ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к новому способу получения замещенного соединения имидазопиридина, более точно к новому способу получения 2,3-диметилимидазол[1.2-а]пиридина, замещенного в 6-м положении карбоксамидной или карбоксиалкильной группой. Согласно другим аспектам настоящее изобретение относится также к новым полупродуктам, используемым в способе.
УРОВЕНЬ ТЕХНИКИ
Настоящее изобретение относится к новому способу, подходящему для крупнотоннажного производства замещенного имидазопиридина формулы (1),

в котором R1 является C1-C6 алкоксильной или NH2 группой, включающему стадию реакции соединения общей формулы (2)

в котором R1 является C1-C6 алкоксильной или NH2 группой, с 3-галоген-2-бутаноном в циклогексаноне.
Аналогичная реакция описана в ЕР 33094, ЕР 204285, ЕР 228 006, ЕР 308917 и WO 99/55706, в которых замещенное соединение аминопиридина общей формулы (X)

вступает в реакцию с соединением формулы
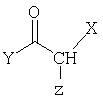
в котором Х - например, Н, СН3 или сложноэфирная группа, такая как СООСН3 или СООС2Н5,
Y - например, СН3, СН2СН3, и
Z - это уходящая группа, такая как галогеновая, мезильная или тозильная, для получения соединения общей структуры

в котором Х и Y такие, как описано выше.
Реакцию осуществляют в инертном растворителе, таком как ацетон, спирты, бензол, N,N-диметилформамид, тетрагидрофуран, хлороформ или диэтиловый эфир, предпочтительно при повышенной температуре и, возможно, в присутствии неорганического или органического основания.
Реакция отличается продолжительным временем реакции, например от 16 до 84 часов, высокими температурами реакции и относительно низким выходом, например от 22% до 55%. Поэтому реакция не подходит для крупнотоннажного производства замещенных соединений имидазопиридина.
Авторы неожиданно обнаружили, что при проведении процесса согласно настоящему изобретению так, как описано в настоящем описании, время реакции может быть укорочено, температура реакции может быть снижена, и выход увеличен.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В настоящем изобретении предложен новый способ крупнотоннажного получения замещенного имидазопиридина общей формулы (1)
в котором R1 означает C1-C6 алкокси или NH2, путем взаимодействия соединения общей формулы (2)
с 3-галоген-2-бутаноном в среде инертного органического растворителя при температуре 80-100°С, при этом в качестве инертного органического растворителя используют циклогексанон.
При температуре реакции от 80°С до 100°С полное превращение происходит в течение нескольких часов, например от 1 до 4 часов. Конверсия обычно составляет выше 95%, а выход выделенного продукта - обычно выше 70%.
Указанный 3-галоген-2-бутанон может представлять собой 3-бром-2-бутанон или 3-хлор-2-бутанон, последний из которых является предпочтительным. Количество 3-галоген-2-бутанона может составлять от 1,1 до 5 молярных эквивалентов. Количество 3-галоген-2-бутанона не является критическим для осуществления настоящего изобретения. С практической и экономической точки зрения предпочтительно добавлять от 1,1 до 5 молярных эквивалентов, предпочтительно от 1,1 до 2 эквивалентов.
Циклогексанон может быть разбавлен инертным растворителем. Количество циклогексанона не является важным для осуществления настоящего изобретения и поэтому в практических условиях его можно отрегулировать в зависимости от необходимости и используемого оборудования. Можно также смешивать циклогексанон с инертными растворителями, такими как простые эфиры. Примером подходящего инертного растворителя является, не ограничиваясь им, тетрагидрофуран (ТГФ). Количество инертного растворителя может составлять примерно до 50 объемных %, не вызывая уменьшения выхода.
В первом варианте выполнения настоящего изобретения соединение формулы (2)
в котором R1 является C1-C6 алкоксильной группой, приводят в реакцию с 3-галоген-2-бутаноном в циклогексаноне, для получения соединения формулы (1),
в котором R1 является C1-C6 алкоксильной группой.
Во втором варианте выполнения настоящего изобретения соединение формулы (2),
в котором R1 является NH2 группой, приводят в реакцию с 3-галоген-2-бутаноном в циклогексаноне для получения соединения формулы (1),
в котором R1 является NH2 группой.
Способ согласно настоящему изобретению осуществляют растворением или суспендированием соединения формулы (2),
в котором R1 является C1-C6 алкоксильной или NH2 группой, в циклогексаноне и добавлением 3-галоген-2-бутанона, нагреванием реакционной смеси в течение нескольких часов и затем выделением соединения формулы (1),
в котором R1 является С1-С6 алкоксильной или NH2 группой, с высоким выходом.
Исходный материал для использования в настоящем изобретении можно приготовить так, как изложено в WO 99/55706, или альтернативно, как описано ниже в Схеме 1.

Схема 1
Стадия i
Соединение (3) в Схеме 1 обрабатывают тионилхлоридом или любым эквивалентным ему реагентом при повышенной температуре в соответствующем растворителе в течение нескольких часов, чтобы получить соответствующее хлорное соединение. Реакцию осуществляют, используя примерно от 1 до 5 эквивалентов тионилхлорида, предпочтительно от 1 до 2,5 эквивалентов, в толуоле при температуре примерно 100°С в течение времени от 2 до 8 часов. Соответствующее хлорное соединение затем обрабатывают 2-25 эквивалентами аммиака, предпочтительно от 3 до 12 эквивалентов, в таком же растворителе, как указано выше, примерно при комнатной температуре, чтобы получить соединение (4).
Стадия ii
Соединение (4) согласно Схеме 1 гидрируют в водно-спиртовом растворе, используя катализатор, с получением соединения (5). Примеры подходящего катализатора включают, не ограничиваясь ими, палладий, рутений или их смеси. Паста Pd-Ru/C является предпочтительным катализатором. Примеры спиртов включают, но не ограничиваясь ими, метанол, этанол и пропанол, из них предпочтительным является метанол.
Замещенное соединение имидазопиридина формулы (1),
в котором R1 является C1-C6 алкоксильной или NH2 группой, приготовленное в соответствии с настоящим изобретением, впоследствии можно использовать для приготовления некоторых замещенных производных имидазопиридина, которые особенно эффективны как ингибиторы желудочно-кишечной Н+, К+-АТФ-азы (аденозинтрифосфатазы) и, следовательно, как ингибиторы выделения желудочного сока.
Соединения формулы (1) можно вводить в реакцию с соединением формулы (6),
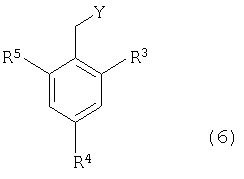
в котором R3 - Н, C1-C6 алкил, гидроксилированный С1-С6 алкил или галоген; R4 - Н, С1-С6 алкил, гидроксилированный C1-C6 алкил или галоген; R5 - Н или галоген; и Y - уходящая группа, такая как галоидная, тозильная или мезильная группа, чтобы получить соединение формулы (7),

в котором R1, R3, R4 и R5 определены выше. Удобно проводить эту реакцию в инертном растворителе, например в ацетоне, ацетонитриле, диметоксиэтане, метаноле, этаноле или диметилформамиде в присутствии основания или без него. Основание представляет собой, например, гидроксид щелочного металла, такой как гидроксид натрия и гидроксид калия, карбонат щелочного металла, такой как карбонат калия и карбонат натрия, или органический амин, такой как триэтиламин.
Соединения формулы (7), в которых R1 является C1-C6 алкоксильной группой, можно в дальнейшем приводить во взаимодействие с аминосодержащим соединением общей формулы (8),
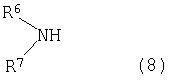
в котором R6 и R7 являются одинаковыми или различными, и они выбраны из группы, состоящей из Н, C1-C6 алкила, C1-C6 алкоксизамещенного С1-С6 алкила, гидроксилированного C1-С6 алкоксизамещенного C1-C6 алкила, арила, чтобы получить соответствующее амидное соединение.
R6 и R7 могут вместе с атомом азота, к которому они присоединены, образовать насыщенное или ненасыщенное кольцо, возможно, содержащее один или более гетероатом, образуя, таким образом, например, морфолин, пиперазин, пирролидин или пиперидин.
Реакцию можно проводить нагреванием реагентов в неразбавленном аминосоединении или разбавленном в инертном растворителе в стандартных условиях.
Альтернативно, соединения формулы (7)
в которых R3, R4 и R5 определены выше, а R1 является NH2 -группой, могут быть гидролизованы при стандартных условиях до соответствующих соединений карбоксильных кислот формулы (9)

в которых R3, R4 и R5 определены выше.
Соединения формулы (9) затем можно ввести в реакцию с аминосоединениями формулы (8),
в которых R6 и R7 определены выше, в присутствии агента сочетания, чтобы получить соответствующее амидное соединение. Реакцию можно проводить в инертном растворителе в стандартных условиях.
ПРИМЕРЫ
Пример 1.1
Приготовление 3-бром-2-бутанона
В реакторе суспендируют бромид натрия (84 кг) в диметилформамиде (125 л). Добавляют 3-хлор-2-бутанон (85 кг) при температуре 15°С-30°С. Перемешивание продолжают в течение 4 часов, а затем фильтруют; отфильтрованный осадок промывают циклогексаноном (38 л). Приготовленный таким образом 3-бром-2-бутанон готов для использования на стадии циклизации.
Пример 1.2
Синтез метил-8-амино-2,3-диметилимидазо [1,2-а]пиридин-6-карбоксилата

К суспензии метилового эфира 5,6-диаминоникотиновой кислоты (1 эквивалент, 5,1 г) в циклогексаноне (50 мл) добавляли 3-бром-2-бутанон (1,2 эквивалента, 3,9 г) в течение 10 минут. Смесь нагревали до 100°С (внутренняя температура) и перемешивали в течение 2,5 часов при этой температуре. Смесь охлаждали до комнатной температуры, а бледно-окрашенное твердое вещество отфильтровывали и промывали ТВМЕ (3×10 мл). Сушку проводили при пониженном давлении при 45°С. Выход составил 6,53 г (75%).
Пример 1.3
Синтез этил-8-амино-2,3-диметилимидазо [1,2-а]пиридин-6-карбоксилата

К суспензии этилового эфира 5,6-диаминоникотиновой кислоты (1 эквивалент, 5,1 г) в циклогексаноне (50 мл) добавляли 3-бром-2-бутанон (1,4 эквивалента, 5,95 г) в течение 15 минут. Темно-коричневую смесь нагревали до 100°С (внутренняя температура) и перемешивали в течение 1,5 часов при этой температуре. Смесь охлаждали до комнатной температуры, а светло-коричневое твердое вещество отфильтровывали и промывали ТВМЕ (20 мл). Сушку проводили при пониженном давлении при 45°С. Выход составил 5,06 г (65%).
Пример 1.4
Синтез изопропил-8-амино-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксилата

К суспензии изопропилового эфира 5,6-диаминоникотиновой кислоты (1 эквивалент, 5,1 г) в циклогексаноне (50 мл) добавляли 3-бром-2-бутанон (1,2 эквивалента, 3,4 мл) в течение 10 минут. Темно-коричневую смесь нагревали до 100°С (внутренняя температура) и перемешивали в течение 1,5 часов при этой температуре. Смесь охлаждали до комнатной температуры, а бледно-желтую твердую фазу отфильтровывали и промывали ТВМЕ (3×10 мл). Сушку проводили при пониженном давлении при 45°С. Выход составил 6,0 г (74%).
Пример 1.5
Синтез 8-амино-2,3-диметилимидазо [1,2-а]пиридин-6-карбоксамида

5,6-Диаминоникотинамид (50 г, 0,313 молей (аналитически: 95,4%), 1,0 эквивалент) суспендировали в циклогексаноне (250 мл). Суспензию нагревали до 100°С. Фильтрат (3-бром-2-бутанон в циклогексаноне) добавляли при 100°С в течение 1 часа 10 минут. Нагревание продолжалось 3 часа, а затем источник нагревания удалили. Реакционную смесь охлаждали до 20°С и перемешивали при этой температуре в течение еще 2 часов. Твердую фазу отфильтровывали, осторожно промывали ТВМЕ (2×330 мл) и сушили до получения 70,3 г указанного в заголовке соединения. Выход составил 70%.
Пример 1.6
Синтез 8-амино-2,3-диметилимидазо [1,2-а]пиридин-6-карбоксамида
NaBr (27,0 г; 0,259 моля; 1,33 эквивалента) суспендировали в циклогексаноне (220 мл) и добавили 3-хлор-2-бутанон (25,7 мл; 0,242 моля; 1,24 эквивалента) за один прием. Смесь нагревали до 80°С и перемешивали в течение 3 часов. Смесь охладили до 50°С, белое твердое вещество отфильтровали и промыли циклогексаноном (60 мл). К фильтрату добавили 5,6-диаминоникотинамид (30 г; 0,1946 молей; 1,0 эквивалент), и смесь нагрели до 100°С в течение 4 часов, после чего 98%-ную конверсию определили с помощью HPLC (жидкостной хроматографии высокого давления). Реакционную смесь охладили до 20°С, перемешивание продолжали в течение 2 часов при 20°С. Твердое вещество отфильтровали, промыли ТВМЕ (220 мл) и высушили с получением 46,6 г указанного в заголовке вещества. Выход: 73%.
Пример 1.7
Синтез 8-амино-2,3-диметилимидазо [1,2-а]пиридин-6-карбоксамида
5,6-Диаминоникотинамид (30,0 г; 0,183 моля; 1,0 эквивалент) суспендировали в циклогексаноне (280 мл). Добавили 3-хлор-2-бутанон (24,мл; 0,22 моля; 1,2 эквивалента), и смесь нагревали до 100°С в течение 4 часов. Реакционную смесь охлаждали до 20°С и перемешивали при этой температуре в течение еще 2 часов. Твердую фазу отфильтровали, осторожно промыли ТВМЕ (200 мл) и сушили до получения 48,4 г указанного в заголовке соединения. Выход составил 78%.
Пример 1.8
Синтез метил-8-амино-2,3-диметил-8-(2,6-диметилбензиламино)-имидазо [1,2-а]пиридин-6-карбоксилата

Метил-8-амино-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксилат (0,8 г, 3,6 ммоля), 2,6-диметилбензилхлорид (0,57 г, 3,7 ммоля), карбонат натрия (1,0 г, 9,4 ммоля) и каталитическое количество иодида калия добавили к ацетонитрилу (10 мл) и подвергли нагреванию с обратным холодильником в течение 20 часов. После фильтрования соли промыли метиленхлоридом, а растворители выпарили при пониженном давлении. Остаток очистили колоночной хроматографией на силикагеле с использованием в качестве элюента смеси метиленхлорид:этилацетат (75:25). Желтый остаток обработали гексаном с получением 0,23 г (19%) указанного в заголовке продукта.
Пример 1.9
Синтез этил-2,3-диметил-8-(2-этил-6-метилбензиламино)-имидазо [1,2-а]пиридин-6-карбоксилата

Этил-8-амино-2,3-диметилимидазо [1,2-а]пиридин-6-карбоксилат (0,7 г, 3,0 ммоля), 2-этил-6-метилбензилхлорид (0,5 г, 3,0 ммоля), карбонат натрия (0,64 г, 6,0 ммоля) и каталитическое количество иодида калия добавили к ацетону (50 мл) и подвергли нагреванию с обратным холодильником в течение 20 часов. После фильтрования ацетон выпарили при пониженном давлении с получением масла. Маслянистый продукт очистили колоночной хроматографией на силикагеле с использованием в качестве элюента смеси диэтиловый эфир:петролейный эфир (1:1), с получением 0,12 г (9%) указанного в заголовке продукта. 1 Н-ЯМР (500 МГц, CDCl3): δ 1.25 (t, 3Н), 1,5 (t, 3H), 2,35 (s, 3Н), 2,42 (s, 3Н), 2,44 (s, 3Н), 2,75 (q, 2H), 4,45-4,5 (m, 4Н), 4,9 (bs, 1H), 6,8 (s, 1Н), 7,05-7,2 (m, 3H), 8,1 (s, 1H).
Пример 1.10
Синтез 2,3-диметил-8-(2-этил-6-метилбензиламино)-N-пропил-имидазо [1,2-а]пиридин-6-карбоксамида

Этил-2,3-диметил-8-(2-этил-6-метилбензиламино)-имидазо[1,2-а]пиридин-6-карбоксилат (0,12 г, 0,33 ммоля), пропиламин (1,0 г, 17 ммоля) и каталитическое количество цианида натрия подвергли нагреванию с обратным холодильником в метаноле (20 мл) в течение 24 часов. Добавили дополнительное количество пропиламина (1,0 г, 17 ммолей), и реакционную смесь подвергли нагреванию с обратным холодильником в течение 24 часов. Растворитель выпарили при пониженном давлении, а остаток очистили колоночной хроматографией на силикагеле с использованием диэтилового эфира в качестве элюента. Кристаллизация из диэтилового эфира дала 0,053 г (42%) указанного в заголовке продукта. 1H-ЯМР (300 МГц, CDCl3): δ 1,0 (t, 3H), 1,2 (t, 3Н), 1,65-1,75 (m, 3Н), 2,3 (s, 3Н), 2,35 (s, 3Н), 2,38 (s, 3Н), 2,7 (q, 2H), 3,4-3,5 (m, 2H), 4,35 (d, 2H), 4,9 (bs, 1H), 6,2 (bs, 1H), 6,357 (s, 1H), 7,0-7,2 (m, 4H), 7,85 (s, 1H).
Пример 1.11
Синтез 2,3-диметил-8-(2-этил-6-метилбензиламино)-имидазо [1,2-а]пиридин-6-карбоксамида

8-Амино-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксамид (3,3 г, 16,2 ммоля), 2-этил-6-метилбензилхлорид (2,73 г, 16,2 ммоля), карбонат калия (8,0 г, 58 ммолей) и иодид калия (1,1 г, 6,6 ммоля) добавили к ацетону (150 мл) и подвергли нагреванию с обратным холодильником в течение 20 часов. Добавили дополнительное количество 2-этил-6-метилбензилхлорида (1,0 г, 5,9 ммоля), и реакционную смесь подвергли нагреванию с обратным холодильником в течение 7 часов. Добавили метиленхлорид (60 мл) и метанол (30 мл). Реакционную смесь отфильтровали, а растворители выпарили при пониженном давлении. Остаток очистили колоночной хроматографией на силикагеле с использованием в качестве элюента смеси метиленхлорид:метанол (100:7). Кристаллизация из этилацетата дала 2,8 г (50%) указанного в заголовке продукта. 1Н-ЯМР (300 МГц, CDCl3): δ 1,2 (t, 3Н), 2,34 (s, 3Н), 2,36 (s, 3Н), 2,38 (s, 3Н), 2,7 (q, 2H), 4,4 (d, 2Н), 4,9 (bs, 1H), 6,0 (bs, 2Н), 6,45 (s, 1H), 7,0-7,2 (m, 3Н), 7,9 (s, 1H).
Пример 1.12
Синтез 2,3-диметил-8-(2-этил-6-метилбензиламино)-имидазо [1,2-а]пиридин-6-карбоновой кислоты

Мезилат (метилсульфонат) 2,3-диметил-8-(2-этил-6-метилбензиламино)-имидазо[1,2-а]пиридин-6-карбоксамида (11,0 г, 0,025 ммоля) и гидроксид натрия (7,0 г, 0,17 ммолей) растворили в 120 мл этанола (95%) и подвергли нагреванию с обратным холодильником в течение 20 часов. Растворитель выпарили при пониженном давлении, а к остатку добавили воду (150 мл). Довели значение рН до 5 добавлением концентрированной HCl и уксусной кислоты, а выпавшее в осадок твердое вещество отделили фильтрованием, промыли водой и ацетоном и высушили, с получением 7,6 г (88%) указанного в заголовке вещества. 1H-ЯМР (500 МГц, DMSO-d6): δ 1,15 (t, 3Н), 2,26 (s, 3Н), 2,34 (s, 3Н), 2,39 (s, 3Н), 2,69 (q, 2H), 4,38 (d, 2Н), 5,2 (bs, 1H), 6,73 (s, 1H), 7,07-7,2 (m, 3H), 8,12 (s, 1H).
Пример 1.13
Синтез 2,3-диметил-8-(2-этил-6-метилбензиламино)-6-(морфолинокарбонил)-имидазо[1,2-а]пиридина

2,3-Диметил-8-(2-этил-6-метилбензиламино)-имидазо[1,2-а]пиридин-6-карбоновую кислоту (0,15 г, 0,44 ммоля) и о-бензотриазол-1-ил-N,N,N',N'-тетраметилурониумтетрафторборат (TBTU) (0,14 г, 0,44 ммоля) добавили к метиленхлориду (10 мл). Добавили морфолин (0,12 г, 1,4 ммоля), и реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов. Реакционную смесь добавили в колонну с силикагелем, и путем очистки при помощи хроматографии с использованием в качестве элюента смеси этилацетат:метиленхлорид (1:1) получили 0,12 г (66%) желаемого продукта. 1H-ЯМР (300 МГц, CDCl3): δ 1,2 (t, 3Н), 2,32 (s, 3Н), 2,35 (s, 3Н), 2,37 (s, 3Н), 2,7 (q, 2H), 3,7 (s, 8Н), 4,35 (d, 2H), 4,95 (bs, 1Н), 6,15 (s, 1H), 7,07-7,2 (m, 3Н), 7,4 (s, 1H).
Пример 1.14
Синтез (2-этил-6-метилбензиламино)-N(2-(2-гидроксиэтокси)этил)-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксамида
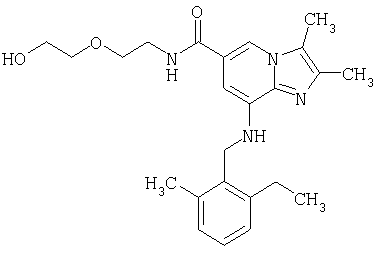
2,3-Диметил-8-(2-этил-6-метилбензиламино)-имидазо[1,2-а]пиридин-6-карбоновую кислоту (0,3 г, 0,88 ммоля) и о-бензотриазол-1-ил-N,N,N',N'-тетраметилурониум тетрафторборат (TBTU) (0,29 г, 0,90 ммоля) добавили к метиленхлориду (10 мл). Добавили 2-(2-аминоэтокси)этанол (0,2 г, 1,9 ммоля), и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Растворитель выпарили при пониженном давлении, и остаток очистили при помощи колоночной хроматографии на силикагеле с использованием в качестве элюента смеси метиленхлорид:метанол (9:1). Кристаллизация из диэтилового эфира дала 0,24 г (80%) желаемого продукта. 1H-ЯМР (500 МГц, CDCl3): δ 1,25 (t, 3Н), 2,25 (s, 3Н), 2,3 (s, 3Н), 2,35 (s, 3Н), 2,75 (q, 2H), 3,4-3,45 (m, 2H), 3,55-3,7 (m, 6H), 4,35 (d, 2H), 5,05 (t, 1H), 6,45 (s, 1H), 7,07-7,2 (m, 4H), 7,5 (s, 1H).
Пример 1.15
Синтез изопропил-8-[(2,6-диметилбензил) амино]-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксилата

Изопропил-8-амино-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксилат (9,85 кг, 1,0 эквивалент, 29,71 моля) суспендировали в изопропаноле (59 л); добавили Nal (0,6 эквивалента, 2,68 кг, 17,88 моля) и К2СО3 (2,5 эквивалента, 10,29 кг, 74,48 моля), и смесь нагрели примерно до 70°С. 2,6-Диметилбензилхлорид (1,1 эквивалент, 5,22 кг, 32,77 моля) растворили в изопропаноле (примерно 60 л), и этот раствор добавили к реакционной смеси. После окончания добавления температуру поддерживали равной 60°С в течение еще 1,5 часов. Дополнительно добавили K2СО3 (9,15 кг), и полученную суспензию перемешивали еще 2 часа при 60°С. Дополнительно медленно добавили при температуре 60°С 2,6-диметилбензилхлорид (2,76 кг) в изопропаноле (22 л); после добавления реакционную смесь перемешивали еще 4 часа при этой температуре. Суспензию разбавили водой (124 л), охладили, перемешали и профильтровали. Осадок на фильтре промыли водой, а затем холодным изопропанолом, высушили при пониженном давлении при 40°С, с получением 11,37 кг влажного материала, выход: 90%.
Пример 1.16
Синтез 8-[(2,6-диметилбензил)амино)]-N-(2-гидроксиэтил)-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксамида

В реактор загрузили изопропил-8-[(2,6-диметилбензил)амино)]-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксилат (11,30 кг, 1 эквивалент, 27,02 моля) и ТГФ (45 л), добавили этаноламин (18,97 кг, 11 эквивалентов, 309,2 моля) при температуре примерно 20°С. Суспензию нагрели примерно до 100°С. Некоторое количество растворителя выпарили, а затем добавили ТГФ (35 л) и продолжили дистилляцию. Процедуру добавления ТГФ и его выпаривания повторяли до полного превращения. К суспензии добавили этанол (140 л) и суспензию нагревали до дефлегмации. Для получения чистого раствора добавили дополнительное количество этанола (13 л). Горячий раствор фильтровали, а затем охлаждали. Белое твердое вещество отфильтровали, промыли этанолом и высушили с получением продукта в виде белого порошка (8271 г).
2. ПРИГОТОВЛЕНИЕ ИСХОДНЫХ МАТЕРИАЛОВ
Пример 2.1
Синтез 6-амино-5-нитро-никотинамида
100 г 6-гидрокси-5-нитро-никотиновой кислоты (0,54 моля; HPLC>98% площади) суспендировали в толуоле (750 мл). Добавили DMF (1 мл, 0,013 моля, 0,024 эквивалента), и смесь нагрели до 110°С (внутренняя температура). В течение 120 минут добавляли тионилхлорид (99 мл, 2,5 эквивалента). Нагревание продолжали в течение 4 часов при 110°С. Реакционную смесь концентрировали до половины объема (отогнали 400 мл растворителя), и добавили к ней толуол (400 мл).
Эту процедуру повторили еще раз (410 мл толуола отогнали, и снова добавили свежий толуол (410 мл)). Затем раствор охладили до 20°С и медленно добавили к водному раствору аммиака (25%, 440 мл, 12 эквивалентов) в течение 40 минут. Немедленно началось осаждение. Во время добавления температуру поддерживали ниже 15°С. После того как добавление было закончено, реакционную смесь оставили нагреваться до комнатной температуры и продолжили перемешивание в течение 16 часов. Твердое вещество отфильтровали, промыли водой (500 мл), этанолом (250 мл), ТВМЕ (250 мл) и высушили (50-10 мбар, температура бани 40°С, 16 часов), с получением 91,3 г указанного в заголовке вещества (0,501 моль, 87%).
Пример 2.2
Синтез 5,6-диаминоникотинамида
44,5 г 6-амино-5-нитроникотинамида (0,24 моля; HPLC: 93% площади) суспендировали в смеси метанол/вода 1:1 (500 мл), добавили 5,0 г катализатора [Pd(4%)-Ru(1%)/C паста (62% Н2О, тип 485; Johnson Matthey); тип 485; Johnson Matthey)]. Гидрирование провели при давлении 5 бар и 30°С в течение 5 часов. После завершения катализатор отфильтровали и промыли смесью метанол/вода 1/1 (50 мл). Отогнали 480 мл растворителя. Полученную суспензию охладили до 20°С и отфильтровали. Твердое вещество промыли метанолом (20 мл) и ТВМЕ (30 мл). После сушки (200-10 мбар; температура бани 40°С, 16 часов) получили 27,3 г указанного в заголовке вещества (0,18 моля, 73%).
Пример 2.3
Синтез 5,6-диаминоникотинамида
42,3 г 6-амино-5-нитро-никотинамида (0,23 моля; HPLC: 93% площади) суспендировали в смеси метанол/вода 1:1 (500 мл). Добавили 5,2 г катализатора [Pd(5%)/C(57,8% H2O; тип: 39, Johnson Matthey]. Провели гидрирование при 5 бар и 30°С в течение 4 часов. После завершения катализатор отфильтровали и промыли смесью метанол/вода 1/1 (100 мл). Отогнали 550 мл растворителя. Полученную суспензию охладили до 20°С и отфильтровали. Твердую фазу промыли метанолом (20 мл) и ТВМЕ (30 мл). После сушки (200-10 мбар; температура бани 40°С, 16 часов) получили 28,5 г указанного в заголовке вещества (0,18 моля, 73%).
Описывается новый способ получения замещенного имидазопиридина общей формулы (1),

где R1 - C1-С6алкокси или NH2, заключающийся во взаимодействии соединения формулы (2)

с 3-галоген-2-бутаноном в циклогексаноне при температуре 80-100°С. Использование циклогексанона в качестве растворителя позволяет сократить длительность процесса и увеличить выход целевого продукта. 8 з.п. ф-лы.
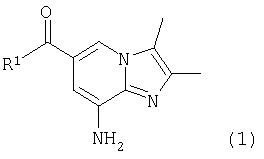
где R1 означает C1-С6 алкокси или NH2,
путем взаимодействия соединения общей формулы (2)

где R1 имеет указанные выше значения,
с 3-галоген-2-бутаноном в среде инертного органического растворителя при температуре 80-100°С, отличающийся тем, что в качестве инертного органического растворителя используют циклогексанон.

в водно-спиртовом растворе в присутствии катализатора.

с тионилхлоридом с последующей обработкой аммиаком полученного соответствующего хлорпроизводного.
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| ЗАПОЛНИТЕЛЬ | 0 |
|
SU308917A1 |
| RU 94033484 A1, 10.08.1996. | |||

