ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Область изобретения
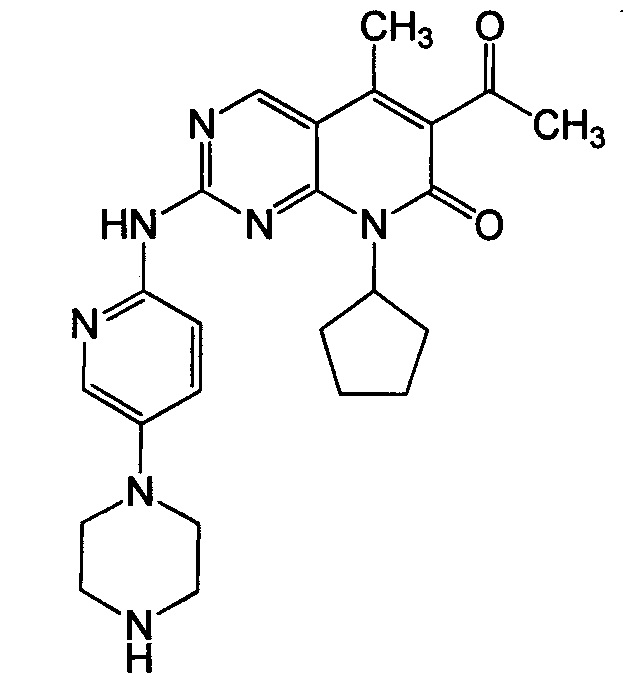
Настоящее изобретение относится к твердым лекарственным формам 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8H-пиридо[2,3-d]пиримидин-7-она (далее - палбоциклиб), имеющим желаемые фармакокинетические характеристики и демонстрирующим надлежащую стабильность при хранении и растворимость.
Описание предшествующего уровня техники
Палбоциклиб является сильным и селективным ингибитором CDK4 и CDK6, имеющим следующую структуру:
Палбоциклиб описан в WHO Drug Information, Vol. 27, No. 2, page 172 (2013). Палбоциклиб и его фармацевтически приемлемые соли раскрыты в международной публикации № WO 2003/062236 и патентах США №№ 6936612, 7208489 и 7456168; международной публикации № WO 2005/005426 и патентах США №№ 7345171 и 7863278; международной публикации № WO 2008/032157 и патенте США № 7781583; и международной публикации № WO 2014/128588. Содержание каждого из приведенных выше источников полностью включено сюда посредством ссылки.
Палбоциклиб утвержден в Соединенных Штатах для лечения поздних стадий рака молочной железы или метастатического рака молочной железы, положительного по рецептору гормонов (HR-положительного) и отрицательного по человеческому эпидермальному фактору роста 2 (НЕК2-отрицательного), в комбинации с летрозолом в качестве начальной эндокринной терапии или в комбинации с фулвестрантом после прогрессирования заболевания на эндокринной терапии. Лекарственное средство имеется в продаже от Pfizer под торговым названием IBRANCE® в форме капсульной лекарственной формы с немедленным высвобождением (IR), содержащей палбоциклиб в форме свободного основания, для перорального введения.
Палбоциклиб является двухосновным соединением и имеет две основные группы с рКа приблизительно 7,3 (вторичный пиперазиновый азот) и 4,1 (пиримидиновый азот). Растворимость свободного основания палбоциклиба зависит от рН. При низком рН (2,1-4,5) палбоциклиб растворим в воде, в то время как при росте рН выше 4,5 растворимость значительно снижается. При рН 7,9 палбоциклиб малорастворим в воде (9 мкг/мл). Одновременный прием агентов, повышающих рН в желудке, может снижать растворимость и всасывание композиций палбоциклиба в форме свободного основания.
На всасывание и биодоступность терапевтического агента при пероральном приеме может влиять множество факторов, включая прием субъектом после еды или натощак и применение определенных лекарственных средств, таких как ингибиторов протонной помпы (ИПП) или антагонисты Н2-рецепторов, а также определенные медицинские состояния. Соединения, растворимость которых зависит от рН, особенно основные соединения, могут демонстрировать нежелательные фармакокинетические свойства, такие как плохое всасывание и/или сниженная биодоступность, которые могут приводить к существенной вариабельности между разными пациентами и у одного и того же пациента.
Сохраняется потребность в разработке улучшенных лекарственных форм палбоциклиба с надлежащими профилями растворимости и фармакокинетики, демонстрирующих при этом хорошую стабильность при хранении. Авторы изобретения неожиданно обнаружили, что твердые лекарственные формы по настоящему изобретению демонстрируют отличную стабильность при хранении и обеспечивают доставку палбоциклиба, почти не зависящую от рН, без существенного влияния приема пищи или нежелательных взаимодействий с ИПП.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В первом аспекте согласно изобретению предложена твердая лекарственная форма, содержащая палбоциклиб, водорастворимую кислоту и фармацевтически приемлемый носитель.
Во втором аспекте согласно изобретению предложена твердая лекарственная форма по любому из воплощений, описанных здесь, где при добавлении лекарственной формы в тестовую среду, содержащую 500 мл 10 мМ ацетатного буфера, рН 5,5, при 37°С в стандартном аппарате 2 согласно Фармакопее США (USP-аппарате 2) с вращающимися лопастями, лопасти которого вращаются с частотой 50 об/мин, происходит растворение: (а) не менее 35% палбоциклиба за 15 минут; (б) не менее 45% палбоциклиба за 30 минут; (в) не менее 55% за 60 минут; или (г) двух или более из (а), (б) и (в).
В третьем аспекте согласно изобретению предложена твердая лекарственная форма по любому из воплощений, описанных здесь, где при добавлении лекарственной формы в тестовую среду, содержащую 500 мл 50 мМ фосфатного буфера, рН 6,5, и 0,1 М NaCl, при 37°С в стандартном USP-аппарате 2 с вращающимися лопастями, лопасти которого вращаются с частотой 50 об/мин, происходит растворение: (а) не менее 15% палбоциклиба за 15 минут; (б) не менее 20% палбоциклиба за 30 минут; (в) не менее 25% палбоциклиба за 60 минут; или (г) двух или более из (а), (б) и (в).
В некоторых воплощениях согласно изобретению предложена твердая лекарственная форма по любому из воплощений, описанных здесь, где при добавлении лекарственной формы в тестовую среду, содержащую 500 мл 50 мМ фосфатного буфера, рН 6,5, и 0,1 М NaCl, при 37°С в стандартном USP-аппарате 2 с вращающимися лопастями, лопасти которого вращаются с частотой 50 об/мин, происходит растворение: (а) не менее 20% палбоциклиба за 15 минут; (б) не менее 30% палбоциклиба за 30 минут; (в) не менее 25% палбоциклиба за 60 минут; или (г) двух или более из (а), (б) и (в).
В других воплощениях согласно изобретению предложена твердая лекарственная форма по любому из воплощений, описанных здесь, где при добавлении лекарственной формы в тестовую среду, содержащую 500 мл 50 мМ фосфатного буфера, рН 6,5, и 0,1 М NaCl, при 37°С в стандартном USP-аппарате 2 с вращающимися лопастями, лопасти которого вращаются с частотой 50 об/мин, происходит растворение: (а) не менее 40% палбоциклиба за 15 минут; (б) не менее 35% палбоциклиба за 30 минут; (в) не менее 25% палбоциклиба за 60 минут; или (г) двух или более из (а), (б) и (в).
В четвертом аспекте согласно изобретению предложена твердая лекарственная форма по любому из воплощений, описанных здесь, имеющая: (а) среднее отношение площади под кривой зависимости концентрации в плазме от времени (AUC) при приеме после еды к AUC при приеме натощак от приблизительно 0,8 до приблизительно 1,25 после введения субъекту однократной пероральной дозы; (б) среднее отношение максимальной концентрации в плазме (Cmax) при приеме после еды к Cmax при приеме натощак от приблизительно 0,8 до приблизительно 1,25 после введения субъекту однократной пероральной дозы; или (в) как (а), так и (б).
В пятом аспекте согласно изобретению предложена твердая лекарственная форма по любому из воплощений, описанных здесь, обеспечивающая: (а) среднюю AUC при приеме натощак в диапазоне от 80% до 125% средней AUC при приеме натощак контрольной капсулы с немедленным высвобождением (IR) для перорального введения, содержащей эквивалентное количество палбоциклиба, после введения субъекту однократной пероральной дозы; или (б) среднюю Cmax при приеме натощак в диапазоне от 80% до 125% средней Cmax при приеме натощак контрольной капсулы с немедленным высвобождением (IR) для перорального введения, содержащей эквивалентное количество палбоциклиба, после введения субъекту однократной пероральной дозы; или (в) как (а), так и (б).
В шестом аспекте согласно изобретению предложена твердая лекарственная форма по любому из воплощений, описанных здесь, обеспечивающая: (а) среднюю AUC в присутствии ингибитора протонной помпы (ИПП) в диапазоне от 80% до 125% средней AUC в отсутствие ИПП после введения субъекту однократной пероральной дозы; (б) среднюю Cmax в присутствии ингибитора протонной помпы (ИПП) в диапазоне от 80% до 125% средней Cmax в отсутствие ИПП после введения субъекту однократной пероральной дозы; или (в) как (а), так и (б). В некоторых воплощениях ИПП представляет собой рабепразол.
В седьмом аспекте согласно изобретению предложена твердая лекарственная форма по любому из воплощений, описанных здесь, демонстрирующая менее 0,3% кислого аддукта по массе после хранения в течение 96 дней при 30°С и относительной влажности (RH) 75%.
В восьмом аспекте согласно изобретению предложена твердая лекарственная форма по любому из воплощений, описанных здесь, демонстрирующая менее 1,0% кислого аддукта по массе после хранения в течение 2 лет при 30°С и RH 75%.
В девятом аспекте согласно изобретению предложена твердая лекарственная форма по любому из воплощений, описанных здесь, демонстрирующая менее 0,05% кислого аддукта по массе после хранения в течение 1 года при 25°С / RH 60%. В некоторых таких воплощениях лекарственная форма упакована в термозапечатанный флакон с влагопоглотителем.
В некоторых воплощениях каждого из аспектов изобретения активный фармацевтический ингредиент (АФИ), палбоциклиб, составляет от приблизительно 10% до приблизительно 35% лекарственной формы по массе. В определенных воплощениях палбоциклиб составляет приблизительно 20% лекарственной формы по массе.
В некоторых воплощениях каждого из аспектов изобретения водорастворимая кислота составляет от приблизительно 5% до приблизительно 40% лекарственной формы по массе. В определенных воплощениях водорастворимая кислота составляет от приблизительно 5% до приблизительно 25% лекарственной формы по массе. В других воплощениях водорастворимая кислота составляет от приблизительно 5% до приблизительно 15% лекарственной формы по массе. В более определенных воплощениях водорастворимая кислота составляет приблизительно 10% лекарственной формы по массе.
В некоторых таких воплощениях водорастворимая кислота выбрана из группы, состоящей из янтарной кислоты, яблочной кислоты и винной кислоты. В определенных воплощениях водорастворимая кислота представляет собой янтарную кислоту. В других воплощениях водорастворимая кислота представляет собой яблочную кислоту. В других воплощениях водорастворимая кислота представляет собой винную кислоту.
В предпочтительном воплощении каждого из аспектов, описанных здесь, твердая лекарственная форма содержит от приблизительно 10 масс. % до приблизительно 35 масс. % палбоциклиба, от приблизительно 5 масс. % до приблизительно 25 масс. % водорастворимой кислоты, выбранной из группы, состоящей из янтарной кислоты, яблочной кислоты и винной кислоты, и фармацевтически приемлемый носитель. В определенных воплощениях водорастворимая кислота представляет собой янтарную кислоту. В некоторых таких воплощениях твердая лекарственная форма содержит приблизительно 20 масс. % палбоциклиба, приблизительно 10 масс. % янтарной кислоты и фармацевтически приемлемый носитель.
В некоторых воплощениях каждого из аспектов, описанных здесь, фармацевтически приемлемый носитель содержит одно или более из следующих фармацевтически приемлемых эксципиентов: разбавители, разрыхлители, связывающие агенты, смазывающие агенты, скользящие агенты и поверхностно-активные агенты. Такие эксципиенты могут быть включены в таблетированные формы интрагранулярно или экстрагранулярно, и таблетки могут содержать одинаковые или разные эксципиенты в форме интрагранулярных или экстрагранулярных компонентов. Например, композиция в форме таблетки может содержать интрагранулярный смазывающий агент, экстрагранулярный смазывающий агент или как интрагранулярный, так и экстрагранулярный смазывающие агенты, которые могут быть одинаковыми или разными.
В некоторых воплощениях каждого из аспектов, описанных здесь, фармацевтически приемлемый носитель содержит по меньшей мере один разбавитель, где разбавитель составляет от приблизительно 50 масс. % до приблизительно 75 масс. % твердой лекарственной формы. В определенных воплощениях носитель содержит по меньшей мере один разбавитель, выбранный из группы, состоящей из микрокристаллической целлюлозы, моногидрата лактозы, маннита, сорбита, ксилита, карбоната магния, двухосновного фосфата кальция и трехосновного фосфата кальция. В определенных воплощениях разбавитель представляет собой микрокристаллическую целлюлозу. В некоторых таких воплощениях разбавитель представляет собой микрокристаллическую целлюлозу.
В других воплощениях каждого из аспектов, описанных здесь, фармацевтически приемлемый носитель содержит смазывающий агент, где смазывающий агент составляет от приблизительно 0,5 масс. % до приблизительно 10 масс. % твердой лекарственной формы. В определенных воплощениях носитель содержит по меньшей мере один смазывающий агент, выбранный из группы, состоящей из стеарата магния, стеарата кальция, стеарата цинка и стеарилфумарата натрия. В определенных воплощениях смазывающий агент представляет собой стеарат магния, который может быть включен интрагранулярно и/или экстрагранулярно. В других воплощениях смазывающий агент представляет собой стеарилфумарат натрия. Другие воплощения включают как стеарат магния, так и стеарилфумарат натрия в качестве смазывающих агентов, которые могут быть включены интрагранулярно и/или экстрагранулярно.
В других воплощениях каждого из аспектов, описанных здесь, фармацевтически приемлемый носитель содержит по меньшей мере один разрыхлитель, где разрыхлитель составляет от приблизительно 5 масс. % до приблизительно 10 масс. % твердой лекарственной формы. В определенных воплощениях носитель содержит по меньшей мере один разрыхлитель, выбранный из группы, состоящей из кросповидона, кроскармеллозы натрия и крахмалгликолята натрия. В определенных воплощениях разрыхлитель представляет собой кросповидон.
В частых воплощениях каждого из аспектов, описанных здесь, твердая лекарственная форма по изобретению имеет форму таблетки. В некоторых воплощениях таблетка имеет пленочную оболочку. В некоторых воплощениях таблетка представляет собой однослойную таблетку. В других воплощениях таблетка представляет собой двухслойную таблетку. В определенных воплощениях таблетки по изобретению содержат палбоциклиб в количестве 25 мг, 75 мг, 100 мг или 125 мг. В определенных воплощениях таблетки по изобретению содержат палбоциклиб в количестве 125 мг.
В частых воплощениях каждого из аспектов изобретения твердая лекарственная форма имеет форму таблетки. В некоторых воплощениях таблетка представляет собой однослойную таблетку. В других воплощениях таблетка представляет собой двухслойную таблетку.
В некоторых воплощениях аспектов и воплощений, описанных здесь, количество палбоциклиба в лекарственной форме составляет 25 мг, 75 мг, 100 мг или 125 мг. В определенных воплощениях количество палбоциклиба в лекарственной форме составляет 125 мг.
Каждое из воплощений настоящего изобретения, описанных здесь, можно сочетать с одним или более чем одним другим воплощением настоящего изобретения, описанным здесь, не являющимся несовместимым с тем воплощением (воплощениями), с которым его сочетают.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Фиг. 1. Данные по in vitro-растворению прототипных композиций, содержащих восемь анализируемых водорастворимых кислот (яблочная, малеиновая, янтарная, фумаровая, винная, n-толуолсульфоновая, бензойная и бензолсульфоновая кислоты) при 37°С в буферном растворе ацетата натрия, 10 мМ, рН 5,5, в USP-аппарате 2 с лопастями, вращающимися с частотой 50 об/мин.
Фиг. 2. Данные по in vitro-растворению прототипных композиций, содержащих янтарную кислоту, яблочную кислоту и винную кислоту при 37°С в буферном растворе ацетата натрия, 10 мМ, рН 5,5, в USP-аппарате 2 с лопастями, вращающимися с частотой 50 об/мин.
Фиг. 3. Данные по in vitro-растворению прототипных композиций, содержащих янтарную кислоту, при 37°С в фосфатном буферном растворе, 50 мМ, рН 6,5, содержащем + 0,1 М NaCl, в USP-аппарате 2 с лопастями, вращающимися с частотой 50 об/мин, при приближении раствора к состоянию насыщения («non-sink in vitro dissolution).
Фиг. 4. Данные по in vitro-растворению композиций A1, А2 и В при 37°С в фосфатном буферном растворе, 50 мМ, рН6,5, содержащем + 0,1 М NaCl, в USP-аппарате 2 с лопастями, вращающимися с частотой 50 об/мин, при приближении раствора к состоянию насыщения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение может быть понятнее со ссылкой на следующее подробное описание предпочтительных воплощений изобретения и включенные сюда Примеры. Следует понимать, что терминология, использованная здесь, приведена лишь с целью описания конкретных воплощений и не является ограничивающей. Также следует понимать, что если здесь конкретно не определено иное, терминологию, используемую здесь, следует использовать в ее традиционном значении, как известно в соответствующей области.
Если не указано иное, при использовании здесь формы единственного числа включают множественное число. Например, «эксципиент» включает один или более чем один эксципиент.
Термин «приблизительно» обозначает значение в пределах приемлемого стандарта ошибки среднего при рассмотрении специалистом в данной области. Часто термин «приблизительно» относится к ± 15%, предпочтительно ± 10% и более предпочтительно ± 5% от значения или диапазона, к которому он относится. Например, «приблизительно 10 масс. %» означает 10 масс. % ± 1,5 масс. %, предпочтительно 10 масс. % ± 1 масс. % и более предпочтительно 10 масс. % ± 0,5 масс. %.
Если здесь не указано иное, палбоциклиб относится к 6-ацетил-8-циклопентил-5-метил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-8H-пиридо[2,3-d]пиримидин-7-ону в форме свободного основания, который может присутствовать в кристаллической или аморфной форме или как смесь аморфной и кристаллической форм.
На всасывание принятых перорально лекарственных средств могут влиять изменения рН по мере прохождения лекарственного средства через желудочно-кишечный тракт (ЖКТ). Всасывание может происходить в разных местах по ходу ЖКТ, например, на внутренней поверхности щеки или в желудке, двенадцатиперстной кишке, тощей кишке, подвздошной кишке или толстой кишке. рН в разных местах всасывания различается, при этом рН желудка (рН 1-3,5) существенно отличается от рН тонкой кишки (рН 4,5-8). Лекарственные средства, растворимость которых зависит от рН, могут выпадать в осадок из раствора при прохождении лекарственного средства через ЖКТ, приводя к существенной вариабельности между разными пациентами или у одного и того же пациента по степени и/или скорости всасывания между разными дозами или пациентами.
рН ЖКТ может также варьировать в зависимости от приема пищи субъектом. Обычно время пребывания лекарственного средства в желудке в присутствии пищи дольше, чем натощак. Если присутствие или отсутствие пищи в ЖКТ существенно влияет на биодоступность лекарственного средства, говорят, что лекарственного средство демонстрирует «эффект приема пищи». Скорость эвакуации из желудка может также влиять на концентрацию лекарственного средства в растворе, доступного для всасывания, в различных местах по ходу ЖКТ.
Одновременный прием определенных лекарственных средств, а также такие медицинские состояния, как ахлоргидрия, могут также влиять на рН ЖКТ. Применение кислотоснижающих агентов, таких как ингибиторы протонной помпы (ИПП) или антагонисты Н2-рецепторов, может приводить к относительно высокому рН в желудке, что может приводить к лишь частичному растворению лекарственных средств, растворимость которых зависит от рН, в желудке. Дальнейшее растворение нерастворенного лекарственного средства может быть ингибировано низкой растворимостью в условиях более высокого рН в проксимальных отделах кишечника. Это может приводить к неравномерному растворению лекарственных средств, растворимость которых зависит от рН, повышая риск лекарственных взаимодействий и потенциально приводя к уменьшению всасывания и снижению полезного терапевтического эффекта.
Исследование, проведенное у здоровых добровольцев, продемонстрировало, что экспозиция при приеме палбоциклиба (125 мг, один раз в день в форме капсулы со свободным основанием) была несколько больше при приеме субъектами после еды (AUCinf, 23%-27%; Cmax, 21%-24%), по сравнению с приемом натощак (AUCinf, 39%; Cmax, 73%), и при приеме после еды вариабельность фармакокинетических параметров была значительно снижена. Ruiz-Garcia et al., Annals of Oncology (2014) 25 (suppl_4): iv146-iv164. 10.1093/annonc/mdu331. Ввиду снижения вариабельности между разными пациентами, наблюдаемой при приеме после еды, в США в листке-вкладыше рекомендовано принимать имеющиеся в продаже капсулы со свободным основанием вместе с пищей.
При включении соединения в таблетку или другую твердую лекарственную форму желательно разрабатывать композицию, сохраняющую стабильность во время хранения при значениях температуры и относительной влажности, превышающих обычные. Также можно пытаться придать композиции другие желательные свойства, такие как быстрое растворение, когда таблетка быстро растворяется, делая лекарственное средство доступным для всасывания. Соответственно, хорошая стабильность при хранении и быстрое растворение были, среди прочего, свойствами, рассматриваемыми как желаемые характеристики для настоящего изобретения.
Растворение лекарственного средства является критическим фактором, влияющим на скорость системного всасывания. Разработано множество in vitro-методов для оценки растворимости фармацевтических композиций, и тесты растворения иногда используют вместо непосредственной оценки биодоступности лекарственных средств. См., например, Emmanuel et al., Pharmaceutics (2010), 2:351-363, и процитированные там ссылки. В тестах растворения определяют процент АФИ, высвобожденного из лекарственной формы (то есть таблетки или капсулы) и растворенного в растворителе в контролируемых условиях теста за определенный период времени. Для поддержания условий неприближения раствора к состоянию насыщения («sink conditions), растворимость лекарственного средства при насыщении должна по меньшей мере в три раза превышать концентрацию лекарственного средства во время теста. В случае соединений с низкой растворимостью растворение можно иногда определять в условиях приближения раствора к состоянию насыщения («non-sink conditions). На растворение оказывают влияние свойства АФИ (например, размер частиц, форма кристаллов, объемная плотность), состав лекарственной формы (например, лекарственное средство, эксципиенты), способ изготовления (например, усилия прессования) и стабильность при условиях хранения (например, температуре, влажности).
Методы оценки химической стабильности твердых лекарственных форм при хранении в условиях ускоренного старения описаны в литературе. См., например, S. Т. Colgan, Т. J. Watson, R. D. Whipple, R. Nosal, J. V. Beaman, D. De Antonis, "The Application of Science and Risk Based Concepts to Drug Substance Stability Strategies" J. Pharm. Innov. 7:205-2013 (2012); Waterman КС, Carella AJ, Gumkowski MJ, et al. Improved protocol and data analysis for accelerated shelf-life estimation of solid dosage forms. Pharm Res 2007; 24(4):780-90; и S. T. Colgan, R. J. Timpano, D. Diaz, M. Roberts, R. Weaver, K. Ryan, K. Fields, G. Scrivens, "Opportunities for Lean Stability Strategies" J. Pharm. Innov. 9:259-271 (2014).
Для твердых лекарственных форм по настоящему изобретению ключевым продуктом деградации, мониторинг которого проводят для оценки стабильности при хранении, является кислый аддукт пиперазинильной группировки палбоциклиба и водорастворимой кислоты.
Как описано здесь далее, согласно настоящему изобретению предложены твердые лекарственные формы, содержащие палбоциклиб, водорастворимую кислоту и фармацевтически приемлемый носитель, и способы их изготовления и применения.
Твердые лекарственные формы включают, без ограничения, таблетки и капсулы с немедленным высвобождением, таблетки и капсулы с контролируемым высвобождением (CR), быстрорастворимые лекарственные формы, жевательные лекарственные формы, саше и так далее. Предпочтительно, лекарственная форма по настоящему изобретению имеет форму таблетки, включая однослойные или двухслойные таблетки.
«Твердая лекарственная форма» по настоящему изобретению представляет собой фармацевтически приемлемую твердую лекарственную форму, безопасную для перорального введения людям, где все эксципиенты в лекарственной форме являются фармацевтически приемлемыми для применения в композициях для перорального применения, иными словами, безопасны при приеме человеком внутрь. В частых воплощениях твердая лекарственная форма представляет собой таблетку.
При использовании здесь термин «стандартная доза» или «стандартная дозировка» относится к физически дискретной единице, содержащей предопределенное количество активного ингредиента, рассчитанное для оказания желаемого терапевтического эффекта. Стандартная доза или стандартная дозировка может иметь форму таблетки, капсулы, саше и так далее, называемых здесь «стандартная лекарственная форма».
При использовании здесь термин «натощак» определяют следующим образом: прием после голодания (приема 0 калорий) в течение ночи на протяжении по меньшей мере 10 часов (то есть 10 или более часов). Субъекты могут принимать лекарственную форму с 240 мл воды. Не следует разрешать прием пищи на протяжении по меньшей мере 4 часов после приема лекарственной формы. Можно разрешить пить воду по желанию, за исключением одного часа до и после приема лекарственного средства.
При использовании здесь термин «после еды» определяют следующим образом: прием после голодания (приема 0 калорий) в течение ночи на протяжении по меньшей мере 10 часов, после которого субъекты начинают рекомендованный прием пищи. Субъектам следует завершать этот прием пищи за 30 минут или менее, однако лекарственную форму следует принимать через 30 минут после начала приема пищи. Лекарственную форму можно принимать с 240 мл воды. Не следует разрешать прием пищи на протяжении по меньшей мере 4 часов после приема лекарственной формы. Можно разрешить пить воду по желанию, за исключением одного часа до и после приема лекарственного средства.
Для оценки отношения показателей при приеме после еды к показателям при приеме натощак может быть проведено однократное пероральное введение палбоциклиба: через 30 минут после приема жирной высококалорийной пищи (приблизительно 800-1000 калорий со 150, 250, и 500-600 калориями от белков, углеводов и жиров, соответственно); через 30 минут после приема маложирной низкокалорийной пищи (приблизительно 400-500 калорий со 120, 250, и 28-35 калориями от белков, углеводов и жиров, соответственно); или в промежутке между приемами пищи (через 1 час после / за 2 часа до) в случае умеренно жирной и калорийной пищи (приблизительно 500-700 калорий, состоящих из 15% белка, 50% углеводов и 35% жиров).
Жирную высококалорийную пищу можно использовать в качестве тестовой пищи для условий после еды. Примером жирной тестовой пищи могут быть два яйца, жаренные в масле, две полоски бекона, два поджаренных кусочка хлеба с маслом, четыре унции (113,4 г) картофельных оладий и восемь унций (226,8 г) цельного молока.
Расчет средней площади под кривой зависимости концентрации в сыворотке от времени (AUC) является методикой, хорошо известной в области фармацевтики, и описан, например, в Welling, "Pharmacokinetics Processes and Mathematics," ACS Monograph 185 (1986). При использовании здесь AUC включает площадь под кривой зависимости концентрации от времени с момента времени ноль в экстраполяции на бесконечность после однократного введения или площадь под кривой зависимости концентрации от времени с момента времени ноль до окончания интервала введения после достижения стационарного состояния / нескольких введений.
Кроме того, расчеты Cmax, Cmin,ss, Tmax и периода полувыведения 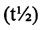 также известны специалистам в данной области и описаны, например, в Shargel, Wu-Pong, and Yu, Applied Biopharmaceutics and Pharmacokinetics (2005).
также известны специалистам в данной области и описаны, например, в Shargel, Wu-Pong, and Yu, Applied Biopharmaceutics and Pharmacokinetics (2005).
Для определения среднего отношения AUC при приеме после еды к AUC при приеме натощак сначала рассчитывают отдельные отношения средней площади под кривой зависимости концентрации палбоциклиба в плазме от времени (например, AUC0-inf) при приеме после еды к средней площади под кривой зависимости концентрации палбоциклиба в плазме от времени (например, AUC0-inf) при приеме натощак, после чего определяют среднее соответствующих отдельных отношений. В этом смысле, определяют среднее всех соответствующих индивидуальных отношений.
Ингибиторы протонной помпы (ИПП) являются хорошо известным классом лекарственных средств, снижающих образование кислоты желудочного сока и изменяющих посредством этого рН в желудке. Типичные ИПП включают, например, рабепразол, омепразол, (включая S- и В-формы, натриевые и магниевые соли), лансопразол, пантопразол, эзомепразол и тому подобное.
При использовании здесь термин «контрольная капсула с немедленным высвобождением (IR) для перорального введения» относится к имеющейся в продаже композиции палбоциклиба в форме IR-капсулы, как описано в Примере 11. В качестве контрольных здесь могут быть упомянуты эта композиция, а также композиция с изетионатной солью (ISE) и лекарственная форма в виде таблетки со свободным основанием без водорастворимой кислоты, в остальном по существу идентичная композиции по Примеру 1.
«Тест растворения 1» относится к следующему анализу лекарственных форм палбоциклиба. Тест растворения проводят в стандартном аппарате 2 согласно Фармакопее США (USP-аппарате 2) с вращающимися лопастями, как раскрыто в United States Pharmacopoeia (USP) Dissolution Test Chapter 711, Apparatus 2. Частота вращения лопастей составляет 50 об/мин, и лекарственную форму помещают в 500 мл ацетатного буфера, 10 мМ, рН5,5, при 37°С. В соответствующие моменты времени после начала теста (например, помещения лекарственной формы в аппарат) фильтрованные аликвоты (обычно 1,5 мл) тестовой среды анализируют на предмет палбоциклиба высокоэффективной жидкостной хроматографией (ВЭЖХ). Результаты растворения представляют как долю всей анализируемой дозы палбоциклиба, растворенную в различные моменты времени, в процентах.
«Тест растворения 2» относится к следующему анализу лекарственных форм палбоциклиба. Тест растворения проводят в стандартном USP-аппарате 2 с вращающимися лопастями, как раскрыто в United States Pharmacopoeia (USP) Dissolution Test Chapter 711, Apparatus 2. Частота вращения лопастей составляет 50 об/мин, и лекарственную форму помещают в 500 мл фосфатного буфера, 50 мМ, рН 6,5, и 0,1 М NaCl при 37°С. В соответствующие моменты времени после начала теста (например, помещения лекарственной формы в аппарат) фильтрованные аликвоты (обычно 1,5 мл) тестовой среды анализируют на предмет палбоциклиба высокоэффективной жидкостной хроматографией (ВЭЖХ). Результаты растворения представляют как долю всей анализируемой дозы палбоциклиба, растворенную в различные моменты времени, в процентах.
Термином «сухое гранулирование» обозначают процесс смешивания нерасфасованного активного продукта с по меньшей мере одним эксципиентом. Затем смесь прессуют или уплотняют с получением спрессованного или «уплотненного» материала. Этот материал можно разделять на части с образованием гранул посредством дробления, измельчения или разрезания, получая сухие гранулированные частицы. Возможно, частицы могут быть дополнительно обработаны. Процессы дробления, измельчения или разрезания включают действия, приводящие к уменьшению размера спрессованного материала, например, посредством размалывания или другими действиями, известными специалистам в данной области.
Термин «водорастворимый», использованный здесь в связи с кислотой, присутствующей в композиции, относится к кислоте, растворимость которой в воде при 25°С составляет по меньшей мере 0,2% по массе. Водорастворимая кислота может представлять собой органическую или неорганическую кислоту и предпочтительно представляет собой органическую кислоту, имеющую по меньшей мере одно значение рКа, по меньшей мере на одну единицу (предпочтительно по меньшей мере на две единицы) рК ниже наивысшего рКа основных групп, присутствующих в лекарственном средстве. В случае палбоциклиба, имеющего значения рКа приблизительно 4,1 и 7,3, кислота предпочтительно имеет рКа менее 6,3 и более предпочтительно рКа менее 5,3. Водорастворимые органические кислоты включают, например, С2-С8 или С2-С6 алифатические моно- или поликарбоновые кислоты и предпочтительно С4-С6 алифатические моно- или поликарбоновые кислоты. Особенно предпочтительны С4-С6 дикарбоновые кислоты, которые могут быть насыщенными или ненасыщенными.
Твердые лекарственные формы по изобретению могут содержать одну водорастворимую кислоту или могут содержать комбинацию двух или более таких кислот. В отдельных воплощениях изобретения водорастворимая кислота выбрана из группы, состоящей из янтарной кислоты, яблочной кислоты и винной кислоты. В определенных предпочтительных воплощениях изобретения водорастворимая кислота представляет собой янтарную кислоту.
Водорастворимую кислоту можно объединять с лекарственным средством до гранулирования, или она может быть включена в лекарственную форму вместе с экстрагранулярными эксципиентами. В двухслойной таблетке водорастворимая кислота может присутствовать в активном слое, содержащем палбоциклиб, может быть включена в отдельный кислотный слой или водорастворимые кислоты (которые могут быть одинаковыми или разными) могут быть включены как в активный слой, так и в кислотный слой.
Без ограничения теорией, полагают, что присутствие кислоты в твердой лекарственной форме в тесном контакте с лекарственным средством усилит солюбилизацию посредством взаимодействия палбоциклиба с кислотой. Посредством этого твердые лекарственные формы по изобретению обеспечивают более высокую местную концентрацию лекарственного средства в растворе после перорального приема субъектом по сравнению с введением композиций палбоциклиба без водорастворимой кислоты.
В некоторых воплощениях твердая лекарственная форма по любому из воплощений, описанных здесь, в условиях теста растворения 1 (ацетатный буфер, рН 5,5, 37°С) обеспечивает растворение: (а) не менее 35% палбоциклиба за 15 минут; (б) не менее 45% палбоциклиба за 30 минут; (в) не менее 55% за 60 минут; или (г) двух или более из (а), (б) и (в).
В других воплощениях твердая лекарственная форма по изобретению в условиях теста растворения 1 обеспечивает растворение: (а) не менее 25%, 30%, 35%, 40%, 45% или 50% или более 50% палбоциклиба за 15 минут; (б) не менее 35%, 40%, 45%, 50%, 55% или 60% или более 60% палбоциклиба за 30 минут; и/или (в) не менее 45%, 50%, 55%, 60%, 65% или 70% или более 70% палбоциклиба за 60 минут.
В некоторых воплощениях твердая лекарственная форма по любому из воплощений, описанных здесь, в условиях приближения раствора к состоянию насыщения в тесте растворения 2 (фосфатный буфер, рН6,5, и 0,1 М NaCl, 37°С) обеспечивает растворение: (а) не менее 40% палбоциклиба за 15 минут; (б) не менее 35% палбоциклиба за 30 минут; (в) не менее 25% палбоциклиба за 60 минут; или (г) двух или более из (а), (б) и (в).
В других воплощениях твердая лекарственная форма по любому из воплощений, описанных здесь, в условиях приближения раствора к состоянию насыщения в тесте растворения 2 (фосфатный буфер, рН6,5, и 0,1 М NaCl, 37°С) обеспечивает растворение: (а) не менее 15% палбоциклиба за 15 минут; (б) не менее 20% палбоциклиба за 30 минут; (в) не менее 25% палбоциклиба за 60 минут; или (г) двух или более из (а), (б) и (в).
В других воплощениях в тесте растворения 2 происходит растворение: (а) не менее 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45% или 50% или более 50% палбоциклиба за 15 минут; (б) не менее 15%, 20%, 25%, 30%, 35%, 40%, 45% или 50% или более 50% палбоциклиба за 30 минут; и/или (в) не менее 15%, 20%, 25%, 30%, 35% или 40% или более 40% палбоциклиба за 60 минут.
В других воплощениях в тесте растворения 2 происходит растворение: (а) не менее 30%, 35%, 40%, 45% или 50% или более 50% палбоциклиба за 15 минут; (б) не менее 25%, 30%, 35%, 40%, 45% или 50% или более 50% палбоциклиба за 30 минут; и/или (в) не менее 15%, 20%, 25%, 30%, 35% или 40% или более 40% палбоциклиба за 60 минут.
В некоторых воплощениях согласно изобретению предложена твердая лекарственная форма, содержащая палбоциклиб, водорастворимую кислоту и фармацевтически приемлемый носитель, обеспечивающая: (а) среднее отношение площади под кривой зависимости концентрации в плазме от времени (AUC) при приеме после еды к AUC при приеме натощак от приблизительно 0,8 до приблизительно 1,25 после введения субъекту однократной пероральной дозы; (б) среднее отношение максимальной концентрации в плазме (Cmax) при приеме после еды к Cmax при приеме натощак от приблизительно 0,8 до приблизительно 1,25 после введения субъекту однократной пероральной дозы; или (в) как (а), так и (б).
В некоторых воплощениях согласно изобретению предложена твердая лекарственная форма, содержащая палбоциклиб, водорастворимую кислоту и фармацевтически приемлемый носитель, обеспечивающая: (а) среднюю AUC при приеме натощак в диапазоне от 80% до 125% средней AUC при приеме натощак контрольной капсулы с немедленным высвобождением (IR) для перорального введения, содержащей эквивалентное количество палбоциклиба, после введения субъекту однократной пероральной дозы; (б) среднюю Cmax при приеме натощак в диапазоне от 80% до 125% средней Cmax при приеме натощак контрольной капсулы с немедленным высвобождением (IR) для перорального введения, содержащей эквивалентное количество палбоциклиба, после введения субъекту однократной пероральной дозы; или (в) как (а), так и (б).
В других воплощениях согласно изобретению предложена твердая лекарственная форма, содержащая палбоциклиб, водорастворимую кислоту и фармацевтически приемлемый носитель, обеспечивающая: (а) среднюю AUC в присутствии ингибитора протонной помпы (ИПП), предпочтительно рабепразола, в диапазоне от 80% до 125% средней AUC в отсутствие ИПП после введения субъекту однократной пероральной дозы; (б) среднюю Cmax в присутствии ингибитора протонной помпы (ИПП), предпочтительно рабепразола, в диапазоне от 80% до 125% средней Cmax в отсутствие ИПП после введения субъекту однократной пероральной дозы; или (в) как (а), так и (б).
В других воплощениях согласно изобретению предложена твердая лекарственная форма, содержащая палбоциклиб, водорастворимую кислоту и фармацевтически приемлемый носитель, демонстрирующая менее 0,4%, 0,35%, 0,3%, 0,25%, 0,2%, 0,15% или 0,1% кислого аддукта по массе после хранения в течение 96 дней при 30°С и RH 75%.
В других воплощениях согласно изобретению предложена твердая лекарственная форма, содержащая палбоциклиб, водорастворимую кислоту и фармацевтически приемлемый носитель, демонстрирующая менее 1,5%, 1,4%, 1,3%, 1,2%, 1,1%, 1,0%, 0,9%, 0,8%, 0,7%, 0,6%, 0,5%, 0,4% или 0,3% кислого аддукта по массе после хранения в течение 2 лет при 30°С и RH 75%.
В некоторых воплощениях каждого из аспектов и воплощений изобретения водорастворимая кислота выбрана из группы, состоящей из янтарной кислоты, яблочной кислоты и винной кислоты. В определенных воплощениях каждого из аспектов и воплощений, указанных здесь, водорастворимая кислота представляет собой янтарную кислоту. В других воплощениях водорастворимая кислота представляет собой яблочную кислоту. В других воплощениях водорастворимая кислота представляет собой винную кислоту.
В некоторых воплощениях каждого из аспектов изобретения водорастворимая кислота составляет от приблизительно 5% до приблизительно 40% лекарственной формы по массе. В определенных воплощениях водорастворимая кислота составляет от приблизительно 5% до приблизительно 25% лекарственной формы по массе. В некоторых воплощениях водорастворимая кислота составляет приблизительно 5%, 10%, 15%, 20%, 25%, 30%, 35% или 40% лекарственной формы по массе.
В некоторых воплощениях каждого из аспектов изобретения палбоциклиб составляет от приблизительно 10% до приблизительно 35% лекарственной формы по массе. В определенных воплощениях палбоциклиб составляет приблизительно 20% лекарственной формы по массе. В некоторых воплощениях палбоциклиб составляет приблизительно 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45% или 50% лекарственной формы по массе.
В частых воплощениях каждого из аспектов и воплощений, указанных здесь, твердая лекарственная форма представляет собой таблетку, предпочтительно таблетку, изготовленную сухим гранулированием. В некоторых таких воплощениях таблетка представляет собой двухслойную таблетку. В определенных воплощениях двухслойная таблетка содержит: (а) активный слой, содержащий палбоциклиб и фармацевтически приемлемый носитель; и (б) кислотный слой, содержащий водорастворимую кислоту и фармацевтически приемлемый носитель. В некоторых воплощениях двухслойная таблетка содержит: (а) активный слой, содержащий палбоциклиб, водорастворимую кислоту и фармацевтически приемлемый носитель; и (б) кислотный слой, содержащий водорастворимую кислоту и фармацевтически приемлемый носитель; где водорастворимая кислота в активном слое может быть такой же, как водорастворимая кислота в кислотном слое, или отличаться от нее. В определенном воплощении водорастворимая кислота в активном слое представляет собой янтарную кислоту и водорастворимая кислота в кислотном слое представляет собой винную кислоту.
В другом аспекте согласно изобретению предложен способ лечения рака, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества твердой лекарственной формы по любому из аспектов и воплощений, описанных здесь. В определенных воплощениях рак представляет собой рак молочной железы. В некоторых таких воплощениях рак молочной железы представляет собой рак молочной железы, положительный по рецептору гормонов (HR+). В некоторых таких воплощениях рак молочной железы представляет собой рак молочной железы, положительный по рецептору эстрогенов (ER+). В некоторых таких воплощениях рак молочной железы представляет собой рак молочной железы, отрицательный по рецептору человеческого эпидермального фактора роста 2 (HER-). В других таких воплощениях рак молочной железы представляет собой рак молочной железы, положительный по рецептору человеческого эпидермального фактора роста 2 (HER+). В других воплощениях рак молочной железы представляет собой поздние стадии рака молочной железы или метастатический рак молочной железы, которые могут быть охарактеризованы как HR+, HER2- или ER+, HER2-.
Палбоциклиб может быть введен сам по себе или в комбинации с другими лекарственными средствами, в частности ингибиторами ароматазы, например, летрозолом, фулвестрантом или экземестаном, и его будут обычно вводить в форме композиции вместе с одним или более чем одним фармацевтически приемлемым эксципиентом. Термином «эксципиент» описывают любой ингредиент, не являющийся палбоциклибом или его солью.
Палбоциклиб может быть введен перорально. Пероральное введение может включать прием внутрь, в результате которого соединение проникает в желудочно-кишечный тракт, или может быть применено трансбуккальное или сублингвальное введение, посредством которого соединение проникает в кровоток непосредственно из полости рта.
Терапевтически эффективное количество лекарственной формы по изобретению может быть введено субъекту, нуждающемуся в таком лечении. При использовании здесь термин «терапевтически эффективное количество» относится к такому количеству вводимого соединения, которое будет в некоторой степени облегчать один или более чем один симптом расстройства, по поводу которого проводят лечение. Применительно к лечению рака терапевтически эффективное количество относится к такому количеству, которое оказывает эффект (1) уменьшения размера опухоли, (2) ингибирования (то есть, некоторого замедления, предпочтительно прекращения) метастазирования опухоли, (3) некоторого ингибирования (то есть, некоторого замедления, предпочтительно прекращения) роста опухоли или инвазии опухоли или (4) некоторого облегчения (или, предпочтительно, устранения) одного или более чем одного признака или симптома, связанного с раком.
При использовании здесь «субъект» относится к субъекту-человеку или субъекту-животному. В определенных предпочтительных воплощениях субъект представляет собой человека. В некоторых воплощениях субъект представляет собой пациента, страдающего от болезненного состояния. В других воплощениях субъект может представлять собой здорового добровольца.
Если не указано иное, при использовании здесь термин «осуществление лечения» подразумевает обратное развитие, уменьшение интенсивности, ингибирование прогрессирования или предотвращение расстройства или состояния, к которому относится такой термин, или одного или более чем одного симптома такого расстройства или состояния. Если не указано иное, при использовании здесь термин «лечение» относится к действиям по проведению лечения, как определено выше. Термин «лечение» также включает адъювантное и неоадъювантное лечение субъекта. Палбоциклиб может быть введен сам по себе или в комбинации с ингибитором ароматазы, таким как летрозол, фулвестрант или экземестан.
При использовании здесь «рак» относится к любому злокачественному и/или инвазивному росту или опухоли, вызванной аномальным ростом клеток. При использовании здесь «рак» относится к солидным опухолям, названным по типу образующих их клеток (например, рак молочной железы) и к опухолям крови, костного мозга или лимфатической системы. Примеры солидных опухолей включают, без ограничения, саркомы и карциномы. Примеры опухолей крови включают, без ограничения, лейкозы, лимфомы и миелому. Термин «рак» включает, без ограничения, первичный рак, возникший в определенном месте в организме, метастатический рак, распространившийся из места, где он начался, на другие части тела, рецидив исходного первичного рака после ремиссии и второй первичный рак, представляющий собой новый первичный рак у человека с анамнезом предшествующего рака другого типа.
Конкретнее, примеры рака в связи с настоящим изобретением включают, среди прочего, рак молочной железы, предпочтительно в комбинации с ингибитором ароматазы. Например, рак может представлять собой рак молочной железы, положительный по рецептору гормонов (HR+) и, в частности, рак молочной железы, положительный по рецептору эстрогенов (ER+). В некоторых воплощениях указанный ER+ рак молочной железы отрицателен по человеческому эпидермальному фактору роста 2 (HER2). В других воплощениях рак представляет собой ER+, HER2- метастатический рак молочной железы на поздней стадии, где лекарственное средство вводят в комбинации с ингибитором ароматазы для лечения метастатического заболевания.
Композиции, подходящие для перорального применения, включают твердые композиции, такие как таблетки, капсулы, порошки, пастилки (включая заполненные жидкостью), саше и тому подобное. В предпочтительном аспекте изобретения твердая лекарственная форма, предложенная здесь, представляет собой таблетку. В некоторых таких воплощениях таблетка имеет пленочную оболочку. В других таких воплощениях таблетка представляет собой двухслойную таблетку.
В случае лекарственных форм в виде таблеток палбоциклиб может, в зависимости от дозы, составлять от 1 масс. % до 80 масс. % лекарственной формы, обычно от 5 масс. % до 60 масс. %, чаще от приблизительно 10 масс. % до приблизительно 35 масс. % или, еще чаще, от приблизительно 15 масс. % до приблизительно 25 масс. % лекарственной формы. В определенных воплощениях палбоциклиб составляет приблизительно 20 масс. % лекарственной формы по массе.
В твердых лекарственных формах по изобретению носитель может содержать множество фармацевтически приемлемых эксципиентов, включая, например, разбавители, разрыхлители, связывающие агенты, смазывающие агенты, скользящие агенты и поверхностно-активные агенты. Композиции могут также содержать такие эксципиенты, как консерванты, антиоксиданты, корригенты и красители, а также другие эксципиенты, известные в данной области.
Твердые лекарственные формы, такие как таблетки, обычно содержат разбавители, например, лактозу (моногидрат, моногидрат, высушенный распылительной сушкой, безводную и тому подобное), маннит, ксилит, декстрозу, сахарозу, сорбит, прессуемый сахар, микрокристаллическую целлюлозу, порошкообразную целлюлозу, крахмал, прежелатинизированный крахмал, декстраты, декстран, декстрин, декстрозу, мальтодекстрин, карбонат кальция, двухосновный фосфат кальция, трехосновный фосфат кальция, сульфат кальция, карбонат магния, оксид магния, полоксамеры, полиэтиленоксид, гидроксипропилметилцеллюлозу и их смеси. Для использования в композициях, описанных здесь, могут быть подходящими различные типы микрокристаллической целлюлозы. Примеры микрокристаллической целлюлозы включают типы Avicel® РН101, РН102, РН103, РН105, РН 112, РН113, РН200, РН301 и другие типы микрокристаллической целлюлозы, такие как силикатированная микрокристаллическая целлюлоза (SMCC). В некоторых воплощениях разбавитель выбран из группы, состоящей из микрокристаллической целлюлозы, моногидрата лактозы, маннита, сорбита, ксилита, карбоната магния, двухосновного фосфата кальция, трехосновного фосфата кальция или их смесей. В определенных воплощениях разбавитель содержит микрокристаллическую целлюлозу. В некоторых воплощениях разбавитель содержит один или более чем один тип микрокристаллической целлюлозы, например, Avicel® РН105, Avicel® PH200 или их смеси. В некоторых таких воплощениях разбавитель не содержит моногидрата лактозы. В других таких воплощениях разбавитель содержит микрокристаллическую целлюлозу и дополнительно содержит моногидрат лактозы. Разбавители часто составляют от приблизительно 25 масс. % до приблизительно 75 масс. % твердой лекарственной формы и предпочтительно от приблизительно 50 масс. % до приблизительно 75 масс. % лекарственной формы.
Твердые лекарственные формы часто содержат разрыхлители. Примеры разрыхлителей включают крахмалгликолят натрия, карбоксиметилцеллюлозу натрия, карбоксиметилцеллюлозу кальция, кроскармеллозу натрия, кросповидон, поливинилпирролидон, метилцеллюлозу, микрокристаллическую целлюлозу, гидроксипропилцеллюлозу, замещенную низшими алкилами, крахмал, прежелатинизированный крахмал и альгинат натрия. В некоторых воплощениях разрыхлитель представляет собой кросповидон. Может быть использован кросповидон любой марки, например, для использования в композициях, описанных здесь, подходит кросповидон марок CL, CL-SF и XL. Конкретные примеры включают Kollidon, KollidonCL®, Kollidon CL-M®, Polyplasdone XL®, Polyplasdone XL-10® и Polyplasdone INF-10®. В некоторых воплощениях носитель содержит по меньшей мере один разрыхлитель, выбранный из группы, состоящей из кросповидона, кроскармеллозы натрия и крахмалгликолята натрия. В определенных воплощениях разрыхлитель представляет собой кросповидон. Разрыхлители часто составляют от приблизительно 1 масс. % до приблизительно 25 масс. %, предпочтительно от приблизительно 5 масс. % до приблизительно 20 масс. %, более предпочтительно от приблизительно 5 масс. % до приблизительно 10 масс. % лекарственной формы.
Связывающие агенты могут быть использованы для придания композиции в форме таблетки когезионных свойств. Подходящие связывающие агенты включают микрокристаллическую целлюлозу, желатин, сахара, полиэтиленгликоль, природные и синтетические камеди, поливинилпирролидон, прежелатинизированный крахмал, гидроксипропилцеллюлозу и гидроксипропилметилцеллюлозу. В некоторых воплощениях связывающий агент выбран из группы, состоящей из микрокристаллической целлюлозы, гидроксипропилцеллюлозы и гидроксипропилметилцеллюлозы. В определенных воплощениях связывающий агент представляет собой микрокристаллическую целлюлозу, например, Avicel® PH105. Когда присутствуют, связывающие агенты могут составлять от приблизительно 0 масс. % до приблизительно 15 масс. % или от приблизительно 0,2 масс. % до приблизительно 10 масс. % лекарственной формы. В некоторых воплощениях связывающий агент составляет от приблизительно 5 масс. % до приблизительно 10 масс. % лекарственной формы. В определенных воплощениях связывающий агент составляет приблизительно 10 масс. % лекарственной формы.
Твердые лекарственные формы часто содержат один или более чем один смазывающий агент. Примеры смазывающих агентов включают стеарат магния, стеарат кальция, стеарат цинка, стеарилфумарат натрия, смеси стеарата магния с лаурилсульфатом натрия или смеси двух или более из указанного выше. В некоторых воплощениях смазывающий агент представляет собой стеарат магния и/или стеарилфумарат натрия. В некоторых воплощениях смазывающий агент представляет собой стеарат магния. В некоторых таких воплощениях твердая лекарственная форма представляет собой таблетку, содержащую интрагранулярный и экстрагранулярный стеарат магния. В других воплощениях твердая лекарственная форма представляет собой таблетку, содержащую интрагранулярный стеарат магния и экстрагранулярный стеарилфумарат натрия. Когда присутствуют, смазывающие агенты часто составляют от приблизительно 0,25 масс. % до приблизительно 10 масс. %, предпочтительно от приблизительно 0,5 масс. % до приблизительно 6 масс. % лекарственной формы.
Таблетки могут также содержать скользящие агенты, например, диоксид кремния, коллоидный диоксид кремния, силикат магния, трисиликат магния, тальк и другие формы диоксида кремния, такие как агрегированные силикаты и гидратированный диоксид кремния. В некоторых воплощениях скользящий агент представляет собой диоксид кремния. Когда присутствуют, скользящие агенты могут составлять от приблизительно 0 масс. % до приблизительно 10 масс. %, предпочтительно от приблизительно 0,2 масс. % до приблизительно 5 масс. % или от приблизительно 0,5 масс. % до приблизительно 2 масс. % таблетки.
Таблетки могут, возможно, содержать поверхностно-активные агенты, такие как лаурилсульфат натрия и полисорбат 80. Когда присутствуют, поверхностно-активные агенты могут составлять от 0 масс. % до 10 масс. % или, предпочтительно, от 0,2 масс. % до 5 масс. % таблетки.
В целом, твердые лекарственные формы по изобретению изготавливают способами, обычно применяемыми в фармацевтической химии. Выбранные эксципиенты и АФИ могут быть включены в экстрагранулярный и/или интрагранулярный компартмент.
Типичные композиции в форме таблеток содержат от приблизительно 10 масс. % до приблизительно 35 масс. % палбоциклиба, обычно от приблизительно 15 масс. % до приблизительно 25 масс. % палбоциклиба; от приблизительно 5 масс. % до приблизительно 15 масс. % водорастворимой кислоты; от приблизительно 25 масс. % до приблизительно 75 масс. % разбавителя; от приблизительно 5 масс. % до приблизительно 10 масс. % разрыхлителя; от приблизительно 0,5 масс. % до приблизительно 6 масс. % смазывающего агента; и, возможно, от приблизительно 0 масс. % до приблизительно 5 масс. % скользящего агента и от приблизительно 0 масс. % до приблизительно 15 масс. % связывающего агента.
Другие типичные композиции в форме таблеток содержат приблизительно 20 масс. % палбоциклиба; приблизительно 10 масс. % водорастворимой кислоты, предпочтительно янтарной кислоты; от приблизительно 50 масс. % до приблизительно 75 масс. % разбавителя, предпочтительно микрокристаллической целлюлозы; от приблизительно 5 масс. % до приблизительно 10 масс. % разрыхлителя, предпочтительно кросповидона; от приблизительно 0,5 масс. % до приблизительно 6 масс. % смазывающего агента, предпочтительно стеарата магния и/или стеарилфумарата натрия; возможно, от приблизительно 0 масс. % до приблизительно 5 масс. % скользящего агента; и, возможно, от приблизительно 0 масс. % до приблизительно 15 масс. % связывающего агента. Когда присутствует, скользящий агент предпочтительно представляет собой диоксид кремния и связывающий агент предпочтительно представляет собой микрокристаллическую целлюлозу подходящего типа (например, Avicel® PH105) в форме сухого связывающего агента.
Твердые композиции для перорального применения могут быть изготовлены для немедленного и/или модифицированного высвобождения. Композиции с модифицированным высвобождением включают замедленное, длительное, импульсное, контролируемое, целевое и программируемое высвобождение. См. общее описание композиций с модифицированным высвобождением в патенте США №6106864.
Лекарственные средства в виде твердых таблеток заданной формы обычно изготавливают прессованием веществ, составляющих конечный продукт, с получением таблеток желаемой формы. Такие вещества могут включать активные фармацевтические ингредиенты, а также фармацевтически неактивные эксципиенты, придающие продукту необходимые или полезные свойства во время и после процесса изготовления. Твердость таблеток или предел прочности на разрыв могут быть использованы в качестве меры когезионной способности ингредиентов таблетки. Если таблетка не обладает достаточными когезионными свойствами, она может утратить целостность при использовании. Конечная композиция может содержать один или более чем один слой и может иметь покрытие или не иметь покрытия.
Как известно в данной области, гранулирование является способом, применяемым для улучшения свойств композиции при использовании и изготовлении, например, посредством увеличения размера частиц для улучшения сыпучести. Гранулирование практически не меняет физическую форму лекарственного средства, такую как его кристаллический или аморфный характер. Для изготовления лекарственных форм в виде таблеток специалисты в данной области применяют различные способы. Примеры таких способов включают сухое гранулирование, влажное гранулирование, гранулирование в псевдоожиженном слое и прямое прессование. Тип применяемого способа может зависеть от таких факторов, как физические свойства активных фармацевтических ингредиентов в композиции, типы используемых эксципиентов и желаемые физические свойства готового продукта. Каждый из этих способов включает стадии, включающие смешивание ингредиентов лекарственной формы. В определенных воплощениях настоящего изобретения предпочтительно сухое гранулирование.
Для получения однородного и целостного конечного продукта обычно необходима некоторая степень перемешивания ингредиентов лекарственной формы. Тем не менее, при изготовлении фармацевтических таблеток влажным и сухим гранулированием было обнаружено, что степень и интенсивность перемешивания ингредиентов перед прессованием связаны со снижением сжимаемости и когезионной способности композиции, приводящим к снижению прочности таблеток.
Сходный результат можно наблюдать при использовании роликового пресса, например, в способах сухого гранулирования. Роликовый пресс может быть использован для получения гранул, которые затем прессуют с получением таблеток. Использование роликового пресса может впоследствии снижать сжимаемость и когезионную способность лекарственной формы.
Сухое гранулирование представляет собой способ, при котором грануляты получают посредством стадии уплотнения, после которой частицам уплотненного материала придают размер, облегчающий дальнейшую обработку. Его часто используют для улучшения свойств сыпучести и/или уплотнения композиции, что позволяет облегчить дальнейший процесс изготовления, такой как таблетирование, инкапсулирование и заполнение порошком. Уплотненный материал получают непосредственно из порошкообразных смесей, обычно содержащих активный ингредиент и другие эксципиенты, включая смазывающий агент.
Применение методик сухого гранулирования может быть предпочтительнее способов влажного гранулирования, благодаря меньшему времени обработки и меньшей стоимости. Тем не менее, сухое гранулирование обычно ограничено теми ситуациями, в которых физические свойства лекарственного средства или активного ингредиента являются подходящими для получения фармацевтически приемлемых гранулятов и лекарственных форм, таких как таблетки.
Обычно в композицию необходимо добавлять по меньшей мере один эксципиент, что приводит к увеличению размера таблеток конечного продукта. Поскольку для использования таблеток в качестве подходящей лекарственной формы их размеры необходимо сохранять в пределах определенных параметров, существует граница, после которой увеличение размера таблеток для включения все больших количеств эксципиентов для повышения прессуемости становится нецелесообразным. В результате, изготовители часто вынуждены ограничивать применение способа сухого гранулирования низкими дозами активного ингредиента в каждой прессованной таблетке, когда композиция может вместить достаточное количество эксципиентов, позволяющее проводить сухое гранулирование.
При разработке фармацевтических лекарственных форм важно соблюдать баланс нескольких разных задач. Важно изготовить фармацевтическую лекарственную форму максимально экономичным, по возможности, способом. Желательно, чтобы способ изготовления был простым и включал немного стадий обработки. Лекарственная форма также должна обеспечивать оптимальную доступность присутствующего в ней активного соединения для пациента. Кроме того, лекарственная форма должна быть такой, чтобы ее было легко принимать внутрь. Пациенты лучше воспринимают лекарственные формы меньшего размера, что повышает приверженность пациентов к лечению.
Конечную фармацевтическую композицию обрабатывают с получением стандартной лекарственной формы (например, таблетки или капсулы) и затем упаковывают для реализации. Стадия обработки будет варьировать в зависимости от конкретной стандартной лекарственной формы. Например, таблетки обычно прессуют под давлением с приданием желаемой формы, а в случае капсул применяют простое заполнение. Способы, применяемые для изготовления различных стандартных лекарственных форм, хорошо известны специалистам в данной области.
Таблетки обычно изготавливают, оказывая давление на материал, подлежащий таблетированию, на таблеточном прессе. Композиция должна иметь хорошие свойства сыпучести для точного объемного дозирования материала в полость матрицы и подходящую сжимаемость, прессуемость и эжекционные свойства для получения таблеток.
Существует много таблеточных прессов, различающихся по производительности, но сходных по основным функциям и действию. Все обеспечивают сжатие таблетируемой композиции в полости матрицы посредством давления, оказываемого между двух стальных пуансонов, нижним пуансоном и верхним пуансоном. Таблеточные прессы обычно снабжены загрузочной воронкой для размещения и подачи композиции, подающим механизмом для подачи композиции в полость матрицы, механизмом для установки пуансонов и матриц и, в роторных таблеточных прессах, механизмом, направляющим движение пуансонов. Двумя типами таблеточных прессов являются одностанционный или однопуансонный пресс и многостанционный роторный пресс. Некоторые таблеточные прессы обеспечивают большее время нахождения в полости матрицы, чем другие, обеспечивая более прочное связывание. Другие прессы могут обеспечивать предварительную компрессию.
Для получения гранул фармацевтической композиции могут также быть применены способы влажного гранулирования. Способы влажного гранулирования описаны в Remington: The Science and Practice of Pharmacy, Mack Publishing Company, Easton, Pa., 19th Edition 1995. Эти и другие способы обычно известны специалистам в данной области. При применении влажного гранулирования летучий агент может быть включен в смесь до, во время или после смешивания ингредиентов, но до получения гранул. Например, твердый летучий агент можно смешивать с порошками смеси до, во время или после добавления растворов связывающих агентов. Другие твердые лекарственные формы могут быть изготовлены с применением методик, включающих роторное гранулирование или дисперсию, высушенную распылением (SDD).
Изобретение будет проиллюстрировано в следующих неограничивающих примерах.
ПРИМЕРЫ
Пример 1
Свободное основание с янтарной кислотой
Таблетку, содержащую АФИ (палбоциклиб) в форме свободного основания после сухого гранулирования с янтарной кислотой, изготавливали с применением следующей методики. Состав таблетки указан в Таблице 1.
Таблица 1
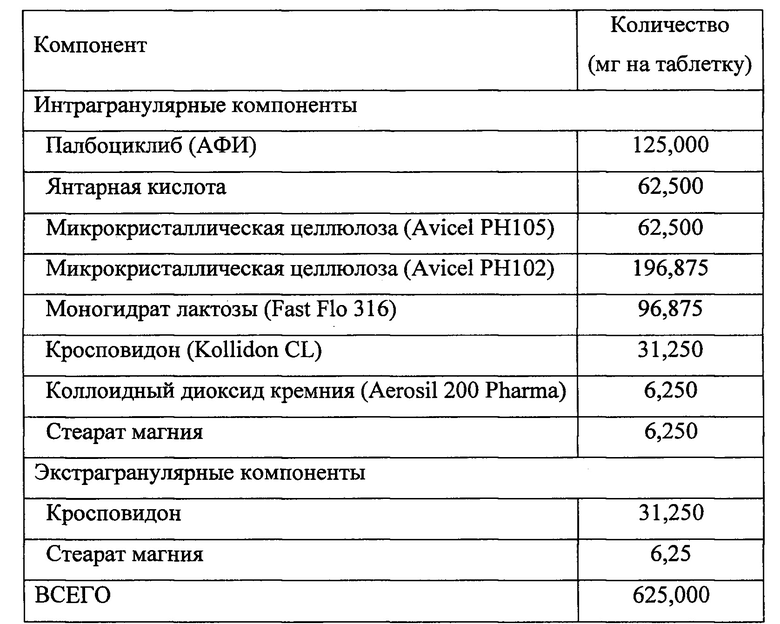
Микрокристаллическую целлюлозу (Avicel PH105) помещали в смеситель (бункерный смеситель или его эквивалент) и перемешивали на небольшой скорости в течение приблизительно 25 оборотов (2 минуты при 12 об/мин). В смеситель добавляли АФИ, промывая контейнер с АФИ порцией моногидрата лактозы, и проводили медленное перемешивание. В смеситель добавляли янтарную кислоту, моногидрат лактозы, кросповидон и коллоидный диоксид кремния в количествах, рассчитанных на одну партию, и проводили перемешивание на небольшой скорости в течение приблизительно 180 оборотов (15 минут при 12 об/мин).
Измельчитель и мешок предварительно обрабатывали микрокристаллической целлюлозой (Avicel РН102) в количестве 50% от количества, рассчитанного на одну партию. Смесь пропускали сверху через измельчитель. Измельчитель промывали оставшейся частью микрокристаллической целлюлозы (Avicel РН102), измельченный материал переносили из мешка в смеситель (бункерный смеситель или его эквивалент) и проводили перемешивание на небольшой скорости в течение приблизительно 180 оборотов (15 минут при 12 об/мин). Интрагранулярный стеарат магния просеивали через сито с отверстиями подходящего размера и добавляли в смеситель, использованный на предыдущей стадии. Смесь перемешивали на небольшой скорости в течение приблизительно 60 оборотов (5 минут при 12 об/мин). Смесь прессовали роликовым прессом (Gerteis Minipactor или его эквивалент) без сбора или повторного использования мелких фрагментов. Если измельчение не проводили сразу, прессованную роликовым прессом смесь пропускали через Comil U5 или U10, оборудованный импеллером 1601 с размером отверстий сита 050G при скорости 1000 или 700 об/мин, соответственно.
Экстрагранулярный кросповидон добавляли в смеситель, использованный на предыдущей стадии. Смесь перемешивали на небольшой скорости в течение приблизительно 180 оборотов (15 минут при 12 об/мин). Экстрагранулярный стеарат магния просеивали через сито с отверстиями подходящего размера. Его добавляли в смеситель, использованный на предыдущей стадии, и проводили перемешивание в течение приблизительно 60 оборотов (5 минут при 12 об/мин).
Для изготовления таблеток использовали одностанционный пресс (Korsch ХР 1 или его эквивалент) или роторный пресс.
Пример 2
Композиция в форме двухслойных таблеток
Изготавливали двухслойные таблетки, содержащие активный слой и кислотный слой. Активный слой состоял из гранулированной смеси по Примеру 1. Состав активного и кислотного слоя указан в Таблице 2.
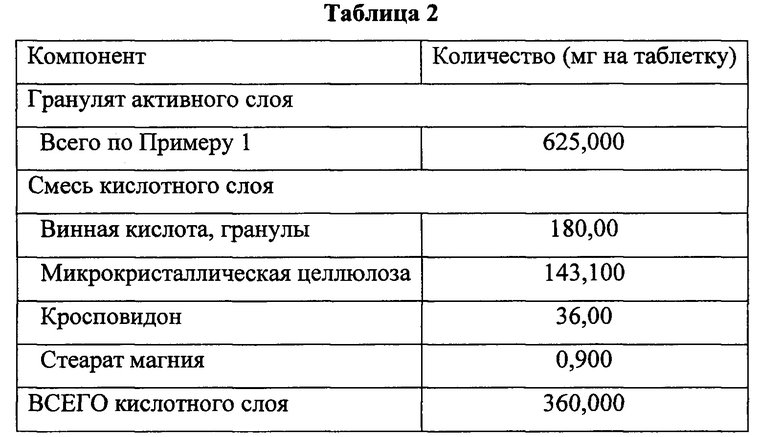
Смесь кислотного слоя получали смешиванием микрокристаллической целлюлозы, винной кислоты и кросповидона в контейнере подходящего размера. Смесь перемешивали в течение приблизительно 120 оборотов и затем пропускали через Comil U5 или U10, оборудованный импеллером 1601 с размером отверстий сита 018R при скорости 1000 или 700 об/мин, соответственно.
Измельченный материал переносили в смеситель, после чего компоненты перемешивали в течение приблизительно 120 оборотов. Стеарат магния просеивали через сито с отверстиями подходящего размера (1 мм сито, US Sieve #20) и затем добавляли в смеситель, проводя перемешивание в течение приблизительно 50 оборотов.
Двухслойные таблетки изготавливали с применением следующих методик с использованием подходящего роторного пресса для изготовления двухслойных таблеток, такого как Korsch ХР 1. Гранулят активного слоя (полученный согласно методикам, описанным в Примере 1) прессовали до целевой массы активного слоя 625 мг и рекомендованной толщины активного слоя 6,66 мм. Добавляли смесь кислотного слоя и кислотный слой и активный слой прессовали до желаемой массы содержимого 985 мг.
Пример 3
Гранулирование в псевдоожиженном слое
Аппарат для нанесения покрытий в псевдоожиженном слое (Niro МР-2 Fluid Bed Coater с чашей МР-1 и конфигурацией для гранулирования распылением сверху) предварительно разогревали с применением следующих условий до стабилизации точки росы поступающего воздуха (12°С) и температуры в чаше с продуктом (45°С или выше) на целевых значениях. Распылительная форсунка представляла собой Schlick970 с отверстием 0,8 мм при расстоянии от форсунки до дна чаши 33 см.
Для получения гранулята в псевдоожиженном слое янтарную кислоту сначала измельчали вручную с использованием ступки и пестика. Периодически измельченный порошок помещали на сито с размером ячеек 60 меш и вручную встряхивали его для пропускания материала в сборный контейнер. Материал, оставшийся на сите, помещали обратно в ступку и добавляли неизмельченный материал. Измельчение продолжали до прохождения желаемого количества через сито.
Помимо янтарной кислоты, необходимое количество каждого из последующих компонентов сухого слоя по отдельности просеивали через сито с размером ячеек 30 меш в отдельные сборные контейнеры.

Суспензию со связывающим агентом получали, наливая воду в контейнер подходящего размера и добавляя затем маннит и гидроксипропилцеллюлозу в указанный контейнер. Раствор перемешивали в течение по меньшей мере 2 часов, и в нем не было видимых агломератов.
Затем в раствор медленно добавляли АФИ, получая суспензию со связывающим агентом. Суспензию со связывающим агентом перемешивали в процессе обработки до ее использования. Суспензия со связывающим агентом содержала следующие компоненты, указанные в Таблице 4.
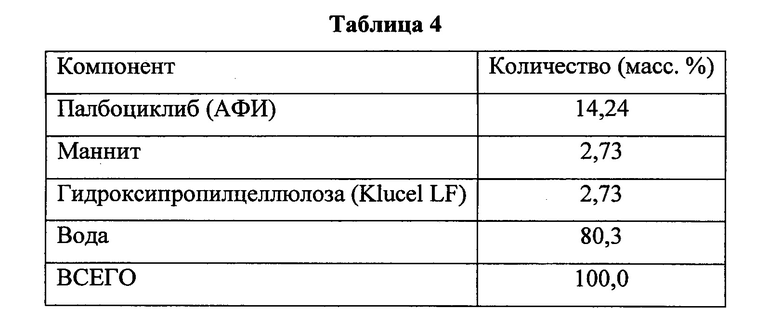
После стабилизации температуры в чаше продукта с псевдоожиженным слоем сухие вещества вносили в псевдоожиженный слой в следующем порядке: маннит, микрокристаллическая целлюлоза, янтарная кислота и кросповидон.
Затем начинали гранулирование в псевдоожиженном слое при температуре слоя 29-31°С, скорости распыления 12-30 г/мин, потоке воздуха 70-115 куб.м/час (м3/ч) и давлении распыления 1,1 бар, получая гранулят в псевдоожиженном слое.
Пример 4
Таблетки с гранулятом, полученным в псевдоожиженном слое
Таблетки изготавливали из гранулята, полученного в псевдоожиженном слое (FBG), по Примеру 3 с применением следующих методик. Сначала размер гранул уменьшали, пропуская их через Comil U5 с импеллером 1601 и размером отверстий сита 018R при скорости 1500 об/мин. Гранулы вносили в Comil настолько равномерно, насколько это возможно при визуальном контроле (от 20 до 25 минут на 2 кг гранулята). Измельченные гранулы пропускали через сито с размером ячеек 60 меш и материал, прошедший через сито, собирали в мешок и откладывали. Материал, оставшийся на сите с размером ячеек 60 меш, измельчали в Comil второй раз. Измельченные гранулы пропускали через сито с размером ячеек 60 меш и материал собирали в мешок. Материал, оставшийся на сите с размером ячеек 60 меш после второго просеивания осторожно продавливали через сито с использованием шпателя/скребка до тех пор, пока весь он не проходил через сито. Материал добавляли в мешок.
Состав конечной смеси для изготовления FBG-таблеток указан в Таблице 5.
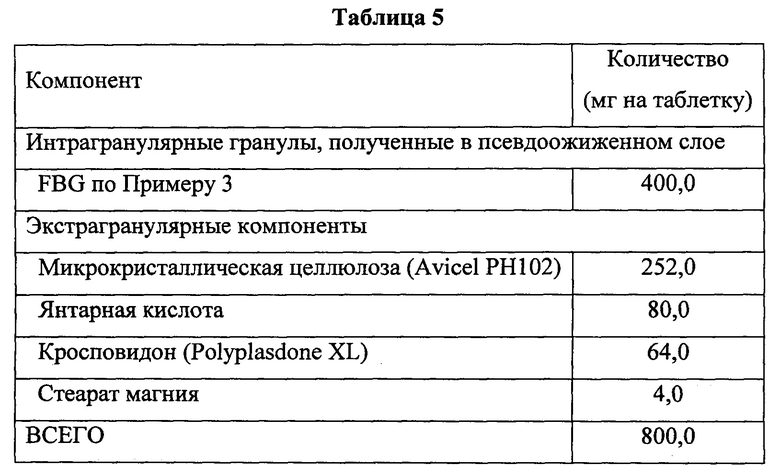
По необходимости, проводили расчет скорректированной массы экстрагранулярных компонентов на основании общей массы гранул, измельченных на предшествующей стадии.
Янтарную кислоту измельчали вручную (с использованием ступицы и пестика) небольшими аликвотами. Использовали количество, достаточное для получения достаточного количества измельченного материала. Измельченный материал пропускали через сито с размером ячеек 60 меш и материал собирали в новый мешок. Любой материал, оставшийся на сите, помещали обратно в ступку со следующей аликвотой. Необходимое количество измельченной янтарной кислоты разделяли на части.
Затем микрокристаллическую целлюлозу и кросповидон пропускали через сито с размером ячеек 30 меш и добавляли в смеситель. Измельченную янтарную кислоту и измельченные гранулы, полученные в псевдоожиженном слое, помещали в смеситель подходящего размера. Объемная плотность составляла 0,39 г/см3. Смесь перемешивали в течение 11,25 минуты при 16 об/мин (180 оборотов)
Затем стеарат магния смешивали с 3-10-кратным объемом (по визуальной оценке) смеси, полученной на предыдущей стадии, в емкости подходящего размера. Смесь перемешивали вручную, осторожно встряхивая ее в течение 30 секунд, и затем пропускали через сито с размером ячеек 30 меш. Содержимое помещали в смеситель и перемешивали в течение 3,75 минуты при 16 об/мин (60 оборотов).
Партию прессовали до целевых значений с использованием подходящего таблеточного пресса, такого как Korsch ХМ 12.
Пример 5
Дисперсия, высушенная распылением
Раствор гипромеллозы (НРМС Е3 Prem) получали растворением 3,25 масс. % НРМС Е3 в смеси растворителей из 90/10 метанола/воды (масс/масс.) с получением раствора НРМС, 3,25 масс %. Достаточное количество палбоциклиба (АФИ) добавляли в этот раствор с получением суспензии для распыления со следующим составом: 1,75 масс. % АФИ, 3,25 масс. % гипромеллозы, 85,5 масс. % метанола и 9,5 масс. % воды. Затем суспензию постоянно перемешивали для предотвращения осаждения АФИ в емкости с суспензией.
Распылительную сушилку предварительно разогревали с использованием нагретого сушильного газа (азот) при скорости потока 1850 г/мин и температуре сушильного газа на входе 130°С. После предварительного разогрева проводили распыление смеси из 90/10 (масс/масс.) метанола/воды до достижения стационарного термодинамического состояния. По достижении стационарного состояния в распылительной сушилке суспензию для распыления вносили в распылительную сушилку импульсным распылением при скорости подачи 130 г/мин, температуре раствора 130°С и давлении 250 ф/кв.дюйм (1723,75 кПа). Для предотвращения засорения форсунки использовали вторичный поток газообразного азота вокруг форсунки при давлении 60 ф/кв.дюйм (413,7 кПа) Сбор частиц проводили при температуре 45°С на выходе из распылительной сушилки.
После сбора частицы помещали в конвекционную полочную сушилку при 40°С и относительной влажности 15% по меньшей мере на 6 часов. Это приводило к снижению содержания остаточного растворителя в частицах до 0,3 масс. % остаточного метанола или менее.
Размер вторичных высушенных частиц измеряли анализатором размера частиц Malvern Particle Size Analyzer, доступным от Malvern Instruments Ltd., Фремингем, Массачусетс, с применением низкого дисперсионного давления от 0,5 до 1,0 бар (от 50 до 100 кПа), где D(4,3) представляет собой средний объемный диаметр; DV10 представляет собой объемный диаметр, равный или превышающий объемный диаметр 10% всех частиц; DV50 представляет собой объемный диаметр, равный или превышающий объемный диаметр 50% всех частиц; и DV90 представляет собой объемный диаметр, равный или превышающий объемный диаметр 90% всех частиц. Размер частиц указан в Таблице 6.

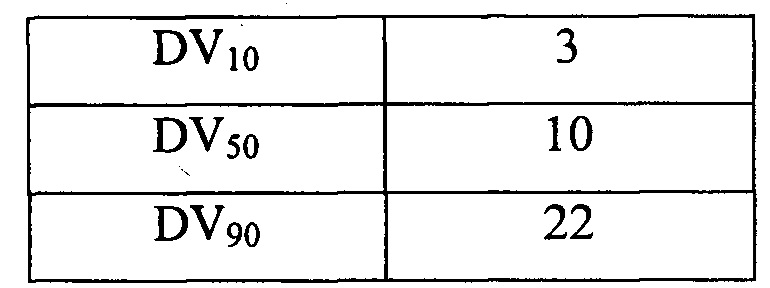
Разброс размера частиц (DV90-DV10)/DV50 составил 1,96. Удельный насыпной объем частиц составлял 5,6 см3 /г до уплотнения и 3,0 см3 /г после уплотнения.
Температура стеклования (Tg) частиц, измеренная при относительной влажности менее 5%, составила 117,5°С, как определено дифференциальной сканирующей калориметрией (DSC). Дифракция рентгеновских лучей на порошке (PXRD) продемонстрировала аморфность АФИ без поддающейся выявлению кристалличности. Морфология частиц, как определено сканирующей электронной микроскопией (SEM), продемонстрировала частицы в форме сфер и спавшихся сфер.
Пример 6
Сравнительная таблетка с дисперсией, высушенной распылительной сушкой
Из дисперсии, высушенной распылительной сушкой (SDD) по Примеру 5 изготавливали таблетки без водорастворимой кислоты с применением следующей методики. Половину количества микрокристаллической целлюлозы помещали в смеситель (бункерный смеситель или его эквивалент) и перемешивали на небольшой скорости в течение приблизительно 25 оборотов (2 минуты при 12 об/мин). В смеситель добавляли SDD. Смеситель промывали порцией хлорида натрия и проводили перемешивание.
В смеситель добавляли хлорид натрия, кроскармеллозу натрия и коллоидный диоксид кремния в количествах, рассчитанных на одну партию, и проводили перемешивание на небольшой скорости в течение приблизительно 180 оборотов (15 минут при 12 об/мин). Состав конечной смеси указан в Таблице 7. 

Измельчитель и мешок предварительно обрабатывали с использованием 25 масс. % количества микрокристаллической целлюлозы. Смесь, полученную на предыдущей стадии, пропускали через измельчитель с использованием Comil U5 с импеллером 1601 и размером отверстий сита 032R при скорости 1000 об/мин. Измельчитель промывали оставшейся частью количества микрокристаллической целлюлозы, рассчитанного на одну партию, измельченный материал переносили из мешка в смеситель (бункерный смеситель или его эквивалент) и проводили перемешивание на небольшой скорости в течение приблизительно 180 оборотов (15 минут при 12 об/мин).
Интрагранулярный стеарат магния просеивали через сито с отверстиями подходящего размера и добавляли в смеситель, использованный на предыдущей стадии, после чего проводили перемешивание на небольшой скорости в течение приблизительно 60 оборотов (5 минут при 12 об/мин).
Затем смесь прессовали с использованием Korsch ХР 1 или его эквивалента.
Прессованную смесь пропускали через измельчитель с использованием Comil U5 с импеллером 1601 и размером отверстий сита 050G при скорости 1000 об/мин.
Гранулят переносили из мешка в смеситель (бункерный смеситель или его эквивалент). Рассчитывали количество экстрагранулярного коллоидного диоксида кремния и добавляли его в смеситель, использованный на предыдущей стадии, после чего проводили перемешивание в течение приблизительно 180 оборотов (15 минут при 12 об/мин). Рассчитывали необходимое количество экстрагранулярного стеарата магния, просеивали его через сито с отверстиями подходящего размера и добавляли в смеситель, использованный на предыдущей стадии, после чего проводили перемешивание на небольшой скорости в течение приблизительно 60 оборотов (5 минут при 12 об/мин).
Таблетки прессовали с использованием одностанционного пресса (Korsch ХР 1 или его эквивалента).
Пример 7
Таблетка с дисперсией, высушенной распылительной сушкой
Таблетки изготавливали из SDD по Примеру 5 с применением следующих методик. Половину количества микрокристаллической целлюлозы помещали в смеситель (бункерный смеситель или его эквивалент) и перемешивали на небольшой скорости в течение приблизительно 25 оборотов (2 минуты при 12 об/мин). В смеситель добавляли SDD, промывали его порцией хлорида натрия и проводили перемешивание. В смеситель добавляли янтарную кислоту, хлорид натрия, кроскармеллозу натрия и коллоидный диоксид кремния в количествах, рассчитанных на одну партию, и проводили перемешивание на небольшой скорости в течение приблизительно 180 оборотов (15 минут при 12 об/мин). Конечный состав смеси указан в Таблице 8.
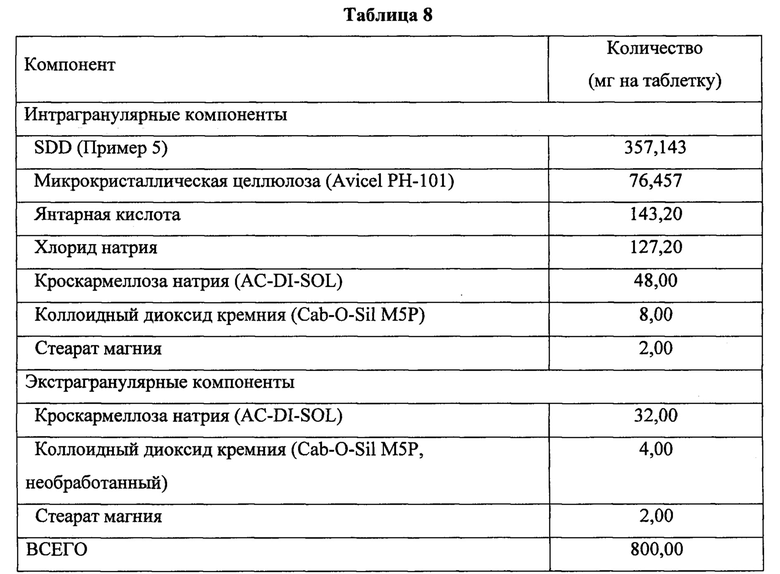
Измельчитель и мешок предварительно обрабатывали с использованием 25 масс. % количества микрокристаллической целлюлозы. Смесь, полученную на предыдущей стадии, пропускали через измельчитель с использованием Comil U5 с импеллером 1601 и размером отверстий сита 018R при скорости 1000 об/мин. Измельчитель промывали оставшейся частью количества микрокристаллической целлюлозы, рассчитанного на одну партию, измельченный материал переносили из мешка в смеситель (бункерный смеситель или его эквивалент) и проводили перемешивание на небольшой скорости в течение приблизительно 180 оборотов (15 минут при 12 об/мин).
Интрагранулярный стеарат магния просеивали через сито с отверстиями подходящего размера и добавляли в смеситель, использованный на предыдущей стадии, после чего проводили перемешивание на небольшой скорости в течение приблизительно 60 оборотов (5 минут при 12 об/мин).
Затем смесь прессовали с использованием Korsch ХР 1 или его эквивалента.
Прессованную смесь пропускали через измельчитель с использованием Comil U5 с импеллером 1601 и размером отверстий сита 050G при скорости 1000 об/мин. Рассчитывали необходимое количество экстрагранулярного стеарата магния, просеивали его через сито с отверстиями подходящего размера и добавляли в смеситель, использованный на предыдущей стадии. Смесь перемешивали в течение приблизительно 60 оборотов (5 минут при 12 об/мин).
Таблетки прессовали с использованием одностанционного пресса (Korsch ХР 1 или его эквивалента).
Пример 8
Тест растворения 1 при рН 5,5
Для анализа растворения изготавливали тестовые композиции в форме таблеток, содержащие палбоциклиб в форме свободного основания (АФИ), водорастворимую кислоту, микрокристаллическую целлюлозу (Avicel PH102), моногидрат лактозы (Fast Flo 316), кросповидон (Kollidon CL) и стеарат магния. С применением метода сухого гранулирования (СГ), описанного в Примере 1, были изготовлены тестовые таблетки со следующими кислотами: яблочная кислота, малеиновая кислота, янтарная кислота, фумаровая кислота, винная кислота, n-толуолсульфоновая кислота, бензойная кислота и бензолсульфоновая кислота. Прямым прессованием (ПП) (без сухого гранулирования) были изготовлены тестовые таблетки с яблочной кислотой и лимонной кислотой.
Растворение таблеток анализировали в USP-аппарате 2 с лопастями, вращающимися с частотой 50 об/мин, в 500 мл ацетатного буфера, 10 мМ, рН 5,5, при температуре 37°С. В каждой контрольной точке отбирали образец объемом 6 мл и пропускали его через полнопоточный фильтр с размером пор 10 мкм. Анализ проводили в автономном режиме при длине волны в ультрафиолетовом (УФ) диапазоне 367 нм.
Сравнительные данные по растворению получали с использованием капсулы палбоциклиба с изетионатной солью (ISE) и таблетки с АФИ в форме свободного основания, изготовленной с применением метода сухого гранулирования по Примеру 1 и смеси без водорастворимой кислоты. Результаты теста растворения для таблеток, содержащих янтарную кислоту, малеиновую кислоту, яблочную кислоту, фумаровую кислоту и винную кислоту, показаны на Фиг. 1. Таблетки, содержащие янтарную кислоту, яблочную кислоту и винную кислоту, продемонстрировали лучшее растворение, при этом за 30 минут происходило растворение более 50% лекарственного средства, как показано на Фиг. 2.
Пример 9
Химическая стабильность композиций
Тестовые таблетки, полученные в Примере 8, хранили при 70°С / RH 75% в течение 8 дней. Таблетки измельчали и анализировали на предмет примесей с применением метода высокоэффективной жидкостной хроматографии (ВЭЖХ) следующим образом: колонка Waters CSH С18, 2,1×100 мм, 1,7 мкм; подвижная фаза (градиентное элюирование) А - 0,03%-ная трифторуксусная кислота, подвижная фаза В - 0,03%-ная трифторуксусная кислота в ацетонитриле; температура колонки 45°С; скорость потока 0,5 мл/мин; УФ-детекция при 234 нм; вводимый объем 2 мкл; и время анализа 10,72 минуты. Результаты представлены в Таблице 9.
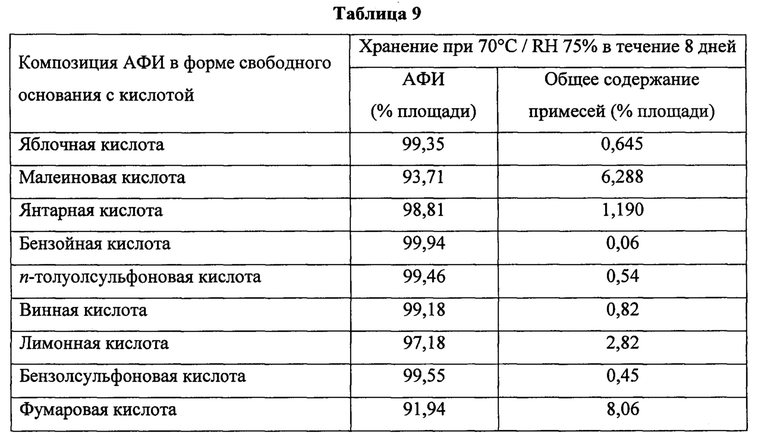
Таблетки, содержащие яблочную кислоту, янтарную кислоту, бензойную кислоту, n-толуолсульфоновую кислоту, винную кислоту и бензолсульфоновую кислоту, имели приемлемое общее содержание примесей после хранения при 70°С / RH 75% в течение 8 дней. На основании лучших результатов экспериментов растворения и стабильности для дальнейшей разработки были выбраны композиции, содержащие янтарную кислоту, яблочную кислоту и винную кислоту.
Пример 10
Тест растворения 2 с приближением раствора к состоянию насыщения
Для анализа композиций был разработан метод растворения in vitro с приближением раствора к состоянию насыщения. В этом методе таблетки помещали в USP-аппарат 2 (с лопастями) с перемешиванием при 50 об/мин и 500 мл 50 мМ фосфатного буфера + 0,1 М NaCl (рН 6,5) при температуре 37°С. Периодически отбирали образцы и фильтровали их через фильтры с размером пор 10 мкм. Концентрацию АФИ измеряли в автономном режиме при УФ 367 нм. Получали данные о растворении таблеток, изготовленных в Примере 1 (свободное основание (FB) + янтарная, таблетка), Примере 2 (FB + янтарная, двухслойная таблетка), Примере 4 (FB + янтарная, FBG-таблетка), Примере 6 (SDD, таблетка) и Примере 7 (SDD + янтарная, таблетка). Профили растворения показаны на Фиг. 3. Также показаны данные о растворении имеющихся в продаже капсул палбоциклиба в форме свободного основания (капсулы со свободным основанием), состав которых указан в Таблице 10.
Как показано на Фиг. 3, помещение лекарственных форм по Примерам 1, 2 и 4 в условия растворения с приближением раствора к состоянию насыщения приводит к растворению (а) не менее 40% палбоциклиба за 15 минут; (б) не менее 35% палбоциклиба за 30 минут; (в) не менее 25% палбоциклиба за 60 минут; или (г) двух или более из (а), (б) и (в).
Пример 11
Уровни экспозиции лекарственного средства при использовании имеющихся в продаже капсул со свободным основанием
У здоровых добровольцев проводили рандомизированное, открытое, перекрестное исследование с однократным введением, 4 последовательностями и 4 периодами. Каждый из двадцати восьми (28) субъектов принимал 125 мг палбоциклиба однократно в 4 различных условиях или вариантах приема (утром натощак [А], после приема жирной пищи [В], после приема нежирной пищи [С] и с приемом умеренно жирной пищи за 1 час до и через 2 часа после приема палбоциклиба [D]).
Последовательность вариантов приема на протяжении периодов 1-4 указана в Таблице 11. Периоды исследования были разделены периодами выведения продолжительностью по меньшей мере 10 дней. На протяжении 144 часов после приема лекарственного средства у субъектов отбирали образцы для анализа фармакокинетики (ФК). Значения фармакокинетических параметров указаны в Таблице 12. 
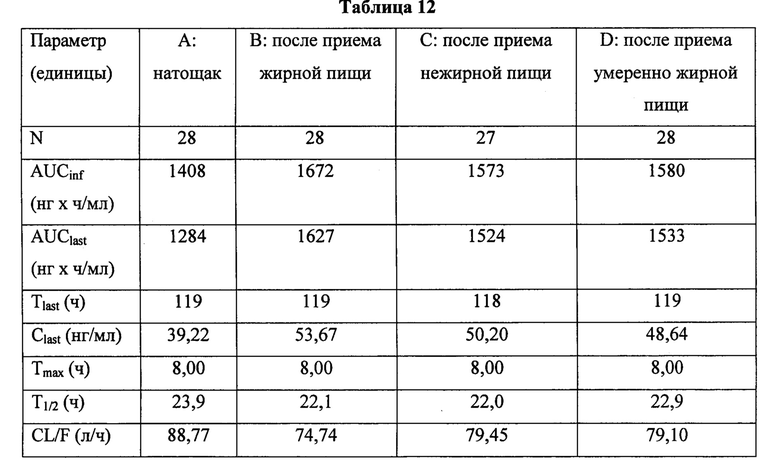

При приеме натощак имеющаяся в продаже композиция в форме капсул со 125 мг палбоциклиба продемонстрировала меньшие AUC и Cmax, по сравнению с уровнями экспозиции в трех условиях после еды. Соответственно, было рекомендовано принимать имеющиеся в продаже капсулы с пищей.
Пример 12
Эффект антацида на биодоступность палбоциклиба
Задачей данного исследования было изучение возможного эффекта повышения рН в желудке, достигаемого многократным приемом ингибитора протонной помпы (ИПП, конкретно, рабепразола), на фармакокинетику (ФК) при однократном пероральном приеме имеющейся в продаже капсулы со 125 мг палбоциклиба натощак.
Результаты представлены в Таблице 13 ниже. В присутствии рабепразола имеющаяся в продаже композиция в форме капсулы со 125 мг палбоциклиба продемонстрировала существенно меньшие AUC и Cmax, чем при введении палбоциклиба самого по себе.
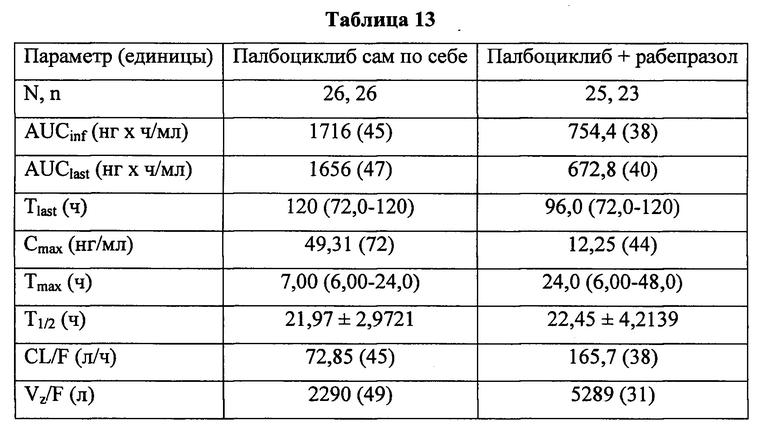
Пример 13
Эффект антацида на биодоступность тестовых композиций
У здоровых добровольцев проводили перекрестное, открытое, нерандомизированное фармакокинетическое исследование для оценки эффекта приема антацида на биодоступность 125 мг таблетки (однократное введение) шести экспериментальных композиций палбоциклиба в присутствии рабепразола, по сравнению с введением палбоциклиба самого по себе натощак. В Таблице 14(A)-(F) показаны результаты для групп 1-6: Пример 1; Пример 6; Пример 7; Пример 2; Пример 4; и раствор палбоциклиба, 125 мг, для перорального введения, соответственно.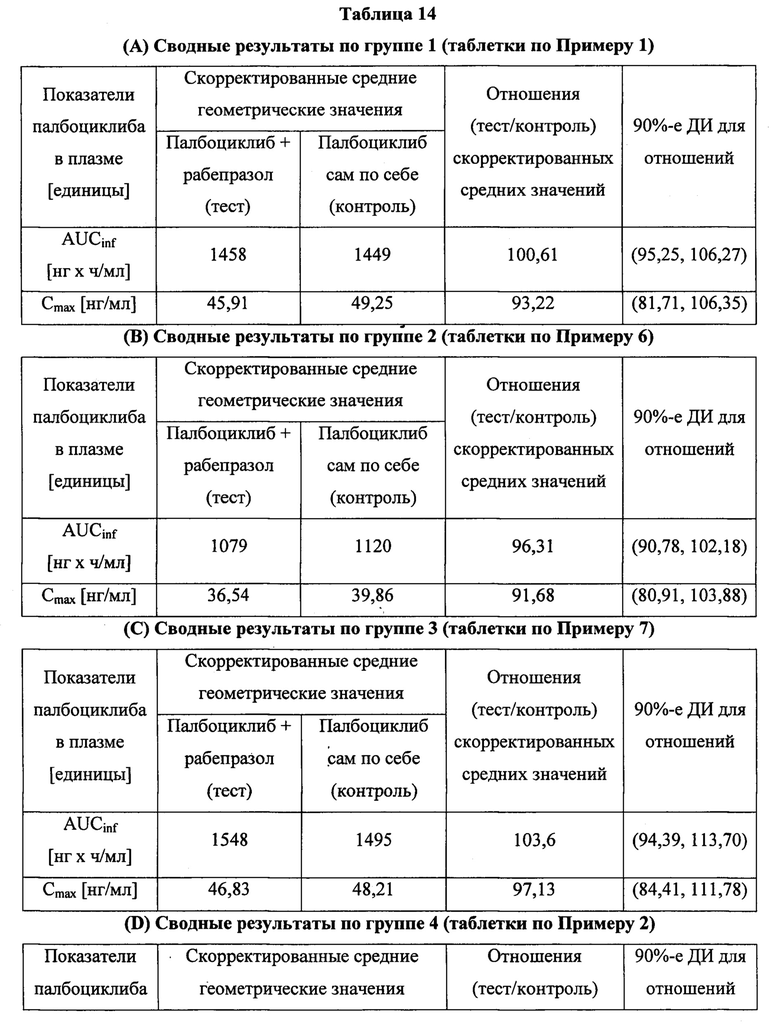
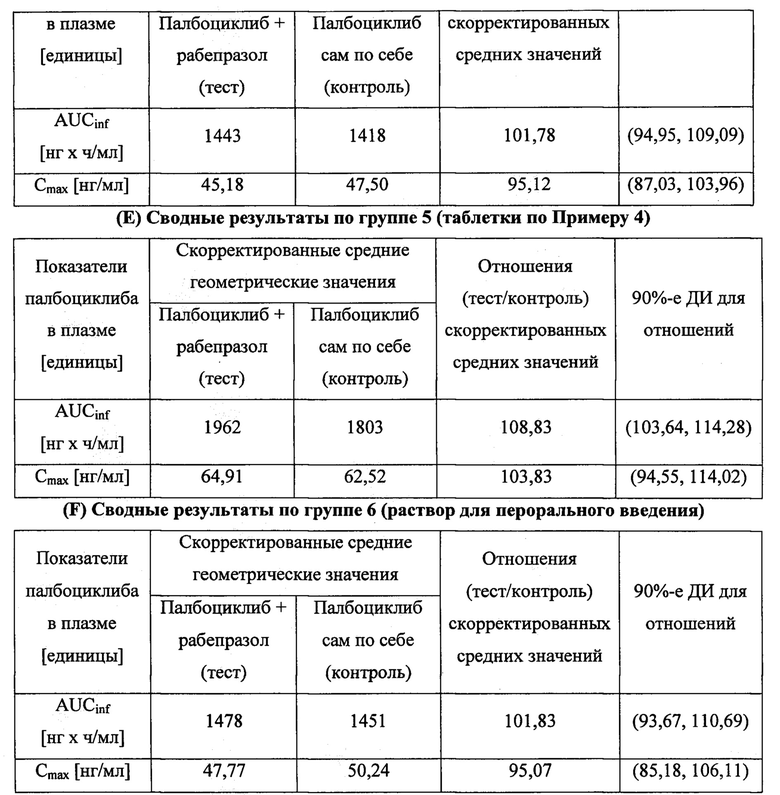
Пример 14
Ускоренный тест на стабильность
Стабильность таблеток по Примеру 1 (сухой гранулят + янтарная кислота) при ускоренном старении определяли в следующих условиях. Таблетки помещали в лабораторные стаканы с указанными ниже условиями. Контроль влажности осуществляли посредством термостатного контроля или насыщенных солевых растворов. Периодичность забора образцов в указанных условиях показана в Таблице 15 ниже. До анализа образцы (включая интактные контроли) хранили в холодильнике.
Указанную относительную влажность поддерживали с использованием следующих насыщенных солевых растворов: RH 22% контролировали с использованием ацетата калия; RH 50% контролировали с использованием бромида натрия для условий 30°С и 40°С; и RH 75% - с использованием термостатов с контролируемой влажностью.

Продуктом деградации является сукциниловый аддукт АФИ, имеющий структуру:
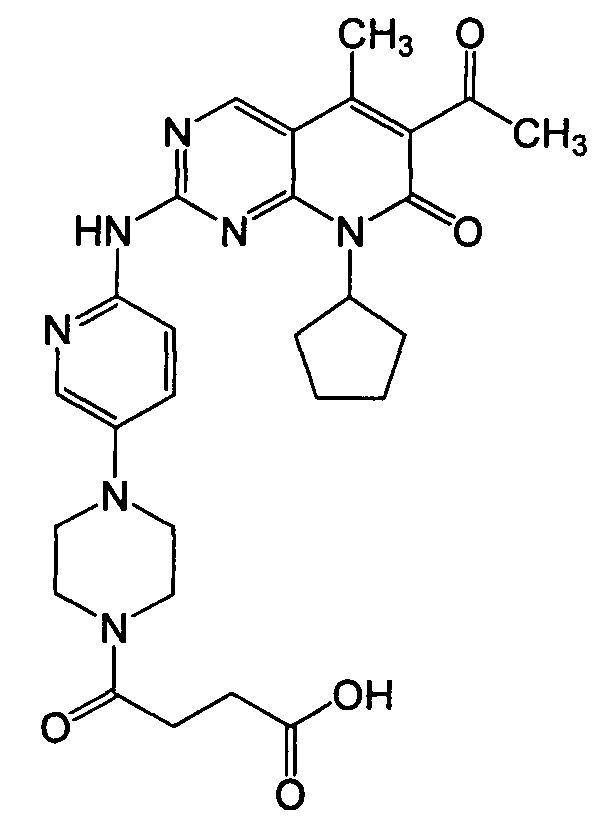
Подготовку образцов проводили, помещая каждую таблетку в мерную колбу объемом 100 мл с якорем магнитной мешалки и приблизительно 100 мл растворителя (1440 мл воды: 400 мл ацетонитрила: 160 мл 1 Н HCl; 0,1 Н HCl: ацетонитрил 80:20). Колбу помещали на магнитную мешалку и образец перемешивали в течение одного часа при наибольшей возможной скорости якоря. Затем отбирали аликвоту и центрифугировали ее в полипропиленовой пробирке при 3000 об/мин в течение 5 минут. Супернатант разводили 1:5 растворителем и отбирали образец в пробирку для анализа посредством ВЭЖХ. Каждая таблетка содержала 125 мг АФИ, что приводило к концентрации конечного раствора 0,25 мг/мл. Образцы анализировали с применением метода ВЭЖХ, описанного ниже. Результаты представлены в Таблице 15 выше и демонстрируют отличную долгосрочную стабильность.
Растворитель (разбавитель):
1440 мл воды: 400 мл ацетонитрила: 160 мл 1 Н HCl /// (0,1 Н HCl: ацетонитрил, 80:20).
Подвижная фаза:
подвижная фаза А: 0,03%-ная трифторуксусная кислота (0,6 мл трифторуксусной кислоты в 2000 мл воды);
подвижная фаза В: 0,03%-ная трифторуксусная кислота в ацетонитриле (0,6 мл трифторуксусной кислоты в 2000 мл ацетонитрила);
0,5 мл/мин.
Градиент
Исходно 88% А 12% В
0,22 мин 88% А 12% В
8,72 мин 15% А 85% В
8,82 мин 88% А 12% В
10,72 мин Общее время анализа
234 нм, % площади против палбоциклиба (основная полоса).
Введение образцов (подготовленных, как описано ниже) по 2 мкл. В камере с образцами поддерживали температуру 5°С.
Подготовка образцов
Подготовку образцов проводили, помещая каждую таблетку в мерную колбу объемом 100 мл с якорем магнитной мешалки и приблизительно 100 мл растворителя. Колбу помещали на магнитную мешалку и образец перемешивали в течение одного часа при наибольшей возможной скорости якоря. Затем отбирали аликвоту и центрифугировали ее в полипропиленовой пробирке при 3000 об/мин в течение 5 минут. Супернатант разводили 1:5 растворителем и отбирали образец в пробирку для анализа посредством ВЭЖХ. Каждая таблетка содержала 125 мг палбоциклиба, что приводило к концентрации конечного раствора 0,25 мг/мл.
Кроме того, аналогичным образом анализировали таблетки по Примеру 4 (гранулят, полученный в псевдоожиженном слое + янтарная кислота). Результаты представлены в следующей Таблице 16 и демонстрируют, что концентрация продукта деградации была неприемлемой для долгосрочной стабильности. 

Пример 15
Стабильность при длительном хранении
Таблетки по Примеру 1 упаковывали в термозапечатанный флакон с влагопоглотителем. После хранения в течение 1 года при 25°С / RH 60% уровень сукцинилового аддукта в таблетках составил менее 0,05%. Таблетки по Примеру 4 упаковывали в термозапечатанный HDPE-флакон с влагопоглотителем. После хранения в течение 6 месяцев при 25°С / RH60% уровень сукцинилового аддукта в таблетках составил 0,23%.
Пример 16
Композиции в форме таблеток, покрытых пленочной оболочкой
В Таблице 17 описаны оптимизированные композиции A1, А2 и В, продемонстрировавшие приемлемые производственные характеристики для изготовления твердых лекарственных форм в коммерческих целях. Таблетки были изготовлены с применением следующей методики. Микрокристаллическую целлюлозу РН102, коллоидный диоксид кремния, интрагранулярный кросповидон CL, и АФИ смешивали и пропускали через Comil для гомогенизации. В смесь добавляли интрагранулярный стеарат магния. Проводили сухое гранулирование смеси с применением роликового пресса и измельчения гранул.
С гранулами смешивали просеянную янтарную кислоту, микрокристаллическую целлюлозу РН-200 и экстрагранулярный кросповидон (CL или CL-SF). В конечную смесь добавляли экстрагранулярный стеарат магния или стеарилфумарат натрия в качестве смазывающего агента для изготовления таблеток. Таблетки изготавливали с использованием роторного таблеточного пресса с предварительным прессованием. В композиции А2 была использована система наружной смазки (ELS) с нанесением номинального количества смазывающего агента непосредственно на концы таблеточного пресса.
Таблетки покрывали пленочной оболочкой до прибавки массы от 2 до 4% с использованием Opadry Pink (03К140024) и очищенной воды до содержания твердых веществ 12% масс/масс.
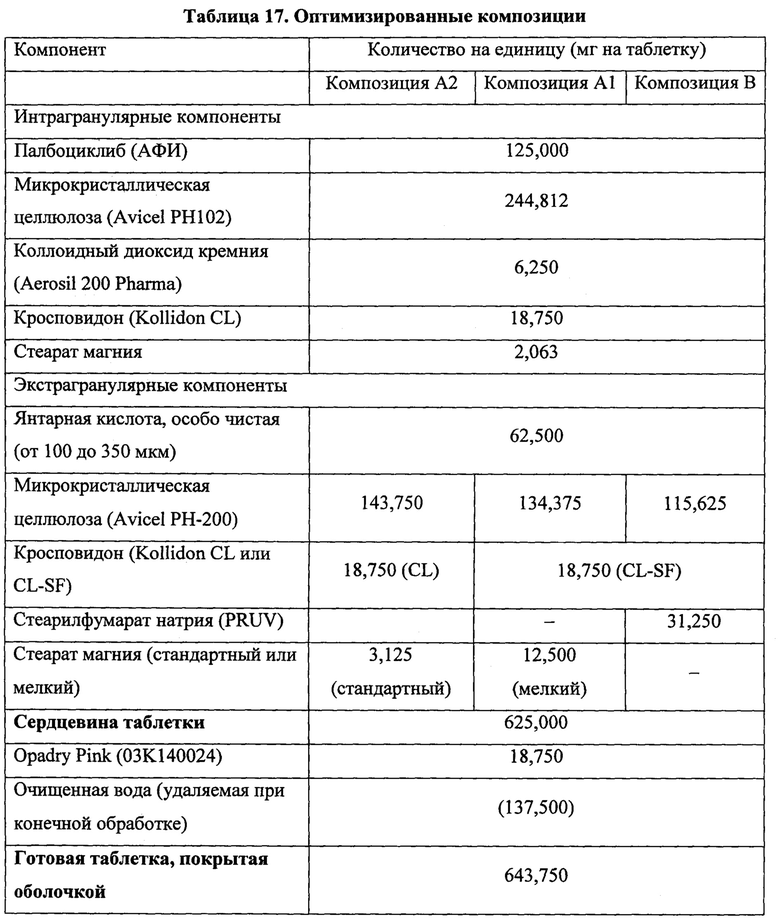
Пример 17
Оптимизация уровня смазывающего агента
Композиции изготавливали, как описано для композиции А2 в Примере 16, с применением разных уровней интрагранулярного и экстрагранулярного смазывающего агента стеарата магния, как показано в Таблице 17. Для каждой композиции измеряли твердость (прочность таблеток на разрыв, USP<1217>), истираемость (USP<1216>, за 100 оборотов), расширенную истираемость (USP<1216>, за 375 оборотов) и адгезивность к пуансону (Hutchins, MacDonald, Mullarney, Assessing Tablet-Sticking Propensity, Pharmaceutical Technology, Volume 36, Issue 1, 2012), как указано в Таблице 18. Для USP-методов использовали образцы меньшего размера.
Снижение уровней как интрагранулярного, так и экстрагранулярного смазывающего агента существенно повышало твердость таблеток и уменьшало истираемость. Адгезивность таблеток была более чувствительна к уровню экстрагранулярного смазывающего агента, чем к уровню интрагранулярного смазывающего агента. Адгезивность была существенно снижена при повышении уровня экстрагранулярного стеарата магния. Различий адгезивности к рифленому/гладкому ролику при различных уровнях интрагранулярного стеарата магния не наблюдали. Как показано в Таблице 18, уменьшение отношения интрагранулярного смазывающего агента к экстрагранулярному с 1 масс. % / 0 масс. % до 0,33 масс. % / 0,5 масс. % обеспечивало хорошую прочность таблеток, снижало истираемость и снижало адгезивность таблеток композиции А2. Уровни смазывающих агентов приблизительно 5 масс. % стеарилфумарата натрия или 2 масс. % мелкого стеарата (CaSt 2249) уменьшали адгезивность к пуансону и дефекты таблеток.
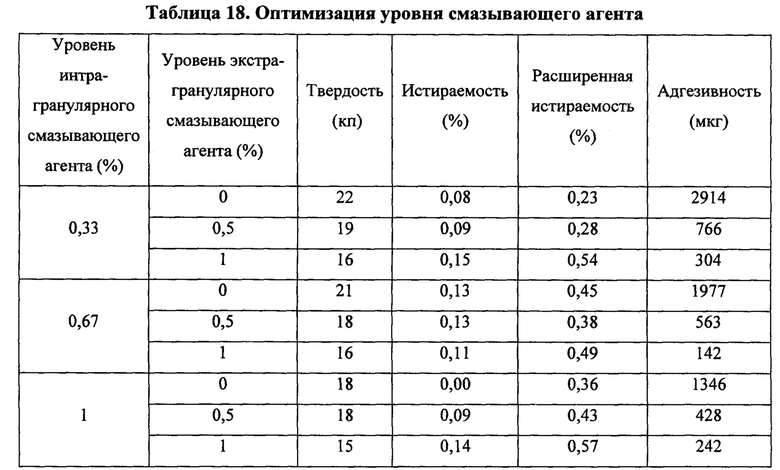
Пример 18
Оптимизация твердости и истираемости таблеток
В целом, композиции изготавливали, как описано для композиции А2 в Примере 16, изменяя порядок добавления кислоты, добавления моногидрата лактозы, добавления сухих связывающих агентов (Kollidon SF, Avicel PH105, Klucel EXF) и изменяя отношение интрагранулярной микрокристаллической целлюлозы к экстрагранулярной. Определяли свойства этих разных композиций: объемную плотность исходной смеси (USP<616>), прочность ленты, полученной с использованием роликового пресса, на разрыв (Zinchuk AV, Mullarney MP, Hancock ВС.Simulation of roller compaction using a laboratory scale compaction simulator. Int J Pharm. 2004 Jan 28;269(2):403-15.), адгезивность к пуансону (Hutchins, MacDonald, Mullarney, Assessing Tablet-Sticking Propensity, Pharmaceutical Technology, Volume 36, Issue 1, 2012), прочность таблеток (прочность таблеток на разрыв, USP<1217>), истираемость (USP<1216>, за 375 оборотов) и время распадаемости (USP<701>). Для USP-методов использовали образцы меньшего размера.
Композиции без лактозы продемонстрировали более высокую твердость таблеток и меньшую истираемость. Кроме того, добавление Kollidon SF (высший сорт) способствовало уменьшению времени распадаемости с одновременным повышением твердости таблеток. Было обнаружено, что экстрагранулярное добавление кислоты предпочтительно с точки зрения уменьшения времени распадаемости.
Удаление лактозы из композиции приводило к получению таблеток с меньшей истираемостью, сохранявших быструю распадаемость. Включение сухих связывающих агентов не обеспечивало дополнительных преимуществ по снижению истираемости, но несколько снижало адгезивность. Тем не менее, включение связывающих агентов может, в некоторых случаях, отрицательно влиять на распадаемость/набухание. 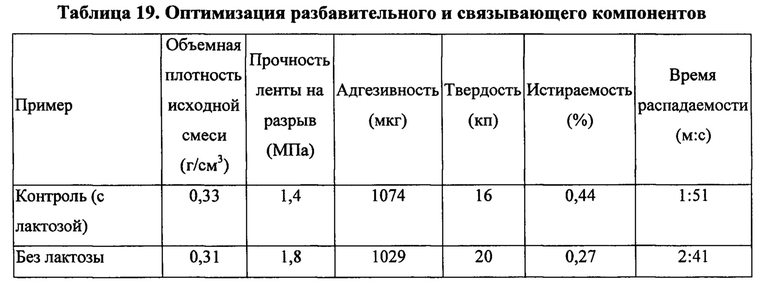
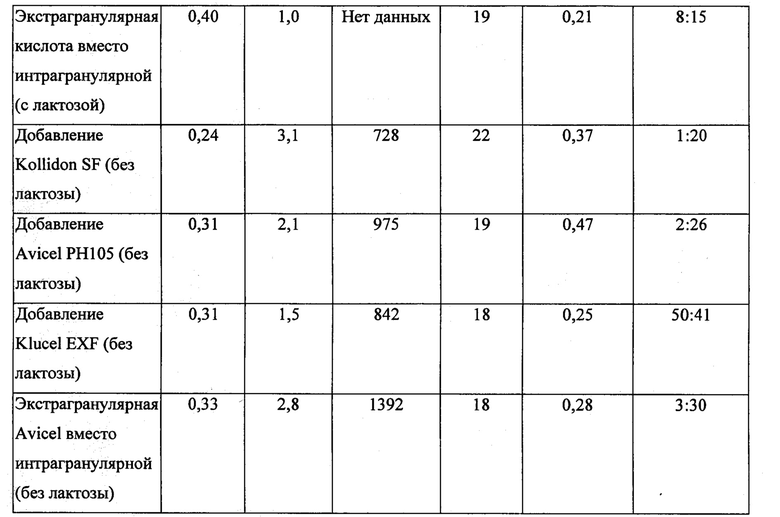
Пример 19
In vitro-растворение композиций A1, А2 и В
In vitro-растворение композиций A1, А2 и В определяли с применением теста растворения 2 в условиях приближения раствора к состоянию насыщения (фосфатный буфер, 50 мМ, рН6,5, 0,1 М NaCl), описанного в Примере 10. Профили растворения этих композиций сравнивали с таблетками, изготовленными согласно Примерам 1-7. Таблетка по Примеру 1 продемонстрировала отсутствие лекарственного взаимодействия с ингибитором протонной помпы при приеме здоровыми добровольцами натощак, и ее использовали как минимальный целевой профиль растворения. Сравнительные данные по растворению показаны на Фиг. 4. Все композиции A1, А2 и В превосходили как имеющиеся в продаже капсулы со свободным основанием (изготовленные, как описано в Примере 10), так и капсулы с изетионатной солью палбоциклиба (ISE), использованные на ранних этапах разработки, состав которых указан в Таблице 20. После 10 минут все композиции A1, А2 и В превосходили таблетки по Примерам 1-7 и, таким образом, соответствовали целевому растворению для твердых лекарственных форм.
Таблица 20
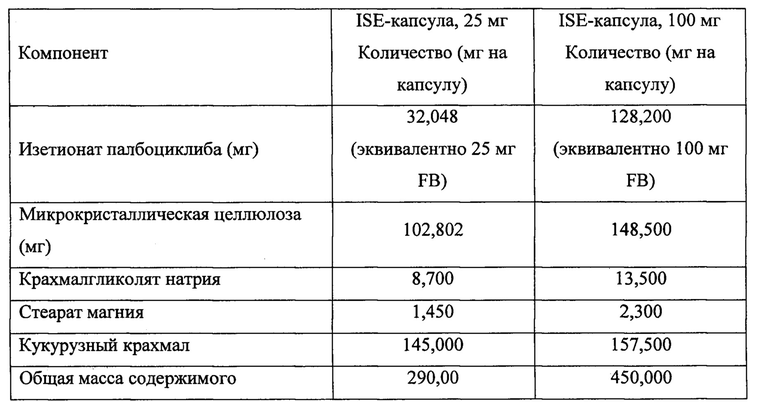
Пример 20
Влияние распределения размера частиц янтарной кислоты
Влияние распределения размера частиц янтарной кислоты на скорость образования сукцинилового аддукта оценивали для кислоты непосредственно после получения (широкое распределение размера частиц диаметром до 1 мм) и трех вариантов просеянной кислоты. Меньший размер частиц кислоты приводил к большей скорости образования примеси ввиду большей удельной поверхности, приводящей к более высокой частоте контактов кислоты с АФИ в форме свободного основания. Таким образом, для улучшения химической стабильности в составе лекарственной формы была предпочтительна кислота с размером частиц более приблизительно 100 мкм. 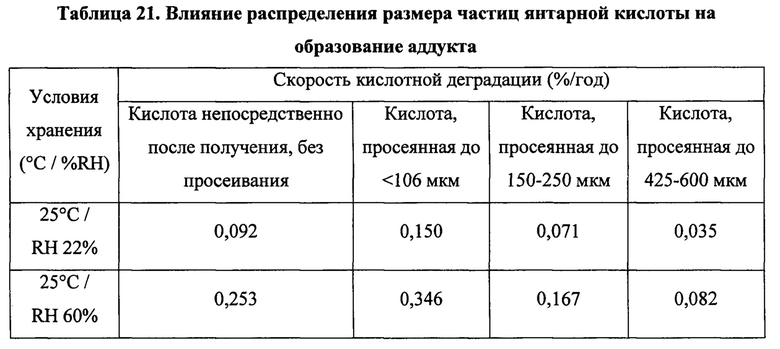

Пример 21
Анализ физической стабильности
Для оценки физической стабильности композиций был разработан метод количественной рамановской спектроскопии. Для рамановского метода использовали PhAT-зонд от Kaiser Optical Systems, и модель количественного анализа выстраивали с использованием набора калибровочных стандартов, полученных из АФИ, сукцинатного комплекса АФИ и эксципиентов. Относительную стабильность композиций А1 и В оценивали, определяя количество сукцинатного комплекса АФИ в композиции после хранения таблеток при 30-50°С и RH до 75% на протяжении периода продолжительностью до 1 месяца.
Композиция В продемонстрировала меньшее, по сравнению с композицией А1, преобразование в сукцинатный комплекс. Степень преобразования нарастала с течением времени, и его скорость увеличивалась при хранении в условиях повышенной температуры и влажности.
Количество сукцинатного комплекса АФИ, образованного в условиях разной температуры и влажности в композициях А1 и В, показано в Таблице 22 как функция времени, где «LOD» относится к пределу обнаружения и «LOQ» относится к пределу количественного определения.
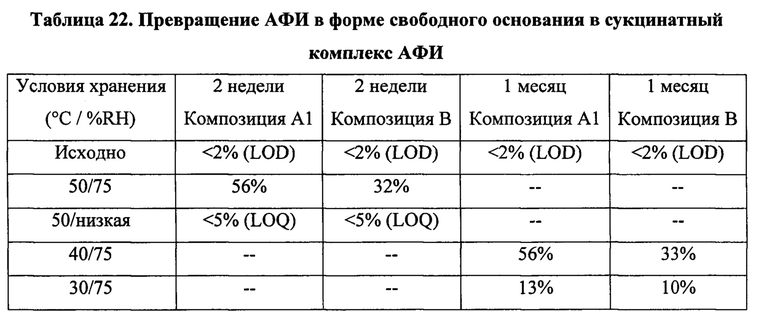
Оценивали физическую стабильность прототипной композиции А1 в форме таблетки без оболочки в открытых условиях. Таблетки подвергали повышенным температурам и влажности. Превращение в сукцинатный комплекс в анализируемых образцах наблюдали с применением рамановской спектроскопии.
Проводили сравнение химической чистоты композиций А1 и В после хранения в течение 1 месяца при 40°С / RH 75%. Как показано в Таблице 23, в композиции В степень образования примесей была существенно меньше, чем в композиции А1.
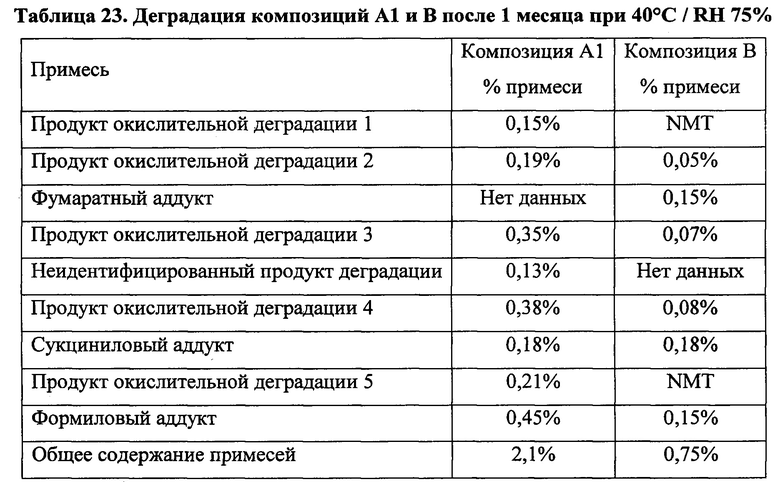
На основании этих данных было определено, что влажность (%RH) оказывает существенное влияние на скорость превращения АФИ из свободного основания в сукцинатный комплекс. Повышение температуры также влияло на превращение в комплекс, но оказывало менее сильный эффект, чем влажность.
Таблетки, предварительно уравновешенные при RH 60% перед упаковкой в фольгу, продемонстрировали быстрое образование сукцинатного комплекса. Данные по стабильности на протяжении 9 недель показали, что для минимизации образования сукцинатного комплекса палбоциклиба в лекарственной форме необходимо контролировать активность воды в таблетках. В условиях повышенной влажности (RH75%) композиция В (содержащая экстрагранулярный стеарилфумарат натрия в качестве смазывающего агента) продемонстрировала меньшее превращение в сукцинатный комплекс по сравнению с композицией А1 (содержащей стеарат магния в качестве экстрагранулярного смазывающего агента).
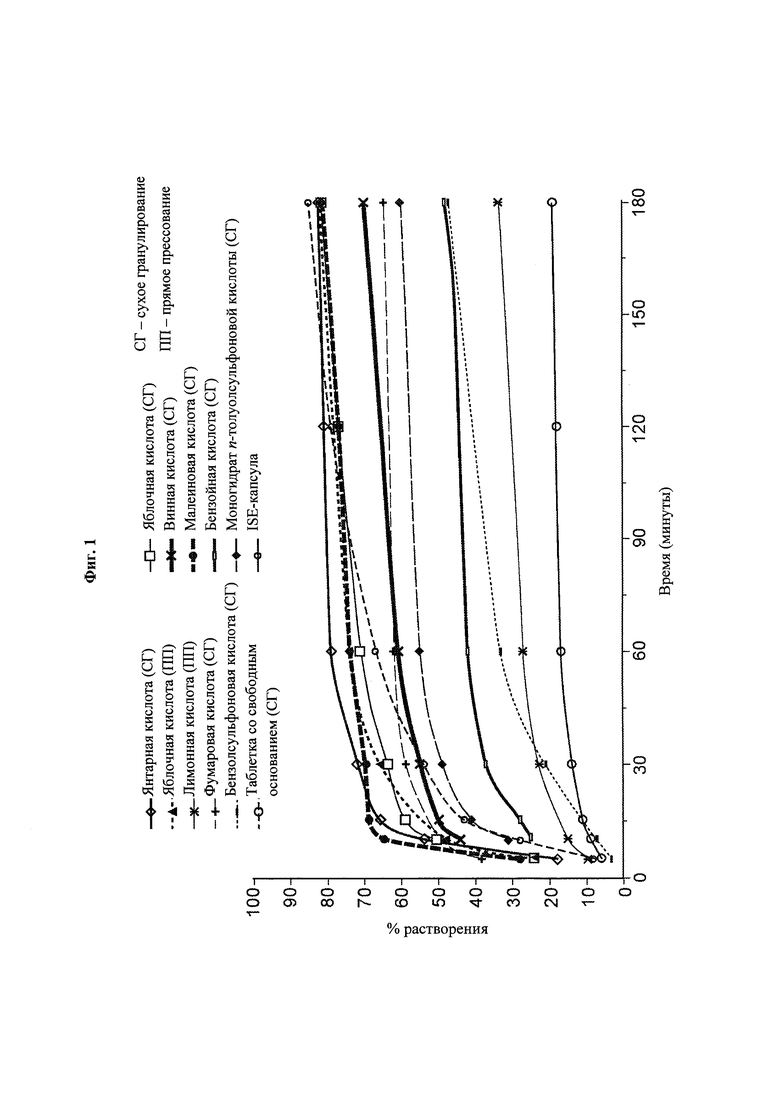
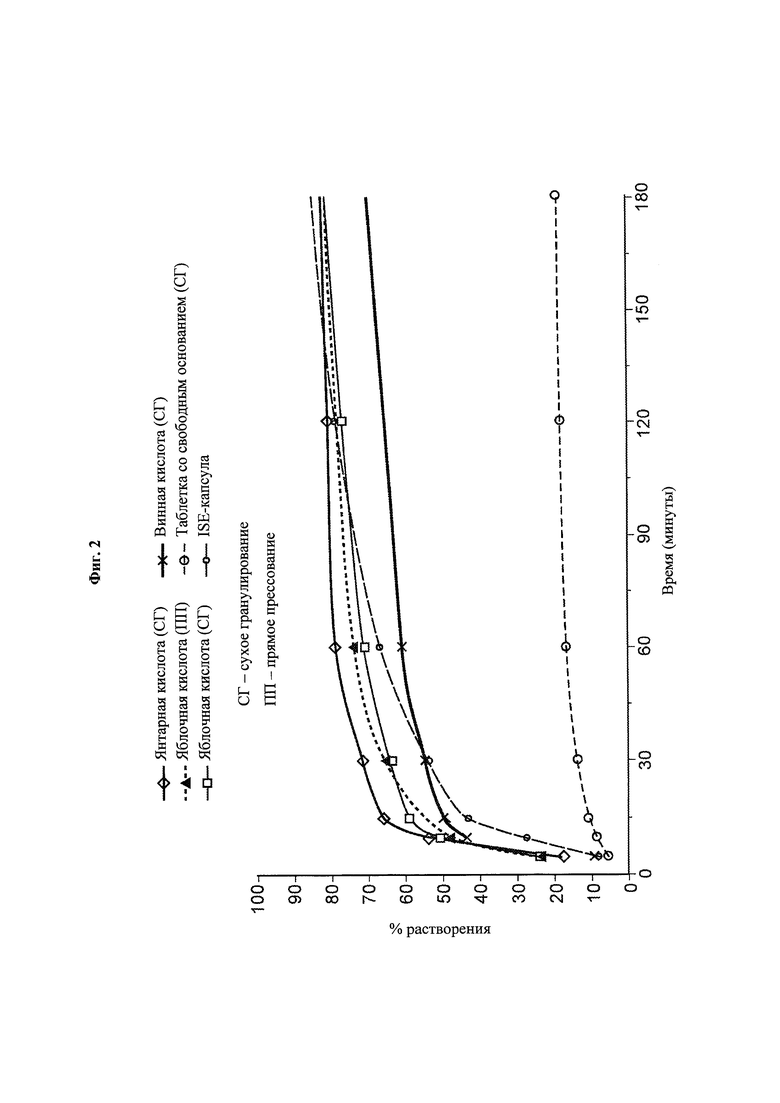
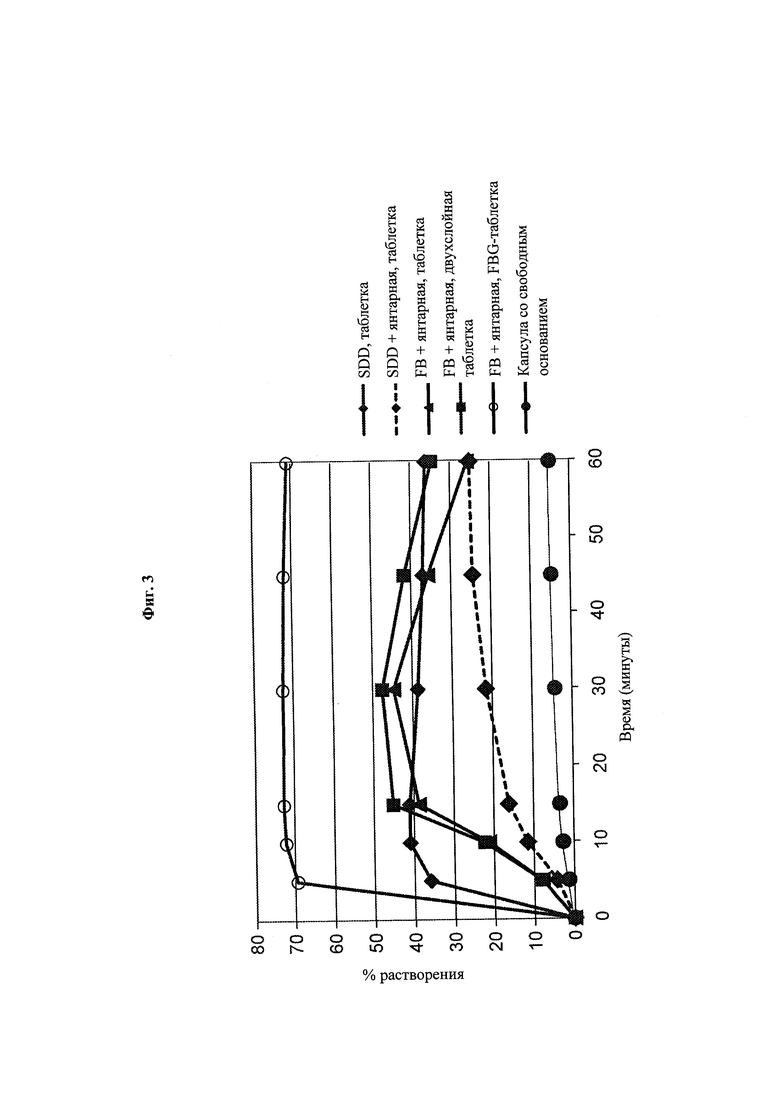
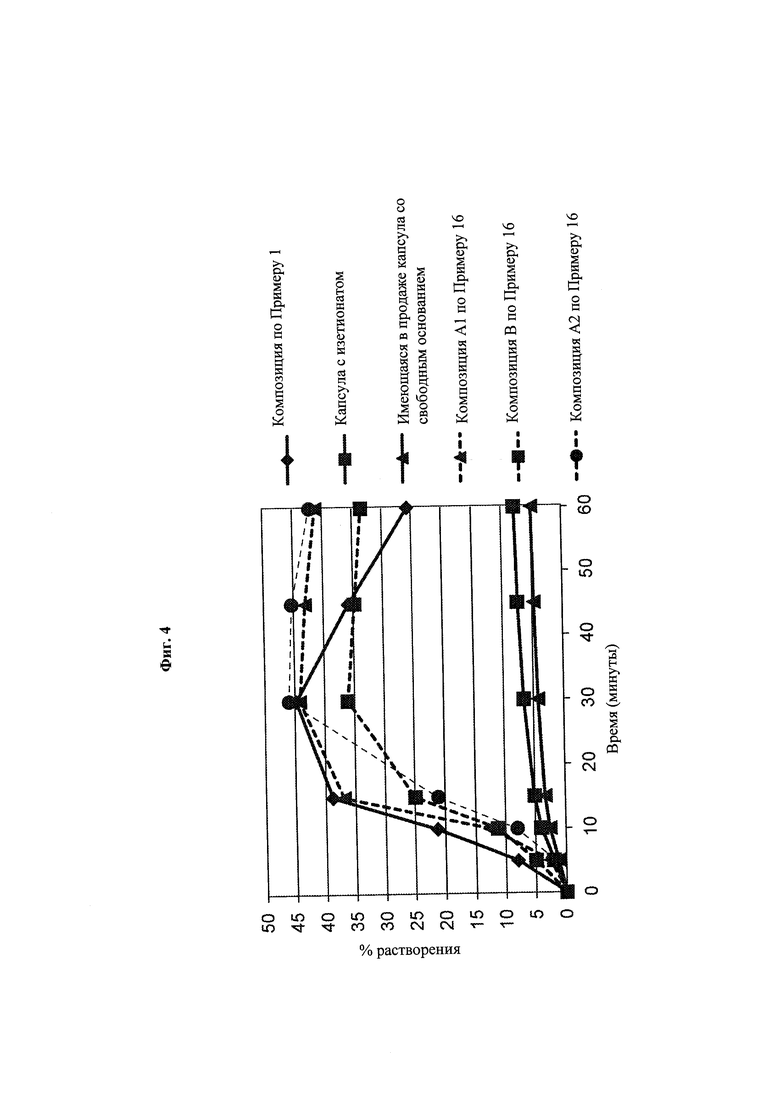
Группа изобретений относится к твердым лекарственным формам палбоциклиба. Твердая лекарственная форма палбоциклиба в форме таблетки содержит от 10 до 35 мас.% палбоциклиба, от 5 до 25 мас.% водорастворимой кислоты, выбранной из группы, состоящей из янтарной кислоты, яблочной кислоты и винной кислоты, и фармацевтически приемлемый носитель. В частности, таблетка содержит 20 мас.% палбоциклиба, 10 мас.% янтарной кислоты, от 50 до 63 мас.% микрокристаллической целлюлозы, от 5 до 10 мас.% кросповидона и от 0,5 до 6 мас.% стеарата магния. Группа изобретений обеспечивает стабильность при хранении твердой лекарственной формы палбоциклиба и его доставку, не зависящую от рН, без существенного влияния приема пищи или нежелательных взаимодействий с ингибиторами протонной помпы. 2 н. и 12 з.п. ф-лы, 4 ил., 23 табл., 21 пр.
1. Твердая лекарственная форма палбоциклиба в форме таблетки, содержащая от 10 до 35 мас.% палбоциклиба, от 5 до 25 мас.% водорастворимой кислоты, выбранной из группы, состоящей из янтарной кислоты, яблочной кислоты и винной кислоты, и фармацевтически приемлемый носитель.
2. Таблетка по п. 1, где водорастворимая кислота представляет собой янтарную кислоту.
3. Таблетка по п. 1, где фармацевтически приемлемый носитель содержит по меньшей мере один разбавитель и где разбавитель составляет от 50 до 75 мас.% твердой лекарственной формы.
4. Таблетка по п. 3, где разбавитель выбран из группы, состоящей из микрокристаллической целлюлозы, моногидрата лактозы, маннита, сорбита, ксилита, карбоната магния, двухосновного фосфата кальция и трехосновного фосфата кальция.
5. Таблетка по п. 1, где фармацевтически приемлемый носитель содержит по меньшей мере один смазывающий агент и где смазывающий агент составляет от 0,5 до 10 мас.% твердой лекарственной формы.
6. Таблетка по п. 5, где смазывающий агент выбран из группы, состоящей из стеарата магния, стеарата кальция, стеарата цинка и стеарилфумарата натрия.
7. Таблетка по п. 1, где фармацевтически приемлемый носитель содержит по меньшей мере один разрыхлитель и где разрыхлитель составляет от 5 до 10 мас.% твердой лекарственной формы.
8. Таблетка по п. 7, где разрыхлитель выбран из группы, состоящей из кросповидона, кроскармеллозы натрия и крахмалгликолята натрия.
9. Таблетка по любому из пп. 1-8, где таблетка имеет пленочную оболочку.
10. Таблетка по любому из пп. 1-8, где таблетка представляет собой двухслойную таблетку.
11. Таблетка по любому из пп. 1-8, где количество палбоциклиба в таблетке составляет 25 мг, 75 мг, 100 мг или 125 мг.
12. Таблетка по п. 11, где количество палбоциклиба в таблетке составляет 125 мг.
13. Твердая лекарственная форма палбоциклиба в форме таблетки, содержащая 20 мас.% палбоциклиба, 10 мас.% янтарной кислоты, от 50 до 63 мас.% микрокристаллической целлюлозы, от 5 до 10 мас.% кросповидона и от 0,5 до 6 мас.% стеарата магния.
14. Таблетка по п. 13, дополнительно содержащая 1 мас.% диоксида кремния.
| КЛАПАН ДЛЯ ДОЗИРУЮЩЕЙ МАШИНЫ И СПОСОБ | 2011 |
|
RU2523999C1 |
| US 20150140036 A1, 21.05.15 | |||
| Чуешов В.И | |||
| и др | |||
| Промышленная технология лекарств | |||
| Электронный учебник / Национальный фармацевтический университет, кафедра заводской технологии лекарств, Харьков, 2010. | |||

