ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к способам очистки соединений из ферментационного бульона. В частности, данное изобретение относится к выделению статинов из ферментационного бульона в кристаллической форме.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Осложнения сердечно-сосудистых заболеваний, как например, инфаркт миокарда, внезапные приступы и периферические сосудистые заболевания, составляют половину смертей в Соединенных Штатах. Высокий уровень липопротеинов низкой плотности (low density lipoprotein, LDL) в токе крови приводит к образованию коронарных повреждений, которые препятствуют течению крови и могут разрываться и вызывать тромбоз (Goodman. Gilman. The Pharmacological Basis of Therapeutics 879 (Joel G.Hardman et al., eds. 9th ed. 19%)). Было показано, что понижение уровней LDL в плазме уменьшает риск клинических случаев у больных с сердечно-сосудистыми заболеваниями и у больных, которые не имеют сердечно-сосудистых заболеваний, но у которых гиперхолестеринемия (Scandinavian Simvastatin Survival Study Groups, 1994; Lipid Research Clinics Program, 1984a, 1984b).
Препараты статинов являются в настоящее время наиболее терапевтически эффективными лекарственными препаратами, пригодными для уменьшения уровня LDL в токе крови больных при повышенном риске сердечно-сосудистых заболеваний. Этот класс лекарственных препаратов включает inter alia, компактин, ловастатин, симвастагин, правастатин и флувастатин. Механизм действия препаратов статина объяснен в некоторых деталях. Они нарушают синтез холестерина и других стеринов в печени при конкурентном ингибировании фермента 3-гидрокси-3-метил-глутарил-коэнзима А редуктазы (3-hydroxy-3-methyl-glutaryl-coenzyme A reductase, "HMG-CoA редуктаза"). HMG-CoA редуктаза катализирует превращение HMG-CoA в мевалонат, что является скоростьопределяющей стадией в биосинтезе холестерина. Следовательно, это ингибирование приводит к уменьшению скорости образования холестерина в печени.
Компактин является общим медицинским названием химического соединения 1,2,3,7,8,8а-гексагидро-7-метил-8-[2-(тетрагидро-4-гидрокси-6-оксо-2Н-пиран-2-ил)-этил-1-нафталенилового эфира 2-метилбутановой кислоты с формулой

Было показано, что компактин, также называемый мевастатином является первым препаратом статина - ингибитором HMG-CoA редуктазы. Компактин был очищен от Penicillium citrinum и Penicillium adametzioides (см. патенты США №№3983140, 4049495 и 5691173, которые приведены в данном изобретении в качестве ссылки).
Ловастатин является общим медицинским названием химического соединения 1,2,3,7,8,8а-гексагидро-3,7-диметил-8-[2-(тетрагидро-4-гидрокси-6-оксо-2Н-пиран-2-ил)-этил]-1-нафталенилового эфира 2-метилбутановой кислоты с формулой
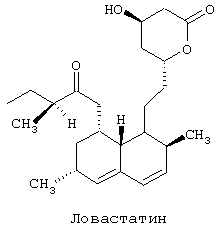
Ловастатин, также называемый мевинолином, отличается от компактина только наличием метильной группы и может выделяться из Aspergillus terreus (см. патенты США №№4294926, 4420491, 4319039 и 4294896, которые приведены в данном изобретении в качестве ссылки). Ловастатин также был выделен из некоторых других микроорганизмов (см. британский патент № 2046737, немецкий патент №4402591, канадский патент №2129416 и венгерский патент № 208997).
Компактин и ловастатин, а также другие статины существуют в виде оксикислот с открытым кольцом и в виде лактона, как показано:

Равновесие между лактоном и оксикислотой делает очистку затруднительной, поскольку формы свободной кислоты и лактона соединений статина имеют различные полярности. Способ очистки одной формы, очевидно, заключается в удалении другой формы, что тем самым уменьшает общий выход. Следовательно, большую осторожность обычно необходимо проявлять, когда очищают соединения статина, чтобы выделить их с большим выходом.
Патент США №5202029 относится к способу очистки лактонной формы соединений статина с помощью ВЭЖХ (HPLC). Неочищенный ферментационный бульон (например, ловастатин, симвастатин, правастатин, флувастатин и мевастатин) растворяют в органическом растворителе и элюируют через ВЭЖХ-колонку (HPLC-колонку). Статин элюируют из колонки в виде растворенного в элюенте вещества. Элюент частично упаривают и впоследствии прибавляют воду, чтобы вызвать кристаллизацию. Основной недостаток производства в промышленном масштабе заключается в большой стоимости хроматографических колонок.
Патент США №5616595 относится к непрерывному способу выделения широкого ряда нерастворимых в воде соединений из ферментационного бульона с помощью тангенциальной фильтрации. Способ может применяться к лактонной форме ловастатина. правастатина и симвастатина. Способ включает периодическое пропускание ферментационного бульона через фильтр, который удерживает нерастворимое соединение. Соединение растворяют в растворителе и раствор фильтруют. Раствор нужного соединения собирают в виде фильтрата и нужное соединение затем можно подвергнуть дополнительной очистке. Из-за нерастворимости соединений в воде способ требует растворителя для растворения и требуются многочисленные фильтрационные мембраны. Повторная фильтрация делает способ весьма дорогостоящим для крупномасштабного производства.
Способ выделения ловастатина в форме лактона описан в патенте США №5712130. В указанном способе ловастатин экстрагируют из ферментационного бульона бутилацетатом. Полученный раствор далее центрифугируют и водную фазу отбрасывают. Органическую фазу перегоняют в вакууме при 40°С, что помимо концеитрирования раствора способствует образованию лактона при удалении воды. Кристаллы лактона ловастатина образуются при охлаждении и их перекристаллизуют до степени чистоты в 90% или выше. Однако до применения в качестве лекарственного препарата кристаллизованный ловастатин должен дополнительно очищаться, что уменьшит общий выход и добавит дополнительные расходы для осуществления способа.
ЦЕЛИ И СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Известные способы выделения статина из ферментационного бульона не достигают фармацевтически приемлемого уровня чистоты с помощью способа, экономичного в промышленном масштабе, или требуют хроматографического разделения для достижения высокой чистоты. Цель данного изобретения состоит в разработке простого, быстрого способа с высоким выходом выделения статинов из ферментационного бульона при фармацевтически приемлемом уровне чистоты (т.е. не менее 98,5%).
Другой целью данного изобретения является способ выделения соединения статина из ферментационного бульона, где соединение статина содержит карбоновую кислоту. способную образовать лактон и конденсированное бициклическое кольцо. Способ включает стадии: экстракция соединения статина из ферментационного бульона при обработке его экстракционным растворителем и экстракция соединения статина из ферментационного бульона в экстракционный растворитель, где экстракционный растворитель является гидрофобным органическим экстракционным растворителем; отделение гидрофобного органического экстракционного растворителя от ферментационного бульона; концентрирование раствора гидрофобного органического экстракционного растворителя, содержащего экстрагированное соединение статина; и очистка экстрагированного соединения статина кристаллизацией.
Другой целью данного изобретения является способ выделения соединения статина из ферментационного бульона, где соединение статина содержит карбоновую кислоту, способную образовать лактон и конденсированное бициклическое кольцо, способ включает стадии:
(а) предварительную обработку ферментационного бульона в щелочных условиях для удаления неполярных примесей;
(б) экстракцию соединения статина из ферментационного бульона в гидрофобный органический экстракционный растворитель;
(в) отделение гидрофобного органического экстракционного растворителя из ферментационного бульона;
(г) концентрирование раствора гидрофобного органического экстракционного растворителя, содержащего экстрагированное соединение статина;
(д) промывание концентрированного раствора гидрофобного органического экстракционного растворителя водным раствором, содержащим основание для очистки лактона; и
(е) очистку экстрагированного соединения статина кристаллизацией.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Данное изобретение относится к процессу получения высокоочищенных кристаллов соединений статина из ферментационного бульона, как проиллюстрировано при очистке компактна и ловастатина. Однако специалистам понятно, что способ, описанный в данном изобретении, можно использовать для очистки других соединений, которые получают микробиологическим или ферментативным способом и которые содержат лактон, как например, симвастатин, правастатин и флувастатин. Специалистам также понятно, что оптимальные условия очистки соединения статина могут изменяться в зависимости от данного выделяемого соединения статина, ферментационного бульона и микроорганизма, продуцирующего соединение.
Предпочтительный вариант данного изобретения включает стадии:
(I) предварительную обработку ферментационного бульона в щелочных условиях для удаления неполярных примесей;
(II) экстракцию статина в кислых условиях в виде оксикислоты или лактона из ферментационного бульона в гидрофобный органический экстракционный растворитель;
(III) отделение гидрофобного органического экстракционного растворителя от ферментационного бульона;
(IV) концентрирование гидрофобного органического экстракционного растворителя, содержащего экстрагированное соединение статина, с образованием лактона; и
(V) очистку экстрагированного соединения статина кристаллизацией.
В другом варианте изобретения стадию предварительной обработки ферментационного бульона в щелочных условиях опускают.
В другом варианте до кристаллизации концентрированный гидрофобный органический растворитель промывают водным раствором, содержащим основание, для увеличения выхода. Предпочтительные щелочные растворы содержат гидроокись аммония (NH4OH, рН от 7,5 до 10) или 1-5% (вес./вес.) раствор бикарбоната натрия (NaHCO3) или карбоната натрия (Na2СО3).
Предварительная обработка ферментационного бульона в щелочных условиях
Предварительная обработка ферментационного бульона включает обработку ферментационного бульона в щелочных условиях с последующей экстракцией в гидрофобный органический экстракционный растворитель или смесь растворителей. Щелочные условия стадии предварительной обработки гидролизуют лактон статина в соответствующую оксикислоту статина (Andrew Streitweiser, Jr. Clayton Heathcock, Introduction to Organic Chemistry. 858-60 MacMillan Publishing Co., 2d Ed. 1981). Оксикислоту статина удерживают в водном ферментационном бульоне, в то время как жирные и маслянистые вещества, а также другие неполярные органические примеси из ферментационного бульона распределяются в гидрофобном органическом экстракционном растворителе или смеси растворителей предварительной обработки. Таким образом, предварительная обработка ферментационного бульона в щелочных условиях приводит к полной очистке оксикислоты статина путем отделения ее or примесей.
Удаление неполярных примесей улучшает общий выход кристаллизации, поскольку полагают, что присутствие неполярных примесей в ферментационном бульоне уменьшает общий выход при кристаллизации. Таким образом. предварительная обработка ферментационного бульона в щелочных условиях удаляет неполярные примеси из ферментационного бульона, тем самым существенно увеличивает общий выход на стадии кристаллизации.
Во время предварительной обработки рН ферментационного бульона или смеси ферментационного бульона и гидрофобного органического экстракционного растворителя доводят до щелочного рН. Предпочтительно, когда рН доводят прибавлением неорганического основания или органического амина. Предпочтительные неорганические основания или органические амины включают NaOH, КОН, LiOH, Ca(OH)2, NH4OH и триэтиламин. Наиболее предпочтительным основанием является NaOH, рН - от 7,0 до 13,9. Предпочтительно, когда рН для компактина и ловастатина регулируют в пределах от 8,5 до 10,0. Наиболее предпочтительно, когда рН для компактина и ловастатина составляет от 9,0 до 9,6,
Предварительное инкубирование в щелочных условиях может осуществляться при температурах от 15°С до 100°С. Температуры 15-20°С приводят к пониженному общему выходу очищенного статина.
При 80-100°С предпочтительные условия предварительного инкубирования - 15 минут при рН от 12,0 до 13,9. При 55-65°С предпочтительные условия предварительного инкубирования - 2 часа при рН от 9,0 до 9,6. При 15-25°С предпочтительные условия предварительного инкубирования - 48 часов при рН от 9,0 до 9,6.
Гидрофобные органические экстракционные растворители предварительной обработки включают, но не ограничиваются, изобутилацетатом, н-бутилацетатом, трет-бутилацетатом, этилацетатом, пропилацетатом, этилформиатом, бутилметилкетоном, дихлорметаном, хлороформом, четыреххлористым углеродом, дихлорэтаном, толуолом, ацетонитрилом, метилформиатом, метанолом, этанолом, изопропанолом, н-пропанолом, н-бутанолом, изобутанолом, трет-бутанолом, амиловым спиртом и бензиловым спиртом.
Или же гидрофобный органический экстракционный растворитель предварительной обработки может включать смесь любых упомянутых выше растворителей. Органические растворители для щелочной экстракции включают, но не ограничиваются, изобутилацетатом, н-бутилацетатом, трет-бутилацетатом, этилацетатом, пропилацетатом, этилформиатом, бутилметилкетоном, дихлорметаном, хлороформом, четыреххлористым углеродом, дихлорэтаном, толуолом и бензиловым спиртом. Предпочтительным гидрофобным органическим экстракционным растворителем предварительной обработки является изобутилацетат, этилацетат и толуол. Наиболее предпочтительным гидрофобным органическим экстракционным растворителем является изобутилацетат. Спирты работают (оправдывают себя), но не предпочтительны из-за регенерации растворителя. Тем не менее спирты присутствуют в случае ацетатов при щелочном рН. Спирты помогают раскрытию лактонового кольца в щелочных условиях. Предварительную обработку также можно проводить без экстракционных растворителей.
В данном предпочтительном варианте после предварительной обработки гидрофобный органический экстракционный растворитель контактирует с ферментационным бульоном в щелочных условиях до тех пор, пока жирные и маслянистые вещества, а также другие неполярные органические примеси практически не будут извлечены из ферментационного бульона. Для оценки извлечения неполярных примесей из ферментационного бульона может использоваться тонкослойная хроматография или любой другой способ, включая субъективное мнение. Для оптимального удаления примесей могут проводиться многократные экстракции. Однако высокоэффективны одна-две экстракции, когда применяют изобутилацетат для щелочной экстракции. Предпочтительно, когда щелочную экстракцию проводят объемом растворителя, который составляет от 20% до 50% (об./об.) от объема ферментации.
рН регулирование до экстракции ферментационного бульона в кислых условиях
Предпочтительно, когда рН очищенного ферментационного бульона доводят до рН 1,0-6,4 сильной кислотой до экстракции соединения статина в гидрофобный органический экстракционный растворитель. Предпочтительный интервал рН для ловастатина и компактина составляет от 2,0 до 4,5. Предпочтительный интервал рН для правастатина составляет от 4,5 до 6,0. Предпочтительными кислотами для регулирования рН являются серная и фосфорная кислоты. Или же экстракцию соединения статина можно проводить при доведении рН гидрофобного экстракционного растворителя до интервала 1,0-6,4.
Экстракция соединения статина в кислых условиях в виде оксикислоты или лактона
Гидрофобный органический экстракционный растворитель контактирует с очищенным ферментационным бульоном в кислых условиях до тех пор, пока оксикислота и лактон практически полностью будут извлечены из ферментационного бульона. Для оценки извлечения лактона в гидрофобный органический экстракционный растворитель может использоваться тонкослойная хроматография или любой другой способ, включая субъективное мнение. Для оптимального извлечения могут проводиться многократные экстракции. Однако высокоэффективны две-три экстракции, когда гидрофобным органическим экстракционным растворителем является изобутилацетат и рН находится в оптимальном диапазоне, как изложено выше. Предпочтительно, когда экстракцию проводят объемом органического экстракционного растворителя, который в два раза меньше объема ферментационного бульона.
Наиболее предпочтительным гидрофобным органическим экстракционным растворителем для экстракции компактина является изобутилацетат. Другие пригодные гидрофобные органические экстракционные растворители включают, но не ограничиваются, н-бутилацетатом, трет-бутилацетатом, этилацетатом, пропилацетатом, этилформиатом, бутилметилкетоном, дихлорметаном, хлороформом, четыреххлористым углеродом, дихлорэтаном и толуолом. Предпочтительными экстракционными растворителями являются изобутилацетат, н-бутилацетат, этилацетат и бутилметилкетон. При кислотной экстракции смеси растворителей не применяются.
Разделение фаз ферментационного бульона и гидрофобного органического экстракционного растворителя
Ферментационный бульон может отделяться от гидрофобного органического экстракционного растворителя известными способами. Предпочтительные способы разделения включают противоточную экстракцию. Аппараты для декантации представляют собой известное для этого оборудование. После разделения фаз чистоту растворителя можно улучшить промыванием водой.
Концентрирование гидрофобного органического экстракционного растворителя
Объем гидрофобного органического экстракционного растворителя после разделения фаз уменьшают. Уменьшение объема может достигаться упариванием при пониженном давлении при температуре от 30 до 90°С. Предпочтительно, когда упаривание проводят при пониженном давлении и температуре от 40 до 70°С. Или же упаривание гидрофобного растворителя проводят при повышенной температуре (ниже 90°С) при пониженном или при атмосферном давлении.
Очистка экстрагированного соединения статина кристаллизацией
Гидрофобный органический экстракционный растворитель упаривают до тех пор, пока концентрация соединения статина при кристаллизации не достигнет 50-250 г/л. Ловастатин оптимально кристаллизуют при концентрации 80-100 г/л, компактин оптимально кристаллизуют при концентрации 130-170 г/л, а правастатин оптимально кристаллизуют при концентрации 80-120 г/л.
Кристаллизацию можно проводить при комнатной температуре в течение ночи. Предпочтительным температурным интервалом является (-10)-5°С. Кристаллизацию наиболее предпочтительно проводить при -10°С в течение 20 часов.
Кристаллизацию можно проводить в любом из следующих растворителей или комбинации следующих растворителей: этанол, изопропанол, н-пропанол, изобутанол, н-бутанол, третбутанол, этилацетат, ацетон, метанол, ацетонитрил, этилформиат, изобутилацетат, третбутилацетат, н-бутилацетат, толуол, пропилацетат и бутилметилкетон. Предпочтительные растворители включают толуол, изопропанол, изобутилацетат или смесь этанол-вода. Наиболее предпочтительными растворителями являются ичобутилацетаг и смесь этанол-вода.
Кристаллы, полученные с использованием предпочтительного варианта, содержат менее 3,6% (вес./вес.) примесей и получают с выходом выше 75% (вес./вес.). Для очистки статина, полученного перекристаллизацией из смеси вода:этанол, предпочтительное соотношение воды к этанолу составляет от 0,8 до 2,0. Наиболее предпочтительное соотношение воды к этанолу составляет от 0,9 до 1,2. Кристаллы, полученные с использованием предпочтительного варианта, имеют чистоту, по крайней мере, 98,5% (вес./вес.).
Данное изобретение будет дополнительно объяснено на следующих примерах. Если не указано особо, все выходы приведены в % (вес./вес.) и они представляют общин выход или выхода всех стадий. Однако данное изобретение этими примерами не ограничивается. Специалистам понятно, как изменить приведенные в примерах способы, чтобы получить желаемые результаты.
ПРИМЕРЫ
ПРИМЕР 1: Выделение Компактина
Ферментационный бульон (70 м3), содержащий 350 кг компактина, получали известным способом (См., например, патенты США №№3983140, 4049495 и 5691173). К водному ферментационному бульону (70 м3) непрерывно прибавляли изобутилацетат (35 м3) и воду (35 м3). рН доводили до 9,0-9,6 прибавлением концентрированной NaOH. Смесь затем нагревали до 60°С и выдерживали при данной температуре в течение 2 часов. Полученную органическую и водную фазы затем разделяли, применяя противоточную экстракцию. Чтобы во время разделения помочь разрушению эмульсии, прибавляли лаурилсульфат натрия.
Очищенный ферментационный бульон подкисляли до рН 2,0-4,5 с помощью серной кислоты. Затем непрерывно прибавляли изобутилацетат (35 м3) и после перемешивания in situ изобутилацетатную фазу, содержащую компактин, непрерывно отделяли.
Изобутилацетатную фазу концентрировали под вакуумом до объема около 2150 л. Концентрированный раствор оставляли стоять при 0-5°С в течение ночи, при этом компактин кристаллизовали с 78% выходом (вес./вес.) и 93% чистотой (вес./вес.). Неочищенный компактин затем перекристаллизовывали из смеси этанол:вода, равной 1,2:0,9, с 75% (вес./вес.) выходом при чистоте 99,0% (вес./вес.).
ПРИМЕР 2: Выделение Компактина без стадии предварительной очистки ферментационного бульона в щелочных условиях
Ферментационный бульон, содержащий компактин, получали как в примере 1. Ферментационный бульон (50 л) подкисляли до рН 2,0-4,5 с помощью серной кислоты. Затем прибавляли изобутилацетат (25 л) и воду (25 л) и после перемешивания в течение 0,5 часа отделяли изобутилацетатную фазу, содержащую компактин. Экстракцию повторяли с чистым изобутилацетатом (25 л). Объединенную изобутилацетатную фазу концентрировали под вакуумом до объема около 1,5 л. Концентрированный раствор оставляли стоять при 0-5°С в течение ночи, при этом компактин кристаллизовали с 33% (вес./вес.) выходом при чистоте 94,5% (вес./вес.). В то время как пример 1 служит примером применения щелочной экстракции, пример 2 отличается тем, что он иллюстрирует использование технологий без щелочной экстракции.
ПРИМЕР 3: Выделение Компактина: предварительная обработка ферментационного бульона в щелочных условиях концентрированным изобутилацетатом и промывание NaHCO3
Ферментационный бульон получали, как в примере 1. К водному ферментационному бульону (50 л) прибавляли изобутилацетат (25 л) и воду (25 л). рН доводили до 9,0-9,6 прибавлением концентрированной NaOH. Смесь затем нагревали до 60°С и выдерживали при данной температуре в течение 2 часов. Затем разделяли фазы. Чтобы во время разделения помочь разрушению эмульсии, прибавляли додецилтриметиламмоний хлорид. Очищенный ферментационный бульон подкисляли до рН 2,0-4,5 с помощью серной кислоты. Затем прибавляли изобутилацетат (25 л) и после перемешивания в течение 0,5 часа отделяли изобутилацетатную фазу, содержащую компактин. Кислую экстракцию повторяли с чистым изобутилацетатом (25 л).
Объединенную изобутилацетатную фазу концентрировали под вакуумом до объема около 1,5 л. Концентрированный раствор разбавляли до объема примерно 8 л и промывали насыщенным раствором бикарбоната натрия. Промытый раствор снова концентрировали до 1,5 л и оставляли стоять при 0-5°С в течение ночи, при этом компактин кристаллизовали с 76,5% выходом (вес./вес.) и 98,9% чистотой (вес./вес.).
В отсутствии предварительной щелочной экстракции компактин получали только с 39% (вес./вес.) выходом при чистоте 99,1% (вес./вес.).
ПРИМЕР 4: Выделение Компактина: влияние пониженной температуры во время предварительной обработки ферментационного бульона в щелочных условиях
Ферментационный бульон получали, как в примере 1. К водному ферментационному бульону (50 л) прибавляли изобутилацетат (25 л) и воду (25 л). рН доводили до 9,0-9,6 прибавлением концентрированной NaOH. Смесь выдерживали при температуре 15-20°С в течение 2 часов. Затем разделяли фазы. Чтобы во время разделения помочь разрушению эмульсии, прибавляли додецилтриметиламмоний хлорид. Очищенный ферментационный бульон подкисляли до рН 2,0-4,5 с помощью серной кислоты. Затем прибавляли изобутилацетат (25 л) и после перемешивания в течение 0,5 часа отделяли изобутилацетатную фазу, содержащую компактин. Экстракцию повторяли с чистым изобутилацетатом (25 л). Объединенную изобутилацетатную фазу концентрировали под вакуумом до объема около 1,5 л. Концентрированный раствор оставляли стоять при 0-5°С в течение ночи, при этом компактин кристаллизовался. Неочищенный компактин затем перекристаллизовывали из смеси этанол:вода =1,2:0,9. Общий выход составлял 67% (вес./вес.) при чистоте 99.1% (вес./вес.).
ПРИМЕР 5: Выделение Компактина: влияние кислого рН 1,0-2,0 при кислотной экстракции и пониженной температуре во время кристаллизации
Ферментационный бульон получали, как в примере 1. К водному (ферментационному бульону (50 л) прибавляли изобутилацетат (25 л) и воду (25 л). рН доводили до 9,0-9,6 прибавлением концентрированной NaOH. Смесь выдерживали при температуре 60°С в течение 2 часов. Затем разделяли фазы. Чтобы во время разделения помочь разрушению эмульсии, прибавляли додецилтриметиламмоний хлорид. Ферментационный бульон подкисляли до рН 1,0-2,0 с помощью серной кислоты. Затем прибавляли изобутилацетат (25 л) и после перемешивания в течение 0,5 часа отделяли изобутилацетатную фазу, содержащую компактин. Экстракцию повторяли с чистым изобутилацетатом (25 л).
Объединенную изобутилацетатную фазу концентрировали под вакуумом до тех пор, пока концентрация компактна составила примерно 150 г/л. Концентрированный раствор выдерживали при температуре около - 10°С в течение 20 часов. Кристаллы (фильтровали и промывали изобутилацетатом. Неочищенный компактин затем перекристаллизовали из смеси этанол:вода =1,2:0,9. Компактам получали с 67% (вес./вес.) выходом при чистоте 98,8% (вес./вес.). Кислая экстракция (при рН 1,0-2,0) снижала выход.
Если способ, описанный в данном примере, с предварительной обработкой ферментационного бульона в щелочных условиях проводили при 15°С, общий выход компактина уменьшался до 60% (вес./вес.) при чистоте 98,7% (вес./вес.).
ПРИМЕР 6: Выделение Ловастатина
Ферментационный бульон, содержащий ловастатин, получали известным способом. (См., например, патенты США №№5403728, 4420491, 4342767, 4319039 и 4294846). К водному ферментационному бульону (50 л) прибавляли изобутилацетат (25 л) и воду (25 л). рН доводили до 9,0-9,6 прибавлением концентрированной NaOH. Смесь затем нагревали до 60±5°С и выдерживали при данной температуре в течение 2 часов. Затем (фазы разделяли. Чтобы во время разделения помочь разрушению эмульсии, прибавляли додецилтриметиламмоний хлорид.
Очищенный ферментационный бульон подкисляли до рН 2,0-4,5 с помощью серной кислоты. Затем прибавляли изобутилацетат (25 л) и после перемешивания в течение 2 часов отделяли изобутилацетатную фазу, содержащую ловастатин. Экстракцию повторяли с чистым изобутилацетатом (25 л).
Объединенную изобутилацетатную фазу концентрировали под вакуумом до объема примерно 2,7 л. Концентрированный раствор оставляли стоять при температуре - 10°С в течение 36 часов, при этом ловастатин кристаллизовался с 76% выходом (вес./вес.) и 96,4% чистотой (вес./вес.). Неочищенный ловастатин затем перекристаллизовывали из смеси этанол:вода =1,2:0,9 с 71,4% (вес./вес.) выходом при чистоте 98,7% (вес./вес.).
ПРИМЕР 7: Выделение Ловастатина без предварительной очистки ферментационного бульона в щелочных условиях
Ферментационный бульон, содержащий ловастатин, получали, как в примере 6. Ферментационный бульон (50 л) подкисляли до рН 2,0-4,5 с помощью серной кислоты. Затем прибавляли изобутилацетат (25 л) и воду (25 л) и после перемешивания в течение 2 часов отделяли изобутилацетатную фазу, содержащую ловастатин. Экстракцию повторяли с чистым изобутилацетатом (25 л). Объединенную изобутилацетатную фазу концентрировали под вакуумом до объема около 2,7 л. Концентрированный раствор оставляли стоять при - 10°С в течение 36 часов, при этом ловастатин кристаллизовался с 26% выходом (вес./вес.) при чистоте 88,0% (вес./вес.).
ПРИМЕР 8: Выделение Ловастатина: применение промывания NaHCO3
Ферментационный бульон получали, как в примере 6. К водному ферментационному бульону (50 л) прибавляли изобутилацетат (25 л) и воду (25 л). рН доводили до 9,0-9,6 прибавлением концентрированной NaOH. Смесь затем нагревали до 60°С и выдерживали при данной температуре в течение 2 часов. Затем разделяли фазы. Чтобы во время разделения помочь разрушению эмульсии, прибавляли додецилтриметиламмоний хлорид. Очищенный ферментационный бульон подкисляли до рН 2,0-4,5 с помощью серной кислоты. Затем прибавляли изобутилацетат (25 л) и после перемешивания в течение 2 часов отделяли изобутилацетатную фазу, содержащую ловастатин. Экстракцию повторяли с чистым изобутилацетатом (25 л).
Изобутилацетатную фазу концентрировали под вакуумом до объема примерно 2,7 л. Концентрированный раствор разбавляли до объема примерно 11 л и промывали насыщенным раствором бикарбоната натрия. Промытый раствор снова концентрировали до 2,7 л и его оставляли стоять при - 10°С в течение 36 часов, при этом ловастатин получали с 73,4% (вес./вес.) выходом при чистоте 98,8% (вес./вес.).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ НАТРИЕВЫХ СОЛЕЙ СТАТИНОВ | 2000 |
|
RU2246481C2 |
| ПРОМЫШЛЕННЫЙ СПОСОБ ПОЛУЧЕНИЯ КОМПАКТИНА | 2013 |
|
RU2585233C2 |
| МИКРОБНЫЙ СПОСОБ ПОЛУЧЕНИЯ ПРАВАСТАТИНА | 2000 |
|
RU2252258C2 |
| СПОСОБ ПОЛУЧЕНИЯ КОМПАКТИНА | 2013 |
|
RU2585234C2 |
| ГИДРОКСИЛИРОВАНИЕ КОМПАКТИНА ДО ПРАВАСТАТИНА С ПОМОЩЬЮ MICROMONOSPORA | 2000 |
|
RU2235780C2 |
| СПОСОБ ВЫДЕЛЕНИЯ И СПОСОБ ОЧИСТКИ ИНГИБИТОРА HMG-COA-РЕДУКТАЗЫ | 1999 |
|
RU2235130C2 |
| ШТАММ ГРИБА ASPERGILLUS TERREUS № 44-62 - ПРОДУЦЕНТ ЛОВАСТАТИНА, ПРОМЫШЛЕННЫЙ СПОСОБ ВЫДЕЛЕНИЯ ЛОВАСТАТИНА И СПОСОБ ЛАКТОНИЗАЦИИ СТАТИНОВ | 2003 |
|
RU2261901C2 |
| СПОСОБ ВЫДЕЛЕНИЯ ЛОВАСТАТИНА | 1994 |
|
RU2114912C1 |
| СПОСОБ ОЧИСТКИ ПРАВАСТАТИНА | 2001 |
|
RU2260582C2 |
| СПОСОБ ПОЛУЧЕНИЯ КЛАВУЛАНОВОЙ КИСЛОТЫ ИЛИ ЕЕ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ ИЛИ ЭФИРОВ | 1995 |
|
RU2088586C1 |
Изобретение относится к биотехнологии. Соединения статина из ферментационного раствора очищают с помощью экстракции и кристаллизации. Ферментационный бульон подвергают процедуре предварительной обработки, которая включает щелочную предварительную обработку с последующей экстракцией неполярных примесей. После процедуры предварительной обработки соединение статина экстрагируют в гидрофобный растворитель, который затем отделяют. Далее раствор гидрофобного органического растворителя концентрируют, при необходимости промывают водным раствором, содержащим основание. Затем экстрагированное соединение статина очищают кристаллизацией. Данное изобретение позволяет выделять статины из ферментационного бульона с высокой степенью эффективности при фармацевтически приемлемом уровне чистоты. 2 н. и 26 з.п. ф-лы.
а) предварительную обработку ферментационного бульона для удаления неполярных примесей, причем эта предварительная обработка включает обработку ферментационного бульона в щелочных условиях с последующей экстракцией неполярных примесей в гидрофобный органический экстракционный растворитель предварительной обработки,
б) экстракцию соединения статина из ферментационного бульона при обработке ферментационного бульона экстракционным растворителем и последующую экстракцию соединения из ферментационного бульона в экстракционный растворитель, являющийся гидрофобным органическим экстракционным растворителем,
в) отделение гидрофобного органического экстракционного растворителя от ферментационного бульона,
г) концентрирование раствора гидрофобного органического экстракционного растворителя, содержащего экстрагированное соединение статина, и
д) очистку экстрагированного соединения статина кристаллизацией.
(а) температуре от 15 до 100°С,
(б) рН от 7,0 до 13,9 и
(в) в течение периода времени от 0 до 48 ч.
(а) температуре от 55 до 65°С,
(б) рН от 9,0 до 9,6 и
(в) в течение периода времени около 2 ч.
а) предварительную обработку ферментационного бульона для удаления неполярных примесей, причем эта предварительная обработка включает обработку ферментационного бульона в щелочных условиях с последующей экстракцией неполярных примесей в гидрофобный органический экстракционный растворитель предварительной обработки.
б) экстракцию соединения статина из ферментационного бульона при обработке ферментационного бульона экстракционным растворителем и последующую экстракцию соединения из ферментационного бульона в экстракционный растворитель, являющийся гидрофобным органическим экстракционным растворителем,
в) отделение гидрофобного растворителя от ферментационного бульона,
г) концентрирование раствора гидрофобного растворителя, содержащего экстрагированное соединение статина,
д) промывание концентрированного раствора гидрофобного растворителя водным раствором, содержащим основание, и
е) очистку экстрагированного соединения статина кристаллизацией.
| US 5989877 А, 23.11.1999 | |||
| US 5712130 A, 27.11.1998 | |||
| RU 96101191 А, 10.10.1998. |

