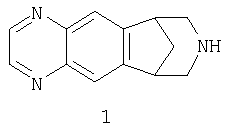
Настоящее изобретение относится к лекарственным формам фармацевтического препарата на основе соединения 1, 5,8,14-триазатетрацикло[10.3.1.02,11.04,9]гексадека-2(11),3,5,7,9-пентаена, контролируемого высвобождения для перорального введения и к родственным соединениям, а также к способам их применения для снижения привыкания к никотину или для способствования прекращению употребления табака или снижению его употребления при одновременном уменьшении тошноты, проявляющейся в качестве нежелательного действия лекарственного средства. Настоящее изобретение также относится к композиции с низкой дозой лекарственного вещества немедленного высвобождения, имеющей устойчивый состав с однородным распределением лекарственного вещества и действенности.
Предшествующий уровень техники
Соединение 1, известное также как 7,8,9,10-тетрагидро-6,10-метан-6Н-пиразин[2,3-h][3]бензазепин, связывается с никотиновыми ацетилхолиновыми рецепторами нейронов и используется при модуляции холинергической функции. Соответственно, это соединение является применимым при лечении воспалительных заболеваний кишечника (включая, но без ограничения, неспецифический язвенный колит, гангренозную приодермию и болезнь Крона), синдрома раздраженной толстой кишки, спастической дистонии, хронической боли, острой боли, брюшной спру, pouchitis, сужения кровеносных сосудов, тревоги, панического состояния, депрессии, биполярного расстройства, аутизма, нарушений сна, расстройства нормального циркадного ритма, проявляющегося повышенной утомляемостью, бокового амиотрофического склероза (ALS), нарушения познавательной способности, гипертензии, булимии, анорексии, ожирения, сердечной аритмии, повышенной секреции желудочного сока, язв, феохромоцитомы, прогрессирующего наднуклеарного паралича, лекарственных зависимостей и привыканий (например, никотиновой зависимости или привыкания к никотину (и/или к табачным изделиям), алкоголю, бензодиазепинам, барбитуратам, опиоидам или кокаину), головной боли, мигрени, удара, черепно-мозговой травмы (TBI), обсессивно-компульсивного состояния (OCD), психоза, хореи Гентингтона, поздней дискинезии, гиперкинезии, дислексии, шизофрении, слабоумия, вызванного повторными инфарктами, снижения познавательной способности, обусловленного возрастом, эпилепсии, включая эпилептический статус, старческого слабоумия типа болезни Альцгеймера (AD), болезни Паркинсона (PD), повышенной активности, обусловленной недостатком внимания (ADHD), и синдрома Туретта.
Соединение 1 и его фармацевтически приемлемые аддитивные соли с кислотой описаны в международной заявке на патент WO 99/35131, опубликованный 15 июля 1999, которая включена во всей полноте в настоящее описание в качестве ссылки.
Поскольку лекарственные формы вышеуказанного соединения немедленного высвобождения (IR), то есть лекарственные формы, предназначенные для обеспечения доступности лекарственного средства в растворенной форме при проглатывании в течение менее приблизительно 30 минут, обеспечивают терапевтически пригодные уровни лекарственного вещества в крови и головном мозге, наблюдалось, что больные часто страдают тошнотой, в особенности при достаточно высоких дозах, являющихся терапевтически необходимыми для некоторых больных. Поскольку тошнота может привести к нарушению схемы приема больным лекарственного средства, имеется потребность в получении соединения 1 в форме, уменьшающей частоту появления тошноты.
Соответственно, настоящее изобретение относится к лекарственным формам соединения 1 CR, которые уменьшают или устраняют тошноту при одновременном поддержании терапевтического уровня лекарственного вещества в крови и центральной нервной системе (ЦНС). Хотя в данной области имеются примеры, предполагающие, что лекарственные формы CR могут в некоторых случаях обеспечивать уменьшение таких побочных действий, как тошнота (например, оксикодон (J.R.Caldwell et al., J. of Rheumatology 1999, 26, 862-869), венлафаксин (R.Entsuah и R.Chitra, Psychopharmacology Bulletin, 1997, 33, 671-676) и пароксетин (R.N.Golden et al., J. Clin. Psychiatry, 2002, 63, 577-584), существуют также противоположные примеры, которые показывают, что лекарственные формы CR иногда уменьшают тошноту не лучше, чем лекарственные формы немедленного высвобождения, и поэтому в них не указывается на полезность использования лекарственной формы CR в качестве средства, уменьшающего побочные действия. Противоположные примеры включают использование морфинсульфата (T.D.Walsh et al., J. Clin. Oncology, 1992, 15, 268-272), гидроморфона (H. Hays et al., Cancer, 1994, 74, 1808-1816), тартрата дигидрокодеина (G. Xu et al., Zhongguo Yaowu Yilaixing Zazhi, 1999, 8, 52-57) и карбидопы/леводопы (G. Block et al., European Neurology, 1997, 37, 23-27). Кроме того, во многих случаях лекарственные формы CR вызывают уменьшение биодоступности по сравнению с лекарственными формами IR, что влечет за собой увеличение дозы или даже делает использование лекарственной формы CR невозможным. Следовательно, остается невозможным предсказать a priori, какие лекарственные вещества, вызывающие тошноту, будут в действительности выгодными при использовании их в виде лекарственных форм CR. Кроме того, скорость, при которой лекарственное вещество становится доступным, то есть скорость его растворения, может значительно изменяться от слегка более низкой, чем скорость лекарственной формы IR, до доставки в течение продолжительного периода времени (примерно до 24 часов). Авторы обнаружили, что лекарственные формы соединения 1 CR с определенным диапазоном скоростей доставки лекарственного вещества будут обеспечивать терапевтические уровни лекарственного вещества в крови и ЦНС при одновременном уменьшении частоты появления тошноты по сравнению с лекарственной формой IR. Авторы также обнаружили конкретные предпочтительные способы формулирования соединения 1 для достижения требуемых скоростей введения лекарственного средства. Авторы также обнаружили предпочтительные схемы введения лекарственного средства, которые обеспечивают терапевтические уровни лекарственного вещества при одновременном сохранении низких уровней вероятности появления тошноты.
Высокая действенность соединения 1 в качестве лиганда никотинового рецептора обеспечивает возможность использования низких доз для введения. Для облегчения манипулирования, изготовления и удобства для больного лекарственные средства с низкой дозой активного вещества часто получают при значительном разбавлении эксципиентами. Однако при приготовлении и хранении таких разбавленных препаратов могут иметь место нежелательные процессы. Во-первых, высокое разбавление может дать возможность эксципиентам или даже содержащимся в эксципиенте примесям вызвать значительное разложение лекарственного вещества во время хранения. Примеры свойств эксципиента, которые могут влиять на разложение лекарственного вещества, включают влагосодержание и подвижность влаги (см. J.T.Carstensen, Drug Stability: Principles and Practices, 2nd Ed. Marcel Dekker, NY, 1995, 449-452) и кислотность эксципиента, оказывающую неблагоприятное влияние на рН местных микросред (см. K.Waterman et al., Pharm. Dev. Tech., 2002, 7 (2), 113-146). Примеры содержащихся в эксципиентах примесей, оказывающих влияние на разложение лекарственного средства, включают следовые количества металлов, пероксиды и муравьиную кислоту (см. K.Waterman et al., Pharm. Dev. Tech., 2002, 7 (1), 1-32). Хотя для толкования возможных путей разложения во внимание принимаются химическая структура и идентификация химически активных групп, предсказание a priori будет ли конкретный эксципиент образовывать приемлемо устойчивый препарат с данным лекарственным веществом остается невозможным. Кроме того, наблюдалось, что соединение 1 взаимодействует со многими обычными эксципиентами и содержащимися в эксципиентах примесями. Поэтому остается потребность в нахождении эксципиента и комбинации эксципиентов, которые могут обеспечить приемлемые составы (для таких свойств, как способность к образованию таблеток) при одновременном обеспечении подходящей устойчивости соединения 1. Авторы также обнаружили конкретные предпочтительные способы формулирования соединения 1 для достижения требуемой устойчивости. Более конкретно, авторы обнаружили конкретные составы и способы, предназначенные для достижения требуемой устойчивости таблеток с пленочным покрытием.
Второй предмет для обсуждения, иногда рассматриваемый в отношении действенных лекарственных средств, полученных при высоком разбавлении, включает изменчивость действенности вследствие сегрегации и прилипания к оборудованию во время производства. Было обнаружено, что это явление является также проблемой при получении препаратов на основе соединения 1. Один метод достижения однородного распределения лекарственного вещества в смеси, содержащей низкую дозу лекарственного вещества, о котором сообщалось недавно, включает использование эксципиента-носителя (лактозы) для образования упорядоченной смеси, содержащей тонкоизмельченное лекарственное вещество (L. Wu et al., AAPS PharmSciTech, 2000, 1 (3), статья 26). Хотя можно эффективно осуществить стадию ручной очистки щеткой для извлечения активного ингредиента, сегрегированного флюидизацией или прилипшего к металлическим поверхностям в мелкомасштабном оборудовании, стадия ручной очистки является как неэффективной, так и нежелательной в среде производственного масштаба. Процессы, осуществляемые в жидкой среде, могут свести к минимуму потерю лекарственного вещества во время производства готовой лекарственной формы; однако соединения, которые подвергаются изменениям формы (например, за счет образования полиморфных, гидратных или сольватных форм), существенно затрудняют процессы в жидкой среде с одновременным сохранением устойчивости лекарственного вещества (физической и химической). Хотя для решения указанных общих проблем использовалось много методов, предсказание того, какие конкретные методы будут эффективными для данного выбора лекарственных веществ и эксципиентов, остается невозможным. Поэтому вследствие высокого разбавления, необходимого для соединения 1, остается потребность в способе, подходящем для коммерциализации соединения 1, посредством которого может быть сохранена адекватная однородность действенности от лекарственной формы (например, таблетки) к лекарственной форме и от партии к партии. Авторы также обнаружили предпочтительные способы получения препаратов соединения 1 для достижения требуемой однородной действенности лекарственного вещества и однородного распределения лекарственного вещества.
Сущность изобретения
Настоящее изобретение относится к определенным лекарственным формам фармацевтического препарата на основе соединения 1 или его фармацевтически приемлемых солей контролируемого высвобождения (CR) для перорального введения нуждающемуся пациенту, при этом лекарственная форма CR содержит указанное соединение или его фармацевтически приемлемую соль и средство для доставки соединения или его фармацевтически приемлемой соли указанному пациенту со скоростью менее приблизительно 6 мгА/час (где мгА относится к миллиграммам активного лекарственного вещества в расчете на свободную основу), вследствие чего происходит введение по меньшей мере около 0,1 мгА соединения или его фармацевтически приемлемой соли в течение 24 часов. Для некоторых пациентов может быть выгодным введение раскрытой в данном описании лекарственной формы немедленного высвобождения (IR), содержащей соединение или его фармацевтически приемлемую соль, после введения лекарственной формы CR последовательными дозами.
Настоящее изобретение в особенности относится к способам лечения с использованием лекарственных форм CR фармацевтического препарата на основе соединения 1, которые приводят к уменьшению тошноты, проявляющейся в качестве нежелательного действия лекарственного средства. Такие лекарственные формы CR характеризуются поступлением лекарственного вещества в желудочно-кишечный тракт (ЖКТ) в растворенной форме со скоростью в диапазоне от около 0,03 мгА/час до около 6 мгА/час, более предпочтительно от около 0,06 мгА/час до около 3 мгА/час и наиболее предпочтительно от около 0,10 мгА/час до около 1 мгА/час. Настоящее изобретение также относится к лекарственным формам CR, которые обеспечивают достижение уменьшения средней максимальной концентрации лекарственного вещества (Смакс.) в крови при первом введении дозы пациенту на величину от 10 до 80% от средней Смакс. относительно первоначального введения лекарственной формы немедленного высвобождения; более предпочтительно на величину от 30 до 70%. В настоящем изобретении также предлагаются лекарственные формы, которые обеспечивают увеличение времени, требуемого для достижения указанного максимального уровня концентрации в крови Тмакс. В частности, было найдено, что увеличение среднего Тмакс. на 50% по сравнению со средним временем, полученным для болюсов немедленного высвобождения, приводит к уменьшению тошноты. В настоящем изобретении также предлагается лекарственная форма, с помощью которой скорость высвобождения соединения 1, определенная методом растворения II типа фармакопеи США, составляет менее 6 мгА/час и время растворения 50 мас./мас.% указанного лекарственного вещества находится в диапазоне от 1 до 15 часов, более предпочтительно в диапазоне от 2 до 10 часов.
Настоящее изобретение также относится к фармацевтическим композициям, предназначенным для достижения указанных скоростей доставки лекарственного вещества. Настоящее изобретение в особенности относится к лекарственным формам соединения 1, которые включают такие средства доставки, как гидрофильные матрицы, гидрофобные матрицы, таблетки CR с покрытием и мультичастицы, трансбуккальные системы, чрескожные системы, суппозитории и депо. Из таблеток с покрытием в особенности предпочтительной лекарственной формой является система, полученная асимметричной мембранной технологией (которая раскрыта в патентах США №№5612059 и 5698220, содержание которых включено в данное описание в качестве ссылок).
В настоящем изобретении также предлагается такая лекарственная форма контролируемого высвобождения, которая представляет собой форму с комбинацией отсроченного и пролонгированного высвобождения, показывающую период замедления до 8 часов перед началом пролонгированного высвобождения, из которой пентаен высвобождается со скоростью не более приблизительно 0,1 мгА/час во время периода замедления и где период замедления регулируется во времени или пространстве положением в желудочно-кишечном тракте.
Задачей настоящего изобретения является также обеспечение уменьшения тошноты в том случае, когда соединение 1 вводят больным, начиная курс лечения с введения лекарственной формы CR, и затем проводя курс лечения с использованием лекарственной формы IR.
Использованный в данном описании термин «контролируемого высвобождения» (CR) относится к лекарственным формам, которые медленно высвобождают лекарственное вещество или доставляют лекарственное вещество больному с такой скоростью, чтобы по меньшей мере часть лекарственного вещества была недоступной в первый час. Система CR может высвобождать лекарственное вещество с постоянной скоростью (нулевого порядка), при равномерном уменьшении скорости (первого порядка) или даже с неровной или пульсирующей скоростью. Доставка лекарственного вещества может также включать латентный период при первоначальном высвобождении лекарственного вещества. Латентный период может иметь временный характер или может зависеть от положения лекарственного средства в организме пациента. Так, например, лекарственная форма CR может быть получена с использованием энтеросолюбильного покрытия, при этом лекарственное вещество высвобождается из нее при достижении рН кишечника после перорального введения.
Подходящая лекарственная форма соединения 1 CR может быть идентифицирована в настоящем изобретении одним или двумя из следующих методов.
(1) Первый метод включает определение поведения лекарственного вещества в лекарственной форме отбором проб и анализом крови после первоначального введения лекарственного средства пациенту (создание фармакокинетического профиля). Первоначальное введение относится к введенному пациенту лекарственному средству или в первый раз, или по меньшей мере через четыре дня после предыдущего введения любой формы соединения 1. Было обнаружено, что особое значение для уменьшения тошноты при использовании соединения 1 имеет максимальный уровень соединения 1 в крови, достигнутый после первоначального введения лекарственного средства (Смакс.), и время, требуемое для достижения такого максимума (Тмакс.). При измерении Смакс. и Тмакс. специалистами в данной области было признано существование значительной изменчивости, зависящей от доз и пациентов. Для достижения адекватного сравнения Смакс. и Тмакс. и, вследствие этого, для определения того, будет ли данная лекарственная форма обеспечивать достижение требуемого уменьшения тошноты, необходимо измерить указанные параметры для по меньшей мере 10 пациентов в опыте, осуществляемом двойным слепым методом (то есть каждый пациент получает обе лекарственные формы IR и CR), в течение по меньшей мере 7 дней между экспериментальными латентными периодами. В частности, было обнаружено, что для уменьшения тошноты является необходимым среднее уменьшение первоначальной Смакс. для достижения значения от 10 до 80% от значения, достигнутого при среднем первоначальном введении лекарственной формы IR; более предпочтительно для достижения значения от 30 до 70%. Что касается Тмакс., то увеличение среднего первоначального Тмакс. для лекарственной формы CR по сравнению с лекарственной формой IR должно составлять по меньшей мере 50% (то есть число часов для средней дозы лекарственной формы CR по сравнению со средней дозой лекарственной формы IR в 1,5 раза больше).
(2) Второй метод анализа лекарственной формы CR для определения того, будет ли происходить уменьшение тошноты, включает испытание in vitro. Авторы обнаружили, что построение графической зависимости процентного содержания растворенного соединения 1 от времени лучше всего использовать для определения времени, необходимого для растворения 50% лекарственного вещества. Данные, необходимые для построения указанной графической зависимости, получают с использованием стандартного прибора для растворения II типа (50 об/мин, 500 мл 0,01 н. соляная кислота, 37°С), используемого в USP (Фармакопея Соединенных Штатов), такого как прибор Хансона модели SR8. Анализ проб осуществляют с использованием ВЭЖХ с обращенной фазой. Было обнаружено, что тошнота уменьшается, когда лекарственная форма показывает растворение 50 мас./мас.% от общей дозы в течение времени от около 1 до около 15 часов, более предпочтительно в течение времени от 2 до 10 часов.
Соответственно, настоящее изобретение дополнительно относится к лекарственным формам немедленного высвобождения, подходящим для введения пациенту, которые являются устойчивыми лекарственными формами с однородным распределением лекарственного вещества и действенности, включающими сердцевину, содержащую соединение формулы 1 или его фармацевтически приемлемую соль, и фармацевтически приемлемый разбавитель, по существу не содержащий редуцирующий карбогидрат. Изобретение в особенности относится к такой лекарственной форме немедленного высвобождения, где лекарственная форма IR содержит L-тартратную или цитратную соль 5,8,14-триазатетрацикло[10.3.1.02,11.04,9]гексадека-2(11),3,5,7,9-пентаена.
Использованное в данном описание выражение «по существу не содержащий редуцирующий карбогидрат» означает содержание редуцирующего сахара менее примерно 20 мас./мас.% (включающего, но без ограничения, лактозу). Лекарственные формы, полученные в соответствии с настоящим изобретением, предпочтительно будут содержать менее 10 мас./мас.% редуцирующего сахара и более предпочтительно менее 5 мас./мас.%.
Лекарственная форма немедленного высвобождения согласно изобретению может дополнительно содержать средство для скольжения, средство, способствующее распадаемости лекарственной формы, и/или смазывающее вещество. Настоящее изобретение также относится к способам получения указанных лекарственных форм немедленного высвобождения.
Лекарственная форма немедленного высвобождения по изобретению может дополнительно включать пленочное покрытие. Настоящее изобретение также относится к способам получения указанных лекарственных форм немедленного высвобождения, покрытых пленкой.
Настоящее изобретение также относится к составу, подходящему для покрытия пленкой лекарственных форм соединения 1 немедленного высвобождения, в котором полимерное связующее для таких покрытий включает по существу полимер целлюлозы. В особенности предпочтительный полимер целлюлозы представляет собой гидроксипропилметилцеллюлозу (НРМС). Указанное покрытие дополнительно содержит глушитель (в особенности диоксид титана), пластификатор и/или средство для скольжения, все из которых содержат менее около 20 мас./мас.% редуцирующих карбогидратов. В особенности предпочтительные составы для покрытия содержат НРМС, диоксид титана и триацетин или ПЭГ (PEG).
Настоящее изобретение также относится к способам, которые обеспечивают высокую действенность и однородность содержимого вышеуказанных смесей. Такие способы включают способ геометрического разбавления лекарственного вещества эксципиентами до осуществления таблетирования. Такие способы также включают использование при смешивании компонентов умеренного сдвигающего усилия. В предпочтительном способе смешивания используется «бункерный смеситель», однако, также применимы другие смесители, которые дают подобные сдвиги.
Раскрытые способы лечения с использованием лекарственных форм фармацевтического препарата на основе соединения 1 CR, которые приводят к уменьшению тошноты, проявляющейся в качестве нежелательного действия лекарственного средства, характеризуются обеспечением лекарственного вещества в желудочно-кишечном тракте (ЖКТ) в растворенной форме со скоростью в диапазоне от около 0,03 мгА/час до около 8 мгА/час, более предпочтительно от около 0,06 мгА/час до около 3 мгА/час и наиболее предпочтительно от 0,10 мгА/час до около 1 мгА/час.
Настоящее изобретение в особенности относится к способу снижения привыкания к никотину или способствования прекращению употребления табака или снижению его употребления пациентом, включающему введение пациенту некоторого количества или лекарственной формы соединения 1 контролируемого высвобождения, или лекарственной формы соединения 1 немедленного высвобождения, которое является эффективным для снижения привыкания к никотину или для способствования прекращению употребления табака или снижению его употребления. Изобретение в особенности относится к такому способу, в котором лекарственная форма CR или IR содержит или L-тартратную, или цитратную соль 5,8,14-триазатетрацикло[10.3.1.02,11.04,9]гексадека-2(11),3,5,7,9-пентаена.
Настоящее изобретение также относится к способу лечения расстройства или состояния, выбранного из воспалительного заболевания кишечника, неспецифического язвенного колита, гангренозной приодермии, болезни Крона, синдрома раздраженной толстой кишки, спастической дистонии, хронической боли, острой боли, брюшной спру, pouchitis, сужения кровеносных сосудов, тревоги, панического состояния, депрессии, биполярного расстройства, аутизма, нарушений сна, расстройства нормального циркадного ритма, проявляющегося повышенной утомляемостью, бокового амиотрофического склероза (ALS), нарушения познавательной способности, гипертензии, булимии, анорексии, ожирения, сердечной аритмии, повышенной секреции желудочного сока, язв, феохромоцитомы, прогрессирующего наднуклеарного паралича, лекарственных зависимостей и привыканий; никотиновой зависимости или привыкания к никотину, табачным изделиям, алкоголю, бензодиазепинам, барбитуратам, опиоидам или кокаину; головной боли, удара, черепно-мозговой травмы (ЧМТ), обсессивно-компульсивного состояния (OCD), психоза, хореи Гентингтона, поздней дискинезии, гиперкинезии, дислексии, шизофрении, слабоумия, вызванного повторными инфарктами, снижения познавательной способности, обусловленного возрастом, эпилепсии, включая эпилептический статус, старческого слабоумия типа болезни Альцгеймера (AD), болезни Паркинсона (PD), повышенной активности, обусловленной недостатком внимания (ADHD), и синдрома Туретта, включающему введение пациенту количества или лекарственной формы соединения 1 контролируемого высвобождения или лекарственной формы соединения 1 немедленного высвобождения, которое является эффективным при лечении такого расстройства или состояния. Изобретение в особенности относится к такому способу, в котором лекарственная форма CR или IR содержит или L-тартратную, или цитратную соль 5,8,14-триазатетрацикло[10.3.1.02,11.04,9]гексадека-2(11),3,5,7,9-пентаена.
Настоящее изобретение также относится к фармацевтическим композициям, предназначенным для достижения указанных скоростей введения. Настоящее изобретение в особенности относится к лекарственным формам соединения 1, которые включают такие средства введения, как гидрофильные матрицы, гидрофобные матрицы, осмотические системы, мультичастицы, лекарственные формы контролируемого высвобождения, имеющие проницаемое покрытие, суппозитории, трансбуккальные системы, чрескожные системы и имплантируемые системы. Из осмотических систем в особенности предпочтительной лекарственной формой является система, полученная асимметричной мембранной технологией (которая раскрыта в патентах США №№5612059 и 5698220, содержание которых включено в данное описание в качестве ссылок).
Настоящее изобретение также относится к способам введения лекарственного средства, которые приводят к уменьшению тошноты, проявляющейся в качестве нежелательного действия, когда соединение 1 вводят больным, начиная курс лечения с введения лекарственной формы CR и затем проводя курс лечения с использованием лекарственной формы IR.
Использованное в данном описании выражение лекарственная форма «немедленного высвобождения» (IR) относится к лекарственной форме, которая при пероральном введении обеспечивает по существу лекарственное вещество в форме, доступной для всасывания, в течение около одного часа.
Выражение «матричная» система относится к особой лекарственной форме CR, в которой лекарственное вещество смешано с эксципиентами, часто в прессованной или экструдированной форме с тем, чтобы высвобождение лекарственного вещества из лекарственной формы регулировалось комбинацией из эрозии и диффузии. Эрозионный контроль доставки лекарственного вещества включает медленное удаление материала матрицы жидкостью ЖКТ для постепенного обнажения и высвобождения лекарственного вещества из матрицы. Диффузионный контроль доставки лекарственного вещества включает диффузию растворимого лекарственного вещества через сетку матрицы эксципиентов регулируемым путем. На практике многие матричные лекарственные формы включают в некоторой степени комбинацию двух механизмов.
«Гидрофильная матрица» представляет собой матрицу лекарственной формы CR, в которой водорастворимые или водонабухаемые полимеры образуют сетку, содержащую лекарственное вещество. Скорость, с которой лекарственное вещество диффундирует к поверхности лекарственной формы, и скорость, с которой матрица устраняется, регулируют скорость, которая делает лекарственное вещество доступным для системы ЖКТ.
«Гидрофобная матрица» представляет собой матрицу лекарственной формы CR, в которой водорастворимые или только частично водорастворимые материалы замедляют скорость, с которой лекарство подвергается текучей среде системы ЖКТ, вследствие чего становится возможным регулирование скорости всасывания лекарственного вещества.
«Проницаемое покрытие» системы CR относится к различным покрытиям на таблетках или частицах, которые действуют в качестве барьеров для лекарственного вещества, высвобождающегося из таблетки, или для воды, доходящей до лекарственного вещества. Такие покрытия включают энтеросолюбильные покрытия, которые становятся проницаемыми, так как рН возрастает, когда лекарственная форма поступает в желудок. Примеры таких покрытий включают Eudragits™, выпускаемый фирмой Rohm GmbH Pharma Polymers (Darmstadt, Германия), и кислый фталат ацетата целлюлозы (CAP), выпускаемый Eastman Chemical (Kingsport, TN). Одна группа таких покрытий систем CR включает осмотические системы. Такие лекарственные формы CR включают полупроницаемую мембрану, окружающую сердцевину лекарственного вещества, оказывающую осмотическое давление, достаточное для приведения в движение воды через мембрану в систему ЖКТ. Осмотическое давление может затем вытолкнуть лекарственное вещество из сердцевины через предварительно образованные или образованные in situ отверстия или поры в покрытии. Такие системы часто включают добавление средств (осмотических средств), предназначенных для повышения осмотического давления в сердцевине. Обзор, описывающий указанные системы, найден в G.Santus and R.W.Baker, J. Control. Rel., 1995, 35, 1-21.
«Асимметричная мембранная технология» AMT описывает специфическую осмотическую систему CR, где покрытие выполнено пористым за счет процесса разделения фаз во время операции нанесения покрытия, как описано в патентах США №№5612059 и 5698220, содержание которых включено в данное описание в качестве ссылок.
«Чрескожные системы доставки» представляют собой приспособления для доставки лекарственного средства, предназначенные для системного обеспечения лекарственного вещества больному через кожу. Такие системы обычно включают слой материала, содержащего лекарственное вещество на подкладке с клеем, предназначенным для прикрепления материала к коже пациента.
«Трансбуккальные системы доставки» представляют собой лекарственные формы, которые обеспечивают процесс всасывания лекарственного вещества через щечную ткань (внутренняя поверхность щеки).
«Депо» представляет собой лекарственную форму лекарственного вещества контролируемого высвобождения, при этом лекарственное вещество и соответствующие эксципиенты вводят или подкожно, или внутримышечно и таким образом образуют массу (матрицу), которая обеспечивает медленное поступление лекарственного вещества в большой круг кровообращения.
Лекарственное вещество 1 для целей настоящего изобретения относится к исходному лекарственному веществу и ко всем его фармацевтически приемлемым солям и пролекарствам.
Термин «мгА» относится к числу миллиграммов активного лекарственного вещества в расчете на свободную основную форму лекарственного вещества.
Выражение «фармацевтически приемлемый» указывает, что вещество или композиция должны быть химически, физически и/или токсикологически совместимы с другими ингредиентами, образующими препарат, и/или с подвергнутым лечению млекопитающим.
Термин «активный ингредиент» относится к терапевтически активному соединению, а также к любым его пролекарствам и фармацевтически приемлемым солям, гидратам и сольватам соединения и пролекарств.
Выражение «соответствующий период времени» или «подходящий период времени» относится к периоду времени, необходимому для достижения требуемого эффекта или результата. Так, например, смесь может быть перемешана до достижения эффективного распределения, которое находится в приемлемом качественном диапазоне для данного применения или использования перемешанной смеси.
Использованный в данном описании термин «стандартная доза» или «стандартная дозировка» относится к физически дискретной единице, которая содержит заданное количество активного ингредиента, рассчитанное на получение требуемого терапевтического действия. Стандартная доза или стандартная дозировка может быть в форме таблетки, капсулы, саше и т.д., которые в данном описании отнесены к «стандартной лекарственной форме».
Подробное описание изобретения
Методики получения соединения 1 описаны в патенте США №6410550, содержание которого включено в данное описание в качестве ссылки, и разделение его рацемических смесей описано в WO 01/62736. В соответствии с настоящим изобретением фармацевтические композиции соединения 1 CR могут быть в случае необходимости введены дозами в диапазоне от около 0,1 до около 6 мгА в день, более предпочтительно от около 0,5 до 4 мгА в день и наиболее предпочтительно от около 1 до 4 мгА в день однократной дозой или дробными дозами, хотя в зависимости от массы тела пациента и его состояния неизбежны отклонения от указанных доз. В зависимости от индивидуальных реакций могут быть более адекватными уровни доз, которые меньше нижнего предела вышеуказанного диапазона, тогда как в других случаях могут быть использованы еще более высокие уровни доз, не вызывающие вредных побочных действий.
Хотя в настоящем изобретении может быть использована любая фармацевтически приемлемая форма соединения 1, предпочтительно использовать солевую форму лекарственного вещества. В особенности предпочтительной солевой формой лекарственного вещества является L-тартратная соль.
Для борьбы с тошнотой с использованием лекарственной формы соединения 1 CR скорость высвобождения лекарственного вещества должна быть такой, чтобы лекарственное вещество дозировалось в систему ЖКТ в форме, доступной для всасывания со скоростью, значительно более медленной, чем скорость всасывания лекарственной формы IR. При использовании в клинических испытаниях дробных дозировок IR было обнаружено, что если лекарственное вещество высвобождается со скоростью, соответствующей примерно 12 мгА/час (при суммарной дозе 3 мгА), частота появления тошноты у испытуемых пациентов превышала 50%. И наоборот, при скорости дозирования, соответствующей около 8 мгА/час (при суммарной дозе 2 мгА) частота появления тошноты падала до величины около 13%. Поэтому верхний предел скорости введения лекарственного средства, необходимый для уменьшения тошноты при использовании лекарственной формы CR, составляет 8 мгА/час. Принимая во внимание настоящее изобретение, можно предвидеть, что при использовании более медленных скоростей высвобождения лекарственного средства будет достигнуто еще большее уменьшение тошноты. Ожидается, что лекарственные формы CR для перорального введения могут обычно подвергаться примерно не более 18 часам всасывания лекарственного вещества в зависимости от подвижности лекарственной формы в организме пациента. Основываясь на уровнях лекарственного вещества в крови пациента, необходимых для его действенности, ожидается, что необходимая суммарная доза лекарственного вещества составляет примерно от 0,5 до 6 мгА в день. С учетом вышесказанного нижний предел скорости введения лекарственного средства составляет примерно 0,03 мгА/час. Хотя данные пределы будут несомненно обеспечивать преимущества, указанные в настоящем изобретении, авторы обнаружили, что для достижения требуемых терапевтических уровней лекарственного вещества в крови пациента с одновременным сохранением уменьшения тошноты, лекарственное средство вводится со скоростью от около 0,06 до 3 мгА/час и более предпочтительно от 0,1 до 1 мгА/час.
Для достижения требуемой скорости введения лекарственного средства был найден ряд средств, обеспечивающих получение такой системы CR. Одним таким средством является матрица. В соответствии с настоящим изобретением могут быть, в частности, получены таблетки соединения 1 с матрицей или мультичастицы соединения 1 с матрицей. В случае мультичастиц конечной лекарственной формой могут быть частицы в капсуле или саше, или другая такая форма. Указанные матричные лекарственные формы могут быть образованы с использованием традиционных методик, таких как прессование с применением таблетирующего пресса, или посредством таких процессов, как экструзия или застывание расплава. Для соединения 1 являются подходящими два типа матричных лекарственных форм: гидрофильные и гидрофобные. Гидрофильная матрица матричного состава обычно состоит из смесей водорастворимых полимеров с высокой и низкой молекулярной массой. Указанные матричные материалы в особенности состоят из комбинаций гидроксипропилметилцеллюлозы (НРМС), полиэтиленоксида (РЕО), гидроксипропилцеллюлозы (НРС), полиакрилатов, альгината, ксантановой смолы и других таких полимеров с различными молекулярными массами. В особенности предпочтительные полимеры включают НРМС и РЕО. В особенности предпочтительный состав состоит из смеси НРМС, выпускаемой под товарным наименованием Methocel™ К4М (Dow Corp., Midland, MI), и вторичного кислого фосфата кальция, выпускаемого под товарным наименованием D-tab™ (Rhodia Inc., Cranbury, NJ). Гидрофобные матричные препараты соединения 1 могут быть получены с использованием гидрофобных материалов, замедляющих соответственно скорость, с которой вода вступает в контакт с соединением 1. В особенности предпочтительные гидрофобные материалы включают карнаубский воск, глицерилбегенат и стеариновую кислоту. Однако специалистам в данной области понятно, что другие подобные воскообразные материалы будут действовать эквивалентным образом.
Требуемый уровень скорости высвобождения соединения 1 могут также обеспечить осмотические лекарственные формы. Примеры таких лекарственных форм описаны в G.Santus и R.W.Baker, J. Control. Rel., 1995, 35, 1-21, включенном в данное описание в качестве ссылки. В особенности предпочтительная осмотическая лекарственная форма соединения 1 находится в форме системы, полученной AMT, которая описана, например, в патентах США №№5612059 и 5698220 (см., например, S.M.Herbig, J. Control. Rel., 1995, 35, 127-136). Такие системы обеспечивают высокий контроль высвобождения лекарственного вещества в системе ЖКТ. Авторы также обнаружили, что предпочтительные препараты состоят из сердцевин, включающих L-тартратную соль лекарственного вещества, маннит, микрокристаллическую целлюлозу, вторичный кислый фосфат кальция и стеарат магния. Такие сердцевины могут быть получены прямым прессованием, мокрым гранулированием (с использованием мокрого гранулятора с высоким или низким сдвигающим усилием или гранулятора с псевдоожиженным слоем), гранулированием выдавливанием, ротационным гранулированием или уплотнением с помощью валков. Уплотнение с помощью валков является в особенности предпочтительным вследствие его способности предотвращать сегрегацию лекарственного вещества с одновременным сохранением устойчивости лекарственного вещества (в отличие от водного мокрого гранулирования, которое может привести к образованию гидрата лекарственного вещества). Таблетки могут быть получены на стандартных таблетирующих прессах (вращательных). Сердцевины таблеток покрывают затем с использованием машины с чашей для нанесения покрытия. Покрытие предпочтительно состоит из смеси ацетата целлюлозы (СА) и полиэтиленгликоля (PEG), нанесенной из ацетона и воды. Соотношение компонентов выбирают таким образом, чтобы комбинация СА/PEG давала пористое, полупроницаемое покрытие, которое вводит лекарственное вещество через поры в ЖКТ с требуемой скоростью. Наиболее предпочтительно отношение СА к PEG выбирают таким образом, чтобы PEG находился в одной фазе с СА, поскольку было обнаружено, что PEG, находящийся в отдельной фазе, приводит к разложению лекарственного вещества в конечной лекарственной форме при повышенных температурах. Для цели настоящего изобретения совместимость фаз может быть определена с использованием стандартного дифференциального сканирующего калориметра при требуемом отношении СА к PEG в смеси. Отсутствие перехода расплава PEG при температуре между 30 и 50°С является показателем единой фазы и, следовательно, показателем того, что такое отношение будет давать предпочтительную пленку. Поэтому наиболее предпочтительно, чтобы отношение СА/PEG оставалось выше приблизительно 4.
Системы CR для неперорального введения могут также обеспечивать уменьшение тошноты с одновременным сохранением эффективности при введении соединения 1. Такие системы включают суппозитории, чрескожные системы, трансбуккальные системы, депо и имплантируемые приспособления. Для функционирования с целью уменьшения тошноты такие приспособления должны обеспечивать контролируемое высвобождение, как описано выше. В особенности предпочтительной лекарственной формой для неперорального введения является чрескожная лекарственная форма.
При использовании всех лекарственных форм CR лекарственное вещество предпочтительно доставляется со скоростью от около 0,06 до 3 мгА/час и более предпочтительно от 0,1 до 1 мгА/час. Пригодность для настоящего изобретения может быть определена испытанием или in vivo, или in vitro. В особенности предпочтительно, чтобы средняя первоначальная Смакс. уменьшалась до достижения значения от 10 до 80% от Смакс., достигнутой при среднем первоначальном введении лекарственной формы IR, более предпочтительно до достижения значения от 30 до 70%. Что касается Тмакс., то увеличение среднего первоначального Тмакс. для лекарственной формы CR по сравнению со средним первоначальным Тмакс. для лекарственной формы IR должно предпочтительно составлять по меньшей мере 50%. Предпочтительные лекарственные формы согласно изобретению обеспечивают в растворе 50 мас./мас.% суммарной дозы в течение времени от около 1 до 15 часов, более предпочтительно в течение времени от 2 до 10 часов.
Системы CR согласно изобретению могут включать период задержки или латентный период между периодом введения дозы и периодом доступности лекарственного вещества для всасывания. Такие задержки могут быть временными или могут зависеть от положения лекарственного средства в желудочно-кишечном тракте. Указанные системы будут эффективными для целей настоящего изобретения при условии, что с момента начала всасывания лекарственного вещества скорость попадает в пределы указанного диапазона. В особенности предпочтительные системы замедленного действия представляют собой таблетки или мультичастицы, покрытые энтеросолюбильным покрытием. Предпочтительные системы с энтеросолюбильным покрытием, распадающиеся в кишечнике, могут быть получены покрытием таблеток или мультичастиц такими материалами, как фталат ацетата целлюлозы, или энтеросолюбильными полиакриловыми соединениями, такими которые выпускаются под маркой Eudragit (Rohm Pharmaceuticals).
Составы, пригодные для настоящего изобретения, могут быть получены с использованием широкого диапазона материалов и способов, известных в данной области. Однако авторы обнаружили, что присутствие редуцирующих карбогидратов является неблагоприятным для устойчивости лекарственного средства при его хранении. В особенности предпочтительны препараты CR, содержащие менее 20 мас./мас.% редуцирующих карбогидратов, более предпочтительными являются препараты CR, содержащие менее 10 мас./мас.% редуцирующих карбогидратов, и наиболее предпочтительны препараты CR, содержащие менее 5 мас./мас.% редуцирующих карбогидратов. Особый редуцирующий сахар, которого предпочтительно следует избегать, представляет собой лактозу.
Для приготовления лекарственных форм контролируемого высвобождения и немедленного высвобождения активный ингредиент может быть использован как таковой или в форме его фармацевтически приемлемой соли, сольвата и/или гидрата. Активный ингредиент может быть использован как таковой или в форме его фармацевтически приемлемой соли, сольвата и/или гидрата. Термин «фармацевтически приемлемая соль» относится к нетоксичным аддитивным солям кислоты, полученным из неорганических и органических кислот. Подходящие солевые производные включают галогениды, тиоцианаты, сульфаты, бисульфаты, сульфиты, бисульфиты, арилсульфонаты, алкилсульфаты, фосфонаты, вторичные кислые фосфаты, первичные кислые фосфаты, метафосфаты, пирофосфонаты, алканоаты, циклоалкилалканоаты, арилалконаты, адипаты, альгинаты, аспартаты, бензоаты, фумараты, глюкогептаноаты, глицерофосфаты, лактаты, малеаты, никотинаты, оксалаты, пальмитаты, пектинаты, пикраты, пивалаты, сукцинаты, тартраты, цитраты, камфораты, камфорсульфонаты, диглюконаты, трифторацетаты и подобные соли.
Конечную фармацевтическую композицию перерабатывают в стандартную лекарственную форму (например, таблетку, капсулу или саше) и затем упаковывают для продажи. Стадия переработки зависит от конкретной стандартной лекарственной формы. Так, например, таблетку обычно прессуют под давлением с приданием ей требуемой формы и для получения капсулы или саше используют обычную операцию заполнения. Специалистам в данной области хорошо знакомы методики, используемые для изготовления различных стандартных лекарственных форм.
Активная смесь лекарственной формы немедленного высвобождения обычно включает один или несколько фармацевтически приемлемых эксципиентов, носителей или разбавителей. Конкретный выбор носителя, разбавителя или эксципиента для использования будет зависеть от средств и цели, для которой применяется активный ингредиент. Обычно таблетированный препарат немедленного высвобождения включает такие средства, как разбавители, связующие, смазывающие вещества, средства для скольжения, средства, способствующие распадаемости лекарственной формы, и их смеси. Хотя специалистам в данной области известно множество таких эксципиентов, авторы обнаружили, что только подгруппа из указанных веществ обеспечивает наиболее устойчивые составы. Авторы в особенности обнаружили, что предпочтительные композиции содержат менее приблизительно 20 мас./мас.% редуцирующих карбогидратов. Редуцирующие карбогидраты представляют собой сахара и их производные, которые содержат свободную альдегидную или кетоновую группу, способную взаимодействовать в качестве восстановителя через передачу электронов. Примеры редуцирующих карбогидратов включают моносахариды и дисахариды и, более конкретно, включают лактозу, глюкозу, фруктозу, мальтозу и другие подобные сахара. Авторы также обнаружили, что композиции, содержащие вторичный кислый фосфат кальция, являются в особенности устойчивыми. Более конкретно, устойчивые композиции получают с использованием более приблизительно 20 мас./мас.% вторичного кислого фосфата кальция. Другие приемлемые эксципиенты включают крахмал, маннит, каолин, сульфат кальция, неорганические соли (например, хлорид натрия), порошкообразные производные целлюлозы, фосфат кальция, сульфат кальция, карбонат магния, оксид магния, полоксамеры, такие как полиэтиленоксид и гидроксипропилметилцеллюлоза. Для обеспечения однородности содержимого смеси в особенности предпочтительно используются частицы лекарственного вещества со средним объемным диаметром, меньшим или равным 30 микронам. Предпочтительными разбавителями являются микрокристаллическая целлюлоза (например, Avicel® PH200, PH102 или PH101, FMC Pharmaceutical, Philadelphia, PA) и вторичный кислый фосфат кальция или дикальцийфосфат (например, A-Tab®, Rhodia, Chicago Heights, IL). Средний размер частиц микрокристаллической целлюлозы обычно находится в диапазоне от около 90 мкм до около 200 мкм. Подходящие сорта вторичного кислого фосфата кальция включают безводный (средний размер частиц от 135 до 180 мкм, PenWest Pharmaceuticals Co., Patterson, NY, или Rhodia, Cranbury, NJ) и дигидрат (размер частиц около 180 мкм, PenWest Pharmaceuticals Co., Patterson, NY, или Rhodia, Cranbury, NJ). Обычно микрокристаллическая целлюлоза присутствует в количестве от около 10 мас.% до около 70 мас.% и вторичный кислый фосфат кальция присутствует в количестве от около 10 мас.% до около 50 мас.%, более предпочтительно микрокристаллическая целлюлоза присутствует в количестве около 30-70 мас.% и вторичный кислый фосфат кальция присутствует в количестве около 20-40 мас.%.
В случае необходимости может быть добавлено связующее. Подходящие связующие включают такие вещества, как целлюлозы (например, целлюлозу, метилцеллюлозу, этилцеллюлозу, гидроксипропилцеллюлозу и гидроксиметилцеллюлозу), полипропилпирролидон, поливинилпирролидон, желатин, гуммиарабик, полиэтиленгликоль, крахмал, природные и синтетические смолы (например, акациевую, альгинаты и гуммиарабик) и воски.
Для предотвращения прилипания таблеток и пуансонов в штампе таблетирующей машины в композиции для изготовления таблеток обычно используют смазывающее вещество. Подходящие смазывающие вещества включают стеарат кальция, глицерилмоностеарат, глицерилпальмитостеарат, гидрированное растительное масло, легкое минеральное масло, стеарат магния, минеральное масло, полиэтиленгликоль, бензоат натрия, лаурилсульфат натрия, стеарилфумарат натрия, стеариновую кислоту, тальк и стеарат цинка. Предпочтительное смазывающее вещество представляет собой стеарат магния. Стеарат магния обычно присутствует в количестве от около 0,25 мас.% до около 4,0 мас.%.
Для распада лекарственной формы и высвобождения активного соединения к композиции могут быть добавлены средства, способствующие распадаемости лекарственной формы. Подходящие средства, способствующие распадаемости лекарственной формы, включают гликолят натриевого крахмала, натриевую соль карбоксиметилцеллюлозы, кальциевую соль карбоксиметилцеллюлозы, натрийкросскармеллозу, поливинилпирролидон, метилцеллюлозу, микрокристаллическую целлюлозу, порошкообразную целлюлозу, низший-алкил-замещенную гидроксипропилцеллюлозу, калийполакрилин, крахмал, предварительно желатинированный крахмал и альгинат натрия. Из указанных средств предпочтительными являются натрийкросскармеллоза и гликолят натриевого крахмала, при этом наиболее предпочтительной является натрийкросскармеллоза. Натрийкросскармеллоза обычно присутствует в количестве от около 0,5 мас.% до около 6 мас.%. Количество средства, способствующего распадаемости, включенного в лекарственную форму, зависит от нескольких факторов, включающих дисперсионные свойства, свойства порообразователя (обсужденные ниже) и свойства выбранного средства, способствующего распадаемости лекарственной формы. Обычно средство, способствующее распадаемости лекарственной формы, составляет от 1 до 15 мас.%, предпочтительно от 1 до 10 мас.% от массы лекарственной формы.
Примеры средств для скольжения включают диоксид кремния, тальк и кукурузный крахмал.
Пленочное покрытие на лекарственной форме немедленного действия может обеспечивать легкость проглатывания, уменьшение неприятного вкуса или запаха во время введения, повышенную фотохимическую устойчивость во время использования глушителя, большую элегантность, уменьшенное трение во время высокоскоростной упаковки или оно может служить в качестве барьера между несовместимыми веществами (G.Cole, J.Hogan и M.Aulton, Pharmaceutical Coating Technology, Taylor and Francis Ltd, Ch.1, 1995). Авторы обнаружили, что при использовании покрытия, содержащие большинство полимеров целлюлозы, обеспечивают высокую химическую устойчивость лекарственного средства. Производные целлюлозы представляют собой полимеры, полученные из целлюлозы. Примеры полимеров включают такие соединения целлюлозы, как гидроксипропилметилцеллюлоза, гидроксипропилцеллюлоза, гидроксиэтилцеллюлоза, метилгидроксиэтилцеллюлоза, метилцеллюлоза и натриевая соль карбоксиметилцеллюлозы. Предпочтительным полимером является гидроксипропилметилцеллюлоза. Покрытия по изобретению включают полимер, глушитель, пластификатор, фармацевтически приемлемый разбавитель/наполнитель и необязательно краситель. Глушитель является эксципиентом, помогающим уменьшить пропускание света через покрытие к сердцевине таблетки. Примеры глушителей включают диоксид титана и тальк. Предпочтительным глушителем является диоксид титана. Пластификатор является материалом, снижающим температуру стеклования полимера, вследствие чего обычно улучшаются физические свойства. Примеры пластификаторов включают многоатомные спирты, такие как глицерин и полиэтиленгликоли, и сложные эфиры уксусной кислоты, такие как глицерилтриацетат (триацетин) и триэтилцитрат. Композиции согласно изобретению необязательно могут включать краситель. Такие красители доступны от ряда поставщиков и хорошо известны специалистам в данной области. В особенности предпочтительные составы покрытий содержат НРМС, триацетин и диоксид титана или НРМС, PEG и диоксид титана.
Для достижения однородного распределения лекарственного вещества в смеси перед изготовлением таблеток или капсул изобретены два способа. В первом способе используется процесс геометрического разбавления. В данном способе приготавливают предварительно смесь лекарственного вещества и части эксципиента и затем смесь дополнительно разбавляют оставшимися эксципиентами во время 2-5 дополнительных стадий. На первой стадии разбавления лекарственное вещество смешивают с 10-30 мас.% эксципиента(ов). На второй стадии разбавления первую предварительно приготовленную смесь дополнительно разбавляют 10-40 мас.% эксципиента(ов). На третьей - пятой стадиях разбавления смесь, полученную после второй стадии разбавления, дополнительно разбавляют 10-75 мас.% эксципиента(ов) с образованием конечной смеси. Предпочтительная схема разбавления включает первое разбавление лекарственного вещества дикальцийфосфатом с двумя приращениями и затем объединение со смесью оставшегося(ихся) эксципиента(ов).
Второй способ достижения однородного распределения лекарственного вещества включает смешение композиции при определенном уровне сдвигающего усилия. Авторы неожиданно обнаружили, что требуемое сдвигающее усилие достигается с использованием или бункерного смесителя, или смесителя с высоким сдвигающим усилием, работающего при низкой скорости сдвига (менее 200 об/мин). Типичное время перемешивания для смешения в бункерном смесителе составляет от около 20 минут до около 30 минут. Хотя может быть использовано время перемешивания более 30 минут, следует заботиться о том, чтобы смесь не разделялась на составные части. После первоначальной стадии перемешивания активная смесь может быть просеяна с использованием конической мельницы (Comil 197, Quadro Engineering Inc., Waterloo, Ontario, Canada), снабженной ситом с размером ячеек 0,8 мм. Затем к активной смеси добавляют смазывающее вещество и перед сухим гранулированием перемешивают в течение около 3 минут в смесителе со сдвоенными статорами «V» или в бункерном смесителе.
Описанные выше способы обеспечивают эффективное смешение и более однородное распределение активного ингредиента без существенного разложения активного ингредиента; однако потеря активного ингредиента, вследствие сегрегации или прилипания соединения к металлическим поверхностям оборудования (например, к ситам и поверхностям емкости), представляет дополнительное препятствие, в особенности для композиций, содержащих низкие дозы активного вещества (например, менее 4 мг в стандартной дозе). Авторы нашли третий путь достижения приемлемой действенности смеси, который включает использование абразивного эксципиента, такого как дикальцийфосфат. Более конкретно, предпочтительные композиции содержат 10-50 мас.% дикальцийфосфата.
Фармацевтическая композиция может быть использована для получения стандартных лекарственных форм, содержащих от около 0,1 мг до около 10,0 мг активного ингредиента в разовой дозе, предпочтительно от около 0,2 мг до около 5,0 мг активного ингредиента в разовой дозе. Масса таблетки (то есть стандартной лекарственной формы) обычно составляет от около 100 мг до 600 мг.
Таблетки обычно получают прессованием на роторном прессе. Однако конкретный способ, используемый для формования таблеток, не является ограничительным и хорошо известен специалистам в данной области. После формования таблетки часто покрывают одним или несколькими покрытиями. Таблетка может быть покрыта покрытием для маскировки вкуса, в качестве герметика и/или в качестве поверхности при печатании логотипа или товарного знака на поверхности таблетки. Альтернативно таблетка может быть покрыта пленкообразующим(и) защитным(и) средством(ами) для модифицирования растворимости таблетки. Так, например, таблетка может быть покрыта пленкообразующим покрытием, которое препятствует растворению в течение заданного периода времени, что приводит к замедленному или пролонгированному высвобождению активного ингредиента. Подходящие пленкообразующие защитные средства включают целлюлозы (например, гидроксипропилметилцеллюлозу, гидроксипропилцеллюлозу, метилцеллюлозу), поливинилпирролидон и сополимеры этилакрилата и метилметакрилата. Составы для нанесения покрытия могут также включать добавки, такие как пластификаторы (например, полиэтиленгликоль или триацетин), консерванты, подсластители, корригенты вкуса, красители и другие известные добавки, обеспечивающие элегантность лекарственного средства. Предпочтительный состав для нанесения покрытия содержит 40-70 мас.% полимера(ов) целлюлозы. Водное покрытие лекарственной формы немедленного высвобождения согласно изобретению содержит Opadry® (YS-1-18202-A) и Opadry Clear® (YS-2-19114-A), производимые Colorcon, West Point, Pennsylvania. Opadry®, применимое в качестве непрозрачного покрытия, содержит гидроксипропилметилцеллюлозу, диоксид титана и полиэтиленгликоль или триацетин. Opadry Clear®, применимое в качестве полировального покрытия, содержит гидроксипропилметилцеллюлозу и триацетин.
Авторы также обнаружили, что предпочтительные композиции состоят из сердцевин, включающих L-тартратную соль лекарственного вещества, маннит, микрокристаллическую целлюлозу, вторичный кислый фосфат кальция и стеарат магния. Более предпочтительные композиции состоят из сердцевин, включающих L-тартратную соль лекарственного вещества, микрокристаллическую целлюлозу, вторичный кислый фосфат кальция и стеарат магния. Еще более предпочтительные композиции состоят из сердцевин, включающих L-тартратную соль лекарственного вещества, микрокристаллическую целлюлозу, вторичный кислый фосфат кальция, натрийкросскармеллозу, диоксид кремния и стеарат магния. Указанные сердцевины могут быть получены прямым прессованием, мокрым гранулированием (с использованием мокрого гранулятора с высоким или низким сдвигающим усилием или гранулятора с псевдоожиженным слоем), гранулированием выдавливанием, ротационным гранулированием или уплотнением с помощью валков. Уплотнение с помощью валков является в особенности предпочтительным, вследствие его способности предотвращать сегрегацию лекарственного вещества с одновременным сохранением устойчивости лекарственного вещества (в отличие от водного мокрого гранулирования, которое может привести к образованию гидрата лекарственного вещества). Таблетки могут быть получены на стандартных таблетирующих прессах (роторных). Сердцевины таблеток затем покрывают с использованием машины с чашей для нанесения покрытия. Предпочтительное покрытие состоит из смеси гидроксипропилметилцеллюлозы, диоксида титана, полиэтиленгликоля или триацетина и необязательно красителя.
Альтернативно активная фармацевтическая смесь может быть заключена в твердые оболочковые капсулы, которые отнесены в данном описании к капсулам, заполненным сухой смесью (DFC). Состав капсул и способ их изготовления подобны сообщенным выше составу для сердцевины таблетки и способу ее получения. Твердая оболочковая капсула может состоять из желатина и воды или гидроксипропилметилцеллюлозы, воды и гелеобразователя (гелеобразующей смолы или каррагенана).
Фармацевтическая композиция (или препарат) может быть упакована множеством способов. Обычно изделие для распределения лекарственной формы включает контейнер, который содержит фармацевтическую композицию в соответствующей форме. Подходящие контейнеры хорошо известны специалистам в данной области и включают такие материалы, как бутылочки (пластмассовые и стеклянные), саше, вытяжные упаковки из фольги и подобные. Контейнер может также включать конструкцию для предотвращения неосторожного доступа к содержимому упаковки. Кроме того, контейнер обычно имеет прикрепленную к нему этикетку, которая описывает содержимое контейнера и соответствующие предостережения или инструкции.
Фармацевтические композиции, содержащие указанное в данном описании соединение 1, являются применимыми для лечения или профилактики, среди прочих заболеваний, заболевания кишечника (включающего, но без ограничения, неспецифический язвенный колит, гангренозную приодермию и болезнь Крона), синдрома раздраженной толстой кишки, спастической дистонии, хронической боли, острой боли, брюшной спру, pouchitis, сужения кровеносных сосудов, тревоги, панического состояния, депрессии, биполярного расстройства, аутизма, нарушений сна, расстройства нормального циркадного ритма, проявляющегося повышенной утомляемостью, бокового амиотрофического склероза (ALS), нарушения познавательной способности, гипертензии, булимии, анорексии, ожирения, сердечной аритмии, повышенной секреции желудочного сока, язв, феохромоцитомы, прогрессирующего наднуклеарного паралича, лекарственных зависимостей и привыканий (например, никотиновый зависимости или привыкания к никотину (и/или к табачным изделиям), алкоголю, бензодиазепинам, барбитуратам, опиоидам или кокаину), головной боли, мигрени, удара, черепно-мозговой травмы (ЧМТ), обсессивно-компульсивного состояния (OCD), психоза, хореи Гентингтона, поздней дискинезии, гиперкинезии, дислексии, шизофрении, слабоумия, вызванного повторными инфарктами, снижения познавательной способности, обусловленного возрастом, эпилепсии, включая эпилептический статус, старческого слабоумия типа болезни Альцгеймера (AD), болезни Паркинсона (PD), повышенной активности, обусловленной недостатком внимания (ADHD) и синдрома Туретта.
Соответственно, фармацевтические композиции, содержащие соединение 1, и способы их получения, раскрытые в данном описании, могут быть использованы в производстве лекарственного препарата для указанных выше терапевтических применений.
Терапевтически эффективное количество изготовленного лекарственного препарата может быть введено человеку в случае необходимости такого лечения или для профилактики. Используемый в данном описании термин «терапевтически эффективное количество» относится к количеству активного ингредиента, способному ингибировать или предотвращать различные патологические состояния или их симптомы и осложнения. Термины «ингибировать» или «ингибирование» относятся к предупреждению, лечению, смягчению, уменьшению интенсивности, приостановлению, сдерживанию, замедлению или обратимости прогрессирования болезни или к уменьшению тяжести патологического состояния или симптома, связанного с соответствующим состоянием больного, подвергаемого лечению, или являющегося результатом такого состояния. Фармацевтические препараты как таковые могут быть использованы для консервативного лекарственного (острого или хронического) и/или профилактического (превентивного) введения соответствующим образом. Доза, частота введения и продолжительность лечения зависят от таких факторов, как происхождение и тяжесть состояния подвергаемого лечению, возраст и общее состояние здоровья хозяина, и от переносимости хозяином активного ингредиента. Фармацевтическая композиция или лекарственный препарат могут быть назначены в виде одной суточной дозы, многократных доз в течение дня или даже недельной дозы. Лекарственное лечение может продолжаться в течение времени от около 2-3 дней до нескольких недель или более. Обычно препарат вводят больному человеку один или два раза в день разовой дозой от около 0,25 мг до около 10,0 мг, но вышеуказанная доза может быть должным образом изменена в зависимости от возраста, массы тела, общего состояния больного и типа введения лекарственного средства.
Следующие примеры представлены с целью иллюстрации и их не следует истолковывать как примеры, ограничивающие объем настоящего изобретения.
Следующий список материалов, использованных в примерах, может быть получен или приобретен из соответствующего источника.
Соединение 1 (L-тартратная соль) может быть получено способами, описанными в заявках на патент WO 9935131 А1 или WO 162736 А1, включенных в данное описание в качестве ссылок.
Микрокристаллическая целлюлоза (Avicel™ PH200), поставляемая FMC Pharmaceutical (Philadelphia, PA).
Маннит (гранулированный 2080), поставляемый SPI Polyols, Inc. (New Castle, DE).
Вторичный фосфат кальция безводный (A-tab™), поставляемый Rhodia Inc. (Chicago Heights, IL).
Натрийкросскармеллоза (Ac-Di-Sol™), поставляемая FMC BioPolymer (Philadelphia, PA).
Гликолят натриевого крахмала (Explotab™), поставляемый Penwest (Patterson, NJ).
Диоксид кремния коллоидный (Cab-O-Sil™), поставляемый Cabot Corporation (Boston, MA).
Силикатированная микрокристаллическая целлюлоза (ProSolv™), поставляемая Penwest (Patterson, NJ).
Гидроксипропилцеллюлоза (Klucel™), поставляемая Hercules, Inc. (Hopewell, VA).
Лактоза безводная, поставляемая Quest International (Norwich, NY).
Стеарат магния животного или растительного происхождения, поставляемый Mallinckrodt (St. Louis, MO).
Пленочные покрытия, Opadry™, поставляемые (Colorcon, West Point, PA).
Ацетат целлюлозы (398-10NF), поставляемый Eastman Chemicals (Kingsport, TN).
Полиэтиленгликоль (PEG 3350), поставляемый Union Carbide Corp. (субсидируемый Dow Chemical Co., Midland, MI).
Гидроксипропилметилцеллюлоза (НРМС, К4М, methocel™), поставляемая Dow Chemical Co., Midland, MI.
Пример 1
Получение лекарственной формы L-тартратной соли соединения 1 CR с использованием АМТ
Загрузку 3 кг таблетируемого гранулометрического состава получали следующим образом: 450 г микрокристаллической целлюлозы и 1602 г вторичного кислого фосфата кальция смешивали в V-смесителе емкостью 8 кварт в течение 10 минут. Половину смеси выгружали в полиэтиленовый мешок, оставляя при этом половину смеси в смесителе. В стеклянный сосуд емкостью 1250 см3 добавляли 450 г маннита и 10,3 г лекарственного вещества. Смесь перемешивали с использованием смесителя Turbula™ (доступного от Geln Mills Inc., Clifton, NJ). Полученную смесь добавляли в V-смеситель, содержащий вышеперечисленные материалы. В стеклянную бутыль дополнительно добавляли 450 г маннита и затем в течение 5 минут перемешивали в смесителе Turbula для вымывания из бутыли лекарственного вещества. Полученную смесь также добавляли в V-смеситель и смесь перемешивали в течение 20 минут. Смесь, которая была выгружена в полиэтиленовый мешок, затем добавляли в V-смеситель и смесь перемешивали в течение еще 20 минут. Затем в V-смеситель добавляли 22,5 г (аликвоту) стеарата магния и смесь перемешивали в течение 5 мин. Смесь уплотняли с использованием валкового уплотнителя TF-Mini (доступного от Vector Corp., Marion IA) с валками DSP при давлении валков 30 кг/см2, скорости вращения валков 4,0 об/мин и скорости вращения шнека 15,6 об/мин. Образованные ленты дробили с использованием мельницы M5A (доступной от Fitzpatrick Corp., Elmhurst, IL) с шероховальным ситом Conidur с размером ячеек 18 меш, вращающимся со скоростью 300 об/мин. Затем порошок опять помещали в V-смеситель и добавляли еще 15 г стеарата магния, после чего осуществляли перемешивание в течение 5 минут.
Гранулометрический состав таблетировали с использованием таблетирующего пресса Kilian T100 (доступного от Kilian & Co. Inc., Horsham, PA) с режущим инструментом SRC 9/32 дюйма (11 мм), при этом получали таблетки массой 250 мг (0,5 мгА). Использованное предварительное сжимающее усилие составляло 2,8 кН, основное сжимающее усилие 8 кН при пропускании через валки со скоростью 74 об/мин, скорость вращения лопасти 20 об/мин. Полученные таблетки показывали твердость 7-9 кп, при этом хрупкость была неизмеримой.
Таблетки покрывали с приготовлением сначала раствора для нанесения покрытия, состоящего из 538 г ацетата целлюлозы и 134,5 г PEG в 4506 г ацетона и 1547 г воды. Нанесение покрытий осуществляли с использованием Hicoater HCT-30EX (доступной от Vector Corp., Marion, IA). Скорость напыления, равную 20,0 г/мин, поддерживали с помощью температуры на выходе 28°С до достижения прироста массы целевого покрытия 27,5%. Затем таблетки сушили на поддоне в печи при 40°С в течение 24 часов.
Таблетки показывали независимый рН растворения in vitro при использовании растворения II типа фармакопеи США (37°С, вращение лопастей со скоростью 50 об/мин, анализ действенности лекарственного вещества с использованием ВЭЖХ). Процент растворенного лекарственного вещества в растворяющей среде в виде функции времени был следующим: через 2 час 1%; через 5 час 8%; через 8 час 35%; через 10 час 52%; через 12 час 65%; через 16 час 81%; через 24 час 95%. Таким образом, композиция доставляет 0,03 мг/час лекарственного вещества после задержки, равной 5 часам.
Пример 2
Результаты клинических испытаний лекарственной формы АМТ из примера 1 на возникновение тошноты
При использовании соединения 1 в клиническом испытании однократной дозы лекарственной формы IR, вводимой стойким некурильщикам, тошнота возникала у 50% пациентов (2/4) при дозе 1 мгА и у 75% пациентов (3/4) при дозе 3 мгА. При введении многократными дозами 1 мгА в день наблюдалась высокая толерантность к лекарственному средству; однако постоянная тошнота была достаточно сильной (7/12 пациентов) при дозе 2 мгА/день, поэтому такое исследование прекращали. При введении однократной дозы здоровым курильщикам тошнота или жалобы возникали у 2/16 пациентов, получивших максимальную дозу 2 мгА IR. И наоборот, дозы 3 и 4 мгА вышеуказанной лекарственной формы АМТ приводили к таким же уровням тошноты, которые наблюдались при введении пониженной дозы лекарственной формы IR (2/16 для каждого случая). В исследованиях, проводимых с введением многократных доз, уровни тошноты, вызываемые введением 3 мгА таблеток АМТ, были сравнимы с уровнями тошноты, вызываемыми введением 1 мгА таблеток IR дважды в день, и значительно превосходили уровни тошноты, вызываемые введением 2 мгА таблеток IR один раз в день.
Пример 3
Получение предпочтительной лекарственной формы L-тартратной соли соединения 1 CR АМТ
Загрузку 7 кг таблетируемого гранулометрического состава получали следующим образом: 1050 г микрокристаллической целлюлозы и 3340 г вторичного кислого фосфата кальция смешивали в V-смесителе емкостью 16 кварт в течение 20 минут. В V-смеситель емкостью 8 кварт добавляли 2450 г маннита и 71,8 г лекарственного вещества. Смесь перемешивали в течение 30 минут. Полученную смесь добавляли в V-смеситель емкостью 16 кварт (к смеси из первого процесса смешивания) и смесь перемешивали в течение 30 мин (смесь может быть использована для ополаскивания смесителя с целью обеспечения надежных полных переносов). Затем в V-смеситель добавляли 52,5 г аликвоту стеарата магния и смесь перемешивали в течение 5 минут. Смесь уплотняли с использованием валкового уплотнителя TF-Mini с валками DSP при давлении валков 30 кг/см2, скорости вращения валков 4,0 об/мин и скорости вращения шнека 15 об/мин, что приводило к получению лент толщиной от 0,06 до 0,08 дюймов. Ленты дробили с использованием мельницы М5А (доступной от Fitzpatrick Corp., Elmhurst, IL) с шероховальным ситом Conidur с размером ячеек 18 меш, вращающимся со скоростью 300 об/мин. Затем порошок опять помещали в V-смеситель и добавляли еще 35 г стеарата магния, после чего осуществляли перемешивание в течение 5 минут.
Гранулометрический состав таблетировали с использованием таблетирующего пресса Kilian T100 с режущим инструментом SRC 9/32 дюйма (11 мм), при этом получали таблетки массой 250 мг (1,5 мгА). Использованное предварительное сжимающее усилие составляло 1,2 кН, основное усилие 8 кН при пропускании через валки со скоростью 74 об/мин, скорость вращения лопасти 20 об/мин. Полученные таблетки показывали твердость 5-8 кп, при этом хрупкость была неизмеримой.
Таблетки покрывали с приготовлением сначала раствора для нанесения покрытия, состоящего из 4095 г ацетата целлюлозы и 405 г PEG в 30,6 кг ацетона и 9,9 кг воды. Нанесение покрытий на таблетки в количестве от 40000 до 48000 штук в партии осуществляли с использованием Hicoater HCT-60 (доступной от Vector Corp., Marion, IA). Скорость напыления, равную 180 г/мин, поддерживали с помощью температуры на выходе 27°С до достижения прироста массы целевого покрытия 13%. Затем таблетки сушили на поддоне в печи при 40°С в течение 16 часов.
Таблетки показывали независимый рН растворения in vitro при использовании растворения II типа фармакопеи США (37°С, вращение лопаток со скоростью 50 об/мин, анализ действенности лекарственного вещества с использованием ВЭЖХ). Процент растворенного лекарственного вещества в растворяющей среде в виде функции времени был следующим: через 2 час 5%; через 5 час 30%; через 7 час 50%; через 10 час 70%; через 12 час 80%; через 24 час 97%. Таким образом, композиция доставляет 0,1 мг/час лекарственного вещества после задержки, равной 2 часам.
Пример 4
Получение лекарственной формы L-тартратной соли соединения 1 CR с гидрофильной матрицей
НРМС К4М (45,000 г) и 50,575 г вторичного кислого фосфата кальция смешивали в бутыли в смесителе Turbula в течение 10 минут. Примерно 10 г полученной смеси объединяли с 3,425 г L-тартратной соли соединения 1 и перемешивали в смесителе Turbula в течение 10 мин. Оставшийся от первой смеси порошок затем добавляли к смеси, содержащей лекарственное вещество и полученную комбинированную смесь перемешивали в смесителе Turbula в течение 20 минут. Затем добавляли стеарат магния (1,000 г) и комбинированную смесь перемешивали в течение еще 3 минут. Таблетки получали с использованием F-пресса Manesty™ (таблетирующая машина с одним пуансоном, доступная от Manesty Corporation, Liverpool, UK), с режущим инструментом SRC 1/4 дюйма. Средняя масса таблетки составляла 102 мг, что соответствовало 0,5 мгА, и твердость таблеток была равна 5-7 кп. Эксперименты с растворением in vitro осуществляли с использованием моделирующей кишечной жидкости (рН 6,8) при 37°С, с применением корзин и таблеток с грузилами и лопастей, вращающихся со скоростью 50 об/мин. Количество лекарственного вещества, растворенного с течением времени, измеряли с использованием анализа действенности лекарственного вещества ВЭЖХ следующим образом: через 2 час 59%; через 4 час 85%; через 8 час 94%; через 16 час 97%. Таким образом, композиция доставляла 0,10 мг/час лекарственного вещества.
Пример 5
Получение лекарственной формы L-тартратной соли соединения 1 CR с гидрофобной матрицей
Смесь 0,86 г соединения 1 и 42,25 г маннита пропускали через сито №36 и затем перемешивали в смесителе Turbula в течение 2 минут. В химический стакан добавляли карнаубский воск (6,04 г) и стеариновую кислоту (0,61 г) и плавили с использованием водяной бани при 90°С. К расплавленной смеси воска и стеариновой кислоты при перемешивании добавляли маннит и лекарственное вещество. После этого теплую смесь просеивали через сито №20 и затем оставляли для охлаждения в течение ночи. Смесь объединяли с 0,09 г диоксида кремния и перемешивали в смесителе Turbula в течение 2 минут. Добавляли стеарат магния (0,17 г) и затем еще перемешивали в смесителе Turbula в течение 0,5 мин. С использованием режущего инструмента SRC 5/16 дюйма и F-пресса получали таблетки массой 200 мг (2 мгА).
Пример 6
Выбор способа производства на основе устойчивости таблеток и эксплуатационных свойств
В этом примере сравниваются традиционные способы прямого прессования и мокрого гранулирования с сухим гранулированием как предпочтительным способом производства. Способ сухого гранулирования представлен с использованием как двойного, так и тройного состава разбавителя.
Сухое гранулирование
В бункерный смеситель добавляли следующие ингредиенты, приведенные в таблице 1, при этом лекарственное вещество размещали слоями между эксципиентами.
Таблица 1
Смесь перемешивали в течение 30 минут. К смеси добавляли стеарат магния и затем перемешивали в течение 3 минут. Смазанную смесь затем уплотняли с помощью валков в ленты при давлении валков 30 кг/см2, скорости вращения валков 4 об/мин и скорости вращения шнека 15 об/мин (с использованием валкового пресса TF-Mini, доступного от Vector Corp., Marion, IA). Ленты дробили через сито с размером ячеек 20 меш (вращающийся гранулятор от Vector) с получением гранулометрического состава. Гранулометрический состав перемешивали в течение 10 минут. К гранулометрическому составу добавляли вторую порцию стеарата магния и перемешивали в течение 3 минут. Конечную смесь прессовали в таблетки массой 200 мг с использованием таблетирующего пресса Kilian T100 (Kilian & Co., Inc., Horsham, PA), снабженного стандартными кругловогнутыми пуансонами размером 5/16 дюйма.
Прямое прессование (сравнительный способ)
Двойной состав разбавителя (то есть микрокристаллическая целлюлоза и вторичный фосфат кальция) получали с уровнями концентраций соединений, перечисленных ниже:
Приготавливали две разные смеси, называемые «преварительной смесью эксципиента» и «активной предварительной смесью». «Предварительная смесь эксципиента» состояла из микрокристаллической целлюлозы, диоксида кремния и натрийкросскармеллозы. Указанные ингредиенты добавляли в V-смеситель и смешивали в течение 20 минут. Активная предварительная смесь состояла из лекарственного вещества и одной половины вторичного кислого фосфата кальция. Ингредиенты активной предварительной смеси добавляли в V-смеситель и смешивали в течение 30 минут и выгружали. Одну половину «предварительной смеси эксципиента» добавляли в смеситель подходящего размера и затем добавляли всю «активную предварительную смесь» и перемешивали в течение 20 минут. В пустой смеситель, использованный для перемешивания «активной предварительной смеси», добавляли вторую часть вторичного кислого фосфата кальция и перемешивали в течение 5 минут. Ее и вторую половину «предварительной смеси эксципиента» добавляли в смеситель, содержащий активную предварительную смесь. Смесь перемешивали в течение 20 минут. К смеси добавляли стеарат магния (5,00 г) и затем перемешивали в течение 5 минут. Конечную смесь прессовали в таблетки массой 200 г с использованием таблетирующего пресса Kilian T100 (Kilian & Co., Inc., Horsham, PA), снабженного стандартными кругловогнутыми пуансонами размером 5/16 дюйма.
Мокрое гранулирование (сравнительные композиции и способы)
Способ мокрого гранулирования оценивали с использованием двух различных гранулирующих средств, включающих воду и изопропиловый спирт. Композиции, приготовленные для каждого из гранулирующих средств, перечислены в таблице 2.
Таблица 2
Химически неактивные ингредиенты, перечисленные над гранулирующим средством (вода или изопропиловый спирт) в таблице 2 композиции, добавляли в смеситель с высоким сдвигающим усилием и перемешивали в сухом виде в течение 1 минуты при скорости вращения импеллера 100 об/мин. Одну половину смеси эксципиента удаляли из контейнера и в смеситель добавляли все количество L-тартрата соединения 1 и покрывали удаленной смесью. Полученную смесь перемешивали в течение 1 минуты со скоростью 100 об/мин. При продолжении перемешивания смеси в течение 1 минуты добавляли гранулирующее средство со скоростью вращения рубильной машины 1000 об/мин и скоростью вращения импеллера 300 об/мин. Мокрый гранулометрический состав перемешивали в течение еще 15 секунд и затем добавляли воду или изопропиловый спирт. Мокрую массу сушили в печи с температурой 50°С до уровня содержания влаги 1% относительно первоначального значения перед гранулированием. Высушенный гранулометрический состав дробили в конической мельнице (Comil, Quadro Engineering, Inc., Waterloo, Ontario, Canada), снабженной ситом с размером ячеек 0,050 дюйма и импеллером с закругленными краями, вращающимся со скоростью 1770 об/мин. К указанному гранулометрическому составу добавляли коллоидный диоксид кремния и перемешивали в V-смесителе в течение 20 минут. В смеситель добавляли стеарат магния и перемешивали в течение 5 минут. Конечную смесь прессовали в таблетки массой 300 мг с использованием таблетирующего пресса Kilian T100 (Kilian & Co., Inc. Horsham, PA), снабженного стандартными кругловогнутыми пуансонами размером 11/32 дюйма.
Однородность смеси способов прямого прессования и сухого гранулирования сравнивали ниже. Для загрузок использовали одну и ту же поступающую насыпью партию лекарственного вещества, загрузку лекарственного вещества (0,868%) и одну и ту же массу таблеток (200 мг). Данные действенности лекарственного средства и изменчивости сведены в таблицу 3 для способов прямого прессования и сухого гранулирования. Влияние сухого гранулирования композиции на однородность смеси иллюстрируется уменьшением изменчивости смеси RSD с 8% до 1,8%.
(% RSD)
Высокая изменчивость (8% RSD) действенности в конечной смеси перед прямым прессованием таблеток была основой для выбора сухого гранулирования в качестве предпочтительного способа.
Способы мокрого и сухого гранулирования сравнивали по эксплуатационным свойствам с точки зрения действенностей и изменчивостей гранулированных смесей и таблеток (процент относительно стандартного отклонения или %RSD). Для загрузок использовали одну и ту же поступающую насыпью партию лекарственного вещества, загрузку лекарственного вещества (0,57%) и одну и ту же массу таблеток (300 мг). Данные действенности и изменчивости сведены в таблицу 4 для трех оцененных способов гранулирования.
В начале
В начале
Значения действенности гранулометрического состава и таблеток являются наиболее близкими к желаемым 100% в способе мокрого гранулирования, в котором в качестве гранулирующего средства использовалось вода. Способы сухого гранулирования и мокрого гранулирования с изопропиловым спиртом приводили к получению подобных результатов в отношении эксплуатационных свойств.
В представленной таблице 5 показаны результаты устойчивости таблеток, хранимых при ускоренных условиях старения в течение 6 недель и подвергнутых анализу ВЭЖХ в способах мокрого и сухого гранулирования.
Было обнаружено, что мокрое гранулирование с использованием в качестве гранулирующего средства воды является физически нестойким вследствие превращения состояния L-тартрата соединения 1 из безводного в гидратное. Впоследствии гидратная форма утрачивалась во время стадии сушки и образовывалась безводная лекарственная форма. Указанные изменения физической стойкости во время процесса мокрого гранулирования с водой и сушки помогали определиться при выборе предпочтительного способа. Сухое гранулирование и мокрое гранулирование с изопропиловым спиртом являются предпочтительными способами получения таблеток на основе L-тартрата соединения 1. Способом, обеспечивающим наименьший общий уровень примесей, был способ сухого гранулирования, после которого следовало мокрое гранулирование с водой и затем мокрое гранулирование с изопропиловым спиртом.
Следовательно, наиболее предпочтительным способом гранулирования для изготовления таблеток L-тартрата соединения 1, с точки зрения устойчивости, однородности смеси и эксплуатационных свойств, является сухое гранулирование.
Пример 7
Выбор разбавителя на основе устойчивости таблеток
Разбавители, используемые при изготовлении таблеток на основе L-тартрата соединения 1, выбирали на основе химической стойкости и эксплуатационных свойств. Оценивали три разбавителя (вторичный кислый фосфат кальция, микрокристаллическую целлюлозу и маннит) с использованием предпочтительного способа сухого гранулирования и в композицию включали два (бинарный разбавитель) или три (трехкомпонентный разбавитель) разбавителя (см. таблицу 6).
Таблица 6
В представленной ниже таблице 7 приведены результаты устойчивости таблеток, полученных способом сухого гранулирования с использованием или трехкомпонентного, или бинарного (без вторичного кислого фосфата кальция) состава разбавителя, хранимых в течение 3 месяцев при ускоренных условиях старения и анализированных ВЭЖХ.
В композиции, полученной сухим гранулированием, обеспечивающей самый низкий общий уровень примесей, использовали вторичный кислый фосфат кальция. Предпочтительные композиции, полученные сухим гранулированием, содержат бинарные или трехкомпонентные разбавители, включающие вторичный кислый фосфат кальция, микрокристаллическую целлюлозу и маннит. Наиболее предпочтительные композиции, полученные сухим гранулированием, содержат вторичный кислый фосфат кальция в качестве одного из главных разбавителей.
В представленной ниже таблице 8 приведены результаты устойчивости таблеток, хранимых при ускоренных условиях старения в течение 6-12 недель и анализованных ВЭЖХ, при этом таблетки получены предпочтительным способом сухого гранулирования с использованием или трех бинарных составов разбавителя, или трехкомпонентного состава разбавителя.
Было обнаружено, что бинарный состав разбавителя, содержащий лактозу/вторичный кислый фосфат кальция, является менее устойчивым при ускоренных условиях старения, включающих температуру/влажность. Как показано в таблице 8, таблетки с бинарным разбавителем микрокристаллическая целлюлоза/вторичный кислый фосфат кальция и маннит/вторичный кислый фосфат кальция показывали общий уровень содержания примесей, подобный таковому в таблетках с трехкомпонентным составом разбавителя. Поэтому трехкомпонентные составы и двухкомпонентные составы МСС (микрокристаллическая целлюлоза)/Dical (вторичный кислый фосфат кальция) и маннит/Dical являются предпочтительными вариантами изобретения.
Пример 8
Выбор разбавителя на основе эксплуатационных свойств и однородности содержимого таблеток
Если основываться только на химической устойчивости, то подходящими композициями L-тартрата соединения 1 являются композиции, содержащие два бинарных состава разбавителя (MCC/Dical и маннит/Dical), описанные в примере 7. Для выбора наиболее предпочтительной композиции осуществляли производственную оценку на прессе Kilian T100 при трех положениях режущего инструмента SRC 5/16 дюйма. Таблетки прессовали при усилии 4, 8, 12, 16 и 20 кН и проводили испытания для определения массы, толщины, твердости, времени распадаемости и % хрупкости при каждом состоянии. Указанные данные перечислены в таблице 9.
Для бинарного состава маннит/вторичный кислый фосфат кальция требовались жесткие условия нанесения покрытия и он не мог быть таблетирован до твердости выше 3 кП, тогда как целевой диапазон для данного размера инструмента составляет 6-9 кП. При указанных твердостях таблетки имели плохую механическую целостность, принимая во внимание высокий % хрупкости (требуемая величина менее 0,2%). Альтернативно таблетки, содержащие бинарный разбавитель МСС/вторичный кислый фосфат кальция, имели значения твердости и хрупкости в пределах целевых диапазонов. Поэтому более предпочтительным бинарным составом разбавителя на основе производственной оценки является состав микрокристаллическая целлюлоза/вторичный кислый фосфат кальция. Трехкомпонентный состав является предпочтительным составом на основе устойчивости и производственной оценки и также является вариантом данного изобретения.
Пример 9
Выбор средства, способствующего распадаемости лекарственной формы, на основе устойчивости таблеток
Таблетки, содержащие гликолят натриевого крахмала (SSG) в качестве средства, способствующего распадаемости лекарственной формы, анализировали на степень чистоты и сравнивали с таблетками, содержащими натрийкросскармеллозу (CS). Таблетки помещали в сосуды емкостью 60 см3, изготовленные из ПЭВП (HDPE)/HIS, при 5°С/RH 75%, 40°С/RH 75% и 50°С/RH 20% для осуществления анализа через 6 и 12 недель. Результаты определения степени чистоты через 6 и 12 недель показаны в таблице 10.
Разложение таблеток, содержащих SSG (от 0,3 до 1,1%), является более высоким, чем разложение, наблюдаемое для таблеток, содержащих в качестве средства, способствующего распадаемости лекарственной формы CS. Общая степень разложения таблеток, содержащих CS, никогда не превышала значения 0,3% при отсутствии в таблетках лактозы в любом состоянии через 6 или 12 недель. По этой причине натрийкросскармеллоза выбрана как более желательное средство, способствующее распадаемости таблеток на основе L-тартрата соединения 1, что было установлено на основе повышенной химической устойчивости по сравнению с гликолятом натриевого крахмала.
Пример 10
Средство для скольжения, включенное в композицию для уменьшения когезионной способности смеси
Влияние добавления средства для скольжения, в данном случае коллоидного диоксида кремния, в композицию для получения таблеток оценивали с использованием испытания на осыпание стандартного порошка для охарактеризовывания характеристик текучести. Для указанной оценки использовали бинарный состав плацебо, поскольку загрузка лекарственного вещества была менее 1%. Составы перечислены в таблице 11. Указанные таблетки получали способом сухого гранулирования, описанным в примере 6.
Непосредственно перед каждой из стадий смазывания отбирали пробы смеси и гранулометрического состава для анализа. Оценивали когезионную способность, изменчивость текучести и размер частиц, полученные результаты представлены в таблице 12. Размер частиц двух партий гранулометрического состава был очень подобным и поэтому не должен был оказывать влияние на результаты осыпания порошка. Когезионная способность и изменчивость текучести усовершенствовались за счет присутствия диоксида кремния. Его добавление уменьшало когезионную способность до степени от «низкой» до «очень низкой» для смесей и до степени от «высокой» до «низкой» для гранулометрических составов. Присутствие 0,50% диоксида кремния также уменьшало степень изменчивости текучести гранулометрического состава от умеренной до низкой.
Во время таблетирования усилие извлечения из формы регулировали как функцию усилия прессования. В таблице 13 перечислены усилия извлечения из формы, полученные в результате усилий прессования в диапазоне 5-20 кН композиций, содержащих диоксид кремния в диапазоне от 0 до 0,5%.
Таблица 13
8,9
27,47
18,6
21,56
9,1
25,40
18,6
23,56
Таблетки, содержащие 0,50% Cab-O-Sil, показывали слегка более низкое усилие извлечения из формы в большей части указанного диапазона прессования. На основе требуемых свойств (уменьшенной когезионной способности, изменчивости текучести и усилий извлечения из формы) было установлено, что таблетки, содержащие средство для скольжения, являются более предпочтительным препаратом.
Пример 11
Выбор пленочного покрытия на основе устойчивости таблеток
Предпочтительное белое пленочное покрытие для таблеток L-тартрата соединения 1 выбирали на основе химической устойчивости с использованием ускоренных условий старения. Четыре состава для нанесения белого пленочного покрытия Opadry наносили на одну из более предпочтительных сухих гранулированных таблеточных композиций.
Сердцевины таблеток получали с использованием схемы смешивания, включающей геометрическое разбавление, перед уплотнением с помощью валков, содержащиеся в сердцевинах компоненты перечислены ниже:
Приготавливали две разные смеси, называемые «предварительной смесью эксципиента» и «активной предварительной смесью». «Предварительная смесь эксципиента» состояла из микрокристаллической целлюлозы, диоксида кремния и натрийкросскармеллозы. Указанные ингредиенты добавляли в V-смеситель и смешивали в течение 20 минут. «Активная предварительная смесь» состояла из лекарственного вещества и одной половины вторичного кислого фосфата кальция. Ингредиенты «активной предварительной смеси» добавляли в V-смеситель и смешивали в течение 30 минут и выгружали. Одну половину «предварительной смеси эксципиента» добавляли в V-смеситель подходящего размера и затем добавляли всю «активную предварительную смесь» и перемешивали в течение 20 минут. В пустой смеситель, использованный для перемешивания «активной предварительной смеси», добавляли вторую часть вторичного кислого фосфата кальция и перемешивали в течение 5 минут. Ее и вторую половину «предварительной смеси эксципиента» добавляли в смеситель, содержащий активную предварительную смесь. Смесь перемешивали в течение 20 минут. К смеси добавляли первую порцию стеарата магния и затем перемешивали в течение 5 минут. Смазанную смесь уплотняли с помощью валков в ленты с использованием давления валков 30 кг/см2, скорости вращения валков 4 об/мин и скорости вращения шнека 15 об/мин (валковый уплотнитель TF-Mini от Vector). Ленты измельчали пропусканием через сито с размером ячеек 20 меш (вращающийся гранулятор от Vector) с получением гранулометрического состава. Гранулометрический состав перемешивали в течение 10 минут. К гранулометрическому составу добавляли вторую часть стеарата магния и перемешивали в течение 5 минут. Конечную смесь прессовали в таблетки массой 200 мг с использованием таблетирующего пресса Kilian T100 (Kilian & Co., Inc., Horsham, PA), снабженного стандартными кругловогнутыми пуансонами размером 5/16 дюйма.
Качественные композиции четырех составов для нанесения покрытия перечислены в таблице 14. Композиция для нанесения покрытия, указанная в виде партии номер А, состояла из лактозы, гидроксипропилметилцеллюлозы или НРМС, диоксида титана и триацетина. Основное различие между составами для нанесения покрытия В-D, не содержащими лактозу, заключалось в типе полимера (гидроксипропилметилцеллюлоза или НРМС в сравнении с поливиниловым спиртом или PVA) и типе пластификатора (полиэтиленгликоль или PEG и триацетин). PVA покрытие содержало также тальк. Конечные лекарственные формы покрывали 4 мас.% белого покрытия и 0,5 мас.% прозрачного покрытия. Покрытые пленкой таблетки помещали на 10 дней в сосуды емкостью 60 см3, изготовленные из ПЭВП (HDPE)/HIS, и вызывали старение при 5°С и 70°С/RH 75% и затем оценивали степень чистоты. Для сравнения оценивали также непокрытые сердцевины таблеток. Получали таблетки плацебо и анализировали на степень чистоты в первоначальный момент времени в качестве контроля. Установленные значения чистоты показаны в таблице 15.
Было обнаружено, что содержащие НРМС таблетки (В и С), покрытые пленкой, не содержащей лактозу, являются более химическими устойчивыми, чем таблетки, покрытые пленкой из лактозы/НРМС (А) или PVA (D). Было обнаружено, что общая степень разложения партий таблеток с покрытием, содержащим НРМС, и таблеток с покрытием, содержащим PEG и триацетиновый пластификатор, составляла 0,4-1,2% и 0,5-1,0% соответственно. Между тем общая степень разложения контрольных партий с лактозой и партий с PVA была высокой и составляла соответственно 3,5 и 2,9%. На основе повышенной химической устойчивости установлено, что предпочтительные пленочные покрытия состояли из НРМС, диоксида титана и или триацетина, или PEG в составах В и С соответственно.
Пример 12
Выбор способа, обеспечивающего однородность содержимого сухого гранулометрического состава
Этот пример иллюстрирует более предпочтительный способ смешения для обеспечения действенности и однородности смеси и таблеток. Оценивали V-смешивание (с геометрическим разбавлением и без него), бункерное смешивание (с отражательными перегородками и без них и с прямым вращением в сравнении с угловым вращением) и смешивание с высоким сдвигающим усилием. Композиция содержала бинарный состав разбавителя, содержащий вторичный кислый фосфат кальция и микрокристаллическую целлюлозу, компоненты композиции перечислены ниже.
V-Смешивание с геометрическим разбавлением
Описание состава для сердцевины таблеток и способа ее получения представлено в примере 11.
V-Смешивание за одну стадию
Смесь (без смазывающего вещества) перемешивали в течение 30 минут. К смеси добавляли первую порцию стеарата магния и затем перемешивали в течение 5 минут. Смазанную смесь уплотняли валками в ленты при давлении валков 30 кг/см2, скорости вращения валков 4 об/мин и скорости вращения шнека 15 об/мин (валковый уплотнитель TF-Mini от Vector). Ленты дробили через сито с размером ячеек 20 меш (вращающийся гранулятор от Vector) с получением гранулометрического состава. Гранулометрический состав перемешивали в течение 10 минут. К гранулометрическому составу добавляли вторую порцию стеарата магния и перемешивали в течение 5 минут. Конечную смесь прессовали в таблетки массой 200 мг с использованием таблетирующего пресса Kilian T100 (Kilian & Co., Horsham, PA), снабженного стандартными кругловогнутыми пуансонами.
Бункерное смешивание
В бункерный смеситель с лекарственным веществом, расположенным слоями в середине, добавляли ингредиенты (без смазывающего вещества). Устанавливали конфигурацию смесителя (с отражательными перегородками или без них и с прямым или угловым вращением). Смесь перемешивали в течение 30 минут, добавляли первую порцию смазывающего вещества и перемешивали в течение 5 минут. Смазанную смесь уплотняли валками в ленты при давлении валков 30 кг/см2, скорости вращения валков 4 об/мин и скорости вращения шнека 15 об/мин (валковый уплотнитель TF-Mini от Vector). Ленты дробили через сито с размером ячеек 20 меш (вращающийся гранулятор от Vector) с получением гранулометрического состава. Гранулометрический состав перемешивали в бункерном смесителе в течение 10 минут. К гранулометрическому составу добавляли вторую порцию стеарата магния и перемешивали в течение 5 минут. Конечную смесь прессовали в таблетки массой 200 мг с использованием таблетирующего пресса Kilian T100 (Kilian & Co., Horsham, PA), снабженного стандартными кругловогнутыми пуансонами размером 5/16 дюйма.
Смешивание с высоким сдвигающим усилием
В смеситель с высоким сдвигающим усилием с лекарственным веществом, расположенным слоями в середине, добавляли ингредиенты (без смазывающего вещества). Смесь перемешивали в течение 10 минут со скоростью вращения импеллера 200 об/мин и скоростью рубильной машины 0 об/мин. Добавляли первую порцию смазывающего вещества и перемешивали в течение 5 минут. Смазанную смесь уплотняли валками в ленты при давлении валков 30 кг/см2, скорости вращения валков 4 об/мин и скорости вращения шнека 15 об/мин (валковый уплотнитель TF-Mini от Vector). Ленты дробили через сито с размером ячеек 20 меш (вращающийся гранулятор от Vector) с получением гранулометрического состава. Гранулометрический состав перемешивали в V-смесителе в течение 10 минут. К гранулометрическому составу добавляли вторую порцию стеарата магния и перемешивали в течение 5 минут. Конечную смесь прессовали в таблетки массой 200 мг с использованием таблетирующего пресса Kilian T100 (Kilian & Co., Horsham, PA), снабженного стандартными кругловогнутыми пуансонами размером 5/16 дюйма.
Результаты действенности и однородности гранулометрического состава и таблеток представлены в таблице 16. Способы V-смешивания за одну стадию и смешивания с высоким сдвигающим усилием приводили к получению самых низких значений действенности гранулометрического состава. Более предпочтительным способом смешивания, как было установлено на основе результатов действенности и однородности гранулометрического состава и таблеток, является смешивание с геометрическим разбавлением и бункерное смешивание с любой конфигурацией отражательных перегородок и любым вращением. Смеситель с высоким сдвигающим усилием, работающий при низких скоростях вращения импеллера (от низкого до умеренного сдвига в данном смесителе), является также предпочтительным вариантом настоящего изобретения.
Пример 13
Выбор разбавителя на основе однородности содержимого гранулометрического состава
Предпочтительный разбавитель, используемый в «активной предварительной смеси» в способе смешивания с геометрическим разбавлением, выбирали на основе действенности и однородности гранулометрического состава и таблеток. Исследовали два основных разбавителя (вторичный кислый фосфат кальция и маннит) на их способность быть эксципиентом-носителем для способствования перемешиванию L-тартрата соединения 1 в композиции. Ингредиенты, используемые в таблеточной композиции, содержащей трехкомпонентный разбавитель (такая же композиция, как в примере 7), перемешивали в соответствии со схемой геометрического разбавления, описанной в примере 11. В «активной предварительной смеси» использовали или одну половину маннита (13А), или вторичного кислого фосфата кальция (13В). В этом примере лекарственное вещество измельчали в струйной мельнице примерно до половины первоначального среднего размера частиц перед смешиванием с эксципиентами.
Таблица 17
Для каждой таблеточной композиции приготавливали две различные смеси, называемые в настоящем описании «предварительной смесью эксципиента» и «активной предварительной смесью». «Предварительная смесь эксципиента» состояла из микрокристаллической целлюлозы, диоксида кремния, натрийкросскармеллозы и вторичного кислого фосфата кальция или маннита. Указанные ингредиенты добавляли в V-смеситель и смешивали в течение 20 минут. «Активная предварительная смесь» состояла из лекарственного вещества и примерно одной половины или маннита (12А), или вторичного кислого фосфата кальция (12В). Ингредиенты «активной предварительной смеси» добавляли в V-смеситель, смешивали в течение 30 минут и выгружали. Одну половину «предварительный смеси эксципиента» добавляли в V-смеситель подходящего размера, затем добавляли всю «активную предварительную смесь» и перемешивали в течение 20 минут. В пустой смеситель, использованный для перемешивания «активной предварительной смеси», добавляли вторую часть маннита или вторичного кислого фосфата кальция и перемешивали в течение 5 минут. Ее и вторую половину «предварительной смеси эксципиента» добавляли в смеситель, содержащий активную предварительную смесь. Смесь перемешивали в течение 20 минут. К смеси добавляли первую порцию стеарата магния и затем перемешивали в течение 5 минут. Смазанную смесь уплотняли валками в ленты при давлении валков 30 кг/см2, скорости вращения валков 4 об/мин и скорости вращения шнека 15 об/мин (валковый уплотнитель TF-Mini Vector). Ленты дробили через сито с размером ячеек 20 меш (вращающийся гранулятор от Vector) с получением гранулометрического состава. К гранулометрическому составу добавляли вторую порцию стеарата магния и перемешивали в течение 5 минут. Конечную смесь прессовали в таблетки массой 300 мг с использованием таблетирующего пресса Kilian T100 (Kilian & Co., Horsham, PA), снабженного стандартными кругловогнутыми пуансонами размером 11/32 дюйма. Значения действенности и изменчивости (в виде % RSD) конечного гранулометрического состава и таблеток перечислены в таблице 18.
Значения действенности гранулометрических составов с маннитом и вторичным кислым фосфатом кальция, которые являются разбавителями «активной предварительной смеси», подобны. Однако значения действенности таблеток увеличивались от 95,1 до 97,2%, когда в качестве разбавителя «активной предварительной смеси», использованной в способе смешивания с геометрическим разбавлением, вместо маннита использовали вторичный кислый фосфат кальция. Следовательно, более предпочтительным разбавителем, используемым в «активной предварительной смеси» способа смешивания с геометрическим разбавлением, является вторичный кислый фосфат кальция.
| название | год | авторы | номер документа |
|---|---|---|---|
| Твёрдые лекарственные формы палбоциклиба | 2016 |
|
RU2686840C1 |
| СУХИЕ ГРАНУЛИРОВАННЫЕ КОМПОЗИЦИИ АЗИТРОМИЦИНА | 2003 |
|
RU2283092C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ АТОРВАСТАТИНА | 2004 |
|
RU2325903C2 |
| НОВАЯ КОМПОЗИЦИЯ ЛАПАТИНИБА В ВИДЕ ТВЕРДОЙ ЛЕКАРСТВЕННОЙ ФОРМЫ ДЛЯ ПЕРОРАЛЬНОГО ПРИМЕНЕНИЯ И СПОСОБ ЕЕ ИЗГОТОВЛЕНИЯ | 2019 |
|
RU2821950C2 |
| СТАБИЛЬНЫЙ ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ, СОДЕРЖАЩИЙ АМЛОДИПИНА БЕЗИЛАТ И АТЕНОЛОЛ | 1998 |
|
RU2188636C2 |
| ТАБЛЕТКИ ТАМЗУЛОСИНА | 2002 |
|
RU2311903C2 |
| МАТРИЧНАЯ ТАБЛЕТКА С МОДИФИЦИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ НЕРАМЕКСАНА | 2006 |
|
RU2422135C2 |
| ПЕРОРАЛЬНАЯ ФАРМАЦЕВТИЧЕСКАЯ ЛЕКАРСТВЕННАЯ ФОРМА С ПРОДОЛЖИТЕЛЬНЫМ ВЫСВОБОЖДЕНИЕМ | 1998 |
|
RU2214232C2 |
| ЛЕКАРСТВЕННЫЕ ФОРМЫ АЗИТРОМИЦИНА С КОНТРОЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ | 1995 |
|
RU2130311C1 |
| СПОСОБЫ МОКРОЙ ГРАНУЛЯЦИИ АЗИТРОМИЦИНА | 2002 |
|
RU2283651C2 |
Настоящее изобретение относится к лекарственной форме контролируемого высвобождения, предназначенной для уменьшения привыкания к никотину. Лекарственная форма содержит 5,8,14-триазатетрацикло[10.3.1.02,11.04,9]гексадека-2(11),3,5,7,9-пентаен или его фармацевтически приемлемую соль и вводится пациенту со скоростью менее 6 мг/час, что соответствует введению по меньшей мере около 0,1 мг указанного соединения в течение 24 часов. Изобретение также относится к лекарственной форме немедленного высвобождения, содержащей низкую дозу указанного лекарственного вещества. Способ применения заключается в оральном введении на первой стадии лекарственной формы контролируемого высвобождения за период времени от около 1 дня до около 30 дней с последующим введением на второй стадии лекарственной формы немедленного высвобождения. Изобретение обеспечивает уменьшение привыкания к никотину с одновременным уменьшением частоты появления тошноты в качестве нежелательного действия. 3 н. и 7 з.п. ф-лы, 18 табл.
| WO 9935131 A1, 15.07.1999 | |||
| 0 |
|
SU162736A1 | |
| СПОСОБ ВЫДЕЛЕНИЯ БЛАГОРОДНЫХ МЕТАЛЛОВ ИЗ ОТХОДОВ ПИРОМЕТАЛЛУРГИИ | 1994 |
|
RU2092597C1 |
| ВАННА ПАСТЕРИЗАЦИИ | 1995 |
|
RU2092089C1 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКОГО АМИДА ИЛИ ИХ ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, ОБЛАДАЮЩАЯ АНТИАЦЕТИЛХОЛИНЭСТЕРОДНОЙ АКТИВНОСТЬЮ | 1991 |
|
RU2010794C1 |
| RU 94032792 A1, 10.09.1996. | |||

