Настоящее изобретение относится к новым соединениям, которые являются избирательными в отношении мускариновых ацетилхолиновых субтипов рецепторов, а также лекарственному препарату, способствующему активации мускариновых рецепторов и для лечения или облегчения заболеваний, при которых положительной является модификация активности мускариновых рецепторов.
Мускариновые ацетилхолиновые рецепторы играют ведущую роль в центральной нервной системе в высшей познавательной функции, а также в периферической парасимпатической нервной системе. В результате клонирования было установлено наличие пяти отдельных субтипов мускариновых рецепторов (обозначенных m1-m5) (подтверждено T.I. Bonner et al., Science 237, 1987, pp. 527-532; T.I. Bonner et al., Neuron 1, 1988, pp. 403-410). Было установлено, что m1 является преобладающим субтипом в коре головного мозга, и полагается, что он участвует в регуляции познавательной функции, m2 преобладает в сердце и считается, что он участвует в регуляции сердечного ритма, по всей вероятности, m3 участвует в стимуляции желудочно-кишечного тракта и мочевых путей, а также потоотделения и саливации, m4 присутствует в мозге, и m5 находится в мозге, и могут выполнять определенные функции в центральной нервной системе, связанные с дофанергической системой.
В опытах на животных по изучению различных мускариновых лиганд (подтверждено S. Iversen, Life Sciences 60 (Nos. 13/14), 1997, pp. 1145-1152) было показано, что мускариновые соединения имеют выраженный эффект на познавательную функцию, например обучение и память. На основании этого можно предположить о потенциальной применимости мускариновых агонистов в улучшении познавательной функции при заболеваниях, характеризующихся нарушением познавательной функции, как связанных с возрастом (таких как болезнь Альцгеймера или другие виды слабоумия) и не связанных с возрастом (таких как дефицитное гиперактивное нарушение внимания).
Основываясь на наличии субтипов мускариновых рецепторов в различных тканях, становится очевидным, что рецепторы m1-субтипа более распространены в коре головного мозга, базальном ганглии и гиппокампусе, где их число достигает 35-60% от всех связывающих сайтов мускариновых рецепторов (подтверждено A. Levey, Proc. Natl. Acad. Sci. USA 93, 1996, pp. 13541-13546). Было высказано предположение, что m1-субтип (и, возможно, m4) играет главную роль в качестве постсинаптических мускариновых рецепторов (расположенных в холинорецептивных нейронах в неокортексе и гиппокампусе) в различных видах познавательной и двигательной функций и, по-видимому, вносит основной вклад в ответные реакции, осуществляемые через m1, в этих участках мозга.
Ранее было установлено, что заболевания, связанные с нарушением познавательной функции, такие как болезнь Альцгеймера, сопровождаются селективной потерей ацетилхолина в мозге. Полагается, что это является результатом дегенерации холинергетических нейронов в базальной части переднего мозга, которые иннервируют (возбуждают) ассоциативные зоны коры головного мозга и гиппокампуса, участвующих в высшей нервной деятельности (подтверждено S.Iversen, выше). На основании этого обнаруженного факта предполагается, что такие состояния можно лечить или, по меньшей мере, ослаблять лекарственными препаратами, которые усиливают холинергическую функцию в пораженных участках мозга.
Лечение ингибиторами ацетилхолинэстеразы (AChE), такими как 9-амино-1,2,3,4-тетрагидроакридин (такрин), приводит к увеличению ацетилхолина в мозге, которое опосредованно вызывает стимуляцию мускариновых рецепторов. Лечение такрином приводит к умеренному и временному улучшению познавательной функции у больных с болезнью Альцгеймера (подтверждено Kasa et al., выше). С другой стороны, было установлено, что такрин обладает холинергическими побочными эффектами, как результат периферической стимуляции ацетилхолина. Они включают спазмы в брюшной области, тошноту, рвоту, диарею, анорексию, потерю в весе, миопатию и депрессию. Побочные эффекты со стороны желудочно-кишечного тракта наблюдали примерно у трети пациентов, которые подвергались лечению. Было также установлено, что такрин вызывает значительную гепатотоксичность, примерно у 30% пациентов наблюдали повышенный уровень трансаминаз (подтверждено P.Taylor, «Anticholinergic Agents», Chapter 8 in Goodman and Gilman: The Pharmacological Basis of Therapeutics, 9th Ed., 1996, pp. 161-176). Побочные эффекты такрина в значительной степени ограничивают его клиническую применимость. Другой ингибитор AChE (R,S)-1-бензил-4-[5,6-диметокси-1-инданон-2-ил]метил пиперидин·HCl (донепезил) недавно был разрешен для лечения симптомов болезни Альцгеймера в легкой и средней форме (подтверждено P.Kasa et al., выше). Для этого соединения не наблюдали повреждающего действия на печень, но оно обладает эффектами в отношении желудочно-кишечного тракта, аналогичными таковым для такрина, возможно, в результате стимуляции m3-рецептора, вызванной повышенным парасимпатическим тонусом.
Ранее было высказано предположение, что поскольку мускариновые m1-рецепторы в лобной части коры и гиппокампуса являются интактными, становится возможным лечить или, по меньшей мере, ослаблять потерю ацетилхолина у пациентов с болезнью Альцгеймера назначением лекарственных препаратов, действующих в качестве агонистов этих мускариновых рецепторов (подтверждено J.H.Brown and P.Taylor, «Muscarinic Receptor Agonists and Antagonists», Chapter 7 in Goodman and Gilman: The Pharmacological Basis of Therapeutics, 9th Ed., 1996, pp. 147).
До настоящего времени мускариновые агонисты (по всей вероятности, избирательные для m1), предложенные для лечения болезни Альцгеймера, такие как ареколин, не показывают более высокой эффективности в клинических испытаниях, чем ингибиторы AChE (подтверждено S.V.P.Jones et al., выше). В одном исследовании (подтверждено T.Sunderland et al., Brain Res. Rev. 13, 1988, pp. 371-389) было установлено, что ареколин не обладает столь значительным усилением познавательной функции в отношении поведенческих изменений, часто наблюдаемых у пациентов с болезнью Альцгеймера, таких как значительное увеличение двигательной активности, значительное повышение настроения и значительное снижение анэргии. Однако позднее было установлено, что предполагаемые агонисты m1 являются слабыми неполностью избирательными агонистами для рецепторов m2- и/или m3-субтипов (H.Bruner-Osborne et al., J. Med. Chem. 38, 1995, pp. 2188-2195). Как указывалось выше, предполагается, что избирательность для m2-рецепторов ответственна за сердечно-сосудистые эффекты, наблюдаемые для этих агонистов, например, тахикардию и брадикардию, и полагают, что активность для m3-рецепторов ответственна за побочные эффекты агонистов со стороны желудочно-кишечного тракта.
Следовательно, до настоящего времени активность для m2- и/или m3-рецепторов является существенным недостатком для мускариновых агонистов, предложенных для лечения болезни Альцгеймера, в значительной степени снижая дозы лекарственных препаратов, которые можно назначать пациентам, и которые, следовательно, вынуждены получать субоптимальные дозы. Кроме того, отсутствие избирательности в отношении субтипов и низкая эффективность имеющихся в настоящее время холинергических соединений способствует проявлению отрицательных побочных эффектов и имеет ограниченное действие на познавательную функцию вследствие слабых и/или противоположных эффектов в мозге. Следовательно, будет большим преимуществом разработать соединения, которые обладают улучшенной избирательностью в отношении субтипа m1, но которые обладают низкой или отсутствием активности для субтипов m2 и m3.
Настоящее изобретение относится к соединениям с активностью мускаринового агониста общей формулы (I):

где
Х1, Х2, Х3, Х4 и Х5 выбираются из С, N и О;
k равно 0 или 1;
t равно 0, 1 или 2;
R1 является нормальным или разветвленным С1-8 алкилом, С2-8 алкенилом, С2-8 алкинилом, С1-8 алкилиденом, С1-8 алкокси, С1-8 гетероалкилом, С1-8 аминоалкилом, С1-8 галогеналкилом, С1-8 алкокси карбонилом, С1-8 гидроксиалкокси, С1-8 гидроксиалкилом, -SH, С1-8 алкилтио, -О-СН2-С5-6 арилом, -С(О)-С5-6 арилом, замещенным С1-3 алкилом или галогеном; С5-6 арилом или С5-6 циклоалкилом, необязательно включающими 1 или более гетероатомов, выбранных из N, S и О; -С(О)NR3R4, -NR3R4, -NR3C(O)NR4R5, -CR3R4, -OC(O)R3,-(O)(CH2)sNR3R4 или -(CH2)sNR3R4, где R3, R4 и R5 являются одинаковыми или разными, каждый независимо выбирается из Н, С1-6 алкила; С5-6 арила, необязательно включающего 1 или более гетероатомов, выбранных из N, O и S, и необязательно замещенного галогеном или С1-6 алкилом; С3-6 циклоалкила, или R3 и R4 вместе с атомом N, когда имеются, образуют циклическую кольцевую структуру, включающую 5-6 атомов, выбранных из C, N, S и О; и s равно целому числу от 0 до 8;
А является С5-12 арилом или С5-7 циклоалкилом, каждый необязательно включающий 1 или более гетероатомов, выбранных из N, S и О;
R2 является Н, амино, гидроксилом, галогеном или нормальным или разветвленным С1-6 алкилом, С2-6 алкенилом, С2-6 алкинилом, С1-6 алкокси, С1-6 гетероалкилом, С1-6 аминоалкилом, С1-6 галогеналкилом, С1-6 алкилтио, С1-6 алкоксикарбонилом, -CN, -CF3, -OR3, -COR3, NO2, -NHR3, -NHC(O)R3, -C(O)NR3R4, -NR3R4, -NR3C(O)NR4R5, -OC(O)R3, -C(O)R3R4, -O(CH2)qNR3, -CNR3R4 или -(CH2)qNR3R4, где q равно целому числу от 1 до 6;
n равно 0, 1, 2, 3 или 4, радикалы R2, где n > 1, являются одинаковыми или разными;
р равно 0 или целому числу от 1 до 5;
Y является O, S, CHOH, -NHC(O)-, -C(O)NH-, -C(O)-, -OC(O)-, NR7 или -CH=N-, и R7 является Н или С1-4 алкилом или отсутствует; и
Z является CR8R9, где R8 и R9 независимо выбираются из Н и нормального или разветвленного С1-8 алкила; или
их фармацевтически приемлемая соль, эфир и пролекарство.
Настоящее изобретение также относится к лекарственному препарату, включающему эффективное количество соединения формулы (I), пригодное для
лечения симптомов заболевания или состояния, связанного с пониженными уровнями ацетилхолина, причем указанное лечение включает назначение терапевтически эффективного количества препарата, включающего соединение формулы (I).
Лекарственный препарат также пригоден для лечения симптомов заболевания или состояния, связанного с повышенным внутриглазным давлением такого, например, как глаукома, где способ включает назначение терапевтически эффективного количества препарата, включающего соединение формулы (I).
Краткое описание чертежей
Фигура 1 является графиком, показывающим необработанные данные по скринингу 35000, небольших органических молекул с использованием 96-луночного планшета в тесте, описанном в примере XVI.
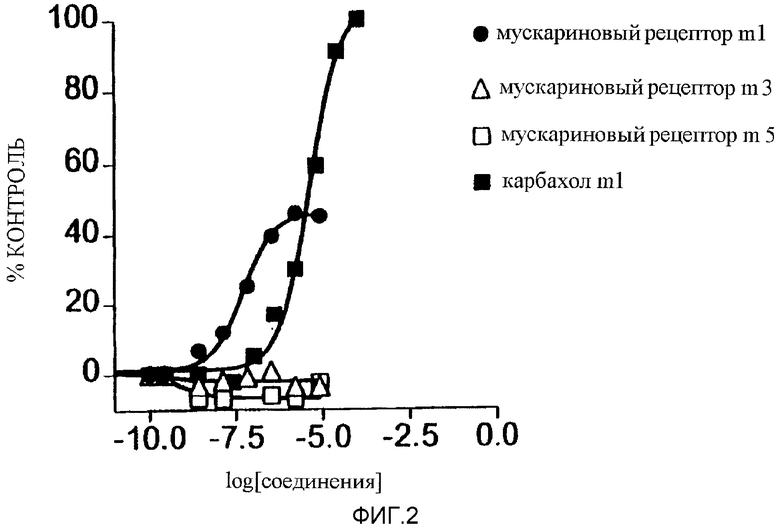
Фигура 2 является графиком, показывающим данные по сравнению профиля стандартного антагониста атропина с клетками, трансфектированными мускариновым рецептором m1, стимулированные либо карбахолом (открытые треугольники), либо соединением А (пример I) (закрытые треугольники).
Детальное описание изобретения
Настоящее изобретение относится к соединениям, предпочтительно показывающим относительно высокую избирательность по отношению к рецепторам субтипа m1 в сравнении с другими мускариновыми субтипами, которые могут оказывать положительное воздействие в лечении нарушения познавательной функции, такого как болезнь Альцгеймера или другие состояния, связанные с возрастным падением познавательной функции, одновременно избегая побочных эффектов лекарственных препаратов, предложенных к настоящему времени для этой цели. С удивлением в результате скрининга против рецепторов m1-m5-субтипов были выделены соединения, проявляющие это свойство.
Согласно одному воплощению, настоящее изобретение обеспечивает соединения формулы (I), гдеХ1, Х2, Х3, Х4 и Х5 являются С; или один из Х1, Х2, Х3, Х4 и Х5 является О или N и другие являются С;
k равно 0 или 1;
t равно 1;
R1 является нормальным или разветвленным С1-8 алкилом, С2-8 алкенилом, С2-8 алкинилом, С1-8 алкилиденом, С1-8 алкокси, С1-8 аминоалкилом, С1-8 галогеналкилом, С1-8 алкоксикарбонилом, -C(O)NR3R4, -NR3R4, -NR3C(O)NR4R5, -OC(O)R3 или -(CH2)sNR3R4, где R3, R4 и R5 являются одинаковыми или разными, каждый независимо выбирается из Н и С1-6 алкила; и s равно целому числу от 1 до 8;
n равно 1, 2 или 3; и
А является фенилом или нафтилом;
где R2 является нормальным или разветвленным С1-6 алкилом, С2-6 алкенилом, С2-6 алкинилом, С1-6 алкокси, С1-6 аминоалкилом, С1-6 галогеналкилом, С1-6 алкоксикарбонилом, -CN, -CF3, -OH, -COR3, -NHR3, -NHC(O)R3, -C(O)NR3R4, -NR3R4, -NR3C(O)NR4R5, -OC(O)R3 или -(CH2)qNR3R4; где q равно целому числу от 1 до 6; или
А является арилом, включающим 1 или более гетероатомов, выбранных из N, S и O;
R2 является Н, галогеном, нормальным или разветвленным С1-6 алкилом, С2-6 алкенилом, С2-6 алкинилом, С1-6 алкокси, С1-6 гетероалкилом, С1-6 аминоалкилом, С1-6 галогеналкилом, С1-6 алкоксикарбонилом, -CN, -CF3, -OH, -COR3, -NHR3, -NHC(O)R3, -C(O)NR3R4, -NR3R4, -NR3C(O)NR4R5, -OC(O)R3 или -(CH2)qNR3R4; или
их фармацевтически приемлемая соль, эфир или пролекарства.
В одном предпочтительном воплощении соединение имеет формулу (II):

Предпочтительные варианты соединений формулы (II) включают соединения формул (IIa) и (IIb):


Предпочтительно, в соединениях формул I, II, IIa и IIb t равно 1, и Y является -C(O)-, -NHC(O)-, S, O или -OC(O)-. В другом варианте Х3 является С. Предпочтительно R1 является алкилом, где предпочтительно R2 является алкилом, аминоалкилом, алкокси или гидроксилом. В одном воплощении р равно 3. В другом, R1 является С2-8 алкилом, и R2 является метилом, гидроксилом или алкокси.
В одном воплощении n равно 1 или 2; Y является -C(O)- или О, и t равно 1. Предпочтительно R2 является галогеном. Согласно другим воплощениям t равно 0, или R1 является алкокси, бензилом или фенилом.
Х3 также может быть N, где согласно одному воплощению R1 является алкилом или алкокси; или R1 является бензилом или фенилом, где R2 является алкилом или алкокси.
Согласно другому воплощению Х3 является О, где t может быть равно, например, 0. Предпочтительно R2 является алкилом или алкокси; или R2 является галогеном.
Предпочтительные воплощения изобретения включают:
4-Метокси-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-Этокси-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-Пропокси-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-Бутокси-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-Метоксиметил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-Этоксиметил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-Пропоксиметил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-(2-Метоксиэтил)-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-(2-Этоксиэтил)-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-Метокси-4-метил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-Метокси-4-этил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-Метокси-4-пропил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-Метокси-4-н-бутил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-Этокси-4-метил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-Этокси-4-этил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-Этокси-4-пропил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-Этокси-4-н-бутил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-Пропокси-4-метил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-Пропокси-4-этил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-Пропокси-4-пропил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-Пропокси-4-н-бутил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутокси-4-метил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутокси-4-этил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутокси-4-пропил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутокси-4-н-бутил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
2-[3-(4-н-Бутилпиперидин)пропокси]толуол;
2-[3-(4-н-Бутилпиперидин)пропансульфанил]толуол;
2-[3-(4-н-Бутилпиперидин)пропансульфинил]толуол;
3-(4-н-Бутилпиперидин)-о-толил-бутан-1-тион;
3-(4-н-Бутилпиперидинпропил)-о-толиламин;
N-(4-(4-н-Бутилпиперидин)-1-о-толилбутил)гидроксиламин;
4-н-Бутил-1-[4-(2-хлорфенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(2-бромфенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(2-фторфенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(2-меркаптофенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(2-сульфанилметилфенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(2-сульфанилэтилфенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(2-аминофенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(2-метиламинофенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(2-этиламинофенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(2-диметиламинофенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(2-диэтиламинофенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(1-Н-имидазол-2-ил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(1-имидазол-1-ил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(1-тиазол-2-ил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-([1,2,3]триазол-1-ил)-4-оксо-1-бутил]пиперидин;
2-[4-н-бутил-пиперидин-1-этил]-8-метил-3,4-дигидро-2Н-нафтален-1-он;
2-[4-н-бутил-пиперидин-1-этил]-7-метил-индан-1-он;
3-[4-н-бутил-пиперидин-1-этил]-хроман-4-он;
2-[4-н-бутил-пиперидин-1-этил]-1Н-бензоимидазол;
4-н-Бутил-1-[4-(4-фтор-2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(2-гидроксифенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(2-метоксифенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(1-тиофен-2-ил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(2-этилфенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(2-этоксифенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(2,4-диметилфенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(2,3-диметилфенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(3-метоксифенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(2-бензилоксифенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(4-метилфенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-N-фенилбутирамид;
4-Метил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(нафтален-1-ил)-4-оксо-1-бутил]пиперидин;
4-Бензил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
1-[4-(2-метилфенил)-4-оксо-1-бутил]пирролидин;
4-Бензил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперазин;
2-Пропил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
2-Этил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-н-Пропил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперазин;
3,5-Диметил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-Метил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперазин;
4-н-Гексил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперазин;
4-Гидроксиэтил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперазин;
4-Этил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперазин;
4-Бензил-1-[4-(4-фторфенил)-4-оксо-1-бутил]пиперидин;
4-Бензил-1-[4-(4-бромфенил)-4-оксо-1-бутил]пиперидин;
4-Фенил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперазин;
3-Гидроксиметил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-Метил-1-[4-(4-бромфенил)-4-оксо-1-бутил]пиперидин;
1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
2-Гидроксиметил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
4-Бензил-1-[4-(2-метилфенил)-4-оксо-1-пентил]пиперазин;
4-н-Гексил-1-[4-(2-метилфенил)-4-оксо-1-пентил]пиперазин;
4-(Пиперидин-1-ил)-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин;
1-[4-(2-метилфенил)-4-оксо-1-бутил]-2,3-дигидро-1Н-индол;
4-Бензил-1-[5-(2-метилфенил)-5-оксо-1-пентил]пиперидин;
4-н-Бутил-1-[5-(2-метилфенил)-5-оксо-1-пентил]пиперидин;
4-н-Бутил-1-[4-(2,6-диметилфенил)-4-оксо-1-бутил]пиперидин;
4-н-Бутил-1-[4-(2-метоксиметилфенил)-4-оксо-1-бутил]пиперидин;
1-(2-Метилфенил)-2-(4-бензилпиперазин-1-ил)этанон;
3,5-Диметил-1-[5-(2-метилфенил)-5-оксо-1-пентил]пиперидин;
3,5-Диметил-1-[4-(4-фторфенил)-4-оксо-1-бутил]пиперидин;
1-[4-(4-Фторфенил)-4-оксо-1-бутил]пирролидин;
4-Бензил-1-[6-(2-метилфенил)-6-оксо-1-гексил]пиперазин;
3,5-Диметил-1-[6-(2-метилфенил)-6-оксо-1-бутил]пиперидин;
4-Бензил-1-[5-(2-метоксифенил)-5-оксо-1-пентил]пиперазин;
4-Бензил-1-[3-фенил-3-оксо-1-пропил]пиперазин;
4-н-Бутил-1-[5-(2-метоксифенил)-5-оксо-1-пентил]пиперидин;
3,5-Диметил-1-[4-(4-фтор-2-метилфенил)-4-оксо-1-бутил]пиперидин;
3-н-Бутил-1-[4-(2-метилфенил)-4-оксо-1-бутил]азетидин;
4-н-Бутил-1-[4-(2-метилфенил)-4-оксо-2-метил-1-бутил]пиперидин;
4-н-Бутил-1-[4-(2-метилфенил)-4-оксо-2,2-диметил-1-бутил]пиперидин;
4-н-Бутил-1-[4-(2-метилфенил)-4-оксо-2-этил-1-бутил]пиперидин;
4-н-Бутил-1-[4-(2-метилфенил)-4-оксо-2-пропил-1-бутил]пиперидин; и
4-н-Бутил-1-[4-(2-метилфенил)-4-оксо-2,2-диэтил-1-бутил]пиперидин.
Соединениями per se, специально исключенными из объема формулы I, являются 4-н-бутил-1-[4-фенил-4-оксо-1-бутил]пиперидин; 4-н-бутил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперазин; 2-[3-(3-н-бутилпиперидин)пропансульфанил]толуол и 4-пропилокси-1-[4-(4-фторфенил)-4-оксо-1-бутил]пиперидин (т.е. соединения, где (СН2)р-Y- является -(СН2)3-С(О)- или -(СН2)3-S-; и Х1 до Х5 являются С; так, что -А-(R2)n и R1 не являются вместе о-метилфенилом и н-бутилом соответственно; фенилом и н-бутилом, соответственно или п-фторфенилом и -О-(СН2)2СН3, соответственно).
Настоящее изобретение дополнительно относится к способу воздействия агонистом на мускариновый рецептор, включающий контактирование рецептора с эффективным количеством соединения формулы (I), включая все соединения в объеме формулы (I) (т.е. включая 4-н-бутил-1-[4-фенил-4-оксо-1-бутил]пиперидин, 4-н-бутил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперазин, 2-[3-(3-н-бутилпиперидин)пропансульфанил]толуол и 4-пропилокси-1-[4-(4-фторфенил)-4-оксо-1-бутил]пиперидин).
Настоящее изобретение дополнительно относится к лекарственным препаратам, включающим эффективное количество соединения формулы (I), включая все соединения в объеме формулы (I) (т.е. включая 4-н-бутил-1-[4-фенил-4-оксо-1-бутил]пиперидин, 4-н-бутил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперазин, 2-[3-(3-н-бутилпиперидин)пропансульфанил]толуол и 4-пропилокси-1-[4-(4-фторфенил)-4-оксо-1-бутил]пиперидин).
Настоящее изобретение также дополнительно относится к способам лечения симптомов заболевания или состояния, связанного с пониженными уровнями ацетилхолина, способ включает назначение терапевтически эффективного количества композиции, описанной здесь. Примеры заболеваний или состояний включают нейродегенеративное заболевание, нарушение познавательной функции, возрастное падение познавательной функции или слабоумие.
Было также показано, что соединения по настоящему изобретению обладают способностью снижать внутриглазное давление и, следовательно, могут использоваться для лечения таких заболеваний, как глаукома. Глаукома представляет собой заболевание, при котором наблюдается аномалия в механизме регуляции, циркуляции водянистой влаги, заполняющей переднюю камеру, т.е. пространства, образованного между роговицей и хрусталиком. Это приводит к увеличению объема водянистой влаги и повышению внутриглазного давления, приводя соответственно к дефекту поля зрения и даже потере зрения в результате компульсии и сокращения сосочков оптического нерва.
Соединения по настоящему изобретению предпочтительно показывают избирательную активность агониста в отношении m1-рецептора. Подобный агонист определяется как соединение, которое увеличивает активность мускаринового рецептора m1, когда оно контактирует с рецептором. Избирательность определяется как свойство агониста, мускаринового рецептора m1 посредством количества агониста эффективного для усиления активности m1-рецептора, вызывать незначительное или вовсе отсутствие усиления активности рецепторов m3- и m5-субтипов и предпочтительно m2- и m4-субтипов.
Как здесь использовано, термин «алкил» означает нормальный или разветвленный алкановый радикал с 1-6 атомами углерода в цепи, например, метил, этил, пропил, изопропил, н-бутил, сек-бутил, трет-бутил и т.д. Термин «гетероалкил» предназначен для обозначения алканового радикала, содержащего 1 или 2 гетероатома, выбранных из O, S или N.
Как здесь использовано, термин «алкенил» означает нормальный или разветвленный алкеновый радикал с 2-6 атомами углерода в цепи; термин «алкинил» предназначен для обозначения нормального или разветвленного алкинового радикала с 2-6 атомами углерода в цепи.
Как здесь использовано, термин «арил» и «циклоалкил» предпочтительно относится к моно- и бициклическим кольцевым структурам, включающим от 5 до 12 атомов углерода, более предпочтительно к моноциклическим кольцам, включающим от 5 до 6 атомов углерода. Там, где такие кольца включают один или более гетероатомов, выбранных из N, S и О (т.е. гетероциклические кольца), такие кольца включают в целом от 5 до 12 атомов, более предпочтительно от 5 до 6 атомов. Гетероциклические кольца включают, но не ограничиваются фурилом, пирролилом, пиразолилом, тиенилом, имидазолилом, изоксазолилом, оксазолилом, тиазолилом, изотиазолилом, пиридилом, пиперидинилом, пиперазинилом, пиридазинилом, пиримидинилом, пиразинилом, морфолинилом, оксадиазолилом, тиадиазолилом, имидазолинилом, имидазолидинилом и тому подобное. Кольцо может быть замещено одним или более радикалами, включенными в определение R2 выше. Понятно, что заместители С1-6 алкил, С1-6 алкенил, С1-6 алкинил, С1-6 алкокси, С1-6 гетероалкил, С1-6 аминоалкил, С1-6 галогеналкил или С1-6 алкоксикарбонил могут, если присутствуют, быть замещены одним или более гидроксилом, С1-4 алкокси, галогеном, циано, амино или нитро.
Как здесь использовано, термин «галоген» или «гало» включает хлор, фтор, йод или бром.
Понятно, что кольцо, представленное структурой
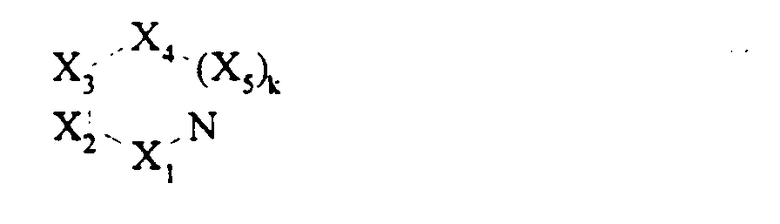
может быть насыщенным или ненасыщенным.
Соединения по настоящему изобретению можно получить способами, аналогичными способам, раскрытым в патенте Великобритании 1142143 и патенте США 3816433. Пути модификации этих способов, заключающиеся во включении других реагентов и т.д., будут поняты специалистами в этой области. Так, например, соединения формулы I можно получить, как представлено на следующей реакционной схеме.

Исходные соединения, имеющие формулу (X), можно получить общими способами органического синтеза. Для общих способов получения соединений формулы (Х) следует обратиться к Fuller R.W. et al., J. Med. Chem. 14:322-325 (1971); Foye W.O. et al., J. Pharm Sci. 68:591-595 (1979); Bossier J.R. et al., Chem. Abstr. 66:46195h and 67:21527a (1967); Aldous F.A.B., J. Med. Chem. 17:1100-1111 (1974); Fuller R.W. et al., J. Pharm. Pharmacol. 25:828-829 (1973); Fuller R.W. et al., Neuropharmacology 14:739-746 (1975); Conde S. et al., J. Med. Chem. 21:978-981 (1978); Lukovits I. et al., Int. J. Quantum Chem. 20:429-438 (1981); and Law B., J. Cromatog. 407:1-18 (1987), которые включаются в качестве ссылки во всей их полноте. Можно получить меченые изотопами соединения, имеющие формулы (ХХ), например, используя меченый тритием восстанавливающий агент для проведения восстановительного аминирования или при использовании 14С-меченого исходного вещества.
Альтернативно там, где исходное вещество включает карбонильную группу, соединение формулы (XXII) можно восстановить, например, AlH3, дибораном:метилсульфидом или другими стандартными восстанавливающими агентами для карбонильной группы для получения лиганда формулы (ХХХ).

Лиганды рецепторов формулы (XXXII) можно получить нуклеофильным замещенным электрофила (Е) аминопроизводным (XXXI). Примеры электрофилов, которые можно использовать для этой цели включают галиды, такие как I, Cl, Br, тозилат или мезилат.

Когда Y в формуле (XXXII) является -С(О)-, то это соединение можно получить окислением вторичного спирта, например, хлорхроматом пиридиния или N-хлор сукцинимидом или CrO3-H2SO4 или перекисью никеля или металлом (Al, K) или DCC-ДМСО.
Когда Y в формуле (XXXII) является -О-, то это соединение можно получить алкилированием спирта арилгалидами, например, при катализе Cu.
Когда Y в формуле (XXXII) является -S-, то это соединение можно получить алкилированием тиола арилгалидами, например, при катализе Cu.
Когда Y в формуле (XXXII) является -СНОН-, то это соединение можно получить восстановлением соответствующего кетона каталитическим гидрированием или при использовании NaBH4 или при использовании LiAlH4.
Пригодные фармацевтически приемлемые соли соединений по этому изобретению включают кислотные аддитивные соли, которые можно получить, например, смешением раствора соединения по изобретению с раствором фармацевтически приемлемой кислоты, такой как соляная кислота, серная кислота, фумаровая кислота, малеиновая кислота, янтарная кислота, уксусная кислота, бензойная кислота, щавелевая кислота, лимонная кислота, винная кислота, угольная кислота или фосфорная кислота. Кроме того, там, где соединения по изобретению имеют кислотную группу, их пригодные фармацевтически приемлемые соли могут включать соли щелочных металлов, например, соли натрия или калия; соли щелочноземельных металлов, например, соли кальция или магния; и соли, образованные пригодными органическими лигандами, например, четвертичные соли аммония. Примеры фармацевтически приемлемых солей включают ацетат, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид кальция, карбонат, хлорид, клавуланат, цитрат, дигидрохлорид, фумарат, глюконат, глутамат, гидробромид, гидрохлорид, гидроксинафтоат, йодид, изотионат, лактат, лактобионат, лаурат, малеат, манделат, мезилат, метилбромид, метилнитрат, метилсульфат, нитрат, N-метил глюкаминовая соль аммония, олеат, оксалат, фосфат/дифосфат, салицилат, стеарат, сульфат, сукцинат, таннат, тартрат, тозилат, триэтйодид и валерат.
В объеме настоящего изобретения включают пролекарства соединений по этому изобретению. В основном, подобные пролекарства являются неактивными производными соединений согласно настоящему изобретению, которые легко превращаются in vivo в необходимое соединение. Обычные способы выбора и получения пригодных пролекарств описываются, например, в «Design of Prodrugs», ed. H.Bundgaard, Elsevier, 1985. Метаболиты этих соединений включают активные виды, образующиеся при введении соединений по изобретению в биологическую среду.
В случае, если соединения по изобретению имеют, по меньшей мере, один хиральный центр, они могут находиться в виде рацемата или в виде энантиомеров. Следует отметить, что все такие изомеры и их смеси включены в объем настоящего изобретения. Кроме того, некоторые кристаллические формы соединений по настоящему изобретению могут находиться в виде полиморфных форм, и они также предназначаются для включения в настоящее изобретение. Кроме того, некоторые соединения по настоящему изобретению могут образовывать сольваты с водой (т.е. гидраты) или обычные органические сольваты. Такие сольваты также включаются в объем настоящего изобретения.
В случае, если способы получения соединений по изобретению дают смеси стереизомеров, такие изомеры можно разделить обычными способами, такими как препаративная хиральная хроматография. Соединения можно получить в рацемической форме или отдельные энантиомеры можно получить стереоселективным синтезом или разделением. Соединения можно, например, разделить на их компоненты-энантиомеры стандартными способами, такими как образование диастереоизомерных пар при образовании соли с оптически активной кислотой, такой как (-)-ди-п-толуоил-d-винная кислота и/или (+)-ди-п-толуоил-L-винная кислота, с последующей фракционной кристаллизацией и регенерацией свободного основания. Соединения можно также разделить при образовании диастереоизомерных эфиров или амидов с последующим хроматографическим разделением или удалением хирального вспомогательного вещества.
При проведении любого из способов получения соединений по настоящему изобретению может быть необходимым и/или желательным защитить чувствительные или реакционноспособные группы в любой из имеющих отношение молекул. Это можно достичь с помощью обычных защитных групп таких, как описанные в «Protective Groups in Organic Chemistry», ed. J.F.W.McOmie, Plenum Press, 1973; and T.W.Greene & P.G.M.Wuts, «Ptotective Groups in Organic Synthesis», John Wiley & Sons, 1991. Защитные группы можно удалить на соответствующей последующей стадии, используя способы, известные в этой области.
Соединения по настоящему изобретению можно вводить в любой из вышеупомянутых препаратов и соответственно схемам доз, установленным в этой области, в тех случаях, когда требуется специфическая фармакологическая модификация активности мускариновых рецепторов.
Настоящее изобретение также относится к лекарственным препаратам, включающим одно или более соединений по изобретению вместе с фармацевтически приемлемым разбавителем или наполнителем. Предпочтительно такие препараты находятся в единичных лекарственных формах, таких как таблетки, пилюли, капсулы (включая композиции с непрерывным высвобождением или замедленным высвобождением), порошки, гранулы, эликсиры, настойки, сиропы и эмульсии, стерильные растворы или суспензии для парентерального введения, аэрозоли или жидкие спреи, капли, ампулы, устройство для самовведения или суппозитории; для перорального, парентерального (например, внутривенного, внутримышечного или подкожного), интраназального, сублингвального или ректального введения, или для введения путем ингаляций или инсуффляций, и могут быть составлены соответствующим образом и в соответствии с принятой практикой, как описано в Remington's Pharmaceutical Sciences, Gennaro, Ed., Mack Publishing Co., Easton PA, 1990. Альтернативно лекарственные препараты могут быть в форме с непрерывным высвобождением, пригодной для введения раз в неделю или раз в месяц; например, нерастворимую соль активного соединения, такую как деканоат, можно адаптировать для обеспечения препарата-депо для внутримышечного введения. Настоящее изобретение также относится к препаратам, пригодным для местного применения, например, в глаз или на кожу или слизистую.
Например, для перорального введения в форме таблетки или капсулы, активный компонент лекарственного препарата можно объединить с нетоксичным фармацевтически приемлемым инертным носителем для перорального введения, таким как этанол, глицерин, вода и тому подобное. Более того, когда желательно или необходимо, то в смесь можно также включить пригодные связующие вещества, смазывающие вещества, разрыхлители, ароматизаторы и окрашивающие агенты. Пригодные связующие вещества включают, без ограничения, крахмал, желатин, натуральные сахара, такие как глюкоза или бета-лактоза, натуральные или синтетические камеди, такие как аравийская камедь, трагакант или альгинат натрия, карбоксиметилцеллюлоза, полиэтиленгликоль, воска и тому подобное. Смазывающие вещества, используемые в этих лекарственных формах, включают, без ограничения, олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и тому подобное. Разрыхляющие вещества включают, без ограничения, крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и тому подобное.
Для получения твердых препаратов, таких как таблетки, активный ингредиент смешивается с пригодным фармацевтическим наполнителем, например, как описанные выше, и другими фармацевтическими разбавителями, например водой, для получения твердой композиции для последующего формирования, содержащей гомогенную смесь соединения по настоящему изобретению или его фармацевтически приемлемой соли. Под термином «гомогенный» понимается, что активный ингредиент равномерно распределяется в препарате так, что его можно легко разделить на равноценно эффективные единичные лекарственные формы, такие как таблетки, пилюли и капсулы. Твердый препарат для последующего формирования можно затем разделить на единичные лекарственные формы типа, описанного выше, содержащие от 0,1 до примерно 50 мг активного ингредиента по настоящему изобретению. Таблетки или пилюли по настоящему изобретению можно покрыть оболочкой или иначе составить для получения лекарственной формы пролонгированного действия. Например, таблетка или пилюля может включать внутреннее ядро, содержащее активное соединение, и внешний слой в виде оболочки, покрывающей ядро. Внешняя оболочка может быть кишечным слоем, который служит для защиты от распада в желудке, и позволяет внутреннему ядру проходить интактным в двенадцатиперстную кишку или медленно высвобождаться. Для подобных кишечных слоев или оболочек можно использовать разнообразные вещества, включая ряд полимерных кислот и смеси полимерных кислот с обычными веществами, такими как шеллак, цетиловый спирт и ацетат целлюлозы.
Жидкие формы, в которые можно включать настоящие лекарственные препараты для перорального или инъекционного введения, включают водные растворы, соответствующим образом ароматизированные сиропы, водные или масляные суспензии, и ароматизированные эмульсии с пищевыми маслами, такими как масло семян хлопчатника, кунжутное масло, кокосовое масло или арахисовое масло, а также эликсиры и подобные фармацевтические носители. Пригодные диспергирующие или суспендирующие агенты для водных суспензий включают синтетические и натуральные камеди, такие как трагакант, аравийская камедь, альгинат, декстран, натриевую соль карбоксиметилцеллюлозы, желатин, метилцеллюлозу или поливинилпирролидон. Другие диспергирующие агенты, которые можно использовать, включают глицерин и тому подобное. Для парентерального введения желательны стерильные суспензии и растворы. Когда желательно внутривенное введение, используются изотонические препараты, которые обычно содержат пригодные консерванты. Препараты также можно составлять в виде глазных растворов или суспензий, т.е. глазных капель, для внутриглазного введения.
Следовательно, настоящее изобретение также относится к облегчению или лечению заболевания или состояния, при котором положительное воздействие оказывает модификация активности мускаринового рецептора, в частности, активности m1-рецептора, назначением терапевтически эффективного количества соединения по настоящему изобретению субъекту при необходимости такого лечения. Подобные заболевания или состояния могут, например, возникнуть в результате неадекватной стимуляции или активации мускариновых рецепторов. Ожидается, что при использовании соединений, которые являются избирательными для определенного субтипа мускариновых рецепторов, в частности m1, можно в значительной степени избежать проблем с побочными эффектами, наблюдаемыми с известными мускариновыми препаратами, такими как тахикардия или брадикардия, или желудочно-кишечные эффекты.
Термин «субъект», использованный здесь, относится к животному, предпочтительно млекопитающему, наиболее предпочтительно человеку, который является объектом лечения, наблюдения или эксперимента.
Термин «терапевтически эффективное количество», использованный здесь, означает количество активного вещества или фармацевтического агента, которое вызывает ответную биологическую или лечебную реакцию в ткани, системе, животном или человеке, которой добивается исследователь, ветеринар, медицинский врач или другой клиницист, которая включает ослабление симптомов заболевания, которое подлежит лечению.
Предпочтительно, соединения общей формулы I проявляют субтиповую избирательность для мускариновых рецепторов m1-субтипа. Также соединения проявляют избирательность к мускариновым рецепторам m1-субтипа по сравнению с другими тестированными, связанными с G-протеином рецепторами, включая серотониновые, гистаминовые, дофаминовые или адренергические рецепторы. Одним важным значением этой избирательности является то, что эти соединения могут быть эффективными для лечения или ослабления ряда заболеваний или нарушений центральной нервной системы без проявления нежелательных побочных эффектов, ранее наблюдавшихся с неизбирательными соединениями.
Способность соединений по настоящему изобретению проявлять избирательность для мускариновых рецепторов m1-субтипа делает их потенциально очень полезными для лечения ряда заболеваний или нарушений, характеризующихся нарушением познавательной функции, такой как дефицитное нарушение внимания, или нейродегенеративные заболевания, такие как болезнь Альцгеймера, другие формы возрастного снижения познавательной функции, например, старческое слабоумие, или связанные со слабоумием симптомы, такие как пониженная двигательная активность, изменения настроения, анэргия, апатия, беспокойство и агрессивное поведение. В настоящее время полагают, что мускариновый m1-рецептор может также участвовать в регуляции внутриглазного давления и, следовательно, мускариновые m1-агонисты можно использовать для лечения или облегчения глазных заболеваний, таких как глаукома.
Преимущественно соединения по настоящему изобретению можно вводить в одной суточной дозе или общую суточную дозу можно назначать в разделенных дозах два, три или четыре раза в день. Кроме того, соединения по настоящему изобретению можно вводить в интраназальной форме посредством местного использования пригодных носителей для интраназального введения или через кожу, используя такие формы лепешек на кожу для трансдермального прохождения, хорошо известных специалистам в этой области. При назначении в форме системы для доставки через кожу введение будет в большей степени непрерывным, чем прерывистым во время курса введения.
Курс введения, в котором используются соединения по настоящему изобретению, выбирается в соответствии с разнообразными факторами, включая тип, вид, возраст, массу, пол и медицинское состояние пациента; тяжесть заболевания, которое лечится, путь введения, состояние функции почек и печени у пациента и конкретное используемое вещество. Врач или ветеринар обычной квалификации может легко определить или назначить эффективное количество лекарственного препарата, необходимое для профилактики, противодействия или остановки прогрессирования заболевания или нарушения, которое лечится.
Ежедневное введение продуктов может колебаться в широких пределах от 0,01 до 100 мг на взрослого человека в день. Для перорального введения предпочтительны композиции в форме таблеток, содержащих 0,01, 0,05, 0,1, 0,5, 1,0, 2,5, 5,0, 10,0, 15,0, 25,0 или 50,0 мг активного ингредиента для симптоматической регуляции дозировки для пациента, который подвергается лечению. Единичная доза обычно содержит примерно от 0,001 мг до 50 мг активного ингредиента, предпочтительно примерно от 1 мг до 10 мг активного ингредиента. Эффективное количество лекарственного препарата обычно доставляется в дозе примерно от 0,0001 мг/кг до примерно 25 мг/кг массы тела в день. Предпочтительно, пределы составляют примерно от 0,001 до 10 мг/кг массы тела в день и особенно примерно от 0,001 мг/кг до 1 мг/кг массы тела в день. Соединения можно вводить курсом 1-4 раза в день.
Соединения по настоящему изобретению можно использовать per se в соответствующих дозах, определенных обычным тестированием для получения оптимального фармакологического эффекта на мускариновый рецептор, в частности, мускариновый рецептор m1-субтипа, в то же время сводя до минимума любые потенциальные токсические или иные нежелательные эффекты. Кроме того, в некоторых случаях может быть желательным совместное или последовательное введение других агентов, которые улучшают эффект соединения.
Фармакологические свойства и избирательность соединений по этому изобретению для мускариновых рецепторов определенного субтипа можно продемонстрировать с помощью ряда методов анализа с использованием рекомбинантных рецепторных субтипов, предпочтительно рецепторов человека, если таковые имеются в распоряжении, например, обычными способами вторичного переносчика или связывания. Особенно удобной функциональной системой для теста является селекция и амплификация рецепторов, раскрытые в патенте США 5707798, в котором описывается способ скрининга биоактивных соединений при использовании способности клеток, трансфектированных ДНК рецептора, например, кодирующей различные мускариновые субтипы, амплифицировать в присутствии лиганда рецептора. Клеточная амплификация детектируется по повышенным уровням маркера, также экспрессируемого клетками.
Изобретение далее раскрывается в следующих примерах, которые никоим образом не предназначаются для ограничения объема изобретения в соответствии с формулой изобретения.
ПРИМЕРЫ
Пример I - 4-н-Бутил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин (5)
1-Бензил-4-н-бутилиденпиперидин (2). В 3-горлую колбу емкостью 500 мл, снабженную мешалкой, загрузили гидрид натрия (1,61 г, 67 ммоль) и ДМСО (40 мл). Полученную суспензию нагревали до 90°С в течение 30 мин, пока не закончилось выделение водорода. Суспензию охладили на ледяной бане в течение 20 мин с последующим добавлением суспензии бромида бутилтрифенилфосфония (26,6 г, 67 ммоль) в ДМСО (70 мл). Красную смесь перемешивали в течение 15 мин при комнатной температуре. В течение 30 мин медленно добавили 1-бензил-4-пиперидон 1 (14,0 г, 74 ммоль) и смесь перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавили Н2О (200 мл) с последующей экстракцией гептаном (4 × 100 мл) и этилацетатом (2 × 100 мл). Объединенные органические фазы высушивали и выпаривали досуха, получив 38,1 г желтого масла. Масло перегнали, получив 14,9 г (88%) 2, т.кип. 101-105°С (0,1 мм рт.ст.). 1Н ЯМР (CDCl3) 0,90-0,95 (т, 3Н), 1,25-1,41 (м, 2Н), 1,90-2,20 (м, 2Н), 2,18-2,30 (м, 4Н), 2,40-2,45 (м, 4Н), 2,50 (с, 2Н), 5,17 (т, 1Н), 7,20-7,42 (м, 5Н).
4-н-Бутилпиперидин (3). В колбу емкостью 500 мл, снабженную мешалкой, добавили суспензию 2 (13,2 г, 58 ммоль) и 10% палладий на угле (1,2 г) в этаноле (70 мл) с последующим добавлением концентрированной соляной кислоты (1,5 мл). Из реакционной колбы откачали воздух и ввели в нее водород. В целом израсходовано 2,5 дм3 водорода. Реакционную смесь профильтровали и выпаривали и остаток растворили в Н2О (40 мл) и NaOH (20 мл, 2М) с последующей экстракцией этилацетатом (3 × 100 мл). Объединенные органические фазы промыли рассолом (30 мл) и выпаривали досуха, получив 7,1 г сырого 3. Сырой продукт подвергали колоночной хроматографии [элюент:гептан:EtOAc (4:1)], получив чистое 3 (2,7 г, 33%). 1Н ЯМР (CDCl3) 0,85 (т, 3Н), 1,0-1,38 (м, 9Н), 1,65 (дд, 2Н), 2,38 (с, 1Н), 2,55 (дт, 2Н), 3,04 (дт, 2Н).
4-(4-н-Бутилпиперидин-1-ил)бутанитрил (4). В колбу с магнитной мешалкой емкостью 100 мл поместили 3 (2,3 г, 16,4 ммоль), 4-бромбутиронитрил (2,4 г, 16,4 ммоль), порошкообразный карбонат калия (2,5 г, 18 ммоль) в ацетонитриле (20 мл). Реакционную смесь перемешивали при комнатной температуре в течение 5 ч с последующим добавлением Н2О (15 мл). Смесь экстрагировали этилацетатом (3 × 30 мл) и объединенные органические фазы выпаривали досуха, получив 3,9 г сырого 4. Сырой продукт подвергали колоночной хроматографии [элюент:гептан:EtOAc (1:1)], получив чистое 4 (2,3 г, 87%). 1Н ЯМР (CDCl3) 0,82 (т, 3Н), 1,19-1,37 (м, 9Н), 1,64-1,75 (д, 2Н), 1,84-2,01 (м, 4Н), 2,39-2,54 (м, 4Н), 2,89-2,97 (д, 2Н).
4-н-Бутил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин (5). В высушенную в сушильном шкафу колбу емкостью 25 мл загрузили Mg стружки (125 мг, 5,2 ммоль), которые активировали при использовании теплового металлизатора. В инертной атмосфере добавили суспензию 2-йоданизола (1,13 г, 5,2 ммоль) в Et2O (4 мл) и реакционную смесь выдерживали при комнатной температуре в течение 1 ч. Добавили соединение 4 (720 мг, 3,4 ммоль), растворенное в Et2O (4 мл), и смесь кипятили с обратным холодильником в течение ночи. Добавили ТГФ (15 мл) и серную кислоту (4 мл, 2 М) и реакционную смесь перемешивали в течение 4 ч с последующим добавлением NaOH (6 мл, 2М). Реакционную смесь экстрагировали этилацетатом (3 × 50 мл) и объединенные органические фазы выпаривали досуха, получив 1,2 г сырого 5. Сырой продукт подвергали колоночной хроматографии [элюент:CH2Cl2:CH3OH (99:1)], получив чистое 5 (0,42 г, 26%). 1Н ЯМР (CDCl3) 0,83 (т, 3Н), 1,20-1,42 (м, 9Н), 1,65-1,73 (д, 2Н), 1,96-2,20 (м, 4Н), 2,53 (т, 2Н), 3,02-3,17 (м, 4Н), 3,89 (с, 3Н), 6,95-7,01 (м, 2Н), 7,44 (т, 1Н), 7,65 (д, 1Н).
Пример II - 3-Гидроксиметил-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин (7)
4-(3-Гидроксиметил-пиперидин-1-ил)-бутиронитрил (6). В высушенную в сушильном шкафу колбу емкостью 25 мл добавили пиперидин-3-ил-метанол (1,12 г, 10 ммоль) в ацетонитриле (10 мл) с последующим добавлением карбоната калия (1,38 г, 10 ммоль) и 4-бромбутиронитрила (0,90 мл, 9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 12 ч. Смесь профильтровали и выпаривали досуха. После добавления Н2О (20 мл) экстрагировали этилацетатом (3 × 20 мл) и объединенные органические фазы высушивали (MgSO4) и выпаривали досуха, получив 1,50 г сырого 6, которое использовали без дополнительной очистки в синтезе соединения 7.
3-Гидроксиметил-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин (7). В высушенную в сушильном шкафу колбу емкостью 50 мл добавили Mg стружки (780 мг, 32 ммоль), которые активировали при использовании теплового металлизатора в вакууме с последующим добавлением безводного ТГФ (7 мл). В инертной атмосфере добавили суспензию 2-йодтолуола (5,3 г, 24 ммоль) в ТГФ (10 мл) и реакционную смесь кипятили с обратным холодильником в течение 4 ч. Через шприц добавили суспензию соединения 6 (1,50 г, 8 ммоль) в ТГФ (5 мл) с последующим добавлением CuBr (23 мг, 0,16 ммоль, 2 моль%) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь погасили добавлением Н2SO4 (20 мл, 2 М) и перемешивали при комнатной температуре в течение 2 ч с последующим добавлением NaOH (8 мл, 2 М). После добавления ТГФ (15 мл) экстрагировали СН2Cl2 (3 × 20 мл) и органические фазы высушивали (MgSO4) и выпаривали досуха, получив 0,41 г сырого 7. Сырой продукт подвергали препаративной ВЭЖХ [элюент:буфер А:0,1% ТФК; буфер В:80% СН3CN + 0,1% ТФК], получив аналитически чистую пробу соединения 7. ЖХ-МС [М+Н]+ 275 (высчит. 275,2).
Пример III - 2-Пропил-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин (9)
4-(2-пропил-пиперидин-1-ил)-бутиронитрил (8). Смесь 2-пропилпиперидина (550 мг, 4,3 ммоль), 4-бромбутиронитрила (430 мг, 3,0 ммоль) и карбоната калия (550 мг, 4,0 ммоль) в ацетонитриле (5 мл) перемешивали при комнатной температуре в течение 12 ч с последующим добавлением насыщенного рассола (25 мл). Реакционную смесь экстрагировали этилацетатом (3 × 25 мл) и объединенные органические фазы высушивали (MgSO4) и выпаривали досуха, получив сырое 8. Сырой продукт подвергали колоночной хроматографии [элюент:СН2Cl2:МеОН (99:1)], получив чистое 8 (0,48 г, 83%); ЖХ-МС [М+Н]+ 194 (высчит. 194,2).
2-Пропил-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин (9). В высушенную в сушильном шкафу колбу добавили Mg стружки (97 мг, 4,1 ммоль), которые активировали при использовании теплового металлизатора в вакууме. В инертной атмосфере добавили суспензию 2-йодтолуола (380 мг, 2,8 ммоль) в Et2O (3 мл) и реакционную смесь кипятили с обратным холодильником в течение 1 ч. Через шприц добавили смесь соединения 8 (0,43 г, 2,2 ммоль) в СН2Cl2 (3 мл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь погасили добавлением Н2SO4 (10 мл, 2 М) и перемешивали при комнатной температуре в течение 12 ч с последующим добавлением NaOH (10 мл, 2 М). После добавления ТГФ (15 мл) экстрагировали этилацетатом (3 × 50 мл) и объединенные органические фазы промывали рассолом (10 мл) и NaOH (10 мл, 2 М), высушивали (MgSO4) и выпаривали досуха, получив 0,43 г сырого 9. Сырой продукт подвергали препаративной ВЭЖХ [элюент:буфер А:0,1% ТФК; буфер В:80% СН3CN + 0,1% ТФК], получив аналитически чистую пробу соединения 9; ЖХ-МС [М+Н]+287 (высчит. 287,2).
Пример IV -1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперидин (11)
В высушенную в сушильном шкафу колбу емкостью 10 мл добавили Mg стружки (97 мг, 4,1 ммоль), которые активировали при использовании теплового металлизатора в вакууме. В инертной атмосфере добавили суспензию 2-йодтолуола (380 мг, 3,0 ммоль) в Et2O (3 мл) и реакционную смесь кипятили с обратным холодильником в течение 1 ч. Посредством шприца добавили суспензию 4-пиперидин-1-ил-бутанитрила (10) (Dahlbom et al., Acta. Chem. Scand. 1951, 5, 690-697) (0,305 мг, 2,0 ммоль) в СН2Cl2 (3 мл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь погасили добавлением Н2SO4 (10 мл, 2 М) и перемешивали при комнатной температуре в течение 12 ч с последующим добавлением NaOH (12 мл, 2 М). После добавления ТГФ (15 мл) экстрагировали этилацетатом (3 × 50 мл) и объединенные органические фазы промывали рассолом (10 мл) и NaOH (10 мл, 2 М) и высушивали (MgSO4) и выпаривали досуха, получив 0,21 г сырого 11. Сырой продукт подвергали препаративной ВЭЖХ [элюент:буфер А:0,1% ТФК; буфер В:80% СН3CN + 0,1% ТФК], получив аналитически чистую пробу соединения 11; ЖХ-МС [М+Н]+ 245 (высчит. 245,2).
Пример V - 4-Метил-1-[4-(4-бромфенил)-4-оксо-1-бутил]пиперидин (12)
В высушенную в колбу емкостью 10 мл добавили 4-метилпиперидин (719 мг, 6 ммоль), диоксан (5 мл) с последующим добавлением карбоната калия (0,30 г, 2,18 ммоль), йодида калия (10 мг) и 4-бром-4-хлорбутирофенона (785 мг, 2,76 ммоль). Реакционную смесь выдержали при 110°С в течение 12 ч с последующим растворением в Н2О (10 мл). Реакционную смесь экстрагировали Et2O (3 × 15 мл) и объединенные органические фазы высушивали (MgSO4) и выпаривали досуха, получив 0,50 г сырого 12. Сырой продукт подвергали препаративной ВЭЖХ [элюент:буфер А:0,1% ТФК; буфер В:80% СН3CN + 0,1% ТФК], получив аналитически чистую пробу соединения 12; ЖХ-МС [М+Н]+ 322 (высчит. 323,1).
Пример VI - 1-[4-(2-метилфенил)-4-оксо-1-бутил]пирролидин (13)
В высушенную в сушильном шкафу колбу емкостью 10 мл загрузили Mg стружки (30 мг, 1,2 ммоль), которые активировали при использовании теплового металлизатора в вакууме. В инертной атмосфере добавили суспензию 2-йодтолуола (0,22 г, 1,0 ммоль) в Et2O (2 мл) и реакционную смесь кипятили с обратным холодильником в течение 1 ч. Посредством шприца добавили 4-пирролидин-1-ил-бутиронитрил (Burckhalter et al., J. Org. Chem. 1961, 26, 4070-4076) (0,14 г, 1,0 ммоль) в СН2Cl2 (2 мл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь погасили добавлением Н2SO4 (10 мл, 2 М) и перемешивали при комнатной температуре в течение 2 ч с последующим добавлением NaOH (10 мл, 2 М). После добавления ТГФ (15 мл) экстрагировали этилацетатом (3 × 20 мл), и органические фазы высушивали (MgSO4) и выпаривали досуха, получив 0,12 г сырого 13. Сырой продукт подвергали препаративной ВЭЖХ [элюент:буфер А:0,1% ТФК; буфер В:80% СН3CN + 0,1% ТФК], получив аналитически чистую пробу соединения 13; ЖХ-МС [М+Н]+ 231 (высчит. 231,3).
Пример VII - 4-Метил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперазин (15)
4-(4-Метил-пиперазин-1-ил)бутиронитрил (14). В колбу емкостью 25 мл внесли 1-метилпиперазин (0,52 г, 5,1 ммоль), 4-бромбутиронитрил (0,78 г, 5,3 ммоль) и карбонат калия (0,71 г, 5,3 ммоль), суспендированные в ацетонитриле (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч с последующим добавлением Н2O (20 мл) и экстрагировали этилацетатом (3 × 25 мл). Объединенные органические фазы промывали рассолом (25 мл), высушивали (MgSO4) и выпаривали досуха, получив 0,72 г сырого 14, который использовали без дополнительной очистки в синтезе соединения 15.
4-Метил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперазин (15). В высушенную в сушильном шкафу колбу емкостью 10 мл добавили Mg стружки (116 мг, 4,0 ммоль), которые активировали при использовании теплового металлизатора в вакууме. В инертной атмосфере добавили смесь 2-йодтолуола (0,65 г, 3,0 ммоль) в Et2O (3 мл) и реакционную смесь кипятили с обратным холодильником в течение 1 ч. Посредством шприца добавили раствор соединения 14 (0,33 г, 2,0 ммоль) в СН2Cl2 (3 мл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь погасили добавлением H2SO4 (6 мл, 2 М) и перемешивали при комнатной температуре в течение 2 ч с последующим добавлением NaOH (8 мл, 2 М). После добавления ТГФ (15 мл) экстрагировали CH2Cl2 (3 × 20 мл). Органические фазы высушивали (MgSO4) и выпаривали досуха, получив 0,26 г сырого 15. Сырой продукт подвергли препаративной ВЭЖХ [элюент:буфер А:0,1% ТФК; буфер В:80% СН3CN + 0,1% ТФК], получив аналитически чистую пробу соединения 15; ЖХ-МС [М+Н]+ 260 (высчит. 260,4).
Пример VIII - 4-н-Бутил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперазин (17)
4-(4-Бутил-пиперазин-1-ил)бутиронитрил (16). В колбу емкостью 25 мл внесли 1-бутилпиперазин (712 мг, 5,0 ммоль), 4-бромбутиронитрил (779 мг, 5,3 ммоль) и карбонат калия (687 мг, 5,0 ммоль), суспендированные в ацетонитриле (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 12 ч с последующим добавлением Н2O (20 мл) и экстрагировали этилацетатом (3 × 25 мл). Объединенные органические фазы промывали рассолом (25 мл), высушивали (MgSO4) и выпаривали досуха, получив 0,89 г сырого 16, которое использовали без дополнительной очистки в синтезе соединения 17.
4-н-Бутил-1-[4-(2-метилфенил)-4-оксо-1-бутил]пиперазин (17).
В высушенную в сушильном шкафу колбу емкостью 10 мл загрузили Mg стружки (100 мг, 4,0 ммоль), которые активировали при использовании теплового металлизатора в вакууме. В инертной атмосфере добавили смесь 2-йодтолуола (0,66 г, 3,0 ммоль) в Et2O (3 мл) и реакционную смесь кипятили с обратным холодильником в течение 1 ч. Через шприц добавили суспензию соединения 16 (0,43 г, 2,0 ммоль) в СН2Cl2 (3 мл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь погасили добавлением Н2SO4 (6 мл, 2 М) и перемешивали при комнатной температуре в течение 2 ч с последующим добавлением NaOH (8 мл, 2 М). После добавления ТГФ (15 мл) экстрагировали CH2Cl2 (3 × 20 мл) и органические фазы высушивали (MgSO4) и выпаривали досуха, получив 0,50 г сырого 17. Сырой продукт подвергали препаративной ВЭЖХ [элюент:буфер А:0,1% ТФК; буфер В:80% СН3CN + 0,1% ТФК], получив аналитически чистую пробу соединения 17; ЖХ-МС [М+Н]+ 302 (высчит. 302,5).
Пример IX - 4-н-Бутил-1-[4-(2-этоксифенил)-4-оксо-1-бутил]пиперидин (18)
В высушенную в сушильном шкафу колбу емкостью 10 мл добавили Mg стружки (94 мг, 3,8 ммоль), которые активировали при использовании теплового металлизатора в вакууме. В инертной атмосфере добавили суспензию 1-этокси-2-йодбензола (0,71 г, 2,9 ммоль) в Et2O (3 мл) и реакционную смесь кипятили с обратным холодильником в течение 3 ч. Соединение 4 (0,40 г, 1,9 ммоль) растворили в СН2Cl2 (3 мл) и смесь перемешивали при 40°С в течение еще 3 ч. Реакционную смесь погасили добавлением H2SO4 (10 мл, 2 М) и оставили при перемешивании при комнатной температуре на ночь с последующим добавлением NaOH (20 мл, 2 М) до щелочной реакции. Реакционную смесь экстрагировали этилацетатом (3 × 50 мл) и объединенные органические фазы промывали рассолом (10 мл) и NaOH (10 мл, 2 М) и объединенные органические фазы высушивали (MgSO4) и выпаривали досуха, получив 0,60 г сырого 18. Сырой продукт подвергали колоночной хроматографии [элюент:тол:EtOAc (1:1)], получив чистое 18 (0,32 г, 34%); ЖХ-МС [М+Н]+ 331 (высчит. 331,5).
Пример Х - 4-н-Бутил-1-[4-(2,3-диметилфенил)-4-оксо-1-бутил]пиперидин (19)
В высушенную в сушильном шкафу колбу емкостью 10 мл добавили Mg стружки (94 мг, 3,8 ммоль), которые активировали при использовании теплового металлизатора в вакууме. В инертной атмосфере добавили суспензию 1-йод-2,3-диметилбензола (0,69 г, 3,0 ммоль) в Et2O (5 мл) при самопроизвольном закипании и реакционную смесь кипятили с обратным холодильником в течение 4 ч. Суспензию соединения 4 (0,41 г, 2,0 ммоль) в СН2Cl2 (2 мл) добавили в реакционную смесь и оставили при комнатной температуре на ночь. Реакционную смесь погасили добавлением H2SO4 (7 мл, 2 М) и перемешивали при комнатной температуре в течение 3 ч с последующим добавлением NaOH (20 мл, 2 М) до щелочной реакции. Реакционную смесь экстрагировали этилацетатом (3 × 50 мл) и объединенные органические фазы промывали рассолом (10 мл) и NaOH (10 мл, 2М) и органические фазы высушивали (MgSO4) и выпаривали досуха, получив 0,69 г сырого 19. Сырой продукт подвергали колоночной хроматографии [элюент:СН2Cl2:MeOH (99:1)], получив чистое 19 (0,40 г, 64%); ЖХ-МС [М+Н]+ 315 (высчит. 315,5).
Пример XI - 4-н-Бутил-1-[4-(2,4-диметилфенил)-4-оксо-1-бутил]пиперидин (20)
В высушенную в сушильном шкафу колбу емкостью 10 мл загрузили Mg стружки (95 мг, 3,9 ммоль), которые активировали при использовании теплового металлизатора в вакууме. В инертной атмосфере добавили суспензию 1-йод-2,4-диметилбензола (0,69 г, 2,9 ммоль) в Et2O (4,5 мл) при самопроизвольном закипании, и реакционную смесь кипятили с обратным холодильником в течение 3 ч. Соединение 4 (0,41 г, 2,0 ммоль), растворенное в СН2Cl2 (2 мл), добавили в инертной атмосфере в реакционную смесь и оставили при перемешивании при комнатной температуре на ночь. Реакционную смесь погасили добавлением H2SO4 (8 мл, 2 М) и перемешивали при комнатной температуре в течение 4 ч, затем реакционную смесь подщелачивали добавлением NaOH (20 мл, 2 М). После добавления ТГФ (20 мл) экстрагировали этилацетатом (3 × 50 мл) и объединенные органические фазы промывали рассолом (10 мл) и NaOH (10 мл, 2 М), и органические фазы высушивали (MgSO4) и выпаривали досуха, получив 0,61 г сырого 20. Сырой продукт подвергали колоночной хроматографии [элюент:СН2Cl2:MeOH (99:1)], получив чистое 20 (0,21 г, 35%); ЖХ-МС [М+Н]+ 315 (высчит. 315,5).
Пример XII - 4-н-Бутил-1-[4-(2-метоксифенил)-4-оксо-1-бутил]пиперидин (21)
В высушенную в сушильном шкафу колбу емкостью 10 мл загрузили Mg стружки (0,12 г, 4,9 ммоль), которые активировали при использовании теплового металлизатора в вакууме. В инертной атмосфере добавили суспензию 1-бром-2-этилбензола (0,66 г, 3,6 ммоль) в Et2O (2 мл) и реакционную смесь кипятили с обратным холодильником в течение 2 ч. Через шприц добавили суспензию соединения 4 (0,50 г, 2,4 ммоль) в СН2Cl2 (2 мл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь погасили добавлением H2SO4 (14 мл, 2 М) и перемешивали при комнатной температуре в течение 2 ч с последующим добавлением NaOH (20 мл, 2 М). После добавления ТГФ (20 мл) экстрагировали этилацетатом (3 × 50 мл) и объединенные органические фазы промывали рассолом (10 мл) и NaOH (10 мл, 2 М) и органические фазы высушивали (MgSO4) и выпаривали досуха, получив 0,75 г сырого 21. Сырой продукт подвергали колоночной хроматографии [элюент:СН2Cl2:MeOH (99:1)], получив чистое 21 (0,68 г, 90%); ЖХ-МС [М+Н]+ 315 (высчит. 315,5).
Пример XIII - 4-н-Бутил-1-[4-(2,4-диметилфенил)-4-оксо-1-бутил]пиперидин (22)
В высушенную в сушильном шкафу колбу емкостью 10 мл загрузили Mg стружки (88 мг, 3,6 ммоль) и активировали при использовании теплового металлизатора в вакууме. В инертной атмосфере добавили суспензию 1-йод-2-метоксиметилбензола (0,67 г, 2,7 ммоль) в Et2O (4 мл), и реакционную смесь кипятили с обратным холодильником в течение 1 ч. Посредством шприца добавили суспензию соединения 8 (0,38 г, 1,8 ммоль) в СН2Cl2 (4 мл), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь погасили добавлением H2SO4 (10 мл, 2 М) и перемешивали при комнатной температуре в течение 2 ч с последующим добавлением NaOH (10 мл, 2 М). После добавления ТГФ (15 мл) экстрагировали этилацетатом (3 × 50 мл) и объединенные органические фазы промывали рассолом (10 мл) и NaOH (10 мл, 2 М) и органические фазы высушивали (MgSO4) и выпаривали досуха, получив 0,51 г сырого 22. Сырой продукт подвергали колоночной хроматографии [элюент:СН2Cl2:MeOH (99:1)], получив чистое 22 (0,14 г, 23%); ЖХ-МС [М+Н]+ 331 (высчит. 331,5).
Пример XIV - 4-н-Бутил-1-[4-(2-пиридинил)-4-оксо-1-бутил]пиперидин (24)
Метиловый эфир 4-(4-бутил-пиперидин-1-ил)масляной кислоты (23). В реакционную колбу емкостью 25 мл добавили метиловый эфир 4-бром-масляной кислоты (2,04 г, 11,2 ммоль), соединение 3 (1,51 г, 10,8 ммоль) и карбонат калия (1,63 г, 11,8 ммоль), суспендированные в СН3CN (10 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре с последующим фильтрованием и выпариванием досуха. После добавления Н2О (50 мл) экстрагировали этилацетатом (3 × 100 мл). Объединенные органические фазы высушивали (MgSO4) и выпаривали досуха, получив 2,84 г сырого 23. Сырой продукт подвергали колоночной хроматографии [элюент:СН2Cl2:MeOH (99:1)], получив чистое 23 (1,93 г, 75%); ЖХ-МС [М+Н]+ 241 (высчит. 241,2).
4-н-Бутил-1-[4-(2-пиридинил)-4-оксо-1-бутил]пиперидин (24). В сухую реакционную колбу емкостью 25 мл добавили 2-бромпиридин (200 мг, 1,3 ммоль), растворенный в СН2Cl2 (3 мл), и температуру довели до -78°С. После перемешивания в течение 20 мин добавили н-BuLi (0,84 мл, 1,4 ммоль) в инертной атмосфере. Еще через 30 мин добавили раствор 23 в СН2Cl2 (2 мл). Реакционной смеси дали нагреться до комнатной температуры в течение ночи перед погашением Н2SO4 (5 мл, 1 М). Реакционную смесь экстрагировали этилацетатом (6 × 25 мл) и объединенные органические фазы высушивали (MgSO4) и выпаривали досуха, получив 0,31 г сырого 24. Сырой продукт подвергали колоночной хроматографии [элюент:СН2Cl2:MeOH (10:1)], получив чистое 24 (75 мг, 12%); ЖХ-МС [М+Н]+ 288 (высчит. 288,2).
Пример XV - 4-н-Бутил-1-[4-(2-гидроксифенил)-4-оксо-1-бутил]пиперидин (27)
1-бензилокси-2-йод-бензол (25). В высушенной в сушильном шкафу колбе емкостью 25 мл растворили 2-йодфенол (1,03 г, 4,7 ммоль) и карбонат калия (0,71 г, 5,2 ммоль) в сухом ацетоне (10 мл). Смесь перемешивали в течение 15 мин с последующим добавлением бензилбромида (0,61 мл, 5,2 ммоль) и оставили на ночь при комнатной температуре. После добавления Н2О (50 мл) экстрагировали этилацетатом (3 × 50 мл) и объединенные органические фазы высушивали (MgSO4) и выпаривали досуха, получив 1,7 г сырого 25. Сырой продукт подвергали колоночной хроматографии [элюент:гептан:EtOAc (9:1)], получив чистое 25 (1,2 г, 81%); ЖХ-МС [М+Н]+ 310 (высчит. 310,0).
4-н-Бутил-1-[4-(2-бензилоксифенил)-4-оксо-1-бутил]пиперидин (26). В высушенную в сушильном шкафу колбу емкостью 25 мл добавили Mg стружки (123 мг, 5,1 ммоль), которые активировали при использовании теплового металлизатора в вакууме. В инертной атмосфере добавили раствор 1-бензилокси-2-йод-бензола (25) (1,18 г, 3,8 ммоль) в Et2O (10 мл) и реакционную смесь кипятили с обратным холодильником в течение 3,5 ч. Раствор 4-(4-н-бутилпиперидин-1-ил)бутанитрила 4 (0,53 г, 2,5 ммоль) в СН2Cl2 (3 мл), добавили к реакционной смеси и перемешивали при 40°С в течение ночи. Реакционную смесь погасили добавлением Н2SO4 (10 мл, 2 М) и оставили перемешиваться в течение 1 ч с последующим добавлением NaOH (20 мл, 2 М) до щелочной реакции. Реакционную смесь экстрагировали этилацетатом (3 × 50 мл) и объединенные органические фазы промывали рассолом (10 мл) и NaOH (10 мл, 2 М) и объединенные органические фазы высушивали (MgSO4) и выпаривали досуха, получив 1,28 г сырого 26. Сырой продукт подвергали колоночной хроматографии [элюент:тол:EtOAc (1:1)], получив чистое 26 (0,51 г, 51%); ЖХ-МС [М+Н]+ 393 (высчит. 393,7).
4-н-Бутил-1-[4-(2-гидроксифенил)-4-оксо-1-бутил]пиперидин (27). В реакционную колбу емкостью 25 мл добавили раствор 4-н-бутил-1-[4-(2-бензилоксифенил)-4-оксо-1-бутил]пиперидина (26) (49 мг, 1,2 ммоль) в сухом EtOH (10 мл) и концентрированной HCl (0,1 мл) с последующим добавлением палладия на угле (40 мг). В реакционную колбу ввели Н2 при использовании баллона и оставили перемешиваться при комнатной температуре на ночь в атмосфере Н2. Реакционную смесь подщелачивали добавлением NaOH (2 мл, 2,0 М) и профильтровали через целит. Водную фазу экстрагировали этилацетатом (3 × 50 мл) и объединенные органические фазы промывали рассолом (10 мл) и NaOH (10 мл, 2 М), высушивали (MgSO4) и выпаривали досуха, получив 0,42 г сырого 27. Сырой продукт подвергали колоночной хроматографии [элюент:СН2Cl2:MeOH (99:1)], получив чистое 27 (0,21 мг, 58%); ЖХ-МС [М+Н]+ 303 (высчит. 303,2).
Пример XVI - Скрининг испытываемых соединений в тесте с использованием мускариновых рецепторов субтипов m1, m2, m3, m4 и m5
Трансфекция клеток ДНК мускариновых рецепторов (общая методика). Клетки NIH 3Т3 (получены от Американской Типовой культуральной коллекции под номером АТСС CRL 1658) выращивали при 37°С в термостате с увлажненной атмосферой (5% СО2) в модифицированной по способу Дульбекко среде Игла (ДМЕМ) с добавлением 4,5 г/л глюкозы, 4 мМ глутамина, 50 Е/мл пенициллина, 50 Е/мл стрептомицина (получены от Advanced Biotechnologies, Inc., Gaithersburg, MD) и 10% сыворотки теленка (получена от Sigma, St. Louis, MI). Клетки обработали трипсином-ЭДТА, быстро вылили и посеяли в концентрации 2×106 на чашку размером 15 см в 20 мл ДМЕМ, содержащей 10% сыворотки теленка.
Мускариновые рецепторы m1-m5-субтипов клонировали в основном, как описано Bonner et al., Science 237, 1987, p. 527, and Bonner et al., Neuron 1, 1988, p. 403. Для рецепторов m2 и m4 клетки совместно трансфектировали с ДНК, кодирующей химеры белка Gq и пяти аминокислот с концевой карбоксильной группой белка Gi (конструкция Gq-i5 описывается Conklin et al., Nature 363, 1993, p. 274).
В первый день клетки трансфектировали с использованием реагента для трансфекции Superfect (получен от из Qiagen, Valencia, CA) в соответствии с инструкциями изготовителя. На чашку внесли ДНК рецептора, ДНК β-gal (pSI-β-галактозидаза, получена от Promega, Madison, WI), химерную ДНК Gq-i5 в отношении рецепторов субтипов m2 и m4 и ДНК спермы семги (получена от Sigma, St. Louis, MI) в качестве наполнителя, в целом 20 мкг ДНК. Перед внесением в чашки к ДНК добавили 60 мкл Superfect и тщательно перемешали путем набора и спуска пипеткой жидкости несколько раз. Смесь инкубировали при комнатной температуре в течение 10-15 мин. Среду аспирировали и в чашки добавили 12 мл свежей ДМЕМ, содержащей 10% сыворотки теленка и 50 Е/мл пенициллина/стрептомицина. Раствор ДНК-Superfect еще раз перемешали пипеткой и внесли в чашки, которые переворачивали для равномерного распределения смеси ДНК по поверхности. Клетки инкубировали в течение ночи при 37°С и 5% СО2.
После инкубации среду аспирировали и чашки промыли один раз 15 мл забуференного солевого раствора Хэнкса. Чашки переворачивали для гарантии тщательного промывания. В чашки добавили 20 мл свежей среды ДМЕМ, обогащенной 10% сыворотки теленка и 50 Е/мл пенициллина/стрептомицина. Клетки инкубировали в течение 24-28 ч, пока на чашках не образовался 100% слитый слой.
Анализ клеток NIH 3Т3, трансфектированных рецепторами мускариновых субтипов (общая методика). ДМЕМ, содержащую 2% Cyto-SF3, нагревали при 37°С на водяной бане в стерильных условиях. Стерильные рабочие маточные растворы тестируемых соединений, которые подверглись анализу, готовили разведением соединений в ДМЕМ до 8× конечной концентрации для тестирования. В тесте в качестве положительного контроля использовали соединение (карбахол) и его также развели в ДМЕМ до 8× конечной концентрации. 50 мкл ДМЕМ, содержащей 2% Cyto-SF3, внесли в каждую лунку 96-луночного планшета для микротитрования в стерильных условиях. Затем в верхние лунки планшетов добавили 16 мкл растворов соединений и разводили растворы, отбирая 16 мкл растворов соединений из верхних лунок и внося их пипеткой в следующий ряд лунок. Эту процедуру повторили с каждым последующим рядом лунок, за исключением того, что 50 мкл одной среды внесли в лунки основного контроля (лунки, которые содержали среду и клетки, но не тестируемые соединения) и лунки контроля на планшет (лунки, содержащие среду, но не тестируемые соединения и клетки). Планшеты поместили в термостат при 37°С для уравновешивания температуры и рН.
Когда культуры клеток достигали 100% сливаемости, среду аспирировали и каждую чашку промывали 15 мл забуференного солевого раствора Хэнкса (HBS). Клетки оставили в термостате примерно на 10-15 мин, пока HBS не стал слегка желтым. Затем HBS аспирировали и в каждую чашку добавили 1 мл трипсина и переворачивали так, чтобы полностью покрыть чашки. Чашки мягко встряхнули несколько раз для отделения клеток. После того как клетки сместились с поверхности, добавили 8 мл ДМЕМ, содержащей 10% сыворотки теленка и 50 Е/мл пенициллина и 50 Е/мл стрептомицина для ингибирования трипсина. Чашки промыли этой средой и клетки перенесли пипеткой в пробирку. Клетки отцентрифурировали при 1000 об/мин в течение 5-10 мин на центрифуге IEC Centra CL2 (изготовлена Sorvall). После этого среду осторожно аспирировали так, чтобы не сместить клетки. Клеточный осадок суспендировали в 1600 мкл ДМЕМ, содержащей 10% сыворотки теленка и 50 Е/мл пенициллина и 50 Е/мл стрептомицина, после чего внесли 20 мл ДМЕМ, с добавлением 2% Cyto-SF3. В лунки 96-луночного планшета для микротитрования, подготовленного, как описано выше, внесли 50 мкл этой клеточной суспензии (за исключением лунок контроля на планшет). Планшеты инкубировали в течение 4 суток при 37°С и 5% СО2.
После инкубации среду удалили при переворачивании планшетов для микротитрования и мягком их встряхивании, после чего их промокнули абсорбирующей бумагой. В каждую лунку внесли хромогенный субстрат (3,5 мМ о-нитрофенил-β-D-галоктопиранозида, 0,5% нонидет NP-40 в забуференном фосфатом физиологическом растворе) и планшеты инкубировали при 30°С, пока не получили максимальное поглощение при 405 нм. Из всех значений вычитали поглощение лунок основного контроля и контроля на планшет.
Результаты. Используя общую методику, описанную выше, клетки NIH 3Т3 совместно трансфектировали ДНК, кодирующей рецепторы m1-, m3- и m5-субтипов. Библиотека соединений, содержащая примерно 35000 небольших органических соединений (1 на лунку) была подвергнута скринингу против рецепторов по описанной выше методике. На фигуре 1 представлены данные, полученные с одного 96-луночного планшета в скрининге. На этом планшете два соединения были активны в отношении одного или более трансфектированных рецепторов. В общем скрининге было идентифицировано четыре относящихся соединения, проявивших активность. Для того чтобы определить, который из рецепторов был активирован в скрининге, соединение тестировали, как описано выше, против каждого из рецепторов, трансфектированных в отдельных клеточных культурах. Соединение А активировало только рецептор m1-субтипа, для которого оно было сильным неполным агонистом, индуцируя более слабую ответную реакцию, чем стандартное соединение карбахол.
В последующих опытах было установлено четыре соединения, которые избирательно активировали m1-рецептор без наличия значительной активности к мускариновым рецепторам m2, m3, m4 или m5. Наиболее активное соединение, соединение А, не было антагонистом индуцированных карбахолом ответных реакций мускариновых рецепторов пяти субтипов.
Соединение А дополнительно тестировали на активность агониста в отношении нескольких других рецепторов на α-адренергических рецепторах субтипов 1D, 1В, 1А, 2А, 2В и 2С, гистаминовом Н1 и серотониновых субтипов 5-НТ1А и 5-НТ2А. Соединение в этих тестах не проявило значительной активности. В опытах по оценке антагонистической активности соединение А не ингибировало ответные реакции α-адренергических рецепторов субтипов 2А, 2В или 2С, или серотониновых рецепторов субтипов 5-НТ1А или 5-НТ2А. Как показано на фигуре 2, ответные реакции, индуцированные соединением А, блокировались мускариновым антагонистом атропином с той же эффективностью, как ответные реакции, индуцированные мускариновым агонистом карбахолом.
Пример XVII - Тест R-SAT
Поставили тесты R-SAT (смотри патент США № 5707798, включенный здесь в качестве источника литературы), в которых клетки, трансфектированные рецепторами m1, m3 или m5, подвергали воздействию семи соединений в концентрации 1,5 мкМ. Реакцию клеток выражали в виде процента от максимальной ответной реакции (определялась в виде ответной реакции на 10 мкМ карбахола). Результаты представлены в следующей таблице.
1,5 M
1,5 M
1,5 M
(пример I)
(пример IX)
(пример XV)
(пример X)
(пример XI)
(пример XII)
Как указывается выше, соединения являются избирательными агонистами для m1-рецептора.
Описанное и заявленное здесь изобретение не ограничивается в объеме раскрытыми здесь определенными воплощениями, поскольку эти воплощения предназначаются в качестве иллюстрации нескольких аспектов изобретения. Любые равноценные воплощения включены в объем изобретения. Действительно различные модификации изобретения в дополнении к представленным и описанным здесь будут понятны специалистам в этой области из вышеприведенного описания. Подобные модификации также предназначаются для попадания в объем приложенной формулы изобретения.
Здесь цитируются различные источники литературы, раскрытие которых приводится в качестве источника литературы полностью.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1999 |
|
RU2230740C2 |
| МУСКАРИНОВЫЕ АГОНИСТЫ | 2001 |
|
RU2269523C2 |
| АГОНИСТЫ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2015 |
|
RU2678835C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СПИРОСОЕДИНЕНИЯ | 2010 |
|
RU2506266C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛИДИНОНА В КАЧЕСТВЕ АГЕНТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2002 |
|
RU2288919C2 |
| Бициклические азотсодержащие соединения как агонисты М1 мускариновых рецепторов | 2015 |
|
RU2685230C2 |
| АЗАЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ДЛЯ ПРИМЕНЕНИЯ ПРИ ЛЕЧЕНИИ ОПОСРЕДОВАННЫХ СЕРОТОНИНОМ ЗАБОЛЕВАНИЙ | 2001 |
|
RU2398765C1 |
| ДИАМИДНЫЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ АНТАГОНИСТА МУСКАРИНОВОГО РЕЦЕПТОРА И АКТИВНОСТЬЮ АГОНИСТА β АДРЕНЕРГИЧЕСКОГО РЕЦЕПТОРА | 2010 |
|
RU2676686C2 |
| СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ МУСКАРИНОВЫХ РЕЦЕПТОРОВ И АГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ БЕТА2-АДРЕНОРЕЦЕПТОРОВ | 2012 |
|
RU2606121C2 |
| ПИПЕРИДИНИЛПИРАЗОЛОПИРИМИДИНОНЫ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2713937C2 |
Изобретение относится к новым производным азотсодержащих гетероциклических соединений общей формулы I

где X1-X5 являются СН2 или один из них является NH, а другие X1-X5 являются СН2,
k равно 0 или 1; t равно 0, 1 или 2; радикалы R1, когда t равно 2, являются одинаковыми или разными;
R1 является нормальным или разветвленным C1-8 алкилом или C1-8 гидроксиалкокси;
А является фенилом или пиридинилом;
R2 является Н, гидроксилом, галогеном, C1-6 алкилом, С1-6 алкокси; n равно 0, 1-4, радикалы R2, где n>1, являются одинаковыми или разными;
р равно 0 или 1-5;
Y является -ОС(O)-,
Z является CH2, или
к их фармацевтически приемлемым солям.
Соединения формулы I обладают агонистической активностью в отношении мускариновых рецепторов и могут найти применение в медицине в качестве лекарственных препаратов для лечения нейродегенеративных заболеваний или заболеваний, связанных с повышением внутриглазного давления. 4 н. и 2 з.п. ф-лы, 1 табл., 2 ил.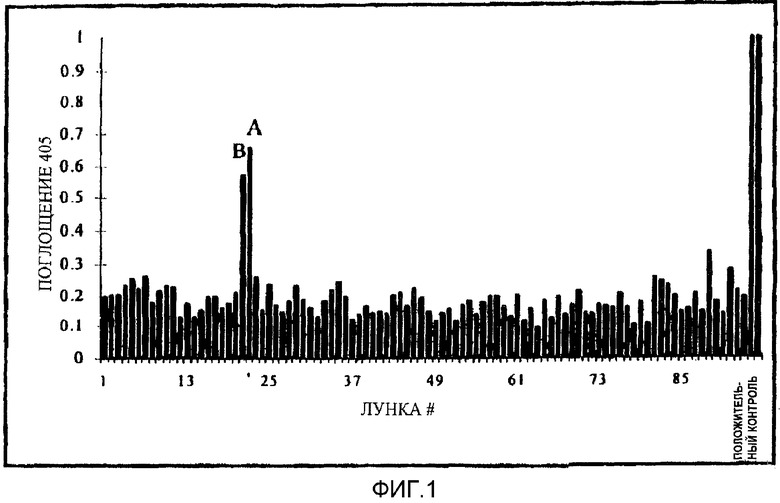

где X1, X2, Х3, Х4 и Х5 являются СН2 или один из них является NH, а другие X1-X5 являются СН2;
k равно 0 или 1;
t равно 0, 1 или 2, радикалы R1, когда t равно 2, являются одинаковыми или разными;
R1 является нормальным или разветвленным C1-8 алкилом или C1-8 гидроксиалкокси;
А является фенилом или пиридинилом;
R2 является Н, гидроксилом, галогеном или нормальным или разветвленным C1-6 алкилом, C1-6 алкокси;
n равно 0, 1, 2, 3 или 4, радикалы R2, где n>1, являются одинаковыми или разными;
р равно 0 или целому числу от 1 до 5;
Y является -ОС(O)-;
Z является СН2,
или его фармацевтически приемлемая соль.
| RU 94031753 A1, 20.07.1996 | |||
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |

