (м, 1H); 4,15 (уш.с, 1H, NH); 3,75 (кв, 1H, J=7 Гц); 1,55 (д, 3H, J=7 Гц).
К раствору 2-[3-(2-хлорофениламино)фенил]пропионитрила (0,64 г, 2,5 ммоль) в диоксане (10 мл) добавляли 37% HCl (2 мл). Смесь перемешивали при 70°C на 4 ч. После охлаждения до комнатной температуры диоксан упаривали, и добавляли к остатку холодную воду (10 мл). Водную фазу нейтрализовали 2N NaOH и экстрагировали (3×10 мл) CHCl3. После осушки над Na2SO4 и упаривания растворителя при пониженном давлении получали чистую 2-[3-(2-хлорофениламино)фенил]пропионовую кислоту в виде порошка слегка белого цвета (0,67 г, 2,45 ммоль). Выход 98%.
[α]D 25 (c=1, MeOH): -42° (30%), 1H-ЯМР (DMSO-d6): δ 7,22 (д, 1H, J=3 Гц); 7,09 (м, 1H); 7,05 (м, 1H); 6,72 (м, 2H); 6,64 (м, 2H); 6,57 (м, 1H); 4,15 (уш.с, 1H, NH); 3,85 (кв, 1H, J=7 Гц); 1,62 (д, 3H, J=7 Гц).
В соответствии с такими же экспериментальными процедурами, и используя коммерчески доступный 2-бромоанизол в качестве исходного реагента, следующее вещество было синтезировано:
(R)-2-[3-(2-метоксифениламино]фенил]пропионовая кислота (XIII)
[α]D 25 (c=1, MeOH): -27°, 1H-ЯМР (DMSO-d6): δ 7,52 (д, 1H, J=7 Гц); 7,25 (м, 1H); 7,08 (м, 1H); 6,80 (м, 2H); 6,62 (м, 2H); 6,50 (м, 1H); 4,15 (уш.с, 1H, NH); 3,80 (с, 3H); 3,72 (кв, 1H, J=7 Гц); 1,52 (д, 3H, J=7 Гц).
В соответствии с теми же экспериментальными процедурами, и используя коммерчески доступный 2-бромопиридин в качестве исходного реагента, следующее соединение было получено:
(R)-2-[3-(2-пиридин-2-иламино]фенил]пропионовая кислота (XIV)
[α]D 25 (c=1, MeOH): -31°, 1H-ЯМР (DMSO-d6): δ 8,15 (уш.с, 1H, CONH); 7,50 (м, 1H); 7,15-6,98 (м, 3H); 6,90 (м, 1H); 6,82 (м, 2H); 6,75 (м, 1H); 3,55 (кв, 1H, J=7 Гц); 1,50 (д, 3H, J=7 Гц).
(R)-2-(3-оксазол-2-илфенил)пропионовая кислота (XV)
Реакция проводилась по следующей процедуре, описанной в Suzuki A. et al., Syn. Commun. 1981, 11, 513-519.
К раствору 2-(3-йодофенил)пропионитрила (0,6 г, 2,33 ммоль) в сухом THF (10 мл), были последовательно добавлены в атмосфере азота Pd(PPh3)4 (4% моль, 0,108 мг) и Na2CO3 (0,493 г, 4,66 ммоль). После перемешивания в течение 20 мин добавляли коммерчески доступную 1,3-оксазол-2-бороновую кислоту (0,289 г, 2,56 ммоль). Реакционную смесь кипятили в течение 4 ч. После охлаждения до комнатной температуры THF упаривали при пониженном давлении, EtOAc (10 мл) добавляли к сырому продукту, органическую фазу промывали водой (3×10 мл) и рассолом (3×10 мл). После осушки над Na2SO4 и упаривания растворителя получали остаток, который после флэш-хроматографии (элюирующая смесь n-гексан/EtOAc 8:2) давал 2-(3-оксазол-2-илфенил)пропионитрил в виде масла желтого цвета (0,360 г, 1,82 ммоль). Выход 78%.
1H-ЯМР (CDCl3): δ 8,09 (с, 1H); 7,98-7,93 (м, 1H); 7,70 (с, 1H); 7,45 (м, 2H); 7,25 (с, 1H); 3,85 (кв, 1H, J=7 Гц); 1,58 (д, 3H, J=7 Гц).
К раствору 2-(3-оксазол-2-илфенил)пропионитрила (0,360 г, 1,82 ммоль) в 5 мл диоксана добавляли 37% HCl (5 мл). Смесь перемешивали при 70°C на 4 ч. После охлаждения до комнатной температуры диоксан упаривали и добавляли к остатку холодную воду (10 мл) и EtOAc (10 мл). Две фазы встряхивали и разделяли, и органическую фазу экстрагировали 1N NaOH (2×5 мл). Смесь подкисляли до pH=1 2N HCl, сырой продукт экстрагировали CH2Cl2 (3×10 мл). Собранные органические экстракты осушили над Na2SO4, после упаривания растворителя при пониженном давлении получили чистую 2-(3-оксазол-2-илфенил)пропионовую кислоту в виде бесцветного масла (0,360 г, 1,66 ммоль). Выход 92%.
[α]D 25 (c=1, EtOH): -33° (38%), 1H-ЯМР (CDCl3): δ 8,07 (с, 1H); 7,95-7,90 (м, 1H); 7,70 (с, 1H); 7,44 (м, 2H); 7,23 (с, 1H); 3,82 (кв, 1H, J=7 Гц); 1,55 (д, 3H, J=7 Гц).
В соответствии с такими же экспериментальными процедурами, и используя 2-фуранбороновую кислоту в качестве исходного реагента, было синтезировано следующее соединение:
(R)-2-(3-фуран-2-илфенил)пропионовая кислота (XVI)
[α]D 25 (c=1, EtOH): -32°, 1H-ЯМР (CDCl3): δ 7,68-7,58 (м, 2H); 7,48 (с, 1H); 7,35-7,25 (м, 2H); 6,68 (д, 1H, J=4 Гц); 6,48 (дд, 1H, J1=4 Гц, J2=2 Гц); 3,80 (кв, 1H, J=7 Гц); 1,55 (д, 3H, J=7 Гц).
(R)-2-(2-оксо-1,2,3,4-тетрагидрохинолин-7-ил]пропионовая кислота (XVII)
К раствору 2-(3-аминофенил)пропионитрила (0,500 г, 3,38 ммоль), приготовленного, как описывалось ранее, в CH2Cl2 (8 мл) добавляли раствор Et3N (0,515 мл, 3,72 ммоль) и 3-хлоропропионил хлорида (0,355 мл, 3,72 ммоль) в CH2Cl2 (4 мл). Реакционную смесь перемешивали при кипячении в течение 5 ч. После охлаждения до комнатной температуры смесь разбавили CH2Cl2 (10 мл), органическую фазу промывали KH2PO4 буферным раствором (pH=5) (3×10 мл) и рассолом (2×10 мл). После осушки над Na2SO4 и упаривания растворителя при пониженном давлении получали чистый 2-[3-(3-хлоропропиониламино)фенил]пропионитрил в виде бесцветного масла (0,654 г, 2,77 ммоль). Выход 82%.
1H-ЯМР (CDCl3): δ 8,00 (уш.с, 1H, CONH); 7,50-7,46 (м, 2H); 7,20 (м, 1H); 7,05 (д, 1H, J=7 Гц); 3,95 (кв, 1H, J=7 Гц); 3,75 (м, 2H); 2,50 (м, 2H); 1,60 (д, 3H, J=7 Гц).
К раствору 2-[3-(3-хлоропропиониламино)фенил]пропионитрила (0,654 г, 2,77 ммоль) в CH2Cl2 (8 мл) при 0°C добавляли порциями AlCl3 (1,10 г, 8,31 ммоль). Реакционную смесь перемешивали 5 мин, затем кипятили с обратным холодильником 8 ч. После охлаждения до 0°C смесь промывали 6N HCl раствором (3×10 мл), водой (3×10 мл) и рассолом (2×10 мл). После осушки над Na2SO4 и упаривания растворителя получали сырой остаток, который после флэш-хроматографии (элюирующая смесь n-гексан/EtOAc 85:15) давал 2-(2-оксо-1,2,3,4-тетрагидрохинолин-7-ил]пропионитрил в виде масла желтого цвета (0,345 г, 1,72 ммоль). Выход 62%.
1H-ЯМР (CDCl3): δ 8,00 (уш.с, 1H, CONH); 7,46 (с, 1H); 7,18 (д, 1H, J=7 Гц); 7,05 (д, 1H, J=7 Гц); 3,90 (кв, 1H, J=7 Гц); 2,90 (м, 2H); 2,56 (м, 2H); 1,58 (д, 3H, J=7 Гц).
К раствору 2-(2-оксо-1,2,3,4-тетрагидрохинолин-7-ил]пропионитрила (0,345 г, 1,72 ммоль) в 5 мл диоксана добавляли 37% HCl (5 мл). Смесь перемешивали при 40°C в течение ночи. После охлаждения до комнатной температуры диоксан упаривали, добавляли к остатку холодную воду (10 мл) и EtOAc (15 мл). Две фазы встряхивали и разделяли, органическую фазу экстрагировали 1N NaOH (2×5 мл). К собранным основным водным экстрактам добавляли 37% HCl для осаждения требуемой кислоты. По окончании осаждения, после фильтрования получали чистую 2-(2-оксо-1,2,3,4-тетрагидрохинолин-7-ил]пропионовую кислоту в виде твердого вещества белого цвета (0,293 г, 1,34 ммоль). Выход 78%.
[α]D 25 (c=1, EtOH): -40° (35%), 1H-ЯМР (CDCl3): δ 8,02 (уш.с, 1H, CONH); 7,46 (с, 1H); 7,18 (д, 1H, J=7 Гц); 7,05 (д, 1H, J=7 Гц); 3,86 (кв, 1H, J=7 Гц); 2,90 (м, 2H); 2,56 (м, 2H); 1,55 (д, 3H, J=7 Гц).
(R)-2-[3-(бензолсульфонил))фенил]пропионовая кислота (XVIII)
Реакция была проведена по следующей процедуре, описанной в H. Suzuki et al., Tetrahedron Letters 1995, 36, 6239-6242.
К раствору 2-(3-йодофенил)пропионитрила (0,6 г, 2,33 ммоль) в DMF (8 мл) добавляли в атмосфере азота CuI (0,658 г, 3,45 ммоль) и коммерчески доступную натриевую соль бензолсульфоновой кислоты (0,612 г, 3,73 ммоль). Смесь перемешивали 6 ч при 110°C. Конверсию контролировали по ТСХ (тонкослойная хромотография, TLC). После охлаждения до комнатной температуры воду (15 мл) и Et2O (12 мл) добавляли к раствору, органическую фазу отделяли, промывали рассолом (3×10 мл) и осушили над Na2SO4. Удаление (упаривание) растворителя при пониженном давлении давало маслянистый остаток, который очищали хроматографически, используя n-гексан/EtOAc 9:1 для выделения 2-[3-бензолсульфонил)фенил]пропионитрила в виде масла желтоватого цвета (0,38 г, 1,40 ммоль). Выход 60%.
1H-ЯМР (CDCl3): δ 7,98-7,75 (м, 4H); 7,60-7,35 (м, 5H); 3,55 (кв, 1H, J=7 Гц); 1,55 (д, 3H, J=7 Гц).
К раствору 2-[3-бензолсульфонил)фенил]пропионитрила (0,38 г, 1,40 ммоль) в 5 мл диоксана добавляли 37% HCl (5 мл). Смесь перемешивали при 70°C в течение 4 ч. После охлаждения до комнатной температуры диоксан упаривали и добавляли к остатку холодную воду (10 мл) и этилацетат (10 мл). Две фазы встряхивали и разделяли, органическую фазу экстрагировали 1N NaOH (2×5 мл). К собранным основным водным экстрактам добавляли 37% HCl для осаждения требуемой кислоты. По окончании осаждения, после фильтрации получали чистую 2-[3-бензолсульфонил)фенил]пропионовую кислоту в виде твердого вещества белого цвета (0,324 г, 1,12 ммоль). Выход 80%.
[α]D 25 (c=1, EtOH): -29°, 1H-ЯМР (CDCl3): δ 7,96-7,75 (м, 4H); 7,62-7,38 (м, 5H); 3,50 (кв, 1H, J=7 Гц); 1,50 (д, 3H, J=7 Гц).
Синтез амидов формулы (I)
Пример 1
(R)-2-[3-(изобутирил)фенил]пропионамид
(R)-2-(3-изобутирилфенил)пропионовая кислота (I) (0,61 г, 2,78 ммоль) растворяли в SOCl2 (5 мл) и полученный раствор перемешивали при кипячении 3 ч. После охлаждения до комнатной температуры смесь упаривали при пониженном давлении; сырой ацилхлорид растворяли в сухом THF (5 мл) и охлаждали до 0-5°C. Сухой газообразный аммиак был в избытке пропущен через смесь при интенсивном перемешивании. Конверсия определяли по ТСХ; после полного исчезновения исходного реагента растворитель упаривали при пониженном давлении, остаток разбавляли CHCl3 (10 мл) и водой (10 мл); две фазы встряхивали и разделяли, органическую фазу промыли насыщенным раствором NaHCO3 (3×10 мл) и водой (2×10 мл), осушали над Na2SO4 и упаривали в вакууме, что давало чистый (R)-2-(3-изобутирилфенил)пропионамид (0,56 г, 2,58 ммоль) в виде бесцветного масла. Выход 93%.
[α]D 25 (c=1, EtOH): -35°, 1H-ЯМР (CDCl3): δ 7,90 (с, 1H); 7,86 (д, 1H, J=7 Гц); 7,52-7,45 (м, 2H); 5,50 (уш.с, 2H, CONH 2); 3,80 (кв, 1H, J=7 Гц); 3,45 (м, 1H); 1,50 (д, 3H, J=7 Гц); 1,1 (д, 6H, J=7 Гц).
В соответствии с такой же экспериментальной процедурой, и используя соответствующие 2-арилпропионовые кислоты, описанные выше, как исходные реагенты, были синтезированы следующие соединения:
Пример 2
(R)-2-[3-(циклопентанкарбонил)фенил]пропионамид
[α]D 25 (c=1, EtOH): -28°, 1H-ЯМР (CDCl3): δ 7,86 (с, 1H); 7,76 (д, 1H, J=7 Гц); 7,45-7,35 (м, 2H); 5,60-5,50 (уш.с, 2H, CONH 2); 3,75 (кв, 1H, J=7 Гц); 3,70 (м, 1H); 2,23 (м, 2H); 2,05 (м, 3H); 1,85 (м, 3H); 1,45 (д, 3H, J=7 Гц).
Пример 3
(R)-2-[(3-(фуран-2-карбонил)фенил]пропионамид
[α]D 25 (c=1, MeOH): -41°,1H-ЯМР (CDCl3): δ 8,10 (д, 1H, J=3 Гц); 7,86 (м, 1H); 7,82 (д, 1H, J=7 Гц); 7,64 (с, 1H); 7,49 (м, 2H); 7,41 (м, 1H); 5,80 (уш.с, 2H, CONH 2); 3,79 (кв, 1H, J=7 Гц); 1,41 (д, 3H, J=7 Гц).
Пример 4
(R)-2-[3-(2-бензофуран-2-карбонил)фенил]пропионамид
[α]D 25 (c=1, EtOH): -48°, 1H-ЯМР (CDCl3): δ 8,30 (с, 1H); 8,15 (д, 1H, J=8 Гц); 7,51 (д, 1H, J=8 Гц); 7,42 (д, 2H, J=8 Гц); 7,28 (т, 2H, J=8 Гц); 7,11 (т, 2H, J=8 Гц); 5,25 (уш.с, 2H, CONH 2); 3,65 (кв, 1H, J=7 Гц); 1,36 (д, 3H, J=7 Гц).
Пример 5
(R)-2-[3-(тиазол-2-илкарбонил)фенил]пропионамид
[α]D 25 (c=1, MeOH): -30°,1H-ЯМР (CDCl3): δ 8,40 (м, 2H); 8,08 (д, 1H, J=3 Гц); 7,75 (д, 1H, J=3 Гц); 7,63 (д, 1H, J=7 Гц); 7,51 (т, 1H, J=7 Гц); 5,55 (уш.с, 2H, CONH 2); 3,88 (кв, 1H, J=7 Гц); 1,63 (д, 3H, J=7 Гц).
Пример 6
(R)-2-[3-(1,3-оксазол-2-илкарбонил)фенил]пропионамид
[α]D 25 (c=1, EtOH): -39°, 1H-ЯМР (CDCl3): δ 8,45 (м, 2H); 7,90 (с, 1H); 7,68 (д, 1H, J=7 Гц); 7,50 (т, 1H, J=7 Гц); 7,38 (с, 1H); 5,66 (уш.с, 2H, CONH 2); 3,90 (кв, 1H, J=7 Гц); 1,56 (д, 3H, J=7 Гц).
Пример 7
3-((R)-1-карбамоилэтил)-N-(2,6-дихлорофенил)бензамид
[α]D 25 (c=1, EtOH): -27°, 1H-ЯМР (DMSO-d6): δ 10,4 (уш.с, 1H, CONH); 8,22-8,12 (м, 2H); 7,75-7,60 (м, 5H); 6,60 (уш.с, 2H, CONH2); 3,95 (кв, 1H, J=7 Гц); 1,50 (д, 3H, J=7 Гц).
Пример 8
3-((R)-1-карбамоилэтил)-N-(2,6-диметилфенил)бензамид
[α]D 25 (c=1, EtOH): -34°, 1H-ЯМР (DMSO-d6): δ 9,75 (уш.с, 1H, CONH); 8,00-7,90 (м, 2H); 7,60-7,40 (м, 3H); 7,10 (с, 2H); 5,80 (уш.с, 2H, CONH 2); 3,70 (кв, 1H, J=7 Гц); 2,15 (с, 6H, J=7 Гц); 1,35 (д, 3H, J=7 Гц).
Пример 9
3-((R)-1-карбамоилэтил)-N-(3-хлоропиридин-2-ил)бензамид
[α]D 25 (c=1, EtOH): -30°, 1H-ЯМР (CDCl3): δ 8,70 (уш.с, 1H, CONH); 8,20 (д, 1H, J=9 Гц); 7,80-7,68 (м, 3H); 7,40-7,18 (м, 3H); 6,12 (уш.с, 2H, CONH 2); 3,80 (кв, 1H, J=7 Гц); 1,58 (д, 3H, J=7 Гц).
Пример 10
(R)-2-[3-(2-метоксифенокси)фенил]пропионамид
[α]D 25 (c=1, EtOH): -38°, 1H-ЯМР (CDCl3): δ 7,22-7,12 (м, 2H); 7,00-6,85 (м, 5H); 6,72 (д, 1H, J=7 Гц); 5,50-5,20 (уш.с, 2H, CONH 2); 3,75 (с, 3H); 3,55 (кв, 1H, J=7 Гц); 1,50 (д, 3H, J=7 Гц).
Пример 11
(R)-2-[3-(2-хлорофениламино)фенил]пропионамид
[α]D 25 (c=1, MeOH): -37°, 1H-ЯМР (DMSO-d6): δ 7,22 (д, 1H, J=3 Гц); 7,09 (м, 1H); 7,05 (м, 1H); 6,72 (м, 2H); 6,64 (м, 2H); 6,57 (м, 1H); 5,60-5,35 (уш.с, 2H, CONH 2); 4,15 (уш.с, 1H, NH); 3,85 (кв, 1H, J=7 Гц); 1,62 (д, 3H, J=7 Гц).
Пример 12
(R)-2-[3-(2-метоксифениламино)фенил]пропионамид
[α]D 25 (c=1, MeOH): -31°, 1H-ЯМР (DMSO-d6): δ 7,50 (д, 1H, J=7 Гц); 7,28 (м, 1H); 7,10 (м, 1H); 6,78 (м, 2H); 6,60 (м, 2H); 6,50 (м, 1H); 5,58 (уш.с, 2H, CONH 2); 4,15 (уш.с, 1H, NH); 3,80 (с, 3H); 3,70 (кв, 1H, J=7 Гц); 1,50 (д, 3H, J=7 Гц).
Пример 13
(R)-2-[3-(пиридин-2-иламино)фенил]пропионамид
[α]D 25 (c=1, MeOH): -36°, 1H-ЯМР (DMSO-d6): δ 8,15 (уш.с, 1H, CONH); 7,50 (м, 1H); 7,15-6,98 (м, 3H); 6,88 (м, 1H); 6,82 (м, 2H); 6,75 (м, 1H); 5,58-5,38 (уш.с, 2H, CONH 2); 3,58 (кв, 1H, J=7 Гц); 1,52 (д, 3H, J=7 Гц).
Пример 14
(R)-2-(3-оксазол-2-ил)фенил]пропионамид
[α]D 25 (c=1, EtOH): -29°, 1H-ЯМР (CDCl3): δ 8,00 (с, 1H); 7,95-7,92 (м, 1H); 7,68 (с, 1H); 7,42 (м, 2H); 7,20 (с, 1H); 5,20 (уш.с, 2H, CONH 2); 3,60 (кв, 1H, J=7 Гц); 1,55 (д, 3H, J=7 Гц).
Пример 15
(R)-2-(3-фуран-2-ил)фенил]пропионамид
[α]D 25 (c=1, EtOH): -36°, 1H-ЯМР (CDCl3): δ 7,68-7,58 (м, 2H); 7,48 (с, 1H); 7,35-7,25 (м, 2H); 6,70 (д, 1H, J=4 Гц); 6,50 (дд, 1H, J1=4 Гц, J2=2 Гц); 5,35 (уш.с, 2H, CONH 2); 3,65 (кв, 1H, J=7 Гц); 1,58 (д, 3H, J=7 Гц).
Пример 16
(R)-2-(оксо-1,2,3,4-тетрагидрохинолин-7-ил)пропионамид
[α]D 25 (c=1, EtOH): -43° 1H-ЯМР (CDCl3): δ 8,00 (уш.с, 1H, CONH); 7,46 (с, 1H); 7,18 (д, 1H, J=7 Гц); 7,05 (д, 1H, J=7 Гц); 5,70-5,58 (уш.с, 2H, CONH 2); 3,90 (кв, 1H, J=7 Гц); 2,90 (м, 2H); 2,56 (м, 2H); 1,58 (д, 3H, J=7 Гц).
Пример 17
(R)-2-(3-бензолсульфонилфенил)пропионамид
[α]D 25 (c=1, EtOH): -36°, 1H-ЯМР (CDCl3): δ 7,96-7,75 (м, 4H); 7,62-7,38 (м, 5H); 5,65 (уш.с, 2H, CONH 2); 3,50 (кв, 1H, J=7 Гц); 1,50 (д, 3H, J=7 Гц).
Пример 18
2-(3-ацетиламино)фенилпропионамид
К раствору 2-(3-амино)фенилпропионамида (0,2 г, 1,26 ммоль) (приготовленного из 2-(3-амино)фенилпропионитрила, как описано в Erdelmeier I. et al., J. Org. Chem., 2000, 65, 8152-8157) в 10 мл сухого CH2Cl2 добавляли триэтиламин (0,19 мл, 1,39 ммоль) и ацетилхлорид (90 мкл, 1,26 ммоль). Смесь перемешали при комнатной температуре в течение 4 ч, промыли водой H2O (3×15 мл) и осушили над Na2SO4. После упаривания растворителя при пониженном давлении получали остаток, который после очистки посредством флэш-хроматографии давал 2-(3-ацетиламино)фенилпропионамид в виде прозрачного масла (0,202 г, 1,01 ммоль). Выход 80%.
1H-ЯМР (CDCl3): δ 8,59 (уш.с, 1H, CONH); 7,46 (м, 2H); 7,20 (т, 1H, J=8 Гц); 6,97 (д, 1H, J=8 Гц); 5,55 (уш.с, 2H, CONH 2); 3,53 (кв, 1H, J=7 Гц); 2,09 (с, 3H); 1,43 (д, 3H, J=7 Гц).
В соответствии с той же экспериментальной процедурой, и используя бензоилхлорид в качестве исходного реагента, было синтезировано следующее соединение:
Пример 19
2-(3-бензоиламино)фенилпропионамид
1H-ЯМР (CDCl3): δ 8,59 (уш.с, 1H, CONH); 8,15 (м, 2H); 7,62 (м, 1H); 7,45 (м, 2H); 7,40 (м, 2H); 7,22 (т, 1H, J=8 Гц); 6,94 (д, 1H, J=8 Гц); 5,55 (уш.с, 2H, CONH 2); 3,53 (кв, 1H, J=7 Гц); 1,43 (д, 3H, J=7 Гц).
Пример 20
N-[(R)-2-(3-циклопентанкарбонилфенил)пропионил]метансульфонамид
Реакция была проведена, как описано в Uehling D.E. et al., J. Med. Chem., 2002, 45(3), 567-583.
1,1'-Карбонилдиимидазол (0,5 г, 3,06 ммоль) добавляли к раствору (R)-2-[3-циклопентаноил)фенил]пропионовой кислоты (II) (0,68 г, 2,78 ммоль) в сухом CH2Cl2 (8 мл) и полученную смесь перемешивали при комнатной температуре в течение 90 мин. Добавляли метансульфонамид (0,26 г, 2,78 ммоль) и DBU (0,43 мл, 2,78 ммоль), смесь перемешивали последующие 16 ч при комнатной температуре. Органическую фазу промывали 0,5N HCl (2×10 мл), 5% NaH2PO4 (3×10 мл) и водой (2×10 мл). После осушки Na2SO4 растворитель упаривали в вакууме и сырой продукт очищали флэш-хроматографией (элюирующая смесь CH2Cl2/MeOH 95:5). Чистый N-[(R)-2-(3-циклопентанкарбонилфенил)пропионил]метансульфонамид 22 выделяли в виде бесцветного масла (0,67 г, 2,09 ммоль). Выход 79%.
[α]D 25 (c=1, EtOH): -48°, 1H-ЯМР (CDCl3): δ 7,80 (м, 2H); 7,42 (м, 2H); 3,68 (м, 2H); 3,15 (с, 3H); 1,88 (м, 4H); 1,62 (м, 4H); 1,43 (д, 3H, J=7 Гц).
В соответствии с такой же экспериментальной процедурой, и используя соответствующие вышеописанные арилпропионовые кислоты, были синтезированы следующие соединения:
Пример 21
N-{[(R)-2-[3-(фуран-2-карбонил)фенил]пропионил}метансульфонамид
[α]D 25 (c=1, EtOH): -23,5°. 1H-ЯМР (CDCl3): δ 7,95 (м, 1H); 7,85 (с, 1H); 7,71 (с, 1H); 7,50 (м, 2H); 7,28 (д, 1H, J=2 Гц); 6,60 (д, 1H, J=2 Гц); 3,82 (кв, 1H, J=7 Гц); 3,20 (с, 3H); 1,55 (д, 3H, J=7 Гц).
Пример 22
N-{[(R)-2-[3-(5-метилфуран-2-карбонил)фенил]пропионил}метансульфонамид
[α]D 25 (c=1, EtOH): -15°, 1H-ЯМР (CDCl3): δ 7,95 (м, 1H); 7,84 (м, 2H); 7,48 (уш.с, 1H+CONH); 7,10 (д, 1H, J=2 Гц); 6,21 (д, 1H, J=2 Гц); 3,80 (кв, 1H, J=7 Гц); 3,25 (с, 3H); 2,42 (с, 3H); 1,60 (д, 3H, J=7 Гц).
Пример 23
N-{[(R)-2-[3-(тиофен-2-карбонил)фенил]пропионил}метансульфонамид
[α]D 25 (c=1, EtOH): -37°, 1H-ЯМР (CDCl3): δ 7,80 (м, 1H); 7,71 (м, 2H); 7,58 (м, 1H); 7,40 (м, 2H); 7,10 (м, 1H); 3,75 (кв, 1H, J=7 Гц); 3,18 (с, 3H); 1,54 (д, 3H, J=7 Гц).
Пример 24
N-{[(R)-2-[3-(бензофуран-2-карбонил)фенил]пропионил}метансульфонамид
[α]D 25 (c=1, EtOH): -62,5°, 1H-ЯМР (CDCl3): δ 8,05 (м, 1H); 7,95 (с, 1H); 7,75 (м, 1H); 7,69 (м, 1H); 7,55 (м, 4H); 7,30 (м, 1H); 3,85 (кв, 1H, J=7 Гц); 3,29 (с, 3H); 1,65 (д, 3H, J=7 Гц).
Пример 25
N-{[(R)-2-[3-(оксазол-2-кабонил)фенил]пропионил}метансульфонамид
[α]D 25 (c=1, EtOH): -83°, 1H-ЯМР (CDCl3): δ 8,48 (м, 1H); 8,35 (с, 1H); 8,05 (уш.с, 1H, CONH); 7,95 (с, 1H); 7,66 (м, 2H); 7,40 (с, 1H); 3,82 (кв, 1H, J=7 Гц); 3,25 (с, 3H); 1,60 (д, 3H, J=7 Гц).
Пример 26
(R)-2-[3-(фуран-2-карбонил)фенил]-N-пирид-2-илпропионамид
Тионилхлорид (0,2 мл, 2,7 ммоль) добавляли к раствору (R)-2-[3-(2-фуран-2-карбонил)фенил]пропионовой кислоты (V) (0,065 г, 0,27 ммоль) в сухом CH2Cl2 (5 мл), и полученный раствор кипятили в течение 2 ч. После охлаждения до комнатной температуры толуол и тионилхлорид удаляли упариванием в вакууме и остаток растворяли в CH2Cl2 (2 мл); 2-аминопиридин (0,05 г, 0,54 ммоль) добавляли и раствор перемешивали в течение ночи при комнатной температуре. Органический раствор промывали водой (2×10 мл), после осушки Na2SO4 растворитель удаляли упариванием в вакууме, сырой продукт очищали хроматографически на силикагеле (элюирующая смесь n-гексан/EtOAc 8:2), что давало чистый 28 в виде бесцветного масла (0,07 г, 0,22 ммоль). Выход 80%.
[α]D 25 (c=0,6, MeOH): -69°, 1H-ЯМР (CDCl3): δ 8,22 (м, 2H); 8,00 (с, 1H); 7,88 (м, 2H); 7,80 (уш.с, 1H, CONH); 7,70 (с, 2H); 7,61 (м, 1H); 7,52 (м, 1H); 7,00 (м, 1H); 6,62 (м, 1H); 3,82 (кв, 1H, J=7 Гц); 1,65 (д, 3H, J=7 Гц).
По той же экспериментальной методике, и используя соответствующие 2-арилпропионовые кислоты и амин, были синтезированы следующие соединения:
Пример 27
(R)-2-[3-(фуран-2-карбонил)фенил]-N-(2H-тиазол-2-ил)пропионамид
[α]D 25 (c=0,5, MeOH): -7°, 1H-ЯМР (CDCl3): δ 8,05 (с, 1H); 7,90 (м, 1H); 7,75 (с, 1H); 7,60 (м, 1H); 7,52 (м, 2H); 7,22 (д, 1H, J=2 Гц); 7,02 (д, 1H, J=2 Гц); 6,68 (д, 1H, J=2 Гц); 3,95 (кв, 1H, J=7 Гц); 1,70 (д, 3H, J=7 Гц).
Пример 28
(R)-2-[3-(фуран-2-карбонил)фенил]-N-(4-трифторметил-2H-тиазол-2-ил)пропионамид
Реагент -амин - 2-амино-4-трифторметилтиазол получали, как описано в Moazzam M. et al., Indian J. Chem., 1988, 27B(11), 1051-1053.
[α]D 25 (c=0,6, MeOH): -11°, 1H-ЯМР (CDCl3): δ 9,35 (уш.с, 1H, CONH); 7,95 (м, 2H); 7,75 (с, 1H); 7,58-7,39 (м, 2H); 7,30 (с, 1H); 7,25 (с, 1H); 6,55 (с, 1H); 3,96 (кв, 1H, J=7 Гц); 1,65 (д, 3H, J=7 Гц).
По той же экспериментальной методике, и используя арилпропионовую кислоту VI и амин - 2-амино-4-трифторметилтиазол, следующее соединение было получено:
Пример 29
(R)-2-[3-(бензофуран-2-карбонил)фенил]-N-(4-трифторметил-2H-тиазол-2-ил)пропионамид
[α]D 25 (c=1, EtOH): -55°, 1H-ЯМР (CDCl3): δ 8,85 (уш.с, 1H, CONH); 8,15 (м, 1H); 8,05 (с, 1H); 7,78 (д, 1H, J=7 Гц); 7,65-7,58 (м, 5H); 7,40 (с, 1H); 7,35 (т, 1H, J=7 Гц); 4,05 (кв, 1H, J=7 Гц); 1,80 (д, 3H, J=7 Гц).
По той же экспериментальной методике, и используя арилпропионовую кислоту II и амин - 2-аминопиридин, было синтезировано следующее соединение:
Пример 30
(R)-2-(3-циклопентанкарбонилфенил)-N-пиридин-2-илпропионамид
[α]D 25 (c=1, EtOH): -55°, 1H-ЯМР (CDCl3): δ 8,70 (уш.с, 1H, CONH); 8,10 (с, 1H); 7,98 (д, 1H, J=3 Гц); 7,84 (м, 1H); 7,80 (д, 1H, J=7 Гц); 7,45 (д, 1H, J=7 Гц); 7,37 (м, 1H); 7,10 (д, 1H, J=3 Гц); 6,95 (м, 1H); 3,75 (кв, 1H, J=7 Гц); 3,70 (м, 1H); 2,20 (с, 2H); 2,0 (м, 3H); 1,80 (м, 3H); 1,55 (д, 3H, J=7 Гц).
Пример 31
(R)-2-[3-(фуран-2-карбонил)фенил]-N-гидроксипроптонамид
Тионилхлорид (1,6 мл, 27 ммоль) добавляли к раствору (R)-2-[3-(2-фураноил)фенил]пропионовой кислоты (V) (0,53 г, 2,15 ммоль) в сухом толуоле (10 мл), полученный раствор кипятили в течение 3 ч. После охлаждения до комнатной температуры толуол и тионилхлорид были удалены упариванием в вакууме, остаток растворяли в сухом CH2Cl2 (30 мл) и прикапывали к раствору гидроксиламин солянокислый (0,179 г, 2,57 ммоль) и триэтиламин (0,71 мл, 5,14 ммоль) в сухом CH2Cl2 (10 мл). Полученный раствор перемешивали в течение ночи при комнатной температуре. Органический раствор разбавляли 1N HCl (20 мл), после разделения фаз органическую фазу промывали водой (2×20 мл). После осушки Na2SO4 растворитель упаривали в вакууме, и сырой продукт очищали хроматографически (элюирующая смесь CHCl3/CH3OH 95:5), что давало чистый 33 в виде масла желтоватого цвета (0,65 г, 2,53 ммоль). Выход 85%.
[α]D 25 (c=1, MeOH): -44°, 1H-ЯМР (CDCl3): δ 7,92 (м, 2H); 7,75 (с, 1H); 7,57 (м, 1H); 7,50 (т, 1H, J=7 Гц); 7,25 (д, 1H, J=2 Гц); 6,61 (м, 1H); 3,85 (кв, 1H, J=7 Гц); 1,95 (уш.с, 1H, NHOH); 1,62 (д, 3H, J=7 Гц).
По той же экспериментальной методике, и используя арилпропионовую кислоту IV, было синтезировано следующее соединение:
Пример 32
(R)-2-[3-(тиазол-2-карбонил)фенил]-N-гидроксипропионамид
[α]D 25 (c=1, MeOH): -28°, 1H-ЯМР (CDCl3): δ 8,44 (м, 2H); 8,12 (д, 1H, J=3 Гц); 7,73 (д, 1H, J=2 Гц); 7,65 (д, 1H, J=7 Гц); 7,50 (т, 1H, J=7 Гц); 3,87 (кв, 1H, J=7 Гц); 1,90 (уш.с, 1H, NHOH); 1,70 (д, 3H, J=7 Гц).
Пример 33
2-{(R)-2-[3-(фуран-2-карбонил)фенил]пропиониламино}пропионовая кислота
К раствору (R)-2-[3-(2-фураноил)фенил]пропионовой кислоты (V) (2 г, 8,2 ммоль) в диоксане (5 мл) добавляли тионилхлорид (0,92 мл, 12,3 ммоль), полученный раствор кипятили в течение 3 ч. После охлаждения до комнатной температуры растворитель упаривали, сырой ацилхлорид растворяли в DMF (5 мл) при 0°C, DCC (1,69 г, 8,2 ммоль) и HOBT (1,01 г, 7,5 ммоль) добавляли при перемешивании. Через 30 мин раствор метилового эфира D,L-аланингидрохлорида (1,08 г, 7,5 ммоль) и триэтиламина (1,01 мл) в DMF (2 мл) добавляли. Полученную смесь перемешивали в течение 2 ч при 0°C и в течение ночи при комнатной температуре. Осадившийся DCU отфильтровывали; фильтрат разбавляли EtOAc (15 мл), органическую фазу промывали 10% лимонно-кислотным буфером (2×10 мл), насыщенным раствором NaHCO3 (2×10 мл) и далее рассолом (10 мл). После осушки Na2SO4, растворитель упаривали, что давало сырой продукт, который суспендировали в n-гексане (20 мл) и перемешивали в течение ночи при комнатной температуре. Метиловый эфир 2-[(R)-2-[3-(фуран-2-карбонил)фенил]пропиониламино]пропионовой кислоты выделяли фильтрованием в виде порошка белого цвета (1,66 г, 5,7 ммоль). Выход 69%. К раствору полученного метилового эфира в диоксане (3 мл) добавляли 1N NaOH (5,7 мл), смесь перемешивали в течение ночи при комнатной температуре. Смесь лед/вода (40 мл) добавляли, полученную смесь подкисляли концентрированной H2SO4 до pH 2. Водную фазу экстрагировали CH2Cl2 (4×15 мл), собранные органические экстракты промывали рассолом (15 мл), осушали Na2SO4 и упаривали в вакууме, что давало маслянистый остаток. 37 выделяли кристаллизацией из диэтилового эфира (10 мл) в виде твердого вещества белого цвета (0,72 г, 2,28 ммоль). Выход 40%.
[α]D 25 (c=1, MeOH): -21°, 1H-ЯМР (CDCl3) δ 7,86 (м, 1H), 7,80 (д, 1H, J=7 Гц), 7,64 (с, 1H); 7,47 (м, 1H); 7,35 (м, 1H); 7,16 (д, 1H, J=7 Гц); 6,53 (м, 1H); 5,95 (уш.с, 1H, CONH); 4,50 (кв, 1H, J=7 Гц); 3,65 (кв, 1H, J=7 Гц); 1,53 (д, 3H, J=7 Гц), 1,35 (д, 3H, J=7 Гц).
По той же экспериментальной методике, и используя метиловый эфир глицингидрохлорида, было синтезировано следующее соединение:
Пример 34
2-{(R)-2-[3-(фуран-2-карбонил)фенил]пропиониламино}уксусная кислота
[α]D 25 (c=1, MeOH): -13,5°, 1H-ЯМР (CDCl3) δ 7,80 (м, 1H), 7,82 (д, 1H, J=7 Гц), 7,64 (с, 1H); 7,47 (м, 1H); 7,33 (м, 1H); 7,15 (д, 1H, J=7 Гц); 6,51 (м, 1H); 5,90 (уш.с, 1H, CONH); 4,05 (с, 2H); 3,61 (кв, 1H, J=7 Гц); 1,53 (д, 3H, J=7 Гц).
Соединения, не обладающие активностью (ингибирующей), направленной на PMNs C5a индуцированный хемотаксис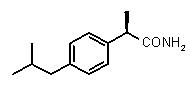
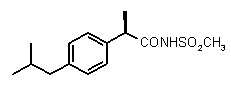
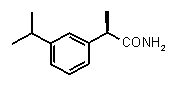
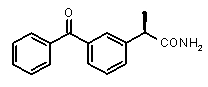
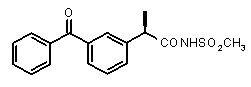
Соединения, обладающие активностью, (ингибирующей) направленной на PMNs C5a индуцированный хемотаксис 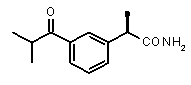
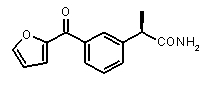
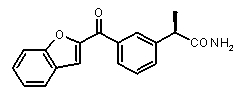
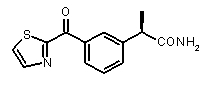
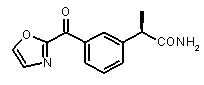
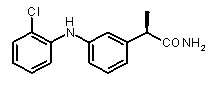
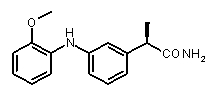
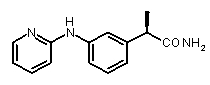
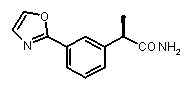
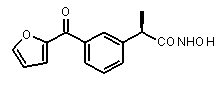
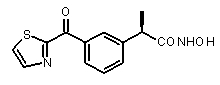
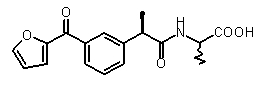
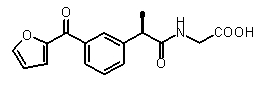
b концентрация препарата: 10-8 M
| название | год | авторы | номер документа |
|---|---|---|---|
| (R)-АРИЛАЛКИЛАМИНОПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2006 |
|
RU2458051C2 |
| ЧЕТВЕРТИЧНЫЕ АММОНИЕВЫЕ СОЛИ ОМЕГА-АМИНОАЛКИЛАМИДОВ R-2-АРИЛПРОПИОНОВЫХ КИСЛОТ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ | 2002 |
|
RU2291857C2 |
| (R)-4-(ГЕТЕРОАРИЛ)ФЕНИЛЭТИЛЬНЫЕ ПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2008 |
|
RU2475486C2 |
| 2-АРИЛПРОПИОНОВЫЕ КИСЛОТЫ И ПРОИЗВОДНЫЕ, И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ УКАЗАННЫЕ СОЕДИНЕНИЯ | 2009 |
|
RU2520212C2 |
| ОМЕГА-АМИНОАЛКИЛАМИДЫ R-2-АРИЛПРОПИОНОВЫХ КИСЛОТ В КАЧЕСТВЕ ИНГИБИТОРОВ ХЕМОТАКСИСА ПОЛИМОРФНОЯДЕРНЫХ И ОДНОЯДЕРНЫХ КЛЕТОК, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2272024C2 |
| АМИДИНЫ И ИХ ПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2004 |
|
RU2375346C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ ДИПЕПТИДИЛПЕПТИДАЗЫ IV | 2004 |
|
RU2353617C2 |
| 3-Имино-N-арил-2-хлорофуро[2,3-b]пиридин-2-карбоксамиды в качестве антидотов 2,4-Д на подсолнечнике и способ их получения | 2024 |
|
RU2829740C1 |
| (2R)-2-[(4-СУЛЬФОНИЛ)АМИНОФЕНИЛ]ПРОПАНАМИДЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2007 |
|
RU2457201C2 |
| СУЛЬФОНОВЫЕ КИСЛОТЫ, ПРОИЗВОДНЫЕ УКАЗАННЫХ КИСЛОТ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2004 |
|
RU2345063C2 |
Настоящее изобретение относится к производным (R)-2-арилпропионамидам общей формулы I, в которой Ar - это фенильная группа, замещенная в 3 (мета) положении группой R1, выбираемой из: линейного или разветвленного C1-C8-алканоила, C3-C6-цнклоалканоила, гетероарилкарбонила, С1-С6-алкиламинокарбонила, ариламинокарбонила, С1-C6-алкиламино, C1-С6-ациламино, ариламино, бензоиламино, арилокси, гетероарила, C1-С6-алкоксикарбонила, С6-арилоксикарбонила, C1-C8-алкансульфонила, арилсульфонила, или 3,4-дигидро-1H-хинолил-2-он; R выбирается из: -Н, ОН; - гетероарильная группа выбирается из: пиридина, пиримидина, пиррола, тиофена, фурана, индола, тиазола, оксазола; - α или β карбоксиалкильный остаток может состоять из прямого или разветвленного С1-С6-алкила, С3-С6-циклоалкила, необязательно, замещенных другой карбоксильной (СООН) группой; - остаток с формулой SO2Rd, в которой Rd - это C1-С6-алкил, С3-С6-циклоалкил, С2-С6-алкенилили пиридил, при условии, что соединения формулы (I) не являются следующими соединениями: (R)-2-(3-феноксифенил)-пропаноилфенилглицин; (R)-2-(3-феноксифенил)-пропаноилглицин; (R)-2-[(3′-ацетил)фенил]-N-(4″-пиримидил)пропионамид. Также изобретение относится к способу получения соединения формулы I и применению соединения формулы I для приготовления медикаментов для лечения заболеваний, включающих С5а индуцированный хемотаксис PMNs человека. Технический результат: получены новые производные (R)-2-арилпропионамида, обладающие полезными биологическими свойствами. 5 н. и 4 з.п. ф-лы, 2 ил., 2 табл.
1. (R)-2-арилпропионамиды с общей формулой (I)
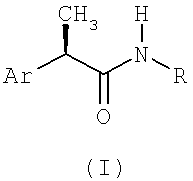
в которой Ar - это фенильная группа, замещенная в 3 (мета)положении группой R1, выбираемой из:
линейного или разветвленного C1-C8-алканоила, С3-С6-циклоалканоила, гетероарилкарбонила, C1-С6-алкиламинокарбонила, ариламинокарбонила, С1-С6-алкиламино, С1-С6-ациламино, ариламино, бензоиламино, арилокси, гетероарила, C1-С6-алкоксикарбонила, С6-арилоксикарбонила, C1-C8-алкансульфонила, арилсульфонила, или 3,4-дигидро-1Н-хинолил-2-он;
R выбирается из Н, ОН;
гетероарильная группа выбирается из пиридина, пиримидина, пиррола, тиофена, фурана, индола, тиазола, оксазола;
α или β карбоксиалкильный остаток может состоять из прямого или разветвленного С1-С6-алкила, С3-С6-циклоалкила, необязательно замещенных другой карбоксильной (СООН) группой;
остаток с формулой SO2Rd, в которой Rd - это С1-С6-алкил, С3-С6-циклоалкил, С2-С6-алкенил или пиридил,
при условии, что соединения формулы (I) не являются следующими соединениями:
(R)-2-(3-феноксифенил)-пропаноил-фенилглицин;
(R)-2-(3-феноксифенил)-пропаноил-глицин;
(R)-2-[(3′-ацетил)фенил]-N-(4″-пиримидил)пропионамид.
2. Соединения по п.1, в которых
Ar - это фенильная группа, замещенная в положении 3 (мета)группой R1, выбираемой из:
линейного или разветвленного C1-C8-алканоила; 2-фурила, 2-оксазолила, 3-изоксазолила, 2-бензоксазолила, 3-бензоизоксазолила, 2-тиазолила, 2-пиридила; фуранкарбонила; бензофуранкарбонила; тиофенкарбонила; пиридинкарбонила; С1-С6-ациламино; бензоиламино; арилокси; ариламино, или 3,4-дигидро-1H-хинолил-2-он;
R выбирается из Н, ОН; 2-пиридила, 2-тиазолила;
карбоксиалкильная группа может состоять из прямого или разветвленного С1-С6-алкила;
остаток с формулой SO2Rd, в которой Rd - это C1-С6-алкил.
3. Соединения по п.1 или 2, выбранные из:
(R)-2-(3-изобутирилфенил)пропионамида,
(R)-2-(3-циклопентанкарбонилфенил)пропионамида,
(R)-2-[(3-(фуран-2-карбонил)фенил]пропионамида,
(R)-2-[(3-(тиазол-2-карбонил)фенил]пропионамида,
(R)-2-[(3-(оксазол-2-карбонил)фенил]пропионамида,
(R)-2-[(3-(бензофуран-2-карбонил)фенил]пропионамида,
3-((R)-1-карбамоилэтил)-N-(2,6-дихлорофенил)бензамида,
3-((R)-1-карбамоилэтил)-N-(2,6-диметилфенил)бензамида,
3-((R)-1-карбамоилэтил)-N-(3-хлоропиридин-2-ил)бензамида,
(R)-2-[3-(2-метоксифенокси)фенил)пропионамида,
(R)-2-[3-(2-хлорофениламино)фенил]пропионамида,
(R)-2-[3-(2-метоксифениламино)фенил]пропионамида,
(R)-2-[3-(пиридин-2-иламино)фенил]пропионамида,
(R)-2-(3-оксазол-2-ил)фенил]пропионамида,
(R)-2-[(3-фуран-2-ил)фенил]пропионамида,
(R)-2-(оксо-1,2,3,4-тетрагидрохинолин-7-ил)пропионамида,
(R)-2-(3-бензолсульфонилфенил)пропионамида,
2-(3-ацетиламинофенил)пропионамида,
2-(3-бензоиламинофенил)пропионамида,
N-[(R)-2-(3-циклопентанкарбонилфенил)пропионил]метансульфонамида,
N-{(R)-2-[3-(фуран-2-карбонил)фенил]пропионил}метансульфонамида,
N-{(R)-2-[3-(5-метилфуран-2-карбонил)фенил]пропионил}метансульфонамида,
N-{(R)-2-[(3-(тиофен-2-карбонил)фенил]пропионил}метансульфонамида,
N-{(R)-2-[(3-(бензофуран-2-карбонил)фенил]пропионил)метансульфонамида,
N-{(R)-2-[(3-(оксазол-2-карбонил)фенил]пропионил}метансульфонамида,
(R)-2-[3-(фуран-2-карбонил)фенил]-N-пирид-2-илпропионамида,
(R)-2-[3-(фуран-2-карбонил)фенил]-N-(2Н-тиазол-2-ил)пропионамида,
(R)-2-[3-(фуран-2-карбонил)фенил]-N-(4-трифторметил-2Н-тиазол-2-ил)пропионамида,
(R)-2-[(3-(бензофуран-2-карбонил)фенил]-N-(4-трифторметил-2Н-тиазол-2-ил)пропионамида,
(R)-2-(3-циклопентанкарбонилфенил)-N-пирид-3-илпропионамида,
(R)-2-[3-(фуран-2-карбонил)фенил]-N-гидроксипропионамида,
(R)-2-[3-(тиазол-2-карбонил)фенил]-N-гидроксипропионамида,
2-{(R)-2-[3-(фуран-2-карбонил)фенил]пропиониламино}пропионовой кислоты,
2-{(R)-2-[3-(фуран-2-карбонил)фенил]пропиониламино}уксусной кислоты.
4. Способ получения соединений формулы (I) по п.1, включающий реакцию соединения формулы (II)
в которой Ar - фенильная группа, замещенная по 3 (мета)положению посредством R1, в которой R1 аналогичен таковому в п.1, с амином формулы NHR, где R аналогичен таковому в п.1.
5. Соединения по любому из пп.1 и 2 для использования в качестве медикаментов, ингибирующих С5а индуцированную PNM миграцию.
6. Соединения по п.3 для использования в качестве медикаментов, ингибирующих С5а индуцированную PNM миграцию.
7. Применение соединений по пп.1-3 для приготовления медикаментов для лечения заболеваний, включающих С5а индуцированный хемотаксис PMNs человека.
8. Применение соединений по любому из пп.1-3 для приготовления медикаментов для лечения сепсиса.
9. Применение соединений по любому из пп.1-3 для приготовления медикаментов для лечения псориаза, пузырчатки, ревматоидного артрита, неспецифического язвенного колита, синдрома острой дыхательной недостаточности, идиопатического фиброза, муковисцидоза, ХОЗЛ, гломерулонефрита и в предупреждении и лечении поражений, вызванных ишемией и реперфузией.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| WO 00/24710 А, 04.05.2000 | |||
| ЛЕВИТ Г.Л | |||
| и др | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ЖОрХ, 1998, т.34, вып.3, с.378-382. | |||

