Изобретение относится к биоорганической химии и медицине, к пептидным производным гемина, обладающим противоопухолевым действием, а также к фармкомпозиции, включающей геминпептиды как основной действующий компонент. Изобретение является основой для получения лекарственного средства, эффективного в терапии рака и малотоксичного.
Недостаточный терапевтический индекс, ограниченная эффективность и побочные эффекты существующих противораковых химиотерапевтических агентов представляет важную фармакологическую проблему.
Известны соединения пептидной природы, используемые в медицине в качестве противоопухолевых средств. К ним относятся пептиды из группы блеомицина, оказывающие прямое цитотоксическое действие, обусловленное захватом блеомицином ионов железа и генерацией активных форм кислорода образовавшимся комплексом. Однако их применение ограничено выраженными побочными эффектами, главным образом в отношении легких и почек [Переводчикова Н.И. // Клиническая химиотерапия опухолевых заболеваний. М.: Медицина, 1976. С.100-103].
Известны порфирины для терапии рака. К ним относятся неприродные синтетические порфирины и металлопорфирины для фотодинамической и темновой терапии рака, с прямым противоопухолевым действием, опосредованным, в частности, генерацией активных форм кислорода ("фотогем", "терафтал"). Их применение также сопровождается значительными побочными эффектами, в частности длительной остаточной кожной чувствительностью [Российский биотерапевтический журнал. 2004. Т.3, №2. С.26, 70].
Задачей настоящего изобретения является создание эффективного лекарственного средства для терапии рака с прямым противоопухолевым действием, повышенной селективностью в отношении здоровых и опухолевых клеток; с новыми механизмами действия и без побочных эффектов.
Поставленная задача решается путем применения новых и известных соединений - конъюгатов двух природных эндогенных веществ - металлопорфирина гемина и пептидов.
Природное происхождение составных частей геминпептидов обеспечивает их низкую токсичность и естественный метаболизм.
Известно, что некоторые геминпептиды обладают пероксидазной активностью [Желтухина Г.А., Евстигнеева Р.П., Рожкова Е.А. // Кинетика и катализ, Т.40, №2, 1999, С.256-260] и способностью катализировать окисление органических субстратов [Желтухина Г.А., Евстигнеева Р.П., Лукашова Е.А., Соловьева А.Б., Лубсандоржиева Л.К. // Ж. физ. химия, Т.69, №11, 1995. С.1972-1979], а также ингибировать протеиназы [Р.П. Евстигнеева, Г.А. Желтухина, Т.В. Зарубина, В.Е. Небольсин, Д.Н. Носик, Н.Н. Носик. // Патент №2238950 БИ 0430 2002 г.]. Однако совсем не была известна их способность индуцировать апоптоз клеток и противоопухолевая активность.
Объектом настоящего изобретения являются геминпептиды общей формулы I
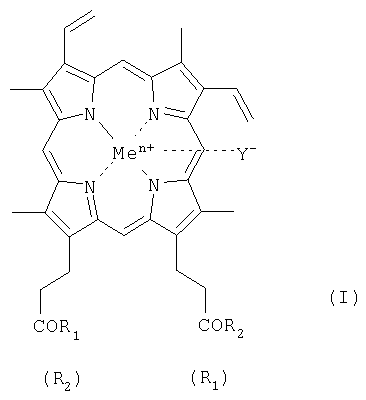
где R1 и R2 - заместители, представляющие собой пептид с числом составляющих его аминокислот от одной до семи; при этом карбоксильные группы пептида могут быть модифицированы C1-C8 алкиловым эфиром, а боковые функции пептида могут быть защищены, причем возможно, что R1=R2 или R1≠R2, в частности, при R1=-ArgTrpHisArgLeuLysGlu(OMe)OH, R2=-OH (II), или R1=-TrpHisArgLeuLysGlu(OMe)OH, R2=-OH (III); или R1=R2=-SerOMe (IV); или R1=R2=-LysOMe (V), или R1=R2=-AspOH (VI), за исключением R1=R2=-ArgOMe (VII); карбоксильная группа гемина может быть модифицирована метиловым или другим C1-C8 эфиром или физиологически приемлемой солью; или его фармацевтически приемлемые соли; Y- представляет собой Cl-; Me представляет собой Zn, Cu, Fe, Mn. Кроме того, поставленная задача решается применением известного геминпептида, и его фармацевтически приемлемых солей, где R1=R2=-ArgOMe (VII).
Предлагаемые геминпептиды (II, III) могут быть синтезированы твердофазным методом по разработанной нами методологии [Евстигнеева Р.П., Желтухина Г.А., Зарубина Т.В., Небольсин В.Е., Антоненкова Т.Н., Костанян И.А., Драницына С.М., Астапова М.В., Сурина Е.А. // ДАН, 2003, т.388, №1, с.1-5] на полимере с 2-хлортритилхлоридной якорной группой в сочетании с Fmoc-стратегией с последующим одновременным отщеплением от полимера целевого продукта и боковых защитных групп пептидов при действии кислоты в мягких условиях.
После очистки колоночной гель-хроматографией на сефадексе геминпептиды (II, III) были получены с высокими выходами 84% и 56%, соответственно, считая на исходную аминокислоту на полимере.
Геминпептиды (II-III) могут быть получены в виде фармацевтически приемлемых солей, например ацетатов, хлоридов, сульфатов, лактатов и т.д., путем варьирования природы кислоты при отщеплении геминпептидов от полимера или превращены в таковую путем применения ионообменной хроматографии.
Геминпептиды (IV-VI) получали с применением методов классического пептидного синтеза в растворе. В качестве активированного производного гемина использовался N-оксисукцинимидный эфир.
Синтез производных гемина отражен в примерах 2, 4-7. Таким образом, предлагаемые геминпептиды (II, III) получены с высокими выходами, высокой степенью чистоты и охарактеризованы электронной и ИК-спектроскопией, масс-спектрометрией, ТСХ.
Список сокращений
DCC - N,N'-дициклогексилкарбодиимид
DMF - N,N'-диметилформамид
ОМе - метиловый эфир
ТСХ - хроматография в тонком слое
Fmoc - 9-флуоренилметилоксикарбонил
FmocONsu - 9-флуоренилметилсукцинимидокарбонат
1-НОВТ - 1-гидроксибензотриазол
TFE - трифторэтанол
DCM - хлористый метилен
Mtt - метилтритил
mTrt - метокситритил
TFA - трифторуксусная кислота
ВЭЖХ - высокоэффективная жидкостная хроматография
HEPES - N-2-гидроксиэтилпиперазин-N'-этансульфоновая кислота
Экспериментальная часть
В синтезе использовали L-аминокислоты и их производные фирмы Reanal (Венгрия), Bachem (Швейцария), а также синтезированные нами по известным методикам [Гершкович А.В., Кибирев В.К. Химический синтез пептидов. Киев: Наука думка, 1992] с использованием FmocONSu. Индивидуальность полученных соединений подтверждали с помощью ТСХ на пластинах Kieselgel 60 F254 "Мегск" (Германия) в системах: н-бутанол - пиридин - уксусная кислота - вода 63:36:6:45 (1), н-бутанол - пиридин - уксусная кислота - вода 30:20:6:24 (2), хлороформ-метанол 9:1 (3). Аминокислоты и пептиды на хроматограммах обнаруживали с помощью раствора нингидрина и хлор-толидинового реагента, а также реактива Паули и реагента Эрлиха. ВЭЖХ осуществляли на хроматографе Breeze (Waters) (детектор Waters 2487) в условиях: колонка 4.6-250 мм, Symmetry С 8,5 mkm; элюция градиентом 10→60% фазы Б в течение 15 мин (буфер А - 0.1%-ная водная TFA; буфер Б - 0.09%-ная TFA в смеси ацетонитрил-вода 60:40), детекция при 280 нм. Гидролиз пептидилполимеров, пептидов и геминпептидов проводили в смеси 12 н. соляной и пропионовой кислот (1:1) при 140°С в течение 2 ч. Аминокислотный состав определяли на автоматическом аминокислотном анализаторе Biotronic IC5001 (Германия). Величины углов удельного оптического вращения измеряли на спектрополяриметре Perkin Elmer 341 (США). Температуры плавления веществ определяли на термоплавильном столике Boetius (Германия). Электронные и ИК-спектры регистрировали, соответственно, на приборах Jasco model UV/VS 7800 Spectrophotometer (Япония) и Perkin ELMER FT-IR Spectrometer 1725X (Швеция). Масс-спектры высокого разрешения регистрировали на времяпролетном масс-спектрометре с лазерной десорбционной ионизацией (TOF MALDI) Vision 2000 (Thermo Bioanalysis, Англия).
В качестве полимерного носителя в синтезе пептидов и геминпептидов использовали сополимер стирола с 1% дивинилбензола с 2-хлортритилхлоридной якорной группой с содержанием хлора 1.43 ммоль/г полимера (Bachem, Швейцария). Твердофазный синтез пептидов и геминпептидов осуществляли в соответствии с Fmoc-стратегией [Barlos К., Chatzi О., Gamos D., Stavropouls G. // Int. J. Peptide Protein. Res 1991. V.37. P.513-520]. Нагрузку стартовой аминокислоты на носителе определяли спектрофотометрическим методом [Dryland A., Sheppard R.C. // J.Chem. Soc. Perkin Trans. I. 1986. N1. P.125-137] и по данным количественного аминокислотного анализа. Деблокирование Fmoc-пептидилполимеров проводили по следующей схеме: 1) промывка DMF 3 мин × 2; 2) обработка 20% раствором пиперидина в DMF 3 мин × 1, 11 мин × 1; 3) промывка DMF 1 мин × 5. Полноту протекания реакций пептидообразования на полимере проверяли с помощью полуколичественного нингидринового теста [Kaiser E., Colescott R.L., Rossinger G.D., Cook P.J. // Anal. Biochem. 1970. V.34. N2. P.595-598].
Пример 1. H-Arg-Trp-His-Arg-Leu-Lys-Glu(OMe)-OH (VIII)
К 1 г полимера с содержанием Р-С1 1.43 ммоль Cl-, предварительно набухшего в 10 мл дихлорэтана, добавляли свежеприготовленный раствор 0.548 г (1.43 ммоль) Fmoc-Glu(OMe)-OH и 0.50 мл (3.58 ммоль) триэтиламина в 6 мл дихлорэтана, перемешивали воздухом в течение 25 мин при 20°С. Реакцию останавливали прибавлением 20 мл смеси метанол-триэтиламин (9:1), перемешивали 1 мин. Аминоацилполимер отфильтровывали, промывали дихлоэтаном (2 мин × 3), DMF (2 мин × 2), изопропанолом (2 мин × 2), DMF (2 мин × 2), изопропанолом (2 мин × 2), метанолом (2 мин × 1), диэтиловым эфиром (2 мин × 2). Объем одной порции промывочного раствора 3 мл. Содержание γ-метилового эфира глутаминовой кислоты в аминоацилполимере составило 0.45 ммоль/г.
К раствору 0.864 г (1.35 ммоль) Fmoc-Lys(mTrt)-OH в 6 мл DMF при охлаждении и перемешивании добавляли 0.18 г (1.35 ммоль) 1-НОВТ и 0.28 г (1.35 ммоль) DCC. Смесь перемешивали воздухом в течение 20 мин, прибавляли к деблокированному по вышеописанной методике аминоацилполимеру и перемешивали в течение 30 мин, оставляли на 24 ч при комнатной температуре, отфильтровывали, промывали DMF (1 мин × 5). Нингидриновый тест пробы дипептидилполимера отрицательный.
К раствору 0.48 г (1.35 ммоль) Fmoc-Leu-OH в 6 мл DMF при охлаждении и перемешивании добавляли 0.18 г (1.35 ммоль) 1-НОВТ и 0.28 г (1.35 ммоль) DCC. Смесь перемешивали воздухом в течение 20 мин, прибавляли к деблокированному дипептидилполимеру и перемешивали в течение 30 мин, оставляли на 24 ч при комнатной температуре. Трипептидилполимер отфильтровывали, промывали аналогично описанному для дипептидилполимера. Нингидриновый тест отрицательный.
К раствору 0.535 г (1.35 ммоль) Fmoc-Arg-OH в 6 мл DMF при охлаждении и перемешивании добавляли 0.415 мл (1.35 ммоль) 3.25 N HCl в диоксане. Смесь перемешивали в течение 20 мин при 0°С, добавляли 0.18 г (1.35 ммоль) 1-НОВТ и 0.28 г (1.35 ммоль) DCC, перемешивали в течение 20 мин, прибавляли к деблокированному трипептидилполимеру и перемешивали в течение 30 мин, оставляли на 24 ч при комнатной температуре. Тетрапептидилполимер отфильтровывали, промывали аналогично описанному для дипептидилполимера. Нингидриновый тест положительный. Реакцию повторяли с 1.2 эквивалентом карбоксильного компонента. Оставляли на 2 ч. Нингидриновый тест отрицательный.
К раствору 0.856 г (1.35 ммоль) Fmoc-His(Mtt)-OH в 6 мл DMF при охлаждении и перемешивании добавляли 0.18 г (1.35 ммоль) 1-НОВТ и 0.28 г (1.35 ммоль) DCC. Смесь перемешивали воздухом в течение 20 мин, прибавляли к деблокированному тетрапептидилполимеру и перемешивали в течение 30 мин, оставляли на 24 ч при комнатной температуре. Пентапептидилполимер отфильтровывали, промывали аналогично описанному для дипептидилполимера. Нингидриновый тест отрицательный.
К раствору 0.576 г (1.35 ммоль) Fmoc-Trp-OH в 3 мл DMF при охлаждении и перемешивании добавляли 0.18 г (1.35 ммоль) 1-НОВТ и 0.28 г (1.35 ммоль) DCC. Смесь перемешивали воздухом в течение 20 мин, прибавляли к деблокированному пентапептидилполимеру и перемешивали в течение 30 мин, оставляли на 24 ч при комнатной температуре. Гексапептидилполимер отфильтровывали, промывали аналогично описанному для дипептидилполимера. Нингидриновый тест отрицательный.
К раствору 0.369 г (0.93 ммоль) Fmoc-Arg-OH в 6 мл DMF при охлаждении и перемешивании добавляли 0.29 мл (0.93 ммоль) 3.25 N HCl в диоксане. Смесь перемешивали в течение 20 мин при 0°С, добавляли 0.13 г (0.93 ммоль) 1-НОВТ и 0.19 г (0.93 ммоль) DCC, перемешивали в течение 20 мин, прибавляли к деблокированному гексапептидилполимеру и перемешивали в течение 30 мин, оставляли на 24 ч при комнатной температуре. Гептапептидилполимер отфильтровывали, промывали аналогично описанному для дипептидилполимера. Нингидриновый тест положительный. Реакцию повторяли с 1.5 эквивалентом карбоксильного компонента. Оставляли на 2 ч. Нингидриновый тест положительный. Реакцию повторяли с 1.5 эквивалентом карбоксильного компонента. Оставляли на 2 ч. Нингидриновый тест отрицательный.
Получили 0.680 г гептапептидилполимера Fmoc-Arg-Trp-His(Mtt)-Arg-Leu-Lys(mTrt)-Glu(OMe)-OP (II). Отщепление проводили по методике [Barlos К., Chatzi O., Gamos D., Stavropouls G.// Int. J. Peptide Protein. Res. 1991. V.37. P.513-520]. К 0.13 г деблокированного гептапептидилполимера прибавляли 4 мл смеси СН3СООН-TFE-DCM. Суспензию перемешивали 2 ч при 20°С, затем смолу отделяли, промывали смесью СН3СООН-TFE-DCM. Растворители из фильтрата удаляли в вакууме. Остаток растирали с эфиром, выпавший осадок отделяли, сушили в вакууме над Р2O5. Целевое вещество переосаждали 4 мл медицинского эфира из 1 мл н-бутилового спирта. Выход 0.037 г (62%), считая на исходную аминокислоту на полимере, Rf 0.17 (1). Масс-спектр, m/z: [М]+ 1038.5. ВЭЖХ: индивидуальный пик, время удерживания 11.386 мин.
Пример 2. Nα-[6(7)-(Протогемин IX)-ил]-Arg-Trp-His-Arg-Leu-Lys-Glu(OMe)-OH (II)
Деблокировали 0.172 г гептапептидилполимера и прибавляли к нему раствор (0.23 ммоль) 6(7)-моно-N-оксисукцинимидного эфира протогемина IX в 7 мл DMF, перемешивали в течение 30 мин и оставляли на 24 ч при комнатной температуре. Гемингептапептидилполимер отделяли, промывали DMF (1 мин × 7). Нингидриновый тест отрицательный. К геминпептидилполимеру прибавляли 6 мл смеси СН3СООН-TFE-DCM, перемешивали 2 ч. Полимер отделяли, промывали смесью СН3СООН-TFE-DCM, 1 мин × 4. Растворители из фильтрата удаляли в вакууме. Остаток растирали с эфиром, выпавший осадок отделяли, сушили в вакууме над P2O5. Продукт очищали колоночной гель-хроматографией. Выход 0.109 г (84%). Rf 0.34 (2). Электронный спектр (метанол), λмах, нм (ε·10-3): 398.40 (67.42), 499.80 (5.62), 536.0 (4.39), 620.20 (1.46). Масс-спектр, m/z: [M]+ 1636.5. Аминокислотный анализ: Glu 1.42 (1), Leu 0.76 (1), Lys 0.60 (1), His 1.34 (1), Arg 1.89 (2). Содержание пептидного материала в геминпептиде (II), определенное количественным аминокислотным анализом, составляет 83% от теоретического.
Пример 3. H-Trp-His-Arg-Leu-Lys-Glu(OMe)-OH (IX)
К 0.125 г деблокированного гексапептидилполимера прибавляли 3 мл смеси СН3СООН-TFE-DCM. Суспензию перемешивали 2 ч при 20°С, затем смолу отделяли, промывали смесью СН3СООН-TFE-DCM. Растворители из фильтрата удаляли в вакууме. Остаток растирали с эфиром, выпавший осадок отделяли, сушили в вакууме над P2O5. Целевое вещество переосаждали 4 мл медицинского эфира из 1 мл н-бутилового спирта. Выход 0.028 г (56%). Rf 0.16 (1). Масс-спектр, m/z: [M]+882.8. ВЭЖХ: индивидуальный пик, время удерживания 8.349 мин.
Пример 4. Nα-[6(7)-(Протогемин IX)-ил]-Trp-His-Arg-Leu-Lys-Glu(OMe)-OH (III)
Деблокировали 0.19 г гексапептидилполимера и прибавляли к нему раствор (0.3 ммоль) 6(7)-моно-N-оксисукцинимидного эфира протогемина IX (XVII) в 5 мл DMF, перемешивали в течение 4 ч и оставляли на 24 ч при комнатной температуре. Гемингексапептидилполимер отделяли, промывали DMF (1 мин × 7). Нингидриновый тест отрицательный. К гемингексапептидилполимеру прибавляли 6 мл смеси СН3СООН-TFE-DCM. Суспензию перемешивали 2 ч при 20°С, затем смолу отделяли, промывали смесью СН3СООН-TFE-DCM. Растворители из фильтрата удаляли в вакууме. Остаток растирали с эфиром, выпавший осадок отделяли, сушили в вакууме над Р2O5. Продукт очищали колоночной гель-хроматографией. Выход 0.071 г (56%). Rf 0.42 (1). Электронный спектр (метанол), λмах, нм (ε·10-3): 397.80 (90.9), 499.80 (4.15), 538.40 (2.47), 625.20 (1.23). Масс-спектр, m/z: [M]+ 1479.3. Аминокислотный анализ: Glu 1.37 (1), Leu 1.17 (1), Lys 0.77 (1), His 0.99 (1), Arg 2.7 (3). Содержание пептидного материала в геминпептиде (I), определенное количественным аминокислотным анализом, составляет 84% от теоретического.
Пример 5. Nα-[6 (7)-(Протогемин IX)-ил]-Ser(ОМе) (IV)
К раствору 0.055 мл (0.40 ммоль) Et3N в 1 мл DMF прибавляли 0.062 г (0.40 ммоль) HCl·H-Ser-OMe, оставляют на 15 мин при 20°С. К раствору полученного аминокомпонента добавляли раствор 6,7-бис-N-оксисукцинимидного эфира протогемина IX в 2 мл DMF, синтезированного из 0.13 г (0.20 ммоль) гемина. Реакционную смесь перемешивают в течение 5 ч, оставляют на 72 ч при 20°С. Продукт очищали колоночной гель-хроматографией. Растворитель из реакционной смеси удаляют в вакууме. Для введения противоиона раствор геминпептида (IV) в 5 мл н-бутанола обрабатывают 0.5 н. раствором соляной кислоты, промывают водой до нейтральной реакции. Органический слой сушат над безводным Na2SO4, растворитель удаляют в вакууме, остаток сушат над безводным CaCl2. Выход 0.11 г (63%). ИК-спектр, ν, см-1, вазелиновое масло: 1745 (СО сл. эф.), 1632 (амид I), 1558 (амид II). Масс-спектр, m/z: [M]+818.7. Количественный аминокислотный анализ: Ser, содержание аминокислоты составляет 89% от теоретического.
Пример 6. Nα-[6(7)-(Протогемин IX)-ил]-Lys(OMe) (V)
К раствору 0.055 мл (0.40 ммоль) Et3N в 1 мл DMF прибавляли 0.1 68 г (0.40 ммоль) HCl·H-Lys(Fmoc)-OMe, оставляют на 15 мин. при 20°С. К раствору полученного аминокомпонента добавляли раствор 6,7-бис-N-оксисукцинимидного эфира протогемина IX в 2 мл DMF, синтезированного из 0.13 г (0.20 ммоль) гемина. Реакционную смесь перемешивали в течение 5 ч, оставляют на 72 ч при 20°С. Fmoc-защиту отщепляли 20% раствором пиперидина в DMF. Продукт очищали колоночной гель-хроматографией. Растворитель из реакционной смеси удаляли в вакууме. Для введения противоиона раствор геминпептида (V) в 5 мл н-бутанола обрабатывали 0.5 н. раствором соляной кислоты, промывали водой до нейтральной реакции. Органический слой сушили над безводным Na2SO4, растворитель удаляли в вакууме, остаток сушили над безводным CaCl2. Выход 0.10 г (53%). ИК-спектр, ν, см-1, вазелиновое масло: 1748 (СО сл. эф.), 1630 (амид I), 1556 (амид II). Масс-спектр, m/z: [М]+ 900.9. Количественный аминокислотный анализ: Lys, содержание аминокислоты составляет 91% от теоретического.
Пример 6. Nα-[6(7)-(Протогемин IX)-ил]-Asp-OH (VI)
К раствору 0.055 мл (0.40 ммоль) Et3N в 1 мл DMF прибавляли 0.079 г (0.40 ммоль) HCl·H-Asp(OMe)-OMe, оставляют на 15 мин при 20°С. К раствору полученного аминокомпонента добавляли раствор 6,7-бис-N-оксисукцинимидного эфира протогемина IX в 2 мл DMF, синтезированного из 0.13 г (0.20 ммоль) гемина. Реакционную смесь перемешивали в течение 5 ч, оставляют на 72 ч при 20°С. К твердому остатку прибавляли 1.5 мл смеси 0.5 н. водного раствора NaOH и диоксана (1:1). Постепенно растворяющуюся суспензию перемешивали в течение 0.5 часа, после чего при охлаждении подкисляли уксусной кислотой до рН 6. Растворители удаляли в вакууме досуха. Из остатка экстрагировали геминпептид (VI) безводным спиртом и очищали колоночной гель-хроматографией. Растворитель из реакционной смеси удаляли в вакууме. Для введения противоиона раствор геминпептида (VI) в 5 мл н-бутанола обрабатывают 0.5 н. раствором соляной кислоты, промывали водой до нейтральной реакции. Органический слой сушили над безводным Na2SO4, растворитель удаляли в вакууме. Выход 0.09 г (49%). ИК-спектр, ν, см-1, вазелиновое масло: 1702 (СООН), 1634 (амид I), 1559 (амид II). Масс-спектр, m/z: [M]+ 846.7. Количественный аминокислотный анализ: Asp, содержание аминокислоты составляет 90% от теоретического.
Пример 7. Исследование проникновения геминпептидов общей формулы I в опухолевые и нормальные клетки
Для оценки проникновения пептидных производных гемина, соответствующих общей формуле I, в живые нормальные клетки использовали взвесь тимоцитов (из тимуса крысы) в среде инкубации (HEPES, рН 7.5, 80 ммоль/л NaCl, 1.6 ммоль/л KCl, 1 ммоль/л MgCl2, 2 ммоль/л CaCl2). Проникновение геминпептидов по изобретению в опухолевые клетки исследовали на клетках карциномы Эрлиха в среде инкубации (HEPES, рН 7.5, 0.154 ммоль/л NaCl). Жизнеспособность клеток проверяли путем окрашивания мазка из клеток раствором трипанового синего с концентрацией 5 мг/мл в 0.9% NaCl. Оценку проникновения заявляемых соединений в клетки проводили с помощью световой микроскопии на приборе фирмы "ЛОМО", г.Ленинград.
Кинетику проникновения геминпептидов по изобретению в тимоциты определяли спектрофотометрически (измеряли оптическую плотность при длине волны 400 нм в 1 см кювете суспензии тимоцитов (контроль)), затем к суспензии добавляли заявляемые соединения и измеряли оптическую плотность во времени (от 0 до 30 минут) при той же длине волны. Затем исследуемый образец центрифугировали 3 минуты при 600 об/мин. Надосадок помещали в спектрофотометрическую кювету и измеряли оптическую плотность при 400 нм. В качестве контроля использовали 1 мл среды инкубации. Концентрацию соединений в надосадке рассчитывали по закону Ламберта-Бугера-Бера. Разность между начальной концентрацией геминпептидов в исследуемой смеси и в надосадке позволяет получить процентное содержание соединений в клетках.
Параллельно пробы клеток исследовали с помощью световой микроскопии. Оказалось, что исследуемые соединения способны проникать в тимоциты и опухолевые клетки. Так, соединение (II) значительно проникает в тимоциты (свыше 50%) и весьма выраженно проникает в опухолевые клетки карциномы Эрлиха (более 90%). При этом наружная мембрана клеток карциномы Эрлиха из светло-блестящей (в контроле) становилась темно-коричневой, почти черной. При этом цитоплазма клеток карциномы Эрлиха становилась золотистой (в контроле цитоплазма светлая), и в ней были видны черные точки. В опытах с нормальными клетками наружная мембрана не выглядит черной каймой, внутри клетки не видно черных точек и не возникает золотистого окрашивания цитоплазмы. Соединение (VII) слабо проникает в тимоциты (10-15%) и заметно проникает в опухолевые клетки (степень проникновения около 40%).
Пример 8. Изучение противоопухолевого действия геминпептидов, соответствующих общей формуле I
Исследование противоопухолевой активности заявляемых соединений проводили на культуре клеток асцитной карциномы Эрлиха, культивируемых на мышах линии NMR-1 в сравнении с нормальными клетками, в качестве которых применяли взвесь тимоцитов (из тимуса крысы). Асцитную суспензию разбавляли вдвое ледяной средой HENKS, рН 7.4. Действие геминпептидов по изобретению изучали при концентрации 10-4 М. Цитотоксичность определяли через 30 мин. Подсчет клеток проводили в камере Горяева после окрашивания трипановым синим. Оценка результатов проводилась в проходящем свете при помощи светового микроскопа МБИ-1 с иммерсионным объективом ЛОМО АПО-ИМ-90-1.30 (г. Ленинград) при оптическом увеличении × 900.
Оказалось, что исследованные соединения, в частности геминпептиды (II, VII) обладают цитотоксической активностью по отношению к клеткам асцитной карциномы Эрлиха, однако проявления их цитолитического действия различны. Соединение (II) оказывает избирательное цитопатогенное действие только на опухолевые клетки и не влияет на нормальные тимоциты, тогда как соединение (VII) несколько менее селективно и эффективно (табл.1.).
Результаты исследования цитотоксического действия соединений (II, VII), соответствующих общей формуле I
Пример 9. Фармацевтические композиции настоящего изобретения могут быть использованы в виде фармацевтических препаратов (например, в твердой, полутвердой или жидкой формах), содержащих предлагаемые в настоящем изобретении соединения в качестве активных ингредиентов в смеси с органическим или неорганическим носителем или наполнителем, приемлемым для внутримышечного, внутривенного, интраназального, перорального, сублингвального, ингаляционного и интраректального введения. Активный ингредиент может быть включен в фармацевтическую композицию вместе с обычно используемыми нетоксичными, фармацевтически приемлемыми носителями, пригодными для изготовления растворов, таблеток, пилюль, капсул, суппозиториев, эмульсий, суспензий, мазей и любых других лекарственных форм. В качестве носителей могут быть использованы вода, глюкоза, лактоза, аравийская камедь, желатин, крахмал, триксилит магния, тальк, кукурузный крахмал, мочевина и другие носители, пригодные для изготовления твердых, мягких или жидких препаратов. При этом в качестве добавок могут быть использованы стабилизаторы, загустители, красители и отдушки.
Активное целевое соединение вводят в фармацевтическую композицию в количестве, достаточном для получения нужного эффекта, в зависимости от нозологии, течения и тяжести заболевания.
При изготовлении разовой лекарственной формы, количество активного ингредиента, используемого в комбинации с носителем, может варьировать в зависимости от реципиента, подвергающегося лечению, от конкретного способа введения лекарственного средства.
Так, например, при использовании соединений настоящего изобретения в виде растворов для инъекций содержание действующего начала в них составляет 1-10%, в том числе в виде комплекса с белками плазмы. В качестве разбавителя вещества могут быть использованы 0,9% раствор натрия хлорида, дистиллированная вода, раствор новокаина для инъекций, раствор Рингера, раствор глюкозы. При использовании соединения общей формулы (I) в виде таблеток и суппозиториев количество вещества составляет 1,0-100,0 мг на единичную дозированную форму. Для таблеток и суппозиториев в качестве фармацевтического наполнителя используют любую фармацевтически пригодную основу.
Примеры лекарственных форм.
А. Желатиновые капсулы.
Состав вводимого в капсулу порошка:
Указанные выше ингредиенты смешивают, и смесь вводят в твердые желатиновые капсулы в количестве 151-285 мг.
Б. Таблетированная форма.
Таблетированную форму получают, используя приведенные ниже ингредиенты:
Компоненты смешивают и прессуют для образования таблеток весом 200 мг каждая.
В. Аэрозольная форма.
Состав аэрозольной смеси, рассчитанной на 10 приемов:
Соединение смешивают с наполнителями и помещают в специальное устройство для распыления.
Г. Суппозитории.
В качестве суппозиторной основы могут быть использованы:
нерастворимые в воде основы - масло какао;
растворимые в воде или смешиваемые с водой основы - желатино-глицериновые или полиэтиленоксидные;
комбинированные основы - мыльно-глицериновые.
Пример состава суппозитория:
При необходимости возможно изготовление ректальных, вагинальных и уретральных суппозиториев с соответствующими наполнителями.
Д. Мази.
В качестве мазевой основы могут быть использованы:
углеводородные мазевые основы - вазелин белый и желтый (Vaselinum album, Vaselinum flavum), вазелиновое масло (Oleum Vaselini), мазь белая и жидкая (Unguentum album, Unguentum flavum), а в качестве добавок для придания более плотной консистенции твердый парафин и воск;
абсорбтивные мазевые основы - гидрофильный вазелин (Vaselinum hydrophylicum), ланолин (Lanolinum), кольдкрем (Unguentum leniens);
мазевые основы, смываемые водой - гидрофильная мазь (Unguentum hydrophylum); водорастворимые мазевые основы - полиэтиленгликолевая мазь (Unguentum Glycolis Polyaethyleni), бентонитовые основы и другие.
Пример состава мази:
Мази изготавливают по соответствующей технологии.
Е. Раствор для инъекций.
В качестве растворителя при приготовлении раствора для инъекций могут быть использованы 0,9% раствор натрия хлорида, дистиллированная вода, раствор новокаина. Форма выпуска - ампулы, флаконы, шприц-тюбики, «insert».
Состав раствора для инъекций:
Возможно изготовление различных лекарственных форм для инъекций - стерильных растворов, стерильных порошков и таблеток.
Заключение
Производные гемина, соответствующие общей формуле I, в свободной форме или в виде фармацевтически приемлемой соли; в эффективном количестве в фармацевтически приемлемом носителе, в том числе в виде комплекса с белком плазмы, обладают полезными фармакологическими свойствами и могут использоваться в качестве основы для создания фармацевтических препаратов с противоопухолевым действием.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕМИНПЕПТИД, ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОТИВОВИРУСНОГО И ВИРУЛЕЦИДНОГО АГЕНТА | 2004 |
|
RU2296131C2 |
| ПРОИЗВОДНЫЕ ГЕМИНА И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ, ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2002 |
|
RU2238950C2 |
| ПРОИЗВОДНЫЕ ГЕМИНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ, КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2007 |
|
RU2404191C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ГЕМИНПЕПТИДОВ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ НУКЛЕОЛИТИЧЕСКИХ АГЕНТОВ | 2003 |
|
RU2250906C2 |
| ПРОИЗВОДНЫЕ ГЕМИНА, ОБЛАДАЮЩИЕ АНТИМИКРОБНОЙ АКТИВНОСТЬЮ, ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ, ФАРМКОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2009 |
|
RU2415868C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ ГЕМИНА С АНТИБАКТЕРИАЛЬНОЙ И ПРОТИВОВИРУСНОЙ АКТИВНОСТЬЮ | 2011 |
|
RU2475498C1 |
| ЦИКЛИЧЕСКИЙ ОКТАПЕПТИД, РАДИОФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО НА ЕГО ОСНОВЕ И СПОСОБ ПРИМЕНЕНИЯ РАДИОФАРМАЦЕВТИЧЕСКОГО СРЕДСТВА ДЛЯ ПОЛУЧЕНИЯ ЛЕКАРСТВЕННЫХ (ФАРМАЦЕВТИЧЕСКИХ) СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ НОВООБРАЗОВАНИЙ, ЭКСПРЕССИРУЮЩИХ СОМАТОСТАТИНОВЫЕ РЕЦЕПТОРЫ | 2013 |
|
RU2528414C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2004 |
|
RU2355418C2 |
| АНАЛОГИ ОКСИТОЦИНА | 2009 |
|
RU2496788C2 |
| ЦИКЛИЧЕСКИЕ ПЕПТИДЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2095368C1 |
Геминпептиды общей формулы I:
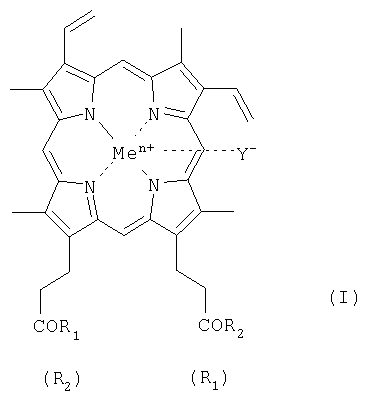
где R1 и R2 заместители, которые могут представлять собой пептид, состоящий из 1-7 аминокислотного остатка, где карбоксильные группы пептида могут быть модифицированы С1-С8 алкиловым эфиром, а боковые группы аминокислотных остатков пептида могут быть защищены, и выбраны из группы, где: при R1 = -ArgTrpHisArgLeuLysGlu(OMe)OH, R2 = -OH, при R1 = -TrpHisArgLeuLysGlu(OMe)OH, R2 = -OH, при R1 = -SerOMe, R2 = -SerOMe, при R1 =-AspOH, R2 =-AspOH , Me представляет собой Fe, Y представляет собой Cl, при этом карбоксильная группа гемина может быть модифицирована метиловым или С2-С8 эфиром или физиологически приемлемой солью; или его фармацевтически приемлемые соли. Геминпептиды обладают способностью индуцировать апоптоз клеток и противоопухолевым действием и могут использоваться для биомедицинских целей. 3 н. и 2 з.п. ф-лы, 1 табл.
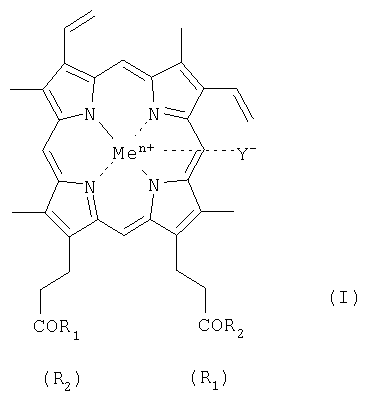
где R1 и R2 заместители, которые могут представлять собой пептид, состоящий из 1-7 аминокислотного остатка, где карбоксильные группы пептида могут быть модифицированы С1-С8 алкиловым эфиром, а боковые группы аминокислотных остатков пептида могут быть защищены, и выбраны из группы, где при R1 = -ArgTrpHisArgLeuLysGlu(OMe)OH, R2 = -OH,
при R1 = -TrpHisArgLeuLysGlu(OMe)OH, R2 = -OH,
при R1 = -SerOMe, R2 = -SerOMe,
при R1 =-AspOH, R2 =-AspOH ,
Me представляет собой Fe,
Y представляет собой Cl,
при этом карбоксильная группа гемина может быть модифицирована метиловым или С2-С8 эфиром или физиологически приемлемой солью,
или их фармацевтически приемлемые соли.
где R1 и R2 заместители, которые могут представлять собой пептид, состоящий из 1-7 аминокислотного остатка, где карбоксильные группы пептида могут быть модифицированы С1-С8 алкиловым эфиром, а боковые группы аминокислотных остатков пептида могут быть защищены, при этом R1= R2 или R1≠R2= -ОН, Me представляет собой Fe, Y представляет собой Cl, и карбоксильная группа гемина может быть модифицирована метиловым или С2-С8 эфиром или физиологически приемлемой солью,
или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
при R1 = -ArgTrpHisArgLeuLysGlu(OMe)OH, R2 = -OH,
при R1 = -TrpHisArgLeuLysGlu(OMe)OH, R2 = -OH,
при R1 = -SerOMe, R2 = -SerOMe,
при R1=-LysOMe, R2 = -LysOMe,
при R1 =-AspOH, R2 =-AspOH и
при R1 =-ArgOMe, R2 =-ArgOMe.
,
где R1 и R2 заместители, которые могут представлять собой пептид, состоящий из 1-7 аминокислотного остатка, где карбоксильные группы пептида могут быть модифицированы С1-С8 алкиловым эфиром, а боковые группы аминокислотных остатков пептида могут быть защищены, при этом R1= R2 или R1≠R2= -ОН, Me представляет собой Fe,Y представляет собой Cl, и карбоксильная группа гемина может быть модифицирована метиловым или С2-С8 эфиром или физиологически приемлемой солью,
или его фармацевтически приемлемой соли в качестве агента, индуцирующего апоптоз клеток и обладающего противоопухолевой активностью.
| Ru 2002111028 A, 27.10.2003.Su 717039A, 25.02.1980. |

