Изобретение относится к новым промежуточным соединениям и их использованию в способе получения оксабиспидиновых соединений.
Предшествующий уровень техники
Количество документально подтвержденных соединений, включающих структуру 9-окса-3,7-диазабицикло-[3.3.1]нонана (оксабиспидина), весьма незначительно. Как результат, известно очень мало способов, которые адаптированы конкретно для получения оксабиспидиновых соединений.
Некоторые оксабиспидиновые соединения описаны в Chem. Ber., 96 (11), 2827 (1963) как промежуточные соединения в синтезе 1,3-диаза-6-окса-адамантанов.
Гемиацетали (и родственные соединения), имеющие структуру оксабиспидинового кольца, описаны в J. Org. Chem. 31, 277 (1966), там же 61 (25), 8897 (1996), там же 63 (5), 1566 (1998) и там же 64 (3), 960 (1999) как непредвиденные продукты окисления 1,5-диазациклооктан-1,3-диолов или восстановления 1,5-диазациклооктан-1,3-дионов.
1,3-Диметил-3,7-дитосил-9-окса-3,7-диазабицикло[3.3.1]нонан описан в J. Org. Chem. 32, 2425 (1967) как продукт попытки ацетилирования транс-1,3-диметил-1,5-дитосил-1,5-диазабициклооктан-1,3-диола.
В международной заявке на патент WO 01/28992 описывается синтез огромного ряда оксабиспидиновых соединений, при этом показано, что данные соединения полезны в лечении сердечных аритмий. Среди описанных соединений есть ряд соединений, несущих N-2-(трет-бутоксикарбониламино)этильный заместитель.
В международной заявке на патент WO 02/083690 описывается inter alia способ получения соединения формулы I

где R1 представляет собой Н или аминозащитную группу, и R2 представляет собой C1-6алкил (возможно замещенный и/или оканчивающийся одним или более чем одним заместителем, выбранным из -ОН, галогено, циано, нитро и арила) или арил, где каждая арильная и арилоксигруппа, если не оговорено особо, возможно замещена;
включающий взаимодействие соединения формулы II
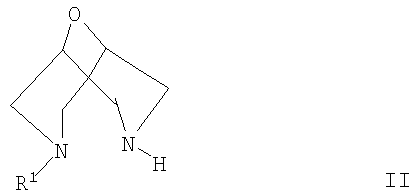
где R1 такой, как он определен выше, с:
(1) соединением формулы III

где R16 представляет собой незамещенный С1-4алкил, С1-4перфторалкил или фенил, причем последняя группа возможно замещена одним или более чем одним заместителем, выбранным из С1-6алкила, галогено, нитро и C1-6алкокси, и R2 такой, как он определен выше; или
(2) акриламидом с последующим взаимодействием полученного промежуточного соединения формулы IV

где R1 такой, как он определен выше, со спиртом формулы R2-OH и агентом, стимулирующим (или агентами, в комбинации стимулирующими) перегруппировку и окисление соединения формулы IV до промежуточного изоцианата, который затем может взаимодействовать со спиртом формулы R2-ОН, где R2 такой, как он определен выше.
В приведенной выше заявке также описывается способ получения соединения формулы I, где R1 представляет собой Н, который включает получение соответствующего соединения формулы I, в котором R1 представляет собой аминозащитную группу, описанными в данной заявке способами с последующим удалением аминозащитной группы из этого соединения. Кроме того, в Примере 3, Альтернатива II, описано, что соль присоединения 2,4,6-триметилбензолсульфоновой кислоты [2-(7-бензил-9-окса-3,7-диазабицикло[3.3.1]-нон-3-ил)этил]карбаминовой кислоты трет-бутилового эфира превращали в свободное основание с помощью водного гидроксида натрия. Полученный [2-(7-бензил-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)этил]карбаминовой кислоты трет-бутиловый эфир гидрировали в присутствии лимонной кислоты и 5%-ного Pd/C с получением [2-(9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)этил]карбаминовой кислоты трет-бутилового эфира, который подвергали взаимодействию непосредственно без дальнейшей очистки с получением (2-{7-[3-(4-цианоанилино)пропил]-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил}-этил)карбаминовой кислоты трет-бутилового эфира.
Сейчас обнаружены некоторые новые твердые соли, которые имеют преимущества по сравнению с известными способами.
Описание изобретения
Согласно первому аспекту изобретения предложены соли присоединения кислот соединений формулы I
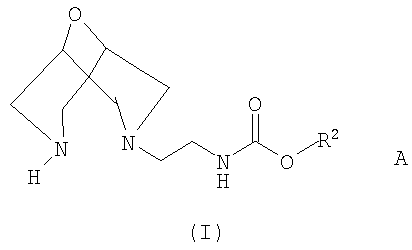
где R2 представляет собой C1-6алкил (возможно замещенный и/или оканчивающийся одним или более чем одним заместителем, выбранным из -ОН, галогено, циано, нитро и арила) или арил,
где каждая арильная группа, если не оговорено особо, возможно замещена одним или более чем одним заместителем, включая -ОН, циано, галогено, нитро, C1-6алкил, C1-6алкокси, -N(R14a)R14b, -C(O)R14c, -C(O)OR14d, -C(O)N(R14e)R14f, -N(R14g)C(O)R14h, -N(R14m)S(O)2R13b, -S(O)2R13c и/или -OS(O)2R13d, где радикалы от R13b до R13d независимо представляют собой C1-6алкил; R14a и R14b независимо представляют собой Н, C1-6алкил, или совместно представляют собой С3-6алкилен, давая в результате четырех-семичленное азотсодержащее кольцо; радикалы от R14c до R14m независимо представляют собой Н или C1-6алкил;
А представляет собой

где R16 представляет собой незамещенный С1-4алкил, С1-4перфторалкил или фенил, причем последняя группа возможно замещена одним или более чем одним заместителем, выбранным из С1-6алкила, галогено, нитро и C1-6алкокси. Конкретные соли, которые могут быть упомянуты, включают толуолсульфонат, бензолсульфонат, нозилат, бросилат, бесилат и мезитилат.
В одном аспекте данные соли находятся в твердой форме.
В другом аспекте соль представляет собой соль присоединения 2,4,6-триметилбензолсульфоновой кислоты [2-(9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)этил]карбаминовой кислоты трет-бутилового эфира.
В следующем аспекте согласно настоящему изобретению предложен способ получения соединения формулы II

где R1 представляет собой структурный фрагмент формулы Ia

в котором А представляет собой СН2 и R3 представляет собой -ОН или -N(H)R7;
R4 представляет собой Н, C1-6алкил или совместно с R3 представляет собой =O;
R5 представляет собой фенил или пиридил, причем обе эти группы возможно замещены одним или более чем одним заместителем, выбранным из -ОН, циано, галогено, нитро, C1-6алкила (возможно оканчивающегося группой -N(H)C(O)OR13a), C1-6алкокси, -N(R14a)R14b, -C(O)R14c, -C(O)OR14d, -C(O)N(R14e)R14f, -N(R14g)C(O)R14h, -N(R14i)C(O)N(R14j)R14k, -N(R14m)S(O)2R13b, -S(O)2R13c и/или -OS(O)2R13d;
R7 представляет собой Н, C1-6алкил, -E-арил, -Е-Het1, -C(O)R9a, -C(O)OR9b, -S(O)2R9c, -[С(O)]pN(R10a)10b или -C(NH)NH2;
радикалы от R9a до R9c независимо представляют собой в каждом случае, когда использованы здесь, C1-6алкил (возможно замещенный и/или оканчивающийся одним или более чем одним заместителем, выбранным из галогено, арила и Het2), арил, Het3, или радикалы R9a и R9c независимо представляют собой Н;
R10a и R10b независимо представляют собой в каждом случае, когда использованы здесь, Н или C1-6алкил (возможно замещенный и/или оканчивающийся одним или более чем одним заместителем, выбранным из галогено, арила и Het4), арил, Het5, либо совместно представляют собой С3-6алкилен, возможно прерванный атомом О;
Е представляет собой в каждом случае, когда использован здесь, прямую связь или С1-4алкилен;
В представляет собой -Z-, -Z-N(R12)-, -N(R12)-Z-, -Z-S(O)n- или -Z-O- (причем в последних двух группах Z присоединен к углеродному атому, несущему R3 и R4);
Z представляет собой прямую связь или С1-4алкилен;
R12 независимо представляет собой Н или C1-6алкил;
радикалы от R13a до R13d независимо представляют собой C1-6алкил;
R14a и R14b независимо представляют собой Н, С1-6алкил, или совместно представляют собой С3-6алкилен, давая в результате четырех-семичленное азот-содержащее кольцо;
радикалы от R14c до R14m независимо представляют собой Н или С1-6алкил; и
n представляет собой 0, 1 или 2;
р представляет собой 1 или 2;
радикалы от Het1 до Het5 независимо представляют собой в каждом случае, когда использованы здесь, пяти-двенадцатичленные гетероциклические группы, содержащие один или более чем один гетероатом, выбранный из кислорода, азота и/или серы, которые возможно замещены одним или более чем одним заместителем, выбранным из =O, -ОН, циано, галогено, нитро, C1-6алкила, С1-6алкокси, арила, арилокси, -N(R15a)R15c, -C(O)R15c, -C(O)OR15d, -C(O)N(R15e)R15f, -N(R15g)C(O)R15h и -N(R15i)S(O)2R15j;
радикалы от R15a до R15j независимо представляют собой C1-6алкил, арил или радикалы от R15a до R15j независимо представляют собой Н;
и R2 представляет собой С1-6алкил (возможно замещенный и/или оканчивающийся одним или более чем одним заместителем, выбранным из -ОН, галогено, циано, нитро и арила) или арил, где каждая арильная и арилоксигруппа, если не оговорено особо, возможно замещена одним или более чем одним заместителем, включая -ОН, циано, галогено, нитро, С1-6алкил, C1-6алкокси, -N(R14a)R14b, -C(O)R14c, -С(O)OR14d, -C(O)N(R14e)R14f, -N(R14g)C(O)R14h, -N(R14m)S(O)2R13b, -S(O)2R13c и/или -OS(O)2R13d;
при котором соль соединения формулы I
где R2 такой, как он определен ранее, и А представляет собой
где R16 представляет собой незамещенный С1-4алкил, С1-4перфторалкил или фенил, причем последняя группа возможно замещена одним или более чем одним заместителем, выбранным из С1-6алкила, галогено, нитро и C1-6алкокси, подвергают взаимодействию с соединением формулы III
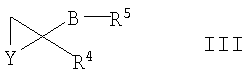
где Y представляет собой О или N(R7), и R4, R5, R7 и В такие, как они определены ранее, при температуре в интервале от 0 до 100°С, например при повышенной температуре (например от 60°С до температуры флегмы), в присутствии воды и в присутствии основания, например карбоната натрия.
В первом аспекте данная соль была выделена в твердой форме перед этой стадией способа.
Во втором аспекте предложен способ получения трет-бутил-2-{7-[(2S)-3-(4-цианофенокси)-2-гидроксипропил]-9-окса-3,7-диаза-бицикло[3.3.1]-нон-3-ил}этилкарбамата, включающий взаимодействие соли [2-(9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)-этил]-карбаминовой кислоты трет-бутилового эфира с 4-[(2S)-оксиранилметокси]бензнитрилом при температуре в интервале от 0 до 100°С в присутствии воды и в присутствии основания, например карбоната натрия.
В другом относящемся к способу аспекте используют выделенную соль [2-(9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)-этил]-карбаминовой кислоты трет-бутилового эфира, в частности соль 2,4,6-триметилбензолсульфоновой кислоты.
Использование воды в качестве реакционной среды в способе имеет важные преимущества в плане удаления отходов и последствий для окружающей среды.
Термин «арил», когда использован в данном описании, включает С6-10арильные группы, такие как фенил, нафтил и им подобные. Термин «арилокси», когда использован в данном описании, включает С6-10арилоксигруппы, такие как фенокси, нафтокси и им подобные. Во избежание неопределенности подразумевается, что упомянутые здесь арилоксигруппы присоединены к остатку молекулы через атом О оксигруппы. Если не оговорено особо, арильные и арилоксигруппы могут быть замещены одним или более чем одним заместителем, включая -ОН, циано, галогено, нитро, C1-6алкил, C1-6алкокси, -N(R14a)R14b, -C(O)R14c, -С(O)OR14d, -C(O)N(R14e)R14f, -N(R14g)C(O)R14h, -N(R14m)S(O)2R13b, -S(O)2R13c и/или -OS(O)R13d (где радикалы от R13b до R13d и радикалы от R14a до R14m такие, как они определены в данном описании ранее). Если арильные и арилоксигруппы являются замещенными, то они предпочтительно замещены заместителями в количестве от одного до трех.
Термин «галогено», когда использован в данном описании, включает фторо, хлоро, бромо и йодо.
Het-группы (от Het1 до Het5), которые могут быть упомянуты, включают группы, содержащие от 1 до 4 гетероатомов (выбранных из кислорода, азота и/или серы), в которых общее количество атомов в кольцевой системе составляет от пяти до двенадцати. По своему характеру Het-группы (от Het1 до Het5) могут быть полностью насыщенными, целиком ароматическими, частично ароматическими и/или бициклическими. Гетероциклические группы, которые могут быть упомянуты, включают бенздиоксанил, бенздиоксепанил, бенздиоксолил, бензофуранил, бензимидазолил, бензморфолинил, бензоксазинонил, бензотиофенил, хроманил, циннолинил, диоксанил, фуранил, имидазолил, имидазо[1,2-а]пиридинил, индолил, изохинолинил, изоксазолил, морфолинил, оксазолил, фталазинил, пиперазинил, пиперидинил, пуринил, пиранил, пиразинил, пиразолил, пиридинил, пиримидинил, пирролидинонил, пирролидинил, пирролинил, пирролил, хиназолинил, хинолинил, тетрагидропиранил, тетрагидрофуранил, тиазолил, тиенил, тиохроманил, триазолил и им подобные. Заместители на Het-группах (от Het1 до Het5) могут, как целесообразно, быть локализованы на любом атоме в кольцевой системе, включая гетероатом. Точка присоединения Het-групп (от Het1 до Het5) может быть через любой атом в кольцевой системе, включая (где целесообразно) гетероатом или атом на любом конденсированном карбоциклическом кольце, который может быть представлен как часть кольцевой системы. Кроме того, Het-группы (от Het1 до Het5) могут быть в N- или S-окисленной форме.
Использование защитных групп со всей полнотой описано в «Protective Groups in Organic Chemistry», edited by J.W.F. McOmie, Plenum Press (1973) и «Protective Groups in Organic Synthesis», 3rd edition, T.W. Greene & P.G.M. Wutz, Wiley-lnterscience (1999). Способ по изобретению имеет неожиданное преимущество, заключающееся в том, что соединения формулы I могут быть получены удобным образом из твердых (в противоположность, например, маслянистым или полутвердым) предшественников, причем эти предшественники могут быть очищены с использованием простых методик (например, перекристаллизацией).
Кроме того, способ по изобретению может иметь преимущество, состоящее в том, что соединения формулы I могут быть получены с более высокими выходами, за меньшее количество стадий, за меньшее время, более удобным образом и с более низкой стоимостью, по сравнению с их получением согласно способу, описанному в международной заявке на патент WO 01/28992.
Данное изобретение проиллюстрировано, но никоим образом не ограничено, следующими далее примерами.
Примеры
ОБЩИЕ ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДИКИ
Масс-спектры регистрируют на одном из следующих приборов: единичный квадрупольный масс-спектрометр с электрораспылением (S/N mc350) Waters ZMD; спектрометр Perkin-Elmer SciX API 150ex; строенный квадрупольный масс-спектрометр VG Quattro II; единичный квадрупольный масс-спектрометр VG Platform II или единичный квадрупольный масс-спектрометр Micromass Platform LCZ (последние три прибора были оборудованы пневматически управляемым интерфейсом электрораспыления (LC-MS)). 1H ЯМР- и 13С ЯМР-измерения выполняли на спектрометрах Varian 300, 400 и 500, работающих на 1H-частотах 300, 400 и 500 МГц, соответственно, и на 13С-частотах 75,5, 100,6 и 125,7 МГц, соответственно.
Ротамеры могут различаться или могут не различаться в спектре в зависимости от легкости интерпретации спектра. Если не оговорено особо, химические сдвиги дают в миллионных долях (м.д.) с растворителем в качестве внутреннего стандарта.
Сокращения
Префиксы н-, втор-, изо- и трет- имеют свои обычные значения: нормальный, вторичный, изо- и третичный.
Пример 1
а) Соль 2.4.6-триметилбензолсульфоновой кислоты и [2-(9-окса-3.7-диазабицикло[3.3.1]нон-3-ил)-этил]-карбаминовой кислоты трет-бутилового эфира
Соль 2,4,6-триметилбензолсульфоновой кислоты и [2-(7-бензил-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)-этил]-карбаминовой кислоты трет-бутилового эфира (150 г; полученная, как описано ниже), 4-метил-2-пентанол (MIBC) (300 мл) и метанол (300 мл) объединяли в металлическом сосуде для гидрирования. Добавляли твердый катализатор 5%-ный Pd/C (4,5 г; 61%-ной влажности; тип 440L; Johnson Matthey). Затем смесь гидрировали под давлением водорода 2,5 бар (250 кПа) и одновременно нагревали до 55°С. Измерение поглощения газа показало, что реакция завершилась через 2 часа. После охлаждения до 40°С катализатор удаляли фильтрованием через стекловолокнистую фильтровальную бумагу. Катализатор промывали на фильтре с помощью MIBC (300 мл) и промывки добавляли к основному фильтрату. Растворитель (185 мл) удаляли перегонкой при атмосферном давлении. Затем удаляли большее количество растворителя (243 мл) перегонкой при пониженном давлении (менее 100 мм рт.ст. (менее 13,33 кПа)). Быстро добавляли при 70°С изопропиловый эфир (IPE) (1050 мл), что приводило к падению температуры до 45°С. В реакционном сосуде образовывался неперемешиваемый осадок. Смесь нагревали повторно, растворитель отгоняли и собирали (268 мл). Добавляли MIBC (150 мл) и при 80°С все вещество растворялось. Теперь отношение MIBC:IPE составляло приблизительно 4:5. Раствор оставляли охлаждаться и вносили затравку (86 мг) при 70°С. Реакционную смесь оставляли охлаждаться в течение ночи при температуре окружающей среды. Смесь охлаждали до 8°С и затем твердый продукт собирали фильтрованием. Твердое вещество промывали на фильтре с помощью IPE (450 мл) и затем сушили отсасыванием. Последующая сушка in vacuo при 60°С дала указанное в заголовке соединение в виде твердого вещества белого цвета (115,0 г; 91%).
Т.пл. 147-9°С.
б) трет-Бутил-2-{7-[(2S)-3-(4-цианофенокси)-2-гидроксипропил]-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил}этилкарбамат
Водный раствор карбоната натрия (1М, 53 мл) добавляли к раствору соли 2,4,6-триметилбензолсульфоновой кислоты и [2-(9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)-этил]-карбаминовой кислоты трет-бутилового эфира (50,0 г) в воде (100 мл). Добавляли твердый 4-[(2S)-оксиранилметокси]бензонитрил (19,1 г) и выливали в реакционную колбу воду (50 мл). Реакционную смесь нагревали до 75°С в течение 3 часов и затем оставляли перемешиваться при температуре окружающей среды в течение ночи. Добавляли толуол (350 мл), затем водный гидроксид натрия (2М, 90 мл). Смесь перемешивали в течение 5 минут и затем разделяли фазы. Водную фазу отбрасывали, а толуоловую фазу промывали водной лимонной кислотой (10% масс./об., 180 мл). Толуоловую фазу отбрасывали. К фазе лимонной кислоты добавляли MIBC (240 мл) и водный гидроксид натрия (5М, 180 мл). После основательного перемешивания фазы разделяли и водную фазу отбрасывали. MIBC-фазу промывали водным хлоридом натрия (20% масс./об., 50 мл). MIBC-фазу концентрировали под вакуумом при температуре ниже 55°С. Растворитель собирали (13 мл воды, 29 мл MIBC). MIBC-раствор охлаждали до температуры окружающей среды и фильтровали, промывая с использованием MIBC (50 мл). Растворитель (152 мл) отгоняли под вакуумом при температуре ниже 66°С и затем отгонку прекращали. Добавляли IPE (360 мл), что приводило к снижению температуры от 65 до 37°С. После перемешивания в течение 15 минут температура понижалась на 2°С до 35°С и начиналась кристаллизация. Смесь оставляли охлаждаться с перемешиванием до температуры окружающей среды в течение ночи. Смесь охлаждали до 5°С и продукт собирали фильтрованием. Твердое вещество промывали на фильтре с использованием IPE (150 мл) и сушили отсасыванием. Последующая сушка in vacua при 55°С дала указанное в заголовке соединение в виде твердого вещества белого цвета (41,2 г; 87%).
Получение соли 2,4,6-триметилбензолсульфоновой кислоты и [2-(7-бензил-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)этил]-карбаминовой кислоты трет-бутилового эфира
а) 2-(трет-Бутилоксикарбониламино)этил-2,4,6-триметилбензолсульфонат
Триэтиламин (65 мл; 465,3 ммоль; 1,5 экв.) добавляли в виде одной порции к раствору трет-бутил-N-(2-гидроксиэтил)карбамата (50,11 г; 310,2 ммоль; 1,0 экв.) в дихлорметане (250 мл, 5 объемов). Раствор охлаждали до -10°С и добавляли триметиламин гидрохлорид (14,84 г; 155,1 ммоль; 0,5 экв.) в виде одной порции. Полученную смесь охлаждали дальше до -15°С, перемешивали в течение 5 минут, затем обрабатывали раствором мезитиленсульфонилхлорида (74,74 г; 341,2 ммоль; 1,1 экв.) в дихлорметане (250 мл, 5 объемов) в продолжение 28 минут так, чтобы внутренняя температура оставалась ниже -10°С. После завершения добавления образовывался осадок, и смесь перемешивали при -10°С в течение дополнительных 30 минут. Добавляли воду (400 мл, 8 объемов), и весь осадок растворялся. Смесь быстро перемешивали в течение 5 минут и затем разделяли два слоя. Замену растворителя с дихлорметана на IPA осуществляли путем отгонки при пониженном давлении. Растворитель удаляли (450 мл) и заменяли на IPA (450 мл) (начальное давление составляло 450 мбар (45 кПа), т.кип. 24°С; конечное давление составляло 110 мбар (11 кПа), т.кип. 36°С). В конце отгонки удаляли растворитель (150 мл), что приводило к уменьшению объема до 350 мл (7 объемов относительно использованного количества трет-бутил-N-(2-гидроксиэтил)карбамата). Раствор охлаждали до 25°С, затем медленно при перемешивании добавляли воду (175 мл), что приводило к постепенному помутнению раствора. На этой стадии твердое вещество не выпадало в осадок. Добавляли дополнительное количество воды (125 мл), и после добавления 75 мл начинал образовываться твердый осадок. Внутренняя температура поднималась от 25 до 31°С. Смесь медленно перемешивали и охлаждали до 7°С. Твердое вещество собирали фильтрованием, промывали смесью IPA/вода (1:1, 150 мл) и сушили in vacuo при 40°С в течение 21 часа с получением указанного в заголовке соединения в виде кристаллического твердого вещества белого цвета (92,54 г; 87%).
Т.пл. 73,5°С.
1H-ЯМР (300 МГц, CDCl3) δ 1.42 (9Н, s), 2.31 (3H, s), 2.62 (6H, s), 3.40 (2H, q), 4.01 (2H, t), 4.83 (1H, bs), 6.98 (2H, s).
б) 3-Бензил-9-окса-3,7-диазабицикло[3.3.]нонан
б(1) N,N-Бис(2-оксиранилметил)бензолсульфонамид
Воду (2,5 л; 10 объемов), затем эпихлоргидрин (500 мл, 4 экв.) добавляли к бензолсульфонамиду (250 г, 1 экв.). Реагенты нагревали до 40°С. Добавляли водный гидроксид натрия (130 г в 275 мл воды) так, чтобы температура реакции оставалась между 40 и 43°С. Это заняло приблизительно 2 часа. (Скорость добавления гидроксида натрия должна быть меньше в начале добавления, чем в конце для того, чтобы поддерживать температуру в установленном интервале.) После того, как добавление гидроксида натрия было завершено, реакционную смесь перемешивали при 40°С в течение 2 часов, затем при температуре окружающей среды в течение ночи. Избыток эпихлоргидрина удаляли в виде азеотропной смеси с водой путем вакуумной перегонки (приблизительно 4 кПа (40 мбар), внутренняя температура 30°С) до тех пор, пока не прекращалась отгонка эпихлоргидрина. Добавляли дихлорметан (1 л) и смесь быстро перемешивали в течение 15 минут. Оставляли разделяться фазы (для этого потребовалось 10 минут, хотя полностью прозрачные фазы получали после отстаивания в течение ночи). Фазы разделяли и дихлорметановый раствор использовали в последующей стадии, приведенной ниже.
1H ЯМР (400 МГц, CDCl3): δ 2.55-2.65 (2Н, m), 2.79 (2H, t, J 4.4), 3.10-3.22 (4H, m), 3.58-3.73 (2H, m), 7.50-7.56 (2H, m), 7.58-7.63 (1H, m), 7.83-7.87 (2H, m).
б(2) 5-Бензил-3,7-дигидрокси-1-фенилсульфонил-1,5-диазациклооктан
К дихлорметановому раствору со стадии (1), приведенной выше, добавляли IMS (2,5 л; 10 объемов). Раствор перегоняли до тех пор, пока внутренняя температура не достигала 70°С. Собирали приблизительно 1250 мл растворителя. Дополнительно добавляли IMS (2,5 л; 10 объемов), затем бензиламин (120 мл; 0,7 экв.) в виде одной порции (выделения тепла не наблюдали) и реакционную смесь нагревали при температуре флегмы в течение 6 часов (никаких изменений по сравнению с двухчасовой контрольной точкой). Еще раз добавляли бензиламин (15 мл) и раствор нагревали в течение дополнительных 2 часов. Отгоняли IMS (приблизительно 3,25 л) и добавляли толуол (2,5 л). Еще раз отгоняли растворитель (приблизительно 2,4 л) и затем еще раз добавляли толуол (1 л). Температура погона теперь составляла 110°С. Собирали дополнительные 250 мл растворителя при 110°С. Теоретически продукт оставался приблизительно в 2,4 л толуола при 110°С. Этот раствор использовали на следующей стадии.
1H ЯМР (400 МГц, CDCl3): δ 7.83-7.80 (4H, m, ArH), 7.63-7.51 (6Н, m, ArH), 7.30-7.21 (10Н, ArH), 3.89-3.80 (4H, m, CH(a)+СН(б)), 3.73 (2H, s, CH2Ph(a)), 3.70 (2H, s, СН2Ph(б)), 3.59 (2H, dd, CHHNSO2Ar(a)), 3.54 (2H, dd, CHHNSO2Ar(6)), 3.40 (2H, dd, CHHNSO2Ar(6)), 3.23 (2H, dd, CHHNSO2Ar(a)), 3.09-2.97 (4H, m, CHHNBn(a)+CHHNBn(6)), 2.83 (2H, dd, CHHNBn(6)), 2.71 (2H, dd, CHHNBn(a)).
(Данные приведены для очищенного вещества, содержащего смесь 1:1 транс- (а) и цис-диола (б))
б(3) 3-Бензил-7-(фенилсульфонил)-9-окса-3,7-диазабицикло[3.3.1]нонан
Толуоловый раствор со стадии (2), приведенной выше, охлаждали до 50°С. Добавляли безводную метансульфоновую кислоту (0,2 л). Это приводило к повышению температуры от 50 до 64°С. Через 10 минут добавляли метансульфоновую кислоту (1 л) и реакционную смесь нагревали до 110°С в течение 5 часов. Затем толуол отгоняли из реакционной смеси; собирали 1,23 л. (Внимание: внутренняя температура не должна подниматься выше 110°С на каждой стадии, иначе выход будет снижен). Затем реакционную смесь охлаждали до 50°С и для удаления остатка толуола использовали вакуум. Нагревание до 110°С и 65 кПа (650 мбар) позволили удалить дополнительно 0,53 л. (Если толуол может быть удален при более низких температуре и давлении, тогда это выгодно.) Затем реакционную смесь оставляли охлаждаться до 30°С и добавляли деионизованную воду (250 мл). Это приводило к повышению температуры от 30 до 45°С. Добавляли еще воду (2,15 л) в продолжение промежутка времени суммарно в 30 минут, так чтобы температура оставалась ниже 54°С. Раствор охлаждали до 30°С и затем добавляли дихлорметан (2 л). С использованием внешнего охлаждения и быстрого перемешивания реакционную смесь подщелачивали посредством добавления водного гидроксида натрия (10 М, 2 л) со скоростью, при которой внутренняя температура сохранялась ниже 38°С. Это потребовало 80 минут. Перемешивание останавливали, и фазы разделялись в течение 3 минут. Проводили разделение слоев. К дихлорметановому раствору добавляли IMS (2 л) и начинали перегонку. Растворитель (2,44 л) собирали до тех пор, пока температура погона не достигала 70°С. Теоретически продукт оставался приблизительно в 1,56 л IMS. Затем раствор оставляли охлаждаться до температуры окружающей среды в течение ночи при медленном перемешивании. Твердый продукт, выпавший в осадок, отфильтровывали и промывали, используя IMS (0,5 л), с получением продукта желтовато-коричневой окраски, который после сушки при 50°С в вакууме давал 50,8 г (8,9% за 3 стадии).
20 г этого продукта растворяли в ацетонитриле (100 мл) при температуре флегмы с получением бледно-желтого раствора. После охлаждения до температуры окружающей среды образовавшиеся кристаллы собирали фильтрованием и промывали ацетонитрилом (100 мл). Продукт сушили in vacuo при 40°С в течение 1 часа с получением 17,5 г (87%) указанного в подзаголовке соединения.
1H ЯМР (400 МГц, CDCl3): 5 7.18-7.23 (10Н, m), 3.86-3.84 (2H, m), 3.67 (2H, d), 3.46 (2H, s), 2.91 (2H, d), 2.85 (2H, dd), 2.56 (2H, dd).
б(4) 3-Бензил-9-окса-3,7-диазабицикло[3.3.]нонан дигидрохлорид
Концентрированную бромистоводородную кислоту (1,2 л; 3 отн. об.) добавляли к твердому 3-бензил-7-(фенилсульфонил)-9-окса-3,7-диазабицикло[3.3.1]нонану (400 г, см. приведенную выше стадию (3)) и смесь нагревали до температуры флегмы в атмосфере азота. Твердое вещество растворялось в кислоте при 95°С. После нагревания реакционной смеси в течение 8 часов ВЭЖХ-анализ показал, что реакция завершилась. Содержимое охлаждали до комнатной температуры. Добавляли толуол (1,2 л; 3 отн. об.) и смесь интенсивно перемешивали в течение 15 минут. Перемешивание останавливали и производили разделение фаз. Толуоловую фазу отбрасывали вместе с небольшим количеством межфазного вещества. Кислотную фазу возвращали в первоначальный реакционный сосуд и добавляли гидроксид натрия (10 M; 1,4 л; 3,5 отн. об.) в виде одной порции. Внутренняя температура поднималась от 30 до 80°С. Осуществляли контроль рН для того, чтобы убедиться, что его величина превышала 14. Добавляли толуол (1,6 л, 4 отн. об.), и температура опускалась от 80 до 60°С. После интенсивного перемешивания в течение 30 минут производили разделение фаз. Водный слой отбрасывали вместе с небольшим количеством межфазного вещества. Толуоловую фазу возвращали в первоначальный реакционный сосуд и добавляли 2-пропанол (4 л, 10 отн. об.). Температуру устанавливали между 40 и 45°С. Добавляли концентрированную соляную кислоту (200 мл) в продолжение 45 минут, так чтобы температура сохранялась между 40 и 45°С. Образовывался осадок белого цвета. Смесь перемешивали в течение 30 минут и затем охлаждали до 7°С. Продукт собирали фильтрованием, промывали 2-пропанолом (0,8 л; 2 отн. об.), сушили отсасыванием и затем дополнительно сушили в вакуумном шкафу при 40°С. Выход = 297 г (91%).
1H ЯМР (CD3OD + 4 капли D2O): δ 2.70 (br d, 2H), 3.09 (d, 2H), 3.47 (br s, 4H), 3.60 (s, 2H), 4.12 (br s, 2H), 7.30-7.45 (m, 5H).
API MS; m/z=219 [С13Н18N2O+Н]+.
б(5) 3-Бензил-9-окса-3,7-диазабицикло[3.3.1]нонан
Все объемы и эквиваленты определяли относительно использованного количества 3-бензил-9-окса-3,7-диазабицикло[3.3.1]нонана дигидрохлорида (см. приведенную выше стадию (4)). К 3-бензил-9-окса-3,7-диазабицикло[3.3.1]нонану дигидрохлориду (60,07 г; 206,03 ммоль; 1,0 экв.; см. приведенную выше стадию (4)) добавляли толуол (420 мл, 7 объемов) и водный раствор гидроксида натрия (2М, 420 мл, 7 объемов; 4,0 экв.). Смесь перемешивали в атмосфере азота, нагревали до 60°С и выдерживали при этой температуре в течение 30 минут, в течение которых образовывались два прозрачных слоя. Нижний, водный слой, удаляли, а толуоловый раствор указанного в заголовке соединения (свободного основания) азеотропно сушили при атмосферном давлении (суммарный объем удаленного растворителя = 430 мл; суммарный объем добавленного толуола = 430 мл), затем концентрировали до объема 240 мл (4 объема). Анализ по Карлу Фишеру на этой стадии показал содержание воды в растворе 0,06%. Высушенный раствор указанного в заголовке соединения (теоретически 44,98 г; 206,03 ммоль; 1,0 экв.) использован как таковой на последующей стадии.
в) Соль 2,4,6-триметилбензолсульфоновой кислоты и [2-(7-бензил-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)этил]карбаминовой кислоты трет-бутилового эфира
Теплый (28°С) раствор 2-(трет-бутилоксикарбониламино)этил 2,4,6-триметилбензолсульфоната (70,93 г; 206,03 ммоль; 1,0 экв.; см. Получение (а), выше) в толуоле (240 мл, 4 объема) добавляли к раствору 3-бензил-9-окса-3,7-диазабицикло[3.3.1]нонана (44,98 г; 206,03 ммоль; 1,0 экв.) в толуоле (240 мл, 4 объема) (см. Получение б(5), выше). Полученный раствор быстро перемешивали в атмосфере азота с нагреванием при 68°С в течение 8 часов. Реакционную смесь оставляли перемешиваться при температуре окружающей среды в течение 84 часов. Образовывался плотный осадок твердого вещества белого цвета в растворе бледно-желтой окраски. Смесь охлаждали до +9°С и указанное в заголовке соединение собирали фильтрованием. Реакционный сосуд промывали толуолом (100 мл) и добавляли на фильтр. Осадок на фильтре промывали толуолом (150 мл). Твердый продукт белого цвета сушили отсасыванием в течение 15 минут, затем сушили до постоянного веса in vacua при 40°С в течение 23 часов. Выход полученного указанного в заголовке соединения составил 79,61 г (141,7 ммоль; 69%). Объединенный фильтрат и промывки (670 мл) промывали водным раствором гидроксида натрия (2М, 200 мл; 3,3 объема). Смесь нагревали до 60°С и выдерживали при этой температуре в течение 20 минут при быстром перемешивании. Затем разделяли два слоя. Толуоловый раствор концентрировали до объема 200 мл посредством вакуумной перегонки (т.кип. 50-54°С при 650-700 мбар (65-70 кПа); т.кип. 46°С при 120 мбар (12 кПа) в конце). В ходе перегонки раствор становился мутным вследствие образования указанного в заголовке соединения. Предположительно 20% первоначального количества 3-бензил-9-окса-3,7-диазабицикло[3.3.1]нонана оставалось в фильтрате, и поэтому дополнительно добавляли 2-(трет-бутилоксикарбониламино)этил-2,4,6-триметилбензолсульфонат (14,20 г; 41,21 ммоль; 0,2 экв.) в виде одной порции (загрузка в виде твердого вещества предпочтительнее, чем в виде раствора в толуоле). Мутный раствор нагревали при 67°С в течение 8 часов при быстром перемешивании и затем оставляли перемешиваться при температуре окружающей среды в течение 11 часов. Смесь охлаждали до +8°С и указанное в заголовке соединение собирали фильтрованием. Реакционный сосуд промывали дополнительно толуолом (2×30 мл) и добавляли на фильтр. Твердый продукт белого цвета сушили отсасыванием в течение 15 минут, затем сушили до постоянной массы in vacuo при 40°С в течение 7 часов. Выход указанного в заголовке соединения составил 23,25 г (41,39 ммоль; 20%). Объединенный выход указанного в заголовке соединения (твердого вещества белого цвета) составил 102,86 г (183,11 ммоль; 89%).
Т.пл. 190-190,5°С.
1H-ЯМР (300 МГц, CDCl3) δ 1.43 (9Н, s), 2.17 (3Н, s), 2.51 (6Н, s), 2.73-2.80 (2H, m), 2.90-2.94 (4H, m), 3.14-3.22 (4H, m), 3.37 (2H, bm), 3.89 (2H, bs), 4.13 (2H, bs), 6.74 (2H, s), 7.12 (1H, bt), 7.42-7.46 (5H, m).
| название | год | авторы | номер документа |
|---|---|---|---|
| 3,7-ДИАЗАБИЦИКЛО[3.3.0]ОКТАНЫ И ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ СЕРДЕЧНЫХ АРИТМИЙ | 2002 |
|
RU2293085C2 |
| НОВЫЕ 5,6-ДИГИДРОПИРИДИН-2-ОНОВЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТРОМБИНА | 2004 |
|
RU2335492C2 |
| НОВЫЕ БИСПИДИНОВЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ СЕРДЕЧНЫХ АРИТМИЙ | 2000 |
|
RU2250903C2 |
| НОВЫЕ ОКСАБИСПИДИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ СЕРДЕЧНЫХ АРИТМИЙ | 2005 |
|
RU2379311C9 |
| Новый ингибитор на основе производного хинолина | 2019 |
|
RU2802283C2 |
| ИНГИБИТОР, СОДЕРЖАЩИЙ БИЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2820948C2 |
| МОРФОЛИНО-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ БИЦИКЛИЧЕСКИХ ПИРИМИДИНМОЧЕВИНЫ ИЛИ КАРБАМАТА В КАЧЕСТВЕ ИНГИБИТОРОВ mTOR | 2012 |
|
RU2609208C2 |
| НОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ МОДУЛЯТОРОВ ОПИОИДНЫХ РЕЦЕПТОРОВ | 2003 |
|
RU2332411C2 |
| НОВЫЙ СПОСОБ СИНТЕЗА СОЕДИНЕНИЙ ТИАЗОЛИДИНДИОНА | 2011 |
|
RU2593370C2 |
| СИНТЕЗ СОЕДИНЕНИЙ ТИАЗОЛИДИНДИОНА | 2011 |
|
RU2594725C2 |
Изобретение относится к новым солям присоединения кислоты соединения формулы I

где R2 представляет собой C1-6алкил (возможно замещенный и/или оканчивающийся одним или более чем одним заместителем, выбранным из -ОН, галогено, циано, нитро и арила) или арил, где каждая арильная группа, если не оговорено особо, возможно замещена одним или более чем одним заместителем, включая -ОН, циано, галогено, нитро, C1-6алкил, C1-6алкокси, -N(R14a)R14b, -C(O)R14c, -C(O)OR14d, -C(O)N(R14e)R14f, -N(R14g)C(O)R14h, -N(R14m)S(O)2R13b, -S(O)2R13c и/или -OS(O)2R13d, где радикалы от R13b до R13d независимо представляют собой C1-6алкил; R14a и R14b независимо представляют собой Н, C1-6алкил, или совместно представляют собой С3-6алкилен, давая в результате четырех-семичленное азотсодержащее кольцо; радикалы от R14c до R14m независимо представляют собой Н или C1-6алкил; А представляет собой

где R16 представляет собой незамещенный С1-4алкил, С1-4перфторалкил или фенил, причем последняя группа возможно замещена одним или более чем одним заместителем, выбранным из C1-6алкила, галогено, нитро и C1-6алкокси. Изобретение также относится к способу получения соединения формулы II. Технический результат - получение новых промежуточных соединений и их использование в способе получения оксабиспидиновых соединений формулы II, которые полезны в лечении сердечных аритмий. 2 н. и 7 з.п. ф-лы.
 ,
,
где R2 представляет собой C1-6алкил (возможно замещенный и/или оканчивающийся одним или более чем одним заместителем, выбранным из -ОН, галогено, циано, нитро и арила) или арил,
где каждая арильная группа, если не оговорено особо, возможно замещена одним или более чем одним заместителем, включая -OH, циано, галоцено, нитро, C1-6алкил, C1-6алкокси, -N(R14a)R14b, -C(O)R14c, -C(O)OR14d, -C(O)N(R14e)R14f, -N(R14g)C(O)R14h, -N(R14m)S(O)2R13b, -S(O)2R13c и/или -OS(O)2R13d, где радикалы от R13b до R13d независимо представляют собой C1-6алкил; R14a и R14b независимо представляют собой Н, С1-6алкил, или совместно представляют собой С3-6алкилен, давая в результате четырех-семичленное азотсодержащее кольцо; радикалы от R14c до R14m независимо представляют собой Н или C1-6алкил;
А представляет собой
 ,
,
где R16 представляет собой незамещенный С1-4алкил, С1-4перфторалкил или фенил, причем последняя группа возможно замещена одним или более чем одним заместителем, выбранным из С1-6алкила, галогено, нитро и С1-6алкокси.

где R1 представляет собой структурный фрагмент формулы Ia

в котором А представляет собой СН2, и R3 представляет собой -ОН или -N(H)R7;
R4 представляет собой Н, С1-6алкил или совместно с R3 представляет собой =O;
R5 представляет собой фенил или пиридил, причем обе эти группы возможно замещены одним или более чем одним заместителем, выбранным из -ОН, циано, галогено, нитро, C1-6алкила (возможно оканчивающегося группой -N(H)C(O)OR13a), C1-6алкокси, -N(R14a)R14b, -C(O)R14c, -C(O)OR14d, -C(O)N(R14e)R14f, -N(R14g)C(O)R14h, -N(R14i)C(O)N(R14j)R14k, -N(R14m)S(O)2R13b, -S(O)2R13c и/или -OS(O)2R13d;
R7 представляет собой Н, С1-6алкил, -Е-арил, -E-Het1, -C(O)R9a, -C(O)OR9b, -S(O)2R9c, -[С(О)]pN(R10a)R10b или -C(NH)NH2;
радикалы от R9a до R9c независимо представляют собой в каждом случае, когда использованы здесь, C1-6алкил (возможно замещенный и/или оканчивающийся одним или более чем одним заместителем, выбранным из галогено, арила и Het2), арил, Het3, или радикалы R9a и R9c независимо представляют собой Н;
R10a и R10b независимо представляют собой в каждом случае, когда использованы здесь, Н или C1-6алкил (возможно замещенный и/или оканчивающийся одним или более чем одним заместителем, выбранным из галогено, арила и Het4), арил, Het5, либо совместно представляют собой С3-6алкилен, возможно прерванный атомом О;
Е представляет собой в каждом случае, когда использован здесь, прямую связь или С1-4алкилен;
В представляет собой -Z-, -Z-N(R12)-, -N(R12)-Z-, -Z-S(O)n- или -Z-O- (причем в последних двух группах Z присоединен к углеродному атому, несущему R3 и R4);
Z представляет собой прямую связь или С1-4алкилен;
R12 независимо представляет собой Н или С1-6алкил;
радикалы от R13a до R13d независимо представляют собой C1-6алкил;
R14a и R14b независимо представляют собой Н, С1-6алкил, или совместно представляют собой С3-6алкилен, давая в результате четырех-семичленное азотсодержащее кольцо;
радикалы от R14c до R14m независимо представляют собой Н или C1-6алкил; и
n представляет собой 0, 1 или 2;
р представляет собой 1 или 2;
радикалы от Het1 до Het5 независимо представляют собой в каждом случае, когда использованы здесь, пяти-двенадцатичленные гетероциклические группы, содержащие один или более чем один гетероатом, выбранный из кислорода, азота и/или серы, которые возможно замещены одним или более чем одним заместителем, выбранным из =O, -ОН, циано, галогено, нитро, С1-6алкила, С1-6алкокси, арила, арилокси, -N(R15a)R15b, -C(O)R15c, -C(O)OR15d, -C(O)N(R15e)R15f, -N(R15g)C(O)R15h, и -N(R15i)S(O)2R15j;
радикалы от R15a до R15j независимо представляют собой C1-6алкил, арил, или радикалы от R15a до R15j независимо представляют собой Н;
и R2 представляет собой C1-6алкил (возможно замещенный и/или оканчивающийся одним или более чем одним заместителем, выбранным из -ОН, галогено, циано, нитро и арила) или арил, где каждая арильная и арилоксигруппа, если не оговорено особо, возможно замещена одним или более чем одним заместителем, включая -ОН, циано, галогено, нитро, C1-6алкил, C1-6алкокси, -N(R14a)R14b, -C(O)R14c, -C(O)OR14d, -C(O)N(R14e)R14f, -N(R14g)C(O)R14h, -N(R14m)S(O)2R13b, -S(O)2R13c и/или -OS(O)2R13d;
при котором соль соединения формулы I
,
где R2 такой, как он определен ранее, и А представляет собой
,
где R16 представляет собой незамещенный С1-6алкил, С1-4перфторалкил или фенил, причем последняя группа возможно замещена одним или более чем одним заместителем, выбранным из C1-6алкила, галогено, нитро и С1-6алкокси, подвергают взаимодействию с соединением формулы III

где Y представляет собой О или N(R7), и R4, R5, R7 и В такие, как они определены здесь ранее, при температуре в интервале от 0 до 100°С, например при повышенной температуре (например от 60°С до температуры флегмы), в присутствии воды и в присутствии основания.
включающий взаимодействие соли [2-(9-окса-3,7-диазабицикло [3.3.1]нон-3-ил)-этил]-карбаминовой кислоты третбутилового эфира с 4-[(2S)-оксиранилметокси]бензнитрилом при температуре в интервале от 0 до 100°С в присутствии воды и в присутствии основания.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 6-ОКСИ-7-АЗА- 1-ОКСАБИЦИКЛО- | 0 |
|
SU355172A1 |

