Настоящая заявка истребует приоритет на основании предварительной заявки на патент США № 60/295 429, поданной 1 июня 2001 года и включенной в него полностью в качестве ссылки.
Предпосылки создания изобретения
Настоящее изобретение относится к различным пираноиндазолам. Указанные новые соединения полезны для снижения и контроля (регуляции) нормального или повышенного внутриглазного давления (ВГД) и для лечения глаукомы.
Болезненное состояние, относящееся к глаукоме, характеризуется наличием постоянной потери зрительной функции из-за необратимого изменения зрительного нерва. Несколько морфологически или функционально различных типов глаукомы обычно характеризуются повышенным ВГД, которое рассматривается как причина, связанная с патологическим развитием заболевания. Глазная гипертензия представляет собой состояние, при котором внутриглазное давление повышается, но без очевидной потери зрительной функции, и такие пациенты рассматриваются как имеющие высокий риск потенциального развития потери зрения, связанного с глаукомой. Если глаукому или внутриглазную гипертензию выявить на ранней стадии и подвергнуть соответствующему лечению лекарственными препаратами, которые способны эффективно снижать повышенное внутриглазное давление, то потеря зрительной функции или ее прогрессивное ухудшение могут быть в основном ослаблены. Лекарственная терапия, которая считается эффективной для снижения внутриглазного давления, включает средства, которые снижают образование глазной жидкости, а также средства, которые повышают отток (способность к выведению жидкости). Указанная терапия осуществляется в основном посредством введения лекарственных средств одним из двух возможных способов, а именно путем местного введения (непосредственное введение в глаз) или путем перорального приема.
Однако имеются некоторые индивидуумы, которые не способны соответствующим образом реагировать на лечение, проводимое в рамках имеющихся способов терапии глаукомы. В этой связи имеется потребность в разработке других местных терапевтических средств, которые могли бы контролировать ВГД.
Имеются сообщения о том, что серотонинергические агонисты 5-HT1A оказывают на моделях животных нейропротекторное действие, и многие из указанных средств были испытаны, в числе других показаний для их применения, для лечения острого нарушения мозгового кровообращения. Указанный класс соединений отмечался в качестве средств для лечения глаукомы (снижения и контроля ВГД), в частности в WO 98/18458 (DeSantis, et al.) и EP 0771563 A2 (Mano, et al.), в работе Осборна с соавт. (Osborne, et al., Ophthalmologica, Vol.210:308-314, 1996), в которых раскрывается, что 8-гидроксидипропиламинотетралин (8-OH-ДПАТ) (агонист 5-HT1A) снижает ВГД у кроликов. Ванг с соавт. (Wang, et al., Current Eye Research, Vol. 16(8):769-775, август 1997 и IVOS, Vol. 39(4), S488, март 1998) указывают, что 5-метилурапидил, α1А антагонист и 5-HT1A агонист снижают ВГД у обезьян, но благодаря их активности в отношении α1А рецептора. Указывается также, что антагонисты 5-HT1A полезны для лечения глаукомы (повышенного ВГД) (например, WO 92/0338, McLees)). Кроме того, Десаи с соавт. (DeSai et al., WO 97/35579) и Макор с соавт. (Macor et al., US 5 578 612) рассматривают использование 5-HT1 и 5-HT1-подобных агонистов для лечения глаукомы (повышенного ВГД). Указанные соединения с противомигреневой активностью, например суматриптан и наратриптан, как и родственные им соединения, являются агонистами 5-HT1B,D,E,F.
Было показано, что серотонинергические соединения, которые обладают агонистической активностью в отношении 5-HT2 рецепторов, эффективно снижают и контролируют нормальное и повышенное ВГД и полезны при лечении глаукомы, смотри одновременно рассматриваемую заявку PCT/US99/19888, включенную в настоящее описание полностью в качестве ссылки. Соединения, которые действуют как агонисты 5-HT2 рецептора, хорошо известны и продемонстрировали множество полезных свойств, прежде всего в случае заболеваний или состояний, связанных с центральной нервной системой (ЦНС). Патент США № 5 494 928 относится к некоторым производным 2-(индол-1-ил)этиламина, которые являются агонистами 5-HT2С, для лечения неврозов навязчивых состояний и других расстройств личности центрального генеза. Патент США № 5571833 относится к производным триптамина, которые являются агонистами 5-HT2, для лечения портальной гипертензии и мигрени. Патент США № 5874477 относится к способу лечения малярии с использованием агонистов 5-HT2A/2С. Патент США № 5902815 относится к использованию агонистов 5-HT2A для предупреждения неблагоприятных (побочных) эффектов, связанных с гипофункцией NMDA рецептора. WO 98/31354 относится к агонистам 5-HT2B, применяемым для лечения депрессий и других состояний ЦНС. WO 00/12475 относится к производным индолина, WO 00/12510 и WO 00/44753 относятся к некоторым производным индола, являющимся агонистами 5-HT2В и5-HT2С рецептора, применяемым для лечения различных расстройств центральной нервной системы, в особенности для лечения ожирения. WO 00/35922 относится к некоторым производным пиразино[1,2-а]хиноксалина, являющимся агонистами 5-HT2С, которые применяются для лечения неврозов навязчивых состояний, депрессии, расстройств аппетита и других расстройств, в которых задействована ЦНС. WO 00/77002 и WO 00/77010 относятся к некоторым замещенным тетрациклическим пиридо[4,3-b]индолам, являющимся агонистами 5-HT2С, с точки зрения их применения для лечения расстройств ЦНС, включающих, в числе других, ожирение, тревожность, депрессию, расстройства сна, головную боль и социальные фобии. Указывается, что реакция агониста на 5-HT2A рецептор является основной активностью, ответственной за галлюциногенное действие, при этом участию 5-HT2С рецептора отводится меньшая роль [Psychopharmacology, Vol. 121:357, 1995].
Описаны несколько конденсированных индазолов, содержащих фуран или пиран. Химический синтез 7-метил- и 1,7-диметил-1Н-фуро[2,3-g]индазола [Gazz. Chim. Ital. 106, 1083 (1976)], а также 3-метил- и 1-(4-аминофенил)-3-метил-1Н-бензо[b]фуро[2,3-g]индазола [An. Asoc. Quim. Argent. 59, 69 (1971)] описаны без обсуждения возможностей их применения. Заявка на европейский патент EP 990650 (Номер международной публикации WO 98/56768) относится к замещенным 2-(фуро[2,3-g]индазол-1-ил)этиламинам, таким как (S)-2-(фуро[2,3-g]индазол-1-ил)-1-метилэтиламин, которые, по данным авторов, обладают высокой селективностью и афинностью в отношении 5-HT2С рецепторов и являются потенциально полезными для лечения различных расстройств центральной нервной системы. Химический синтез 9-метил-1Н-пирано[2,3-g]индазол-7-она и соответствующего неметилированного соединения описан в литературе [Indian J. Chem. 26B, (1987)] без указаний возможностей его применения.
Патенты США №№ 5561150 и 5646173 относятся к некоторым производным соединениям трициклического пиразола, которые идентифицированы как агонисты 5-HT2С, пригодные для лечения заболеваний ЦНС и преимущественно направлены на их липофильные аналоги, обладающие высокой вероятностью поступления в мозг. Аналогично, WO 98/56768 относится к трициклическим агонистам 5-HT2С для лечения заболеваний ЦНС. Все указанные выше, равно как и приведенные далее патенты и публикации включены в настоящее описание в полном объеме в качестве ссылок.
5-Гидрокситриптамин (серотонин) не проходит через гематоэнцефалический барьер и не поступает в мозг. Однако для повышения уровня серотонина в мозге можно вводить 5-гидрокситриптофан. Транспорт 5-гидрокситриптофана в мозг легко осуществляется и уже в мозге 5-гидрокситриптофан легко декарбоксилируется с образованием серотонина.
Соответственно, имеется потребность в разработке новых соединений, которые были бы свободны от указанных выше недостатков и которые бы обеспечивали повышенную химическую стабильность и желательную длительность терапевтической активности в случае, например, применения их для снижения внутриглазного давления и лечения глаукомы.
Краткое описание сущности изобретения
Объектом настоящего изобретения является разработка новых соединений, которые являются агонистами 5-НТ2.
Другим объектом настоящего изобретения является разработка соединений, которые обладают повышенной химической стабильностью и которые полезны с точки зрения снижения или контроля (регуляции) нормального или повышенного внутриглазного давления и/или лечения глаукомы.
Другим объектом настоящего изобретения является разработка соединений, которые обеспечивают желательный уровень терапевтической активности по снижению или контролю нормального или повышенного внутриглазного давления и/или лечению глаукомы.
Дополнительные объекты и преимущества настоящего изобретения будут указаны частично в приведенном ниже описании и частично они будут очевидны из приведенного описания или же они могут быть выявлены на практике в ходе осуществления настоящего изобретения. Цели и другие преимущества настоящего изобретения можно оценить и достигнуть с помощью элементов и их сочетаний, конкретно указанных в описании и в прилагаемой формуле изобретения.
Для достижения указанных и других преимуществ и в соответствии с целями настоящего изобретения, которые составляют его суть и широко представлены в настоящем описании, данное изобретение относится к соединению, имеющему формулу I:
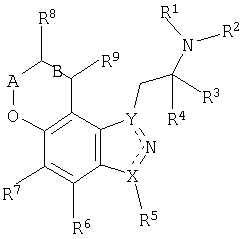
или его фармацевтически приемлемые соли или сольваты или пролекарственные формы соединений формулы I. В указанной формуле
R1 и R2 независимо выбирают из водорода или алкильной группы, такой как С1-4алкил;
R3 и R4 независимо выбирают из водорода или алкильной группы, такой как С1-4алкил, или R3 и R4 и атом углерода, к которому они присоединяются, могут образовывать циклоалкильное кольцо или далее,
R2 и R3 вместе могут представлять собой (CH2)m с образованием насыщенного гетероцикла;
R5 выбирают из водорода, галогена, алкильной группы, такой как С1-6алкил или С1-4алкил, замещенный галогеном;
R6 и R7 независимо выбирают из водорода, галогена, циано, алкилтио, такого как С1-4алкилтио, алкила, такого как С1-4алкил, или замещенного алкила, такого как С1-4алкил, замещенный галогеном;
R8 и R9 независимо выбирают из водорода, гидроксила, алкила, такого как С1-6алкил, алкокси, такого как С1-6алкокси, =O, NR10R11, OC(=O)NR1R2, OC(=O)С1-4алкила, алкилтиола, такого как С1-6алкилтиол, замещенного алкила, такого как С1-6алкил, замещенный галогеном, гидроксилом или NR10R11;
R10 и R11 независимо выбирают из водорода, алкильной группы, такой как С1-4алкил, C(=O)С1-4алкил, C(=O)OC1-4алкил, C(=O)NR1R2, или замещенного алкила, такого как С1-6алкил, замещенный галогеном, гидроксилом, NR1R2 или R10 и R11 вместе могут составлять насыщенное 5- или 6-членное гетероциклическое кольцо, которое может включать дополнительный гетероатом, выбранный из N, O или S, в случае 6-членного кольца;
А обозначает (CH)n, С=О или CHC1-4алкил;
B обозначает одинарную или двойную связь, в том случае, когда B обозначает двойную связь, R8 и R9 выбирают из водорода или насыщенной или ненасыщенной алкильной группы;
m=2-4;
n=0-2;
X и Y обозначают N или C, при этом X и Y отличаются друг от друга; и
пунктирная линия обозначает соответствующую одинарную или двойную связь.
Настоящее изобретение также относится к фармацевтическим композициям, содержащим по меньшей мере одно соединение формулы I.
Настоящее изобретение также относится к способам снижения или контроля нормального или повышенного внутриглазного давления путем введения эффективного количества композиции, содержащей соединение, имеющее описанную выше формулу I.
Настоящее изобретение также относится к способу лечения глаукомы, который включает введение эффективного количества композиции, содержащей соединение, имеющее описанную выше формулу I.
Следует понимать, что приведенные далее как общее описание, так и подробное описание изобретения являются лишь иллюстративными и предназначены для пояснения рассматриваемого изобретения, суть которого выражена в прилагаемой формуле изобретения.
Подробное описание настоящего изобретения
Настоящее изобретение относится к множеству соединений, полезных при их использовании согласно настоящему изобретению. Указанные соединения в основном описываются формулой I
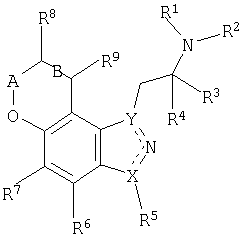
В указанной формуле
R1 и R2 независимо выбирают из водорода или алкильной группы, такой как С1-4алкил;
R3 и R4 независимо выбирают из водорода или алкильной группы, такой как С1-4алкил, или R3 и R4 и атом углерода, к которому они присоединяются, могут образовывать циклоалкильное кольцо (например, циклопропильное кольцо) или далее,
R2 и R3 вместе могут представлять собой (CH2)m с образованием насыщенного гетероцикла;
R5 выбирают из водорода, галогена, замещенной или незамещенной алкильной группы, такой как С1-6алкил или С1-4алкил, замещенный галогеном;
R6 и R7 независимо выбирают из водорода, галогена, циано, алкилтио, такого как С1-4алкилтио, алкила, такого как С1-4алкил, или замещенного алкила, такого как С1-4алкил, замещенный галогеном;
R8 и R9 независимо выбирают из водорода, гидроксила, алкила, такого как С1-6алкил, алкокси, такого как С1-6алкокси, =O, NR10R11, OC(=O)NR1R2, OC(=O)С1-4алкила, алкилтиола, такого как С1-6алкилтиол, замещенного алкила, такого как С1-6алкил, замещенный галогеном, гидроксилом или NR10R11;
R10 и R11 независимо выбирают из водорода, алкильной группы, такой как С1-4алкил, C(=O)С1-4алкил, C(=O)OC1-4алкил, C(=O)NR1R2, или замещенной алкильной группы, такой как С1-6алкил, замещенный галогеном, гидроксилом, NR1R2, или R10 и R11 вместе могут составлять насыщенное 5- или 6-членное гетероциклическое кольцо, которое может включать дополнительный гетероатом, выбранный из N, O или S, в случае 6-членного кольца;
А обозначает (CH2)n или CHC1-4алкил;
B обозначает одинарную или двойную связь, в том случае, когда B обозначает двойную связь, R8 и R9 выбирают из водорода, алкильной группы, такой как С1-4алкил, или замещенной алкильной группы, такой как С1-4алкил, замещенный галогеном, гидроксилом или NR10R11;
m=2-4;
n=0-2;
X и Y обозначают N или C, при этом X и Y отличаются друг от друга; и пунктирная линия обозначает соответствующую одинарную или двойную связь.
Фармацевтически приемлемые соли и сольваты и пролекарственные формы соединения формулы I также составляют часть настоящего изобретения.
Предпочтительными соединениями являются:
Такие, в которых R1 и R2 независимо выбирают из водорода или С1-4алкила;
R3 и R4 независимо выбирают из водорода, С1-4алкила или R2 и R3 вместе могут представлять собой (CH2)m с образованием насыщенного гетероцикла;
R5 выбирают из водорода, галогена или С1-6 алкила;
R6 и R7 независимо выбирают из водорода, галогена, циано, С1-4алкилтио, С1-4алкила или С1-4алкила, замещенного галогеном;
R8 и R9 независимо выбирают из водорода, гидроксила, С1-6алкила, С1-6алкокси, NR10R11 или С1-6 алкила, замещенного галогеном, гидроксилом или NR10R11;
R10 и R11 независимо выбирают из водорода, С1-4алкила, C(=O)С1-4алкила, C(=O)OC1-4алкила, C(=O)NR1R2 или R10 и R11 вместе могут составлять насыщенное 6-членное гетероциклическое кольцо, которое может включать дополнительный гетероатом, выбранный из N, O или S;
А обозначает (CH2)n или CHC1-4алкил;
B обозначает одинарную или двойную связь, в том случае, когда B обозначает двойную связь, R8 и R9 выбирают из водорода, С1-4алкила или С1-4алкила, замещенного галогеном, гидрокси или NR10R11;
m=3-4;
n=1-2;
X и Y обозначают N или C, при этом X и Y отличаются друг от друга; и
пунктирная линия обозначает соответствующую одинарную или двойную связь.
Наиболее предпочтительными соединениями являются:
Такие, в которых R1 и R2 независимо выбирают из водорода или С1-4алкила;
R3 обозначает С1-2алкил или R2 и R3 вместе могут представлять собой (CH2)3 с образованием пирролидина;
R4 обозначает водород;
R5 выбирают из водорода или С1-6алкила;
R6 и R7 независимо выбирают из водорода, галогена или С1-4алкила;
R8 и R9 независимо выбирают из водорода, гидроксила, С1-6алкокси, NR10R11 или С1-6алкила, замещенного гидроксилом или NR10R11;
R10 и R11 независимо выбирают из водорода, С1-4алкила, C(=O)С1-4алкила или R10 и R11 вместе могут составлять насыщенное 6-членное гетероциклическое кольцо, которое может включать дополнительный гетероатом, выбранный из N, O или S;
А обозначает (CH2)n;
B обозначает одинарную связь;
n=1;
X обозначает C и Y обозначает N; и
пунктирная линия обозначает соответствующую одинарную или двойную связь.
Репрезентативные примеры предпочтительных соединений формулы I включают:
1-(2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
(R)-1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
(S)-1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
1-((S)-2-аминопропил)-3-метил-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
1-(S)-1-пирролидин-2-илметил-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
1-((S)-2-аминопропил)-5-фтор-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
(R)-1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-иламин;
[1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ил]диметиламин;
[1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ил]метанол;
1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8,9-диол;
1-((S)-2-аминопропил)-9-метокси-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
1-(2-аминопропил)-3,7,8,9-тетрагидропирано[3,2-е]индазол-8-ол;
1-(пирролидин-2-илметил)-3,7,8,9-тетрагидропирано[3,2-е]индазол-8-ол;
1-((S)-2-аминопропил)-3,7,8,9-тетрагидропирано[3,2-е]индазол-8-ол;
1-((S)-2-аминопропил)-3-метил-3,7,8,9-тетрагидропирано[3,2-е]индазол-8-ол;
или их сочетания.
Некоторые соединения формулы I могут содержать один или более хиральных центров. Настоящее изобретение включает все энантиомеры, диастереомеры и их смеси.
В приведенных выше определениях общее число атомов углерода в замещающей группе указано префиксом Ci-j, где числа i и j обозначают количество атомов углерода. Указанное определение охватывает линейные цепи, разветвленные цепи и циклические или (циклоалкильные) алкильные группы. Заместитель может быть представлен в единственном или во множественном числе, когда он включается в указанную структурную единицу. Так, например, в случае галогена-заместителя, который представляет собой фтор, хлор, бром или йод, имеется в виду, что структура, к которой он присоединяется, может быть замещена одним или более атомами галогена, которые могут быть одинаковыми или различными.
Синтез
Соединения формулы I могут быть получены с использованием одной из нескольких синтетических методик. Так, например 1-(2-аминометил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-олы могут быть получены из соответствующим образом защищенного 1-(6-гидроксииндазол-1-ил)пропан-2-ола 1, как показано на схеме 1. Pg обозначает соответствующую защитную группу, которая вводится для гарантии того, что конкретный атом не будет модифицирован в ходе рассматриваемой химической реакции.
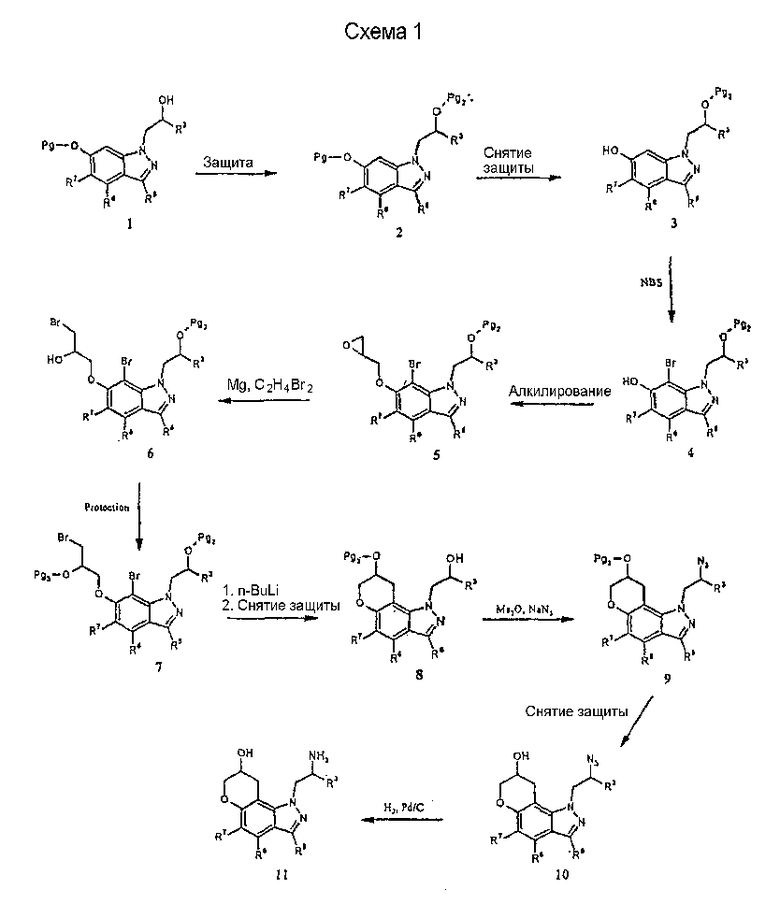
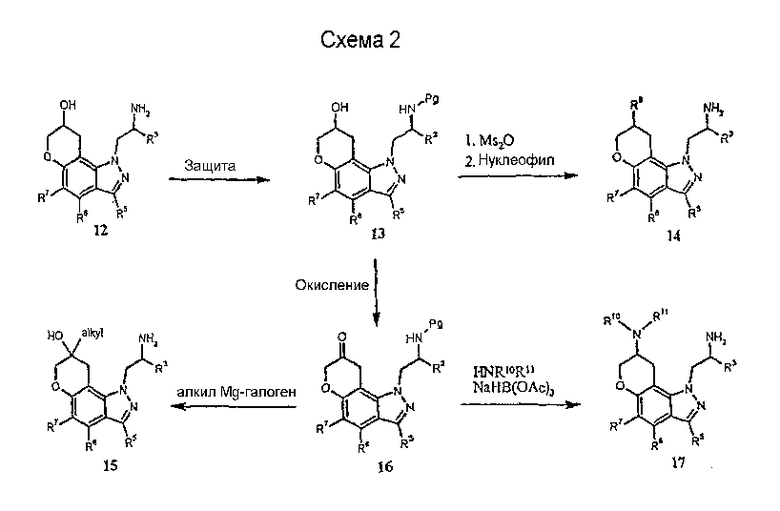
Другие соединения формулы I могут быть получены из соединения 12 путем известных в технике трансформаций выбранной функциональной группы. Например, осуществляя вначале защиту первичной аминогруппы с последующей активацией гидроксильной группы в ходе образования сульфонатного эфира, например метансульфонила, и последующая реакция с желательным нуклеофилом, таким как алкиламины, диалкиламины, арил- или алкилтиолы и т.п., приведет к получению соединения 14 формулы I. Кроме того, прямое окисление 13 подходящим окислителем, например гипервалентным йодным реагентом, таким как О-йодоксибензойная кислота [J.Org. Chem.60, 7272 (1995)], дает кетон 16, который может быть функционализирован с получением других соединений формулы I, таких как 17, посредством восстановительного алкилирования, и 15 - посредством реакции присоединения Гриньяра.
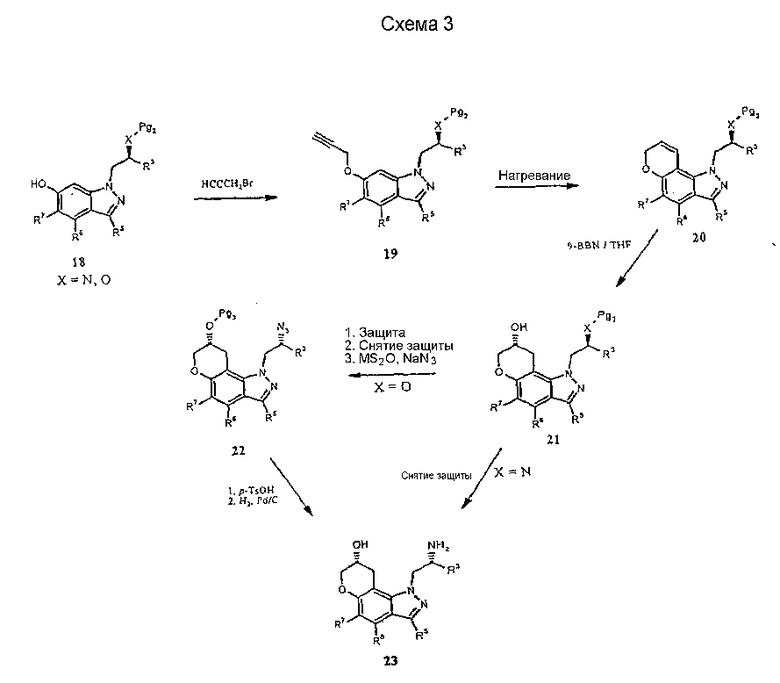
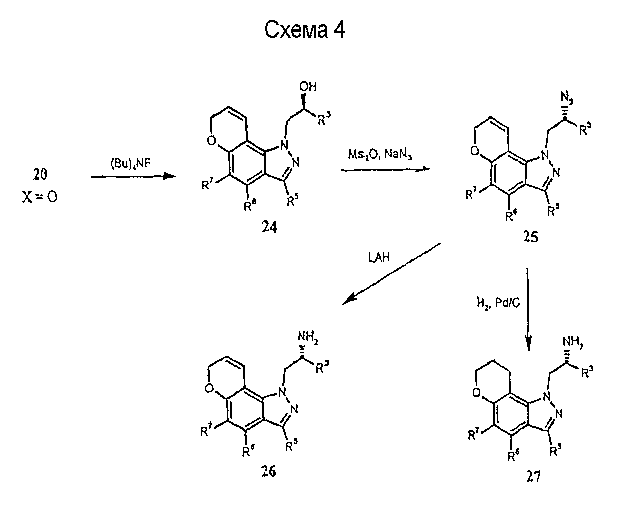
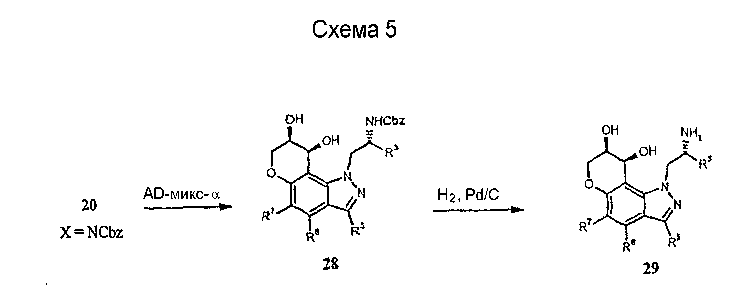
Альтернативно, соединения формулы I могут быть получены из соответствующим образом замещенных 5-пропаргилоксииндазолов (19) через исходные реакции перегруппировки Кляйзена [Tetrahedron Lett. 33, 2179 (1992), ibid. 35, 45 (1994) ibid. 41, 3541 (2000)] с образованием промежуточных замещенных пирано[2,3-g]индазолов 20 (схема 3). Другие процедуры синтеза с использованием 20, как показано, например, на схемах 3-5, с использованием хорошо известных трансформаций функциональных групп, дают другие желательные соединения формулы I.
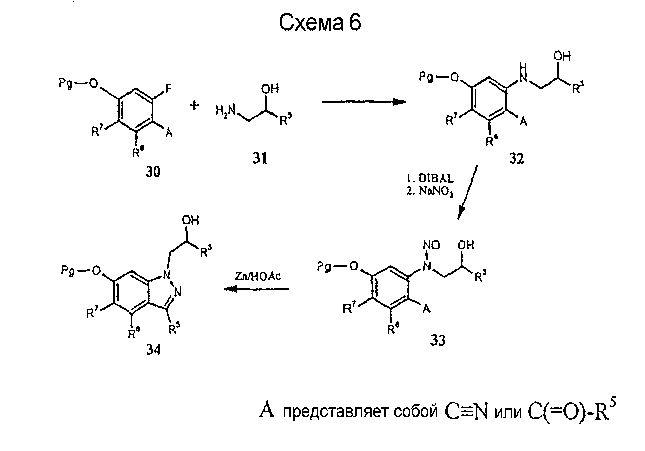
1-(Гидроксиалкил)индазолы, представляющие интерес для получения соединений формулы I, могут быть синтезированы, как показано на схеме 6, и в совместно рассматриваемой заявке на патент США 60/295 427, включенной в настоящее описание полностью в качестве ссылки. Реакция активированного фторфенола 30 с соответствующим аминоспиртом 31, в том случае, когда А обозначает нитрил, приводит к восстановлению с образованием соответствующего альдегида 32. Нитрозирование дает соединение 33, которое после восстановительной циклизации приводит к получению 1-(гидроксиалкил)индазолов 34.
Промежуточные пираноиндазолы 34 могут быть получены путем алкилирования соответствующего О-защищенного 6-гидроксииндазола (35), в котором соответствующие О-защитные группы представляют собой, например, метил или бензил, с помощью способов, известных в уровне техники и проиллюстрированных на схеме 7 [патент США 5494928 (1997), WO 98/30548 (1998)], с использованием желательного эпоксида, например пропиленоксида. Альтернативно, для целей получения некоторых соединений может быть удобно провести реакцию алкилата 35 с использованием хлорацетона с последующим восстановлением, например, с помощью NaBH4, промежуточного кетона, получая интермедиат 34.
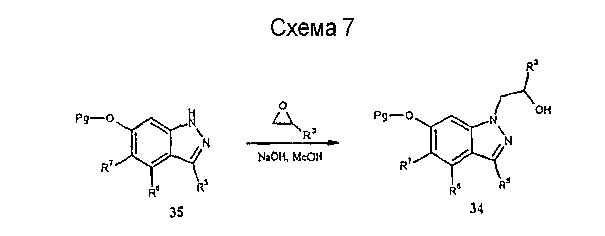
Может быть также удобно для получения некоторых соединений формулы I использовать соответствующим образом замещенный 1,7,8,9-тетрагидропирано[2,3-g]индазол, такой как 36, показанный на схеме 8. Так, например, алкилирование соединения 36 в условиях, описанных выше на схеме 4, с последующим соответствующим активированием гидроксильной группы до достижения нуклеофильного аминирования путем образования сульфонатного эфира [J.Chem. Soc., Perkins Vol. 1:1479, 1981], например метансульфонила, толуолсульфонила, бромфенилсульфонила или нитрофенилсульфонила, и последующая реакция с желательным амином приводят к получению соединения 38 формулы I.
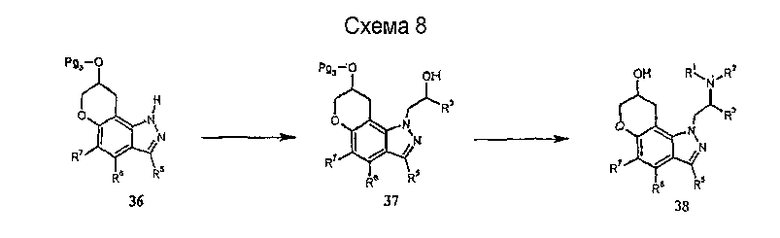
Далее реакция индазолов 36 с активированным аланинолом 39 дает соединение 40, снятие защиты с которого дает соединение 38 формулы I, как показано на схеме 9. Замещение 39 на схеме 9, например, активированным сульфонатным эфиром или соответствующим галогенидом или N-защищенным (например, с помощью трет-бутилоксикарбонила, бензилоксикарбонила) пирролидин-3-метанолом приводит, после удаления аминозащитной группы, к получению другого соединения формулы I. Далее замещение 39 на схеме 9 активированным сульфонатным эфиром N-(2-гидрокси-1,1-диметил-этил)фталимида [J. Amer. Chem. Soc., Vol. 108:3811, 1986], 2-[(т-бутилоксикарбонил)амино]-2-метилпропанола [J. Amer. Chem. Soc., Vol. 113:8879, 1991], 1-[(т-бутилоксикарбонил)амино]циклопропил-1-метанола [J. Med. Chem., Vol. 31:1694, 1988] или 2-метил-2-нитро-пропан-1-ола [J. Amer. Chem. Soc., Vol. 68:12, 1946] приводит, после удаления N-защитных групп в первых трех случаях или после восстановления нитрогруппы, в последнем случае, к другим вариантам получения соединения 38 формулы I.
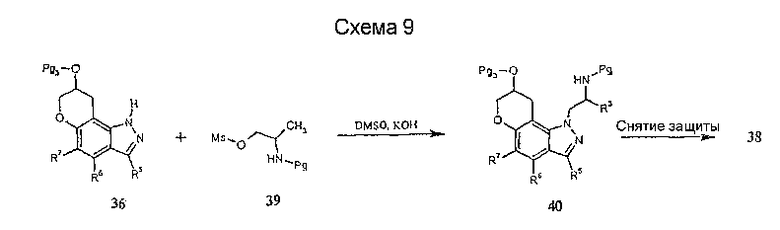
Некоторые желательные замещенные 1,7,8,9-тетрагидропирано[2,3-g]индазолы могут быть получены из соответствующим образом замещенного 1Н-индазол-6-ола (41), как описано в методике синтеза, приведенной на схеме 10. Алкилирование индазолов 41 аллилиодидом с последующей обработкой в условиях реакции перегруппировки Кляйзена дает соединение 43. Защита гидроксильной группы от других реакций путем превращения, например, в сложный эфир, такой как ацетильный, и аналогичное включение защитной группы по атому азота, например, путем реакции с соответствующим изоцианидом с образованием уреида, дает желательный аллилиндазол 45. Эпоксидирование олефина с использованием, например, 3-хлор-пербензойной кислоты и последующая циклизация в щелочных условиях приводят к получению пираноиндазольного интермедиата 47. Превращение гидроксильной группы пираноиндазола 47 в другие функциональные группы, соответствующие формуле I, может быть осуществлено с помощью известных в технике трансформаций функциональной группы.
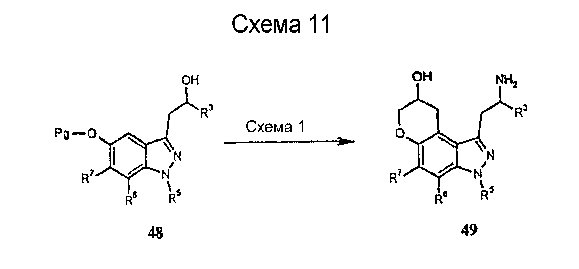
Желательные пирано[3,2-e]индазол-3-этиламины 49 (схема 11) формулы I могут быть получены из соответствующим образом замещенного 3-(2-гидроксипропил)1Н-индазол-5-ола 48 с использованием способов, проиллюстрированных на схемах 1 и 3 и описанных в заявке на международный патент № PCT/US00/31143, включенной в настоящее описание полностью в качестве ссылки.
Соединения согласно настоящему изобретению могут использоваться для снижения контроля ВГД, включая ВГД, связанное с глаукомой, развивающейся при нормальном давлении, при глазной гипертензии, и с глаукомой у теплокровных животных, включая человека. Поскольку лечение глаукомы предпочтительно проводят с использованием соединений, которые не проходят в ЦНС, относительно полярные соединения, являющиеся агонистами 5-НТ2, представляют особый интерес. Соединения предпочтительно вводят в состав фармацевтических композиций, которые предпочтительно являются пригодными для местной доставки в глаз пациента.
Соединения согласно настоящему изобретению формулы I могут быть включены в состав фармацевтических композиций различных типов, таких как офтальмологические препараты для введения в глаз (например, для местного введения, внутрь камеры глаза или путем имплантирования). Соединения предпочтительно включают в местные офтальмологические композиции для введения в глаз. Соединения могут быть объединены с офтальмологически приемлемыми консервантами, усилителями вязкости, усилителями проницаемости, буферами, хлоридом натрия и водой с получением водных стерильных глазных суспензий или растворов. Композиции раствора для офтальмологического применения могут быть получены при растворении соединения в физиологически приемлемом изотоническом водном буфере. Далее офтальмологический раствор может включать офтальмологически приемлемое поверхностно-активное вещество для облегчения растворения соединения. Кроме того, офтальмологический раствор может содержать средство, способствующее увеличению вязкости, такое как гидроксиметилцеллюлоза, гидроксиэтилцеллюлоза, гидроксипропилметилцеллюлоза, метилцеллюлоза, поливинилпирролидон и т.п., для улучшения удерживания композиции в конъюнктивальном мешке. Могут также использоваться желирующие средства, которые включают, не ограничиваясь приведенными средствами, gellan и ксантановую камедь. Для получения стерильных офтальмологических композиций мазей активный ингредиент объединяют с консервантом в соответствующем носителе, таком как минеральное масло, жидкий ланолин или белый вазелин. Стерильные офтальмологические гелевые композиции могут быть получены при суспендировании активного ингредиента в гидрофильной основе, полученной при сочетании, например, с карбополом-974 или аналогичным соединением в соответствии с опубликованными данными по получению аналогичных офтальмологических препаратов, могут быть также включены консерванты и средства для усиления тонуса.
Соединения предпочтительно готовятся в виде местных глазных суспензий или растворов, имеющих pH примерно 5-8. В норме указанные соединения входят в состав рассматриваемых композиций в количестве от 0,01 мас.% до 5 мас.%, предпочтительно в количестве от 0,25 мас.% до 2 мас.% по весу. Таким образом, в случае местной формы композиции (1-2 капли указанных композиций) вводят на поверхность глаза от 1 до 4 раз в день, согласно предписанию лечащего врача.
Соединения могут также использоваться в сочетании с другими средствами, применяемыми для лечения глаукомы, такими как, но не ограничеваясь ими, β-блокаторы (например, тимолол, бетаксолол, левобетаксолол, картеолол, левобунолол, пропранолол), ингибиторы карбоангидразы (например, бринзоламид и дорзоламид), антагонисты α1 (например, нипрадолол), антагонисты α2 (например, йопидин и бримонидин), миотические средства (например, пилокарпин и эпинефрин), аналоги простагландинов (например, латанопрост, травапрост, унопростон и соединения, указанные в патентах США №№ 5889052, 5296504, 5422368 и 5151444, "гипотензивные липиды" (например, лумиган и соединения, указанные в патенте № 5352708) и нейропротекторы (например, соединения, указанные в патенте США № 6690931, в частности элипродил и R-элипродил, указанные в совместно рассматриваемой заявке U.S.S.N. 06/203350, и соответствующие соединения из WO 94/13275, включая мемантин.
В описанных выше формулах алкильная группа может быть линейно-цепочечной, разветвленной или циклической и т.п. Галоген включает Cl, Br, F или I. Алкокси обозначает алкильную группу, соединенную связью через атом кислорода.
Соединения согласно настоящему изобретению предпочтительно функционируют как 5-НТ2 агонисты и предпочтительно не проходят в ЦНС. Более детально, конкретные соединения согласно настоящему изобретению включают в свою структуру фенольный гидроксил, который, как считается, сравним с таковым у серотонина, и, таким образом, соединения согласно настоящему изобретению предпочтительно не пересекают гематоэнцефалический барьер и не поступают в мозг. Соединения, обладающие способностью действовать как 5-НТ2 агонисты, полезны для контроля ВГД, а также для лечения глаукомы, как было показано в публикации заявки на международный патент № WO 00/16761, включенной в настоящее описание полностью в качестве ссылки. Соединения согласно настоящему изобретению предпочтительно обладают повышенной химической стабильностью и предпочтительно демонстрируют желательный уровень терапевтической активности, которая предусматривает снижение или контроль ВГД.
Соединения согласно настоящему изобретению могут использоваться для контроля или снижения ВГД у теплокровных животных, включая человека. Предпочтительно, пациенту вводят эффективное количество соединения, так чтобы достигался контроль или снижение ВГД до приемлемых уровней. Кроме того, соединения согласно настоящему изобретению могут использоваться для лечения глаукомы у теплокровных животных, включая человека, путем введения пациенту эффективного для лечения глаукомы количества соединения при необходимости такого лечения. Примеры подходящих количеств фармацевтически приемлемых соединений согласно настоящему изобретению включают те количества, которые приведены в примерах.
Другой вариант осуществления настоящего изобретения представляет способ активизации или связывания с рецепторами серотонина, включающий введение пациенту эффективного количества по меньшей мере одного соединения согласно настоящему изобретению с использованием количеств, эффективных для активизации или связывания с рецепторами серотонина, таких как, не ограничиваясь указанными значениями, приведенные в настоящем описании дозировки.
Показанные ниже примеры даны для иллюстрации способов получения соединений, которые являются объектом настоящего изобретения, но они не должны рассматриваться как включающие какие-либо ограничения пунктов формулы изобретения. Предпочтительные соединения формулы I описаны в примерах 4 и 5. Наиболее предпочтительное соединение описано в примере 4. Спектр протонного магнитного резонанса каждого соединения, приведенного в примерах, соответствует предполагаемой структуре.
Способ 1
Тест на связывание с 5-НТ2 рецептором
Для оценки афинности серотонинергических соединений к 5-НТ2 рецепторам определяют их способность конкурировать с агонистом радиолигандом [125I]DOI за связывание с 5-HT2 рецепторами головного мозга, как описано ниже, по приведенной в литературе методике, включающую незначительные модификации (Neuropharmology, 26, 1803 (1987)]. Аликвоты гомогенатов коры головного мозга крысы или человека, взятых после смерти (400 мкл), диспергируют в 50 мМ Трис-HCl буфера (pH 7,4) и инкубируют с [125I]DOI (конечная концентрация 80 пМ) в отсутствие или в присутствии метиотепина (конечная концентрация 10 мкМ) в общем объеме 0,5 мл, для определения общего и неспецифического связывания, соответственно. Смесь для анализа инкубируют в течение 1 часа при 23°С в полипропиленовых пробирках и реакцию останавливают быстрым фильтрованием в вакууме на стекловолокнистых фильтрах Whatman GF/B, предварительно вымоченных в 0,3% полиэтиленимине с использованием ледяного буфера. Вместо метиотепина вводят исследуемые соединения (в различных концентрациях). Связанную с фильтром радиоактивность определяют методом сцинтилляционной спектрометрии на бета-счетчике. Для определения показателей афинности соединения полученные данные анализируют с использованием нелинейного итеративного графика, построенного с помощью компьютерной программы [Trends Pharmacol. Sci., 16, 413 (1995)]. Концентрацию соединения, необходимую для ингибирования связывания [125I]DOI на 50% от максимального значения, обозначают как величину ИК50 или Кi.
Способ 2
Функциональный тест на 5-НТ2: мобилизация [Ca2+]I
Изучают опосредованную рецептором мобилизацию внутриклеточного кальция ([Ca2+]I) с использованием флуоресцентного планшет-ридера (счетчика пластин) (FLIPR). Гладкомышечные клетки сосудов крысы А7r5 выращивают в нормальной среде DMEM/10% ФСТ и 10 мкг /мл гентамицина. Монослои слившихся клеток обрабатывают трипсином, получают осадок и ресуспендируют в нормальной среде. Клетки высевают в объеме 50 мкл с плотностью 20000 клеток/ячейку в 96-ячеечные планшеты для культуры ткани с черной стенкой и выращивают в течение 2 дней.
В день эксперимента одну ампулу с красителем из набора для определения кальция FLIPR ресуспендируют в 50 мл FLIPR буфера, состоящего из сбалансированного солевого раствора Хэнкса (HBSS), 20 мМ HEPES и 2,5 мМ пробенецида, pH 7,4. Клетки обрабатывают чувствительным к кальцию красителем, добавляя равный объем (по 50 мкл) к каждой ячейке 96-луночного планшета и инкубируют с красителем в течение 1 часа при 23°С.
В типичном случае исследуемое соединение хранят в концентрации 25 мкМ в растворителе, включающем 50% ДМСО/50% этанол. Соединение разбавляют в соотношении 1:50 смесью 20% ДМСО/20% этанола. Для «хит-скрининга» соединение далее разбавляют в соотношении 1:10 в FLIPR буфере и исследуют в конечной концентрации 10 мкМ. Для экспериментов по типу «доза-ответ» соединение разбавляют в соотношении 1:50 в FLIPR буфере и делают серийные разбавления 1:10 с получением кривых доза-ответ с 5 или 8 точками.
Планшет с соединением и планшет с клетками помещают в анализатор FLIPR. В начале эксперимента проводят сигнальный тест для определения базального флуоресцентного сигнала от клеток, содержащих краситель, и для подтверждения однородности прохождения сигнала через планшет. Базальную флуоресценцию доводят до значения 8000-12000 импульсов за счет изменения времени экспозиции, значения F-stop камеры или мощности лазера. Устанавливают следующие параметры прибора для проведения типичного анализа: мощность лазера - 0,3-0,6 Вт, камера F-stop F/2 и время экспозиции - 0,4 секунды. Добавляют аликвоту (25 мкл) исследуемого соединения к 100 мкл клеток с красителем со скоростью разлива 50 мкл/сек. Данные флуоресценции собирают в режиме реального времени с интервалами 1,0 секунда в течение первых 60 секунд и с интервалами 6,0 секунд еще в течение 120 секунд. Ответные реакции измеряют по пику интенсивности флуоресценции за вычетом базальной флуоресценции и там, где это приемлемо, выражают в виде процента от максимального значения 5-НТ-индуцированного ответа. В том случае, когда соединения исследуют в качестве агонистов против 10 мкМ 5-НТ, их инкубируют с клетками в течение 15 минут перед добавлением 5-НТ.
Использованные выше методики привели к получению данных, показанных в Таблицах 1-2.
Данные по связыванию с 5-НТ2А рецептором и функциональные характеристики
(Еmax, %)
ВГД реакция у находящихся в сознании обезьян Cynomolgus
Часы после введения дозы
Способ получения 1
1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол
Стадия А: 6-аллилокси-1Н-индазол
К раствору 1Н-индазол-6-ола (20,0 г, 150 ммоль) в ацетоне (450 мл) добавляют измельченный карбонат калия (22,4 г, 162 ммоль), карбонат цезия (2,00 г, 5,7 ммоль) и аллилиодид (14,63 мл, 160 ммоль) и смесь перемешивают в течение 18 часов при температуре окружающей среды. Добавляют дополнительное количество карбоната калия (5,00 г, 36 ммоль) и аллилиодида (1,4 мл, 15 ммоль) и смесь перемешивают еще 2 часа и затем фильтруют. К фильтрату добавляют воду (200 мл) и объем смеси уменьшают в вакууме до половины (вдвое) и экстрагируют дихлорметаном (2х100 мл). Объединенные экстракты сушат (MgSO4) и выпаривают с получением остатка, который очищают хроматографией (силикагель, 20% - 50% EtOAc/гексан) с получением желтого твердого вещества (14,7 г, 56%): Тпл - 110-112°С; LC/MS (+APCI) m/z 175 (M+H). Непрореагировавший исходный материал отбрасывают (4,71 г).
Стадия В: 7-аллил-1Н-индазол-6-ол
Раствор продукта, полученного на стадии А (14,2 г, 82 ммоль), в 1,2-дихлорбензоле (90 мл) кипятят с обратным холодильником в течение 6 часов и реакционную смесь выпаривают до получения остатка, который очищают хроматографией (силикагель, EtOAc) с получением рыжевато-коричневого твердого вещества (8,59 г, 60%), которое используют на следующей стадии: LC/MS (+APCI) m/z 175 (M+H).
Стадия С: 7-аллил-1Н-индазол-6-иловый эфир уксусной кислоты
Раствор продукта, полученного на стадии В (6,35 г, 37 ммоль) в ТГФ (100 мл), содержащий триэтиламин (7,6 мл, 55 ммоль), перемешивают в течение 5 минут при температуре окружающей среды, охлаждают до 0°С (ледяная баня) и добавляют ацетилхлорид (2,63 мл, 37 ммоль). Смесь перемешивают при 0°С в течение 2 часов, вносят дополнительное количество ацетилхлорида (0,26 мл, 3,7 ммоль) и смесь перемешивают в течение 10 минут, после чего вносят еще порцию ацетилхлорида (0,26 мл, 3,7 ммоль) и перемешивание продолжают в течение 15 минут. Реакцию гасят добавлением триэтиламина (1 л) и насыщенного водного раствора бикарбоната натрия (100 мл) и проводят экстракцию этилацетатом (100 мл). Экстракты сушат (MgSO4) и выпаривают до получения масла (9,27 г), которое очищают хроматографией (силикагель, 10% - 50% EtOAc/гексан) с получением белого твердого вещества (3,5 г, 44%): LC/MS (+APCI) m/z 217 (M+H).
Стадия D: 7-аллил-1-этилкарбамоил-1Н-индазол-6-иловый эфир уксусной кислоты
К раствору продукта, полученного на стадии С (2,5 г, 11,6 ммоль), в ТГФ (10 мл) добавляют этилизоцианат (1,01 мл, 13 ммоль) и смесь нагревают при 70°С в течение 18 часов. Реакционную смесь выпаривают с получением остатка, который очищают хроматографией (силикагель, 10% - 50% EtOAc/гексан) с получением бесцветного масла (2,7 г, 81%): LC/MS (+APCI) m/z 288 (M+H).
Стадия Е: 1-этилкарбамоил-7-оксиранилметил-1Н-индазол-6-иловый эфир уксусной кислоты
К раствору продукта, полученного на стадии D (2,70 г, 9,4 ммоль), в дихлорметане (15 мл) добавляют 3-хлор-пербензойную кислоту (2,31 г, 10,3 ммоль, 77% чистоты) и смесь перемешивают при температуре окружающей среды в течение 1 часа. Вносят дополнительное количество 3-хлор-пербензойной кислоты (0,2 г, 0,9 ммоль) и реакцию проводят еще в течение 3 часов. Далее реакцию гасят добавлением насыщенного водного раствора бикарбоната натрия (100 мл) и проводят экстракцию дихлорметаном (50 мл). Экстракт сушат (MgSO4) и выпаривают с получением белого твердого вещества (1,59 г, 56%): Тпл. - 110-111°С; LC/MS (+APCI) m/z 304 (M+H).
Стадия F: 1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол
К раствору продукта, полученного на стадии Е (1,44г, 4,75ммоль), в метаноле (100 мл) добавляют насыщенный водный раствор карбоната калия (10 мл) и смесь перемешивают в течение 18 часов при температуре окружающей среды. К реакционной смеси добавляют воду (200 мл) и значение pH доводят до 7 с помощью концентрированной HCl, после чего проводят экстракцию этилацетатом (5х100 мл). Объединенные экстракты сушат (MgSO4) и выпаривают с получением рыжевато-коричневого твердого вещества (0,84 г, 93%): 1H ЯМР(ДМСО-d6) δ 12,77 (с, 1H), 7,94 (с, 1H), 7,49 (д, J =6,0 Гц, 1H), 6,66 (д, J = 6,0 Гц, 1H) 5,02 (т, J=6,0 Гц, 1H), 4,85-4,95 (м, 1H), 3,62 (м, 2H), 2,9-3,4 (м, 2H); LC/MS(+APCI) m/z 191 (M+H).
Способ получения 2
1-(6-бензилоксииндазол-1-ил-пропан-2-ол)
Стадия А: (6-бензилоксииндол-1-ил-пропан-2-ол)
К перемешанной охлажденной (10°С) суспензии гидрида натрия (80,7 г 60% дисперсии в минеральном масле, 2,02 моль) в безводном ТГФ (1,9 л) добавляют раствор 6-бензилоксииндола (375 г, 1,68 моль) в безводном ТГФ (1,9 л), поддерживая температуру ниже 25°С. Через 2 часа при 10°С добавляют по каплям пропиленоксид (140 мл, 2,0 моль), поддерживая температуру ниже 25°С. Через 48 часов при 10°С добавляют пропиленоксид (71 мл, 1,0 моль). Через 96 часов при 10°С осторожно добавляют насыщенный водный раствор дигидрофосфата калия (3,8 л) и этилацетат (3,8 л), слои разделяют и водный слой экстрагируют 3,8 л этилацетата. Объединенные органические экстракты сушат над сульфатом натрия и концентрируют в вакууме с получением твердого вещества (520 г, 110%, с содержанием минерального масла).
Стадия В: N-(5-бензилокси-2-формилфенил)-N-(2-гидроксипропил)формамид
Раствор продукта, полученного на стадии А (172 г), в 1,5 л дихлорметана охлаждают до -78°С и озонируют (4% озона в кислороде). Избыток озона замещают кислородом в течение 5 минут и затем добавляют диметилсульфид (78 мл) и нагревают до 25°С. Раствор концентрируют до половинного объема, проводят элюирование через флорисил (Florisil), промытый смесью этиловый эфир - этилацетата, и концентрируют в вакууме. Процедуру повторяют четыре раза: один раз с использованием порции 172 г и три раза порциями по 58 г. Объединенные продукты пропускают через силикагель (2,5 кг) с использованием градиента 10%-80% ацетата-гексана, получая после концентрирования в вакууме масло (351 г, 70%).
Стадия С: 4-бензилокси-2-(2-гидроксипропиламино)бензальдегид
Охлажденный льдом раствор продукта, полученного на стадии В (298 г, 0,95 моль), в ТГФ (3 л) обрабатывают 1 М водным раствором гидроксида натрия (1,95 л, 1,9 моль), поддерживая температуру ниже 8°С. После потребления исходного материала смесь разбавляют насыщенным раствором соли и дважды экстрагируют этиловым эфиром. Органический раствор промывают водой до нейтральной реакции и затем насыщенным раствором соли, сушат над сульфатом натрия, обрабатывают древесным углем и пропускают через силикагель (1 кг) с использованием эфира и смеси этилацетата-гексана (1:1) с получением после концентрирования в вакууме желтого твердого вещества (207 г, 76%).
Стадия D: 1-(6-бензилоксииндазол-1-ил)пропан-2-ол
Продукт, полученный на стадии С (202,7 г, 0,71 моль), обрабатывают, как описано для стадий С и D в способе получения 3. После превращения нитрозаминового промежуточного продукта в смесь желательного индазольного продукта и непрореагировавшего исходного материла (5:1) добавляют нитрит натрия (29,5 г, 0,43 моль) для повторного нитрозирования исходного материала. Затем порциями при охлаждении, как описано выше, добавляют цинковую пыль (84 г, 1,28 моль). После потребления исходного материала реакционную смесь обрабатывают, как описано выше, объединяют с продуктом из другой партии, получение которого велось со 176 г продукта со стадии С. Объединенные неочищенные продукты очищают хроматографией (Biotage Kiloprep-250) с получением твердого вещества (226 г, 60%): 99% чистоты по данным ВЭЖХ.
Способ получения 3
(R)-1-(6-бензилоксииндазол-1-ил)пропан-2-ол
Стадия А: 4-бензилокси-2-фторбензонитрил
К раствору 2-фтор-4-гидроксибензонитрила (490 г, 3,57 моль) в ацетоне (3,4 л) добавляют бензилбромид (467 мл, 3,93 моль) и карбонат калия (1,4 кг, 10,1 моль). Перемешанную смесь нагревают при 60°С в течение 20 часов, затем охлаждают и фильтруют. Фильтрат концентрируют и полученное твердое вещество растирают со смесью 10% этилацетата/гексана (5 л) и сушат в вакууме при 35°С получением целевого продукта (787 г, 97%).
Стадия В: 4-бензилокси 2-((R)-2-гидроксипропиламино)бензонитрил
Раствор (R)-(-)-1-аминопропан-2-ола (389 г, 5,19 моль) в диметилсульфоксиде (600 мл) добавляют к раствору продукта со стадии А (786 г, 3,46 моль), основному глинозему (786 г) и молекулярных сит 4А (131 г). Перемешанную смесь нагревают при 110-140°С в течение 24 часов, охлаждают и фильтруют, фильтр (устройство) промывают 10 л смеси эфира-этилацетата (4:1) и затем 4 л смеси этилацетат-гексана (3:2). Органические промывные жидкости экстрагируют водой (5 л) и водную фазу экстрагируют смесью 25% этилацетата-гексана (4 х 2 л). Объединенные органические фазы промывают водой и насыщенным раствором соли, сушат над безводным сульфатом натрия, концентрируют примерно до 3 л и оставляют на 48 часов. Осажденный твердый материал собирают фильтрованием, промывают гексаном и сушат в вакууме с получением целевого продукта в виде двух партий (619 г и 86 г). Концентрированный супернатант наносят на 5 кг слоя силикагеля и элюируют градиентом 10-50% этилацетата-гексана с получением после концентрирования в вакууме дополнительного количества продукта (119 г): общий выход составляет 791 г (81%).
Стадия С: 4-бензилокси-2-((R)-2-гидроксипропиламино)бензальдегид
К раствору продукта, полученного на стадии В (790 г, 2,8 моль), в смеси пиридина-уксусной кислоты-воды (2:1:1) (7 л) добавляют гидрат гипофосфита натрия (986 г, 11,2 моль) и никелевый катализатор Ренея (500 г 50% водной суспензии). Смесь перемешивают при 45°С в течение 7 часов, затем охлаждают до 25°С в течение ночи и фильтруют через фильтровальное устройство, промывая его водой и этилацетатом. Фильтрат промывают насыщенным гидрофосфатом натрия до pH 5, затем водой и насыщенным раствором соли, сушат над сульфатом натрия и концентрируют. В ходе концентрирования добавляют 4 л гептана для азеотропного удаления пиридина. После удаления 8 л растворителя продукт отвердевает. Добавляют гептан (5 л) и растирают твердое вещество, отделяют его фильтрованием и сушат в вакууме при 35°С с получением целевого продукта (722 г, 90%).
Стадия D: (R)-1-(6-бензилоксииндазол-1-ил)пропан-2-ол
В течение 25 минут добавляют нитрит натрия (209 г, 3,03 моль) к перемешанному раствору продукта, полученного на стадии С (720 г, 2,53 моль), в уксусной кислоте (5,6 л) и воде (1,4 л), поддерживая температуру ниже 25°С. Полученный раствор нитрозаминового интермедиата охлаждают в ледяной бане и добавляют цинковую пыль (595 г, 9,10 моль) порциями по 25 г в течение 3,5 часа, поддерживая температуру ниже 35°С. Добавляют этилацетат (7 л) и густую суспензию фильтруют через воронку со спекшимся стеклом, промывая ее этилацетатом (7,5 л). К фильтрату, содержащему смесь желательного индазольного продукта и восстановленного исходного материала (5:1), добавляют реагент Т Жирара (Girard's Reagent Т) (98 г, 0,58 моль). После перемешивания при 25°С в течение 1 дня добавляют другую порцию реагента Т Жирарда 150 г (0,90 моль). Через 3 дня весь исходный материал потребляется. Смесь экстрагируют дважды водой, водным раствором гидрофосфата натрия для удаления уксусной кислоты, водой и насыщенным раствором соли, сушат над сульфатом натрия, фильтруют через флорисил и концентрируют. Остаток пропускают через 5 кг силикагеля с использованием смеси этилацетат-гексана (1:1). Соответствующие фракции объединяют и концентрируют и добавляют гептан (4 л) для осаждения индазольного продукта. Твердое вещество собирают фильтрованием, промывают смесью этилацетат-гексан (1:1) и сушат в вакууме при 35°С с получением желтого твердого вещества (417 г, 58%): ВЭЖХ анализ: (R) - 96,7%; (S) - 0,3%, исходный материал - 3%. Концентрирование супернатанта приводит к получению дополнительно 141 г (20%) целевого продукта.
Способ получения 4
(S)-1-(6-бензилоксииндазол-1-ил)пропан-2-ол
Способ 1. Указанный S стереоизомер получают, как описано выше в способе получения рацемического 1-(6-бензилоксииндазол-1-ил)пропан-2-ола, но используют (S)-1-амино-2-пропанол вместо рацемического аминоспирта.
Способ 2. Стадия А: 4-бензилокси-2-фторбензонитрил
Смесь 2-фтор-4-гидроксибензонитрила (15,0 г, 109 ммоль), карбоната калия (21,0 г, 152 ммоль) и бензилбромида (19,6 г, 115 ммоль) в ацетоне (150 мл) в атмосфере азота нагревают в течение ночи при 50°С. Твердый материал удаляют фильтрованием и фильтрат выпаривают до получения остатка, который смешивают с этилацетатом (500 мл). Указанный раствор промывают насыщенным раствором соли, сушат и выпаривают с получением аморфного твердого вещества (24,9 г, 100%).
Стадия В: 4-бензилокси-2-((S)-2-гидроксипропиламино)бензонитрил
Смесь продукта со стадии А (24,8 г, 109 ммоль), (S)-1-амино-2-пропанола (12,3 г, 164 ммоль), молекулярных сит 4А (4,0 г) и основного глинозема (32 г) в безводном диметилсульфоксиде (100 мл) в атмосфере азота нагревают при 95°С в течение 40 часов. Суспензию охлаждают до температуры окружающей среды, фильтруют через фильтрующее устройство, которое промывают этилацетатом (2 х 300 мл) и водой (300 мл). Водный слой фильтрата экстрагируют этилацетатом (2 х 300 мл) и объединенный органический материал промывают насыщенным раствором соли (200 мл), сушат (MgSO4) и очищают хроматографией (силикагель, EtOAc/гексан) с получением вязкого масла (24,9 г, 81%).
Стадия С: 4-бензилокси-2-((S)-2-гидроксипропиламино)бензальдегид
К раствору продукта, полученного на стадии В (19,3 г, 68,3 ммоль), в смеси безводного циклогексана и ТГФ (200 мл, 40 мл) при 0°С в атмосфере азота в течение 30 минут добавляют гидрид диизобутиламмония (1 М раствор в гексане, 239 мл, 239 ммоль). Смесь перемешивают в течение 18 часов при температуре окружающей среды, вносят дополнительное количество гидрида диизобутиламмония (40 мл, 40 ммоль) и смесь перемешивают еще в течение 24 часов. Реакционную смесь охлаждают в ледяной бане и реакцию гасят добавлением MeOH (экзотермическая реакция) и 2 н. HCl для поддержания pH 1. Далее смесь экстрагируют EtOAc (3 х 300 мл) и экстракты сушат и концентрируют до коричневого масла (18,5 г). Неочищенное масло растирают с использованием EtOAc/гексана и фильтруют с получением масла (16,1 г, 83% выход неочищенного материала). Небольшую порцию указанного материала очищают хроматографией (силикагель, 20%-50% EtOAc/гексана) с получением твердого материала, Тпл. 68-69°С.
Стадия D: (S)-1-(6-бензилоксииндазол-1-ил)пропан-2-ол (4)
К смеси продукта, полученного на стадии С (16,0 г, 56,1 ммоль), в смеси уксусной кислоты/воды (150 мл/30 мл) при 0°С порциями в течение 40 минут добавляют нитрит натрия (7,75 г, 112 ммоль). Смесь перемешивают в течение 50 минут, охлаждают (ледяная баня) и добавляют порциями цинк (14,7 г, 224 ммоль). Через 1 час суспензию нагревают до комнатной температуры и добавляют еще цинк (14,7 г, 224 ммоль). Смесь перемешивают в течение 1 часа, концентрируют и экстрагируют EtOAc (2 х 300 мл). Экстракты фильтруют через фильтрующее устройство, фильтрат промывают насыщенным водным раствором гидрофосфата динатрия (до pH 8) и насыщенным раствором соли, сушат и очищают хроматографией (силакагель, 25% EtOAc/гексан) с получением масла (7,01 г, 44%).
Пример 1
1-(2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол
Стадия А: 6-бензилокси-1-[2-(трет-бутилдиметилсиланилокси)пропил]-1Н-индазол
К смеси 1-(6-бензилоксииндазол-1-ил)пропан-2-ола (10 г, 35,5 ммоль) в ТГФ (100 мл) и имидазоле (3,4 г, 50 ммоль) добавляют трет-бутилхлордиметил-силан (6,42 г, 42,6 ммоль) и смесь перемешивают еще в течение 15 минут при температуре окружающей среды. Реакционную смесь вливают в насыщенный водный раствор ацетата аммония (300 мл) и экстрагируют этилацетатом (2 х 150 мл). Очистка хроматографией (силикагель, EtOAc/гексан) дает твердый желтый материал (10,6 г, 76%): Тпл. 56-58°С; LC/MS (+APCI) m/z 397 (M+H).
Стадия В: 1-[2-(трет-бутилдиметилсиланилокси)пропил]-1Н-индазол-6-ол
Смесь продукта, полученного на стадии А (10,6 г, 27 ммоль), и палладия на угле (10% г, 0,26 г) в метаноле (250 мл) перемешивают в атмосфере водорода в течение 6 часов, добавляют дихлорметан (100 мл) и смесь фильтруют. Выпаривание фильтрата дает желтовато-белое твердое вещество (7,0 г, 85%): Тпл. 169-174°С; LC/MS (+APCI) m/z 307 (M+H).
Стадия С: 7-бром-1-[2-(трет-бутилдиметилсиланилокси)пропил]-1Н-индазол-6-ол
К раствору продукта, полученного на стадии В (6,00 г, 19,6 ммоль), в безводном ТГФ (300 мл) при 0°С добавляют N-бромсукцинимид (3,49 г, 19,6 ммоль) в виде 10 порций в течение 20 минут. Смесь вливают в насыщенный водный раствор бисульфита натрия (300 мл) и экстрагируют EtOAc (3 х 100 мл). Объединенные экстракты сушат (MgSO4) и выпаривают до получения остатка (6,39 г). Хроматография (силикагель, 10% EtOAc/гексан) дает твердый материал (4,95 г, 66%): LC/MS (+APCI) m/z 385, 387 (M+H).
Стадия D: 7-бром-1-[2-(трет-бутилдиметилсиланилокси)пропил]-6-оксиранилметокси-1Н-индазол
Суспензию продукта, полученного на стадии С (4,47 г, 11,6 ммоль), карбонат калия (2,25 г, 16 ммоль) и эпибромгидрин (1,59 мл, 19 ммоль) в ацетоне (230 мл) нагревают до температуры кипения с обратным холодильником в течение 20 часов. Вносят дополнительное количество эпибромгидрина (1,5 мл, 17,9 ммоль) и смесь нагревают при температуре кипения с обратным холодильником в течение 18 часов. Твердый материал удаляют фильтрованием и фильтрат концентрируют до получения масла, которое растворяют в EtOAc (150 мл). Указанный раствор промывают насыщенным водным раствором ацетата аммония (150 мл), сушат (MgSO4) и выпаривают до получения остатка, который очищают хроматографией (силикагель, 2%-10% EtOAc/гексан) с получением масла (3,77 г, 74%): LC/MS (+APCI) m/z 441, 443 (M+H).
Стадия Е: 1-бром-3-[7-бром-1-[2-(трет-бутилдиметилсиланилокси)пропил]-1Н-индазол-6-илокси]пропан-2-ол
К суспензии порошка магния (0,81 г, 33,5 ммоль) в безводном ТГФ (150 мл) в атмосфере азота добавляют по каплям дибромэтан (1,09 мл, 12,6 ммоль). В ходе указанного процесса смесь нагревают примерно до 50°С до выделения газа и затем позволяют охладиться до 40°С. Вносят дополнительное количество дибромэтана (0,05 мл) и смесь нагревают при температуре кипения с обратным холодильником в течение 20 минут и помещают в ледяную баню. К охлажденной смеси добавляют раствор продукта, полученного на стадии D (3,70 г, 8,38 ммоль), в ТГФ (50 мл). После перемешивания смеси в течение 20 минут при температуре окружающей среды реакцию гасят добавлением насыщенного водного раствора хлорида аммония (200 мл) и смесь экстрагируют EtOAc (2 х 150 мл). Выпаривание экстрактов дает неочищенное масло (3,78 г, 86%): LC/MS (+APCI) m/z 521, 523, 525 (M+H).
Стадия F: 7-бром-6-[3-бром-2-(1-этоксиэтокси)пропокси]-1-[2-(трет-бутилдиметилсиланилокси)пропил]-1Н-индазол
К раствору продукта, полученного на стадии Е (3,78 г, 7,2 ммоль), и п-толуолсульфоновой кислоты (0,14 г) в дихлорметане (50 мл) при 0°С добавляют этилвиниловый эфир (2,75 мл, 28,8 ммоль). Через 30 минут реакцию гасят добавлением насыщенного раствора бикарбоната натрия (50 мл) и смесь экстрагируют дихлорметаном (3 х 80 мл). Выпаривание и очистка хроматографией (силикагель, 1%-8% EtOAc/гексан) дает вязкое масло (3,30 г, 77%): LC/MS (+APCI) m/z 593, 595, 597 (M+H).
Стадия G: 1-[8-(1-этоксиэтокси)-8,9-дигидро-7Н-пирано[2,3-g]индазол-1-ил]пропан-2-ол
К раствору продукта, полученного на стадии F (3,3 г, 5,5 ммоль), в безводном ТГФ (100 мл) при -78°С в атмосфере азота добавляют н-бутиллития (2 М в гексане, 2,76 мл, 6,90 ммоль). Через 30 минут реакцию гасят добавлением насыщенного раствора бикарбоната натрия (200 мл) и смесь экстрагируют EtOAc (2 х 150 мл). Объединенные экстракты сушат (MgSO4) и выпаривают до получения остатка, который хроматографируют (силикагель, 10% EtOAc/гексан) с получением масла (1,06 г), которое растворяют в ТГФ (50 мл) и затем к указанному раствору добавляют фторид тетрабутиламмония (1 М в ТГФ, 3,84 мл, 13,8 ммоль). Смесь перемешивают в течение ночи при температуре окружающей среды, вливают в насыщенный раствор бикарбоната натрия (200 мл) и экстрагируют EtOAc (2 х 100 мл). Объединенные экстракты сушат (MgSO4), выпаривают и хроматографируют (силикагель, 20%-50% EtOAc/гексан) с получением масла (0,52 г, 29%): LC/MS (+APCI) m/z 321 (M+H).
Стадия H: 1-(2-азидопропил)-8-(1-этоксиэтокси)-1,7,8,9-тетрагидропирано[2,3-g]индазол
К раствору продукта, полученного на стадии G (0,52 г, 1,6 ммоль), и триэтиламина (1,12 мл, 8,1 ммоль) в безводном ТГФ (75 мл) при 0°С добавляют метансульфоновый ангидрид (0,71 г, 4,05 ммоль). Смесь перемешивают в течение 20 минут и добавляют азид натрия (2,11 г, 32,4 ммоль) вместе с ДМСО (20 мл). Удаляют ТГФ (в вакууме) и реакционную смесь нагревают при 90°С в течение 3 часов. Смесь охлаждают, вливают в насыщенный раствор бикарбоната натрия (150 мл) и смесь экстрагируют EtOAc (3 х 100 мл). Объединенные экстракты промывают насыщенным раствором соли (100 мл), сушат (MgSO4) и выпаривают до получения остатка, который очищают хроматографией (силикагель, 10% EtOAc/гексан) с получением масла (0,40 г, 72%): LC/MS (+APCI) m/z 346 (M+H).
Стадия I: 1-(2-азидопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол
К смеси продукта, полученного на стадии Н (0,40г, 1,1 ммоль), в ТГФ (60 мл) добавляют 1 н. HCl (26 мл). После перемешивания в течение 40 минут добавляют насыщенный раствор бикарбоната натрия (150 мл) и смесь экстрагируют EtOAc (3 х 100 мл). Объединенные экстракты сушат и выпаривают до получения остатка, который очищают хроматографией (силикагель, 50% EtOAc/гексан) с получением масла (0,29 г, 92%): LC/MS (+APCI) m/z 374 (M+H).
Стадия J: Гидрохлорид 1-(2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ола
Смесь продукта, полученного на стадии I (0,27г, 0,99 ммоль), и палладия на угле (10%, 0,03 г) в EtOHc (20 мл) перемешивают в атмосфере водорода в течение 18 часов при температуре окружающей среды. Смесь фильтруют и фильтрат выпаривают до получения остатка, который растворяют в смеси EtOAc/гексан (1:1) (10 мл). После выстаивания в течение 18 часов собирают образовавшийся в виде бесцветного твердого вещества осадок (0,14 г, 57%): Тпл 124-125°С: LC/MS (+APCI) m/z 248 (M+H). Аналитические данные для C13H17N3O2 х 0,33 H2O: Расчет: C - 61,64: H - 7,03; N - 16,59. Найдено: С - 61,62; Н - 6,83; N - 16,43.
Пример 2
Гидрохлорид 1-((R)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол
Стадия А: 6-бензилокси-1-[(S)-2-(трет-бутилдиметилсиланилокси)пропил]-1Н-индазол
К смеси (S)-1-(6-бензилоксииндазол-1-ил)пропан-2-ола (2,03 г, 7,20 ммоль) в безводном ТГФ/ДМФ (100 мл/35 мл) в атмосфере азота добавляют гидрид натрия (60% в минеральном масле, 0,40 г, 10,0 ммоль). Через 30 минут добавляют трет-бутилхлордиметилсилан (1,52 г, 10 ммоль) и каталитическое количество NaH (5 ммоль) и смесь перемешивают в течение ночи при температуре окружающей среды. Вносят дополнительное количество NaH (5 ммоль) и трет-бутилхлордиметилсилан (5 ммоль) и реакционную смесь перемешивают в течение 6 часов. Далее реакционную смесь выпаривают до получения остатка, который смешивают с насыщенным водным раствором бикарбоната натрия и экстрагируют этилацетатом (2 х 200 мл). Очистка хроматографией (силикагель, EtOAc/гексан) дает масло (2,81 г, 99%).
Стадия В: 1-[(S)-2-(трет-бутилдиметилсиланилокси)пропил]-1Н-индазол-6-ол
Смесь продукта, полученного на стадии А (5,44 г, 13,7 ммоль), и палладия на угле (10% г, 0,50 г) в метаноле (200 мл) перемешивают в атмосфере водорода в течение 18 часов, фильтруют и выпаривают с получением желтовато-белого твердого вещества (3,80 г, 90%): Тпл. 171-172°С.
Стадия С: 7-бром-1-[(S)-2-(трет-бутилдиметилсиланилокси)пропил]-1Н-индазол-6-ол
К раствору продукта, полученного на стадии В (3,79 г, 12,4 ммоль), в безводном ТГФ (100 мл) при 0°С добавляют N-бромсукцинимид (2,20 г, 12,4 ммоль) в виде 3 порций в течение 10 минут. Через 20 минут смесь вливают в насыщенный водный раствор бисульфита натрия (100 мл) и экстрагируют EtOAc (3 х 100 мл). Объединенные экстракты сушат (MgSO4), и выпаривают до получения остатка (4,79 г). Хроматография (силикагель, 10% EtOAc/гексан) дает твердый материал (3,66 г, 77%): Тпл. 103-105°С.
Стадия D: 7-бром-1-[(S)-2-(трет-бутилдиметилсиланилокси)пропил]-6-оксиранилметокси-1Н-индазол
Суспензию продукта, полученного на стадии С (3,66 г, 9,51 ммоль), карбоната калия (1,92 г, 1,46 ммоль) и эпибромгидрина (1,32 г, 1,60 ммоль) в ацетоне (200 г) нагревают при температуре кипения с обратным холодильником в течение 30 часов. Твердый материал удаляют фильтрованием и фильтрат концентрируют до получения масла, которое очищают хроматографией (силикагель, 2%-10% EtOAc/гексан) с получением масла (3,33 г, 79%): LC/MS (+APCI) m/z 441, 443 (M+H).
Стадия Е: 1-бром-3-[7-бром-1-[(S)-2-(трет-бутилдиметилсиланилокси)пропил]-1Н-индазол-6-илокси]пропан-2-ол
К суспензии порошка магния (0,73 г, 30,2 ммоль) в безводном ТГФ (50 мл) в атмосфере азота добавляют дибромэтан (2,13 г, 0,98 мл, 11,3 ммоль) порциями в течение примерно 30 минут. В ходе указанного процесса смесь нагревают до температуры примерно 50°С до момента выделения газа. Далее смесь перемешивают еще в течение 1 часа, помещают в ледяную баню и добавляют раствор продукта, полученного на стадии D (3,33 г, 7,55 ммоль), в ТГФ (10 мл). После перемешивания в течение 1 часа при температуре окружающей среды смесь гасят добавлением насыщенного водного раствора хлорида аммония (100 мл) и экстрагируют EtOAc (3 х 100 мл). Выпаривание экстрактов дает неочищенное масло (3,76 г, 95%): LC/MS (+APCI) m/z 441, 443 (M+H-HBr).
Стадия F: 7-бром-6-[3-бром-2-(1-этоксиэтокси)пропокси]-1-[(S)-2-(трет-бутилдиметилсиланилокси)пропил]-1Н-индазол
К раствору продукта, полученного на стадии Е (1,85 г, 3,54 ммоль), и п-толуолсульфоновой кислоты (0,01 г) в дихлорметане (50 мл) при 0°С добавляют этилвиниловый эфир (1 мл, 10,5 ммоль). Через 30 минут реакцию гасят добавлением насыщенного раствора бикарбоната натрия (50 мл) и экстрагируют EtOAc (3 х 80 мл). Выпаривание и очистка остатка хроматографией (силикагель, 1%-8% EtOAc/гексан) дает масло (1,79 г, 81%): LC/MS (+APCI) m/z 595 (M+H).
Стадия G: (S)-1-[8-(1-этоксиэтокси)-8,9-дигидро-7Н-пирано[2,3-g]индазол-1-ил]пропан-2-ол
К раствору продукта, полученного на стадии F (0,90 г, 2,07 ммоль), в безводном ТГФ (50 мл) при -78°С в атмосфере азота в течение 3 минут добавляют н-бутиллития (2,5 М в гексанах, 1,56 мл, 3,90 ммоль). Через 30 минут реакцию гасят добавлением насыщенного раствора бикарбоната натрия (80 мл) и смесь экстрагируют EtOAc (3 х 100 мл). Объединенные экстракты сушат (MgSO4) и выпаривают с получением масла (1,30 г), которое растворяют в ТГФ (50 мл), и затем к указанному раствору добавляют фторид тетрабутиламмония (1 М раствор в ТГФ). Смесь перемешивают в течение ночи при температуре окружающей среды, добавляют порциями бикарбонат натрия (80 мл) и экстрагируют EtOAc (3 х 100 мл). Объединенные экстракты выпаривают и очищают хроматографией (силикагель, 10%-40% EtOAc/гексан) с получением масла (0,35 г, 53 %): LC/MS (+ES) m/z 321 (M+H).
Стадия H: 1-((R)-2-азидопропил)-8-(1-этоксиэтокси)-1,7,8,9-тетрагидропирано[2,3-g]индазол
К раствору продукта, полученного на стадии G (0,35 г, 1,09 ммоль), и триэтиламина (0,55 г, 5,47 ммоль) в безводном ТГФ (50 мл) при 0°С добавляют метансульфоновый ангидрид (0,47 г, 2,73 ммоль). Смесь перемешивают в течение 1 часа и добавляют азид натрия (0,71 г, 10,9 ммоль). Реакционную смесь выпаривают и полученный остаток растворяют в безводном ДМФ (80 мл) и нагревают при 95°C в течение 3 часов. Смесь охлаждают, выливают в насыщенный раствор бикарбоната натрия (80 мл) и экстрагируют EtOAc (3 х 80 мл). Объединенные экстракты сушат (MgSO4) и выпаривают до получения остатка, который очищают хроматографией (силикагель, 10% EtOAc/гексан) с получением масла (0,30 г, 80%): LC/MS (+APCI) m/z 346 (M+H).
Стадия I: 1-((R)-2-азидопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол
К смеси продукта, полученного на стадии Н (0,30 г, 0,87 ммоль), в ТГФ (50 мл) добавляют 1 н. HCl (20 мл). После перемешивания в течение 40 минут добавляют насыщенный раствор бикарбоната натрия (80 мл) и смесь экстрагируют EtOAc (3 х 50 мл). Объединенные экстракты сушат и выпаривают до получения остатка, который очищают хроматографией (силикагель, 20%-50% EtOAc/гексан) с получением масла (0,22 г, 92%).
Стадия J: Гидрохлорид 1-((R)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ола
Смесь продукта, полученного на стадии I (0,21г, 0,77 ммоль), и палладия на угле (10%, 0,02 г) в MeOH (30 мл) перемешивают в атмосфере водорода в течение 18 часов при температуре окружающей среды. Смесь фильтруют и фильтрат объединяют с раствором хлористого водорода в этаноле (2 N, 1 мл), далее указанный раствор выпаривают при 60°С в высоком вакууме с получением твердого вещества (0,13 г, 59%): Тпл 82-86°С: LC/MS (+APCI) m/z 248 (M+H). Аналитические данные для C13H17N3O2 х HCl х 0,1 H2O х 0,4 С3H7NO: Расчет: C - 54,17: H - 6,72; N - 15,12. Найдено: С - 54,06; Н - 6,76; N - 14,98.
Пример 3
1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол
Стадия А: 1-((S)-2-азидопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол
Данное соединение синтезируют, следуя процедуре, описанной в примере 2, стадия I, но с использованием 1-((S)-2-азидопропил)-8-(1-этоксиэтокси)-1,7,8,9-тетрагидропирано[2,3-g]индазола, который получают из (S)-1-(6-бензилоксииндазол-1-ил)пропан-2-ола вместо (R)-1-6-бензилоксииндазол-1-ил)пропан-2-ола, приведенного выше: при этом получают масло (0,45 г, 79%); LC/MS (+APCI) m/z 274 (M+H).
Стадия В: 1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол
Раствор продукта, полученного на стадии А, в метаноле обрабатывают, как было описано в примере 2, стадия J, с получением свободного основания в виде аморфного твердого вещества (0,36 г, 88%): Тпл 46-51°С; GCMS(Cl+) m/z 248 (M+H). Аналитические данные для C13H17N3O2 х 0,1 H2O: Расчет: C - 62,68: H - 6,96; N - 16,97. Найдено: С - 62,55; Н - 7,03; N - 16,64.
Пример 4
(R)-1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол
Стадия А: 1-[2-(R)-(трет-бутилдиметилсиланилокси)пропил]-6-проп-2-инилокси-1Н-индазол
К раствору 1-[(R)-2-трет-бутилдиметилсиланилокси)пропил]-1Н-индазол-6-ола (10,93 г, 35,7 ммоль), полученному из (R)-1-(6-бензилоксииндазол-1-ил)пропан-2-ола по методике примера 2, стадия В, в ацетоне (250 мл) добавляют карбонат калия (6,90 г, 35,7 ммоль) и пропаргилбромида (5,19 мл, 46,4 ммоль) и смесь нагревают при температуре кипения с обратным холодильником в течение 18 часов. Вносят дополнительное количество карбоната калия (1,97 г, 14 ммоль) и пропаргилбромида (1,2 мл, 10,7 ммоль) и смесь нагревают при температуре кипения с обратным холодильником в течение 2 часов. Удаляют твердый материал фильтрованием и фильтрат концентрируют до масла, которое очищают хроматографией (силикагель, 5% этилацетат в гексане) с получением масла (1,40 г, 96%): LC/MS (+APCI) m/z 345 (M+H).
Стадия В: 1-[(R) -2-(трет-бутилдиметилсиланилокси)пропил]-1,7-дигидро-пирано[2,3-g]индазол
Раствор продукта, полученного на стадии А (10,9 г, 31,8 ммоль), в мезитилене (60 мл) помещают в пробирку, находящуюся под давлением, и дегазируют в вакууме. Далее пробирку запаивают и нагревают при 190°в течение 20 часов. Раствор охлаждают и очищают хроматографией (силикагель, 10% этилацетат в гексане) с получением твердого вещества (9,53 г, 87%): Тпл. 58-59°С; LC/MS (+APCI) m/z 345 (M+H).
Стадия С: (R)-1-[(R)-2-(трет-бутилдиметилсиланилокси)пропил]-1,7,8,9-тетрагидропирано[2,3-g]индазол-8 ол
К продукту, полученному на стадии В (1,00 г, 2,91 ммоль), добавляют при перемешивании в атмосфере азота 9-BBN (0,5 М в ТГФ, 13 мл, 6,4 ммоль). Раствор нагревают при 70°С в течение 2 часов, охлаждают до температуры окружающей среды и реакцию гасят добавлением метанола (5 мл) и пероксида водорода (30%, 5 мл). После перемешивания в течение 30 минут смесь выпаривают до получения остатка, который объединяют с насыщенным раствором бикарбоната натрия (50 мл), после чего указанную смесь экстрагируют этилацетатом (3 х 50 мл). Объединенные экстракты выпаривают до получения остатка, который очищают хроматографией (силикагель, 10%-30% этилацетата в гексане) с получением желательного диастереомера в виде вязкого масла (0,56 г, 75%) и смеси (0,22 г) неразделяемых диастереомеров. LC/MS (+APCI) m/z 363 (M+H).
Стадия D: (R)-1-((S)-2-азидопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8 ол
Раствор продукта, полученного на стадии С (0,56 г, 1,55 ммоль), п-толуолсульфоната пиридиния (50 мг) и этилвинилового эфира (1 мл) в безводном дихлорметане (50 мл) перемешивают в атмосфере азота при 0°С в течение 1 часа. Удаляют охлаждающую баню, смесь перемешивают в течение 1 часа и добавляют триэтиламин (1 мл). Указанную смесь выпаривают до получения остатка, который объединяют с ТГФ (10 мл) и фторидом тетрабутиламмония (1 М раствор в ТГФ, 3,1 мл, 3,1 ммоль) и перемешивают в течение 1 часа. Реакционную смесь выпаривают до получения остатка, который очищают хроматографией (силикагель, 10%-30% этилацетата в гексане) с получением гидроксиэфирного интермедиата в виде масла (0,47 г, 96%). К раствору указанного масла (0,46 г, 1,44 ммоль) в безводном ТГФ (50 мл) при 0°С добавляют триэтиламин (0,726 г, 7,19 ммоль) и метансульфоновый ангидрид (0,50 г, 2,88 ммоль) и смесь перемешивают в течение 30 минут. Добавляют азид натрия (0,936 г, 14,4 ммоль) и растворитель удаляют выпариванием. Затем добавляют ДМФ (50 мл) и суспензию нагревают при 100°С в течение 4 часов, охлаждают и экстрагируют этилацетатом (3 х 80 мл). Объединенные экстракты промывают водой, сушат и очищают хроматографией (1%-10% этилацетата в гексане) с получением азидоэфира в виде масла (0,38 г, 77%). Масло растворяют в метаноле, добавляют п-толуолсульфоновую кислоту (50 мг) и перемешивают полученный раствор в течение 1 часа. Добавляют триэтиламин (0,1 мл) и смесь выпаривают до получения остатка, который очищают хроматографией (10%-35% этилацетата в гексане) с получением желательного азидоспирта в виде твердого вещества (0,27 г, 99%). LC/MS (+APCI) m/z 274 (M+H).
Стадия Е: (R)-1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8 ол
Продукт, полученный на стадии D, обрабатывают, как было описано в примере 2, стадия J, с получением желательного соединения в виде желтоватого твердого вещества (0,26 г, 65%): Тпл 126-128°С; [α]D=+47,7°С (с 0,352, CH3OH); [α]405=+115°С (с 0,352, CH3OH); LC/MS (+APCI) m/z 248 (M+H); Аналитические данные для C13H17N3O2: Расчет: C - 63,14: H - 6,93; N - 16,99. Найдено: С - 63,37; Н - 6,79; N - 16,93.
Пример 5
(S)-1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол
Стадия А: (S)-1-((S)-2-азидопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол
Продукт, полученный в примере 3 на стадии А (1,35 г), наносят на хроматографическую колонку, заполненную хиральным адсорбентом (Chiracel OJ). Элюирование проводят смесью гексана и 2-пропанола (9:1), которая позволяет разделить смесь на два диастереомера: S,8S-диастереомера (0,68 г) и S,8R-дистереомера (0,65 г).
Стадия В: (S)-1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8 ол
Раствор S,8S-диастереомера (0,23 г), полученного на стадии А, в метаноле обрабатывают, как было описано в примере 3, стадия В, с получением масла (0,18 г, 87%); [α]D=-6,21°С (с 0,467, CH3OH); [α]405=-3,5°С (с 0,467, CH3OH). Аналитические данные для C13H17N3O2 х 0,2 H2O: Расчет: C - 62,22: H - 7,03; N - 16,74. Найдено: С - 62,36; Н - 7,06; N - 16,93.
Пример 6
Тригидрохлорид (R)-1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-иламина
Стадия А: (S)-8-азидо-1-((R)-2-азидопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол
К раствору S,8S-диастереомера, полученного в примере 5 на стадии А (0,44 г, 1,61 ммоль), и триэтиламина (0,90 мл 6,44 ммоль) в безводном ТГФ (50 мл) при 0°С добавляют метансульфоновый ангидрид (0,56 г, 3,22 ммоль) и указанную смесь перемешивают в течение 30 минут, затем удаляют ледяную баню и смесь перемешивают еще в течение 20 минут. Реакционную смесь выпаривают до получения остатка, к которому добавляют ДМСО (50 мл) и азид натрия (1,05 г, 16,1 ммоль) и затем нагревают смесь при 90°С в течение 5 часов. Далее смесь охлаждают, выливают в воду и экстрагируют этилацетатом (3 х 60 мл). Объединенные экстракты сушат, фильтруют и выпаривают досуха. Очистка хроматографией (силикагель, 5%-25% этилацетата в гексане) дает масло (0,19 г 16,1 ммоль): LC/MS (+APCI) m/z 299 (M+H).
Стадия В: Тригидрохлорид (R)-1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-иламина
Раствор продукта, полученного на стадии А, в метаноле обрабатывают, как описано на стадии J в примере 2, с получением желтоватого твердого вещества (0,16 г, 78%); Тпл. >300°С; LC/MS (+APCI) m/z 247 (M+H). Аналитические данные для C13H21Cl3N4O х 0,33 C2H5OH х 0,5 H2O: Расчет: C - 43,14: H - 6,35; N - 14,72. Найдено: С - 43,52; Н - 6,39; N - 14,51.
Пример 7
(8R*,9S*)-1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8,9-диол
Стадия А: Бензил (S)-2-(6-гидрокси-1Н-индазол-1-ил)метилэтилкарбамат
К суспензии 1-((S)-2-аминопропил)-1Н-индазол-6-ола (2,00 г, 10,5 ммоль) в ТГФ (20 мл) добавляют насыщенный водный раствор бикарбоната натрия (10 мл) и бензилхлорформиат (1,50 мл 1,5 ммоль). Смесь перемешивают до растворения всего твердого исходного амина. После перемешивания при температуре окружающей среды в течение 45 минут добавляют насыщенный раствор бикарбоната натрия (150 мл) и смесь экстрагируют этилацетатом (3 х 150 мл). Объединенные экстракты сушат (MgSO4) и выпаривают с получением рыжевато-коричневой пены (2,65 г 78%): LC/MS (+APCI) m/z 326 (M+H).
Стадия В: Бензил (S)-1-метил-2-(6-проп-2-инилоксииндазол-1-ил)этилкарбамат
Раствор продукта, полученного на стадии А (2,88 г, 8,86 ммоль), в ацетоне (100 мл) дегазируют при пониженном давлении в атмосфере азота. Добавляют порошок карбоната калия (1,35 г 9,75 ммоль) и пропаргилбромид (80 мас.% в толуоле, 0,99 мл, 8,86 ммоль) и смесь кипятят с обратным холодильником в течение 24 часов. Охлажденную реакционную смесь фильтруют и фильтрат выпаривают до желтого масла (3,15 г), которое очищают хроматографией (силикагель, 20%-50% этилацетата в гексане) с получением белого твердого вещества (2,37 г, 74%): Тпл 104-106°С; LC/MS (+APCI) m/z 364 (M+H).
Стадия С: Бензил (S)-1-метил-2-(7Н-пирано[2,3-g]индазол-1-ил)этилкарбамат
Продукт, полученный на стадии В (2,37 г, 6,53 ммоль), нагревают в мезитилене (40 мл) по процедуре, аналогичной описанной в примере 4, стадия В, с получением после очистки (силикагель, 20%-50% этилацетата в гексане) масла (1,01 г, 43%): LC/MS (+APCI) m/z 364 (M+H).
Стадия D: Бензил (S)-2-((8R*,9S*)-8,9-дигидрокси-8,9-дигидро-7Н-пирано[2,3-g]индазол-1-ил)-1-метилэтилкарбамат
Раствор продукта, полученного на стадии С (1,01 г, 2,78 ммоль), в смеси трет-бутилового спирта (20 мл) и воды (20 мл) добавляют при комнатной температуре к смеси трет-бутилового спирта (25 мл), воды (25 мл), AD-микс-α (4,2 г) и метансульфонамида (0,26 г, 2,8 ммоль). Реакционную смесь перемешивают в течение 24 часов и затем добавляют порошок сульфита натрия (5 г) и смесь перемешивают еще в течение часа. Далее к смеси добавляют насыщенный водный раствор бикарбоната натрия (150 мл) и экстрагируют ее этилацетатом (3 х 150 мл). Объединенные экстракты сушат (MgSO4) и выпаривают до масла (1,03 г), которое очищают хроматографией (силикагель, 50% этилацетат в гексане - этилацетат) с получением двух продуктов. Продукт А получают в виде белой пены (0,16 г, 15%): соотношение диастереомеров 4:1; LC/MS (+APCI) m/z 398 (M+H). Продукт В представляет собой бесцветное аморфное твердое вещество (0,18 г, 16%): Тпл.64-67°С; соотношение диастереомеров 1:4; LC/MS (+APCI) m/z 398 (M+H).
Стадия Е: (8R*,9S*)-1-[(2S)-2-аминопропил]-1,7,8,9-тетрагидропирано[2,3-g]индазол-8,9-диол
Раствор продукта А, полученного на стадии D (0,14 г, 0,354 ммоль), в ТГФ (20 мл) обрабатывают по методике стадии J примера 2 с получением свободного основания в виде бесцветного твердого вещества (50 мг, 54%): Тпл.115-117°С; [α]D=-79,3°С (с 0,27, ТГФ), соотношение диастереомеров 4:1; LC/MS (+APCI) m/z 264 (M+H) и 264 (M+H-H2O).
Пример 8
Дигидрохлорид (S)-2-(8,9-дигидро-7Н-пирано[2,3-g]индазол-1-ил)-1-метилэтиламина
Стадия А: (R)-1-(7Н-пирано[2,3-g]индазол-1-ил)пропан-2-ол
Смесь продукта, полученного на стадии В примера 4,(0,26 г, 0,76 ммоль), и фторида тетрабутиламмония (1 М 1,52 ммоль) в ТГФ (3 мл) перемешивают при температуре окружающей среды в течение 4 часов. Реакционную смесь добавляют к насыщенному водному раствору бикарбоната натрия (10 мл) и указанную смесь экстрагируют этилацетатом (3 х 5 мл). Объединенные экстракты сушат и выпаривают до остатка, который очищают хроматографией (силикагель, 10%-40% этилацетата в гексане) с получением спирта в виде масла (0,14 г, 81%): LC/MS (+APCI) m/z 231 (M+H).
Стадия В: 1-((S)-2-азидопропил)-1,7-дигидропирано[2,3-g]индазол
К раствору продукта, полученного на стадии А (0,14 г, 0,61 ммоль), и триэтиламина (0,18 мл, 1,8 ммоль) в безводном ТГФ (50 мл) при 0°С добавляют метансульфоновый ангидрид (0,16 г, 0,92 ммоль). Смесь перемешивают в течение 10 минут и добавляют азид натрия (0,40 г, 6,1 ммоль), растворитель выпаривают и добавляют безводный ДМФ (50 мл) и затем нагревают при 110°С в течение 3 часов. Смесь охлаждают, выливают в насыщенный раствор бикарбоната натрия (80 мл) и экстрагируют этилацетатом (3 х 60 мл). Объединенные экстракты сушат, фильтруют и выпаривают до остатка, который очищают хроматографией (силикагель, 10% этилацетат в гексане) с получением масла (0,06 г, 39%): LC/MS (+APCI) m/z 256 (M+H).
Стадия С: Дигидрохлорид (S)-2-(8,9-дигидро-7Н-пирано[2,3-g]индазол-1-ил)-1-метилэтиламина
Раствор продукта, полученного на стадии В, в метаноле обрабатывают по методике стадии J примера 2 с получением желтоватого твердого вещества (0,07 г, 77%): Тпл.>120°С; LC/MS (+APCI) m/z 232 (M+H). Продукт в виде свободного основания представляет собой бесцветное твердое вещество: Тпл. 95-98°С; [α]D=-66,7°С (с 0,445, ТГФ). Аналитические данные для C13H17N3O: Расчет: C - 67,51: H - 7,41; N - 18,17. Найдено: С - 67,47; Н - 7,51; N - 17,91.
Пример 9
(S)-1-метил-2-(7Н-пирано[2,3-g]индазол-1-ил)этиламин
К раствору продукта, полученного в примере 8 на стадии В (0,10 г, 0,39 ммоль), в сухом ТГФ (20 мл) при 0°С добавляют раствор алюмогидрида лития (0,39 мл, 1,56 ммоль 1М раствора в ТГФ) и смеси позволяют нагреться до комнатной температуры (1 час) при перемешивании. К реакционной смеси добавляют водный раствор гидроксида калия (2 М, 0,02 мл) и образовавшийся твердый материал удаляют фильтрованием. Фильтрат выпаривают до желтого масла (0,05 г, 56%): LC/MS (+APCI) m/z 230. Аналитические данные для C13H15N3O х 0,17 H2O: Расчет: C - 67,26: H - 6,60; N - 18,10. Найдено: С - 67,35; Н - 6,45; N - 17,76.
Пример 10
Трифторацетат 1-(S)-2-аминопропил)-1Н-пирано[2,3-g]индазол-7-она
Суспензию 1-((S)-2-аминометил)-1Н-индазол-6-ола (1,00 г, 5,23 ммоль) и яблочной кислоты (0,74 г, 5,5 ммоль) в концентрированной серной кислоте (3 мл) нагревают при 80°С в течение 48 часов и при 90°С в течение 24 часов. Реакционную смесь нейтрализуют фосфатом натрия (двуосновным, до pH 7) и добавляют насыщенный раствор соли (100 мл), после чего проводят экстракцию указанной смеси ТГФ (3 х 100 мл). Объединенные экстракты сушат (MgSO4) и выпаривают до желтого остатка (0,54 г), который очищают хроматографией с обращением фаз (С-18, вода/ацетонитрил, 0,1% трифторуксусная кислота) с получением желтого масла (70 мг): LC/MS (+APCI) m/z 244 (M+H).
Пример 11
9-амино-1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол
Стадия А: Бензил (S)-2-(8-бром-9-гидрокси-8,9-дигидро-7Н-пирано[2,3-g]индазол-1-ил)-1-метилэтилкарбамат
К раствору продукта, полученного в примере 7 на стадии C (1,35 г, 3,71 ммоль), в смеси диметилсульфоксида и воды (20 мл:2 мл) при 0°С добавляют N-бромсукцинимид (0,69г, 3,89 ммоль). Полученную смесь перемешивают в течение 2 часов, объединяют с водой (100 мл) и экстрагируют этилацетатом (3 х 100 мл). Объединенные экстракты сушат и концентрируют до остатка, который очищают хроматографией (силикагель, 20% этилацетат в гексане) с получением масла (1,18 г, 69%): LC/MS m/z 460, 462 (М+Н).
Стадия В: Бензил (S)-2-(9-азидо-8-бром-8,9-дигидро-7Н-пирано[2,3-g]индазол-1-ил)-1-метилэтилкарбамат
Смесь продукта, полученного на стадии А (0,31 г, 0,67 ммоль), и азида натрия (0,65 г, 10 ммоль) в диметилсульфоксиде (20 мл) нагревают при 80°С в течение 3 часов, охлаждают до комнатной температуры и экстрагируют этилацетатом. Объединенные экстракты сушат и концентрируют до остатка, который очищают хроматографией (силикагель, 30% этилацетат в гексане) с получением масла (0,26 г, 68%): LC/MS (+APCI) m/z 423 (M+H).
Стадия С: 9-амино-1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол
Продукт, полученный на стадии В (0,26 г, 0,30 ммоль), и Pd/C (10%, 0,026 г) смешивают с метанолом (5 мл) и помещают в атмосферу водорода на 18 часов. Смесь фильтруют и выпаривают с получением желтоватого твердого вещества (0,155 г, 96%): Тпл. 84-88°С; LC/MS (+APCI) m/z 263 (M+H).
Пример 12
1-((S)-2-аминопропил)-9-метокси-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол
Стадия А: Бензил (S)-2-(8-гидрокси-9-метокси-8,9-дигидро-7Н-пирано[2,3-g]индазол-1-ил)-1-метилэтилкарбамат
К раствору продукта, полученного в примере 11 на стадии А (0,46 г, 1,12 ммоль), в тетрагидрофуране (50 мл) добавляют 2 н. гидроксид натрия (6 мл). После перемешивания в течение 10 минут добавляют метанол (10 мл) и смесь перемешивают в течение 1 часа, после чего выпаривают до остатка, который смешивают с водой (50 мл) и экстрагируют этилацетатом (3 х 50 мл). Объединенные экстракты сушат над сульфатом магния и выпаривают до остатка, который очищают хроматографией (силикагель, 20%-35% этилацетата в гексане) с получением масла (0,45 г); LC/MS (+APCI) m/z 378 (M+H).
Стадия В: 1-((S)-2-аминопропил)-9-метокси-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол
К раствору продукта, полученного на стадии А (0,22 г, 0,58 ммоль), в метаноле (5 мл) добавляют трифторуксусную кислоту (5 мл) и смесь перемешивают при температуре окружающей среды в течение 2 дней. Далее смесь выпаривают до остатка, который перемешивают с 2 н. раствором хлористого водорода в этаноле (5 мл) и выпаривают с получением твердого вещества (0,21 г): Тпл.105-108°С; LC/MS (+APCI) m/z 278 (M+H). Приведенные ниже местные глазные композиции применимы согласно настоящему изобретения при введении 1-4 раза в день по рекомендации лечащего врача.
Пример 13
Пример 14
Пример 15
Пример 16
Другие варианты осуществления настоящего изобретения будут понятны специалисту в данной области на основе приведенного описания и практики осуществления настоящего изобретения, рассмотренной в настоящем материале. Следует иметь в виду, что настоящее описание и примеры следует рассматривать только как иллюстративные, поскольку сущность и объем настоящего изобретения определены формулой изобретения и ее эквивалентами.
Пример 17
(S)-1-метил-2-(3,7,8,9-тетрагидропирано-[3,2-е]-индазол-1-ил)этиламин
Стадия А: 3-((R)-2-гидроксипропил)-1Н-индазол-5-ол
К раствору (R)-1-(5-метокси-1Н-индазол-3-ил)пропан-2-ола (2,31 г, 11,2 ммоль) в безводном дихлорметане (50 мл), охлажденному при -78°С, добавляют 1 н. раствор трибромида бора в дихлорметане (33,6 мл, 33,6 ммоль). После завершения введения смеси позволяют нагреться до температуры окружающей среды и перемешивают в течение 2 ч. Смесь охлаждают (на бане со льдом) и гасят 20 мл воды. Дихлорметан выпаривают и остаток смешивают с этилацетатом (100 мл). К данному раствору добавляют небольшими порциями карбонат калия до тех пор, пока не прекратится выделение газа, и рН не станет примерно 8. Смесь экстрагируют этилацетатом (3×100 мл) и объединенные экстракты сушат и выпаривают досуха. Хроматографией на диоксиде кремния (градиент, 50-90% этилацетата в гексане) получают масло (1,50 г, 70%): LC/MS (+APCI) m/z 193 (М+Н).
Стадия В: (R)-1-(5-проп-2-инилокси-1Н-индазол-3-ил)-пропан-2-ол
К перемешиваемой смеси продукта со стадии А (1,50 г, 7,81 ммоль) в смеси этанола в воде (50 мл/10 мл) добавляют карбонат калия (1,62 г, 11,7 ммоль) и пропаргилбромид (80% в толуоле, 1,74 г, 11,7 ммоль). Смесь нагревают при 75°С в течение 2 ч в атмосфере азота, выпаривают досуха, смешивают с насыщенным водным раствором бикарбоната натрия (50 мл) и экстрагируют этилацетатом. Хроматографией на диоксиде кремния (градиент, 25-95% этилацетата в гексане) получают масло (1,25 г, 69%): LC/MS (+APCI) m/z 231 (М+Н).
Стадия С: 3-[(R)-2-(трет-бутилдиметилсиланилокси)пропил]-5-проп-2-инилокси-1Н-индазол
К перемешиваемому раствору продукта со стадии В (1,25 г, 5,43 ммоль) в безводном ТГФ (тетрагидрофуране) (50 мл) добавляют трет-бутилдиметилсилилхлорид (1,23 г, 8,15 ммоль), имидазол (0,74 г, 10,9 ммоль) и каталитическое количество DMAP (0,1 г). Смесь перемешивают в течение 6 ч и вводят дополнительное количество каждого из реагентов. Смесь перемешивают до утра и объединяют с насыщенным водным раствором бикарбоната натрия (100 мл) и экстрагируют этилацетатом. Хроматографией на диоксиде кремния (градиент, 2-30% этилацетата в гексане) получают масло (0,98 г, 52%). Данный продукт медленно разлагается при выдерживании во влажных условиях с получением желаемого продукта. Хроматографией смеси получают дополнительный желаемый продукт в виде масла (0,68 г, 36%, суммарно 88%): LC/MS (+APCI) m/z 345 (М+Н).
Стадия D: 1-[(R)-2-(трет-бутилдиметилсиланилокси)пропил]-3,7-дигидропирано-[3,2-е]-индазол
Смесь продукта со стадии С (1,66 г, 4,82 ммоль) в мезитилене (8 мл) помещают в реакционный сосуд, продувают газообразным азотом в течение 5 мин и герметизируют. Сосуд нагревают при 210°С в течение 1 ч, охлаждают до температуры окружающей среды и хроматографируют на диоксиде кремния (градиент, 2-15% этилацетата в гексане) с получением масла (1,31 г, 79%): LC/MS (+APCI) m/z 345 (М+Н).
Стадия Е: (R)-1-(3,7-дигидропирано-[3,2-е]-индазол-1-ил)пропан-2-ол
Смесь продукта со стадии D (0,30 г, 0,87 ммоль) и тетрабутиламмонийфторида (0,5 М, 2,62 мл, 1,31 ммоль) в ТГФ (30 мл) перемешивают при температуре окружающей среды в течение ночи и выпаривают досуха. Хроматографией на диоксиде кремния (градиент, 30-100% этилацетата в гексане) получают масло (0,165 г, 83%).
Стадия F: (S)-1-(2-азидопропил)-3,7-дигидропирано-[3,2-е]-индазол
К перемешиваемому раствору продукта со стадии Е (0,16 г, 0,70 ммоль) и триэтиламина (0,28 г, 0,39 мл, 7,51 ммоль) в безводном ТГФ (50 мл) при 0°С добавляют ангидрид метансульфокислоты (0,24 г, 1,4 ммоль). Смесь перемешивают в течение примерно 30 мин и вводят дополнительный 1 экв. ангидрида метансульфокислоты. Через 30 мин к реакционной смеси добавляют азид натрия (1,10 г, 7,0 ммоль). ТГФ выпаривают и безводный диметилсульфоксид (20 мл) добавляют к остатку, который нагревают при 90°С в течение 1 ч. Реакционную смесь охлаждают, выливают в насыщенный раствор бикарбоната натрия (80 мл) и экстрагируют этилацетатом (3×50 мл). Объединенные экстракты сушат (над MgSO4), фильтруют и выпаривают до масла. Хроматографией на диоксиде кремния (градиент, 10-20% этилацетата в гексане) получают масло (0,09 г, 51%): LC/MS (+APCI) m/z 256 (М+Н).
Стадия G: (S)-1-метил-2-(3,7,8,9-тетрагидропирано-[3,2-е]-индазол-1-ил)этиламиндигидрохлорид
Смесь продукта со стадии F (0,09 г) и палладия-на-углероде (10%, 0,01 г) в метаноле (20 мл) помещают в атмосферу водорода и перемешивают в течение ночи. Смесь очищают ВЭЖХ (HPLC) (высокоэффективная жидкостная хроматография) с обращенной фазой (градиент, ацетонитрил/вода, 0-55% с 0,1% трифторуксусной кислоты). Главные фракции объединяют, выпаривают, обрабатывают 2 н. HCl в этаноле (5 мл) и выпаривают досуха с получением аморфного твердого вещества (0,053 г, 58%): температура плавления 235-240°С; LC/MS (+APCI) m/z 232 (М+Н).
Пример 18
[1-((S)-2-аминопропил)-7,8-дигидро-1Н-фуро-[2,3-g]-индазол-7-ил]метанол
Стадия А: Трет-бутиловый эфир [1-[(R)-2-(трет-бутилдиметилсиланилокси)пропил]-7-гидроксиметил-1Н-индазол-6-ил-окси]уксусной кислоты
К смеси 1-[(R)-2-(трет-бутилдиметилсиланилокси)пропил]-1Н-индазол-6-ола (3,06 г, 10 ммоль) в ДМФА (DMF) (диметилформамид) (60 мл) добавляют формальдегид (37%, 2,43 мл, 30 ммоль) и 2 н. NaOH (10 ммоль). Смесь перемешивают до утра и добавляют трет-бутилбромацетат (2,42 мл, 2,93 г, 15 ммоль). Через 1 ч смесь смешивают с насыщенным водным раствором бикарбоната натрия (100 мл) и экстрагируют этилацетатом.
Хроматографией на диоксиде кремния (градиент, 5-25% этилацетата в гексане) получают масло (2,87 г, 64%): LC/MS (+APCI) m/z 451 (М+Н).
Стадия В: Трет-бутиловый эфир [1-(R)-2-(трет-бутилдиметилсиланилокси)пропил]-7-хлорметил-1Н-индазол-6-илокси]-уксусной кислоты
К перемешиваемому раствору продукта со стадии А (2,78 г, 6,18 ммоль) в безводном дихлорметане (100 мл) добавляют тионилхлорид (0,68 г, 9,27 ммоль). Через 1,5 ч добавляют избыток тионилхлорида (0,23 мл) и перемешивание продолжают в течение 20 мин.
Выпаривание данной смеси при высоком вакууме дает масло (2,90 г), которое используют в последующей реакции без дополнительной очистки.
Стадия С: Трет-бутиловый эфир [1-(R)-2-(трет-бутилдиметилсиланилокси)пропил]-7,8-дигидро-1Н-фуро-[2,3-g]-индазол-7-карбоновой кислоты
К раствору продукта со стадии В (2,90 г, 6,18 ммоль) в безводном 1-метил-2-пирролидоне (30 мл) добавляют гидрид натрия (60%, 0,371 г, 9,27 ммоль). Смесь нагревают при 50°С в течение ночи и выдерживают при температуре окружающей среды в течение 2 дней, охлаждают и выливают в насыщенный водный раствор бикарбоната натрия (100 мл) и экстрагируют этилацетатом.
Хроматографией на диоксиде кремния (градиент, 2-25% этилацетата в гексане) получают масло (0,48 г, 18%): LC/MS (+APCI) m/z 433 (М+Н).
Стадия D: Трет-бутиловый эфир 1-[(R)-2-гидроксипропил]-7,8-дигидро-1Н-фуро-[2,3-g]-индазол-7-карбоновой кислоты
К смеси продукта со стадии С (0,46 г, 1,06 ммоль) в ТГФ (50 мл) добавляют тетрабутиламмонийфторид (1 н. в ТГФ, 2,13 мл, 2,13 ммоль). Смесь объединяют с насыщенным водным раствором бикарбоната натрия (100 мл) и экстрагируют этилацетатом.
Хроматографией на диоксиде кремния (градиент, 20-70% этил ацетата в гексане) получают масло (0,39 г): LC/MS (+ESI) m/z 319 (М+Н).
Стадия Е: Трет-бутиловый эфир 1-[(S)-2-азидопропил]-7,8-дигидро-1Н-фуро-[2,3-g]-индазол-7-карбоновой кислоты
К раствору продукта со стадии D (0,39 г, 1,23 ммоль) и триэтиламина (0,49 г, 4,90 ммоль) в безводном ТГФ (30 мл) при 0°С добавляют ангидрид метансульфокислоты (0,34 г, 3,38 ммоль). Смесь перемешивают в течение 30 мин и вводят дополнительный ангидрид метансульфокислоты (0,17 г). Через 30 мин летучие компоненты выпариваются и к остатку добавляют азид натрия (1,22 г, 18,8 ммоль, 10 экв.) и безводный диметилсульфоксид (50 мл). Смесь нагревают при 80°С в течение 4 ч, охлаждают, выливают в насыщенный водный раствор бикарбоната натрия (80 мл) и экстрагируют этилацетатом (3×60 мл). Объединенные экстракты сушат (над MgSO4), фильтруют и выпаривают с получением масла.
Хроматографией на диоксиде кремния (градиент, 0-10% этилацетата в гексане) получают масло (0,22 г, 52%): LC/MS (+ESI) m/z 344 (М+Н).
Стадия F: [1-((S)-2-аминопропил)-7,8-дигидро-1Н-фуро-[2,3-g]-индазол-7-ил]метанолдигидрохлорид
К смеси продукта со стадии Е (0,21 г, 0,61 ммоль) в безводном ТГФ (50 мл) добавляют 1 М раствор литийалюминийгидрида в ТГФ (3,0 мл, 3,0 ммоль) с перемешиванием в атмосфере азота. Смесь перемешивают в течение 3 ч, охлаждают на льду и гасят добавлением метанола и затем 2 н. HCl. Выпаривание данной смеси дает остаток, который очищают ВЭЖХ с обращенной фазой (градиент ацетонитрила и воды, 0-50% с 0,1% трифторуксусной кислоты). Желаемые фракции объединяют, концентрируют, обрабатывают 2 н. HCl в этаноле (10 мл) и выпаривают с получением не совсем белого твердого вещества (0,13 г, 59%), которое сушат в высоком вакууме при 78°С до утра: LC/MS (+APCI) m/z 248 (М+Н).
Анализ: Рассчитано для C13H19Cl2N3O2·0,5 C2H5OH·H2O: С, 46,92; Н, 6,72; N, 11,73. Определено: С, 47,25; Н, 6,41; N, 11,50.
Схема 11
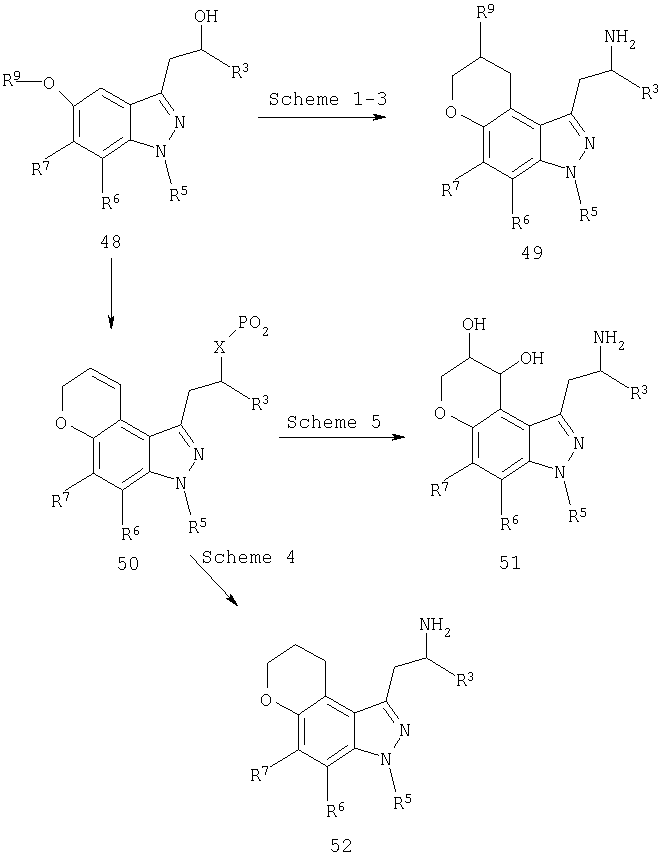
Некоторые желаемые замещенные 7,8-дигидро-фурано-[2,3-g]-индазолы формулы I могут быть получены из соответственно замещенного 1H-индазол-6-ола (53), как описано в последовательности синтеза, представленной на схеме 12. Кроме того, применение стандартных превращений функциональных групп, хорошо известных в технике, например, при модификации соединения 60, может дать еще другие соединения формулы I.
Схема 12
При использовании методик, описанных на вышеприведенных схемах, в примерах, и хорошо известных методик специалист в данной области техники может получить соединения, рассмотренные здесь.
AL-39695A: Пример 20
AL-40105A: Пример 21
AL-38035: Пример 22
Пример 20
(8R)-1-[(2S)-2-аминопропил]-1,7,8,9-тетрагидропирано-[2,3-g]-индазол-8-илацетат
Стадия A: (8R)-1-[(2S)-2-азидопропил]-1,7,8,9-тетрагидропирано-[2,3-g]-индазол-8-илметилкарбамат
К перемешиваемому раствору продукта из примера 4, стадия D (0,22 г, 0,81 ммоль) и триэтиламина (0,224 мл, 1,6 ммоль) в безводном дихлорметане (30 мл) в атмосфере азота при 0°С добавляют с помощью шприца ацетилхлорид (0,069 мл, 0,97 ммоль, 1,2 экв.). Через 30 мин баню со льдом удаляют и реакционную смесь перемешивают при температуре окружающей среды в течение 18 ч. Летучие продукты выпаривают и остаток растворяют в этилацетате, фильтруют и концентрируют с получением масла (0,12 г, 55%), которое превращается в тускло-белое твердое вещество при выстаивании: температура плавления 88-89°С; данные 1H-ЯМР соответствуют желаемому ацетату.
Стадия В: (8R)-1-[(2S)-2-аминопропил]-1,7,8,9-тетрагидропирано-[2,3-g]-индазол-8-илацетат
Смесь продукта со стадии А (0,12 г, 0,38 ммоль) и Pd/C (10%, 0,012 г) в метаноле (10 мл) перемешивают в атмосфере азота в течение 18 ч, фильтруют и выпаривают досуха с получением масла. Масло растворяют в дихлорметане и обрабатывают 2 н. HCl в этаноле. Выпаривание смеси дает желтоватый порошок (0,12 г): LC/MS (+APCI) m/z 290 (М+Н). Анализ. Рассчитано для С15Н19N3О3·HCl·1,2 Н2O: Н 6,50; С 51,86; N 12,21.
Определено: Н 6,58; С 51,88; N, 11,93.
Пример 21
(8R)-1-[(2S)-2-аминопропил]-1,7,8,9-тетрагидропирано-[2,3-g]-индазол-8-илметилкарбамат
Стадия А: Бензиловый эфир [(S)-2-((R)-8-гидрокси-8,9-дигидро-7Н-пирано-[2,3-g]-индазол-1-ил)-1-метилэтил]карбаминовой кислоты
К перемешиваемому, раствору продукта из примера 4 (16,2 г, 65,6 ммоль) в этилацетате (100 мл) при 0°С добавляют насыщенный водный раствор бикарбоната натрия (150 мл) и бензилхлорформиата (95%, 9,32 мл, 65,6 ммоль). Смесь перемешивают при температуре окружающей среды в течение 3 ч и органический слой отделяют, сушат и концентрируют с получением масла. Тонкослойной хроматографией (градиент, 30-50% этилацетата в гексане) получают твердое вещество (16,2 г, 65%): LC/MS (+APCI) m/z 382 (М+Н).
Стадия В: (R)-1-((S)-2-бензилоксикарбониламинопропил)-1,7,8,9-тетрагидропирано-[2,3-g]-индазол-8-ил-овый эфир имидазол-1-карбоновой кислоты
К перемешиваемому раствору продукта со стадии А (2,0 г, 5,2 ммоль) в безводном дихлорметане (100 мл) добавляют карбонилдиимидазол (1,1 г, 6,8 ммоль, 1,3 экв.) при температуре окружающей среды. Через 1 ч добавляют водный раствор бикарбоната натрия, органическую часть выпаривают, а водную часть экстрагируют этилацетатом. Экстракт выпаривают с получением белого твердого вещества: (2,4 г, 98%): LC/MS (+ESI) m/z 476 (М+Н).
Стадия С: Бензиловый эфир [(S)-1-метил-2-((R)-8-метилкарбамоилокси-8,9-дигидро-7Н-пирано-[2,3-g]-индазол-1-ил)-этил]карбаминовой кислоты
К раствору соединения со стадии В (0,30 г, 0,63 ммоль) в безводном тетрагидрофуране (10 мл) добавляют метиламин (2 М в МеОН, 0,47 мл, 0,95 ммоль) и смесь нагревают при 40°С в течение 1 ч. Реакционную смесь концентрируют и очищают тонкослойной хроматографией (градиент, 20-80% этилацетат/гексан) с получением твердого вещества (0,13 г, 47%): температура плавления 186-188°С; LC/MS (+APCI) m/z 439 (М+Н).
Стадия D: (8R)-1-[(2S)-2-аминопропил]-1,7,8,9-тетрагидропирано-[2,3-g]-индазол-8-илметилкарбамат
Смесь продукта со стадии С (0,13 г, 0,30 ммоль) и Pd/C (10%, 0,02 г) в смеси метанола (20 мл) и этилацетата (20 мл) перемешивают в атмосфере азота в течение 3 ч, фильтруют и выпаривают с получением масла. Масло растворяют в этаноле и обрабатывают 2 н. HCl в этаноле. Выпаривание данной смеси дает неочищенное твердое вещество. Твердое вещество растворяют в метаноле (1 мл) и добавляют этилацетат (около 10 мл); данный раствор оставляют стоять до утра при температуре окружающей среды. Твердое вещество собирают при фильтрации и сушат с получением желаемого продукта (0,089 г, 88%): LC/MS (+APCI) m/z 305 (М+Н). Анализ. Рассчитано для C15H19N4O3·HCl: С 52,86; Н 6,21; N 16,44. Определено: С 52,77; Н 6,25; N 16,21.
Пример 22
1-[(2S)-2-аминопропил]-1,7,8,9-тетрагидропирано-[2,3-g]-индазол-8-илкарбамат
Стадия А: 1-[(2S)-2-азидопропил]-1,7,8,9-тетрагидропирано-[2,3-g]-индазол-8-илкарбамат
Раствор хлорсульфурилизоцианата (0,064 мл, 0,73 ммоль) в диэтиловом эфире (3 мл) при комнатной температуре обрабатывают раствором продукта из примера 3, стадия А (100 мг, 0,35 ммоль) в диэтиловом эфире (4 мл). Реакционную смесь перемешивают при температуре окружающей среды в течение одного часа и реакцию гасят водой (10 мл), перемешивают в течение 10 мин и нейтрализуют водным раствором NaH2PO4. Смесь экстрагируют этилацетатом (3×10 мл) и объединенные органические слои сушат (над сульфатом магния) и выпаривают до желтого твердого вещества (110 мг). После очистки тонкослойной хроматографией (градиент, диоксид кремния, гексан/этилацетат) получают бесцветное твердое вещество (60 мг, 52%): температура плавления 141-145°С; LC/MS m/z 317.
Стадия В: 1-[(2S)-2-аминопропил]-1,7,8,9-тетрагидропирано-[2,3-g]-индазол-8-илкарбамат
Раствор продукта со стадии А (90 мг, 0,28 ммоль) в метаноле (10 мл) и ТГФ (2 мл) обрабатывают 10% палладия на углероде (10 мг) и водородом (1 ати) при перемешивании при комнатной температуре в течение 18 ч. Реакционную смесь фильтруют и выпаривают с получением желтого масла (70 мг, 86%): LC/MS m/z 291. Данные 1H-ЯМР соответствуют структуре.
5-HT2A рецепторное связывание и функциональные данные
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ N-(1H-ИНДАЗОЛ-4-ИЛ) ИМИДАЗОЛ [1,2-А]ПИРИДИН-3- КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ РЕЦЕПТОРНОЙ ТИРОЗИНКИНАЗЫ III ТИПА | 2011 |
|
RU2591195C2 |
| АЗАИНДОЛЫ В КАЧЕСТВЕ АКТИВАТОРОВ ГЛЮКОКИНАЗЫ | 2010 |
|
RU2593369C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОАРИЛОМ ПИРИДИНЫ И СПОСОБЫ ПРИМЕНЕНИЯ | 2017 |
|
RU2756743C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ГИДАНТОИНА В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕИНАЗ | 2005 |
|
RU2376301C2 |
| БИЦИКЛИЧЕСКИЕ АРИЛЬНЫЕ МОНОБАКТАМОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2017 |
|
RU2733402C2 |
| ПРОИЗВОДНЫЕ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ И ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНЫХ ПУТЕЙ | 2001 |
|
RU2265011C2 |
| БЕНЗОПРОИЗВОДНЫЕ С ШЕСТИЧЛЕННЫМ КОЛЬЦОМ В КАЧЕСТВЕ ИНГИБИТОРА DPP-4 И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2702644C2 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ 637: ПИРИДОПИРИМИДИНДИОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE4 | 2008 |
|
RU2479584C2 |
| МОДУЛЯТОРЫ ПИРУВАТКИНАЗЫ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2797518C2 |
| БИАРИЛЬНЫЕ МОНОБАКТАМНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2016 |
|
RU2746129C2 |
Изобретение относится к новым пираноиндазолам формулы I:
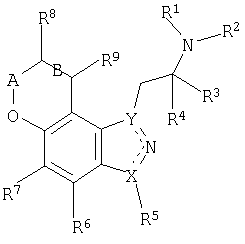
где R1 и R2 независимо выбирают из водорода или алкильной группы; R3 и R4 представляют собой независимо водород или алкильную группу; R5, R6 и R7 обозначают водород, R8 и R9 обозначают водород, гидроксил, алкокси, NR10R11, OC(=O)NR1R2, ОС(=O)С1-4алкил или алкилтиол; R10 и R11 обозначают водород, А обозначает (СН2)n, С=O; В обозначает одинарную или двойную связь; n=0-2; Y обозначают N, Х обозначает С; и пунктирная линия обозначает соответствующую одинарную или двойную связь. Изобретение также относится к фармацевтической композиции на основе соединений формулы I, способу регулирования нормального или повышенного внутриглазного давления, способу лечения глаукомы и способу блокирования или связывания серотониновых рецепторов. Технический результат - получения новых пираноиндазолов, обладающих ценным фармацевтическим действием. 5 н. и 9 з.п. ф-лы, 3 табл.
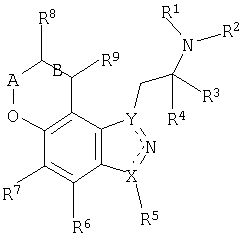
где R1 и R2 независимо выбирают из водорода или алкильной группы;
R3 и R4 представляют собой независимо водород или алкильную группу или
R5, R6 и R7 обозначают водород;
R8 и R9 обозначают водород, гидроксил, алкокси, NR10R11, ОС(=O)NR1R2, ОС(=O)С1-4алкил или алкилтиол, где R10 и R11 обозначают водород;
А обозначает (СН2)n или С=O;
B обозначает или одинарную или двойную связь;
n=0-2;
Y обозначают N, Х означает С; пунктирная линия обозначает соответствующую одинарную или двойную связь.
R3 и R4 выбирают независимо из водорода или С1-4алкила;
R8 и R9 выбирают из водорода, гидроксила, С1-6алкокси или NR10R11,
А обозначает (СН2)n.
R3 обозначает C1-2алкил;
R4 обозначает водород;
R8 и R9 выбирают независимо из водорода, гидроксила, C1-6алкокси или NR10R11;
А обозначает (СН2)n,
В обозначает одинарную связь;
n=1.
1-(2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
(R)-1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
(S)-1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
1-((S)-2-аминопропил)-3-метил-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
1-(S)-1-пирролидин-2-илметил-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
1-((S)-2-аминопропил)-5-фтор-1,7,8,9-тетоагидропирано[2,3-g]индазол-8-ол;
(R)-1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-иламин;
[1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ил]диметиламин;
1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8,9-диол;
или их сочетания.
R3 и R4 выбирают независимо из водорода или С1-4алкила;
R8 и R9 выбирают из водорода, гидроксила, C1-6алкокси или NR10R11;
А обозначает (СН2)n.
1-(2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
(R)-1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
(S)-1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
1-((S)-2-аминопропил)-3-метил-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
1-(S)-1-пирролидин-2-илметил-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
1-((S)-2-аминопропил)-5-фтор-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
(R)-1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-иламин;
[1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ил]диметиламин;
1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8,9-диол;
или их сочетания.
1-(2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
(R)-1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
(S)-1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
1-((S)-2-аминопропил)-3-метил-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
1-(S)-1-пирролидин-2-илметил-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
1-((S)-2-аминопропил)-5-фтор-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ол;
(R)-1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-иламин;
[1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8-ил]диметиламин;
1-((S)-2-аминопропил)-1,7,8,9-тетрагидропирано[2,3-g]индазол-8,9-диол;
или их сочетания.
| ПРОИЗВОДНЫЕ ПИРАНО-ХИНОЛИНОВ, СПОСОБ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2125573C1 |
| US 5561150 А, 01.10.1996 | |||
| WO 9818458 A1, 07.05.1998 | |||
| US 5646173 А, 08.07.1997 | |||
| US 5494928 А, 27.02.1996. | |||

