ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новым биарильным монобактамным соединениям, способам их получения и к их применению в качестве терапевтических агентов. Более конкретно, настоящее изобретение относится к биарильным монобактамным соединениям и к их применению в качестве антибиотиков для лечения бактериальных инфекций.
УРОВЕНЬ ТЕХНИКИ
Использование антибиотиков для лечения бактериальных инфекций является одним из самых больших медицинских достижений 20-го века. Однако за последние несколько десятилетий во всем мире стали появляться бактерии, устойчивые ко многим антибиотикам, угрожая эффективности антибактериальной терапии. Только в Соединенных Штатах по меньшей мере 23000 человек каждый год умирают непосредственно в результате инфекций, вызванных устойчивыми к антибиотикам бактериями, а также многие другие люди умирают от обычных патологий, осложненных подобными инфекциями (Antibiotic Resistance Threats in the United States, 2013, Centers for Disease Control, Atlanta, Georgia). Новые антибиотики необходимы для борьбы с текущей и будущей угрозой со стороны бактерий с множественной лекарственной устойчивостью.
Для лечения тяжелых бактериальных инфекций наиболее часто используются β-лактамные антибиотики. Они включают карбапенемы, цефалоспорины, пенициллины и монобактамы. Как было отмечено для других классов антибиотиков, существует устойчивость к β-лактамам. Для большинства грамотрицательных бактерий, эта устойчивость в основном связана с экспрессией β-лактамаз, ферментов, которые гидролизуют β-лактамные соединения. Существует 4 различных класса β-лактамаз (A, B, C и D), способных гидролизовать перекрывающиеся, но различные подклассы β-лактамов (Drawz & Bonomo, Clin. Micro. Rev., 2010, 23:160-201). Хотя β-лактамазы класса В, также известные как металло-β-лактамазы (MBL), не относятся к наиболее распространенным β-лактамазам, найденных в клинике, однако частота и случаи их экспрессии возрастают, что представляют собой значительную медицинскую угрозу, поскольку (I) MBL обладают способностью гидролизовать все β-лактамы, кроме монобактамов, и (II) в отличие от β-лактамаз классов A и C, не известно каких-либо ингибиторов, действующих в отношении MBL.
Монобактам, такой как азтреонам, был впервые одобрен в США в 1986 году для лечения аэробных грамотрицательных бактериальных инфекций, и он остается единственным из монобактамов, используемых в США в настоящее время. Тем не менее, азтреонам обладает неудовлетворительной активностью против штаммов Pseudomonas и Acinetobacter. Поскольку монобактамы по своей природе устойчивы к гидролизу под действием MBL, несколько компаний приступили к разработке новых монобактамных соединений для лечения инфекций, вызванных грамотрицательными бактериями. Монобактамное соединение, содержащее фрагмент сидерофора, раскрыто в WO 2007/065288, WO 2012/073138, J. Medicinal Chemistry 56: 5541-5552 (2013) и Bioorganic and Medicinal Chemstry Letters 22:5989 (2012).
Публикация патентной заявки США US 2014/0275007 раскрывает оксамазиновые монобактамы и их применение в качестве антибактериальных агентов. Публикация патентной заявки США US 2015/0266867 также раскрывает новые монобактамные соединения, используемые в качестве антибактериальных агентов. Публикация международной патентной заявки WO 2013/110643 раскрывает новые замещенные амидиновые монобактамные производные и их применение в качестве антимикробных агентов.
Для преодоления множественной лекарственной устойчивости сохраняется потребность в новых антибиотиках. Соединения, описанные в данном изобретении, были разработаны в качестве ответа на эту медицинскую потребность, где такие соединения можно вводить отдельно или в сочетании с подходящим ингибитором β-лактамазы.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Изобретение относится к области разработки и синтеза ряда биарильных монобактамных аналогов, которые представляют собой новый класс высокоактивных антибиотиков, эффективных против широкого спектра грамотрицательных бактерий. Эти соединения и их фармацевтически приемлемые соли могут быть использованы в клинике в качестве терапевтических средств для лечения различных инфекций, вызванных грамотрицательными бактериями, включая штаммы, которые устойчивы ко многим лекарственным средствам. Соединения могут быть использованы отдельно или в комбинации с соответствующим ингибитором β-лактамазы. Более конкретно, настоящее изобретение включает соединение формулы I:
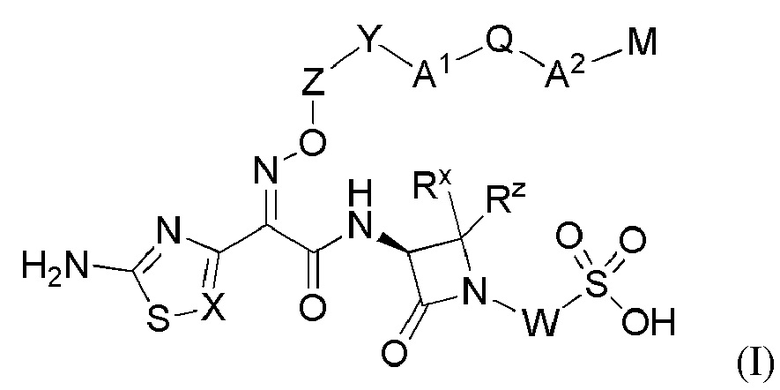
или его фармацевтически приемлемую соль, где:
W представляет собой связь или O;
Rx и Rz независимо представляют собой водород, -SC1-C3алкил, C1-C3алкил, -(C1-C3алкилен)nOC1-C3алкил или -(C1-C3алкилен)nNC1-C3алкил, где -SC1-C3алкил, C1-C3алкил, -(C1-C3алкилен)nOC1-C3алкил и -(C1-C3алкилен)nNC1-C3алкил необязательно замещены 1-7 атомами фтора;
или, альтернативно, Rx и Rz, вместе с атомом углерода, к которому они присоединены, образуют моноциклический C4-C7циклоалкил или моноциклический C4-C7гетероциклоалкил с 1, 2 или 3 гетероатомами в кольце, независимо выбранными из N, O и S, где указанный C4-C7циклоалкил и указанный C4-C7гетероциклоалкил необязательно замещены 1-3 заместителями, независимо выбранными из -F, -OH и -OC1-C3алкила;
X представляет собой N или CR1;
R1 представляет собой водород, C1-C3алкил, или галоген; где указанный C1-C3алкил необязательно замещен 1-3 Ra;
в каждом случае Ra независимо представляет собой водород, галоген, C1-C3алкил, -NRcRd или -ORe;
Z представляет собой C1-C3алкилен, необязательно замещенный 1-3 Rb;
в каждом случае Rb независимо представляет собой -C1-C8алкил, -C3-C7циклоалкил, -C(O)ORe, -C(O)NRcRd, тетразолил, оксадиазолонил, HetA, AryA, -S(O)mRe, -S(O)mNRcRd или -P(O)(Re)p, где указанный -C1-C8алкил и указанный -C3-C7циклоалкил необязательно замещены 1-3 Ra;
HetA представляет собой 4-6-членное насыщенное или мононенасыщенное моноциклическое кольцо с 1, 2 или 3 гетероатомами в кольце, независимо выбранными из N, N в виде четвертичной соли, O и S, необязательно замещенное 1-4 R4;
AryA представляет собой 5-6-членное моноциклическое ароматическое кольцо с 0, 1, 2 или 3 кольцевыми гетероатомами, независимо выбранными из N, N в виде четвертичной соли, O и S, необязательно замещенное 1-4 R4;
Y представляет собой связь, O, NR2, S или CH2;
R2 представляет собой водород, -C1-C3алкил, -C(O)Re, -C(O)NRcRd, -S(O)mRe или -S(O)mNRcRd, где указанный -C1-C3алкил необязательно замещен 1-3 Ra;
A1 представляет собой AryA;
A2 представляет собой -(CH2)nN(R3)2-, C3-C7циклоалкил, AryC или HetC, где указанный C3-C7циклоалкил необязательно замещен 1-4 R4;
AryC представляет собой 5-6-членное моноциклическое ароматическое кольцо с 0, 1, 2 или 3 кольцевыми гетероатомами, независимо выбранными из N, N в виде четвертичной соли, O и S, необязательно замещенное 1-4 R4;
HetC представляет собой 4-7-членное насыщенное или мононенасыщенное моноциклическое кольцо с 1, 2 или 3 гетероатомами в кольце, независимо выбранными из N, N в виде четвертичной соли, O и S, необязательно замещенное 1-4 R4;
в каждом случае R4 независимо представляет собой: -C1-C8алкил, -C2-C8алкенил, -C2-C8алкинил, галоген, -ORe, -S(O)mRe, -S(O)mNRcRd, -C(O)Re, -OC(O)Re, -C(O)ORe, -CN, -C(O)NRcRd, -NRcRd, -(CH2)nNRcRd; -NRcC(O)Re, -NRcC(O)ORe, -NRcC(O)NRcRd, -NRcS(O)mRe, =NH, -CF3, -OCF3, -OCHF2, -C3-C6циклоалкил, -O-C3-C6циклоалкил, -C1-C10алкилен-C3-C6циклоалкил, -O-C1-C10 алкилен-C3-C6циклоалкил, HetB, -O-HetB, -C1-C10алкилен-HetB, -O-C1-C10алкилен-HetB, AryA, -O-AryA, -C1-C10алкилен-AryA или -O-C1-C10алкилен-AryA, где каждый R4 является незамещенным, или он замещен 1-4 зместителями, выбранными из галогена, -C1-C6алкила и -(CH2)nNRcRd, или где R4 и M, вместе с атомами, к которым они присоединены, образуют 4-7-членный циклогетероалкил, необязательно содержащий 1-2 дополнительных гетероатома, независимо выбранных из O, S и -NRg;
HetB представляет собой 3-6-членное насыщенное или мононенасыщенное моноциклическое кольцо с 1, 2 или 3 гетероатомами в кольце, независимо выбранными из N, N в виде четвертичной соли, O и S, необязательно замещенное 1-3 Ra;
Q представляет собой связь, CH2, O, S, -(CH2)nNR3- или -NR3(CH2)n-, где каждый CH2 является незамещенным, или он замещен 1-2 заместителями, выбранными из галогена, -C1-C6алкила, ORe и -(CH2)nNRcRd;
R3 представляет собой водород или -C1-C3алкил, где указанный -C1-C3алкил необязательно замещен 1-3 Ra;
M представляет собой R5, -NHR5, -N(R5)2, -OR5, -(CH2)nR5, -C(O)R5, -C(NH)R5 или -S(O)mR5;
R5 представляет собой H, C2-C10алкил, C3-C7циклоалкил, C1-C6алкил-C3-C7циклоалкил, HetB, AryB или -NH(C1-C6алкил), где указанный C1-C6алкил, указанный C2-C10алкил и указанный C3-C7циклоалкил необязательно замещены 1-4 R6;
AryB представляет собой 5-6-членное моноциклическое ароматическое кольцо с 0, 1, 2 или 3 кольцевыми атомами, независимо выбранными из N, O и S, необязательно замещенное 1-4 R4;
в каждом случае R6 независимо выбран из группы, состоящей из: галогена, -ORe, -S(O)mRe, -S(O)mNRcRd, -C(O)Re, -OC(O)Re, -C(O)ORe, -CN, -C(O)NRcRd, -C(NH)NRcRd, -NRcRd, -(CH2)nNRcRd, -N(Rc)(C(O)Re), -N(Rc)(C(O)ORe), -N(Rc)(C(O)NRcRd), -N(Rc)(S(O)mRe) и HetB;
в каждом случае Rc и Rd независимо представляют собой: водород, -C1-C10алкил, -C2-C10алкенил, -C3-C6циклоалкил, -C1-C10алкилен-C3-C6 циклоалкил, HetA, -C1-C10алкилен-HetB, AryB, -C1-C10алкилен-AryB и -C1-C10алкилен-HetB, или, альтернативно, Rc и Rd вместе с атомом азота, к которому они присоединены, образуют 4-7-членный циклогетероалкил, необязательно содержащий 1-2 дополнительных гетероатома, независимо выбранных из O, S и -NRg, и где каждый Rc и Rd необязательно замещен 1-3 Rf;
в каждом случае Re независимо представляет собой: водород, -C1-C10алкил, -C2-C10алкенил, -OH, -OC1-C4алкил, -C3-C6циклоалкил, -C1-C10алкилен-C3-C6циклоалкил, HetB, -C1-C10алкилен-HetB, AryB, -C1-C10алкилен-AryB или -C1-C10алкилен-HetB; где каждый Re необязательно замещен 1-3 Rh;
в каждом случае Rf независимо представляет собой: галоген, -C1-C10алкил, -OH, -OC1-C4алкил, -S(O)mC1-C4алкил, -CN, -CF3, -OCHF2, -OCF3, или NH2, где указанный -C1-C10алкил необязательно замещен 1-3 заместителями, независимо выбранными из -OH, галогена, циано и -S(O)2CH3;
в каждом случае Rg независимо представляет собой: водород, -C(O)Re и -C1-C10 алкил, где указанный -C1-C10алкил необязательно замещен 1-5 атомами фтора;
в каждом случае Rh независимо представляет собой: галоген, -C1-C10алкил, -OH, -OC1-C4алкил, -S(O)mC1-C4алкил, -CN, -CF3, -OCHF2 или -OCF3; где указанный -C1-C10алкил необязательно замещен 1-3 заместителями, независимо выбранными из -OH, галогена, циано или -S(O)2CH3;
каждый n независимо равен 0, 1, 2, 3 или 4;
каждый m независимо равен 0, 1 или 2, и
каждый p независимо равен 1 или 2.
Настоящее изобретение также включает соединение формулы I:
или его фармацевтически приемлемую соль, где:
W представляет собой связь или O;
Rx и Rz независимо представляют собой водород, -SC1-C3алкил, C1-C3алкил, -(C1-C3алкилен)nOC1-C3алкил или -(C1-C3алкилен)nNC1-C3алкил, где -SC1-C3алкил, C1-C3алкил, -(C1-C3алкилен)nOC1-C3алкил и -(C1-C3алкилен)nNC1-C3алкил необязательно замещены 1-7 атомами фтора;
или, альтернативно, Rx и Rz, вместе с атомом углерода, к которому они присоединены, образуют моноциклический C4-C7циклоалкил или моноциклический C4-C7гетероциклоалкил с 1, 2, или 3 гетероатомами в кольце, независимо выбранными из N, O и S, где указанный C4-C7циклоалкил и указанный C4-C7гетероциклоалкил необязательно замещены 1-3 заместителями, независимо выбранными из -F, -OH и -OC1-C3алкила;
X представляет собой N или CR1;
R1 представляет собой водород, C1-C3алкил, или галоген; где указанный C1-C3алкил необязательно замещен 1-3 Ra;
в каждом случае Ra независимо представляет собой водород, галоген, C1-C3алкил, -NRcRd или -ORe;
Z представляет собой C1-C3алкилен, необязательно замещенный 1-3 Rb;
в каждом случае Rb независимо представляет собой -C1-C8алкил, -C3-C7циклоалкил, -C(O)ORe, -C(O)NRcRd, тетразолил, оксадиазолонил, HetA, AryA, -S(O)mRe, -S(O)mNRcRd или -P(O)(Re)p, где указанный -C1-C8алкил и указанный -C3-C7циклоалкил необязательно замещены 1-3 Ra;
HetA представляет собой 4-6-членное насыщенное или мононенасыщенное моноциклическое кольцо с 1, 2 или 3 гетероатомами в кольце, независимо выбранными из N, N в виде четвертичной соли, O и S, необязательно замещенное 1-4 R4;
AryA представляет собой 5-6-членное моноциклическое ароматическое кольцо с 0, 1, 2 или 3 кольцевыми атомами, независимо выбранными из N, N в виде четвертичной соли, O и S, необязательно замещенное 1-4 R4;
Y представляет собой связь, O, NR2, S или CH2;
R2 представляет собой водород, -C1-C3алкил, -C(O)Re, -C(O)NRcRd, -S(O)mRe или -S(O)mNRcRd, где указанный -C1-C3алкил необязательно замещен 1-3 Ra;
A1 представляет собой AryA;
A2 представляет собой C3-C7циклоалкил, AryC или HetC, где указанный C3-C7циклоалкил необязательно замещен 1-4 R4;
AryC представляет собой 5-6-членное моноциклическое ароматическое кольцо с 0, 1, 2 или 3 кольцевыми атомами, независимо выбранными из N, N в виде четвертичной соли, O и S, необязательно замещенное 1-4 R4;
HetC представляет собой 4-7-членное насыщенное или мононенасыщенное моноциклическое кольцо с 1, 2 или 3 гетероатомами в кольце, независимо выбранными из N, N в виде четвертичной соли, O и S, необязательно замещенное 1-4 R4;
в каждом случае R4 независимо представляет собой: -C1-C8алкил, -C2-C8алкенил, -C2-C8алкинил, галоген, -ORe, -S(O)mRe, -S(O)mNRcRd, -C(O)Re, -OC(O)Re, -C(O)ORe, -CN, -C(O)NRcRd, -NRcRd, -NRcC(O)Re, -NRcC(O)ORe, -NRcC(O)NRcRd, -NRcS(O)mRe, =NH, -CF3, -OCF3, -OCHF2, -C3-C6циклоалкил, -O-C3-C6циклоалкил, -C1-C10алкилен-C3-C6циклоалкил, -O-C1-C10 алкилен-C3-C6циклоалкил, HetB, -O-HetB, -C1-C10алкилен-HetB, -O-C1-C10алкилен-HetB, AryA, -O-AryA, -C1-C10алкилен-AryA или -O-C1-C10алкилен-AryA;
HetB представляет собой 3-6-членное насыщенное или мононенасыщенное моноциклическое кольцо с 1, 2 или 3 гетероатомами в кольце, независимо выбранными из N, N в виде четвертичной соли, O и S, необязательно замещенное 1-3 Ra;
Q представляет собой связь, CH2, O, S, -(CH2)nNR3- или -NR3(CH2)n-;
R3 представляет собой водород или -C1-C3алкил, где указанный -C1-C3алкил необязательно замещен 1-3 Ra;
M представляет собой R5, -NR5, -OR5, -(CH2)nR5, -C(O)R5, -CH(NH)R5 или -S(O)mR5;
R5 представляет собой C2-C10алкил, C3-C7циклоалкил, HetB, или AryB, где указанный C2-C10алкил и указанный C3-C7циклоалкил необязательно замещены 1-4 R6;
AryB представляет собой 5-6-членное моноциклическое ароматическое кольцо с 0, 1, 2 или 3 кольцевыми атомами, независимо выбранными из N, O и S, необязательно замещенное 1-4 R4;
в каждом случае R6 независимо выбран из группы, состоящей из: галогена, -ORe, -S(O)mRe, -S(O)mNRcRd, -C(O)Re, -OC(O)Re, -C(O)ORe, -CN, -C(O)NRcRd, -C(NH)NRcRd, -NRcRd, -N(Rc)(C(O)Re), -N(Rc)(C(O)ORe), -N(Rc)(C(O)NRcRd) и -N(Rc)(S(O)mRe);
в каждом случае Rc и Rd независимо представляют собой: водород, -C1-C10алкил, -C2-C10алкенил, -C3-C6циклоалкил, -C1-C10алкилен-C3-C6циклоалкил, HetA, -C1-C10алкилен-HetB, AryB, -C1-C10алкилен-AryB и - C1-C10алкилен-HetB, или, альтернативно, Rc и Rd, вместе с атомом азота, к которому они присоединены, образуют 4-7-членный циклогетероалкил, необязательно содержащий 1-2 дополнительных гетероатома, независимо выбранных из O, S и -NRg, и где каждый Rc и Rd необязательно замещен 1-3 Rf;
в каждом случае Re независимо представляет собой: водород, -C1-C10алкил, -C2-C10алкенил, -OH, -OC1-C4алкил, -C3-C6циклоалкил, -C1-C10алкилен-C3-C6циклоалкил, HetB, -C1-C10алкилен-HetB, AryB, -C1-C10алкилен-AryB или -C1-C10алкилен-HetB; где каждый Re необязательно замещен 1-3 Rh;
в каждом случае Rf независимо представляет собой: галоген, -C1-C10алкил, -OH, -OC1-C4алкил, -S(O)mC1-C4алкил, -CN, -CF3, -OCHF2 или -OCF3; где указанный -C1-C10алкил необязательно замещен 1-3 заместителями, независимо выбранными из -OH, галогена, циано и -S(O)2CH3;
в каждом случае Rg независимо представляет собой: водород, -C(O)Re и -C1-C10алкил, где указанный -C1-C10алкил необязательно замещен 1-5 атомами фтора;
в каждом случае Rh независимо представляет собой: галоген, -C1-C10алкил, -OH, -OC1-C4алкил, -S(O)mC1-C4алкил, -CN, -CF3, -OCHF2, или -OCF3; где указанный -C1-C10алкил необязательно замещен 1-3 заместителями, независимо выбранными из -OH, галогена, циано или -S(O)2CH3;
каждый n независимо равен 0, 1, 2, 3 или 4;
каждый m независимо равен 0, 1 или 2, и
каждый p независимо равен 1 или 2.
Настоящее изобретение также относится к фармацевтической композиции для лечения бактериальной инфекции у субъекта, в том числе инфекции грамотрицательных бактериальных штаммов с множественной лекарственной устойчивостью, содержащей биарильное монобактамное соединение по изобретению и фармацевтически приемлемый носитель, разбавитель или эксципиент.
Соединения формулы (I) (также называемые здесь как «биарильное монобактамное соединение») и их фармацевтически приемлемые соли могут быть полезными, например, для ингибирования роста грамотрицательных бактериальных штаммов, в том числе, но без ограничения, штаммов Pseudomonas и Acinetobacter, и/или для лечения или профилактики их клинических проявлений у пациента.
Настоящее изобретение также относится к способам лечения грамотрицательных бактериальных инфекций у субъекта, нуждающегося в таком лечении, включающим введение субъекту эффективного количества биарильного монобактамного соединения по изобретению. В конкретных вариантах осуществления настоящего изобретения, способ включает введение соединения, которое является ингибитором β-лактамазы.
Варианты осуществления, частные варианты осуществления и признаки настоящего изобретения, будут либо дополнительно описаны здесь или они будут очевидны из последующего описания, примеров и прилагаемой формулы изобретения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Изобретение относится к новым биарильным монобактамным аналогам, относящимся к классу высокоактивных антибиотиков, эффективных против широкого спектра грамотрицательных бактерий. Эти соединения применяются в качестве терапевтических агентов для клинического лечения различных инфекций, вызванных грамотрицательными бактериями, включая штаммы, которые устойчивы ко многим лекарственным средствам, а также для лечения или профилактики клинических патологий, связанных с ними.
В каждом из различных вариантов осуществления изобретения, описанных здесь, каждая переменная, в том числе и для соединений формул I, II-1, II-2, II-3, III-1, III-2 и III-3, и их различных вариантов, выбрана независимо от других, если не указано иное.
Настоящее изобретение охватывает все соединения формул I, II-1, II-2, II-3, III-1, III-2 и III-3, а также их различные варианты, например, любые сольваты, гидраты, стереоизомеры и таутомеры указанных соединений, и их любые фармацевтически приемлемые соли.
Соединения формулы (I)
В одном аспекте настоящее изобретение включает соединение формулы I:
или его фармацевтически приемлемая соль, где X, Y, Z, A1, Q, А2, М, W, RX и RZ являются такими, как определено в настоящем описании для соединений формулы (I); и где эти соединения могут быть полезными для лечения бактериальных инфекций.
Первый вариант осуществления настоящего изобретения (вариант осуществления Е1) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где X, Y, Z, A1, Q, A2, M, W, RX и RZ имеют значения, которые определены для формулы I в разделе "Сущность изобретения".
Второй вариант осуществления изобретения (вариант осуществления Е2) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W представляет собой связь, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Третий вариант осуществления изобретения (вариант осуществления Е3) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W представляет собой O, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Четвертый вариант осуществления изобретения (вариант осуществления Е4) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X представляет собой N, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Пятый вариант осуществления изобретения (вариант осуществления Е5) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X представляет собой CR1, и R1 представляет собой водород, галоген или C1-C3 алкил, необязательно замещенный 1-3 Ra, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
В частном варианте осуществления варианта Е5 (вариант осуществления Е5-A), R1 представляет собой водород.
В другом частном варианте осуществления варианта Е5 (вариант осуществления Е5-B), R1 представляет собой галоген.
В другом дополнительном частном варианте осуществления варианта Е5 (вариант осуществления Е5-C), R1 представляет собой хлор.
В еще одном дополнительном частном варианте осуществления варианта Е5 (вариант осуществления Е5-D), R1 представляет собой фтор.
В еще одном частном варианте осуществления варианта Е5 (вариант осуществления Е5-E), R1 представляет собой C1-C3 алкил, необязательно замещенный 1-3 Ra, где в каждом случае Ra независимо представляет собой водород, галоген, C1-C3алкил, -NRcRd или - ORe.
Шестой вариант осуществления изобретения (вариант осуществления Е6) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z представляет собой C1-C3алкилен, необязательно замещенный 1-3 Rb, где в каждом случае Rb независимо представляет собой водород, C1-C8алкил, C3-C7циклоалкил, -C(O)ORe, -C(O)NRcRd, тетразолил, оксадиазолонил, HetA, AryA, -S(O)mRe, -S(O)mNRcRd или -P(O)(Re)p, где указанный C1-C8алкил и указанный C3-C7циклоалкил необязательно замещены 1-3 Ra; и все другие переменные имеют определения, представленные в варианте осуществления Е1.
В частном варианте осуществления варианта Е6, Z представляет собой C1-C3алкилен, замещенный одним Rb. В другом частном варианте осуществления варианта Е6, Z представляет собой C1-C3алкилен, замещенный двумя Rb. В дополнительном частном варианте осуществления варианта Е6, Z представляет собой C1-C3алкилен, замещенный тремя Rb.
Седьмой вариант осуществления изобретения (вариант осуществления Е7) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z представляет собой C1-C3алкилен, замещенный -C(O)ORe; и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Восьмой вариант осуществления изобретения (вариант осуществления Е8) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z представляет собой C1-C3алкилен, замещенный -C(O)OH; и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Девятый вариант осуществления изобретения (вариант осуществления Е9) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z представляет собой C1-C3алкилен, замещенный тетразолилом; и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Десятый вариант осуществления изобретения (вариант осуществления Е10) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z представляет собой C1-C3алкилен, замещенный C1-C8алкилом, необязательно замещенным 1-3 Ra, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Одиннадцатый вариант осуществления изобретения (вариант осуществления Е11) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z представляет собой C1-C3алкилен, замещенный метилом; и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Двенадцатый вариант осуществления изобретения (вариант осуществления Е12) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z представляет собой C1-C3алкилен, замещенный метилом и -C(O)OH; и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Тринадцатый вариант осуществления изобретения (вариант осуществления Е13) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z представляет собой -CH(C(O)OH)CH2-, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Четырнадцатый вариант осуществления изобретения (вариант осуществления Е14) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z представляет собой C1-C3алкилен, замещенный оксадиазолонилом, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Пятнадцатый вариант осуществления изобретения (вариант осуществления Е15) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y представляет собой связь, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Шестнадцатый вариант осуществления изобретения (вариант осуществления Е16) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y представляет собой O, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Семнадцатый вариант осуществления изобретения (вариант осуществления Е17) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y представляет собой NR2, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
В частном варианте осуществления варианта Е17, R2 представляет собой водород. В другом частном варианте осуществления варианта Е17, R2 представляет собой C1-C3алкил, необязательно замещенный 1-3 Ra. В дополнительном частном варианте осуществления варианта E17, R2 представляет собой C(O)Re. В еще одном частном варианте осуществления варианта E17, R2 представляет собой -C(O)NRcRd. В еще одном дополнительном частном варианте осуществления варианта Е17, R2 представляет собой -S(O)mRe. В дополнительном частном варианте осуществления варианта E17, R2 представляет собой -S(O)mNRcRd.
Восемнадцатый вариант осуществления изобретения (вариант осуществления Е18) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y представляет собой S, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Девятнадцатый вариант осуществления изобретения (вариант осуществления Е19) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y представляет собой CH2, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Двадцатый вариант осуществления изобретения (вариант осуществления Е20) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 представляет собой 5-6-членное моноциклическое ароматическое кольцо с 0, 1, 2 или 3 кольцевыми атомами, независимо выбранными из N, N в виде четвертичной соли, O и S, необязательно замещенное 1-4 R4, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Двадцать первый вариант осуществления изобретения (вариант осуществления Е21) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 представляет собой 5-членное моноциклическое ароматическое кольцо с 0, 1, 2 или 3 кольцевыми атомами, независимо выбранными из N, N в виде четвертичной соли, O и S, необязательно замещенное 1-4 R4, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Двадцать второй вариант осуществления изобретения (вариант осуществления Е22) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 представляет собой 6-членное моноциклическое ароматическое кольцо с 0, 1, 2 или 3 кольцевыми атомами, независимо выбранными из N, N в виде четвертичной соли, O и S, необязательно замещенное 1-4 R4, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
В частном варианте осуществления варианта Е21 или E22, A1 имеет 1 кольцевой атом, независимо выбранный из N, N в виде четвертичной соли, O и S. В дополнительном частном варианте осуществления варианта E21 или E22, A1 имеет 2 кольцевых атома, независимо выбранных из N, N в виде четвертичной соли, O и S. В другом частном варианте осуществления варианта Е21 или E22, A1 имеет 2 кольцевых атома, независимо выбранных из N, N в виде четвертичной соли, O и S. В еще одном частном варианте осуществления варианта E21 или E22, A1 имеет 3 кольцевых атома, независимо выбранных из N, N в виде четвертичной соли, O и S.
Двадцать третий вариант осуществления изобретения (вариант осуществления Е23) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 представляет собой 6-членное моноциклическое ароматическое кольцо, необязательно замещенное 1-4 R4, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Двадцать четвертый вариант осуществления изобретения (вариант осуществления Е24) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 представляет собой незамещенное 6-членное моноциклическое ароматическое кольцо, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Двадцать пятый вариант осуществления изобретения (вариант осуществления Е25) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 представляет собой:
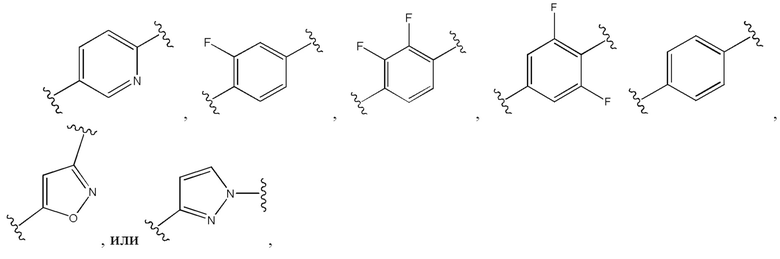
и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Двадцать шестой вариант осуществления изобретения (вариант осуществления Е26) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q представляет собой связь, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Двадцать седьмой вариант осуществления изобретения (вариант осуществления Е27) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q представляет собой CH2, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Двадцать восьмой вариант осуществления изобретения (вариант осуществления Е28) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q представляет собой O, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Двадцать девятый вариант осуществления изобретения (вариант осуществления Е29) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q представляет собой S, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Тридцатый вариант осуществления изобретения (вариант осуществления Е30) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q представляет собой -(CH2)nNR3-, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Тридцать первый вариант осуществления изобретения (вариант осуществления Е31) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q представляет собой -NR3(CH2)n-, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Тридцать второй вариант осуществления изобретения (вариант осуществления Е32) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q представляет собой NH, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Тридцать третий вариант осуществления изобретения (вариант осуществления Е33) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q представляет собой N(C1-C3алкил), необязательно замещенный 1-3 Ra, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Тридцать четвертый вариант осуществления изобретения (вариант осуществления Е34) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой C3-C7циклоалкил, необязательно замещенный 1-4 R4; и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Тридцать пятый вариант осуществления изобретения (вариант осуществления Е35) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой AryC; и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Тридцать шестой вариант осуществления изобретения (вариант осуществления Е36) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой 5-членное моноциклическое ароматическое кольцо с 0, 1, 2 или 3 кольцевыми атомами, независимо выбранными из N, N в виде четвертичной соли, O и S, необязательно замещенное 1-4 R4; и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Тридцать седьмой вариант осуществления изобретения (вариант осуществления Е37) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой 6-членное моноциклическое ароматическое кольцо с 0, 1, 2 или 3 кольцевыми атомами, независимо выбранными из N, N в виде четвертичной соли, O и S, необязательно замещенных 1-4 R4, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Тридцать восьмой вариант осуществления изобретения (вариант осуществления Е38) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой HetC; и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Тридцать девятый вариант осуществления изобретения (вариант осуществления Е39) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой 4-7-членное насыщенное или мононенасыщенное моноциклическое кольцо с 1 гетероатомом в кольце, выбранным из N, N в виде четвертичной соли, O и S, необязательно замещенное 1-4 R4; и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Сороковой вариант осуществления изобретения (вариант осуществления Е40) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой 4-7-членное насыщенное или мононенасыщенное моноциклическое кольцо с 2 гетероатомами в кольце, независимо выбранными из N, N в виде четвертичной соли, O и S, необязательно замещенное 1-4 R4; и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Сорок первый вариант осуществления изобретения (вариант осуществления Е41) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой 4-7-членное насыщенное или мононенасыщенное моноциклическое кольцо с 3 гетероатомами в кольце, независимо выбранными из N, N в виде четвертичной соли, O и S, необязательно замещенное 1-4 R4; и все другие переменные имеют определения, представленные в варианте осуществления Е1.
В частных вариантах осуществления вариантов E34-E41, A2 является незамещенным. В других частных вариантах осуществлений A2 замещен одним R4. В других частных вариантах осуществлений A2 замещен двумя R4. В других дополнительных частных вариантах осуществлений A2 замещен тремя R4. В еще других частных вариантах осуществлений A2 замещен четырьмя R4.
В частных вариантах осуществления вариантов E34-E41, A2 замещен метилом. В дополнительных вариантах осуществлений, A2 замещен -NH2. В других дополнительных частных вариантах осуществлений, A2 замещен =NH. В других дополнительных частных вариантах осуществлений, A2 замещен 1-4 заместителями, выбранными из: =NH, -NH2, и -CH3.
Сорок второй вариант осуществления изобретения (вариант осуществления Е42) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой:
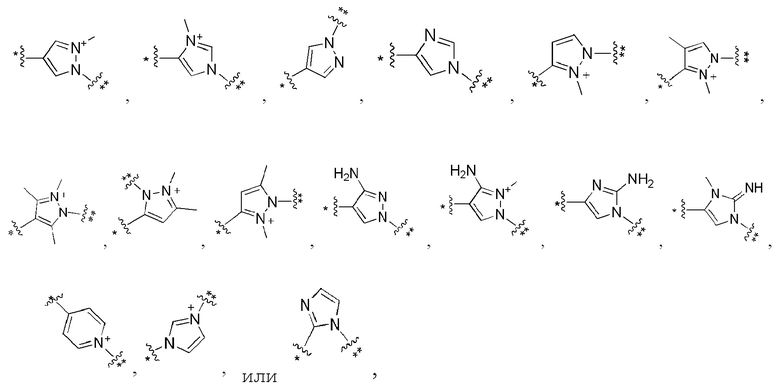
где * указывает на присоединение к Q, и ** указывает на присоединение к M, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Сорок третий вариант осуществления изобретения (вариант осуществления Е43) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой:
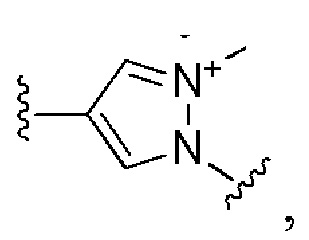
и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Сорок четвертый вариант осуществления изобретения (вариант осуществления Е44) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой:
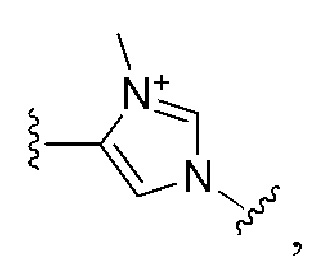
и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Сорок пятый вариант осуществления изобретения (вариант осуществления Е45) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой:
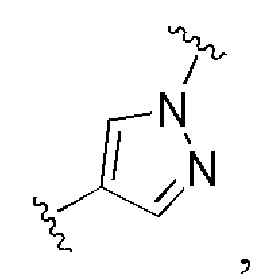
и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Сорок шестой вариант осуществления изобретения (вариант осуществления Е46) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой:
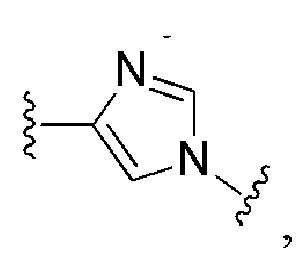
и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Сорок седьмой вариант осуществления изобретения (вариант осуществления Е47) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой:
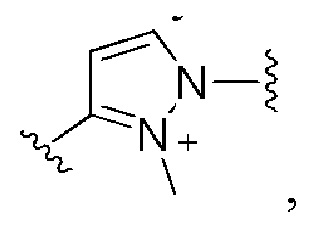
и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Сорок восьмой вариант осуществления изобретения (вариант осуществления Е48) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой:
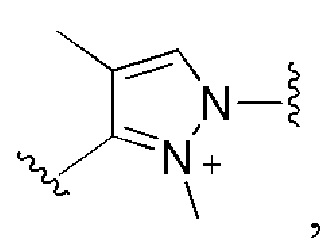
и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Сорок девятый вариант осуществления изобретения (вариант осуществления Е49) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой:
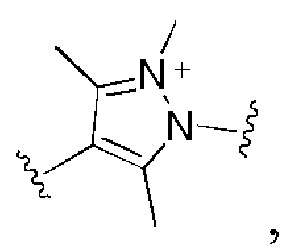
и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Пятидесятый вариант осуществления изобретения (вариант осуществления Е50) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой:
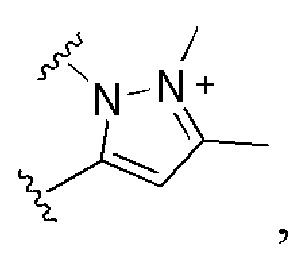
и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Пятьдесят первый вариант осуществления изобретения (вариант осуществления Е51) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой:
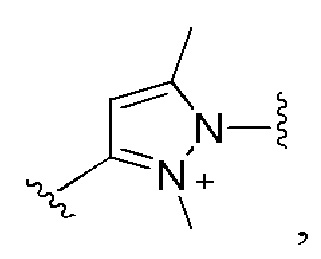
и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Пятьдесят второй вариант осуществления изобретения (вариант осуществления Е52) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой:
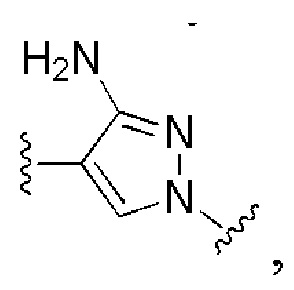
и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Пятьдесят третий вариант осуществления изобретения (вариант осуществления Е53) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой:
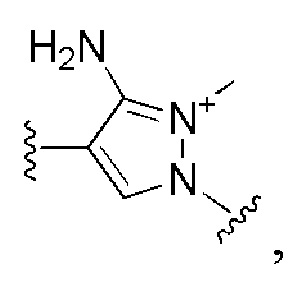
и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Пятьдесят четвертый вариант осуществления изобретения (вариант осуществления Е54) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой:
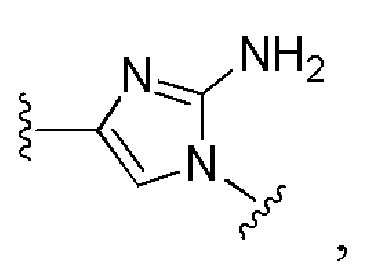
и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Пятьдесят пятый вариант осуществления изобретения (вариант осуществления Е55) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой:
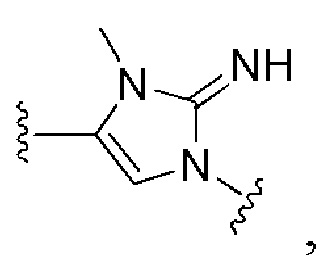
и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Пятьдесят шестой вариант осуществления изобретения (вариант осуществления Е56) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой:
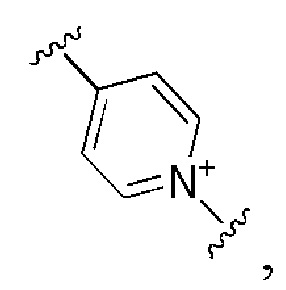
и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Пятьдесят седьмой вариант осуществления изобретения (вариант осуществления Е57) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой:
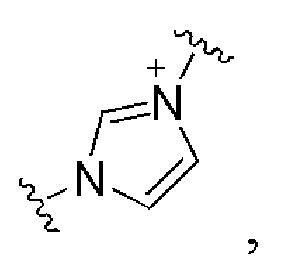
и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Пятьдесят восьмой вариант осуществления изобретения (вариант осуществления Е58) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 представляет собой:
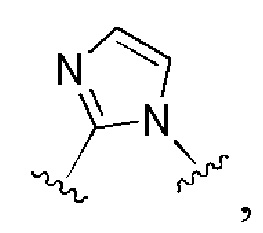
и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Пятьдесят девятый вариант осуществления изобретения (вариант осуществления Е59) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 имеет определение, представленное в любом из вариантов осуществлений E34-E58, M представляет собой R5, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Шестидесятый вариант осуществления изобретения (вариант осуществления Е60) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 имеет определение, представленное в любом из вариантов осуществлений E34-E58, M представляет собой C2-C10алкил, необязательно замещенный 1-4 R6, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Шестьдесят первый вариант осуществления изобретения (вариант осуществления Е61) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 имеет определение, представленное в любом из вариантов осуществлений E34-E58, M представляет собой C3-C7циклоалкил, необязательно замещенный 1-4 R6, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Шестьдесят второй вариант осуществления изобретения (вариант осуществления Е62) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 имеет определение, представленное в любом из вариантов осуществлений E34-E58, M представляет собой HetB, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Шестьдесят третий вариант осуществления изобретения (вариант осуществления Е63) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 имеет определение, представленное в любом из вариантов осуществлений E34-E58, M представляет собой AryB, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Шестьдесят четвертый вариант осуществления изобретения (вариант осуществления Е64) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 имеет определение, представленное в любом из вариантов осуществлений E34-E58, M представляет собой -NR5, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Шестьдесят пятый вариант осуществления изобретения (вариант осуществления Е65) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 имеет определение, представленное в любом из вариантов осуществлений E34-E58, M представляет собой -OR5, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Шестьдесят шестой вариант осуществления изобретения (вариант осуществления Е66) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 имеет определение, представленное в любом из вариантов осуществлений E34-E58, M представляет собой -(CH2)nR5, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Шестьдесят седьмой вариант осуществления изобретения (вариант осуществления Е67) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 имеет определение, представленное в любом из вариантов осуществлений E34-E58, M представляет собой -C(O)R5, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Шестьдесят восьмой вариант осуществления изобретения (вариант осуществления Е68) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 имеет определение, представленное в любом из вариантов осуществлений E34-E58, M представляет собой -CH(NH)R5, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Шестьдесят девятый вариант осуществления изобретения (вариант осуществления Е69) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 имеет определение, представленное в любом из вариантов осуществлений E34-E58, M представляет собой -S(O)mR5, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Семидесятый вариант осуществления изобретения (вариант осуществления Е70) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 имеет определение, представленное в любом из вариантов осуществлений E34-E58, M представляет собой -(CH2)3NH2, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Семьдесят первый вариант осуществления изобретения (вариант осуществления Е71) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 имеет определение, представленное в любом из вариантов осуществлений E34-E58, M представляет собой -(CH2)1-2HetB, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Семьдесят второй вариант осуществления изобретения (вариант осуществления Е72) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 имеет определение, представленное в любом из вариантов осуществлений E34-E58, M представляет собой -CH2AryB, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Семьдесят третий вариант осуществления изобретения (вариант осуществления Е73) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 имеет определение, представленное в любом из вариантов осуществлений E34-E58, M имеет определение, представленное в любом из вариантов осуществлений E59-E72, RX и RZ представляют собой метил, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Семьдесят четвертый вариант осуществления изобретения (вариант осуществления Е74) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 имеет определение, представленное в любом из вариантов осуществлений E34-E58, M имеет определение, представленное в любом из вариантов осуществлений E59-E72, RX представляет собой водород и RZ представляет собой метил, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Семьдесят пятый вариант осуществления изобретения (вариант осуществления Е75) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 имеет определение, представленное в любом из вариантов осуществлений E34-E58, M имеет определение, представленное в любом из вариантов осуществлений E59-E72, один из RX и RZ представляет собой SCH3, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Семьдесят шестой вариант осуществления изобретения (вариант осуществления Е76) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 имеет определение, представленное в любом из вариантов осуществлений E34-E58, M имеет определение, представленное в любом из вариантов осуществлений E59-E72, один из RX и RZ представляет собой SC1-C3алкил, необязательно замещенный 1-7 атомами фтора, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Семьдесят седьмой вариант осуществления изобретения (вариант осуществления Е77) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 имеет определение, представленное в любом из вариантов осуществлений E34-E58, M имеет определение, представленное в любом из вариантов осуществлений E59-E72, один из RX и RZ представляет собой SCH3, необязательно замещенный 1-3 атомами фтора, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Семьдесят восьмой вариант осуществления изобретения (вариант осуществления Е78) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 имеет определение, представленное в любом из вариантов осуществлений E34-E58, M имеет определение, представленное в любом из вариантов осуществлений E59-E72, один из RX и RZ представляет собой C1-C3алкил, необязательно замещенный 1-7 атомами фтора, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Семьдесят девятый вариант осуществления изобретения (вариант осуществления Е79) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 имеет определение, представленное в любом из вариантов осуществлений E34-E58, M имеет определение, представленное в любом из вариантов осуществлений E59-E72, один из RX и RZ представляет собой (C1-C3алкилен)nOC1-C3алкил, необязательно замещенный 1-7 атомами фтора, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Восьмидесятый вариант осуществления изобретения (вариант осуществления Е80) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 имеет определение, представленное в любом из вариантов осуществлений E34-E58, M имеет определение, представленное в любом из вариантов осуществлений E59-E72, один из RX и RZ представляет собой (C1-C3алкилен)nNC1-C3алкил, необязательно замещенный 1-7 атомами фтора, и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Восемьдесят первый вариант осуществления изобретения (вариант осуществления Е81) представляет собой соединение формулы I или его фармацевтически приемлемую соль, где W имеет определение, представленное в варианте осуществления Е2 или E3, X имеет определение, представленное в любом из вариантов осуществлений Е4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z имеет определение, представленное в любом из вариантов осуществлений E6-E13, Y имеет определение, представленное в любом из вариантов осуществлений E15-E19, A1 имеет определение, представленное в любом из вариантов осуществлений E20-E25, Q имеет определение, представленное в любом из вариантов осуществлений E26-E33, A2 имеет определение, представленное в любом из вариантов осуществлений E34-E58, M имеет определение, представленное в любом из вариантов осуществлений E59-E72, RX и RZ, вместе с атомом углерода, к которому они присоединены, образуют вместе моноциклический C4-C7циклоалкил или моноциклический C4-C7гетероциклоалкил с 1, 2 или 3 гетероатомами в кольце, независимо выбранными из N, O и S, где указанный C4-C7циклоалкил и указанный C4-C7гетероциклоалкил необязательно замещены 1-3 заместителями, независимо выбранными из -F, -OH и -OC1-C3алкила; и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Восемьдесят второй вариант осуществления изобретения (вариант осуществления Е82) представляет собой соединение формулы II-1, II-2, II-3, III-1, III-2 или III-3, или его фармацевтически приемлемую соль,
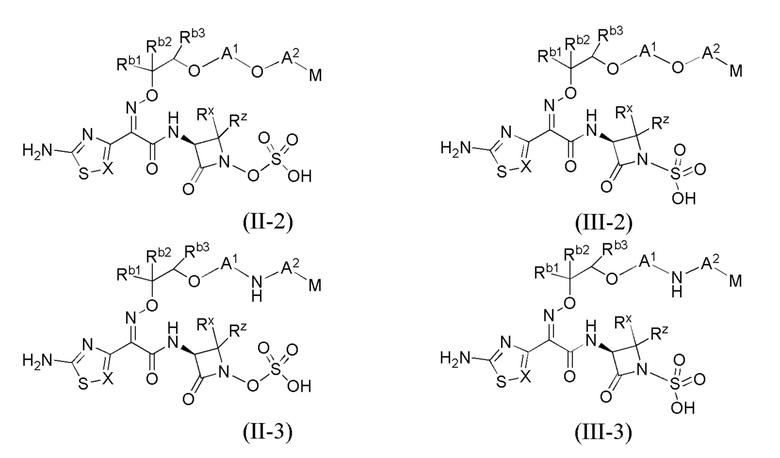
где X имеет определение, представленное в любом из вариантов осуществлений E1, E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E;
A1 имеет определение, представленное в любом из вариантов осуществлений E1, E20-E25;
A2 имеет определение, представленное в любом из вариантов осуществлений E1, E34-E58;
M имеет определение, представленное в любом из вариантов осуществлений E1, E59-E72;
RX и RZ имеют определения, представленные в любом из вариантов осуществлений E1, E73-E81;
Rb1, Rb2 и Rb3 независимо представляют собой водород, C1-C8алкил, C3-C7циклоалкил, -C(O)ORe, -C(O)NRcRd, тетразолил, оксадиазолонил, HetA, AryA, -S(O)mRe, -S(O)mNRcRd или -P(O)(Re)p, где указанный C1-C8алкил и указанный C3-C7циклоалкил необязательно замещены 1-3 Ra; и все другие переменные имеют определения, представленные в варианте осуществления Е1.
Восемьдесят третий вариант осуществления изобретения (вариант осуществления Е83) представляет собой соединение формулы II-1, II-2, II-3, III-1, III-2 или III-3, или его фармацевтически приемлемую соль, где Rb1 и Rb2 независимо представляют собой водород, C1-C3алкил, тетразолил, оксадиазолонил или -C(O)ORe; и Rb3 представляет собой водород, и все другие переменные имеют определения, представленные в варианте осуществления Е82.
Восемьдесят четвертый вариант осуществления изобретения (вариант осуществления Е84) представляет собой соединение, имеющее структуру:
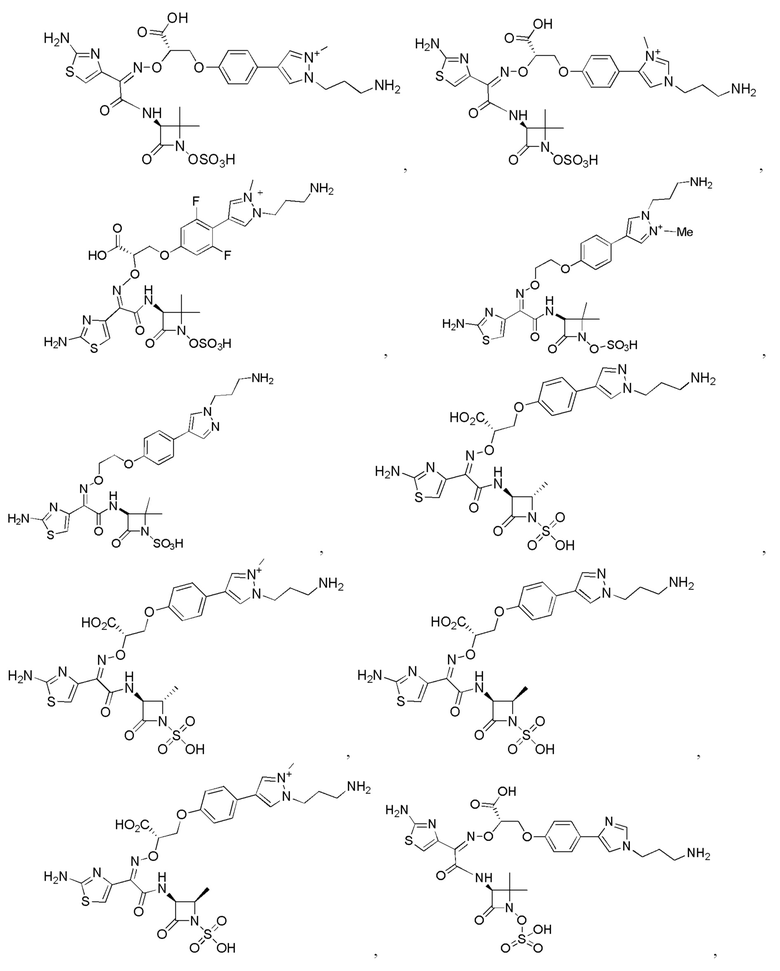
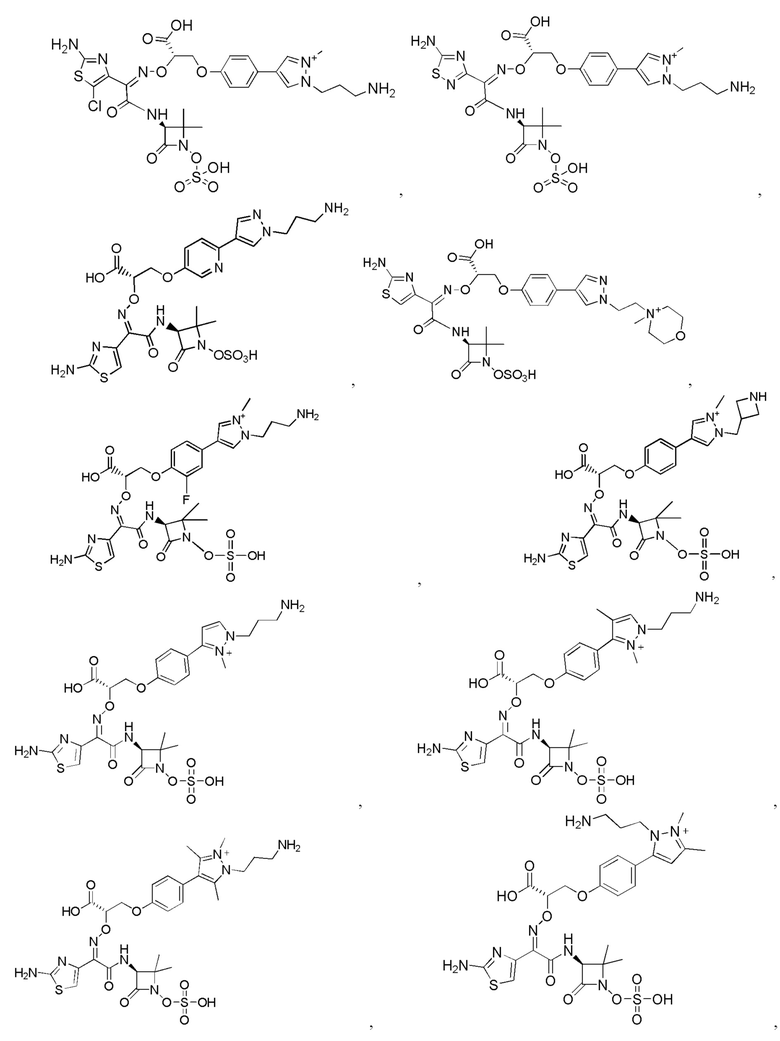
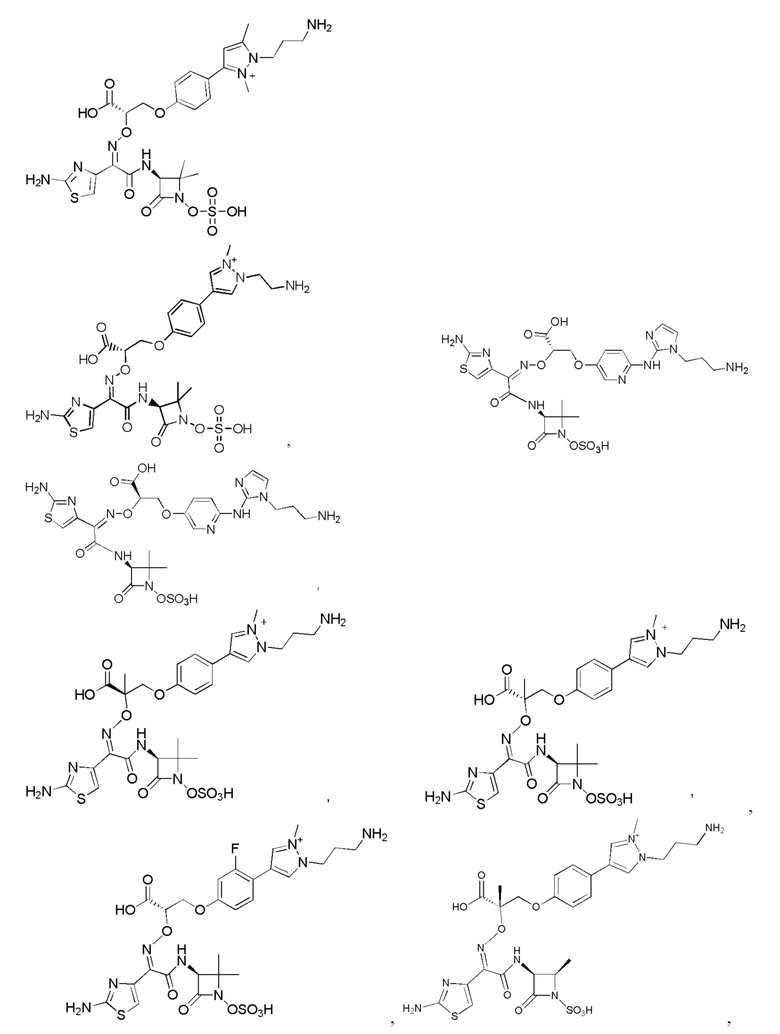
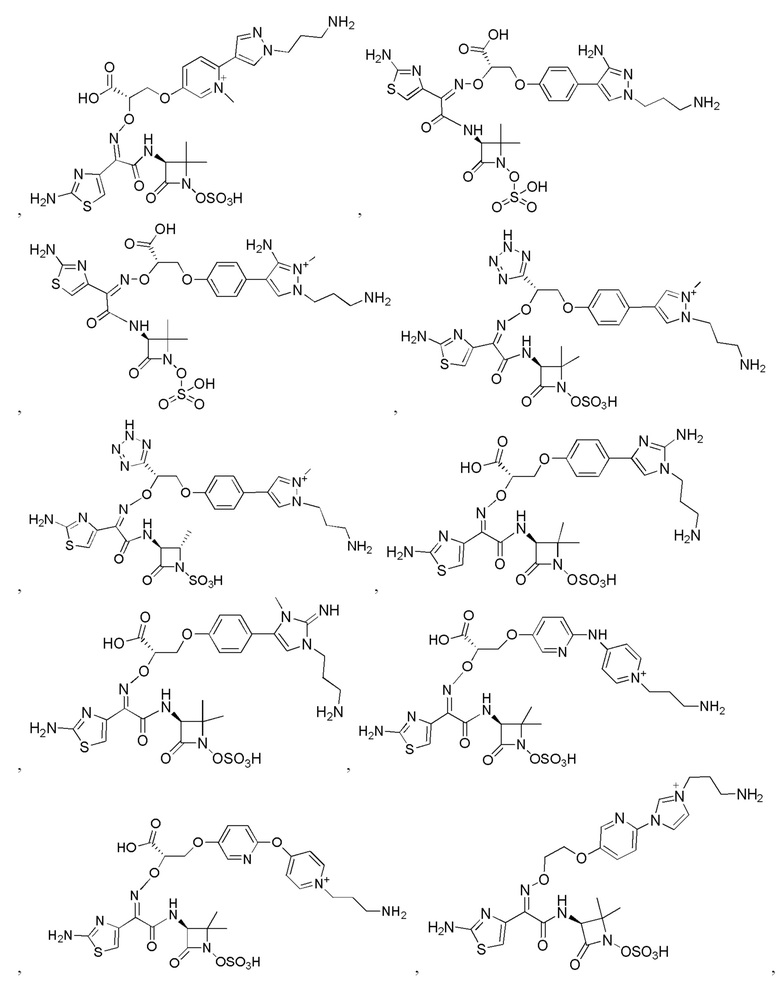
или
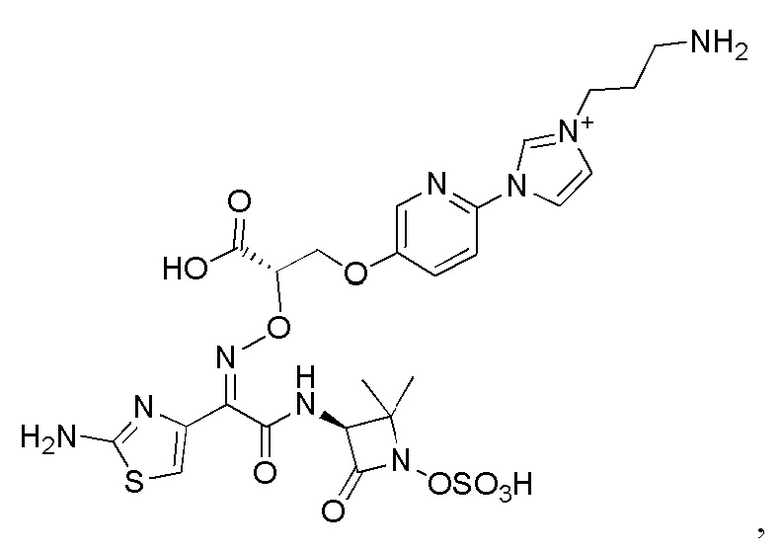
или его фармацевтически приемлемую соль.
В восемьдесят пятом варианте осуществления, в каждом случае R4 независимо представляет собой: -C1-C8алкил, -C2-C8алкенил, -C2-C8алкинил, галоген, -ORe, -S(O)mRe, -S(O)mNRcRd, -C(O)Re, -OC(O)Re, -C(O)ORe, -CN, -C(O)NRcRd, -NRcRd, -(CH2)nNRcRd, -NRcC(O)Re, -NRcC(O)ORe, -NRcC(O)NRcRd, -NRcS(O)mRe, =NH, -CF3, -OCF3, -OCHF2, -C3-C6циклоалкил, -O-C3-C6циклоалкил, -C1-C10алкилен-C3-C6циклоалкил, -O-C1-C10алкилен-C3-C6циклоалкил, HetB, -O-HetB, -C1-C10алкилен-HetB, -O-C1-C10алкилен-HetB, AryA, -O-AryA, -C1-C10алкилен-AryA, или -O-C1-C10алкилен-AryA, где каждый R4 является незамещенным, или он замещен 1-4 заместителями, выбранными из галогена, -C1-C6алкила и -(CH2)nNRcRd.
В восемьдесят шестом варианте осуществления R5 представляет собой C2-C10алкил, C3-C7циклоалкил, C1-C6алкил-C3-C7 циклоалкил, HetB, AryB, или -NH(C1-C6алкил), где указанный C1-C6алкил, указанный C2-C10алкил и указанный C3-C7циклоалкил необязательно замещены 1-4 R6.
Наряду с вариантами осуществления изобретения, связанными с соединениями формулы I, изобретение также включает и другие варианты осуществления, которые связаны с соединениями формулы II и формулы III, в частности, соединениями формул II-1, II-2, II-3, III-1, III-2 и III-3, частные варианты осуществления, связанные с соединениями формул I, II-1, II-2, II-3, III-1, III-2 и III-3, и иные варианты осуществления, описанные здесь, а также варианты, связанные с конкретными соединениями, описанными здесь.
Другие варианты осуществления настоящего изобретения включают следующие объекты.
(a) Фармацевтическая композиция, содержащая эффективное количество соединения формулы I, II-1, II-2, II-3, III-1, III-2 или III-3, как определено здесь, или его фармацевтически приемлемой соли, и фармацевтически приемлемый носитель.
(b) Фармацевтическая композиция (а), дополнительно содержащая второе соединение, где второе соединение представляет собой ингибитор β-лактамазы.
(c) Фармацевтическая композиция (b), где второе соединение выбрано из группы, состоящей из: релебактама, тазобактама, клавулановой кислоты, сульбактама и авибактама.
(d) Фармацевтическая композиция, содержащая (i) соединение формулы I, II-1, II-2, II-3, III-1, III-2 или III-3, или его фармацевтически приемлемую соль, и (ii) второе соединение, где второе соединение является ингибитором β-лактамазы, при этом соединение формулы I, II-1, II-2, II-3, III-1, III-2 или III-3, и второе соединение, каждое, используют в количестве, которое делает сочетание эффективным для лечения или профилактики бактериальной инфекции.
(e) Сочетание (d), где второе соединение выбрано из группы, состоящей из: релебактама, тазобактама, клавулановой кислоты, сульбактама и авибактама.
(f) Способ лечения бактериальной инфекции у субъекта, который включает введение субъекту, нуждающемуся в таком лечении, эффективного количества соединения формулы I, II-1, II-2, II-3, III-1, III-2 или III-3, или его фармацевтически приемлемой соли.
(g) Способ профилактики и/или лечения бактериальной инфекции, который включает введение субъекту, нуждающемуся в этом, фармацевтической композиции, содержащей эффективное количество соединения формулы I, II-1, II-2, II-3, III-1, III-2 или III-3, или его фармацевтически приемлемой соли, и фармацевтически приемлемый носитель.
(h) Способ лечения бактериальной инфекции, который включает введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества композиции (a), (b), (с), (d) или (е).
(i) Способ лечения бактериальной инфекции, как указано в (f), (g) или (h), где бактериальная инфекция вызвана грамотрицательной бактерией.
(j) Способ лечения бактериальной инфекции, как указано в (f), (g), (h) или (i), в котором бактериальная инфекция вызвана Pseudomonas aeruginosa или Acinetobacter baumannii.
Настоящее изобретение также включает соединение формулы I, II-L, II-2, II-3, III-1, III-2 или III-3, или его фармацевтически приемлемую соль, (i) для применения в (II) для применения в качестве лекарственного средства для, или (III) для применения при получении (или производстве) лекарственного средства для терапии или лечения бактериальной инфекции, в том числе инфекции, вызванной бактериальным штаммом с множественной лекарственной устойчивостью. В этих применениях соединения по настоящему изобретению необязательно могут быть использованы в комбинации с одним или несколькими вторыми терапевтическими агентами, включая релебактам, тазобактам, клавулановую кислоту, сульбактам и авибактам.
Дополнительные варианты осуществления настоящего изобретения включают фармацевтические композиции, комбинацию и способы, как изложено выше в пунктах (а)-(j), и применения, как изложено в предыдущем абзаце, где используемое соединение по изобретению представляет собой соединение одного из вариантов, частных вариантов, классов или подклассов, из числа описанных выше. В этих вариантах осуществления изобретения соединение может быть необязательно использовано в виде фармацевтически приемлемой соли.
В вариантах осуществления, касающихся соединений и солей, приведенных выше, следует понимать, что каждый вариант осуществления может быть объединен с одним или несколькими другими вариантами осуществления изобретения, в той степени, когда такое сочетание обеспечивает стабильное соединение или соль в соответствии с описанием вариантов. Также следует понимать, что варианты осуществления композиций и способов, представленных выше как (а)-(j), включают все варианты осуществления соединений и/или солей, включая варианты, которые представляют собой комбинации вариантов осуществления изобретения.
Дополнительные варианты осуществления настоящего изобретения включают фармацевтические композиции, комбинации, способы и применения, изложенные в предыдущих абзацах описания, где соединение по изобретению или его соль, используемая в этих вариантах осуществления, является по существу чистым. Что касается фармацевтической композиции, содержащей соединение формулы I, II-1, II-2, II-3, III-1, III-2 или III-3 или его соль, фармацевтически приемлемый носитель и, необязательно, один или несколько эксципиентов, следует понимать, что термин "по существу чистый" относится к соединению формулы I, II-1, II-2, II-3, III-1, III-2 или III-3 или его соли per se; то есть он характеризует чистоту этого активного ингредиента в композиции.
Определения и сокращения
Термины, используемые здесь, имеют свои обычные значения, и смысловое значение таких терминов не зависит от случая их использования. Несмотря на это, и за исключением случаев, когда указано иное, в описании и формуле изобретения используются следующие определения. Для описания одинаковых структур могут быть использованы взаимозаменяемо химические названия, обычные наименования и химические структуры. Если химическое соединение описывается с помощью химической структуры и с помощью химического названия, и имеет место разночтение между структурой и названием, то структура считается преобладающей. Эти определения применяются независимо от того, используется ли термин сам по себе или в сочетании с другими терминами, если не указано иное. Таким образом, определение «алкил» относится к «алкилу», а также к «алкильной» части «гидроксиалкил», «галогеналкил, «-O-алкил и т.п.
Следующие термины, используемые здесь и по всему тексту описания, если не указано иное, имеют следующие значения.
Термин «ингибитор β-лактамазы» относится к соединению, которое способно ингибировать ферментативную активность фермента, относящегося к β-лактамазам. Как используется здесь, ингибировании активности β-лактамазы означает ингибирование активности β-лактамаз класса А, С и/или D. Для противомикробных применений ингибирования при ингибирующей концентрации 50% предпочтительно достигается на уровне или ниже величины, приблизительно равной 100 мкг/мл, или на уровне или ниже величины, приблизительно равной 50 микрограммов/мл, или на уровне или ниже величины, приблизительно равной 25 мкг/мл. Термины β-лактамазы «класса А», «класса B», «класса C» и «класса D» понятны специалистами в данной области техники и описаны в S.G. Waley, β-lactamase: mechanisms of action, в Chemistry of β-Lactams, M.I. Page, Ed.; Chapman и Hall, London, (1992) 198-228.
Термин «металло-β-лактамаза» обозначает металлопротеины, способные инактивировать β-лактамный антибиотик. β-лактамаза может быть ферментом, который катализирует гидролиз β-лактамного кольца β-лактамного антибиотика. Особый интерес здесь представляют микробные металло-β-лактамазы. Металло-β-лактамаза может быть, например, цинковой металло-β-лактамазой. β-лактамазы, представляющие интерес, включают те, которые описаны, например, в S.G. Waley, β lactamase: mechanisms of action, в Chemistry of β-Lactams, M.I. Page, Ed.; Chapman и Hall, London, (1992) 198-228. b-лактамазы, представляющие здесь особый интерес, включают металло-β-лактамазы Escherichia coli (например, New Delhi металло-β-лактамаза, NDM), Serratia marcescens (например, IMP) и Klebsiella spp. (например, интегрон-кодированная металло-β-лактамаза Verona, VIM). Дополнительные металло-β-лактамазы, представляющие здесь интерес, включают ферменты типа SPM, GIM, SIM, KHM, AIM, DIM, SMB, TMB и FIM.
Термин «антибиотик» относится к соединению или композиции, которая уменьшает жизнеспособность микроорганизма, или которая ингибирует рост или пролиферацию микроорганизма. Фраза «ингибирует рост или пролиферацию» означает увеличение времени генерации (т.е., времени, необходимого для разделения бактериальной клетки или для увеличения популяции клеток в два раза) по меньшей мере приблизительно в 2 раза. Предпочтительными антибиотиками являются те, которые могут увеличить время генерации по меньшей мере приблизительно в 10 раз или более (например, по меньшей мере приблизительно в 100 раз или даже до бесконечности, как в случае полной гибели клеток). Как используется в данном описании, термин «антибиотик» также подразумевает, что антибиотик включает антимикробный, бактериостатический или бактерицидный агент. Примеры антибиотиков включают пенициллины, цефалоспорины и карбапенемы.
Термин «β-лактамный антибиотик» относится к соединению с антибиотическими свойствами, который содержит функциональный β-лактам. Неограничивающие примеры β-лактамных антибиотиков включают пенициллины, цефалоспорины, пенемы, карбапенемы и монобактамы.
Термин «приблизительно», при указании количества (например, кг, л или их эквиваленты) вещества или композиции, или физического показателя, или параметра, характеризующего стадию процесса (например, температуры, при который проводят стадию процесса), или тому подобное, относится к изменению численного значения, которое может иметь место, например, при обычных измерениях, операциях и выборках, проводимых при получении, характеризации и/или при использовании вещества или композиции; как показатель случайной ошибки в этих процедурах; как различие при изготовлении, использовании различных источников получения ингредиентов или чистоты ингредиентов, используемых для изготовления или использования композиций или выполнения операций; и т.п. В некоторых вариантах осуществления изобретения термин «приблизительно» может означать изменение, составляющее ± 0,1, 0,2, 0,3, 0,4, 0,5, 1,0, 2,0, 3,0, 4,0 или 5,0 соответствующих единиц измерения. В некоторых вариантах осуществления изобретения термин "приблизительно" может означать изменение, составляющее ± 1%, 2%, 3%, 4%, 5%, 10% или 20%.
Другой вариант осуществления настоящего изобретения представляет соединение формулы I, II-1, II-2, II-3, III-1, III-2 или III-3, или его фармацевтически приемлемую соль, которые первоначально определены или определены в любом из указанных выше вариантов осуществления, частных вариантах осуществления изобретения и его аспектах, классах или подклассах, где соединение или его соль представлены по существу в чистом виде. Используемый здесь термин «по существу чистый» означает, соответственно, что продукт, содержащий соединение формулы I, II-1, II-2, II-3, III-1, III-2 или III-3 или его соль (например, продукт, выделенный из реакционной смеси, полученное соединение или соль) состоит из соединения или соли в количестве, составляющем по меньшей мере приблизительно 60 мас.%, обычно по меньшей мере приблизительно 70 мас.%, предпочтительно по меньшей мере приблизительно 80 мас.%, более предпочтительно по меньшей мере приблизительно 90 мас.% (например, от приблизительно 90 мас.% до приблизительно 99 мас.%), еще более предпочтительно по меньшей мере приблизительно 95 мас.% (например, от приблизительно 95 мас.% до приблизительно 99 мас.%, или от приблизительно 98 мас.% до 100 мас.%) и наиболее предпочтительно по меньшей мере приблизительно 99 мас.% (например, 100 мас.%). Уровень чистоты соединений и солей может быть определен с использованием стандартного метода анализа, такого как тонкослойная хроматография, гель-электрофорез, высокоэффективная жидкостная хроматография и/или масс-спектрометрия. Если используют более чем один метод анализа, и методы обеспечивают экспериментально значимые различия уровня определенной чистоты, то метод, который показывает более высокий уровень чистоты, признают определяющим. Соединение или соль, имеющее 100%-й уровень чистоты, представляет собой соединение или соль, которая свободна от детектируемых примесей, определяемых стандартным методом анализа.
В отношении соединения по изобретению, которое имеет один или несколько асимметрических центров, и которое может существовать в виде смеси стереоизомеров, по существу чистое соединение может быть либо по существу чистой смесью стереоизомеров или по существу чистым индивидуальным диастереомером или энантиомером, если явно не указано иное. Настоящее изобретение охватывает все стереоизомерные формы соединений формулы I, II-1, II-2, II-3, III-1, III-2 или III-3. Если конкретная стереохимия не указана, то настоящее изобретение предусматривает, что охватываются все изомерные формы этих соединений. Центры асимметрии, которые присутствуют в соединениях формулы I, II-1, II-2, II-3, III-1, III-2 или III-3, могут все независимо друг от друга иметь (R) конфигурацию или (S) конфигурацию. Когда в структурных формулах соединений настоящего изобретения связи у хирального атома углерода изображены в виде прямых линий, то следует понимать, что подразумеваются обе (R) и (S) конфигурации хирального атома углерода, и, следовательно, этой формулой охватываются оба энантиомера и их смеси. Точно так же, когда название соединения указано без хирального обозначения для хирального атома углерода, то следует понимать, что этим названием охватываются как (R), так и (S) конфигурации хирального атома углерода, и, следовательно, отдельные энантиомеры и их смеси. Описание получения конкретных стереоизомеров или их смесей могут быть найдены в примерах, где такое стереоизомеры или смеси были получены, но это никоим образом не ограничивает включение всех стереоизомеров и их смесей в объем настоящего изобретения.
Изобретение включает все возможные энантиомеры и диастереомеры, а также смеси двух или более стереоизомеров, например, смеси энантиомеров и/или диастереомеров в любых соотношениях. Таким образом, энантиомеры являются объектом изобретения в энантиомерно чистой форме, как левовращающие, так и правовращающие антиподы, в форме рацематов и в виде смесей двух энантиомеров в любых соотношениях. В случае цис/транс-изомерии, изобретение включает как цис-форму, так и транс-форму, а также смеси этих форм в любых соотношениях. Получение индивидуальных стереоизомеров может быть осуществлено, если желательно, путем разделения смеси обычными методами, например путем хроматографии или кристаллизации, с использованием стереохимически однородных исходных материалов для синтеза или путем стереоселективного синтеза. Перед разделением стереоизомеров может быть осуществлена необязательная дериватизация. Разделение смеси стереоизомеров можно проводить на промежуточной стадии в процессе синтеза соединений формулы I, II-II, II-2, II-3, III-1, III-2 или III-3, или это может быть сделано в отношении конечного рацемического продукта. Абсолютная стереохимия может быть определена с помощью рентгеновской кристаллографии кристаллических продуктов или кристаллических промежуточных соединений, которые модифицируют, в случае необходимости, реагентом, содержащим стереогенный центр с известной конфигурацией. Там, где соединения по изобретению способны к таутомеризации, все отдельные таутомеры, а также их смеси включены в объем настоящего изобретения. Если указан конкретный изомер, соль, сольват (в том числе гидрат) или сольватированная соль рацемата, энантиомера, диастереомера или таутомера, то настоящее изобретение включает все такие изомеры, а также соли, сольваты (в том числе гидраты) и сольватированные соли рацематов, энантиомеров, диастереомеров и таутомеров и их смесей.
«Ас» означает ацетил, который представляет собой СН3С(=O)-.
«Алкил» означает насыщенные углеродные цепи, которые могут быть линейными или разветвленными, или быть их комбинацией, если углеродная цепь не определена иным образом. Другие группы, имеющие префикс «алк», такие как алкокси и алканоил, также могут быть линейными или разветвленными, или быть их комбинацией, если углеродная цепь не определена иным образом. Примеры алкильных групп включают метил, этил, пропил, изопропил, бутил, втор- и трет-бутил, пентил, гексил, гептил, октил, нонил и т.п.
«Алкилен», как используется здесь, относится к алкильной группе, определенной выше, в которой один из атомов водорода алкильной группы заменен связью. Не ограничивающие примеры алкиленовых групп включают -CH2-, -CH2CH2-, -CH2CH2CH2-, -CH2CH2CH2CH2-, -CH(CH3)CH2CH2-, -CH(CH3)- и -CH2CH(CH3)CH2-. В одном варианте осуществления изобретения алкиленовая группа включает от 1 до приблизительно 6 атомов углерода. В одном варианте осуществления алкиленовая группа имеет от 1 до приблизительно 3 атомов углерода. В другом варианте осуществления изобретения алкиленовая группа является разветвленной. В другом варианте осуществления изобретения алкиленовая группа является линейной. В одном варианте осуществления изобретения алкиленовая группа представляет собой -CH2-. Термин «C1-C6 алкилен» относится к алкиленовой группе, имеющей от 1 до 6 атомов углерода.
«Алкенил» означает углеродные цепи, которые содержат по меньшей мере одну двойную углерод-углеродную связь, и которая может быть линейной или разветвленной, или представлять их комбинацию, если иное не указано. Примеры алкенила включают винил, аллил, изопропенил, пентенил, гексенил, гептенил, 1-пропенил, 2-бутенил, 2-метил-2-бутенил и тому подобное.
Термин «алкинил» означает углеродные цепи, которые содержат по меньшей мере одну тройную углерод-углеродную связь, и которая может быть линейной или разветвленной, или представлять их комбинацию, если иное не указано. Примеры алкинила включают этинил, пропаргил, 3-метил-1-пентинил, 2-гептинил и тому подобное.
«Ароматическая кольцевая система» или «ароматическая» со ссылкой на кольцо, означает моноциклическое, бициклическое или трициклическое ароматическое кольцо или кольцевую систему, содержащую 5-14 атомов в кольце, где по меньшей мере одно из колец является ароматическим. Термин может быть использован для описания насыщенного или мононенасыщенного карбоциклического кольца, конденсированного с арильной группой. Так, например, 5-7-членный циклоалкил может быть слит через два соседних кольцевых атома с 5-6-членным гетероарилом, содержащим 1, 2 или 3 гетероатома в кольце, выбранных из N, О и S. В другом примере гетеромоноциклическое кольцо слито с помощью двух кольцевых атомов с фенилом или 5-6-членным гетероарилом, содержащим 1, 2 или 3 гетероатома, выбранных из N, O и S. в случае гетеромоноциклического кольца, содержащего один или несколько атомов N, N может быть в виде четвертичного амина. В некоторых вариантах осуществления атом N в кольце может быть в форме N-оксида.
«Арил» означает моноциклическое, бициклическое или трициклическое карбоциклическое ароматическое кольцо или кольцевую систему, содержащую 6-14 атомов углерода, где по меньшей мере одно из колец является ароматическим. Примеры арила включают фенил и нафтил. В одном из вариантов осуществления настоящего изобретения, арил представляет собой фенил.
«Циклоалкил» означает насыщенное моноциклическое, бициклическое или мостиковое карбоциклическое кольцо, имеющее определенное количество атомов углерода. Примеры циклоалкила включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, инданил и тому подобное. В одном из вариантов осуществления настоящего изобретения, циклоалкил выбран из циклопропана, циклобутана, циклопентана и циклогексана.
«Циклоалкенил» означает неароматическое моноциклическое или бициклическое карбоциклическое кольцо, содержащее по меньшей мере одну двойную связь. Примеры включают циклоалкенил циклопропенил, циклобутенил, циклопентенил, циклогексенил, циклогептенил, циклоокстенил и тому подобное.
Термин «гетероциклоалкил», используемый в данном описании, относится к неароматической насыщенной моноциклической или полициклической кольцевой системе, содержащей от 3 до 11 кольцевых атомов, где от 1 до 4 кольцевых атомов независимо представляют собой Н, NН, S (включая SO и SO2) и O, а остальные атомы в кольце являются атомами углерода. Группа гетероциклоалкила может быть соединена с помощью кольцевого атома углерода или атома азота в кольце (если он присутствует). Там, где кольцо или кольцевая система содержит один или несколько атомов N, то N может быть в виде четвертичного амина. В одном варианте осуществления изобретения гетероциклоалкильная группа является моноциклической и имеет от приблизительно 3 до приблизительно 7 кольцевых атомов. В другом варианте осуществления изобретения гетероциклоалкильная группа является моноциклической и имеет от приблизительно 4 до приблизительно 7 кольцевых атомов. В другом варианте осуществления изобретения, гетероциклоалкил группа является бициклической и имеет от приблизительно 7 до приблизительно 11 атомов в кольце. Когда гетероциклоалкил содержит два кольца, кольца могут конденсированными или спироциклическими. В еще одном варианте осуществления изобретения гетероциклоалкильная группа является моноциклической и имеет 5 или 6 кольцевых атомов. В одном варианте осуществления изобретения гетероциклоалкильная группа является моноциклической. В другом варианте осуществления изобретения гетероциклоалкильная группа является бициклической. В кольцевой системе нет соседних атомов кислорода и/или серы. Любая -NH группа в гетероциклоалкильном кольце может быть защищена, например, -N(Вос), -N(Cbz), -N(Tos) и тому подобное; такие защищенные гетероциклоалкильные группы считаются частью настоящего изобретения. Гетероциклоалкильная группа может быть необязательно замещена одним или несколькими «заместителями кольцевой системы», которые могут быть одинаковыми или различными, и они являются такими, как определено ниже в настоящем описании. Атом азота или атом серы в гетероциклоалкиле (если он присутствует) может быть необязательно окислен до соответствующего N-оксида, S-оксида или S,S-диоксида. Не ограничивающие примеры моноциклических гетероциклоалкильных колец включают оксетанил, пиперидил, пирролидинил, пиперазинил, морфолинил, тиоморфолинил, тиазолидинил, 1,4-диоксанил, тетрагидрофуранил, дельта-лактам, дельта-лактон, силациклонентан, силапирролидин и тому подобное, и все их изомеры.
«Лекарственно устойчивый», в связи с грамотрицательным бактериальным штаммом, означает штамм, на который больше не действует по меньшей мере одно из ранее эффективных лекарственных средств; который приобрел способность противостоять антибиотическому действию по меньшей мере одного из ранее эффективных лекарственных средств. «Множественная лекарственная устойчивость», в связи со штаммом бактерий, означает штамм, которое больше не восприимчив к двум или более лекарственным средствам, которые ранее были эффективны; который приобрел способность противостоять антибиотическому действию двух или более ранее эффективных лекарственных средств. Штамм с лекарственной устойчивостью может предавать эту способность противостояния своему потомству. Указанная устойчивость может возникать из-за случайных генетических мутаций в бактериальной клетке, которая изменяет его чувствительность к одному лекарственному средству или к различным лекарственным средствам.
«Гетероциклоалкенил» означает неароматическое моноциклическое, бициклическое или мостиковое карбоциклическое кольцо или кольцевую систему, содержащую по меньшей мере одну двойную связь, и содержащую по меньшей мере один гетероатом, выбранный из N, NH, S и O.
«Гетероарил» означает моноциклическое, бициклическое или трициклическое кольцо или кольцевую систему, содержащую 5-14 атомов углерода, и содержащее по меньшей мере один кольцевой гетероатом, выбранный из N, NH, S (включая SO и SO2) и О, где по меньшей мере одно из содержащих гетероатом колец является ароматическим. В случае гетероарильной кольцевой системы, где одно или несколько колец являются насыщенными и содержащими один или несколько атомов N, то N может быть в виде четвертичного амина. В одном варианте осуществления изобретения гетероарильная группа содержит в кольце от 5 до 10 атомов. В другом варианте осуществления изобретения гетероарильная группа является моноциклической, и содержит 5 или 6 кольцевых атомов. В другом варианте осуществления изобретения, гетероарильная группа является бициклической. Гетероарильная группа может быть необязательно замещена одним или несколькими заместителями кольцевой системы, которые могут быть одинаковыми или различными. Любой атом азота в гетероариле может быть необязательно окислен до соответствующего N-оксида. Термин «гетероарил» охватывает также гетероарильную группу, как определено выше, которая конденсирована с бензольным кольцом. Примеры гетероарила включают пирролил, изоксазолил, изотиазолил, пиразолил, пиридил, оксазолил, оксадиазолил, тиадиазолил, тиазолил, имидазолил, триазолил, тетразолил, фуранил, триазинил, тиенил, пиримидил, пиридазинил, пиразинил, бензизоксазолил, бензоксазолил, бензотиазолил, бензимидазолил, бензопиразолил, бензофуранил, бензотиофенил (включая S-оксид и диоксид), бензотриазолил, фуро(2,3-b)пиридил, хинолил, индолил, изохинолил, хиназолинил, дибензофуранил и тому подобное. В одном из вариантов осуществления изобретения гетероарил выбран из пиридина, пиримидина, тиазола, бензимидазола, бензотиазола, бензоксазола и бензизоксазола. В другом варианте осуществления изобретения гетероарил представляет собой пиридин.
Примеры бициклических колец включают:
«Гетероцикл» означает моноциклическую или бициклическую насыщенную, частично ненасыщенную или ненасыщенную кольцевую систему, содержащую 5-10 атомов, и содержащую по меньшей мере один кольцевой гетероатом, выбранный из N, S и O. В некоторых вариантах осуществления изобретения кольцевая система содержит 1-4 гетероатома, выбранных из N, S и O. Когда гетероцикл содержит два кольца, то кольца могут быть конденсированными, мостиковыми или спироциклическими. Примеры моноциклических гетероциклических колец включают пиперазин, пиперидин и морфолин. Примеры бициклических гетероциклических колец включают 1,4-диазабицикло[2.2.2]октан и 2,6-диазаспирогептан.
Термин «галоген» включает фтор, хлор, бром и йод.
«Оксо» означает атом кислорода, связанный двойной связью с другим атомом, и он может быть представлен как «=O».
Когда любая переменная (например, R1, Ra и т.п.) встречается более чем один раз в любой структуре или в формуле I, II-1, II-2, II-3, III-1, III-2 или III-3, ее определение в каждом случае не зависит от ее определения в каждом другом случае. Кроме того, комбинации заместителей и/или переменных допустимы, если только такие комбинации приводят к получению стабильных соединений. Волнистая линия поперек связи в переменном или заместителе представляет собой точку присоединения.
«Стабильное» соединение представляет собой соединение, которое может быть получено и выделено, и структура и свойства которого остаются или могут оставаться по существу неизменными в течение периода времени, достаточного, чтобы позволить применить соединение для целей, описанных здесь (например, для терапевтического введения субъекту). Соединения настоящего изобретения ограничены стабильными соединениями, которые охватываются формулой I, II-1, II-2, II-3, III-1, III-2 или III-3.
По стандартной номенклатуре, используемой в данном описании, концевая часть указанной боковой цепи описана последней, а перед ней описана присоединенная к ней предшествующая функциональная группа по направлению к точке присоединения.
При выборе соединений настоящего изобретения, обычному специалисту в данной области техники понятно, что различные заместители, т.е. R1, R2 и т.п., должны быть выбраны в соответствии с хорошо известными принципами химической связи и стабильностью структуры.
Термин «замещенный» включает множество уровней замещения указанными заместителем. Когда несколько фрагментов заместителей раскрыты или заявлены, замещенное соединение может быть независимо замещено одним или несколькими из раскрытых или заявленных замещающих фрагментов, по-отдельности или в комбинации. Замещение независимо друг от друга означает, что (два или более) заместителя могут быть одинаковыми или различными. Когда группа, например, C1-С8 алкил, указана как замещенная, такие замещения могут также иметь место, когда такая группа является частью более крупного заместителя, например, C1-С6алкил-C3-С7циклоалкила и C1-C8алкил-арила.
В соединениях формулы I, II-1, II-2, II-3, III-1, III-2 или III-3, атомы могут иметь свои природные изотопные формы, или один или несколько атомов могут быть искусственно обогащены конкретным изотопом, имеющий тот же атомный номер, но атомная масса или массовое число которого отличается от атомной массы или массового числа изотопа, преимущественно встречающегося в природе. Настоящее изобретение включает все подходящие изотопные варианты соединений формулы I, II-1, II-2, II-3, III-1, III-2 или III-3. Например, различные изотопные формы водорода (H) включают протий (1Н) и дейтерий (2Н или D). Протий является преобладающим изотопом водорода в природе. Обогащение дейтерием может дать определенные терапевтические преимущества, такие как увеличение времени полужизни соединения в условиях in vivo, или снижение требуемой дозировки, или это может обеспечить соединение, используемое в качестве стандарта для определения характеристик биологических образцов. Изотопно-обогащенные соединения формулы I, II-1, II-2, II-3, III-1, III-2 или III-3 могут быть получены без излишних экспериментов с помощью обычных методик, хорошо известных специалистам в данной области техники, или способами, аналогичными тем, которые описаны в схемах и примерах в данном описании, с использованием соответствующих изотопно-обогащенных реагентов и/или промежуточных соединений.
Если в конкретном контексте специально не указано иное, то любое из различных циклических колец и кольцевых систем, описанных в настоящей заявке, могут быть присоединены к остальной части соединения при любом кольцевом атоме (т.е., при любом атоме углерода или при любом гетероатоме), при условии, что соединение будет устойчивым.
Если специально не указано иное, все диапазоны и интервалы, указанные здесь, являются включительными. Например, если указано гетероароматическое кольцо, содержащее от «1 до 4 гетероатомов», то это означает, что кольцо может содержать 1, 2, 3 или 4 гетероатома. Кроме того, следует понимать, что любой диапазон или интервал, указанный здесь, включает в свой объем все поддиапазоны в пределах этого диапазона. Так, например, гетероциклическое кольцо, указанное как содержащее от «1 до 4 гетероатомов» предусматривает, что включены его аспекты, т.е. гетероциклические кольца, содержащие от 2 до 4 гетероатомов, 3 или 4 гетероатома, от 1 до 3 гетероатомов, 2 или 3 гетероатома, 1 или 2 гетероатома, 1 гетероатом, 2 гетероатома, 3 гетероатома и 4 гетероатома. Аналогично, указание C1-C6, при использовании с цепью, например, с алкильной цепью, означает, что цепь может содержать 1, 2, 3, 4, 5 или 6 атомов углерода. Это указание также включает все диапазоны, охватываемые этим определением, включая C1-C5, C1-C4, C1-C3, C1-C2, C2-C6, C3-C6, C4-C6, C5-C6 и все другие возможные комбинации.
Следует также отметить, что предполагается, что у любого атома углерода, а также гетероатома с ненасыщенными валентностями, из числа указанных в тексте, схемах, примерах и таблицах настоящего документа, имеют достаточное количество атомов водорода, чтобы удовлетворить требования валентности.
Соединения настоящего изобретения имеют по меньшей мере один асимметричный центр и могут иметь один или несколько дополнительных центров в результате присутствия определенных заместителей и/или комплекса заместителей. Соответственно, соединения по настоящему изобретению могут существовать в виде смеси стереоизомеров или в виде отдельных диастереомеров или энантиомеров. Все изомерные формы этих соединений, по-отдельности или в смесях, входят объем настоящего изобретения.
Термин «соединение» относится к соединению в свободном виде, и, при условии, что они являются стабильными, к любому его гидрату или сольвату. Гидрат представляет собой соединение в виде комплекса с водой, и сольват представляет собой соединение в виде комплекса с органическим растворителем.
Как указано выше, соединения по изобретению могут быть использованы в виде фармацевтически приемлемых солей. Следует понимать, что, как использовано здесь, соединения по настоящему изобретению могут также включать фармацевтически приемлемые соли, а также соли, которые не являются фармацевтически приемлемыми, когда они используются в качестве предшественников свободных соединений или их фармацевтически приемлемых солей, или в процессах синтеза.
Термин «фармацевтический приемлемая соль» относится к соли, которая обладает эффективностью исходного соединения и которая не является биологически или иным образом нежелательной (например, не является токсичным или иным образом вредным для пациента). Термин «фармацевтически приемлемая соль» относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований или кислот, включая неорганические или органические основания и неорганические или органические кислоты. Соли основных соединений, охватываемых термином «фармацевтически приемлемая соль», относятся к нетоксичным солям соединений по изобретению, которые обычно получают путем взаимодействия свободного основания с подходящей органической или неорганической кислотой. Иллюстративные соли основных соединений по изобретению, включают, но без ограничения, следующие: ацетат, аскорбат, адипат, альгинат, аспират, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, бутират, камфорат, камфорсульфонат, камсилат, карбонат, хлорид, клавуланат, цитрат, циклопентанпропионат, диэтилацетат, диглюконат, дигидрохлорид, додецилсульфонат, эдетат, эдизилат, эстолат, эзилат, этансульфонат, формиат, фумарат, глюцептат, глюкогептаноат, глюконат, глутамат, глицерофосфат, гликоллиларсанилат, гемисульфат, гептанат, гексаноат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, 2-гидроксиэтансульфонат, гидроксинафтоат, иодид, изоникотинат, изотионат, лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилбромид, метилнитрат, метилсульфат, метансульфонат, мукат, 2-нафталинсульфонат, напсилат, никотинат, нитрат, соль N-метилглюкаминаммония, олеат, оксалат, памоат (эмбонат), пальмитат, пантотенат, пектинат, персульфат, фосфат/дифосфат, пимелинат, фенилпропионат, полигалактуронат, пропионат, салицилат, стеарат, сульфат, субацетат, сукцинат, таннат, тартрат, теоклат, тиоцианат, тозилат, триэтиодид, трифторацетат, ундеканоат, валерат и тому подобное. Кроме того, если соединения по изобретению несут кислотную группу, подходящие фармацевтически приемлемые соли включают, но без ограничения, соли, полученные из неорганических оснований, в том числе соли алюминия, аммония, кальция, меди, трехвалентного железа, лития, магния, марганца, двухвалентного марганца, калия, натрия, цинка и тому подобное. Особенно предпочтительными являются соли аммония, кальция, магния, калия и натрия. Соли, полученные из фармацевтически приемлемых нетоксичных органических оснований, включают соли первичных, вторичных и третичных аминов, циклических аминов, дициклогексиламинов и основных ионообменных смол, и включают такие соли, как соли аргинина, бетаина, кофеина, холина, N,N-дибензилэтилендиамина, диэтиламина, 2-диэтиламиноэтанола, 2-диметиламиноэтанола, этаноламина, этиламина, этилендиамина, N-этилморфолина, N-этилпиперидина, глюкамина, глюкозамина, гистидина, гидрабамина, изопропиламина, лизина, метилглюкамина, морфолина, пиперазина, пиперидина, полиаминных смол, прокаина, пурина, теобромина, триэтиламина, триметиламина, трипропиламина, трометамина и тому подобное. Кроме того, также включены основные азотсодержащие группы, которые могут быть кватернизованы такими агентами, как низшие алкилгалогениды, такие как метил, этил, пропил и бутил хлориды, бромиды и иодиды; диалкилсульфаты, такие как диметил, диэтил, дибутил и диамил сульфаты, галогениды с длинной цепью, такие как децил, лаурил, миристил и стеарил хлориды, бромиды и иодиды, аралкил галогениды, такие как бензил и фенэтил бромиды, и другие.
Эти соли могут быть получены известными способами, например, путем смешивания соединения по изобретению, с эквивалентным количеством и раствором, содержащим требуемую кислоту, основание или тому подобное, с последующим получением нужной соли путем фильтрования соли или отгонкой растворителя. Соединения по изобретению и их соли могут образовывать сольваты с растворителем, таким как вода, этанол или глицерин. Соединения по изобретению могут образовывать кислотно-аддитивную соль и соль с основанием, в зависимости от типа заместителя в боковой цепи.
Соединение по изобретению также может быть использовано в виде пролекарства. Любой известный в данной области техники предшественник пролекарства может быть использован для получения пролекарства по изобретению. В некоторых аспектах этого варианта осуществления изобретения, водород в -СООН в формуле I может быть заменен на любую из следующих групп: C1-6алкил, C3-6циклоалкил, -C1-6алкилен-C3-6циклоалкил, C3-7циклогетероалкил, -C1-6алкилен-C3-7циклогетероалкил, арил, -C1-10алкилен-арил, гетероарил и -C1-10алкилен-гетероарил. В некоторых аспектах этого варианта осуществления изобретения, C1-6алкил, C3-6циклоалкил или C3-7циклогетероалкил может быть замещенным. В других аспектах этого варианта осуществления изобретения каждый арил и гетероарил может быть замещенным.
Как указано выше, настоящее изобретение включает фармацевтические композиции, содержащие соединение формулы I по изобретению, необязательно, один другой активный компонент (например, ингибитор β-лактамазы) и фармацевтический приемлемый носитель. Характеристики носителя будут зависеть от пути введения. Под указанием «фармацевтически приемлемый» понимают, что ингредиенты фармацевтической композиции должны быть совместимы друг с другом, не мешать эффективности активного ингредиента(ов), а также не быть вредными (например, токсичными) для пациента. Таким образом, композиции согласно изобретению могут, в дополнение к ингибитору, содержать разбавители, наполнители, соли, буферы, стабилизаторы, солюбилизаторы и другие материалы, хорошо известные в данной области техники.
Кроме того, как указано выше, настоящее изобретение относится к способу лечения бактериальной инфекции, где способ включает введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы I, II-1, II-2, II-3, III-1, III-2 или III-3, или его фармацевтически приемлемой соли, необязательно в комбинации с ингибитором β-лактамазы. Термин «субъект» (или, в качестве альтернативы, «пациент»), используемый здесь, относится к животному, предпочтительно млекопитающему, наиболее предпочтительно человеку, который является объектом лечения, наблюдения или эксперимента. Термин «введение» и его варианты (например, «вводить») по отношению к соединению формулы I, II-1, II-2, II-3, III-1, III-2 или III-3, означает, что обеспечивается доставка соединения или его фармацевтически приемлемой соли человеку, нуждающемуся в лечении. Когда соединение или его соль используется в сочетании с одним или несколькими другими активными агентами (например, ингибитором β-лактамазы), то под термином «введение» и его вариантами понимается доставка соединения или его соли, а также других агентов, в одно и то же время или в разное время. Когда одновременно вводят агенты в комбинации, то их можно вводить вместе в составе одной композиции, или они могут быть введены по-отдельности. Следует понимать, что «комбинация» активных агентов может быть как одна композиция, содержащая все активные агенты, или как несколько композиций, каждая из которых содержит один или несколько активных агентов. В случае двух активных агентов, комбинация может быть либо одной композицией, содержащей оба агента, или двумя отдельными композициями, каждая из которых содержит один из агентов; в случае трех активных агентов комбинация может быть либо одной композицией, содержащей все три вещества, либо тремя отдельными композициями, каждая из которых содержит один из агентов, или двумя композициями, одна из которых содержит два агента, а другой содержит третий агент; и т.д.
Композиции и комбинации по изобретению предпочтительно вводят в эффективных количествах. Термин «эффективное количество», используемое в настоящем описании, означает количество активного вещества, достаточное для ингибирования роста бактерий, и тем самым вызвать требуемый ответ (т.е., «ингибирующий эффект») в клетке, ткани, системе, у животного или у человека. В одном варианте осуществления изобретения, эффективное количество представляет собой «терапевтически эффективное количество», облегчающее симптомы заболевания или состояния, подлежащего лечению (например, излечение состояний, связанных с бактериальной инфекцией и/или с бактериями, устойчивыми к лекарственному средству). В другом варианте осуществления изобретения, эффективное количество представляет собой «профилактически эффективное количество» для профилактики симптомов заболевания или состояния, возникновение или развитие которого предотвращается. Когда активное соединение (т.е. активный ингредиент) вводят в виде соли, указания, касающиеся количества активного ингредиента, относятся к соединению в виде свободной кислоты или в виде свободного основания.
Композиции по настоящему изобретению применимы для парентерального, перорального, сублингвального, трансдермального, местного, интраназального, трахеального, внутриглазного или ректального введения, при этом композиция подходящим образом изготовлена для введения выбранным путем с использованием методов, хорошо известных в данной области техники, в том числе, например, способами получения и введения композиций, описанных в главах 39, 41, 42, 44 и 45 справочника Remington - Science and Practice of Pharmacy, 21st edition, 2006. В одном варианте осуществления изобретения, введение соединения по изобретению осуществляют внутривенно в условиях стационара. В другом варианте осуществления изобретения введение осуществляют перорально, при этом композиция представлена в форме таблетки или капсулы, или тому подобного. При системном введении, терапевтическую композицию, например, соответствующим образом вводят в дозе, достаточной для достижения в крови уровня ингибитора, составляющего по меньшей мере приблизительно 1 мкг/мл, а в еще одном варианте осуществления изобретения - по меньшей мере приблизительно 10 мкг/мл, и по меньшей мере приблизительно 25 мкг/мл. Для локализованного введения могут быть эффективны гораздо более низкие концентрации, чем указанные, и гораздо более высокие концентрации могут быть переносимыми.
Внутривенное введение соединения по изобретению может быть выполнено с восстановлением порошкообразной формы соединения приемлемым растворителем. Подходящие растворители включают, например, солевые растворы (например, раствор 0,9% хлорида натрия для инъекций) и стерильную воду (например, стерильную воду для инъекций, бактериостатическую воду для инъекций с метилпарабеном и пропилпарабеном, или бактериостатическую воду для инъекций с 0,9% бензилового спирта). Порошкообразную форму соединения можно получить с использованием гамма-облучения соединения или путем лиофилизации раствора соединения, после чего порошок можно хранить (например, в герметичном флаконе) при комнатной температуре или ниже до тех пор, пока он не будет восстановлен. Концентрация соединения в восстановленном растворе для внутривенного введения (IV) может быть, например, в диапазоне от приблизительно 0,1 мг/мл до приблизительно 20 мг/мл.
Настоящее изобретение также включает способ ингибирования роста бактерий, где способ включает введение в культуру бактериальных клеток или в инфицированную бактериями культуру клеток, ткань или организм, эффективное ингибирующее количество соединения формулы I, II-1, II-2, II-3, III-1, III-2 или III-3. Дополнительные варианты осуществления настоящего изобретения включают способ ингибирования роста бактерий, как указано выше, где соединение по изобретению, используемое в этом способе, представляет собой соединение согласно одному из вариантов осуществления, частных вариантов или классов, как описано выше. В этих вариантах осуществления изобретения, соединение необязательно может быть применено в форме фармацевтически приемлемой соли. Способ может включать введение соединения формулы I, II-1, II-2, II-3, III-1, III-2 или III-3 в экспериментальную культуру клеток in vitro, чтобы предотвратить рост бактерий, устойчивых к β-лактамам. Способ может альтернативно включать введение соединения формулы I животному, включая человека, для предотвращения роста бактерий, устойчивых к β-лактамам, в условиях in vivo. В этих случаях соединение формулы I, II-1, II-2, II-3, III-1, III-2 или III-3, как правило, вводят совместно с ингибитором β-лактамазы.
Способы, раскрытые в заявленных объектах, являются полезными для лечения этих состояний тем, что они ингибируют начало, рост или развитие состояния, вызывают регрессию состояния, излечивают состояние, или иным образом улучшают общее состояние субъекта, страдающего или подвергающегося опасности возникновения такого состояния. Таким образом, в соответствии с раскрытиями заявленных объектов, термины «лечить», «лечение» и их грамматические варианты, а также выражение «способ лечения», предназначены для охвата любого желаемого терапевтического вмешательства, в том числе, но без ограничения, способа лечения существующей инфекции у субъекта, а также к способу профилактики (т.е., предотвращению) инфекции у субъекта, такого как у субъекта, который подвергся воздействию микроба, как описано здесь, или в отношении которого ожидается возможность воздействия микроба, как описано здесь.
Соединения по изобретению могут быть использованы для лечения, профилактики инфекций или для ингибирования роста бактерий, которые являются устойчивыми к β-лактамным антибиотикам. Более конкретно, бактерии могут быть металло-β-лактамазными положительными штаммами, которые обладают высокой устойчивостью к β-лактамным антибиотикам. Термины «слабоустойчивые» и «высокоустойчивые» хорошо известны и понятны специалистам в данной области (см., например, Payne et al., Antimicrobial Agents and Chemotherapy 38:767-772 (1994); Hanaki et al., Antimicrobial Agents and Chemotherapy 30:11.20-11.26 (1995)). Для целей настоящего изобретения, к бактериальным штаммам, которые обладают высокой устойчивостью к имипенему, относятся штаммы, у которых MIC к имипенему составляет > 16 мкг/мл, а к бактериальным штаммам, которые обладают слабой устойчивостью к имипенему, относятся штаммы, у которых MIC к имипинему составляет > 4 мкг/мл.
Соединения по изобретению могут быть использованы в комбинации с ингибитором β-лактамазы для лечения инфекций, вызванных штаммами, продуцирующими β-лактамазу, в дополнение к тем инфекциям, которые входят спектр антибактериального действия антибиотика. Примеры бактерий, продуцирующих β-лактамазу, являются Pseudomonas aeruginosa, Pseudomonas putida, Enterobacter cloacae, Klebsiella pneumoniae, Klebsiella oxytoca, Escherichia coli, Serratia marcescens, Enterobacter aerogenes, Enterobacter asburiae, Citrobacter freundii, Proteus mirabilis, Morganella morganii, Providencia rettgeri, Stenotrophomonas maltophilia и Acinetobacter baumannii.
Как правило, целесообразно использовать соединение формулы I, II-1, II-2, II-3, III-1, III-2 или III-3 в смеси или сочетании с ингибитором β-лактамазы или его пролекарства. Полезно использовать соединение формулы I в сочетании с ингибитором β-лактамазы класса А и C, из-за устойчивости β-лактамаз класса В к соединениям по изобретению. Кроме того, предпочтительно использовать соединение формулы I в комбинации с одним или несколькими ингибиторами β-лактамаз классов A, C или D для дополнительного ограничения стойкости к β-лактамам. Как уже отмечалось, соединение формулы I и ингибитор β-лактамазы могут быть введены раздельно (одновременно или в разное время), либо в виде одной композиции, содержащей оба активных ингредиента.
Релебактам, тазобактам, клавулановая кислота, сульбактам, авибактам и другие ингибиторы β-лактамазы и металло-β-лактамаз, пригодные для использования в настоящем изобретении, включают те, в отношении которых известно, что они обладают ингибирующей активностью в отношении β-лактамаз.
Сокращения и аббревиатуры, используемые в настоящем документе, включают следующее. Водн. (aq.)=водный; ACN=ацетонитрил; BLI=ингибитор β-лактамазы; Bn=бензил; BOC (или Boc)=t-бутилоксикарбонил или трет-бутилоксикарбонил; BOC2O=ди-трет-бутилдикарбонат; CAN= церий-аммоний нитрат; CBZ (или Cbz)=карбобензокси (альтернативно, бензилоксикарбонил); CDCl3= дейтерированный хлороформ; CH3CN=ацетонитрил; Co-катализатор=(R,R')-N,N'-бис(3,5-ди-трет-бутилсалицилиден)-1,2-циклогександиамино-кобальт(III)-1,1,1,3,3,3-гексафтор-2-(трифторметил)пропан-2-олат; cv=объем колонки; DBU=1,8-диазабицикло[5.4.0]ундец-7-ен; DCC=дициклогексил карбодиимид; DCE=дихлорэтан; DCM=дихлорметан; DEAD= диэтил азодикарбоксилат; DIAD= диизопропил азодикарбоксилат; DIPEA=ди-изопропилэтиламин; DMA=диметилацетамид; DMAP=4-диметиламинопиридин или N,N-диметиламино-пиридин; DMF=N,N-диметилформамид; DMSO=диметилсульфоксид; EDC=1-этил-3-(3-диметиламинопропил) карбодиимид; eq. (экв.) или equiv.=эквивалент(ы); Et=этил; Et3N=триэтиламин; Et2O=этиленоксид; EA или EtOAc=этилацетат; EtOH=этанол; г=грамм(ы); час или ч=час(ы); hex=гексан; HiVac=высокий вакуум; HMDS=гексаметил-дисилазид; HOBT=1-гидроксибензотриазол; HPLC=высокоэффективная жидкостная хроматография; IPA=изопропиловый спирт; iPrMgCl=изопропил магний хлорид; IPAc=изопропилацетат; л=литр(ы); LC/MS или LC-MS=жидкостная хроматография/масс-спектрометрия; LDA=литий диизопропиламид; мин=минута(ы); мг=миллиграмм(ы); мл=миллилитр(ы); m-CPBA=м-хлорпероксибензойная кислота; MBL=металло-β-лактамаза; Me=метил; MeCN=ацетонитрил; MeOH=метанол; MeI=метилиодид; MITC=минимальная порговая ингибирующая концентрация; MOPS=3-(N-морфолино)пропансульфоновая кислота; MPLC=жидкостная хроматография среднего давления; MTBE=метил-трет-бутиловый эфир; NBS=N-бромсукцинимид; NCS=N-хлорсукцинимид; ЯМР=ядерный магнитный резонанс; MS=масс-спектрометрия; MW=молекулярная масса; Pd/c=палладий на угле; PdCl2(dppf)=[1,2'-бис(дифенил-фосфино)-ферроцен] дихлорпалладий(II); ди-t-BuDPPF-PdCl2=дихлорид 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладия; PE=петролейный эфир; PG=защитная группа; Ph=фенил; PPTS=пиридиний п-толуолсульфонат; RP-HPLC= высокоэффективная жидкостная хроматография с обращенной фазой; r.t. или RT=комнатная температура; нас.=насыщенный; tBu=трет-бутил; TBAI=тетрабутиламмоний иодид; TBAF=тетрабутиламмоний фторид; TBS= трет-бутилдиметилсилил; TBS-Cl=трет-бутилдиметилсилилхлорид; TBDMS-Cl=трет-бутилдиметилсилилхлорид; t-BuOH=трет-бутанол; TBSO=трет-бутилдиметилсилил; TEA=триэтиламин; TEMPO=(2,2,6,6-тетраметилпиперидин-1-ил)оксил; TFA=трифторуксусная кислота; THF=тетрагидрофуран; TLC=тонкослойная хроматография; TMS= триметилсилил; TMS-Cl=триметилсилил хлорид; TMS-I=триметилсилил иодид; и TMS-N3=триметилсилил азид.
Способы получения соединений формулы (I)
Соединения, описанные здесь, могут быть получены и испытаны в соответствии со следующими схемами реакций и примерами, или их модификациями, с использованием легкодоступных исходных материалов, реагентов и обычных методик синтеза. В этих реакциях также можно использовать их варианты, которые известны специалистам в данной области техники, но которые не упомянуты здесь более подробно. Кроме того, другие способы получения соединений, описанных здесь, являются очевидными для обычного специалиста в данной области техники, в свете приведенных схем реакций и примеров. Если не указано иное, все переменные являются такими, как определено выше.
Схема 1
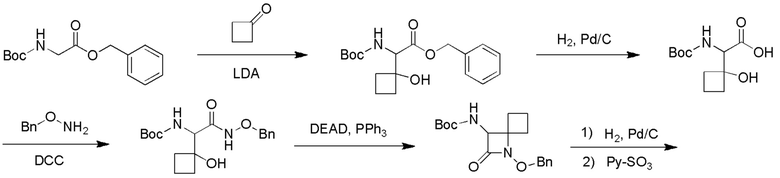
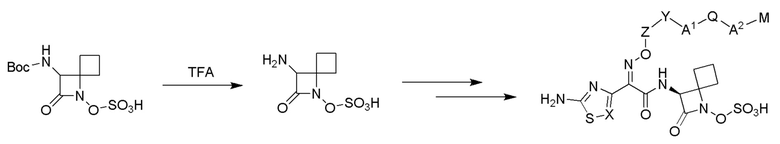
β-лактамное промежуточное соединение может быть либо приобретено из коммерческих источников или синтезировано по схеме, где показан синтез β-лактамных аналогов, где Rx и Rz вместе образуют 4-членное спироциклическое кольцо. Схема синтеза была подробно описана ранее в литературе (см. EP 0229012). Этот амин может быть превращен в конечные монобактамные соединения процедурой, аналогичной той, которая показана в следующих примерах.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 1
Трет-бутил оксиран-2-карбоксилат
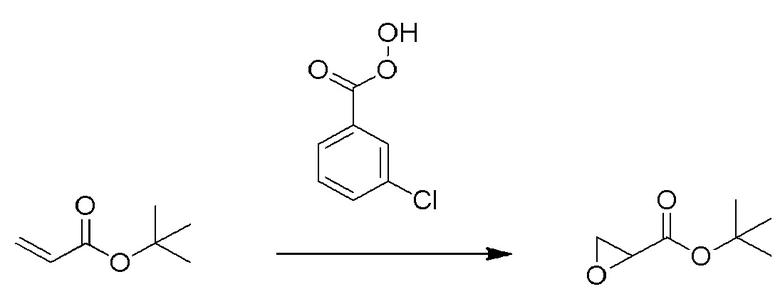
В 1-литровую 2-горлую колбу с круглым дном, снабженную холодильником, содержащую раствор трет-бутилакрилата (20 г, 156 ммоль) в DCM (200 мл) порциями добавляли m-CPBA (48,5 г, 281 ммоль). Полученный раствор нагревали до 58-60°C на масляной бане и кипятили с обратным холодильником в течение 2 с половиной дней. Затем смесь охлаждали до комнатной температуры, добавляли небольшими порциями насыщенный раствор тиосульфата натрия (приблизительно 40 мл, экзотермический эффект, добавляли небольшими порциями, пока не прекращалось выделение тепла). После перемешивания в течение приблизительно 1 часа, образовывалось большое количество осадка. Для разбавления эмульсии и образования систему из двух фаз добавляли приблизительно 60-100 мл воды и 100-200 мл DCM. Водную фазу отделяли и органическую фазу промывали насыщенным раствором NaHCO3 (2×200 мл) и солевым раствором (100 мл), сушили над MgSO4 и концентрировали досуха (температура воды в ванне 35°С). Твердое вещество суспендировали в 150 мл гексана и оставляли стоять при комнатной температуре в течение 1 часа. Затем смесь фильтровали, и фильтрат концентрировали удалением гексана в роторном испарителе (< 35°C), получая указанное в заголовке соединение.
1H-ЯМР (500 МГц, CDCl3) δ 3,35 (м, 1H), 2,86 (м, 2H), 1,52 (с, 9H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 2
(R,R)-Co-катализатор
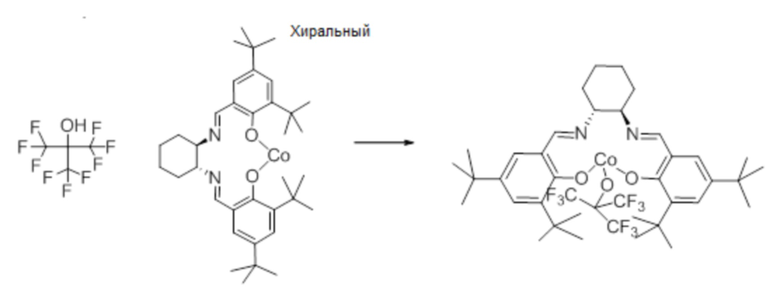
Ссылка: J. Am. Chem. Soc. 1999, 121, 6086-6087.К раствору перфтор-трет-бутанола (1,96 г, 8,28 ммоль) в DCM (97 мл) добавляли раствор (R,R)-(-)-N,N'-бис(3,5-ди-трет-бутилсалицилиден)-1,2-циклогександиаминокобальт(II) (5 г, 8,28 ммоль). Раствор перемешивали при 30°С в течение 45 минут при доступе воздуха. Реакционную смесь концентрировали, сушили в высоком вакууме, получая указанное в заголовке соединение.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 3
Трет-бутил-(R)-3-(4-бромфенокси)-2-гидроксипропаноат
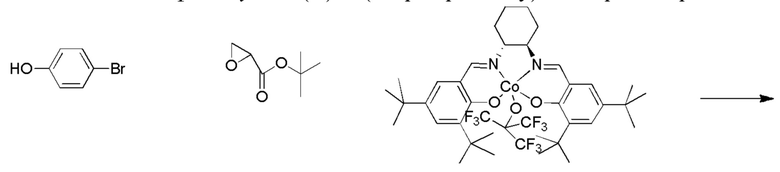
К смеси 4-бромфенола (2 г, 11,56 ммоль), молекулярного сита 4Å (4 г) и трет-бутил-оксиран-2-карбоксилата (3,67 г, 25,4 ммоль) в атмосфере N2 добавляли трет-бутилметиловый эфир (50 мл) и (R,R)-Co-катализатор (0,97 г, 1,15 ммоль). Полученную суспензию перемешивали при комнатной температуре в атмосфере N2 в течение уикенда. LC-MS показала конверсию приблизительно 60%. Добавляли дополнительное количество эпоксида промежуточного соединения 1 (0,5 г) и Co-катализатора (100 мг), и перемешивали в течение ночи. LC-MS показала конверсию приблизительно 80%. Добавляли еще эпоксид (0,5 г) и катализатор (100 мг), и перемешивали в течение ночи. Смесь фильтровали и остаток промывали EtOAc. Фильтрат концентрировали, и остаток очищали на колонке с силикагелем (240г) с использованием 0-100% EtOAc/Hex, получая указанное в заголовке соединение.
1H-ЯМР (500 МГц, CDCl3) δ 7,31 (д, J=7,2 Гц, 2H), 6,83 (д, J=7,2 Гц, 2H), 4,45 (м, 1H), 4,28 (м, 2H), 1,52 (с, 9H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 4
Трет-бутил-(R)-2-гидрокси-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)пропаноат

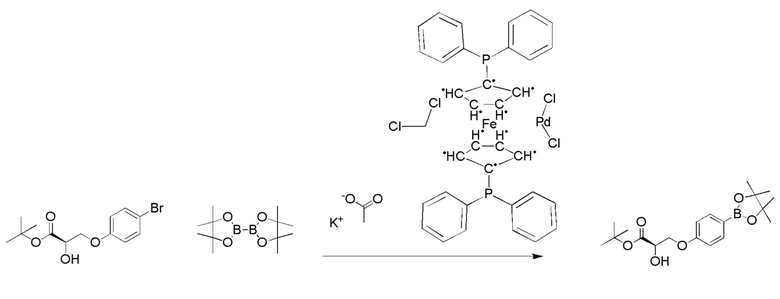
К раствору (R)-трет-бутил-3-(4-бромфенокси)-2-гидроксипропаноата (1,95 г, 6,15 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (2,342 г, 9,22 ммоль), и комплекса дихлорида 1,1'-бис(дифенилфосфино)ферроцен-палладия (II) с дихлорметаном (0,309 г, 0,378 ммоль) в 1,4-диоксане (22 мл) добавляли ацетат калия (1,81 г, 18,4 ммоль). Смесь дегазировали, продували азотом и нагревали при 85°С в течение ночи. Смесь фильтровали и концентрировали досуха в вакууме. Остаток очищали с помощью хроматографии на колонке с силикагелем (120 г) и элюировали смесью гексан/этилацетат (0-50%), получая указанное в заголовке соединение.
LC-MS [M+H+Na]: m/z 387,00.
1H-ЯМР (500 МГц, CDCl3) δ 7,67 (д, J=7,2 Гц, 2H), 6,82 (д, J=7,2 Гц, 2H), 4,33 (м, 1H), 4,16 (м, 2H), 1,41 (с, 9H), 1,29 (с, 12H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 5
2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусная кислота
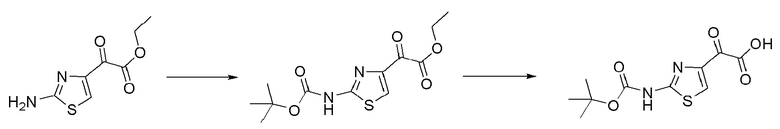
Стадия A: этил-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоацетат
К раствору этил-2-(2-аминотиазол-4-ил)-2-оксоацетата (10 г, 49,9 ммоль) в ацетонитриле (250 мл) добавляли Вос-ангидрид (23,2 мл, 100 ммоль), затем N,N,N',Ν'-тетраметилэтилендиамин (9,80 мл, 64,9 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов. Растворитель удаляли, и остаток разделяли между EtOAc и 1Н HCl. Органический слой промывали NaHCO3 (насыщенный водный раствор), насыщенным раствором соли, сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (redi flash 220г), элюировали смесью EtOAc/гексан (0-30%, 5 объемов колонки (cv); 30%, 10 cv), получая указанное в заголовке соединение. LC-MS [М+Н]+: m/z 301.
Стадия B: 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусная кислота
Этил-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоацетат (10,2 г, 34,1 ммоль) растворяли в THF (140 мл)/MeOH (50 мл) и добавляли гидроксид натрия (68,3 мл, 68,3 ммоль, 1М). Раствор перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь выливали в воду (1 л) и экстрагировали EtOAc (3×200 мл). Водный слой подкисляли раствором HCl (1Н) и повторно экстрагировали EtOAc (3×200 мл). Органический слой промывали насыщенным раствором соли и сушили над Na2SO4 и концентрировали, получая указанное в заголовке соединение.
LC-MS [M+H] +: m/z 273. 1H-ЯМР (500 МГц, CDCl3) δ 8,35 (с,1H), 1,55 (с, 9H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 6
2-(2-((трет-бутоксикарбонил)амино)-5-хлортиазол-4-ил)-2-оксоуксусная кислота
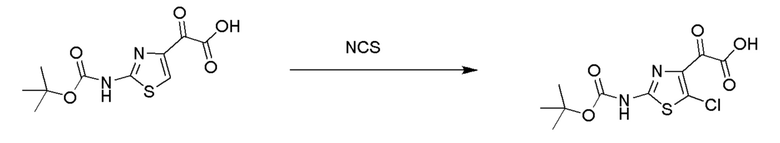
К суспензии 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусной кислоты (промежуточное соединение 5) (1 г, 3,67 ммоль) в DMF (10,0 мл) добавляли NCS (0,589 г, 4,41 ммоль). Смесь нагревали при 50°С в течение ночи. Затем смесь разбавляли EtOAc (100 мл), промывали водой (3×30 мл) и насыщенным раствором соли. Органический слой сушили над Na2SO4. Растворитель удаляли в вакууме, получая указанное в заголовке соединение. LC-MS [М+Н]+: m/z 307.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 7
2-(5-((трет-бутоксикарбонил)амино)-1,2,4-тиадиазол-3-ил)-2-оксоуксусная кислота
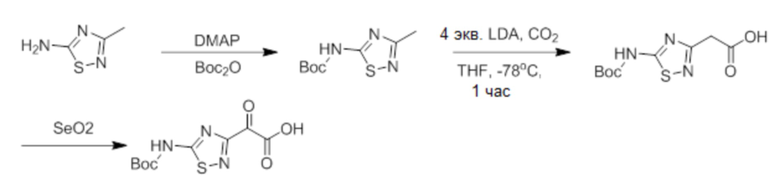
Стадия A: трет-бутил (3-метил-1,2,4-тиадиазол-5-ил)карбамат
5-литровую 4-горлую круглодонную колбу продували и создавали в ней инертную атмосферу азота. В нее помещали 3-метил-1,2,4-тиадиазол-5-амин (167 г, 1,45 моль, 1,00 экв.), 4-диметиламино-пиридин (17,7 г, 144,88 ммоль, 0,10 экв.), ди-трет-бутилдикарбонат (348 г, 1,59 моль, 1,10 экв.) и бутан-1-ол (1670 мл). Полученный раствор перемешивали в течение 1 часа при 40°C. Полученную смесь концентрировали в вакууме. Остаток промывали гексаном. В результате получали указанное в заголовке соединение.
Стадия B: 2-(5-((трет-бутоксикарбонил)амино)-1,2,4-тиадиазол-3-ил)уксусная кислота
5-литровую 4-горлую круглодонную колбу продували и создавали в ней инертную атмосферу азота. В нее помещали раствор трет-бутил-N-(3-метил-1,2,4-тиадиазол-5-ил)карбамата (128 г, 595 ммоль, 1,00 экв.) в тетрагидрофуране (640 мл). Полученный раствор перемешивали при температуре -78°С,, добавляли LDA (1190,69 мл, 4,00 экв.). Через 30 минут вводили СO2 (газ) в течение 30 минут при -30°C. Затем реакционную смесь гасили добавлением 1280 мл воды. Полученный раствор экстрагировали 640 мл этилацетата и водные слои объединяли. Значение рН раствора доводили до 2 с помощью НСl (2 моль/л). Полученный раствор экстрагировали 2,5 л этилацетата и органические слои объединяли. Полученную смесь промывали 2000 мл насыщенного солевого раствора. Смесь сушили над безводным сульфатом натрия и концентрировали в вакууме, в результате чего получая указанное в заголовке соединение.
Стадия C: 2-(5-((трет-бутоксикарбонил)амино)-1,2,4-тиадиазол-3-ил)-2-оксоуксусная кислота
2-литровую 4-горлую круглодонную колбу продували и создавали в ней инертную атмосферу азота. В нее помещали раствор 2-(5-[[(трет-бутокси)карбонил]амино]-1,2,4-тиадиазол-3-ил)уксусной кислоты (76 г, 293,12 ммоль, 1,00 экв.) в диоксане (1520 мл) и SeO2 (65,14 г, 587 ммоль, 2,00 экв.). Полученный раствор перемешивали в течение 3 часов при 80°С в масляной бане, а затем концентрировали. Неочищенный продукт очищали с помощью флэш-препаративной HPLC при следующих условиях (IntelFlash-1): колонка - силикагель; подвижная фаза - ACN/ H2O (0,5% НСl)=10/90-30/70 с увеличением до ACN/H2O (0,5% HCl)=90/10 до 70/30 в течение 20 минут; детектор - УФ 254 нм. В результате получали указанное в заголовке соединение.
LC-M: (ES, m/z): [M+H]+=274. 1H-ЯМР (300МГц, DMSO, м.д.): δ 1,523-1,502 (с, 9H), 12,806 (с, 1H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 8
трет-бутил 2-((трет-бутилдиметилсилил)окси)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)пропаноат
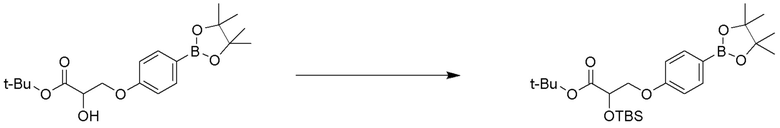
К раствору трет-бутил-2-гидрокси-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)пропионата (0,5 г, 1,37 ммоль) (промежуточное соединение 4), имидазола (0,467 г, 6,86 ммоль) и TBS-С1 в DCM (3,43 мл, 3,43 ммоль) в ацетонитриле (5 мл) добавляли DMAP (0,017 г, 0,137 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 3 часов. После концентрирования остаток очищали на колонке с силикагелем (40 г) с использованием EtOAc/гексан с градиентом 0-10%, получая указанное в заголовке соединение.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 9
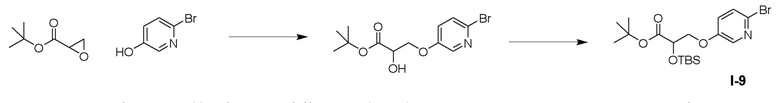
Стадия A: трет-бутил 3-((6-бромпиридин-3-ил)окси)-2-гидроксипропаноат
Карбонат калия (7,94 г, 57,5 ммоль) добавляли к перемешиваемой смеси 6-бромпиридин-3-ола (5 г, 28,7 ммоль) и трет-бутил-оксиран-2-карбоксилата (20,7 г, 144 ммоль) в CH3CN (60 мл), и смесь перемешивали при 90°С в течение 3 часов. Смесь охлаждали и фильтровали через тонкий слой целита, а затем промывали EtOAc. Растворитель удаляли при пониженном давлении, и остаток очищали на колонке ISCO (gold, 120 г), элюируя смесью EtOAc/гексан с градиентом 0-60%, получая указанное в заголовке соединение.
LC-MS [M+H]+: m/z 318,2.
Стадия B: трет-бутил 3-((6-бромпиридин-3-ил)окси)-2-((трет-бутилдиметил-силил)окси)пропаноат
К раствору трет-бутил-3-((6-бромпиридин-3-ил)окси)-2- гидроксипропаноата (6700 мг, 21,1 ммоль), имидазола (3440 мг, 50,5 ммоль) и TBDMS-Сl (3800 мг, 25,3 ммоль) в ацетонитриле (100 мл), добавляли DMAP (257 мг, 2,11 ммоль). Полученный раствор перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали, и остаток очищали на колонке ISCO (gold, 120 г), элюируя смесью EtOAc/гексан с градиентом 0-25%, получая указанное в заголовке соединение. LC-MS [М+Н]+: m/z 432,4.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 10
2-(дифторметил)-6-(1-(трифторметил)циклопропил)бензойная кислота
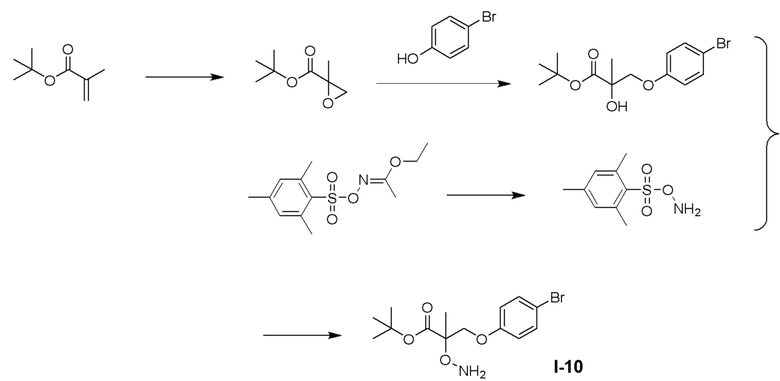
Стадия А: трет-бутил-2-метилоксиран-2-карбоксилат
В 2-литровую 3-горлую круглодонную колбу, снабженную обратным холодильником, содержащую раствор трет-бутилметакрилат (44 г, 309 ммоль) в CH2Cl2 (1000 мл) добавляли порциями m-СРВА (120 г, 557 ммоль). Полученный раствор перемешивали при 30°С в течение 20 часов, в результате чего получали большое количество осадка. Суспензию фильтровали, к фильтрату добавляли небольшими порциями насыщенный раствор тиосульфата натрия (приблизительно 100 мл, добавляли небольшими порциями, пока не прекращалось выделение тепла). Водную фазу отделяли и органическую фазу промывали насыщенным NaHCO3 (2×400 мл) и насыщенным раствором соли (400 мл), сушили над Na2SO4, фильтровали и концентрировали, получая указанное в заголовке соединение.
1H-ЯМР (400 МГц, хлороформ-d) δ 3,02 (д, J= 5,95 Гц, 1H), 2,68 (д, J= 6,17 Гц, 1H), 1,51 (с, 3H), 1,46 (с, 9H).
Стадия B: трет-бутил-3-(4-бромфенокси)-2-гидрокси-2-метилпропаноат
Смесь 4-бромфенола (15 г, 87 ммоль), K2CO3 (17,97 г, 130 ммоль) и трет-бутил-2-метилоксиран-2-карбоксилата (20,6 г, 130 ммоль) в DMF (200 мл) нагревали и перемешивали при 80°C в течение 16 часов. ТСХ показал полную конверсию. Смесь разбавляли водой (100 мл) и экстрагировали CH2Cl2 (3×100 мл), промывали насыщенным солевым раствором (2×40 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии (SiO2, PE:этилацетат=1:0-9:1), получая указанное в заголовке соединение.
1H-ЯМР (400 МГц, хлороформ-d) δ 7,37 (д, J= 8,61 Гц, 2H), 6,77 (д, J= 9,00 Гц, 2H), 4,09 (д, J= 9,00 Гц, 1H), 3,95 (д, J= 9,00 Гц, 1H), 3,60 (с, 1H), 1,44-1,47 (м, 12H).
Стадия C: O-(мезитилсульфонил)гидроксиламин
Раствор (E)-этил-N-(мезитилсульфонил)оксиацетимидата (20 г, 70,1 ммоль) в 1,4-диоксае (20 мл) охлаждали до 0°С. Добавляли по каплям (медленно) хлорную кислоту (8,45 г, 84 ммоль). После перемешивания в течение 15 минут смесь затвердевала. Содержимое реакции переносили в 200 мл ледяной воды, и колбу промывали смесью воды (100 мл) и трет-бутилметилового эфира (100 мл). Содержимое переносили в отдельно воронку и экстрагировали трет-бутилметиловым эфиром (2×100 мл). Органический слой нейтрализовали и частично сушили над безводным карбонатом калия, а затем фильтровали. Фильтрат концентрировали до менее чем 80 мл общего объема, и затем выливали в 400 мл охлажденного льдом гексана, и оставляли кристаллизоваться в течение 30 минут. Белые кристаллы выделяли фильтрованием, получая указанное в заголовке соединение, которое помещали в пластиковую пробирку Falcon и хранили при -20°С.
1H-ЯМР (400 МГц, хлороформ-d) δ 7,01 (с, 2H), 2,65 (с, 6H), 2,33 (с, 3H).
Стадия D: трет-бутил 2-(аминоокси)-3-(4-бромфенокси)-2-метилпропаноат
Трет-бутил-3-(4-бромфенокси)-2-гидрокси-2-метилпропаноат (9 г, 27,2 ммоль) растворяли в безводном THF (120 мл) в атмосфере азота, и охлаждали до 0°C. Добавляли 3-я порциями NaH (1,96 г, 48,9 ммоль, (60% в парафине)). Смесь перемешивали при 0°С в течение 30 минут. Затем в смесь добавляли O-(мезитилсульфонил)гидроксиламин (6,43 г, 29,9 ммоль). Смесь перемешивали при 0°C в течение 2 часов. Анализ LCMS показал, что исходный материал израсходован и получено целевое соединение. Смесь гасили насыщенным раствором NH4Cl (20 мл), экстрагировали EtOAc (3×45 мл). Объединенный органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии (SiO2, PE:этилацетат=1:0-10:1), получая указанное в заголовке соединение.
1H-ЯМР (400 МГц, хлороформ-d) δ 7,38 (д, J= 8,61 Гц, 2H), 6,84 (д, J= 8,61 Гц, 2H), 4,34 (д, J= 9,78 Гц, 1H), 4,07 (д, J= 9,78 Гц, 1H), 1,47-1,52 (м, 12H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 11
Этил-(R)-3-(4-бромфенокси)-2-гидроксипропаноат
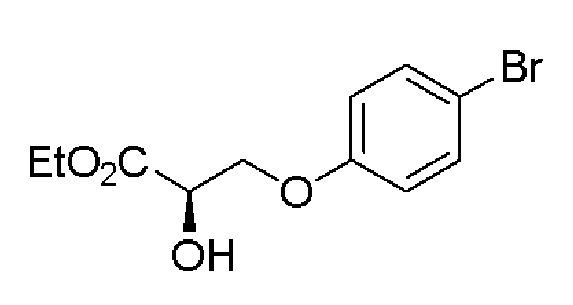
Указанное в заголовке соединение получали, выполняя процедуры, аналогичные процедурам, выполненным при получении промежуточного соединения 3, с заменой трет-бутил-оксиран-2-карбоксилата на этил-оксиран-2-карбоксила.
LC-MS [М+Н]: m/z 289,1.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 12
Трет-бутил-(R)-3-((6-бромпиридазин-3-ил)окси)-2-гидроксипропаноат
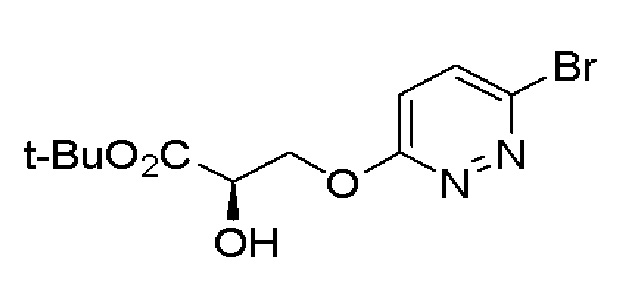
Указанное в заголовке соединение получали, выполняя процедуры, аналогичные процедурам, выполненным при получении промежуточного соединения 3, с заменой 4-бромфенола на 3-бром-6-гидроксипиридазин.
LC-MS [M+Na]: m/z 341,0.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 13
Этил-(R)-2-гидрокси-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)пропаноат
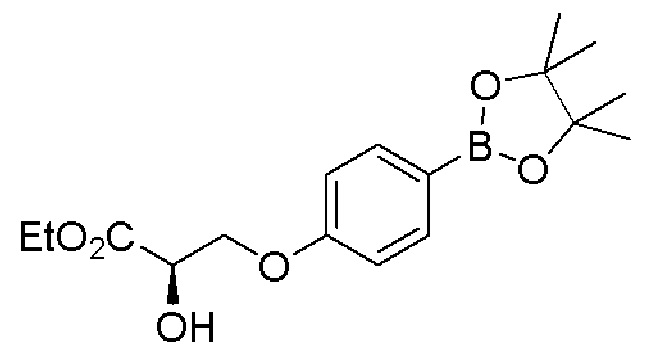
Указанное в заголовке соединение получали, выполняя процедуры, аналогичные процедурам, выполненным при получении промежуточного соединения 8, с заменой трет-бутил-оксиран-2-карбоксилата на этил-оксиран-2-карбоксилат.
LC-MS [М+Н]: m/z 337,3.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 14
трет-бутил-(S)-2-(аминоокси)-3-(4-бром-3-фторфенокси)-2-метилпропаноат
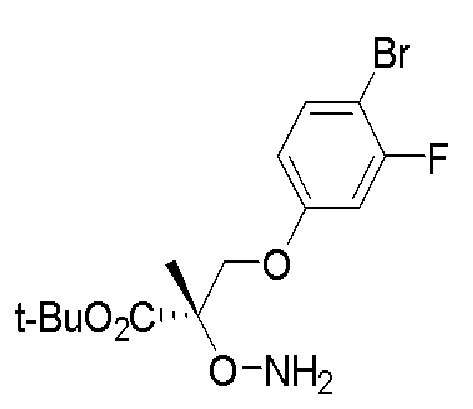
Стадия A: 1-бром-2-фтор-4-((2-метилаллил)окси)бензол
3-Бром-2-метилпроп-1-ен (5,3 мл, 52 ммоль) добавляли к перемешиваемой суспензии 4-бром-3-фторфенола (10 г, 52 ммоль) и K2CO3 (8,7 г, 63 ммоль) в DMF (105 мл). Реакционную смесь перемешивали при комнатной температуре (rt) в течение 1,5 часов, а затем разделяли между эфиром и водой. Слои разделяли, органический слой промывали водой и насыщенным раствором соли, сушили (MgSO4), фильтровали и концентрировали в вакууме, получая указанное в заголовке соединение, которое использовали далее без дополнительной очистки.
Стадия B: ( R )-3-(4-бром-3-фторфенокси)-2-метилпропан-1,2-диол
AD-смесь α (12 г, 0,035 ммоль осмия) добавляли к суспензии 1-бром-2-фтор-4-((2-метил-аллил)окси)бензола (2,1 г, 8,5 ммоль) в трет-бутаноле (41 мл) и воды (41 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, затем распределяли между EtOAc и водой. Слои разделяли, и водный слой экстрагировали EtOAc. Объединенные органические слои промывали солевым раствором, сушили (MgSO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (0-100% EtOAc в гексане в качестве элюента) получая указанное в заголовке соединение.
LC-MS [М+Н]: m/z 279,0.
Стадия C: (S)-3-(4-бром-3-фторфенокси)-2-гидрокси-2-метилпропановая кислота
Смесь NaH2РO4 (2,43 г, 20,2 ммоль) в воде (41 мл) добавляли к перемешиваемому раствору (R)-3-(4-бром-3-фторфенокси)-2-метилпропан-1,2-диола (2,26 г, 8,10 ммоль) в THF (41 мл). Затем добавляли ТЕМПО (0,127 г, 0,81 ммоль), с последующим добавлением растворов хлорита натрия (1,83 г, 16,2 ммоль) и гипохлорита натрия (0,83 мл, 0,81 ммоль) в воде (1,5 мл для каждого). Реакционную смесь перемешивали при комнатной температуре в течение 24 часов, затем дополнительными порциями добавляли TEMPO (0,127 г, 0,81 ммоль) и гипохлорит натрия (0,83 мл, 0,81 ммоль). Через 2 часа реакционную смесь распределяли между EtOAc и насыщенным водным раствором тиосульфата натрия. Слои разделяли, и органический слой промывали солевым раствором, сушили (MgSO4), фильтровали и концентрировали в вакууме, получая указанное в заголовке соединение. LC-MS [М+2Na]: m/z 336,9.
Стадия D: трет-бутил-(S)-3-(4-бром-3-фторфенокси)-2-гидрокси-2-метилпропаноат
Трет-бутил-N,N'-диизопропилкарбамидат (1,87 мл, 8,0 ммоль) добавляли к перемешиваемому раствору (S)-3-(4-бром-3-фторфенокси)-2-гидрокси-2-метилпропановой кислоты (1,17 г, 4,0 ммоль) в THF (20 мл). Полученную смесь нагревали до 60°С. Через 3 часа реакционную смесь охлаждали до комнатной температуры, фильтровали через слой целита (CeliteTM) и концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (0-80% EtOAc в гексане в качестве элюента), получая указанное в заголовке соединение.
LC-MS [М+(Na-Tbu)]: m/z 314,6.
Стадия E: трет-бутил-(S)-2-(аминоокси)-3-(4-бром-3-фторфенокси)-2-метилпропаноат
Указанное в заголовке соединение получали в соответствии с процедурой, аналогичной процедуре стадии D получения промежуточного соединения 10.
LC-MS [М+Н]: m/z 364,2.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 15
Метил-(R)-3-(4-бромфенокси)-2-гидроксипропаноат
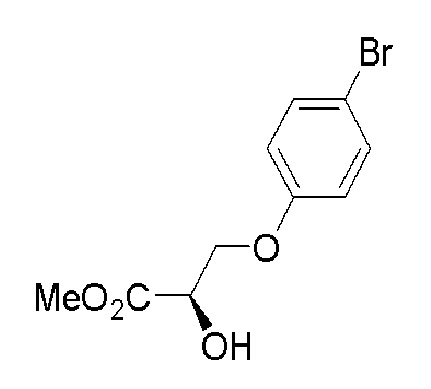
Стадия A: ( R )-3-(4-бромфенокси)-2-гидроксипропановая кислота
TFA (850 мкл, 11,0 ммоль) добавляли к перемешиваемому раствору промежуточного соединения 3 (350 мг, 1,10 ммоль) в DCM (5,5 мл). Реакционную смесь перемешивали при комнатной температуре. Через 1,5 часа реакционную смесь концентрировали в вакууме, и полученное твердое вещество использовали далее без дополнительной очистки.
LC-MS [M+Na]: m/z 283,2.
Стадия B: Метил ( R )-3-(4-бромфенокси)-2-гидроксипропаноат
Триметилсилилдиазометан (2,2 мл 2,0М раствор в гексане, 4,41 ммоль) добавляли к смеси DCM:СН3ОН (1:1) (2 мл). Полученную смесь добавляли к перемешиваемому раствору (R)-3-(4-бромфенокси)-2-гидроксипропановой кислоты (288 мг, 1,10 ммоль) в смеси DCM:СН3ОН (1:1) (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Затем реакционную смесь концентрировали в вакууме, получая указанное в заголовке соединение, которое использовали без дополнительной очистки.
LC-MS [М+Н]: m/z 275,2.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 16
Трет-бутил-3-((4-бром-1H-пиразол-1-ил)метил)-3-фторазетидин-1-карбоксилат
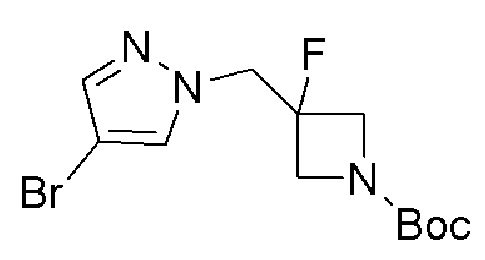
Стадия A: трет-бутил-3-фтор-3-((тозилокси)метил)азетидин-1-карбоксилат
п-толуолсульфонил хлорид (1,02 г, 5,32 ммоль) добавляли к перемешиваемому раствору трет-бутил-3-фтор-3-(гидроксиметил)азетидин-1-карбоксилата (993 мг, 4,84 ммоль) и триэтиламина (742 мкл, 5,32 ммоль) в DCM (20 мл) при 0°C. Реакционную смесь оставляли нагреваться до комнатной температуры, затем добавляли дополнительное количество триэтиламина (337 мкл, 2,66 ммоль). После перемешивания в течение ночи реакционную смесь концентрировали в вакууме, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (0-60% EtOAc в гексане в качестве элюента), получая указанное в заголовке соединение.
LC-MS [M- (tBu+H)]: m/z 304,2.
Стадия B: трет-бутил 3-((4-бром-1H-пиразол-1-ил)метил)-3-фторазетидин-1-карбоксилат
Гидрид натрия (116 мг 60%-ная дисперсия в минеральном масле, 2,89 ммоль) добавляли несколькими порциями к перемешиваемому раствору трет-бутил-3-фтор-3-((тозилокси)метил)азетидин-1-карбоксилата (1,04 г, 2,89 ммоль) и 4-бром-1H-пиразола (425 мг, 2,89 ммоль) в DMF (13 мл) при 0°C. Реакционную смесь оставляли нагреваться до комнатной температуры, и перемешивали в течение ночи. Затем реакционную смесь разбавляли этилацетатом и выливали на лед. Слои разделяли, и органический слой промывали солевым раствором, сушили (MgSO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (0-100% EtOAc в гексане в качестве элюента), получая указанное в заголовке соединение. LC-MS [М+Н]: m/z 334,0.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 17
3-азидопропил-трифторметансульфонат

Указанное в заголовке соединение получали по процедуре, аналогичной процедуре, описанной для стадии А получения промежуточного продукта 16, с заменой трет-бутил-3-фтор-3-(гидроксиметил)-азетидин-1-карбоксилата на 3-азидопропан-1-ол, и с заменой п-тозилхлорида на трифлатный ангидрид.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 18
Трет-бутил-(2-((5-бромпиридин-2-ил)амино)этил)карбамат
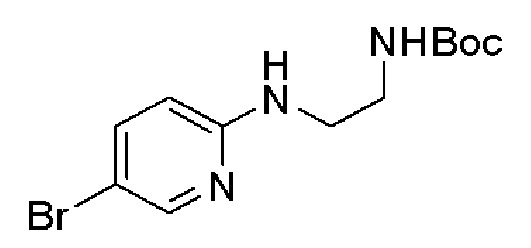
Стадия A: N1-(5-бромпиридин-2-ил)этан-1,2-диамин
Этилендиамин (25,4 мл, 380 ммоль) добавляли к перемешиваемому раствору 2,5-дибромпиридина (5,00 г, 21,1 ммоль). Реакционную смесь нагревали до 100°С в течение 16 часов, затем охлаждали до комнатной температуры и концентрировали в вакууме. Полученный остаток разбавляли в DCM. Органические слои промывали водой, сушили (MgSO4), фильтровали и концентрировали в вакууме, получая указанное в заголовке соединение, которое использовали на следующей стадии без очистки.
LC-MS [М+Н]: m/z 215,8.
Стадия B: трет-бутил-(2-((5-бромпиридин-2-ил)амино)этил)карбамат
Ди-трет-бутил-ди-карбонат (5,53 г, 25,3 ммоль) добавляли к перемешиваемой суспензии N1-(5-бромпиридин-2-ил)этан-1,2-диамина (4,56 г, 21,1 ммоль) и карбоната натрия (2,35 г, 22,2 ммоль) в растворе смеси диоксан:вода (1:1) (общий объем 64 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем реакционную смесь экстрагировали с помощью EtOAc. Органический слой промывали солевым раствором, сушили (MgSO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (0-30% EtOAc в гексане в качестве элюента), получая указанное в заголовке соединение.
LC-MS [М+Н]: m/z 316,3.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 19
Трет-бутил-(3-((6-хлорпиридазин-3-ил)амино)пропил)карбамат
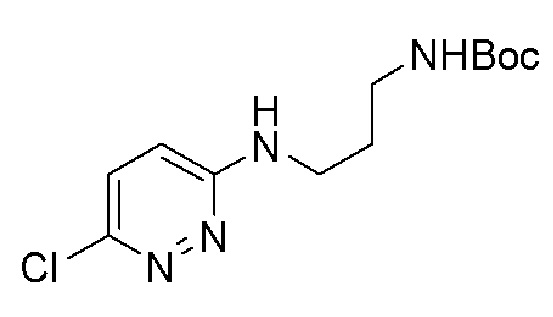
Стадия A: трет-бутил-(3-((6-хлорпиридазин-3-ил)амино)пропил)карбамат
К перемешиваемому раствору 3,6-дихлорпиридазина (1,00 г, 6,71 ммоль) в DMSO (10,0 мл) во флаконе для микроволновой печи добавляли трет-бутил-(3-аминопропил)карбамат (1,23 г, 7,05 ммоль), с последующим добавлением триэтиламина (1,50 мл, 10,8 ммоль). Микроволновый флакон герметизировали, и реакционную смесь нагревали в микроволновом реакторе до 130°С в течение 15 минут. После охлаждения до комнатной температуры реакционную смесь распределяли между EtOAc и насыщенным водным раствором NH4Cl. Слои разделяли, и органический слой промывали водой и насыщенным раствором соли. Органический слой сушили (Na2SO4), фильтровали и концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (0-50% (3:1 EtOAc:EtOH) в гексане в качестве элюента), получая указанное в заголовке соединение.
LC-MS [М+Н]: m/z 287,2.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 20
Трет-бутил-(3-(4-бром-1H-пиразол-1-ил)-2-фторпропил)карбамат

Указанное в заголовке соединение получали в соответствии с процедурой, аналогичной процедуре, описанной для стадии А получения промежуточного соединения 16, с заменой трет-бутил-3-фтор-3-(гидроксиметил)азетидин-1-карбоксилата на трет-бутил-(2-фтор-3-гидроксипропил)карбамат.
LC-MS [M+Na]: m/z 370,2.
Стадия B: трет-бутил (3-(4-бром-1H-пиразол-1-ил)-2-фторпропил)карбамат
Указанное в заголовке соединение получали в соответствии с процедурой, описанной для стадии B получения промежуточного соединения 16.
LC-MS [М+Н]: m/z 322,2.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 21
Трет-бутил-(3-(1H-имидазол-4-ил)пропил)карбамат
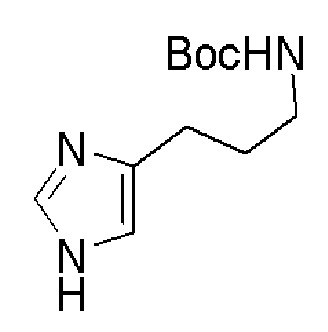
Стадия A: 3-(1-тритил-1 H -имидазол-4-ил)пропан-1-ол
Литий-алюминий гидрид (2,5 мл раствора в THF, 1М, 2,5 ммоль) добавляли к раствору метил-3-(1-тритил-1Н-имидазол-4-ил)пропионата (500 мг, 1,26 ммоль) в THF (4,0 мл) при 0°С. Полученную смесь нагревали до 70°С и оставляли для перемешивания при 70°С в течение 2 часов. Затем реакционную смесь охлаждали до комнатной температуры и гасили добавлением воды, а затем 2Н NaOH. Полученную суспензию фильтровали через колонку с целитом (Celite™), которую промывали EtOAc. Фильтрат концентрировали в вакууме, получая указанное в заголовке соединение, которое использовали без дополнительной очистки. LC-MS [М+Н]: m/z 369,4.
Стадия B: 2-(3-(1-тритил-1 H -имидазол-4-ил)пропил)изоиндолин-1,3-дион
Фталимид (278 мг, 1,89 ммоль) добавляли к перемешиваемому раствору 3-(1-тритил-1Н-имидазол-4-ил)пропан-1-ола (465 мг, 1,26 ммоль) и трифенилфосфина (496 мг, 1,89 ммоль) в THF (12 мл). Реакционную смесь охлаждали до 0°С, затем медленно по каплям добавляли DIAD (0,37 мл, 1,89 ммоль), и полученную смесь оставляли нагреваться до комнатной температуры. После перемешивания в течение ночи реакционную смесь фильтровали, и твердые вещества промывали THF, получая указанное в заголовке соединение.
LC-MS [М+Н]: m/z 498,5.
Стадия C: 3-(1-тритил-1H-имидазол-4-ил)пропан-1-амин
Метиламин (11 мл раствора 33% (вес./об.) этанола, 88 ммоль) добавляли к перемешиваемому раствору 2-(3-(1-тритил-1Н-имидазол-4-ил)пропил)изоиндолин-1,3-диона (1,92 г, 3,86 ммоль) в смеси этанола (40 мл) и воды (3,5 мл). Полученную смесь перемешивали при комнатной температуре в течение ночи, затем реакционную смесь концентрировали в вакууме, и подвергали несколько раз азеотропной перегонке с толуолом, получая указанное в заголовке соединение, которое использовали без дальнейшей обработки или очистки.
LC-MS [М+Н]: m/z 368,4.
Стадия D: трет-бутил-(3-(1-тритил-1H-имидазол-4-ил)пропил)карбамат
Ди-трет-бутил-ди-карбонат (1,69 г, 7,72 ммоль) добавляли к перемешиваемому раствору 3-(1-тритил-1Н-имидазол-4-ил)пропан-1-амина (1,42 г, 3,86 ммоль) и триэтиламина (1,08 мл, 7,72 ммоль) в DCM (30 мл). Реакционную смесь перемешивали при комнатной температуре. По завершении реакции реакционную смесь концентрировали в вакууме на слое целита, и полученную суспензию очищали с помощью колоночной хроматографии на силикагеле (0-100% EtOAc в гексане в качестве элюента), получая указанное в заголовке соединение.
LC-MS [М+Н]: m/z 468,4.
Стадия E: трет-бутил-(3-(1 H -имидазол-4-ил)пропил)карбамат
Анизол (377 мг, 3,49 ммоль) добавляли к раствору трет-бутил-(3-(1-тритил-имидазол-1Н-4-ил)пропил)карбамата (1,63 г, 3,49 ммоль) в уксусной кислоте (5,0 мл), и реакционную смесь нагревали до 60°С. Через 24 часа реакционную смесь концентрировали в вакууме, и подвергали последовательно азеотропной перегонке с толуолом и ацетонитрилом. Полученный остаток суспендировали в EtOAc и DCM, и смесь адсорбировали на целите. Полученную суспензию очищали с помощью колоночной хроматографии на силикагеле (0-100% (3:1 EtOAc:EtOH) в гексане в качестве элюента), получая указанное в заголовке соединение.
LC-MS [М+Н]: m/z 226,2.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 22
(S)-3-(2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоацетамидо)-2,2-диметил-4-оксоазетидин-1-ил гидросульфат
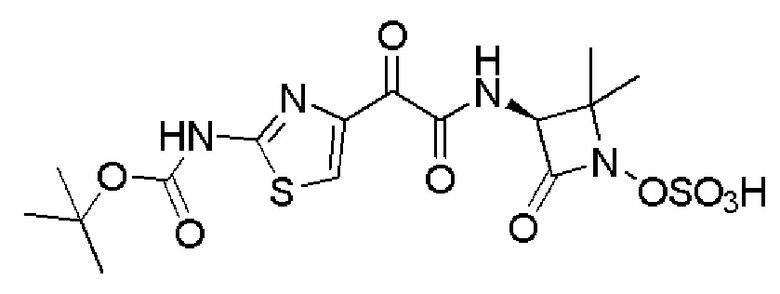
К раствору 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусной кислоты (промежуточное соединение 5, 2 г, 7,4 ммоль), (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил-гидросульфата (3,1 г, 14,7 ммоль, из коммерческого источника CAS: 102507-49-3) и пиридина (1,8 мл, 22 ммоль) в MeCN (37 мл) добавляли EDC (3,5 г, 18 ммоль) при 0°C. Реакционную смесь оставляли нагреваться до комнатной температуры в течение ночи. Через 16 часов реакционную смесь выливали в насыщенный раствор соли (100 мл) и экстрагировали с помощью MeCN (50 мл). Органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Полученный остаток очищали с помощью флэш-хроматографии на SiO2, элюируя смесью гексан/(3:1 EtOAc/EtOH) 0-100%, получая указанное в заголовке соединение.
LC-MS [М+Н]: m/z 465,2.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 23
Трет-бутил-(S)-(3-(4-бром-1H-пиразол-1-ил)-2-((третбутилдиметилсилил)окси)пропил)карбамат
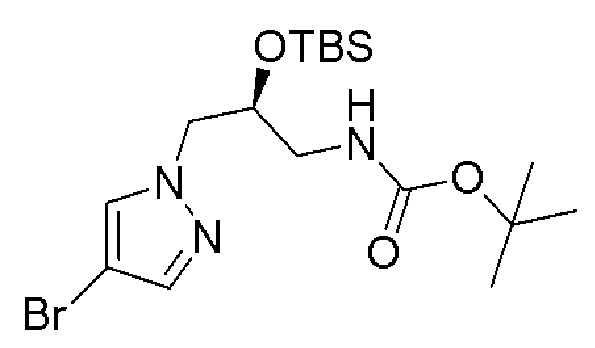
Стадия A: трет-бутил N-[(2S)-2,3-дигидроксипропил]карбамат
В 500-мл 4-горлую круглодонную колбу помещали раствор (2S)-3-аминопропан-1,2-диола (20 г, 220 ммоль, 1 экв.) в метаноле (200 мл) и ди-трет-бутилдикарбоната (57 г, 261 ммоль, 1,2 экв.). Реакционную смесь перемешивали в течение ночи при комнатной температуре, а затем концентрировали в вакууме. Полученный остаток очищали на колонке с силикагелем с использованием смеси дихлорметан/метанол (10:1), получая указанное в заголовке соединение.
Стадия B: трет-бутил (S)-(2,3-бис((трет-бутилдиметилсилил)окси)пропил)карбамат
В 500-мл 3-горлую круглодонную колбу помещали раствор трет-бутил-N-[(2S)-2,3-дигидрокси-пропил]карбамата (20 г, 105 ммоль, 1 экв.) в N,N-диметилформамиде (200 мл) и имидазола (32 г, 470 ммоль, 4,50 экв.). Затем несколькими порциями добавляли трет-бутил-(хлор)диметилсилан (40,8 г, 271 ммоль, 2,6 экв.) при 0°С. Реакционную смесь перемешивали в течение ночи при комнатной температуре, а затем разбавляли этилацетатом (500 мл). Полученную смесь промывали водой (2×400 мл) и насыщенным раствором соли (400 мл), а затем концентрировали под вакуумом. Полученный остаток наносили на колонку с силикагелем, используя в качестве элюента смесь этилацетат/петролейный эфир (1:20), получая указанное в заголовке соединение.
Стадия C: трет-бутил-(S)-(2-((трет-бутилдиметилсилил)окси)-3-гидроксипропил)карбамат
В 5-л 4-горлую круглодонную колбу помещали раствор трет-бутил-N-[(2S)-2,3-бис[(трет-бутилдиметилсилил)окси]пропил]карбамата (200 г, 476 ммоль, 1 экв.) в этаноле (2 л) и PPTS (110 г, 1 экв.). Реакционную смесь перемешивали в течение ночи при комнатной температуре, а затем гасили путем добавления TEA (100 мл). Полученную смесь концентрировали в вакууме. Полученный раствор разбавляли 2 л этилацетата, затем промывали H2O (2 л). Смесь сушили над безводным сульфатом магния и концентрировали в вакууме. Полученный остаток очищали на колонке с силикагелем с использованием смеси этилацетат/петролейный эфир (1:3), получая указанное в заголовке соединение.
Стадия D: (S)-3-((трет-бутоксикарбонил)амино)-2-((трет-бутилдиметилсилил)окси)пропил метансульфонат
Раствор трет-бутил-N-[(2S)-2-[(трет-бутилдиметилсилил)окси]-3-гидроксипропил]карбамата (40 г, 131 ммоль, 1 экв.) в дихлорметане (400 мл), TEA (36,5 мл, 2 экв.), и метансульфонилхлорида (14,5 мл, 1,50 экв.) перемешивали в течение 2 часов при комнатной температуре. Затем реакционную смесь гасили путем добавления бикарбоната натрия (водный раствор, 200 мл). Полученный раствор экстрагировали этилацетатом (300 мл). Органические слои объединяли, сушили над безводным сульфатом магния и концентрировали в вакууме, получая указанное в заголовке соединение.
Стадия E: трет-бутил-(S)-(3-бром-2-((трет-бутилдиметилсилил)окси)пропил)карбамат
Раствор трет-бутил-N-[(2S)-2-[(трет-бутилдиметилсилил)окси]-3-(метансульфонилокси)пропил]карбамата (47 г, 123 ммоль, 1 экв.) в тетрагидрофуране (1600 мл) и бромлитий (53 г, 610 ммоль, 5 экв.) перемешивали в течение 2 дней при 70°С. Затем реакционную смесь разбавляли H2O (500 мл), и полученный раствор экстрагировали 500 мл этилацетата. Объединенные органические слои сушили над безводным сульфатом натрия и концентрировали в вакууме. Полученный остаток очищали на колонке с силикагелем с использованием смеси этилацетат/петролейный эфир (1:30), получая указанное в заголовке соединение.
Стадия F: трет-бутил-(S)-(3-(4-бром-1H-пиразол-1-ил)-2-((третбутилдиметилсилил)окси)-пропил)карбамат
Раствор трет-бутил-N-[(2S)-3-бром-2-[(трет-бутилдиметилсилил)окси]пропил]карбамата (210 г, 570 ммоль, 1,20 экв.), N,N-диметилформамида (2 л), 4-бром-1Н-пиразола (70 г, 476 ммоль, 1 экв.) и Cs2СО3 (313 г, 957 ммоль, 2 экв.) перемешивали в течение ночи при 70°С. Затем реакционную смесь разбавляли 2 л EA. Полученную смесь промывали Н2О (2×4 л). Смесь сушили над безводным сульфатом магния и концентрировали в вакууме. Полученный остаток наносили на колонку с силикагелем с использованием смеси этилацетат/петролейный эфир (1:10), получая указанное в заголовке соединение.
LC-MS: (ES, m/z): 434 [M-H]-.
1H-ЯМР: (400 МГц, d-DMSO, м.д.): δ -0,32 (3H, с), 0,05 (3H, с), 0,76 (9H, с), 1,38 (9H, с), 2,93-3,00(2H, м), 3,91-3,96 (2H, м), 4,10-4,15 (2H, м), 6,93-6,96 (1H, т), 7,54(1H, с), 7,87 (1H, с).
ПРИМЕР 1
(S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-2-(4-(1-(3-аммонийпропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат 2,2,2-трифторацетат (C1)
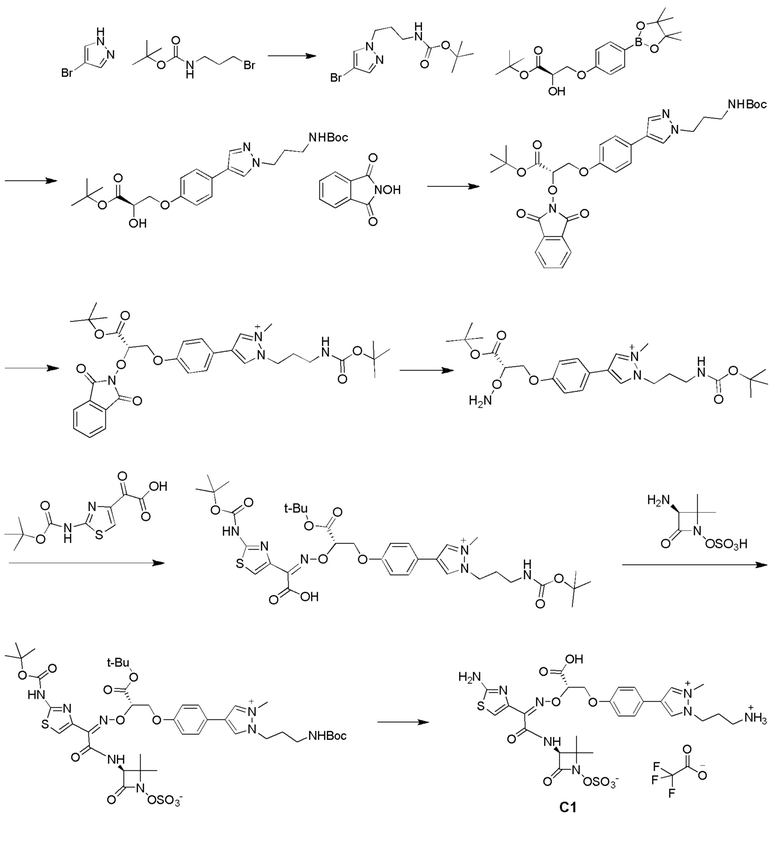
Стадия A: трет-бутил-(3-(4-бром-1H-пиразол-1-ил)пропил)карбамат
Раствор 4-бром-1H-пиразола (3 г, 20,4 ммоль), трет-бутил (3-бромпропил)карбамата (4,86 г, 20,4 ммоль) и карбоната цезия (9,98 г, 30,6 ммоль) в DMF (20 мл) перемешивали при комнатной температуре в течение ночи. Смесь разбавляли водой (30 мл) и экстрагировали EtOAc (3×10 мл). Экстракты объединяли, сушили над MgSO4, и концентрировали досуха. Остаток очищали с помощью колоночной флэш-хроматографиий (колонка с силикагелем Biotage, 80 г gold), элюируя с градиентом DCM/MeOH (0-10%), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 304,19.
1H-ЯМР (500 МГц, CDCl3) δ 7,45 (с, 2H), 4,15 (м, 2H), 3,10 (м, 2H), 1,99 (м, 2H), 1,27 (с, 9H).
Стадия B: трет-бутил-(R)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-2-гидроксипропаноат
В 25-мл флакон к раствору (S)-трет-бутил-2-гидрокси-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)пропионату (5 г, 13,7 ммоль), трет-бутил-(3-(4-бром-1H-пиразол-1-ил)пропил)карбамата (4,18 г, 13,7 ммоль) и дихлорида 1,1'-бис(ди-трет-бутилфосфино)ферроценпалладия (0,895 г, 1,37 ммоль) в THF (150 мл) добавляли 1М водный раствор трехосновного фосфата калия (41,2 мл, 41,2 ммоль). Флакон укупоривали, дегазировали (3x) и заполняли азотом, и перемешивали при 60°С в течение ночи. Анализ LC-MS показал полную конверсию. Реакционную смесь разбавляли насыщенным раствором NH4Cl (100 мл). Органическую фазу отделяли, и водную фазу экстрагировали EtOAc (2×60 мл). Органические экстракты объединяли, сушили над MgSO4 и концентрировали досуха. Остаток очищали на флэш-SiO2 колонке (120 г gold) с EtOAc/гексан (0-100%), получая указанное в заголовке соединение.
LC-MS [M+H]+: m/z 462,56.
1H-ЯМР (500 МГц, CDCl3) δ 7,78 (с, 1H), 7,61 (с, 1H), 7,40 (д, J=7,5 Гц, 2H), 6,96 (д, J=7,5 Гц, 2H),4,92 (с, 1H), 4,40 (м, 2H), 4,23 (м, 2H), 3,16 (м, 2H), 2,01 (м, 2H), 1,48 (с, 9H).
Стадия C: трет-бутил-(S)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноат
К раствору (S)-трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-7Н-пиразол-4-ил)фенокси)-2-гидроксипропаноата (2,2 г, 4,77 ммоль), 2-гидроксиизоиндолин-1,3-диона (0,933 г, 5,72 ммоль) и трифенилфосфина (1,50 г, 5,72 ммоль) в DCM (100 мл) добавляли DIAD (1,11 мл, 5,72 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов. Анализ LC-MS показал полную конверсию. Смесь концентрировали досуха и очищали с помощью флэш-хроматографии на SiO2 (120 г gold), элюировали смесью гексан/EtOAc 0-100%), получая указанное в заголовке соединение.
LC-MS [M+H]+: m/z 607,59.
1H-ЯМР (500 МГц, CDCl3) δ 7,82-7,92 (м, 4H), 7,78 (с, 1H), 7,61 (с, 1H), 7,41 (д, J=7,5 Гц, 2H), 6,92 (д, J=7,5 Гц, 2H),5,12 (с, 1H), 4,54 (м, 2H), 4,25 (с, 2H), 3,18 (м, 2H), 2,12 (м, 2H), 1,48 (с, 9H)
Стадия D: (S)-4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий
К раствору (S)-трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1Н-пиразол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропионата (1,8 г, 2,97 ммоль) в ацетонитриле (100 мл) добавляли иодистый метил (4,21 г, 29,7 ммоль). Смесь герметично укупоривали в 250-мл флаконы под давлением, и перемешивали при 70°С в течение 16 часов. Анализ LC-MS показал полную конверсию. Затем смесь охлаждали до комнатной температуры, и концентрировали досуха в вакууме, получая указанное в заголовке соединение, которое непосредственно использовали на следующей стадии без дополнительной очистки.
LC-MS [М]+: m/z 621,61.
Стадия E: (S)-4-(4-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий
К раствору (S)-4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий иодида (2 г, 2,67 ммоль) в ацетонитриле (20 мл) добавляли гидразин (0,092 мл, 2,94 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов. Анализ LC-MS показал полную конверсию. Смесь затем концентрировали досуха в вакууме, получая указанное в заголовке соединение, которое непосредственно использовали на следующей стадии без дополнительной очистки.
LC-MS [М]+: m/z 491,35.
Стадия F: (S,Z)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)-тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий
К раствору (S)-4-(4-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)-пропил)-2-метил-1H-пиразол-2-ий иодида (1,6 г, 2,59 ммоль) в этаноле (100 мл) и ClCH2CH2Cl (20 мл) добавляли 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусную кислоту (0,775 г, 2,85 ммоль, промежуточное соединение 5). Смесь перемешивали при комнатной температуре в течение 4 часов. Анализ LC-MS показал полную конверсию. Смесь концентрировали, получая указанное в заголовке соединение, которое непосредственно использовали на следующей стадии без дополнительной очистки.
LC-MS [М]+: m/z 745,43.
Стадия G: (S)-3-((Z)-2-((((S)-1-(трет-бутокси)-3-(4-(1-(3-((трет-бутокси-карбонил)амино)пропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
К раствору (S,Z)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)-амино)пропил)-2-метил-1H-пиразол-2-ия (1,9 г, 2,17 ммоль) в DMF (20 мл) добавляли DCC (0,898 г, 4,35 ммоль) и HOBt (0,667 г, 4,35 ммоль). Cмесь перемешивали при комнатной температуре в течение 30 минут, с последующим добавлением (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата (0,915 г, 4,35 ммоль, из коммерческого источника CAS: 102507-49-3) и бикарбоната натрия (1,10 г, 13,1 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи (16 часов). Анализ LC-MS показал полную конверсию. Раствор в DMF сушили в токе азота в течение ночи. Остаток повторно растворяли в смеси MeCN/вода (2:1, 2,5 мл), и очищали с помощью обращенно-фазовой хроматографии на колонке ISCO (130 г), элюируя смесью MeCN/H2O/0,05% TFA (0-100%). Фракции собирали и лиофилизовали, получая указанное в заголовке соединение.
LC-MS [М]+: m/z 938,00.
1H-ЯМР (500 МГц, CDCl3) δ 9,53 (с, 1H), 8,94 (м, 2H), 7,68 (м, 2H), 7,14 (м, 2H), 4,95 (с, 1H), 4,60 (м, 2H), 4,42 (с, 2H), 3,08 (м, 2H), 2,14 (м, 2H), 1,47 (с, 9H), 1,40 (с, 9H), 1,37 (с, 9H).
Стадия H: Моно 4-(4-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)фенил)-1-(3-аммонийпропил)-2-метил-1H-пиразол-2-ий) моно(2,2,2-трифторацетат)
К раствору 4-(4-((S)-3-(трет-бутокси)-2-(((E)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксо-пропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ия (40 мг, 0,043 ммоль) в DCM (1 мл) добавляли 1 мл TFA. Смесь перемешивали при комнатной температуре в течение 30 минут. Анализ LC-MS показал полную конверсию. Смесь концентрировали досуха в вакууме (без нагрева), добавляли DCM (10 мл) и концентрировали три раза для удаления TFA. Остаток промывали сухим MeCN (2×2 мл) для удаления примесей. Остаток растворяли в H2O и очищали с помощью препаративной высокоэффективной жидкостной хроматографии с использованием в качестве элюента смесь MeCN/H2O (0,05% TFA в обоих) с градиентом 0-40% в течение 10 минут. Фракции продукта собирали и лиофилизировали, получая указанное в заголовке соединение.
LC-MS [М]+: m/z 681,48.
1H-ЯМР (500 МГц, CDCl3) δ 8,72 (с, 1H), 8,68 (с, 1H), 7,78 (д, J=6,8 Гц, 2H), 7,16 (д, J=6,8, 2H), 7,08 (с, 1H), 5,18 (с, 1H), 4,63 (м, 2H), 4,58 (м, 2H), 4,19 (с, 3H), 3,16 (м, 2H), 2,39 (м, 2H).
ПРИМЕР 2
(S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-2-(4-(1-(3-аммонийпропил)-3-метил-1H-имидазол-3-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат (C2)
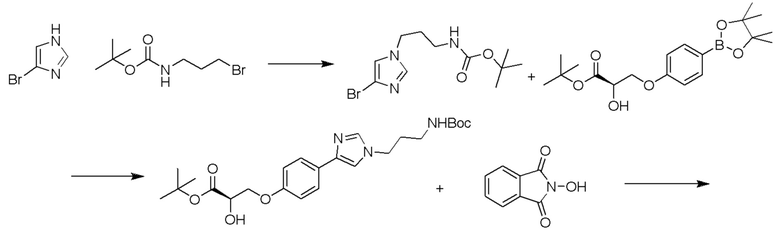
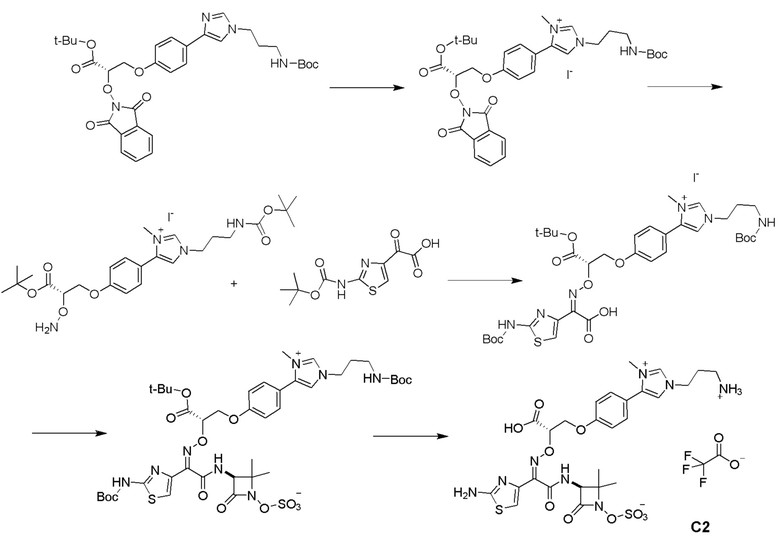
Стадия A: трет-бутил-(3-(4-бром-1H-имидазол-1-ил)пропил)карбамат
К раствору 4-бром-1H-имидазола (1 г, 6,80 ммоль) в DMF (20 мл) при 0°С добавляли NaH (0,272 г, 6,80 ммоль). Смесь перемешивали в течение 10 минут, добавляли трет-бутил-(3-бромпропил)карбамат (1,62 г, 6,80 ммоль). Раствор перемешивали при комнатной температуре в течение 2 часов, а затем нагревали при 50°С в течение 1 часа. Смесь гасили водой (50 мл) и экстрагировали EtOAc (3×20 мл). Органический слой промывали водой (30 мл), насыщенным раствором соли (30 мл) и сушили над Na2SO4. Растворитель удаляли в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (колонка Redi, 40 г gold) и элюировали с EtOAc/гексан (0-100%, 6 cv; 100%, 10 cv), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 304,19.
1H-ЯМР (500 МГц, CDCl3) δ 7,40 (с, 1H); 6,94 (с, 1H); 4,67 (с, 1H); 3,98 (т, J=7,0 Гц, 2H); 3,17 (д, J=7,5 Гц, 2H); 1,98 (п, J=6,8 Гц, 2H); 1,47 (с, 9H).
Стадия B: трет-бутил-(R)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-4-ил)фенокси)-2-гидроксипропаноат
Применяли процедуру, аналогичную процедуре на стадии B примера 1, с использованием в качестве реагентов трет-бутил-(3-(4-бром-7Н-имидазол-1-ил)пропил)карбамата, (R)-трет-бутил-2-гидрокси-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)пропаноата (300 мг, 0,824 ммоль), трет-бутил-(3-(4-бром-7Н-имидазол-1-ил)пропил)карбамата (276 мг, 0,906 ммоль) (промежуточное соединение 4), дихлорида 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладия (53,7 мг, 0,082 ммоль), THF (4 мл) и фосфата калия (2,471 мл, 2,471 ммоль, 1М, водн.).
LC-MS [М+Н]+: m/z 462.
Стадия C: трет-бутил-(S)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноат
Применяли процедуру, аналогичную процедуре на стадии С примера 1, с использованием в качестве реагентов трет-бутил-(R)-3-(4-(1-(3-((трет-бутоксикарбонил)-амино)пропил)-1H-имидазол-4-ил)фенокси)-2-гидроксипропаноата (0,25 г, 0,542 ммоль), 2-гидроксиизоиндолин-1,3-диона (0,097 г, 0,596 ммоль), трифенилфосфина (0,170 г, 0,650 ммоль), DIAD (0,126 мл, 0,650 ммоль) и THF (2 мл). LC-MS [М+Н]: m/z 607.
Стадия D: (S)-4-(4-(3-(трет-Бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-3-метил-1H-имидазол-3-ий иодид
Применяли процедуру, аналогичную процедуре на стадии D примера 1, с использованием в качестве реагентов трет-бутил-(S)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноата (0,17 г, 0,280 ммоль), MeI (0,105 мл, 1,681 ммоль) в MeCN (2 мл). LC-MS [М]+: m/z 621.
Стадия E: (S)-4-(4-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-3-метил-1H-имидазол-3-ий иодид
Применяли процедуру, аналогичную процедуре на стадии Е примера 1, с использованием в качестве реагентов (S)-4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-3-метил-1H-имидазол-3-ий иодида (0,210 г, 0,28 ммоль), гидразина (8,79 мкл, 0,280 ммоль) в этаноле (4 мл). LC-MS [М]+: m/z 491.
Стадия F: (S,Z)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)-тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутокси-карбонил)амино)пропил)-3-метил-1H-имидазол-3-ий иодид
Применяли процедуру, аналогичную процедуре на стадии F примера 1, с использованием в качестве реагентов (S)-4-(4-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-3-метил-1H-имидазол-3-ий иодида (0,173 г, 0,28 ммоль), 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусной кислоты (0,080 г, 0,294 ммоль) (промежуточное соединение 5) в этаноле (4 мл) и ClCH2СН2Сl (2 мл).
LC-MS [М]+: m/z 745.
Стадия G: (S)-3-((Z)-2-((((S)-1-(трет-Бутокси)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-3-метил-1H-имидазол-3-ий-4-ил)фенокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
Применяли процедуру, аналогичную процедуре на стадии G примера 1, с использованием в качестве реагентов (S,Z)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-3-метил-1H-имидазол-3-ий иодида (0,244 г, 0,280 ммоль) в DMF (4 мл), DCC (0,144 г, 0,699 ммоль), НОВТ (0,107 г, 0,699 ммоль), S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата (0,118 г, 0,559 ммоль) и бикарбоната натрия (0,117 г, 1,398 ммоль). После завершения реакции, неочищенную смесь фильтровали и очищали с помощью RP-HPLC (Gilson) на колонке (С-18), элюируя смесью ACN/H2O/0,05% TFA (20-100%, 10 мин). Фракцию продукта лиофилизировали, получая указанное в заголовке соединение.
LC-MS: [M]+ m/z 937,85.
Стадия H: Моно(4-(4-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)фенил)-1-(3-аммонийпропил)-3-метил-1H-имидазол-3-ий) моно(2,2,2-трифторацетат)
К раствору 4-(4-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-3-метил-1H-имидазол-3-ия (0,17 г, 0,162 ммоль) в CH2Cl2 (0,5 мл) добавляли TFA (0,5 мл). Раствор перемешивали при комнатной температуре в течение часа. Растворитель удаляли под вакуумом. Остаток дважды промывали Et2О и сушили в вакууме, с получением неочищенного твердого вещества. Неочищенный продукт растворяли в воде (2 мл) и DMSO (0,5 мл), а затем очищали с помощью RP-HPLC (Gilson) на колонке (С-18), элюируя смесью MeCN/вода (0,05% муравьиной кислоты в обоих), с градиентом 0-60% за 15 минут. Фракции продукта собирали и лиофилизировали, получая указанное в заголовке соединение.
LC-MS [M]+: m/z 681,38.
1H ЯМР (500 МГц, MeOD): δH 8,97 (д, J=1,8 Гц, 1H); 7,73 (с, 1H); 7,49 (д, J=8,4 Гц, 2H); 7,15-7,17 (м, 2H); 6,93 (с, 1H); 5,15-5,16 (м, 1H); 4,64-4,67 (м, 1H); 4,53-4,57 (м, 2H); 4,38 (т, J=7,0 Гц, 2H); 3,86 (с, 3H); 3,07-3,10 (м, 2H); 2,30-2,33 (м, 2H); 1,53 (с, 3H); 1,18 (с, 3H).
Таблица 1. При использовании в основном тех же процедур, как и в примере 2, с заменой соответствующих регентов, синтезировали и охарактеризовывали с помощью LC/MS следующее соединение.
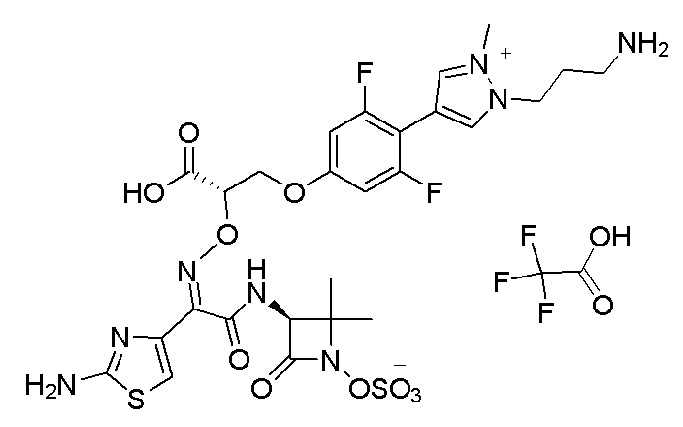
ПРИМЕР 4
(S,Z)-3-(2-((2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)этокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат (C4)
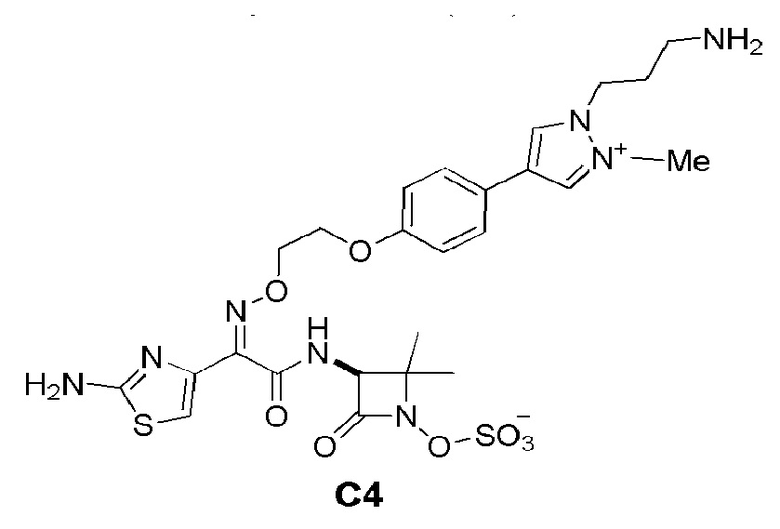
Стадия A: трет-бутил-(3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропил)карбамат
Раствор 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (2 г, 10,3 ммоль), трет-бутил-(3-бромпропил)карбамата (2,45 г, 10,3 ммоль) и карбоната цезия (3,36 г, 10,3 ммоль) в DMF (20 мл) перемешивали при 60°C в течение ночи. Смесь распределяли между EtOAc и водой. Слои разделяли, и органический слой промывали водой. Органический слой промывали солевым раствором, сушили (Na2SO4) и концентрировали досуха, получая в результате указанное в заголовке соединение, которое использовали на следующей стадии без очистки.
LC-MS [М+Н]+: m/z 352,6.
Стадия B: 1-бром-4-(2-бромэтокси)бензол
1,2-дибромэтан (4,26 мл, 49,4 ммоль) и карбонат цезия (6,83 г, 49,4 ммоль) добавляли к перемешиваемому раствору 4-бромфенола (1,40 г, 8,09 ммоль) в ацетоне (10 мл). Реакционную смесь нагревали до 80°С в течение 8 часов, после чего реакционную смесь охлаждали до комнатной температуры и фильтровали. Органический фильтрат концентрировали, и полученный неочищенный остаток очищали хроматографией на колонке с силикагелем (10-100% градиент EtOAc:гексан в качестве элюента), получая указанное в заголовке соединение.
1H-ЯМР (500 МГц, CDCl3) δ 7,42 (ш, 2H), 6,90 (ш, 2H), 4,31 (м, 2H), 3,71 (м, 2H).
Стадия C: трет-бутил-(3-(4-(4-(2-бромэтокси)фенил)-1H-пиразол-1-ил)пропил)карбамат
2,0 М водный раствор карбоната натрия (1,60 мл, 3,20 ммоль) добавляли во флакон для СВЧ, содержащий перемешиваемый раствор 1-бром-4-(2-бромэтокси)бензола (300 мг, 1,07 ммоль), трет-бутил-(3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиразол-1-ил)пропил)карбамата (565 мг, 1,61 ммоль) и дихлорида 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладия (78 мг, 0,1 1 ммоль) в 1,4-диоксане (4,3 мл). Смесь дегазировали с помощью потока азота в течение приблизительно 5 минут, затем флакон герметично закрывали и нагревали в микроволновом реакторе при 120°С в течение 20 минут. После охлаждения до комнатной температуры смесь фильтровали через слой диатомовой земли (Celite™), и твердый слой промывали EtOAc. Объединенные органические слои концентрировали, и полученный неочищенный остаток очищали с помощью колоночной хроматографии на силикагеле (0-60% EtOAc:гексан в качестве элюента), получая указанное в заголовке соединение.
LC-MS [M+Na]+: m/z 446,3.
Стадия D: трет-бутил-(3-(4-(4-(2-((1,3-диоксоизоиндолин-2-ил)окси)этокси)фенил)-1H-пиразол-1-ил)пропил)карбамат
N-гидроксифталимид (185 мг, 1,13 ммоль) добавляли к раствору трет-бутил-(3-(4-(4-(2-бромэтокси)фенил)пиразол-1-ил)пропил)карбамата (320 мг, 0,754 ммоль) и DBU (138 мг, 0,905 ммоль) в DMF (3,8 мл). Полученную смесь нагревали до 50°С. Через 16 часов реакционную смесь охлаждали до комнатной температуры и добавляли EDC (117 мг, 0,754 ммоль). После перемешивания при комнатной температуре в течение 30 минут реакционную смесь распределяли между EtOAc и водой. Слои разделяли, и органический слой промывали водой, а затем насыщенным раствором соли. Органический слой сушили (Na2SО4), фильтровали и концентрировали, получая неочищенный остаток, который очищали с помощью колоночной хроматографии на силикагеле (0-50% градиент EtOAc:гексан в качестве элюента), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 507,3.
Вышеуказанный промежуточный продукт (продукт стадии D) превращали в (S,Z)-3-(2-((2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)этокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат по процедурам, аналогичным тем, которые описаны на стадиях D-H примера 1.
LC-MS [М+Н]+: m/z 637,2.
ПРИМЕР 5
(S,Z)-3-(2-((2-(4-(1-(3-аминопропил)-1H-пиразол-4-ил)фенокси)этокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат (C5)
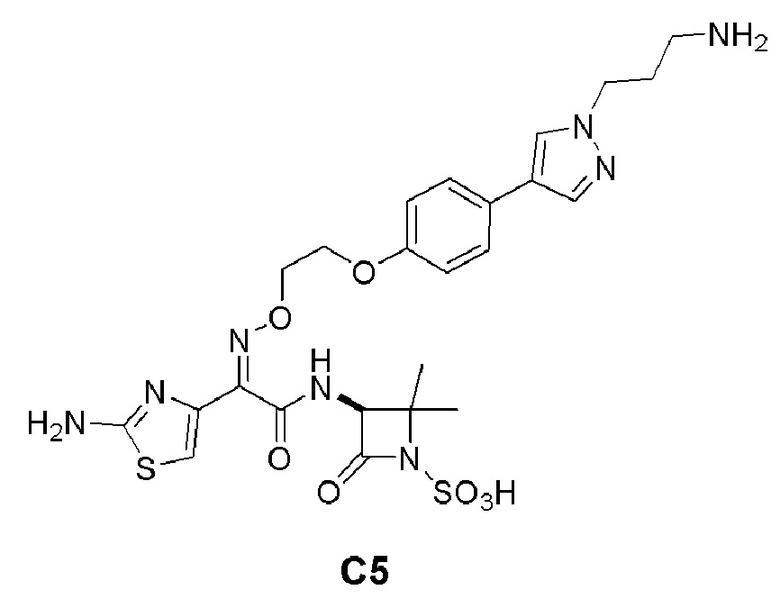
Соединение 5 получали из трет-бутил-(3-(4-(4-(2-((1,3-диоксоизоиндолин-2-ил)окси)этокси)фенил)-1H-пиразол-1-ил)пропил)карбамата по процедурам, аналогичным тем, которые описаны на стадиях E-H примера 1.
LC-MS [М+Н]+: m/z 623,3.
ПРИМЕР 6
(S)-3-(4-(1-(3-аминопропил)-1H-пиразол-4-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((2S,3S)-2-метил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота (C6)
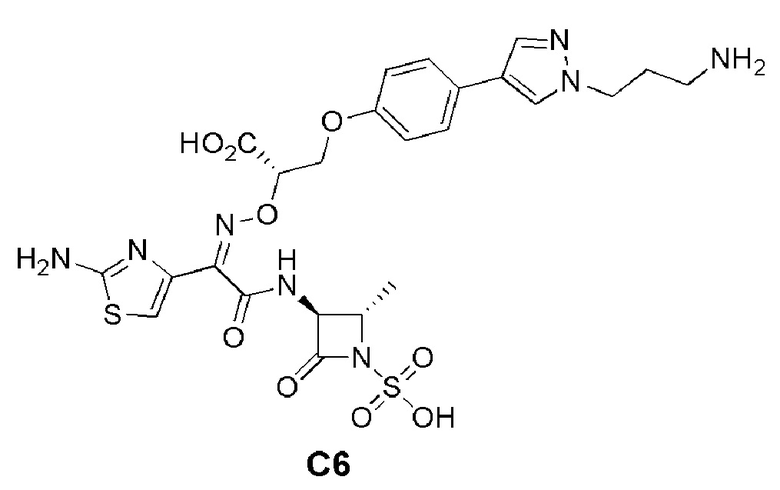
Соединение 6 получали по процедурам, аналогичным тем, которые описаны выше в примере 1, путем исключения стадии D и замены (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата на (2S,3S)-3-амино-2-метил-4-оксоазетидин-1-сульфоновую кислоту (из коммерческого источника CAS: 80080-65-1), где необходимо. LC-MS [М+Н]+: m/z 637,4.
ПРИМЕР 7
(2S,3S)-3-((Z)-2-(((S)-2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2-метил-4-оксоазетидин-1-сульфонат (C7)
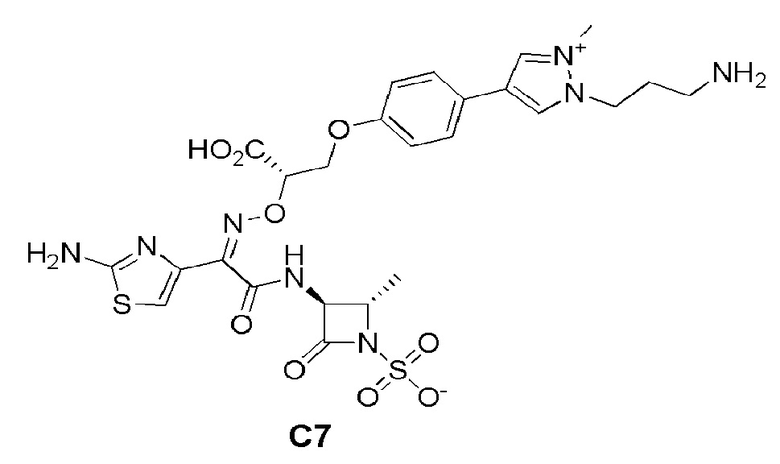
Соединение 7 получали по процедурам, аналогичным тем, которые описаны выше в примере 1, с заменой (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата на (2S,3S)-3-амино-2-метил-4-оксоазетидин-1-сульфоновую кислоту (CAS: 80582-09-8).
LC-MS [M+H]+: m/z 651,4.
ПРИМЕР 8
(S)-3-(4-(1-(3-аминопропил)-1H-пиразол-4-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((2R,3S)-2-метил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота (C8)
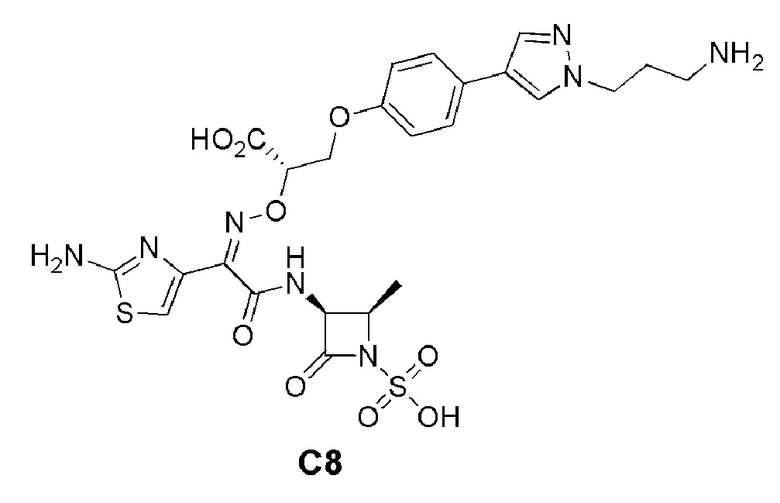
Соединение 8 получали по процедурам, аналогичным тем, которые описаны выше в примере 1, путем исключения стадии D и замены (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата на (2R,3S)-3-амино-2-метил-4-оксоазетидин-1-сульфоновую кислоту, где необходимо.
LC-MS [М+Н]+: m/z 637,4.
ПРИМЕР 9
(2R,3S)-3-((Z)-2-(((S)-2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2-метил-4-оксоазетидин-1-сульфонат (C9)
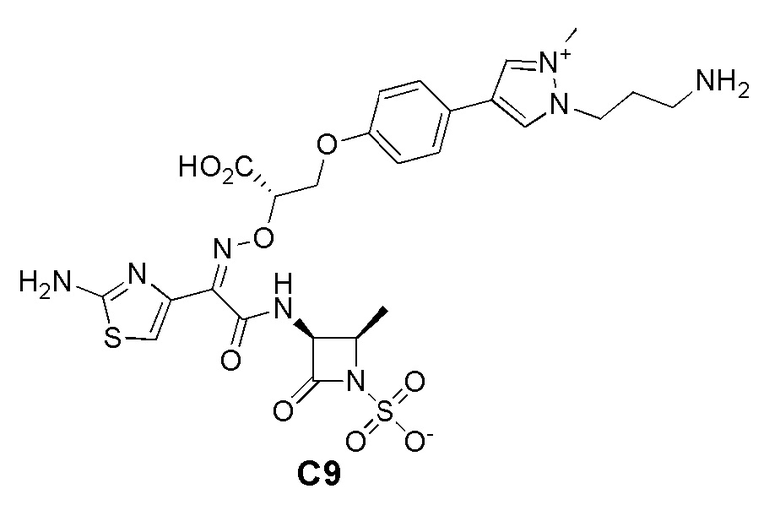
Соединение 9 получали по процедурам, аналогичным тем, которые описаны выше в примере 1, путем замены (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата на (2R,3S)-3-амино-2-метил-4-оксоазетидин-1-сульфоновую кислоту.
LC-MS [М+Н]+: m/z 651,3.
ПРИМЕР 10
(S)-3-(4-(1-(3-Аминопропил)-1H-имидазол-4-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота (C10)
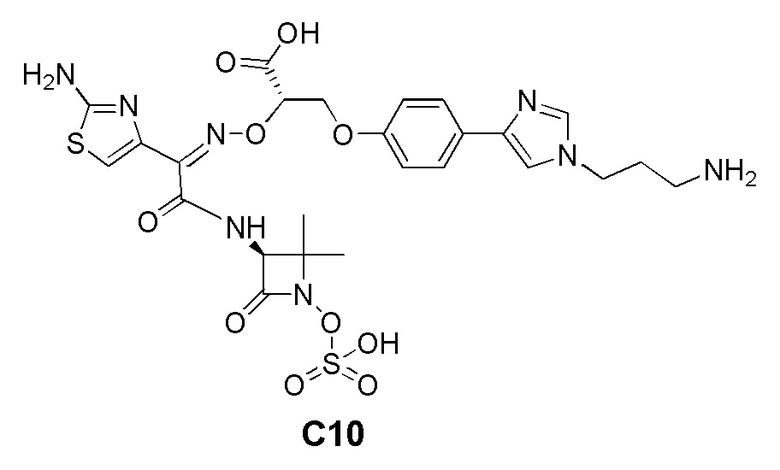
Стадия A: трет-бутил-(S)-2-(аминоокси)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-4-ил)фенокси)пропаноат
Применяли процедуру, аналогичную процедуре, используемой на стадии Е примера 1, с использованием в качестве реагентов (S)-трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноата (80 мг, 0,109 ммоль) (со стадии C примера 2), гидразина (3,42 мкл, 0,109 ммоль) в EtOH (4 мл).
LC-MS [M+H]+: m/z 477,26.
Стадия B: (S,Z)-2-(((1-(трет-бтокси)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-4-ил)фенокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)уксусная кислота
Применяли процедуру, аналогичную процедуре, используемой на стадии F примера 1, с использованием в качестве реагентов 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусной кислоты (42,0 мг, 0,154 ммоль) (промежуточное соединение 6) и (S)-трет-бутил-2-(аминоокси)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-4-ил)фенокси)пропаноата (70 мг, 0,147 ммоль) в этаноле (4 мл) и ClCH2CH2Cl (2 мл).
LC-MS [M+H]+: m/z 731,34.
Стадия C: трет-бутил-(S)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-4-ил)фенокси)-2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропаноат
Применяли процедуру, аналогичную процедуре, используемой на стадии G примера 1, с использованием в качестве реагентов (S,Z)-2-(((1-(трет-бутокси)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-4-ил)фенокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)уксусной кислоты (107 мг, 0,146 ммоль), DCC (76 мг, 0,366 ммоль), HOBt (56,1 мг, 0,366 ммоль), (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата (61,6 мг, 0,293 ммоль) и бикарбоната натрия (61,5 мг, 0,732 ммоль) в DMF (4 мл). Использовали способ очистки как на стадии G примера 2.
LC-MS [М+НМ+Н]+: m/z 923,48.
Стадия D: (S)-3-(4-(1-(3-аминопропил)-1H-имидазол-4-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота
Применяли процедуру, аналогичную процедуре, используемой на стадии H примера 2 с использованием в качестве реагентов (S)-трет-бутил 3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-4-ил)фенокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропаноата (36 мг, 0,035 ммоль) в CH2Cl2 (0,5 мл) и TFA (0,5 мл, 6,49 ммоль).
LC-MS [M]+: m/z 667,24.
1H ЯМР (500 МГц, DMSO): δH 9,42 (д, J=8,0 Гц, 1H); 7,76 (с, 1H); 7,27 (с, 2H); 7,00 (д, J=8,4 Гц, 2H); 6,79 (с, 1H); 4,93 (с, 1H); 4,61 (д, J=7,9 Гц, 1H); 4,34 (д, J=32,5 Гц, 2H); 4,14 (с, 2H); 2,78 (с, 2H); 2,06 (ш.с. 2H); 1,36 (с, 3H); 1,12 (с, 3H).
ПРИМЕР 11
(S)-3-((Z)-2-(2-амино-5-хлортиазол-4-ил)-2-(((S)-2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат (C11)
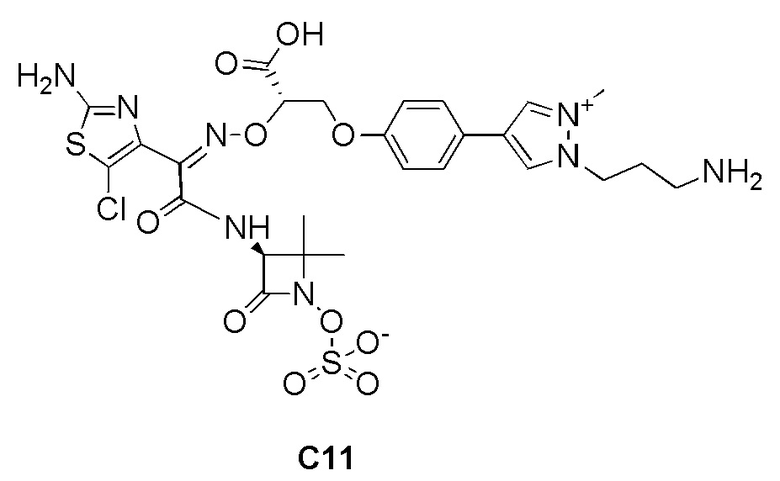
Стадия A: (S,Z)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)-5-хлортиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)-амино)пропил)-2-метил-1H-пиразол-2-ий иодид
Применяли процедуру, аналогичную процедуре, используемой на стадии F примера 1, с использованием в качестве реагентов 2-(2-((трет-бутоксикарбонил)амино)-5-хлортиазол-4-ил)-2-оксоуксусной кислоты (0,052 г, 0,170 ммоль, промежуточное соединение 6) и (S)-4-(4-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий иодида (0,1 г, 0,162 ммоль) (со стадии Е примера 1), этанола (4 мл) и ClCH2CH2Cl (2 мл).
LC-MS [M]+: m/z 779,30.
Стадия B: (S)-3-((Z)-2-((((S)-1-(трет-бутокси)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)-5-хлортиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
Применяли процедуру, аналогичную процедуре, используемой на стадии G примера 2, с использованием в качестве реагентов (S,Z)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)-5-хлортиазол-4-ил)(карбокси)-метилен)амино)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий иодида (0,14 г, 0,154 ммоль), DCC (0,080 г, 0,386 ммоль), НОВТ (0,059 г, 0,386 ммоль), (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата (0,065 г, 0,309 ммоль), бикарбоната натрия (0,065 г, 0,772 ммоль) в DMF (4 мл).
LC-MS [M]+: m/z 971,43.
Стадия C: (S)-3-((Z)-2-(2-амино-5-хлортиазол-4-ил)-2-(((S)-2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
Применяли процедуру, аналогичную процедуре, используемой на стадии H примера 2, с использованием в качестве реагентов 4-(4-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)-5-хлор-тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксо-этилиден)амино)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ия (36 мг, 0,033 ммоль), CH2Cl2 (0,5 мл) и TFA (1мл, 6,49 ммоль).
LC-MS [M]+: m/z 715,22.
1H ЯМР (500 МГц, DMSO): δH 8,77 (ш.с., 1H); 8,65 (ш.с., 1H); 7,34 (ш.с., 4H); 6,97 (с, 2H); 4,82 (с, 1H); 4,72 (ш.с., 1H); 4,57 (ш.с., 2H); 4,50 (ш.с., 2H); 4,27 (ш.с., 1H); 4,19 (с, 3H); 3,01 (ш.с., 2H); 2,21 (ш.с., 1H); 2,11 (ш.с., 1H); 1,42 (с, 3H); 1,30 (ш.с., 3H).
ПРИМЕР 12
(S)-3-((Z)-2-(5-амино-1,2,4-тиадиазол-3-ил)-2-(((S)-2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат (C12)
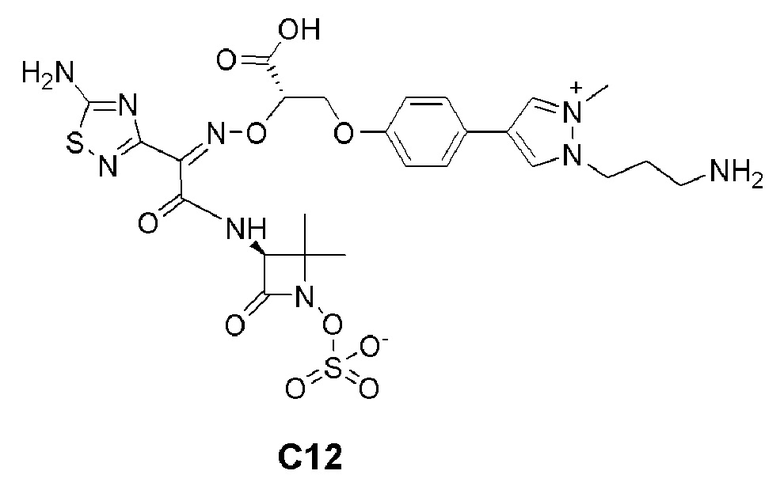
Стадия A: (S,Z)-4-(4-(3-(трет-бутокси)-2-((((5-((трет-бутоксикарбонил)амино)-1,2,4-тиадиазол-3-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)-амино)пропил)-2-метил-1H-пиразол-2-ий иодид
Применяли процедуру, аналогичную процедуре, используемой на стадии F примера 1, с использованием в качестве реагентов 2-(5-((трет-бутоксикарбонил)амино)-1,2,4-тиадиазол-3-ил)-2-оксоуксусной кислоты (0,046 г, 0,170 ммоль) (промежуточное соединение 7) и (S)-4-(4-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий иодида (0,1 г, 0,162 ммоль) в EtOH (4 мл) и ClCH2CH2Cl (2 мл).
LC-MS [M]+: m/z 746,31.
Стадия B: (S)-3-((Z)-2-((((S)-1-(трет-бутокси)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-оксопропан-2-ил)окси)имино)-2-(5-((трет-бутоксикарбонил)амино)-1,2,4-тиадиазол-3-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
Применяли процедуру, аналогичную процедуре, используемой на стадии G примера 2, с использованием в качестве реагентов (S,Z)-4-(4-(3-(трет-бутокси)-2-((((5-((трет-бутоксикарбонил)амино)-1,2,4-тиадиазол-3-ил)(карбокси)-метилен)амино)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий иодида (0,14 г, 0,160 ммоль), DCC (0,083 г, 0,401 ммоль), НОВТ (0,061 г, 0,401 ммоль), S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата (0,067 г, 0,320 ммоль) и бикарбоната натрия (0,067 г, 0,801 ммоль) в DMF (4 мл).
LC-MS [М]+: m/z 938,61.
Стадия C: (S)-3-((Z)-2-(5-амино-1,2,4-тиадиазол-3-ил)-2-(((S)-2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
Применяли процедуру, аналогичную процедуре, используемой на стадии H примера 2, с использованием в качестве реагентов 4-(4-((S)-3-(трет-бутокси)-2-(((Z)-(1-(5-((трет-бутоксикарбонил)амино)-1,2,4-тиадиазол-3-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ия (0,14 г, 0,133 ммоль) в CH2Cl2 (0,5 мл) и TFA (0,5 мл, 6,49 ммоль).
LC-MS [M]+: m/z 682,27.
1H ЯМР (500 МГц, DMSO): δH 8,72 (с, 1H); 8,61 (с, 1H); 8,11 (с, 2H); 7,29 (с, 2H); 6,93 (д, J=8,2 Гц, 2H); 4,83 (с, 1H); 4,73 (д, J=7,6 Гц, 1H); 4,60 (ш.с., 1H); 4,51 (с, 2H); 4,24 (д, J=10,3 Гц, 1H); 4,20 (с, 3H); 3,02 (д, J=23,8 Гц, 2H); 2,21 (ш.с., 1H); 2,09 (ш.с., 1H); 1,44 (с, 3H); 1,30 (с, 3H).
Таблица 2. При использовании в основном тех же процедур, как и в примере 12, без использования стадии D, в примере 13 синтезировано и охарактеризовано с помощью LC/MS следующее соединение.
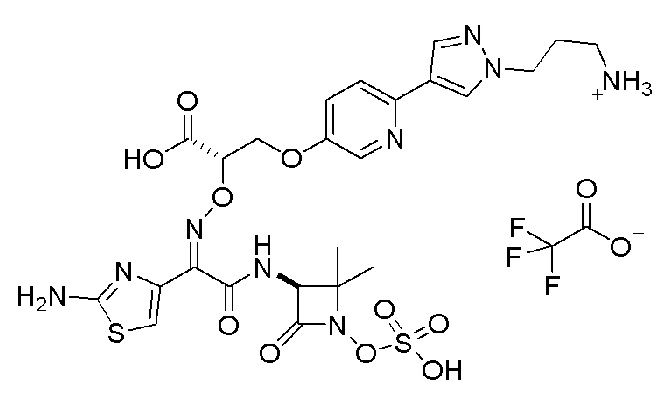
ПРИМЕР 14
4-(2-(4-(4-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)фенил)-1H-пиразол-1-ил)этил)-4-метилморфолин-4-ий 2,2,2-трифторацетат (C14)
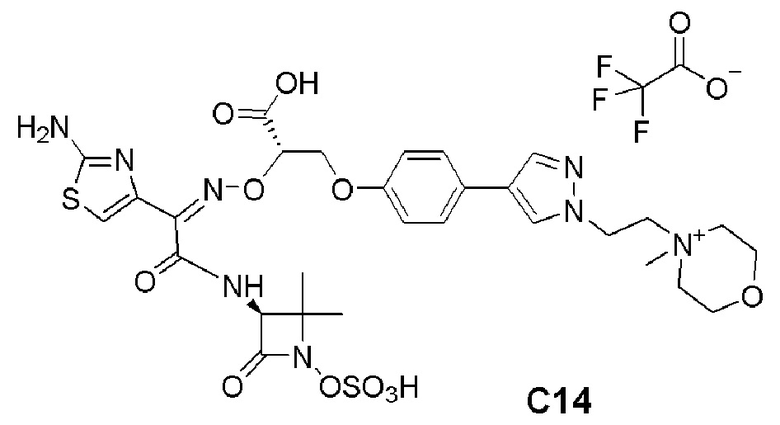
Стадия A: трет-бутил-(R)-2-гидрокси-3-(4-(1-(2-морфолиноэтил)-1H-пиразол-4-ил)фенокси)пропаноат
К раствору промежуточного продукта 3 ((R)-трет-бутил 3-(4-бромфенокси)-2-гидроксипропаноата) (500 мг, 1,58 ммоль), 4-(2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)этил)морфолина (726 мг, 2,36 ммоль) и дихлорида 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладия (103 мг, 0,158 ммоль) в THF (10 мл ) добавляли водный раствор 1 М трехосновного фосфата калия (4,73 мл, 4,73 ммоль). 20-мл флакон для СВЧ укупоривали, дегазировали (3 х) и заполняли N2, и перемешивали при 60°С в течение ночи. Анализ LC-MS показал полную конверсию. Реакционную смесь разбавляли насыщенным раствором NH4Cl, экстрагировали EtOAc (2×50 мл). Органические экстракты объединяли, сушили над MgSO4 и концентрировали досуха. Остаток очищали флэш-хроматографией на колонке SiO2 (40 г gold) с MeOH в DCM (0-15%), получая указанное в заголовке соединение.
LC-MS [M+H]+: m/z 418,42.
Стадия B: трет-бутил-(S)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-(4-(1-(2-морфолиноэтил)-1H-пиразол-4-ил)фенокси)пропаноат
Применяли процедуру, аналогичную процедуре, используемой на стадии C примера 1, с использованием в качестве реагентов трет-бутил-(R)-2-гидрокси-3-(4-(1-(2-морфолиноэтил)-1H-пиразол-4-ил)фенокси)пропаноата (0,480 г, 1,15 ммоль), 2-гидроксиизоиндолин-1,3-диона (0,225 г, 1,38 ммоль), трифенилфосфина (0,362 г, 1,38 ммоль), DIAD (0,272 мл, 1,380 ммоль) и THF (4 мл).
LC-MS [М+Н]+: m/z 563,47.
Стадия C: (S)-4-(2-(4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-фенил)-1H-пиразол-1-ил)этил)-4-метилморфолин-4-ий иодид
К раствору (S)-трет-бутил-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-(4-(1-(2-морфолиноэтил)-1H-пиразол-4-ил)-фенокси)пропаноата (250 мг, 0,444 ммоль) в ацетонитриле (4 мл) добавляли иодметан (0,167 мл, 2,67 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи и концентрировали в высоком вакууме, получая указанное в заголовке соединение.
LC-MS [M]+: m/z 577,56.
Стадия D: (S)-4-(2-(4-(4-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)фенил)-1H-пиразол-1-ил)этил)-4-метилморфолин-4-ий иодид
Применяли процедуру, аналогичную процедуре, используемой на стадии Е примера 1, с использованием в качестве реагентов (S)-4-(2-(4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1H-пиразол-1-ил)этил)-4-метилморфолин-4-ий иодида (310 мг, 0,440 ммоль), гидразина (14,0 мкл, 0,440 ммоль) в этаноле (4 мл).
LC-MS [M]+: m/z 447,50.
Стадия E: (S,Z)-4-(2-(4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)фенил)-1H-пиразол-1-ил)этил)-4-метилморфолин-4-ий иодид
Применяли процедуру, аналогичную процедуре, используемой на стадии F примера 1, с использованием в качестве реагентов 2-(5-((трет-бутоксикарбонил)амино)-1,2,4-тиадиазол-3-ил)-2-оксоуксусной кислоты (0,118 г, 0,435 ммоль) (промежуточное соединение 7) и (S)-4-(2-(4-(4-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)фенил)-1H-пиразол-1-ил)этил)-4-метилморфолин-4-ий иодида (0,250 г, 0,435 ммоль) в этаноле (6 мл) и ClCH2CH2Cl (2 мл).
LC-MS [M]+: m/z 701,00.
Стадия F: 4-(2-(4-(4-((S)-3-(трет-бутокси)-2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)фенил)-1H-пиразол-1-ил)этил)-4-метилморфолин-4-ий иодид
Применяли процедуру, аналогичную процедуре, используемой на стадии F примера 2, с использованием в качестве реагентов (S,Z)-4-(2-(4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)фенил)-1H-пиразол-1-ил)этил)-4-метилморфолин-4-ий иодида (0,250 г, 0,302 ммоль), DCC (0,124 г, 0,603 ммоль), НОВТ (0,092 г, 0,603 ммоль, (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата (0,127 г, 0,603 ммоль), бикарбоната натрия (0,101 г, 1,207 ммоль) в DMF (4 мл).
LC-MS [M]+: m/z 894,00.
Стадия G: (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-1-карбокси-2-(4-(1-(2-(4-метил-морфолино-4-ий)этил)-1H-пиразол-4-ил)фенокси)этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
Применяли процедуру, аналогичную процедуре, используемой на стадии Н примера 2, с использованием в качестве реагентов 4-(2-(4-(4-((S)-3-(трет-бутокси)-2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)-амино)-окси)-3-оксопропокси)фенил)-1H-пиразол-1-ил)этил)-4-метилморфолин-4-ий иодида (0,070 г, 0,069 ммоль) в CH2Cl2 (0,5 мл) и TFA (0,5 мл, 6,49 ммоль).
LC-MS [М+Н]+: m/z 737,43.
Таблица 3. Используя те же процедуры получения, как в примере 1, были синтезированы и охарактеризованы с помощью LC/MS соединения примеров 15-22
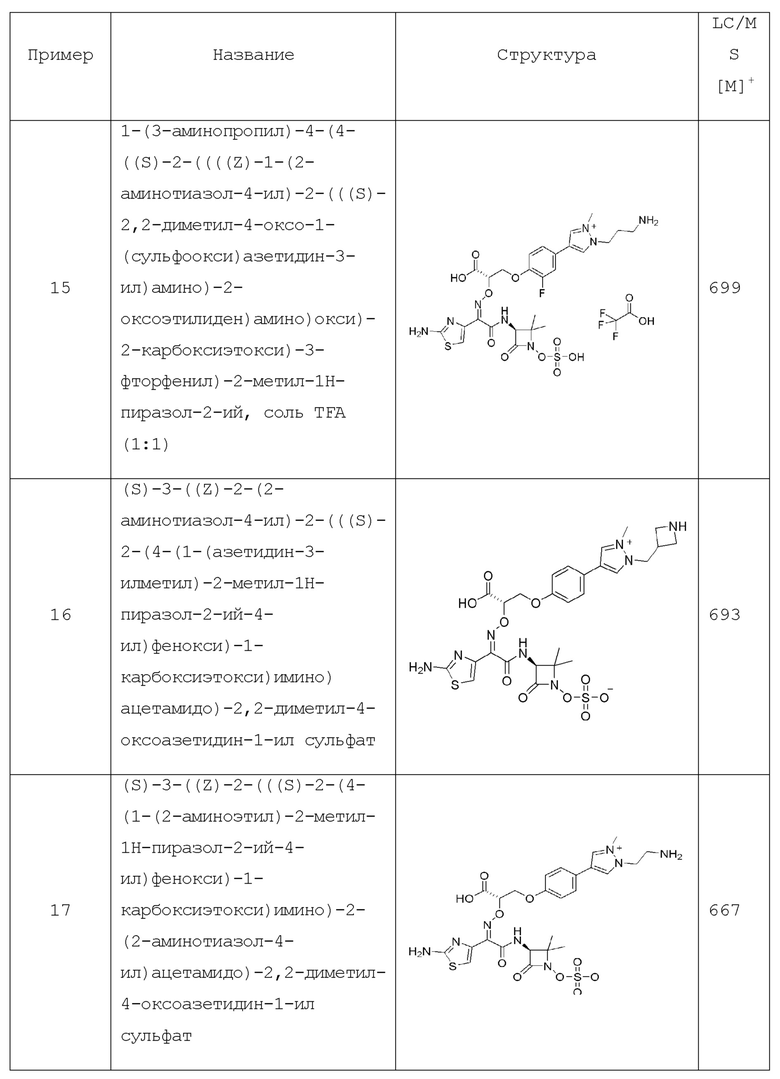
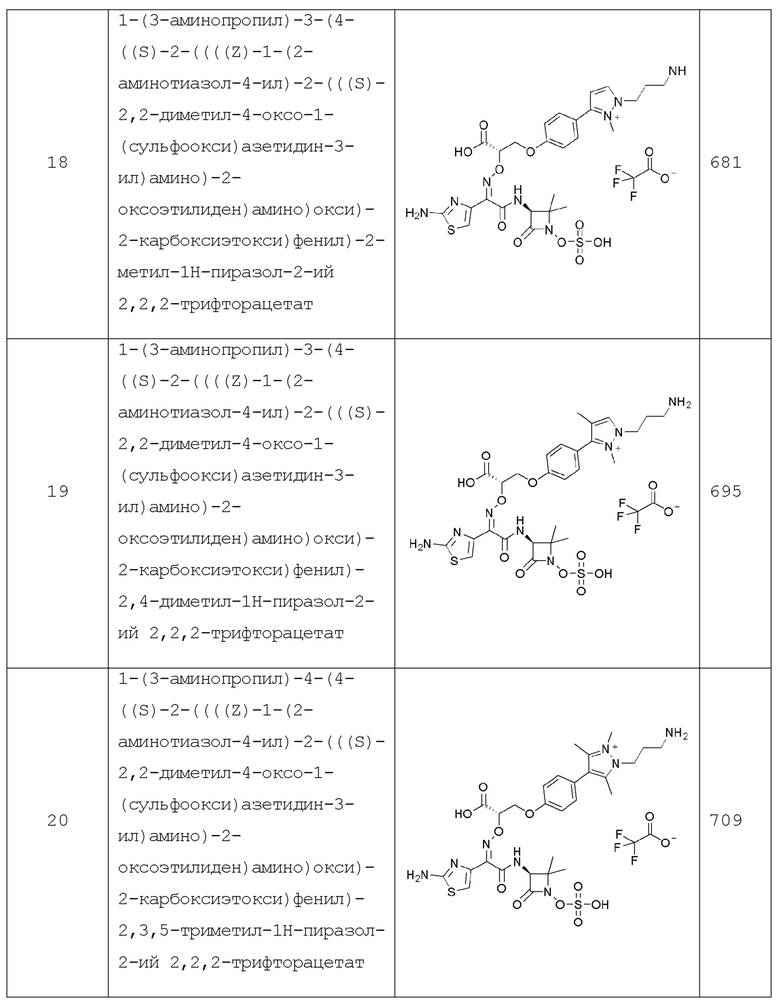
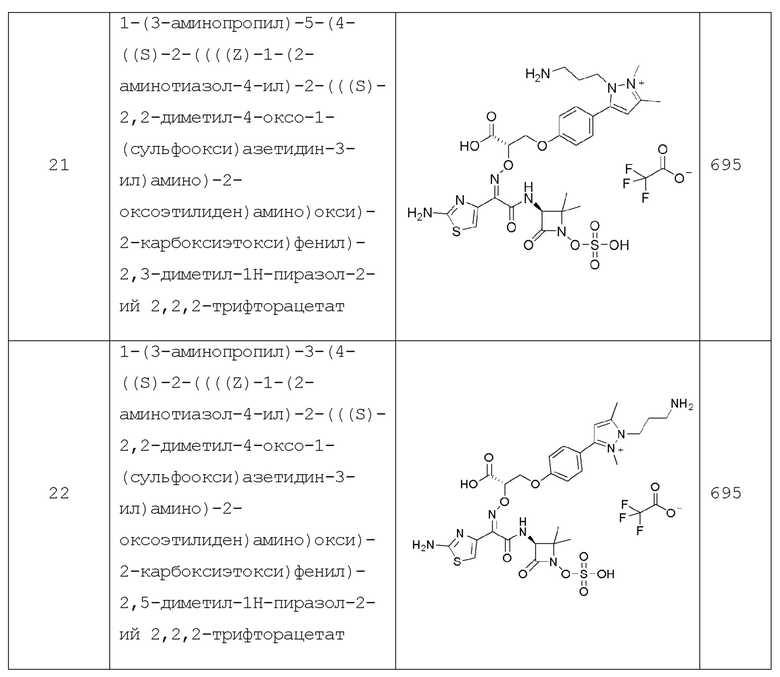
ПРИМЕРЫ 23A и 23B
23A: 1-(3-аминопропил)-4-(4-(2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксипропокси)фенил)-2-метил-1H-пиразол-2-ий 2,2,2-трифторацетат
23B: 1-(3-аминопропил)-4-(4-(2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксипропокси)фенил)-2-метил-1H-пиразол-2-ий 2,2,2-трифторацетат
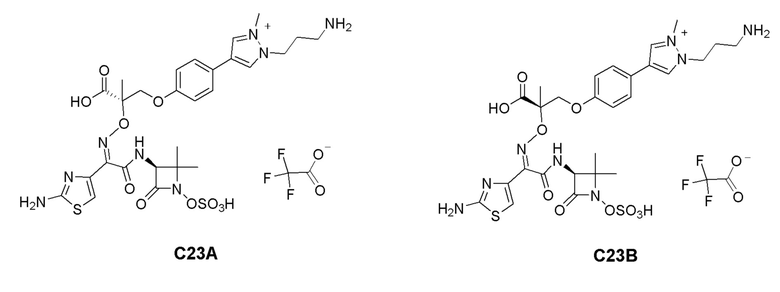
Стадия A: трет-бутил-(3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропил)карбамат
Карбонат цезия (12,2 г, 37,5 ммоль) добавляли при комнатной температуре к смеси 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (4,85 г, 25,00 ммоль), трет-бутил-(3-бромпропил)карбамата (5,95 г, 25,0 ммоль) в 40 мл DMF, и смесь перемешивали при комнатной температуре в течение ночи. Анализ LC-MS показал, что реакция завершена. Смесь разбавляли EtOAc и водой, экстрагировали EtOAc. Объединенный органический слой промывали насыщенным раствором соли и сушили над MgSO4, отфильтровывали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью EtOAc/изогексан (10-80% градиент), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 352,33.
Стадия B: трет-бутил 3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-2-метилпропаноат
К раствору трет-бутил-2-(аминоокси)-3-(4-бромфенокси)-2-метилпропаноата (100 мг, 0,289 ммоль), трет-бутил-(3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропил)карбамата (101 мг, 0,289 ммоль) и дихлорида 1,1'-бис(ди-трет-бутилфосфино)ферроценпалладия (18,82 мг, 0,029 ммоль) в диоксане (1 мл) добавляли водный раствор 1 М фосфата калия (0,867 мл, 0,867 ммоль). Флакон закупоривали, дегазировали и заполняли азотом, и перемешивали при 70°С в течение 1 часа. Анализ LC-MS показал полную конверсию. Реакционную смесь разбавляли водой и экстрагировали EtOAc х 2. Органический слой сушили над MgSO4, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью CH2Cl2/MeOH (100-90%), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 491,43.
Стадия C: трет-бутил 3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-2-метилпропаноат
К смеси трет-бутил-2-(аминоокси)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-2-метилпропаноата (540 мг, 1,10 ммоль), молекулярных сит (200 мг) в толуоле при комнатной температуре добавляли 4-метилбензол-сульфоновую кислоту (12% в уксусной кислоте) (63,2 мг, 0,044 ммоль), и полученную смесь перемешивали при 100°С в течение 4 часов. Анализ LC-MS показал, что реакция практически завершена. Отфильтровывали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью CH2Cl2/MeOH (100-90%), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 622,56.
Стадия D: 4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-2-метил-3-оксопропокси)-фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий иодид
Процедура получения аналогична процедуре, используемой на стадии D примера 1, с использованием в качестве реагентов третбутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-2-метилпропаноата (0,495 г, 0,797 ммоль), MeI (0,399 мл, 6,38 ммоль) в MeCN (3 мл).
LC-MS [М]+: m/z 635,58.
Стадия E: (Z)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-2-метил-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий иодид
К перемешиваемой при комнатной температуре смеси 4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-2-метил-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ия (485 мг, 0,763 ммоль) в EtOH/DCM добавляли гидразин (0,024 мл, 0,763 ммоль) в 0,3 мл DCM, и смесь перемешивали при комнатной температуре в течение 4 часов. Анализ LC-MS показал, что промежуточное соединение получено. Затем к реакционной смеси добавляли 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусную кислоту (228 мг, 0,839 ммоль) и перемешивали при комнатной температуре в течение ночи. Анализ LC-MS показал, что реакция завершена. Раствор фильтровали, и фильтрат концентрировали. Остаток очищали с помощью препаративной высокоэффективной жидкостной хроматографии, элюируя смесью ацетонитрил/вода+0,05% TFA (0-100%), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 759,58.
Стадия F: 4-(4-(3-(трет-бутокси)-2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((2R,3S)-2-метил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-метил-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий
К раствору (Z)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-2-метил-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий иодида (80 мг, 0,105 ммоль) в DMF (1 мл) добавляли DCC (87 мг, 0,421 ммоль) и НОВТ (60,5 мг, 0,316 ммоль). Затем смесь перемешивали при комнатной температуре в течение 30 минут, с последующим добавлением (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфат (76 мг, 0,421 ммоль) и бикарбоната натрия (35,4 мг, 0,421 ммоль). Конечную смесь перемешивали при комнатной температуре в течение ночи (16 часов). Анализ LC-MS показал полную конверсию. Реакционную смесь фильтровали, и фильтрат очищали с помощью обращенно-фазовой хдоматографией на колонке ISCO (130 г, 0-100%, Н2О/MeCN/0,05% TFA), фракции собирали и лиофилизировали, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 921,76.
Стадия G: (S)-3-((Z)-2-((((S)-1-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-2-карбоксипропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат объединенный с 2,2,2-трифторуксусной кислотой (1:1)
К раствору 4-(4-(3-(трет-бутокси)-2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-метил-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ия (110 мг, 0,116 ммоль) в DCM добавляли 2 мл TFA. Смесь перемешивали при комнатной температуре в течение 90 минут. Анализ LC-MS показал полную конверсию. Смесь концентрировали досуха в вакууме (без нагрева). Добавляли DCM (10 мл), и смесь концентрировали три раза для удаления TFA. Остаток промывали сухим MeCN (2×2 мл) для удаления примесей. Остаток растворяли в DMSO и очищали с помощью препаративной высокоэффективной жидкостной хроматографии с использованием смеси MeCN/H2O (0,05% TFA в обоих) с градиентом 2-25% за 10 минут. Быстро элюированный изомер представлял собой соединение 23А, а медленно элюироемое соединение представляло собой соединение 23B. Фракции продуктов собирали и лиофилизовали, получая указанные в заголовке соединения.
Соединение 23А: LC-MS [М+Н]: m/z 695,33.
Соединение 23B: LC-MS [М+Н]: m/z 695,16.
ПРИМЕР 24
(S)-3-((Z)-2-(((S)-2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)-3-фторфенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат (C24)
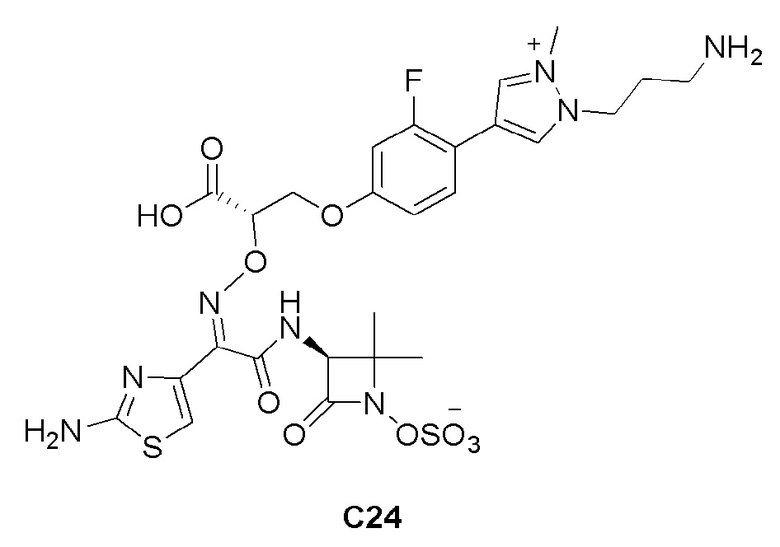
Стадия A: трет-бутил-(R)-3-(4-бром-3-фторфенокси)-2-гидроксипропаноат
Процедура получения аналогична процедуре, используемой для получения промежуточного соединения 3, с использованием в качестве реагентов трет-бутил-оксиран-2-карбоксилата (4,19 г, 29,1 ммоль), (R,R)-CO-катализатора (0,595 г, 0,709 ммоль), 4-бром-3-фторфенола (2,71 г, 14,19 ммоль).
LC-MS [M+Na]+: m/z 358,92.
Стадия B: трет-бутил-(R)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)-3-фторфенокси)-2-гидроксипропаноат
Процедура получения аналогична процедуре, используемой на стадии B примера 1, с использованием в качестве реагентов трет-бутил-(R)-3-(4-бром-3-фторфенокси)-2-гидроксипропаноата (470 мг, 1,40 ммоль), трет-бутил-(3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропил)карбамата (512 мг, 1,458 ммоль), дихлорида 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладия (91 мг, 0,140 ммоль), 1,4-диоксана (4 мл) и фосфата калия (4,21 мл, 4,21 ммоль, 1 М водн.).
LC-MS [М+Н]+: m/z 480,84.
Стадия C: трет-бутил-(S)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)-3-фторфенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноат
Процедура получения аналогична процедуре, используемой на стадии C примера 1, с использованием в качестве реагентов трет-бутил-(R)-3-(4-(1-(3-((трет-бутоксикарбонил)-амино)пропил)-1H-пиразол-4-ил)-3-фторфенокси)-2-гидроксипропаноата (0,451 г, 0,94 ммоль), 2-гидроксиизоиндолин-1,3-диона (0,153 г, 0,94 ммоль), трифенилфосфина (0,271 г, 1,04 ммоль), DEAD (0,164 мл, 1,04 ммоль) и THF (4 мл).
LC-MS [М+Н]+: m/z 625,51.
Стадия D: (S)-4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-фторфенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий иодид
Процедура получения аналогична процедуре, используемой на стадии D примера 1, с использованием в качестве реагентов трет-бутил-(S)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)-3-фторфенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноата (0,576 г, 0,922 ммоль), MeI (0,461 мл, 7,38 ммоль) в MeCN (3 мл).
LC-MS [М]+: m/z 639,55.
Стадия E: (S,Z)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-(карбокси)метилен)амино)окси)-3-оксопропокси)-2-фторфенил)-1-(3-((трет-бутокси-карбонил)амино)пропил)-2-метил-1H-пиразол-2-ий иодид
Процедура получения аналогична процедуре, используемой на стадии E примера 1, с использованием в качестве реагентов гидразина (0,028 мл, 0,89 ммоль), S)-4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-фторфенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий иодида (712 мг, 0,89 ммоль), 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусной кислоты (267 мг, 0,979 ммоль).
LC-MS [М+Н]+: m/z 763,53.
Стадия F: 4-(4-((S)-3-(трет-бутокси)-2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2-фторфенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий
Процедура получения аналогична процедуре, используемой на стадии F примера 1, с использованием в качестве реагентов (S,Z)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)-амино)окси)-3-оксопропокси)-2-фторфенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий иодида (0,679 г, 0,889 ммоль), DCC (0,550 г, 2,67 ммоль), НОВТ (0,510 г, 2,67 ммоль), (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата (0,280 г, 1,33 ммоль) и бикарбоната натрия (0,299 г, 3,56 ммоль) в DMF (3 мл).
LC-MS [M]+: m/z 955,69.
Стадия G: (S)-3-((Z)-2-(((S)-2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)-3-фторфенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат объединенный с муравьиной кислотой (1:1)
Процедура получения аналогична процедуре, используемой на стадии G примера 1, с использованием в качестве реагентов 4-(4-((S)-3-(трет-бутокси)-2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2-фторфенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ия (0,13 г, 0,136 ммоль), TFA (2 мл, 26 ммоль).
LC-MS [M]+: m/z 699,69/
ПРИМЕР 25
1-(3-аминопропил)-4-(4-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((2R,3S)-2-метил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксипропокси)фенил)-2-метил-1H-пиразол-2-ий 2,2,2-трифторацетат (C25)
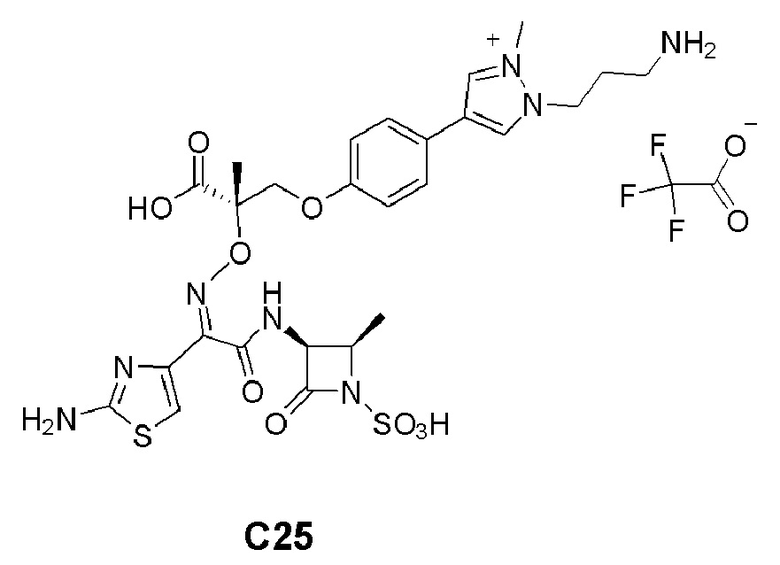
Стадия A: 4-(4-(3-(трет-бутокси)-2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((2R,3S)-2-метил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-метил-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий
Процедура получения аналогична процедуре, используемой на стадии F примера 1, с использованием в качестве реагентов (Z)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-2-метил-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ия (0,080 г, 0,105 ммоль), DCC (0,087 г, 0,421 ммоль), НОВТ (0,0605 г, 0,316 ммоль), (2R,3S)-3-амино-2-метил-4-оксоазетидин-1-сульфоновой кислоты (0,076 г, 1,05 ммоль) и бикарбоната натрия (0,0354 г, 0,421 ммоль).
LC-MS [M]+: m/z 921,76.
Стадия B: (2R,3S)-3-((Z)-2-((((S)-1-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-2-карбоксипропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2-метил-4-оксоазетидин-1-сульфонат объединенный с 2,2,2-трифторуксусной кислотой (1:1)
Процедура получения аналогична процедуре, используемой на стадии G примера 1, с использованием в качестве реагентов 4-(4-(3-(трет-бутокси)-2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((2R,3S)-2-метил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-метил-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ия (0,086 г, 0,093 ммоль), TFA (2,5 мл, 32,4 ммоль).
LC-MS [M]: m/z 665,20.
ПРИМЕР 26
2-(1-(3-аминопропил)-1H-пиразол-4-ил)-5-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксо-этилиден)амино)окси)-2-карбоксиэтокси)-1-метилпиридин-1-ий 2,2,2-трифторацетат (C26)
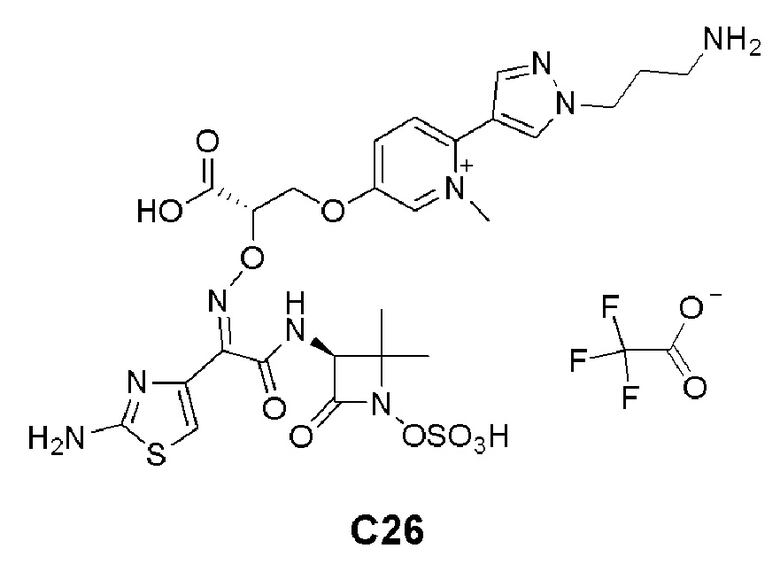
Стадия A: трет-бутил 3-((6-бромпиридин-3-ил)окси)-2-гидроксипропаноат
K2CO3 (6,35 г, 46,0 ммоль) добавляли к перемешиваемой при комнатной температуре смеси трет-бутил-оксиран-2-карбоксилата (6,63 г, 46,0 ммоль), 6-бромпиридин-3-ола (4 г, 22,99 ммоль) в ацетонитриле, и полученную смесь перемешивали при 90°С в течение 3 часов, охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью EtOAc/изогексан (0-50%), получая указанное в заголовке соединение.
Пример 26 (S)-3-((Z)-2-(((S)-2-((6-(1-(3-аминопропил)-1H-пиразол-4-ил)-1-метилпиридин-1-ий-3-ил)окси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат объединенный с 2,2,2-трифторуксусной кислотой (1:1) получали из трет-бутил 3-((6-бромпиридин-3-ил)окси)-2-гидроксипропаноата процедурой из 6 стадий. Процедуры этих стадий аналогичны процедурам стадий B, C, D, E, F, G примера 2.
LC-MS [М]+: m/z 682,20.
ПРИМЕР 27
(S)-3-(4-(3-амино-1-(3-аминопропил)-1H-пиразол-4-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота (C27)
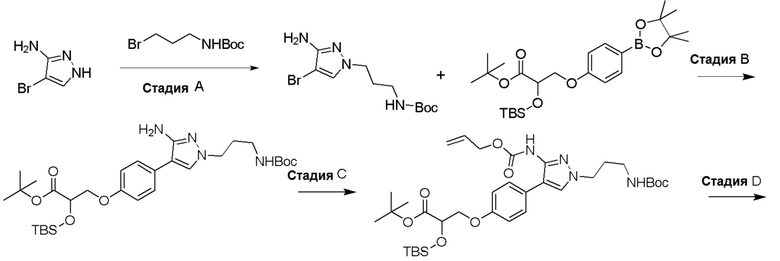
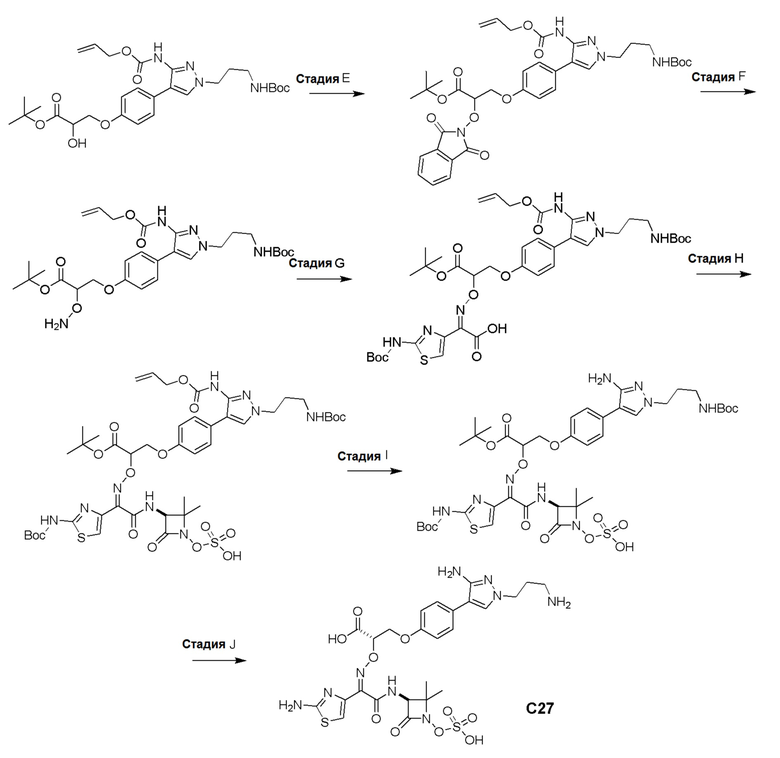
Стадия A: трет-бутил-(3-(3-амино-4-бром-1H-пиразол-1-ил)пропил)карбамат
Процедура получения аналогична процедуре, используемой на стадии А примера 1, с использованием в качестве реагентов 4-бром-1H-пиразол-3-амина и трет-бутил-(3-бромпропил)карбамата.
Стадия B: трет-бутил-(S)-3-(4-(3-амино-1-(3-((трет-бутоксикарбонил)амино)-пропил)-1H-пиразол-4-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноат
Процедура получения аналогична процедуре, используемой на стадии В примера 1, с использованием в качестве реагентов трет-бутил-2-((трет-бутилдиметилсилил)окси)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)-пропаноата (0,21 г, 0,439 ммоль) (промежуточное соединение 8), трет-бутил-(3-(3-амино-4-бром-1H-пиразол-1-ил)пропил)карбамата (0,17 г, 0,533 ммоль) и дихлорида 1,1'-бис(ди-трет-бутилфосфино)-ферроцен палладия (0,029 г, 0,044 ммоль) в THF (3 мл).
LC-MS [М+Н]+: m/z 592,00.
Стадия C: трет-бутил-3-(4-(3-(((аллилокси)карбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноат
К раствору трет-бутил-3-(4-(3-амино-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноата (0,54 г, 0,914 ммоль) в CH2Cl2 (10 мл) добавляли DIPEA (0,319 мл, 1,83 ммоль) с последующим добавлением аллилкарбонхлоридата (0,194 мл, 1,828 ммоль) при комнатной температуре. Смесь перемешивали в течение 4 часов, затем растворитель удаляли. Остаток очищали с помощью колоночной хроматографии на силикагеле (колонка Redi 40 г gold), элюируя смесью EtOAc/гексан (0-50%, 6 cv; 50%, 10 cv), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 676,71.
Стадия D: трет-бутил-3-(4-(3-(((аллилокси)карбонил)амино)-1-(3-((трет-бутоксикарбонил)-амино)пропил)-1H-пиразол-4-ил)фенокси)-2-гидроксипропаноат
К раствору трет-бутил-3-(4-(3-(((аллилокси)карбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноата (0,54 г, 0,80 ммоль) при комнатной температуре добавляли TBAF в THF (0,80 мл, 0,80 ммоль, 1 М). Смесь перемешивали в течение 1 часа, растворитель удаляли. Остаток очищали с помощью колоночной хроматографии на силикагеле (колонка Redi 24 г gold), элюировали смесью EtOAc/гексан (0-80%, 6 cv; 80%, 10 cv), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 561,1.
Стадия E: трет-бутил 3-(4-(3-(((аллилокси)карбонил)амино)-1-(3-((трет-бутоксикарбонил)-амино)пропил)-1H-пиразол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноат
Процедура получения аналогична процедуре, используемой на стадии С примера 1, с использованием в качестве реагентов трет-бутил-3-(4-(3-(((аллилокси)карбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-2-гидроксипропаноата (0,3 г, 0,535 ммоль), 2-гидроксиизоиндолин-1,3-диона (0,096 г, 0,589 ммоль), трифенилфосфина (0,168 г, 0,642 ммоль), DIAD (0,125 мл, 0,642 ммоль) в THF (1 мл).
LC-MS [М+Н]+: m/z 706.
Стадия F: трет-бутил-3-(4-(3-(((аллилокси)карбонил)амино)-1-(3-((трет-бутоксикарбонил)-амино)пропил)-1H-пиразол-4-ил)фенокси)-2-(аминоокси)пропаноат
Процедура получения аналогична процедуре, используемой на стадии E примера 1, с использованием в качестве реагентов трет-бутил-3-(4-(3-(((аллилокси)карбонил)-амино)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноата (35 мг, 0,050 ммоль) и гидразина (3,1 1 мкл, 0,099 ммоль) в THF (2 мл).
LC-MS [М+Н]+: m/z 576,47.
Стадия G: (Z)-2-(((3-(4-(3-(((аллилокси)карбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)-пропил)-1H-пиразол-4-ил)фенокси)-1-(трет-бутокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)уксусная кислота
Процедура получения аналогична процедуре, используемой на стадии F примера 1, с использованием в качестве реагентов 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусной кислоты (0,028 г, 0,101 ммоль) и трет-бутил 3-(4-(3-(((аллилокси)карбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-2-(аминоокси)пропаноата (0,053 г, 0,092 ммоль) в этаноле (2 мл).
LC-MS [М+Н]+: m/z 830,64.
Стадия H: трет-бутил-3-(4-(3-(((аллилокси)карбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)-пропил)-1H-пиразол-4-ил)фенокси)-2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2 оксоэтилиден)амино)окси)-пропаноат
Процедура получения аналогична процедуре, используемой на стадии G примера 1, с использованием в качестве реагентов Z)-2-(((3-(4-(3-(((аллилокси)карбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-1-(трет-бутокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)уксусной кислоты (76 мг, 0,092 ммоль), DCC (56,7 мг, 0,275 ммоль), НОВТ (42,1 мг, 0,275 ммоль), (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата (77 мг, 0,366 ммоль) и бикарбоната натрия (77 мг, 0,916 ммоль) в DMF (4 мл). Реакционную смесь фильтровали и переносили на следующую стадию без дополнительной очистки.
LC-MS [М+Н]+: m/z 1023,31.
Стадия I: трет-бутил-3-(4-(3-амино-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропаноат
К неочищенной реакционной смеси трет-бутил-3-(4-(3-(((аллилокси)карбонил)амино)-1-(3-((трет-бутокси-карбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропаноата (70 мг, 0,068 ммоль) в DMF (2 мл) добавляли тетракис (23,7 мг, 0,021 ммоль) и фенилсилан (74,1 мг, 0,685 ммоль). Смесь перемешивали при комнатной температуре в течение 10 минут. Смесь фильтровали и очищали методом RP-HPLC (Gilson) (колонка С-18), элюировали смесью ACN/вода с 0,05% TFA (20-100%, 8 мин; 100% 10 мин). Фракцию продукта лиофилизировали, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 938,88.
Стадия J: (S)-3-(4-(3-амино-1-(3-аминопропил)-1H-пиразол-4-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота
Процедура получения аналогична процедуре, используемой на стадии H примера 1, с использованием в качестве реагентов трет-бутил-3-(4-(3-амино-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропаноата (40 мг, 0,038 ммоль), TFA (0,5 мл, 6,49 ммоль) в DCM (0,5 мл).
LC-MS [М+Н]+: m/z 682,45.
1H-ЯМР (500 МГц, D2O) δ 7,62 (с, 1H); 7,30 (д, J=8,4 Гц, 2H); 6,96 (т, J=4,4 Гц, 3H); 4,94-4,96 (м, 1H); 4,40-4,43 (м, 2H); 4,04-4,07 (м, 2H); 2,87-2,90 (м, 2H); 2,07-2,10 (м, 2H); 1,33 (с, 3H); 1,07 (с, 3H).
ПРИМЕР 28
(S)-3-((Z)-2-(((S)-2-(4-(3-амино-1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат (C28)
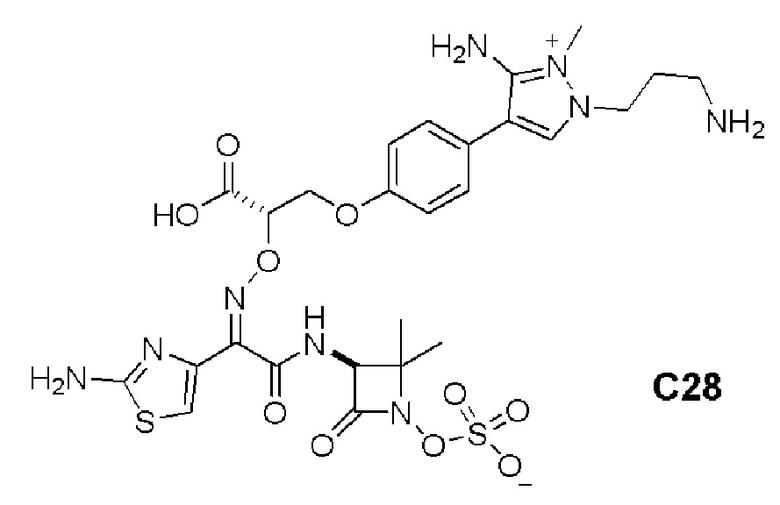
Стадия A: (S)-3-(((Аллилокси)карбонил)амино)-4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий иодид
Процедура получения аналогична процедуре, используемой на стадии D примера 1, с использованием в качестве реагентов (S)-трет-бутил 3-(4-(3-(((аллилокси)карбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноата (110 мг, 0,156 ммоль), MeI (0,078 мл, 1,247 ммоль) в CAN (1,5 мл).
LC-MS [M]+: m/z 720,09.
Процедуры на стадиях B-F аналогичны процедурам стадий F-J примера 28, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 696,41.
1H-ЯМР (500 МГц, D2O) δ 7,97 (1H, с), 7,38 (2H, д, J=8,3 Гц), 7,01-7,08 (3H, м), 4,99 (1H, д, J=5,8 Гц), 4,43-4,51 (2H, м), 4,33 (2H, т, J=7,2 Гц), 3,77 (3H, с), 3,06 (2H, т, J=7,9 Гц), 2,14-2,20 (2H, м), 1,44 (3H, с), 1,10 (3H, с).
ПРИМЕР 29
1-(3-аминопропил)-4-(4-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-(2H-тетразол-5-ил)этокси)фенил)-2-метил-1H-пиразол-2-ий 2,2,2-трифторацетат (C29)
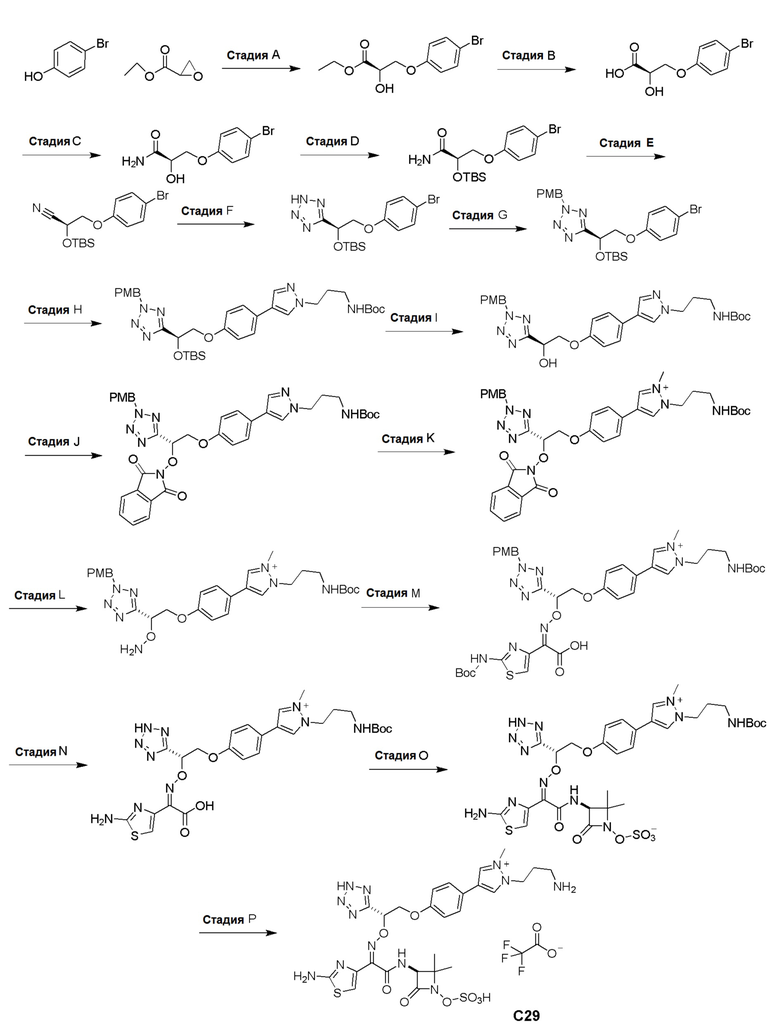
Стадия A: (R)-этил-3-(4-бромфенокси)-2-гидроксипропаноат
К смеси 4-бромфенола (2 г, 11,6 ммоль), R,R-Co-катализатора (III) (0,485 г, 0,578 ммоль) (J. Am. Chem. Soc, 1999, 121, 6086-6087), молекулярных сит 4Å (4 г), этил оксиран-2-карбоксилата (2,95 г, 25,4 ммоль) под азотом добавляли трет-бутиловый эфир (6 мл), и полученную суспензию перемешивали при комнатной температуре в атмосфере азота в течение выходных дней. Фильтрат концентрировали после фильтрования, и остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюента, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 288,9, 290,9.
Стадия B: (R)-3-(4-бромфенокси)-2-гидроксипропановая кислота
К раствору (R)-этил-3-(4-бромфенокси)-2-гидроксипропаноата (3,13 г, 10,8 ммоль) в THF (60 мл) и МеОН (20 мл) при 0°С добавляли NaOH (21,6 мл, 21,6 ммоль), и полученный раствор перемешивали при 0°С в течение 4 часов. Раствор затем подкисляли 4 Н HCl в диоксане (5,5 мл), и полученный раствор концентрировали. Остаток распределяли между этилацетатом и водой, водную фазу экстрагировали с помощью EtOAc, объединенные органические фазы сушили над Na2SO4 и концентрировали, получая указанное в заголовке соединение.
1H ЯМР (CDCl3, 500 МГц): δ 7,42-7,40 (д, J=9,0 Гц, 2H), 6,84-6,82 (д, J=9,0 Гц, 2H), 4,63-4,62 (т, J=3,5Гц, 1H), 4,32-4,31 (д, J=3,5Гц, 2H).
Стадия C: (R)-3-(4-бромфенокси)-2-гидроксипропанамид
К суспензии (R)-3-(4-бромфенокси)-2-гидроксипропановой кислоты (2,81 г, 10,8 ммоль) в DCM (80 мл) добавляли моногидрат 1-гидрокси-2,5-пирролидиндиона (1,50 г, 11,3 ммоль) и EDC (3,10 г, 16,1 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Смесь распределяли между водой и DCM, органическую фазу промывали водой, насыщенным раствором соли, сушили над Na2SO4 и концентрировали. Остаток растворяли в диоксане (80 мл) и охлаждали до 0°С. К вышеуказанному раствору добавляли гидроксид аммония (15,0 мл, 108 ммоль), полученный раствор перемешивали при 0°С в течение 30 минут, затем концентрировали и остаток обрабатывали с помощью DCM. Твердое вещество собирали фильтрованием. Фильтрат концентрировали и остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей, объединяя с твердым веществом, полученным выше, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 260,0; 262,0.
Стадия D: (R)-3-(4-бромфенокси)-2-((трет-бутилдиметилсилил)окси)-пропанамид
К раствору (R)-3-(4-бромфенокси)-2-гидроксипропанамида (1,8 г, 6,92 ммоль) в ацетонитриле (20 мл) добавляли имидазол (2,36 г, 34,6 ммоль), TBS-Cl (2,61 г, 17,3 ммоль) и DMAP (0,085 г, 0,692 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь распределяли между EtOAc и насыщ. NaHCO3. Водную фазу экстрагировали три раза EtOAc. Объединенную органическую фазу сушили над Na2SO4, концентрировали и остаток очищали на колонке с силикагелем, используя EtOAc/гексан в качестве элюирующих растворителей, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 374,1; 376,1.
Стадия E: (S)-3-(4-бромфенокси)-2-((трет-бутилдиметилсилил)окси)-пропаннитрил
К раствору (R)-3-(4-бромфенокси)-2-((трет-бутилдиметил-силил)окси)пропанамида (2,51 г, 6,71 ммоль) в DMF (8 мл) при 0°С добавляли цианурхлорид (0,663 г, 3,60 ммоль), и полученный раствор перемешивали при 0°С в течение 1 часа. Смесь гасили добавлением насыщенного NaHCO3. Смесь разбавляли EtOAc и промывали насыщ. NaHCO3 три раза, сушили над Na2SO4, концентрировали, и остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей, получая указанное в заголовке соединение.
1H ЯМР (CDCl3, 500 МГц): δ 7,43-7,42 (д, J=8,9 Гц, 2H), 6,83-6,81 (д, J=8,9 Гц, 2H), 4,82-4,79 (дд, J=5,6Гц, 1H), 4,18-4,15 (дд, J=5,6Гц, 2H), 0,95 (с, 9H), 0,25 (с, 3H), 0,20 (с, 3H).
Стадия F: (S)-5-(2-(4-бромфенокси)-1-((трет-бутилдиметилсилил)окси)этил)-2H-тетразол
К раствору (S)-3-(4-бромфенокси)-2-((трет-бутилдиметилсилил)окси)пропаннитрила (1,82 г, 5,11 ммоль) в толуоле (30 мл) (0,381 г, 1,53 ммоль) и ТМС-N3 (2,03 мл, 15,3 ммоль). Полученную смесь нагревали при 110°С в течение ночи. После концентрирования остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 399,1; 401,1.
Стадия G: (S)-5-(2-(4-бромфенокси)-1-((трет-бутилдиметилсилил)окси)этил)-2-(4-метоксибензил)-2H-тетразол
Смесь K2CO3 (1,13 г, 8,19 ммоль), гидрата тетрабутиламмония хлорида, (0,162 г, 0,546 ммоль), 4-метоксибензилхлорида (0,407 мл, 3,00 ммоль) и (S)-5-(2-(4-бромфенокси)-1-((трет-бутилдиметилсилил)окси)-этил)-2H-тетразола (1,09 г, 2,73 ммоль) в воде (4 мл) и ClCH2CH2Cl (16 мл) нагревали при 40°С в течение ночи. Смесь распределяли между EtOAc/водой. Полученную смесь трижды экстрагировали с помощью DCM, объединенную органическую фазу сушили над Na2SO4, концентрировали, и остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей, получаям указанное в заголовке соединение.
LC/MS [М+Н]+: 519,2; 521,2.
Стадия H: (S)-трет-бутил-(3-(4-(4-(2-((трет-бутилдиметилсилил)окси)-2-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)фенил)-1H-пиразол-1-ил)пропил)карбамат
К раствору (S)-5-(2-(4-бромфенокси)-1-((трет-бутилдиметилсилил)окси)этил)-2-(4-метоксибензил)-2H-тетразола (0,57 г, 1,10 ммоль) и трет-бутил-(3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропил)карбамата (0,424 г, 1,21 ммоль) в диоксане (20 мл) добавляли Na2CO3 (1,646 мл, 3,29 ммоль). Полученный раствор продували азотом в течение 1 часа перед добавлением аддукта PdCl2(dppf)-CH2Cl2 (0,090 г, 0,110 ммоль). Полученную смесь продолжали барботировать азотом в течение 20 минут, а затем нагревали при 100°С в течение ночи. После фильтрации через целит, фильтрат концентрировали и остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 664,6.
Стадия I: (S)-трет-бутил-(3-(4-(4-(2-гидрокси-2-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)фенил)-1H-пиразол-1-ил)пропил)карбамат
К раствору (S)-трет-бутил-(3-(4-(4-(2-((трет-бутилдиметилсилил)окси)-2-(2-(4-метоксибензил)-2H-тетразол-5-ил)-этокси)фенил)-1H-пиразол-1-ил)пропил)карбамата (0,25 г, 0,377 ммоль) в THF (15 мл) при 0°С добавляли по каплям TBAF (1 M в THF) (0,377 мл, 0,377 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 0,5 часа, раствор концентрировали и остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующего растворителя, получая указанное в заголовке соединение.
LC/MS [ М+Н]+: 550,5.
Стадия J: (R)-трет-бутил-(3-(4-(4-(2-((1,3-диоксоизоиндолин-2-ил)окси)-2-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)фенил)-1H-пиразол-1-ил)пропил)карбамат
К раствору (S)-трет-бутил-(3-(4-(4-(2-гидрокси-2-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)фенил)-1H-пиразол-1-ил)пропил)карбамата (0,19 г, 0,346 ммоль) в THF (10 мл) добавляли 2-гидроксиизоиндолин-1,3-дион (0,068 г, 0,415 ммоль), трифенилфосфин (0,136 г, 0,519 ммоль) и DEAD (0,082 мл, 0,519 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 2 часов. Затем раствор концентрировали, и остаток очищали на колонке с силикагелем, используя в качестве элюента смесь EtOAc/гексан, получая указанное в заголовке соединение.
LC/MS [M+H]+: 695,5.
Стадия K: (R)-1-(3-((трет-бутоксикарбонил)амино)пропил)-4-(4-(2-((1,3-диоксоизоиндолин-2-ил)окси)-2-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)фенил)-2-метил-1H-пиразол-2-ий иодид
К смеси R)-трет-бутил-(3-(4-(4-(2-((1,3-диоксоизоиндолин-2-ил)окси)-2-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)фенил)-1H-пиразол-1-ил)пропил)карбамата (240 мг, 0,345 ммоль) в ацетонитриле (3 мл) добавляли MeI (0,108 мл, 1,73 ммоль). Полученную смесь нагревали при 60°С в течение ночи. Добавляли дополнительное количество MeI (0,108 мл, 1,727 ммоль), и полученный раствор нагревали при 60°С в течение 24 часов. Раствор концентрировали, получая указанное в заголовке соединение.
LC/MS [М]+: 709,6.
Стадия L: (R)-4-(4-(2-(аминоокси)-2-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий
К раствору (R)-1-(3-((трет-бутоксикарбонил)амино)пропил)-4-(4-(2-((1,3-диоксоизоиндолин-2-ил)окси)-2-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)фенил)-2-метил-1H-пиразол-2-ий иодида (301 мг, 0,360 ммоль) в этаноле (4 мл) и CH2Cl2 (4 мл) при 0°С добавляли гидразин (0,011 мл, 0,360 ммоль). Полученный раствор перемешивали при 0°С в течение 30 минут. Затем раствор концентрировали, получая указанное в заголовке соединение.
LC/MS [М]+: 579,5.
Стадия M: (R,Z)-1-(3-((трет-бутоксикарбонил)амино)пропил)-4-(4-(2-((((2-((трет-бутокси-карбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-2-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)фенил)-2-метил-1H-пиразол-2-ий
К суспензии (R)-4-(4-(2-(аминоокси)-2-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ия (209 мг, 0,361 ммоль) в этаноле (6 мл) и CH2Cl2 (6 мл) добавляли 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусную кислоту (196 мг, 0,721 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 часа, затем, после концентрирования, остаток очищали при помощи обращенно-фазовой MPLC, с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05% TFA) в качестве подвижной фазы, получая указанное в заголовке соединение.
LC/MS [М]+: 833,6.
Стадия N: (R,Z)-4-(4-(2-((((2-аминотиазол-4-ил)(карбокси)метилен)-амино)окси)-2-(2H-тетразол-5-ил)этокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий
В колбу, содержащую (R,Z)-1-(3-((трет-бутоксикарбонил)-амино)пропил)-4-(4-(2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)-метилен)амино)окси)-2-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)фенил)-2-метил-1H-пиразол-2-ий (230 мг, 0,276 ммоль), добавляли TFA (4 мл, 51,9 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 30 минут. Раствор концентрировали, и остаток растворяли в TFA (4 мл). Полученный раствор нагревали при 70°С в течение 45 минут. Раствор концентрировали, и остаток растворяли в DMFА (16 мл). К полученному раствору при 0°С добавляли TEA (0,231 мл, 1,655 ммоль) и BOC2O (0,077 мл, 0,331 ммоль). Полученный раствор перемешивали при 0°С в течение 1 часа, затем очищали при помощи обращенно-фазовой MPLC, с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05% TFA) в качестве подвижной фазы, получая указанное в заголовке соединение.
LC/MS [М]+: 613,5.
Стадия O: (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((R)-2-(4-(1-(3-((трет-бутоксикарбонил)-амино)пропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-(2H-тетразол-5-ил)этокси)-имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
К раствору (R,Z)-4-(4-(2-((((2-аминотиазол-4-ил)(карбокси)метилен)амино)окси)-2-(2H-тетразол-5-ил)этокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ия (47 мг, 0,077 ммоль) в DMF (4 мл) добавляли DCC (79 мг, 0,383 ммоль) и HOBT (58,6 мг, 0,383 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 30 минут, а затем добавляли (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфат (64,4 мг, 0,306 ммоль) и бикарбонат натрия (77 мг, 0,919 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Дополнительно добавляли DCC (79 мг, 0,383 ммоль) и (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил-гидросульфат (64,4 мг, 0,306 ммоль), и полученный раствор перемешивали при комнатной температуре в течение 1 часа. После фильтрации реакционной смеси фильтрат очищали с помощью обращенно-фазовой HPLC, с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05% TFA) в качестве подвижной фазы, получая указанное в заголовке соединение.
LC/MS [М]+: 805,4.
Стадия P: 1-(3-аминопропил)-4-(4-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-(2H-тетразол-5-ил)этокси)фенил)-2-метил-1H-пиразол-2-ий 2,2,2-трифторацетат
К раствору (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((R)-2-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-(2H-тетразол-5-ил)этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата (30 мг, 0,037 ммоль) в CH2Cl2 (2 мл) добавляли TFA (2 мл, 26,0 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 30 минут. После концентрирования остаток очищали с помощью обращенно-фазовой HPLC, с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05% TFA) в качестве подвижной фазы, получая указанное в заголовке соединение.
LC/MS [М]+: 705,4.
1H ЯМР (DMSO-d6, 500 МГц): δ 9,48-9,46 (д, J=7,7 Гц, 1H), 8,98 (с, 1H), 8,94 (с, 1H), 7,77 (с, 1H), 7,66-7,63 (д, J=8,5 Гц, 2H), 7,31 (с, 1H), 7,11-7,09 (д, J=8,6 Гц, 2H), 5,92 (с, 1H), 4,59-4,52 (м, 6H), 4,12 (с, 3H), 2,94-2,90 (м, 2H), 2,20-2,17 (м, 2H), 1,38 (с, 3H), 1,15 (с, 3H).
ПРИМЕР 30
1-(3-аминопропил)-4-(4-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((2S,3S)-2-метил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-(2H-тетразол-5-ил)этокси)фенил)-2-метил-1H-пиразол-2-ий 2,2,2-трифторацетат (C30)
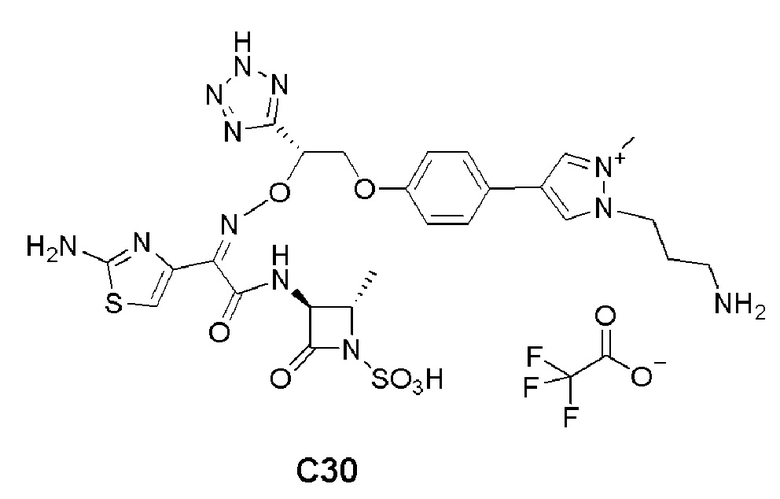
Стадия A: 4-(4-((R)-2-(((Z)-(1-(2-аминотиазол-4-ил)-2-(((2S,3S)-2-метил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-(2H-тетразол-5-ил)этокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий
К раствору (R,Z)-4-(4-(2-((((2-аминотиазол-4-ил)(карбокси)метилен)амино)окси)-2-(2H-тетразол-5-ил)этокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ия (18,8 мг, 0,031 ммоль) в DMF (2 мл) добавляли DCC (63,2 мг, 0,306 ммоль) и НОВТ (23,46 мг, 0,153 ммоль) Полученный раствор перемешивали при комнатной температуре в течение 30 минут, затем добавляли (2S,3S)-3-амино-2-метил-4-оксоазетидин-1-сульфоновую кислоту (22,1 мг, 0,123 ммоль) и бикарбонат натрия (30,9 мг, 0,368 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 1 часа. После фильтрации реакционной смеси фильтрат очищали с помощью обращенно-фазовой HPLC, с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05% TFA) в качестве подвижной фазы, получая указанное в заголовке соединение.
LC/MS [М]+: 775,6.
Стадия B: 1-(3-аминопропил)-4-(4-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((2S,3S)-2-метил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-(2H-тетразол-5-ил)этокси)фенил)-2-метил-1H-пиразол-2-ий 2,2,2-трифторацетат
К раствору 4-(4-((R)-2-(((Z)-(1-(2-аминотиазол-4-ил)-2-(((2S,3S)-2-метил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-(2H-тетразол-5-ил)этокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ия (11 мг, 0,014 ммоль) в CH2Cl2 (2 мл) добавляли TFA (2 мл, 26,0 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 30 минут. Летучие компоненты выпаривали, и остаток очищали с помощью обращенно-фазовой HPLC, с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05% TFA) в качестве подвижной фазы, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 675,4.
1H ЯМР (DMSO-d6, 500 МГц): δ 9,29-9,27 (д, J=6,7 Гц, 1H), 8,98 (с, 1H), 8,95 (с, 1H), 7,78 (м, 3H), 7,66-7,63 (д, J=10,2 Гц, 2H), 7,31 (с, 2H), 7,15-7,13 (д, J=9,6 Гц, 2H), 6,82 (с, 1H), 5,91 (т, J=7,3 Гц, 1H), 4,62-4,59 (м, 2H), 4,55-4,53 (т, J=7,0 Гц, 2H), 4,43-4,41 (д, J=9,7 Гц, 1H), 4,12 (с, 3H), 3,72-3,71 (м, 1H), 2,94-2,90 (м, 2H), 2,21-2,17 (т, J=7,8 Гц, 2H), 1,30-1,28 (д, J=6,1 Гц, 3H).
ПРИМЕР 31
3-(2-амино-4-(4-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)фенил)-1H-имидазол-1-ил)пропан-1-аминий 2,2,2-трифторацетат (C31)
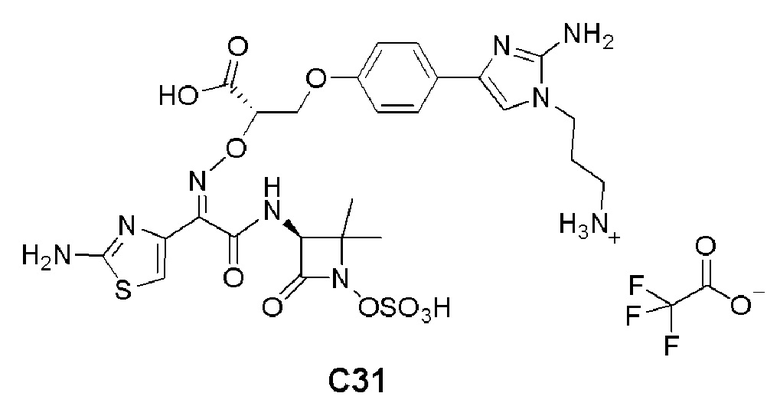
 Стадия A: трет-бутил-(3-(2-нитро-1H-имидазол-1-ил)пропил)карбамат
Стадия A: трет-бутил-(3-(2-нитро-1H-имидазол-1-ил)пропил)карбамат
К перемешиваемой смеси 3-(ВОС-амино)пропилбромида (901 мг, 3,78 ммоль) и 2-нитро-1Н-имидазола (389 мг, 3,44 ммоль) в DMF (20 мл) добавляли карбонат цезия (2240 мг, 6,88 ммоль), и смесь перемешивали при 60°С в течение ночи. Смесь разбавляли этилацетатом, три раза промывали водой, а затем насыщенным раствором соли, сушили над NaSО4, фильтровали, и растворитель выпаривали при пониженном давлении, с получением неочищенного продукта. Неочищенный продукт очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 271,1.
Стадия B: трет-бутил-(3-(4-бром-2-нитро-1H-имидазол-1-ил)пропил)карбамат
К раствору трет-бутил-(3-(2-нитро-1H-имидазол-1-ил)пропил)карбамата (1,17 г, 4,33 ммоль) в DMF (8 мл) добавляли NBS (0,847 г, 4,76 ммоль). Полученный раствор перемешивали при 60°С в течение 3 часов. Раствор распределяли между EtOAc и насыщ. NaHCO3. Органическую фазу промывали три раза насыщенным раствором NaHCO3, сушили над Na2SO4, концентрировали, и остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 349,1, 351,1.
Стадия C: (R)-трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-2-нитро-1H-имидазол-4-ил)фенокси)-2-гидроксипропаноат
К раствору трет-бутил-(3-(4-бром-2-нитро-1H-имидазол-1-ил)пропил)карбамата (0,82 г, 2,348 ммоль) в диоксане (20 мл) добавляли (R)-трет-бутил-2-гидрокси-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)пропаноат (1,112 г, 3,05 ммоль) и Na2CО3 (3,52 мл, 7,05 ммоль). Полученную смесь продували азотом в течение 20 минут перед добавлением аддукта PdCl2(dppf)-CH2Cl2 (0,192 г, 0,235 ммоль). Смесь дополнительно продували азотом в течение 15 минут, затем нагревали при 100°С в течение ночи. После фильтрования через целит фильтрат концентрировали, и остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 507,4.
Стадия D: (R)-трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-2-нитро-1H-имидазол-4-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноат
К раствору (R)-трет-бутил 3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-2-нитро-1H-имидазол-4-ил)фенокси)-2-гидроксипропаноата (2460 мг, 4,86 ммоль) в DMF (20 мл) добавляли имидазол (1653 мг, 24,28 ммоль), TBS-Cl (1464 мг, 9,71 ммоль) и DMAP (59,3 мг, 0,486 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 часа, затем распределяли между EtOAc и насыщенным раствором NaHCO3. Органическую фазу три раза промывали насыщенным раствором NaHCO3, сушили над Na2SO4, концентрировали, и остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 621,5.
Стадия E: (R)-трет-бутил-3-(4-(2-амино-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-4-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноат
К раствору (R)-трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-2-нитро-1H-имидазол-4-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноата (2,4 г, 3,87 ммоль) в MeOH (50 мл) добавляли 10% Pd/C (0,411 г, 0,387 ммоль). Полученную смесь гидрировали при давлении 42 фунта на кв. дюйм (2,896 бар) в течение ночи. После фильтрования через целит фильтрат концентрировали, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 591,5.
Стадия F: (R)-трет-бутил-3-(4-(2-(бис(трет-бутоксикарбонил)амино)-1-(3-((трет-бутоксикарбонил)-амино)пропил)-1H-имидазол-4-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноат
К раствору (R)-трет-бутил-3-(4-(2-амино-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-4-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноата (2,53 г, 4,28 ммоль) в CH2Cl2 (40 мл) добавляли BOC2О (2,98 мл, 12,85 ммоль), TEA (1,194 мл, 8,56 ммоль) и DMAP (0,052 г, 0,428 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 30 минут. После концентрирования остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 791,6.
Стадия G: (R)-трет-бутил 3-(4-(2-(бис(трет-бутоксикарбонил)амино)-1-(3-((трет-бутокси-карбонил)амино)пропил)-1H-имидазол-4-ил)фенокси)-2-гидроксипропаноат
К раствору (R)-трет-бутил 3-(4-(2-(бис(трет-бутоксикарбонил)амино)-1-(3-((трет-бутоксикарбонил)-амино)пропил)-1H-имидазол-4-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноата (1,80 г, 2,28 ммоль) в THF (20 мл) при 0°С добавляли TBAF (1 М в THF) (2,275 мл, 2,275 ммоль). Полученный раствор перемешивали при 0°С в течение 30 минут. После концентрирования раствора остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 677,5.
Стадия H: (S)-трет-бутил 3-(4-(2-(бис(трет-бутоксикарбонил)амино)-1-(3-((трет-бутокси-карбонил)амино)пропил)-1H-имидазол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)-окси)пропаноат
К раствору (R)-трет-бутил 3-(4-(2-(бис(трет-бутоксикарбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-4-ил)фенокси)-2-гидроксипропаноата (1,39 г, 2,05 ммоль) в THF (20 мл) добавляли 2-гидроксиизоиндолин-1,3-дион (0,402 г, 2,46 ммоль), трифенилфосфин (0,808 г, 3,08 ммоль) и DEAD (0,488 мл, 3,08 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1,5 часов. После концентрирования реакционного раствора остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 822,5.
Стадия I: (S)-трет-бутил 2-(аминоокси)-3-(4-(2-(бис(трет-бутоксикарбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-4-ил)фенокси)пропаноат
К раствору (S)-трет-бутил 3-(4-(2-(бис(трет-бутоксикарбонил)амино)-1-(3-((трет-бутоксикарбонил)-амино)пропил)-1H-имидазол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноата (0,58 г, 0,706 ммоль) в этаноле (8 мл) и CH2Cl2 (8,00 мл) при 0°C добавляли гидразин (0,022 мл, 0,706 ммоль). Полученный раствор перемешивали при 0°С в течение 0,5 часа. После концентрирования остаток растворяли в DCM (10 мл) и перемешивали при комнатной температуре в течение 5 минут, а затем фильтровали. Фильтрат концентрировали, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 692,5.
Стадия J: (S,Z)-2-(((3-(4-(2-(бис(трет-бутоксикарбонил)амино)-1-(3-((трет-бутоксикарбонил)-амино)пропил)-1H-имидазол-4-ил)фенокси)-1-(трет-бутокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)уксусная кислота
К раствору (S)-трет-бутил-2-(аминоокси)-3-(4-(2-(бис(трет-бутоксикарбонил)амино)-1-(3-((трет-бутоксикарбонил)-амино)-пропил)-1H-имидазол-4-ил)фенокси)пропаноата (0,61 г, 0,882 ммоль) в этаноле (8 мл) и DCM (8 мл) добавляли 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусную кислоту (0,240 г, 0,882 ммоль). Полученный раствор перемешивали при комнатной температуре в течение ночи. Раствор концентрировали, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 946,6.
Стадия K: (S)-трет-бутил-3-(4-(2-(бис(трет-бутоксикарбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-4-ил)фенокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропаноат
К раствору (S,Z)-2-(((3-(4-(2-(бис(трет-бутоксикарбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-4-ил)фенокси)-1-(трет-бутокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)уксусной кислоты (0,42 г, 0,444 ммоль) в DMF (4 мл) добавляли DCC (0,275 г, 1,33 ммоль) и НОВТ (0,204 г, 1,332 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут перед добавлением (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата (0,187 г, 0,888 ммоль) и бикарбоната натрия (0,298 г, 3,55 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 5 часов. После фильтрации смеси фильтрат очищали при помощи обращенно-фазовой MPLC, с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05% TFA) в качестве подвижной фазы, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 1139,5.
Стадия L: 3-(2-амино-4-(4-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)фенил)-1H-имидазол-1-ил)пропан-1-аминий 2,2,2-трифторацетат
К раствору (S)-трет-бутил-3-(4-(2-(бис(трет-бутоксикарбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-4-ил)фенокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропаноата (184 мг, 0,162 ммоль) в CH2Cl2 (2 мл) добавляли TFA (4 мл, 51,9 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 часа. После концентрирования раствора на роторном испарителе при комнатной температуре остаток обрабатывали Et2О и снова концентрировали. Твердый остаток растворяли в DMSO и очищали при помощи обращенно-фазовой HPLC, с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05% TFA) в качестве подвижной фазы, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 682,3.
1H ЯМР (D2O, 500 МГц): δ 7,29-7,27 (д, J=10,8 Гц, 2H), 6,99 (с, 1H), 6,89 (с, 1H), 6,89-6,87 (д, J=10,2 Гц, 2H), 5,04-5,02 (м, 1H), 4,54 (с, 1H), 4,43-4,41 (м, 2H), 3,83-3,81 (д, J=9,0 Гц), 3,00-2,98(м, 2H), 2,08-2,04 (м, 2H), 1,33 (с, 3H), 1,02 (с, 3H).
ПРИМЕР 32
3-(4-(4-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)-азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)фенил)-2-имино-3-метил-2,3-дигидро-1H-имидазол-1-ил)пропан-1-аминий 2,2,2-трифторацетат (C32)
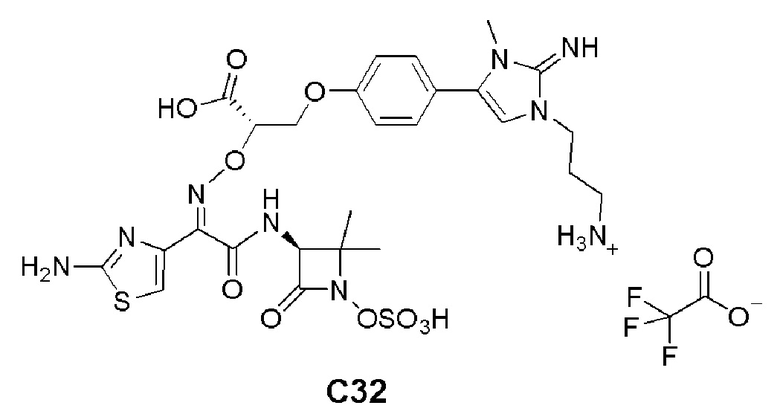
Стадия A: (S,E)трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноат
К раствору (S)-трет-бутил-3-(4-(2-(бис(трет-бутоксикарбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноата (157 мг, 0,191 ммоль) в ацетонитриле (2 мл) добавляли MeI (0,036 мл, 0,573 ммоль). Полученный раствор нагревали при 60°С в течение ночи. Раствор концентрировали, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 736,5.
Стадия B: (S,E)трет-бутил-2-(аминоокси)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)пропаноат
К раствору (S,E)трет-бутил 3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-((1,3-диоксо-изоиндолин-2-ил)окси)пропаноата (0,18 г, 0,245 ммоль) в этаноле (4,00 мл) и CH2Cl2 (4 мл) при 0°С добавляли гидразин (0,015 мл, 0,489 ммоль). Полученный раствор перемешивали при 0°С в течение 2 часов, а затем концентрировали, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 606,5.
Стадия C: (Z)-2-((((S)-1-(трет-бутокси)-3-(4-((E)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-1-оксо-пропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)уксусная кислота
К раствору (S,E)трет-бутил 2-(аминоокси)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)пропаноата (126 мг, 0,208 ммоль) в этаноле (8 мл) и DCM (8 мл) добавляли 2-(2-((трет-бутокси-карбонил)амино)тиазол-4-ил)-2-оксоуксусную кислоту (142 мг, 0,520 ммоль) Полученный раствор перемешивали при комнатной температуре в течение 3 часов. Затем раствор концентрировали, и остаток очищали при помощи обращенно-фазовой MPLC, с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05% TFA) в качестве подвижной фазы, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 860,4.
Стадия D: (S)-трет-бутил-3-(4-((E)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропаноат
К раствору (Z)-2-((((S)-1-(трет-бутокси)-3-(4-((E)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)уксусной кислоты (152 мг, 0,177 ммоль) в DMF (5 мл) добавляли DCC (292 мг, 1,41 ммоль) и НОВТ (122 мг, 0,795 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 30 минут перед добавлением (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата (130 мг, 0,619 ммоль) и бикарбоната натрия (297 мг, 3,53 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 28 часов, а затем фильтровали. Фильтрат очищали при помощи обращенно-фазовой MPLC, с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05% TFA) в качестве подвижной фазы, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 1052,6.
Стадия E: 3-(4-(4-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)фенил)-2-имино-3-метил-2,3-дигидро-1H-имидазол-1-ил)пропан-1-аминий 2,2,2-трифторацетат
К раствору (S)-трет-бутил-3-(4-((E)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропаноата (140 мг, 0,133 ммоль) в CH2Cl2 (2 мл) добавляли TFA (4 мл, 51,9 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 40 минут, затем концентрировали. Остаток обрабатывали Et2O и снова концентрировали. Твердый сухой остаток очищали при помощи обращенно-фазовой HPLC, с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05% TFA) в качестве подвижной фазы, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 696,3.
1H ЯМР (D2O, 500 МГц): δ 7,29-7,27 (д, J=10,3 Гц, 2H), 7,04(с, 1H), 6,99-6,97 (д, J=10,3 Гц, 2H), 6,80 (с, 1H), 5,08 (с, 1H), 4,66 (м, 1H), 4,63 (с, 1H), 4,48-4,43 (м, 2H), 3,91-3,89 (т, J=7,1 Гц, 2H), 3,29 (с, 3H), 3,0-2,96 (м, 2H), 2,09-2,05 (м, 2H), 1,35 (с, 3H), 0,97 (с, 3H).
ПРИМЕР 33
1-(3-аминопропил)-4-((5-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)пиридин-2-ил)амино)пиридин-1-ий 2,2,2-трифторацетат (C33)
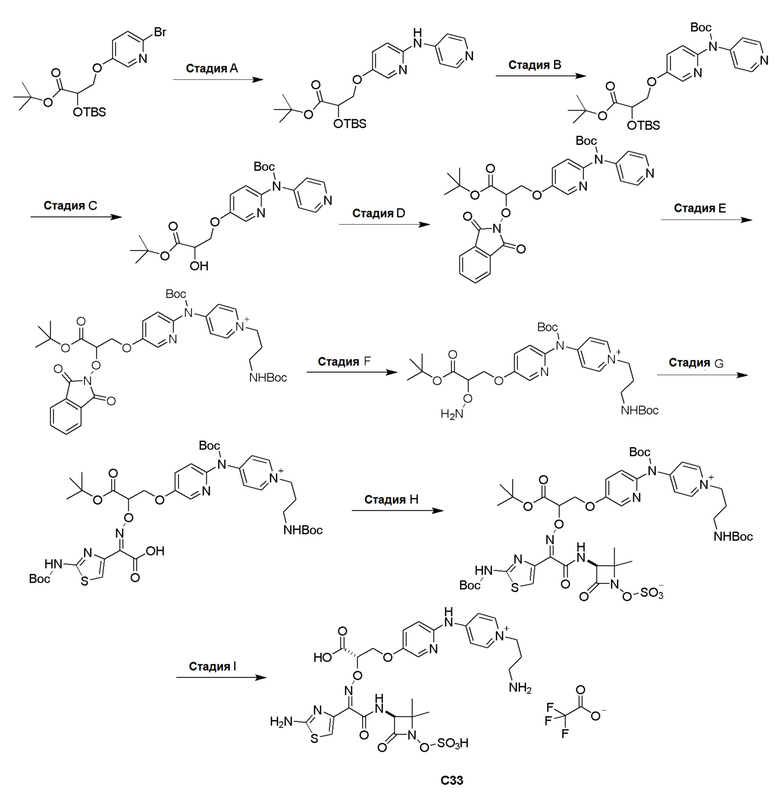
Стадия A: трет-бутил 2-((трет-бутилдиметилсилил)окси)-3-((6-(пиридин-4-иламино)пиридин-3-ил)окси)пропаноат
Смесь пре-катализатора J009 G3 (Aldrich, кат. # 88755, 250 мг, 0,27 ммоль), Cs2СО3 (2,64 г, 8,12 ммоль), трет-бутил-3-((6-бромпиридин-3-ил)окси)-2-((трет-бутилдиметилсилил)окси)пропаноата (1170 мг, 2,71 ммоль) и пиридин-4-амина (306 мг, 3,25 ммоль) в запаянной трубке дегазировали три раза в режиме вакуум/азот перед добавлением диоксана (12 мл). Полученную смесь дополнительно дегазировали три раза в режиме вакуум/азот, а затем нагревали при 100°С в течение 16 часов. Смесь фильтровали через целит, фильтрат концентрировали и остаток очищали на колонке с силикагелем, используя смесь МеОН/DCM в качестве элюента, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 446,36.
 Стадия B: трет-бутил-3-((6-((трет-бутоксикарбонил)(пиридин-4-ил)амино)пиридин-3-ил)окси)-2-((трет-бутилдиметилсилил)окси)пропаноат
Стадия B: трет-бутил-3-((6-((трет-бутоксикарбонил)(пиридин-4-ил)амино)пиридин-3-ил)окси)-2-((трет-бутилдиметилсилил)окси)пропаноат
К раствору трет-бутил-2-((трет-бутилдиметил-силил)окси)-3-((6-(пиридин-4-иламино)пиридин-3-ил)окси)пропаноата (0,61 г, 1,37 ммоль) в CH2Cl2 (25 мл) добавляли TEA (0,382 мл, 2,74 ммоль), Boc2О (0,381 мл, 1,64 ммоль) и DMAP (0,167 г, 1,37 ммоль). Полученный раствор перемешивали при комнатной температуре в течение ночи. После концентрирования остаток очищали на колонке с силикагелем используя смесь EtOAc/гексан в качестве элюента, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 546,47.
Стадия C: трет-бутил-3-((6-((трет-бутоксикарбонил)(пиридин-4-ил)амино)пиридин-3-ил)окси)-2-гидроксипропаноат
К раствору трет-бутил-3-((6-((трет-бутоксикарбонил)(пиридин-4-ил)амино)пиридин-3-ил)окси)-2-((трет-бутилдиметилсилил)окси)пропаноата (0,28 г, 0,513 ммоль) в THF (10 мл) при 0°C добавляли TBAF (0,513 мл, 0,513 ммоль). Полученный раствор перемешивали при 0°С в течение 0,5 часа. После концентрирования остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей, получая указанное в заголовке соединение.
LC/MS: [М+Н]+: 432,32.
Стадия D: трет-бутил-3-((6-((трет-бутоксикарбонил)(пиридин-4-ил)амино)пиридин-3-ил)окси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноат
К раствору трет-бутил 3-((6-((трет-бутоксикарбонил)(пиридин-4-ил)амино)пиридин-3-ил)окси)-2-гидроксипропаноата (0,23 г, 0,533 ммоль) в THF (6 мл) добавляли 2-гидроксиизоиндолин-1,3-дион (0,104 г, 0,640 ммоль), трифенилфосфин (0,210 г, 0,80 ммоль) и DEAD (0,127 мл, 0,800 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 3 часов. После концентрирования раствора остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 577,41.
Стадия E: 4-((5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)пиридин-2-ил)(трет-бутоксикарбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)пропил)пиридин-1-ий
К раствору трет-бутил-3-((6-((трет-бутоксикарбонил)(пиридин-4-ил)амино)пиридин-3-ил)окси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноата (0,25 г, 0,434 ммоль) в ацетонитриле (4 мл) добавляли трет-бутил-(3-иодпропил)карбамат (0,148 г, 0,520 ммоль). Полученный раствор нагревали при 60°С в течение трех дней. Добавляли бикарбонат натрия (0,146 г, 1,73 ммоль) и дополнительное количество трет-бутил-(3-иодпропил)карбамата (0,296 г). Полученный раствор нагревали при 60°С в течение ночи. Раствор фильтровали, фильтрат концентрировали и остаток очищали с помощью обращенно-фазовой жидкостной хроматографии среднего давления (RP-MPLC), с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05% TFA) в качестве подвижной фазы, получая указанное в заголовке соединение.
LC/MS [М]+: 734,41.
Стадия F: 4-((5-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)пиридин-2-ил)(трет-бутокси-карбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)пропил)пиридин-1-ий
К раствору 4-((5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)пиридин-2-ил)(трет-бутоксикарбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)пропил)пиридин-1-ия (110 мг, 0,150 ммоль) в CH2Cl2 (4 мл) и этаноле (4,00 мл) при 0°С добавляли гидразин (5,64 мкл, 0,180 ммоль). Полученный раствор перемешивали при 0°С в течение 1 часа, затем концентрировали. Остаток обрабатывали CH2Cl2 (4 мл), фильтровали, и фильтрат концентрировали, получая указанное в заголовке соединение.
LC/MS [М]+: 604,54.
Стадия G: (Z)-4-((5-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-(карбокси)метилен)амино)окси)-3-оксопропокси)пиридин-2-ил)(трет-бутоксикарбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)пропил)пиридин-1-ий
К раствору 4-((5-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)пиридин-2-ил)(трет-бутоксикарбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)пропил)пиридин-1-ия (100 мг, 0,165 ммоль) в этаноле (4 мл) и CH2Cl2 (4 мл) добавляли 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусную кислоту (58,5 мг, 0,215 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 часа. Раствор концентрировали, и остаток очищали с помощью обращенно-фазовой жидкостной хроматографии среднего давления, с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05% TFA) в качестве подвижной фазы, получая указанное в заголовке соединение.
LC/MS [M]+: 858,52.
Стадия H: 4-((5-(3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)пиридин-2-ил)(трет-бутоксикарбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)-пропил)пиридин-1-ий
К раствору (Z)-4-((5-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)пиридин-2-ил)(трет-бутоксикарбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)пропил)пиридин-1-ия (16 мг, 0,019 ммоль) в DMF (1 мл) добавляли DCC (30,7 мг, 0,149 ммоль) и НОВТ (11,41 мг, 0,075 ммоль). Полученный в результате раствор перемешивали при комнатной температуре в течение 30 минут перед добавлением (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата (19,58 мг, 0,093 ммоль) и бикарбоната натрия (31,3 мг, 0,373 ммоль). Полученный раствор перемешивали при комнатной температуре в течение ночи. После фильтрации реакционной смеси остаток очищали с помощью обращенно-фазовой HPLC, с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05% TFA), получая указанное в заголовке соединение.
LC/MS [М]+: 1050,41.
Стадия K: 1-(3-аминопропил)-4-((5-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-пиридин-2-ил)амино)пиридин-1-ий 2,2,2-трифторацетат
К раствору 4-((5-(3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)пиридин-2-ил)(трет-бутоксикарбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)пропил)пиридин-1-ия (22 мг, 0,021 ммоль) в CH2Cl2 (1 мл) добавляли TFA (2,5 мл, 32,4 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1,5 часа. Реакционный раствор концентрировали упариванием на роторном испарителе при комнатной температуре, и остаток дополнительно сушили в высоком вакууме. Твердое вещество очищали с помощью обращенно-фазовой HPLC с использованием ацетонитрила (0,05% TFA)/вода (0,05% TFA), получая указанное в заголовке соединение (более полярный диастереомер).
LC/MS [М+Н]+: 694,17.
1H ЯМР (D2O, 500 МГц): δ 8,13-8,11 (д, J=8,8 Гц, 2H), 8,01-8,00 (д, J=1,6 Гц, 1H), 7,50-7,42 (м, 3H), 7,08-7,07 (д, J=8,8 Гц, 1H), 7,02 (с, 1H), 5,05 (с, 1H), 4,66 (с, 1H), 4,48 (с, 2H), 4,27-4,24(т, J=7,1 Гц, 2H), 2,99-2,96 (т, J=7,4 Гц, 2H), 2,21-2,18 (т, J=7,4 Гц, 2H), 1,35 (с, 3H), 1,05 (с, 3H).
ПРИМЕР 34
1-(3-аминопропил)-4-((5-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)пиридин-2-ил)окси)пиридин-1-ий 2,2,2-трифторацетат (C34)
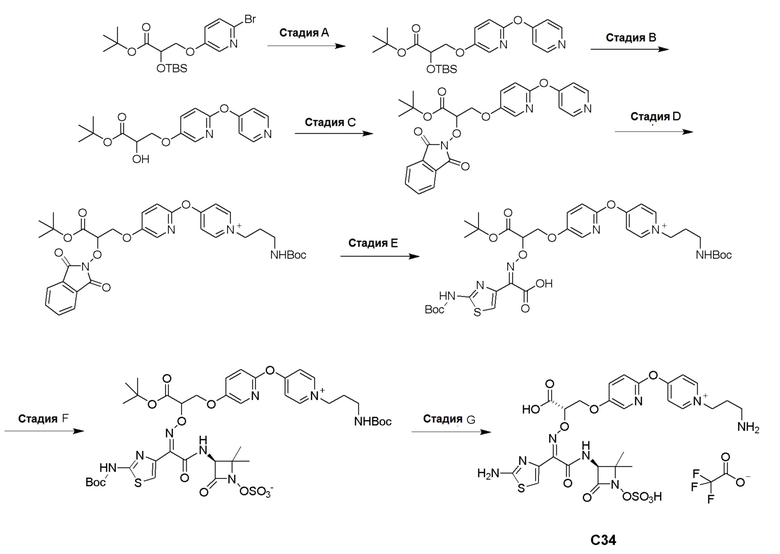
Стадия A: трет-бутил-2-((трет-бутилдиметилсилил)окси)-3-((6-(пиридин-4-илокси)пиридин-3-ил)окси)пропаноат
Смесь пре-катализатора J009 G3 (Aldrich, кат. # 88755, 2,7 мг, 0,079 ммоль), Cs2CO3 (1025 мг, 3,15 ммоль), трет-бутил-3-((6-бромпиридин-3-ил)окси)-2-((трет-бутилдиметилсилил)окси)пропаноата (680 мг, 1,57 ммоль) и пиридин-4-ола (179 мг, 1,887 ммоль) в запаянной трубке дегазировали три раза в режиме вакуум/азот перед добавлением диоксана (12 мл). Полученную смесь дополнительно дегазировали три раза в режиме вакуум/азот, а затем нагревали при 90°С в течение 16 часов. Смесь фильтровали через целит, фильтрат концентрировали и остаток очищали на колонке с силикагелем, использу EtOAc/гексан, получая указанное в заголовке соединение (менее полярный региоизомер).
LC/MS [М+Н]+: 447,71.
Стадия B: трет-бутил-2-гидрокси-3-((6-(пиридин-4-илокси)пиридин-3-ил)окси)пропаноат
К раствору трет-бутил-2-((трет-бутилдиметилсилил)окси)-3-((6-(пиридин-4-илокси)пиридин-3-ил)окси)пропаноата (220 мг, 0,493 ммоль) в THF (6 мл) при 0°С добавляли TBAF (0,493 мл, 0,493 ммоль). Полученный раствор перемешивали при 0°С в течение 30 минут. После концентрирования раствора остаток очищали на колонке с использованием силикагеля и EtOAc/гексан в качестве элюирующих растворителей, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 333,21.
Стадия C: трет-бутил-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-((6-(пиридин-4-илокси)пиридин-3-ил)окси)пропаноат
К раствору трет-бутил-2-гидрокси-3-((6-(пиридин-4-илокси)пиридин-3-ил)окси)пропаноата (331 мг, 0,996 ммоль) в THF (6 мл) добавляли 2-гидроксиизоиндолин-1,3-дион (195 мг, 1,20 ммоль), трифенилфосфин (392 мг, 1,49 ммоль) и DEAD (0,237 мл, 1,494 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 2 часов, затем концентрировали на роторном испарителе. Остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 478,26.
Стадия D: 4-((5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)пиридин-2-ил)окси)-1-(3-((трет-бутоксикарбонил)амино)пропил)пиридин-1-ий
К раствору трет-бутил-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-((6-(пиридин-4-илокси)пиридин-3-ил)окси)пропаноата (0,81 г, 1,696 ммоль) в ацетонитриле (10 мл) добавляли трет-бутил-(3-иодпропил)карбамат (1,451 г, 5,09 ммоль). Полученный раствор нагревали при 60°С в течение ночи. После концентрирования реакционного раствора остаток очищали обращенно-фазовой MPLC с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05% TFA) в качестве подвижной фазы, получая указанное в заголовке соединение.
LC/MS [М+Н]+: 635,50.
Стадия E: (Z)-4-((5-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-(карбокси)метилен)амино)окси)-3-оксопропокси)пиридин-2-ил)окси)-1-(3-((трет-бутокси-карбонил)амино)пропил)пиридин-1-ий
К раствору 4-((5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)пиридин-2-ил)окси)-1-(3-((трет-бутоксикарбонил)-амино)пропил)пиридин-1-ия (100 мг, 0,157 ммоль) в CH2Cl2 (4 мл) и этаноле (4,00 мл) при 0°С добавляли гидразин (8,89 мкл, 0,283 ммоль). Полученный раствор перемешивали при 0°С в течение 1 часа, а затем к указанному раствору добавляли 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусную кислоту (86 мг, 0,315 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 часа, затем смесь концентрировали и остаток очищали обращенно-фазовой MPLC с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05% TFA) в качестве подвижной фазы, получая указанное в заголовке соединение.
LC/MS [М]+: 759,49.
Стадия F: 4-((5-(3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)пиридин-2-ил)окси)-1-(3-((трет-бутоксикарбонил)амино)пропил)пиридин-1-ий
К раствору (Z)-4-((5-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)пиридин-2-ил)окси)-1-(3-((трет-бутокси-карбонил)амино)пропил)пиридин-1-ия (28 мг, 0,037 ммоль) в DMF (20 мл) добавляли DCC (60,8 мг, 0,295 ммоль) и НОВТ (22,57 мг, 0,147 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 30 минут перед добавлением (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата (38,7 мг, 0,184 ммоль) и бикарбоната натрия (61,9 мг, 0,737 ммоль). Полученный раствор перемешивали при комнатной температуре в течение ночи. После фильтрации реакционной смеси остаток очищали с помощью обращенно-фазовой HPLC на колонке Gilson с использованием 20-100% ацетонитрила (0,05% TFA), с получением указанного в заголовке соединения.
LC/MS [М]+: 951,51.
Стадия G: 1-(3-аминопропил)-4-((5-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-пиридин-2-ил)окси)пиридин-1-ий 2,2,2-трифторацетат
К раствору 4-((5-(3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)пиридин-2-ил)окси)-1-(3-((трет-бутоксикарбонил)амино)пропил)пиридин-1-ия (28 мг, 0,029 ммоль) в CH2Cl2 (1 мл) добавляли TFA (2 мл, 26,0 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 75 минут. Раствор концентрировали, и остаток обрабатывали Et2O (10 мл). Et2O удаляли, и твердый остаток сушили под высоким вакуумом. Сухой остаток растворяли в DMSO (2 мл) и очищали с помощью обращенно-фазовой HPLC, с использованием ацетонитрил (0,05% TFA)/вода (0,05% TFA) в качестве подвижной фазы, получая указанное в заголовке соединение (более полярный диастереомер).
LC/MS [М+Н]+: 695,19.
1H ЯМР (D2O, 500 МГц): δ 8,64-8,62 (д, J=7,1 Гц, 2H), 7,98-7,97 (д, J=2,4 Гц, 1H), 7,61-7,58 (м, 1H), 7,46-7,45 (д, J=7,4 Гц, 2H), 7,21-7,20 (д, J=8,3 Гц, 1H), 7,02 (с, 1H), 5,03 (с, 1H), 4,75 (с, 1H), 4,52-4,46 (м, 4H), 3,03-3,00 (т, J=7,9 Гц, 2H), 2,30-2,26 (т, J=7,9 Гц, 2H), 1,43 (с, 3H), 1,12 (с, 3H).
ПРИМЕР 35
(S,Z)-3-(2-((2-((6-(3-(3-аминопропил)-1H-имидазол-3-ий-1-ил)пиридин-3-ил)окси)этокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат 2,2,2-трифторацетат (C35)
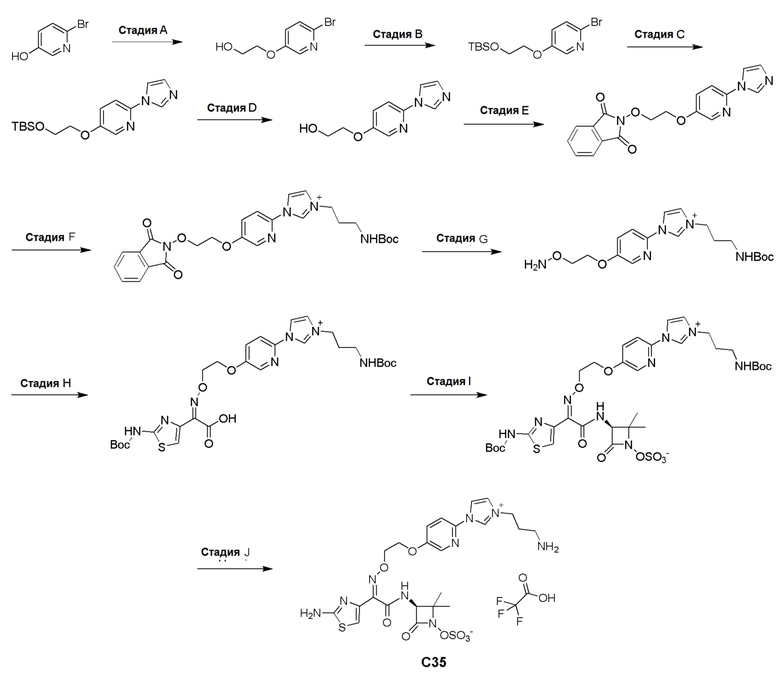
Стадия A: 2-((6-бромпиридин-3-ил)окси)этан-1-ол
Объединяли 6-бромпиридин-3-ол (700 мг, 4,02 ммоль), 2-хлорэтанол (972 мг, 12,1 ммоль) и K2CO3 (1,39 г, 10,1 ммоль), и суспендировали в ацетонитриле (18 мл). Поток газообразного N2 пропускали через мембрану в раствор в течение 10 минут, и полученный раствор нагревали при температуре 80°С в течение 18 часов. После выдерживания при температуре окружающей среды в течение двух дней, смесь фильтровали, и фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (24 г) на колонке, используя градиент смеси 0-100% EtOAc/гексан в качестве элюента, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 218,03.
1H ЯМР (CHCl3-d, 500 МГц): δ 8,11 (1H, д, J=3,2 Гц), 7,41 (1H, д, J=8,7 Гц), 7,16 (1H, дд, J=8,7, 3,2 Гц), 4,14 (2H, т, J=4,5 Гц), 4,03-4,00 (2H, м), 2,07-2,03 (1H, м).
Стадия B: 2-бром-5-(2-((трет-бутилдиметилсилил)окси)этокси)пиридин
К раствору 2-((6-бромпиридин-3-ил)окси)этан-1-ола (315 мг, 1,445 ммоль) и имидазола (492 мг, 7,22 ммоль) в ацетонитриле (9 мл) добавляли 1 М раствор трет-бутилдиметилхлорсилана в CH2Cl2 (3,2 мл, 3,2 ммоль) с последующим добавлением DMAP (17,65 мг, 0,144 ммоль). После перемешивания в течение 3 часов при комнатной температуре смесь концентрировали в вакууме. Остаток разбавляли EtOAc, промывали 2 раза насыщенным раствором NaHCO3 (водный раствор), насыщенным раствором соли (1 х). Органические экстракты сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (24 г) на колонке, с использованием градиента смеси 0-40% EtOAc/гексан в качестве элюента, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 332,10.
1H ЯМР (CHCl3-d, 500 МГц): δ 8,09 (1H, д, J=3,1 Гц), 7,38 (1H, д, J=8,7 Гц), 7,15 (1H, дд, J=8,7, 3,2 Гц), 4,09 (2H, т, J=4,9 Гц), 3,99 (2H, т, J=4,9 Гц), 0,93 (9H, д, J=8,8 Гц), 0,12 (6H, д, J=5,7 Гц).
Стадия C: 5-(2-((трет-бутилдиметилсилил)окси)этокси)-2-(1H-имидазол-1-ил)пиридин
Объединяли 2-бром-5-(2-((трет-бутилдиметилсилил)окси)этокси)пиридин (480 мг, 1,44 ммоль), имидазол (148 мг, 2,17 ммоль), L-пролин (27 мг, 0,231 ммоль), K2CO3 (399 мг, 2,89 ммоль) и CuI (13,8 мг, 0,072 ммоль), и суспендировали в DMSO (3 мл). Поток N2 пропускали через мембрану в раствор в течение 10 минут, и полученный раствор нагревали при 90°С в течение ночи. Смесь охлаждали, разбавляли этилацетатом и промывали H2O (1 х) и насыщенным раствором соли (1 х). Органические экстракты сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (12 г), с использованием градиента смеси 0-100% (3:1 EtOAc/MeOH)/гексан в качестве элюента, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 320,33.
1H ЯМР (CHCl3-d, 500 МГц): δ 8,24 (1H, с), 8,19 (1H, д, J=2,9 Гц), 7,58 (1H, с), 7,40 (1H, дд, J=8,8, 3,0 Гц), 7,31-7,28 (1H, м) 7,21 (1H, с), 4,15 (2H, т, J=4,9 Гц), 4,02 (2H, т, J=4,9 Гц), 0,93 (9H, с), 0,13 (6H, с).
Стадия D: 2-((6-(1H-имидазол-1-ил)пиридин-3-ил)окси)этан-1-ол
К раствору 5-(2-((трет-бутилдиметилсилил)окси)этокси)-2-(1H-имидазол-1-ил)пиридина (140 мг, 0,438 ммоль) в THF (5 мл) добавляли 1 М раствор TBAF в CH2Cl2 (0,438 мл, 0,438 ммоль). После перемешивания при комнатной температуре в течение 45 минут смесь концентрировали в вакууме, и остаток очищали с помощью колоночной хроматографии на силикагеле (4 г) на колонке, используя градиент смеси 0-100% (3:1 EtOAc/MeOH)/гексан в качестве элюента, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 205,90.
Стадия E: 2-(2-((6-(1H-имидазол-1-ил)пиридин-3-ил)окси)этокси)изоиндолин-1,3-дион
К раствору 2-((6-(1H-имидазол-1-ил)пиридин-3-ил)окси)этан-1-ола (80 мг, 0,390 ммоль) в THF (4 мл) добавляли N-гидроксифталимид (76 мг, 0,468 ммоль) и трифенилфосфин (123 мг, 0,468 ммоль). По каплям добавляли DIAD (0,091 мл, 0,468 ммоль) и полученный раствор перемешивали при комнатной температуре в течение ночи. Смесь концентрировали в вакууме, и остаток очищали с помощью колоночной хроматографии на силикагеле (12 г), с использованием градиента смеси 0-100% (3:1 EtOAc/MeOH)/гексан в качестве элюента, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 350,98.
1H ЯМР (CHCl3-d, 500 МГц): δ 8,24 (1H, с), 8,14 (1H, д, J=2,9 Гц), 7,89-7,86 (2H, м), 7,82-7,79 (2H, м), 7,58 (1H, с), 7,40 (1H, дд, J=8,8, 3,0 Гц), 7,33-7,30 (1H, м) 7,21 (1H, с), 4,65-4,63 (2H, м), 4,48-4,46 (2H, м).
Стадия F: 3-(3-((трет-бутоксикарбонил)амино)пропил)-1-(5-(2-((1,3-диоксоизоиндолин-2-ил)окси)этокси)пиридин-2-ил)-1H-имидазол-3-ий
К раствору 2-(2-((6-(1H-имидазол-1-ил)пиридин-3-ил)окси)этокси)изоиндолин-1,3-диона (131 мг, 0,374 ммоль) в диоксане (3 мл) добавляли 3-(boc-амино)пропилбромид (214 мг, 0,89 ммоль) и DMAP (5 мг, 0,041 ммоль), и полученную смесь перемешивали при 60°С в течение ночи. Добавляли дополнительное количество DMAP (5 мг, 0,041 ммоля) и каталитическое количество йодида натрия. Нагревание при 60°С продолжали в течение 4 часов, после чего смесь концентрировали под вакуумом. Остаток растирали с EtOАс для удаления исходных материалов и примесей, получая указанное в заголовке соединение.
LC-MS [M]+: m/z 508,29.
Стадия G: 1-(5-(2-(аминоокси)этокси)пиридин-2-ил)-3-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-3-ий
В MeOH (3 мл) суспендировали 3-(3-((трет-бутоксикарбонил)амино)пропил)-1-(5-(2-((1,3-диоксоизоиндолин-2-ил)окси)этокси)пиридин-2-ил)-1H-имидазол-3-ий (0,19 г, 0,374 ммоль), и добавляли 1 М раствор гидразина в MeOH (0,41 мл, 0,41 ммоль). После перемешивания при комнатной температуре в течение 2 часов смесь концентрировали в вакууме, удаляли избыток гидразина с помощью азеотропной перегонки с MeOH с последующей перегонкой с CH2Cl2. Остаток использовали на следующей стадии без дополнительной очистки.
LC-MS [М]+: m/z 378,25.
Стадия H: (Z)-3-(3-((трет-бутоксикарбонил)амино)пропил)-1-(5-(2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)этокси)пиридин-2-ил)-1H-имидазол-3-ий
К раствору 1-(5-(2-(аминоокси)этокси)пиридин-2-ил)-3-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-3-ия (0,141 г, 0,373 ммоль) в этаноле (2 мл) и в EtOH (2 мл) и ClCH2CH2Cl (4 мл) добавляли промежуточное соединение 6 (0,107 г, 0,392 ммоль). После перемешивания при комнатной температуре в течение ночи растворители удаляли в вакууме, и остаток использовали на следующей стадии без дополнительной очистки.
LC-MS [M]+: m/z 632,35.
Стадия I: (S,Z)-3-(3-((трет-бутоксикарбонил)амино)пропил)-1-(5-(2-(((1-(2-((трет-бутокси-карбонил)амино)тиазол-4-ил)-2-((2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)этокси)пиридин-2-ил)-1H-имидазол-3-ий
К смеси (Z)-3-(3-((трет-бутоксикарбонил)амино)пропил)-1-(5-(2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)этокси)пиридин-2-ил)-1H-имидазол-3-ия (237 мг, 0,374 ммоль) и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиний хлорида (176 мг, 0,636 ммоль) добавляли (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфат (102 мг, 0,486 ммоль) в виде раствора в DMF (4 мл). После перемешивания при температуре окружающей среды в течение ночи, добавляли дополнительное количество (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата (102 мг, 0,486 ммоль) и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метил-морфолиний хлорида (176 мг, 0,636 ммоль). После перемешивания при температуре окружающей среды в течение ночи, смесь разбавляли H2O и очищали с помощью обращенно-фазовой хроматографии (колонка Gilson SunFire С-18 30X150 мм) с использованием градиента 10-90% CH3CN/H2O (с 0,5% TFA) в качестве элюента. При лиофилизации получали указанное в заголовке соединение.
LC-MS [M+H]+: m/z 824,78.
Стадия J: (S,Z)-3-(2-((2-((6-(3-(3-аминопропил)-1H-имидазол-3-ий-1-ил)пиридин-3-ил)окси)этокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат 2,2,2-трифторацетат
К раствору (S,Z)-3-(3-((трет-бутоксикарбонил)амино)-пропил)-1-(5-(2-(((1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-((2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)этокси)пиридин-2-ил)-1H-имидазол-3-ия (56 мг, 0,068 ммоль) в CH2Cl2 (1 мл) добавляли TFA (1 мл, 12,98 ммоль). После перемешивания при комнатной температуре в течение 30 минут смесь концентрировали в вакууме, а избыток TFA удаляли путем азеотропной перегонки из CH2Cl2. После растирания с Et2O остаток очищали с помощью обращенно-фазовой хроматографии (колонка Gilson SunFire С-18 30X150 мм), с использованием градиента 5-40% CH3CN/H2O (с 0,5% TFA) в качестве элюента, продукт лиофилизировали, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 624,36.
ПРИМЕР 36
(S)-3-((Z)-2-(((S)-2-((6-(3-(3-аминопропил)-1H-имидазол-3-ий-1-ил)пиридин-3-ил)окси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат объединенный с 2,2,2-трифторуксусной кислотой (1:1) (C36)
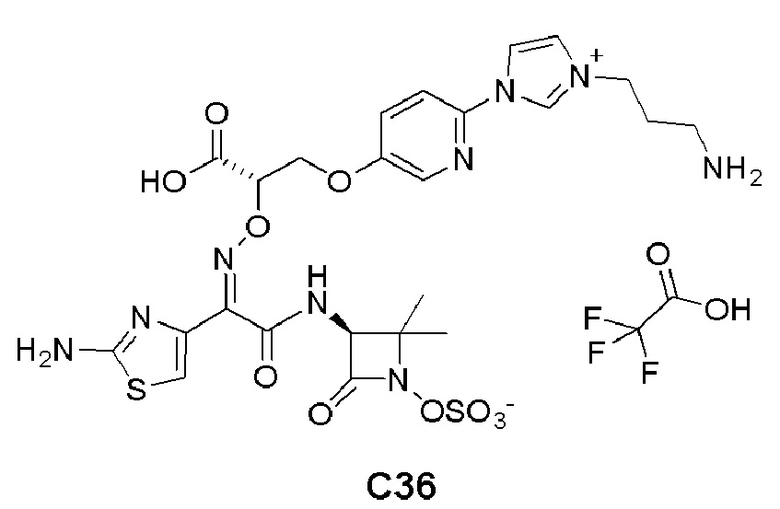
Стадия A: Этил-(R)-3-((6-бромпиридин-3-ил)окси)-2-гидроксипропаноат
Этил-оксиран-2-карбоксилат (1,47 г, 12,6 ммоль) суспендировали в МТВЕ (25 мл), и добавляли 6-бромпиридин-3-ол (1,0 г, 5,75 ммоль) и молекулярные сита 4Å (2 г). Добавляли промежуточное соединение 2 (0,482 г, 0,575 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 3-х дней. Твердые вещества удаляли фильтрацией, фильтрат концентрировали в вакууме, и остаток очищали с помощью колоночной хроматографии на силикагеле (колонка 40 г) с использованием градиента 0-60% EtOAc/гексан в качестве элюента, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 290,10.
1H ЯМР (CHCl3-d, 500 МГц): δ 8,10 (1H, д, J=3,1 Гц), 7,40 (1H, д, J=8,7 Гц), 7,16 (1H, дд, J=8,7, 3,2 Гц), 4,53 (1H, т, J=3,2 Гц), 4,35-4,31 (4H, м), 3,24 (1H, ш.с.),1,32 (3H, т, J=7,1 Гц).
Стадия B: (R)-3-((6-бромпиридин-3-ил)окси)-2-гидроксипропановая кислота
К раствору этил (R)-3-((6-бромпиридин-3-ил)окси)-2-гидроксипропаноата (513 мг, 1,77 ммоль) в THF (14 мл) при 0°С добавляли 1 М NaOH (3,54 мл, 3,54 ммоль). После перемешивания в течение 3 часов при 0°С рН доводили до 3 с помощью 1 Н HCl, и смесь распределяли между EtOAc и водой. Слои разделяли, и органическую фазу промывали солевым раствором (3 раза), сушили над MgSO4, фильтровали и концентрировали в вакууме, получая указанное в заголовке соединение, которое использовали на следующей стадии без дополнительной очистки.
LC-MS [М+Н]+: m/z 261,76.
Стадия C: трет-бутил-(R)-3-((6-бромпиридин-3-ил)окси)-2-гидроксипропаноат
К раствору (R)-3-((6-бромпиридин-3-ил)окси)-2-гидроксипропановой кислоты (464 мг, 1,77 ммоль) в THF (6 мл) добавляли 2-трет-бутил-1,3-диизопропилизомочевину (886 мг, 4,43 ммоль), и полученную смесь нагревали при 60°С в течение 2 часов 45 минут. Твердые вещества удаляли фильтрованием, фильтрат концентрировали под вакуумом, и остаток очищали с помощью колоночной хроматографии на силикагеле (12 г), с использованием градиента 0-60% EtOAc/гексан в качестве элюента, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 318,17.
1H ЯМР (CHCl3-d, 500 МГц): δ 8,09 (1H, д, J=3,1 Гц), 7,40 (1H, д, J=8,7 Гц), 7,16 (1H, дд, J=8,7, 3,1 Гц), 4,41 (1H, с), 4,32-4,26 (2H, м), 3,26 (1H, ш.с.), 1,51 (9H, с).
Стадия D: трет-бутил-(R)-3-((6-бромпиридин-3-ил)окси)-2-((трет-бутилдиметилсилил)окси)-пропаноат
К раствору трет-бутил-(R)-3-((6-бромпиридин-3-ил)окси)-2-гидрокси-пропаноата (265 мг, 0,833 ммоль) в CH2Cl2 (6 мл) при 0°С добавляли триэтиламин (0,348 мл, 2,50 ммоль), и после перемешивания при 0°С в течение 30 минут добавляли по каплям трет-бутилдиметилсилил трифторметансульфонат (0,287 мл, 1,25 ммоль). Смесь распределяли между CH2Cl2 и Н2О. Слои разделяли, и органический слой промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (12 г), с использованием градиента 0-30% EtOAc/гексан в качестве элюента, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 432,07.
1H ЯМР (CHCl3-d, 500 МГц): δ 8,09 (1H, д, J=3,1 Гц), 7,39 (1H, д, J=8,7 Гц), 7,15 (1H, дд, J=8,7, 3,1 Гц), 4,47 (1H, дд, J=6,8, 3,4 Гц), 4,27 (1H, дд, J=9,7, 3,4 Гц), 4,16 (1H, дд, J=9,7, 6,8 Гц), 1,50 (9H, с), 0,93 (9H, с), 0,16 (3H, с), 0,10 (3H, с).
Стадия E: трет-бутил-(R)-3-((6-(1H-имидазол-1-ил)пиридин-3-ил)окси)-2-((трет-бутилдиметилсилил)окси)пропаноат
Процедура получения была такой же, как описано для стадии С примера 35, с использованием в качестве реагентов (R)-3-((6-бромпиридин-3-ил)окси)-2-((трет-бутилдиметилсилил)окси)пропаноата (318 мг, 0,735 ммоль), имидазола (90 мг, 1,32 ммоль), L-пролина (51 мг, 0,44 ммоль), K2CO3 (102 мг, 0,735 ммоль) и CuI (42 мг, 0,22 ммоль) в DMSO (3 мл).
LC-MS [M+H]: m/z 420,45.
Стадия F: трет-бутил-(R)-3-((6-(1H-имидазол-1-ил)пиридин-3-ил)окси)-2-гидроксипропаноат
Процедура получения была такой же, как описано для стадии D примера 35, с использованием в качестве реагентов(R)-3-((6-(1H-имидазол-1-ил)пиридин-3-ил)окси)-2-((трет-бутилдиметилсилил)окси)пропаноата (80 мг, 0,191 ммоль) в THF (4 мл) и 1M TBAF в THF (0,191 мл, 0,191 ммоль).
LC-MS [M+H]+: m/z 306,27.
Стадия G: трет-бутил-(S)-3-((6-(1H-имидазол-1-ил)пиридин-3-ил)окси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноат
Процедура получения была такой же, как описано для стадии E примера 35, с использованием в качестве реагентов трет-бутил-(R)-3-((6-(1H-имидазол-1-ил)пиридин-3-ил)окси)-2-гидроксипропаноата (76 мг, 0,249 ммоль), N-гидроксифталимида (48,7 мг, 0,299 ммоль), трифенилфосфина (78 мг, 0,299 ммоль) и DIAD (0,058 мл, 0,299 ммоль) в THF (3 мл).
LC-MS [М+Н]+: m/z 451,33.
Стадия H: (S)-3-(3-аминопропил)-1-(5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)пиридин-2-ил)-1H-имидазол-3-ий
К раствору трет-бутил-(S)-3-((6-(1H-имидазол-1-ил)пиридин-3-ил)окси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноата (85 мг, 0,189 ммоль) в диоксане (2 мл) добавляли 3-(Вос-амино)пропилбромид (67,4 мг, 0,283 ммоль) и смесь перемешивали при 60°С в течение ночи. Добавляли йодид натрия (10 мг, 0,067 ммоль) и нагревание при 60°С продолжали в течение 6 часов, затем добавляли дополнительное количество 3-( Вос-амино) пропилбромида (67,4 мг, 0,283 ммоль) и иодида натрия (34 мг, 0,22 ммоль). Добавляли ацетон (0,2 мл), и смесь нагревали при 60°С в течение ночи. Смесь концентрировали в вакууме, и растирали с EtOAc, получая указанное в заголовке соединение в виде остатка, который использовали на следующей стадии без дополнительной очистки.
LC-MS [М]+: m/z 508,39.
Стадия I: (S)-1-(5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)пиридин-2-ил)-3-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-3-ий
К раствору (S)-3-(3-аминопропил)-1-(5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)пиридин-2-ил)-1H-имидазол-3-ия (46 мг, 0,090 ммоль) в CH2Cl2 (3 мл) добавляли Вос-ангидрид (29,6 мг, 0,136 ммоль) и N,N,N',N'-тетраметилэтилендиамин (15 мкл, 0,099 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 3-х дней. После концентрирования в вакууме и растирания с Et2O, остаток лиофилизировали из CH3CN/H2O.
LC-MS [M+H]+: m/z 608,10.
Стадия J: (S)-1-(5-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)пиридин-2-ил)-3-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-3-ий
Процедура получения была такой же, как описано для стадии F примера 35, с использованием в качестве реагентов (S)-1-(5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)пиридин-2-ил)-3-(3-((трет-бутоксикарбонил)-амино)пропил)-1H-имидазол-3-ия (26 мг, 0.043 ммоль) 1 M гидразина в MeOH (47 мкл, 0,047 ммоль) в MeOH (1 мл).
LC-MS [M]+: m/z 478,40.
Стадия K: (S,Z)-1-(5-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)пиридин-2-ил)-3-(3-((трет-бутокси-карбонил)амино)пропил)-1H-имидазол-3-ий
Процедура получения была такой же, как описано для стадии H примера 35, с использованием в качестве реагентов (S)-1-(5-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)пиридин-2-ил)-3-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-3-ия (21 мг, 0,044 ммоль) и промежуточного соединения 6 (12,6 мг, 0,046 ммоль) в этаноле (0,5 мл) и ClCH2CH2Cl (1 мл).
LC-MS [М]+: m/z 732,51.
Стадия L: 1-(5-((S)-3-(трет-бутокси)-2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)пиридин-2-ил)-3-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-3-ий
Процедура получения была такой же, как описано для стадии I примера 35, с использованием в качестве реагентов (S,Z)-1-(5-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)-амино)окси)-3-оксопропокси)пиридин-2-ил)-3-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-3-ия (32 мг, 0,044 ммоль), (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата (11,93 мг, 0,057 ммоль) и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиний хлорида (20,54 мг, 0,074 ммоль) в DMF (1 мл).
LC-MS [М+Н]+: m/z 924,83.
Стадия M: (S)-3-((Z)-2-(((S)-2-((6-(3-(3-аминопропил)-1H-имидазол-3-ий-1-ил)пиридин-3-ил)окси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат объединенный с 2,2,2-трифторуксусной кислотой (1:1)
Процедура получения была такой же, как описано для стадии J примера 35, с использованием в качестве реагентов 1-(5-((S)-3-(трет-бутокси)-2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)пиридин-2-ил)-3-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-3-ия (7 мг, 7,57 мкмоль) и TFA (0,6 мл, 7,79 ммоль) в CH2Cl2 (0,6 мл).
LC-MS [М+Н]+: m/z 668,37.
ПРИМЕР 37A и 37B
3-(2-((5-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)пиридин-2-ил)амино)-1H-имидазол-1-ил)пропан-1-аминий 2,2,2-трифторацетат (C37A); и 3-(2-((5-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)пиридин-2-ил)амино)-1H-имидазол-1-ил)пропан-1-аминий 2,2,2-трифторацетат (C37B)
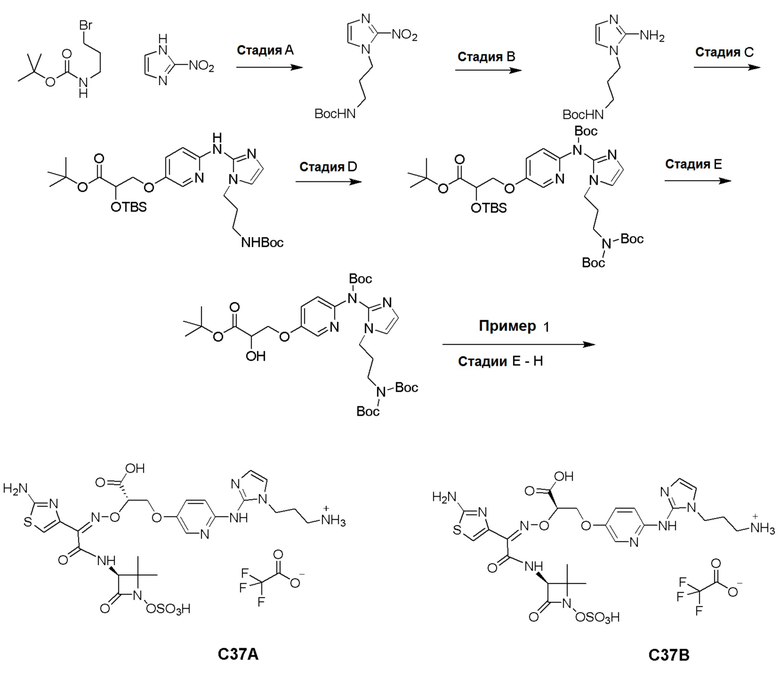
Стадия A: трет-бутил-(3-(2-нитро-1H-имидазол-1-ил)пропил)карбамат
Карбонат цезия (576 мг, 1,77 ммоль) добавляли к перемешиваемой смеси 3-(Вос-амино)пропилбромида (232 мг, 0,973 ммоль) и 2-нитро-1H-имидазола (100 мг, 0,884 ммоль) в DMF (2 мл). Смесь перемешивали при комнатной температуре в течение 1 часа, а затем смесь перемешивали в течение ночи при 60°С. Смесь разбавляли этилацетатом, промывали водой (3 раза) и насыщенным раствором соли, сушили (MgSO4), фильтровали и выпаривали растворитель при пониженном давлении, с получением неочищенного продукта, который очищали с помощью ISCO (gold, 40 г), элюируя смесью этилацетат/гексан с градиентом 0-100%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 271,3.
Стадия B: трет-бутил-(3-(2-амино-1H-имидазол-1-ил)пропил)карбамат
Смесь трет-бутил-(3-(2-нитро-1H-имидазол-1-ил)пропил)карбамата (500 мг, 1,85 ммоль) и 3% Pt/0,6% С (по массе) (100 мг, 0,015 ммоль) в метаноле (10 мл) вакуумировали и продували водородом (3 раза) и перемешивали при комнатной температуре в течение 2,5 часов. Реакционную смесь разбавляли EtOAc и фильтровали через целит. Фильтрат концентрировали при пониженном давлении, получая указанное в заголовке соединение, которое использовали без дополнительной обработки на следующей стадии.
LC-MS [М+Н]: m/z 241,6.
Стадия C: трет-бутил-3-((6-((1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-2-ил)амино)пиридин-3-ил)окси)-2-((трет-бутилдиметилсилил)окси)пропаноат
Смесь трет-бутил-3-((6-бромпиридин-3-ил)окси)-2-((трет-бутилдиметилсилил)окси) пропаноата (178 мг, 0,412 ммоль), трет-бутил-(3-(2-амино-1H-имидазол-1-ил)пропил)карбамата (90 мг, 0,375 ммоль), палладиевого прекатализатора G3 J009 и карбоната цезия (366 мг, 1,12 ммоль) в диоксане (3,5 мл) три раза вакуумировали и продували азотом, а затем смесь нагревали при 100°С в течение ночи. Смесь разбавляли EtOAc, фильтровали через тонкий слой целита и промывали EtOAc. Растворитель удаляли при пониженном давлении, получая указанное в заголовке соединение, которое непосредственно использовали на следующей стадии.
LC-MS [М+Н]+: m/z 592,7.
Стадия D: трет-бутил-3-((6-((1-(3-(бис(трет-бутоксикарбонил)амино)пропил)-1H-имидазол-2-ил)(трет-бутоксикарбонил)амино)пиридин-3-ил)окси)-2-((трет-бутилдиметилсилил)окси)пропаноат
Смесь трет-бутил-3-((6-((1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-2-ил)амино)пиридин-3-ил)окси)-2-((трет-бутилдиметилсилил)окси)пропаноата (120 мг, 0,203 ммоль) в Вос2O (0,5 мл) перемешивали при 120°С в течение 2,5 часов. Реакционную смесь охлаждали и очищали на колонке ISCO (40 г gold), элюируя смесью EtOAc/гексан с градиентом 0-100%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 792,9.
Стадия E: трет-бутил-3-((6-((1-(3-(бис(трет-бутоксикарбонил)амино)пропил)-1H-имидазол-2-ил)(трет-бутоксикарбонил)амино)пиридин-3-ил)окси)-2-гидроксипропаноат
К перемешиваемой смеси трет-бутил-3-((6-((1-(3-(бис(трет-бутоксикарбонил)амино)пропил)-1H-имидазол-2-ил)(трет-бутоксикарбонил)амино)-пиридин-3-ил)окси)-2-((трет-бутилдиметилсилил)окси)пропаноата (320 мг, 0,404 ммоль) и THF (3 мл) добавляли TBAF (1,0 М в THF, 0,606 мл, 0,606 ммоль), и смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель удаляли и остаток очищали на колонке ISCO с силикагелем (40 г gold), элюируя смесью EtOAc/гексан с градиентом 0-100%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 678,8.
Продукт из стадии Е (трет-бутил-3-((6-((1-(3-(бис(трет-бутоксикарбонил)амино)пропил)-1H-имидазол-2-ил)(трет-бутоксикарбонил)амино)пиридин-3-ил)окси)-2-гидроксипропаноат) превращали в соединения C37A и C37B в виде солей TFA, следуя процедуре, аналогичной процедурам стадии C, стадий Е-Н примера 1.
Соединение 37А: 3-(2-((5-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)пиридин-2-ил)амино)-1H-имидазол-1-ил)пропан-1-аминий 2,2,2-трифторацетат.
LC-MS [М+Н]+: m/z 683,6.
1H ЯМР (500 МГц, оксид дейтерия) δ 7,88 (д, J=2,9 Гц, 1H), 7,35 (д, J=8,7 Гц, 1H), 7,04-6,96 (м, 2H), 6,93 (с, 2H), 4,99 (с, 1H), 4,62 (ш.с., 1H), 4,47-4,34 (м, 2H), 4,04 (т, J=7,3 Гц, 2H), 2,96 (т, J=8,0 Гц, 2H), 2,08 (м, 2H), 1,31 (с, 3H), 1,00 (с, 3H).
Соединение 37B: 3-(2-((5-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)пиридин-2-ил)амино)-1H-имидазол-1-ил)пропан-1-аминий 2,2,2-трифторацетат.
LC-MS [М+Н]+: m/z 683,6.
1H ЯМР (500 МГц, оксид дейтерия) δ 7,89-7,85 (с, 1H), 7,39-7,32 (д, 1H), 7,03-6,95 (м, 2H), 6,93 (с, 2H), 4,95 (с, 1H), 4,62 (ш.с., 1H), 4,45-4,35 (м, 2H), 4,04 (т, J=7,3 Гц, 2H), 2,96 (т, J=8,0 Гц, 2H), 2,08 (м, 2H), 1,31 (с, 3H), 1,02 (с, 3H).
ПРИМЕР 38
(S)-3-((Z)-2-(((S)-2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)-3-хлорфенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат

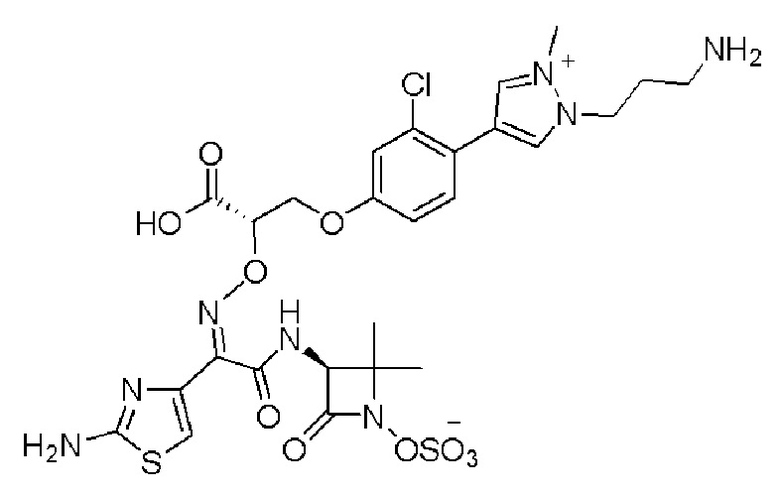
Стадия A: трет-бутил-(R)-3-(4-бром-3-хлорфенокси)-2-гидроксипропаноат
(R,R)-Co-катализатор (0,24 г, 0,29 ммоль) добавляли к перемешиваемой при комнатной температуре смеси трет-бутил-оксиран-2-карбоксилата (1,7 г, 12 ммоль) и 4-бром-3-хлорфенола (1,2 г, 5,8 ммоль) в трет-бутилметиловом эфире. Затем добавляли молекулярные сита (2,3 г), полученную смесь перемешивали при комнатной температуре в течение ночи и затем фильтровали. Фильтрат концентрировали, и полученный остаток очищали на колонке с силикагелем (80 г), элюируя смесью EtOAc/гексан (5-40%), получая указанное в заголовке соединение.
Стадия B: трет-бутил-(R)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)-3-хлорфенокси)-2-гидроксипропаноат
К раствору (R)-трет-бутил-3-(4-бром-3-хлорфенокси)-2-гидроксипропаноата (0,42 г, 1,2 ммоль), трет-бутил-(R)-3-(4-бром-3-хлорфенокси)-2-гидроксипропаноата (0,42 г, 1,2 ммоль) и дихлорида 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладия (0,078 г, 0,12 ммоль) в диоксане (1 мл) добавляли 1 М водный раствор фосфата калия (3,6 мл, 3,6 ммоль). Реакционную колбу герметично закрывали, дегазировали и заполняли N2. Реакционную смесь перемешивали при 72°С в течение 1 часа, а затем разбавляли водой и дважды экстрагировали EtOAc. Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали. Полученный остаток очищали на колонке с силикагелем (25 г), элюируя смесью CH2Cl2/MeOH (100-90%), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 496,34.
Стадия C: трет-бутил-(S)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)-3-хлорфенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноат
(E)-диэтил диазен-1,2-дикарбоксилат (0,20 мл, 1,3 ммоль) добавляли при 0°C к смеси (R)-трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)-3-хлорфенокси)-2-гидроксипропаноата (580 мг, 1,2 ммоль), 2-гидроксиизоиндолин-1,3-диона (190 мг, 1,2 ммоль) и трифенилфосфина (340 мг, 1,3 ммоль) в THF. Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, а затем концентрировали. Полученный остаток очищали хроматографией на колонке с силикагелем (25 г), элюируя смесью EtOAc/изогексан 5-50%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 641,44.
Стадия D: (S)-4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-хлорфенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий иодид
К смеси S)-трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)-3-хлорфенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноата (625 мг, 0,98 ммоль) и CH3CN добавляли MeI (610 мкл, 9,8 ммоль). Реакционную смесь перемешивали при 74°С в течение ночи, а затем охлаждали до комнатной температуры. Реакционную смесь концентрировали, получая указанное в заголовке соединение.
LC-MS [М]+: m/z 655,40.
Стадия E: (S,Z)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-(карбокси)метилен)амино)окси)-3-оксопропокси)-2-хлорфенил)-1-(3-((трет-бутокси-карбонил)амино)пропил)-2-метил-1H-пиразол-2-ий иодид
К смеси (S)-4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-хлорфенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий иодида и CHCl3/этанол добавляли гидразин (28 мг, 0,86 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, а затем добавляли 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусную кислоту (230 мг, 0,86 ммоль) Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, а затем концентрировали, получая указанное в заголовке соединение.
LC-MS [M]+: m/z 779,33.
Стадия F: 4-(4-((S)-3-(трет-бутокси)-2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2-хлорфенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий
К раствору (S,Z)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)-2-хлорфенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ия (810 мг, 1,0 ммоль) в DMF (1 мл) добавляли N,N'-метандиилидендициклогексанамин (DCC) (540 мг, 2,6 ммоль) и 1H-бензо[d][1,2,3]триазол-1-ол гидрат (HOBt) (500 мг, 2,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 20 мин, затем добавляли (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфат (330 мг, 1,6 ммоль) и гидрокарбонат натрия (520 мг, 6,2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, а затем фильтровали. Фильтрат очищали обращенно-фазовой хроматографией на колонке C-18 150 г с использованием 0-75-100% ацетонитрила (с 0,05% TFA) в течение 25 минут, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 971,27.
Стадия G: (S)-3-((Z)-2-(((S)-2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)-3-хлорфенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
TFA (2 мл, 26 ммоль) добавляли к перемешиваемой при комнатной температуре смеси 4-(4-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2-хлорфенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ия (175 мг, 0,18 ммоль) и DCM. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа 20 минут, а затем концентрировали. Полученный неочищенный продукт растворяли в воде и очищали с помощью SFC, используя смесь вода/ацетонитрил (0,1% НСООН), получая указанное в заголовке соединение.
LC-MS [M]+: m/z 715,68.
ПРИМЕР 39
(S)-3-((Z)-2-((((S)-1-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-2-карбоксипропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
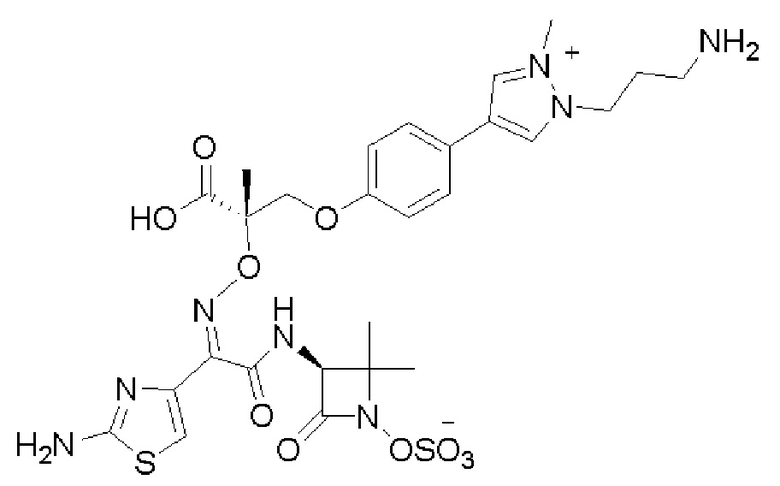
Стадия A: трет-бутил-(3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропил)карбамат
Карбонат цезия (12 г, 38 ммоль) добавляли при комнатной температуре к смеси 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (4,8 г, 25 ммоль) и трет-бутил-(3-бромпропил)карбамата (6,0 г, 25 ммоль) в DMF (40 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем разбавляли EtOAc и водой, и экстрагировали EtOAc. Объединенные органические слои промывали насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на колонке с силикагелем, элюируя смесью EtOAc/изогексан (10-80%), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 352,33.
Стадия B: трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)-фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-2-метилпропаноат
К раствору трет-бутил-2-(аминоокси)-3-(4-бромфенокси)-2-метилпропаноата (100 мг, 0,29 ммоль)), трет-бутил-(3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропил)карбамата (100 мг, 0,29 ммоль) и дихлорида 1,1-бис (ди-трет-бутилфосфино) ферроценпалладия (19 мг, 0,029 ммоль) в диоксане (1 мл) добавляли водный раствор 1 М фосфата калия (0,87 мл, 0,87 ммоль). Сосуд закрывали, дегазировали, заполняли азотом, и смесь перемешивали при 70°С в течение 1 часа. Затем реакционную смесь разбавляли водой и экстрагировали два раза этилацетатом. Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью CH2Cl2/MeOH (100-90%), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 491,43.
Стадия C: трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-2-метилпропаноат
К смеси трет-бутил-2-(аминоокси)-3-(4-(1-(3-((трет-бутоксикарбонил)-амино)пропил)-1H-пиразол-4-ил)фенокси)-2-метилпропаноата (540 мг, 1,1 ммоль) и молекулярных сит (200 мг) в толуоле при комнатной температуре добавляли 4-метил-бензолсульфоновую кислоту (12% в уксусной кислоте, 63 мг, 0,044 ммоль). Реакционную смесь перемешивали при 100°С в течение 4 часов, а затем фильтровали и концентрировали. Полученный остаток очищали хроматографией на колонке с силикагелем, элюируя смесью CH2Cl2/MeOH (100-90%), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 622,56.
Стадия D: 4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-2-метил-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий иодид
Указанное в заголовке соединение получали в соответствии с процедурой стадии D примера 1, используя в качестве реагентов трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-2-метилпропаноат (0,50 г, 0,80 ммоль) и MeI (0,40 мл, 6,4 ммоль) в MeCN (3 мл).
LC-MS [M]+: m/z 635,58.
Стадия E: (Z)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-2-метил-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий иодид
К смеси 4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-2-метил-3-оксопропокси)фенил)-1-(3-((трет-бутокси-карбонил)-амино)пропил)-2-метил-1H-пиразол-2-ия в EtOH/DCM добавляли гидразин (0,024 мл, 0,76 ммоль) в 0,3 мл DCM. Реакционную смесь перемешивали при комнатной температуре в течение ночи, а затем фильтровали. Фильтрат концентрировали, и полученный остаток очищали с помощью препаративной высокоэффективной жидкостной хроматографии, элюируя смесью ацетонитрил/вода+0,05% TFA (0-100%), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 759,58.
Стадия F: 4-(4-(3-(трет-бутокси)-2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-метил-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий
К раствору (Z)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-2-метил-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий иодида (80 мг, 0,105 ммоль) в DMF (1 мл) добавляли DCC (87 мг, 0,42 ммоль) и НОВТ (60 мг, 0,32 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут, с последующим добавлением (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата (76 мг, 0,42 ммоль) и бикарбоната натрия (35 мг, 0,42 ммоль ). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов и фильтровали. Фильтрат очищали с помощью обращенно-фазовой хроматографией на колонке ISCO (130 г, 0-100%, H2O/MeCN/0,05% TFA), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 921,76.
Стадия G: (S)-3-((Z)-2-((((S)-1-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-2-карбоксипропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
К раствору 4-(4-(3-(трет-бутокси)-2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-метил-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ия (110 мг, 0,12 ммоль) в DCM добавляли 2 мл трифторуксусной кислоты. Реакционную смесь перемешивали при комнатной температуре в течение 90 минут, и затем концентрировали в вакууме без нагрева. Добавляли DCM (10 мл), и смесь концентрировали три раза для удаления TFA. Полученный остаток промывали сухим MeCN (2×2 мл). Затем остаток растворяли в DMSO и очищали с помощью препаративной высокоэффективной жидкостной хроматографии используя MeCN/H2O (0,05% TFA в обоих) с градиентом 2-25% в течение 10 минут, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 695,14.
ПРИМЕР 40
(3S)-3-((Z)-2-((2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-фосфоноэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
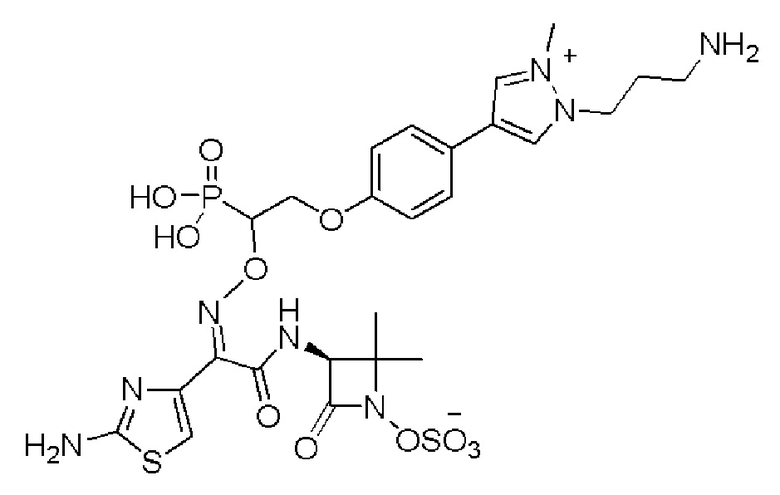
Стадия A: 1-бром-4-(2,2-диэтоксиэтокси)бензол
Карбонат калия (11 г, 78 ммоль) добавляли к перемешиваемой при комнатной температуре смеси 4-бромфенола (5,4 г, 31 ммоль) и 2-бром-1,1-диэтоксиэтана (1,8 мл, 12 ммоль) в DMF. Реакционную смесь перемешивали при 80°С в течение ночи, затем охлаждали до комнатной температуры и разбавляли EtOAc. Смесь промывали водой и насыщенным раствором соли. Органический слой отделяли, сушили над MgSO4, фильтровали и концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью EtOAc/изогексан (0-40%), получая указанное в заголовке соединение.
Стадия B: 2-(4-бромфенокси)ацетальдегид
К перемешиваемой при комнатной температуре смеси 1-бром-4-(2,2-диэтоксиэтокси)бензола (2,6 г, 9,0 ммоль) в THF добавляли HCl (3,7 мл, 22 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, после чего смесь экстрагировали диэтиловым эфиром. Органический слой отделяли, промывали водой и насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали, получая указанное в заголовке соединение.
Стадия C: диметил (2-(4-бромфенокси)-1-гидроксиэтил)фосфонат
К перемешиваемой при 0°C смеси 2-(4-бромфенокси)-ацетальдегида (1,2 г, 5,3 ммоль) и триэтиламина (1,8 мл, 13 ммоль) в THF добавляли по каплям диметил фосфонат (1,20 мл, 13 ммоль). Реакционную смесь перемешивали при 0°С в течение 1 часа, а затем при комнатной температуре в течение 1 часа, и затем реакционную смесь концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на колонке с силикагелем 40 г, элюируя смесью CH2Cl2/MeOH (100-90%), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 325,00.
Стадия D: трет-бутил-(3-(4-(4-(2-(диметоксифосфорил)-2-гидроксиэтокси)фенил)-1H-пиразол-1-ил)пропил)карбамат
Указанное в заголовке соединение получали в соответствии с процедурой стадии B примера 38.
LC-MS [М+Н]+: m/z 470,29.
Стадия E: трет-бутил-(3-(4-(4-(2-(диметоксифосфорил)-2-((1,3-диоксоизоиндолин-2-ил)окси)-этокси)фенил)-1H-пиразол-1-ил)пропил)карбамат
Указанное в заголовке соединение получали в соответствии с процедурой стадии С примера 38.
LC-MS [М+Н]+: m/z 615,29.
Стадия F: 1-(3-((трет-бутоксикарбонил)амино)пропил)-4-(4-(2-(диметоксифосфорил)-2-((1,3-диоксоизоиндолин-2-ил)окси)этокси)фенил)-2-метил-1H-пиразол-2-ий трифторметан-сульфонат
Метилтрифторметансульфонат (0,069 мл, 0,63 ммоль) добавляли к перемешиваемой охлажденной до 0°C смеси трет-бутил-(3-(4-(4-(2-(диметоксифосфорил)-2-((1,3-диоксоизоиндолин-2-ил)окси)этокси)фенил)-1H-пиразол-1-ил)пропил)карбамата (370 мг, 0,60 ммоль) в CH3CN. Смесь перемешивали при 0°С в течение 10 минут, затем при комнатной температуре в течение ночи, и затем концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью CH2Cl2/MeOH (100-80%), получая указанное в заголовке соединение.
LC-MS [М]+: m/z 629,33.
Стадия G: 1-(3-аминопропил)-4-(4-(2-((1,3-диоксоизоиндолин-2-ил)окси)-2-фосфоноэтокси)-фенил)-2-метил-1H-пиразол-2-ий трифторметансульфонат
Бромтриметилсилан (0,77 мл, 5,9 ммоль) добавляли к перемешиваемой при комнатной температуре смеси 1-(3-((трет-бутокси-карбонил)амино)пропил)-4-(4-(2-(диметоксифосфорил)-2-((1,3-диоксоизоиндолин-2-ил)окси)-этокси)фенил)-2-метил-1H-пиразол-2-ий трифторметансульфоната (570 мг, 0,59 ммоль) в DCM. Реакционную смесь перемешивали при комнатной температуре в течение ночи, а затем концентрировали, получая указанное в заголовке соединение.
LC-MS [М]+: m/z 501,26.
Стадия H: 1-(3-((трет-бутоксикарбонил)амино)пропил)-4-(4-(2-((1,3-диоксоизоиндолин-2-ил)окси)-2-фосфоноэтокси)фенил)-2-метил-1H-пиразол-2-ий
Триэтиламин (0,31 мл, 2,2 ммоль) добавляли к перемешиваемой охлаждаемой до комнатной температуре смеси 1-(3-аминопропил)-4-(4-(2-((1,3-диоксоизоиндолин-2-ил)окси)-2-фосфоноэтокси)фенил)-2-метил-1H-пиразол-2-ий трифторметансульфоната (0,28 г, 0,56 ммоль) и ди-трет-бутилдикарбонат (0,24 г, 1,12 ммоль) в DMF. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь очищали с помощью препаративной обращенно-фазовой HPLC (С-18), элюируя смесью CH3CN/вода с 0,05% TFA (2-100%), получая указанное в заголовке соединение.
LC-MS [М]+: m/z 601,32.
Стадия I: 1-(3-((трет-бутоксикарбонил)амино)пропил)-4-(4-(2-((((Z)-1-(2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксо-этилиден)амино)окси)-2-фосфоноэтокси)фенил)-2-метил-1H-пиразол-2-ий
Гидразин (10 мкл, 0,33 ммоль) добавляли к перемешиваемой при комнатной температуре смеси 1-(3-((трет-бутоксикарбонил)амино)пропил)-4-(4-(2-((1,3-диоксоизоиндолин-2-ил)окси)-2-фосфоноэтокси)-фенил)-2-метил-1H-пиразол-2-ия в EtOH/CHCl3. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, а затем добавляли (S)-3-(2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоацетамидо)-2,2-диметил-4-оксоазетидин-1-ил гидросульфат и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь фильтровали и фильтрат концентрировали. Полученный остаток растворяли в DMFА и очищали с помощью обращенно-фазовой препаративной HPLC (С-18), элюируя смесью ацетонитрил/вода+0,1% TFA (2-100%), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 917,24.
Стадия J: (3S)-3-((Z)-2-((2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-фосфоноэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
Указанное в заголовке соединение получали в соответствии с процедурой стадии G примера 2 с использованием в качестве реагентов 1-(3-((трет-бутоксикарбонил)амино)пропил)-4-(4-(2-((((Z)-1-(2-((трет-бутокси-карбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-фосфоноэтокси)фенил)-2-метил-1H-пиразол-2-ия (0,13 г, 0,14 ммоль) и TFA (1,5 мл, 19 ммоль).
LC-MS [М+Н]+: m/z 111,36.
ПРИМЕР 41
(3S)-3-((Z)-2-(((1R)-2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-(гидрокси(метокси)фосфорил)этокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат объединенный с 2,2,2-трифторуксусной кислотой (1:1)
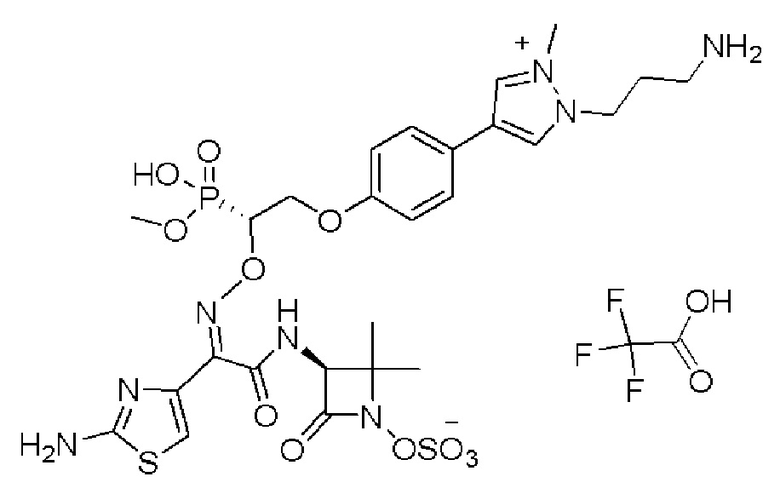
Стадия A: 1-(3-((трет-бутоксикарбонил)амино)пропил)-4-(4-(2-((1,3-диоксоизоиндолин-2-ил)окси)-2-(гидрокси(метокси)фосфорил)этокси)фенил)-2-метил-1H-пиразол-2-ий иодид
MeI (0,145 мл, 2,327 ммоль) добавляли к перемешиваемой при комнатной температуре смеси трет-бутил-(3-(4-(4-(2-(диметоксифосфорил)-2-((1,3-диоксоизоиндолин-2-ил)окси)этокси)фенил)-1H-пиразол-1-ил)пропил)карбамата (пример 40, стадия Е, 143 мг, 0,23 ммоль) в CH3CN. Реакционную смесь перемешивали при 72°С в течение ночи, а затем концентрировали, получая указанное в заголовке соединение.
LC-MS [М]+: m/z 615,36.
Стадия B: 1-(3-((трет-бутоксикарбонил)амино)пропил)-4-(4-((2R)-2-((((Z)-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-2-(гидроксил-(метокси)фосфорил)этокси)фенил)-2-метил-1H-пиразол-2-ий
Указанное в заголовке соединение получали в соответствии с процедурой, описанной на стадии E примера 38.
LC-MS [M]+: m/z 739,46.
Стадия C: 1-(3-((трет-бутоксикарбонил)амино)пропил)-4-(4-(2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-(гидрокси(метокси)фосфорил)этокси)фенил)-2-метил-1H-пиразол-2-ий
Указанное в заголовке соединение получали в соответствии с процедурой, описанной на стадии F примера 38.
LC-MS [M]+: m/z 931,38.
Стадия D: (3S)-3-((Z)-2-(((1R)-2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-(гидрокси(метокси)фосфорил)этокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат объединенный с 2,2,2-трифторуксусной кислотой (1:1)
Указанное в заголовке соединение получали в соответствии с процедурой, описанной на стадии G примера 38.
LC-MS [М+Н]+: m/z 731,68.
ПРИМЕР 42
(3S)-3-((Z)-2-(((1S)-2-(4-(1-(3-амино-2-гидроксипропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
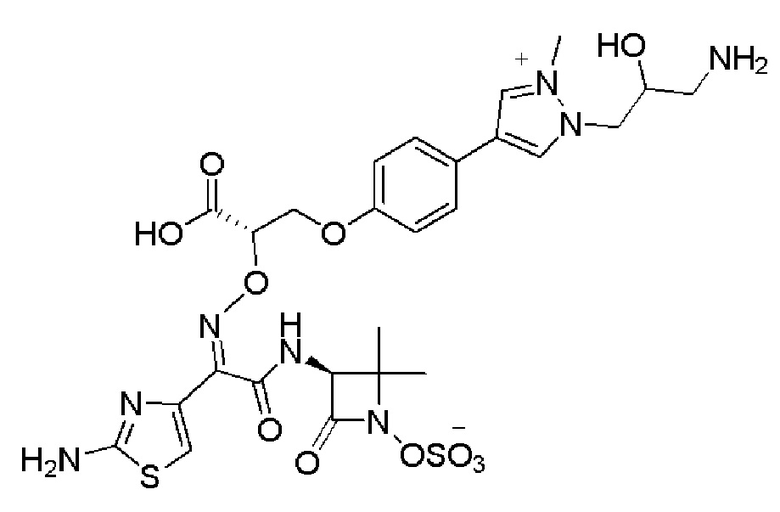
Стадия A: трет-бутил-2,2-диметил-5-((4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)метил)оксазолидин-3-карбоксилат
Карбонат цезия (3,3 г, 10,2 ммоль) добавляли при комнатной температуре к смеси 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (1,3 г, 6,8 ммоль) и трет-бутил 5-(бромметил)-2,2-диметилоксазолидин-3-карбоксилата (2 г, 6,8 ммоль) в DMF (10 мл). Реакционную смесь перемешивали при 60°С в течение 5 часов, затем охлаждали до комнатной температуры и фильтровали. Фильтрат разбавляли EtOAc и водой. Водный слой экстрагировали с помощью EtOAc. Объединенные органические слои промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью EtOAc/изогексан (10-80%), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 408,33.
Стадия B: трет-бутил 5-((4-(4-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1H-пиразол-1-ил)метил)-2,2-диметилоксазолидин-3-карбоксилат
Указанное в заголовке соединение получали в соответствии с процедурой, описанной на стадиях A-C примера 38.
LC-MS [М+Н]+: m/z 663,45.
Стадия C: 4-(4-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1-((3-(трет-бутоксикарбонил)-2,2-диметилоксазолидин-5-ил)метил)-2-метил-1H-пиразол-2-ий трифторметансульфонат
Указанное в заголовке соединение получали в соответствии с процедурой, описанной на стадии D примера 40.
LC-МС [M]+: m/z 677,47.
Стадия D: 1-(3-амино-2-гидроксипропил)-4-(4-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-2-метил-1H-пиразол-2-ий трифторметансульфонат
DL-10-камфорсульфоновую кислоту (0,028 г, 0,122 ммоль) добавляли к перемешиваемой при комнатной температуре смеси 4-(4-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-фенил)-1-((3-(трет-бутоксикарбонил)-2,2-диметилоксазолидин-5-ил)метил)-2-метил-1H-пиразол-2-ий трифторметансульфоната (1,0 г, 1,2 ммоль) в МеОН. Реакционную смесь перемешивали при комнатной температуре в течение ночи, а затем концентрировали, получая указанное в заголовке соединение.
LC-MS [М]+: m/z 537,28.
Стадия E: 4-(4-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)-2-гидроксипропил)-2-метил-1H-пиразол-2-ий 2,2,2-трифторацетат
К перемешиваемой при комнатной температуре смеси 1-(3-амино-2-гидроксипропил)-4-(4-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-2-метил-1H-пиразол-2-ий трифторметансульфоната (830 мг, 1,2 ммоль) и BOC-ангидрида (225 мкл, 0,97 ммоль) в CHCl3 добавляли Et3N (170 мкл, 1,2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали. Полученный остаток очищали с помощью хроматографии на колонке с силикагелем 40 г, элюируя смесью CH2Cl2/MeOH (100-90%), с получением смеси. Смесь растворяли в смеси CH3CN/вода, и очищали с помощью препаративной HPLC с обращенной фазой (С-18), элюируя смесью ацетонитрил/вода+0,1% TFA (5-80-100%), получая указанное в заголовке соединение.
LC-MS [М]+: m/z 637,33.
Стадия F: (3S)-3-((Z)-2-(((1S)-2-(4-(1-(3-амино-2-гидроксипропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
Указанное в заголовке соединение получали в соответствии с процедурой, описанной на стадии E примера 38.
LC-MS [М+Н]+: m/z 691,24.
ПРИМЕР 43
(S)-3-((Z)-2-(((S)-2-(4-(1-((S)-3-амино-2-гидроксипропил)-2-метил-1H-пиразол-2-ий-4-ил)-3-фторфенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат объединенный с муравьиной кислотой (1:1)
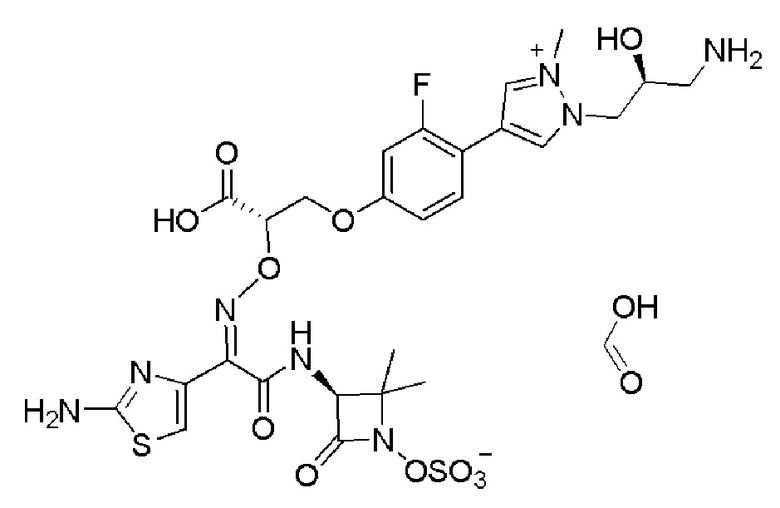
Стадия A: (R)-бензгидрил 3-(4-бром-3-фторфенокси)-2-гидроксипропаноат
К раствору (R)-3-(4-бром-3-фторфенокси)-2-гидроксипропановой кислоты (6 г, 22 ммоль) и Et3N (4,5 мл, 32 ммоль) в ацетонитриле (40 мл) при комнатной температуре в течение 5 минут добавляли по каплям (бромметилен)дибензол (6,9 г, 28 ммоль) в ацетонитриле (20 мл). Реакционную смесь нагревали при 80°С в течение 4 часов, и затем концентрировали. Полученный остаток распределяли между Н2O/DCM (75 мл/75 мл), и водный слой экстрагировали DCM (2×75 мл). Объединенные органические слои промывали насыщенным солевым раствором (200 мл), сушили над MgSO4, фильтровали и концентрировали. Полученный остаток очищали с помощью ISCO (80 г, 0-30% EtOAc/гексан), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 468,91.
Стадия B: бензгидрил (R)-3-(3-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)-2-гидроксипропаноат
Ацетат калия (3,0 г, 31 ммоль) добавляли к перемешиваемой при комнатной температуре смеси (R)-бензгидрил-3-(4-бром-3-фторфенокси)-2-гидроксипропаноата (4,6 г, 9,4 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (3,2 г, 12 ммоль) и ди-трет-BuDPPF-PdCl2 (0,34 г, 0,52 ммоль) в диоксане. Реакционную смесь дегазировали два раза азотом и перемешивали при 70°С в течение 4 часов. Затем реакционную смесь фильтровали, и фильтрат концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (40 г), элюируя смесью EtOAc/изогексан (10-50%), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 493,35.
Стадия C: бензгидрил-(2R)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)-2-((трет-бутилдиметил-силил)окси)пропил)-1H-пиразол-4-ил)-3-фторфенокси)-2-гидроксипропаноат
Водный раствор фосфата калия (2,6 мл, 2,6 ммоль) добавляли при комнатной температуре к смеси (R)-бензгидрил-3-(3-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)-2-гидроксипропаноата (480 мг, 0,97 ммоль), (S)-трет-бутил-(3-(4-бром-1H-пиразол-1-ил)-2-((трет-бутилдиметилсилил)окси)пропил)карбамата (380 мг, 0,88 ммоль) и ди-трет-BuDPPF-PdCl2 (34 мг, 0,053 ммоль) в диоксане. Реакционную смесь дегазировали азотом и перемешивали при 65°С в течение 1,5 часов. Затем реакционную смесь охлаждали до комнатной температуры и экстрагировали EtOAc. Органический слой промывали насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью EtOAc/изогексан (5-80%), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 720,50.
Стадия D: (Z)-2-((((2S)-1-(бензгидрилокси)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)-2-((трет-бутилдиметилсилил)окси)пропил)-1H-пиразол-4-ил)-3-фторфенокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил) уксусная кислота
DEAD (0,12 мл, 0,78 ммоль) добавляли к охлажденной до 0°C перемешиваемой смеси бензгидрил-(2R)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)-2-((трет-бутилдиметилсилил)окси)пропил)-1H-пиразол-4-ил)-3-фторфенокси)-2-гидроксипропаноата (0,51 г, 0,71 ммоль), 2-гидроксиизоиндолин-1,3-диона (116 мг, 0,71 ммоль) и трифенилфосфина (200 мг, 0,78 ммоль) в THF. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, а затем концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (40 г), элюируя смесью гексан/EtOAc (100-60%), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 865,56.
Стадия E: (Z)-2-((((S)-1-(бензгидрилокси)-3-(4-(1-((S)-3-((трет-бутоксикарбонил)амино)-2-((трет-бутилдиметилсилил)окси)пропил)-2-метил-1H-2l4-пиразол-4-ил)-3-фторфенокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)уксусная кислота, трифторметансульфонатная соль
Указанное в заголовке соединение получали в соответствии с процедурой, описанной на стадии D примера 40.
LC-MS [M]+: m/z 1003,60.
Стадия F: 4-(4-((S)-3-(бензгидрилокси)-2-((((Z)-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)-2-фторфенил)-1-(3-((трет-бутоксикарбонил)амино)-2-гидроксипропил)-2-метил-1H-пиразол-2-ий
TBAF (0,28 г, 1,06 ммоль) добавляли к охлажденной до 0°C перемешиваемой смеси трифторметансульфонатной соли (Z)-2-((((S)-1-(бензгидрилокси)-3-(4-(1-((S)-3-((трет-бутоксикарбонил)амино)-2-((трет-бутилдиметил-силил)окси)пропил)-2-метил-1H-2l4-пиразол-4-ил)-3-фторфенокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)уксусной кислоты (0,80 г, 0,53 ммоль) в THF. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, а затем разбавляли EtOAc. Органический слой промывали водой и насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали. Полученный остаток очищали с помощью препаративной HPLC с обращенной фазой (с-18), элюируя смесью ацетонитрил/вода+0,1% TFA, получая указанное в заголовке соединение.
LC-MS [M]+: m/z 889,45.
Стадия G: 4-(4-((S)-3-(бензгидрилокси)-2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)-амино)окси)-3-оксопропокси)-2-фторфенил)-1-((S)-3-((трет-бутоксикарбонил)амино)-2-гидроксипропил)-2-метил-1H-пиразол-2-ий
К смеси 4-(4-((S)-3-(бензгидрилокси)-2-((((Z)-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)-2-фторфенил)-1-(3-((трет-бутоксикарбонил)амино)-2-гидроксипропил)-2-метил-1H-пиразол-2-ия (220 мг, 0,24 ммоль) и (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата (102 мг, 0,48 ммоль) в ацетонитриле (12 мл) в атмосфере азота при -10°C добавляли пиридин (0,059 мл, 0,73 ммоль). Затем добавляли N1-((этилимино)метилен)-N3,N3-диметилпропан-1,3-диамин гидрохлорид (102 мг, 0,53 ммоль). Реакционную смесь перемешивали при -10-0°С в течение 1 часа, и затем концентрировали. Полученный остаток очищали с помощью обращенно-фазовой препаративной хроматографии (С-18), элюируя смесью ацетонитрил/вода+0,1% TFA, получая указанное в заголовке соединение.
Стадия H: ((3S)-3-((Z)-2-(((1S)-2-(4-(1-(3-амино-2-гидроксипропил)-2-метил-1H-пиразол-2-ий-4-ил)-3-фторфенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат объединенный с муравьиной кислотой (1:1)
TFA (2 мл) добавляли при комнатной температуре к перемешиваемой смеси 4-(4-((S)-3-(бензгидрилокси)-2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2-фторфенил)-1-((S)-3-((трет-бутоксикарбонил)амино)-2-гидроксипропил)-2-метил-1H-пиразол-2-ия (240 мг, 0,22 ммоль) в DCM. Реакционную смесь перемешивали при комнатной температуре в течение 45 минут, затем концентрировали и дважды обрабатывали эфиром. Полученный остаток растворяли в DMSO (3 мл) и эфире (2 мл), и концентрировали под вакуумом в течение 5 минут. Полученный раствор разбавляли DMSO (4 мл) и очищали обращенно-фазовой хроматографией, получая указанное в заголовке соединение в виде отдельного изомера.
LC-MS [М+Н]+: m/z 715,30.
ПРИМЕР 44
(3S)-3-((Z)-2-(((1S)-2-(4-(1-(3-амино-2-гидроксипропил)-2-метил-1H-пиразол-2-ий-4-ил)-3-фторфенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат объединенный с муравьиной кислотой (1:1)
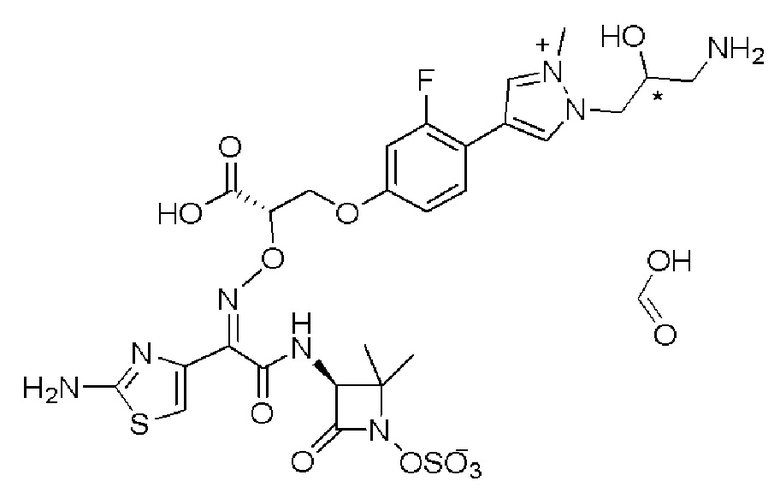
Указанное в заголовке соединение получали в соответствии с процедурой, описанной в примере 43, используя соответствующие исходные материалы.
LC-MS [М+Н]+: m/z 715,28.
ПРИМЕР 45 и ПРИМЕР 46
(S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-1-карбокси-2-(4-(1-((R)-2,3-диаминопропил)-2-метил-1H-пиразол-2-ий-4-ил)-3-фторфенокси)этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат (диастереомерные по амину)
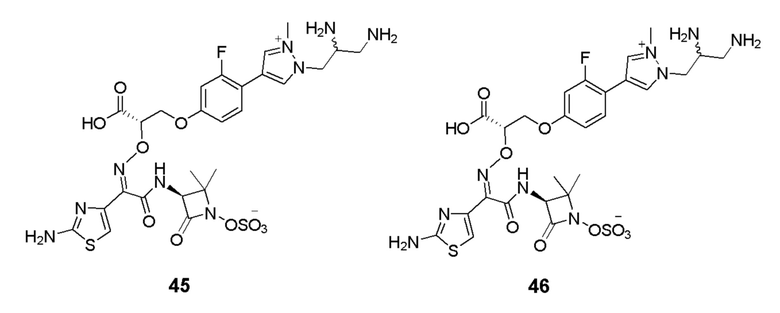
Стадия A: трет-бутил-(2-((трет-бутилдиметилсилил)окси)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропил)карбамат
Ацетат калия (3,48 г, 36 ммоль) добавляли к перемешиваемой при комнатной температуре смеси 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (3,6 г, 14 ммоль), трет-бутил-(3-(4-бром-1H-пиразол-1-ил)-2-((трет-бутилдиметилсилил)окси)пропил)карбамата (5,1 г, 12 ммоль) и ди-трет-BuDPPF PdCl2 (0,38 г, 0,591 ммоль) в диоксане. Реакционную смесь дегазировали 2 раза азотом и перемешивали при 70°С в течение 4 часов. Затем реакционную смесь фильтровали, и фильтрат концентрировали. Полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле (120 г), элюируя смесью EtOAc/изогексан (0-50%), получая указанное в заголовке соединение.
LC-MS [M+H].+: m/z 482,49.
Стадия B: трет-бутил-(2-гидрокси-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропил)карбамат
TBAF (0,61 г, 2,3 ммоль) добавляли к перемешиваемой охлажденной до 0°C смеси трет-бутил-(2-((трет-бутилдиметилсилил)окси)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропил)карбамата (0,93 г, 1,9 ммоль) в THF. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью EtOAc/изогексан (5-80%), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 368,27.
Стадия C: 1-((трет-бутоксикарбонил)амино)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропан-2-ил метансульфонат
К перемешиваемому раствору трет-бутил-(2-гидрокси-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропил)карбамата (480 мг, 1,30 ммоль) и Et3N (0,24 мл, 1,7 ммоль) в дихлорметане (5 мл) при 0°C в атмосфере N2 добавляли одной порцией MsCl (0,104 мл, 1,33 ммоль). Реакционную смесь перемешивали при 0°С в течение 1 часа, затем концентрировали в вакууме и распределяли между водой (50 мл) и этилацетатом (40 мл). Водный слой отделяли и экстрагировали этилацетатом (2×40 мл). Объединенные органические слои промывали солевым раствором (40 мл), сушили (сульфат магния) и концентрировали в вакууме, получая указанное в заголовке соединение.
Стадия D: трет-бутил-(2-азидо-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропил)карбамат
К перемешиваемому раствору 1-((трет-бутоксикарбонил)амино)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропан-2-ил метансульфоната (590 мг, 1,33 ммоль)) в диметилформамиде (5 мл) добавляли одной порцией азид натрия (173 мг, 2,7 ммоль). Реакционную смесь нагревали до 80°С и перемешивали в течение 18 часов, а затем охлаждали до комнатной температуры. Реакционную смесь выливали в воду (20 мл), и экстрагировали этилацетатом (2 раза по 20 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили (сульфат магния) и концентрировали в вакууме, получая указанное в заголовке соединение.
LC-MS [М+Н]+: 393,38.
Стадия E: трет-бутил-(2-амино-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропил)карбамат
К смеси трет-бутил-(2-азидо-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропил)карбамата (525 мг, 1,34 ммоль) в MeOH при комнатной температуре добавляли Pd-C (280 мг, 0,27 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода в течение ночи. Затем смесь фильтровали, и фильтрат концентрировали досуха, получая указанное в заголовке соединение.
LC-MS [М+Н]+: 367,24.
Стадия F: ди-трет-бутил-(3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропан-1,2-диил)дикарбамат
ВОС-ангидрид (0,34 мл, 1,5 ммоль) добавляли к перемешиваемой при комнатной температуре смеси трет-бутил-(2-амино-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропил)карбамата (570 мг, 1,3 ммоль) в THF. Смесь перемешивали при комнатной температуре в течение 1 часа, а затем концентрировали, получая указанное в заголовке соединение.
LC-MS [М+Н]+: 467,36.
Стадия G: бензгидрил (2R)-3-(4-(1-(2,3-бис((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)-3-фторфенокси)-2-гидроксипропаноат
Водный раствор фосфата калия (4,0 мл, 4,0 ммоль) добавляли при комнатной температуре к смеси (R)-бензгидрил-3-(4-бром-3-фторфенокси)-2-гидроксипропаноат (590 мг, 1,3 ммоль), ди-трет-бутил-(3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропан-1,2-диил)дикарбамата (630 мг, 1,3 ммоль) и ди-трет-BuDPPF PdCl2 (52 мг, 0,079 ммоль) в диоксане. Реакционную смесь дегазировали азотом и перемешивали при 65°С в течение 1,5 часов, затем охлаждали до комнатной температуры и экстрагировали EtOAc. Органический слой промывали насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали. Полученный остаток очищали с помощью колоночной хроматографии, элюируя смесью CH2Cl2/MeOH (100-90%), получая указанное в заголовке соединение в виде диастереомерной смеси.
LC-MS [М+Н]+: 705,45.
Диастереомеры разделяли, используя колонку OJ-H в условиях SFC, получая указанные в заголовке соединения. Соединение примера 45 получали быстрым элюированием изомера в соответствии с процедурой стадии B-H примера 1.
LC-MS [М+Н]+: m/z 714,20.
Соединение примера 46 получали более медленным элюированием изомера в соответствии с процедурой стадии B-H примера 1.
LC-MS [М+Н]+: m/z 714,36.
ПРИМЕР 47
(S)-3-((Z)-2-(((S)-2-(4-(3-амино-1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)-3-фторфенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
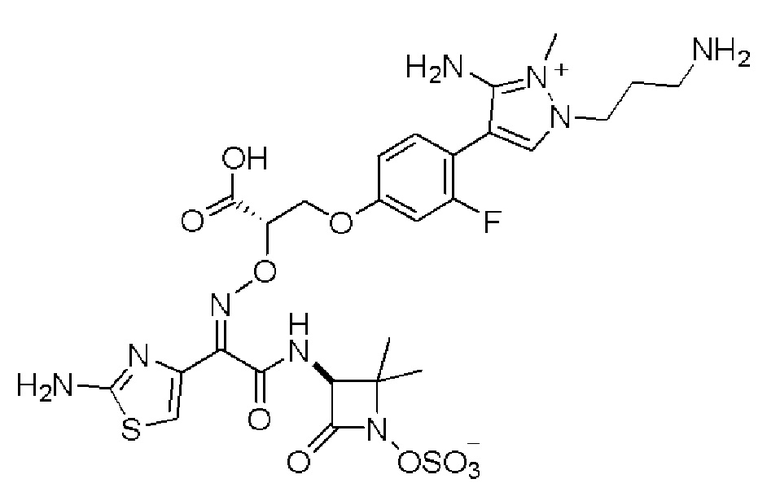
Стадия A: трет-бутил-(R)-3-(4-(3-амино-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)-3-фторфенокси)-2-((трет-бутилдиметилсилил)окси)пропаноат
К раствору во флаконе для СВЧ (R)-трет-бутил-2-((трет-бутилдиметилсилил)окси)-3-(3-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)пропаноата (2,6 г, 5,24 ммоль), трет-бутил-(3-(3-амино-4-бром-1H-пиразол-1-ил)пропил)карбамата (2,0 г, 6,3 ммоль) и дихлорида 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладия (0,34 г, 0,52 ммоль) в THF (15 мл) во флаконе для СВЧ добавляли 1 М водный раствор фосфата калия (7,8 мл, 7,8 ммоль). Флакон для СВЧ герметизировали, три раза дегазировали и заполняли азотом. Реакционную смесь перемешивали при 70°С в течение 2 часов, затем разбавляли насыщенным раствором NH4Cl и экстрагировали EtOAc. Органические слои объединяли, сушили над MgSO4 и концентрировали. Полученный остаток очищали флэш-хроматографией на колонке с SiO2 (80 г gold), элюируя смесью EtOAc/гексан (0-100% 3 cv, 100% 6 cv), получая указанное в заголовке соединение.
LC-МС [M+1]: m/z 609,00.
Стадия B: трет-бутил-(R)-3-(4-(3-(((аллилокси)карбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)-3-фторфенокси)-2-((трет-бутилдиметилсилил)окси)пропаноат
К раствору (R)-трет-бутил-3-(4-(3-амино-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)-3-фторфенокси)-2-((трет-бутилдиметилсилил)окси)пропаноата (2,3 г, 3,8 ммоль) в CH2Cl2 (100 мл) добавляли DIPEA (1,3 мл, 7,6 ммоль), с последующим добавлением аллилкарбонхлоридата (0,80 мл, 7,6 ммоль). Смесь перемешивали при комнатной температуре в течение 4 часов, после чего растворитель удаляли. Полученный остаток очищали с помощью колоночной хроматографией на силикагеле (80 г gold), элюируя смесью EtOAc/гексан (0-50%, 6 cv; 50%, 10 cv), получая указанное в заголовке соединение.
LC-MS [М+1]: m/z 693,48.
Стадия C: трет-бутил-(R)-3-(4-(3-(((аллилокси)карбонил)амино)-1-(3-((трет-бутоксикарбонил)-амино)пропил)-1H-пиразол-4-ил)-3-фторфенокси)-2-гидроксипропаноат
К раствору (R)-трет-бутил-3-(4-(3-(((аллилокси)карбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)-пропил)-1H-пиразол-4-ил)-3-фторфенокси)-2-((трет-бутилдиметилсилил)окси)пропаноата (2,6 г, 3,8 ммоль) в THF (30 мл) добавляли раствор TBAF в THF (3,75 мл, 3,75 ммоль, 1 М). Реакционную смесь перемешивали в течение 1 часа при комнатной температуре, после чего растворитель удаляли. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (40 г gold), элюируя смесью EtOAc/гексан (0-80%, 6 cv; 80%, 10 cv), получая указанное в заголовке соединение.
LC-MS [М+1]: m/z 580,30.
Стадия D: трет-бутил-(S)-3-(4-(3-(((аллилокси)карбонил)амино)-1-(3-((трет-бутоксикарбонил) амино)пропил)-1H-пиразол-4-ил)-3-фторфенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-пропаноат
К раствору (R)-трет-бутил-3-(4-(3-(((аллилокси)карбонил)амино)-1-(3-((трет-бутоксикарбонил) амино)пропил)-1H-пиразол-4-ил)-3-фторфенокси)-2-гидроксипропаноата (1,13 г, 2,0 ммоль) в THF (5 мл) при комнатной температуре добавляли 2-гидроксиизоиндолин-1,3-дион (0,38 г, 2,3 ммоль) и трифенилфосфин (0,62 г, 2,3 ммоль), с последующим добавлением DIAD (0,46 мл, 2,3 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов, после чего растворитель удаляли. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (колонка Redi 40 г gold), элюируя смесью EtOAc/гексан (0-70%, 6 cv; 70%, 10 cv), получая указанное в заголовке соединение.
LC-MS [М+1]: m/z 725,25.
Стадия E: (S)-3-(((аллилокси)карбонил)амино)-4-(4-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)-2-фторфенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий трифлат
К раствору (S)-трет-бутил-3-(4-(3-(((аллилокси)карбонил)амино)-1-(3-((трет-бутоксикарбонил) амино)пропил)-1H-пиразол-4-ил)-3-фторфенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноата (0,7 г, 0,97 ммоль) в ацетонитриле (12 мл) при 0°C добавляли метилтрифторметансульфонат (0,11 мл, 0,97 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем добавляли раствор аммиака в метаноле (1,4 мл, 9,7 ммоль, 7 M). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем растворитель удаляли в вакууме, получая указанное в заголовке соединение.
LC-MS [М+1]: m/z 608,44.
Стадия F: (S,Z)-3-(((Аллилокси)карбонил)амино)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутокси-карбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)-2-фтор-фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий трифлат
Раствор 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусной кислоты (0,16 г, 0,60 ммоль) и (S)-3-(((аллилокси)карбонил)амино)-4-(4-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)-2-фторфенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий трифлата (0,38 г, 0,50 ммоль) в этаноле (1 мл) и CH2ClCH2Cl (0,5 мл) перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь концентрировали, получая указанное в заголовке соединение.
LC-MS [М+1]: m/z 862,39.
Стадия G: (S)-3-((Z)-2-((((S)-3-(4-(3-(((аллилокси)карбонил)амино)-1-(3-((трет-бутокси-карбонил)амино)пропил)-2-метил-1H-пиразол-2-ий-4-ил)-3-фторфенокси)-1-(трет-бутокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
К раствору (S,Z)-3-(((аллилокси)карбонил)амино)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)(карбокси)-метилен)амино)окси)-3-оксопропокси)-2-фторфенил)-1-(3-((трет-бутоксикарбонил)-амино)пропил)-2-метил-1H-пиразол-2-ий трифлата (0,49 г, 0,50 ммоль) в DMF (5 мл) добавляли DCC (0,31 г, 1,50 ммоль) и HOBt (0,23 г, 1,50 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 30 минут, затем добавляли (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфат (0,26 г, 1,25 ммоль) и бикарбонат натрия (0,25 г, 3,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Полученный твердый продукт отфильтровывали. Фильтрат очищали на колонке RP С-18 с силикагелем, элюируя смесью ACN/вода с 0,05% TFA, (20-100% 8 cv, 100% 6 cv), получая указанное в заголовке соединение.
LC-MS [М+1]: m/z 1054,77.
Стадия H: (S)-3-((Z)-2-((((S)-3-(4-(3-амино-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий-4-ил)-3-фторфенокси)-1-(трет-бутокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат, трифторацетат
К раствору (S)-3-((Z)-2-((((S)-3-(4-(3-(((аллилокси)карбонил)амино)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий-4-ил)-3-фторфенокси)-1-(трет-бутокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата TFA (0,5 г, 0,43 ммоль) в DMF (4 мл) при 0°С добавляли тетракис (0,049 г, 0,043 ммоль) и фенилсилан (0,53 мл, 4,3 ммоль). Реакционную смесь перемешивали в течение 10 минут, а затем фильтровали. Фильтрат очищали на RP HPLC (Gilson, колонка С-18), элюируя смесью ACN/воды, содержащей 0,05% трифторуксусной кислоты (TFA) (20-100%, 8 мин; 100% 10 мин), получая указанное в заголовке соединение.
LC-MS [М+1]: m/z 970,48.
Стадия I: (S)-3-((Z)-2-(((S)-2-(4-(3-амино-1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)-3-фторфенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
К раствору (S)-3-((Z)-2-((((S)-3-(4-(3-амино-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий-4-ил)-3-фторфенокси)-1-(трет-бутокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата TFA (170 мг, 0,16 ммоль) в CH2Cl2 (1 мл) добавляли TFA (2 мл, 26 ммоль). Раствор перемешивали при комнатной температуре в течение 40 минут, затем растворитель удаляли. Полученный остаток промывали три раза Et2O, и сушили в вакууме. Полученный твердый продукт растворяли в DMSO, и очищали с помощью RP-HPLC (Gilson, колонка С-18), элюируя смесью ACN/вода с 0,05% TFA (0-40%, 12 мин), получая указанное в заголовке соединение в виде соли трифторуксусной кислоты.
LC-MS [M+1]: m/z 712,30.
1H-ЯМР (500 МГц, D2O) δH 7,96 (1H, с), 7,31 (1H, т, J=8,6 Гц), 7,09 (1H, с), 6,85 (2H, д, J=10,8 Гц), 5,14 (1H, с), 4,51 (2H, т, J=14,7 Гц), 4,32 (2H, т, J=7,2 Гц), 3,74 (3H, с), 3,02 (2H, т, J=7,7 Гц), 2,66 (1H, с), 2,15 (2H, т, J=8,0 Гц), 1,42 (3H, с), 1,08 (3H, с).
ПРИМЕР 48
(S)-3-((Z)-2-(((S)-2-(4-(3-амино-1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)-3-фторфенокси)-1-карбоксиэтокси)имино)-2-(2-амино-5-хлортиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
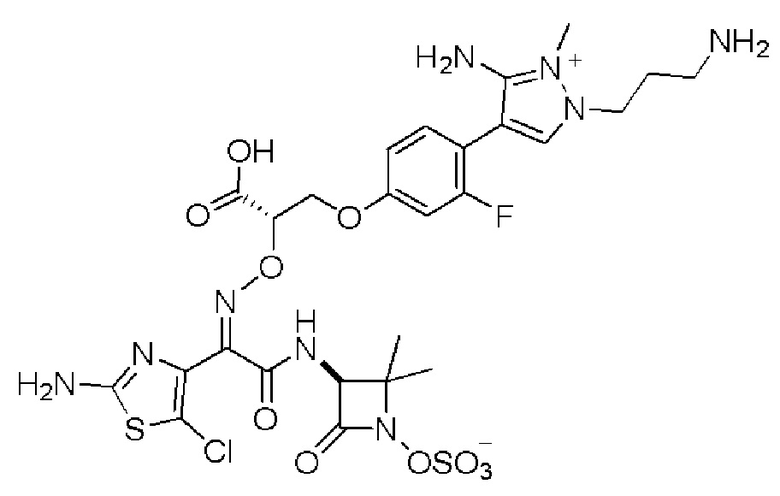
Указанное в заголовке соединение получали с использованием процедуры примера 47 с использованием на стадии F 2-(2-((трет-бутоксикарбонил)амино)-5-хлортиазол-4-ил)-2-оксоуксусной кислоты.
LC-MS [М+1]: m/z 746,26.
1H ЯМР (D2O, 500 МГц): δH 7,97 (1H, с), 7,31 (1H, т, J=8,7 Гц), 6,84-6,87 (2H, м), 5,15 (1H, д, J=5,0 Гц), 4,44-4,53 (2H, м), 4,32 (2H, т, J=7,2 Гц), 3,75 (3H, с), 3,02 (2H, т, J=7,8 Гц), 2,15 (2H, т, J=7,9 Гц), 1,42 (3H, с), 1,07 (3H, с).
ПРИМЕР 49
(S)-3-((Z)-2-(((S)-2-(4-(3-амино-1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-амино-5-хлортиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
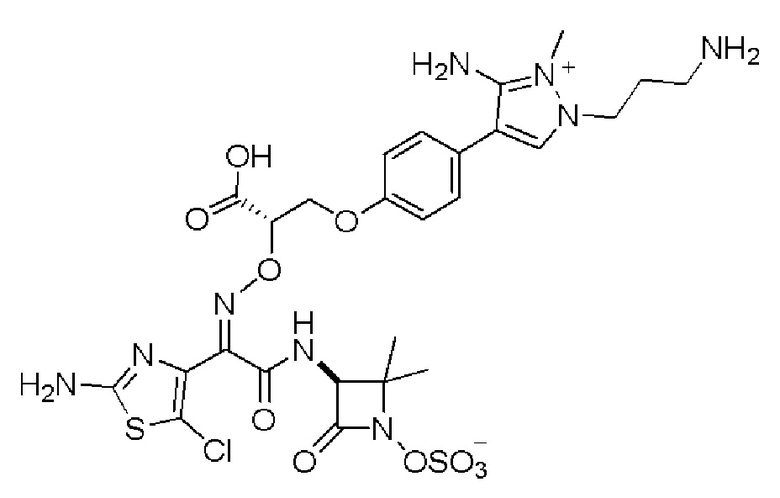
Указанное в заголовке соединение получали процедурой, описанной в примере 47.
LC-MS [М+1]: m/z 728,36.
1H ЯМР (D2O, 500 МГц): δH 7,93 (1H, с), 7,34 (2H, д, J=8,2 Гц), 7,02 (2H, д, J=8,2 Гц), 5,15 (1H, с), 4,29 (2H, т, J=7,2 Гц), 3,73 (3H, с), 3,02 (2H, т, J=7,7 Гц), 2,14 (2H, т, J=8,0 Гц), 1,40 (3H, с), 1,04 (3H, с).
ПРИМЕР 50
(S)-3-((Z)-2-(((S)-2-(4-(5-амино-1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
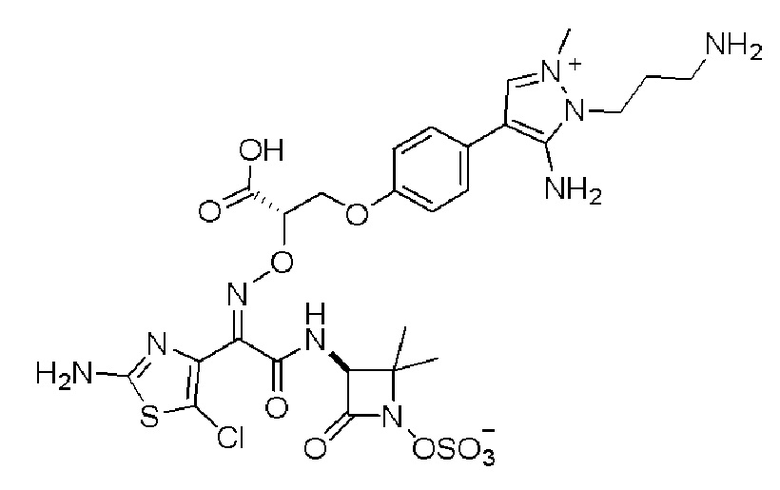
Указанное в заголовке соединение получали процедурой, описанной в примере 47.
LC-MS [М+1]: m/z 696,21.
1H ЯМР (D2O, 500 МГц): δH 7,88 (1H, с), 7,34 (2H, д, J=8,4 Гц), 6,97-7,04 (4H, м), 4,96 (1H, д, J=5,6 Гц), 4,42-4,47 (3H, м), 4,31 (2H, т, J=7,6 Гц), 3,81-3,83 (4H, м), 3,08 (2H, т, J=7,9 Гц), 2,67 (1H, с), 2,15 (2H, т, J=8,1 Гц), 1,42 (3H, с), 1,09 (3H, с).
ПРИМЕР 51A и 51B
(S)-3-((Z)-2-(2-амино-5-хлортиазол-4-ил)-2-((((R)-1-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-2-карбоксипропан-2-ил)окси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат (51A)
(S)-3-((Z)-2-(2-амино-5-хлортиазол-4-ил)-2-((((R)-1-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-2-карбоксипропан-2-ил)окси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат (51B)
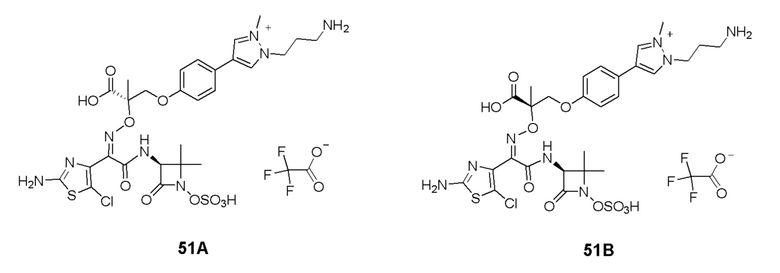
Указанные в заголовке соединения получали с использованием процедуры, описанной в примерах 23А и 23B.
Пример 51А: LC-MS [М+1]: m/z 727,36.
1H-ЯМР (500 МГц, D2O) δH 8,49 (1H, с), 8,43 (1H, с), 7,49 (2H, д), 7,02 (2H, с), 4,57-4,53 (3H, м), 4,48 (1H, д), 4,40 (1H, д), 4,11 (3H, с), 3,12(2H, т), 2,32 (2H, м), 1,60 (3H, с), 1,38 (3H, с), 1,17 (3H, с).
Пример 51B: LC-MS [M+1]: m/z 727,36.
1H-ЯМР (500 МГц, D2O) δH 8,50 (1H, с), 8,44 (1H, с), 7,50 (2H, д), 7,04 (2H, с), 4,59-4,53 (4H, м), 4,37 (1H, д), 4,11 (3H, с), 3,12(2H, т), 2,32 (2H, м), 1,61 (3H, с), 1,39 (3H, с), 1,13 (3H, с).
ПРИМЕР 52
(S)-3-((Z)-2-(((S)-2-(4-(1-((S)-3-амино-2-гидроксипропил)-2-(азетидин-3-илметил)-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
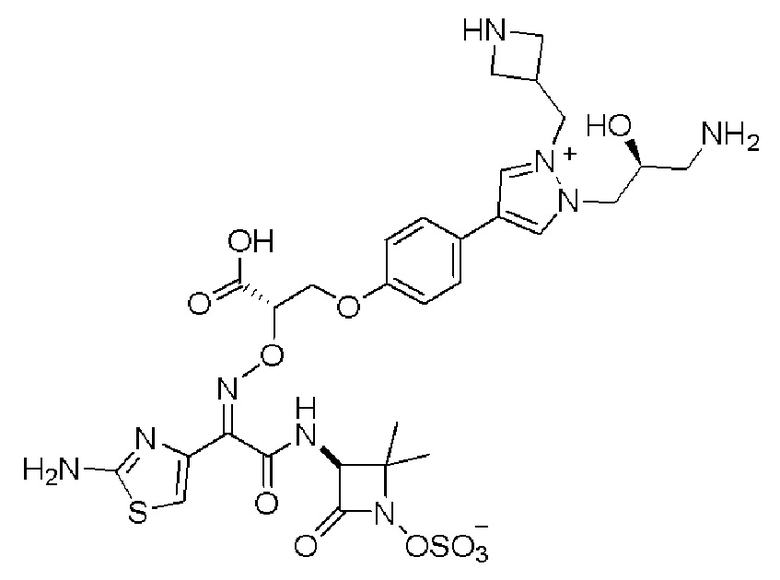
Стадия A: трет-бутил-(R)-3-(4-(1-((S)-3-((трет-бутоксикарбонил)амино)-2-((трет-бутил-диметилсилил) окси)пропил)-1H-пиразол-4-ил)фенокси)-2-((трет-бутилдиметилсилил)-окси)пропаноат
Смесь трет-бутил-(3-(4-бром-1H-пиразол-1-ил)-2-((трет-бутилдиметилсилил)окси) пропил)карбамата (0,53 г, 1,22 ммоль), трет-бутил- 2-гидрокси-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)пропаноата (1 г, 2,8 ммоль), дихлорида 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладия (0,18 г, 0,28 ммоль) и фосфата калия (4,12 мл, 8,24 ммоль, 2 М водный раствор) в THF (15 мл) дегазировали и три раза продували азотом. Реакционную смесь нагревали при 60°С в течение 2 часов, затем разбавляли насыщенным раствором NH4Cl и экстрагировали EtOAc. Органические слои объединяли, сушили над MgSO4 и концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (Redi, 80 г gold), элюируя смесью EtOAc/гексан (0-35%, 6 cv; 35%, 10 cv), получая указанное в заголовке соединение.
LC-MS [M+1]: m/z 592,34.
Стадия B: трет-бутил-(S)-3-(4-(1-((S)-3-((трет-бутоксикарбонил)амино)-2-((трет-бутилдиметил-силил) окси)пропил)-1H-пиразол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноат
К раствору (2R)-трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)-2-((трет-бутил-диметилсилил)окси)пропил)-1H-пиразол-4-ил)фенокси)-2-гидроксипропаноата (1,12 г, 1,9 ммоль) в THF (10 мл) при комнатной температуре добавляли 2-гидроксиизоиндолин-1,3-дион (0,37 г, 2,3 ммоль), трифенилфосфин (0,60 г, 2,3 ммоль), с последующим добавлением DIAD (0,44 мл, 2,3 ммоль). Реакционную смесь перемешивали в течение ночи, затем растворитель удаляли. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (Redi, 80 г gold), элюируя смесью EtOAc/гексан (0-50%, 5 cv, 50% 10 cv), получая указанное в заголовке соединение.
LC-MS [М+1]: m/z 737,41.
Стадия C: 4-(4-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1-((S)-3-((трет-бутоксикарбонил)амино)-2-((трет-бутилдиметилсилил)окси)пропил)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1H-пиразол-2-ий трифторметансульфонат
Раствор трет-бутил-3-(гидроксиметил)азетидин-1-карбоксилата (510 мг, 2,7 ммоль) в CH2Cl2 (5 мл) охлаждали до -78°С, и добавляли трифторметансульфоновый ангидрид (0,56 мл, 3,4 ммоль) и основание Хюнига (0,95 мл, 5,4 ммоль). Реакционную смесь перемешивали при -78°С в течение 20 минут, затем гасили водой. Реакционную смесь нагревали до комнатной температуры и распределяли между EtOAc и водой. Органический слой отделяли, промывали насыщенным раствором NaHCO3 и насыщенным раствором соли, сушили над Na2SO4, и концентрировали в вакууме, получая промежуточное соединение. В колбу, содержащую промежуточное соединение, добавляли раствор трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)-2-((трет-бутилдиметилсилил)окси)пропил)-1H-пиразол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноата (500 мг, 0,68 ммоль) и бикарбоната натрия (570 мг, 6,8 ммоль) в безводном CH3CN (10 мл). Реакционную смесь нагревали при 60°С в течение 1 часа, после чего растворитель удаляли. Полученный остаток растирали с Et2O (2×15 мл), а затем очищали с помощью колоночной хроматографии на силикагеле (Redi, 24 г gold), элюируя смесью MeOH/DCM (0-15%, 8 cv; 15%, 8 cv), получая указанное в заголовке соединение.
LC-MS [М+1]: m/z 906,9.
Стадия D: 4-(4-((S)-2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)фенил)-1-((S)-3-((трет-бутоксикарбонил)амино)-2-((трет-бутилдиметилсилил)окси)пропил)-2-((1-(трет-бутокси-карбонил)азетидин-3-ил)метил)-1H-пиразол-2-ий трифторметансульфонат
К 4-(4-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1-((S)-3-((трет-бутоксикарбонил)амино)-2-((трет-бутилдиметилсилил)окси)пропил)-2-((1-(трет-бутоксикарбонил)-азетидин-3-ил)метил)-1H-пиразол-2-ий трифторметансульфонату (0,16 г, 0,18 ммоль) в MeOH (1 мл) добавляли раствор аммиака в метаноле (0,50 мл, 3,5 ммоль, 7 M). Реакционную смесь перемешивали при комнатной температуре в течение 5 часов, затем растворитель удаляли, получая указанное в заголовке соединение.
LC-MS [M+1]: m/z 776,98.
Стадия E: 4-(4-((S)-3-(трет-бутокси)-2-((((Z)-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил) (карбокси)метилен)амино)окси)-3-оксопропокси)фенил)-1-((S)-3-((трет-бутоксикарбонил)-амино)-2-((трет-бутилдиметилсилил)окси)пропил)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1H-пиразол-2-ий трифторметансульфонат
К раствору 4-(4-((S)-2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)фенил)-1-((S)-3-((трет-бутоксикарбонил)амино)-2-((трет-бутилдиметилсилил)окси)пропил)-2-((1-(трет-бутоксикарбонил) азетидин-3-ил)метил)-1H-пиразол-2-ий трифторметансульфоната (500 мг, 0,64 ммоль) в этаноле (4 мл) и ClCH2CH2Cl (2 мл) при комнатной температуре добавляли 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусную кислоту (175 мг, 0,64 ммоль). Реакционную смесь перемешивали в течение ночи, затем растворитель удаляли, получая указанное в заголовке соединение.
LC-MS [М+1]: m/z 1030,51.
Стадия F: 4-(4-((S)-3-(трет-бутокси)-2-((((Z)-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)фенил)-1-((S)-3-((трет-бутоксикарбонил)амино)-2-гидроксипропил)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1H-пиразол-2-ий
К раствору 4-(4-((S)-3-(трет-бутокси)-2-((((Z)-(2-((трет-бутоксикарбонил)амино)-тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)фенил)-1-((S)-3-((трет-бутоксикарбонил)амино)-2-((трет-бутилдиметилсилил)окси)пропил)-2-((1-(трет-бутоксикарбонил)-азетидин-3-ил)метил)-1H-пиразол-2-ий трифторметансульфоната (0,66 г, 0,64 ммоль) в THF (2 мл) добавляли TBAF в THF (1,9 мл, 1,9 ммоль, 1 М). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем растворитель удаляли, получая указанное в заголовке соединение.
LC-MS [M+1]: m/z 917,10.
Стадия G: (S)-3-((Z)-2-((((S)-1-(трет-бутокси)-3-(4-(1-((S)-3-((трет-бутоксикарбонил)амино)-2-гидроксипропил)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1H-пиразол-2-ий-4-ил)фенокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
К раствору 4-(4-((S)-3-(трет-бутокси)-2-((((Z)-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)-окси)-3-оксопропокси)фенил)-1-((S)-3-((трет-бутоксикарбонил)амино)-2-гидроксипропил)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1H-пиразол-2-ий трифторметансульфоната (290 мг, 0,32 ммоль) в DMF (6 мл) добавляли DCC (198 мг, 0,96 ммоль) и HOBt (147 мг, 0,96 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут перед добавлением (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата (168 мг, 0,80 ммоль) и бикарбоната натрия (134 мг, 1,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем фильтровали. Фильтрат очищали с помощью RP-HPLC (Gilson, колонка C-18), элюируя смесью 20-100% ACN/вода с 0,05% TFA (12 мин), получая указанное в заголовке соединение.
LC-MS [М+1]: m/z 1109,52.
Стадия H: (S)-3-((Z)-2-(((S)-2-(4-(1-((S)-3-амино-2-гидроксипропил)-2-(азетидин-3-илметил)-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат TFA
К раствору (S)-3-((Z)-2-((((S)-1-(трет-бутокси)-3-(4-(1-((S)-3-((трет-бутоксикарбонил)амино)-2-гидроксипропил)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1H-пиразол-2-ий-4-ил)фенокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата (140 мг, 0,13 ммоль) в CH2Cl2 (0,5 мл) добавляли TFA (1 мл, 13 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, после чего растворитель удаляли. Полученный остаток промывали Et2O (3 раза), и затем сушили под вакуумом, получая твердое вещество. Полученное твердое вещество растворяли в DMSO и очищали с помощью RP-HPLC (Gilson, колонка С-18), элюируя смесью ACN/вода с 0,05% TFA (0-40%, 12 мин), получая указанное в заголовке соединение.
LC-MS [М+1]: m/z 750,71.
1H-ЯМР (500 МГц, D2O) δH 8,59 (1H, с), 8,53 (1H, с), 7,49 (1H, д), 7,00 (1H, с), 6,97 (2H, д, J=10,8 Гц), 5,05 (1H, д), 4,83 (2H, д), 4,50 (2H, т), 4,41 (1H, м), 4,29-4,18(2H, м), 4,04 (2H, т), 3,61 (1H, м), 3,26 (1H, м), 2,99 (1H, м), 1,32 (3H, с), 0,91 (3H, с).
ПРИМЕР 53
(S)-3-((Z)-2-(((S)-2-(4-(1-(3-аминопропил)-2-(((R)-пирролидин-3-ил)метил)-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
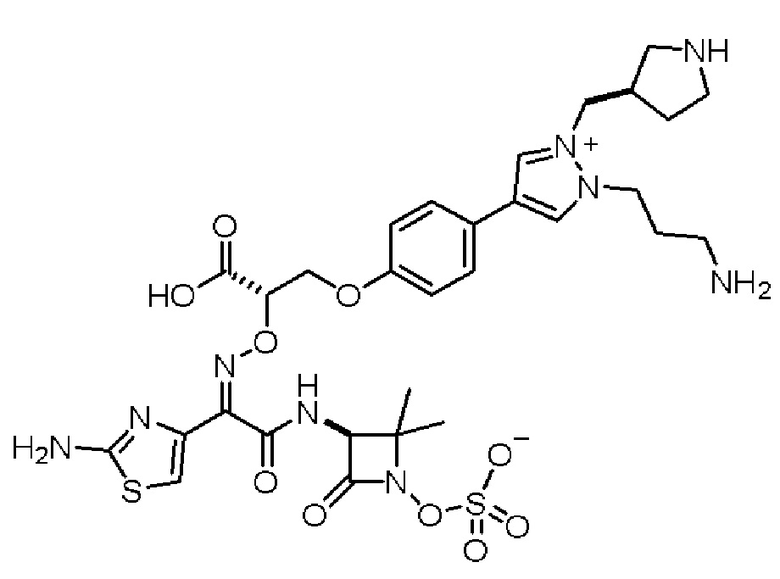
Стадия A: (S)-трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноат
К раствору (R)-трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-2-гидрокси-пропаноата (1,77 г, 3,83 ммоль) в THF (20 мл) добавляли 2-гидроксиизоиндолин-1,3-дион (0,69 г, 4,2 ммоль), трифенилфосфин (1,5 г, 5,8 ммоль) и DEAD (0,91 мл, 5,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, а затем концентрировали. Полученный остаток очищали на силикагеле с использованием смеси EtOAc/гексан в качестве элюирующих растворителей, получая указанное в заголовке соединение.
LC/MS [М+1]+ m/z 607,58.
Стадия B: 4-(4-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-(((R)-1-(трет-бутоксикарбонил)пирролидин-3-ил)метил)-1H-пиразол-2-ий
К раствору (R)-трет-бутил-3-(гидроксиметил)пирролидин-1-карбоксилата (0,60 г, 3,0 ммоль) и основания Хюнига (1,03 мл, 5,9 ммоль) в CH2Cl2 (15 мл) при -78°С в течение 5 минут добавляли трифторметансульфоновый ангидрид (0,61 мл, 3,6 ммоль). Реакционную смесь перемешивали при -78°С в течение 1 часа, а затем гасили насыщенным раствором NaHCO3. Реакционную смесь оставляли нагреваться до 0°C. Органический слой отделяли и промывали насыщенным солевым раствором. Водные слои объединяли, и снова экстрагировали с помощью DCM. Органические слои объединяли, сушили над Na2SO4 и концентрировали в вакууме, с получением (R)-трет-бутил-3-((((трифторметил)сульфонил)окси)метил)пирролидин-1-карбоксилата.
1H-ЯМР(500 МГц, D2O): δ 4,51-4,48 (м, 2H), 3,71-3,57 (м, 2H), 3,21-3,11 (м, 2H), 2,76-2,69 (м, 1H), 2,14-2,08 (м, 1H), 1,81-1,74 (м, 1H).
К раствору (S)-трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)-пропил)-1H-пиразол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноата (0,90 г, 1,5 ммоль) и (R)-трет-бутил-3-((((трифторметил)сульфонил)-окси)метил)пирролидин-1-карбоксилата (0,99 г, 3,0 ммоль) в ацетонитриле (15 мл) добавляли гидрокарбонат натрия (1,2 г, 15 ммоль). Реакционную смесь нагревали при 60°С в течение ночи, а затем фильтровали. Фильтрат концентрировали, получая указанное в заголовке соединение.
LC/MS: [М]+ m/z 790,75.
Стадия C: 4-(4-((S)-2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-(((R)-1-(трет-бутоксикарбонил)пирролидин-3-ил)метил)-1H-пиразол-2-ий
К раствору 4-(4-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-(((R)-1-(трет-бутоксикарбонил)пирролидин-3-ил)метил)-1H-пиразол-2-ия (1,2 г, 1,5 ммоль) в CH2Cl2 (10 мл) и этаноле (10 мл) при 0°С добавляли гидразин (0,056 мл, 1,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, а затем концентрировали. Полученный остаток суспендировали в DCM (20 мл), перемешивали при комнатной температуре в течение 10 минут, а затем фильтровали. Фильтрат концентрировали, получая указанное в заголовке соединение.
LC/MS: [М]+ m/z 660,72.
Стадия D: 4-(4-((S)-3-(трет-бутокси)-2-(((Z)-((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)-амино)пропил)-2-(((R)-1-(трет-бутоксикарбонил)пирролидин-3-ил)метил)-1H-пиразол-2-ий
К раствору 4-(4-((S)-2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-(((R)-1-(трет-бутоксикарбонил)пирролидин-3-ил)метил)-1H-пиразол-2-ия (980 мг, 1,5 ммоль) в MeOH (20 мл) и CH2Cl2 (20 мл) добавляли 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусную кислоту (520 мг, 1,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, и затем концентрировали. Полученный остаток очищали с помощью обращенно-фазовой MPLC, с использованием смеси ацетонитрил (0,1% муравьиная кислота)/вода (0,1% муравьиной кислоты) в качестве элюирующего растворителя, получая указанное в заголовке соединение.
LC/MS:[M]+ m/z 914,84.
Стадия E: 4-(4-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-(((R)-1-(трет-бутоксикарбонил)пирролидин-3-ил)метил)-1H-пиразол-2-ий
К раствору 4-(4-((S)-3-(трет-бутокси)-2-(((Z)-((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)-амино)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-(((R)-1-(трет-бутоксикарбонил)пирролидин-3-ил)метил)-1H-пиразол-2-ия (660 мг, 0,72 ммоль) в ацетонитриле (10 мл) при температуре -5°С добавляли пиридин (0,23 мл, 2,9 ммоль), (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфат (300 мг, 1,4 ммоль) и EDC (300 мг, 1,6 ммоль). Реакционную смесь перемешивали при температуре -5°С в течение 2 часов, а затем концентрировали. Полученный остаток очищали с помощью обращенно-фазовой MPLC, с использованием смеси ацетонитрил (0,1% муравьиная кислота)/вода (0,1% муравьиной кислоты) в качестве элюирующего растворителя, получая указанное в заголовке соединение.
LC/MS: [М]+ m/z 1107,25.
Стадия F: (S)-3-((Z)-2-(((S)-2-(4-(1-(3-аминопропил)-2-((R)-пирролидин-3-илметил)-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
К раствору 4-(4-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)-азетидин-3-ил)амино)-2-оксоэтилиден)амино)-окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-(((R)-1-(трет-бутоксикарбонил)пирролидин-3-ил)метил)-1H-пиразол-2-ия (610 мг, 0,55 ммоль) в CH2Cl2 (3 мл) добавляли TFA (6 мл, 78 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 55 минут, а затем концентрировали. Полученный остаток обрабатывали Et2O (20 мл), собирали осадок и сушили в вакууме. Осадок растворяли в растворе NH4OAc (300 мМ, 2×70 мл) и перемешивали в течение 5 минут, затем очищали на колонке С18, используя смесь ацетонитрил (0,1% муравьиная кислота)/вода (0,1% муравьиной кислоты) в качестве элюирующего растворителя, получая неочищенный продукт. Неочищенный продукт повторно очищали с помощью обращенно-фазовой HPLC с использованием смеси ацетонитрил (0,1% муравьиная кислота)/вода (0,1% муравьиная кислота), получая указанное в заголовке соединение.
LC/MS: [М+1]+ m/z 750,5.
1H-ЯМР (500 МГц, D2O): δ 8,64-8,63 (д, J=5,7 Гц, 2H), 8,41 (с, 1H), 7,53-7,51(д, J=8,6 Гц, 2H), 7,02-7,00 (д, J=9,2 Гц, 2H), 6,91 (с, 1H), 4,92-4,90 (м, 1H), 4,66-4,63 (м, 1H), 4,62-4,59 (т, J=6,8 Гц, 2H), 4,48-4,39 (м, 3H), 3,61-3,56 (м, 1H), 3,54-3,49 (м, 1H), 3,37-3,32 (м, 1H), 3,18-3,12 (м, 3H), 3,08-3,01 (м, 1H), 2,40-2,34 (м, 2H), 2,32-2,25 (м, 1H), 1,91-1,83 (м, 1H), 1,37 (с, 3H), 1,01(с, 3H).
ПРИМЕР 54
(S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-2-(4-(1,2-бис(3-аминопропил)-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
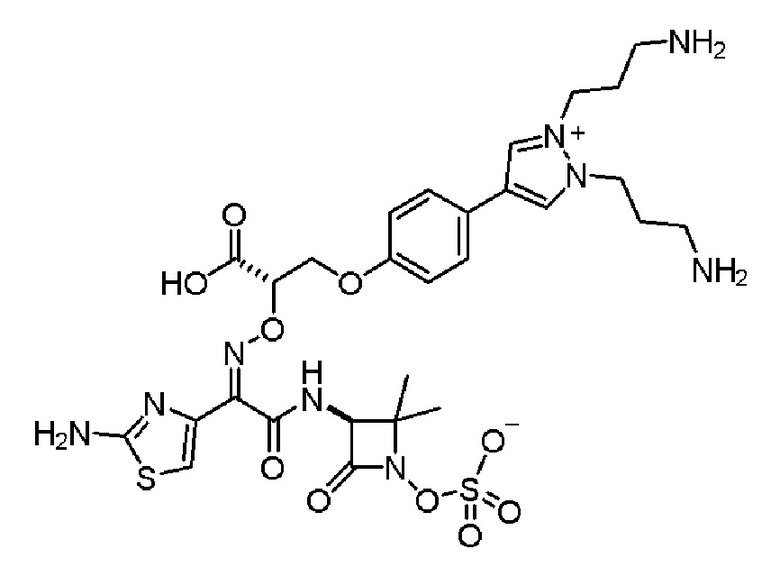
Стадия A: 3-азидопропил-трифторметансульфонат
К раствору 3-азидопропан-1-ола (2,7 г, 26 ммоль) и основания Хюнига (9,2 мл, 52 ммоль) в CH2Cl2 (60 мл) при -78°С добавляли трифторметансульфоновый ангидрид (5,4 мл, 32 ммоль) в течение 5 минут. Реакционную смесь перемешивали при -78°С в течение 1 часа, гасили насыщенным NaHCO3, и оставляли нагреваться до 0°С. Органический слой отделяли и промывали насыщенным солевым раствором. Водные слои объединяли и снова экстрагировали с помощью DCM. Органические слои объединяли, сушили над Na2SO4 и концентрировали в вакууме, получая указанное в заголовке соединение.
1H-ЯМР (500 МГц, D2O): δ 4,67-4,64 (т, J=5,8 Гц, 2H), 3,56-3,54 (т, J=5,8 Гц, 2H).
Стадия B: (S)-2-(3-азидопропил)-4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-2-ий
Смесь (S)-трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноата (6,4 г, 11 ммоль), 3-азидопропил-трифторметансульфоната (5,0 г, 21 ммоль) и бикарбоната натрия (8,9 г, 1 10 ммоль) нагревали при 60°С в течение 3 часов. Затем реакционную смесь фильтровали, и фильтрат концентрировали, получая указанное в заголовке соединение.
LC/MS: [М]+ m/z 690,6.
Стадия C: (S)-4-(4-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)фенил)-2-(3-азидопропил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-2-ий
К раствору (S)-2-(3-азидопропил)-4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-2-ия (7,3 г, 11 ммоль) в CH2Cl2 (30 мл) и этаноле (30,0 мл) при 0°С добавляли гидразин (0,40 мл, 13 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, а затем концентрировали. Полученный остаток обрабатывали DCM (30 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут, затем фильтровали, и фильтрат концентрировали, получая указанное в заголовке соединение.
LC/MS [М]+ m/z 560,6.
Стадия D: (S,Z)-2-(3-азидопропил)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-2-ий
К раствору (S)-4-(4-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)фенил)-2-(3-азидопропил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-2-ия (6,0 г, 11 ммоль) в MeOH (25 мл) и CH2Cl2 добавляли (25 мл) 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусную кислоту (3,8 г, 14 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи и концентрировали. Остаток очищали обращенно-фазовой HPLC с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05% TFA) в качестве элюирующего растворителя, получая указанное в заголовке соединение.
LC/MS: [М]+ m/z 814,7.
Стадия E: (S,Z)-2-(3-аминопропил)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)-тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутокси-карбонил)амино)пропил)-1H-пиразол-2-ий
К раствору (S,Z)-2-(3-азидопропил)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)-амино)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)-амино)пропил)-1H-пиразол-2-ия (7,8 г, 9,6 ммоль) в MeOH (200 мл) добавляли 10% Pd/C (1,5 г, 1,4 ммоль). Реакционную смесь гидрировали при комнатной температуре в течение 40 минут. Реакционную смесь фильтровали через целит, и фильтрат концентрировали, получая указанное в заголовке соединение.
LC/MS [М]+ m/z 788,8.
Стадия F: (S,Z)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)фенил)-1,2-бис(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-2-ий
К раствору (S,Z)-2-(3-аминопропил)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)-амино)пропил)-1H-пиразол-2-ия (7,6 г, 9,6 ммоль) в CH2Cl2 (200 мл) добавляли Вос2O (2,7 мл, 12 ммоль) и TEA (1,3 мл, 9,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, а затем концентрировали. Полученный остаток очищали с помощью обращенно-фазовой MPLC (C18) с использованием ацетонитрила (0,05% TFA) в качестве элюирующего растворителя, получая указанное в заголовке соединение.
LC/MS: [M]+ m/z 888,8.
Стадия G: 4-(4-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)фенил)-1,2-бис(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-2-ий
К раствору (S,Z)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)фенил)-1,2-бис(3-((трет-бутоксикарбонил) амино)пропил)-1H-пиразол-2-ия (2,5 г, 2,8 ммоль) в ацетонитриле (30 мл) при 0°C добавляли (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфат (1,2 г, 5,5 ммоль), пиридин (0,90 мл, 11 ммоль) и EDC (1,3 г, 6,9 ммоль). Реакционную смесь перемешивали при 0°С в течение 1 часа, а затем концентрировали в вакууме при комнатной температуре. Полученный остаток очищали обращенно-фазовой MPLC, с использованием ацетонитрила (0,05% TFA)/вода (0,05% TFA) в качестве элюирующего растворителя, получая указанное в заголовке соединение.
LC/MS: [М]+ m/z 1081,1.
Стадия H: (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-2-(4-(1,2-бис(3-аминопропил)-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
К раствору 4-(4-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)фенил)-1,2-бис(3-((трет-бутоксикарбонил)-амино)пропил)-1H-пиразол-2-ия (400 мг, 0,37 ммоль) в DCM (3 мл) добавляли TFA (6,0 мл, 78 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 50 минут, а затем концентрировали. Полученный остаток промывали Et2O (3×10 мл), сушили в вакууме, и растворяли в ацетате аммония (200 мМ, 80 мл, 16 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 10 минут, а затем очищали обращенно-фазовой MPLC, с использованием смеси ацетонитрил (0,1% муравьиная кислота)/вода (0,1% муравьиная кислота) в качестве элюирующего растворителя, с получением остатка и небольшого количества TFA. Остаток повторно растворяли в 80 мл NH4OAc (200 мМ), и реакционную смесь перемешивали при комнатной температуре в течение 10 минут, а затем очищали обращенно-фазовой MPLC, с использованием смеси ацетонитрил (0,1% муравьиная кислота)/вода (0,1% муравьиная кислота) в качестве элюирующего растворителя, получая указанное в заголовке соединение в виде свободного основания.
LC/MS [М]+ m/z 724,6.
1H-ЯМР(500 МГц, D2O): δ 8,57 (с, 2H), 8,41 (с, 1H), 7,49-7,48 (д, J=8,8 Гц, 2H), 6,99-6,98 (д, J=8,8 Гц, 2H), 6,90(с, 1H), 4,91-4,89 (м, 1H), 4,59-4,56 (т, J=8,0 Гц, 4H), 4,46-4,40 (м, 4H), 3,16-3,12 (м, 4H), 2,39-2,33 (м, 4H), 1,37 (с, 3H), 1,01 (с, 3H).
ПРИМЕР 55
(S)-3-(4-(1-(3-амино-2-(аминометил)пропил)-2-имино-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота
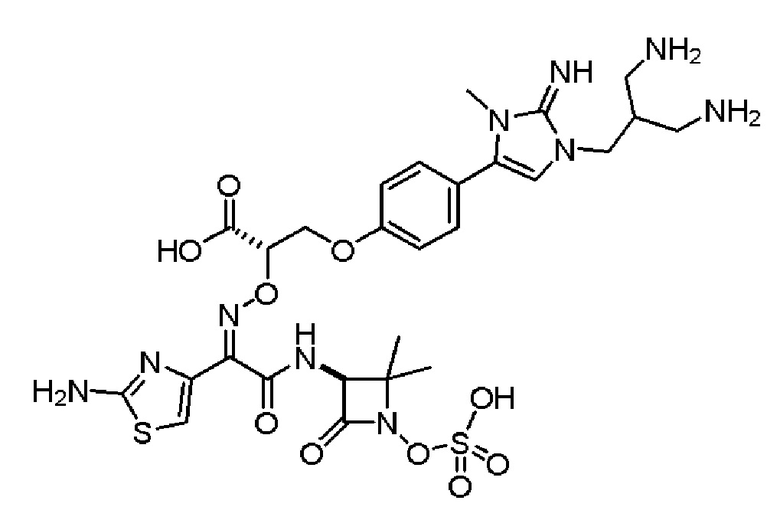
Стадия A: 3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)амино)метил)пропил метансульфонат
К раствору ди-трет-бутил-(2-(гидроксиметил)пропан-1,3-диил)дикарбамата (1,4 г, 4,6 ммоль) в DCM (безводный, 20 мл) при 0°С добавляли TEA (1,3 мл, 9,1 ммоль) и MsCl (0,43 мл, 5,5 ммоль). Реакционную смесь перемешивали при 0°С в течение 1 часа, затем распределяли между DCM (100 мл) и 0,1 М НС1 (на льду). Органический слой промывали 0,1 М НС1 (на льду), сушили над Na2SO4 и концентрировали, получая указанное в заголовке соединение.
LC/MS [М+1]+ m/z 383,4.
Стадия B: ди-трет-бутил-(2-((4-бром-1H-имидазол-1-ил)метил)пропан-1,3-диил)дикарбамат
К раствору 3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)амино)метил)-пропил-метансульфоната (1,8 г, 4,7 ммоль) в DMF (25 мл) добавляли 4-бром-1H-имидазол (0,69 г, 4,7 ммоль) и Cs2CO3 (3,1 г, 9,4 ммоль). Реакционную смесь нагревали при 60°С в течение ночи, затем распределяли между EtOAc (200 мл) и насыщенным раствором NaHCO3 (200 мл). Органический слой отделяли, промывали насыщенным раствором NaHCO3 и насыщенным раствором соли, сушили над Na2SO4 и концентрировали. Полученный остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующего растворителя, получая указанное в заголовке соединение.
LC/MS [М+1]+ m/z 433,3; 435,3.
Стадия C: 4-бром-1-(3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)-амино)метил)пропил)-3-метил-1H-имидазол-3-ий
К раствору ди-трет-бутил-(2-((4-бром-1H-имидазол-1-ил)метил)пропан-1,3-диил)дикарбамата (1,5 г, 3,5 ммоль) в ацетонитриле (10 мл) добавляли MeI (1,08 мл, 17 ммоль). Реакционная смесь нагревали при 60°С в течение ночи, а затем концентрировали. Полученный остаток очищали на колонке с силикагелем с использованием смеси MeOH/DCM в качестве элюирующего растворителя, получая указанное в заголовке соединение.
LC/MS [М]+ m/z 447,4; 449,4.
Стадия D: ди-трет-бутил-(2-((4-бром-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-1-ил)метил)пропан-1,3-диил)дикарбамат
К раствору трет-бутил карбамата (0,87 г, 7,4 ммоль) в CH2Cl2 (10 мл) добавляли трет-бутил гипохлорит (0,97 мл, 8,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут, затем охлаждали до 0°C и добавляли DBU (1,52 мл, 10 ммоль), с последующим добавлением 4-бром-1-(3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)амино)метил)пропил)-3-метил-1H-имидазол-3-ия (1,5 г, 3,4 ммоль) в CH2Cl2 (16 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут, а затем концентрировали. Полученный остаток очищали на колонке с силикагелем с использованием смеси MeOH/DCM в качестве элюирующего растворителя, получая указанное в заголовке соединение.
LC/MS [М+1]+ m/z 562,4; 564,4.
Стадия E: (R)-трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)-амино)метил)пропил)-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-гидроксипропаноат
Смесь трехосновного фосфата калия (3,6 мл, 3,6 ммоль), (R)-трет-бутил-2-гидрокси-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)пропаноата (0,53 г, 1,5 ммоль) и трет-бутил-(4-бром-1-(3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)амино)метил)пропил)-3-метил-1H-имидазол-2(3H)-илиден)карбамата (0,68 г, 1,2 ммоль) в диоксане (12 мл) дегазировали барботированием азота в течение 30 минут, и затем добавляли дихлорид 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладия (0,079 г, 0,12 ммоль). Реакционную смесь дополнительно дегазировали путем пропускания N2 в течение 20 минут, а затем нагревали при 60°С в течение ночи. Реакционную смесь фильтровали через целит, и фильтрат концентрировали. Полученный остаток очищали на колонке с силикагелем с использованием смеси MeOH/DCM в качестве элюирующего растворителя, получая указанное в заголовке соединение.
LC/MS [М+1]+ m/z 720,7.
Стадия F: (S)-трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)-амино)метил)пропил)-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноат
К раствору (R)-трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)амино)метил)пропил)-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-гидроксипропаноата (0,64 г, 0,89 ммоль) в THF (15 мл) добавляли 2-гидроксиизоиндолин-1,3-дион (0,17 г, 1,1 ммоль), трифенилфосфин (0,35 г, 1,3 ммоль) и DEAD (0,21 мл, 1,3 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, а затем концентрировали. Полученный остаток очищали на колонке с силикагелем с использованием смеси MeOH/DCM, получая указанное в заголовке соединение.
LC/MS [М+1]+ m/z 865,8.
Стадия G: (S)-трет-бутил-2-(аминоокси)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)амино)метил)пропил)-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)пропаноат
К раствору (S)-трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)амино)метил)пропил)-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-((1,3-диоксо-изоиндолин-2-ил)окси)пропаноата (0,77 г, 0,89 ммоль) в CH2Cl2 (5 мл) и этаноле (5 мл) при 0°С добавляли гидразин (0,033 мл, 1,1 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, а затем концентрировали. Полученный остаток обрабатывали DCM (10 мл), перемешивали при комнатной температуре в течение 10 минут, а затем фильтровали. Фильтрат концентрировали, получая указанное в заголовке соединение.
LC/MS: [M+1]+ m/z 735,7.
Стадия H: (2Z)-2-((((S)-1-(трет-бутокси)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)амино)метил)пропил)-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)уксусная кислота
К раствору (S)-трет-бутил-2-(аминоокси)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)амино)-метил)пропил)-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)пропаноата (0,65 г, 0,89 ммоль) в CH2Cl2 (10 мл) и MeOH (10 мл) добавляли 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусную кислоту (0,29 г, 1,1 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, а затем концентрировали. Полученный остаток очищали обращенно-фазовой MPLC с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05% TFA) в качестве элюирующего растворителя, получая указанное в заголовке соединение.
LC/MS: [M+1]+ m/z 989,8.
Стадия I: (S)-трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)-амино)метил)пропил)-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропаноат
К раствору (2Z)-2-((((S)-1-(трет-бутокси)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)амино)метил)пропил)-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил) амино)тиазол-4-ил)уксусной кислоты (550 мг, 0,55 ммоль) в ацетонитриле (8 мл) при 0°С добавляли (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфат (230 мг, 1,1 ммоль), пиридин (0,18 мл, 2,2 ммоль) и EDC (260 мг, 1,4 ммоль). Реакционную смесь перемешивали при 0°С в течение 2 часов, а затем концентрировали. Полученный остаток очищали обращенно-фазовой MPLC с использованием смеси ацетонитрил (0,05% TFA)/вода (0,0 5% TFA) в качестве элюирующего растворителя, получая указанное в заголовке соединение.
LC/MS [М+1]+ m/z 1182,0.
Стадия J: (S)-3-(4-(1-(3-амино-2-(аминометил)пропил)-2-имино-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-(((Z)-(1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота
К раствору (S)-трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)-амино)-метил)пропил)-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропаноата (420 мг, 0,36 ммоль) в CH2Cl2 (2 мл) добавляли TFA (4 мл, 52 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 50 минут, а затем концентрировали при комнатной температуре. Полученный остаток обрабатывали Et2O (10 мл) с получением осадка, который три раза промывали Et2O, концентрировали и очищали обращенно-фазовой HPLC с использованием смеси ацетонитрил (0,1% муравьиная кислота)/вода (0,1% муравьиная кислота) в качестве элюирующего растворителя, с получением указанного в заголовке соединения.
LC/MS [М+1]+ m/z 725,7.
1H-ЯМР (500 МГц, D2O): δ 7,35-7,33 (д, J=8,1 Гц, 2H), 7,04-7,02 (д, J=8,1 Гц, 2H), 6,99 (с, 1H), 6,89 (с, 1H), 4,98-4,96 (м, 1H), 4,66 (с, 1H), 4,47-4,41 (м, 2H), 4,08-4,06 (д, J=6,1 Гц, 2H), 3,35 (с, 3H), 3,25-3,19 (м, 2H), 3,14-3,09 (м, 2H), 2,73-2,67 (м, 2H), 1,42 (с, 3H), 1,03 (с, 3H).
ПРИМЕР 56
(2S)-3-(4-(1-(3-амино-2-гидроксипропил)-2-имино-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота
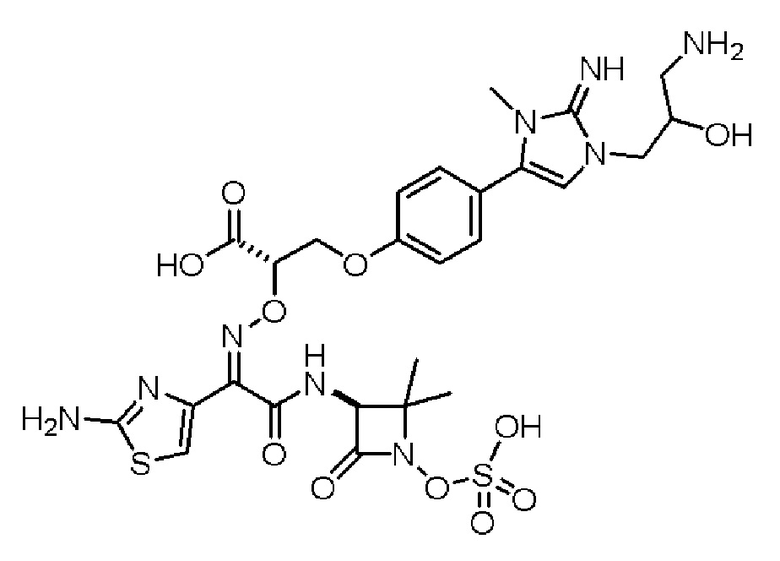
Стадия A: трет-бутил-(3-(4-бром-1H-имидазол-1-ил)-2-((трет-бутилдиметилсилил)окси)-пропил)карбамат
К раствору 5-бром-1H-имидазола (2,8 г, 19 ммоль) и трет-бутил-(3-бром-2-((трет-бутилдиметилсилил)окси)пропил)карбамата (7,4 г, 20 ммоль) в DMF (20 мл) добавляли карбонат цезия (12 г, 38 ммоль). Реакционную смесь нагревали при 60°С в течение ночи, а затем распределяли между водой (200 мл) и EtOAc (200 мл). Органический слой промывали два раза насыщенным раствором NaHCO3, сушили над Na2SO4 и концентрировали. Полученный остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующего растворителя, получая указанное в заголовке соединение.
LC/MS: (M+1)+ m/z 434,2; 436,2.
Стадия B: 4-бром-1-(3-((трет-бутоксикарбонил)амино)-2-((трет-бутилдиметилсилил)окси)пропил)-3-метил-1H-имидазол-3-ий
К раствору трет-бутил-(3-(4-бром-1H-имидазол-1-ил)-2-((трет-бутилдиметилсилил)окси)пропил)карбамата (2,7 г, 6,2 ммоль) в ацетонитриле (10 мл) добавляли метилиодид (1,9 мл, 31 ммоль). Реакционную смесь нагревали при 60°С в течение ночи, а затем концентрировали, получая указанное в заголовке соединение.
LC/MS [М]+ m/z 448,2; 450,2.
Стадия C: трет-бутил-(3-(4-бром-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-1-ил)-2-((трет-бутилдиметилсилил)окси)пропил)карбамат
К раствору трет-бутил карбамата (1,6 г, 14 ммоль) в CH2Cl2 (20 мл) добавляли трет-бутил гипохлорит (1,8 мл, 16 ммоль) и перемешивали при комнатной температуре в течение 30 минут. Затем реакционную смесь охлаждали до 0°C и добавляли DBU (2,8 мл, 19 ммоль), с последующим добавлением 4-бром-1-(3-((трет-бутоксикарбонил)-амино)-2-((трет-бутилдиметил-силил)окси)пропил)-3-метил-1H-имидазол-3-ия (2,8 г, 6,2 ммоль) в CH2Cl2 (16 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут, а затем концентрировали. Полученный остаток очищали хроматографией на колонке с силикагелем с использованием MeOH/EtOAc в качестве элюирующего растворителя, получая указанное в заголовке соединение.
LC/MS [М+1]+ m/z 563,5; 565,5.
Стадия D: (2R)-трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)-2-((трет-бутилдиметил-силил)окси)пропил)-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-гидроксипропаноат (пик A)
Смесь трехосновного фосфата калия (1 мл 1,11 ммоль), (R)-трет-бутил-2-гидрокси-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)пропаноата (1,6 г, 4,4 ммоль) и трет-бутил-(4-бром-1-(3-((трет-бутокси-карбонил)амино)-2-((трет-бутилдиметилсилил)окси)пропил)-3-метил-1H-имидазол-2(3H)-илиден)карбамата (2,1 г, 3,7 ммоль) в диоксане (24 мл) дегазировали барботированием азота в течение 30 минут, а затем добавляли дихлорид 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладия (0,24 г, 0,37 ммоль). Реакционную смесь дополнительно дегазировали барботированием азота в течение 20 минут, а затем нагревали при 75°С в течение 3 часов. Реакционную смесь фильтровали через целит, и фильтрат концентрировали. Полученный остаток очищали на колонке с силикагелем с использованием смеси MeOH/DCM в качестве элюирующего растворителя, получая указанное в заголовке соединение в виде смеси диастереоизомеров.
LC/MS: (М+1)+ m/z 721,5.
Диастереомеры разделяли хиральной SFC (Whelk-01, 4,6×250 мм, IPA+0,1 DIPA) с получением двух отдельных диастереомеров. Быстро элюируемый диастереоизомер использовали на следующей стадии.
Стадия H: (Z)-2-((((2S)-1-(трет-бутокси)-3-(4-((Z)-1-(3-((трет-бутоксикарбонил)амино)-2-гидроксипропил)-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)уксусная кислота
К раствору (2Z)-2-((((2S)-1-(трет-бутокси)-3-(4-(1-(3-((трет-бутокси-карбонил)амино)-2-((трет-бутилдиметилсилил)окси)пропил)-2-((трет-бутокси-карбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)уксусной кислоты (1,2 г, 1,2 ммоль, полученной на стадиях F-H, как описано в примере 55) в THF ( 20 мл) добавляли TBAF (1,13 г, 4,3 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, а затем концентрировали. Полученный остаток очищали обращенно-фазовой MPLC с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05% TFA) в качестве элюирующего растворителя, получая указанное в заголовке соединение.
LC/MS: (М+1)+ m/z 876,5.
Стадия I-J: (2S)-3-(4-(1-(3-амино-2-гидроксипропил)-2-имино-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-(((Z)-(1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота
Указанное в заголовке соединение получали из Z)-2-((((2S)-1-(трет-бутокси)-3-(4-((Z)-1-(3-((трет-бутоксикарбонил)амино)-2-гидроксипропил)-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутокси-карбонил)амино)тиазол-4-ил)уксусной кислоты в соответствии с процедурой, описанной на стадиях I-J примера 55.
LC/MS: (M+1)+ m/z 712,4.
1H-ЯМР(500 МГц, D2O): δ 7,34-7,32 (д, J=8,8 Гц, 2H), 7,04-7,02 (д, J=8,8 Гц, 2H), 6,94(с, 1H), 6,84 (с, 1H), 4,94-4,92 (м, 1H), 4,66 (с, 1H), 4,45-4,39 (м, 2H), 4,24-4,21 (м, 1H), 4,05-4,02 (д, J=4,8 Гц, 1H), 3,92-3,88 (м, 1H), 3,34 (с, 3H), 3,25-3,22 (д, J=4,8 Гц, 1H), 3,00-2,95 (т, J=10,5 Гц, 1H), 1,41 (с, 3H), 1,06 (с, 3H).
ПРИМЕР 57
(S)-3-(4-((Z)-2-((2-аминоэтил)имино)-1-(3-аминопропил)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота
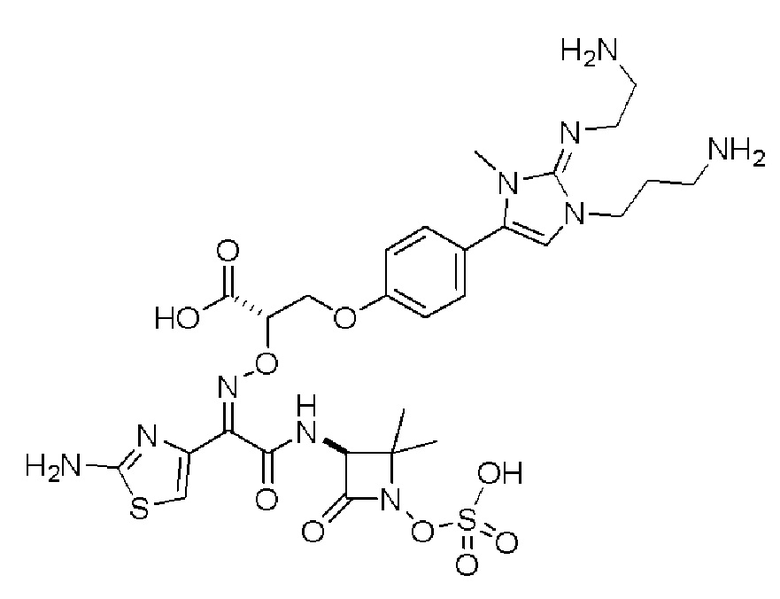
Стадия A: 3-(4-бром-2-имино-3-метил-2,3-дигидро-1H-имидазол-1-ил)пропан-1-амин
К раствору (Z)-трет-бутил-(3-(4-бром-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-1-ил)пропил)карбамата (1,1 г, 2,5 ммоль) в CH2Cl2 (2 мл) добавляли TFA (6,0 мл, 78 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, а затем концентрировали, получая указанное в заголовке соединение.
LC/MS: (M+1)+ m/z 233,1; 235,1.
Стадия B: трет-бутил-(3-(4-бром-2-имино-3-метил-2,3-дигидро-1H-имидазол-1-ил)пропил)-карбамат
К 3-(4-бром-2-имино-3-метил-2,3-дигидро-1H-имидазол-1-ил)пропан-1-амину (0,59 г, 2,5 ммоль) в CH2Cl2 (40 мл) добавляли TEA (1,8 мл, 13 ммоль) и Boc2O (0,59 мл, 2,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем концентрировали. Полученный остаток очищали на колонке с силикагелем с использованием смеси MeOH/DCM в качестве элюирующего растворителя, получая указанное в заголовке соединение.
LC/MS: (М+1)+ m/z 333,1; 335,1.
Стадия C: трет-бутил-(Z)-(3-(4-бром-2-((2-((трет-бутоксикарбонил)амино)этил)имино)-3-метил-2,3-дигидро-1H-имидазол-1-ил)пропил)карбамат
К раствору трет-бутил-(3-(4-бром-2-имино-3-метил-2,3-дигидро-1H-имидазол-1-ил)пропил)карбамата (1,0 г, 3,0 ммоль) и трет-бутил-(2-бромэтил)карбамата (1,3 г, 6,0 ммоль) в ацетонитриле (10 мл) добавляли Cs2СO3 (2,9 г, 9,0 ммоль). Реакционную смесь перемешивали при 50°С в течение 6 часов, а затем концентрировали. Полученный остаток растворяли в DCM (50 мл) и фильтровали. Фильтрат очищали на колонке с силикагелем с использованием смеси MeOH/DCM в качестве элюирующего растворителя, получая указанное в заголовке соединение.
LC/MS: (M+1)+ m/z 476,0; 478,0.
Стадия D: (S)-3-(4-((Z)-2-((2-аминоэтил)имино)-1-(3-аминопропил)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-(((Z)-(1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота
Указанное в заголовке соединение получали из трет-бутил-(Z)-(3-(4-бром-2-((2-((трет-бутоксикарбонил)-амино)этил)имино)-3-метил-2,3-дигидро-1H-имидазол-1-ил)пропил)карбамата в соответствии с процедурой, описанной на стадих E-J примера 55.
LC/MS: (M+1)+ m/z 739,4.
1H-ЯМР (500 МГц, D2O): δ 7,37-7,35 (д, J=8,6 Гц, 2H), 7,15 (с, 1H), 7,06-7,04 (д, J=8,6 Гц, 2H), 6,93 (с, 1H), 4,94-4,93 (м, 1H), 4,63 (с, 1H), 4,47-4,39 (м, 2H), 4,11-4,09 (м, 2H), 3,63-3,61 (м, 2H), 3,52 (с, 3H), 3,29-3,27 (м, 2H), 3,07-3,04 (м, 2H), 2,21-2,16 (м, 2H), 1,41 (с, 3H), 1,02 (с, 3H).
ПРИМЕРЫ 58 и 59
(S)-3-(4-((S)-2-((2-аминоэтил)амино)-4,5-дигидро-1H-имидазол-4-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота (58)
(R)-3-(4-((S)-1-амино-2-((4,5-дигидро-1H-имидазол-2-ил)амино)этил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота (59)
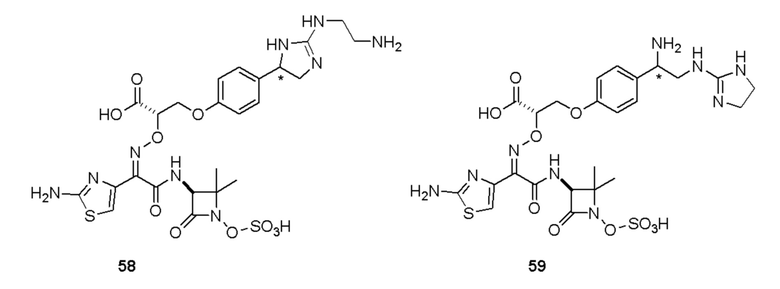
Стадия A: трет-бутил-(R)-2-гидрокси-3-(4-винилфенокси)пропаноат
К раствору (R)-трет-бутил-3-(4-бромфенокси)-2-гидроксипропаноата (2400 мг, 7,6 ммоль ) в этаноле (60 мл) добавляли винилтрифторборат калия (1520 мг, 11 ммоль), Et3N (1,58 мл, 11 ммоль) и аддукт PdCl2(dppf)-CH2Cl2 (310 мг, 0,38 ммоль). Реакционную смесь вакуумировали и продували водородом (3 раза) и нагревали с обратным холодильником в течение ночи, затем охлаждали до комнатной температуры и фильтровали через тонкий слой целита с промывкой CH2Cl2. Фильтрат концентрировали и очищали на колонке ISCO (gold, 40 г), элюируя с градиентом 0-50% EtOAc/гексан, получая указанное в заголовке соединение.
LC-MS [M+Na]+: m/z 287,2.
Стадия B: трет-бутил-(R)-2-((трет-бутилдиметилсилил)окси)-3-(4-винилфенокси)пропаноат
К раствору (R)-трет-бутил-2-гидрокси-3-(4-винилфенокси)пропаноата (1580 мг, 6,0 ммоль), имидазола (980 мг, 14 ммоль) и TBDMS-Сl (1,1 г, 7,2 ммоль) в ацетонитриле (25 мл) добавляли DMAP (73 мг, 0,60 ммоль). Полученный раствор перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь концентрировали при пониженном давлении, и полученный остаток очищали на колонке ISCO Redi-Sep (gold, 40 г), элюируя смесью EtOAc/гексан с градиентом 0-30%, получая указанное в заголовке соединение.
Стадия C: трет-бутил-(2R)-2-((трет-бутилдиметилсилил)окси)-3-(4-(1,2-диазидоэтил)фенокси)-пропаноат
(R)-трет-бутил-2-((трет-бутилдиметилсилил)окси)-3-(4-винилфенокси)пропаноат (1,9 г, 5,0 ммоль) добавляли к перемешиваемой смеси периодата натрия (1,1 г, 5,0 ммоль) и азида натрия (1,0 г, 15 ммоль) в смеси DMSO (15 мл) и уксусной кислоты (5 мл). Реакционную смесь перемешивали при 75°С в течение 2 часов, а затем охлаждали, разбавляли этилацетатом, промывали четыре раза водой и солевым раствором, сушили (MgSO4) и фильтровали. Фильтрат выпаривали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (колонка ISCO, gold, 40 г), элюируя смесью EtOAc/гексан с градиентом 0-20%, получая указанное в заголовке соединение.
Стадия D: трет-бутил-(2R)-2-((трет-бутилдиметилсилил)окси)-3-(4-(1,2-диаминоэтил)-фенокси)пропаноат
Палладий на угле (300 мг, 2,8 ммоль) добавляли к перемешиваемой смеси (2R)-трет-бутил-2-((трет-бутилдиметилсилил)окси)-3-(4-(1,2-диазидоэтил)-фенокси)пропаноата (1500 мг, 3,2 ммоль) в метаноле (30 мл) с продувкой водородом из баллона. Реакционную смесь подвергали вакуумированию с продувкой Н2 (3 раза), а затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли этилацетатом и фильтровали через тонкий слой целита. Фильтрат концентрировали при пониженном давлении, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 411,6.
Стадия E: трет-бутил-(2R)-2-((трет-бутилдиметилсилил)окси)-3-(4-(2-тиоксоимидазолидин-4-ил)фенокси)пропаноат
К перемешиваемой смеси (2R)-трет-бутил-2-((трет-бутилдиметилсилил)окси)-3-(4-(1,2-диаминоэтил)фенокси)пропаноата (800 мг, 1,9 ммоль) в DCM (16 мл) в течение 20 минут медленно добавляли по каплям 1,1'-тиокарбонилдиимидазол (360 мг, 2.0 ммоль) в DCM (8 мл). Реакционную смесь перемешивали при комнатной температуре в течение 15 минут, а затем концентрировали при пониженном давлении. Полученный остаток очищали на колонке с силикагелем (gold, 80 г), используя EtOAc/гексан с градиентом 0-60%, получая указанное в заголовке соединение в виде рацемической смеси. Эту рацемическую смесь разделяли с помощью хиральной SFC (IC (2×15 см+3×15 см); 35% изопропанол/СO2, 100 бар; 50 мл/мин, 220 нм; инжекция: 1,5 мл, 15 мг/мл этанол:DCM), получая указанное в заголовке соединение в виде двух энантиомерно чистых изомеров. Второй изомер (большее время удерживания) использовали на следующей стадии.
LC-MS [М+Н]+: m/z 453,3.
Стадия F: трет-бутил-(R)-2-((трет-бутилдиметилсилил)окси)-3-(4-((S)-2-(метилтио)-4,5-дигидро-1H-имидазол-4-ил)фенокси)пропаноат
Иодметан (0,967 мл, 15,46 ммоль) добавляли к перемешиваемой смеси (R)-трет-бутил-2-((трет-бутилдиметилсилил)окси)-3-(4-(2-тиоксоимидазолидин-4-ил)фенокси)пропаноата (1400 мг, 3,1 ммоль) в MeCN (12 мл). Реакционную смесь перемешивали при 70°С в течение 2 часов, затем охлаждали и концентрировали при пониженном давлении, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 467,3.
Стадия G: трет-бутил-(S)-4-(4-((R)-3-(трет-бутокси)-2-((трет-бутилдиметилсилил)окси)-3-оксо-пропокси)фенил)-2-(метилтио)-4,5-дигидро-1H-имидазол-1-карбоксилат
DMAP (190 мг, 1,5 ммоль) добавляли к перемешиваемой смеси Вос-ангидрида (1,4 мл, 6,2 ммоль), (R)-трет-бутил-2-((трет-бутилдиметилсилил)окси)-3-(4-((S)-2-(метилтио)-4,5-дигидро-1H-имидазол-4-ил)фенокси)пропаноата (1400 мг, 3,1 ммоль) и триэтиламина (1,3 мл, 9,3 ммоль) в CH2Cl2 (14 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, и затем концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (ISCO, gold, 80 г) элюируя смесью этилацетат/гексан с градиентом 0-100%, получая указанное в заголовке соединение в виде смеси Вос-региоизомеров.
LC-MS [M+H]+: m/z 567,4.
Стадия H: трет-бутил-(S)-4-(4-((R)-3-(трет-бутокси)-2-((трет-бутилдиметилсилил)окси)-3-оксопропокси)фенил)-2-((2-((трет-бутоксикарбонил)амино)этил)амино)-4,5-дигидро-1H-имидазол-1-карбоксилат
Уксусную кислоту (0,94 мл) добавляли к перемешиваемой смеси N-Boc-этилендиамина (420 мг, 2,6 ммоль) и смеси Вос-региоизомеров со стадии G (750 мг, 1,3 ммоль) в этаноле (7,5 мл). Реакционную смесь перемешивали при 55°С в течение ночи, затем охлаждали и концентрировали при пониженном давлении. Полученный остаток растворяли в DCM и промывали водным раствором NaHCO3/2 Н NaOH (конечная промывка при рН -10). Водный слой отделяли и 3 раза экстрагировали DCM. Объединенные органические слои промывали насыщенным солевым раствором, сушили (MgSO4) и фильтровали. Фильтрат удаляли при пониженном давлении, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 679,5.
Стадия I: трет-бутил-(S)-4-(4-((R)-3-(трет-бутокси)-2-((трет-бутилдиметилсилил)окси)-3-оксо-пропокси)фенил)-2-((трет-бутоксикарбонил)(2-((трет-бутоксикарбонил)амино)этил)амино)-4,5-дигидро-1H-имидазол-1-карбоксилат
Вос-ангидрид (4,6 мл, 20 ммоль) добавляли к трет-бутил-(S)-4-(4-((R)-3-(трет-бутокси)-2-((трет-бутилдиметилсилил)окси)-3-оксопропокси)фенил)-2-((2-((трет-бутоксикарбонил)амино)этил)амино)-4,5-дигидро-1H-имидазол-1-карбоксилату (900 мг, 1,3 ммоль) и смесь перемешивали при 100°С в течение 1,5 часов. Затем реакционную смесь охлаждали и загружали на колонку ISCO (80 г gold), элюируя смесью EtOAc/гексан с градиентом 0-100%, получая указанное в заголовке соединение в виде смеси региоизомеров.
LC-MS [М+Н]+: m/z 780,5.
Стадия J: трет-бутил-(S)-4-(4-((R)-3-(трет-бутокси)-2-гидрокси-3-оксопропокси)фенил)-2-((трет-бутоксикарбонил)(2-((трет-бутоксикарбонил)амино)этил)амино)-4,5-дигидро-1H-имидазол-1-карбоксилат
TBAF (1,7 мл, 1,7 ммоль) добавляли к перемешиваемой смеси (S)-трет-бутил 4-(4-((R)-3-(трет-бутокси)-2-((трет-бутилдиметилсилил)окси)-3-оксопропокси)фенил)-2-((трет-бутоксикарбонил)(2-((трет-бутоксикарбонил)амино)этил)амино)-4,5-дигидро-1H-имидазол-1-карбоксилата (900 мг, 1,155 ммоль) в THF (8 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, а затем концентрировали в вакууме. Полученный остаток очищали на колонке ISCO с силикагелем (40 г gold), элюируя смесью EtOAc/гексан с градиентом 0-100%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 665,4.
Стадия K: трет-бутил-(S)-4-(4-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-2-((трет-бутоксикарбонил)(2-((трет-бутоксикарбонил)амино)этил)амино)-4,5-дигидро-1H-имидазол-1-карбоксилат
К раствору (S)-трет-бутил-4-(4-((R)-3-(трет-бутокси)-2-гидрокси-3-оксопропокси)фенил)-2-((трет-бутоксикарбонил)(2-((трет-бутокси-карбонил)амино)этил)амино)-4,5-дигидро-1H-имидазол-1-карбоксилата (460 мг смеси, 0,69 ммоль) в THF (5 мл) добавляли 2-гидроксиизоиндолин-1,3-дион (135 мг, 0,83 ммоль) и трифенилфосфин (254 мг, 0,97 ммоль), а затем добавляли по каплям DIAD (0,19 мл, 0,97 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, а затем концентрировали. Полученный остаток очищали на колонке с силикагелем (ISCO, gold, 40 г), элюируя смесью EtOAc/гексан с градиентом 0-100%, получая указанное в заголовке соединение в виде смеси изомеров.
LC-MS [М+Н]+: m/z 810,7.
Стадия L: 2-((((S)-1-(трет-бутокси)-3-(4-((S)-1-(трет-бутоксикарбонил)-2-((трет-бутоксикарбонил)(2-((трет-бутоксикарбонил)амино)этил)амино)-4,5-дигидро-1H-имидазол-4-ил)фенокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)уксусная кислота
К раствору (S)-трет-бутил-4-(4-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-2-((трет-бутоксикарбонил)(2-((трет-бутоксикарбонил)амино)этил)амино)-4,5-дигидро-1H-имидазол-1-карбоксилата (350 мг, 0,432 ммоль) в CH2Cl2 (2 мл) при комнатной температуре добавляли гидразин (0,016 мл, 0,52 ммоль) в этаноле (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут, а затем добавляли 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусную кислоту (190 мг, 0,69 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов, а затем концентрировали, получая указанное в заголовке соединение, которое непосредственно использовали на следующей стадии.
LC-MS [М+Н]+: m/z 934,5.
Стадия M: (S)-трет-бутил-4-(4-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)фенил)-2-((трет-бутоксикарбонил)(2-((трет-бутоксикарбонил)амино)этил)амино)-4,5-дигидро-1H-имидазол-1-карбоксилат
К раствору 2-((((S)-1-(трет-бутокси)-3-(4-((S)-1-(трет-бутоксикарбонил)-2-((трет-бутоксикарбонил)(2-((трет-бутоксикарбонил)амино)этил)амино)-4,5-дигидро-1H-имидазол-4-ил)фенокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)уксусной кислоты (400 мг, 0,43 ммоль) в DMF (3,5 мл) добавляли DCC (260 мг, 1,3 ммоль) и HOBt (200 мг, 1,3 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 30 минут, а затем добавляли (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфат (220 мг, 1,1 ммоль) и бикарбонат натрия (220 мг, 2,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем фильтровали шприцом через спеченный слой и промывали DMFА (1,5 мл). Фильтрат очищали на колонке ISCO С-18 (80 г) с обращенной фазой, элюируя смесью % ACN/вода с 0,05% TFA с градиентом 10-100%, получая указанное в заголовке соединение в виде смеси изомеров.
LC-MS [М+Н]+: m/z 1126,7.
Стадия N: (R)-3-(4-((S)-1-амино-2-((4,5-дигидро-1H-имидазол-2-ил)амино)этил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота (59) и (S)-3-(4-((S)-2-((2-аминоэтил)-амино)-4,5-дигидро-1H-имидазол-4-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота (58)
TFA (5 мл) добавляли к раствору (S)-трет-бутил 4-(4-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)фенил)-2-((трет-бутоксикарбонил)(2-((трет-бутоксикарбонил)амино)этил)амино)-4,5-дигидро-1H-имидазол-1-карбоксилата (200 мг, 0,18 ммоль, смесь изомеров) в CH2Cl2 (4 мл). Смесь перемешивали при комнатной температуре в течение 1,5 часов, а затем концентрировали при пониженном давлении. К полученному остатку добавляли эфир. Затем растворитель удаляли при пониженном давлении. Для разрушения твердого вещества к полученному остатку добавляли эфир, а затем удаляли растворитель. Полученный остаток сушили в вакууме, растворяли в DMSO (5 мл) и очищали с помощью обращенно-фазового разделения (Gilson, используя смесь MeCN/вода с 0,05% TFA с градиентом 5-25%), с получением (2S)-3-(4-(1-амино-2-((4,5-дигидро-1H-имидазол-2-ил)амино)этил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановой кислоты (59); и (S)-3-(4-((S)-2-((2-аминоэтил)амино)-4,5-дигидро-1H-имидазол-4-ил)фенокси)-2-(((Z)-(1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановой кислоты (58).
Пример 58:
LC-MS [M+H]+: m/z 670,4.
1H-ЯМР (500 МГц, D2O, м.д.): δ 7,26 (д, J=10 Гц, 2H), 7,04 (с, 1H), 6,94 (д, J=10 Гц, 2H), 5,03 (м, 1H), 4,74 (с, 1H), 4,64 (м, 1H), 4,38 (м, 3H), 3,66 (дд, J=10, 5 Гц, 1H), 3,55 (дд, J=10, 5 Гц, 1H), 3,48 (с, 3H), 1,37 (с, 3H), 1,04 (с, 3H).
Пример 59:
LC-MS [M+H]+: m/z 670,0.
1H-ЯМР (500 МГц, D2O, м.д.): δ 7,22 (д, J=10 Гц, 2H), 7,02 (с, 1H), 6,88 (д, J=10 Гц, 2H), 5,0 (м, 2H), 4,68 (с, 1H), 4,34 (м, 2H), 3,98 (т, J=10 Гц, 1H), 3,47 (м, 3H), 3,12 (т, J=5 Гц, 2H), 1,34 (с, 3H), 0,97 (с, 3H).
ПРИМЕРЫ 60 и 61
(2S)-3-(4-(2-((2-аминоэтил)амино)-4,5-дигидро-1H-имидазол-5-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота (60)
(2S)-3-(4-(1-амино-2-((4,5-дигидро-1H-имидазол-2-ил)амино)этил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота (61)
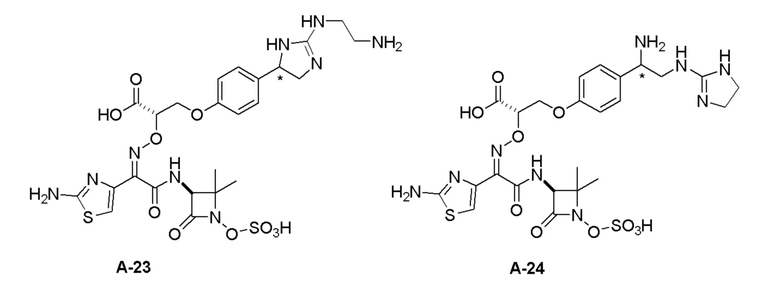
Указанные в заголовке соединения получали в соответствии с процедурой, описанной в примерах 58 и 59, начиная с первого изомера (более короткое время удерживания), полученного на стадии Е.
Пример 60: LC-MS [М+Н]+: m/z 670,4.
Пример 61: LC-MS [М+Н]+: m/z 670,0.
ПРИМЕР 62
(S)-3-((Z)-2-(((S)-2-(4-(2-((2-аминоэтил)амино)-1-метилпиридин-1-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
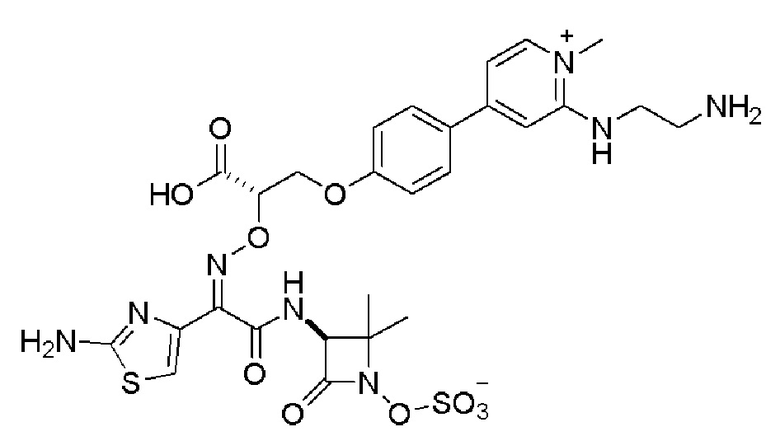
Стадия A: трет-бутил-(2-((4-бромпиридин-2-ил)амино)этил)карбамат Смесь 4-бромпиридин-2-амина (500 мг, 2,9 ммоль), 125 мг молекулярных сит 4Å и трет-бутил-(2-оксоэтил)карбамата (510 мг, 3,2 ммоль) в 1,2-дихлорэтане (10 мл) перемешивали при комнатной температуре в течение 60 мин, с последующим добавлением триацетоксиборгидрида натрия (1200 мг, 5,8 ммоль). Смесь перемешивали в течение ночи, затем разбавляли EtOAc и промывали водным раствором NaHCO3 (3 раза). Органический слой отделяли, промывали водой и насыщенным раствором соли, сушили над Na2SO4 и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали на картридже с силикагелем (ISCO, gold, 40 г) элюируя смесью EtOAc/гексан с градиентом 0-100%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 316,2.
Стадия B: трет-бутил-(R)-3-(4-(2-((2-((трет-бутоксикарбонил)амино)этил)амино)пиридин-4-ил)фенокси)-2-гидроксипропаноат К раствору аддукта PdCl2(dppf)-CH2Cl2 (180 мг, 0,22 ммоль), трет-бутил-(2-((4-бромпиридин-2-ил)амино)этил)карбамата (870 мг, 2,8 ммоль) и (R)-трет-бутил-2-гидрокси-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)-пропаноата (800 мг, 2,2 ммоль) в диоксане (7,5 мл) добавляли раствор Na2CO3 (0,70 г, 6,6 ммоль) в воде (2,5 мл). Реакционную смесь вакуумировали и продували водородом (3 раза), а затем нагревали при 100°С в микроволновой печи в течение 1 часа. Затем смесь очищали на колонке с силикагелем (ISCO, gold, 40 г), элюируя смесью EtOAc/гексан с градиентом 0-100%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 474,5.
Стадия C: (S)-3-((Z)-2-(((S)-2-(4-(2-((2-аминоэтил)амино)-1-метилпиридин-1-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
Соединение трет-бутил-(R)-3-(4-(2-((2-((трет-бутоксикарбонил)амино)этил)-амино)пиридин-4-ил)фенокси)-2-гидроксипропаноат превращали в указанное в заголовке соединение с использованием процедуры, аналогичной процедуре, описанной на стадиях С-Н примера 1.
LC-MS [М+Н]+: m/z 694,0.
1H-ЯМР (500 МГц, D2O, м.д.): δ 7,83 (д, J=5 Гц, 1H), 7,64 (д, J=5 Гц, 2H), 7,13 (д, J=5 Гц, 1H),7,07 (с, 1H), 6,99 (д, J=5 Гц, 2H), 6,96 (с, 1H), 5,01 (м, 1H), 4,49 (с, 1H), 4,43 (м, 2H), 3,75 (т, J=5 Гц, 2H), 3,72 (с, 3H), 3,22 (т, J=5 Гц, 2H), 1,28 (с, 3H), 0,96 (с, 3H).
Таблица 4. Примеры 63-83 были получены в соответствии с процедурой, описанной в примере 62
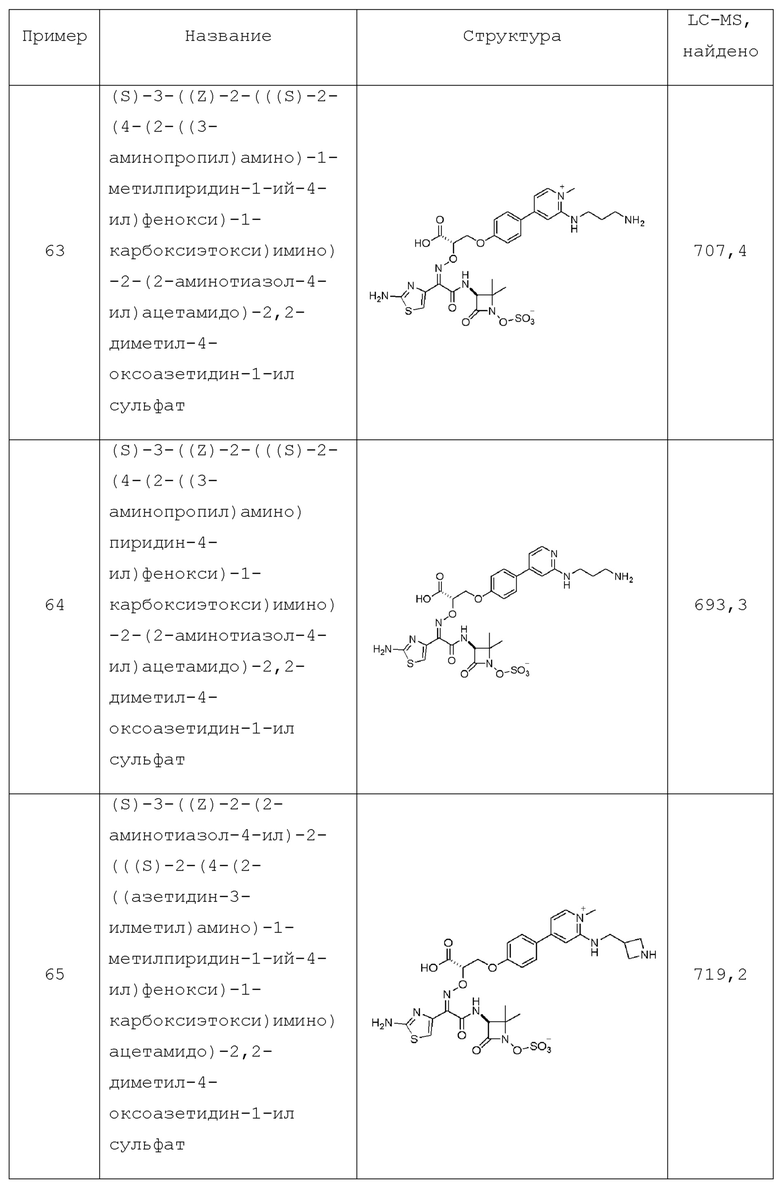
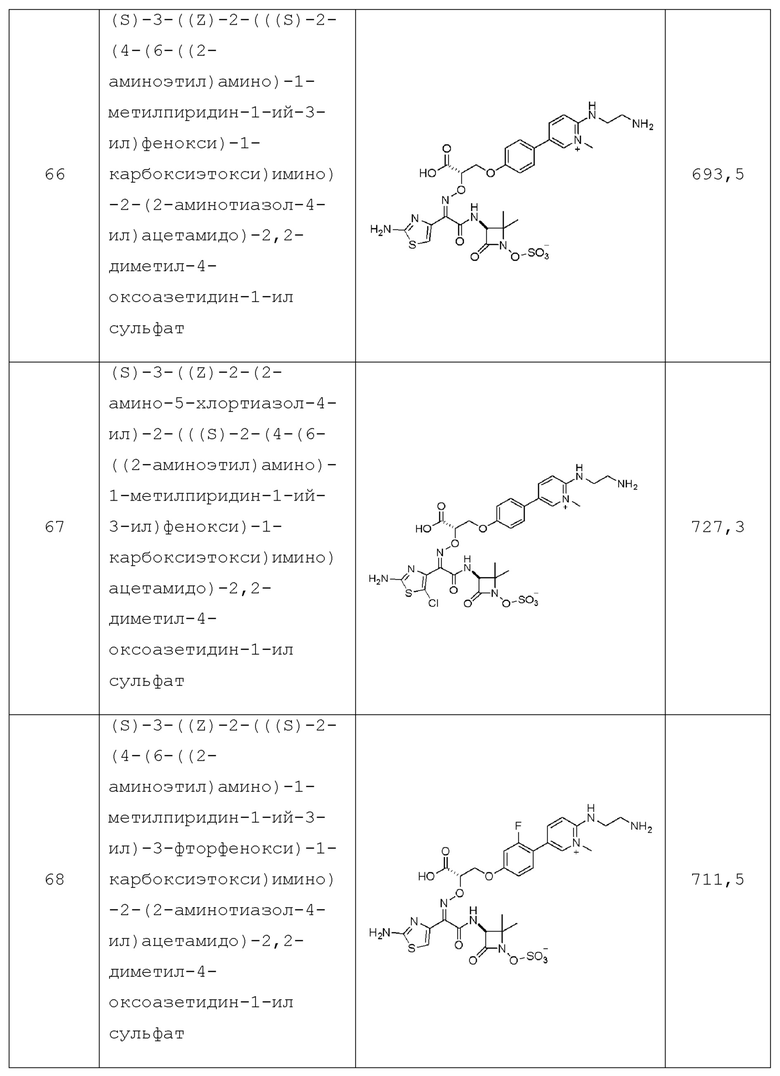
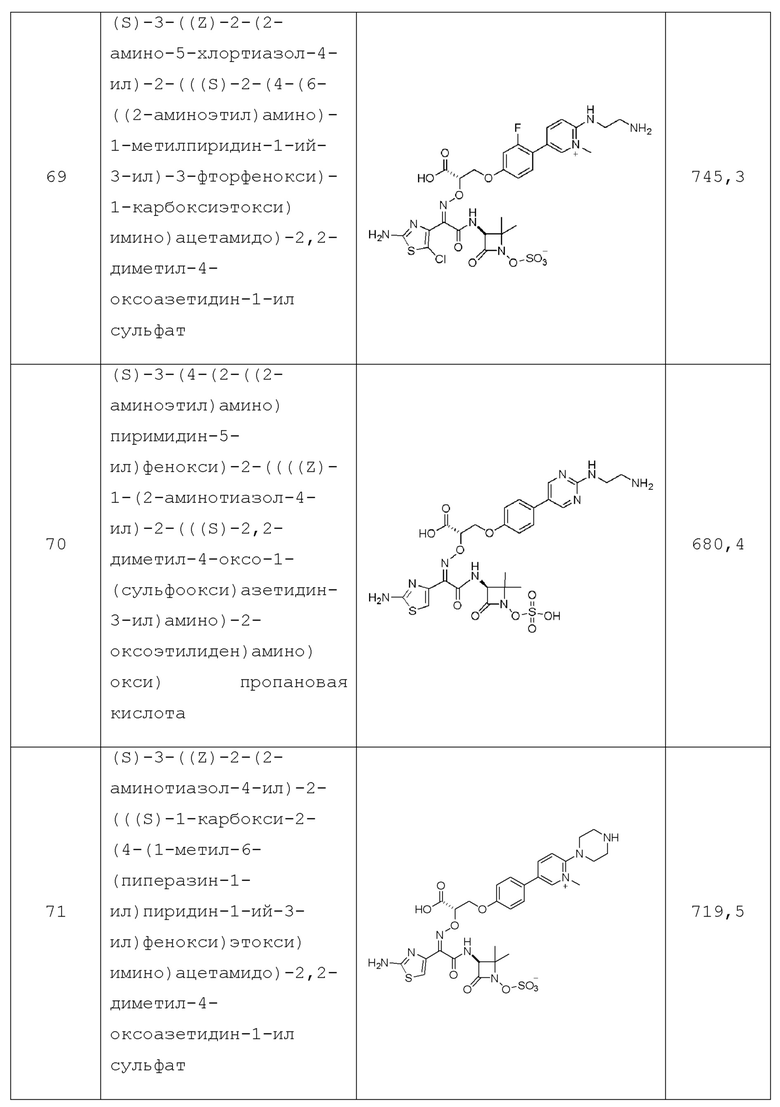
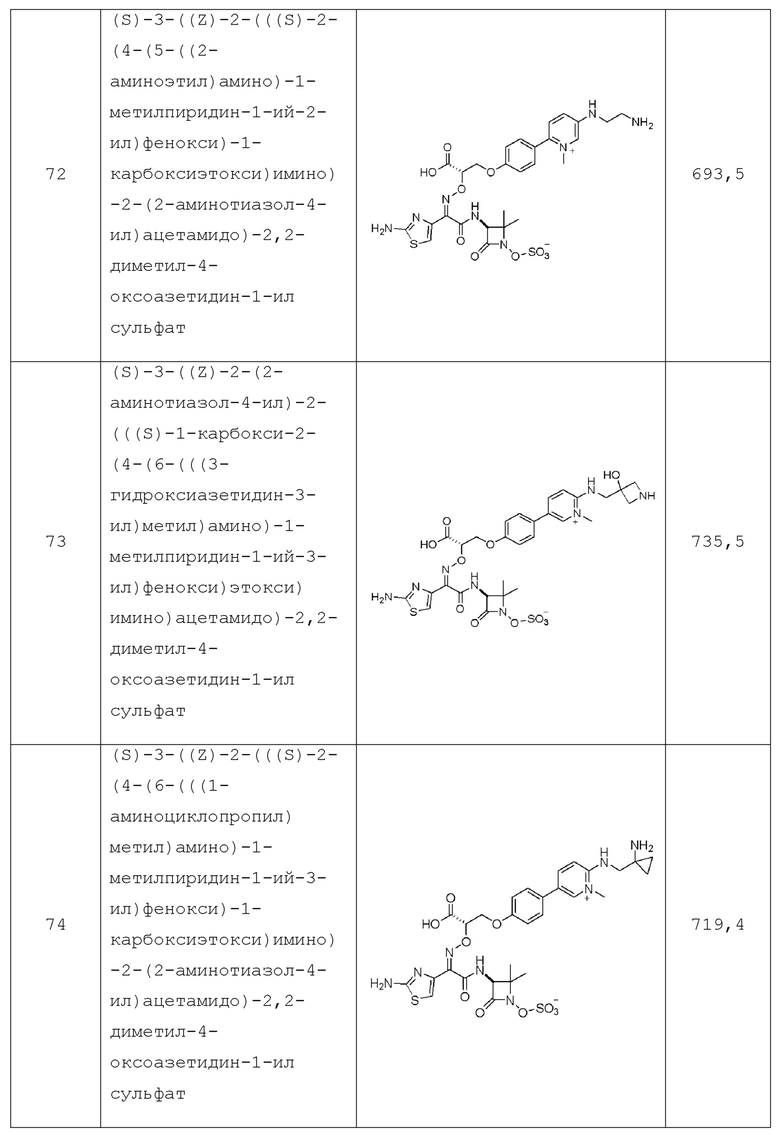
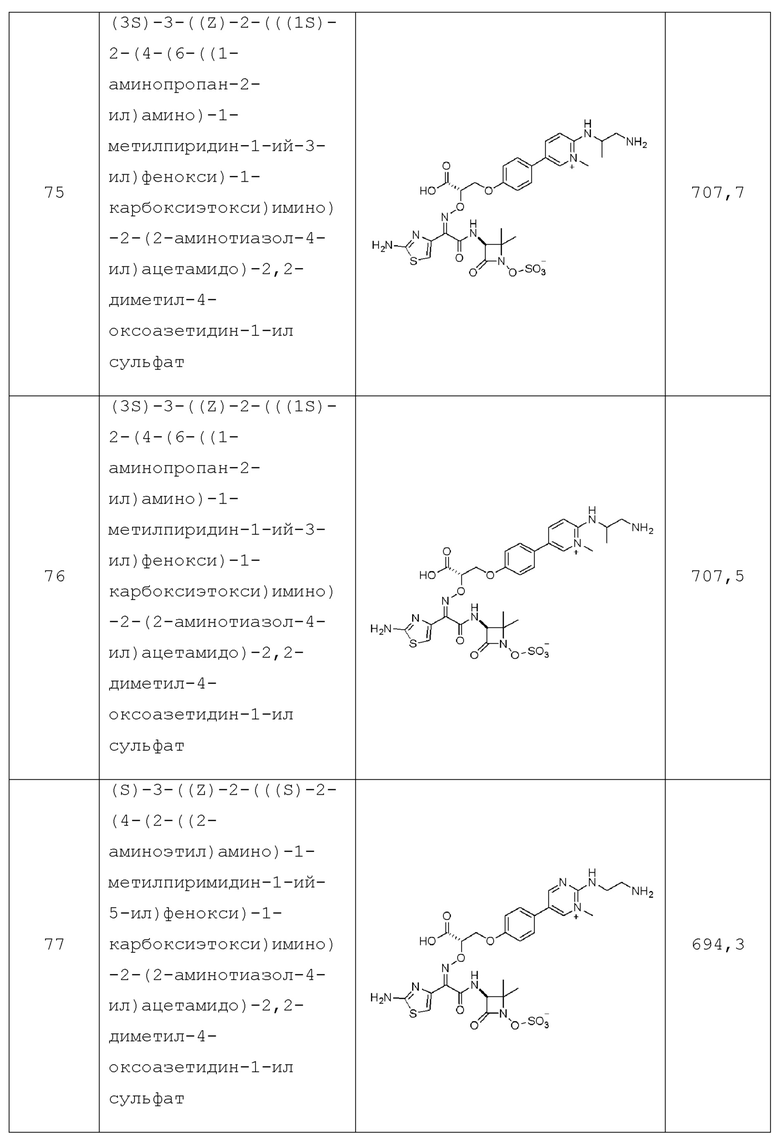

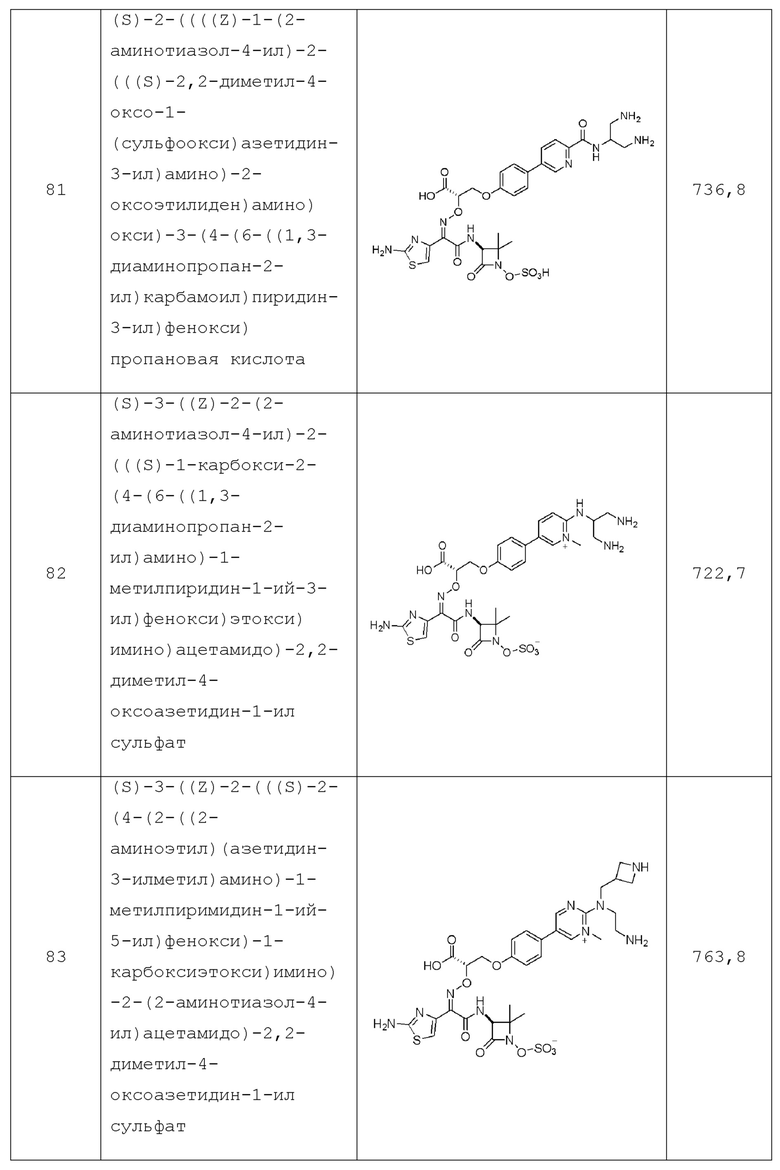
ПРИМЕРЫ 84 и 85
(3S)-3-(2-((2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-2-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат (84) и
(3S)-3-(2-((2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-2-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат (85)
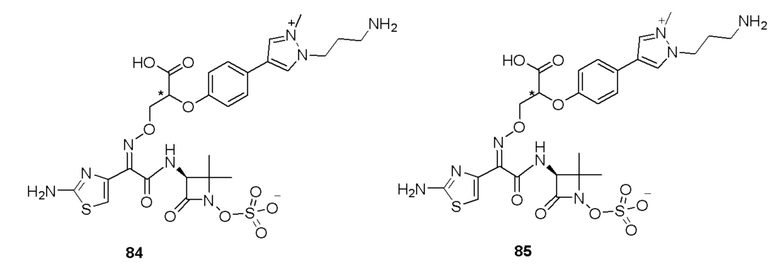
Стадия A: метил 2-(4-бромфенокси)-3-метоксипропаноат
Раствор 4-бромфенола (1,8 г, 10 ммоль), метил-2-бром-3-метоксипропаноата (1,4 мл, 10 ммоль) и гидроксида лития (0,29 г, 12 ммоль) в DMF (25,4 мл) перемешивали при комнатной температуре в течение 3 часов. Эту реакционную смесь распределяли между EtOAc и водой. Отделенный органический слой промывали водой и насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали. Полученный остаток очищали на колонке ISCO с силикагелем (24 г), элюируя смесью этилацетат/гексан с градиентом 0-20%, получая указанное в заголовке соединение.
LC-MS [M+Na]+: m/z 311,2.
Стадия B: метил-2-(4-бромфенокси)-3-гидроксипропаноат
К перемешиваемому раствору метил-2-(4-бромфенокси)-3-метоксипропаноата (1,0 г, 3,6 ммоль) в CHCl3 (18 мл) добавляли TMS-I (3,0 мл, 22 ммоль). Эту смесь перемешивали при комнатной температуре в течение выходных. Затем реакционную смесь гасили с помощью МеОН, и летучие вещества удаляли в вакууме. Полученный остаток затем растворяли в EtOAc, и последовательно промывали водным раствором NaOH (1 Н) и насыщенным раствором соли, сушили над MgSO4, фильтровали и фильтрат концентрировали. Полученный остаток очищали на колонке ISCO с силикагелем (24 г), элюируя смесью этилацетат/гексан с градиентом 0-30%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 275,1.
Стадия C: 2-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-3-гидроксипропановая кислота
К раствору метил-2-(4-бромфенокси)-3-гидроксипропаноата (0,14 г, 0,51 ммоль), трет-бутил-(3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропил)карбамата (0,18 г, 0,51 ммоль)) и дихлорида 1,1'-бис(ди-трет-бутилфосфино)-ферроцен палладия (0,033 г, 0,051 ммоль) в диоксане (2,5 мл) добавляли 1 М водный раствор фосфата калия (1,5 мл, 1,5 ммоль). Флакон для СВЧ герметизировали, дегазировали, заполняли азотом, и перемешивали при 70°С в течение 1 часа в микроволновом реакторе. Смесь фильтровали, фильтрат разбавляли водой и экстрагировали EtOAc. Водный слой подкисляли НС1 и экстрагировали смесью вода/EtOAc. Органический слой сушили над MgSO4, фильтровали и концентрировали, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 406,5.
Стадия D: бензгидрил-2-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-3-гидроксипропаноат
К раствору 2-(4-(1-(3-((трет-бутоксикарбонил)-амино)пропил)-1H-пиразол-4-ил)фенокси)-3-гидроксипропановой кислоты (0,21 г, 0,51 ммоль) в MeOH (5,1 мл) добавляли диазометан (0,50 г, 2,6 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 часа, а затем концентрировали. Полученный остаток очищали на колонке ISCO (12 г, gold) с использованием смеси EtOAc/гексан с градиентом 0-100%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 572,6.
Стадия E: бензгидрил-2-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-3-((1,3-диоксоизоиндолин-2-ил)окси)пропаноат
К раствору бензгидрил-2-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-3-гидроксипропаноата (0,41 г, 0,72 ммоль) в THF (7,2 мл) при комнатной температуре добавляли 2-гидроксиизоиндолин-1,3-дион (0,35 г, 2,2 ммоль), трифенилфосфин (0,23 г, 0,86 ммоль) и DIAD (0,17 мл, 0,86 ммоль). Реакционную смесь перемешивали в течение 2 часов, а затем концентрировали. Полученный остаток очищали на колонке ISCO с силикагелем (40 г), элюируя смесью 0-50% EtOAc в гексане, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 717,7.
Стадия F: 4-(4-((1-(бензгидрилокси)-3-((1,3-диоксоизоиндолин-2-ил)окси)-1-оксопропан-2-ил)окси)-фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий иодид
К раствору бензгидрил-2-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-3-((1,3-диоксоизоиндолин-2-ил)окси)пропаноата (0,12 г, 0,17 ммоль) в ацетонитриле (1,7 мл) добавляли MeI (0,087 мл, 1,4 ммоль). Реакционную смесь перемешивали при 70°С в течение ночи. Затем смесь охлаждали и концентрировали, получая указанное в заголовке соединение.
LC-MS [М]+: m/z 731,4.
Стадия G: 4-(4-((1-(бензгидрилокси)-3-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-1-оксопропан-2-ил)окси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий
К раствору 4-(4-((1-(бензгидрилокси)-3-((1,3-диоксоизоиндолин-2-ил)окси)-1-оксопропан-2-ил)окси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий иодида (0,15 г, 0,17 ммоль) в CH2Cl2 (1,2 мл) при комнатной температуре добавляли гидразин (6,6 мг, 0,21 ммоль) в этаноле (1,2 мл). Полученный раствор перемешивали при комнатной температуре в течение 1,5 часов. К этой смеси добавляли 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусную кислоту (0,085 г, 0,31 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Затем смесь концентрировали, получая указанное в заголовке соединение.
LC-MS [М]+: m/z 855,7.
Стадия H: 4-(4-((1-(бензгидрилокси)-3-(((1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-1-оксопропан-2-ил)окси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий
К раствору 4-(4-((1-(бензгидрилокси)-3-((((2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-1-оксопропан-2-ил)окси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ия (0,15 г, 0,17 ммоль) в DMF (3,5 мл) добавляли DCC (0,090 г, 0,44 ммоль) и HOBt (0,067 г, 0,44 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут, и затем добавляли S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфат (0,073 г, 0,35 ммоль) и гидрокарбонат натрия (0,073 г, 0,87 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи, затем фильтровали шприцом через спеченный слой, и промывали DMF (1,5 мл). Раствор очищали с помощью системы ISCO на колонке с обращенной фазой С-18 (43 г), элюируя смесью 10-100% ACN/вода с 0,05% TFA (25 мин), получая указанное в заголовке соединение.
LC-MS [М]+: m/z 1048,1.
Стадия I: (3S)-3-(2-((2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-2-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат (84)
К раствору (3S)-3-(2-((3-(бензгидрилокси)-2-(4-(1-(3-((трет-бутокси-карбонил)амино)пропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-3-оксопропокси)-имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата (0,10 г, 0,095 ммоль) в CH2Cl2 (3,2 мл) добавляли TFA (3,2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов, и растворитель удаляли при пониженном давлении. К полученному остатку добавляли эфир, и снова удаляли растворитель при пониженном давлении. Полученный остаток сушили в вакууме, а затем растворяли в 4 мл DMSO и очищали на обращенно-фазовой колонке Gilson (MeCN/вода 0,05% TFA, градиент 5-25%), получая указанное в заголовке соединение.
Пример 84: LC-MS [М+Н]+: m/z 681,5.
1H-ЯМР (500 МГц, D2O, м.д.): δ 8,43 (с, 1H), 8,36 (с, 1H), 7,43 (д, J=10 Гц, 2H), 6,97 (с, 1H), 6,99 (д, J=10 Гц, 2H), 4,89 (ш. дд, 1H), 4,63 (м, 1H), 4,59 (с, 1H), 4,55 (м, 1H), 4,47 (т, J=10 Гц, 2H), 4,03 (с, 3H), 3,03 (т, J=5 Гц, 2H), 2,24 (м, 2H), 1,34 (с, 3H), 0,98 (с, 3H).
Пример 85: LC-MS [М+Н]+: m/z 681,5.
ПРИМЕР 86
(S)-3-(2-(2-аминотиазол-4-ил)-2-((2-(4-(4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-2-ил)фенокси)этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил гидросульфат
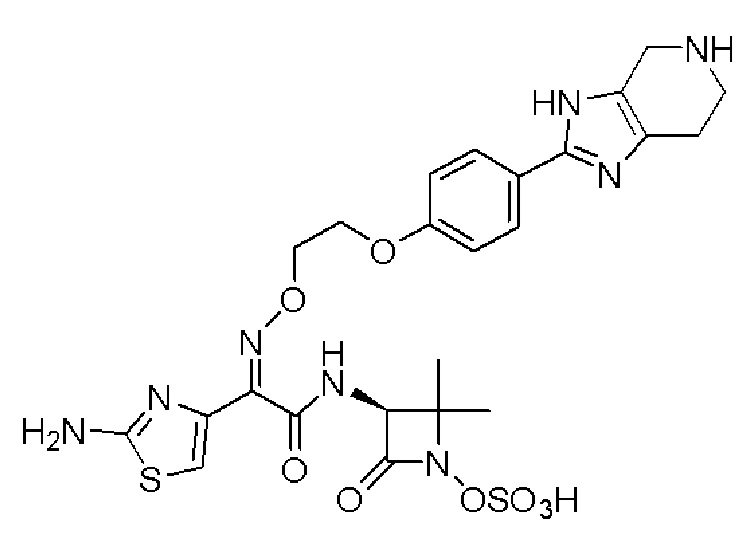
Стадия A: (3S,4R)-трет-бутил-3-азидо-4-(((бензилокси)карбонил)амино)пиперидин-1-карбоксилат
К раствору (3R,4R)-трет-бутил 4-(((бензилокси)карбонил)амино)-3-гидроксипиперидин-1-карбоксилата (2,0 г, 5,7 ммоль) в CH2Cl2 (28 мл) при 0°C добавляли по каплям TEA (1,6 мл, 11 ммоль) и метансульфонилхлорид (0,53 мл, 6,8 ммоль). Реакционную смесь перемешивали в течение 0,5 часа, затем разбавляли CH2Cl2, промывали НСl, насыщенным NaHCO3 и насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали. Полученный остаток растворяли в DMFА (28,5 мл), а затем добавляли азид натрия (1,1 г, 17 ммоль). Реакционную смесь перемешивали в течение ночи при 80°С. Затем реакционную смесь охлаждали, разбавляли EtOAc, промывали водой и насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали. Полученный остаток очищали с использованием колонки ISCO с силикагелем (40 г), элюируя смесью этилацетат/гексан с градиентом 0-30%, получая указанное в заголовке соединение.
Стадия B: (3S,4R)-трет-бутил-3,4-диаминопиперидин-1-карбоксилат
К раствору (3S,4R)-трет-бутил-3-азидо-4-(((бензилокси)карбонил)амино)пиперидин-1-карбоксилата (1,5 г, 4,1 ммоль) в MeOH (20 мл) добавляли Pd-C (0,15 г, 0,14 ммоль). Реакционную смесь перемешивали в течение выходных при комнатной температуре под током водорода из баллона. Затем реакционную смесь фильтровали через целит, промывали с помощью CH2Cl2, и концентрировали, получая указанное в заголовке соединение.
Стадия C: (3aS,7aR)-трет-бутил-2-(4-(бензилокси)фенил)-3a,4,7,7a-тетрагидро-3H-имидазо[4,5-c]пиридин-5(6H)-карбоксилат
К раствору 4-(бензилокси)бензальдегида (0,25 г, 1,2 ммоль) в трет-BuOH (12 мл) при комнатной температуре добавляли (3S,4R)-трет-бутил-3,4-диаминопиперидин-1-карбоксилат (0,30 г, 1,4 ммоль). Реакционную смесь перемешивали в течение 30 минут. Затем добавляли карбонат калия (0,48 г, 3,5 ммоль) и иод (0,37 г, 1,45 ммоль), и реакционную смесь перемешивали при 70°С в течение 3 часов. Реакционную смесь охлаждали, затем добавляли этилацетат, и смесь промывали водой и насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали, получая указанное в заголовке соединение.
LC-MS [М+Н].+: m/z 408,5.
Стадия D: трет-бутил-2-(4-(бензилокси)фенил)-6,7-дигидро-3H-имидазо[4,5-c]пиридин-5(4H)-карбоксилат
К 2 М раствору оксалилхлорида (0,28 мл, 0,55 ммоль) в CH2Cl2 в атимосфере азота при -78°С добавляли по каплям раствор DMSO (0,073 мл, 1,03 ммоль) в CH2Cl2 (0,2 мл). Реакционную смесь перемешивали в течение 5 минут, затем добавляли по каплям раствор (3aS,7aR)-трет-бутил-2-(4-(бензилокси)фенил)-3a,4,7,7a-тетрагидро-3H-имидазо[4,5-c]пиридин-5(6H)-карбоксилата (0,10 г, 0,24 ммоль) в CH2Cl2 (0,5 мл). Реакционную смесь перемешивали в течение 1 часа, затем добавляли по каплям TEA (0,37 мл, 2,6 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 2 часов. Добавляли воду, и смесь экстрагировали с помощью DCM. Органические слои объединяли, промывали насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 406,4.
Стадия E: ди-трет-бутил-2-(4-(бензилокси)фенил)-6,7-дигидро-1H-имидазо[4,5-c]пиридин-1,5(4H)-дикарбоксилат и ди-трет-бутил-2-(4-(бензилокси)фенил)-6,7-дигидро-3H-имидазо[4,5-c]пиридин-3,5(4H)-дикарбоксилат
Смесь трет-бутил-2-(4-(бензилокси)фенил)-6,7-дигидро-3H-имидазо[4,5-c]пиридин-5(4H)-карбоксилата (0,2 г, 0,49 ммоль), BOC-ангидрида (0,23 мл, 0,97 ммоль) и DMAP (0,060 г, 0,49 ммоль) перемешивали при комнатной температуре в течение 1,5 часов. Затем смесь концентрировали и очищали на колонке ISCO с силикагелем (12 г), элюируя смесью этилацетат/гексан с градиентом 0-30%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 506,5.
Стадия F: ди-трет-бутил-2-(4-гидроксифенил)-6,7-дигидро-3H-имидазо[4,5-c]пиридин-3,5(4H)-дикарбоксилат
Смесь ди-трет-бутил-2-(4-(бензилокси)фенил)-6,7-дигидро-3H-имидазо[4,5-c]пиридин-3,5(4H)-дикарбоксилата (0,17 г, 0,34 ммоль), Pd/C (0,36 г, 0,34 ммоль) и формиата аммония (0,10 г, 1,6 ммоль) в MeOH (7 мл) кипятили с обратным холодильником в течение 2 часов. Затем смесь фильтровали через целит с промывкой CH2Cl2, и концентрировали, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 416,3.
Стадия G: ди-трет-бутил-2-(4-(2-((1,3-диоксоизоиндолин-2-ил)окси)этокси)фенил)-6,7-дигидро-3H-имидазо[4,5-c]пиридин-3,5(4H)-дикарбоксилат
К раствору ди-трет-бутил-2-(4-гидроксифенил)-6,7-дигидро-3H-имидазо[4,5-c]пиридин-3,5(4H)-дикарбоксилат, 2-(2-гидроксиэтокси)изоиндолин-1,3-диона (0,061 г, 0,29 ммоль) и PPh3 (0,085 г, 0,32 ммоль) при комнатной температуре добавляли DEAD (0,15 мл, 0,32 ммоль, 40%-ный раствор) в толуоле. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем смесь концентрировали и очищали на колонке ISCO с силикагелем (12 г), элюируя смесью EA/гексан с градиентом 0-100%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 605,4.
Стадия H: (3aS,7aR)-ди-трет-бутил-2-(4-(2-(аминоокси)этокси)фенил)-3a,4,7,7a-тетрагидро-3H-имидазо[4,5-c]пиридин-3,5(6H)-дикарбоксилат
К раствору ди-трет-бутил-2-(4-(2-((1,3-диоксоизоиндолин-2-ил)окси)этокси)фенил)-6,7-дигидро-3H-имидазо[4,5-c]пиридин-3,5(4H)-дикарбоксилата (0,18 г) в CH2Cl2 (1,5 мл) при комнатной температуре добавляли гидразин (11 мкл) в этаноле (1,5 мл). Полученный раствор перемешивали при комнатной температуре в течение 0,5 часа, затем разбавляли DCM и 3 раза промывали насыщенным раствором NaHCO3. Водную фазу экстрагировали с помощью DCM. Объединенные органические слои промывали насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении, получая указанное в заголовке соединение, которое непосредственно использовали на следующей стадии.
LC-MS [М+Н]+: m/z 477,5.
Стадия I: 2-((2-(4-(3,5-бис(трет-бутоксикарбонил)-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-2-ил)фенокси)этокси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)уксусная кислота Раствор ди-трет-бутил-2-(4-(2-(аминоокси)этокси)фенил)-6,7-дигидро-3H-имидазо[4,5-c]пиридин-3,5(4H)-дикарбоксилата (0,10 г, 0,22 ммоль) и 2-(2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)-2-оксоуксусной кислоты (0,072 г, 0,27 ммоль) в смеси этанола (1,5 мл) и СНСl3 (0,74 мл) перемешивали при комнатной температуре в течение 1,5 часа. Затем смесь концентрировали, получая указанное в заголовке соединение, которое непосредственно использовали на следующей стадии.
LC-MS [М+Н]+: m/z 729,5.
Стадия J: (S)-ди-трет-бутил-2-(4-(2-(((1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-((2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)этокси)-фенил)-6,7-дигидро-3H-имидазо[4,5-c]пиридин-3,5(4H)-дикарбоксилат
К раствору 2-((2-(4-(3,5-бис(трет-бутоксикарбонил)-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-2-ил)фенокси)этокси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)уксусной кислоты (0,16 г, 0,22 ммоль) в DMF (2,2 мл) добавляли DCC (0,11 г, 0,55 ммоль) и HOBt (0,085 г, 0,55 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 30 минут, затем добавляли(S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфат (0,093 г, 0,44 ммоль) и гидрокарбонат натрия (0,093 г, 1,1 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи, затем фильтровали через шприц со спеченным слоем и промывали DMF (1,5 мл). Полученный раствор очищали с использованием системы ISCO с обращенно-фазовой колонки C-18 (86 г), элюируя смесью ACN/вода с 0,05% TFA с градиентом 10-100%, получая указанное в заголовке соединение.
LC-MS [М-Вос]+: m/z 821,6.
Стадия K: (S)-3-(2-(2-аминотиазол-4-ил)-2-((2-(4-(4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-2-ил)фенокси)этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил гидросульфат
К раствору (S)-ди-трет-бутил-2-(4-(2-(((1-(2-((трет-бутоксикарбонил)амино)-тиазол-4-ил)-2-((2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)-амино)окси)этокси)фенил)-6,7-дигидро-3H-имидазо[4,5-c]пиридин-3,5(4H)-дикарбоксилата (0,075 г, 0,081 ммоль) в CH2Cl2 (1,357 мл) добавляли TFA (1,4 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1,5 часа, затем растворитель удаляли при пониженном давлении. Полученный остаток сушили в вакууме, затем растворяли в 4 мл DMSO и очищали с разделением на обращенно-фазовой колонке Gilson (MeCN/вода 0,05% TFA, градиент 5-25%), получая указанное в заголовке соединение.
LC-MS [M+H]+: m/z 621,4
1H-ЯМР (500 МГц, D2O, м.д.): δ 7,66 (д, J=10 Гц, 2H), 7,05 (д, J=10 Гц, 2H), 6,91 (с, 1H), 4,63 (с, 1H), 4,50 (м, 2H), 4,35 (с, 2H), 4,33 (м, 2H), 3,55 (т, J=5 Гц, 2H), 3,00 (т, J=5 Гц, 2H), 2,24 (м, 2H), 1,34 (с, 3H), 0,98 (с, 3H).
ПРИМЕР 87
(S)-3-((Z)-2-((((S)-1-(4-(6-((2-аминоэтил)амино)-1-метилпиридин-1-ий-3-ил)фенокси)-2-карбоксипропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат

Стадия A: (S)-трет-бутил-2-(аминоокси)-3-(4-(6-((2-((трет-бутоксикарбонил)амино)этил)амино)пиридин-3-ил)фенокси)-2-метилпропаноат
Смесь (S)-трет-бутил-2-(аминоокси)-3-(4-бромфенокси)-2-метилпропаноата (0,5 г, 1,4 ммоль), бис(пинаколато)-диборона (0,38 г, 1,5 ммоль), ацетата калия (0,42 г, 4,3 ммоль) и дихлорида 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладия (0,14 г, 0,22 ммоль) в диоксане (7,2 мл) трижды дегазировали с помощью вакуума и продувки азотом. Реакционную смесь нагревали при 70°С в течение ночи. Затем реакционную смесь охлаждали, и добавляли дихлорид 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладия (0,094 г, 0,1 экв.), трет-бутил-(2-((5-бромпиридин-2-ил)амино)этил)-карбамат (первый элюент хирального разделения SFC промежуточного соединения 10 с использованием колонки OD-H, 4,6×250 мм, 20% МеОН/СО2, 2,5 мл/мин, 100 бар, 35°С) (0,50 г, 1,6 ммоль) и 1 М раствор трехосновного фосфата калия (4,3 мл, 4,3 ммоль) в воде. Смесь трижды дегазировали с помощью вакуума и продувки водородом, затем нагревали при 70°С в течение 5 часов. Затем реакционную смесь фильтровали через целит, и фильтрат концентрировали. Полученный остаток очищали с помощью ISCO (40 г), используя 0-100% EA/гексан, с получением продукта, который снова очищали с помощью ISCO (40 г), используя 0-30% 3:1 EA/гексан, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 503,6.
Стадия B: (S)-трет-бутил-3-(4-(6-((2-((трет-бутоксикарбонил)амино)этил)амино)пиридин-3-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-2-метилпропаноат
Смесь (S)-трет-бутил-2-(аминоокси)-3-(4-(6-((2-((трет-бутоксикарбонил)амино)этил)амино)пиридин-3-ил)фенокси)-2-метилпропаноата (0,25 г, 0,50 ммоль), изобензофуран-1,3-диона (0,074 г, 0,50 ммоль) и TEA (0,069 мл, 0,50 ммоль) в толуоле (4,97 мл) нагревали при 120°С в течение 1,5 часа. Затем смесь охлаждали до комнатной температуры и концентрировали. Полученный остаток очищали на колонке ISCO с силикагелем (24 г), элюируя смесью ЕА/гексан с градиентом 0-100%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 633,4.
Стадия C: (S)-5-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-2-метил-3-оксо-пропокси)фенил)-2-((2-((трет-бутоксикарбонил)амино)этил)амино)-1-метилпиридин-1-ий иодид
К раствору (S)-трет-бутил-3-(4-(6-((2-((трет-бутоксикарбонил)амино)этил)-амино)пиридин-3-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-2-метилпропаноата (0,13 г, 0,20 ммоль) в ацетонитриле (2,0 мл) добавляли MeI (0,102 мл, 1,6 ммоль). Реакционную смесь перемешивали при 75°С в течение 3 часов, и затем концентрировали, получая указанное в заголовке соединение.
LC-MS [M]+: m/z 648,4.
Стадия D: (S)-3-((Z)-2-((((S)-1-(4-(6-((2-аминоэтил)амино)-1-метилпиридин-1-ий-3-ил)фенокси)-2-карбоксипропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
Соединение (S)-5-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-2-метил-3-оксопропокси)фенил)-2-((2-((трет-бутоксикарбонил)амино)этил)амино)-1-метилпиридин-1-ий иодид превращали в указанное в заголовке соединение в соответствии с процедурой, аналогичной процедуре стадии E-H примера 1.
1H-ЯМР (500 МГц, D2O, м.д.): δ 8,15 (д, J=10 Гц, 1H), 8,11 (с, 1H), 7,40 (д, J=10 Гц, 2H), 7,15 (д, J=10 Гц, 1H), 6,97 (д, J=10 Гц, 2H), 6,90 (с, 1H), 4,50 (с, 1H), 4,44 (д, J=15 Гц, 1H), 4,30 (д, J=15 Гц, 1H), 3,80 (с, 3H), 3,73 (т, J=5 Гц, 2H), 3,20 (т, J=5 Гц, 2H), 1,54 (с, 3H), 1,31 (с, 3H), 1,10 (с, 3H).
LC-MS [M+H]+: m/z 707,4.
ПРИМЕР 88
(S)-3-((Z)-2-(((S)-2-(4-(6-(3-аминопропил)-1-метилпиридин-1-ий-3-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
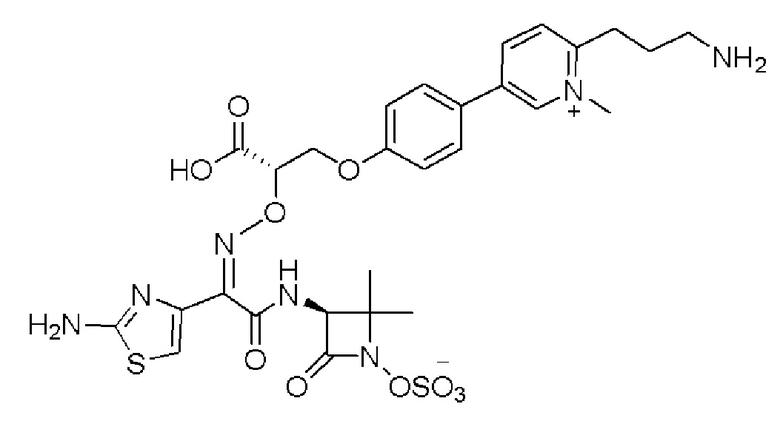
Стадия A: трет-бутил-(3-(5-бромпиридин-2-ил)проп-2-ин-1-ил)карбамат
Смесь 2,5-дибромпиридина (0,76 г, 3,2 ммоль), трет-бутил-проп-2-ин-1-илкарбамата (0,5 г, 3,2 ммоль), дихлорида дифенилфосфин палладия (0,045 г, 0,064 ммоль) и иодида меди (I) (0,012 г, 0,064 ммоль) в TEA (10 мл, 72 ммоль) охлаждали на бане со льдом в течение 1 часа. Затем реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь разбавляли Et2O, промывали водой (4х), насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали. Полученный остаток очищали с помощью ISCO (40 г; 0-30% EA/гексан), получая указанное в заголовке соединение.
Стадия B: (R)-трет-бутил-3-(4-(6-(3-((трет-бутоксикарбонил)амино)проп-1-ин-1-ил)пиридин-3-ил)фенокси)-2-гидроксипропаноат
К раствору (R)-трет-бутил-2-гидрокси-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)пропаноата (0,4 г, 1,1 ммоль), трет-бутил-(3-(5-бромпиридин-2-ил)проп-2-ин-1-ил)карбамата (0,38 г) и и дихлорида 1,1'-бис(ди-трет-бутилфосфино)-ферроцен палладия (0,072 г, 0,11 ммоль) в THF (5,5 мл) во флаконе для СВЧ добавляли 1 М водный раствор фосфата калия (3,3 мл, 3,3 ммоль). Флакон герметизировали, дегазировали (3 х), заполняли азотом, и перемешивали при 70°С в течение 3 часов. Затем реакционную смесь разбавляли насыщенным раствором NH4Cl, и экстрагировали с помощью EtOAc. Органические слои объединяли, сушили над MgSO4 и концентрировали досуха. Полученный остаток очищали на колонке ISCO с силикагелем (24 г), элюируя смесью EtOAc/гексан с градиентом 0-60%, получая указанное в заголовке соединение.
LC-MS [М-Вос]+: m/z 413,3.
Стадия C: (R)-трет-бутил-3-(4-(6-(3-((трет-бутоксикарбонил)амино)пропил)пиридин-3-ил)фенокси)-2-гидроксипропаноат
Смесь (R)-трет-бутил-3-(4-(6-(3-((трет-бутоксикарбонил)-амино)проп-1-ин-1-ил)пиридин-3-ил)фенокси)-2-гидроксипропаноата (0,33 г, 0,71 ммоль) и Pd-C (0,067 г, 0,63 ммоль) в MeOH (3,6 мл) перемешивали при комнатной температуре под током водорода из баллона в течение ночи. Реакционную смесь фильтровали через целит с помощью CH2Cl2, и фильтрат концентрировали, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 473,0.
Стадия D: (S)-3-((Z)-2-(((S)-2-(4-(6-(3-аминопропил)-1-метилпиридин-1-ий-3-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
Соединение (R)-трет-бутил-3-(4-(6-(3-((трет-бутоксикарбонил)амино)пропил)пиридин-3-ил)фенокси)-2-гидроксипропаноат превращали в указанное в заголовке соединение в соответствии с процедурой, аналогичной процедуре стадий C-H примера 1.
1H-ЯМР (500 МГц, D2O, м.д.): δ 8,85 (с, 1H), 8,49 (д, J=5 Гц, 1H), 7,82 (д, J=5 Гц, 1H), 7,56 (д, J=10 Гц, 2H), 7,02 (д, J=10 Гц, 2H), 6,98 (с, 1H), 5,01 (с, 1H), 4,47 (м, 3H), 4,19 (с, 3H), 3,10 (м, 4H), 2,09 (м, 2H), 1,31 (с, 3H), 0,92 (с, 3H).
LC-MS [M+H]+: m/z 692,5.
ПРИМЕР 89
(S)-3-((Z)-2-((((R)-1-(4-(6-((2-аминоэтил)амино)-1-метилпиридин-1-ий-3-ил)фенокси)-3-гидроксипропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат

Стадия A: (R)-4-((4-бромфенокси)метил)-2,2-диметил-1,3-диоксолан
К раствору (R)-(2,2-диметил-1,3-диоксолан-4-ил)метанола (5,0 г, 38 ммоль), трифенилфосфина (13 г, 49 ммоль) и 4-бромфенола (15 г, 87 ммоль) в толуоле (200 мл) добавляли DEAD (6,0 мл, 38 ммоль). Реакционную смесь нагревали до 70°С и перемешивали в течение 18 часов. Затем реакционную смесь упаривали и очищали хроматографией на силикагеле (SiO2, ЕА:PE=от 0% до 5%), получая указанное в заголовке соединение.
Стадия B: (S)-3-(4-бромфенокси)пропан-1,2-диол
К раствору (R)-4-((4-бромфенокси)метил)-2,2-диметил-1,3-диоксолана (9 г, 31 ммоль) в MeOH (150 мл) добавляли Amberlyst-15 (660 мг). Реакционную смесь перемешивали в течение 24 часов при 25°С, после чего отфильтровывали Amberlyst-15, и фильтрат концентрировали. Полученный остаток очищали с помощью хроматографии на силикагеле (SiO2, этилацетат/гексан, от 2:1 до 4:1), получая указанное в заголовке соединение.
Стадия C: (R)-1-(4-бромфенокси)-3-(тритилокси)пропан-2-ол
К раствору (S)-3-(4-бромфенокси)пропан-1,2-диола (5 г, 20 ммоль) и TBAI (2,2 г, 6,1 ммоль) в CH2Cl2 (100 мл) добавляли DIPEA (11 мл, 61 ммоль). Затем при 25°С добавляли (хлорметантриил)трибензол (6,2 г, 22 ммоль), и реакционную смесь перемешивали в течение 18 часов при 25°С. Реакцию гасили добавлением 3 М водного раствора НСl (50 мл). Органический слой отделяли, промывали насыщенным NaHCO3 (50 мл) и насыщенным раствором соли (50 мл), сушили над Na2SO4 и фильтровали. Фильтрат концентрировали, и полученный остаток очищали с помощью хроматографии на силикагеле (SiO2, EtOAc/РЕ, от 1:10 до 1:4), получая указанное в заголовке соединение.
Стадия D: (R)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)-3-(тритилокси)пропан-2-ол
К раствору (R)-1-(4-бромфенокси)-3-(тритилокси)пропан-2-ола (3 г, 6,1 ммоль) в DMF (50 мл) добавляли 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан)(1,7 г, 6,7 ммоль), ацетат калия (1,8 г, 18 ммоль, 3 экв.) и PdCl2(dppf) (0,31 г, 0,43 ммоль). Реакционную смесь продували азотом и нагревали в течение 10 часов при 90°С. Затем реакционную смесь разбавляли 500 мл EtOAc и фильтровали. Фильтрат промывали два раза водой (по 300 мл). Органический слой отделяли, сушили над сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали с помощью хроматографии на силикагеле (SiO2, EA:PE=от 0% до 20%), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 359,2.
Стадия E: (R)-трет-бутил-(2-((5-(4-(2-гидрокси-3-(тритилокси)пропокси)фенил)пиридин-2-ил)амино)этил)карбамат
К раствору PdCl2(dppf)-CH2Cl2 (78 мг, 0,095 ммоль), трет-бутил-(2-((5-бромпиридин-2-ил)амино)этил)карбамата (300 мг) и (R)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)-3-(тритилокси)пропан-2-ола (510 мг, 0,95 ммоль) в диоксане (4,5 мл ) добавляли Na2CO3 (300 мг, 2,8 ммоль) в воде (1,5 мл). Полученную смесь 3 раза вакуумировали и продували азотом, а затем нагревали при 100°С в микроволновой печи в течение 1 часа. Затем реакционную смесь охлаждали и разбавляли EtOAc, сушили (MgSO4), фильтровали и концентрировали. Полученный остаток очищали на колонке с силикагелем (ISCO, gold, 80 г), используя смесь EtOAc/гексан 0-100%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 646,6.
Стадия F: (S)-3-((Z)-2-((((R)-1-(4-(6-((2-аминоэтил)амино)-1-метилпиридин-1-ий-3-ил)фенокси)-3-гидроксипропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
(R)-трет-бутил-(2-((5-(4-(2-гидрокси-3-(тритилокси)- пропокси) фенил)пиридин-2-ил)амино)этил)карбамат превращали в указанное в заголовке соединение в виде соли TFA следуя процедуре, аналогичной процедуре стадий С-H примера 1.
LC-MS [М+Н]+: m/z 679,6.
1H-ЯМР (500 МГц, D2O, м.д.): δ 8,15 (д, J=10 Гц, 1H), 8,11 (с, 1H), 7,41 (д, J=10 Гц, 2H), 7,14 (д, J=10 Гц, 1H), 6,96 (д, J=10 Гц, 2H), 6,82 (с, 1H), 4,55 (с, 1H), 4,33 (д, J=10 Гц, 1H), 4,17 (дд, J=10, 5 Гц, 1H), 3,82 (м, 2H), 3,80 (с, 3H), 3,71 (т, J=5 Гц, 2H), 3,19 ( т, J=5 Гц, 2H), 1,33 (с, 3H), 0,95 (с, 3H).
ПРИМЕР 90
(S)-3-((Z)-2-((((S)-1-(4-(6-((2-аминоэтил)амино)-1-метилпиридин-1-ий-3-ил)фенокси)пропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
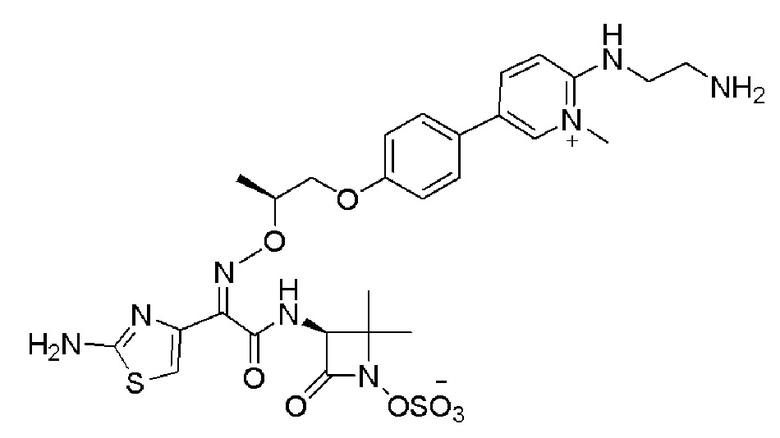
Стадия A: (R)-1-(4-бромфенокси)пропан-2-ол
К раствору 4-бромфенола (1,8 г, 1 1 ммоль) в DMF (25 мл) добавляли K2CO3 (2,9 г, 21 ммоль) и (R)-1-хлорпропан-2-ол (2,0 г, 21 ммоль). Реакционную смесь перемешивали и нагревали при 140°С в течение 48 часов. Анализ TLC показал наличие нового пятна. Реакционную смесь разбавляли водой (80 мл) и экстрагировали EtOAc (100 мл х 3). Объединенные органические слои сушили над Na2SO4, фильтровали, концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии (SiO2, EtOAc/РЕ=от 0% до 15%), получая указанное в заголовке соединение.
Стадия B: (R)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)пропан-2-ол
(R)-1-(4-бромфенокси)пропан-2-ол превращали в (R)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)пропан-2-ол следуя процедуре согласно стадии D примера 89.
Стадия C: (R)-трет-бутил-(2-((5-(4-(2-гидроксипропокси)фенил)пиридин-2-ил)амино)этил)-карбамат
К раствору аддукта PdCl2(dppf)-CH2Cl2 (88 мг, 0,11 ммоль), трет-бутил-(2-((5-бромпиридин-2-ил)амино)этил)карбамата (340 мг) и (R)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)пропан-2-ола (300 мг, 1,1 ммоль) в диоксане (4,5 мл) добавляли Na2CO3 (340 мг, 3,2 ммоль) в воде (1,5 мл). Полученную смесь 3 раза вакуумировали и продували водородом, затем нагревали при 100°С в микроволновой печи в течение 1 часа. Затем реакционную смесь охлаждали и разбавляли EtOAc, сушили (MgSO4), фильтровали и концентрировали. Полученный остаток очищали на колонке с силикагелем (ISCO, gold, 80 г), используя 0-100% EtOAc/гексан, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 388,4.
Стадия D: (S)-трет-бутил-(2-((5-(4-(2-((1,3-диоксоизоиндолин-2-ил)окси)пропокси)фенил)пиридин-2-ил)амино)этил)карбамат
К раствору (R)-трет-бутил-(2-((5-(4-(2-гидроксипропокси)-фенил)пиридин-2-ил)амино)этил)карбамата (210 мг, 0,54 ммоль) в THF (4 мл) добавляли 2-гидроксиизоиндолин-1,3-дион (106 мг, 0,65 ммоль) и трифенилфосфин (200 мг, 0,76 ммоль), с последующим добавлением по каплям DIAD (0,15 мл, 0,76 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 часа, а затем концентрировали. Полученный остаток очищали на колонке с силикагелем (ISCO, gold, 80 г), используя смесь EtOAc/гексан с градиентом 0-100%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 533,3.
Стадия E: (S)-3-((Z)-2-((((S)-1-(4-(6-((2-аминоэтил)амино)-1-метилпиридин-1-ий-3-ил)фенокси)пропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
(S)-трет-бутил-(2-((5-(4-(2-((1,3-диоксоизоиндолин-2-ил)окси)-пропокси)фенил)пиридин-2-ил)амино)этил)карбамат превращали в указанное в заголовке соединение в виде соли муравьиной кислоты следуя процедуре, аналогичной процедуре, описанной на стадии D-H примера 1.
1H-ЯМР (500 МГц, D2O, м.д.): δ 8,06 (д, J=10 Гц, 1H), 8,00 (с, 1H), 7,33 (д, J=10 Гц, 2H), 7,06 (д, J=10 Гц, 1H), 6,95 (д, J=10 Гц, 2H), 6,67 (с, 1H), 4,51 (м, 1H), 4,47 (с, 1H), 4,08 (д, J=10 Гц, 1H), 3,95 (дд, J=10, 5 Гц, 1H), 3,73 (с, 3H), 3,68 (т, J=5 Гц, 2H), 3,17 ( т, J=5 Гц, 2H), 1,21 (с, 3H), 1,18 (д, J=5 Гц, 3H), 0,92 (с, 3H).
LC-MS [М+Н]+: m/z 663,6.
ПРИМЕР 91
(3S)-3-(2-((2-((4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенил)амино)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
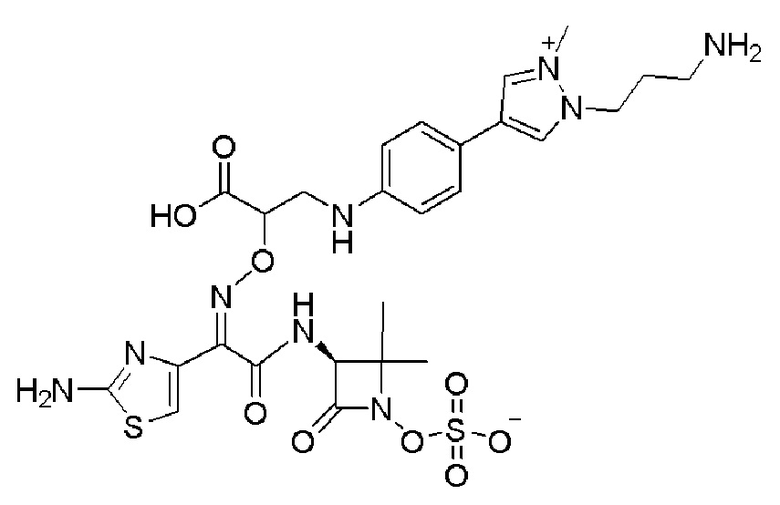
Стадия A: трет-бутил оксиран-2-карбоксилат
К смеси трет-бутил-акрилата (10 г, 78 ммоль) в DCM (100 мл) добавляли 80% m-CPBA (30 г, 140 ммоль) в DCM (140 мл). Реакционную смесь перемешивали при 40°С в течение 48 часов. Затем реакционную смесь охлаждали до 0°С и добавляли по каплям насыщенный водный раствор Na2SО3 (300 мл). Органический слой отделяли, промывали насыщенным водным раствором NaHCO3 (2×200 мл) и насыщенным раствором соли (200 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме, получая указанное в заголовке соединение, которое использовали на следующей стадии без очистки.
Стадия B: трет-бутил-3-((4-бромфенил)амино)-2-гидроксипропаноат
К смеси 4-броманилина (4,0 г, 23 ммоль) в ацетонитриле (80 мл) добавляли трет-бутил-оксиран-2-карбоксилат (5,0 г, 35 ммоль) и трифторметансульфонат лития (5,4 г, 35 ммоль). Реакционную смесь перемешивали при 60°С в атмосфере N2 в течение 15 часов, а затем реакционную смесь разбавляли EtOAc (100 мл) и водой (120 мл). Водный слой отделяли и экстрагировали EtOAc (3×50 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии (SiO2, PE:этилацетат=от 100:1 до 5:1), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 316,2.
Стадия C: трет-бутил-3-((4-бромфенил)амино)-2-((трет-бутилдиметилсилил)окси)пропаноат
К раствору трет-бутил-3-((4-бромфенил)амино)-2-гидроксипропаноата (3,3 г, 10 ммоль) и DBU (3,2 мл, 21 ммоль) в CH3CN (50 мл) добавляли трет-бутилхлордиметилсилан (2,4 г, 16 ммоль). Реакционную смесь перемешивали при 25°С в течение 2 часов, затем концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии (SiO2, PE:этилацетат=от 100:1 до 10:1), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 430,0.
Стадия D: трет-бутил-3-((4-бромфенил)(трет-бутоксикарбонил)амино)-2-((трет-бутилдиметилсилил)-окси)пропаноат
Раствор трет-бутил-3-((4-бромфенил)амино)-2-((трет-бутилдиметил-силил)окси)пропаноата (4,4 г, 10 ммоль) в (Boc)2O (50 мл) перемешивали при 125°С в течение 1 часа. Реакционную смесь охлаждали и растворитель удаляли. Полученный остаток очищали с помощью колоночной хроматографии (SiO2, PE:этилацетат=от 100:1 до 10:1), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 554,1.
Стадия E: трет-бутил-3-((трет-бутоксикарбонил)(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенил)амино)-2-((трет-бутилдиметилсилил)окси)пропаноат
К раствору трет-бутил-3-((4-бромфенил)(трет-бутоксикарбонил)амино)-2-((трет-бутилдиметилсилил)-окси)пропаноата (0,50 г, 0,94 ммоль), трет-бутил-(3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиразол-1-ил)пропил)карбамата (0,33 г, 0,94 ммоль) и дихлорида 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладия (0,061 г, 0,094 ммоль) в диоксане (4,71 мл) во флаконе для СВЧ добавляли водный раствор 1 М фосфата калия (2,8 мл, 2,8 ммоль). Флакон герметизировали, дегазировали и заполняли азотом, и перемешивали при 70°С в течение ночи. Затем реакционную смесь фильтровали, фильтрат разбавляли водой и экстрагировали EtOAc. Органический слой сушили над MgSO4, фильтровали и концентрировали. Полученный остаток очищали на колонке ISCO с силикагелем (12 г), элюируя смесью этилацетат/гексан с градиентом 0-100%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 675,9.
Стадия F: трет-бутил-3-((трет-бутоксикарбонил)(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенил)амино)-2-гидроксипропаноат
К раствору трет-бутил-3-((трет-бутоксикарбонил)(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенил)амино)-2-((трет-бутилдиметилсилил)окси)пропаноата (0,57 г, 0,85 ммоль) в THF (4,25 мл) добавляли 1 М раствор TBAF (0,85 мл, 0,85 ммоль, раствор в THF). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, а затем концентрировали. Полученный остаток очищали на колонке ISCO с силикагелем (12 г), элюируя смесью этилацетат/гексан с градиентом 0-100%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 561,5.
Стадия G: трет-бутил-3-((трет-бутоксикарбонил)(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенил)амино)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноат
К раствору трет-бутил-3-((трет-бутоксикарбонил)(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенил)амино)-2-гидроксипропаноата (0,36 г, 0,65 ммоль) в THF (6,5 мл) добавляли 2-гидроксиизоиндолин-1,3-дион (0,12 г, 0,71 ммоль), трифенилфосфин (0,20 г, 0,78 ммоль) и DIAD (0,15 мл, 0,78 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем концентрировали и очищали на колонке ISCO с силикагелем (12 г), элюируя смесью 0-100% EtOAc в гексане, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 706,6.
Стадия H: (3S)-3-(2-((2-((4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенил)амино)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
Трет-бутил-3-((трет-бутоксикарбонил)(4-(1-(3-((трет-бутокси-карбонил)амино)пропил)-1H-пиразол-4-ил)фенил)амино)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноат превращали в указанное в заголовке соединение в виде соли трифторуксусной кислоты в соответствии с процедурой, аналогичной процедуре, описанной на стадиях D-Н примера 1.
1H-ЯМР (500 МГц, D2O, м.д.): δ 8,39 (с, 1H), 8,35 (с, 1H), 7,35 (д, J=10 Гц, 2H), 6,88 (с, 1H), 6,76 (д, J=10 Гц, 2H), 4,78 (м, 1H), 4,67 (с, 1H), 4,46 (т, J=5 Гц, 2H), 4,03 (с, 3H), 3,61 (м, 2H), 3,03 (т, J=5 Гц, 2H), 2,24 (м, 2H), 1,28 (с, 3H), 1,05 (с, 3H).
LC-MS [M+H]+: m/z 680,6.
ПРИМЕР 92
(3S)-3-(2-((2-((4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенил)(метил)амино)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
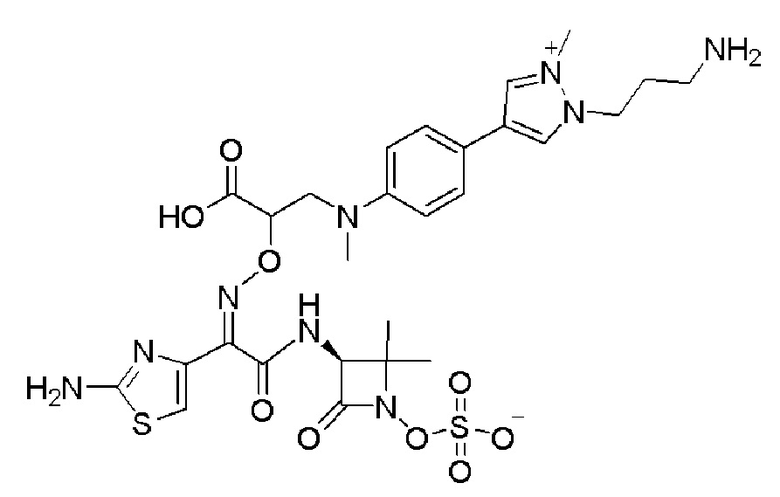
Стадия A: 4-бром-N-метиланилин
К смеси 4-броманилина (5,0 г, 29 ммоль) и Cs2CO3 (9,5 г, 29 ммоль) в DMF (60 мл) добавляли CH3I (2,7 мл, 44 ммоль). Реакционную смесь перемешивали при 25°С в течение 2 часов, затем разбавляли EtOAc (100 мл) и водой (200 мл). Водный слой экстрагировали EtOAc (3×50 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии (SiO2, PE:этилацетат=от 100:1 до 10:1), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 186,0.
Стадия B: трет-бутил-3-((4-бромфенил)(метил)амино)-2-гидроксипропаноат К смеси 4-бром-N-метиланилина (2,2 г, 12 ммоль) в CH3CN (50 мл) добавляли трет-бутил-оксиран-2-карбоксилат (2,6 г, 18 ммоль) и трифторметансульфонат лития (2,8 г, 18 ммоль). Реакционную смесь перемешивали при 65°С в атмосфере N2 в течение 15 часов, а затем разбавляли EtOAc (30 мл) и водой (50 мл). Водный слой экстрагировали с помощью EtOAc (3×30 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии (SiO2, PE:этилацетат=от 100:1 до 5:1), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 330,0.
Стадия C: трет-бутил-3-((4-бромфенил)(метил)амино)-2-((трет-бутилдиметилсилил)окси)-пропаноат
К раствору трет-бутил-3-((4-бромфенил)(метил)амино)-2-гидрокси-пропаноата (3,8 г, 11,51 ммоль) и DBU (2,081 мл, 13,81 ммоль) в CH3CN (50 мл) добавляли трет-бутилхлордиметилсилан (2,60 г, 17,26 ммоль). Реакционную смесь перемешивали при 25°С в течение 2 часов, затем концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии (SiO2, PE:этилацетат=от 100:1 до 10:1), получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 444,1.
Стадия D: (3S)-3-(2-((2-((4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенил)(метил)амино)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
Трет-бутил-3-((4-бромфенил)(метил)амино)-2-((трет-бутилдиметилсилил)окси)-пропаноат превращали в указанное в заголовке соединение в виде соли TFA следуя процедуре, аналогичной процедуре, описанной в примере 91.
1H-ЯМР (500 МГц, D2O, м.д.): δ 8,44 (с, 1H), 8,40 (с, 1H), 7,42 (д, J=10 Гц, 2H), 6,92 (д, J=10 Гц, 2H), 6,80 (с, 1H), 4,86 (м, 1H), 4,48 (т, J=5 Гц, 2H), 4,04 (с, 3H), 4,02 (дд, J=10, 5 Гц, 1H), 3,83 (с, 1H), 3,72 (д, J=10 Гц, 1H), 3,05 (т, J=5 Гц, 2H), 2,89 (с, 3H), 2,26 (м, 2H), 1,28 (с, 3H), 1,05 (с, 3H).
LC-MS [M+H]+: m/z 694,5.
ПРИМЕР 93
(3S)-3-(2-((3-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенил)-1-карбоксипропокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
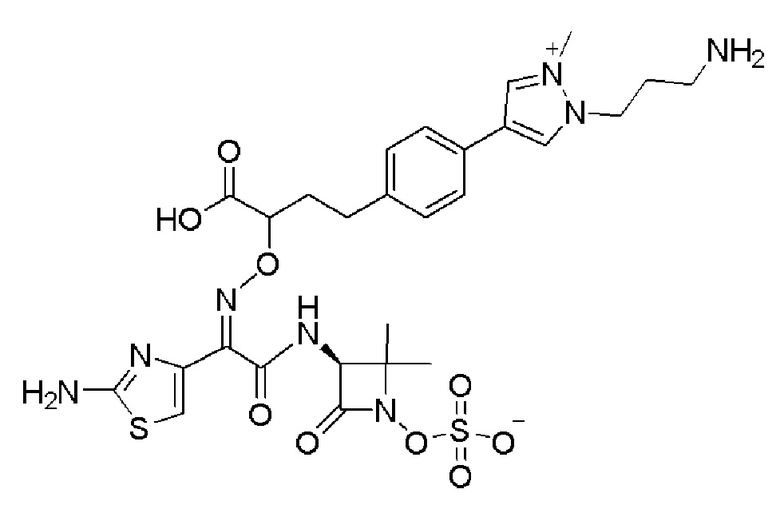
Стадия A: 4-(4-бромфенил)-2-гидроксимасляная кислота
Смесь (4-бромфенил)-метанола (10 г, 54 ммоль)), 2-гидроксипропановой кислоты (5,8 г, 64 ммоль), KOH (5,4 г, 96 ммоль), тетрагидрата ацетата никеля (II) (6,6 г, 27 ммоль) и трифенилфосфина (14 г, 54 ммоль) перемешивали при 160°C в течение 7,5 часов в атмосфере азота. Затем смесь охлаждали до 25°С, и добавляли H2O (40 мл) и 1 Н НС1 до достижения рН < 3. Смесь экстрагировали EtOAc (3×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (SiO2, PE:ЕtOАс=28:72), получая указанное в заголовке соединение.
Стадия B: бензгидрил 4-(4-бромфенил)-2-гидроксибутаноат
К смеси 4-(4-бромфенил)-2-гидроксимасляной кислоты (3,5 г, 6,8 ммоль) в THF (40 мл) добавляли 3,3-дифенил-3H-диазирин (1,3 г, 6,8 ммоль). Реакционную смесь перемешивали при 25°С в течение 16 часов, и затем концентрировали в вакууме. Полученный остаток очищали с помощью препаративной HPLC, получая указанное в заголовке соединение.
Стадия C: бензгидрил-4-(4-бромфенил)-2-гидроксибутаноат
Бензгидрил-4-(4-бромфенил)-2-гидроксибутаноат (4,5 г, 11 ммоль) разделяли с помощью SFC (колонка: OJ (250 мм х 50 мм, 10 мкм), подвижная фаза: А: СО2 В: 0,1% NH3H2O, EtOH, градиент: от 40% до 40% от скорости потока B: 200 мл/мин, длина волны: 220 нм), с получением первого изомера бензгидрил-4-(4-бромфенил)-2-гидроксибутаноата, и второго изомера бензгидрил-4-(4-бромфенил)-2-гидроксибутаноата.
Стадия D: бензгидрил-4-(4-бромфенил)-2-((трет-бутилдиметилсилил)окси)бутаноат
К смеси бензгидрил-4-(4-бромфенил)-2-гидроксибутаноата (2-й изомер, полученный на стадии 3, 2,2 г, 5,2 ммоль) в DMF (15 мл) добавляли 1H-имидазол (0,70 г, 10 ммоль) и трет-бутилхлордиметилсилан (1,9 г, 13 ммоль). Реакционную смесь перемешивали при 25°С в течение 16 часов в атмосфере азота. Затем растворитель удаляли в вакууме, и полученный остаток очищали с помощью колоночной хроматографии (SiO2, PE:этилацетат=20:1-5:1), получая указанное в заголовке соединение.
Стадия E: бензгидрил-4-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенил)-2-((трет-бутилдиметилсилил)окси)бутаноат
К раствору бензгидрил-4-(4-бромфенил)-2-((трет-бутилдиметилсилил)окси)бутаноата, 2-й пик (0,68 г, 1,3 ммоль), трет-бутил-(3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропил)карбамата (0,45 г, 1,3 ммоль) и дихлорида 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладия (0,082 г, 0,13 ммоль) в диоксане (6,3 мл) добавляли 1 M водный раствор фосфата калия (3,8 мл, 3,8 ммоль). Флакон для СВЧ герметизировали, дегазировали и заполняли N2, и перемешивали при 70°С в течение ночи. Затем смесь фильтровали, фильтрат разбавляли водой и экстрагировали EtOAc. Органический слой сушили над MgSO4, фильтровали и концентрировали. Полученный сырой продукт очищали с использованием колонки с силикагелем ISCO (24 г), элюируя смесью этилацетат/гексан с градиентом 0-50%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 684,7.
Стадия F: бензгидрил-4-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенил)-2-гидроксибутаноат
К раствору бензгидрил-4-(4-(1-(3-((трет-бутоксикарбонил)амино)-пропил)-1H-пиразол-4-ил)фенил)-2-((трет-бутилдиметилсилил)окси)бутаноата (0,70 г, 1,03 ммоль) в THF (5,1 мл) добавляли 1 М раствор TBAF (1,03 мл, 1,03 ммоль) в THF. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем концентрировали и очищали на колонке ISCO с силикагелем (24 г), элюируя смесью этилацетат/гексан с градиентом 0-100%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 570,6.
Стадия G: бензгидрил-4-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенил)-2-((1,3-диоксоизоиндолин-2-ил)окси)бутаноат
К раствору бензгидрил-4-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенил)-2-гидроксибутаноата (0,41 г, 0,72 ммоль) в THF (7,2 мл) при комнатной температуре добавляли 2-гидроксиизоиндолин-1,3-дион (0,13 г, 0,79 ммоль), трифенилфосфин (0,23 г, 0,86 ммоль) и DIAD (0,17 мл, 0,86 ммоль). Реакционную смесь перемешивали в течение 1 часа, затем смесь концентрировали и очищали на колонке ISCO с силикагелем (24 г), элюируя смесью этилацетат/гексан с градиентом 0-100%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 715,5.
Стадия H: 4-(4-(4-(бензгидрилокси)-3-((1,3-диоксоизоиндолин-2-ил)окси)-4-оксобутил)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий иодид
К раствору бензгидрил-4-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенил)-2-((1,3-диоксоизоиндолин-2-ил)окси)бутаноата (0,51 г, 0,71 ммоль) в ацетонитриле (7,1 мл) добавляли MeI (0,36 мл, 5,7 ммоль). Реакционную смесь перемешивали при 70°С в течение ночи. Затем смесь охлаждали и концентрировали, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 730,4.
Стадия I: (3S)-3-(2-((3-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенил)-1-карбоксипропокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
Соединение 4-(4-(4-(бензгидрилокси)-3-((1,3-диоксоизоиндолин-2-ил)окси)-4-оксобутил)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-пиразол-2-ий иодид превращали в указанное в заголовке соединение в виде соли с TFA следуя процедуре, аналогичной процедуре стадий Е-H примера 1.
1H-ЯМР (500 МГц, D2O, м.д.): δ 8,52 (с, 1H), 8,47 (с, 1H), 7,44 (д, J=10 Гц, 2H), 7,26 (д, J=10 Гц, 2H), 6,95 (с, 1H), 4,70 (м, 2H), 4,49 (т, J=5 Гц, 2H), 4,08 (с, 3H), 3,06 (т, J=5 Гц, 2H), 2,73-2,60 (м, 2H), 2,27 (т, J=5 Гц, 2H), 2,27-2,15 (м, 2H) 1,43 (с, 3H), 1,22 (с, 3H).
LC-MS [M+H]+: m/z 679,5.
ПРИМЕР 94
(2S)-3-(4-(2-((2-аминоэтил)амино)-1,4,5,6-тетрагидропиримидин-5-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота
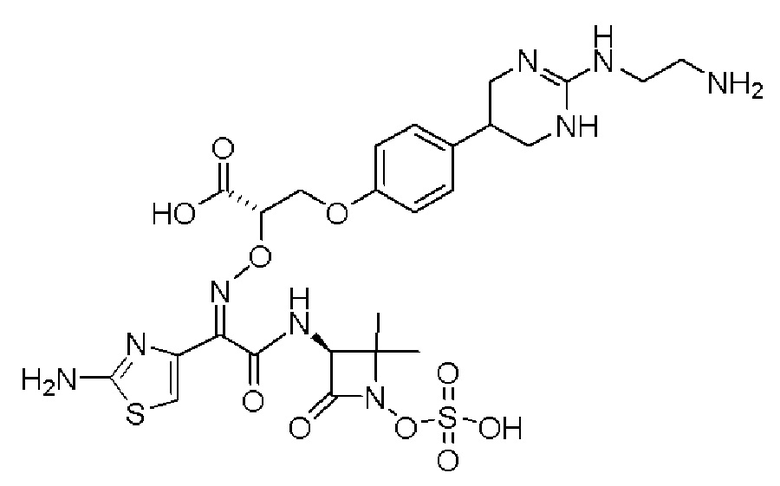
Стадия A: 4-(1,3-динитропропан-2-ил)фенол
К раствору 4-гидроксибензальдегида (5 г, 41 ммоль), бромида бис(трифенилфосфин)никеля (II) (6,1 г, 8,2 ммоль), K2CO3 (23 г, 164 ммоль) и трифенилфосфина (13 г, 49 ммоль) в CH3NO2 (500 мл) добавляли цинк (11 г, 164 ммоль). Смесь перемешивали при 80°С в течение 20 часов, затем смесь фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (SiO2, PE:этилацетат=45:55), получая указанное в заголовке соединение.
Стадия B: (R)-трет-бутил-3-(4-(1,3-динитропропан-2-ил)фенокси)-2-гидроксипропаноат
К смеси 4-(1,3-динитропропан-2-ил)фенола (4 г, 18 ммоль) и молекулярных сит (4Å, 4 г) в трет-бутил-оксиран-2-карбоксилате (10 г, 71 ммоль) добавляли Co-катализатор (CAS: 240489-45-6) (1,2 г, 1,4 ммоль) под N2. Полученную суспензию перемешивали при 25°C под N2 в течение 48 часов, затем смесь фильтровали и фильтрат очищали с помощью колоночной хроматографии (SiO2, PE:этилацетат=45:55), получая указанное в заголовке соединение.
Стадия C: (R)-трет-бутил-2-((трет-бутилдиметилсилил)окси)-3-(4-(1,3-динитропропан-2-ил)фенокси)пропаноат
К раствору (R)-трет-бутил-3-(4-(1,3-динитропропан-2-ил)фенокси)-2-гидроксипропаноата (4 г, 11 ммоль) в DMF (30 мл) добавляли имидазол (2,2 г, 32 ммоль) и трет-бутилхлордиметилсилан (3,3 г, 22 ммоль) при 0°C. Смесь нагревали до 25°С и перемешивали при 25°С в течение 3 часов. Затем к смеси добавляли EtOAc (50 мл) и промывали водой (3×50 мл) и насыщенным раствором соли (3×50 мл). Органический слой сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении, получая указанное в заголовке соединение, которое использовали на следующей стадии без дополнительной очистки.
Стадия D: (R)-трет-бутил-3-(4-(1,3-диаминопропан-2-ил)фенокси)-2-гидроксипропаноат
Раствор (R)-трет-бутил-2-((трет-бутилдиметилсилил)окси)-3-(4-(1,3-динитропропан-2-ил)фенокси)пропаноата (4 г, 8,25 ммоль) в EtOH (150 мл) добавляли оксид платины (II) (0,35 г, 1,7 ммоль). Смесь перемешивали в атмосфере H2 (50 фунтов на кв. дюйм (3,45 бар)) при 25°C в течение 16 часов. Затем смесь фильтровали и концентрировали при пониженном давлении с получением неочищенного (R)-трет-бутил-2-((трет-бутилдиметилсилил)окси)-3-(4-(1,3-динитропропан-2-ил)фенокси)пропаноата, который подвергали десилилированию способом сушки с замораживанием после кислотной очистки препаративной HPLC.
Стадия E: (R)-трет-бутил-2-гидрокси-3-(4-(2-триоксогексагидропиримидин-5-ил)фенокси)пропаноат
К перемешиваемой смеси (R)-трет-бутил-3-(4-(1,3-диаминопропан-2-ил)фенокси)-2-гидроксипропаноата (600 мг, 1,93 ммоль) в DCM (30 мл) добавляли пятью порциями в течение 10 минут 1,1'-тиокарбонилдиимидазол (362 мг, 2,03 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов, затем растворитель удаляли при пониженном давлении. Полученный остаток очищали на колонке с силикагелем (80 г) с использованием смеси EtOAc/гексан с градиентом 0-60%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 353,5.
Стадия F: (R)-трет-бутил-2-((трет-бутилдиметилсилил)окси)-3-(4-(2-триоксогексагидропиримидин-5-ил)фенокси)пропаноат
К раствору (R)-трет-бутил-2-гидрокси-3-(4-(2-триоксогексагидропиримидин-5-ил)фенокси)пропаноата (200 мг, 0,57 ммоль), имидазола (93 мг, 1,36 ммоль) и TBDMS-Cl (100 мг, 0,68 ммоль) в ацетонитриле (4 мл) добавляли DMAP (6,9 мг, 0,057 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь концентрировали. Полученный остаток очищали на колонке ISCO комби-флэш (gold, 40 г), элюируя смесью EtOAc/гексан с градиентом 0-100%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 467,5.
Стадия G: (2R)-трет-бутил-2-((трет-бутилдиметилсилил)окси)-3-(4-(2-(метилтио)-1,4,5,6-тетрагидропиримидин-5-ил)фенокси)пропаноат
Иодметан (0,17 мл, 2,8 ммоль) добавляли к перемешиваемой смеси (R)-трет-бутил-2-((трет-бутилдиметилсилил)окси)-3-(4-(2-триоксогексагидропиримидин-5-ил)фенокси)пропаноата (260 мг, 0,55 ммоль) в MeCN (3 мл). Смесь перемешивали при 70°C в течение 2 часов, затем охлаждали и выпаривали растворитель при пониженном давлении, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 481,0.
Стадия H: трет-бутил-(2R)-3-(4-(2-((2-((трет-бутоксикарбонил)амино)этил)амино)-1,4,5,6-тетрагидропиримидин-5-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноат
Основание Хюнига (0,19 мл, 1,1 ммоль) добавляли к перемешиваемой смеси N-Boc-этилендиамина (130 мг, 0,83 ммоль) и 2R)-трет-бутил-2-((трет-бутилдиметилсилил)окси)-3-(4-(2-(метилтио)-1,4,5,6-тетрагидропиримидин-5-ил)фенокси)пропаноата (260 мг, 0,55 ммоль) в диоксане (3 мл). Смесь перемешивали при 70°С в течение 24 часов, затем растворитель удаляли при пониженном давлении, получая указанное в заголовке соединение, которое использовали непосредственно на следующей стадии.
LC-MS [М+Н]+: m/z 593,5.
Стадия I: трет-бутил 5-(4-((R)-3-(трет-бутокси)-2-((трет-бутилдиметилсилил)окси)-3-оксопропокси)-фенил)-2-((трет-бутоксикарбонил)(2-((трет-бутоксикарбонил)амино)этил)амино)-5,6-дигидропиримидин-1(4H)-карбоксилат
Вос-ангидрид (2100 мкл, 9,3 ммоль) добавляли к (2R)-трет-бутил-3-(4-(2-((2-((трет-бутоксикарбонил)амино)этил)амино)-1,4,5,6-тетра-гидропиримидин-5-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноату (275 мг, 0,46 ммоль), и смесь перемешивали при 100°С в течение 6 часов. Затем реакционную смесь охлаждали и загружали на колонку ISCO (gold, 40 г), элюируя смесью EtOAc/гексан с градиентом 0-100%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 793,6.
Стадия J: трет-бутил 5-(4-((R)-3-(трет-бутокси)-2-гидрокси-3-оксопропокси)фенил)-2-((трет-бутоксикарбонил)(2-((трет-бутоксикарбонил)амино)этил)амино)-5,6-дигидропиримидин-1(4H)-карбоксилат
TBAF (0,79 мл, 0,79 ммоль) добавляли к перемешиваемой смеси трет-бутил-5-(4-((R)-3-(трет-бутокси)-2-((трет-бутилдиметилсилил)окси)-3-оксопропокси)фенил)-2-((трет-бутоксикарбонил)(2-((трет-бутоксикарбонил)амино)этил)амино)-5,6-дигидропиримидин-1(4H)-карбоксилата (420 мг, 0,53 ммоль) в THF (5 мл). Смесь перемешивали при при комнатной температуре в течение 1 часа, после чего растворитель удаляли. Полученный остаток очищали на колонке ISCO с силикагелем (40 г, gold), элюируя смесью EtOAc/гексан с градиентом 0-100%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 679,5.
Стадия K: трет-бутил-5-(4-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-2-((трет-бутоксикарбонил)(2-((трет-бутоксикарбонил)амино)этил)амино)-5,6-дигидропиримидин-1(4H)-карбоксилат
К раствору трет-бутил-5-(4-((R)-3-(трет-бутокси)-2-гидрокси-3-оксопропокси)фенил)-2-((трет-бутоксикарбонил)(2-((трет-бутоксикарбонил)амино)этил)амино)-5,6-дигидропиримидин-1(4H)-карбоксилата (360 мг, 0,53 ммоль) в THF (5 мл) добавляли 2-гидроксиизоиндолин-1,3-дион (104 мг, 0,64 ммоль), трифенилфосфин (195 мг, 0,74 ммоль), а затем по каплям добавляли DIAD (0,144 мл, 0,74 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 часа, затем концентрировали. Полученный остаток очищали на колонке с силикагелем (ISCO, gold, 40 г), используя смесь EtOAc/гексан с градиентом 0-100%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 824,8.
Стадия L: (2S)-3-(4-(2-((2-аминоэтил)амино)-1,4,5,6-тетрагидропиримидин-5-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота
Трет-бутил-5-(4-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-2-((трет-бутоксикарбонил)(2-((трет-бутоксикарбонил)амино)этил)амино)-5,6-дигидропиримидин-1(4H)-карбоксилат превращали в указанное в заголовке соединение в виде соли муравьиной кислоты согласно процедуре, аналогичной к процедуре стадий Е-H примера 1.
1H-ЯМР (500 МГц, D2O, м.д.): δ 7,13 (д, J=10 Гц, 2H), 7,02 (с, 1H), 6,85 (д, J=10 Гц, 2H), 5,05 (м, 1H), 4,68 (с, 1H), 4,36 (м, 2H), 3,45 (дд, J=15, 5 Гц, 2H), 3,38 (т, J=5 Гц, 2H), 3,32 (дд, J=10, 5 Гц, 2H), 3,11 (м, 1H), 3,06 (т, J=5 Гц, 2H), 1,34 (с, 3H), 0,95 (с, 3H).
LC-MS [M+H]+: m/z 684,4.
ПРИМЕР 95
(2S)-3-(4-(2-((2-аминоэтил)амино)-1,4,5,6-тетрагидропиримидин-5-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-метилпропановая кислота
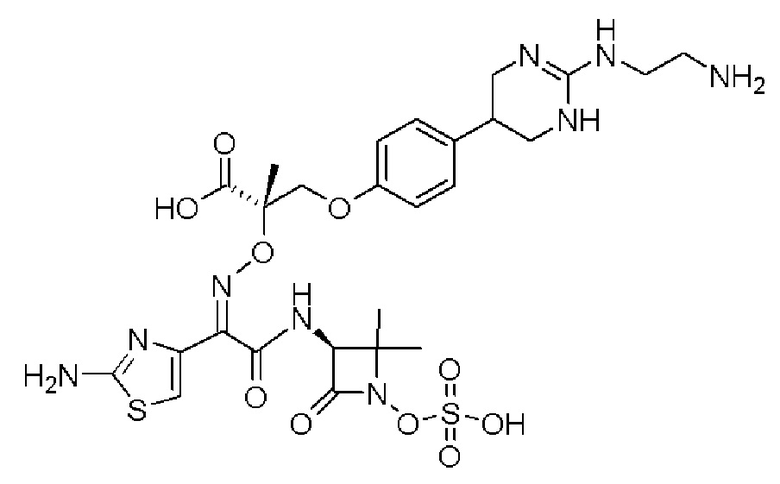
Стадия A: трет-бутил-(2-((5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидин-2-ил)амино)этил)карбамат
К раствору трет-бутил-(2-((5-бромпиримидин-2-ил)-амино)этил)карбамата (700 мг, 2,2 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (840 мг, 3,3 ммоль) и комплекса дихлорида 1,1'-бис(дифенилфосфино)ферроцен-палладия (II) с дихлорметаном (180 мг, 0,22 ммоль) в 1,4-диоксане (10 мл) добавляли ацетат калия (650 мг, 6,6 ммоль). Смесь дегазировали и продували азотом, а затем нагревали при 85°С в течение 4 часов. Затем смесь фильтровали и концентрировали досуха в вакууме, и очищали на колонке ISCO (gold, 40 г, EtOAc/гексан, градиент 0-100%), получая указанное в заголовке соединение.
Стадия B: (S)-трет-бутил-3-(4-бромфенокси)-2-(((трет-бутоксикарбонил)амино)окси)-2-метилпропаноат
Boc-ангидрид (8,38 мл, 36,1 ммоль) добавляли к перемешиваемой смеси (S)-трет-бутил-2-(аминоокси)-3-(4-бромфенокси)-2-метилпропаноата (5 г, 14,44 ммоль) в DCM (10 мл), и смесь перемешивали при 50°C в течение ночи. Затем смесь охлаждали и очищали с помощью ISCO (gold, 120 г), элюируя смесью EtOAc/изогексан с градиентом 0-30%, получая указанное в заголовке соединение.
LC-MS [M+Na]+: m/z 468,2.
Стадия C: (S)-трет-бутил-3-(4-(2-((2-((трет-бутоксикарбонил)амино)этил)амино)пиримидин-5-ил)фенокси)-2-(((трет-бутоксикарбонил)амино)окси)-2-метилпропаноат
К раствору PdCl2(dppf)-CH2Cl2 (75 мг, 0,092 ммоль), трет-бутил-(2-((5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидин-2-ил)амино)этил)карбамата (350 мг, 0,96 ммоль) и (S)-трет-бутил-3-(4-бромфенокси)-2-(((трет-бутоксикарбонил)амино)окси)-2-метилпропаноата (410 мг, 0,92 ммоль) в диоксане (4,5 мл) добавляли Na2CO3 (290 мг, 2,8 ммоль) в воде (1,5 мл). Полученную смесь трижды вакуумировали и продували водородом, а затем нагревали при 100°С в микроволновой печи в течение 1 часа. Смесь охлаждали и разбавляли EtOAc, сушили (MgSO4), фильтровали и концентрировали. Полученный остаток очищали на колонке с силикагелем (ISCO, gold, 80 г), используя смесь EtOAc/гексан с градиентом 0-100%, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 604,5.
Стадия D: (2S)-трет-бутил-3-(4-(2-((2-((трет-бутоксикарбонил)амино)этил)амино)-1,4,5,6-тетрагидропиримидин-5-ил)фенокси)-2-(((трет-бутоксикарбонил)амино)окси)-2-метилпропаноат
К перемешиваемой смеси (S)-трет-бутил-3-(4-(2-((2-((трет-бутоксикарбонил)амино)этил)амино)пиримидин-5-ил)-фенокси)-2-(((трет-бутоксикарбонил)амино)окси)-2-метилпропаноата (100 мг, 0,17 ммоль) в MeOH (5 мл) и хлористоводородной кислоты (0,83 мл, 0,83 ммоль) добавляли палладий на угле (20 мг, 0,19 ммоль). Смесь трижды вакуумировали и продували водородом, а затем перемешивали в атмосфере водорода, подаваемого из баллона, при комнатной температуре в течение 2,5 часов. Смесь разбавляли DCM и фильтровали через воронку из спеченного стекла. Фильтрат разбавляли DCM и промывали водным раствором NaOH (~5 мл). Водную фазу экстрагировали с помощью DCM (2 раза). Объединенные органические фазы промывали солевым раствором, сушили (MgSO4), фильтровали и концентрировали, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 608,5.
Стадия E: (2S)-трет-бутил-3-(4-(2-((2-аминоэтил)амино)-1,4,5,6-тетрагидропиримидин-5-ил)фенокси)-2-(аминоокси)-2-метилпропаноат
TFA (1 мл) добавляли к раствору (2S)-трет-бутил-3-(4-(2-((2-((трет-бутоксикарбонил)амино)этил)амино)-1,4,5,6-тетрагидропиримидин-5-ил)фенокси)-2-(((трет-бутоксикарбонил)амино)окси)-2-метилпропаноата (120 мг, 0,20 ммоль) в CH2Cl2 (1 мл) и смесь перемешивали при комнатной температуре в течение 40 минут. Затем реакцию останавливали путем удаления растворителя при пониженном давлении. К остатку добавляли эфир, и растворитель удаляли при пониженном давлении. К полученному остатку добавляли эфир для разрушения твердого вещества, после чего растворитель удаляли с помощью пипетки. Твердый остаток сушили в вакууме, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 408,4.
Стадия F: (2S)-трет-бутил-3-(4-(2-((2-аминоэтил)амино)-1,4,5,6-тетрагидропиримидин-5-ил)-фенокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-метилпропаноат
К раствору (2S)-трет-бутил-3-(4-(2-((2-аминоэтил)амино)-1,4,5,6-тетрагидропиримидин-5-ил)фенокси)-2-(аминоокси)-2-метилпропаноата (148 мг, 0,20 ммоль) в метаноле (2,5 мл) при комнатной температуре добавляли (S)-3-(2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоацетамидо)-2,2-диметил-4-оксоазетидин-1-ил гидросульфат (110 мг, 0,24 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 4 часов, а затем концентрировали, получая указанное в заголовке соединение.
LC-MS [М+Н]+: m/z 854,5.
Стадия G: (2S)-3-(4-(2-((2-аминоэтил)амино)-1,4,5,6-тетрагидропиримидин-5-ил)фенокси)-2-(((Z)-(1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-метилпропановая кислота
TFA (2 мл) добавляли к раствору (2S)-трет-бутил-3-(4-(2-((2-аминоэтил)амино)-1,4,5,6-тетрагидропиримидин-5-ил)фенокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфо-окси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-метилпропаноата (160 мг, 0,19 ммоль) в CH2Cl2 (1 мл), и смесь перемешивали при комнатной температуре в течение 45 минут. Затем реакцию останавливали путем удаления растворителя при пониженном давлении. К остатку добавляли эфир, и растворитель удаляли при пониженном давлении. К полученному остатку добавляли эфир для разрушения твердого вещества, после чего растворитель удаляли с помощью пипетки. Твердый остаток сушили в вакууме, и затем растворяли в 5 мл DMSO для очистки HTP в стандартных условиях с муравьиной кислотой, получая указанное в заголовке соединение в виде соли муравьиной кислоты.
1H-ЯМР (500 МГц, D2O, м.д.): δ 7,10 (д, J=10 Гц, 2H), 6,94 (с, 1H), 6,85 (д, J=10 Гц, 2H), 4,47 (с, 1H), 4,40 (д, J=10 Гц, 1H), 4,22 (д, J=10, 5 Гц, 1H), 3,42 (дд, J=10, 5 Гц, 2H), 3,37 (т, J=5 Гц, 2H), 3,27 (дд, J=15, 5 Гц, 2H), 3,06 (т, J=5 Гц, 2H), 1,53 (с, 3H), 1,36 (с, 3H), 1,12 (с, 3H).
LC-MS [М+Н]+: m/z 698,7.
ПРИМЕР 96
(S)-3-(4-(1-(3-аминопропил)-2-имино-3-метил-2,3-дигидро-1H-имидазол-4-ил)-3-фторфенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота
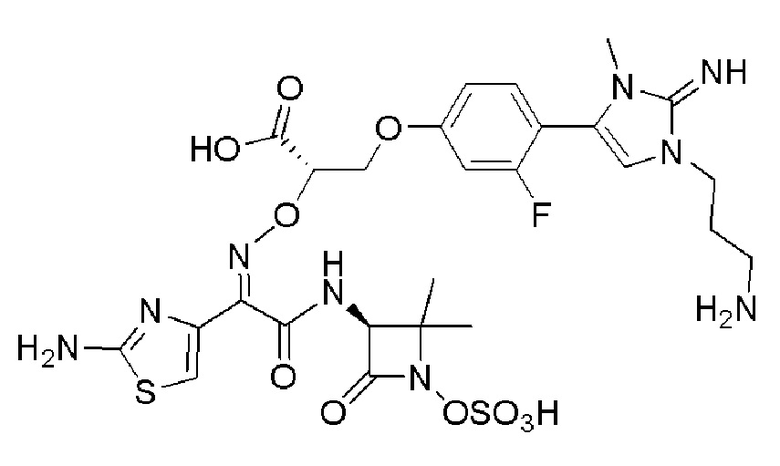
Указанное в заголовке соединение получали, следуя процедуре, описанной в примере 32, с использованием соответствующего исходного материала.
LC-MS [М+Н]+: m/z 714,4.
ПРИМЕР 97
(S)-3-((Z)-2-(((S)-2-(4-(1-(3-аминопропил)-2-(азетидин-3-илметил)-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
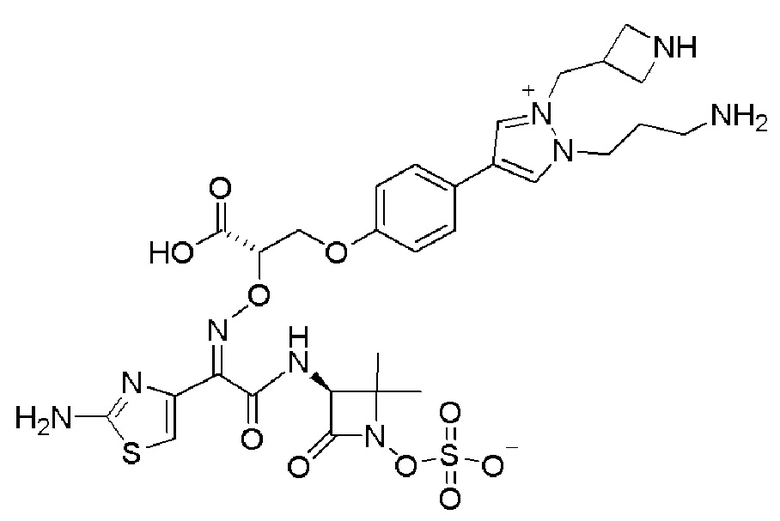
Стадия A: трет-бутил-3-((((трифторметил)сульфонил)окси)метил)азетидин-1-карбоксилат
К раствору трет-бутил-3-(гидроксиметил)азетидин-1-карбоксилата (1,32 г, 7,1 ммоль, пример 1, стадия C) и основания Хюнига (3,1 мл, 18 ммоль) в CH2Cl2 (35,4 мл) при -78°С в течение 5 минут добавляли ангидрид трифторметансульфоновой кислоты (1,8 мл, 11 ммоль). Во время добавления Tf2O внутреннюю температуру поддерживали в диапазоне от -75°С до -65°С. Через 30 мин реакционную смесь гасили насыщенным раствором NaHCO3 (30 мл), и оставляли нагреваться до 0°C. Полученную смесь разделяли, и органический слой промывали насыщенным раствором соли (10 мл). Водные слои повторно экстрагировали CH2Cl2 (15 мл), и органические слои объединяли, сушили над Na2SO4, фильтровали и концентрировали в вакууме, получая указанное в заголовке соединение, которое немедленно использовали далее без дополнительной очистки.
Стадия B: (S)-4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1H-пиразол-2-ий
В колбу, содержащую трет-бутил-3-((((трифторметил)сульфонил)окси)метил)азетидин-1-карбоксилат (2,3 г, 7,1 ммоль) добавляли раствор трет-бутил-(S)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноата (3,03 г, 4,30 ммоль) со стадии С примера 1 в безводном CH3CN (43 мл), с последующим добавлением твердого бикарбоната натрия (3,6 г, 43 ммоль). Полученную смесь нагревали до 60°С в течение 2,5 часов. Затем твердый NaHCO3 удаляли фильтрованием, и фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью флэш-хроматографии на SiO2, и элюировали смесью гексан/(3:1 EtOAc/EtOH) с градиентом 0-100%, получая указанное в заголовке соединение.
LC-MS [М+Н]: m/z 776,6.
Стадия C: (S)-4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1H-пиразол-2-ий
К раствору (S)-4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1H-пиразол-2-ия (1,24 г, 1,6 ммоль) в безводном MeOH (6,4 мл) при температуре окружающей среды добавляли 7 Н раствор аммиака в MeOH (1,8 мл, 12,8 ммоль). Реакционную смесь перемешивали в течение 23 часов, и затем концентрировали в вакууме, получая указанное в заголовке соединение, которое немедленно использовали в следующей реакции.
LC-MS [М+Н]: m/z 647,5.
Стадия D: 4-(4-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((R)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-((1-(трет-бутоксикарбонил)-азетидин-3-ил)метил)-1H-пиразол-2-ий
К смеси (S)-4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1H-пиразол-2-ия (0,12 г, 0,085 ммоль) и (S)-3-(2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоацетамидо)-2,2-диметил-4-оксоазетидин-1-ил гидросульфата (промежуточное соединение 22, 0,071 г, 0,13 ммоль) в безводном DCE (0,36 мл) и безводного MeOH (0,36 мл) добавляли бензолсульфоновую кислоту (4,0 мг, 0,025 ммоль). Полученную смесь перемешивали при комнатной температуре. Через 3 часа реакционную концентрировали в вакууме. Полученный остаток очищали с помощью HPLC на колонке C18 ISCO с обращенной фазой (130 г), и элюировали смесью H2O/AcCN (0-100%). Фракции собирали и лиофилизовали, получая указанное в заголовке соединение.
LC-MS [М+Н]: m/z 1093.
1H ЯМР (500 МГц, DMSO-d6) δ 9,49 (д, J=7,9 Гц, 1H), 9,01 (с, 1H), 8,90 (с, 1H), 7,78-7,62 (м, 2H), 7,34 (с, 1H), 7,10-7,03 (м, 2H), 6,99 (с, 1H), 4,96 (дд, J=4,6, 3,0 Гц, 1H), 4,71 (д, J=7,7 Гц, 2H), 4,62 (дд, J=8,1, 2,3 Гц, 1H), 4,46 (т, J=7,1 Гц, 2H), 4,41 (дд, J=10,8, 4,7 Гц, 1H), 4,33 (дд, J=10,7, 3,0 Гц, 1H), 4,02 (ш.с., 2H), 3,71 (ш.с., 2H), 3,29-3,12 (м, 1H), 3,06 (д, J=6,5 Гц, 2H), 2,06 (дд, J=12,3, 5,4 Гц, 2H), 1,38-1,48 (множ. с., 35H), 1,14 (с, 3H).
Стадия E: (S)-3-((Z)-2-(((S)-2-(4-(1-(3-аминопропил)-2-(азетидин-3-илметил)-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
К раствору 4-(4-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((R)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутокси-карбонил)амино)пропил)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1H-пиразол-2-ия (1,51 г, 1,4 ммоль) в безводном CH2Cl2 (3,5 мл) при внутренней температуре 10°С (баня сухой лед/диоксан) в течение 5 минут добавляли по каплям трифторуксусную кислоту (5,3 мл, 69 ммоль). Во время добавления внутреннюю температуру поддерживали ниже 11°С. Баню удаляли, и реакционную смесь перемешивали при температуре окружающей среды. Через 2 часа реакционную смесь охлаждали до 0°С и в холодный перемешиваемый раствор добавляли пипеткой по каплям МТВЕ (-5°C, 35 мл). Полученное твердое вещество собирали вакуумной фильтрацией и промывали 35 мл МТВЕ, разделенными на 3 порции. Твердое вещество сушили в токе N2 в течение 1,5 часов. Затем неочищенное твердое вещество дважды очищали помощью обращенно-фазовой хроматографии на колонке Waters CSH (50×250 мм, 5 мкм, скорость потока=100 мл/мин, элюент смесь H2O+8% муравьиной кислоты/AcCN+8% муравьиной кислоты (градиент 0%-8%)). Нужные фракции лиофилизировали, получая указанное в заголовке соединение.
LC-MS [М+Н]: m/z 736,3.
1H ЯМР (500 МГц, оксид дейтерия) δ 8,62 (с, 1H), 8,51 (с, 1H), 8,40 (д, J=1,1 Гц, 1H), 7,49 (д, J=8,4 Гц, 2H), 7,00 (д, J=8,5 Гц, 2H), 6,89 (д, J=1,1 Гц, 1H), 4,90 (дд, J=6,3, 2,2 Гц, 1H), 4,82 (д, J=7,6 Гц, 2H), 4,56 (т, J=7,5 Гц, 2H), 4,47 (дд, J=11,7, 2,3 Гц, 1H), 4,44-4,36 (м, 2H), 4,29 (дд, J=9,6, 5,8 Гц, 2H), 4,11 (ддд, J=10,9, 7,3, 2,3 Гц, 2H), 3,64 (п, J=8,0 Гц, 1H), 3,17-3,06 (м, 2H), 2,35 (п, J=7,6 Гц, 2H), 1,36 (с, 3H), 0,99 (с, 3H).
ПРИМЕР 98
(S)-3-((Z)-2-(((S)-2-(4-(6-((2-аминоэтил)амино)-1-(азетидин-3-илметил)пиридин-1-ий-3-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
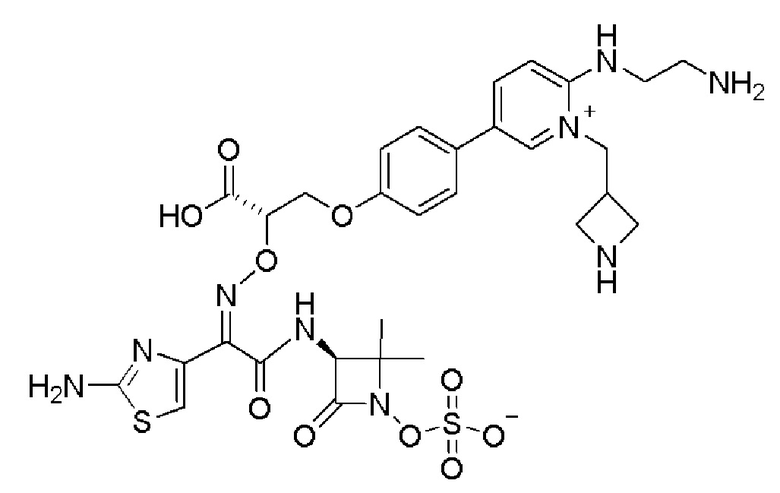
Указанное в заголовке соединение получали, следуя процедуре примера 97.
LC-MS [М+H]: m/z 748,7.
ПРИМЕР 99
(S)-3-((Z)-2-(((S)-2-(4-(2-((2-аминоэтил)амино)-1-(азетидин-3-илметил)пиримидин-1-ий-5-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
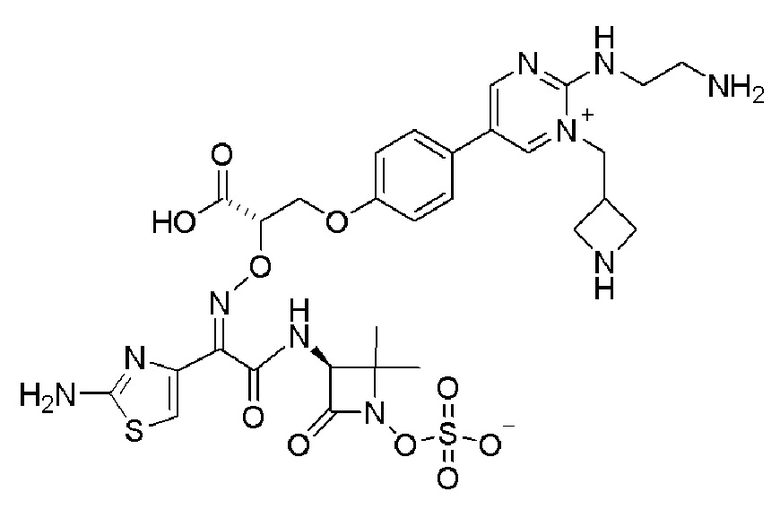
Указанное в заголовке соединение получали, следуя процедуре примера 97.
LC-MS [М+H]: m/z 749,7.
ПРИМЕР 100
(S)-3-((Z)-2-(((S)-2-(4-(2-(2-аминоэтил)-1-(3-аминопропил)-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
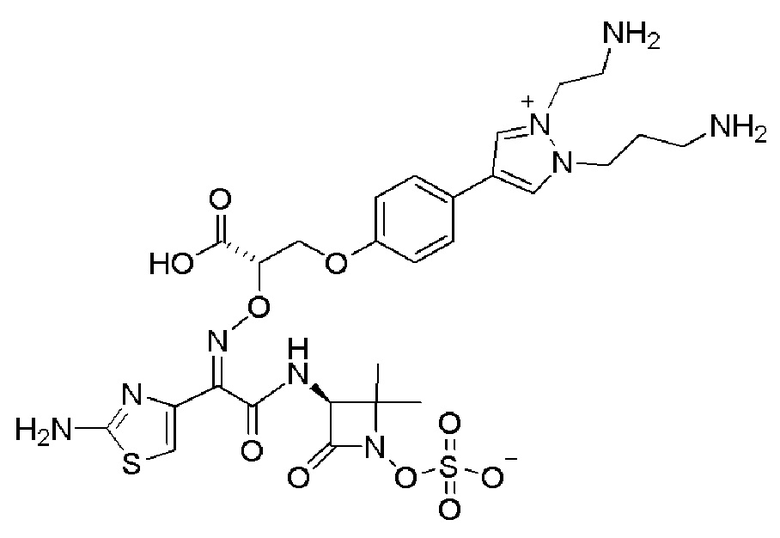
Стадия A: 2-бромэтилтрифторметансульфонат
Согласно процедуре, описанной на стадии А примера 97, 2-бромэтанол (0,41 г, 3,3 ммоль) превращали в соответствующий трифлат, который использовали далее без дополнительной очистки.
Стадия B: (S)-2-(2-бромэтил)-4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-2-ий
Согласно процедуре, описанной на стадии B примера 97, трет-бутил-(S)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноат (0,43 г, 0,61 ммоль) полученного на стадии С примера 1, алкилировали с использованием 2-бромэтилтрифторметансульфоната, получая указанное в заголовке соединение.
LC-MS [М+Н]: m/z 715,04.
Стадия C: (S)-2-(2-азидоэтил)-4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-2-ий
В колбу с мешалкой загружали (S)-2-(2-бромэтил)-4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-2-ий (0,47 г, 0,66 ммоль), азид натрия (0,15 г, 2,4 ммоль) и безводный DMFА (3,3 мл). Реакционную смесь нагревали обычным способом в нагревательном блоке при 50°С в течение 1 часа. Затем реакционную смесь гасили добавлением насыщенного солевого раствора и дважды экстрагировали EtOAc. Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме. Полученный остаток очищали с помощью HPLC на обращенно-фазовой колонке ISCO C18 (86 г), элюируя смесью Н2O/MeCN (0-100%). Нужные фракции собирали и распределяли между EtOAc и насыщенным раствором соли. Водный слой еще два раза экстрагировали EtOAc. Органические слои объединяли, сушили над Na2SO4, фильтровали и концентрировали в вакууме, получая указанное в заголовке соединение.
LC-MS [М+Н]: m/z 677,3.
1H ЯМР (500 МГц, хлороформ-d) δ 8,71 (с, 1H), 8,55 (с, 1H), 7,88 (дд, J=5,4, 3,1 Гц, 2H), 7,80 (дд, J=5,5, 3,1 Гц, 2H), 7,55-7,49 (м, 2H), 6,96 (д, J=8,6 Гц, 2H), 5,38 (с, 1H), 5,08 (дд, J=5,4, 3,5 Гц, 1H), 4,77 (т, J=5,2 Гц, 2H), 4,63 (т, J=6,9 Гц, 2H), 4,60-4,45 (м, 2H), 4,00 (т, J=5,2 Гц, 2H), 3,29 (с, 2H), 2,21 (кв, J=6,5 Гц, 2H), 1,53 (с, 9H), 1,44 (с, 9H).
Стадия D: (S)-4-(4-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)фенил)-2-(2-азидоэтил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-2-ий
Указанное в заголовке соединение получали из (S)-2-(2-азидоэтил)-4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-2-ия (0,31 г, 0,45 ммоль) используя процедуру, описанную на стадии С примера 97.
LC-MS [М+Н]: m/z 547,2.
Стадия E: (S,Z)-2-(2-азидоэтил)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-2-ий К раствору (S)-4-(4-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)фенил)-2-(2-азидоэтил)-1-(3-((трет-бутоксикарбонил)-амино)пропил)-1H-пиразол-2-ия (0,25 г, 0,45 ммоль) в безводном DCE (1,8 мл) и безводном MeOH (1,8 мл) добавляли 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусную кислоту (0,25 г, 0,90 ммоль, промежуточное соединение 5). Реакционную смесь перемешивали при комнатной температуре в течение 21 часа. Затем реакционную смесь концентрировали в вакууме и очищали с помощью HPLC на обращенно-фазовой колонке ISCO C18 (130 г), элюируя смесью Н2O/MeCN (0-100%). Нужные фракции собирали и лиофилизировали, получая указанное в заголовке соединение.
LC-MS [М+Н]: m/z 800,4.
Стадия F: (S,Z)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)фенил)-2-(2-((трет-бутоксикарбонил)-амино)этил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-2-ий
Стадия F: (S,Z)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)фенил)-2-(2-((трет-бутоксикарбонил)-амино)этил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-2-ий
К раствору (S,Z)-2-(2-азидоэтил)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)-амино)пропил)-1H-пиразол-2-ия (0,19 г, 0,24 ммоль) и BOC2O (0,052 г, 0,24 ммоль) в THF (2,4 мл) добавляли 10% Pd/C (0,038 г). Смесь гидрировали водородом при атмосферном давлении в течение 1 часа. Затем катализатор удаляли путем фильтрации через целит, и слой целита тщательно промывали EtOAc. Фильтрат концентрировали в вакууме, получая указанное в заголовке соединение, которое использовали на следующей стадии без дополнительной очистки.
LC-MS [М+Н]: m/z 875,5.
Стадия G: 4-(4-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)фенил)-2-(2-((трет-бутоксикарбонил)амино)этил)-1-(3-((трет-бутоксикарбонил)-амино)пропил)-1H-пиразол-2-ий
К смеси (S,Z)-4-(4-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)фенил)-2-(2-((трет-бутоксикарбонил)амино)этил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-2-ия (0,21 г, 0,24 ммоль) в безводном MeCN (2,1 мл) добавляли (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфат (0,093 г, 0,36 ммоль, из коммерческого источника CAS: 102507-49-3) и пиридин (0,048 мл, 0,59 ммоль) в атмосфере N2 при температуре окружающей среды. Реакционную смесь охлаждали до 0°С и добавляли одной порцией EDC-НСl (0,091 г, 0,47 ммоль). Через 1 час реакционную смесь распределяли между EtOAc и смесью 1:1 0,5 М водного раствора лимонной кислоты и насыщенного раствора соли. Водный слой снова экстрагировали EtOAc. Органические слои объединяли, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Полученный остаток очищали с помощью обращенно-фазовой HPLC на колонке ISCO C18 (86 г), элюируя смесью Н2O/MeCN (0-100%). Нужные фракции собирали и концентрировали в вакууме. Полученный влажный остаток распределяли между EtOAc/AcCN (4:1) и насыщенным солевым раствором, содержащим твердый NaCl. Водный слой снова экстрагировали EtOAc. Органические слои объединяли, сушили над Na2SO4, фильтровали и концентрировали в вакууме, получая указанное в заголовке соединение.
LC-MS [М+Н]: m/z 1066,9.
Стадия H: 2-(2-аминоэтил)-1-(3-аминопропил)-4-(4-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)фенил)-1H-пиразол-2-ий
Согласно процедуре, описанной на стадии Е примера 97, использовали смесь 1:2 DCM/TFA (1,66 мл), и реакционную смесь перемешивали в течение часа при температуре окружающей среды. Из соединения 4-(4-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)фенил)-2-(2-((трет-бутоксикарбонил)амино)этил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-2-ия (0,179 г, 0,168 ммоль) получали целевой продукт путем очистки с помощью обращенно-фазовой хроматографии с использованием колонки Waters CSH (50×250 мм, 5 мкм, скорость потока=100 мл/мин, элюирующая смесь H2O+8% муравьиная кислота/AcCN+8% муравьиная кислота (градиент 0%-10%)), получая указанное в заголовке соединение.
LC-MS [М+1]: m/z 710,2.
1H ЯМР (500 МГц, оксид дейтерия) δ 8,67 (дд, J=14,9, 1,2 Гц, 2H), 8,38 (с, 1H), 7,52 (д, J=8,5 Гц, 2H), 7,00 (д, J=8,6 Гц, 2H), 6,91 (с, 1H), 4,88 (д.т., J=13,2, 5,9 Гц, 3H), 4,60 (т, J=7,5 Гц, 2H), 4,50-4,31 (м, 3H), 3,61 (т, J=6,5 Гц, 2H), 3,22-3,06 (м, 2H), 2,39 (п, J=7,6 Гц, 2H), 1,38 (с, 3H), 1,00 (с, 3H).
ПРИМЕР 101
(S)-3-((E)-2-(2-аминотиазол-4-ил)-2-(((S)-1-карбокси-2-(4-(1-(3-(((S)-2,3-дигидроксипропил)амино)пропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
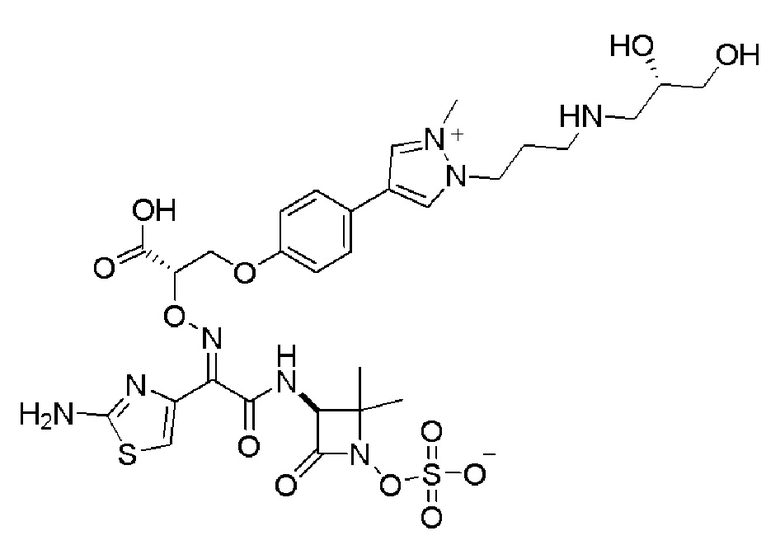
К раствору (S)-3-((E)-2-(((S)-2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата (50 мг, 0,073 ммоль, Пример 1) в DMSO (1 мл) добавляли (R)-2,3-дигидроксипропаналь (9,9 мг, 0,11 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, а затем добавляли триацетоксиборогидрид натрия (23 мг, 0,11 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Затем раствор в DMSO непосредственно вводили для разделения в колонку для препаративной HPLC (Gilson, вода/MeCN/0,05% TFA, 0-40% за 10 мин). Собирали первый пик, объединяли и сушили лиофилизацией, получая указанное в заголовке соединение.
LC-MS [М+Н]: m/z 755,10.
1H-ЯМР (500 МГц, D2O) δ 8,52 (с, 1H); 8,48 (с, 1H); 7,48 (д, J=6,8 Гц, 2H); 6,98 (с, 1H); 6,94 (д, J=6,8, 2H); 5,01 (с, 1H); 4,48 (м, 2H); 4,38 (м, 2H);4,02 (с, 3H); 3,06-3,65 (м, 7H); 2,39 (м, 2H); 1,25 (с, 3H); 0,98 (с, 3H).
ПРИМЕР 102
(S)-3-((E)-2-(((S)-2-(4-(1-(3-((2-аминоэтил)амино)пропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
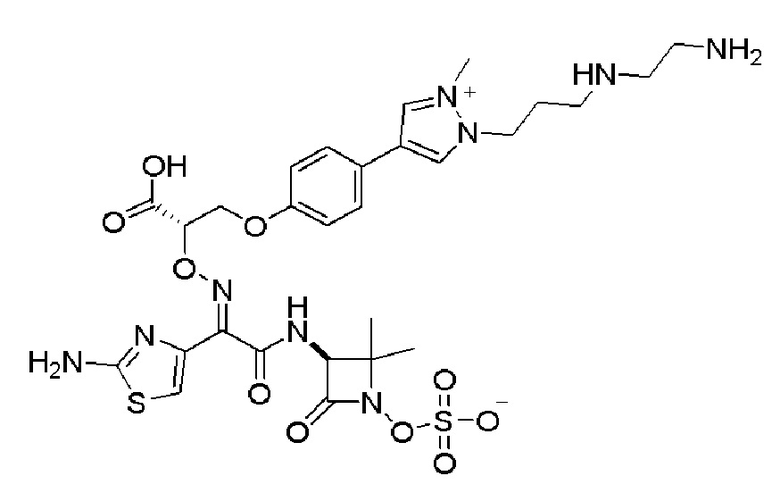
К раствору соединения примера 1 (50 мг, 0,073 ммоль) в DMSO (1 мл) добавляли трет-бутил-(2-оксоэтил)карбамат (18 мг, 0,11 ммоль) и TFA (0,1 мл). Смесь перемешивали при комнатной температуре в течение 1 часа, затем добавляли триацетоксиборгидрид натрия (17 мг, 0,081 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Затем раствор в DMSO непосредственно вводили для разделения в колонку для препаративной HPLC (Gilson, вода/MeCN/0,05% TFA, 0-40% за 10 мин). Собирали первый пик, подвергали сублимационной сушке, получая неочищенный продукт, который растворяли в DCM (1 мл) с 0,2 мл TFA. Полученную смесь перемешивали при комнатной температуре в течение 30 минут, а затем концентрировали досуха. Полученный остаток повторно растворяли в 0,5 мл DMSO и вводили для разделения в колонку для препаративной HPLC (Gilson, вода/MeCN/0,05% TFA, 0-40% в течение 10 мин). Собирали пик продукта и сушили лиофилизацией, получая указанное в заголовке соединение в виде соли TFA.
LC-MS [М+Н]: m/z 724,08.
1H-ЯМР (500 МГц, D2O) δ 8,55 (с, 1H); 8,52 (с, 1H); 7,42 (д, J=8, 2H); 7,07 (с, 2H); 7,00 (д, J=8, 2H); 5,15 (с, 1H); 4,56 (м, 4H); 4,19 (с, 3H); 3,46 (м, 6H); 2,46 (м, 2H); 1,53 (с, 3H); 1,29 (с, 3H).
Таблица 5. Примеры 103 и 104 получали в соответствии с процедурой, описанной в примере 101.
ПРИМЕР 105
(S)-3-((Z)-2-((((S)-1-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)-3-фторфенокси)-2-карбоксипропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
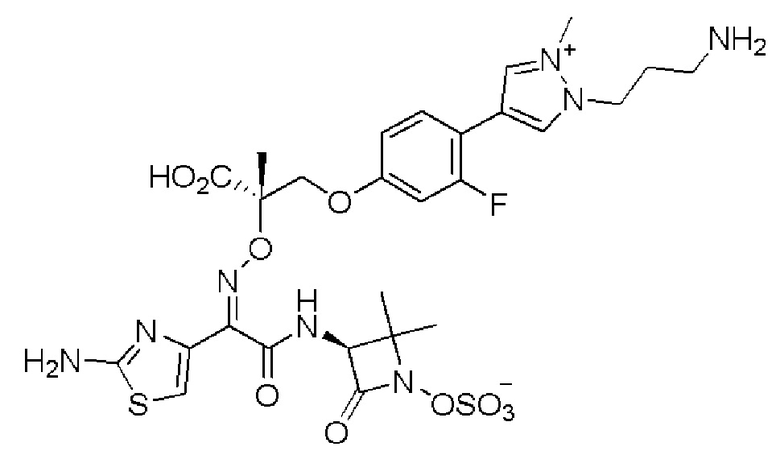
Указанное в заголовке соединение получали, следуя процедурам, которые описаны в примере 39, исходя из промежуточного соединения 14.
LC-MS [М+H]: m/z 713,5.
ПРИМЕР 106
(S)-3-((Z)-2-(((S)-2-((6-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)пиридазин-3-ил)окси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
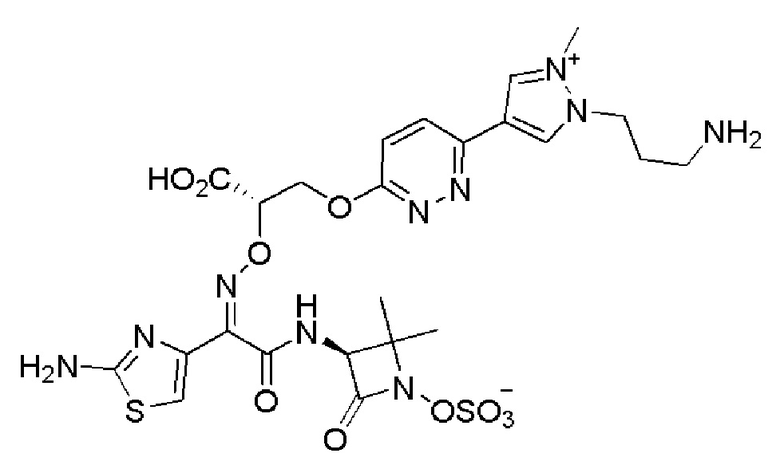
Указанное в заголовке соединение получали, следуя процедурам, которые описаны в примере 1, исходя из промежуточного соединения 12.
LC-MS [М+H]: m/z 683,3.
ПРИМЕР 107
(S)-3-((Z)-2-((((S)-1-(4-(1-(3-аминопропил)-2-имино-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-карбоксипропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
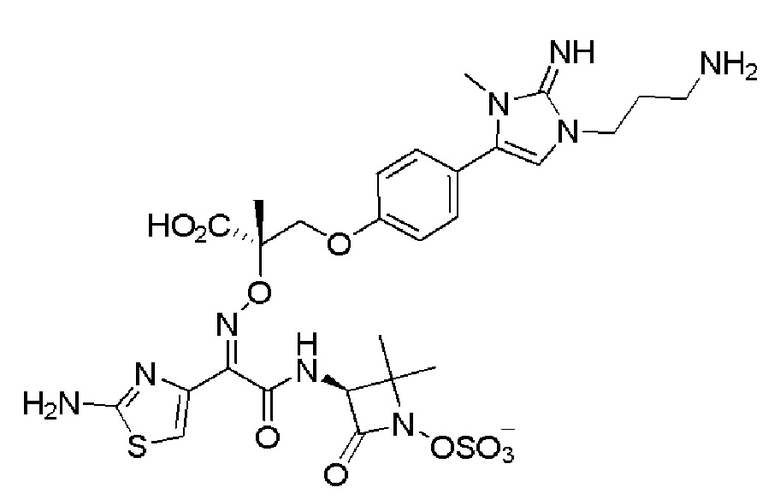
Указанное в заголовке соединение получали, следуя процедурам, которые описаны в примере 39, исходя из промежуточного соединения 10.
LC-MS [М+H]: m/z 728,3.
ПРИМЕР 108
(S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-2-((6-((1-(азетидин-3-илметил)пиридин-1-ий-4-ил)амино)пиридин-3-ил)окси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
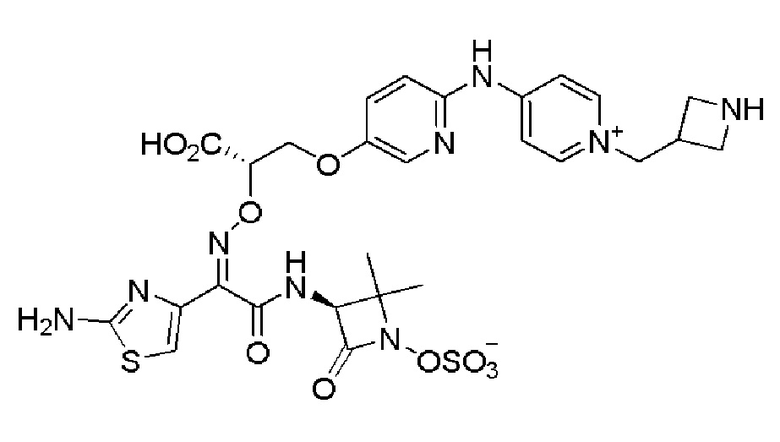
Указанное в заголовке соединение получали, следуя процедурам, описанным в примере 33.
LC-MS [М+H]: m/z 706,6.
Таблица 6. Примеры 109-112 получали процедурами, описанными для синтеза соединения примера 36
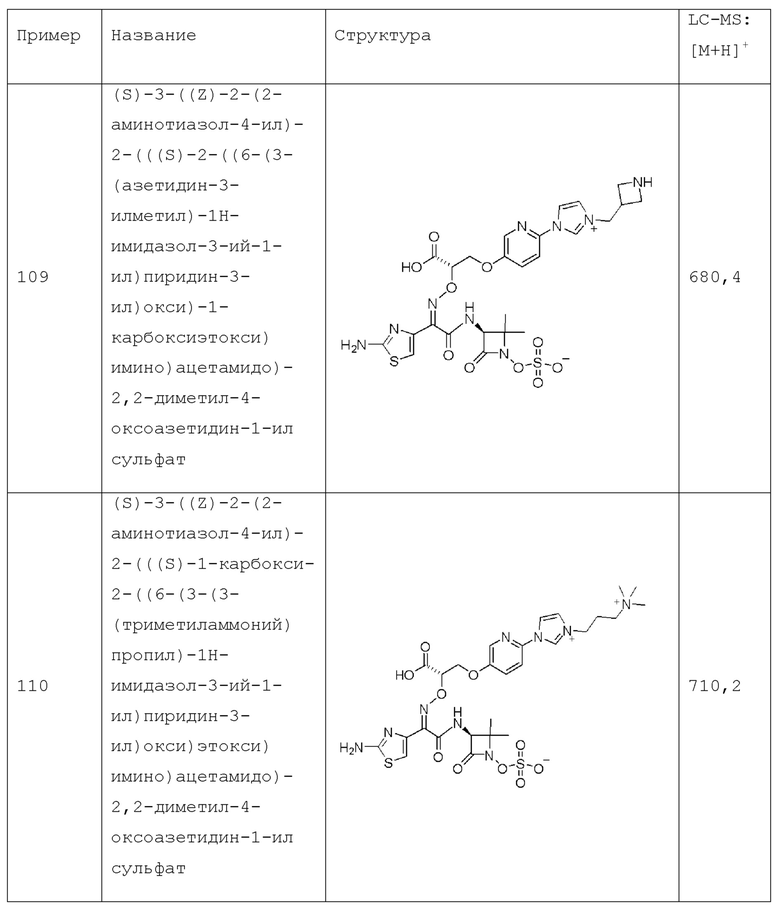
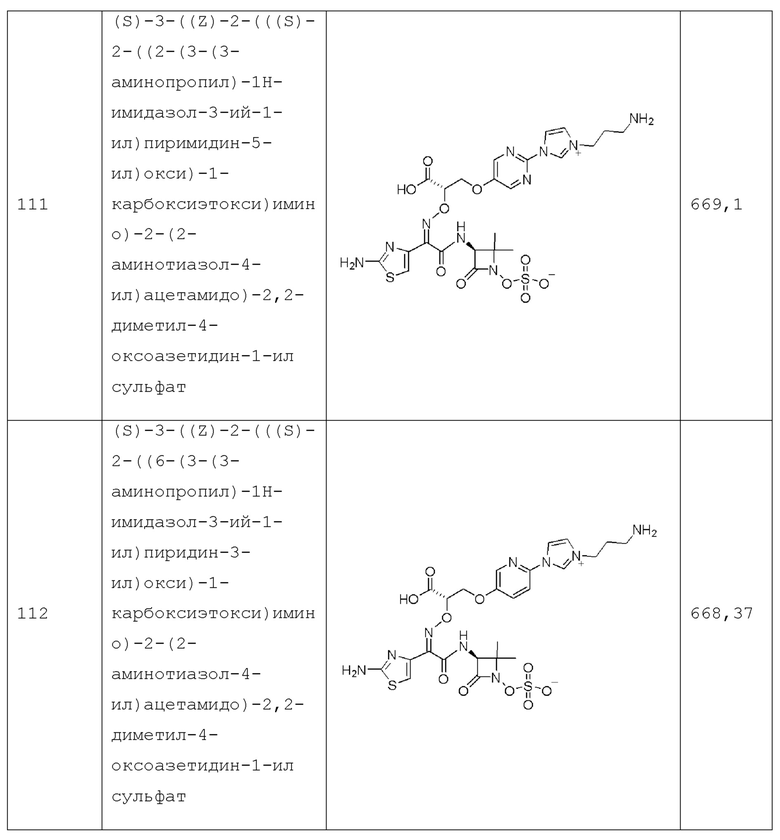
ПРИМЕР 113
(S)-3-((Z)-2-((((S)-1-((6-(3-(3-аминопропил)-1H-имидазол-3-ий-1-ил)пиридин-3-ил)окси)-2-карбоксипропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
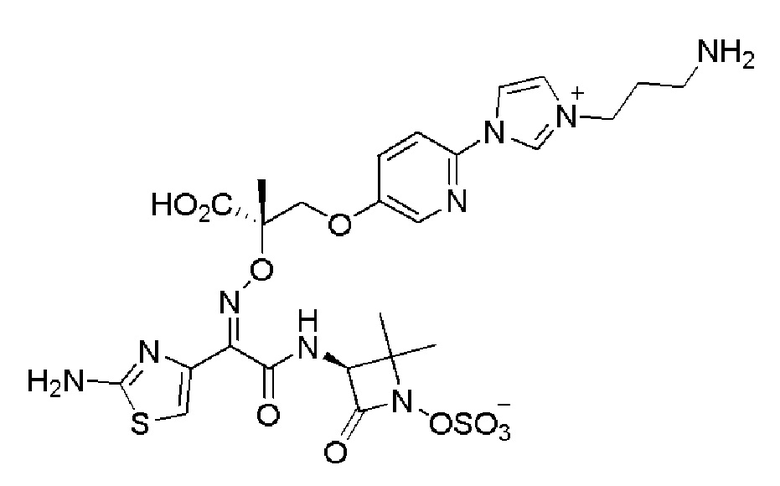
Стадия A: трет-бутил-(S)-3-((6-бромпиридин-3-ил)окси)-2-гидрокси-2-метилпропаноат
К перемешиваемому раствору (S)-3-((6-бромпиридин-3-ил)окси)-2-гидрокси-2-метилпропановой кислоты (180 мг, 0,65 ммоль) в THF (6,5 мл) добавляли трет-бутил-N,N'-диизопропилкарбамидат (380 мкл, 1,63 ммоль). Полученную смесь нагревали до 60°С в течение 1 часа, затем реакционную смесь охлаждали до комнатной температуры и концентрировали в вакууме. Неочищенный остаток очищали с помощью колоночной хроматографии на силикагеле (0-50% EtOAc в гексане в качестве элюента), получая указанное в заголовке соединение.
LC-MS [М+Н]: m/z 332,1.
Стадия B: трет-бутил-(S)-3-((6-(1H-имидазол-1-ил)пиридин-3-ил)окси)-2-гидрокси-2-метилпропаноат
Указанное в заголовке соединение получали, следуя процедурам, описанным на стади E примера 36.
LC-MS [М+Н]: m/z 320,2.
Стадия C: трет-бутил-(S)-3-((6-(1H-имидазол-1-ил)пиридин-3-ил)окси)-2-(аминоокси)-2-метилпропаноат
К перемешиваемому раствору трет-бутил-(S)-3-((6-(1H-имидазол-1-ил)пиридин-3-ил)окси)-2-гидрокси-2-метилпропаноата (100 мг, 0,313 ммоль) в THF (3,1 мл) при 0°С добавляли одной порцией NaH (22,5 мг 60%-ной дисперсии в минеральном масле, 0,56 ммоль). Полученную смесь оставляли перемешиваться при 0°С в течение 15 минут, после чего добавляли O-(мезитилсульфонил)-гидроксиламин (74 мг, 0,344 ммоль), и смесь перемешивали при 0°С в течение часа. Затем реакционную смесь распределяли между EtOAc и водой. Слои разделяли, и органический слой промывали водой и насыщенным раствором соли. Органический слой сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением неочищенного остатка, который очищали с помощью колоночной хроматографии на силикагеле (0-10% CH3OH в DCM в качестве элюента), получая указанное в заголовке соединение.
LC-MS [М+Н]: m/z 335,2.
Стадия D: трет-бутил-(S)-3-((6-(1H-имидазол-1-ил)пиридин-3-ил)окси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-2-метилпропаноат К перемешиваемому раствору трет-бутил-(S)-3-((6-(1H-имидазол-1-ил)пиридин-3-ил)окси)-2-(аминоокси)-2-метилпропаноата (62 мг, 0,185 ммоль) в толуоле (1,9 мл) добавляли фталевый ангидрид (33 мг, 0,223 ммоль), а затем небольшое количество молекулярных сит 3Å. Добавляли полимер п-толуолсульфоновой кислоты (гранулы, 16 мг, 2-3 ммоль/г, 0,032-0,048 ммоль) и полученную смесь нагревали до 100°С. Через 2 часа реакционную смесь охлаждали до комнатной температуры и концентрировали в вакууме с получением суспензии, которую очищали с помощью колоночной хроматографии на силикагеле (0-10% CH3OH в DCM в качестве элюента), получая указанное в заголовке соединение.
LC-MS [М+Н]: m/z 465,3.
Стадия E: (S)-1-(5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-2-метил-3-оксо-пропокси)пиридин-2-ил)-3-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-3-ий
Указанное в заголовке соединение получали, следуя процедурам, аналогичным тем, которые описаны на стадии F примера 35.
LC-MS [М+Н ]: m/z 509,2.
Стадия F: (S)-3-((Z)-2-((((S)-1-((6-(3-(3-аминопропил)-1H-имидазол-3-ий-1-ил)пиридин-3-ил)окси)-2-карбоксипропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат получали процедурами, аналогичными тем, которые описаны на стадиях G-J примера 35.
LC-MS [М+Н]: m/z 682,2.
ПРИМЕР 114
(S)-3-((Z)-2-((((S)-1-амино-3-(4-(1-(3-аминопропил)-2-имино-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-1-оксопропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
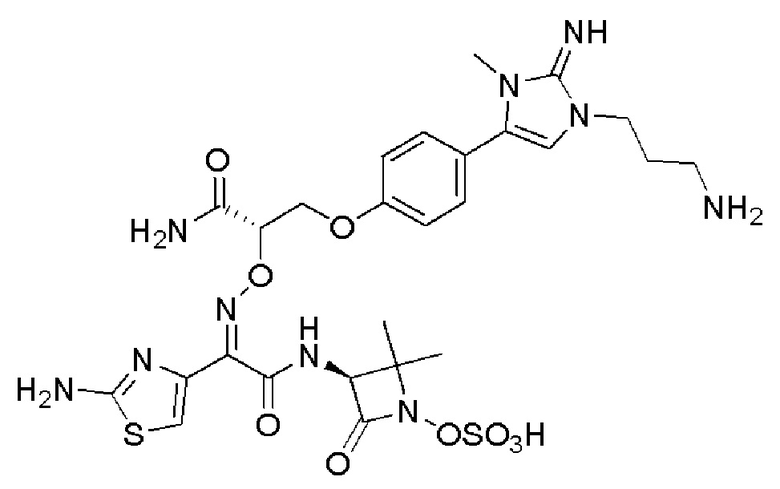
Стадия A: этил-(R,E)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-гидроксипропаноат
Указанное в заголовке соединение получали в соответствии с процедурами, аналогичными тем, которые описаны на стадии B примера 1.
LC-MS [М+Н]: m/z 563,6.
Стадия B: этил-(S,Z)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-2-((трет-бутоксикарбонил)имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноат
Указанное в заголовке соединение получали в соответствии с процедурами, аналогичными тем, которые описаны на стадии C примера 1.
LC-MS [М+Н]: m/z 708,4.
Стадия C: трет-бутил-(S,Z)-(4-(4-(3-амино-2-(аминоокси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-3-метил-1,3-дигидро-2H-имидазол-2-илиден)карбамат
К перемешиваемому раствору этил-(S,Z)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-2-((трет-бутоксикарбонил)-имино)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноата (107 мг, 0,151 ммоль) в CH3OH (2,0 мл) добавляли аммиак (0,65 мл 7,0 M раствора в CH3OH, 4,54 ммоль). Реакционную смесь герметично закрывали и оставляли перемешиваться при комнатной температуре в течение ночи. Затем реакционную смесь концентрировали в вакууме, и полученный неочищенный остаток очищали с помощью хроматографии на силикагеле С-18 с обращенной фазой (0-100% CH3CN в воде в качестве элюента), получая указанное в заголовке соединение.
LC-MS [М+Н]: m/z 549,3.
Стадия D: (S)-3-((Z)-2-((((S)-1-амино-3-(4-(1-(3-аминопропил)-2-имино-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-1-оксопропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)-ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
Указанное в заголовке соединение получали из трет-бутил-(S,Z)-(4-(4-(3-амино-2-(аминоокси)-3-оксопропокси)фенил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-3-метил-1,3-дигидро-2H-имидазол-2-илиден)карбамата в соответствии с процедурами, аналогичными тем, которые описаны на стадиях F-H примера 1.
LC-MS [М+Н]: m/z 695,7.
ПРИМЕР 115
(S)-3-((Z)-2-((((S)-3-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-этокси-1-оксопропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
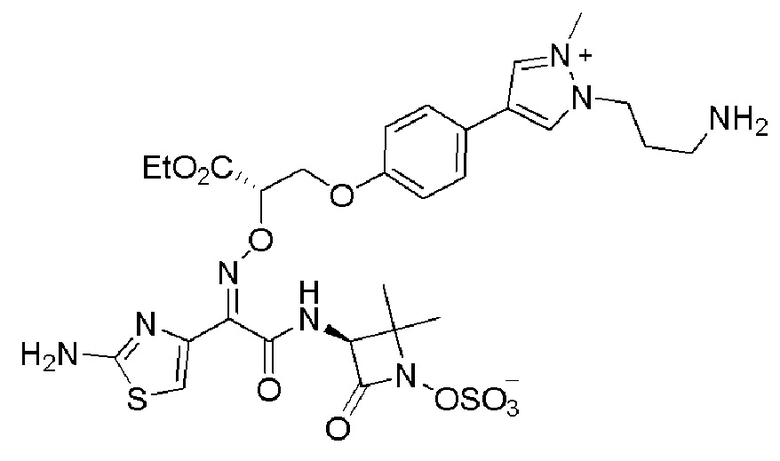
Указанное в заголовке соединение получали из промежуточного соединения 11, следуя процедурам, описанным на стадиях B-H примера 1.
LC-MS [М+Н]: m/z 709,5.
ПРИМЕР 116
Этил-(S)-3-(4-(1-(3-аминопропил)-2-имино-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропаноат
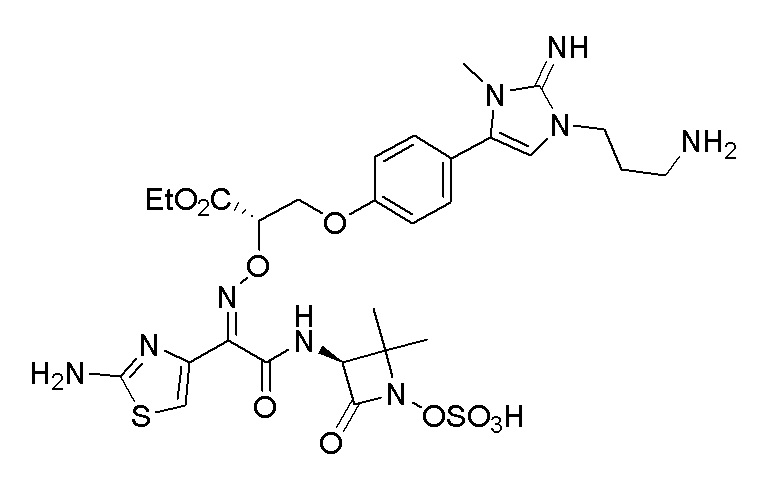
Указанное в заголовке соединение получали из промежуточного соединения 13, следуя процедурам, описанным на стадиях A-B примера 114 и на стадиях F-H примера 1.
LC-MS [М+Н]: m/z 724,6.
ПРИМЕР 117
(S)-3-((Z)-2-((((S)-3-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-4-ил)фенокси)-1-метокси-1-оксопропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат

Указанное в заголовке соединение получали из промежуточного соединения 15 в соответствии с процедурами, описанными на стадиях B-H примера 1.
LC-MS [М+Н]: m/z 695,4.
ПРИМЕР 118
(S)-3-((6-(3-(3-аминопропил)-2-имино-2,3-дигидро-1H-имидазол-1-ил)пиридин-3-ил)окси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота
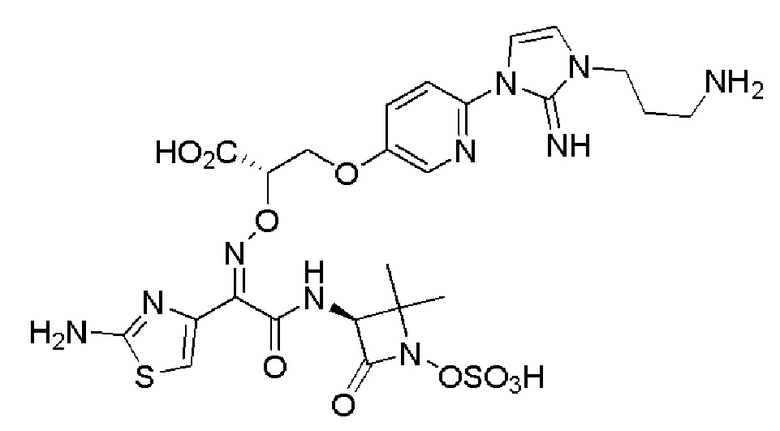
Стадия A: трет-бутил-(S,E)3-((6-(3-(3-((трет-бутоксикарбонил)амино)пропил)-2-((трет-бутоксикарбонил)имино)-2,3-дигидро-1H-имидазол-1-ил)пиридин-3-ил)окси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноат
К смеси трет-бутилкарбамата (229 мг, 1,95 ммоль) в DCM (6,0 мл) добавляли трет-бутилгипохлорит (250 мкл, 2,22 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут, а затем смесь охлаждали до 0°C и добавляли DBU (402 мкл, 2,66 ммоль). К этой перемешиваемой смеси добавляли раствор (S)-1-(5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)пиридин-2-ил)-3-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-3-ия (541 мг, 0,888 ммоль, со стадии F примера 36) в DCM (6,0 мл). Реакционную смесь перемешивали при 0°С в течение 1 часа, затем смесь суспендировали в бензоле. Полученную смесь загружали на колонку с силикагелем и очищали с помощью колоночной хроматографии (0-7% CH3OH в DCM в качестве элюента), получая указанное в заголовке соединение.
LC-MS [М+Н]: m/z 723,6.
Стадии B-E: (S)-3-((6-(3-(3-аминопропил)-2-имино-2,3-дигидро-1H-имидазол-1-ил)пиридин-3-ил)окси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота
Указанное в заголовке соединение получали, следуя процедурам, аналогичным тем, которые описаны на стадиях G-J примера 36.
LC-MS [М+Н]: m/z 683,4.
ПРИМЕР 119
(S)-3-((Z)-2-(((S)-2-(4-(1-(3-аминопропил)-2-((3-фторазетидин-3-ил)метил)-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
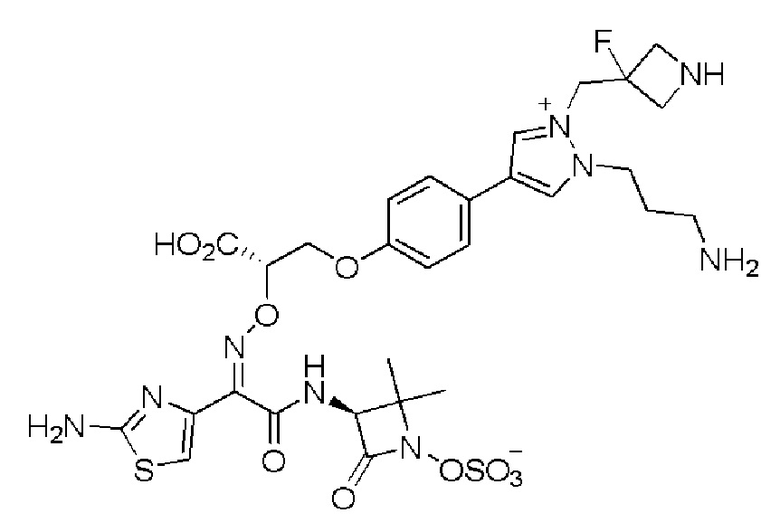
Стадия A: трет-бутил-(R)-3-((4-(4-(3-(трет-бутокси)-2-гидрокси-3-оксопропокси) фенил)-1H-пиразол-1-ил)метил)-3-фторазетидин-1-карбоксилат
Указанное в заголовке соединение получали из промежуточного соединения 4 процедурами, аналогичными тем, которые описаны на стадии B примера 1, заменяя промежуточное соединение 16 (трет-бутил-(3-(4-бром-1H-пиразол-1-ил)пропил)карбамат) на промежуточное соединение 13.
LC-MS [М+Н]: m/z 492,5.
Стадия B: трет-бутил-(S)-3-((4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1H-пиразол-1-ил)метил)-3-фторазетидин-1-карбоксилат
Указанное в заголовке соединение получали, следуя процедурам, аналогичным тем, которые описаны на стадии С примера 1.
LC-MS [М+Н]: m/z 637,6.
Стадия C: (S)-2-(3-азидопропил)-4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1-((1-(трет-бутоксикарбонил)-3-фторазетидин-3-ил)метил)-1H-пиразол-2-ий
К перемешиваемому раствору трет-бутил-(S)-3-((4-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-1H-пиразол-1-ил)метил)-3-фторазетидин-1-карбоксилата (300 мг, 0,471 ммоль) и бикарбоната натрия (396 мг, 4,71 ммоль) в ацетонитриле (5,0 мл) добавляли промежуточное соединение 17 (406 мг, 1,74 ммоль) Полученную суспензию нагревали до 60°С, и через 30 мин реакционную смесь фильтровали через слой целита. Колонку промывали ацетонитрилом, и объединенные органические слои концентрировали в вакууме. Полученный неочищенный остаток растирали с эфиром и DCM, и очищали с помощью колоночной хроматографии на силикагеле (0-100% (3:1 EtOAc:EtOH) в гексане в качестве элюента), получая указанное в заголовке соединение.
LC-MS [М]+: m/z 720,6.
Стадии D-F: (S)-3-((Z)-2-((((S)-3-(4-(1-(3-азидопропил)-2-((1-(трет-бутоксикарбонил)-3-фторазетидин-3-ил)метил)-1H-пиразол-2-ий-4-ил)фенокси)-1-(трет-бутокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
Указанное в заголовке соединение получали, следуя процедурам, описанным на стадиях E-G примера 1.
LC-MS [M+Na]: m/z 1059,8.
Стадия G: (S)-3-((Z)-2-((((S)-3-(4-(1-(3-аминопропил)-2-((1-(трет-бутоксикарбонил)-3-фторазетидин-3-ил)метил)-1H-пиразол-2-ий-4-ил)фенокси)-1-(трет-бутокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
Перемешиваемый раствор (S)-3-((Z)-2-((((S)-3-(4-(1-(3-азидопропил)-2-((1-(трет-бутоксикарбонил)-3-фторазетидин-3-ил)метил)-1H-пиразол-2-ий-4-ил)фенокси)-1-(трет-бутокси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата (50 мг, 0,048 ммоль) в 2,2,2-трифторэтаноле (2,0 мл) трижды дегазировали вакуумом, каждый раз продувая азотом. К этой смеси добавляли 10% палладий на угле (5,0 мг, 4,70 мкмоль), и полученную смесь дегазировали, как описано выше, продувая водородом. Через 2 часа реакционную смесь фильтровали через слой целита и колонку промывали СН3ОН. Объединенные органические слои концентрировали в вакууме, получая указанное в заголовке соединение, которое использовали без дополнительной очистки.
LC-MS [М+]: m/z 1010,9.
Стадия H: (S)-3-((Z)-2-(((S)-2-(4-(1-(3-аминопропил)-2-((3-фторазетидин-3-ил)метил)-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
Указанное в заголовке соединение получали, следуя процедурам, описанным на стадии Н примера 1.
LC-MS [М+Н]: m/z 755,2.
ПРИМЕР 120
(S)-3-((Z)-2-(((S)-2-(4-(2-(3-амино-2-фтор-2λ3-пропил)-1-(3-аминопропил)-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
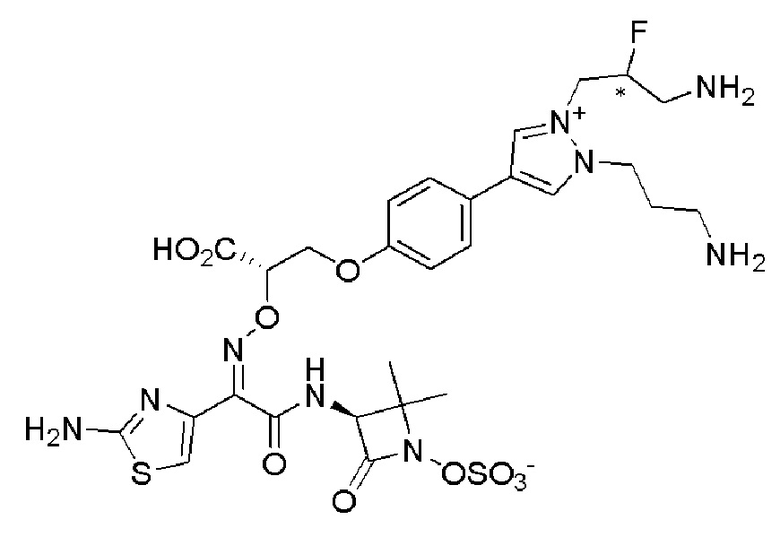
Стадия A: трет-бутил-(2R)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)-2-фторпропил)-1H-пиразол-4-ил)фенокси)-2-гидроксипропаноат
Указанное в заголовке соединение получали в виде смеси стереоизомеров из промежуточного соединения 4 следуя процедурам, аналогичным тем, которые описаны на стадии B примера 1, заменяя трет-бутил-(3-(4-бром-1H-пиразол-1-ил)пропил)карбамат на промежуточное соединение 15.
LC-MS [М+Н]: m/z 480,4.
Стадия B: Разделение стереоизомеров трет-бутил-(2R)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)-2-фторпропил)-1H-пиразол-4-ил)фенокси)-2-гидроксипропаноата
Стереоизомеры трет-бутил-(2R)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)-2-фторпропил)-1H-пиразол-4-ил)фенокси)-2-гидроксипропаноата разделяли с помощью препаративной сверхкритической жидкостной хроматографии (SFC). Раствор трет-бутил-(2R)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)-2-фторпропил)-1H-пиразол-4-ил)фенокси)-2-гидроксипропаноата (260 мг) в метаноле (20 мл) вводили (40×0,5 мл) в полупрепаративную (250×21 мм) колонку Chiralpak® AD-H (от Chiral Technologies, Inc., Exton, Пенсильвания, США) для HPLC (элюент: 40% (этанол+0,2% диизопропиламин)/СO2 при 70 мл/мин, давление на выходе 120 бар; УФ-детектирование при длине волны 215 нм), получая быстро элюируемый энантиомер B-1 (время удерживания=2,4 мин) и более медленно элюируемый энантиомеров B-2 (время удерживания=3,1 мин), условия для аналитического контроля хиральной HPLC: полупрепаративная колонка CHIRALPAK® AD-H (от Chiral Technologies, Inc., Exton, Пенсильвания, США) (250×4,6 мм), элюент: 40% (этанол+0,1% диизопропиламин)/СO2, давление на выходе 120 бар; УФ-детектирование при длине волны 254 нм.
Стадия C: (S)-3-((Z)-2-(((S)-2-(4-(2-(3-амино-2-фтор-2λ3-пропил)-1-(3-аминопропил)-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
Указанное в заголовке соединение получали из промежуточного соединения B-2 стадии B, следуя процедурам, аналогичным тем, которые описаны на стадиях С-Н примера 1.
LC-MS [М+Н]: m/z 754,5.
ПРИМЕР 121
(S)-3-((Z)-2-(((S)-2-(4-(2-(3-амино-2-фтор-2λ3-пропил)-1-(азетидин-3-илметил)-1H-пиразол-2-ий-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
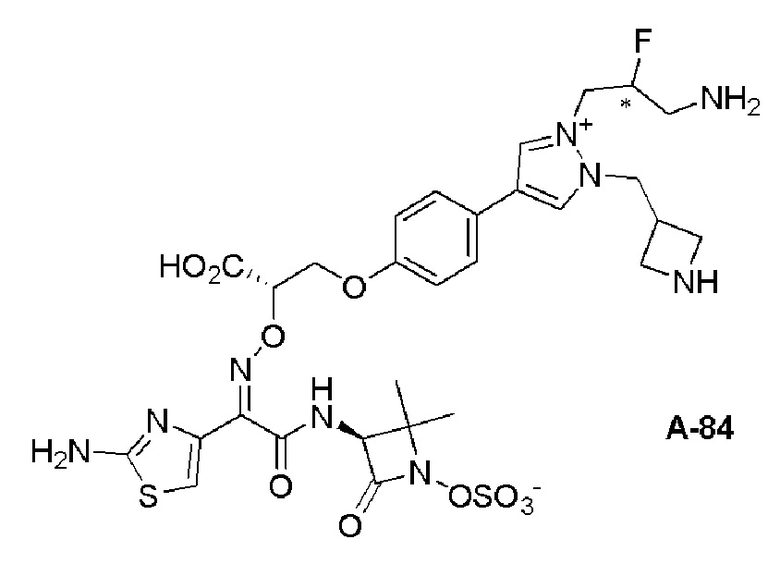
Указанное в заголовке соединение получали из промежуточного соединения 20, следуя процедурам, аналогичным тем, которые описаны на стадиях B-H примера 119.
LC-MS [М+Н]: m/z 742,7.
ПРИМЕРЫ 122 и 123
(S)-3-((Z)-2-(((S)-2-(4-(3-((3-аминопропил)амино)-1-метилпиридазин-1-ий-6-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат (122) и
(S)-3-((Z)-2-(((S)-2-(4-(6-((3-аминопропил)амино)-1-метилпиридазин-1-ий-3-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат (123)
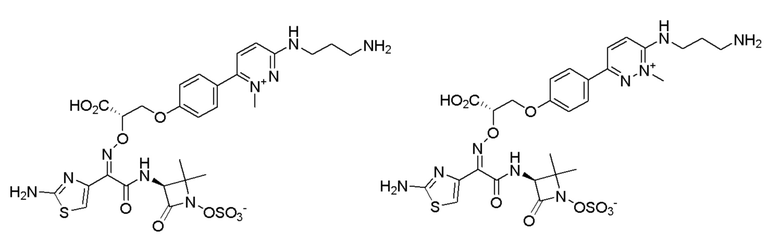
Стадия A: трет-бутил-(R)-3-(4-(6-((3-((трет-бутоксикарбонил)амино)пропил)амино)пиридазин-3-ил)фенокси)-2-гидроксипропаноат
Указанное в заголовке соединение получали из промежуточного соединения 4, следуя процедурам, аналогичным тем, которые описаны на стадии В примера 1.
LC-MS [М+Н]: m/z 489,5.
Стадия B: трет-бутил-(R)-3-(4-(6-((3-((трет-бутоксикарбонил)амино) пропил)амино)пиридазин-3-ил)фенокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноат
Указанное в заголовке соединение получали из трет-бутил-(R)-3-(4-(6-((3-((трет-бутоксикарбонил)амино)пропил) амино)пиридазин-3-ил)фенокси)-2-гидроксипропаноата, следуя процедурам, аналогичным тем, которые описаны на стадии C примера 1.
LC-MS [М+Н]: m/z 634,7.
Стадия C: (R)-6-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-3-((3-((трет-бутоксикарбонил)амино)пропил)амино)-1-метилпиридазин-1-ий (C-1); и
(R)-3-(4-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)фенил)-6-((3-((трет-бутоксикарбонил)амино)пропил)амино)-1-метилпиридазин-1-ий (C-2)
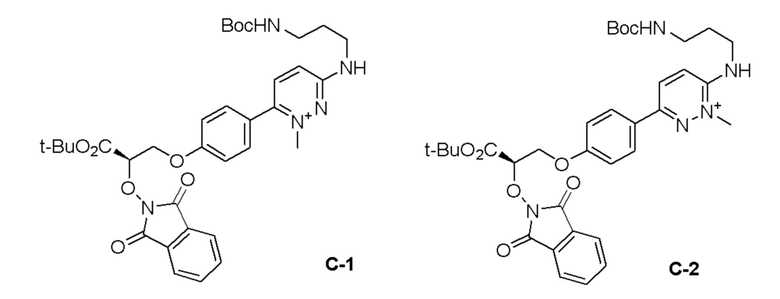
Указанные в заголовке соединения получали в виде смеси региоизомеров из трет-бутил-(R)-3-(4-(6-((3-((трет-бутоксикарбонил)амино)пропил)амино)пиридазин-3-ил)фенокси)-2-((1,3-диоксо-изоиндолин-2-ил)окси)пропаноата, следуя процедурам, аналогичным тем, которые описаны на стадии D примера 1. Неочищенную смесь региоизомеров разделяли с помощью колоночной хроматографии на силикагеле (0-5% СН3ОН в DCM в качестве элюента), с получением промежуточных соединений H1 и Н2.
H1: LC-MS [М+Н]: m/z 648,8.
Н2: LC-MS [М+Н]: m/z 648,8.
(S)-3-((Z)-2-(((S)-2-(4-(3-((3-аминопропил)амино)-1-метилпиридазин-1-ий-6-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат (122)
Указанное в заголовке соединение получали из соединения H1, следуя процедуре, описанной на стадиях E-H примера 1.
LC-MS [М+Н]: m/z 708,8.
(S)-3-((Z)-2-(((S)-2-(4-(6-((3-аминопропил)амино)-1-метилпиридазин-1-ий-3-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат (123)
Указанное в заголовке соединение получали из соединения H2, следуя процедуре, описанной на стадиях E-H примера 1.
LC-MS [М+Н]: m/z 708,7.
ПРИМЕР 124
(S)-3-((Z)-2-(((S)-2-((6-(4-(3-аминопропил)-3-((3-фторазетидин-3-ил)метил)-1H-имидазол-3-ий-1-ил)пиридин-3-ил)окси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
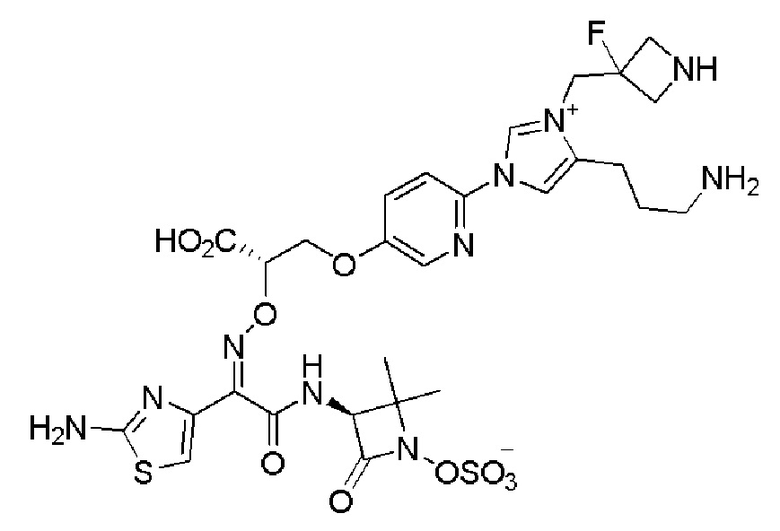
Стадия A: трет-бутил-(R)-3-((6-(4-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-1-ил)пиридин-3-ил)окси)-2-((трет-бутилдиметилсилил)окси)пропаноат
Указанное в заголовке соединение получали из трет-бутил-(R)-3-((6-бромпиридин-3-ил)окси)-2-((трет-бутилдиметилсилил)-окси)пропаноата (пример 36, стадия D), следуя процедуре, описанной на стадии C примера 35.
LC-MS [М+Н]: m/z 577,6.
Стадии B-C: трет-бутил-(S)-3-((6-(4-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-1-ил)пиридин-3-ил)окси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноат
Указанное в заголовке соединение получали из трет-бутил-(R)-3-((6-(4-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-1-ил)пиридин-3-ил)окси)-2-((трет-бутилдиметилсилил)окси)пропаноата, следуя процедуре, описанной на стадиях F и G примера 36.
LC-MS [M+Н]: m/z 608,3.
Стадия D: (S)-1-(5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)пиридин-2-ил)-3-((1-(трет-бутоксикарбонил)-3-фторазетидин-3-ил)метил)-4-(3-((трет-бутоксикарбонил)-амино)пропил)-1H-имидазол-3-ий
Указанное в заголовке соединение получали из трет-бутил-(S)-3-((6-(4-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-1-ил)пиридин-3-ил)окси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноата в соответствии с процедурами, аналогичными описанным на стади Н примера 36.
LC-MS [м+]: m/z 795,8.
4-(3-аминопропил)-1-(5-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)пиридин-2-ил)-3-((3-фторазетидин-3-ил)метил)-1H-имидазол-3-ий
Указанное в заголовке соединение получали из (S)-1-(5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)пиридин-2-ил)-3-((1-(трет-бутоксикарбонил)-3-фторазетидин-3-ил)метил)-4-(3-((трет-бутоксикарбонил)амино)пропил)-1H-имидазол-3-ия в соответствии с процедурами, аналогичными описанным на стадиях E-H примера 1.
LC-MS [М+]: m/z 755,5.
ПРИМЕР 125
(S)-3-(4-(5-(((2-аминоэтил)амино)метил)-1-(3-аминопропил)-1H-пиразол-3-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота
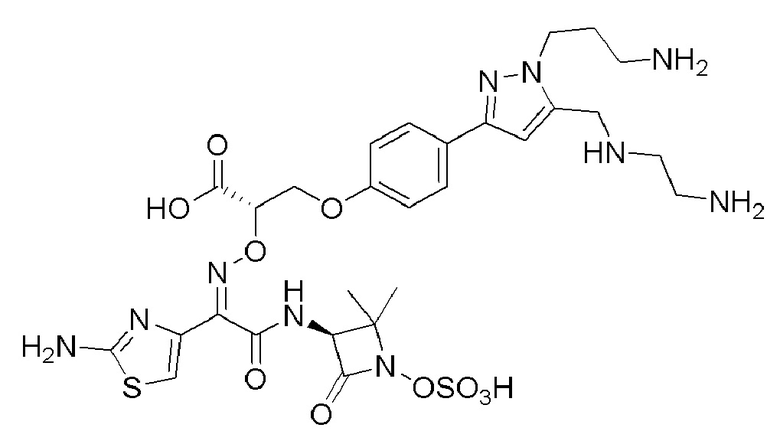
Стадия A: Метил (R)-4-(3-(трет-бутокси)-2-гидрокси-3-оксопропокси) бензоат Смесь трет-бутил оксиран-2-карбоксилата (2,1 г, 14,5 ммоль), метил-4-гидроксибензоата (1 г, 6,6 ммоль), молекулярных сит (0,1 г) и (R,R)-Co-катализатора (0,28 г, 0,33 ммоль) в t-BuOMe (2,2 мл) перемешивали при комнатной температуре в течение 4 дней. Затем реакционную смесь фильтровали через целит, фильтрат концентрировали и полученный остаток очищали на колонке с силикагелем (220 г) с использованием смеси EtOAc/гексан (0-30%), получая указанное в заголовке соединение.
LC/MS: м/е 319,34 [M+Na]+.
Стадия B: Метил (R)-4-(3-(трет-бутокси)-2-((трет-бутилдиметилсилил)окси)-3-оксопропокси)-бензоат К раствору (R)-метил-4-(3-(трет-бутокси)-2-гидрокси-3-оксопропокси)бензоата (1,38 г, 4,7 ммоль) и имидазола (0,79 г, 11,6 ммоль) в DCM (47 мл) добавляли TBDMS-Cl (0,84 г, 5,6 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение ночи и затем распределяли между EtOAc и водой. Органический слой отделяли, промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Полученный остаток очищали на колонке с силикагелем (220 г) с использованием смеси EtOAc/гексан (0-10%), получая указанное в заголовке соединение.
LC/MS: м/е 433,21 [M+Na]+.
Стадия C: трет-бутил-(R)-2-((трет-бутилдиметилсилил)окси)-3-(4-(метокси(метил)карбамоил)-фенокси)пропаноат
К раствору (R)-метил 4-(3-(трет-бутокси)-2-((трет-бутил-диметилсилил)окси)-3-оксопропокси)бензоата и амида Вайнреба (0,96 г, 9,9 ммоль) в THF (47 мл ) добавляли по каплям раствор iPrMgCl (6,5 мл, 19 ммоль, 2,9 М), сохраняя при этом внутреннюю температуру ниже 5°C. Реакционную смесь перемешивали при 0°С в течение 3 часов, а затем гасили водным раствором NH4Cl. Реакционную смесь экстрагировали EtOAc (3 раза), сушили над MgSO4, фильтровали и концентрировали. Полученный остаток очищали на колонке с силикагелем (80 г) с использованием смеси EtOAc/гексан (0-30%), получая указанное в заголовке соединение.
LC/MS: м/е 440,23 [М+Н]+.
Стадия D: трет-бутил-(R)-3-(4-(4-(бензилокси)бут-2-иноил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноат К раствору ((проп-2-ин-1-илокси)метил)бензола (1,87 г, 12,8 ммоль) в THF (12 мл) при -78°C добавляли бутиллитий (4,26 мл, 10,6 ммоль) (2,5 M/гексан). Реакционную смесь перемешивали при -78°С в течение 1 часа, затем добавляли по каплям через шприц к раствору (R)-трет-бутил-2-((трет-бутилдиметилсилил)окси)-3-(4-(метокси-(метил)карбамоил)фенокси)пропаноата (1,56 г, 3,6 ммоль) в THF (5 мл) при -78°С. Реакционную смесь перемешивали при -78°С в течение 30 минут, а затем при 0°С в течение 30 минут. Реакционную смесь гасили насыщенным раствором NH4Cl, и экстрагировали EtOAc (3 х). Объединенные органические слои промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Полученный остаток очищали на колонке с силикагелем (40 г) с использованием смеси EtOAc/гексан (0-30%), получая указанное в заголовке соединение.
LC/MS: м/е 525,23 [М+Н]+.
Стадия E: трет-бутил-(R)-3-(4-(5-((бензилокси)метил)-1H-пиразол-3-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноат
Раствор (R)-трет-бутил-3-(4-(4-(бензилокси)бут-2-иноил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноата (1,16 г, 2,2 ммоль) и гидразина (0,35 мл, 11 ммоль) в EtOH (22 мл) нагревали при 80°С в течение 1 часа. Затем реакционную смесь концентрировали досуха и очищали на колонке с силикагелем (80 г) с использованием смеси EtOAc/гексан (0-30%), получая указанное в заголовке соединение.
LC/MS: м/е 539,25 [М+Н]+.
Стадия F: трет-бутил-(R)-3-(4-(5-((бензилокси)метил)-1-(3-((трет-бутоксикарбонил)амино)-пропил)-1H-пиразол-3-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноат
Раствор (R)-трет-бутил-3-(4-(5-((бензилокси)метил)-1H-пиразол-3-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноата (1,11 г, 2,1 ммоль), трет-бутил-(3-бромпропил)карбамата (0,49 г, 2,1 ммоль) и Cs2CO3 (1,0 г, 3,1 ммоль) в DMF (4,12 мл) перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь разбавляли водой и экстрагировали EtOAc (3 х). Объединенные органические слои промывали насыщенным солевым раствором (3 раза), сушили над MgSO4 и концентрировали. Полученный остаток очищали на колонке с силикагелем (120 г) с использованием смеси EtOAc/гексан (0-30%), получая указанное в заголовке соединение.
LC/MS: м/е 696,37 [М+Н]+.
Стадия G: трет-бутил-(R)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-5-(гидроксиметил)-1H-пиразол-3-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноат Смесь (R)-трет-бутил-3-(4-(5-((бензилокси)метил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-3-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноата (810 мг, 1,16 ммоль) и Pd/C (82 мг, 0,077 ммоль) в MeOH (5,8 мл) перемешивали в токе водорода при комнатной температуре в течение ночи. Анализ TLC показал полноту реакции. Смесь фильтровали через слой целита и концентрировали досуха, получая указанное в заголовке соединение.
LC/MS: м/е 606,32 [М+Н]+.
Стадия H: трет-бутил-(R)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-5-формил-1H-пиразол-3-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноат
К раствору (R)-трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-5-(гидроксиметил)-1H-пиразол-3-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноата (590 мг, 0,97 ммоль) в DCM (9,7 мл) при 0°C добавляли периодинат Десса-Мартина (434 мг, 1,02 ммоль). После добавления реакционную смесь нагревали до комнатной температуры и перемешивали в течение приблизительно 2-х часов. Реакционную смесь гасили 5%-ным раствором Na2S2O3. Смесь экстрагировали EtOAc (3 раза), объединенные органические слои промывали насыщенным NaHCO3 и насыщенным раствором соли, сушили над MgSO4, концентрировали в вакууме и очищали на колонке с силикагелем (40 г) с использованием смеси EtOAc/гексан (0-30%), получая указанное в заголовке соединение.
LC/MS: м/е 604,30 [М+Н]+.
Стадия I: трет-бутил-(R)-3-(4-(5-(((2-((трет-бутоксикарбонил)амино)этил)амино)метил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-3-ил)фенокси)-2-((трет-бутилдиметилсилил)-окси)пропаноат К раствору (R)-трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)-пропил)-5-формил-1H-пиразол-3-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноата (203 мг, 0,34 ммоль) и N-Boc-этилендиамина гидрохлорида (99 мг, 0,50 ммоль) в THF (1,7 мл) при комнатной температуре добавляли триэтиламин (70 мг, 0,50 ммоль) и MgSO4. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, фильтровали и концентрировали. Полученный остаток повторно растворяли в MeOH (1,7 мл), затем добавляли боргидрид натрия (12,7 мг, 0,34 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь распределяли между EtOAc и H2O. Затем водный слой экстрагировали с помощью EtOAc (3 раза), промывали насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме. Полученный остаток очищали на колонке с силикагелем (24 г) с использованием смеси EtOAc/гексан (0-100%), получая указанное в заголовке соединение.
LC/MS: м/е 748,46 [М+Н]+.
Стадия J: трет-бутил-(R)-3-(4-(5-(((трет-бутоксикарбонил)(2-((трет-бутоксикарбонил)амино)-этил)амино)метил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-3-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноат
Раствор (R)-трет-бутил-3-(4-(5-(((2-((трет-бутоксикарбонил)амино)этил)амино)метил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-3-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноата (290 мг, 0,39 ммоль), BOC2O (0,18 мл, 0,78 ммоль) и TEA (0,16 мл, 1,16 ммоль) в THF (10 мл) перемешивали при комнатной температуре в течение ночи, разбавляли водой, экстрагировали EtOAc (3 раза), промывали насыщенным раствором соли, сушили над MgSO4, фильтровали, концентрировали и очищали на колонке с силикагелем (40 г) с использованием смеси EtOAc/гексан (0-80%), получая указанное в заголовке соединение.
LC/MS: м/е 848,47 [М+Н]+.
Стадия K: трет-бутил-(R)-3-(4-(5-(((трет-бутоксикарбонил)(2-((трет-бутоксикарбонил)амино-этил)-амино)метил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-3-ил)фенокси)-2-гидроксипропаноат
К раствору (R)-трет-бутил-3-(4-(5-(((трет-бутоксикарбонил)(2-((трет-бутоксикарбонил)амино)этил)амино)метил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-3-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноата (305 мг, 0,36 ммоль) в THF (3,6 мл) при комнатной температуре добавляли TBAF (430 мг, 0,43 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, а затем концентрировали и очищали на колонка с силикагелем (40 г), используя смесь EtOAc/гексан (0-75%), получая указанное в заголовке соединение.
LC/MS: м/е 734,42 (М+Н)+.
Стадия L: (S)-3-(4-(5-(((2-аминоэтил)амино)метил)-1-(3-аминопропил)-1H-пиразол-3-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)-азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота
Указанное в заголовке соединение получали в соответствии с процедурой, описанной на стадиях C-H примера 1, начиная с трет-бутил-(R)-3-(4-(5-(((трет-бутоксикарбонил)(2-((трет-бутоксикарбонил)амино)этил)амино)метил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-3-ил)фенокси)-2-гидроксипропаноата.
LC/MS: м/е 739,18 [М+Н]+.
1H ЯМР (500 МГц, оксид дейтерия) δ (м.д.) 7,59 (д, J=8,4 Гц, 2H), 7,02-6,86 (м, 3H), 6,74 (с, 1H), 5,07-4,92 (м, 1H), 4,54-4,28 (м, 5H), 4,22 (т, J=6,9 Гц, 2H), 3,43 (т, J=6,9 Гц, 2H), 3,32 (т, J=7,0 Гц, 2H), 2,93 (т, J=7,9 Гц, 2H), 2,10 (п, J=6,8 Гц, 2H), 1,23 (с, 3H), 0,96 (с, 3H).
ПРИМЕР 126
(S)-3-(4-(5-(аминометил)-1-(3-аминопропил)-1H-пиразол-3-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота
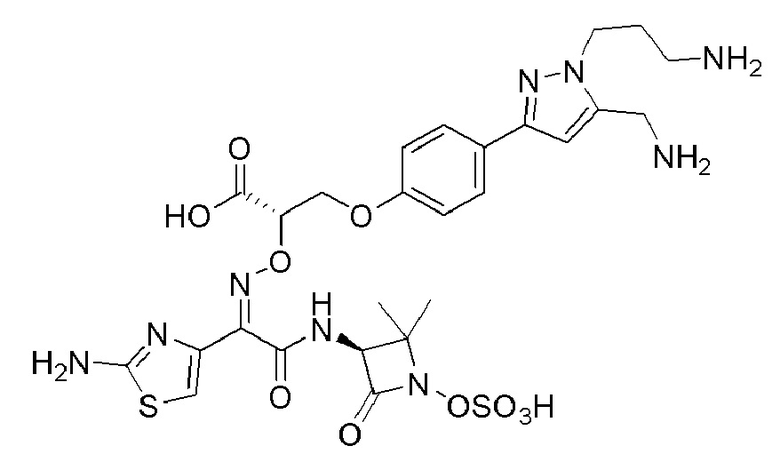
Стадия A: бис(трет-бутоксикарбонил)амид калия
К раствору ди-трет-бутил-иминодикарбоксилата (20 г, 92 ммоль) в EtOH (20 мл) при комнатной температуре добавляли KOH (5,4 г, 97 ммоль) в этаноле (20 мл). Реакционную смесь перемешивали при комнатной температуре в течение 40 минут, а затем добавляли Et2O. Смесь фильтровали и промывали Et2O, получая указанное в заголовке соединение, которое использовали на следующей стадии без дополнительной очистки.
Стадия B: трет-бутил-(R)-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-5-(хлорметил)-1H-пиразол-3-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноат
К раствору (R)-трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)пропил)-5-(гидроксиметил)-1H-пиразол-3-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноата (100 мг, 0,16 ммоль) в DCM (825 мкл) при 0°C добавляли TEA (23 мкл, 0,16 ммоль), а затем метансульфонилхлорид (13 мг, 0,16 ммоль). Реакционную смесь перемешивали при 0°С в течение 1 часа, затем дополнительно добавляли TEA (23,01 мкл, 0,165 ммоль) и метансульфонилхлорид (12,86 мкл, 0,165 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 2 часов, затем распределяли между DCM и водой. Органический слой отделяли, сушили над MgSO4, концентрировали и очищали на колонке с силикагелем (40 г) с использованием смеси EtOAc/гексан (0-30-60%), получая указанное в заголовке соединение.
LC/MS: м/е 624,74 [М+Н]+.
Стадия C: трет-бутил-(R)-3-(4-(5-((бис(трет-бутоксикарбонил)амино)метил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-3-ил)фенокси)-2-((трет-бутилдиметилсилил)-окси)пропаноат
К раствору (R)-трет-бутил-3-(4-(1-(3-((трет-бутоксикарбонил)амино)-пропил)-5-(хлорметил)-1H-пиразол-3-ил)фенокси)-2-((трет-бутилдиметилсилил)-окси)-пропаноата (70 мг, 0,112 ммоль) в DMF (1 мл) при комнатной температуре добавляли бис(трет-бутоксикарбонил)амид калия (32 мг, 0,123 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли дополнительное количество бис(трет-бутоксикарбонил)амида калия (10 мг) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем разбавляли водой и экстрагировали с помощью EtOAc (3 раза). Объединенные органические слои промывали насыщенным солевым раствором (3 раза), сушили над MgSO4, фильтровали и концентрировали. Полученный остаток очищали на колонке с силикагелем (24 г) с использованием EtOAc/гексан (0-30%), получая указанное в заголовке соединение.
LC/MS: м/е 805,45 [М+Н]+.
Стадия D: трет-бутил-(R)-3-(4-(5-((бис(трет-бутоксикарбонил)амино)метил)-1-(3-((трет-бутокси-карбонил)амино)пропил)-1H-пиразол-3-ил)фенокси)-2-гидроксипропаноат
К раствору (R)-трет-бутил-3-(4-(5-((бис(трет-бутоксикарбонил)амино)метил)-1-(3-((трет-бутоксикарбонил)-амино)пропил)-1H-пиразол-3-ил)фенокси)-2-((трет-бутилдиметилсилил)окси)пропаноата (76 мг, 0,094 ммоль) в THF (944 мкл) при комнатной температуре было добавляли TBAF (113 мкл, 0,113 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут, а затем концентрировали. Полученный остаток очищали с помощью ISCO (12 г, EtOAc/гексан (0-60%)), получая указанное в заголовке соединение.
LC/MS: м/е 691,39 [М+Н]+.
Стадия F: (S)-3-(4-(5-(аминометил)-1-(3-аминопропил)-1H-пиразол-3-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота
Указанное в заголовке соединение получали в соответствии с процедурой, описанной на стадиях C-Н примера 1, начиная с (R)-трет-бутил-3-(4-(5-((бис(трет-бутоксикарбонил)амино)метил)-1-(3-((трет-бутоксикарбонил)амино)пропил)-1H-пиразол-3-ил)фенокси)-2-гидроксипропаноата.
LC/MS: м/е 696,27 [М+Н].
1H ЯМР (500 МГц, D2O): δ (м.д.) 7,58 (д, J=8,4 Гц, 2H); 6,93 (д, J=8,3 Гц, 2H); 6,81 (с, 1H); 6,65 (с, 1H); 4,81 (с, 1H); 4,40 (с, 1H); 4,35-4,36 (м, 2H); 4,23 (с, 2H); 4,18 (т, J=6,9 Гц, 2H); 2,94 (т, J=7,8 Гц, 2H); 2,10 (т, J=7,8 Гц, 2H); 1,25 (с, 3H); 1,02 (с, 3H).
БИОЛОГИЧЕСКИЕ АНАЛИЗЫ
Антибактериальная активность: Определение ингибирующей рост концентрации
Концентрации соединений, необходимые для ингибирования роста различных штаммов бактерий, определяли с помощью анализа, в котором оценивали рост бактерий путем измерения оптической плотности при 600 нм (OD600). Тестируемые бактериальные штаммы включали клинические штаммы Escherichia coli, экспрессирующие DM-1 (CLB30016), Klebsiella pneumoniae, экспрессирующие KPC-1 (CL6569), Acinetobacter baumannii, экспрессирующие ТЕМ-1, AmpC и Oxa-24/40 (CL6188) и Pseudomonas aeruginosa, экспрессирующая AmpC (CL5701). Все соединения были протестированы в присутствии ингибитора β-лактамазы (BLi, Релебактам) в 384-луночных микропланшетах. Клинические штаммы хранили в виде замороженных образцов для одноразового использования, оттаивали и разводили в 1,1X скорректированном по катионам бульона Мюллера-Хинтон II, с достижением приблизительно 2×105 КОЕ/мл. Испытуемые соединения растворяли в DMSO, и для анализа серийно разводили в соотношении 1:50, получая конечные концентрации от 100 мкМ до 0,098 мкМ. В день анализа 1 мкл тестируемого соединения добавляли к планшетам, с последующим добавлением 4 мкл раствора BLi (50 мкг/мл) в буфере MOPS и 45 мкл разведения бактерий. Планшеты центрифугировали при 1000 оборотах в минуту в течение 30 секунд, встряхивали при приблизительно 800 оборотах в минуту в течение 1 минуты, и инкубировали при 35±2°С в течение 22 часов. Концентрация BLi, используемая для анализа, составляла 4 мкг/мл. В конце инкубации с помощью спектрофотометра определяли поглощение при 600 нм. Ингибирование количественно оценивали путем определения наименьшей концентрации тестируемого соединения, которое требовалось для ингибирования 95% роста бактерий. Результаты оценки соединений по примерам 1-126 представлены в таблице I, и они выражены как концентрация соединения, которая ингибирует 95% бактериальный рост (минимальная ингибирующая пороговая концентрация; MITC95).
Соединения настоящего изобретения обладают эффектом ингибирования роста бактерий. Например, для типичных соединений по примерам 1-126 ингибирующие рост концентрации определены как 100 мкМ или менее.
Таблица I. Антибактериальная активность соединений примеров 1-126
MITC95
(мкМ)
MITC95
(мкМ)
MITC95
(мкМ)
MITC95
(мкМ)
| название | год | авторы | номер документа |
|---|---|---|---|
| БИЦИКЛИЧЕСКИЕ АРИЛЬНЫЕ МОНОБАКТАМОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2017 |
|
RU2733402C2 |
| ПРОИЗВОДНЫЕ СЕРИНА В КАЧЕСТВЕ АГОНИСТОВ ГРЕЛИНОВЫХ РЕЦЕПТОРОВ | 2015 |
|
RU2695649C2 |
| ТЕРАПЕВТИЧЕСКИЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ | 2015 |
|
RU2733405C2 |
| ТЕРАПЕВТИЧЕСКИЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ | 2019 |
|
RU2817800C2 |
| ИНГИБИТОРЫ TrkA КИНАЗЫ, ОСНОВАННЫЕ НА НИХ КОМПОЗИЦИИ И СПОСОБЫ | 2015 |
|
RU2672583C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2009 |
|
RU2525116C2 |
| АНТИТЕЛА К В7-Н3 И КОНЪЮГАТЫ АНТИТЕЛА И ЛЕКАРСТВЕННОГО СРЕДСТВА | 2017 |
|
RU2764651C2 |
| ФЕНОКСИМЕТИЛЬНЫЕ ПРОИЗВОДНЫЕ | 2016 |
|
RU2746481C1 |
| ЗАМЕЩЕННЫЕ НИКОТИНИМИДНЫЕ ИНГИБИТОРЫ ВТК, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ В ТЕРАПИИ РАКОВЫХ, ВОСПАЛИТЕЛЬНЫХ И АУТОИММУННЫХ ЗАБОЛЕВАНИЙ | 2014 |
|
RU2677884C2 |
| ПРОИЗВОДНЫЕ ИЗОХИНОЛИНОНА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ РАКА | 2015 |
|
RU2690853C2 |
Изобретение относится к биарильным монобактамным соединениям формулы I и их фармацевтически приемлемым солям, где W представляет собой связь или O; Rx и Rz независимо представляют собой водород, C1-C3алкил; X представляет собой N или CR1; R1 представляет собой водород, C1-C3алкил или галоген; в каждом случае Ra независимо представляет собой водород, галоген, C1-C3алкил или -ORe; Z представляет собой C1-C3алкилен, необязательно замещенный 1-3 Rb; в каждом случае Rb независимо представляет собой -C1-C8алкил, -C3-C7циклоалкил, -C(O)ORe, -C(O)NRcRd, тетразолил или -P(O)(Re)p, где указанный -C1-C8алкил и указанный -C3-C7циклоалкил необязательно замещены 1-3 Ra; AryA представляет собой 5-6-членное моноциклическое ароматическое кольцо с 0, 1, 2 или 3 кольцевыми атомами, независимо выбранными из N, N в виде четвертичной соли, необязательно замещенное 1-4 R4; Y представляет собой O; R2 представляет собой водород или -C1-C3алкил, где указанный -C1-C3алкил необязательно замещен 1-3 Ra; A1 представляет собой AryA; A2 представляет собой группы пиразола, имидазола и пиридина (приведены в формуле изобретения), где * указывает на присоединение к Q и ** указывает на присоединение к M; в каждом случае R4 представляет собой галоген; HetB представляет собой 3-6-членное насыщенное или мононенасыщенное моноциклическое кольцо с 1, 2 или 3 гетероатомами в кольце, независимо выбранными из N, N в виде четвертичной соли и O, необязательно замещенное 1-3 Ra; Q представляет собой связь, CH2, O, S, -(CH2)nNR3- или -NR3(CH2)n-, где каждый CH2 является незамещенным, или замещен 1-2 заместителями, выбранными из галогена, -C1-C6алкила, ORe и -(CH2)nNRcRd; R3 представляет собой водород или -C1-C3алкил, где указанный -C1-C3алкил необязательно замещен 1-3 Ra; M представляет собой -(CH2)nR5 или R5; R5 представляет собой H, C2-C10алкил, C3-C7циклоалкил, HetB или -NH(C1-C6алкил), где указанный C2-C10алкил и указанный C3-C7циклоалкил необязательно замещены 1-4 R6; в каждом случае R6 независимо выбран из группы, состоящей из галогена, -ORe, -NRcRd, -(CH2)nNRcRd и HetB; в каждом случае Rc и Rd независимо представляют собой водород, -C1-C10алкил или -C1-C10алкилен-HetB и где каждый Rc и Rd необязательно замещен 1-3 Rf; в каждом случае Re независимо представляет собой водород, -C1-C10алкил, -OH или -OC1-C4алкил, где каждый Re необязательно замещен 1-3 Rh; в каждом случае Rf независимо представляет собой галоген, -C1-C10алкил, -OH, -OC1-C4алкил или NH2, где указанный -C1-C10алкил необязательно замещен 1-3 заместителями, независимо выбранными из -OH, галогена, циано и -S(O)2CH3; каждый n независимо равен 0, 1, 2, 3 или 4; каждый m независимо равен 0, 1 или 2 и каждый p независимо равен 1 или 2. Также изобретение относится к соли трифторуксусной кислоты соединения формулы I, а также к конкретным биарильным соединениям. Технический результат – биарильные соединения, проявляющие антибактериальную активность. 6 н. и 10 з.п. ф-лы, 7 табл., 126 пр.
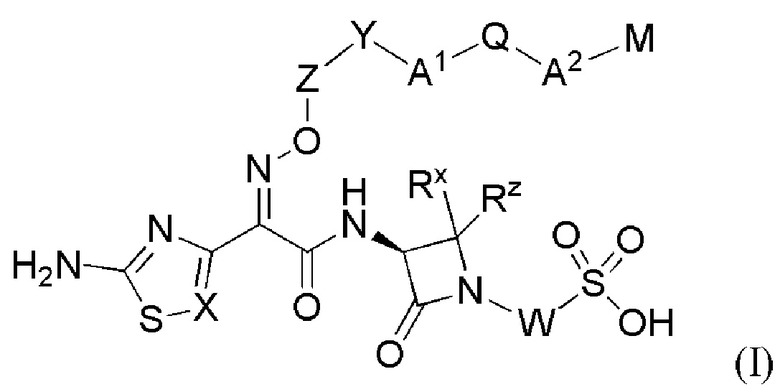
1. Соединение Формулы I
или его фармацевтически приемлемая соль,
где W представляет собой связь или O;
Rx и Rz независимо представляют собой водород, C1-C3алкил;
X представляет собой N или CR1;
R1 представляет собой водород, C1-C3алкил или галоген;
в каждом случае Ra независимо представляет собой водород, галоген, C1-C3алкил или -ORe;
Z представляет собой C1-C3алкилен, необязательно замещенный 1-3 Rb;
в каждом случае Rb независимо представляет собой -C1-C8алкил, -C3-C7циклоалкил, -C(O)ORe, -C(O)NRcRd, тетразолил или -P(O)(Re)p, где указанный -C1-C8алкил и указанный -C3-C7циклоалкил необязательно замещены 1-3 Ra;
AryA представляет собой 5-6-членное моноциклическое ароматическое кольцо с 0, 1, 2 или 3 кольцевыми атомами, независимо выбранными из N, N в виде четвертичной соли, необязательно замещенное 1-4 R4;
Y представляет собой O;
R2 представляет собой водород или -C1-C3алкил, где указанный -C1-C3алкил необязательно замещен 1-3 Ra;
A1 представляет собой AryA;
A2 представляет собой
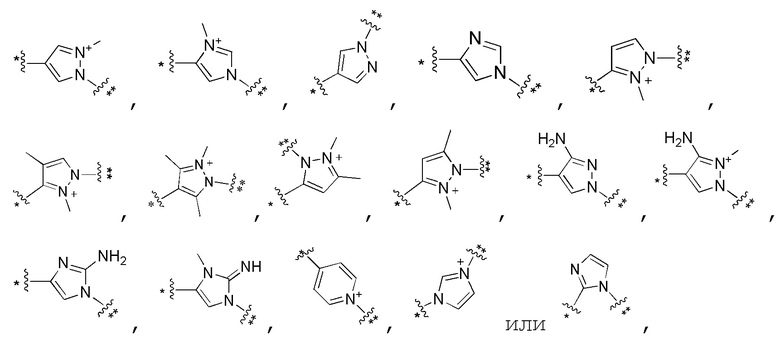
где * указывает на присоединение к Q и ** указывает на присоединение к M;
в каждом случае R4 представляет собой галоген;
HetB представляет собой 3-6-членное насыщенное или мононенасыщенное моноциклическое кольцо с 1, 2 или 3 гетероатомами в кольце, независимо выбранными из N, N в виде четвертичной соли и O, необязательно замещенное 1-3 Ra;
Q представляет собой связь, CH2, O, S, -(CH2)nNR3- или -NR3(CH2)n-, где каждый CH2 является незамещенным или замещен 1-2 заместителями, выбранными из галогена, -C1-C6алкила, ORe и -(CH2)nNRcRd;
R3 представляет собой водород или -C1-C3алкил, где указанный -C1-C3алкил необязательно замещен 1-3 Ra;
M представляет собой -(CH2)nR5 или R5;
R5 представляет собой H, C2-C10алкил, C3-C7циклоалкил, HetB или -NH(C1-C6алкил), где указанный C2-C10алкил и указанный C3-C7циклоалкил необязательно замещены 1-4 R6;
в каждом случае R6 независимо выбран из группы, состоящей из галогена, -ORe, -NRcRd, -(CH2)nNRcRd и HetB;
в каждом случае Rc и Rd независимо представляют собой водород, -C1-C10алкил или -C1-C10алкилен-HetB и где каждый Rc и Rd необязательно замещен 1-3 Rf;
в каждом случае Re независимо представляет собой водород, -C1-C10алкил, -OH или -OC1-C4алкил, где каждый Re необязательно замещен 1-3 Rh;
в каждом случае Rf независимо представляет собой галоген, -C1-C10алкил, -OH, -OC1-C4алкил или NH2, где указанный -C1-C10алкил необязательно замещен 1-3 заместителями, независимо выбранными из -OH, галогена, циано и -S(O)2CH3;
каждый n независимо равен 0, 1, 2, 3 или 4;
каждый m независимо равен 0, 1 или 2, и
каждый p независимо равен 1 или 2.
2. Соединение по п.1 или его фармацевтически приемлемая соль формулы II-1, II-2, II-3, III-1, III-2 или III-3:
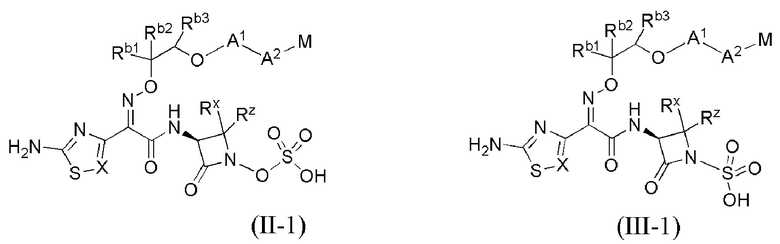
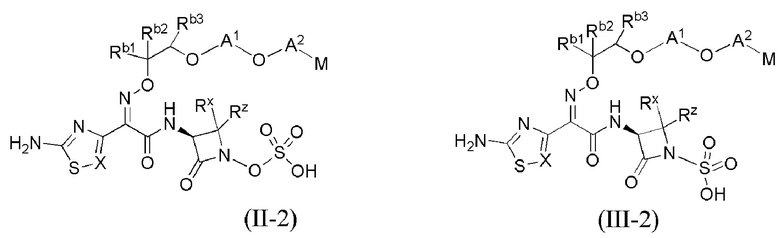
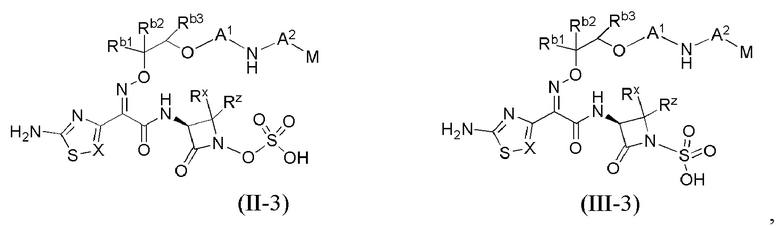
где Rb1 и Rb2 независимо представляют собой водород, C1-C3алкил, тетразолил или -C(O)ORe; и Rb3 представляет собой водород.
3. Соединение по п.1 или 2 или его фармацевтически приемлемая соль, где Х представляет собой N.
4. Соединение по п.1 или 2 или его фармацевтически приемлемая соль, где Х представляет собой CR1.
5. Соединение по любому из пп.1-4 или его фармацевтически приемлемая соль, где А1 представляет собой:
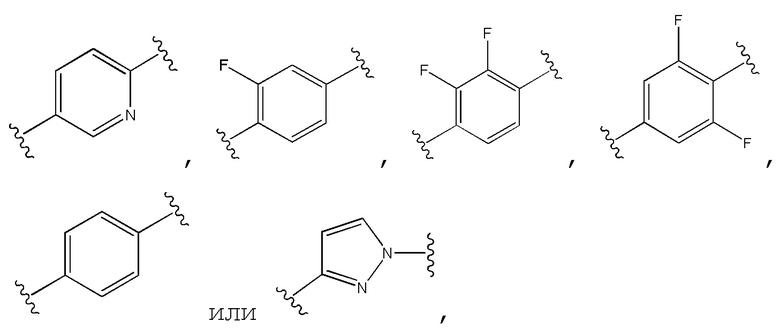
где  представляет собой точку присоединения к остальной части соединения.
представляет собой точку присоединения к остальной части соединения.
6. Соединение по любому из пп.1-5 или его фармацевтически приемлемая соль, где М представляет собой C2-C10алкил, замещенный -NRcRd.
7. Соединение по любому из пп.1-6 или его фармацевтически приемлемая соль, где Rx и Rz представляют собой метил или Rx представляет собой метил и Rz представляет собой водород.
8. Соединение по п.1, имеющее структуру:
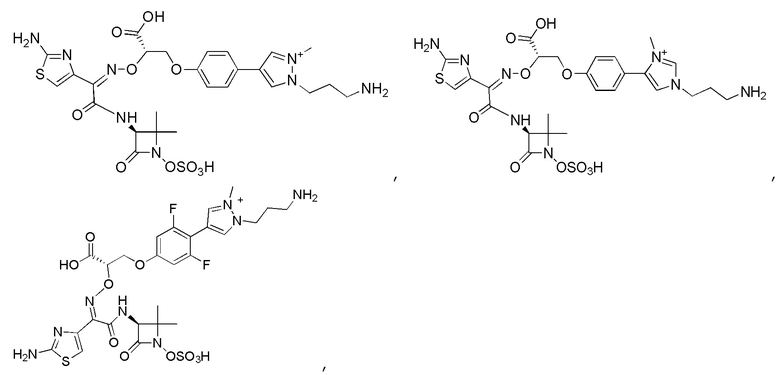
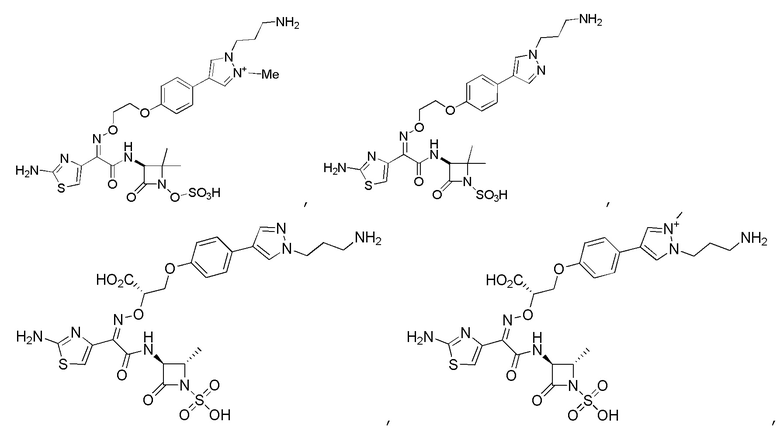
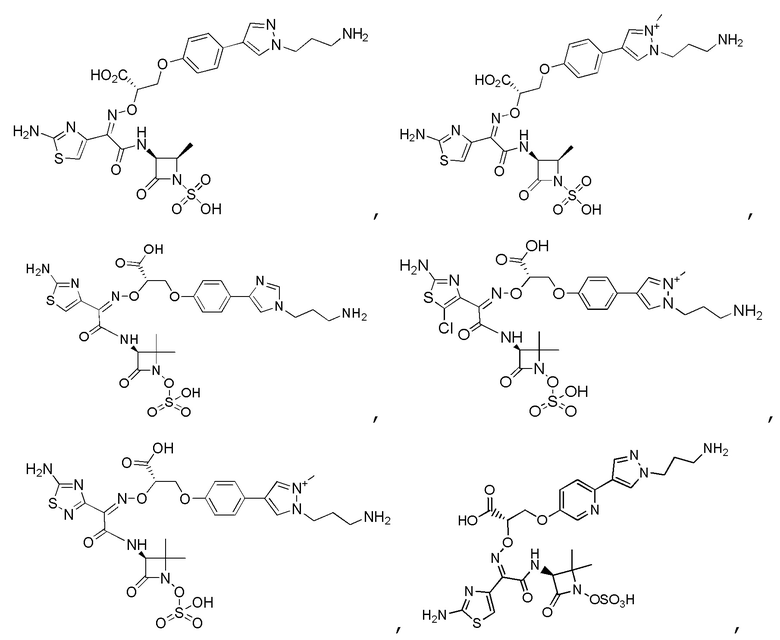
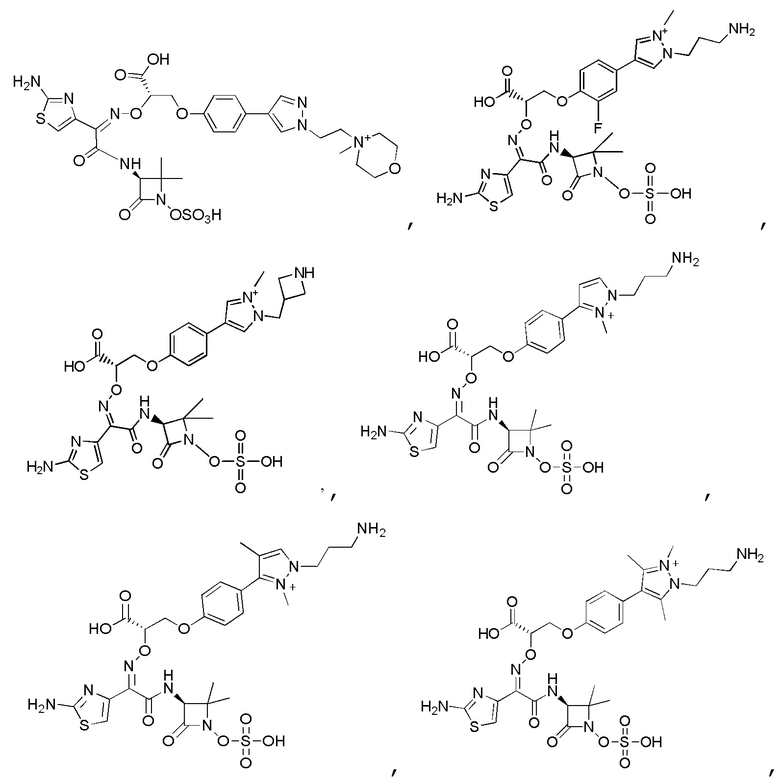
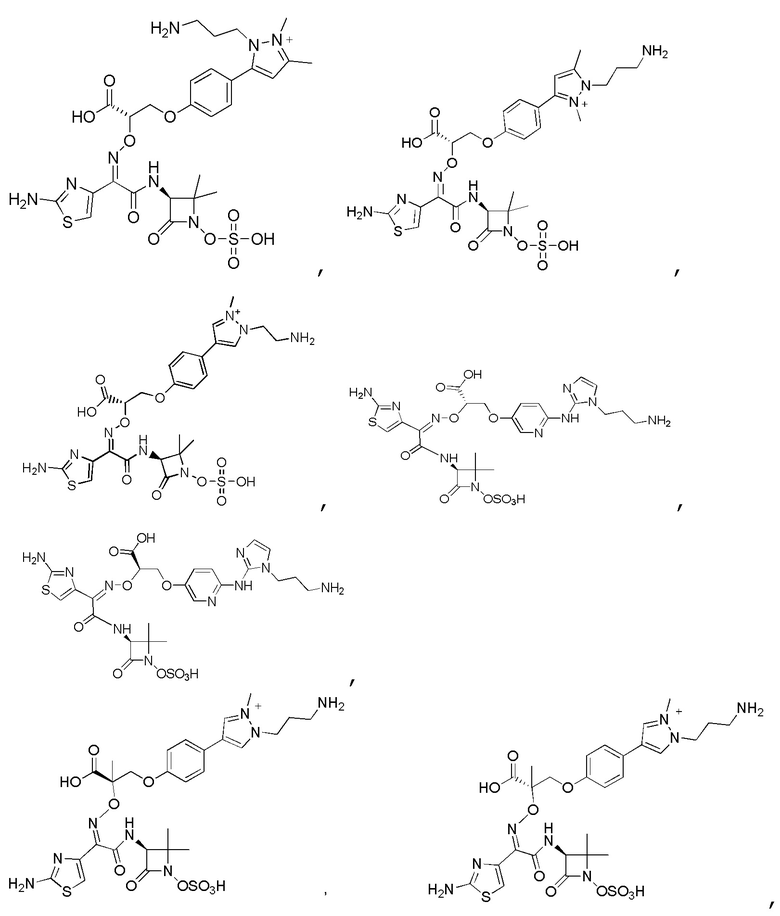
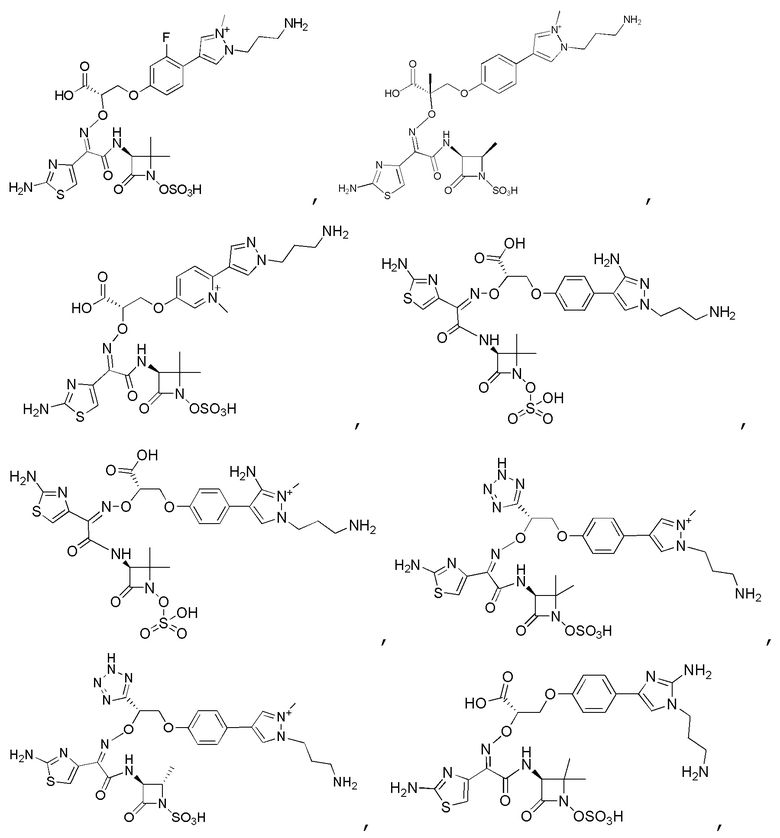
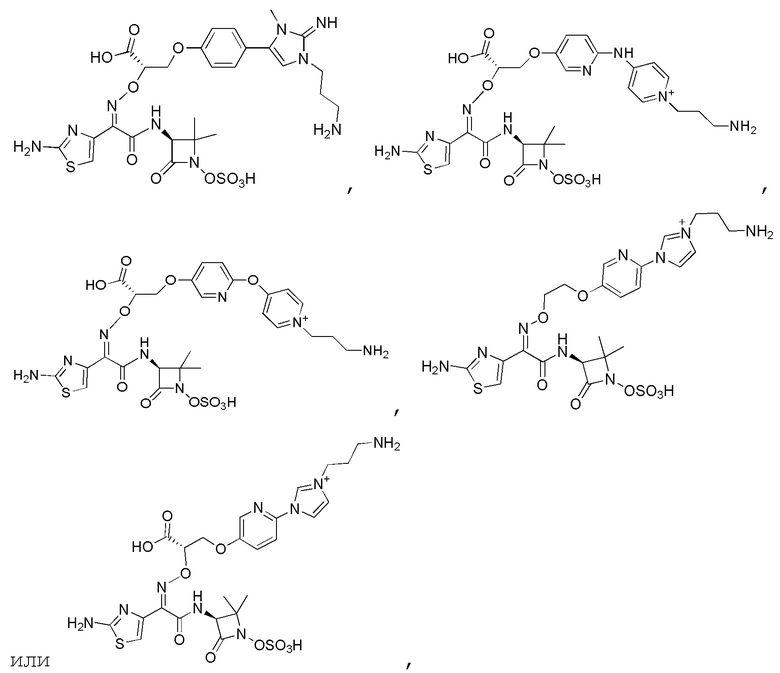
или его фармацевтически приемлемая соль.
9. Соль трифторуксусной кислоты соединения по п.8.
10. Соль трифторуксусной кислоты соединения по п.1.
11. Соединение, выбранное из группы, состоящей из:
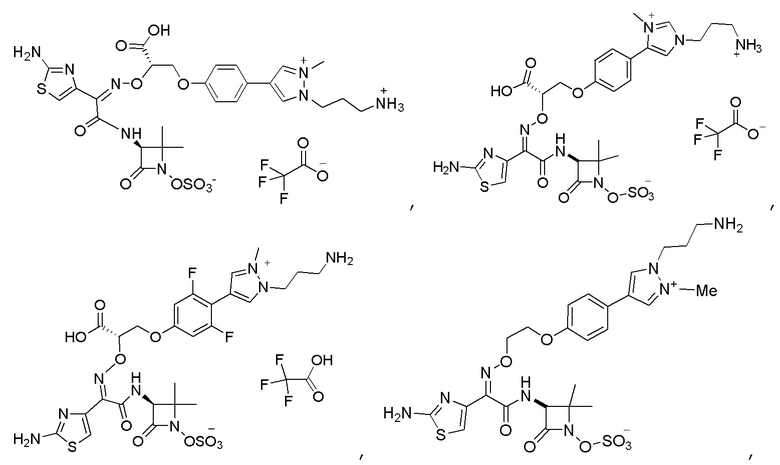
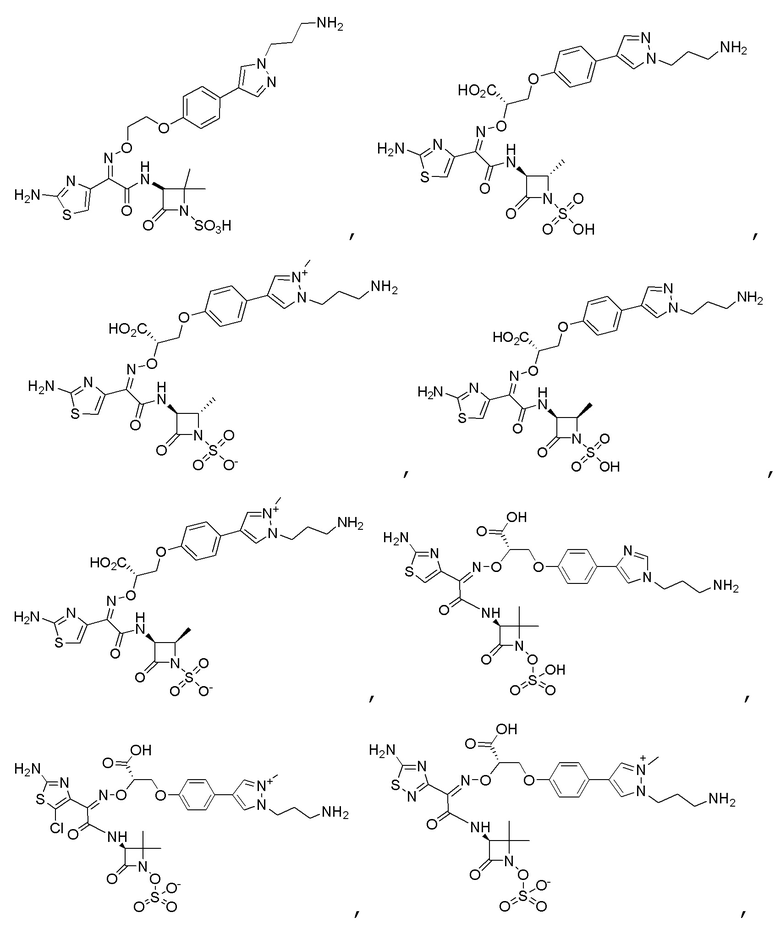
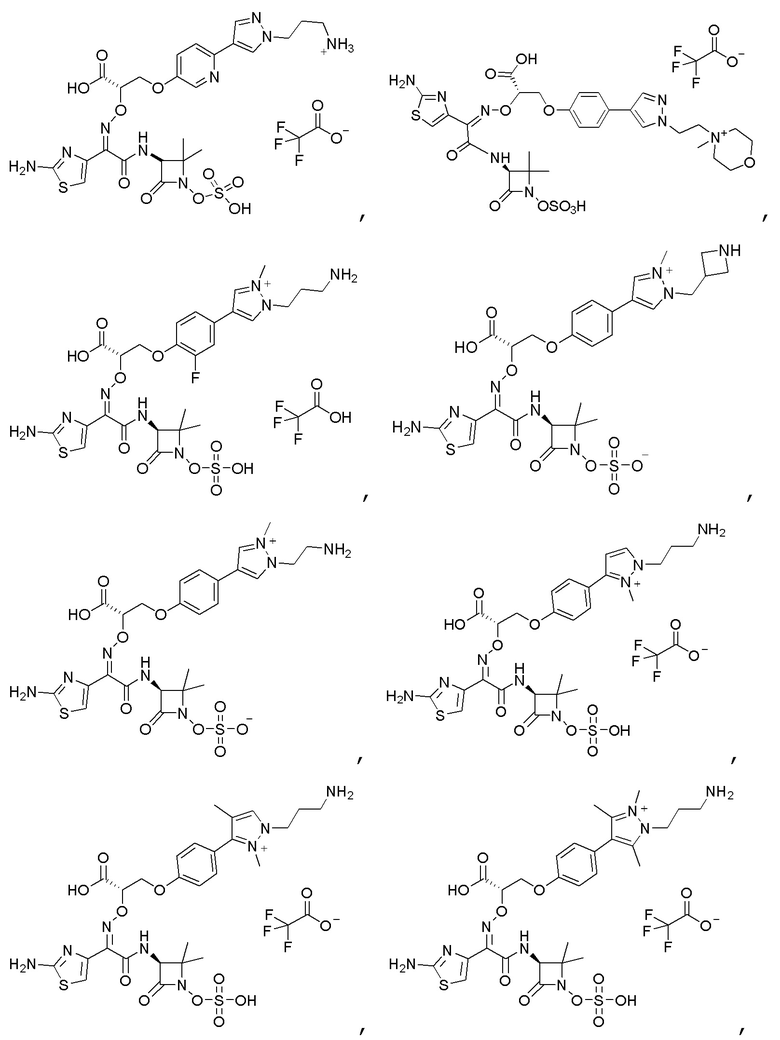
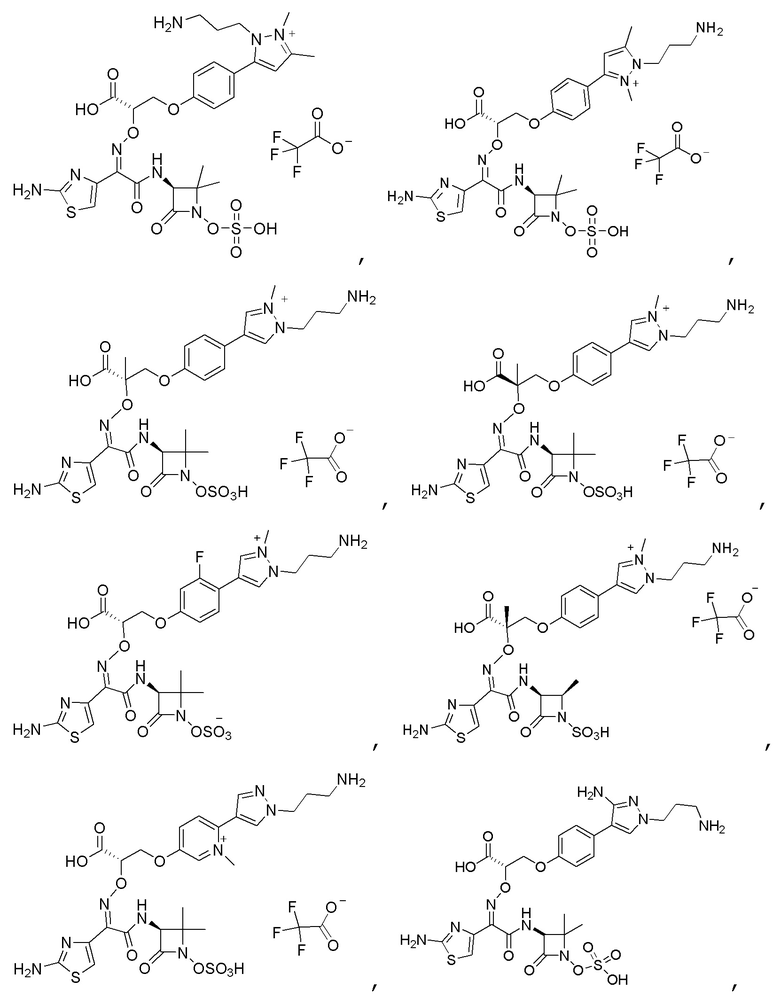
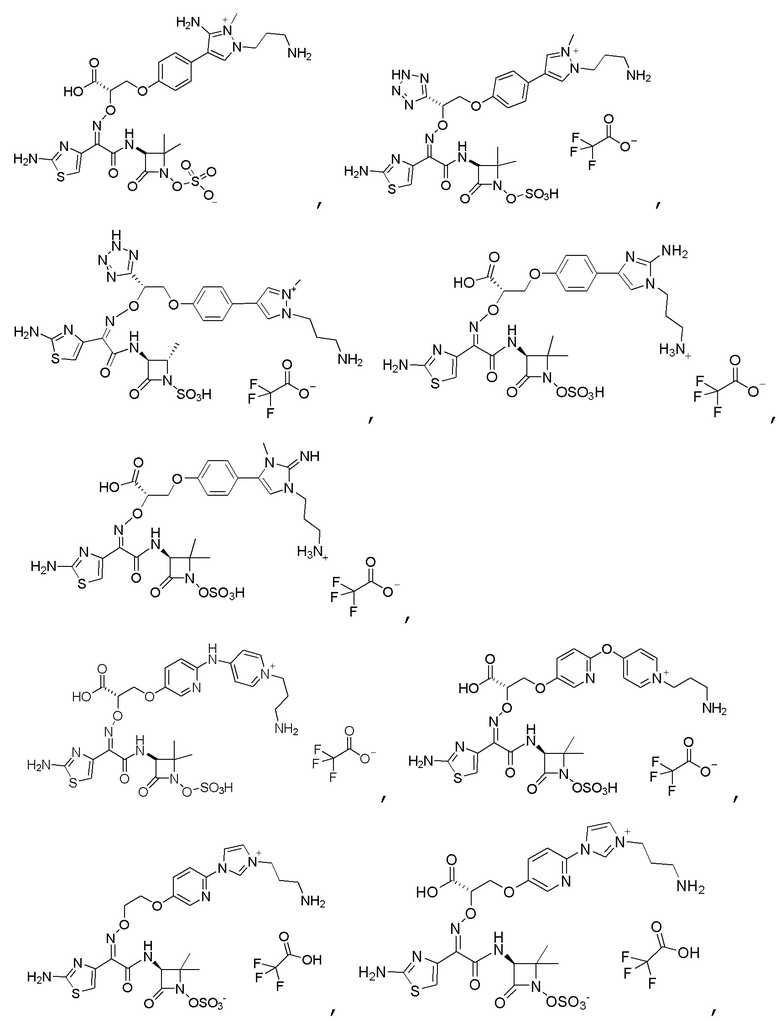
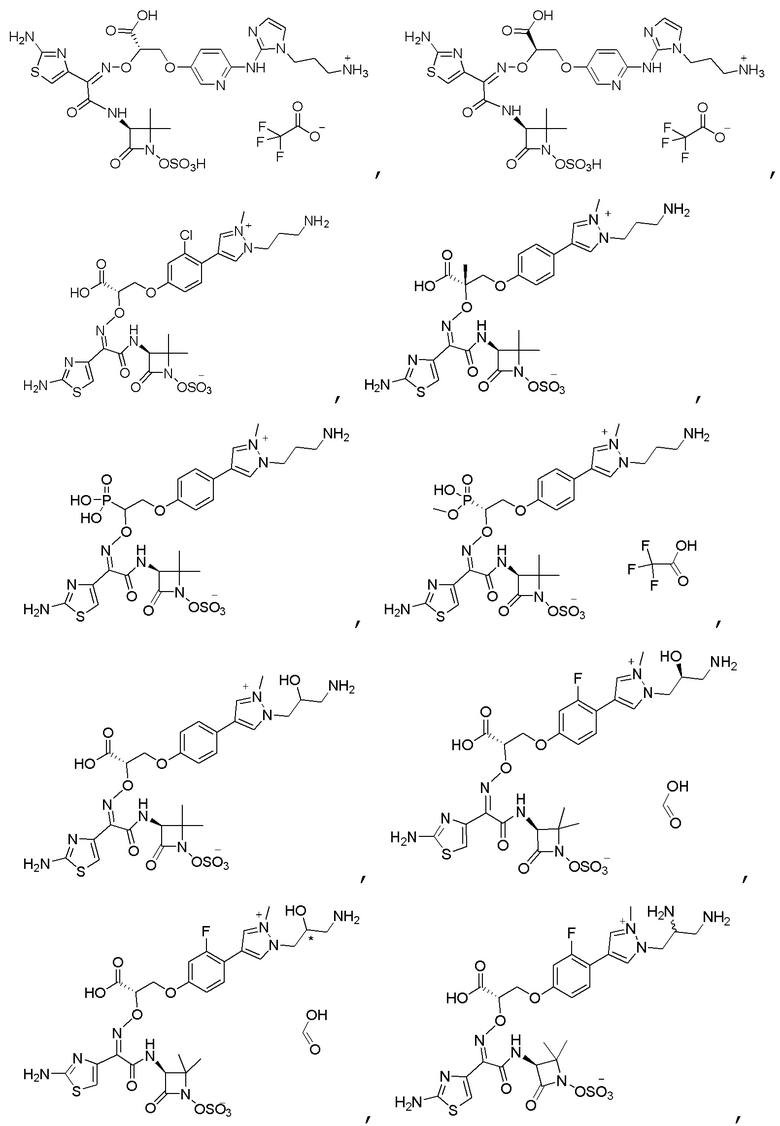
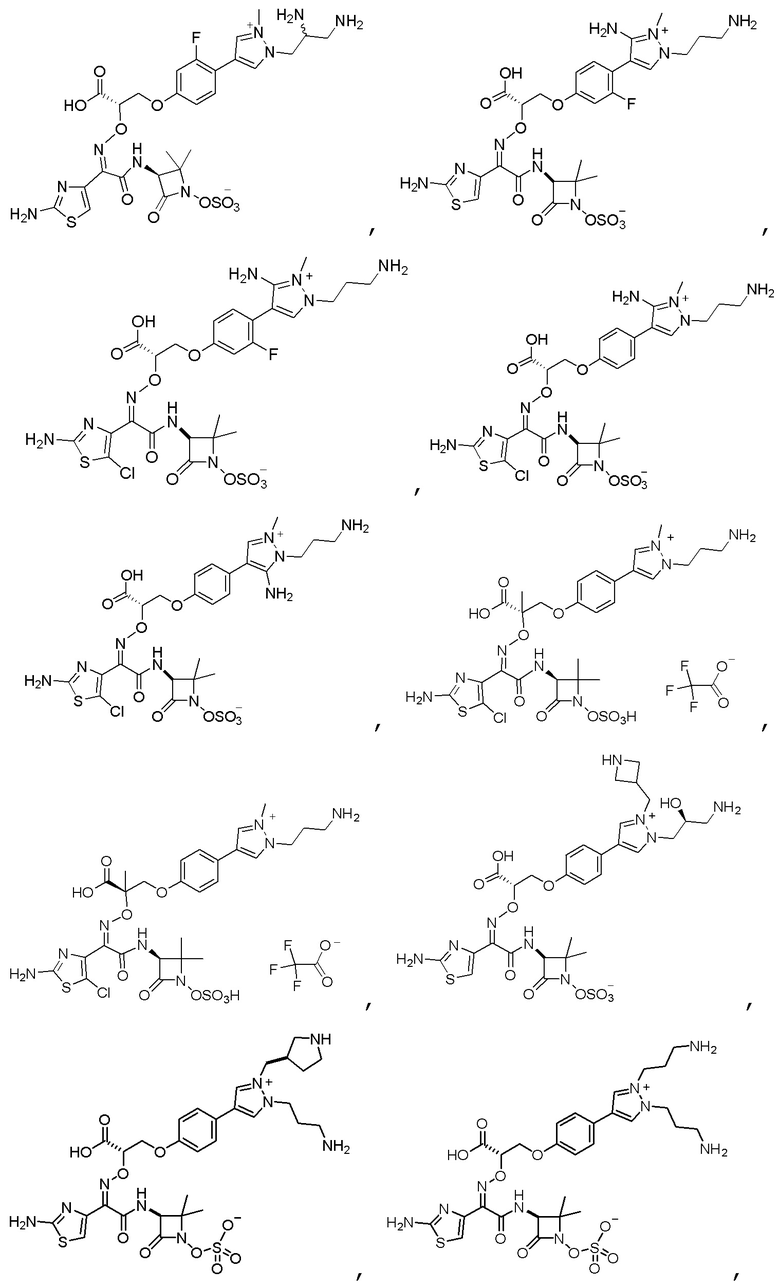
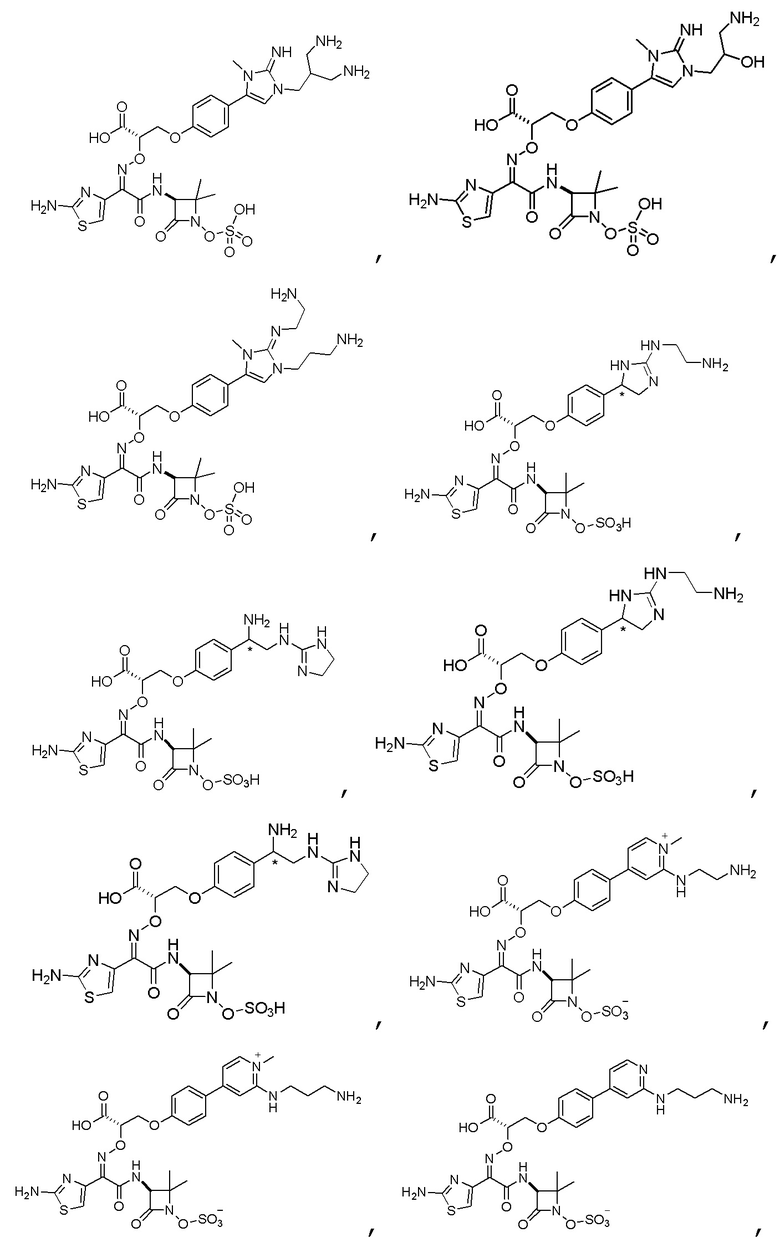
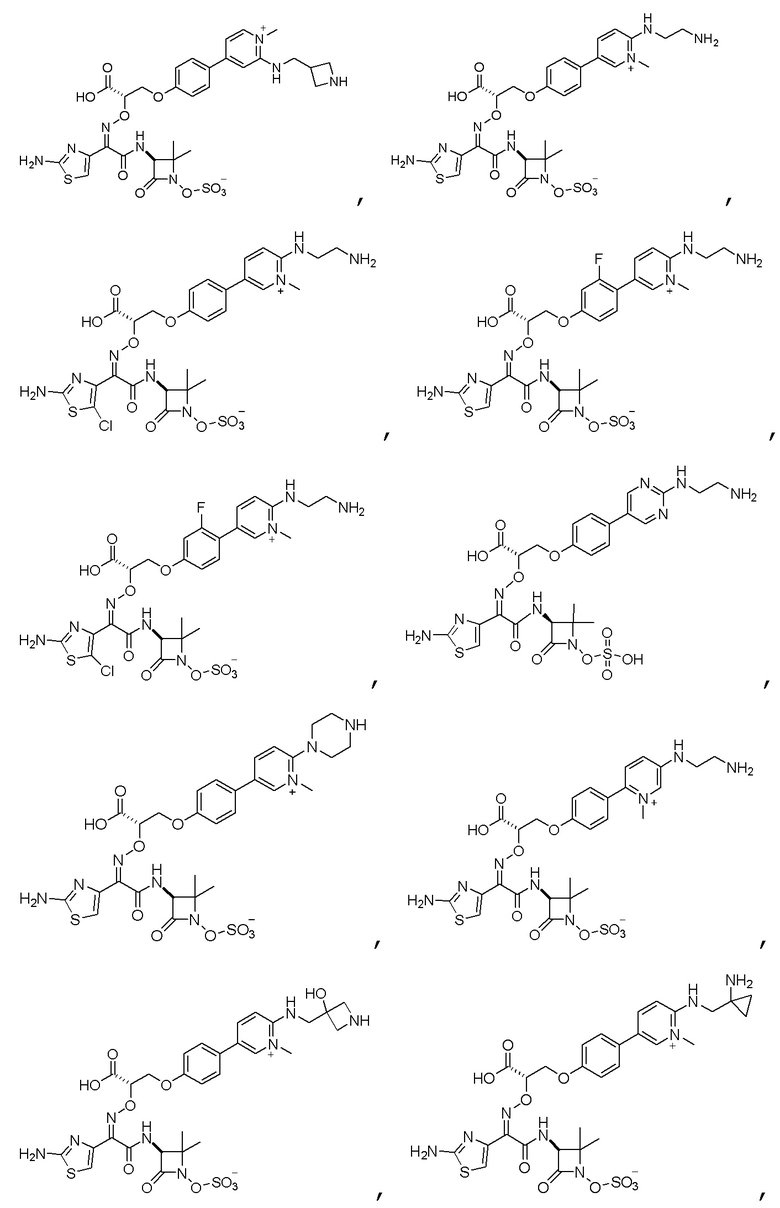
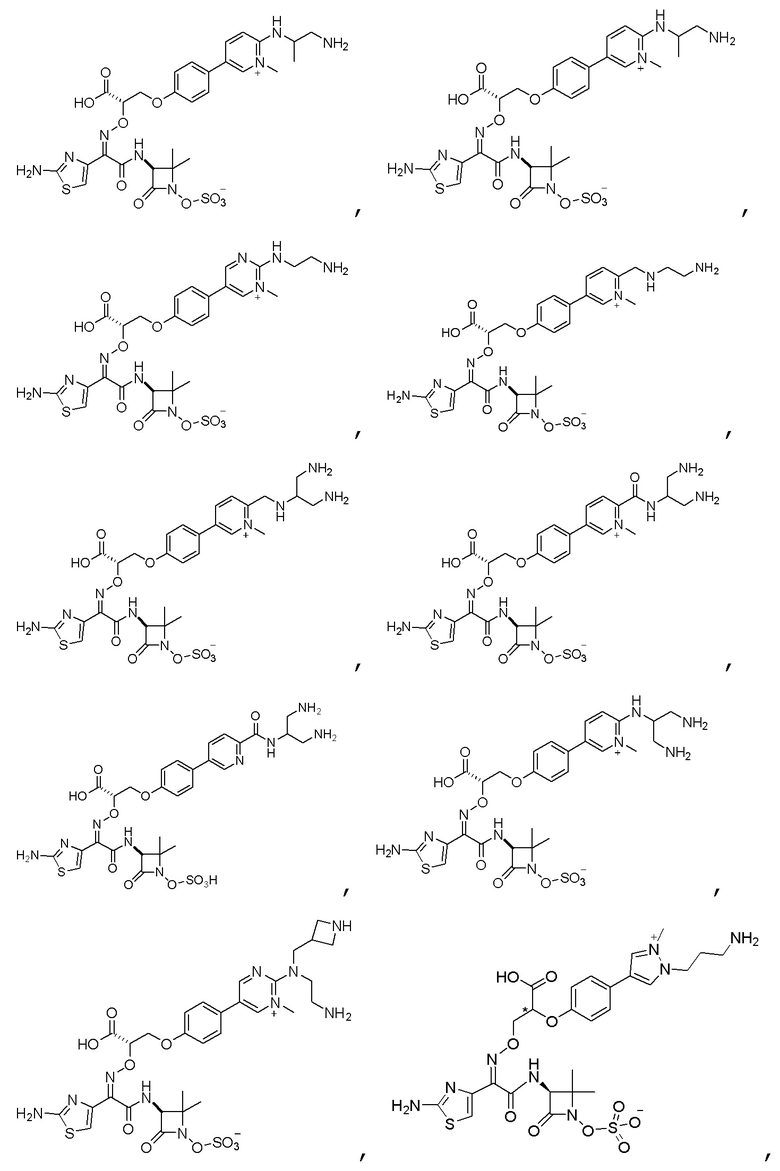
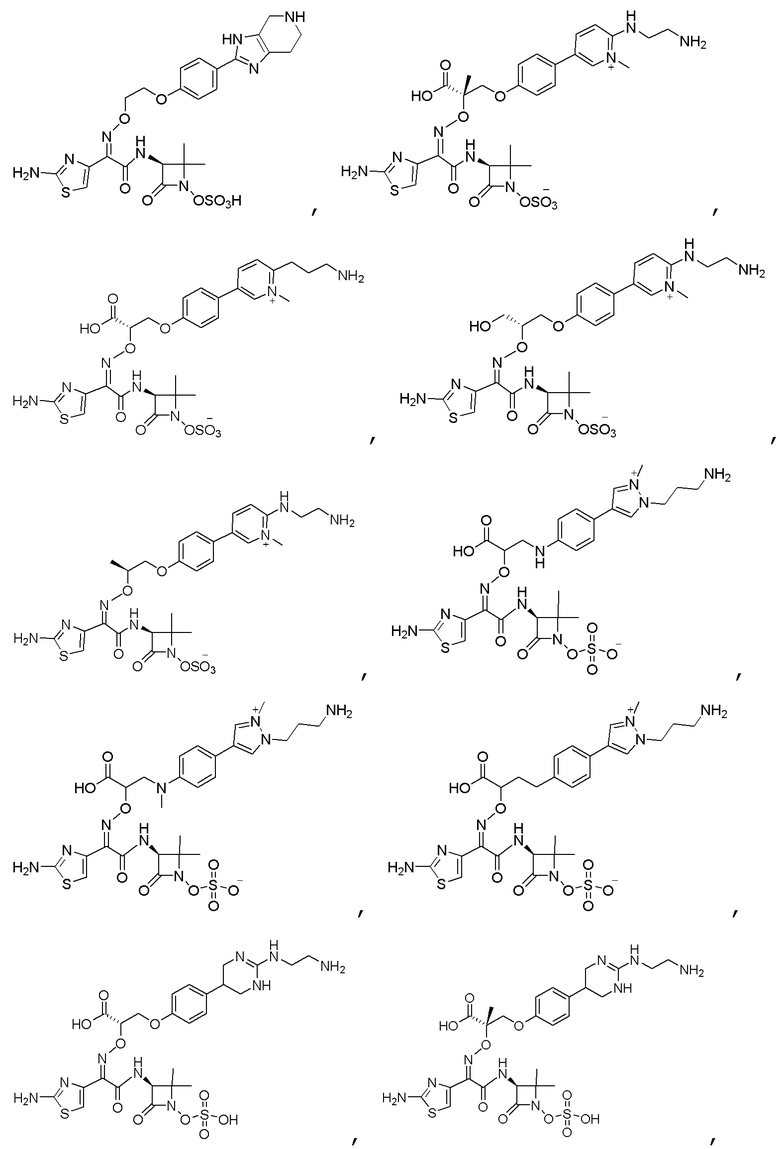
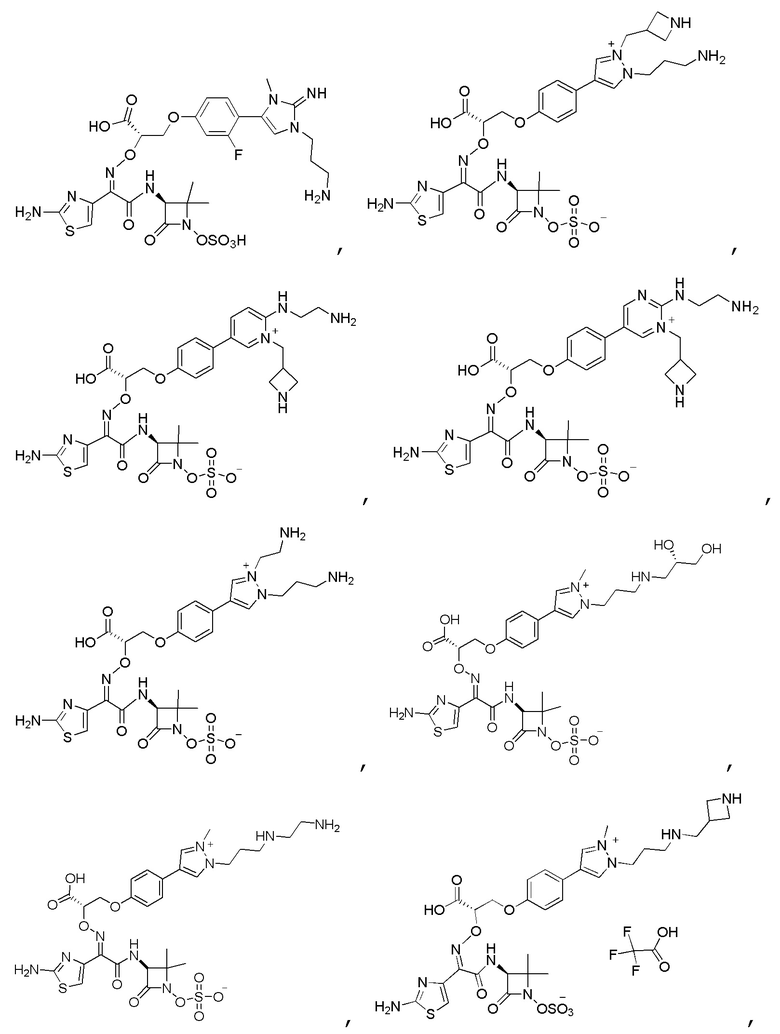
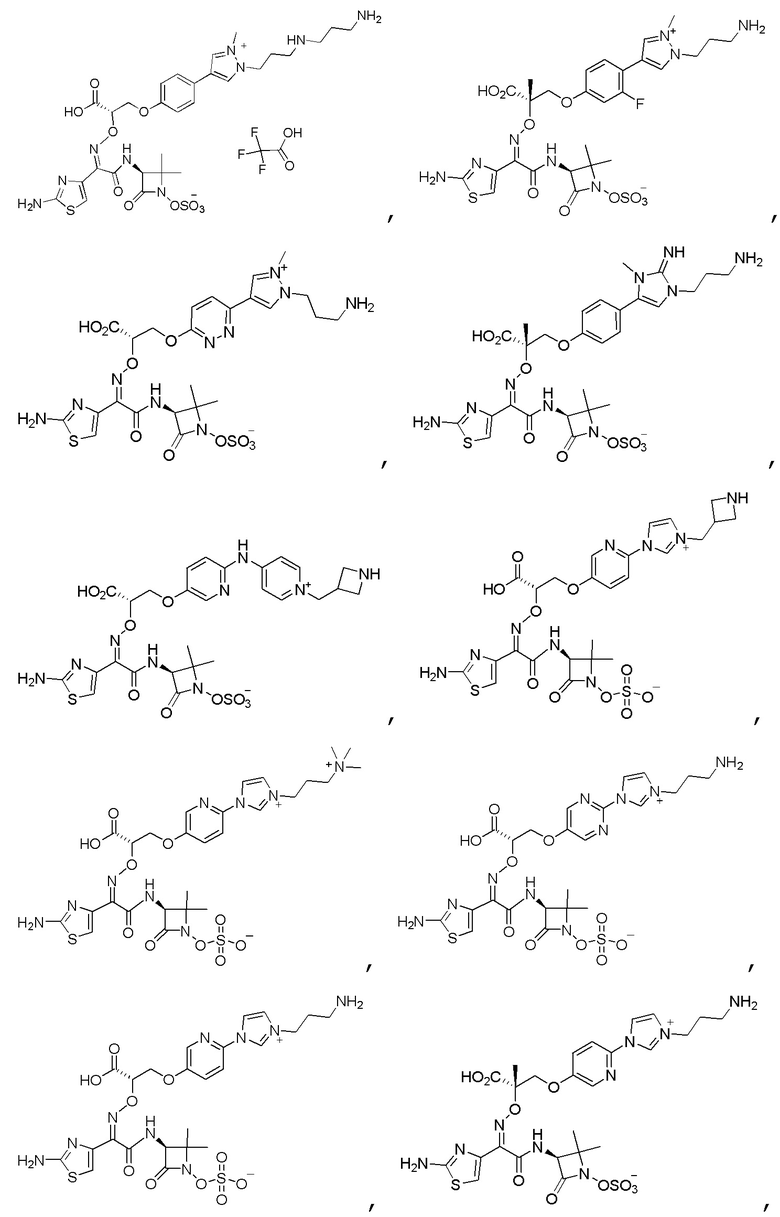
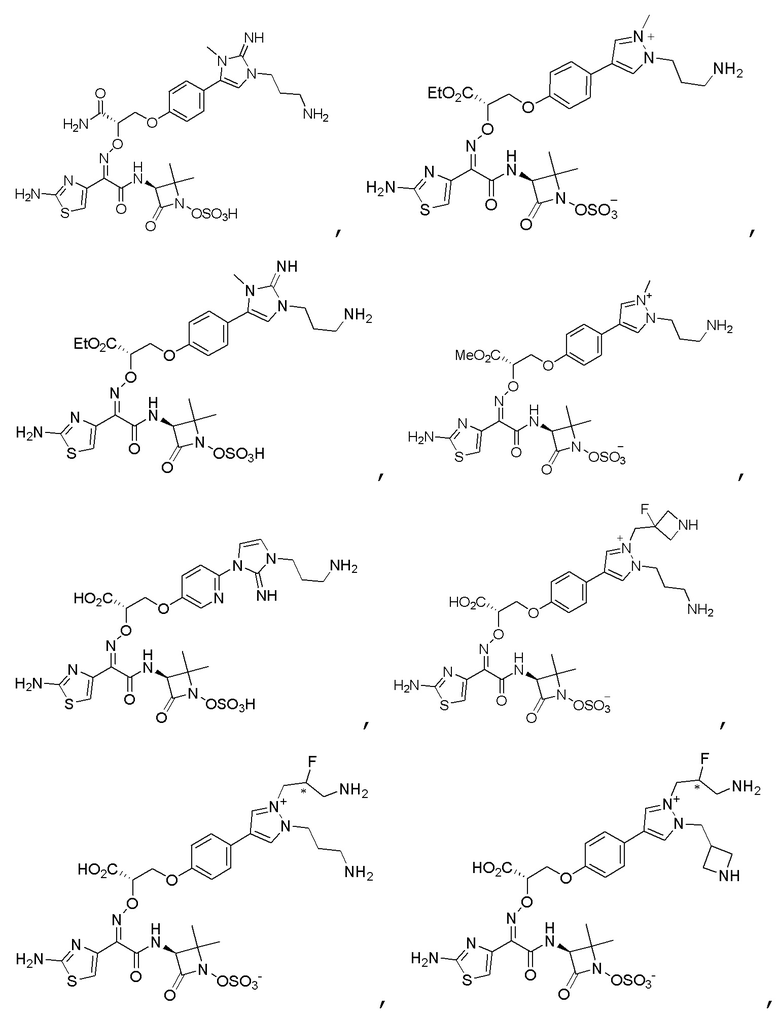
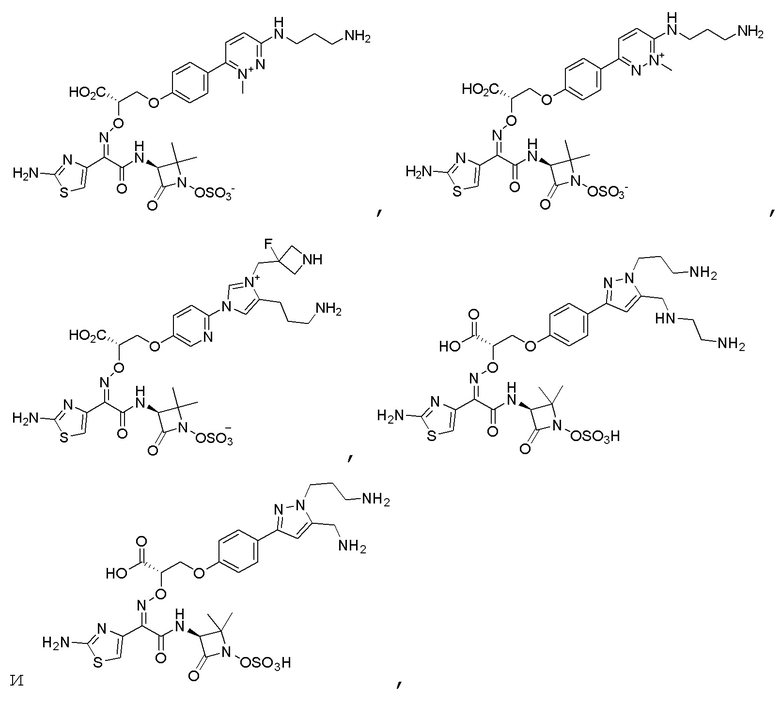
или его фармацевтически приемлемая соль.
12. Соединение по п.11, где фармацевтически приемлемая соль представляет собой соль трифторуксусной кислоты.
13. Соединение, выбранное из группы, состоящей из:
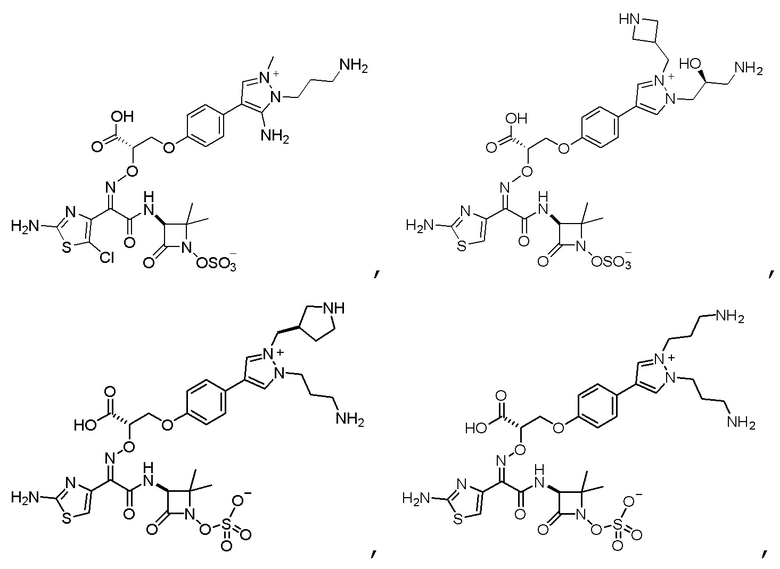
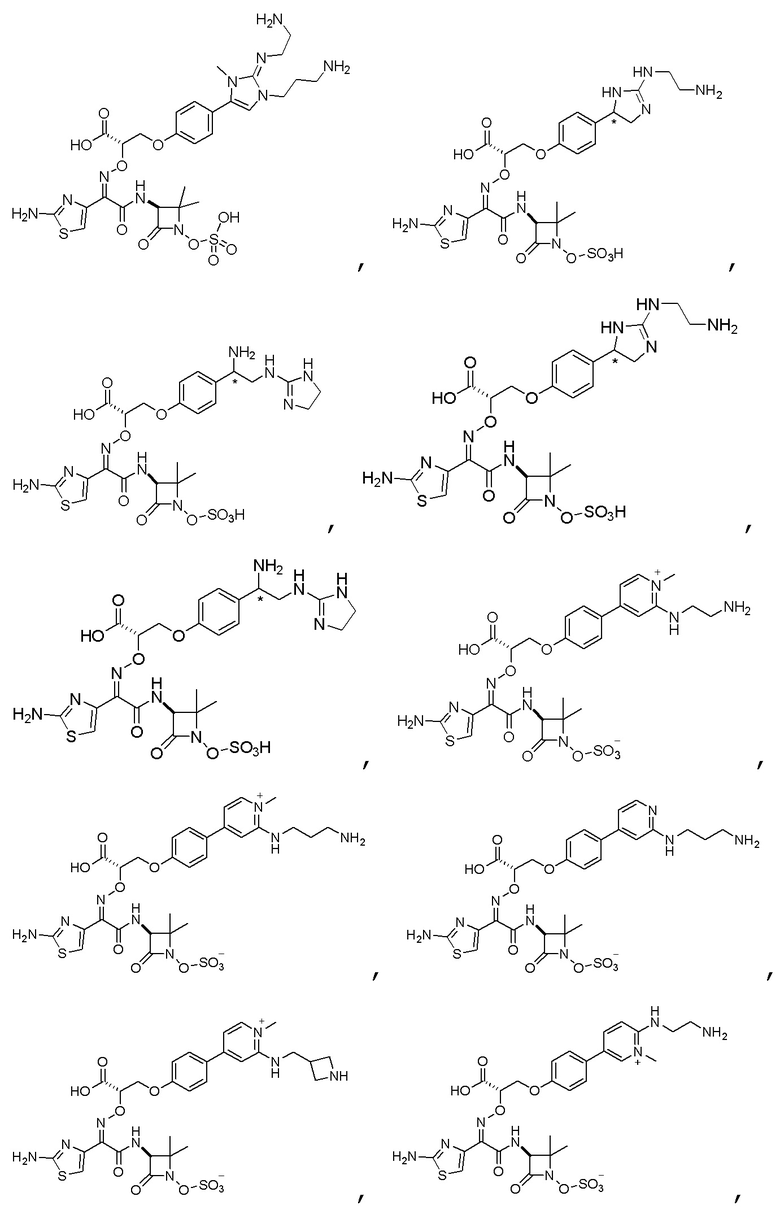
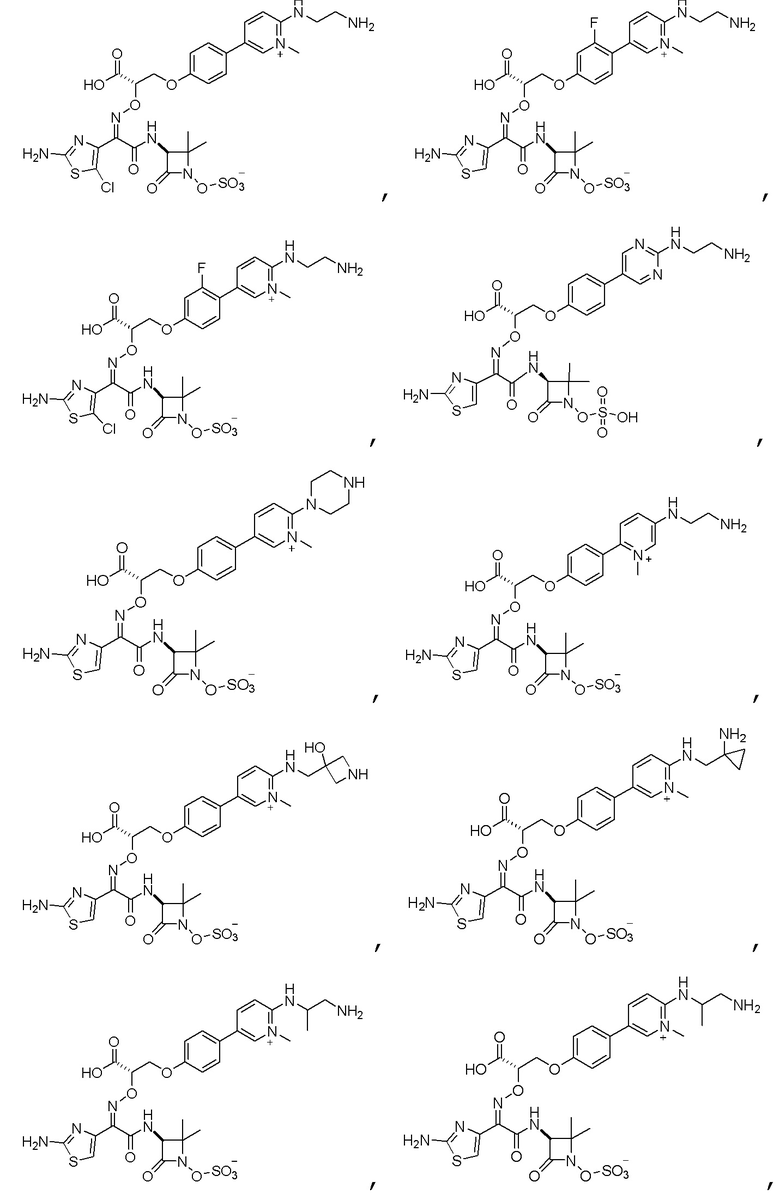
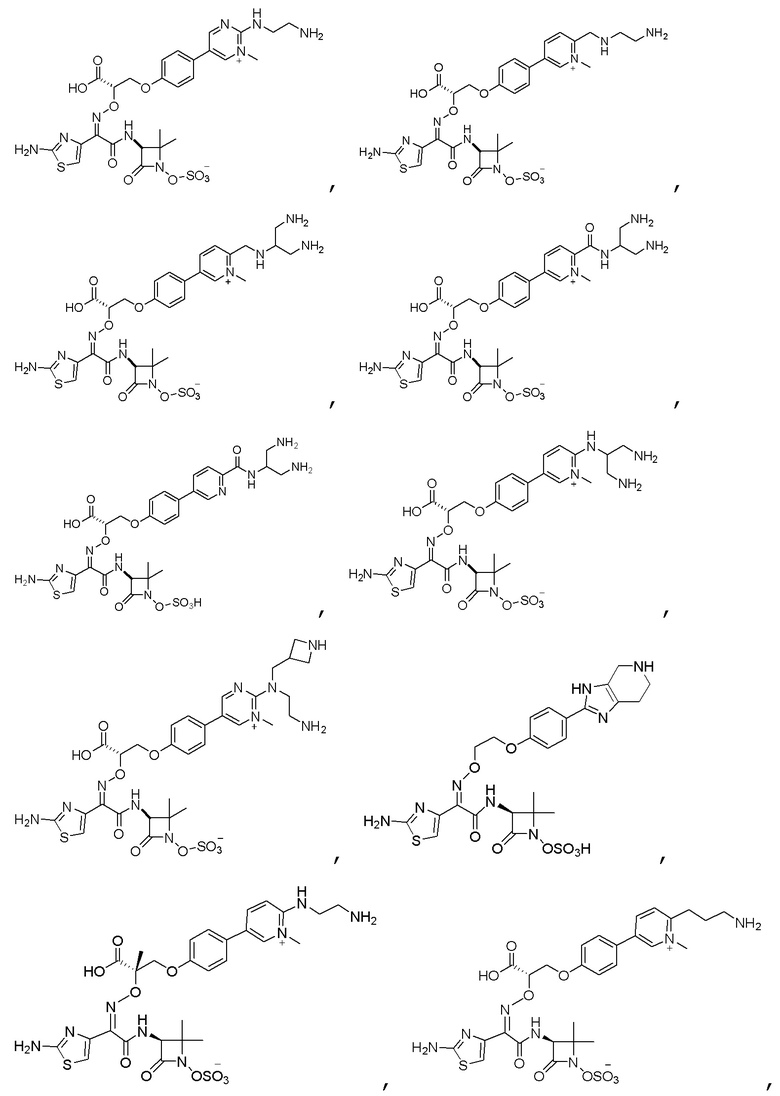
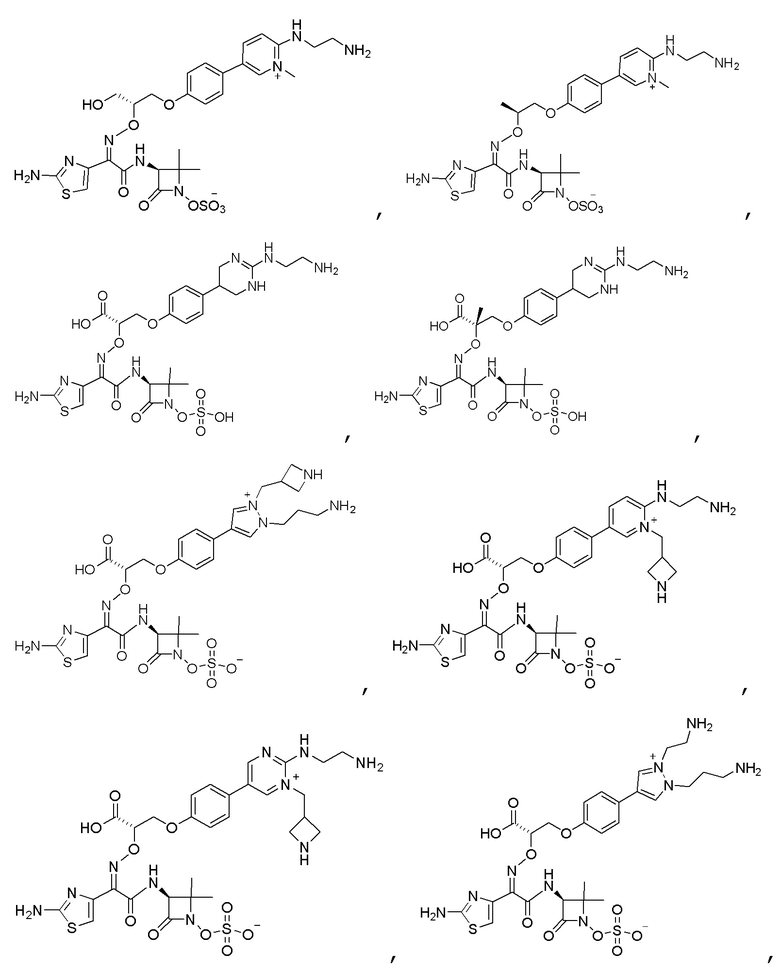
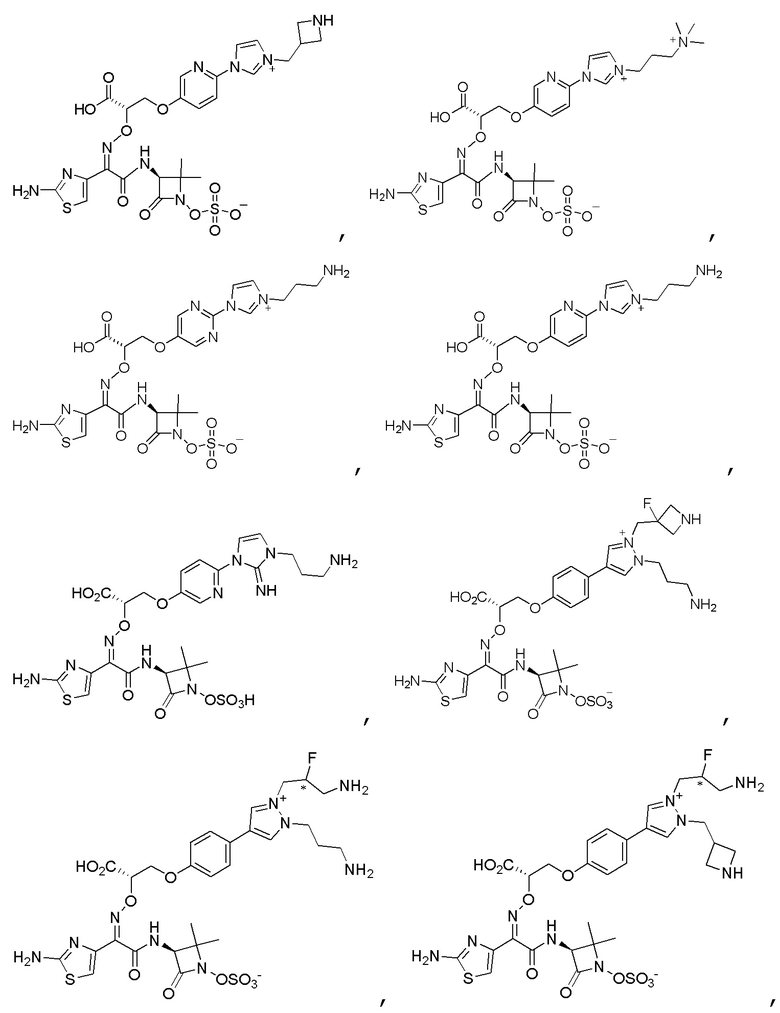
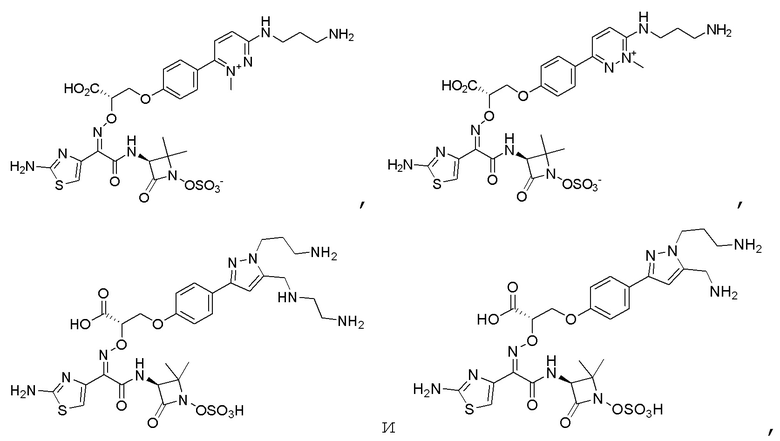
или его фармацевтически приемлемая соль.
14. Соединение по п.13, где фармацевтически приемлемая соль представляет собой соль трифторуксусной кислоты.
15. Соединение, выбранное из группы, состоящей из:
1) (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-2-(4-(1-(3-аммониопропил)-2-метил-1H-пиразол-2-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
2) (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-2-(4-(1-(3-аммониопропил)-3-метил-1H-имидазол-3-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
3) (S)-3-((Z)-2-(((S)-2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)-3,5-дифторфенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
4) (S,Z)-3-(2-((2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)фенокси)этокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
5) (S,Z)-3-(2-((2-(4-(1-(3-аминопропил)-1H-пиразол-4-ил)фенокси)этокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
6) (S)-3-(4-(1-(3-аминопропил)-1H-пиразол-4-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((2S,3S)-2-метил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановой кислоты;
7) (2S,3S)-3-((Z)-2-(((S)-2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2-метил-4-оксоазетидин-1-сульфоната;
8) (S)-3-(4-(1-(3-аминопропил)-1H-пиразол-4-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((2R,3S)-2-метил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановой кислоты;
9) (2R,3S)-3-((Z)-2-(((S)-2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2-метил-4-оксоазетидин-1-сульфоната;
10) (S)-3-(4-(1-(3-аминопропил)-1H-имидазол-4-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановой кислоты;
11) (S)-3-((Z)-2-(2-амино-5-хлортиазол-4-ил)-2-(((S)-2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
12) (S)-3-((Z)-2-(5-амино-1,2,4-тиадиазол-3-ил)-2-(((S)-2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
13) 3-(4-(5-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксо-этилиден)амино)окси)-2-карбоксиэтокси)пиридин-2-ил)-1H-пиразол-1-ил)пропан-1-аминиума;
14) 4-(2-(4-(4-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)фенил)-1H-пиразол-1-ил)этил)-4-метилморфолин-4-иума;
15) 1-(3-аминопропил)-4-(4-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-3-фторфенил)-2-метил-1H-пиразол-2-иума;
16) (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-2-(4-(1-(азетидин-3-илметил)-2-метил-1H-пиразол-2-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
17) (S)-3-((Z)-2-(((S)-2-(4-(1-(2-аминоэтил)-2-метил-1H-пиразол-2-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
18) 1-(3-аминопропил)-3-(4-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)фенил)-2-метил-1H-пиразол-2-иума;
19) 1-(3-аминопропил)-3-(4-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)фенил)-2,4-диметил-1H-пиразол-2-иума;
20) 1-(3-аминопропил)-3-(4-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)фенил)-2,4-диметил-1H-пиразол-2-иума;
21) 1-(3-аминопропил)-5-(4-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)фенил)-2,3-диметил-1H-пиразол-2-иума;
22) 1-(3-аминопропил)-3-(4-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)фенил)-2,5-диметил-1H-пиразол-2-иум 2,2,2-трифторацетата;
23) 1-(3-аминопропил)-4-(4-(2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксипропокси)фенил)-2-метил-1H-пиразол-2-иума;
24) 1-(3-аминопропил)-4-(4-(2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксипропокси)фенил)-2-метил-1H-пиразол-2-иума;
25) (S)-3-((Z)-2-(((S)-2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)-3-фторфенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
26) 1-(3-аминопропил)-4-(4-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((2R,3S)-2-метил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксипропокси)фенил)-2-метил-1H-пиразол-2-иума;
27) 2-(1-(3-аминопропил)-1H-пиразол-4-ил)-5-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксо-этилиден)амино)окси)-2-карбоксиэтокси)-1-метилпиридин-1-иум 2,2,2-трифторацетата;
28) (S)-3-(4-(3-амино-1-(3-аминопропил)-1H-пиразол-4-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановой кислоты;
29) (S)-3-((Z)-2-(((S)-2-(4-(3-Амино-1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
30) 1-(3-аминопропил)-4-(4-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-(2H-тетразол-5-ил)этокси)фенил)-2-метил-1H-пиразол-2-иума;
31) 1-(3-аминопропил)-4-(4-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((2S,3S)-2-метил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-(2H-тетразол-5-ил)этокси)фенил)-2-метил-1H-пиразол-2-иума;
32) 3-(2-амино-4-(4-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)фенил)-1H-имидазол-1-ил)пропан-1-аминиума;
33) 3-(4-(4-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)-азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)фенил)-2-имино-3-метил-2,3-дигидро-1H-имидазол-1-ил)пропан-1-аминиума;
34) 1-(3-аминопропил)-4-((5-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)пиридин-2-ил)амино)пиридин-1-иум 2,2,2-трифторацетата;
35) 1-(3-аминопропил)-4-((5-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)пиридин-2-ил)окси)пиридин-1-иума;
36) (S,Z)-3-(2-((2-((6-(3-(3-аминопропил)-1H-имидазол-3-иум-1-ил)пиридин-3-ил)окси)этокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
37) (S)-3-((Z)-2-(((S)-2-((6-(3-(3-аминопропил)-1H-имидазол-3-иум-1-ил)пиридин-3-ил)окси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
38) 3-(2-((5-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-пиридин-2-ил)амино)-1H-имидазол-1-ил)пропан-1-аминиума;
39) 3-(2-((5-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-пиридин-2-ил)амино)-1H-имидазол-1-ил)пропан-1-аминиума;
40) (S)-3-((Z)-2-(((S)-2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)-3-хлорфенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
41) (S)-3-((Z)-2-((((S)-1-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)фенокси)-2-карбоксипропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
42) (3S)-3-((Z)-2-((2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)фенокси)-1-фосфоноэтилэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
43) (3S)-3-((Z)-2-(((1R)-2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)фенокси)-1-(гидрокси(метокси)фосфорил)этокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
44) (3S)-3-((Z)-2-(((1S)-2-(4-(1-(3-амино-2-гидроксипропил)-2-метил-1H-пиразол-2-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
45) (S)-3-((Z)-2-(((S)-2-(4-(1-((S)-3-амино-2-гидроксипропил)-2-метил-1H-пиразол-2-иум-4-ил)-3-фторфенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
46) (3S)-3-((Z)-2-(((1S)-2-(4-(1-(3-амино-2-гидроксипропил)-2-метил-1H-пиразол-2-иум-4-ил)-3-фторфенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
47) (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-1-карбокси-2-(4-(1-((R)-2,3-диаминопропил)-2-метил-1H-пиразол-2-иум-4-ил)-3-фторфенокси)-этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
48) (S)-3-((Z)-2-(((S)-2-(4-(3-амино-1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)-3-фторфенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
49) (S)-3-((Z)-2-(((S)-2-(4-(3-амино-1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)-3-фторфенокси)-1-карбоксиэтокси)имино)-2-(2-амино-5-хлортиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
50) (S)-3-((Z)-2-(((S)-2-(4-(3-амино-1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-амино-5-хлортиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
51) (S)-3-((Z)-2-(((S)-2-(4-(5-амино-1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
52) (S)-3-((Z)-2-(2-амино-5-хлортиазол-4-ил)-2-((((R)-1-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)фенокси)-2-карбоксипропан-2-ил)окси)имино)-ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
53) (S)-3-((Z)-2-(2-амино-5-хлортиазол-4-ил)-2-((((R)-1-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)фенокси)-2-карбоксипропан-2-ил)окси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
54) (S)-3-((Z)-2-(((S)-2-(4-(1-((S)-3-амино-2-гидроксипропил)-2-(азетидин-3-илметил)-1H-пиразол-2-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
55) (S)-3-((Z)-2-(((S)-2-(4-(1-(3-аминопропил)-2-(((R)-пирролидин-3-ил)метил)-1H-пиразол-2-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
56) (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-2-(4-(1,2-бис(3-аминопропил)-1H-пиразол-2-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
57) (S)-3-(4-(1-(3-амино-2-(аминометил)пропил)-2-имино-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)-амино)окси)пропановой кислоты;
58) (2S)-3-(4-(1-(3-амино-2-гидроксипропил)-2-имино-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановой кислоты;
59) (S)-3-(4-((Z)-2-((2-аминоэтил)имино)-1-(3-аминопропил)-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)-амино)окси)пропановой кислоты;
60) (S)-3-(4-((S)-2-((2-аминоэтил)амино)-4,5-дигидро-1H-имидазол-4-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)-азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановой кислоты;
61) (R)-3-(4-((S)-1-амино-2-((4,5-дигидро-1H-имидазол-2-ил)амино)этил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)-азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановой кислоты;
62) (2S)-3-(4-(2-((2-аминоэтил)амино)-4,5-дигидро-1H-имидазол-5-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановой кислоты;
63) (2S)-3-(4-(1-амино-2-((4,5-дигидро-1H-имидазол-2-ил)амино)этил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановой кислоты;
64) (S)-3-((Z)-2-(((S)-2-(4-(2-((2-аминоэтил)амино)-1-метилпиридин-1-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
65) (S)-3-((Z)-2-(((S)-2-(4-(2-((3-аминопропил)амино)-1-метилпиридин-1-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
66) (S)-3-((Z)-2-(((S)-2-(4-(2-((3-аминопропил)амино)пиридин-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
67) (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-2-(4-(2-((азетидин-3-илметил)амино)-1-метилпиридин-1-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
68) (S)-3-((Z)-2-(((S)-2-(4-(6-((2-аминоэтил)амино)-1-метилпиридин-1-иум-3-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
69) (S)-3-((Z)-2-(2-амино-5-хлортиазол-4-ил)-2-(((S)-2-(4-(6-((2-аминоэтил)амино)-1-метилпиридин-1-иум-3-ил)фенокси)-1-карбоксиэтокси)-имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
70) (S)-3-((Z)-2-(((S)-2-(4-(6-((2-аминоэтил)амино)-1-метилпиридин-1-иум-3-ил)-3-фторфенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
71) (S)-3-((Z)-2-(2-амино-5-хлортиазол-4-ил)-2-(((S)-2-(4-(6-((2-аминоэтил)-амино)-1-метилпиридин-1-иум-3-ил)-3-фторфенокси)-1-карбоксиэтокси)-имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
72) (S)-3-(4-(2-((2-аминоэтил)амино)пиримидин-5-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановой кислоты;
73) (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-1-карбокси-2-(4-(1-метил-6-(пиперазин-1-ил)пиридин-1-иум-3-ил)фенокси)этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
74) (S)-3-((Z)-2-(((S)-2-(4-(5-((2-аминоэтил)амино)-1-метилпиридин-1-иум-2-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
75) (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-1-карбокси-2-(4-(6-(((3-гидроксиазетидин-3-ил)метил)амино)-1-метилпиридин-1-иум-3-ил)фенокси)этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
76) (S)-3-((Z)-2-(((S)-2-(4-(6-(((1-аминоциклопропил)метил)амино)-1-метилпиридин-1-иум-3-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
77) (3S)-3-((Z)-2-(((1S)-2-(4-(6-((1-аминопропан-2-ил)амино)-1-метилпиридин-1-иум-3-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
78) (3S)-3-((Z)-2-(((1S)-2-(4-(6-((1-аминопропан-2-ил)амино)-1-метилпиридин-1-иум-3-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
79) (S)-3-((Z)-2-(((S)-2-(4-(2-((2-аминоэтил)амино)-1-метилпиримидин-1-иум-5-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
80) (S)-3-((Z)-2-(((S)-2-(4-(6-(((2-аминоэтил)амино)метил)-1-метилпиридин-1-иум-3-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
81) (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-1-карбокси-2-(4-(6-(((1,3-диаминопропан-2-ил)амино)метил)-1-метилпиридин-1-иум-3-ил)-фенокси)этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
82) (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-1-карбокси-2-(4-(6-((1,3-диаминопропан-2-ил)карбамоил)-1-метилпиридин-1-иум-3-ил)фенокси)-этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
83) (S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)-азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-(4-(6-((1,3-диаминопропан-2-ил)карбамоил)пиридин-3-ил)фенокси)пропановой кислоты;
84) (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-1-карбокси-2-(4-(6-((1,3-диаминопропан-2-ил)амино)-1-метилпиридин-1-иум-3-ил)фенокси)этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
85) (S)-3-((Z)-2-(((S)-2-(4-(2-((2-аминоэтил)(азетидин-3-илметил)амино)-1-метилпиримидин-1-иум-5-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
86) (S)-3-((Z)-2-(((S)-2-(4-(2-((2-аминоэтил)(азетидин-3-илметил)амино)-1-метилпиримидин-1-иум-5-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
87) (3S)-3-(2-((2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)фенокси)-2-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
88) (3S)-3-(2-((2-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)фенокси)-2-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
89) (S)-3-(2-(2-аминотиазол-4-ил)-2-((2-(4-(4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-2-ил)фенокси)этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил гидросульфата;
90) (S)-3-((Z)-2-((((S)-1-(4-(6-((2-аминоэтил)амино)-1-метилпиридин-1-иум-3-ил)фенокси)-2-карбоксипропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
91) (S)-3-((Z)-2-(((S)-2-(4-(6-(3-аминопропил)-1-метилпиридин-1-иум-3-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
92) (S)-3-((Z)-2-((((R)-1-(4-(6-((2-аминоэтил)амино)-1-метилпиридин-1-иум-3-ил)фенокси)-3-гидроксипропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
93) (S)-3-((Z)-2-((((S)-1-(4-(6-((2-аминоэтил)амино)-1-метилпиридин-1-иум-3-ил)фенокси)пропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
94) (3S)-3-(2-((2-((4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)фенил)-амино)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
95) (3S)-3-(2-((2-((4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)фенил)-(метил)амино)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
96) (3S)-3-(2-((3-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)фенил)-1-карбоксипропокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
97) (2S)-3-(4-(2-((2-аминоэтил)амино)-1,4,5,6-тетрагидропиримидин-5-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)-азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановой кислоты;
98) (2S)-3-(4-(2-((2-аминоэтил)амино)-1,4,5,6-тетрагидропиримидин-5-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-метилпропановой кислоты;
99) (S)-3-(4-(1-(3-аминопропил)-2-имино-3-метил-2,3-дигидро-1H-имидазол-4-ил)-3-фторфенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановой кислоты;
100) (S)-3-((Z)-2-(((S)-2-(4-(1-(3-аминопропил)-2-(азетидин-3-илметил)-1H-пиразол-2-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
101) (S)-3-((Z)-2-(((S)-2-(4-(6-((2-аминоэтил)амино)-1-(азетидин-3-илметил)пиридин-1-иум-3-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
102) (S)-3-((Z)-2-(((S)-2-(4-(2-((2-аминоэтил)амино)-1-(азетидин-3-илметил)пиримидин-1-иум-5-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
103) (S)-3-((Z)-2-(((S)-2-(4-(2-(2-аминоэтил)-1-(3-аминопропил)-1H-пиразол-2-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
104) (S)-3-((E)-2-(2-аминотиазол-4-ил)-2-(((S)-1-карбокси-2-(4-(1-(3-(((S)-2,3-дигидроксипропил)амино)пропил)-2-метил-1H-пиразол-2-иум-4-ил)фенокси)этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
105) (S)-3-((E)-2-(((S)-2-(4-(1-(3-((2-аминоэтил)амино)пропил)-2-метил-1H-пиразол-2-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
106) (S)-3-((E)-2-(2-аминотиазол-4-ил)-2-(((S)-2-(4-(1-(3-((азетидин-3-илметил)амино)-пропил)-2-метил-1H-пиразол- 2-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)-ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
107) (S)-3-((E)-2-(((S)-2-(4-(1-(3-((3-аминопропил)амино)-пропил)-2-метил-1H-пиразол-2-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
108) (S)-3-((Z)-2-((((S)-1-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)-3-фторфенокси)-2-карбоксипропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
109) (S)-3-((Z)-2-(((S)-2-((6-(1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)пиридазин-3-ил)окси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
110) (S)-3-((Z)-2-((((S)-1-(4-(1-(3-аминопропил)-2-имино-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-карбоксипропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
111) (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-2-((6-((1-(азетидин-3-илметил)пиридин-1-иум-4-ил)амино)пиридин-3-ил)окси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
112) (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-2-((6-(3-(азетидин-3-илметил)-1H-имидазол-3-иум-1-ил)пиридин-3-ил)окси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
113) (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-1-карбокси-2-((6-(3-(3-(триметиламмонио)пропил)-1H-имидазол-3-иум-1-ил)пиридин-3-ил)окси)-этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
114) (S)-3-((Z)-2-(((S)-2-((2-(3-(3-аминопропил)-1H-имидазол-3-иум-1-ил)пиримидин-5-ил)окси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
115) (S)-3-((Z)-2-(((S)-2-((6-(3-(3-аминопропил)-1H-имидазол-3-иум-1-ил)пиридин-3-ил)окси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
116) (S)-3-((Z)-2-((((S)-1-((6-(3-(3-аминопропил)-1H-имидазол-3-иум-1-ил)пиридин-3-ил)окси)-2-карбоксипропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
117) (S)-3-((Z)-2-((((S)-1-амино-3-(4-(1-(3-аминопропил)-2-имино-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-1-оксопропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
118) (S)-3-((Z)-2-((((S)-3-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)фенокси)-1-этокси-1-оксопропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
119) этил (S)-3-(4-(1-(3-аминопропил)-2-имино-3-метил-2,3-дигидро-1H-имидазол-4-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропаноата;
120) (S)-3-((Z)-2-((((S)-3-(4-(1-(3-аминопропил)-2-метил-1H-пиразол-2-иум-4-ил)фенокси)-1-метокси-1-оксопропан-2-ил)окси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
121) (S)-3-((6-(3-(3-аминопропил)-2-имино-2,3-дигидро-1H-имидазол-1-ил)пиридин-3-ил)окси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановой кислоты;
122) (S)-3-((Z)-2-(((S)-2-(4-(1-(3-аминопропил)-2-((3-фторазетидин-3-ил)метил)-1H-пиразол-2-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
123) (S)-3-((Z)-2-(((S)-2-(4-(2-(3-амино-2-фтор-2λ3-пропил)-1-(3-аминопропил)-1H-пиразол-2-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
124) (S)-3-((Z)-2-(((S)-2-(4-(2-(3-амино-2-фтор-2λ3-пропил)-1-(азетидин-3-илметил)-1H-пиразол-2-иум-4-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
125) (S)-3-((Z)-2-(((S)-2-(4-(3-((3-аминопропил)амино)-1-метилпиридазин-1-иум-6-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
126) (S)-3-((Z)-2-(((S)-2-(4-(6-((3-аминопропил)амино)-1-метилпиридазин-1-иум-3-ил)фенокси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
127) (S)-3-((Z)-2-(((S)-2-((6-(4-(3-аминопропил)-3-((3-фторазетидин-3-ил)метил)-1H-имидазол-3-иум-1-ил)пиридин-3-ил)окси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата;
128) (S)-3-(4-(5-(((2-аминоэтил)амино)метил)-1-(3-аминопропил)-1H-пиразол-3-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановой кислоты; и
129) (S)-3-(4-(5-(аминометил)-1-(3-аминопропил)-1H-пиразол-3-ил)фенокси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановой кислоты,
или его фармацевтически приемлемая соль.
16. Соединение по п.15, где фармацевтически приемлемая соль представляет собой соль трифторуксусной кислоты.
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| EA 201400838 A1, 30.12.2014 | |||
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| KLINE T | |||
| et al.: "Antimicrobial Effects of Novel Siderophores Linked to beta-Lactam Antibiotics", BIOORGANIC & MEDICINAL CHEMISTRY, 2000, vol.8, no.1, p.73-93 | |||
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| US 4537886 A, 27.08.1985. | |||

