Область изобретения
Настоящее изобретение относится к новому классу (R)-4-(гетероарил)фенилпропионовых производных, которые можно использовать для ингибирования хемотактической активации, индуцированной фракцией C5a комплемента. Указанные соединения можно использовать для лечения патологий, зависимых от хемотактической активации нейтрофилов и моноцитов, индуцированной фракцией C5a комплемента. В частности, соединения по данному изобретению можно использовать для лечения аутоиммунной гемолитической анемии (AIHA), псориаза, буллезного пемфигоида, ревматоидного артрита, язвенного колита, острого респираторного дистресс-синдрома, идиопатического фиброза, гломерулонефрита и для предотвращения и лечения повреждения, вызванного ишемией и реперфузией.
Уровень техники
В качестве ответной реакции на иммунологические и инфекционные события активация системы комплемента опосредует амплификацию воспалительной ответной реакции как за счет непосредственного мембранного действия, так и за счет высвобождения серии пептидных фрагментов, обычно известных как анафилотоксины, образующихся при ферментативном расщеплении фракций C3, C4 и C5 комплемента. Данные пептиды включают C3a и C4a, оба из которых состоят из 77 аминокислот; в свою очередь, C5-конвертаза отщепляет фракцию С5 комплемента с образованием гликопротеина С5а, состоящего из 74 аминокислот.
Пептидный фрагмент C5a комплемента был определен как «полный» провоспалительный медиатор благодаря его хемотактической и воспалительной активности. Действительно другие воспалительные медиаторы, такие как выборочные хемокины (например, IL-8, MCP-1 и RANTES), являются высокоселективными по отношению к самопритягиваемым клеткам, тогда как другие, такие как гистамин и брадикинин, являются лишь слабыми хемотактическими агентами.
Убедительные доказательства поддерживают предположение об участии C5a, in vivo, в ряде патологических состояний, включая ишемию/реперфузию, аутоиммунный дерматит, мембранно-пролиферативный идиопатический гломерулонефрит, невосприимчивость дыхательных путей и хронические воспалительные заболевания, ARDS и CODP, болезнь Альцгеймера, ювенильный ревматоидный артрит (N.P. Gerard, Ann. Rev. Immunol., 12, 755, 1994).
В частности, присутствие повышенных уровней анафилотоксинов C3a и C5a является одним из ряда указаний на гиперактивность системы комплемента у пациентов при ревматоидном артрите (RA). В опубликованной недавно статье (E.P. Grant, J. Exp. Med., 196(11), 1461, 2002) сообщалось, что генетическое устранение C5aR полностью защищает мышей от артрита, индуцированного антиколлагеновыми антителами, указывая на центральную роль C5a-зависимого задействования клеток и их активации на первоначальной фазе артрита. Эти данные увеличивают вероятность того, что новые лекарства и биотерапевтические средства, нацеленные на C5aR, могут обеспечить новые стратегии терапевтического вмешательства для блокирования эффекторной фазы RA.
Также была исследована патологическая значимость C5a и C5aR в развитии заболеваний, связанных с антитело-зависимым аутоиммунитетом типа II, в частности при всплеске аутоиммунной гемолитической анемии (AIHA), заболевания, характеризуемого продуцированием антител, направленных против собственных эритроцитов (RBCs), что вызывает гемолиз. AIHA представляет собой довольно обычное нарушение, при оценке появления 1-3 случая/100000/год. Решающая роль C5a в IgG-зависимой AIHA, независимо от хемотактической функции данного анафилотоксина, была выявлена на экспериментальных животных моделях (V. Kumar, J. Clin. Invest., 116(2), 512, 2006). Действительно, наблюдалось, что мыши с недостатком C5aR частично устойчивы к модели данного заболевания, индуцированного IgG аутоантителом, и взаимное влияние C5aR и активирующих Fcγ рецепторов было выявлено в процессе данного наблюдения, в частности, на макрофагах печени, то есть при введении антиэритроцитных антител повышенное регулирование активирующих FcγR на клетки Купфера отсутствовало у C5aR-дефицитных мышей; параллельно, у мышей с дефицитом FcγR разрушалось продуцирование C5 и C5a. Это является первым подтверждением ранее не установленного FcγR-опосредованного C5a-генерирующего пути, говорящим о роли C5a в развитии антитело-зависимых аутоиммунных заболеваний и о потенциальном терапевтическом выигрыше при блокаде C5a и/или C5aR при AIHA, относящейся к аутоиммунному повреждению типа II.
Контроль синтеза фракций комплемента рассматривается в качестве перспективной терапевтической мишени при лечении шока и для предотвращения отторжения во время трансплантации органа (множественная недостаточность органов и сверхострое отторжение трансплантата) (Issekutz A.C. et al., Int. J. Immunopharmacol, 12, 1, 1990; Inagi R. et at., Immunol. Lett., 27, 49, 1991). Позднее сообщалось, что ингибирование фракций комплемента может быть вовлечено в предотвращение повреждений врожденной и трансплантированной почки, принимая во внимание тот факт, что комплемент вовлечен в патогенез как хронического внутритканевого, так и острого гломерулярного повреждения почки (Sheerin N.S. & Sacks S.H., Curr. Opinion Nephrol. Hypert, 7, 395, 1998).
Характерное накопление нейтрофилов происходит при острых и хронических патологических состояниях, например, в областях псориатических повреждений с высокой воспаленностью и не поддающихся терапевтическому лечению. Нейтрофилы являются хемотактически привлекательными и активируются синергетическим действием хемокинов, таких как CXCL8 и GRO-α, высвобождаемых с помощью стимулированных кератиноцитов, и фракции C5a/C5a-desArg, продуцируемой посредством альтернативной активации пути комплемента (T. Terui et al., Exp. Dermatol., 9, 1, 2000). Авторами настоящего изобретения описан новый класс "омега-аминоалкиламидов R-2-арилпропионовых кислот" в качестве ингибиторов хемотаксиса полиморфоядерных (PMN) и моноядерных клеток" (WO 02/068377). Кроме того, сообщалось о четвертичных аммониевых солях омега-аминоалкиламидов (R)-2-арилпропионовых кислот в качестве селективных ингибиторов C5a-индуцированного хемотаксиса нейтрофилов и моноцитов (WO 03/029187). Позднее авторами настоящего изобретения в качестве эффективных и селективных ингибиторов C5-индуцированного хемотаксиса PMN человека были описаны новые (R)-арилалкиламинопроизводные (PCT/EP2006/068867), принадлежащие к химическим классам сульфонамидов и амидов.
Подробное описание изобретения
Неожиданно, авторами настоящего изобретения был выявлен новый класс (R)-4-(гетероарил)фенилпропионовых производных с высокой селективностью и активностью при ингибировании хемотаксиса нейтрофилов, индуцированного C5a. Новые соединения являются неактивными в отношении ингибирования COX в концентрационном диапазоне от 10-5 до 10-6М.
Новые соединения представляют собой замещенные или незамещенные тетразолы, гидроксиазолы, тиадиазолы, пиразолы и триазолы.
Настоящее изобретение относится к новым соединениям, которые можно использовать для ингибирования хемотактической активации, индуцированной фракцией C5a комплемента. Указанные соединения можно использовать для лечения патологий, зависимых от хемотактической активации нейтрофилов и моноцитов, индуцированной фракцией C5a комплемента. В частности, соединения по данному изобретению можно использовать для лечения аутоиммунной гемолитической анемии (AIHA) и ревматоидного артрита. Более того, их также можно использовать для лечения псориаза, буллезного пемфигоида, язвенного колита, острого респираторного дистресс-синдрома, идиопатического фиброза, гломерулонефрита и для предотвращения и лечения повреждения, вызванного ишемией и реперфузией.
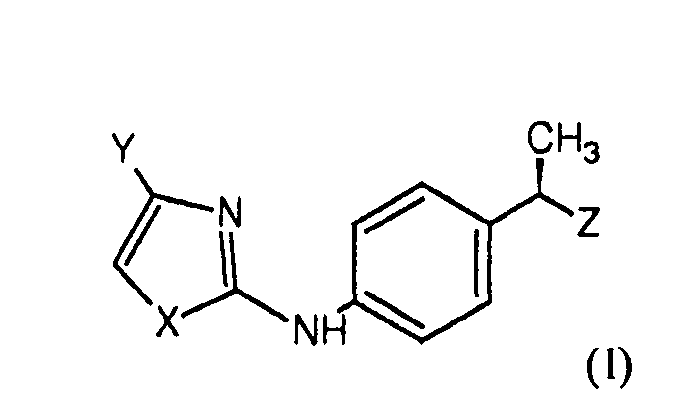
Настоящее изобретение относится к соединениям формулы (I):
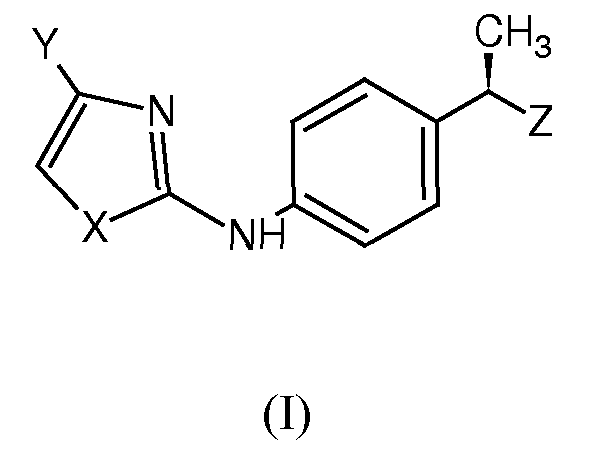
и их фармацевтически приемлемым солям,
где
X представляет собой гетероатом, выбранный из
- S, O и N,
Y представляет собой H или остаток, выбранный из
- галогена, линейного или разветвленного C1-C4-алкила, C2-C4-алкенила, C1-C4-алкокси, гидрокси, -COOH, C1-C4-ацилокси, фенокси, циано, нитро, NH2, C1-C4-ациламино, галоген-C1-C3-алкила, бензоила, линейного или разветвленного C1-C8-алкансульфоната, линейных или разветвленных C1-C8-алкансульфонамидов, линейного или разветвленного C1-C8-алкилсульфонилметила;
Z представляет собой гетероарильное кольцо, выбранное из
незамещенного тетразола и
триазола, пиразола, оксазола, тиазола, изооксазола, изотиазола, тиадиазола и оксадиазола, замещенного одной гидроксильной группой и необязательно дополнительно замещенного одной или несколькими группами, выбранными из группы, состоящей из галогена, линейного или разветвленного C1-C4-алкила, C2-C4-алкенила, C1-C4-алкиламино, C1-C4-алкокси, C1-C4-алкилтио, C1-C4-ацилокси, циано, нитро, NH2, C1-C4-ациламино, галоген-C1-C3-алкила, галоген-C1-C3-алкокси, линейного или разветвленного C1-C8-алкансульфоната и линейных или разветвленных C1-C8-алкансульфонамидов.
В соответствии с предпочтительным вариантом осуществления изобретения, соединения формулы I представляют собой те, в которых
X представляет собой гетероатом, выбранный из
- S и O
Y представляет собой H или остаток, выбранный из
- галогена, линейного или разветвленного C1-C4-алкила и галоген-C1-C3-алкила;
Z представляет собой гетероарильное кольцо, выбранное из группы, состоящей из
незамещенного тетразола и
триазола, пиразола, изооксазола, изотиазола, тиадиазола и оксадиазола, замещенного одной гидроксильной группой и необязательно дополнительно замещенного одной или несколькими группами, выбранными из группы, состоящей из галогена, линейного или разветвленного C1-C4-алкила, C1-C4-алкилтио и галоген-C1-C3-алкила.
Особенно предпочтительными среди вышеуказанных соединений формулы I являются те, где
Y представляет собой H или выбран из группы, состоящей из трифторметила, хлора, метила и трет-бутила,
и/или где указанное триазольное, пиразольное, изооксазольное, изотиазольное, тиадиазольное или оксадиазольное кольцо является замещенным одной гидроксильной группой и необязательно дополнительно замещено одной или несколькими группами, выбранными из группы, состоящей из метила, трифторметила и хлора.
К особенно предпочтительным соединениям формулы (I) относятся
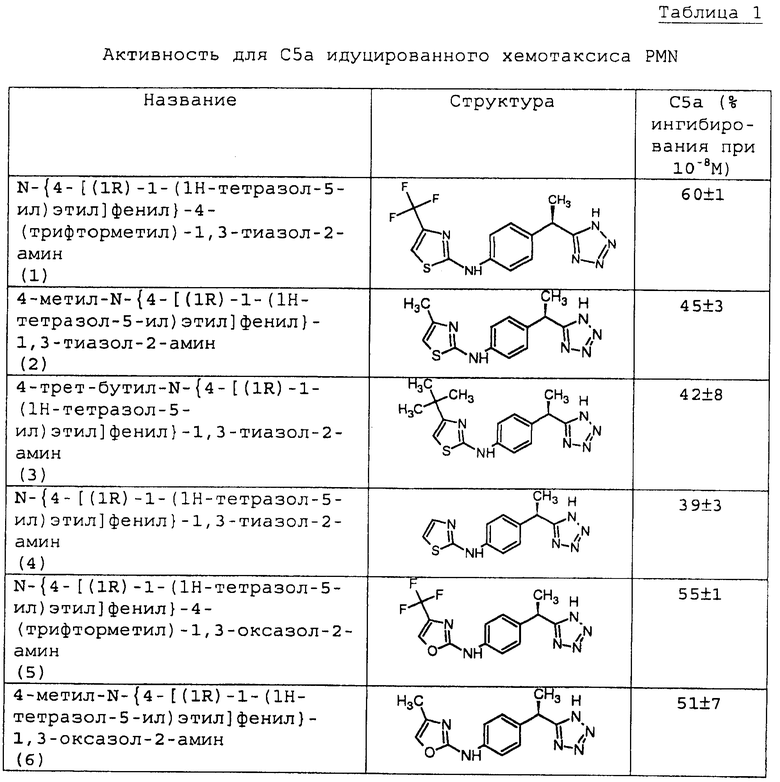
1 - N-{4-[(1R)-1-(1Н-тетразол-5-ил)этил]фенил}-4-(трифторметил)-1,3-тиазол-2-амин;
2 - 4-метил-N-{4-[(1R)-1-(1Н-тетразол-5-ил)этил]фенил}-1,3-тиазол-2-амин;
3 - 4-трет-бутил-N-{4-[(1R)-1-(1Н-тетразол-5-ил)этил]фенил}-1,3-тиазол-2-амин;
4 - N-{4-[(1R)-1-(1Н-тетразол-5-ил)этил]фенил}-1,3-тиазол-2-амин;
5 - N-{4-[(1R)-1-(1Н-тетразол-5-ил)этил]фенил}-4-(трифторметил)-1,3-оксазол-2-амин;
6 - 4-метил-N-{4-[(1R)-1-(1Н-тетразол-5-ил)этил]фенил}-1,3-оксазол-2-амин;
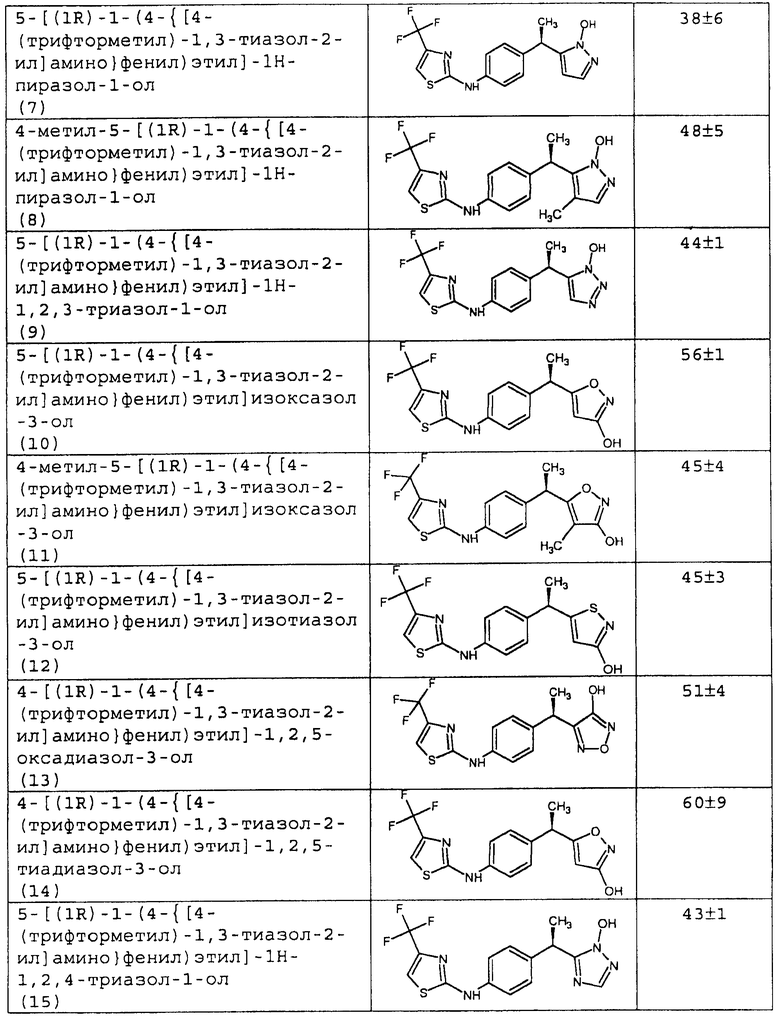
7 - 5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1Н-пиразол-1-ол;
8 - 4-метил-5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1Н-пиразол-1-ол;
9 - 5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1Н-1,2,3-триазол-1-ол;
10 - 5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]изоксазол-3-ол;
11 - 4-метил-5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]изоксазол-3-ол;
12 - 5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]изотиазол-3-ол;
13 - 4-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1,2,5-оксадиазол-3-ол;
14 - 4-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1,2,5-тиадиазол-3-ол;
15 - 5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1Н-1,2,4-триазол-1-ол.
Предпочтительными соединениями являются те, в которых заместитель в 4-положении фенильного кольца представляет собой замещенный или незамещенный 2-аминотиазольный фрагмент.
Наиболее предпочтительным в данном перечне является соединение 1 [N-{4-[(1R)-1-(1Н-тетразол-5-ил)этил]фенил}-4-(трифторметил)-1,3-тиазол-2-амин].
Как будет продемонстрировано в приведенной далее экспериментальной части, соединения формулы (I) являются эффективными ингибиторами хемотаксиса PMN человека, индуцированного C5a. Соответственно, следующая задача настоящего изобретения заключается в создании соединений формулы (I) для применения при лечении заболеваний, в которые вовлечен хемотаксис PMN человека, индуцированный С5а.
Кроме того, также неожиданно было установлено, что соединения формулы (I) не мешают продуцированию PGE2, индуцированному в мышиных макрофагах посредством стимулирования липополисахаридами (LPS, 1 мкг/мл) в концентрационном диапазоне между 10-5 и 10-7М.
Следовательно, следующей задачей настоящего изобретения является применение соединений по изобретению в качестве лекарственных средств. Принимая во внимание обсуждавшиеся выше экспериментальные доказательства и роль, которую выполняет каскад комплемента, а именно его фракция C5a, в процессе, который включает активацию и инфильтрацию нейтрофилов, соединения по изобретению являются особенно полезными при лечении заболеваний, таких как аутоиммунная гемолитическая анемия (AIHA), ревматоидный артрит (М. Selz et al., J. Clin. Invest., 87, 463, 1981), псориаз (R.J. Nicholoff et al., Am. J. Pathol., 138, 129, 1991), буллезный пемфигоид, хронические воспалительные патологии кишечника, такие как язвенный колит (Y. R. Mahida et al., Clin. Sci., 82, 273, 1992), острый респираторный дистресс-синдром и идиопатический фиброз (E. J. Miller, процитировано ранее, и P.C. Carré et al., J. Clin. Invest., 88, 1882, 1991), кистозный фиброз, гломерулонефрит (T. Wada et al., J. Exp. Med., 180, 1135, 1994) и для предотвращения и лечения повреждений, вызванных ишемией и реперфузией.
Следующая задача изобретения заключается в создании соединений формулы (I) для применения для лечения аутоиммунной гемолитической анемии (AIHA), псориаза, буллезного пемфигоида, ревматоидного артрита, язвенного колита, астрого респираторного дистресс-синдрома, идиопатического фиброза, гломерулонефрита и для предотвращения и лечения повреждений, вызванных ишемией и реперфузией.
С этой целью соединения по изобретению формулы (I) обычным образом вводят в состав фармацевтических композиций с использованием общепринятых методов и фармацевтически приемлемых эксципиентов и/или разбавителей, таких как описанные в справочнике "Remington's Pharmaceutical Sciences Handbook" MACK Publishing, New York, 18-е издание, 1990.
Соединения по данному изобретению можно вводить с помощью внутривенной инъекции, в виде болюса, в дерматологических препаратах (кремы, лосьоны, спреи и мази), путем ингаляции, а также перорально в виде капсул, таблеток, сиропа, препаратов контролируемого высвобождения и тому подобного.
Средняя дневная дозировка зависит от ряда факторов, таких как тяжесть заболевания, состояние, возраст, пол и вес пациента. Дозировка обычно будет изменяться от 1 до 1500 мг соединения формулы (I) в день, необязательно будучи разделенной на множество введений.
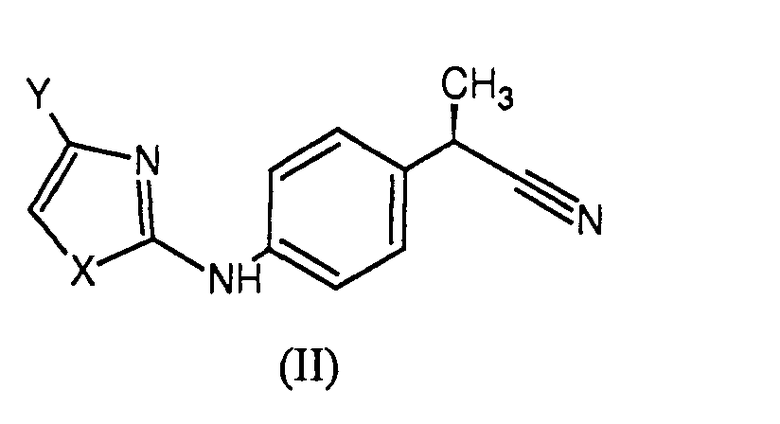
При синтезе соединений формулы (I) использовали различные экспериментальные способы. В той мере, в которой это касается тетразолов, проиллюстрированных в примерах 1-6, их синтезировали обычным способом, исходя из родственной карбоновой кислоты. Кислоты были преобразованы в соответствующие первичные амиды стандартными способами обработки с использованием конденсирующих агентов, таких как 1,1'-карбонилдиимидазол, и последующей реакцией с аммиаком. Преобразование амида в нитрил путем дегидратации, с последующей обработкой нитрила формулы (II)
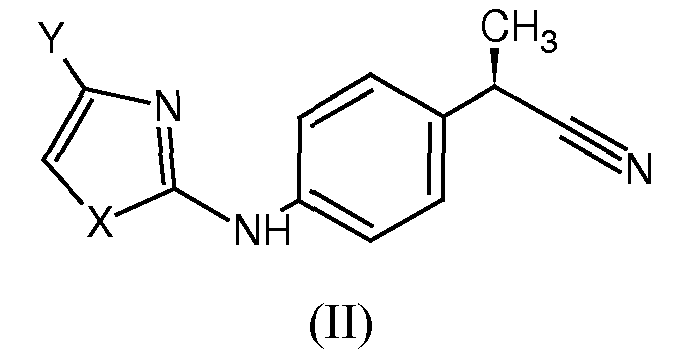
где
X представляет собой гетероатом, выбранный из
- S, O и N,
Y представляет собой H или остаток, выбранный из
- галогена, линейного или разветвленного С1-С4-алкила, C2-C4-алкенила, C1-C4-алкокси, гидрокси, -COOH, C1-C4-ацилокси, фенокси, циано, нитро, -NH2, C1-С4-ациламино, галоген-C1-C3-алкила, бензоила, линейного или разветвленного C1-C8-алкансульфоната, линейных или разветвленных C1-C8-алкансульфонамидов, линейного или разветвленного C1-C8-алкилсульфонилметила;
триметилсилилазидом, приводило к целевым тетразолам. Осуществленные экспериментальные способы получения как для тетразолов, так и для других гетероарильных производных 7-15, были получены на основе опубликованных способов, приспособленных для конкретных субстратов по изобретению (Friederick K. et al. in Rapoport Z., The Chemistry of the Cyano Group, Wiley, NY, 96, 1970; Matzen L. et al., Sisido K. et al., J. Organomet. Chem., 33, 337, 1971; J. Med. Chem., 40, 520, 1997; Stensbøl T. B. et al., J. Med. Chem., 45, 19, 2002; Lolli М. L. et al., J, Med. Chem., 49, 4442, 2006).
Следующие примеры иллюстрируют изобретение.
Экспериментальная часть
Список сокращений:
CH2Cl2: дихлорметан; CH3CN: ацетонитрил; CHCl3: хлороформ; HCl: хлористоводородная кислота; CH3OH: метанол; AcOH: уксусная кислота; EtOAc: этилацетат; DIBAH: диизобутилалюминийгидрид; Et2O: диэтиловый эфир; EtOH: этанол; м-CPBA: мета-хлорпербензойная кислота; CDI: 1,1'-карбонилдиимидазол.
Пример 1: Получение промежуточных соединений
- Метил (2R)-2-[4-(карбамотиоиламино)фенил]пропаноат
Раствор (2R)-2-(4-нитрофенил)пропановой кислоты (25 г, 0,128 моль) в CH3OH (120 мл) обрабатывали при комнатной температуре 37% HCl (5 мл) и нагревали при кипении с обратным холодильником в течение 4 часов. Растворитель удаляли в вакууме и неочищенный метиловый сложный эфир промежуточного соединения использовали на следующей стадии. Порошкообразное железо (71 г, 1,28 моль) суспендировали в смеси CH3OH (250 мл) и воды (20 мл); смесь нагревали, обрабатывали 37% HCl (0,5 мл), затем нагревали при кипении с обратным холодильником в течение 1 часа. После охлаждения до комнатной температуры добавляли по каплям в течение 30 минут раствор неочищенного метилового сложного эфира в CH3OH (25 мл) и полученный раствор нагревали при кипении с обратным холодильником в течение ночи. Суспензию фильтровали в горячем состоянии через слой целита на колонке с коротким путем прохождения раствора и фильтрат упаривали, получая оранжевое масло (20 г), которое разбавляли CH2Cl2 (200 см3) и экстрагировали насыщенным водным раствором NaHCO3 (3×150 мл), сушили над безводным Na2SO4 и упаривали в вакууме, получая чистый метил-(2R)-2-(4-аминофенил)пропаноат (17,5 г, 98 ммоль) в виде оранжевого масла (76%). 1H-ЯМР (CDCl3): δ 7,05 (д, 2H, J=7 Гц), 6,65 (д, 2H, J=7 Гц), 3,80 (м, 1Н), 3,75 (ушир.с, 2H, NH2), 3,60 (с, 3H), 1,45 (д, 3H, J=7 Гц).
К раствору метилового сложного эфира (17,5 г, 98 ммоль) в толуоле (300 мл) медленно добавляли концентрированную H2SO4 (2,6 мл, 0,05 моль). Затем к суспензии добавляли тиоцианат натрия (10,29 г, 0,128 моль) и реакционную смесь нагревали при кипении с обратным холодильником в течение 24 часов. После охлаждения до комнатной температуры смесь промывали насыщенным водным раствором NH4Cl (2×100 мл), сушили над безводным Na2SO4 и упаривали в вакууме, получая неочищенную смесь, из которой после очистки с помощью флэш-хроматографии (н-гексан/EtOAc 1:1) получали метил (2R)-2-[4-(карбамотиоиламино)фенил]пропаноат (10,7 г, 48,4 ммоль) в виде белого твердого вещества (49%). 1H-ЯМР (CDCl3): δ 8,25 (ушир.с, 1Н, CSNH), 7,40 (д, 2H, J=7 Гц), 7,20 (д, 2H, J=7 Гц), 6,20 (ушир.с, 2H, CSNH2), 3,75 (м, 1Н), 3,65 (с, 3H), 1,50 (д, 3H, J=7 Гц).
- (2R)-2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пропановая кислота
Раствор метил-(2R)-2-[4-(карбамотиоиламино)фенил]пропаноата (10,7 г, 0,0484 моль) в диоксане (200 мл) обрабатывали при комнатной температуре 3-бром-1,1,1-трифторпропан-2-оном (5 мл, 0,0484 моль) и полученную смесь нагревали при кипении с обратным холодильником в течение 2 часов. После охлаждения при комнатной температуре растворитель упаривали в вакууме, неочищенную смесь разбавляли CH2Cl2 (200 мл) и промывали насыщенным водным раствором NaHCO3 (3×100 мл), сушили над безводным Na2SO4 и упаривали, получая чистый метил-(2R)-2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пропаноат (12,8 г, 38,7 ммоль) в виде желтого масла (80%). 1H-ЯМР (CDCl3): δ 8,65 (ушир.с, 1Н, NH), 7,30 (м, 4H), 7,05 (с, 1Н), 3,75 (кв, 1Н, J=7 Гц), 3,65 (с, 3H), 1,50 (д, 3H, J=7 Гц).
Раствор метилового сложного эфира (12,8 г, 38,7 ммоль) в AcOH (50 мл) и 37% HCl (17,5 мл) нагревали при кипении с обратным холодильником в течение 12 часов. После охлаждения до комнатной температуры и упаривания растворителей неочищенную смесь разбавляли CH2Cl2 (200 мл) и промывали водой (3×100 мл) и насыщенным раствором соли (3×100 мл). Органический слой сушили над безводным Na2SO4 и растворитель упаривали, получая бледно-желтое масло, из которого после нагревания в н-гексане в течение ночи получали чистую (2R)-2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пропановую кислоту (8,4 г, 26 ммоль) в виде белого твердого вещества (68%). 1H-ЯМР (CDCl3): δ 9,25 (ушир.с, 1Н, NH), 7,40 (д, 2H, J=7 Гц), 7,25 (д, 2H, J=7 Гц), 7,00 (с, 1Н), 3,80 (кв, 1Н, J=7 Гц), 1,55 (д, 3H, J=7 Гц).
- (2R)-2-{4-[(4-метил-1,3-тиазол-2-ил)амино]фенил}пропановая кислота
Кислоту получали в соответствии с тем же способом, который описан для синтеза (2R)-2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пропановой кислоты, исходя из метил-(2R)-2-[4-(карбамотиоиламино)фенил]пропаноата (2,0 г, 8,40 ммоль) и хлор-2-пропанона (0,67 мл, 8,40 ммоль). Последующий кислотный гидролиз давал чистую (2R)-2-{4-[(4-метил-1,3-тиазол-2-ил)амино]фенил}пропановую кислоту (1,65 г, 6,30 моль) в виде желтого масла (75%). 1H-ЯМР (CDCl3): δ 8,15 (ушир.с, 1Н, NH), 7,40 (д, 2H, J=7 Гц), 7,20 (д, 2H, J=7 Гц), 6,35 (с, 1Н), 3,75 (кв, 1Н, J=7 Гц), 2,18 (с, 3H), 1,50 (д, 3H, J=7 Гц).
- (2R)-2-{4-[(4-трет-бутил-1,3-тиазол-2-ил)амино]фенил}пропановая кислота
Кислоту получали в соответствии с тем же способом, который описан для синтеза (2R)-2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пропановой кислоты, исходя из метил-(2R)-2-[4-(карбамотиоиламино)фенил]пропаноата (2,0 г, 8,40 ммоль) и 1-бромпинаколона (1,13 мл, 8,40 ммоль). В результате последующего кислотного гидролиза получали чистую (2R)-2-{4-[(4-трет-бутил-1,3-тиазол-2-ил)амино]фенил}пропановую кислоту (1,41 г, 4,62 ммоль) в виде бледно-желтого масла (55%). 1H-ЯМР (CDCl3): δ 8,30 (ушир.с, 1Н, NH), 7,40 (д, 2H, J=7 Гц), 7,20 (д, 2H, J=7 Гц), 6,40 (с, 1Н), 3,75 (кв, 1Н, J=7 Гц), 1,50 (д, 3H, J=7 Гц), 1,40, (с, 9H).
- (2R)-2-[4-(1,3-тиазол-2-иламино)фенил]пропановая кислота
Кислоту получали в соответствии с тем же способом, который описан для синтеза (2R)-2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пропановой кислоты, исходя из метил-(2R)-2-[4-(карбамотиоиламино)фенил]пропаноата (2,0 г, 8,40 ммоль) и хлорацетальдегида (50% вес. в H2O, 0,54 мл, 8,40 ммоль). В результате последующего кислотного гидролиза получали чистую (2R)-2-{4-[(1,3-тиазол-2-ил)амино]фенил}пропановую кислоту (1,47 г, 5,62 ммоль) в виде бледно-желтого масла (55%). 1H-ЯМР (CDCl3): δ 8,30 (ушир.с, 1Н, NH), 8,10 (д, 1Н, J=2,5 Гц), 7,50 (д, 1Н, J=2,5 Гц) 7,40 (д, 2H, J=7 Гц), 7,20 (д, 2H, J=7 Гц), 3,75 (кв, 1Н, J=7 Гц), 1,50 (д, 3H, J=7 Гц).
- Метил-(2R)-2-[4-(карбамоиламино)фенил]пропаноат
К раствору метил-(2R)-2-(4-аминофенил)пропаноата (3,0 г, 18,1 ммоль) в толуоле (50 мл) медленно добавляли концентрированную H2SO4 (0,47 мл, 50 ммоль). Затем к суспензии добавляли цианистый натрий (1,88 г, 28 ммоль) и реакционную смесь нагревали при кипении с обратным холодильником в течение 24 часов. После охлаждения при комнатной температуре смесь промывали насыщенным водным раствором NH4Cl (2×30 мл), сушили над безводным Na2SO4 и упаривали в вакууме, получая неочищенный продукт, из которого после очистки с помощью флэш-хроматографии (н-гексан/EtOAc 1:1) получали метил (2R)-2-[4-(карбамоиламино)фенил]пропаноат (2,07 г, 9,95 ммоль) в виде белого твердого вещества (55%). 1H-ЯМР (CDCl3): δ 9,35 (ушир.с, 1Н, CONH), 7,45 (д, 2H, J=7 Гц), 7,25 (д, 2H, J=7 Гц), 6,55 (ушир.с, 2H, CONH2), 3,75 (м, 1Н), 1,50 (д, 3H, J=7 Гц).
- (2R)-2-(4-{[4-(трифторметил)-1,3-оксазол-2-ил]амино}фенил)пропановая кислота
Раствор метил (2R)-2-[4-(карбамоиламино)фенил]пропаноата (2,7 г, 9,95 ммоль) в диоксане (50 мл) обрабатывали при комнатной температуре 3-бром-1,1,1-трифторпропан-2-оном (1,03 мл, 10 ммоль) и полученную смесь нагревали при кипении с обратным холодильником в течение 2 ч. После охлаждения при комнатной температуре растворитель упаривали в вакууме, неочищенный продукт разбавляли CH2Cl2 (50 мл) и промывали насыщенным водным раствором NaHCO3 (3×30 мл), сушили над безводным Na2SO4 и упаривали, получая чистый метил (2R)-2-(4-{[4-(трифторметил)-1,3-оксазол-2-ил]амино}фенил)пропаноат (2,5 г, 7,96 ммоль) в виде желтого масла (80%). 1H-ЯМР (CDCl3): δ 10,05 (ушир.с, 1Н, NH), 8,30 (с, 1Н), 7,45 (д, 2H, J=7 Гц), 7,25 (д, 2H, J=7 Гц), 3,75 (кв, 1Н, J=7 Гц), 3,65 (с, 3H), 1,50 (д, 3H, J=7 Гц).
Раствор метилового сложного эфира (2,5 г, 7,96 ммоль) в AcOH (4,1 мл) и 37% HCl (1,42 мл) нагревали при кипении с обратным холодильником в течение 12 часов. После охлаждения до комнатной температуры и упаривания растворителей неочищенный продукт разбавляли CH2Cl2 (20 мл) и промывали водой (3×15 мл) и насыщенным раствором соли (3×15 мл). Органический слой сушили над безводным Na2SO4 и растворитель упаривали, получая бледно-желтое масло, из которого после нагревания в диэтиловом эфире в течение ночи получали чистый (2R)-2-(4-{[4-(трифторметил)-1,3-оксазол-2-ил]амино}фенил)пропановую кислоту (1,86 г, 6,21 ммоль) в виде белого твердого вещества (78%). 1H-ЯМР (CDCl3): δ 9,25 (ушир.с, 1Н, NH), 7,40 (д, 2H, J=7 Гц), 7,25 (д, 2H, J=7 Гц), 7,00 (с, 1Н), 3,80 (кв, 1Н, J=7 Гц), 1,55 (д, 3H, J=7 Гц).
- (2R)-2-{4-[(4-метил-1,3-оксазол-2-ил)амино]фенил}пропановая кислота
Кислоту получали в соответствии с тем же способом, который описан для синтеза (2R)-2-(4-{[4-(трифторметил)-1,3-оксазол-2-ил]амино}фенил)пропановой кислоты, исходя из метил-(2R)-2-[4-(карбамоиламино)фенил]пропаноата (2,0 г, 9,95 ммоль) и хлор-2-пропанона (0,80 мл, 9,95 ммоль). В результате последующего кислотного гидролиза получали чистую (2R)-2-{4-[(4-метил-1,3-тиазол-2-ил)амино]фенил}пропановую кислоту (1,71 г, 6,96 моль) в виде желтого масла (70%). 1H-ЯМР (CDCl3): δ 9,65 (ушир.с, 1Н, NH), 7,95 (с, 1Н), 7,45 (д, 2H, J=7 Гц), 7,25 (д, 2H, J=7 Гц), 3,75 (кв, 1Н, J=7 Гц), 2,20 (с, 3H), 1,50 (д, 3H, J=7 Гц).
Пример 2: Синтез соединения формулы I
N-{4-[(1R)-1-(1Н-тетразол-5-ил)этил]фенил}-4-(трифторметил)-1,3-тиазол-2-амин (1)
1a) (2R)-2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пропанамид
К охлажденной смеси (0-5°C) (2R)-2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пропановой кислоты (1 г, 3,16 ммоль) в CH2Cl2 (20 мл) добавляли 1,1-карбонилдиимидазол (CDI) (0,512 г, 3,16 ммоль). После перемешивания в течение 1 часа при 0-5°C через смесь в течение 4 часов барботировали газообразный аммиак и затем оставляли перемешиваться при комнатной температуре до полного исчезновения исходного вещества. Реакцию гасили, добавляя буферный раствор H3PO4/H2PO4 - (pH=2,0, 5 мл), две фазы отделяли и органическую фазу промывали тем же буфером (3×10 мл) и водой (3×10 мл), сушили над безводным Na2SO4 и упаривали в вакууме, получая (2R)-2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пропанамид (788 мг, 2,5 ммоль) в виде белого твердого вещества (79%), использованного без дополнительной очистки.
1b) (2R)-2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пропаннитрил
К охлажденному (0-5°C) раствору амида в толуоле (10 мл) добавляли по каплям раствор фосгена (1,93М в толуоле, 5,2 мл). Полученную смесь оставляли перемешиваться при комнатной температуре в течение ночи, затем упаривали в вакууме и неочищенный продукт разбавляли CH2Cl2. Органический слой промывали насыщенным раствором NaHCO3 (2×10 мл), водой (3×5 мл) и насыщенным раствором соли (3×5 мл), сушили над безводным Na2SO4 и после упаривания растворителя промежуточное соединение, (2R)-2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пропаннитрил, (639 мг, 2,15 ммоль) выделяли в виде бесцветного масла (86%) и использовали на следующей стадии.
1c) N-{4-[(1R)-1-(1Н-тетразол-5-ил)этил]фенил}-4-(трифторметил)-1,3-тиазол-2-амин
Тригидрат фторида тетрабутиламмония (339 мг, 1,075 ммоль) и триметилсилилазид (0,342 мл, 2,58 ммоль) добавляли к промежуточному нитрилу (639 мг, 2,15 ммоль). Полученную смесь нагревали при интенсивном перемешивании при 85°C в течение 18 часов. После охлаждения до комнатной температуры неочищенную смесь разбавляли EtOAc (20 мл) и промывали 1M HCl (3×5мл), сушили над безводным Na2SO4 и упаривали при пониженном давлении, получая чистый N-{4-[(1R)-1-(1Н-тетразол-5-ил)этил]фенил}-4-(трифторметил)-1,3-тиазол-2-амин (1) (329 мг, 0,97 ммоль) в виде коричневого твердого вещества (45%). [α]D = -36 (c=1; CH3OH); 1H-ЯМР (CD3OD): δ 9,45 (ушир.с, 1Н, NH), 7,45 (д, 2H, J=7 Гц), 7,30 (д, 2H, J=7 Гц), 7,15 (с, 1Н), 3,95 (кв, 1Н, J=7 Гц), 1,65 (д, 3H, J=7 Гц).
4-Метил-N-{4-[(1R)-1-(1Н-тетразол-5-ил)этил]фенил}-1,3-тиазол-2-амин (2)
Соединение 2 получали в соответствии со способом, описанным для синтеза соединения 1, исходя из промежуточной (2R)-2-{4-[(4-метил-1,3-тиазол-2-ил)амино]фенил}пропановой кислоты (4,1 ммоль). Чистый 4-метил-N-{4-[(1R)-1-(1Н-тетразол-5-ил)этил]фенил}-1,3-тиазол-2-амин (2) выделяли (0,65 г, 2,26 ммоль) в виде белого твердого вещества (55%). [α]D = -26 (c=1; CH3OH); 1H-ЯМР (CD3OD): δ 8,20 (ушир.с, 1Н, NH), 7,45 (д, 2H, J=7 Гц), 7,30 (д, 2H, J=7 Гц), 6,25 (с, 1Н), 3,95 (кв, 1Н, J=7 Гц), 2,20 (с, 3H), 1,55 (д, 3H, J=7 Гц).
4-трет-Бутил-N-{4-[(1R)-1-(1Н-тетразол-5-ил)этил]фенил}-1,3-тиазол-2-амин (3)
Соединение 3 получали в соответствии со способом, описанным для синтеза соединения 1, исходя из промежуточной (2R)-2-{4-[(4-трет-бутил-1,3-тиазол-2-ил)амино]фенил}пропановой кислоты (3,5 ммоль). Чистый 4-трет-бутил-N-{4-[(1R)-1-(1Н-тетразол-5-ил)этил]фенил}-1,3-тиазол-2-амин (3) выделяли (0,57 г, 1,75 ммоль) в виде белого твердого вещества (50%). [α]D = -46 (c=1; CH3OH); 1H-ЯМР (CD3OD): δ 9,35 (ушир.с, 1Н, NH), 7,40 (м, 4H), 7,25 (с, 1Н), 3,85 (кв, 1Н, J=7 Гц), 1,55 (д, 3H, J=7 Гц), 1,40 (с, 9H).
N-{4-[(1R)-1-(2H-тетразол-5-ил)этил]фенил}-1,3-тиазол-2-амин (4)
Соединение 4 получали в соответствии со способом, описанным для синтеза соединения 1, исходя из промежуточной (2R)-2-{4-[(1,3-тиазол-2-ил)амино]фенил}пропановой кислоты (3,5 ммоль). Чистый N-{4-[(1R)-1-(2H-тетразол-5-ил)этил]фенил}-1,3-тиазол-2-амин (4) выделяли (0,48 г, 1,75 ммоль) в виде белого твердого вещества (50%). [α]D = -45 (c=1; CH3OH); 1H-ЯМР (CD3OD): δ 8,30 (ушир.с, 1Н, NH), 8,10 (д, 1Н, J=2,5 Гц), 7,50 (д, 1Н, J=2,5 Гц) 7,40 (д, 2H, J=7 Гц), 7,20 (д, 2H, J=7 Гц), 3,75 (кв, 1Н, J=7 Гц), 1,50 (д, 3H, J=7 Гц).
N-{4-[(1R)-1-(1Н-тетразол-5-ил)этил]фенил}-4-(трифторметил)-1,3-оксазол-2-амин (5)
Соединение 5 получали в соответствии со способом, описанным для синтеза соединения 1, исходя из промежуточной (2R)-2-(4-{[4-(трифторметил)-1,3-оксазол-2-ил]амино}фенил)пропановой кислоты (3,5 ммоль). Чистый N-{4-[(1R)-1-(1Н-тетразол-5-ил)этил]фенил}-4-(трифторметил)-1,3-оксазол-2-амин (5) выделяли (0,62 г, 1,92 ммоль) в виде белого твердого вещества (55%). [α]D = -36 (c=1; CH3OH); 1H-ЯМР (CD3OD): δ 10,05 (ушир.с, 1Н, NH), 8,30 (с, 1Н), 7,45 (д, 2H, J=7 Гц), 7,30 (д, 2H, J=7 Гц), 3,95 (кв, 1Н, J=7 Гц), 1,65 (д, 3H, J=7 Гц).
4-Метил-N-{4-[(1R)-1-(1Н-тетразол-5-ил)этил]фенил}-1,3-оксазол-2-амин (6)
Соединение 6 получали в соответствии со способом, описанным для синтеза соединения 1, исходя из промежуточной (2R)-2-{4-[(4-метил-1,3-тиазол-2-ил)амино]фенил}пропановой кислоты (3,5 ммоль). Чистый 4-метил-N-{4-[(1R)-1-(1Н-тетразол-5-ил)этил]фенил}-1,3-оксазол-2-амин (6) выделяли (0,59 г, 2,2 ммоль) в виде белого твердого вещества (50%). [α]D = -19 (c=1; CH3OH); 1H-ЯМР (CD3OD): δ 9,45 (ушир.с, 1Н, NH), 7,95 (с, 1Н), 7,45 (д, 2H, J=7 Гц), 7,30 (д, 2H, J=7 Гц), 3,95 (кв, 1Н, J=7 Гц), 2,20 (с, 3H), 1,65 (д, 3H, J=7 Гц).
5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1Н-пиразол-1-ол (7)
7a) Метил-(4R)-3-оксо-4-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пентаноат
К охлажденной (0-5°C) смеси (2R)-2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пропановой кислоты (3 г, 9,5 ммоль) в сухом CH2Cl2 (70 мл) добавляли ДМФ (0,073 мл, 0,95 ммоль) с последующим добавлением по каплям оксалилхлорида (0,965 мл, 11,4 ммоль). Реакционную смесь перемешивали при 0°C в течение 20 минут и затем оставляли нагреваться до комнатной температуры и перемешивали дополнительно в течение 1,5 часов. После упаривания растворителя (2R)-2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пропаноилхлорид выделяли в виде бледно-желтого масла, достаточно чистого для использования на следующей стадии. К охлажденному раствору перекристаллизованного 2,2-диметил-1,3-диоксан-4,6-диона (кислота Мелдрума) (1,50 г, 10,45 ммоль) в сухом CH2Cl2 (50 мл) добавляли в атмосфере аргона сухой пиридин (1,8 мл, 22,8 ммоль) в течение 10 минут. К полученному прозрачному раствору прикапывали раствор (2R)-2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пропаноилхлорида в сухом CH2Cl2 (10 мл) в течение 20 минут. Полученную реакционную смесь перемешивали в течение 1 часа при 0°C, затем в течение еще одного часа при комнатной температуре. Реакционную смесь разбавляли CH2Cl2 (15 мл) и выливали в 2н HCl (50 мл), содержащую колотый лед. Органическую фазу отделяли и водный слой экстрагировали CH2Cl2 (2×10 мл). Собранные органические экстракты объединяли, промывали 2н HCl (2×10 мл) и насыщенным раствором соли (20 мл), сушили над безводным Na2SO4 и упаривали, получая ацилированное промежуточное соединение Мелдрума в виде бледно-желтого масла. Неочищенный продукт нагревали при кипении с обратным холодильником в сухом CH3OH (30 мл) в течение 2,5 часов. После охлаждения до комнатной температуры и очистки с помощью флэш-хроматографии (н-гексан/EtOAc 8:2) метил-(4R)-3-оксо-4-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пентаноат (1,6 г, 4,3 ммоль) выделяли в виде желтого масла (45%). 1H-ЯМР (CDCl3): δ 9,25 (ушир.с, 1Н, NH), 7,40 (д, 2H, J=7 Гц), 7,25 (д, 2H, J=7 Гц), 7,00 (с, 1Н), 3,90 (кв, 1Н, J=7 Гц), 3,75 (с, 3H); 3,40 (с, 2H); 1,55 (д, 3H, J=7 Гц).
7b) N-{4-[(1R)-1-(1Н-пиразол-5-ил)этил]фенил}-4-(трифторметил)-1,3-тиазол-2-амин
К охлажденному (-78°C) раствору сложного эфира (1,16 г, 3 ммоль) в сухом CH2Cl2 (20 мл) добавляли по каплям в атмосфере аргона DIBAH (1M в гексанах, 3,6 мл) в течение 15 минут с помощью шприца; после завершения прибавления полученный раствор перемешивали при -78°С в течение 1 часа. Реакцию гасили, выливая холодный раствор в насыщенный раствор NH4Cl (10 м). Добавляли 1M HCl (10 мл) и двухфазную смесь интенсивно перемешивали в течение 10 минут. Слои отделяли и органический слой промывали насыщенным раствором соли, тогда как водный слой экстрагировали Et2O (2×10 мл). Собранные органические экстракты сушили над безводным Na2SO4 и концентрировали, получая (4R)-3-оксо-4-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пентаналь (728 мг) в виде белого воскообразного твердого вещества и использовали без дополнительной очистки. К раствору альдегида (728 мг) в смеси EtOH/THF (2:1, 15 мл) добавляли моногидрат гидразина (0,495 мл, 10,2 ммоль) и смесь нагревали при кипении с обратным холодильником в течение 30 минут. После охлаждения до комнатной температуры смесь гасили насыщенным раствором NH4Cl и экстрагировали EtOAc (3×25 мл). Собранные органические экстракты промывали насыщенным раствором соли, сушили над безводным Na2SO4 и упаривали при пониженном давлении, получая N-{4-[(1R)-1-(1Н-пиразол-5-ил)этил]фенил}-4-(трифторметил)-1,3-тиазол-2-амин (421 мг, 1,24 ммоль) в виде бесцветного масла (61%). 1H-ЯМР (CD3OD): δ 9,40 (ушир.с, 1Н, NH); 7,50 (д, 1Н, J=2,5 Гц); 7,40 (д, 2H, J=7 Гц), 7,35 (д, 2H, J=7 Гц), 7,15 (с, 1Н), 6,15 (д, 1Н, J=2,5 Гц), 3,80 (кв, 1Н, J=7 Гц), 1,60 (д, 3H, J=7 Гц).
7c) 5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1Н-пиразол-1-ол
К раствору N-{4-[(1R)-1-(1Н-пиразол-5-ил)этил]фенил}-4-(трифторметил)-1,3-тиазол-2-амина (0,421 г, 1,24 ммоль) в EtOAc (5 мл) добавляли м-CPBA (256 мг, 1,5 ммоль) и полученную смесь перемешивали при комнатной температуре в течение ночи. Неочищенный продукт разбавляли EtOAc (10 мл), промывали водой (2×10 мл) и сушили над безводным Na2SO4. После упаривания растворителя неочищенный продукт очищали с помощью флэш-хроматографии, получая чистый 5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1Н-пиразол-1-ол (7) (0,295 г, 0,65 ммоль) в виде белого твердого вещества (67%). [α]D = -28 (c=0,82; CH3OH); 1H-ЯМР (CD3OD): δ 9,40 (ушир.с, 1Н, NH); 7,55 (д, 1Н, J= 2,5 Гц); 7,40 (д, 2H, J=7 Гц); 7,35 (д, 2H, J=7 Гц); 7,15 (с, 1Н), 6,25 (д, 1Н, J=2,4 Гц); 3,80 (кв, 1Н, J=7 Гц); 1,60 (д, 3H, J=7 Гц).
4-Метил-5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1Н-пиразол-1-ол (8)
Соединение 8 получали в соответствии со способом, описанным для синтеза соединения 7, исходя из промежуточной (2R)-2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пропановой кислоты (0,68 ммоль), путем взаимодействия соответствующего хлорангидрида кислоты с 2,2,5-триметил-1,3-диоксан-4,6-дионом (0,75 ммоль). Чистый 4-метил-5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1Н-пиразол-1-ол (8) выделяли в виде белого твердого вещества (55%). [α]D = -30 (c=1; CH3OH); 1H-ЯМР (CD3OD): δ 9,25 (ушир.с, 1Н, NH), 7,40 (д, 2H, J=7 Гц), 7,35 (д, 2H, J=7 Гц), 7,32 (с, 1Н), 7,15 (с, 1Н), 3,85 (кв, 1Н, J=7 Гц), 2,05 (с, 3H), 1,60 (д, 3H, J=7 Гц).
5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1Н-1,2,3-триазол-1-ол (9)
9a) 2-{4-[(1R)-1-метилпроп-2-ин-1-ил]бензил}-4-(трифторметил)-1,3-тиазол
Диметил-2-оксопропилфосфонат (0,25 мл, 1,2 ммоль) добавляли к суспензии K2CO3 (0,41 г, 3,0 ммоль) и п-толуолсульфонилазида (0,24 г, 1,2 ммоль) в CH3CN (15 мл). После перемешивания в течение 2 часов добавляли раствор (4R)-2-метил-3-оксо-4-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пентаналя (0,34 г, 1,0 ммоль) в CH3OH (5 мл) и полученную смесь перемешивали в течение 8 часов при комнатной температуре. Растворители удаляли в вакууме и остаток разбавляли Et2O (10 мл), промывали водой (2×10 мл) и насыщенным раствором соли (2×5 мл) и сушили над безводным Na2SO4. После упаривания растворителя неочищенный продукт нагревали в н-пентане, получая 2-[4-{(1R)-1-метилпроп-2-ин-1-ил]бензил}-4-(трифторметил)-1,3-тиазол (0,22 г, 0,745 ммоль) в виде бесцветного масла (75%). 1H-ЯМР (CDCl3): δ 8,68 (ушир.с, 1Н, NH); 7,85 (д, 2H, J=7 Гц); 7,55 (д, 2H, J=7 Гц); 7,15 (с, 1Н), 3,50 (кв, 1Н, J=7 Гц); 3,25 (с, 1Н), 1,50 (д, 3H, J=7 Гц).
9b) 4-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]метил}фенил)этил]-1Н-1,2,3-триазол
Охлажденную (0-5°C) смесь 2-{4-[(1R)-1-метилпроп-2-ин-1-ил]бензил}-4-(трифторметил)-1,3-тиазола (0,115 г, 0,4 ммоль), п-толуолсульфонилазида (66 мг, 0,33 ммоль), 2,6-толуидина (48 мг, 0,4 ммоль) и CuI (5% ммоль) в CHCl3 (5 мл) перемешивали в течение 12 часов. Реакцию гасили добавлением буферного раствора (pH=5,4) и продукт экстрагировали CHCl3 (3×5 мл). После упаривания растворителя неочищенный продукт очищали с помощью флэш-хроматографии, получая чистый 1-(4-метилбензолсульфонил)-4-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]метил}фенил)этил]-1Н-1,2,3-триазол (0,95 г, 0,20 ммоль) в виде желтого масла (50%).
9c) N-{4-[(1R)-1-(1Н-1,2,3-триазол-5-ил)этил]фенил}-4-(трифторметил)-1,3-тиазол-2-амин
Соединение добавляли к суспензии магниевых стружек (0,20 ммоль) в CH3OH (3 мл) при комнатной температуре и реакционную смесь перемешивали в течение 2 часов. Реакцию гасили добавлением насыщенного раствора NH4Cl (2 мл). Две фазы отделяли и органическую фазу промывали водой (2×5 мл) и насыщенным раствором соли (2×5 мл) и сушили над безводным Na2SO4. После упаривания растворителя остаток нагревают в н-пентане (5 мл) и выделяют с помощью фильтрования, получая чистый N-{4-[(1R)-1-(1Н-1,2,3-триазол-5-ил)этил]фенил}-4-(трифторметил)-1,3-тиазол-2-амин (0,061 г, 0,18 ммоль) в виде белого твердого вещества (90%). 1H-ЯМР (CD3OD): δ 9,40 (ушир.с, 1Н, NH), 7,40 (д, 2H, J=7 Гц), 7,35 (д, 2H, J=7 Гц), 7,15 (с, 1Н), 7,40 (с, 1Н), 3,70 (кв, 1Н, J=7 Гц), 1,55 (д, 3H, J=7 Гц).
9d) 5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1H-1,2,3-триазол-1-ол
К раствору N-{4-[(1R)-1-(1Н-l,2,3-триазол-5-ил)этил]фенил}-4-(трифторметил)-1,3-тиазол-2-амина (0,06 г, 0,18 ммоль) в EtOAc (10 мл) добавляли м-CPBA (43 мг, 0,25 ммоль) и полученную смесь перемешивали при комнатной температуре в течение ночи. Добавляли EtOAc (10 мл) и органический слой промывали водой (2×10 мл) и сушили над безводным Na2SO4, получая после упаривания растворителя неочищенный продукт, из которого с помощью очистки флэш-хроматографией (EtOAc/CH3OH 7:3) получали чистый 5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1Н-1,2,3-триазол-1-ол (9) (0,025 г, 0,072 ммоль) в виде прозрачного масла (40%). [α]D = -19 (c=1; CH3OH); 1H-ЯМР (CD3OD): δ 9,40 (ушир.с, 1Н, NH), 7,40 (д, 2H, J=7 Гц), 7,35 (д, 2H, J=7 Гц), 7,15 (с, 1Н), 7,40 (с, 1Н), 3,70 (кв, 1Н, J=7 Гц), 1,55 (д, 3H, J=7 Гц).
5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]изоксазол-3-ол (10)
К охлажденному (-30°C) раствору промежуточного соединения 7a, метил-(4R)-3-оксо-4-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пентаноат (372 мг, 1 ммоль), в CH3OH (0,5 мл) добавляли по каплям раствор NaOH (42 мг, 1,05 ммоль) в CH3OH (4 мл). Полученную смесь перемешивали в течение 10 минут, затем добавляли при той же температуре смесь гидрохлорида гидроксиламина (133 мг, 2 ммоль) и NaOH (83 мг, 2 ммоль) в CH3OH/вода (4 мл/0,5 мл). После перемешивания в течение 2 часов при -30°C реакционную смесь выливали в 37% HCl (1,5 мл) и полученную смесь нагревали при 80°C в течение 2 часов. После охлаждения до комнатной температуры и упаривания растворителей неочищенный продукт разбавляли водой и экстрагировали EtOAc (3×10 мл). Объединенные органические экстракты сушили над безводным Na2SO4, упаривали и очищали с помощью флэш-хроматографии (н-гексан/EtOAc 8:2; 1% AcOH), получая чистый 5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]изоксазол-3-ол (10) (202 мг, 0,57 ммоль) в виде бледно-желтого твердого вещества (57%). [α]D = -40 (с=1,4; СН3ОН); 1H-ЯМР (СDСl3): δ 10,70 (ушир. с, 1Н, ОН), 9,15 (ушир. с, 1Н, NH), 7,40 (д, 2Н, J=7 Гц), 7,25 (д, 2Н, J=7 Гц), 7,00 (с, 1Н), 5,70 (с, 1Н), 3,80 (кв, 1Н, J=7 Гц), 1,55 (д, 3Н, J=7 Гц).
4-Метил-5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]изоксазол-3-ол (11)
Соединение получали в соответствии с тем же способом, который описан для синтеза 10, но исходя из промежуточного соединения (4R)-2-метил-3-оксо-4-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пентаноата (0,53 ммоль), описанного для синтеза соединения 8. Чистый 4-метил-5-[(1R)-1-(4-{4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]изоксазол-3-ол (11) (0,11 г, 0,3 ммоль) выделяли в виде бледно-желтого твердого вещества (57%). [α]D=-31 (с=1,4; СН3ОН) ; 1H-ЯМР (СDСl3): δ 10,70 (ушир. с, 1Н, ОН), 9,15 (ушир. с, 1Н, NH), 7,40 (д, 2Н, J=7 Гц), 7,25 (д, 2Н, J=7 Гц), 7,00 (с, 1Н), 5,70 (с, 1Н), 3,80 (кв, 1Н, J=7 Гц), 2,15 (с, 3Н), 1,55 (д, 3Н, J=7 Гц).
5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]изотиазол-3-ол (12)
12а) (4R)-3-оксо-4-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пентановая кислота
Раствор метил-(4R)-3-оксо-4-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пентаноата (372 мг, 1 ммоль) в АсОН (10 мл) и 37% НСl (1,5 мл) нагревали при кипении с обратным холодильником в течение 12 часов. После охлаждения до комнатной температуры и упаривания растворителей неочищенный продукт разбавляли CH2Cl2 (10 мл), промывали водой (3×5 мл) и насыщенным раствором соли (3×5 мл) и сушили над безводным Na2SO4. После упаривания растворителя полученное бледно-желтое масло нагревали в течение ночи в н-гексане. Чистую (4R)-3-оксо-4-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пентановую кислоту (283 мг, 0,79 ммоль) выделяли в виде белого твердого вещества с помощью фильтрования (79%).
12b) (4R)-3-тиоксо-4-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пентанамид
К охлажденной смеси (0-5°C) (4R)-3-оксо-4-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пентановой кислоты (283 мг, 0,79 ммоль) в CH2Cl2 (10 мл) добавляли CDI (0,128 г, 0,79 ммоль). После перемешивания в течение 1 часа при 0-5°C в смесь в течение 2 часов барботировали газообразный аммиак. Смесь перемешивали при комнатной температуре до полного исчезновения исходного вещества. Добавляли буферный раствор H3PO4/H2PO4 - (pH=2,0, 5 мл) и две фазы разделяли; органическую фазу промывали тем же буфером (3×5 мл) и водой (3×5 мл), сушили над безводным Na2SO4 и упаривали в вакууме, получая желтое масло, достаточно чистое для использования на следующей стадии. Безводный EtOH (5 мл) насыщали газообразным HCl и газообразным H2S, пропуская оба газа в течение 30 минут каждый при 0-5°C; добавляли раствор промежуточного соединения 12a в EtOH (5 мл) и газообразный H2S барботировали в раствор дополнительно в течение 10 часов, поддерживая температуру при 0-5°C. После упаривания растворителей и очистки неочищенного продукта с помощью флэш-хроматографии (н-гексан/EtOAc 9:1) (4R)-3-тиоксо-4-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пентанамид (150 мг, 0,40 ммоль) выделяли в виде прозрачного масла (51%).
12c) 5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]изотиазол-3-ол
Раствор йода (135 мг, 0,53 ммоль) в EtOH (5 мл) добавляли по каплям к охлажденной смеси (0-5°C) промежуточного соединения 12b (150 мг, 0,40 ммоль) и K2CO3 (212 мг, 1,53 ммоль) в EtOH (5 мл). Реакционную смесь перемешивали в течение 24 часов при комнатной температуре. Добавляли воду (10 мл) и pH доводили до 3 с использованием 1М HCl. Водный слой экстрагировали EtOAc (3×10 мл); собранные органические экстракты сушили над безводным Na2SO4, и после упаривания растворителя очистка неочищенной смеси с помощью флэш-хроматографии (CH2Cl2/CH3OH 95:5) давала 5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]изотиазол-3-ол (12) (82 мг, 0,22 ммоль) в виде белого твердого вещества (41%). [α]D=-31 (c=1; CH3OH); 1H-ЯМР (CDCl3): δ 10,60 (ушир.с, 1Н, OH), 9,15 (ушир.с, 1Н, NH), 7,40 (д, 2H, J=7 Гц), 7,25 (д, 2H, J=7 Гц), 7,00 (с, 1Н), 5,50 (с, 1Н), 3,80 (кв, 1Н, J=7 Гц), 1,55 (д, 3H, J=7 Гц).
4-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1,2,5-оксадиазол-3-ол (13)
13a) (3R)-2-(Гидроксиамино)-3-{4-[(4-(трифторметил)-1,3-тиазол-2-ил)амино]фенил}бутаннитрил
К охлажденному (0-5°C) раствору цианида калия (0,2 г, 3,66 ммоль) в воде (15 мл) добавляли (2R)-2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пропаналь (1,0 г, 3,33 ммоль) в течение 30 минут. При той же температуре в течение 30 минут добавляли AcOH (3,66 ммоль) и реакционную смесь перемешивали в течение 18 часов. Раствор промежуточного циангидрина медленно добавляли к раствору водного раствора (2 мл) NH4Cl (0,5 г, 9,66 ммоль) вместе с раствором гидроксиламина (50% вес. в H2O; 4,0 ммоль) (5 мл). Полученную реакционную смесь перемешивали при комнатной температуре в течение ночи и затем экстрагировали CH2Cl2 (3×15 мл). Органический слой сушили над безводным Na2SO4 и упаривали в вакууме, получая (3R)-2-(гидроксиамино)-3-{4-[(4-(трифторметил)-1,3-тиазол-2-ил)амино]фенил}бутаннитрил (0,74 г, 2,16 ммоль) в виде красно-коричневого масла, использованного на следующей стадии без дополнительной очистки. 1H-ЯМР (ДМСО-d6): δ 8,25 (ушир.с, 1Н, OH), 7,40 (д, 2H, J=7 Гц), 7,25 (д, 2H, J=7 Гц), 7,05 (с, 1Н), 5,15 (с, 1Н), 3,90 (кв, 1Н, J=7 Гц), 1,75 (д, 3H, J=7 Гц).
13b) 4-[(1R)-1-{4-[(4-(трифторметил)-1,3-тиазол-2-ил)амино]фенил}этил]-1,2,5-оксадиазол-3-амин
Смесь промежуточного соединения 13a (0,738 мг, 2,16 ммоль), гидрохлорида гидроксиламина (83 мг, 2,50 ммоль) и ацетат натрия (410 мг, 5 ммоль) в EtOH (15 мл) нагревали при кипении с обратным холодильником в течение 4 часов. После охлаждения выпавший осадок собирали фильтрованием и сушили. Выпавшее в осадок α-оксимидо-ацетамидоксимное производное ацетата натрия нагревали при кипении с обратным холодильником с избытком PCl5 в сухом Et2O (15 мл) в течение 6 часов. После охлаждения до комнатной температуры реакцию гасили буферным раствором, имеющим pH 8,2 (10 мл), и две фазы отделяли. Водный слой экстрагировали Et2O (2×10 мл) и собранные органические фазы сушили над безводным Na2SO4 и упаривали в вакууме, получая 4-[(1R)-1-{4-[(4-(трифторметил)-1,3-тиазол-2-ил)амино]фенил}этил]-1,2,5- оксадиазол-3-амин (0,57 г, 1,62 ммоль) в виде белого твердого вещества (75%). 1H-ЯМР (ДМСО-d6): δ 8,25 (ушир.с, 1Н, NH), 7,40 (д, 2H, J=7 Гц), 7,25 (д, 2H, J=7 Гц), 7,05 (с, 1Н), 5,15 (ушир.с, 2H, NH2), 3,90 (кв, 1Н, J=7 Гц), 1,75 (д, 3H, J=7 Гц).
13c) 4-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1,2,5-оксадиазол-3-ол
К охлажденному раствору 4-[(1R)-1-{4-[(4-(трифторметил)-1,3-тиазол-2-ил)амино]фенил}этил]-1,2,5-оксадиазол-3-амина (0,2 г, 0,56 ммоль) в AcOH (5 мл) и 37% HCl (3 мл) добавляли по каплям раствор нитрита натрия (44 мг, 0,845 ммоль) в воде (3 мл). Полученную реакционную смесь перемешивали в течение 30 минут, затем добавляли концентрированную H2SO4 (0,5 мл) и реакцию гасили насыщенным раствором NH4Cl (10 мл); полученную смесь экстрагировали Et2O (3×10 мл) и собранные органические экстракты упаривали в вакууме; неочищенный продукт очищали с помощью флэш-хроматографии, получая чистый 4-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1,2,5-оксадиазол-3-ол (13) (0,17 г, 0,48 ммоль) в виде белого твердого вещества (85%). [α]D = -51 (c=1; CH3OH); 1H-ЯМР (ДМСО-d6): δ 8,25 (ушир.с, 1Н, NH), 7,40 (д, 2H, J=7 Гц), 7,25 (д, 2H, J=7 Гц), 7,05 (с, 1Н), 3,90 (кв, 1Н, J=7 Гц), 1,75 (д, 3H, J=7 Гц).
4-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1,2,5-тиадиазол-3-ол (14)
14a) (3R)-2-амино-3-{4-[(4-(трифторметил)-1,3-тиазол-2-ил)амино]фенил}бутаннитрил
К охлажденному (0-5°C) раствору цианида калия (0,2 г, 3,66 ммоль) в воде (15 мл) добавляли в течение 30 минут (2R)-2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пропаналь (1,0 г, 3,33 ммоль) (получен в соответствии со способом, описанным для промежуточного соединения 7b, исходя из соответствующего пропаноата). При той же температуре прикапывали AcOH (3,66 ммоль) и реакционную смесь перемешивали в течение 18 часов. Раствор промежуточного циангидрина медленно добавляли к другому раствору NH4Cl (0,5 г, 9,66 ммоль) в NH4OH (14н в H2O; 4,0 ммоль) (5 мл). Полученную реакционную смесь перемешивали при комнатной температуре в течение 18 часов и затем экстрагировали CH2Cl2 (3×15 мл). Органический слой сушили над безводным Na2SO4 и упаривали в вакууме, получая (3R)-2-амино-3-{4-[(4-(трифторметил)-1,3-тиазол-2-ил)амино]фенил}бутаннитрил (0,705 г, 2,16 ммоль) в виде красного масла, использованный на следующей стадии без дополнительной очистки. 1H-ЯМР (ДМСО-d6): δ 9,25 (ушир.с, 1Н, NH), 7,40 (д, 2H, J=7 Гц), 7,25 (д, 2H, J=7 Гц), 7,00 (с, 1Н), 5,00 (с, 1Н), 3,90 (кв, 1Н, J=7 Гц), 2,35 (ушир.с, 2H), 1,75 (д, 3H, J=7 Гц).
14b) N-{4-[(1R)-1-(4-хлор-1,2,5-тиадиазол-3-ил)этил]фенил}-4-(трифторметил)-1,3-тиазол-2-амин
К охлажденному (0-5°C) раствору монохлорида серы (0,35 мл, 4,32 ммоль) в ДМФ (15 мл) добавляли в течение 1 часа раствор промежуточного соединения 14a (0,7 г, 2,16 ммоль) в ДМФ (5 мл). Реакционную смесь перемешивали в течение 1 часа; добавляли ледяную воду (30 мл) для поддержания температуры ниже 20°C и оставляли смесь для осаждения серы. Смесь фильтровали и маточные растворы разбавляли буферным раствором (pH 8,5, 50 мл). Водный слой экстрагировали CH2Cl2 (2×10 мл) и собранные органические экстракты упаривали, получая неочищенный продукт, из которого после очистки перекристаллизацией из н-гептана получали чистый N-{4-[(1R)-1-(4-хлор-1,2,5-тиадиазол-3-ил)этил]фенил}-4-(трифторметил)-1,3-тиазол-2-амин (1,09 г, 2,80 ммоль) в виде желтого твердого вещества (65%). 1H-ЯМР (CDCl3): δ 9,25 (ушир.с, 1Н, NH), 7,40 (д, 2H, J=7 Гц), 7,25 (д, 2H, J=7 Гц), 7,00 (с, 1Н), 3,85 (кв, 1Н, J=7 Гц), 1,75 (д, 3H, J=7 Гц).
14c) 4-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1,2,5-тиадиазол-3-ол
Промежуточное соединение 14b (1,09 г, 2,80 ммоль) растворяли в растворе NaOH (0,11 г, 2,75 ммоль) в CH3OH (10 мл). Реакционную смесь перемешивали в течение 1 часа при 50°C и затем гасили реакцию насыщенным раствором NH4Cl (10 мл); водный слой экстрагировали CH2Cl2 (2×10 мл) и объединенные органические фазы после сушки над безводным Na2SO4 упаривали в вакууме и кристаллизовали из н-гептана, получая чистый 4-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1,2,5-тиадиазол-3-ол (14) (0,625 г, 1,68 ммоль) в виде белого твердого вещества (60%). [α]D = -33 (c=1; CH3OH); 1H-ЯМР (CDCl3): δ 9,25 (ушир.с, 1Н, NH), 8,35 (ушир.с, 1Н, OH), 7,40 (д, 2H, J=7 Гц), 7,25 (д, 2H, J=7 Гц), 7,00 (с, 1Н), 3,85 (кв, 1Н, J=7 Гц), 1,75 (д, 3H, J=7 Гц).
5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1Н-1,2,4-триазол-1-ол (15)
15a) (2R)-2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пропанимидамид
Газообразный HCl барботировали в течение 5 часов к раствору промежуточного соединения 1b (0,64 г, 2,15 ммоль) в смеси CH3OH/Et2O (1:1, 20 мл) и затем смесь перемешивали в течение ночи при комнатной температуре. Растворитель упаривали и неочищенный продукт, после растворения в CH3OH (10 мл), обрабатывали газообразным NH3 до получения насыщенного раствора. Полученную смесь перемешивали при комнатной температуре в течение ночи. После упаривания остаток растворяли в CH2Cl2 (10 мл) и промывали 1M HCl (3×5 мл). Собранные водные фазы повторно экстрагировали EtOAc (3×10 мл). Объединенные органические экстракты сушили над безводным Na2SO4 и упаривали в вакууме, получая чистый (2R)-2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пропанимидамид (0,4 г, 1,29 ммоль) в виде желтого твердого вещества (60%). 1H-ЯМР (CD3OD): δ 9,45 (ушир.с, 1Н, NH), 9,10 (с, 1Н), 8,80 (с, 2H), 7,45 (д, 2H, J=7 Гц), 7,30 (д, 2H, J=7 Гц), 7,15 (с, 1Н), 3,95 (кв, 1Н, J=7 Гц), 1,55 (д, 3H, J=7 Гц).
15b) N-{4-[(1R)-1-(1Н-1,2,4-триазол-5-ил)этил]фенил}-4-(трифторметил)-1,3-тиазол-2-амин
К раствору промежуточного соединения 15a (0,4 г, 1,29 ммоль) в EtOH (5 мл) добавляли формилгидразин (95 мг, 1,55 ммоль) и смесь нагревали при кипении с обратным холодильником в течение 48 часов. После охлаждения до комнатной температуры растворитель отгоняли и неочищенный продукт растворяли в CH2Cl2 (10 мл), промывали 1M HCl (2×5 мл), сушили над безводным Na2SO4 и упаривали в вакууме, получая неочищенный продукт, из которого, после очистки с помощью флэш-хроматографии, получали чистый N-{4-[(1R)-1-(1Н-l,2,4-триазол-5-ил)этил]фенил}-4-(трифторметил)-1,3-тиазол-2-амин (0,22 мг, 0,645 ммоль) в виде желтого вещества (42%). 1H-ЯМР (CD3OD): δ 9,45 (ушир.с, 1Н, NH), 7,45 (д, 2H, J=7 Гц), 7,30 (д, 2H, J=7 Гц), 7,15 (с, 1Н), 3,95 (кв, 1Н, J=7 Гц), 1,55 (д, 3H, J=7 Гц).
15c) 5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1H-1,2,4-триазол-1-ол
К раствору промежуточного соединения 15b (0,21 г, 0,62 ммоль) в EtOAc (5 мл) добавляли м-CPBA (0,17 г, 0,97 ммоль) и полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь промывали водой (2×10 мл) и безводным Na2SO4 и упаривали в вакууме, получая неочищенный продукт, из которого после очистки с помощью флэш-хроматографии получали чистый 5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1Н-1,2,4-триазол-1-ол (15) в виде белого твердого вещества (67%). [α]D = -35 (c=0,82; CH3OH); 1H-ЯМР (CD3OD): δ 9,40 (ушир.с, 1Н, NH); 7,55 (д, 1Н, J= 2,5 Гц); 7,40 (д, 2H, J=7 Гц); 7,35 (д, 2H, J=7 Гц); 7,15 (с, 1Н), 6,25 (д, 1Н, J=2,4 Гц); 3,80 (кв, 1Н, J=7 Гц); 1,60 (д, 3H, J=7 Гц).
Пример 3: Биологические анализы
3a) Ингибирование хемотактической активности, индуцированной C5a
Для соединений, полученных в примере 2, проводили оценку in vitro их способности ингибировать хемотаксис полиморфоядерных лейкоцитов (далее упоминаемых как PMN) и моноцитов, вызванный фракциями комплемента C5a и C5a-desArg. С этой целью, для выделения PMN из гепаринизированной крови человека, полученной от здоровых взрослых добровольцев, моноядерные клетки удаляли с помощью седиментации на декстране (в соответствии со способом, описанным W.J. Ming et al., J. Immunol., 138, 1469, 1987) и эритроциты с помощью гипотонического раствора. Жизнеспособность клеток рассчитывали по исключению с использованием трипана синего, тогда как соотношение циркулирующих полиморфоядерных клеток оценивали на цитоцентрифугате после окрашивания с помощью Diff Quick.
Рекомбинантные фракции C5a и C5a-desArg человека (Sigma) использовали в качестве стимулирующих агентов в экспериментах по хемотаксису, дающих практически идентичные результаты.
Лиофилизованный C5a растворяли в объеме HBSS, содержащем 0,2% бычьего сывороточного альбумина БСА таким образом, чтобы получить исходный раствор, имеющий концентрацию 10-5М для разбавления в HBSS до концентрации 10-9М для анализа хемотаксиса. В экспериментах по хемотаксису PMN инкубировали с соединения по изобретению формулы (I) в течение 15 минут при 37°С в в атмосфере, содержащей 5% СО2. Хемотактическую активность C5a оценивали на полиморфоядерных клетках крови (PMN), суспендированных в HBSS в концентрации 1,5×106 PMN на мл. В процессе анализа хемотаксиса (в соответствии с публикацией W. Falket et al., J. Immunol. Methods, 33, 239, 1980) использовали не содержащие PVP фильтры с пористостью 5 мкм и микрокамеры, подходящие для репликации.
Соединения примера 2 оценивали в концентрации, колеблющейся в диапазоне от 10-7 до 10-10М; с этой целью их добавляли, в той же концентрации, как в нижние отверстия, так и в верхние отверстия микрокамеры. Лунки в нижней части содержат раствор С5а или простого носителя, лунки в верхней части содержат суспензию PMN.
Ингибирование С5а-индуцированной хемотактической активности с помощью индивидуальных соединений оценивали путем инкубирования микрокамеры для хемотаксиса в течение 60 минут при 37°С в атмосфере, содержащей 5% СО2.
Оценку способности тестируемых соединений ингибировать C5a-индуцированный хемотаксис моноцитов человека проводили в соответствии со способом, описанным Van Damme J. et al. (Eur. J. Immunol., 19, 2367, 1989). Ингибирование С5-индуцированной хемотактической активности индивидуальными соединениями в отношении к моноцитам человека оценивали при концентрациях, колеблющихся от 10-7 до 10-10М путем инкубирования микрокамеры для хемотаксиса в течение 120 минут при 37°С в атмосфере, содержащей 5% СО2.
Полученные данные для ингибирования хемотаксиса PMN (концентрация 10-8М) представлены в таблице 1.
3b) Ингибирование продуцирования PGE 2
Соединения, полученные в примере 2, оценивали ex vivo в цельной крови в соответствии со способом, описанным Patrignani et al. (J. Pharmacol. Exper. Ther., 271, 1705, 1994). Во всех случаях соединения формулы (I) не мешали продуцированию PGE2, индуцированному в мышиных макрофагах при стимулировании липополисахаридами (LPS, 1 мкг/мл) в концентрационном диапазоне от 10-5 до 10-7М. Ингибирование продуцирования PGE2 находится главным образом в пределах статической значимости и обычно находится ниже 15-20% от исходного уровня.
| название | год | авторы | номер документа |
|---|---|---|---|
| (R)-АРИЛАЛКИЛАМИНОПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2006 |
|
RU2458051C2 |
| 2-АРИЛПРОПИОНОВЫЕ КИСЛОТЫ И ПРОИЗВОДНЫЕ, И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ УКАЗАННЫЕ СОЕДИНЕНИЯ | 2009 |
|
RU2520212C2 |
| (2R)-2-[(4-СУЛЬФОНИЛ)АМИНОФЕНИЛ]ПРОПАНАМИДЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2007 |
|
RU2457201C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ 5,7-ДИЗАМЕЩЕННОГО [1,3] ТИАЗОЛО[4,5-D]ПИРИМИДИН-2(3H)-ОНА | 2006 |
|
RU2411245C9 |
| ИНГИБИТОРЫ ФЕРМЕНТА ДИАЦИЛГЛИЦЕРИН-О-АЦИЛТРАНСФЕРАЗЫ ТИПА 1 | 2008 |
|
RU2486186C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ИСПОЛЬЗОВАНИЯ | 2012 |
|
RU2648997C2 |
| КОНДЕНСИРОВАННОЕ 4-ОКСОПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ | 2005 |
|
RU2358969C2 |
| ПРОИЗВОДНЫЕ ИНДОЛА И ИНДАЗОЛА, ОБЛАДАЮЩИЕ КОНСЕРВИРУЮЩИМ ДЕЙСТВИЕМ ПО ОТНОШЕНИЮ К КЛЕТКАМ, ТКАНЯМ И ОРГАНАМ | 2009 |
|
RU2460525C2 |
| ПРОЛЕКАРСТВА СОПРЯЖЕННО-БИЦИКЛИЧЕСКИХ АНТАГОНИСТОВ C5aR | 2019 |
|
RU2794327C2 |
| N-ФЕНИЛАРИЛСУЛЬФОНАМИД, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ УКАЗАННОЕ СОЕДИНЕНИЕ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА, СОЕДИНЕНИЕ, ЯВЛЯЮЩЕЕСЯ ПРОМЕЖУТОЧНЫМ В СИНТЕЗЕ УКАЗАННОГО СОЕДИНЕНИЯ, И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2002 |
|
RU2299202C2 |
Изобретение относится к (R)-4-(гетероарил)фенилэтильным соединениям формулы (I), где Х представляет собой гетероатом, выбранный из -S или О, Y представляет собой Н или остаток, выбранный из группы, состоящей из - линейного или разветвленного C1-C4-алкила, галоген-С1-С3-алкила; Z представляет собой гетероарильное кольцо, выбранное из группы, состоящей из незамещенного тетразола и триазола, пиразола, тиазола, изооксазола, изотиазола, тиадиазола и оксадиазола, замещенного одной гидроксильной группой и необязательно дополнительно замещенного линейным С1-С4-алкилом. (R)-4-(гетероарил)фенилэтильные производные формулы (I), которые можно использовать для применения при лечении заболеваний, в которые вовлечен С5а индуцированный PMN-хемотаксис человека. Соединения формулы (I), где Z представляет собой тетразол, получают путем взаимодействия соединения формулы (II) с триметилсилилазидом. Технический результат - (R)-4-(гетероарил)фенилэтильные производные, обладающие высокой селективностью и активностью при ингибировании хемотаксиса нейтрофилов, индуцированного С5а. 2 н. и 5 з.п. ф-лы, 1 табл., 3 пр.
1. (R)-4-(гетероарил)фенилэтильные соединения формулы (I):
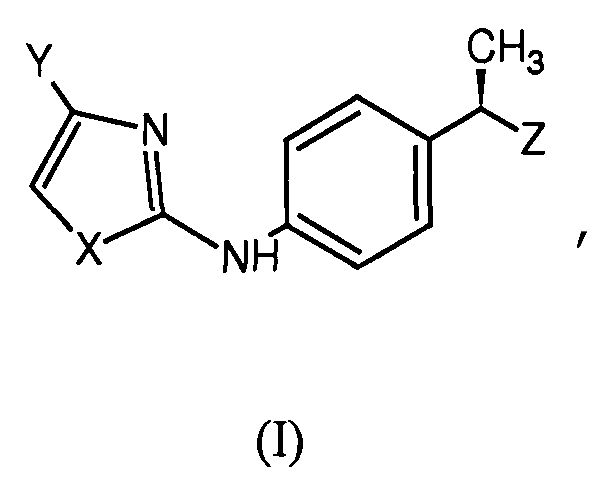
где Х представляет собой гетероатом, выбранный из
- S или О,
Y представляет собой Н или остаток, выбранный из группы, состоящей из
- линейного или разветвленного С1-С4-алкила, галоген-С1-С3-алкила;
Z представляет собой гетероарильное кольцо, выбранное из группы, состоящей из незамещенного тетразола и триазола, пиразола, тиазола, изооксазола, изотиазола, тиадиазола и оксадиазола, замещенного одной гидроксильной группой и необязательно дополнительно замещенного линейным С1-С4-алкилом.
2. Соединения по п.1,
где Y представляет собой Н или остаток, выбранный из группы, состоящей из трифторметила, метила и трет-бутила.
3. Соединения по п.1,
где указанные триазольное, пиразольное, изооксазольное, изотиазольное, тиадиазольное или оксадиазольное кольцо является замещенным одной гидроксильной группой и необязательно дополнительно замещенным метилом.
4. Соединения по п.1, выбранные из группы, состоящей из
N-{4-[(1R)-1-(1Н-тетразол-5-ил)этил]фенил}-4-(трифторметил)-1,3-тиазол-2-амина;
4-метил-N-{4-[(1R)-1-(1Н-тетразол-5-ил)этил]фенил}-1,3-тиазол-2-амина;
4-трет-бутил-N-{4-[(1R)-1-(1Н-тетразол-5-ил)этил]фенил}-1,3-тиазол-2-амина;
N-{4-[(1R)-1-(1Н-тетразол-5-ил)этил]фенил}-1,3-тиазол-2-амина;
N-{4-[(1R)-1-(1Н-тетразол-5-ил)этил]фенил}-4-(трифторметил)-1,3-оксазол-2-амина;
4-метил-N-{4-[(1R)-1-(1Н-тетразол-5-ил)этил]фенил}-1,3-оксазол-2-амина;
5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1Н-пиразол-1-ола;
4-метил-5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1Н-пиразол-1-ола;
5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1Н-1,2,3-триазол-1-ола;
5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]изоксазол-3-ола;
4-метил-5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]изоксазол-3-ола;
5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]изотиазол-3-ола;
4-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1,2,5-оксадиазол-3-ола;
4-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1,2,5-тиадиазол-3-ола;
5-[(1R)-1-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)этил]-1Н-1,2,4-триазол-1-ола.
5. Соединения по любому из пп.1-4 для применения при лечении заболеваний, в которые вовлечен С5а индуцированный PMN-хемотаксис человека.
6. Соединения по п.5 для применения при лечении аутоиммунной гемолитической анемии (AIHA), псориаза, буллезного пемфигоида, ревматоидного артрита, язвенного колита, острого респираторного дистресс-синдрома, идиопатического фиброза, гломерулонефрита и для предотвращения и лечения повреждения, вызванного ишемией и реперфузией.
7. Способ получения соединения по п.1, где Z представляет собой тетразол, включающий взаимодействие соединения формулы (II),
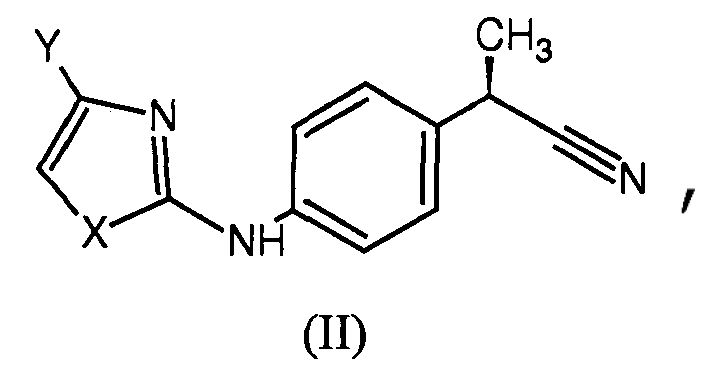
где X и Y имеют такие же значения, как определено в п.1, с триметилсилилазидом, приводящее к получению соответствующих тетразолов формулы (I).
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| N-[4-(ГЕТЕРОАРИЛМЕТИЛ)ФЕНИЛ]-ГЕТЕРОАРИЛАМИНЫ | 1997 |
|
RU2190611C2 |

