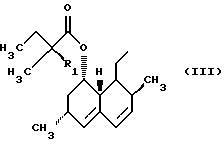
Ловастатин (lovastatin) и его аналоги, например симвастатин (simvastatin), являются сильнодействующими антигиперхолестеринемическими агентами, которые действуют, ограничивая биосинтез холестерина посредством ингибирования фермента HMG-СоА-редуктазы (гипероксиметилглютарил-СоА-редуктазы). Известно, что эти соединения, которые в общем можно обозначить как статины, существуют в форме оксикислот с открытым циклом, а также в форме лактонов. Лактонные формы и формы оксикислот этих соединений имеют следующие общие структурные формулы:

где Z представляет водород, катион металла, такого как натрий или калий, или NH4, и R представляет
где R1 представляет Н или СН3.
Открытая оксикислотная форма статинов (формула II) является биологически активной. Однако обычно статины вводят пациентам в лактонной форме (формула I), которая превращается в организме в активный метаболит - оксикислотную форму.
В способе производства ловастатина и его аналогов, например симвастатина, лактонизация свободной оксикислоты или ее соли в лактонную форму представляет собой существенную стадию.
Известные в литературе способы лактонизации свободной оксикислоты или ее солей проводят либо в условиях жесткого нагревания, а именно при кипячении с инертным растворителем, либо при катализе сильными кислотами, если лактонизацию проводят при комнатной температуре. Способ, раскрытый в патенте США 4820850, включает нагревание способной кислоты или ее соли, например соли аммония, при температуре кипения (обычно 100-110oС) в высококипящих углеводородных растворителях, таких как толуол, в течение 7-8 часов. Считают, что при таких высоких температурах кислотность самой кислоты ответственна за реакцию лактонизации. Кроме того, воду, образующуюся в качестве побочного продукта этой реакции, непрерывно удаляют путем азеотропной перегонки, что позволяет реакции пройти до конца. Способ лактонизации в условиях нагревания при температурах кипения осложняется образованием многих примесей, среди которых образование димера особенно снижает качество конечного лактонного продукта. Димер является трудно удаляемой примесью и присутствует в продукте в количестве от 0,4 до 0,8%. Для того чтобы свести к минимуму димерную примесь, в реакции лактонизации часто используют значительные разбавления за счет эффективности данной реакции и способа, что невыгодно при производстве в промышленном масштабе.
Патент США 4916239 раскрывает другой способ, где реакцию лактонизации проводят при комнатной температуре обработкой аммонийной соли оксикислоты (мевиновой (mevinic) кислоты) в смеси уксусной кислоты и воды и в присутствии сильной кислоты в качестве катализатора. После установления равновесия оксикислота - лактон (реакция протекает до 50% степени конверсии) постепенно добавляют воду, чтобы вызвать кристаллизацию лактонной формы из реакционной среды. Непрерывное удаление лактона сдвигает равновесие в сторону образования лактона, приводя, таким образом, к протеканию реакции до конца. Этот способ имеет некоторые недостатки и также неудобен для проведения в большом масштабе по многим причинам, некоторые из которых обсуждаются ниже.
Использование в качестве катализатора сильной минеральной или органической кислоты, например муравьиной, фосфорной, трифторуксусной, серной, соляной, пара-толуолсульфоновой, метансульфоновой кислот и других, в количестве от 1,2 до 1,5 мольных эквивалентов делает этот способ опасным и непригодным, с точки зрения воздействия на окружающую среду, в промышленном масштабе. Избыток используемого кислотного катализатора необходимо нейтрализовать добавлением сильного основания перед фильтрованием продукта.
Кроме того, реакция лактонизации после достижения равновесия проходит только на 50%. В этот момент любое быстрое или преждевременное добавление воды может привести к серьезным проблемам, связанным с кристаллизацией и фильтрованием. Кроме того, реакция и последующая обработка до завершения занимает примерно 9-12 часов, вследствие чего снижается эффективность процесса.
Указанные выше недостатки делают способ, раскрытый в патенте США 4916239, трудоемким, неэффективным, дорогим и опасным, с точки зрения воздействия на окружающую среду, в промышленном масштабе.
Краткое и подробное описание изобретения
Целью данного изобретения является обеспечение эффективного способа лактонизации статинов, в котором не используют сильных коррозионных кислот и жестких условий нагревания и который дает лактонизованный продукт высокой чистоты и с высоким выходом.
В настоящем изобретении предложен новый способ превращения ингибиторов HMG-CoA-редуктазы, например открытых оксикислотных форм ловастатина, симвастатина и их аналогов, в их лактонные формы, который удобен для проведения в промышленном масштабе. Он позволяет проводить реакцию лактонизации при умеренных температурах без использования небезопасных для применения в промышленности сильных кислот.
Конкретно, способ данного изобретения включает обработку открытых оксикислотных форм статинов, предпочтительно в виде их солей, наиболее предпочтительно в виде их аммонийных солей, уксусной кислотой в отсутствие катализатора - сильной кислоты в инертных безводных условиях и при температурах от комнатной до умеренного нагревания. (Обычно сильной считают кислоту, имеющую рКа<0). Уксусная кислота с мягкими кислотными свойствами (рКа=4,8) служит и в качестве растворителя, и в качестве катализатора, каталитические свойства являются результатом собственной кислотности среды. Реакция лактонизации проходит без добавления сильных кислот в качестве катализатора (как в прототипе), протекает полностью и быстро, предоставляя меньше возможностей для образования примесей. Продукт лактонизации, обычно хорошо растворяющийся в уксусной кислоте, после завершения реакции выделяют в чистом виде, добавляя антирастворитель, который обладает способностью кристаллизовать лактонизованный продукт. Антирастворитель выбирают из воды, гексана, гептана, циклогексана и др., предпочтительны вода, гексан или циклогексан, наиболее предпочтительна вода.
Так как лактонизация является равновесной реакцией, необходимо применять некоторые устройства для удаления побочных продуктов реакции (воды и аммиака) и сдвига равновесия в сторону образования лактона. В выбранных для настоящего процесса условиях побочный продукт реакции аммиак поглощается in situ с уксусной кислотой, которая присутствует в реакционной среде в избытке, с образованием ацетата аммония. Последний, будучи гигроскопичным по своей природе, имеет тенденцию абсорбировать воду, которая также образуется в качестве побочного продукта реакции лактонизации. Таким образом, обеспечен механизм, который позволяет in situ удалять аммиак и воду, что ведет к завершению реакции.
Реакцию лактонизации настоящего изобретения можно представить следующим образом: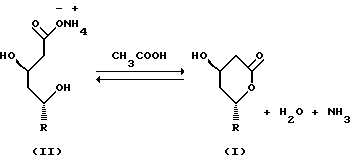

где R является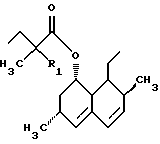
где R1=H или СН3.
Скорость лактонизации очень высока и позволяет завершить реакцию за очень короткий период и получить лактонный продукт с высоким выходом и высокой чистотой. Обычно реакцию проводят при температуре от комнатной до примерно 55oС, предпочтительно примерно 25-45oС, наиболее предпочтительно примерно 35-40oС. Количество уксусной кислоты составляет, по крайней мере, 1 объемную часть на часть исходной соли. Можно использовать большие количества растворителя, обычно до 20 объемных частей. Предпочтительным является количество растворителя в диапазоне примерно от 3 до 7 объемных частей. Как правило, реакцию завершают примерно за 3-5 часов. Однако временной интервал сильно зависит от таких факторов, как общий объем раствора, температура реакции и включенный субстрат.
Основными преимуществами настоящего изобретения по сравнению с известными являются ценовая эффективность, меньшая трудоемкость, высокий выход, повышенная производительность процесса, чистые и благоприятные, с точки зрения окружающей среды, операции.
Уровень образующихся примесей, особенно исходной кислоты и димера, сильно снижен по сравнению с ранее указанными способами. Продукт, который образуется из гомогенной суспензии, делает очень легкой операцию фильтрования в большом масштабе, а обработка не включает стадии нейтрализации перед фильтрованием.
Следующие далее конкретные примеры иллюстрируют способ данного изобретения, но не предназначены для какого-либо ограничения объема данного изобретения.
Пример 1. Получение 6(R)-[2-[8(S)-(2-метилбутирилокси)-2(S), 6(R)-диметил-1,2,6,7,8,8а(R)-гексагидронафтил-1(S)] -этил] -4(R)-гидрокси-3,4,5,6-тетрагидро-2Н-пиран-2-она (формула I, R1=H).
Аммоний-7-[1,2,6,7,8,8a(R)-гексагидро-2(S), 6(R)-диметил-8(3)-(2-метилбутирилокси)-1(S)-нафтил]-3(R), 5(R)-дигидроксигептаноат (формула II, R1= H) (4,5 г, 0,0102 моля) и бутилированный гидрокситолуол (15 мг) растворяют в ледяной уксусной кислоте (27 мл) и перемешивают в атмосфере азота в течение 5 часов при 35-38oС. Через 5 часов, когда реакция почти закончена, добавляют по каплям воду (64 мл) в течение 30 минут при 30-32oС. Затем кристаллизовавшийся продукт перемешивает при этой температуре в течение 30 минут и потом охлаждают до 15oС. Продукт фильтруют и промывают водой (15 мл х 3) для удаления следов уксусной кислоты. Сушка в вакууме при 40-42oС дает указанное в заголовке соединение в виде белого кристаллического вещества (3,82 г) с 92% выходом и чистотой около 98% (определенной способом высокоэффективной жидкостной хроматографии).
Пример 2. Получение 6(R)-[2-[8(S)-[2,2-диметилбутирилокси)-2(S), 6(R)-диметил-1,2,6,7,8,8а(R)-гексагидронафтил-1(S)] -этил] -4(R)-гидрокси-3,4,5,6-тетрагидро-2Н-пиран-2-она (формула I, R1=СН3).
Аммоний-7-[1,2,6,7,8,8а(R)-гексагидро-2(S), 6(R)-диметил-8(S)-(2,2-диметилбутирилокси)-1(3)-нафтил] -3(R), 5(R)-дигидроксигептаноат (формула II, R1= СН3) (10 г, 0,022 моля, 97% чистоты, определенной способом высокоэффективной жидкостной хроматографии) и бутилированный гидрокситолуол (30 мг) растворяют в ледяной уксусной кислоте (40 мл) и перемешивают в атмосфере азота при 35-38oС. Через 6 часов к реакционной смеси добавляют воду (160 мл) в течение 30 минут. Кристаллизовавшийся лактонизованный продукт перемешивают при 30-32oС еще в течение 60 минут. Фильтрование, промывание водой (25 мл х 3) и финальная сушка в вакууме при 40-42oС дают указанное в заголовке соединение в виде белого кристаллического вещества (9,1 г) с чистотой >95% и выходом >98%.
Пример 3. Получение 6(R)-[2[8(S)-(2,2-диметилбутирилокси)-2(S), 6(R)-диметил-1,2,6,7,8,8а(R)-гексагидронафтил-1(S)] -этил] -4(R)-гидрокси-3,4,5,6-тетрагидро-2Н-пиран-2-она (формула I, R1=СН3).
Аммоний-7-[1,2,6,7,8,8a(R)-гексагидро-2(S), 6(R)-диметил-8(S)-(2,2-диметилбутирилокси)-1(S)-нафтил] -3(R), 5(R)-дигидроксигептаноат (формула II, R1 = СН3) (2 г, 0,004 моля, 95% чистоты, определенной способом высокоэффективной жидкостной хроматографии) и бутилированный гидрокситолуол (6 мг) растворяют в ледяной уксусной кислоте (6 мл). Смесь перемешивают в атмосфере азота при 35-40oС в течение 5 часов и затем медленно добавляют гексан (130 мл) для кристаллизации продукта. Реакционную смесь перемешивают еще в течение 60 минут при 30-32oС. Фильтрование, промывание гексаном (5 мл х 2) и сушка в вакууме при 40-42oС дают указанное в заголовке соединение в кристаллическом виде (1,75 г, выход 95%) с чистотой >95%.
Пример 4. Получение 6(R)-[2-[8(S)-(2,2-диметилбутирилокси)-2(S), 6(R)-диметил-1,2,6,7,8,8a(R)-гексагидронафтил-1(S)] -этил]-4(R)-гидрокси-3, 4, 5, 6-тетрагидро-2Н-пиран-2-она (формула I, R1=СН3).
Применяя методику, аналогичную описанной в Примере 3, но используя в качестве антирастворителя циклогексан, получают указанное в заголовке соединение в кристаллическом виде с выходом около 87% и чистотой >97%.
Пример 5. Получение 6(R)-[2-[8(S)-(2-метилбутирилокси)-2(S), 6(R)-диметил-1,2,6,7,8,8a(R)-гексагидронафтил-1(S)] -этил] -4(R)-гидрокси-3,4,5,6-тетрагидро-2Н-пиран-2-она (формула I, R1=H).
Применяя методику, аналогичную описанной в Примере 1, но используя в качестве антирастворителя циклогексан, получают указанное в заголовке соединение в кристаллическом виде с выходом 85% и чистотой >98%.
Изобретение относится к усовершенствованному способу лактонизации при получении статинов, например ингибиторов HMG-СоА-редуктазы ловастатина и симвастатина, в частности, относится к способу получения соединения формулы (I), где R представляет радикал формулы (III), где R1 представляет Н или СН3, включающий (а) обработку соединения формулы (II), где Z представляет водород, катион металла или NH4, и R является таким, как определено выше, слабой органической кислотой в отсутствие сильной кислоты как катализатора и воды при температуре ниже 55oС, и (b) добавление антирастворителя, что вызывает осаждение соединения формулы (I) в виде кристаллического продукта. Способ, в котором не используют сильных коррозионных кислот и жестких условий нагревания, позволяет получить целевой продукт высокой чистоты более 98% и с высоким выходом более 98%. 8 з.п. ф-лы.
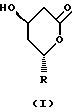


где R представляет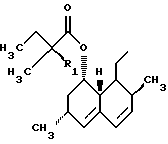
где R1 представляет Н или СН3,
включающий (а) обработку соединения формулы (II)
где Z представляет водород, катион металла или NH4;
R является таким, как определено выше,
слабой органической кислотой в отсутствие сильной кислоты как катализатора и воды при температуре ниже 55oС и (b) добавление антирастворителя, что вызывает осаждение соединения формулы (I) в виде кристаллического продукта.
| Способ гидроочистки асфальтенсодержащего углеводородного сырья | 1972 |
|
SU511867A3 |
| US 4820850 А, 11.04.1989 | |||
| US 4916239 А, 10.04.1990 | |||
| US 5393893 А, 28.02.1995. | |||

