Область техники, к которой относится предлагаемое изобретение
Предлагаемое изобретение относится к способу получения полусинтетических статинов, к промежуточным соединениям, образующимся при осуществлении этого способа, и к способу получения симвастатина высокой степени очистки.
Предпосылки создания предлагаемого изобретения
Лекарства на основе статинов в настоящее время являются наиболее эффективными средствами для понижения уровня липопротеинов низкой плотности в крови пациента, предрасположенного к сердечно-сосудистым заболеваниям. К этому классу лекарств относятся ловастатин (lovastatin), симвастатин (simvastatin), правастатин (pravastatin), компактин (compactin), флувастатин (fluvastatin) и атровастатин (atrovastatin).
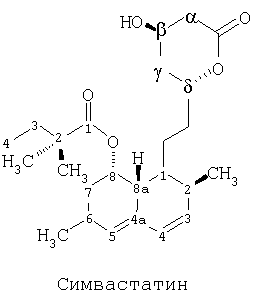
Симвастатин - это общее медицинское наименование для химического соединения, которое полностью называется бутаноикацид,2,2-диметил-1,2,3,7,8,8а-гексагидро-3,7-диметил1-8-[2-(тетрагидро-4-гидрокси-6-оксо-2Н-пиран-2-ил)-этил]-1-нафталинилэфир, [1S*[1а,3а,7b,8b(2S*,4S),-8аb]] (№79902-63-9 в Реестре CAS). Структурная молекулярная формула симвастатина приведена ниже, при этом атомы помечены номерами.
Ловастатин - это общее медицинское наименование для химического соединения, которое полностью называется [1S-[1α(R*),3α,7β(2S*,4S*),8αβ]]-1,2,3,7,8,8а-гексагидро-3,7-диметил-8-[2-(тетрагидро-4-гидрокси-6-оксо-2H-пиран-2-ил)-этил]-1-нафталинил 2-метилбутаноат (№75330-75-5 в Реестре CAS). Структурная молекулярная формула ловастатина приведена ниже, при этом атомы помечены номерами.
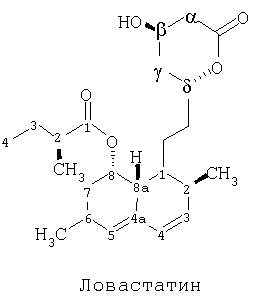
Молекула ловастатина в позиции 8 кольцевой системы гексагидронафталина имеет боковую цепь 2-метилбутирилового эфира. В противоположность этому, молекула симвастатина в позиции 8 кольцевой системы гексагидронафталина имеет боковую цепь 2,2-диметилбутирила. Известно, что симвастатин как средство понижения уровня липопротеинов низкой плотности в крови более эффективен, чем ловастатин.
В предшествующем уровне техники раскрываются способы преобразования ловастатина в симвастатин. В патенте США №4582915, который включен в настоящую заявку по ссылке, раскрываются способы преобразования мевинолина (mevinolin), компактина и их дигидро- и тетрагидропроизводных в более активные ингибиторы редуктазы HMG-CoA путем С-метилирования естественной боковой 2(S)-мелилбутирилокси-цепи с образованием боковой 2,2-димелилбутирилокси-цепи.
В патенте США №5223415, который включен в настоящую заявку по ссылке, раскрывается способ ферментного гидролиза ловастатиновой кислоты путем обработки ловастатиновой кислоты штаммом микроорганизмов Clonostachys compactiuscula ATCC 38009, или АТСС 74178, или же бесклеточным экстрактом из них. Полученный продукт является ингибитором редуктазы HMG-CoA и, следовательно, применим в качестве антигиперхолистеринемического средства. Этот продукт служит также в качестве промежуточного продукта для получения других ингибиторов редуктазы HMG-CoA.
В патенте США №4293496, который включен в настоящую заявку по ссылке, раскрывается способ удаления боковой 2-метилбутириловой цепи путем основного гидролиза сложного эфира ловастатина с помощью гидроокиси щелочного металла, предпочтительно - гидроокиси лития (LiOH). Полученные продукты применимы для синтеза антигиперхолистеринемических средств.
В патенте США №4444784, который включен в настоящую заявку по ссылке, раскрывается способ введения в молекулу гидролизированного ловастатина новой боковой цепи.
В патенте США №5159104, который включен в настоящую заявку по ссылке, раскрывается способ получения симвастатина путем последовательного ацилирования диоллактона (diollactone) с образованием дважды ацилированного промежуточного продукта с последующим выборочным деацилированием и замыканием лактонового кольца с образованием симвастатина.
Краткое описание предлагаемого изобретения
Ближайшим аналогом предлагаемого изобретения является техническое решение по патенту США №5159104.
Способ получения статина, описанный в указанном патенте, не позволяет получать симвастатин высокой степени очистки, практически чистый симвастатин.
Целью предлагаемого изобретения является получение практически чистого симвастатина, содержащего менее чем приблизительно 0,1% (по массе) симва-оксолактона (simva-oxolactone).
Еще одной целью предлагаемого изобретения является получение практически чистого симвастатина, содержащего менее чем приблизительно 0,1% (по массе) ангидросимвастатина (anhydrosimvastatin).
Еще одной целью предлагаемого изобретения является получение практически чистого симвастатина, содержащего менее чем приблизительно 0,1% (по массе) димера симвастатина.
Еще одной целью предлагаемого изобретения является получение практически чистого симвастатина, содержащего менее чем приблизительно 0,1% (по массе) дигидросимвастатина (dihydrosimvastatin).
Еще одной целью предлагаемого изобретения является получение практически чистого симвастатина, содержащего менее чем приблизительно 0,1% (по массе) по меньшей мере одного из соединений из следующего перечня: симва-оксолактон, ангидросимвастатин, димер симвастатина, дигидросимвастатин.
Предлагаемое изобретение относится к способу получения симвастатина высокой степени очистки из ловастатина, включающему следующие стадии: размыкание лактонового кольца путем приведения ловастатина в реакцию с амином для получения амида; защита 1,3-диоловой части с помощью защитной группы; удаление 2-метилбутириловой группы, присоединенной с помощью сложной эфирной связи через посредство кислорода в позиции 8 гексагидронафталинового кольца; присоединение 2,2-диметилбутиратной группы путем образования сложной эфирной связи к гидроксилу в позиции 8; удаление защитной группы; превращение амида в кислую соль; и замыкание лактонового кольца с образованием симвастатина.
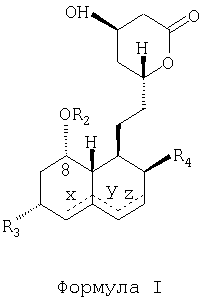
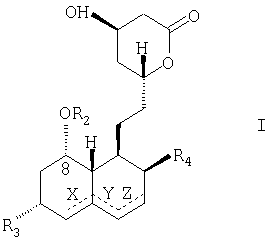
Согласно другому аспекту предлагаемое изобретение относится к способу получения полусинтетического статина, имеющего молекулярную структурную формулу, показанную ниже (Формула I):
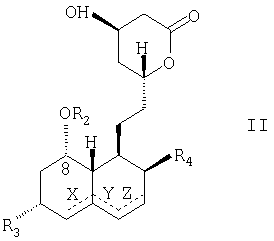
из статина, имеющего следующую молекулярную структурную формулу (Формула II):
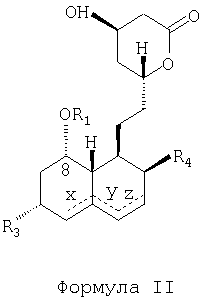
содержащему следующие стадии: размыкание лактонового кольца путем приведения статина, имеющего структурную молекулярную формулу "Формула II", в реакцию с амином для получения амида; защита 1,3-диоловой части с помощью защитной группы; удаление 2-метилбутириловой группы, присоединенной с помощью сложной эфирной связи через посредство кислорода в позиции 8 гексагидронафталинового кольца; присоединение 2,2-диметилбутиратной группы путем образования сложной эфирной связи к гидроксилу в позиции 8; удаление защитной группы; превращение амида в кислую соль; и замыкание лактонового кольца с образованием полусинтетического статина, имеющего структурную молекулярную формулу "Формула I", где R1 и R2 - ацильные группы, присоединенные к кислороду посредством сложной эфирной связи, а R3 и R4 - группы, независимо выбранные из следующего перечня: -Н, -ОН, -С1-10алкил, -С6-14арил, С6-14арил-С1-3.
Подробное описание предлагаемого изобретения
Предметом предлагаемого изобретения является способ получения практически чистого симвастатина, содержащего менее чем приблизительно 0,1% (по массе) симва-оксолактона (simva-oxolactone).
Еще одним предметом предлагаемого изобретения является способ получения практически чистого симвастатина, содержащего менее чем приблизительно 0,1% (по массе) ангидросимвастатина (anhydrosimvastatin).
Еще одним предметом предлагаемого изобретения является способ получения практически чистого симвастатина, содержащего менее чем приблизительно 0,1% (по массе) димера симвастатина.
Еще одним предметом предлагаемого изобретения является способ получения практически чистого симвастатина, содержащего менее чем приблизительно 0,1% (по массе) дигидросимвастатина (dihydrosimvastatin).
Еще одним предметом предлагаемого изобретения является способ получения практически чистого симвастатина, содержащего менее чем приблизительно 0,1% (по массе) по меньшей мере одного из соединений из следующего перечня: симва-оксолактон, ангидросимвастатин, димер симвастатина, дигидросимвастатин.
Способ получения симвастатина высокой степени очистки
Согласно первому аспекту предлагаемое изобретение относится к способу получения симвастатина высокой степени очистки из ловастатина, содержащему следующие стадии: размыкание лактонового кольца путем приведения ловастатина в реакцию с амином для получения амида; защита 1,3-диоловой части с помощью защитной группы; удаление 2-метилбутириловой группы, присоединенной с помощью сложной эфирной связи через посредство кислорода в позиции 8 гексагидронафталинового кольца; присоединение 2,2-диметилбутиратной группы путем образования сложной эфирной связи к гидроксилу в позиции 8; удаление защитной группы; превращение амида в кислую соль; и замыкание лактонового кольца с образованием симвастатина.
Превращение ловастатина в симвастатин по способу по предлагаемому изобретению показано ниже на Схеме I:
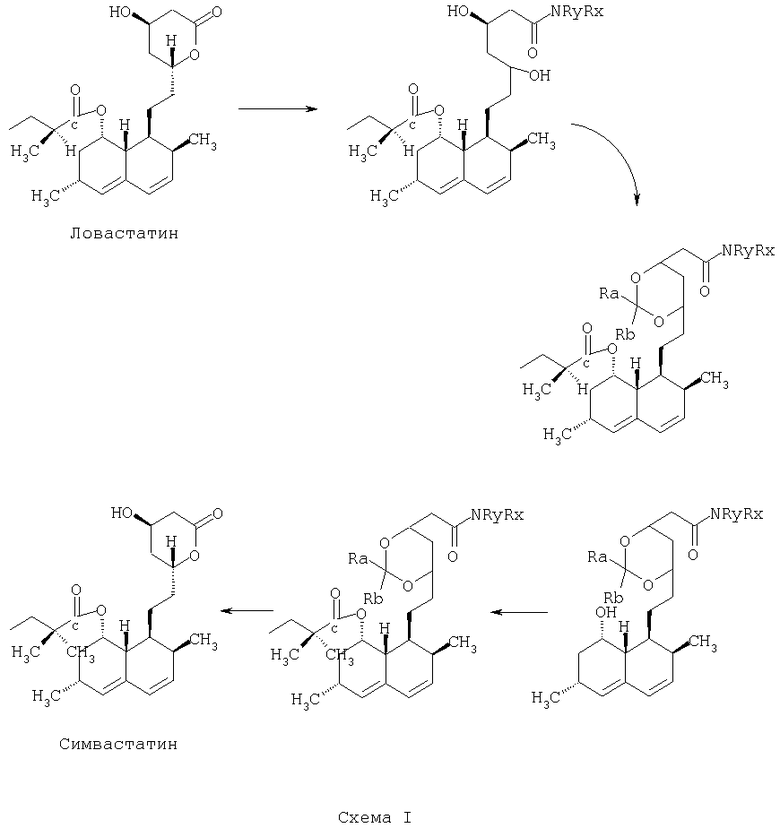
В предпочтительных вариантах осуществления способа по предлагаемому изобретению стадию размыкания лактонового кольца выполняют путем приведения лактона в реакцию с аммиаком, первичным амином или вторичным амином. При выполнении стадии размыкания лактонового кольца путем приведения лактона в реакцию с амином в предпочтительных вариантах осуществления способа по предлагаемому изобретению амин выбирают из следующего перечня: n-бутиламин, циклогексиламин, пиперидин, пирролидин.
Загрязняющие примеси, которые могут образоваться в процессе синтеза симвастатина, показаны ниже на Схеме II:
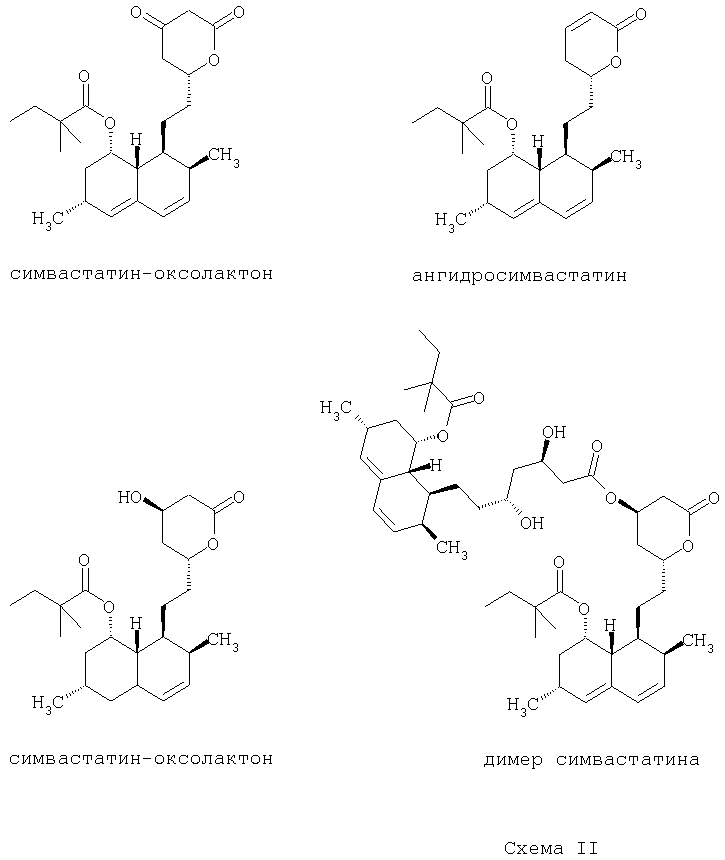
В предпочтительных вариантах осуществления предлагаемого изобретения практически чистый симвастатин, полученный по способу по предлагаемому изобретению, содержит менее чем приблизительно 0,1% (по массе) симваоксолактона (simva-oxolactone).
В предпочтительных вариантах осуществления предлагаемого изобретения практически чистый симвастатин, полученный по способу по предлагаемому изобретению, содержит менее чем приблизительно 0,1% (по массе) ангидросимвастатина.
В предпочтительных вариантах осуществления предлагаемого изобретения практически чистый симвастатин, полученный по способу по предлагаемому изобретению, содержит менее чем приблизительно 0,1% (по массе) дигидросимвастатина.
Кроме того, при применении предлагаемого изобретения обеспечивается получение практически чистого симвастатина, который содержит менее чем 0,1% (по массе) димера симвастатина.
При желании по способу по предлагаемому изобретению практически чистый симвастатин может быть синтезирован из загрязненного ловастатина, содержащего до приблизительно 30% примесей.
Способ получения статина высокой степени очистки
Согласно другому аспекту, предлагаемое изобретение относится к способу получения полусинтетического статина, имеющего молекулярную структурную формулу, показанную ниже (Формула I):
из статина, имеющего следующую молекулярную структурную формулу (Формула II):
содержащему следующие стадии: размыкание лактонового кольца путем приведения статина, имеющего структурную молекулярную формулу "Формула II", в реакцию с амином для получения амида; защита 1,3-диоловой части с помощью защитной группы; удаление 2-метилбутириловой группы, присоединенной с помощью сложной эфирной связи через посредство кислорода в позиции 8 гексагидронафталинового кольца; присоединение 2,2-диметилбутиратной группы путем образования сложной эфирной связи к гидроксилу в позиции 8; удаление защитной группы; превращение амида в кислую соль; и замыкание лактонового кольца с образованием полусинтетического статина, имеющего структурную молекулярную формулу "Формула I", в которой R1 и R2 - ацильные группы, присоединенные к кислороду посредством сложной эфирной связи, а R3 и R4 - группы, независимо выбранные из следующего перечня: -Н, -ОН, -С1-10алкил, -С6-14арил, С6-14арил-С1-3.
Превращение химического соединения, имеющего структурную молекулярную формулу "Формула II", в химическое соединение, имеющее структурную молекулярную формулу "Формула I", по способу согласно предлагаемому изобретению показано ниже на Схеме III:
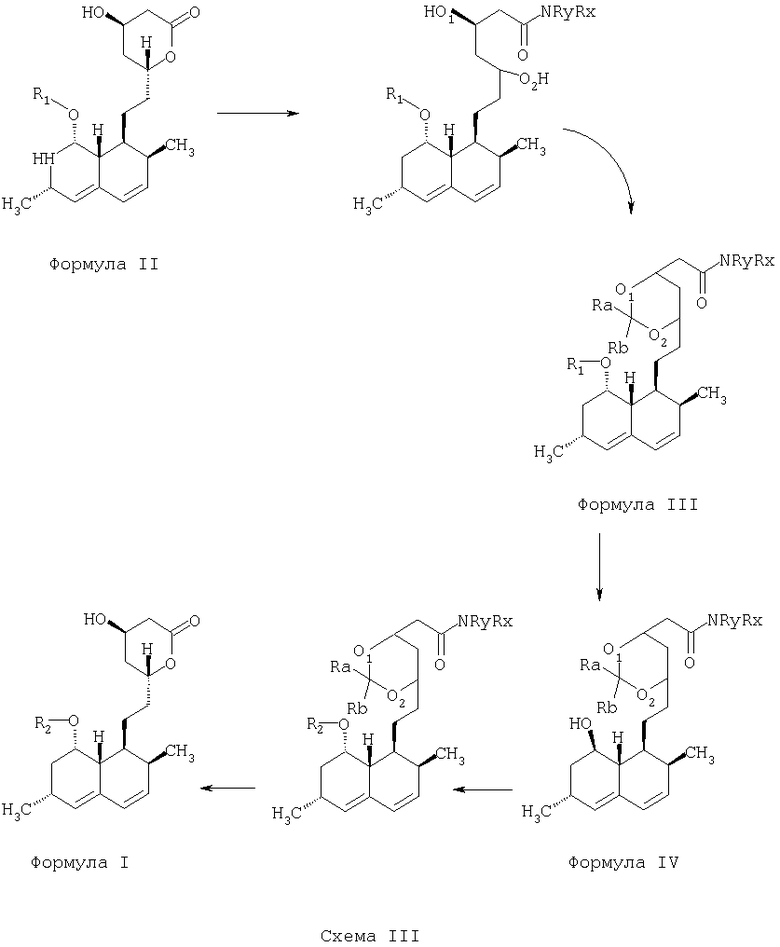
В предпочтительных вариантах осуществления предлагаемого изобретения полусинтетический статин, имеющий структурную молекулярную формулу "Формула I", синтезированный способом по предлагаемому изобретению, содержит менее чем приблизительно 0,1% примесей.
При желании по способу по предлагаемому изобретению полусинтетический статин, имеющий структурную молекулярную формулу "Формула I", может быть синтезирован из загрязненного статина, имеющего структурную молекулярную формулу "Формула II", содержащего до приблизительно 30% примесей.
В предпочтительных вариантах осуществления предлагаемого изобретения R1 - это ацильная группа следующего вида:
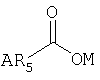
где ОМ - это кислород, который является заместителем гексагидро-нафталинового кольца в позиции 8, R5 - это группа, выбранная из следующего перечня: -С1-15алкил, - -С3-15циклоалкил, -С2-15алкенил, -С2-15алкинил, -фенил, -фенилС1-6алкил, а А - это заместитель R5, выбранный из следующего перечня: водород, галоген, С1-6алкил, С1-6алкокси, С6-14арил.
В предпочтительных вариантах осуществления предлагаемого изобретения R2 - это ацильная группа следующего вида:
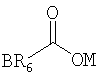
где ОМ - это кислород, который является заместителем гексагидро-нафталинового кольца в позиции 8, R6 - это группа, выбранная из следующего перечня: -С1-15алкил, --С3-15циклоалкил, -С2-15алкенил, -С2-15алкинил, -фенил, -фенилС1-6алкил, а В - это заместитель R6, выбранный из следующего перечня: водород, галоген, С1-6алкил, С1-6алкокси, С6-14арил.
Пунктирными линиями, обозначенными в структурных молекулярных формулах "Формула I" и "Формула II" буквами X, Y и Z, показаны возможные двойные связи, эти двойные связи, когда они присутствуют, находятся или в позициях Х и Z одновременно, или только в одной из позиций X, Y, или Z.
В предпочтительных вариантах осуществления способа по предлагаемому изобретению стадию размыкания лактонового кольца выполняют путем приведения лактонового кольца в реакцию с аммиаком, первичным амином или вторичным амином. Стадия размыкания лактонового кольца может выполняться путем приведения лактонового кольца в реакцию с амином, выбранным из следующего перечня: n-бутиламин, циклогексиламин, пиперидин, пирролидин.
В предпочтительных вариантах осуществления способа по предлагаемому изобретению размыкание лактонового кольца выполняют в органическом растворителе. Этот органический растворитель может быть выбран из следующего перечня: толуол, циклогексан, тетрагидрофуран, ацетонитрил.
В предпочтительных вариантах осуществления способа по предлагаемому изобретению стадию размыкания лактонового кольца выполняют при температуре выше температуры окружающей среды. В предпочтительных вариантах осуществления способа по предлагаемому изобретению стадия размыкания лактонового кольца может выполняться при температуре приблизительно 60°С.
В предпочтительных вариантах осуществления способа по предлагаемому изобретению стадия размыкания лактонового кольца включает удаление непрореагировавшего амина после образования амида. В число способов удаления непрореагировавшего амина входят удаление амина путем испарения и/или промывания органического раствора, содержащего амид, с помощью разбавленной кислоты.
Способ по предлагаемому изобретению включает также стадию защиты 1,3-диоловой части с помощью защитной группы. Способы защиты гидроксильных групп хорошо известны в данной отрасли, они раскрыты, например, в патентах США №№6100407 и 6252091, в Европейском патенте №ЕР 299656 и в Международной патентной заявке WO 95/13283, включенных в настоящую заявку по ссылке. Защитная группа может быть выбрана из следующего перечня: ацетал, кетал, циклический сульфат, циклический фосфат, боратная группа.
В одном из вариантов осуществления предлагаемого изобретения в качестве защитной группы использован кетал. В этом варианте стадия защиты 1,3-диоловой части может выполняться путем получения кетала с помощью кетона. Получение кетала предпочтительно выполняется в органическом растворителе. Этот органический растворитель может быть выбран из следующего перечня: толуол, циклогексан, тетрагидрофуран, ацетонитрил, этилацетат.
В другом варианте осуществления способа по предлагаемому изобретению в качестве защитной группы использован ацетал. В этом варианте стадия защиты 1,3-диоловой части может выполняться путем получения ацетала с помощью альдегида. Получение ацетала предпочтительно выполняется в органическом растворителе. Этот органический растворитель может быть выбран из следующего перечня: толуол, циклогексан, тетрагидрофуран, ацетонитрил, этилацетат.
Еще в одном варианте осуществления способа по предлагаемому изобретению защита 1,3-диоловой части может быть осуществлена путем получения диоксиновой части, посредством которой защита 1,3-диоловой части осуществляется, как показано ниже на Схеме IV:
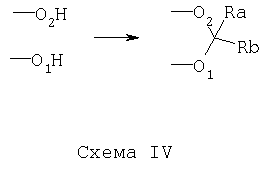
Еще в одном варианте осуществления способа по предлагаемому изобретению защита 1,3-диоловой части может быть осуществлена путем получения ацетала, имеющего следующее строение:
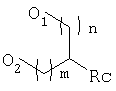
где группа RС может быть выбрана из следующего перечня: водород, галоген, С1-6алкил-, С1-6алкокси, С6-14арил, как, например, фенил или ароматический гетероцикл, a m и n независимо друг от друга могут принимать значения от 0 до 10.
Кроме того, в способе по предлагаемому изобретению предусмотрено также использование других защитных групп, как например:
(1) циклический сульфат
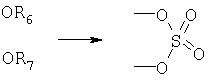
(2) циклический фосфат
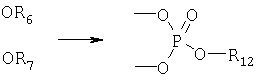
(3) циклический борат
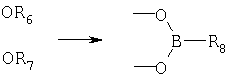
В предпочтительных вариантах осуществления способа по предлагаемому изобретению стадию защиты 1,3-диоловой части выполняют при температуре от приблизительно 5°С до приблизительно 50°С. В еще более предпочтительных вариантах стадию защиты выполняют при температуре от приблизительно 20°С до приблизительно 25°С.
В предпочтительных вариантах осуществления способа по предлагаемому изобретению стадию защиты 1,3-диоловой части выполняют в присутствии каталитического реагента. В качестве такого каталитического реагента предпочтительно использовать кислоту. Эта кислота может быть выбрана из следующего перечня: p-толуоловая сульфокислота, серная кислота.
В одном из вариантов осуществления способа по предлагаемому изобретению стадия удаления R1 включает восстановление статина со структурной молекулярной формулой "Формула III" с помощью восстановителя. Этот восстановитель может быть выбран из следующего перечня: алюмогидрид лития, гидрид алюминия, гидрид диизобутилалюминия. В предпочтительных вариантах стадия восстановления выполняется в инертном растворителе. Этот инертный растворитель может быть выбран из следующего перечня: толуол, тетрагидрофуран. Кроме того, стадия восстановления может дополнительно включать нейтрализацию остаточного восстановителя с помощью воды.
В предпочтительных вариантах осуществления способа по предлагаемому изобретению стадию восстановления выполняют при температуре от приблизительно 0°С до приблизительно 30°С. В еще более предпочтительных вариантах стадию защиты выполняют при температуре от приблизительно 5°С до приблизительно 10°С.
В одном из вариантов осуществления способа по предлагаемому изобретению стадия удаления R1 включает приведение статина со структурной молекулярной формулой "Формула III" в реакцию с металлоорганическим реагентом в среде инертного растворителя.
В качестве металлоорганического реагента может быть использован реактив Гриньяра (магнийгалоидалкил). Температура, при которой статин со структурной молекулярной формулой "Формула III" приводят в реакцию с реактивом Гриньяра, предпочтительно находится в диапазоне от приблизительно -10°С до приблизительно 20°С. Еще более предпочтительный температурный диапазон реакции статина со структурной молекулярной формулой "Формула III" с реактивом Гриньяра составляет от приблизительно -5°С до приблизительно 10°С.
В альтернативном варианте в качестве металлоорганического реагента может быть использовано производное алкиллития. В предпочтительном варианте это производное алкиллития представляет собой n-бутиллитий. Предпочтительный температурный диапазон реакции статина со структурной молекулярной формулой "Формула III" с производным алкиллития составляет от приблизительно -70°С до приблизительно -20°С.
Еще в одном из вариантов осуществления способа по предлагаемому изобретению стадия удаления R1 включает приведение статина со структурной молекулярной формулой "Формула III" в реакцию с амином. В предпочтительных вариантах этот амин представляет собой аммоний или первичный амин. Молярное отношение амина к статину со структурной молекулярной формулой "Формула III" предпочтительно равно 1:1.
Стадия удаления R1 может выполняться в присутствии воды. Кроме того, стадия удаления R1 может выполняться в присутствии органического растворителя.
В предпочтительных вариантах осуществления способа по предлагаемому изобретению стадию удаления R1 выполняют при температуре от приблизительно 100°С до приблизительно 250°С. В еще более предпочтительных вариантах стадию удаления R1 выполняют при температуре от приблизительно 130°С до приблизительно 200°С.
В предпочтительных вариантах осуществления способа по предлагаемому изобретению стадию удаления R1 выполняют при давлении выше атмосферного.
В предпочтительных вариантах осуществления способа по предлагаемому изобретению стадия присоединения R2 включает ацилирование кислорода, который является заместителем гексагидронафталинового кольца в позиции 8. Стадия ацилирования может включать приведение статина со структурной молекулярной формулой "Формула IV" в реакцию с хлорангидридом кислоты. В альтернативном варианте стадия ацилирования может включать приведение статина со структурной молекулярной формулой "Формула IV" в реакцию со свободной кислотой в присутствии карбодиимида. В качестве такого карбодиимида может быть использован 1,3-дициклогексилкарбодиимид. Еще в одном альтернативном варианте стадия ацилирования может включать приведение статина со структурной молекулярной формулой "Формула IV" в реакцию с симметричным ангидридом в присутствии органического растворителя и катализатора. В предпочтительном варианте в качестве катализатора используется 4-диметиламинопиридин.
В предпочтительных вариантах осуществления способа по предлагаемому изобретению стадию ацилирования выполняют при температуре от приблизительно 20°С до приблизительно 110°С. В еще более предпочтительных вариантах стадию удаления R1 выполняют при температуре от приблизительно 80°С до приблизительно 110°С.
В тех вариантах осуществления способа по предлагаемому изобретению, в которых предусмотрена защита гидроксильных групп -O1H и -O2H, способ по предлагаемому изобретению может дополнительно включать удаление защитных групп после стадии присоединения R2. В предпочтительных вариантах стадия удаления защитных групп включает гидролиз в среде смеси воды и органического растворителя в присутствии катализатора. В предпочтительных вариантах в качестве этого органического растворителя используется тетрагидрофуран. В качестве катализатора может быть использован кислотный катализатор. В предпочтительных вариантах в качестве этого кислотного катализатора используется одно из соединений из следующего перечня: хлористый водород, серная кислота, p-толуоловая сульфокислота.
В предпочтительных вариантах осуществления способа по предлагаемому изобретению стадию удаления защитных групп выполняют при температуре от приблизительно 20°С до приблизительно 100°С. В еще более предпочтительных вариантах стадию удаления R1 выполняют при температуре от приблизительно 30°С до приблизительно 70°С.
В предпочтительных вариантах осуществления способа по предлагаемому изобретению стадия превращения амида в кислую соль включает гидролиз. Этот гидролиз может выполняться в среде раствора, содержащего основание, воду и органический растворитель. В предпочтительных вариантах основание выбрано из следующего перечня: гидроокись натрия, гидроокись калия. В предпочтительных вариантах органический растворитель выбран из следующего перечня: метиловый спирт, этиловый спирт, толуол, тетрагидрофуран.
В предпочтительных вариантах осуществления способа по предлагаемому изобретению стадия превращения амида в кислую соль включает образование соли с фармацевтически приемлемым противоионом. Соль с фармацевтически приемлемым противоионом - это предпочтительно соль аммония.
В предпочтительных вариантах осуществления способа по предлагаемому изобретению стадия замыкания лактонового кольца включает образование лактона в среде органического растворителя. В предпочтительных вариантах органический растворитель выбран из следующего перечня: толуол, этилацетат, циклогексан. В предпочтительных вариантах стадию замыкания лактонного кольца выполняют при повышенной температуре. Эта температура находится в диапазоне от приблизительно 60°С до приблизительно 110°С. Еще более предпочтительный температурный диапазон составляет от приблизительно 80°C до приблизительно 110°С.
В одном из альтернативных вариантов осуществления способа по предлагаемому изобретению предусмотрена стадия изолирования статина со структурной молекулярной формулой "Формула I" путем кристаллизации.
Симвастатин высокой степени очистки, получаемый способом по предлагаемому изобретению, в сочетании с по меньшей мере одним фармацевтически приемлемым инертным наполнителем, может найти применение для производства фармацевтических композиций. Такие фармацевтические композиции пригодны для приема млекопитающими пациентами в виде лекарственных форм.
Эти лекарственные формы могут содержать практически чистый симвастатин, или же, в качестве альтернативного варианта, они могут содержать практически чистый симвастатин как часть фармацевтической композиции. Как в чистом виде, так и в составе фармацевтической композиции, практически чистый симвастатин может приниматься в форме порошка, гранул, агрегаций, или в любой другой твердотельной форме. Такие фармацевтические композиции могут включить композиции для таблетирования. В зависимости от примененного способа таблетирования, требуемого выхода продукта и других факторов, эти таблетируемые фармацевтические композиции могут иметь большее или меньшее количество компонентов. Такие фармацевтические композиции могут содержать разбавители, в качестве каковых могут быть использованы, например, такие производные целлюлозы, как порошкообразная целлюлоза, микрокристаллическая целлюлоза, сверхтонкая целлюлоза, метилцеллюлоза, этилцеллюлоза, гидроксиэтилцеллюлоза, гидроксипропилцеллюлоза, соли карбоксиметилцеллюлозы и другие замещенные и незамещенные производные целлюлозы; крахмал; предварительно желатинизированный крахмал; неорганические разбавители, такие как карбонат кальция и дифосфат кальция, а также другие разбавители, хорошо известные среднему специалисту соответствующего профиля. В число разбавителей, пригодных для использования в фармацевтических композициях по предлагаемому изобретению, входят также такие вещества, как воски, сахара (например, лактоза) и "сахарные" (шестиатомные алифатические) спирты (такие как маннит и сорбит), полимеры и сополимеры акрилатов, а также пектиновые вещества, декстрины и желатин.
В число других инертных наполнителей входят связующие вещества, такие как аравийская камедь, предварительно желатинизированный крахмал, альгинат натрия, глюкоза и другие связующие вещества, используемые при мокрой и сухой грануляции и при таблетировании мутем непосредственного сжатия; разрыхлители, такие как крахмалгликолат натрия, кросповидон, низкозамещенная гидроксипропилцеллюлоза и другие; смазывающие агенты, такие как стеарат магния и кальция и стеарилфумарат натрия; ароматизаторы; подсластители; консерванты; фармацевтически приемлемые красители, а также агенты, придающие таблетке скольжение, например двуокись кремния.
Лекарственные формы могут быть приспособлены для перорального, трансбуккального, парентерального, офтальмического, ректального или чрескожного введения. К лекарственным формам для перорального введения относятся таблетки, пилюли, капсулы, лепешки, саше, суспензии, порошки, пастилки, эликсиры и т.п. Симвастатин высокой степени очистки, раскрываемый в настоящем изобретении, может приниматься также в виде суппозиториев, глазных мазей и суспензий, а также в виде парентеральных суспензий, вводимых иными способами. Наиболее предпочтительным способом введения симвастатина, полученного по предлагаемому изобретению, является пероральный.
Капсулированные дозы могут содержать твердую фармацевтическую композицию, заключенную в капсулу, которая может иметь покрытие из желатина. Таблетки и порошки тоже могут быть покрыты энтеросолюбильной оболочкой. Порошковые формы с энтеросолюбильной оболочкой могут иметь в составе этой оболочки такие вещества, как фталевокислая ацетилцеллюлоза, фталат гидроксипропилметилцеллюлозы, фталат поливинилового спирта, карбоксиметилэтилцеллюлоза, сополимер стирола и малеиновой кислоты, сополимер метакриловой кислоты и метилметакрилата и тому подобные вещества, которые, при желании, могут применяться совместно с подходящими пластификаторами и/или наполнителями. Покрытие таблетки может представлять собой поверхностную оболочку, или же таблетка может состоять из частиц порошка или гранул, снабженных энтеросолюбильной оболочкой.
В настоящее время симвастатин присутствует в продаже в виде таблеток 5 мг, 10 мг, 20 мг, 40 мг и 80 мг, в состав которых входят также следующие инертные наполнители: стеарат магния, крахмал, тальк, двуокись титана и другие ингредиенты. В качестве консерванта использован бутилгидроксианизол.
Ловастатин поставляется на рынок в виде таблеток 10 мг, 20 мг и 40 мг для перорального приема. В дополнение к активному ингредиенту, каковым является ловастатин, эти таблетки содержат следующие инертные наполнители: целлюлоза, лактоза, стеарат магния, крахмал. В качестве консерванта использован бутилгидроксианизол.
Функции и преимущества этих и других вариантов осуществления предлагаемого изобретения будут поняты более полно после изучения примеров, рассматриваемых ниже. Следует заметить, что цель этих примеров состоит в том, чтобы проиллюстрировать преимущества предлагаемого изобретения, а не в том, чтобы охватить весь объем предлагаемого изобретения.
Примеры
Условия эксперимента
Анализ по методике жидкостной хроматографии высокого разрешения проводился как описано в источнике А.Хаук и др. (A.Houck et al.), Talanta, том 40(4), 491-494 (1993), "Жидкостно-хроматографическое пределение известных примесей, присутствующих в нерасфасованном ловастатине в малом количестве: применение хроматографии высокого и низкого уровней" ("Liquid Chromatographic determination of the known low level impurities in lovastatin bulk drug: an application of high-low chromatography").
Оборудование для жидкостной хроматографии высокого разрешения:
- Насос/инжектор Alliance Waters;
- Диодная матрица М996 Waters;
- Система обработки данных Millennium Waters;
- Колонка: Prodigy 5 С8 250×4,6 мм (phenomenex).
Условия:
наносимый объем: 10 μл;
градиентный профиль течения (линейный);
А=ацетонитрил;
В=0,1% ортофосфорная кислота (Н3PO4).
Температура колонки: 30°С.
Определение при длинах волн 200 нм и 237 нм.
Образцы подмешивались к ацетонитрилу в концентрации 1,5 мг/мг.
Время удерживания:
Пример 1
Получение пиперидинамида ловастатина:
Смесь, содержащая 1 г (2,5 ммоль) ловастатина, 10 мл (0,1 моль) пиперидина, 100 мг (0,82 ммоль) N,N-диметиламинопиридина и 30 мл толуола была подвергнута дефлегмации в течение 36 часов. Затем смесь была охлаждена до комнатной температуры и дважды промыта 2 N раствором соляной кислоты (HCl), взятого оба раза в объеме 30 мл, и дважды промыта водой, взятой оба раза в объеме 20 мл. Органический слой был высушен с помощью сернокислого натрия, отфильтрован и выпарен. Остаток был перемешан с гексаном, и полученный осадок был отфильтрован, в результате чего было получено 0,87 г пиперидинамида ловастатина в виде твердого вещества белого цвета.
Пример 2
Реакция бутиламида ловастатина с тионилхлоридом
К раствору, содержащему 1,2 г (2,5 ммоль) бутиламида ловастатина в 20 мл толуола, было добавлено 0,76 г (7,5 ммоль) триэтиламина. Затем по каплям было добавлено 0,45 г (3,7 ммоль) тионилхлорида. После выдержки в течение 1 часа при комнатной температуре реакционная смесь была промыта водой, высушена (с помощью сернокислого натрия), профильтрована и выпарена, в результате чего было получено маслянистое вещество коричневого цвета.
Пример 3
Реакция бутиламида ловастатина с фосфорилхлоридом
К раствору, содержащему 1,2 г (2,5 ммоль) бутиламида ловастатина в 20 мл толуола, было добавлено 0,76 г (7,5 ммоль) триэтиламина. Затем по каплям было добавлено 0,58 г (3,8 ммоль) фосфорилхлорида. После выдержки в течение 1 часа при комнатной температуре реакционная смесь была промыта водой, высушена (с помощью сернокислого натрия), профильтрована и выпарена, в результате чего было получено маслянистое вещество коричневого цвета.
Пример 4
А. Получение ацетонида бутиламида ловастатина
Смесь, содержащая 40 г (98 ммоль) ловастатина и 60 мл n-бутиламина была подвергнута дефлегмированию в течение 1 часа, выпарена, а затем дважды выпарена с толуолом, взятым в количестве 100 мл. Полученный неочищенный амид был растворен в 500 мл ацетона, после чего к раствору было добавлено 3 г p-TsOH. Прозрачный раствор был подвергнут перемешиванию при комнатной температуре в течение двух часов, причем за это время произошло образование твердой фазы. Затем смесь была охлаждена до температуры -10°С, при которой была выдержана в течение трех часов, после чего твердая фаза была выделена и высушена до получения 45 г (88%) амида/ацетонида в виде твердого вещества белого цвета. Еще 5 г было получено из маточного раствора частичным выпариванием раствора.
В. Алкилирование амида/ацетонида, полученного на стадии А в качестве промежуточного продукта:
Амид/ацетонид в количестве 19,5 г (37,6 ммоль) в среде смеси тетрагидрофурана и циклогексана (в отношении 4:1), взятой в объеме 280 мл, был охлажден до температуры -40°С, после чего было добавлено 113 мл 1 М литийпирролидина (полученного из пирролидина и n-бутиллития при температуре -15°С), при этом температура поддерживалась на уровне ниже -30°С. После этого раствор в течение двух часов подвергался перемешиванию при температуре -35°С, и к нему было добавлено 5 мл MeI одной порцией. Затем раствор был подвергнут перемешиванию при температуре -30°С в течение одного часа, после чего был допущен подъем температуры до -10°С. После этого к раствору было добавлено 300 мл 1 N раствора соляной кислоты (HCl), и полученная смесь была подвергнута дефлегмированию в течение одного часа. Затем к реакционной смеси был добавлен этилацетат в количестве 300 мл, после чего органический слой был промыт с помощью 3 N раствора соляной кислоты (HCl), взятого в объеме 100 мл, и выпарен. К полученному остатку было добавлено 300 мл метилового спирта и 125 мл 2 N раствора едкого натра (NaOH). Полученная смесь была подвергнута дефлегмированию в течение 12 часов, в результате чего произошло испарение большей части метилового спирта. После этого к реакционной смеси было добавлено 120 мл воды и 300 мл этилацетата, а значение показателя рН было доведено до 5 с помощью 3 N раствора соляной кислоты (HCl). Затем к органическому слою было добавлено 60 мл метилового спирта и 25 мл смеси гидроокиси аммония (NH4OH) и метилового спирта (в соотношении 1:3). После этого полученная смесь подвергалась перемешиванию при комнатной температуре в течение одного часа, после чего она была охлаждена до температуры 10°С. Полученное твердое вещество было собрано и высушено. В результате было получено 13,5 г (80%) аммониевой соли симвастатиновой кислоты.
Пример 5
Способ получения симвастатина из ловастатина путем восстановления сложноэфирной группы R1
А. Получение ацетонида бутиламида ловастатина
Смесь ловастатина, взятого в количестве 40,5 г (100 ммоль), и n-бутиламина, взятого в объеме 75 мл, была подвергнута нагреванию при дефлегмировании в течении 2 часов. Избыток амина был испарен, и реакционная смесь была подвергнута выпариванию совместно с толуолом, взятым в объеме 100 мл. К полученному неочищенному амиду был добавлено 400 мл ацетона и 5 г p-TsOH. Полученная смесь была подвергнута перемешиванию при комнатной температуре в течение 2 часов, после чего подвергалась охлаждению в ванне изо льда/воды в течение 2 часов. Полученное твердое вещество было собрано путем фильтрования и высушено. Затем из маточного раствора была получена еще одна порция продукта, общий выход которого составил 49 г (94-95%).
B1. Восстановление промежуточного продукта, полученного на стадии А, при помощи алюмогидрида лития
Химическое соединение, полученное на стадии А, в количестве 45 г (87 ммоль) было растворено в 200 мл тетрагидрофурана, и полученный раствор добавлялся по каплям к суспензии 7 г (2,1 эквивалента) алюмогидрида лития (LiAlH4) в 100 мл тетрагидрофурана при температуре 10-15°С в течение приблизительно 20 минут. Затем полученная смесь подвергалась перемешиванию в течение 30 минут. После этого реакционная смесь подвергалась обработке 20%-ного раствора гидроокиси калия (КОН) (экзотермическая реакция). Полученные соли были отделены путем фильтрования и промыты тетрагидрофураном, взятым в объеме 200 мл. Комбинированные фильтраты были выпарены до получения сиропообразного вещества, количество которого составило 35,5 г.
B2. Восстановление промежуточного продукта, полученного на стадии А, при помощи хлорметилмагния (реактив Гриньяра)
Раствор 2 г (3,9 ммоль) химического соединения, полученного на стадии А, в 20 мл тетрагидрофурана был охлажден до температуры 0°. Затем к нему в течение 20 минут по каплям было добавлено 12 мл 3 М хлорметилмагния. После выдерживания в течение 18 часов при комнатной температуре было достигнуто полное превращение ацетонида n-бутиламида ловастатина.
В3. Восстановление промежуточного продукта, полученного на стадии А, при помощи n-бутиллития
Раствор 91 г (1,9 ммоль) химического соединения, полученного на стадии А, в 25 мл тетрагидрофурана был охлажден до температуры -50°. Затем к нему в течение 10 минут по каплям было добавлено 2,47 мл 2,5 М хлорметилмагния. После выдерживания в течение 18 часов при комнатной температуре был получен спиртовой промежуточный продукт.
С. Ацилирование промежуточного продукта, полученного на стадии В, и получение аммониевой соли симвастатина
4-диметиламидопиридин в количестве 3 г, разведенный в 300 мл пиридина, добавлялся к раствору 25 г (57 ммоль) промежуточного продукта, полученного на стадии В, и полученная смесь подвергалась нагреванию до температуры 50-55°С, предпочтительно - до температуры 50°С. Затем к охлажденной смеси в виде одной порции добавлялся хлорид 2,2-диметилмасляной кислоты в количестве 50 мл, после чего полученная смесь подвергалась перемешиванию в течение 40 часов (анализ по методике жидкостной хроматографии высокого разрешения показал полное превращение). К реакционной смеси добавлялось 400 мл воды и 400 мл этилацетата (EtOAc). Затем органический слой дважды промывался 10%-ным раствором двууглекислого натрия (NaHCO3), взятым в объеме 400 мл, водой, взятой в объеме 400 мл, и 10%-ным раствором соляной кислоты (HCl), взятым в объеме 400 мл. Органический слой подвергался выпариванию и растворялся в 200 мл тетрагидрофурана, после чего к полученному раствору добавлялось 200 мл воды, а затем 10 г p-TsOH. Затем полученная смесь подвергалась дефлегмированию в течение 2 часов. После этого к реакционной смеси добавлялось 400 мл этилацетата (EtOAc), а затем - 300 мл воды. Органический слой дважды промывался 10%-ным раствором двууглекислого натрия (NaHCO3), взятым в объеме 400 мл, после чего подвергался выпариванию. Полученный остаток подвергался растворению в 300 мл метилового спирта (МеОН), после чего к раствору добавлялось 170 мл 2 N раствора гидроокиси натрия (NaOH). Полученная смесь подвергалась дефлегмированию в течение 3 часов при комнатной температуре, в результате чего просходило испарение большей части метилового спирта (МеОН). После этого к реакционной смеси добавлялось 120 мл воды, а значение показателя рН доводилось до 7 с помощью 3 N раствора соляной кислоты (HCl), после чего добавлялось 300 мл этилацетата (EtOAc). Затем значение показателя рН доводилось до 4, и слои подвергались разделению. К органическому слою добавлялось 100 мл этилацетата (EtOAc), а затем - 40 мл смеси гидроокиси аммония (NH4OH) и метилового спирта (МеОН) (в соотношении 1:3). Полученная смесь подвергалась перемешиванию при температуре -10°С в течение 2 часов, после чего твердое вещество было собрано и промыто этилацетатом (EtOAc) и этиловым спиртом (EtOH) (холодным). Выход продукта составил 16 г (62%), анализ по методике жидкостной хроматографии высокого разрешения показал содержание аммоневой соли симвастатина 98,9%.
D. Превращение аммониевой соли симвастатина в симвастатин
Суспензия 9 г аммониевой соли симвастатина, полученной на стадии С, в 250 мл толуола подвергалась нагреванию до температуры 100°С, при которой выдерживалась в течение 6 часов. Затем суспензия подвергалась дефлегмировагию в течение 30 минут, после чего профильтровывалась и выпаривалась. К остатку добавлялось 100 мл циклогексана, и полученный раствор снова подвергался выпариванию. Из приблизительно 150 мл циклогексанового раствора кристаллизовался неочищенный симвастатин в виде твердого вещества белого цвета. Выход продукта составил 85%, анализ по методике жидкостной хроматографии высокого разрешения показал содержание симвастатина 98,4%.
Пример 6
Способ получения симвастатина из ловастатина путем восстановления сложноэфирной группы R1
А. Получение ацетонида бутиламида ловастатина
Смесь ловастатина, взятого в количестве 950 г (2,4 моль), толуола, взятого в объеме 8 л, и n-бутиламина, взятого в объеме 500 мл (5 моль), была нагрета до температуры 85°С в атмосфере азота. Полученный раствор выдерживался при температуре 85°С-95°С в течение 2 часов, после чего был охлажден до комнатной температуры. Затем к раствору было добавлено 5 л 4 N раствора серной кислоты, после чего полученная смесь подвергалась перемешиванию в течение 5 минут. Нижний слой был удален, а к верхнему слою было добавлено 1,5 л (12 моль) 2,2-диметоксипропана. Затем раствор подвергался перемешиванию при комнатной температуре в течение 30 минут, после чего смесь была подвергнута сгущению путем выпаривания в вакууме при температуре 55-60°С до получения концентрата в количестве 5,4 кг.
В. Восстановление промежуточного продукта, полученного на стадии А, при помощи алюмогидрида лития
5,8 л (5,5 кг, что соответствует 2,4 моль промежуточного продукта, полученного на стадии А) концентрата, полученного на стадии А, было смешано с 2 л толуола. Полученная смесь была охлаждена до температуры 0°С в атмосфере азота. Затем в течение 75 минут к смеси было добавлено 6 л 1 N раствора алюмогидрида лития (LiAlH4) в толуоле (6 моль алюмогидрида лития), при этом температура реакционной смеси поддерживалась ниже 8°С. Полученная смесь подвергалась перемешиванию при температуре 5-10°С в течение 3 часов, после чего в течение 100 минут к смеси было добавлено 5,3 л воды, при этом температура смеси поддерживалась ниже 10-15°С. Затем к смеси (суспензии) было добавлено 5 л 4 N раствора серной кислоты, после чего смесь подвергалась перемешиванию в течение 15 минут. После этого смесь была оставлена в покое для осаждения слоев. Затем нижний молочно-белый слой был удален, а верхний слой был промыт водой, взятой в объеме 6 л, и водным 1 N раствором гидроокиси натрия. После этого 6 л верхнего слоя было удалено путем выпаривания в вакууме (давление от 150 до 300 мм ртутного столба) при температуре 50-60°С.
С. Ацилирование промежуточного продукта, полученного на стадии В, 2,2-диметилбутирилхлорида
К полученному на стадии В раствору спиртового промежуточного продукта, в толуоле, в котором упомянутый промежуточный продукт содержался в количестве 2,4 моль, было добавлено 250 мл толуолового раствора, содержащего 35 г (0,29 моль) 4-(N,N-диметиламино)пиридина, а также 1,6 л (11,4 моль) триэтиламина и 1,5 кг (11 моль) 2,2-диметилбутирилхлорида. Полученный раствор был нагрет до температуры 105-110°С и подвергнут перемешиванию при этой температуре в течение 10 часов в атмосфере азота. Затем полученная суспензия была охлаждена до комнатной температуры, после чего к ней было добавлено 3 л 4 N раствора серной кислоты. Полученная смесь подвергалась перемешиванию в течение 5 минут, после чего она была оставлена в покое для разделения слоев. После этого нижний слой был удален, а верхний слой был промыт 4 N раствором серной кислоты, взятым в количестве 2 л.
D. Получение аммониевой соли симвастатина
Реакционная смесь, полученная на стадии С (в объеме приблизительно 11 л), была смешана с 4,5 л 4 N раствора серной кислоты. Полученная смесь подвергалась нагреванию до температуры 70-75°С в течение 3 часов, при этом через смесь пропускался азот. Затем смесь была оставлена в покое до остывания ее до комнатной температуры, после чего был удален нижний слой. После этого верхний слой был охлажден до температуры 5°С и промыт 2 N раствором гидроокиси натрия, взятого в объеме 2,5 л. После удаления нижнего слоя было добавлено 6 л 2 N раствора серной кислоты, и смесь была подвергнута перемешиванию сначала в течение 3 часов при комнатной температуре, а затем еще в течение 3 часов при температуре 45-55°С. Затем полученная суспензия была охлаждена до температуры 5-10°С, после чего к ней было добавлено 2,75 л 4 N раствора серной кислоты, при этом температура суспензии поддерживалась ниже 10°С. После этого нижний слой был удален, и к остатку был добавлен концентрированный раствор гидроокиси аммония (NH4OH). Затем смесь была подвергнута сгущению путем выпаривания в вакууме при температуре 50-60°С для удаления толуола и воды. После этого к остатку было добавлено 3 л этилацетата, и смесь была подвергнута перемешиванию при температуре 50°С в течение 30 минут, в результате чего была получена однородная суспензия. Эта суспензия была охлаждена до комнатной температуры и профильтрована в вакууме. Полученный кек (осадок на фильтре) был промыт этилацетатом, взятым в объеме 1 л, а затем подвергнут суспендированию в среде этилацетата, взятого в объеме 4 л, после чего полученная суспензия была нагрета до температуры 50°С, при которой выдерживалась в течение 90 минут. После этого нагретая суспензия была профильтрована, а полученный кек (осадок на фильтре) был промыт этилацетатом. В результате была получена кристаллическая аммониевая соль симвастатина в количестве 891 г.
Е. Получение симвастатина
Полученная на стадии D кристаллическая аммониевая соль симвастатина в количестве 570 г была подвергнута суспендированию в среде толуола, взятого в объеме 13 л. Затем к суспензии было добавлено 2 л воды, и значение показателя рН было доведено до 3 путем добавления 4 N раствора серной кислоты. После этого полученная смесь подвергалась перемешиванию в течение 30 минут, а затем был удален нижний слой. Верхний слой был промыт водой, взятой в количестве 2 л, после чего подвергнут сгущению путем выпаривания 4 л толуола при температуре 50-60°С в вакууме. Остальной раствор был подвергнут нагреванию и выдерживался при температуре 85-92°С в атмосфере азота в течение 2,5 часов. Затем раствор был охлажден до температуры 15°С, к нему было добавлено 3 л воды, а значение показателя рН смеси было доведено до 8-8,5 путем добавления 1 N раствора гидроокиси натрия (NaOH). После этого был удален нижний слой, а к верхнему слою было добавлено 3 л воды, после чего значение показателя рН смеси было доведено до 6 путем добавления 6 N раствора серной кислоты. Затем нижний слой был удален, а верхний слой был подвергнут сгущению до объема 1 л путем выпаривания в вакууме при температуре 50-60°С. После этого к полученному концентрату в течение 1 часа при температуре 50-60°С добавлялся n-гексан в количестве 350 мл. Затем полученная смесь подвергалась перемешиванию при температуре 50-60°С в течение 30 минут, после чего она в течение 2 часов подвергалась медленному охлаждению до температуры 15°С. Полученные кристаллы были отфильтрованы и промыты смесью n-гексана и толуола (в соотношении 5:1), взятой в объеме 350 мл. Выход симвастатина составил 440 г.
Пример 7
Способ получения аммониевой соли симвастатина путем восстановления сложноэфирной группы R1 ловастатина
А. Получение циклогексанамида ловастатина
Смесь ловастатина, взятого в количестве 5 г (0,012 моль), циклогексиламина, взятого в объеме 6 мл (0,052 моль), и толуола, взятого в объеме 50 мл, подвергалась дефлегмированию в течение 6 часов. Затем реакционная смесь была охлаждена до комнатной температуры, после чего к ней было добавлено 20 мл этилацетата. После этого смесь была дважды промыта 2 N раствором соляной кислоты (HCl), оба раза взятого в объеме 30 мл, и водой, оба раза взятой в объеме 20 мл. Органический слой был высушен с помощью сернокислого натрия, профильтрован и подвергнут сгущению до объема 15 мл путем выпаривания. После этого к полученному концентрату было добавлено 50 мл гексана, осадок был отфильтрован, в результате чего было получено 5,5 г циклогексанамида ловастатина в виде порошка белого цвета.
В. Получение ацетонида циклогексанамида ловастатина
К раствору 5 г (10 моль) циклогексанамида ловастатина в 25 мл ацетона было добавлено 300 мг (1,6 ммоль) p-TsOH. Полученная смесь подвергалась перемешиванию при комнатной температуре в течение 18 часов, после чего она была влита в смесь, состоящую из 50 мл этилацетата и 50 мл 10%-ного раствора двууглекислого натрия, полученная смесь была высушена с помощью сернокислого натрия, профильтрована и подвергнута выпариванию. Полученный остаток был растворен в толуоле, а полученный раствор был затем выпарен, в результате чего было получено 4,9 г ацетонида циклогексанамида ловастатина.
С. Получение аммониевой соли симвастатина
Суспензия 836 мг (22 ммоль) алюмогидрида лития в среде 15 мл тетрагидрофурана была охлаждена до температуры 0°С, и к ней в течение 15 минут по каплям добавлялся раствор 4,93 г (9,1 ммоль) химического соединения, полученного на стадии В, в 20 мл тетрагидрофурана. Полученная реакционная смесь выдерживалась в течение 18 часов при комнатной температуре, после чего она была охлаждена до температуры 0°С, после чего к ней было добавлено 0,1 мл воды и 10%-ного раствора гидроокиси калия. После этого полученная смесь была профильтрована через целит, а затем был испарен тетрагидрофуран, в результате чего было получено 4,3 г (9 ммоль) спиртового промежуточного продукта.
D. Получение аммониевой соли симвастатина
Смесь, состоящая из 4,3 г (9 ммоль) спиртового промежуточного продукта, полученного на стадии С, 40 мл пиридина, 200 мг N,N-диметиламино-пиридина и 7,2 г (54 ммоль) хлорангидрида 2,2-диметилмасляной кислоты, была подвергнута перемешиванию при температуре 65°С в течение 72 часов. Затем полученная смесь была охлаждена, и к ней было добавлено 100 мл толуола, после чего смесь была дважды промыта 10%-ным раствором двууглекислого натрия, оба раза взятого в объеме 50 мл, и рассолом, взятым в объеме 30 мл. После этого толуоловый слой был высушен с помощью сернокислого натрия, смесь была профильтрована и выпарена. Затем остаток был растворен в 100 мл толуола, который затем был испарен. После этого остаток был растворен в 20 мл тетрагидрофурана и 20 мл воды. Затем к полученному раствору было добавлено 2 г p-TsOH, после чего раствор подвергался дефлегмированию в течение 5 часов. После этого раствор был влит в смесь, состоящую из 70 мл толуола и 50 мл 10%-ного раствора двууглекислого натрия. Органический слой был отделен и промыт 10%-ным раствором двууглекислого натрия, взятым в количестве 30 мл. После этого органический раствор был высушен, отфильтрован и подвергнут выпариванию, в результате чего был получен остаток в количестве 4,8 г. Этот остаток был растворен в 70 мл метилового спирта и 40 мл 2 М раствора гидроокиси натрия (NaOH). Полученная реакционная смесь (раствор) была подвергнута дефлегмированию в течение 72 часов. Метиловый спирт был испарен, а водный слой был охлажден до температуры 0°С. Затем водный слой был подкислен, а именно, значение показателя рН водного слоя было доведено до 5 с помощью 2 N раствора соляной кислоты (HCl). После этого к реакционной смеси было добавлено 75 мл этилацетата, и был отделен органический слой. К этилацетату было добавлено 5 мл 25%-ного раствора аммиака. Затем был отфильтрован осадок, в результате чего было получено 1,1 г аммониевой соли симвастатина, общий выход которого из ацетонида циклогексанамида ловастатина составил 27%.
Пример 8
Получение диацилированного бутиламида ловастатина
А. Введение в бутиламид ловастатина силильной группы
По способу, описанному в литературе (Askin, D.; Verhoeven, T.R.; Liu, T.M.-H.; Shinkai, I., J. Org. Chem., 1991, 56, 4929), был получен t-бутил-диметилсилиловый бутиламид ловастатина, выход которого составил 68% (неочищенный продукт), а определенный по методике жидкостной хроматографии высокого разрешения коэффициент подвижности Rf составил 12,87.
В. Восстановление t-бутилдиметилсилилового бутиламида ловастатина
Раствор t-бутилдиметилсилилового бутиламида ловастатина, взятого в количестве 1,65 г (2,34 ммоль) в 30 мл тетрагидрофурана (THF), был добавлен к 6 мл (2,5 эквивалента) 2 М раствора комплекса алюмогидрид лития - тетрагидрофуран (LiAlH4·2THF) в толуоле при температуре 0°С. Полученная реакционная смесь подвергалась перемешиванию в течение 2 часов, после чего к ней до прекращения газовыделения добавлялся влажный сернокислый натрий (Na2SO4·nH2O). Попытки профильтровать суспензию через стеклянную воронку (Р2) со слоем целита не увенчались успехом. Реакционная смесь была влита в разбавленный (<1 N) раствор соляной кислоты (HCl). Водный слой был удален с помощью диэтилового эфира. Органический слой был промыт рассолом, высушен с помощью сернокислого натрия (Na2SO4) и выпарен. Выход продукта составил 1,07 г (89%). Определенный по методике жидкостной хроматографии высокого разрешения коэффициент подвижности Rf составил 9,27.
С. Ацилирование t-бутилсилилзащищенного ловастатинбутиламидного спирта
К раствору полученного в качестве промежуточного продукта на стадии В ловастатинбутиламидного спирта, взятого в количестве 360 мг (0,58 ммоль) и триэтиламина, взятого в объеме 0,32 мл, в 10 мл толуола было добавлено 0,31 г (4 эквивалента) 2,2-диметилбутирилхлорида. Полученная реакционная смесь подвергалась нагреванию до рефлюкса в течение 10 часов (стандартная процедура). Анализ по методике жидкостной хроматографии высокого разрешения показал наличие смеси химических соединений, среди которых присутствовал и желаемый диацилированный продукт, коэффициент подвижности Rf которого составлял 15,81. Удаление защитных групп было осуществлено по способу, описанному в литературе (Askin, D.; Verhoeven, T.R.; Liu, T.M.-H.; Shinkai, I., J. Org. Chem., 1991, 56, 4929), в результате чего выход продукта составил 68% (неочищенный продукт), а определенный по методике жидкостной хроматографии высокого разрешения коэффициент подвижности Rf составил 12,87.
Пример 9
Получение диацетилбензилиденового производного ловастатина
А. Получение бензилиденового производного бутиламида ловастатина
Бутиламид ловастатина, взятый в количестве 4,77 г (10 ммоль), был растворен в 50 мл толуола. Затем к полученному раствору было добавлено 10,6 г (10 эквивалентов) бензальдегида и 500 мг p-TsOH, после чего полученная смесь подвергалась перемешиванию при комнатной температуре в течение 16 часов. Затем к реакционной смеси был добавлен насыщенный водный раствор двууглекислого натрия (NaHCO3), после чего было произведено разделение слоев. Затем толуоловый слой был промыт насыщенным водным раствором двууглекислого натрия (NaHCO3), насыщенным водным раствором соляной кислоты (HCl), высушен с помощью сернокислого натрия (Na2SO4) и выпарен. После этого остаток был подвергнут очистке с помощью хроматографии на колонке (SiO2) /n-гексан/этилацетат, в результате чего был получен конечный продукт, выход которого составил 2,6 г (46%).
В. Восстановление бензилиденового производного
Бензилиденовое производное соединение в количестве 2,6 г было растворено в 50 мл толуола, и полученный раствор был охлажден до температуры 0°С. Затем к охлажденному раствору по каплям было добавлено 11,5 мл 1 М раствора комплекса алюмогидрид лития - тетрагидрофуран (LiAlH4·2THF) в толуоле, при этом температура реакционной смеси поддерживалась на уровне 10°С. После этого полученный раствор подвергался перемешиванию при температуре 0-5°С. Затем к раствору было добавлено 1,8 мл 30%-ного водного раствора гидроокиси натрия (NaOH), и полученная смесь в течение 16 часов подвергалась перемешиванию при комнатной температуре. Затем смесь была профильтрована сквозь целит, промыта толуолом, взятым в объеме 50 мл, и подвергнута сгущению до объема приблизительно 50 мл.
С. Получение бензилиденового производного симвастатина
К реакционной смеси, полученной на стадии В, было добавлено 1,9 г (4,1 эквивалента) триэтиламина, 2,5 г (4 эквивалента) диметилмасляной кислоты и 50 мг диметиламинопиридина, после чего смесь подвергалась дефлегмированию в течение 16 часов. Затем смесь была влита в систему вода/этилацетат, и была подвергнута разделению на слои. После этого органический слой был промыт сначала водой, а затем насыщенным раствором хлористого натрия, после чего высушен с помощью сернокислого натрия и выпарен, в результате чего было получено 3,3 г неочищенного продукта. Дальнейшее превращение полученного продукта в симвастатин выполняется согласно процедурам, описанным в Примере 5, стадия С и стадия D, вторая часть.
Пример 10
Ловастатиновое восстановление ацетонида пирролидинамида ловастатина
К раствору ацетонида пирролидина бутиамида ловастатина, полученного по способу, описанному в Примере 3 путем реакции между ловастатином и пирролидином, взятого в количестве 1 г (1,94 ммоль) в 20 мл тетрагидрофурана при температуре 0°С было добавлено 40 мг (1,1 ммоль) алюмогидрида лития. Через 18 часов при комнатной температуре превращение составило 50%.
Пример 11
Восстановление бутиламида ловастатина
К суспензии 400 мг (10,5 ммоль) алюмогидрида лития (LiAlH4) в 50 мл тетрагидрофурана при температуре 0°С был добавлен раствор бутиламида ловастатина, взятого в количестве 2,25 г (5 ммоль), в тетрагидрофуране, взятом в объеме 25 мл. Полученная смесь была подвергнута перемешиванию при температуре окружающей среды в течение 16 часов, после чего к ней до прекращения газовыделения добавлялся влажный сернокислый натрий (Na2SO4·nH2O), аналог глауберовой слои. После этого к смеси был добавлен сухой сернокислый натрий (Na2SO4). Затем суспензия была профильтрована через стеклянный фильтр, а полученный фильтрат был выпарен при пониженном давлении до сухого состояния, в результате чего была получен неочищенный продукт в виде густой маслянистой жидкости коричневого цвета, выход которого составил 1,03 г (53%). Определенный по методике жидкостной хроматографии высокого разрешения коэффициент подвижности этого неочищенного продукта Rf составил 12,87 (у исходного материала - 5,79).
Пример 12
Выборочное ацилирующее действие на азот спирта ацетонида бутиламида ловастатина, после чего может быть осуществлено ацилирование гидроксильной (ОН) группы
К раствору спирта ацетонида бутиламида ловастатина, взятого в количестве 2,1 г (5 ммоль), и триэтиламида, взятого в количестве 0,8 мл (5,5 ммоль), в 50 мл толуола при температуре 0°С было добавлено 1,1 эквивалента (0,64 мл, 5,5 ммоль) бензоилхлорида. После этого реакционная смесь подвергалась перемешиванию при комнатной температуре в течение 16 часов. Образец, подвергнутый анализу по методике жидкостной хроматографии высокого разрешения, показал главные пиковые значения коэффициента подвижности Rf=6,16 (исходный материал) и Rf=9,13. Через 21 час появилось пиковое значение Rf=6,67. Анализ по методу ядерного магнитного резонанса показал небольшой пик NH и три других пика в районе 6,5-5 частей на миллион, указывающих на то, что амид ацилирован.
Пример 13
Реакция ловастатина с аммиаком
Суспензия 0,25 г (0,6 ммоль) ловастатина в 15 мл метилового спирта была охлаждена до температуры 5°С в бане лед/вода. Затем метиловый спирт был насыщен аммиаком (газом), после чего реакционная смесь подвергалась термической обработке при температуре 130°С в течение 40 часов в запаянной стеклянной трубке. Анализ по методике жидкостной хроматографии высокого разрешения показал, что полученная в результате этого реакционная смесь содержит 43% соответствующего деацилированного продукта.
Пример 14
Реакция ловастатина с n-бутиламидом
Раствор 0,5 г (1,2 ммоль) ловастатина в 15 мл n-бутиламида был подвергнут термической обработке при температуре 150°С в течение 40 часов в запаянном сосуде. Анализ по методике жидкостной хроматографии высокого разрешения показал, что полученная в результате этого реакционная смесь содержит 12,3% соответствующего деацилированного продукта. Структура деацилированного бутиламида была подтверждена образованием соответствующего ацетонида путем реакции с p-TsOH и ацетоном и сравнением этого ацетонида с другим образцом ацетонида, полученного, как описано в Примере 5, вторая часть стадии А.
Пример 15
Реакция ловастатина с n-гептиламином
Раствор 0,5 г (1,2 ммоль) ловастатина в 10 мл гептиламина подвергался дефлегмированию в течение 70 часов. Анализ по методике жидкостной хроматографии высокого разрешения показал, что полученная в результате этого реакционная смесь содержит 17% соответствующего деацилированного продукта.
Пример 16
Все три деацилированных химических соединения, полученных в Примерах 13, 14 и 15, были превращены в соответствующие ацетониды (путем замыкания кольца) при добавлении к каждому из них 400 мл ацетона и 5 г p-TsOH. Полученные смеси подвергались перемешивании при комнатной температуре в течение 1 часа, после чего охлаждались в ледяной воде в течение 2 часов. Полученное твердое вещество было собрано путем всасывания и высушено.
Пример 17
Три ацетонида, полученных в Примере 16, были каждое по отдельности превращены симвастатин путем ацилирования и с помощью реакций превращения аммониевых солей, как описано в Примере 5, стадии С и D.
Пример 18
Рекристаллизация симвастатина из системы толуол/n-гексан
Неочищенный симвастатин в количестве 35 г был растворен в 140 мл толуола при перемешивании при температуре 60°С. Затем постепенно добавлялся n-гексан при постоянном перемешивании и постепенном понижении температуры до 0-5°С. После перемешивания в течении 1 часа при вышеуказанной температуре был собран осадок, который затем был промыт смесью толуола и n-гексана (в соотношении 1:4 по объему) и высушен, в результате чего был получен рекристаллизированный продукт в количестве 33 г.
Пример 19
Рекристаллизация симвастатина из системы метиловый спирт/вода
Рекристаллизированный симвастатин в количестве 33 г был растворен в 300 мл метилового спирта при комнатной температуре, после чего полученный раствор был подвергнут обработке активированным древесным углем. Затем отработанный древесный уголь был удален фильтрованием, а продукт был осажден путем добавления 450 мл воды. Суспензия была охлаждена до температуры 5-10°С, и продукт был собран, промыт смесью метилового спирта и воды (в соотношении 1:2 по объему) и высушен, в результате чего был получен рекристаллизированный продукт в количестве 31 г.
Изобретение относится к фармацевтике. Получают симвастатин высокой степени очистки из ловастатина, осуществляя следующие стадии: а) размыкание лактонового кольца при введении ловастатина в реакцию с амином для образования амида, b) защита 1,3-диоловой части с помощью защитной группы, с) удаление 2-метилбутириловой группы, присоединенной с помощью сложной эфирной связи через посредство кислорода в позиции 8 гексагидронафталинового кольца, d) присоединение 2,2-диметилбутиратной группы путем образования сложной эфирной связи к гидроксилу в позиции 8, е) удаление защитной группы, f) превращение амида в кислую соль, g) замыкание лактонового кольца с образованием симвастатина. Получают полусинтетический статин из статина, осуществляя следующие стадии: а) размыкание лактонового кольца путем приведения статина в реакцию с амином для получения амида, b) защита 1,3-диоловой части с помощью защитной группы, с) удаление группы R1, присоединенной с помощью сложной эфирной связи через посредство кислорода в позиции 8 гексагидронафталинового кольца, d) присоединение группы R2 путем образования сложной эфирной связи к гидроксилу в позиции 8, е) удаление защитной группы, f) превращение амида в кислую соль и g) замыкание лактонового кольца с образованием полусинтетического статина. Изобретение позволяет повысить чистоту продукта. 2 н. и 15 з.п. ф-лы.
a) размыкание лактонового кольца при введении ловастатина в реакцию с амином для образования амида,
b) защита 1,3-диоловой части с помощью защитной группы,
c) удаление 2-метилбутириловой группы, присоединенной с помощью сложной эфирной связи через посредство кислорода в позиции 8 гексагидронафталинового кольца,
d) присоединение 2,2-диметилбутиратной группы путем образования сложной эфирной связи к гидроксилу в позиции 8,
e) удаление защитной группы,
f) превращение амида в кислую соль,
g) замыкание лактонового кольца с образованием симвастатина.
из статина, имеющего следующую молекулярную структурную формулу II
содержащий следующие стадии:
a) размыкание лактонового кольца путем приведения статина, имеющего структурную молекулярную формулу II, в реакцию с амином для получения амида,
b) защита 1,3-диоловой части с помощью защитной группы,
c) удаление группы R1, присоединенной с помощью сложной эфирной связи через посредство кислорода в позиции 8 гексагидронафталинового кольца,
d) присоединение группы R2 путем образования сложной эфирной связи к гидроксилу в позиции 8,
e) удаление защитной группы,
f) превращение амида в кислую соль, и
g) замыкание лактонового кольца с образованием полусинтетического статина, имеющего структурную молекулярную формулу I,
где R1 и R2 - ацильные группы, присоединенные к кислороду посредством сложной эфирной связи;
R3 и R4 - группы, независимо выбранные из следующего перечня: -Н, -ОН, -С1-10алкил, -С1-10арил, С6-14арил-С1-3.
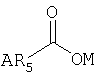
где ОМ - кислород, который является заместителем гексагидронафталинового кольца в позиции 8;
R5 - группа, выбранная из следующего перечня: С1-15алкил, -С3-15циклоалкил, -С2-15алкенил, -C2-15алкинил, -фенил, -фенилС1-6алкил;
A - заместитель R5, выбранный из следующего перечня: водород, галоген, C1-6алкил, C1-6алкокси, С6-14арил.
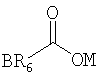
где ОМ - кислород, который является заместителем гексагидронафталинового кольца в позиции 8;
R6 - группа, выбранная из следующего перечня: С1-15алкил, -С3-15циклоалкил, -C2-15алкенил, -C2-15алкинил, -фенил, -фенилС1-6алкил;
В - заместитель R6, выбранный из следующего перечня, водород, галоген, C1-6алкил, C1-6алкокси, С6-14арил.
| US 5159104 А, 27.10.1992 | |||
| US 4444784 A, 24.04.1984 | |||
| US 4582915 А, 15.04.1986. |

