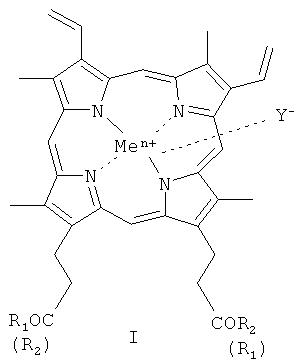
Изобретение относится к области биоорганической химии, а именно к новым производным гемина общей формулы I

где
R1=OH,
R2=-Val-Gly-Ala-(D-Leu)-Ala-(D-Val)-Val-(D-Val)-Trp-(D-Leu)-X-(D-Leu)-Trp-(D-Leu)-Trp-NHCH2CH2OH, где Х=Trp, или Phe, или 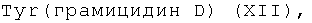


или 

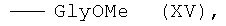


к способу синтеза этих соединений, композиции, в том числе фармацевтической, дезинфицирующей, антисептической композиции, средству для лечения и биомедицинскому применению новых и известных производных гемина формулы I в качестве антимикробных - антибактериальных и противогрибковых, а также вирулицидных агентов. Как известно, многие опасные заболевания человека вызываются бактериями. К ним относятся заболевания эпидемического характера, такие как холера, брюшной тиф, паратифы, чума, дифтерия, туляремия, бруцеллез, а также туберкулез, заражение крови (сепсис), проказа, сифилис и др. У животных бактерии вызывают сап, сибирскую язву, туберкулез и др. Борьба с болезнетворными бактериями основывается на применении дезинфецирующих и антисептических средств, бактериостатических и бактерицидных агентов. У бактерий быстро развивается резистентность по отношению к существующим антибактериальным средствам, главным образом к антибиотикам, поэтому проблема поиска новых нетоксичных, биосовместимых антибактериальных агентов, применение которых не приводило бы к резистентности, весьма актуальна [J.-Y.Maillard. Bacterial resistance to biocides in the healthcare environment: should it be of genuine concern // Journal of Hospital Infection, 2007, V.65(S2) p.60-72].
В последнее время особое внимание уделяется дизайну новых антимикробных агентов на основе антимикробных пептидов (АМП) [А.Giuliani, G.Pirri, S.F.Nicoletto. Antimicrobial peptides: an overview of a promising class of therapeutics// Central European Journal of Biology, 2007, V.2(1), p.1-33]. Резистентность в отношении АМП у микроорганизмов развивается довольно редко [M.N.Melo, D.Dugourd, M.A.Castanho // Recent Patents Anti-Infect Drug Disc, 2006, V.1(2), p.201-207], по-видимому, вследствие того, что основным механизмом действия АМП является деструкция липидной мембраны бактерий, микроскопических грибов и вирусов [Н.Jenssen, P.H., and R.E.W. Hancock. Peptide Antimicrobial Agents // Clin. Microbiol. Rev., 2006, 19(3): 491-511]. В связи с этим антимикробные пептиды рассматриваются как перспективный класс новых антибиотиков [M.R.Yeaman, N.Y.Yount. Mechanisms of Antimicrobial Peptide Action and Resistance // Pharmacol. Rev., 2003, 55(1) p.27-55, A.Giuliani, G.Pirri, S.F.Nicoletto. Antimicrobial peptides: an overview of a promising class of therapeutics. // Central European Journal of Biology, 2007, V.2(1) p.1-33]. Антимикробные пептиды эндогенного происхождения являются частью наиболее древней системы защиты организмов от патогенов. Они обнаружены практически у всех живых организмов - от бактерий до человека [M.R.Yeaman, N.Y.Yount. Mechanisms of Antimicrobial Peptide Action and Resistance // Pharmacol. Rev., 2003, V.55(1) p.27-55]. В процессе эволюции многоклеточные организмы умножали и развивали арсенал АМП, защищающих их от широкого спектра патогенов, в том числе грамположительных и грамотрицательных бактерий [M.R.Yeaman, N.Y.Yount. Mechanisms of Antimicrobial Peptide Action and Resistance // Pharmacol. Rev., 2003, 55(1):27-55], ДНК и РНК-содержащих вирусов [A.Bastian, and H.Schafer. Human alpha-defensin 1 (HNP-1) inhibits adenoviral infection in vitro// Regul. Pept., 2001, V.101, p.157-161. Home, W.S., С.M.Wiethoff, С.Cui, et al. Antiviral cyclic D,L-alpha-peptidestargeting a general biochemical pathway in virus infections// Bioorg. Med. Chem., 2005, V.13, p.5145-5153], микроскопических грибов [De Lucca, A.J., and T.J.Walsh. Antifungal peptides: novel therapeutic compounds against emerging pathogens // Antimicrob. Agents Chemother., 1999, V.43, p.1-11] и простейших [J.Alberola,, A.Rodriguez, O.Francino, et al. Safety and efficacy of antimicrobial peptides against naturally acquired leishmaniasis // Antimicrob. Agents Chemother., 2004, V.48, p.641-643].
Антимикробные пептиды, как правило, являются амфифильными соединениями с ярко выраженными гидрофильной и гидрофобной частями. За редкими исключениями молекулы АМП содержат несколько остатков лизина и/или аргинина и при физиологических значениях рН несут большой положительный заряд. По структуре АМП можно разделить на две большие группы: циклические пептиды, как правило, содержащие цистеиновый мостик, и линейные пептиды [Н.Jenssen, P.Hamill, R.E.W. Hancock. Peptide Antimicrobial Agents// Clinical Microbiology Reveiws, 2006, Vol.19, No.3, p.491-511].
Понимание механизмов узнавания и деструкции мембран клеток патогенов антимикробными пептидами является необходимым условием для создания лекарств на их основе. Наиболее важным фактором селективности действия АМП на бактерии, грибы и вирусы при отсутствии их воздействия на эукариотические клетки является различие в составе и строении мембран [Н.Jenssen, P.Hamill, R.E.W. Hancock. Peptide Antimicrobial Agents// Clinical Microbiology Reveiws, 2006, Vol.19, No.3, p.491-511]. Внешняя поверхность эукариотической мембраны состоит из цвиттер-ионных фосфолипидов, в то время как мембраны бактериальных клеток содержат большое количество отрицательно заряженных фосфолипидов как на внутренней, так и на внешней поверхности липидного бислоя. Кислотный характер внешней стороны прокариотической мембраны обуславливает ее взаимодействие преимущественно с положительно заряженными АМП [M.R. Yeaman, N.Y.Yount. Mechanisms of Antimicrobial Peptide Action and Resistance // Pharmacol. Rev.,2003, Vol.55, p.27-55]. Вторым фактором, обуславливающим селективность АМП, является отсутствие холестерина в мембранах бактериальных клеток. Вследствие этого липидный бислой бактериальных мембран более гибок, и образование пор антимикробными пептидами в нем происходит легче, чем в мембранах эукариотических клеток [Н.Jenssen, P.Hamill, R.E.W. Hancock. Peptide Antimicrobial Agents // Clinical Microbiology Reveiws, 2006, Vol.19, No.3, p.491-511. 3. О.Токе. Antimicrobial Peptides: New Candidates in the Fight Against Bacterial Infections // Biopolymers (Peptide Science), 2005, Vol.80, p.717-735]. При деструкции молекулами АМП мембраны бактерии последняя погибает вследствие утечки ионов и метаболитов, деполяризации мембраны, угнетения клеточного дыхания бактерий и синтеза в них биополимеров [M.R.Yeaman, N.Y.Yount. Mechanisms of Antimicrobial Peptide Action and Resistance // Pharmacol. Rev., 2003, Vol.55, p.27-55]. По последним данным, гибель клеток под действием АМП может происходить и по другим причинам, например в результате взаимодействия антимикробных пептидов с внутриклеточными мишенями [J.-P.S.Powers, R.E.W. Hancock. The relationship between peptide structure and antibacterial activity // Peptides, 2003, V.24, p.1681-1691]. По схожим механизмам АМП воздействуют и на мембраны вирусов, приводя к их дезактивации [Н.Jenssen, P.Hamill, R.E.W. Hancock. Peptide Antimicrobial Agents// Clinical Microbiology Reveiws, 2006, Vol.19, No.3, p.491-511. 3].
Основными препятствиями к применению АМП в клинической практике являются их сравнительно высокая стоимость, чувствительность к действию протеолитических ферментов, а также присущий многим АМП гемолитический эффект [A.Giuliani, G.Pirri, S.F.Nicoletto. Antimicrobial peptides: an overview of a promising class of therapeutics. // Central European Journal of Biology, 2007, V.2(1) 1-33]. Например, антимикробные пептиды грамицидин D и грамицидин S вызывают 100% гемолиз в концентрациях 5 мкМ [С.Н.Rammelkamp and L.Weinstein. Toxic Effects of Tyrothricin, Gramicidin, and Tyrocidine // Jour. Infect. Dis., 1942, V.71, №2, p.166-173] и 62,5 мкМ [G.M.Grotenbreg, M.D.Witte, P.A.V. van Hooft, et al. Synthesis and biological evaluation of gramicidin S dimers // Org. Biomol. Chem., 2005, V.3, p.233-238] соответственно. По-видимому, вследствие этого на рынке пока отсутствуют препараты на основе АМП, состоящие исключительно из аминокислот L-ряда. Большинство АМП отсеиваются на ранних стадиях клинических испытаний. И лишь некоторые препараты на основе АМП в настоящее время проходят завершающие этапы клинических испытаний, например, Омиганан (Omiganan) и соединение MX594AN. Действующее начало этих препаратов представляет собой синтетические аналоги антимикробного пептида индолицидина с измененной аминокислотной последовательностью. В ходе фазы III клинических испытаний в качестве препарата, понижающего колонизацию венозных катетеров микроорганизмами, вызывающими заболевания кровеносной системы в разных группах пациентов, омиганан продемонстрировал эффекты, не позволяющие сделать однозначный вывод о возможности его применения в клинической практике [Y.J.Gordon, A.M.McDermott. A Review of Antimicrobial Peptides and Their Therapeutic Potential as Anti-Infective Drugs // Curr Eye Res., 2005, V.30(7), p.505-515]. Соединение MX594AN прошло клинические испытания фазы IIb, и в настоящее время проходит клинические испытания фазы III в качестве средства для лечения угревой сыпи [Обзор по АМП - Гордон]. Кроме того, несколько препаратов на основе АМП находятся на более ранних стадиях клинических испытаний [Y.J. Gordon, A.M. McDermott. A Review of Antimicrobial Peptides and Their Therapeutic Potential as Anti-Infective Drugs // Curr Eye Res., 2005, V.30(7), p.505-515. A.Giuliani, G.Pirri, S.F.Nicoletto. Antimicrobial peptides: an overview of a promising class of therapeutics. // Central European Journal of Biology, 2007, V.2(1) 1-33].
В настоящее время антимикробные пептиды и их аналоги получают в основном твердофазным синтезом, что делает эти вещества чрезвычайно дорогостоящими, а их применение экономически нецелесообразным.
В последнее время все большее внимание уделяется другим стратегиям дизайна модифицированных АМП [D.Knappe, A.Nimptsch, A.Jr. Kolobov, et al. Chemical modifications of short antimicrobial peptides from insects and vertebrates to fight multi-drug resistant bacteria // Adv. Exp. Med. Biol., 2009, V.611, p.395-396], лишенных перечисленных выше недостатков. Основными стратегиями оптимизации структуры АМН с целью получения новых биоцидных агентов являются следующие.
- Синтез циклических аналогов АМП. В ряде случаев [A.Wessolowski, М.Bienert and M.Dathe. Antimicrobial activity of arginine-and tryptophan-rich hexapeptides: the effects of aromatic clusters, D-amino acid substitution and cyclization. J. Pept. Res., 2004, V.64, p.159-169. M.Dathe, H.Nikolenko, J.Klose, et al. Cyclization Increases the Antimicrobial Activity and Selectivity of Arginine- and Tryptophan-Containing Hexapeptides // Biochemistry, 2004, V.43(28), p.9140-9150] циклизация АМП приводит к увеличению антибактериальной активности и повышению устойчивости к протеиназам [A.Rozek, J-P.S.Powers, C.L.Friedrich, et al. Structure-Based Design of an Indolicidin Peptide Analogue with Increased Protease Stability // Biochemistry, 2003, V.42 (48), p.14130-14138], однако при этом может значительно (в 4 раза) возрастать гемолитический эффект [A.Wessolowski, M.Bienert and M.Dathe. Antimicrobial activity of arginine-and tryptophan-rich hexapeptides: the effects of aromatic clusters, D-amino acid substitution and cyclization // J. Pept. Res., 2004, V.64, p.159-169]. Кроме того, циклизация приводит к заметному повышению себестоимости продукта.
- Введение в молекулу АМП атома фтора или трифторметильной группы. Этот подход приводит к снижению МБК (минимальной бактерицидной концентрации) антимикробных пептидов, однако при этом увеличивается время воздействия на микроорганизм, необходимое для проявления бактерицидного действия [D.Gimenez, С.Andreu, M. del Olmo, et al. The introduction of fluorine atoms or trifluoromethyl groups in short cationic peptides enhances their antimicrobial activity // Bioorg. Med. Chem., 2006, V.14, p.6971-6978].
- Иммобилизация АМП на различных полимерных матрицах [М.Bagheri, М.Beyermann, M.Dathe. Immobilization reduces the activity of surface-bound cationic antimicrobial peptides with no influence upon the activity spectrum // Antimicrob Agents Chemother., 2009, V.53(3), p.1132-41]. Этот сравнительно новый подход пока не привел к успешным результатам, т.к. активность иммобилизованных на полимерах пептидов оказалась существенно ниже, чем у свободных АМП. Кроме того, авторами [М.Bagheri, M.Beyermann, M.Dathe. Immobilization reduces the activity of surface-bound cationic antimicrobial peptides with no influence upon the activity spectrun // Antimicrob Agents Chemother., 2009, V.53(3), p.1132-41] не установлено, какое влияние оказывает иммобилизация на гемолитический эффект пептидов.
Таким образом, химическая модификация антимикробных пептидов является перспективным путем получения новых, более эффективных антимикробных агентов без побочных эффектов с антибактериальным, противогрибковым и вирулицидным действием. При этом остается актуальным поиск новых подходов к модификации АМП, позволяющих повысить возможности их практического применения.
Известны антимикробные пептиды - линейный грамицидин D (II),
представляющий собой смесь пептидов формулы:
Val-Gly-Ala-(D-Leu)-Ala-(D-Val)-Val-(D-Val)-Trp-(D-Leu)-X-(D-Leu)-Trp-(D-Leu)-TrpNHCH2CH2OH, где X=Trp, Tyr, Phe
[W.E.Herrelland D.Heilman. Experimental and Clinical Studies on Gramicidin // J.Clin. Invest., 1941, V.20, 583-591. Annual Reports on NMR Spectroscopy, Volume 60, p 215 London, Academic Press, 2007], циклический грамицидин S (III) [G.Nagamurthi and S.Rambhav. Gramicidin-S: Structure-activity relationship // J.Biosci., 1985, V.7, №3-4, p.323-329], содержащие D-аминокислоты, непротеиногенные аминокислоты (Orn). Соединение II эффективно против грамположительных бактерий [W.E.Herrell and D.Heilman. Experimental and Clinical Studies on Gramicidin // J. Clin. Invest., 1941, V.20, №583] и некоторых вирусов, в частности герпеса [US Patent 6001808, 1999]. Соединение III эффективно главным образом против грамположительных бактерий в концентрациях 5-15 мкМ [Jingbo Xiao, Bernard Weisblum, and Peter Wipf. Electrostatic versus Steric Effects in Peptidomimicry: Synthesis and Secondary Structure Analysis of Gramicidin S Analogues with (E)-Alkene Peptide Isosteres //J. Am. Chem. Soc, 2005, 127 (16), рр 5742-5743]. Соединения II и III используются в медицинской практике только для наружного применения [М.Д.Машковский. Лекарственные средства, 15 изд. Москва, изд-во «Новая волна», 2005, с.822. J.R.Gibson. Trimethoprin-polymyxin В ophthalmic solution in the treatment of presumptive bacterial conjunctivitis-A multicentre trial of its efficacy versus neomycin-polymyxin B-gramicidin and chloramphenicol ophthalmic solutions // J.Antimicrob. Chemother., 1983, V.11, p.217-221]. Недостатками этих антимикробных препаратов являются относительно большая длина этих пептидов и соответственно сравнительно высокая стоимость, недостаточная широта антибактериального, противогрибкового и вирулицидного действия (активность лишь в отношении грамположительных бактерий и вируса герпеса). Но главными их недостатками являются побочные эффекты при применении, в основном гемолиз эритроцитов и аллергические реакции [Heilman, D., and Herrell, W.E.: "Hemolytic Effect of Gramicidin," Proc. Soc. Exper. Biol. and Med., 46: 182 (Jan.) 1941. М.Д.Машковский. Лекарственные средства, 15 изд. Москва, изд-во «Новая волна», 2005, с.822].
Известный пептид 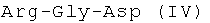 в настоящее время исследуется в различных направлениях. В частности, он представляет интерес для создания средств для лечения инфекционных заболеваний [A.J.Kastin. Handbook of biologically active peptides // Elsevier, Academic Press, USA, 2006, p.576]. Пептид IV является фрагментом антимикробных пептидов семейства цекропинов, многих микробных белков и фибронектина поверхности клеток млекопитающих [M.Naito, N.Ohara, S.Matsumoto, T.Yamada. The novel fibronectin binding motif and key residues of micobacteria //J. Biol. Chem., 1998, V.273, p.2905-9].
в настоящее время исследуется в различных направлениях. В частности, он представляет интерес для создания средств для лечения инфекционных заболеваний [A.J.Kastin. Handbook of biologically active peptides // Elsevier, Academic Press, USA, 2006, p.576]. Пептид IV является фрагментом антимикробных пептидов семейства цекропинов, многих микробных белков и фибронектина поверхности клеток млекопитающих [M.Naito, N.Ohara, S.Matsumoto, T.Yamada. The novel fibronectin binding motif and key residues of micobacteria //J. Biol. Chem., 1998, V.273, p.2905-9].
Известно, что гемин обладает антимикробной активностью, в частности, в отношении золотистого стафилококка. Авторами [Y.Nitzan, H.Ladan, S.Gozansky, Z.Malik. Characterization of hemin antibacterial action on Staphylococcus aureus // FEMS Microbiol. Lett. V.48(3), p.401-406] было показано, что гемин в концентрации 20·10-5 М оказывает сильное, не зависящее от света воздействие на S.aureus, в то время как другие металлопорфирины не проявляли антибактериального эффекта в темноте. Заметное воздействие гемина на инактивацию S.aureus проявилось в немедленной индукции потоков ионов, что было определено рентгеновским микроанализом быстрозамороженных клеток. Авторы [Y.Nitzan, H.Ladan, S.Gozansky, Z.Malik. Characterization of hemin antibacterial action on Staphylococcus aureus // FEMS Microbiol. Lett. V.48(3), p.401-406] полагают, что такое выраженное антибактериальное действие гемина обусловлено тем, что в S.aureus находится множество клеточных мишеней для окислительного воздействия гемина. Однако использование гемина в качестве антибактериального средства затруднено вследствие его нерастворимости в воде, гемолитической активности, а также кратковременности антибактериального эффекта.
Для некоторых пептидных производных гемина общей формулы I, a именно при R1=OH 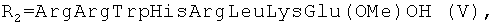 или
или  установлена нуклеазная (нуклеолитическая) активность, проявляющаяся в способности разрушать плазмидную ДНК [Желтухина Г.А., Небольсин В.Е., Лобанова Т.Н // Патент РФ №2250906; Желтухина Г.А., Лобанова Т.Н., Небольсин В.Е., М.О.Галлямов, Драницына С.М., Костанян И.А. // Биоорган, химия. 2006. Т.32, №2, С.198-210]. Отдельные аминокислотные и пептидные производные гемина общей формулы I, а именно при R1=OH,
установлена нуклеазная (нуклеолитическая) активность, проявляющаяся в способности разрушать плазмидную ДНК [Желтухина Г.А., Небольсин В.Е., Лобанова Т.Н // Патент РФ №2250906; Желтухина Г.А., Лобанова Т.Н., Небольсин В.Е., М.О.Галлямов, Драницына С.М., Костанян И.А. // Биоорган, химия. 2006. Т.32, №2, С.198-210]. Отдельные аминокислотные и пептидные производные гемина общей формулы I, а именно при R1=OH, 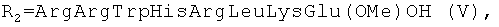 или
или 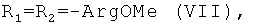 способны ингибировать протеиназу ВИЧ и оказывать вследствие этого противовирусное анти-ВИЧ действие [Р.П.Евстигнеева, Г.А.Желтухина, Т.В.Зарубина, В.Е.Небольсин, Д.Н.Носик, Н.Н.Носик // Патент РФ №2238950].
способны ингибировать протеиназу ВИЧ и оказывать вследствие этого противовирусное анти-ВИЧ действие [Р.П.Евстигнеева, Г.А.Желтухина, Т.В.Зарубина, В.Е.Небольсин, Д.Н.Носик, Н.Н.Носик // Патент РФ №2238950].
Показано, что некоторые известные производные гемина общей формулы I, а именно 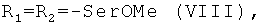
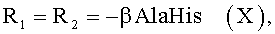
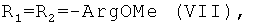
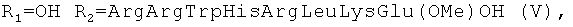
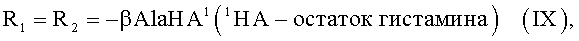
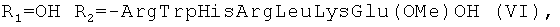 обладают вирулицидной активностью [заявка на изобретение RU 2007125604/04(027891)], однако антибактериальная, антигрибковая активности для них не были известны.
обладают вирулицидной активностью [заявка на изобретение RU 2007125604/04(027891)], однако антибактериальная, антигрибковая активности для них не были известны.
Композиции для предотвращения бактериальных и грибковых инфекций, включающие конъюгаты гемина с аминокислотами, пептидами и пептидомиметиками, неизвестны.
Нами предложено провести конъюгацию двух природных соединений (гемина и антимикробного пептида), обладающих антимикробной активностью с различным механизмом действия с целью повышения эффективности действия, снижения токсичности и приобретения новых полезных свойств, в частности водорастворимости и устойчивости к протеолизу.
Задачей настоящего изобретения является создание новых биосовместимых мембраноактивных конъюгатов гемина с АМП общей формулы I, и их фармацевтически приемлемых солей, обладающих деструктирующим действием по отношению к липидной мембране, эффективных против бактерий, микроскопических грибов и вирусов, при этом нетоксичных, без гемолитической активности и без побочных эффектов; создание на их основе противоинфекционного лекарственного средства, обладающего антибактериальной, противогрибковой и вирулицидной активностью; композиций, в том числе фармацевтических, дезинфецирующих и антисептических, а также способов, позволяющих получать заявленные соединения с достаточными выходами и чистотой.
Еще одной задачей настоящего изобретения является способ синтеза соединения 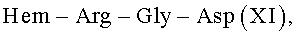 включающий получение входящего в его состав трипептида
включающий получение входящего в его состав трипептида 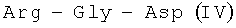 с более высоким выходом, по более простой и экономически целесообразной методике. Известен [Patent GB 2207922А. Tripeptides with pharmacological properties. В.Brunetti, M.Prada, 1989] способ синтеза пептида IV по схеме [1+2] с использованием дикарбобензокси-аргинина и дибензилового эфира аспарагиновой кислоты. Недостатком метода является применение в качестве исходного реагента указанного производного аргинина. Известно, что в таком производном гуанидиновая группа защищена недостаточно, что может приводить к образованию побочных продуктов в синтезах с его использованием [Дж.Гринштейн, М.Виниц. Химия аминокислот и пептидов // М: Мир, 1966, с.558]. Выход целевого трипептида IV, полученного этим способом, был достаточно низким и составил 21%, считая на исходный реагент.Известен другой способ синтеза этого трипептида в растворе [М.Abo-Ghalia, S.Abd El-Rahman, A.El-Kafrawy, and A.Kalomuch. Optimized conventional synthesis of "RGD" and "RGDS" peptides and their sarcosine mimics as integrin GP IIb/IIIa antagonists // Amino Acids, 2003, V.24, p.405-411] по схеме [2+1] с использованием исходных реагентов H-Gly-OEt, Z-Arg(NO2)-OH и Н-Asp(OBzl)-OBzl. Данный подход позволил повысить суммарный выход целевого пептида до 41%. В то же время, существенным недостатком этого метода является использование производного аргинина, известного неполным блокированием гуанидиновой функции аргинина нитрогруппой. Кроме того, известно, что отщепление гидрогенолизом NG-нитрогруппы не всегда бывает удовлетворительным [Дж.Гринштейн, М.Виниц. Химия аминокислот и пептидов // М: Мир, 1966, с.555]. Предусмотренное в данной схеме синтеза омыление Z-Arg(NO2)-GlyOEt представляет собой нежелательное жесткое воздействие на дипептид, которое может приводить к рацемизации и частичной деструкции пептидной связи. Применение переносного гидрирования в присутствии палладиевой черни по данному способу удлиняет и удорожает стадию конечного деблокирования целевого трипептида IV.
с более высоким выходом, по более простой и экономически целесообразной методике. Известен [Patent GB 2207922А. Tripeptides with pharmacological properties. В.Brunetti, M.Prada, 1989] способ синтеза пептида IV по схеме [1+2] с использованием дикарбобензокси-аргинина и дибензилового эфира аспарагиновой кислоты. Недостатком метода является применение в качестве исходного реагента указанного производного аргинина. Известно, что в таком производном гуанидиновая группа защищена недостаточно, что может приводить к образованию побочных продуктов в синтезах с его использованием [Дж.Гринштейн, М.Виниц. Химия аминокислот и пептидов // М: Мир, 1966, с.558]. Выход целевого трипептида IV, полученного этим способом, был достаточно низким и составил 21%, считая на исходный реагент.Известен другой способ синтеза этого трипептида в растворе [М.Abo-Ghalia, S.Abd El-Rahman, A.El-Kafrawy, and A.Kalomuch. Optimized conventional synthesis of "RGD" and "RGDS" peptides and their sarcosine mimics as integrin GP IIb/IIIa antagonists // Amino Acids, 2003, V.24, p.405-411] по схеме [2+1] с использованием исходных реагентов H-Gly-OEt, Z-Arg(NO2)-OH и Н-Asp(OBzl)-OBzl. Данный подход позволил повысить суммарный выход целевого пептида до 41%. В то же время, существенным недостатком этого метода является использование производного аргинина, известного неполным блокированием гуанидиновой функции аргинина нитрогруппой. Кроме того, известно, что отщепление гидрогенолизом NG-нитрогруппы не всегда бывает удовлетворительным [Дж.Гринштейн, М.Виниц. Химия аминокислот и пептидов // М: Мир, 1966, с.555]. Предусмотренное в данной схеме синтеза омыление Z-Arg(NO2)-GlyOEt представляет собой нежелательное жесткое воздействие на дипептид, которое может приводить к рацемизации и частичной деструкции пептидной связи. Применение переносного гидрирования в присутствии палладиевой черни по данному способу удлиняет и удорожает стадию конечного деблокирования целевого трипептида IV.
Поставленная задача решается новыми соединениями общей формулы I, где R1=OH,
R2=-Val-Gly-Ala-(D-Leu)-Ala-(D-Val)-Val-(D-Val)-Trp-(D-Leu)-X-(D-Leu)-Trp-(D-Leu)-Trp-NHCH2CH2OH, где Х=Trp, или Phe, или 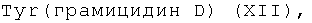
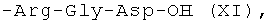
или 

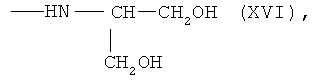
и/или их фармацевтически приемлемыми солями, обладающими способностью деструктировать липидную мембрану;
- средством, обладающим антибактериальной активностью, в том числе против резистентных бактерий, противогрибковой и вирулицидной активностью, представляющим собой новое соединение общей формулы I;
- средством, обладающим антибактериальной активностью, в том числе против резистентных бактерий, и противогрибковой активностью, представляющим собой известное соединение общей формулы I;
- фармацевтической, дезинфицирующей и антисептической композицией, включающей соединения общей формулы I для профилактики и/или лечения у человека и животных различных заболеваний, вызванных бактериями, в том числе резистентными, микроскопическими грибами или вирусами с использованием их антибактериальных, противогрибковых и вирулицидных свойств и содержащей одно или несколько из соединений общей формулы I в эффективном количестве;
- способами синтеза новых производных гемина общей формулы I или их фармацевтически приемлемых солей, включающими ацилирование аминогруппы аминокомпонентов бис-N-оксисукцинимидным эфиром гемина, или включающими ацилирование α-аминогруппы пептида 6(7)-моно-N-окси-5-норборнен-2,3-дикарбоксиимидным эфиром гемина, или включающими ацилирование аминогруппы пептида активированным по одной карбоксильной группе производным гемина, причем в качестве активирующего агента используют ди-трет-бутилпирокарбонат в присутствии пиридина;
- способом получения конъюгата гемина с Arg-Gly-Asp, включающим синтез трипептида Arg-Gly-Asp. В соответствии с настоящим изобретением, способ синтеза Arg-Gly-Asp в растворе включает получение дипептида Z3Arg-Gly-ОН методом смешанных ангидридов с использованием силилированного производного глицина с последующим присоединением к упомянутому дипептиду H-Asp(OtBu)2 также методом смешанных ангидридов; далее деблокирование защищенного трипептида проводится в 2 стадии - сначала трет-бутильных защит - действием трифторуксусной кислоты, с последующим отщеплением Z-групп гидрогенолизом над 10÷20% палладием на угле. Предложенный способ позволяет повысить выход целевого продукта - трипептида до 57%, упростить процесс, в частности исключить стадию жесткого воздействия - омыления дипептида, уменьшить время гидрогенолиза и сделать процесс более экономически целесообразным в результате замены палладиевой черни на 10÷20% палладий на угле.
Список сокращений
DCC - N,N′-дициклогексилкарбодиимид
DMSO - диметилсульфоксид
DOPC - диолеоилфосфатидилхолин
GrD - грамицидин D
НА - гистамин
Hem - остаток гемина
МеОН - метиловый спирт
MES - 2-(N-морфолино)этансульфокислота
МН - среда Mueller-Hilton
ОМе - метиловый эфир
ONb - N-окси-5-норборнен-2,3-дикарбоксиимидный эфир
OtBu - трет-бутиловый эфир
Z - карбобензоксигруппа
ДМСО - диметилсульфоксид
ДМФА-N,N′ - диметилформамид
ИК - инфракрасная спектроскопия
ИР - ингибирование роста
КФ - карбоксифлуоресцеин
МБК - минимальная бактерицидная концентрация
МИК - минимальная ингибирующая концентрация
ОФ-ВЭЖХ - обращенно-фазная высокоэффективная жидкостная хроматография
Tris - трис-(гидроксиметил)аминометан
ТСХ - хроматография в тонком слое
ХЛФ - хлороформ
ЭА -этилацетат
Следующие примеры иллюстрируют предлагаемое изобретение.
В работе использовались аминокислоты и их производные L-ряда фирмы «Bachem» (Германия), «Reanal» (Венгрия), DOPC (Sigma-Aldrich), карбоксифлуоресцеин (Fluka, Германия), MES (Sigma-Aldrich), Tris (Sigma-Aldrich), грамицидин D (Sigma-Aldrich), грамицидин S (Красфарма, Россия) хлорид калия ч.д.а. (Химмед, Россия)
Все растворители безводные, за исключением тех, которые использовались для экстракции из водных растворов. Индивидуальность полученных соединений проверяли методом ТСХ на пластинах Kieselgel 60 F254 (Merck, Германия) в системах: хлороформ - метанол 9:1 (1), хлороформ -метанол 8:2 (2), хлороформ - метанол 5:3 (3), хлороформ - метанол - уксусная кислота 5:3:1 (4), метанол - уксусная кислота - вода 4:1:1 (5), бутанол - уксусная кислота - вода 4:1:1 (6). Хроматограммы проявляли хлортолидиновым реактивом, нингидрином, по свечению в УФ-свете.
Масс-спектры высокого разрешения получали на время-пролетном масс-спектрометре «Ultraflex» («Bruker», Германия) методом матриксной лазерно-десорбционной ионизации (TOF MALDI), в качестве матрицы использовалась 2,5-дигидроксибензойная кислота.
ИК-спектры регистрировали на Фурье-спектрометре: «Magna 750» («Nicolet», США).
Электронные спектры снимали на спектрофотометре «Jasco» модель UV/VS 7800 (Япония).
ОФ-ВЭЖХ проводили на приборе Gilson 305 (Gilson, Франция).
Препаративную ОФ-ВЭЖХ проводили на колонке Luna 10 µ, обращенная фаза С5, размеры 250×21.2 мм, (Phenomenex, США) в следующих условиях.
Изократическая элюция 0.1% раствором TFA в воде, скорость потока 4 мл/мин. Поглощение регистрировали при длине волны 220 нм (7).
Динамику выхода КФ из липосом контролировали на флуориметре Сагу Eclipse (Varian, США).
Липосомы получали с помощью мини-экструдера Avanti (Avanti Polar Lipids, США)
Пример 1
6,7-бис-(метиловый эфир Nα-глицил)-протогемина (IX) (XV)
К суспензии 0.030 г (0.236 ммоль) H-GlyOMe·HCl в 1.5 мл DMF прибавляли 0.033 мл (0.236 ммоль) Et3N и перемешивали при комнатной температуре 3 мин. К полученному раствору прибавляли раствор 0.100 г (0.118 ммоль) 6,7-бис-N-оксисукцинимидного эфира протогемина IX (I) в 5 мл DMF и перемешивали 2 часа при комнатной температуре. Контроль за протеканием реакции осуществляли методом ТСХ в условиях (1) и (2). Раствор концентрировали в вакууме до 0.5 мл и прибавляли 6 мл диэтилового эфира. Полученный остаток растворяли в ХЛФ, при этом наблюдалось выпадение белых кристаллов Et3N·HCl, от которых избавлялись декантацией раствора. Растворитель удаляли в вакууме. Для введения противоиона Cl- остаток растворяли в 15 мл смеси ХЛФ-МеОН (8:2), два раза встряхивали с 7.5 мл 0.5 N раствора соляной кислоты и промывали водой до нейтральной реакции. Растворитель удаляли в вакууме. Вещество очищали на колонке (20×2 см) с силикагелем Kieselgel 60 F254 (Merck, Германия), элюировали смесью ХЛФ-МеОН (8:2). Фракцию, содержащую вещество с Rf 0.71 (2), собирали. Растворитель удаляли в вакууме. Выход 0.063 г (67%), Rf 0.26 (1), 0.71 (2). Электронный спектр, λmax, нм, ХЛФ:МеОН (8:2), (ε·10-3): 400 (115.9), 508 (9.38), 640 (3.81). ИК-Фурье спектр, ν, см-1, таблетка KBr: 1737 (СО сл.эф.), 1648 (амид I), 1539 (амид II). Масс-спектр, m/z: [M]+ 758.5.
Пример 2
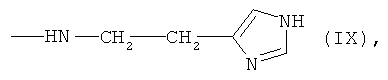
6,7-бис-гистаминил протогемин (IX) (XIX)
К раствору 0.026 г (0.236 ммоль) гистамина в 1.5 мл DMF прибавляли раствор 0.100 г (0.118 ммоль) 6,7-бис-N-оксисукцинимидного эфира протогемина IX (I) в 4 мл DMF и перемешивали 20 мин при комнатной температуре. Контроль за протеканием реакции осуществляли методом ТСХ в условиях (2) и (3). Раствор концентрировали в вакууме до 1 мл и прибавляли 10 мл диэтилового эфира. Для введения противоиона Cl- остаток растворяли в 4 мл МеОН и прибавляли 0.093 мл (0.354 ммоль) 3.8N HCl/МеОН до достижения рН 4. Растворитель удаляли в вакууме при температуре 30°С. Вещество очищали на колонке (31×2 см) с силикагелем Kieselgel 60 F254 (Merck, Германия), элюировали смесью ХЛФ-МеОН (5:3). Фракцию, содержащую вещество с Rf 0.64 (3), собирали. Растворитель удаляли в вакууме. Выход 0.049 г (51%), Rf 0.25 (2), 0.64 (3). Электронный спектр, λmax, нм, ХЛФ:МеОН (8:2), (ε·10-3): 388 (49.1), 508 (4.18), 640 (1.76). ИК-Фурье спектр, ν, см-1, таблетка KBr: 1644 (амид I), 1543 (амид II). Масс-спектр, m/z: [М]+ 802.3.
Пример 3
6,7-бис-[(бис-метиловый эфир Nα-L-аргинил)-L-глутамил]-протогемина IX (XVIII)
1. Бис-(метиловый эфир Nα-аргинил)-L-глутаминовой кислоты
К суспензии 0.069 г (0.266 ммоль) H-ArgOMe·2 HCl в 2 мл DMF прибавляли 0.074 мл (0.531 ммоль) Et3N и перемешивали при комнатной температуре 5 мин. К полученному раствору прибавляли 0.077 г (0.133 ммоль) бис-пентафторфенилового эфира Вос-глутаминовой кислоты, полученного ранее, и перемешивали 4 часа при комнатной температуре. Контроль за протеканием реакции осуществляли методом ТСХ в условиях (5). Раствор концентрировали в вакууме до 1 мл и прибавляли 10 мл диэтилового эфира. Маслообразный осадок отделяли от растворителя декантированием, остатки растворителя удаляли в вакууме. Вещество очищали на колонке (23×1 см) с силикагелем Kieselgel 60 F254 (Merck, Германия), элюировали смесью метанол - уксусная кислота - вода 4:0.5:0.5. Фракцию, содержащую вещество с Rf 0.67 (5), собирали. Растворитель удаляли в вакууме. Выход 0.073 г (77%), Rf 0.67 (5). [α]D 25 - 8.53° (С 0.38; МеОН). ИК-Фурье спектр, ν, см-1, таблетка KBr: 1735 (СО сл.эф.), 1653 (амид I), 1542, 1558 (амид II). Масс-спектр, m/z: [M]+ 588.31.
К 0.073 г (0.0946 ммоль) бис-(метилового эфира Nα-аргинил)-Boc-L-глутаминовой кислоты (V) приливали 6 мл ~ 3 н. HCl/MeOH. Полученную суспензию перемешивали 2 часа при комнатной температуре. Контроль процесса осуществляли ТСХ в условиях (6). После достижения полной конверсии в аминосвободный дипептид растворитель удаляли в вакууме при 30°С. Маслообразный остаток кристаллизовали под безводным диэтиловым эфиром, растворитель отделяли декантацией.
2. 6,7-бис-[(бис-метиловый эфир Nα-L-аргинил)-L-глутамил]-протогемина IX (XVIII)
К суспензии полученного H-Glu(ArgOMe)2-3 HCl в 1.5 мл DMF прибавляли 0.047 мл (0.335 ммоль) Et3N и перемешивали при комнатной температуре 2 мин. Наблюдали выпадение осадка Et3N·HCl. К полученной суспензии прибавляли раствор 0.047 г (0.0558 ммоль) 6,7-бис-N-оксисукцинимидного эфира протогемина IX (I) в 2 мл DMF и перемешивали 22 часа при комнатной температуре. Контроль за протеканием реакции осуществляли ТСХ в условиях (6). Раствор концентрировали в вакууме до 1 мл и прибавляли 10 мл диэтилового эфира. Для введения противоиона Cl- остаток растворяли в 4 мл МеОН и прибавляли 3.8 N HCl/МеОН до достижения рН 4. Растворитель удаляли в вакууме при температуре 30°С. Вещество трижды очищали на колонке (13×1 см) с сефадексом LH-20, элюировали МеОН. Фракцию, содержащую вещество с Rf 0.07 (6), собирали. Растворитель удаляли в вакууме. Выход 0.030 г (34%). Rf 0.07 (6), Rf(на окиси алюминия) 0.70 (6). Электронный спектр, λmax, нм, ХЛФ:МеОН (8:2), (ε·10-3): 400 (92.2), 508 (6.31), 636 (2.39). ИК-Фурье спектр, ν, см-1, таблетка KBr: 1737 (СО сл.эф.), 1649 (амид I), 1528 (амид II). Масс-спектр, m/z: [M]+ 1554.97.
Пример 4
6,7-бис-N-(2-гидроксиэтил)амид-протогемина (IX) (XIV)
К раствору 0.050 г (0.0591 ммоль) 6,7-бис-N-оксисукцинимидного эфира протогемина IX (I) в 3 мл DMF прибавляли 0.010 мл (0.118 ммоль) аминоэтанола и перемешивали 20 мин при комнатной температуре. Контроль за протеканием реакции осуществляли методом ТСХ в условиях (2) и (3). Раствор концентрировали в вакууме до 1 мл и прибавляли 10 мл диэтилового эфира. Для введения противоиона Cl- остаток растворяли в 3 мл МеОН и прибавляли 3.8 N HCl/МеОН до достижения рН 4. Растворитель удаляли в вакууме при температуре 30°С. Вещество очищали на колонке (24×1 см) с силикагелем Kieselgel 60 F254 (Merck, Германия), элюировали смесью ХЛФ-МеОН (5:3). Фракцию, содержащую вещество с Rf 0.52 (3), собирали. Растворитель удаляли в вакууме. Выход 0.036 г (82.5%), Rf 0.35 (2), 0.52 (3). Электронный спектр, λmax, нм, ХЛФ:МеОН (8:2), (ε·10-3): 400 (183.0), 508 (5.23), 640 (2.08). ИК-Фурье спектр, ν, см-1, таблетка KBr: 1632 (амид I), 1549 (амид II). Масс-спектр, m/z: [M]+ 702.5.
Пример 5
6,7-бис-N-(1,3-дигидроксипропан-2-ил)амид протогемина (IX) (XVI)
К раствору 0.050 г (0.0591 ммоль) 6,7-бис-N-оксисукцинимидного эфира протогемина IX (I) в 3 мл DMF прибавляли раствор 0.012 г (0.118 ммоль) 2-амино-1,3-пропандиола в 0.5 мл DMF и перемешивали 20 мин при комнатной температуре. Контроль за протеканием реакции осуществляли методом ТСХ в условиях (4). Раствор концентрировали в вакууме до 0.5 мл и прибавляли 6 мл диэтилового эфира. Для введения противоиона Cl- остаток растворяли в 9 мл смеси ХЛФ-МеОН (8:2), один раз встряхивали с 4 мл 0.06 N раствора соляной кислоты, насыщенного NaCl, и промывали водой до нейтральной реакции. Растворитель удаляли в вакууме. Вещество очищали на колонке (21×2 см) с сефадексом LH-20, элюировали МеОН. Фракцию, содержащую вещество с Rf 0.56 (4), собирали. Растворитель удаляли в вакууме. Выход 0.018 г (36%), Rf 0.56 (4). Электронный спектр, λmax, нм, ХЛФ:МеОН (8:2), (ε·10-3): 400 (40.5), 508 (2.68), 640 (1.02). ИК-Фурье спектр, ν, см-1, таблетка KBr: 1629 (амид I), 1550 (амид II). Масс-спектр, m/z: [M]+ 762.0.
Пример 6
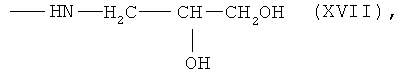
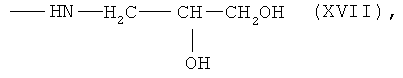
6,7-бис-N-(2,3-дигидроксипропил)амид протогемин (IX) (XVII)
К раствору 0.050 г (0.0591 ммоль) 6,7-бис-N-оксисукцинимидного эфира протогемина IX (I) в 3 мл DMF прибавляли 0.009 мл (0.118 ммоль) 3-амино-1,2-пропандиола и перемешивали 20 мин при комнатной температуре. Контроль за протеканием реакции осуществляли методом ТСХ в условиях (3) и (4). Раствор концентрировали в вакууме до 0.5 мл и прибавляли 6 мл диэтилового эфира. Для введения противоиона Cl- остаток растворяли в 3 мл МеОН и прибавляли 3.8 N HCl/МеОН до достижения рН 4. Растворитель удаляли в вакууме при температуре 30°С. Вещество очищали на колонке (30×2 см) с силикагелем Kieselgel 60 F254 (Merck, Германия), элюировали смесью ХЛФ-МеОН:АсОН (5:3:0.5). Фракцию, содержащую вещество с Rf 0.69 (4), собирали. Растворитель удаляли в вакууме. Выход 0.023 г (46%), Rf 0.33 (3), 0.69 (4). Электронный спектр, λmax, нм, ХЛФ:МеОН (8:2), (ε·10-3): 396-400 (24.2), 488 (2.14), 600 (1.40). ИК-Фурье спектр, ν, см-1, таблетка KBr: 1634 (амид I), 1565 (амид II). Масс-спектр, m/z: [M]+ 762.0.
Пример 7
6(7)-Монометиловый эфир протогемина (IX) (XX)
К раствору 50 мг (0.077 ммоль) гемина в 0.5 мл ДМФА прибавляли 100 мкл пиридина и 17 мг (0.077 ммоль) ди-трет-бутилпирокарбоната и перемешивали 30 мин. К полученному смешанному ангидриду добавляли 2,5 мкл (0.077 ммоль) безводного метанола и перемешивали 6 часов. Растворитель удаляли в вакууме при 30°С. Остаток растворяли в хлороформе. Вещество очищали флэш-хроматографией на колонке (10×2 см) с силикагелем Kieselgel 60 F254 (Merck, Германия), элюировали смесью ХЛФ-МеОН 9:1. Выход 36 мг (65%). Электронный спектр, λmax, нм, хлороформ, (ε·10-3): 387 (130), 511 (2.6) 540 (2.4), 640 (0.11). Масс-спектр, m/z: [M]+ 666.0.
Пример 8
6,7-Диметиловый эфир протогемина (IX) (XXI)
К раствору 50 мг (0.077 ммоль) гемина в 0.5 мл ДМФА прибавляли 100 мкл пиридина и 92 мг (0.420 ммоль) ди-трет-бутилпирокарбоната и перемешивали 30 мин. К полученному смешанному ангидриду добавляли 10 мкл (0.308 ммоль) безводного метанола и перемешивали 6 часов. Растворитель удаляли в вакууме при 30°С. Остаток растворяли в хлороформе. Вещество очищали флэш-хроматографией на колонке (10×2 см) с силикагелем Kieselgel 60 F254 (Merck, Германия), элюировали смесью ХЛФ-МеОН 9:1. Выход 31 мг (60%). Электронный спектр, λmax, нм, хлороформ, (ε·10-3): 387 (125), 538 (1.74), 640 (0.74). Масс-спектр, m/z: [M]+ 680.0.
Пример 9
6(7)-моно-(-Val-Gly-Ala-D-Leu-Ala-D-Val-Val-D-Val-Trp-D-Leu-X-D-Leu-Trp-D-Leu-Trp-NH-CH2CH2OH)амид протогемина (IX) (XII),
где Х=Trp, Tyr, Phe
К раствору 32 мг (0.017 ммоль) грамицидина D в 0.650 мл сухого ДМФА прибавляли 48 мкл 1 н. HCl в метаноле. Реакционную смесь перемешивали 72 ч в темноте при комнатной температуре. Растворитель удаляли в вакууме при 30°С. Выход 30 мг (98%). Масс-спектр, m/z: [M]+ 1854.0
К раствору 30 мг (0.016 ммоль) деформилированного грамицидина D в 0.2 мл ДМФА прибавляли 13.2 мг (0.016 ммоль) 6(7)-моно-N-окси-5-норборнен-2,3-дикарбоксиимидного эфира гемина и перемешивали 1,5 ч при комнатной температуре. Растворитель удаляли в вакууме при 30°С. Остаток растворяли в хлороформе. Остаток очищали на колонке (30×2 см) с силикагелем Kieselgel 60 F254 (Merck, Германия), элюировали смесью ХЛФ-МеОН 9:1. Выход 23.5 мг (59%). ИК-Фурье спектр, ν, см-1, таблетка KBr: 1634 (амид I), 1565 (амид II). Rf 0.65 (1)
Пример 10
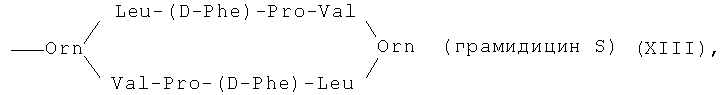
6(7)-моно-[цикло-(Orn-Leu-D-Phe-Pro-Val)2])амид протогемин (IX) (XIII)
К раствору 50 мг (0.077 ммоль) гемина в 0.5 мл сухого ДМФА прибавляли 1 мл пиридина и 25,3 мг (0.116 ммоль) ди-трет-бутилпирокарбоната и перемешивали 15 мин при комнатной температуре. Одновременно к суспензии 93.5 мг (0.077 ммоль) дигидрохлорида грамицидина S в 0.5 мл ДМФА прибавляли 22 мкл (0,154 ммоль) триэтиламина и перемешивали 2 мин.
К полученному смешанному ангидриду гемина прибавляли раствор аминосвободного грамицидина S и перемешивали 3 ч. Растворитель удаляли в вакууме при температуре 30°С. Остаток растворяли в хлороформе. Остаток очищали на колонке (30×2 см) с силикагелем Kieselgel 60 F254 (Merck, Германия), элюировали смесью ХЛФ-МеОН 12:1. Выход 20 мг (15%). Масс-спектр, m/z: [М]+ 1739.5. Электронный спектр, λmax, нм, хлороформ:метанол (4:1), (ε·10-3): 400 (105), 492 (10,6), 640 (6).
Пример 11
Arg-Gly-AspCH3COOHCF3COOH (IV)
а) синтез трикарбобензокси-аргинин-глицина
Смесь 0.043 г (0.573 ммоль) глицина, 0.4 мл (1.6 ммоль) бис-(O,N-триметилсилил)ацетамида и 1 мл сухого хлористого метилена интенсивно перемешивали в плотно закрытой колбе при 40°С до полного растворения глицина. Одновременно к раствору 300 мг (0.52 ммоль) Z-Arg(Z)2-OH и 80 мкл (0.6 ммоль) триэтиламина в 2 мл сухого хлористого метилена, охлажденному до -15°С, прибавляли 55 мкл (0.57 ммоль) этилхлорформиата и смесь перемешивали при указанной температуре 15 минут.
К полученному смешанному ангидриду прибавляли бис-(триметилсилил)глицина, и реакционную массу размешивали при -15°С 1.5 ч. Затем растворитель удаляли в вакууме, осадок растворяли в метаноле. При этом карбоксильная группа дипептида деблокируется. Органический растворитель удаляли в вакууме. Выход 224 мг (68%). Rf 0.25 (1).
б) синтез трикарбобензокси-аргинин-глицицил-ди-третбутилового-аспарагиновой кислоты
К раствору 200 мг (0.32 ммоль) карбобензокси-аргинин-глицина в 1 мл сухого хлороформа прибавляли при -10°С 45 мкл (0.32 ммоль) триэтиламина и 30 мкл (0.32 ммоль) этилхлорформиата. Через 15 мин в реакционную массу вносили 90 мг (0.32 ммоль) гидрохлорида Asp(OtBu)2 в 1 мл DMF, содержащего 45 мкл (0.32 ммоль) триэтиламина. Смесь перемешивали 2 часа. Осадок солей удаляли фильтрованием, растворитель отгоняли в вакууме. Остаток растворяли в этилацетате и промывали водой. Органическую фазу сушили сульфатом натрия, этилацетат отделяли и отгоняли досуха на роторном испарителе, продукт растирали с петролейным эфиром до образования аморфной массы. После кристаллизации из смеси этилацетат-петролейный эфир выход продукта 270 мг (98%). Rf 0.8 (5).
в) деблокирование боковых функций трипептида Arg-Gly-Asp
1) Отщепление трет-бутильных защитных групп остатка аспарагиновой кислоты
К 200 мг (0.23 ммоль) пептида приливали 2 мл смеси TFA:H2O (95:5). Раствор перемешивали 4 часа. Продукт высаживали 10-кратным избытком охлажденного диэтилового эфира, отфильтровывали и промывали дважды эфиром. Выход 169 мг (98%). Rf 0.8 (6).
2) Отщепление Z-защитных групп от остатка аргинина
К 150 мг (0.2 ммоль) N-карбобензокси-Arg-Gly-Asp приливали 0.08 мл смеси метанол-уксусная кислота-вода (6:1:1). К раствору прибавляли палладиевый катализатор 20% Pd/C и гидрировали 1 ч при комнатной температуре, контролируя ход реакции ТСХ в системе (6). После окончания реакции катализатор отфильтровывали, промывали водой, растворитель из маточников удаляли в вакууме при 40°С. Выход 60 мг (87%), Rf 0.4 (6), Масс-спектр, m/z: [M]+ 347.0. ВЭЖХ, время удерживания 1,83 мин в условиях (7).
Пример 12
6(7)-моно-(Arg-Gly-Asp)-протогемин (IX) (XI)
К суспензии 20 мг (0.038 ммоль) Arg-Gly-AspCH3COOH·CF3COOH в 0.5 мл ДМФА добавляли 5 мкл (0.038 ммоль) триэтиламина и перемешивали 2 мин. Затем добавляли 62 мкл (0,251 ммоль) бис-(O,N-триметилсилил)ацетамида и перемешивали при комнатной температуре 30 мин. В полученную суспензию вносили 32 мг (0.038 ммоль) 6,7-бис-N-оксисукцинимидного эфира протогемина IX и реакционную массу перемешивали 6 часов. Затем растворитель удаляли в вакууме, осадок растворяли в метаноле для десилилирования карбоксильных групп трипептида. Целевой продукт перекристаллизовывали из метанола. Выход 20 мг (54%). Масс-спектр, m/z: [M]+ 981,3.
Пример 13
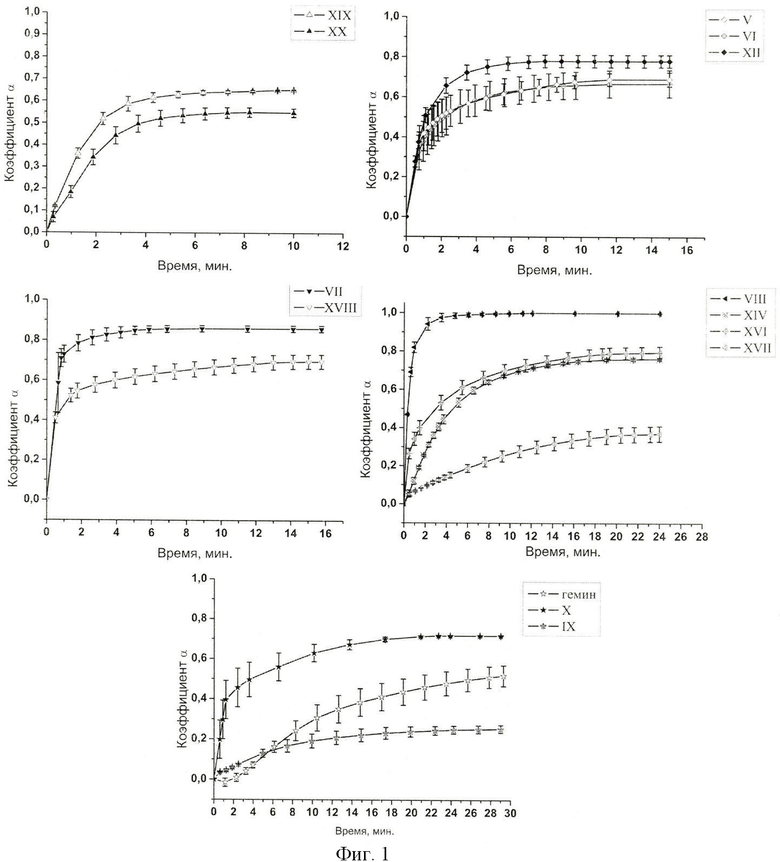
Исследование деструктурирующего действия производных гемина общей формулы I в отношении липидных мембран.
Для изучения влияния производных гемина на проницаемость липидной мембраны нами была использована методика измерения индуцированного этими веществами выхода карбоксифлуоресцеина (КФ) из нагруженных им липосом по методике, описанной в [Y.N.Antonenko, T.B.Stoilova, S.I.Kovalchuk, et al. Large unselective pore in lipid bilayer membrane formed by positively charged peptides containing a sequence of gramicidin A // FEBS Letters, 2005, V.579, p.5247-5252]. Для приготовления липосом использовали стоковый раствор DOPC в хлороформе с концентрацией 20 мг/мл. Стоковый раствор объемом 200 мкл упаривали в токе азота, добавляли 0.4 мл буферного раствора, содержащего 10 мМ Трис, 10 мМ MES, 100 мМ KCl 100 мМ КФ, встряхивали 2 мин и подвергали трем циклам замораживания-оттаивания, встряхивая смесь после каждого цикла. Полученную смесь мультиламеллярных липосом продавливали через поликарбонатный фильтр с порами диаметром 0.1 мкм, используя мини-экструдер Avanti (Avanti Polar Lipids, США). Невключившийся в липосомы КФ отделяли гель-хроматографией на сефадексе G-50. Сефадекс оставляли на ночь в воде для набухания. Колонку объемом 20 мл заполняли набухшим сефадексом, после чего уравновешивали 60 мл буфера, содержащего 10 мМ Tris, 10 мМ MES, 100 мМ KCl (буфер А). Объем суспензии липосом, содержащих КФ, составлял 500 мкл (концентрация липида 0.045 мг/мл). Суспензию помещали в кювету, объем доводили до 2 мл буфером А. К полученной суспензии липосом добавляли 10 мкл 10-4 М раствора производного гемина общей формулы I в ДМСО. Динамику выхода КФ контролировали флуориметрически. Флуоресценцию карбоксифлуоресцеина возбуждали при длине волны 490 нм, регистрировали при 520 нм (ширина обеих щелей 5 нм). Относительное количество вышедшего из липосом красителя в конкретный момент времени рассчитывали по формуле:
α=(Ff-F0)/(Fm-F0),
где F0 и Ff являются уровнем флуоресценции до и после добавления пептида соответственно, a Fm - значение флуоресценции после полного разрушения липосом детергентом Triton Х-100, добавленным до конечной концентрации 2.4% (по массе).
На фиг.1 представлена динамика выхода карбоксифлуоресцеина из липосом под действием производных гемина общей формулы I при соотношении липид/производное гемина 10:1 (моль/моль).
Пример 14
Исследование антибактериальной активности соединений общей формулы I
Для определения антибактериальной активности веществ были использованы штаммы Escherichia coli КМ МГУ С-600 (получен из коллекции кафедры микробиологии биологического факультета МГУ им.М.В.Ломоносова), Bacillus subtilis ВКМ В-501 (получен из Всероссийской коллекции микроорганизмов Института биохимии и физиологии микроорганизмов РАН). Основные параметры, которые характеризуют антибактериальную активность, - это минимальная ингибирующая концентрация (МИК) и минимальная бактерицидная концентрация (МБК). МИК- это наименьшая концентрация исследуемого соединения, полностью ингибирующая размножение бактерий в жидкой среде. МБК - это наименьшая концентрация, вызывающая гибель всех клеток.
МИК оценивали методом ингибирования роста культуры в жидкой среде с серийными разведениями веществ по модифицированной методике [Amsterdam, D. 1996. Susceptibility testing of antimicrobials in liquid media, pp.52-111. In Loman, V., ed. Antibiotics in laboratory medicine, 4 th ed. Williams and Wilkins, Baltimore].
Бактерии культивировали и тестирование проводили в жидкой среде МН (среда Mueller-Hinton: сухой экстракт говяжьего бульона 4 г/л, крахмал 1,5 г/л, гидролизат казеина 17,5 г/л; Sigma-Fluka каталожный номер 70192) при 37°С, 100% влажности и перемешивании. Для тестирования использовали культуру (с 4 по 7 пересев от разморозки) в экспоненциальной фазе роста.
Все исследуемые соединения кроме грамицидина D поглощают свет на длине волны 595 нм, используемой для оценки роста бактериальной культуры. Поэтому при оценке оптической плотности суспензии бактерий учитывали поправку на поглощение каждого соединения с учетом его концентрации в лунке. Ингибирование роста (ИР) бактерий рассчитывали в процентах через 20 часов инкубации клеток с веществами по оптической плотности (А), измеряемой в каждой лунке на длине волны 595 нм, используя формулу:
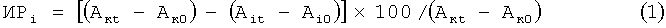
где индекс i обозначает номер лунки, к - контрольная лунка с бактериями, в которую исследуемое соединение не вносится, 0 - измерение проводится сразу же после внесения исследуемого вещества в лунку, t - измерение через 20 часов после внесения вещества.
Для определения антибактериальной активности исследуемых соединений использовали следующий экспериментальный протокол. Криопробирку с культурой тестируемого штамма (Escherichia coli КМ МГУ С600, Bacillus subtilis BKM B-501) в среде с 7% ДМСО, хранившуюся в жидком азоте, быстро размораживали, 100 мкл суспензии клеток добавляли к 1,5 мл свежей среды МН. Клетки растили в течение суток при температуре 37°С и перемешивании на орбитальном шейкере со скоростью 150 об/мин. Морфологические признаки штамма и отсутствие контаминации посторонними бактериями проверяли (а) путем посева на агаризованную (15 г/л агара) среду МН по форме и цвету образующихся колоний, (б) под микроскопом (Микмед-2, ЛОМО, Россия) с 40× объективом по характерным морфологическим признакам клеток. Далее бактерии культивировали в 1 мл жидкой среды МН при температуре 37°С и перемешивании. Клетки пересевали каждые сутки. Начиная с 3-его и заканчивая 6-м пересевом, культуру клеток использовали для постановки тестов.
Для постановки теста 5 мкл бактериальной суспензии в стационарной фазе роста переносили в 1 мл стерильной среды МН и инкубировали до достижения экспоненциальной фазы роста (3-5 ч, 37°С при перемешивании со скоростью 150 об/мин). Чтобы оценить концентрацию микроорганизмов, измеряли оптическую плотность (А) полученной бактериальной культуры на длине волны 595 нм. Считали, что А=0,2, измеренная от 200 мкл суспензии клеток в 96-луночном планшете с учетом поглощения среды, соответствует 4×108 клеток/мл для обоих используемых штаммов. С учетом измерения концентрации клеток суспензию разбавляли средой МН до 5×104-1×105 клеток/мл и переносили стерильный 96-луночный планшет по 100 мкл на лунку. Затем к клеткам вносили исследуемые соединения и делали серийные двукратные разведения этих соединений в лунках планшета. Максимальная концентрация веществ в серии составляла 10-4М, минимальная - 1,6×10-6 М. Исследование антибактериальной активности выполняли в 2 повторах для каждого соединения, а результат усредняли.
В качестве контролей использовали 100 мкл бактериальной культуры без добавления каких-либо веществ (4 лунки); бактериальную культуру, в которую добавлен 1% ДМСО или вода в объеме, как в лунках с максимальной концентрацией тестируемых соединений (4 лунки); 100 мкл стерильной среды МН без бактерий и без исследуемых веществ для контроля случайной контаминации в планшете (4 лунки).
Сразу после внесения соединений с помощью планшетного фотометра "Униплан" (Пикон, Россия) в каждой лунке измеряли Ai0, а в контрольных лунках - Ак0 необходимые для расчета по формуле (1). Планшет инкубировали в течение 20 ч при температуре 37°С и перемешивании со скоростью 150 об/мин. Затем в каждой лунке измеряли Ait, а в контрольных лунках - Aкt и рассчитывали ингибирование роста бактерий по формуле (1). МИК определяли как минимальную концентрацию исследуемого соединения, при которой ингибирование роста составляет 100%.
Для определения МБК среду из лунок, где концентрация исследуемого соединения равнялась МИК, МИКХ2 и МИКХ4, переносили на чашки Петри с агаризованной средой МН (15 г/л агара) и равномерно растирали по площади чашек стерильными шпателями. Чашки инкубировали 2 суток. МБК определяли как наименьшую концентрацию исследуемого соединения, при которой колонии на чашке Петри не вырастают.
Все тесты на антибактериальную активность повторили с перерывом в 2 дня, чтобы оценить изменчивость устойчивости бактериальных клеток к действию исследуемых соединений.



Таким образом, соединения VII, XVIII и VIII подавляют рост грамположительных бактерий S.aureus в концентрациях до 50 мкМ (табл.2).
Бактерии М.luteus высокочувствительны к действию данных соединений. Соединения VII, VIII, гемин, GrD, VI, V, IX и грамицидин S ингибируют рост М.luteus в концентрациях до 30 мкМ. При этом для соединений VII, VIII, GrD, V, IX и грамицидин S (табл.4) МИК достаточно низка - меньше 10 мкМ. Соединения Н и VI несколько менее активны (МИК=12,5 и 25 мкМ соответственно). Все исследованные соединения проявили бактерицидную активность в отношении М. luteus, причем МБК не превышает 30 мкМ.
Энтерококки Е.faecalis в среднем более устойчивы к действию данных соединений, чем микрококки М.luteus и стафилококки S.fureus. Наиболее эффективным соединением в отношении Е.faecalis оказалось соединение XVIII: МИК=12,5 мкМ.
Все исследованные соединения проявляют активность в отношении грамположительных бактерий В. subtilis. Наиболее активными оказались вещества VII, и XVIII (МИК меньше 10 мкМ).
Пример 15.
Определение специфической активности производных гемина в отношении штаммов резистентных бактерий.
Материалы и методы
Испытуемые препараты производные гемина, растворимые в воде (VII, XVIII) и растворимые в ДМСО (VIII, XV, XIV). В качестве препарата сравнения был взят ванкомицин. Каждое вещество испытывалось в трех повторах.
В работе использовались одноразовые стерильные 96-луночные плоскодонные планшеты, чашки Петри, пипетки, наконечники и пробирки (Пан-Эко, Москва).
Питательные среды: бульон Мюллера-Хинтон для работы готовили из сухих сред (Mueller Hinton broth, Acumedia, Baltimore) и стеризовали автоклавированием при 121°С в течение 15 минут.
Для культивирования Staphylococcus aureus использовали готовую сухую среду - Триптиказо-соевый агар (Tripticase Soy Agar, BBL). Для культивирования Enterococcus faecalis использовали готовую сухую среду - Колумбийский агар (Columbia Agar Base, BBL). Среды стерилизовали автоклавированием при 121°С в течение 15 минут.
Исследуемые резистентные бактериальные штаммы
Грамположительные:
Staphylococcus aureus № 25923 АТСС (American Type Culture Collection);
Staphylococcus aureus № 100 КС;
Staphylococcus epidermidis № 533;
Enterococcus faecalis № 559;
Enterococcus faecium № 569.
Бактериальный инокулюм был постоянный и составлял 5×105 КОЕ/мл.
Полученные результаты представлены в таблицах (Мср).
Постановка эксперимента.
Для водорастворимых соединений с 2 по 8 лунки вносили растворитель (вода) по 15 мкл, затем в 1 лунку вносили 30 мкл стокового раствора исследуемого соединения в воде с концентрацией 1×103 М и последовательным двукратным разведением доводили его концентрацию до 0,007×103 М. Из каждой лунки отбирали по 10 мкл и добавляли по 190 мкл бактериальной культуры (105 КОЕ).
Для растворимых в ДМСО соединений с 2 по 8 лунки вносили растворитель (ДМСО) по 10 мкл, затем в 1 лунку вносили 20 мкл стокового раствора исследуемого соединения в воде с концентрацией 5×103 М и послдовательным двукратным разведением доводили его концентрацию до 0,039×103 М. Из каждой лунки отбирали по 2 мкл и добавляли по 198 мкл бактериальной культуры (105 КОЕ).
В качестве контроля включали лунки, не содержащие тестируемых препаратов (контроль роста культуры). Кроме того, ставился контроль чистоты питательных сред и растворителей. Планшеты инкубировали в термостате при 36°С в течение 24 часов.
Оценку роста культур проводили визуально, сравнивая рост микроорганизмов в присутствии изучаемых тест-соединений с ростом культуры без них. За МПК - минимальную подавляющую концентрацию принимали последнее разведение испытуемых препаратов с подавлением роста бактериальной культуры.
Получнные результаты представлены в таблицах.
25923
100 КС
533
559
569
Таким образом, производные гемина общей формулы I активны (в разной степени в отношении Гр+штаммов резистентных бактерий.
Пример 16
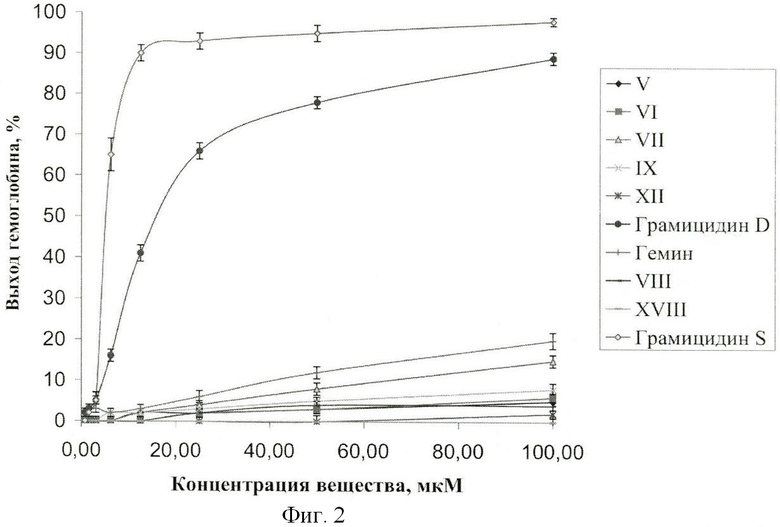
Методика определения гемолитической активности соединений общей формулы I
Для исследования гемолитической активности использовали свежую капиллярную кровь человека. Эффективность гемолиза определяли по выходу гемоглобина из эритроцитов в среде RPMI-1640 (без фенолового красного, с добавлением 10% фетальной телячьей сывотки и 20 мМ L-глутамина) при начальной плотности эритроцитов (1,0±0,1)×107 клеток/мл через 3 часа инкубации с агентом (37°С, 5% СО2, 100% влажность, перемешивание на орбитальном шейкере).
В качестве характеристики гемолитической активности использовали долю гемоглобина, вышедшего из эритроцитов во внешнюю среду. Эритроциты, тени и промежуточные формы, содержащие не вышедший гемоглобин, отделяли центрифугированием. Гемоглобин определяли по поглощению супернатанта на длине волны 414 нм (вблизи максимума полосы Соре гема).
Поскольку все исследуемые соединения кроме GrD поглощают свет на данной длине волны, при расчете доли вышедшего гемоглобина учитывали поправку на поглощение каждого соединения с учетом его концентрации. Выход гемоглобина (ВГ) рассчитывали в процентах по оптической плотности (А) на длине волны 414 нм, используя формулу:
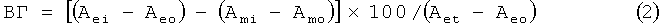
где индекс е обозначает супернатант образца с эритроцитами, i - добавленную концентрацию соединения, о - супернатант образца без добавления исследуемых соединений, m - раствор без эритроцитов, t - супернатант образца со 100% лизированными эритроцитами. Доля вышедшего гемоглобина равна доле лизированных эритроцитов при условии, что лизис происходит полностью, то есть при лизисе из эритроцита выходит весь содержащийся в нем гемоглобин.
Исследование гемолитической активности выполняли в 2 гюзторах для каждого соединения, а результат усредняли.
Для определения гемолитической активности соединений использовали следующий экспериментальный протокол. Кровь (100 мкл) отбирали из пальца здорового донора в пробирку, содержащую 0,9 мл среды RPMI-1640 (без фенолового красного) и гепарин (10 ед/мл). Клетки осаждали центрифугированием в течение 5 мин при 200×g и переносили в 10 мл среды RPMI-1640 (без фенолового красного, с добавлением 10% фетальной телячьей сывотки и 20 мМ L-глутамина, далее - полная среда). Плотность эритроцитов в суспензии определяли подсчетом в камере Горяева с помощью микроскопа "Микмед-2" (ЛОМО, Россия) и разбавляли полной средой до (2±0,2)×107 клеток/мл.
Методом последовательных разведений с шагом в 2 раза готовили серии растворов соединений в полной среде (максимальная концентрация 200 мкМ, минимальная - 1,6 мкМ, объем 75 мкл). К приготовленным растворам быстро добавляли 75 мкл суспензии эритроцитов с плотностью (2±0,2)×107 клеток/мл.
Для отрицательных контролей к 75 мкл суспензии эритроцитов с плотностью (2±0,2)×107 клеток/мл добавляли: а) 75 мкл полной среды (4 образца); б) 60 мкл полной среды и 15 мкл воды (4 образца); или в) 73,5 мкл полной среды и 1,5 мкл ДМСО (4 образца). Для определения 100%-го выхода гемоглобина (положительный контроль, 4 образца) эритроциты из 75 мкл суспензии осадждали центрифугированием (5 мин при 200×g), осадок ресуспендировли в деионизированной воде (50 мкл), после полного лизиса объем пробы доводили полной средой до 150 мкл.
Все образцы (кроме положительного контроля) инкубировали в течение 3 ч при температуре 37°С, 5% СО2, 100% влажности и перемешивании со скоростью 150 об/мин. Затем все образцы центрифугировали в течение 5 мин при 2700×g. В лунки 96-луночного планшета переносили по 130 мкл супернатанта из каждого образца. После чего с помощью фотометрического анализатора "Униплан" (Пикон, Россия) измеряли оптическую плотность в лунках планшета на длине волны 414 нм. Выход гемоглобина рассчитывали по формуле (2). На фиг.2 представлена гемолитическая активность производных гемина и соединений сравнения - грамицидина S, грамицидина D и гемина. Данные представлены в виде среднего по 2-м повторам ± стандартное отклонение.
Соединение XII не проявляет гемолитическую активность вплоть до 100 мкМ. Вещества V, VI, VIII, IX, XVIII имеют низкую гемолитическую активность (Рис.2), причем эффективность гемолиза всех синтезированных соединений значительно ниже, чем у гемина, и весьма низка даже при максимальной концентрации соединений (100 мкМ).
Пример 17
Исследование цитотоксической активности соединений общей формулы I в отношении лейкоцитов человека
Суммарную фракцию лейкоцитов выделяли из крови человека методом свободной седиментации, для чего использовали свежеотобранную венозную кровь здорового донора.
Кровь (10 мл) с добавкой 10 ед./мл гепарина выдерживали в течение 2,5 ч при температуре 15-18°С в темноте. Отбирали светлый верхний слой жидкости (4 мл) над столбом оседающих эритроцитов, трижды отмывали сбалансированным солевым раствором Хенкса и ресуспендировали в среде RPMI-1640 (с добавлением 8% эмбриональной бычьей сыворотки, 2 мМ L-глутамина и 10 мМ HEPES, далее - полная среда).
Концентрацию лейкоцитов и эритроцитов в суспензии определяли подсчетом в камере Горяева, после чего суспензию доводили полной средой до концентрации лейкоцитов (1,0±0,1)×106 клеток/мл. При этом концентрация эритроцитов в используемой для измерений суспензии клеток крови не превышала 30% от концентрации лейкоцитов.
Для определения цитотоксичности исследуемых соединений для лейкоцитов человека суспензию клеток переносили в лунки стерильных 96-луночных планшетов, методом последовательных разведений с шагом в 2 раза вносили исследуемые вещества (диапазон тестируемых концентраций: 3,2-100 мкМ для соединений GrD и грамицидина S 8-500 мкМ для соединения XIII; 8-1000 мкМ для соединений VII, V, VI, VIII, IX, Н, X, XIX, XV, XI, XIV, XX и XXI) и аккуратно перемешивали. Эксперимент выполняли в двух параллельных повторах.
В качестве контролей использовали клетки, к которым добавляли эквивалентное количество растворителя, в котором были приготовлены исследуемые вещества: воду для соединений VII, и X; водный раствор ДМСО для соединения VIII, V, VI, IX; ДМСО для GrD, грамицидин S, XIII, Н, XIX, XV, XI, XIV, XX и XXI. Дополнительный контроль - суспензия лейкоцитов без всяких добавок.
Клетки инкубировали с исследуемыми соединениями 3 ч (37°С, 5% СО2, 100% влажность). Гибель лейкоцитов оценивали при помощи флуоресцентной микроскопии. Для выявления мертвых и живых клеток использовали окрашивание йодистым пропидием (проникает только в ядра мертвых клеток) и Hoechst 33342 (окрашивает все ядра). Клетки инкубировали в планшетах 15 мин одновременно с йодистым пропидием и Hoechst 33342 (37°С, 5% СО2), а затем планшеты помещали под микроскоп для анализа.
Долю погибших клеток определяли по флуоресцентным изображениям клеток в синей (Hoechst 33342) и красной (йодистый пропидий) областях спектра, полученным при помощи флуоресцентного микроскопа Axio Observer (Zeiss, Германия) с 10× объективом ("Plan-Neofluar" 10×/0.3). Использовали следующие наборы флуоресцентных фильтров. Для йодистого пропидия: полосовой фильтр на возбуждение 530-585 нм; барьерное дихроичное зеркало 600 нм, длинноволновый барьерный анализирующий фильтр 615 нм. Для Hoechst 33342: полосовой фильтр на возбуждение 359-372 нм; барьерное дихроичное зеркало 395 нм, длинноволновый барьерный анализирующий фильтр 397 нм.
С помощью цифровой фотокамеры регистрировали три типа изображений клеток: а) в проходящем белом свете; б) флуоресцентное изображение в синей области спектра с ультрафиолетовым возбуждением; в) флуоресцентное изображение в красной области спектра с возбуждением зеленым светом. Фотографировали не менее 4 полей зрения на лунку в разных ее областях. Результат анализа усредняли по 1000-1500 клеток для каждой концентрации исследуемых соединений.
Соединения 20b(VII) X, VIII, IX и V в концентрации до 1 мМ не вызывают гибель лейкоцитов из крови человека. При этом небольшой токсический эффект растворов грамицидина S, XI, XX и XXI в ДМСО полностью обусловлен токсическим действием растворителя (ДМСО). Для соединений VI, XIII, XIX, XV и XIV обнаружен слабый токсический эффект, который проявляется при концентрациях, близких к 1 мМ, для соединений XIX, XV и XIV, при концентрациях больше 125 мкМ для соединений XIII и VI. Соединение сравнения - гемин обладает существенно более высокой токсичностью в отношении лейкоцитов по сравнению с заявляемыми соединениями (40% против 17÷3%, табл.6).
Полученные результаты свидетельствуют о перспективности заявляемых веществ для создания на их основе нетоксичных, биосовместимых антимикробных агентов, в том числе для профилактики и лечения заболеваний, вызванных различными микроорганизмами.
Пример 1
Методика изучения вирулицидного действия препаратов в культуре клеток на интактные вирусные частицы.
Вируссодержащую жидкость с титром 4,0 lg для вируса гриппа и 5,0 lg для вируса герпеса инкубировали при комнатной температуре в присутствии и в отсутствии препаратов. Экспозицию с препаратами вируса гриппа проводили в течение часа, вируса герпеса 30 и 60 минут.
Вирусодержащий материал инкубировали в присутствии препарата в исследуемой концентрации и определяли величину снижения инфекционного титра вируса по сравнению с контролем - вирусом, инкубируемым в тех же условиях, но без препарата.
Инфекционный титр вирусов определяли путем заражения культур клеток.
Перед инфицированием клетки дважды промывали средой без сыворотки для снижения неспецифической реакции. Инфицирование проводили 10-кратными разведениями проб вирусов с препаратами и без них на соотвествующей среде с добавлением трипсина (ТРСК treated,Sigma) в случае исследования вируса гриппа. Адсорбцию вирусов проводили в течение 40-60 минут при 37°С. Несорбировавшийся вирус удаляли 3-кратной промывкой средой без сыворотки. Контроли вирусов и клеток культивировали в соотвествующей клеткам среде. Далее планшеты инкубировали в термостате в течение 48-72 часов при 37°С.
Учет результатов проводили по определению гемагглютинирующей активности вируса гриппа в надосадочной жидкости в реакции гемагглютинации с человеческими эритроцитами 0 (1) группы.
Инфекционный титр вируса герпеса ВПГ-1 определяли в реакции иммуноферментного анализа, используя коммерческие тест-системы и ПНР фирмы «Амплисенс».

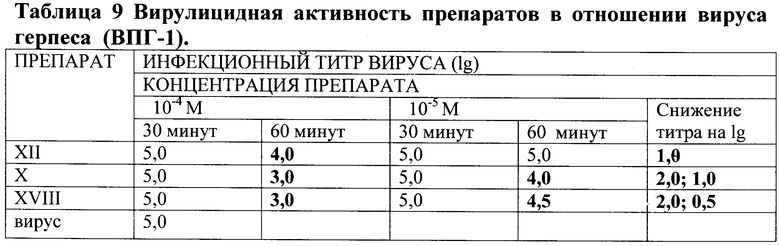
Таким образом, заявляемые соединения проявляют не только антибактериальную, но и заметную вирулицидную активность в отношении вирусов гриппа и герпеса, что повышает их потенциальную практическую ценность как противоинфекционных агентов.
Пример 19
Исследование противогрибковой активности соединений общей формулы I.
Объектом воздействия являлись клетки музейного штамма Cryptococcus neoformans № 3465, а также клетки музейного штамма Candida albicans №927, выращенных на глюкозо-пептон-дрожжевой среде при 32°С в течение 1 суток. В качестве соединения-сравнения использовались антимикотики пимафуцин, амфотерицин, флуконазол.
Эксперимент проводили в круглодонных 96-луночных планшетах фирмы "Ленмедполимер» в двух повторностях. В первый столбец (0) вносили 10 мкл стокового раствора водорастворимых препаратов с концентрацией 10-2÷10-3 моль/л, во второй также 10 мкл плюс 10 мкл растворителя, соответствующего данному соединению, суспендировали и переносили 10 мкл в следующую ячейку и т.д. до 8 ячейки ряда. Стоковые растворы водонерастворимых соединений общей формулы 1 (V, VIII, XII, XIII, XIV, гемин) готовили в ДМСО, разбавляли ДМСО и средой. Концентрация ДМСО в реакционной ячейке составляла ≤ 5%.
Затем в каждую ячейку вносили 190 мкл суспензии клеток культуры Cr. neoformans в синтетической питательной среде (конечная концентрация клеток примерно 103 КОЕ/мл), содержащей рН-индикатор бромкрезоловый синий (рН среды 5,5, цвет индикатора - голубой). При понижении рН до 4.5 цвет индикатора в среде изменяется на желтый, что является признаком роста клеток гриба. Синтетическая среда состояла из следующих компонентов: соли, аминокислоты, микроэлементы, витамины, антибиотик и глюкоза (всего 30 компонентов) [Yarrow D. Methods for the isolation, maintenance and identification of yeasts // In: The Yeasts, A Taxonomic Study. - 1998. - Elsevier. - Ed. by Kurtsman C.P, Fell J.W. - P.77-100]. После внесения суспензии клеток Cr. neoformans планшет в течение 1 часа инкубировали при 32°С с перемешиванием, затем переносили в стационарный термостат и инкубировали 2 и 5 суток при 27°С. После внесения суспензии клеток С. albicans планшеты инкубировали в течение 1 часа при 32°С с перемешиванием, затем переносили в стационарный термостат и инкубировали 1 и 4 суток при 27°С. Результаты приведены в таблицах 10 и 11.
Контрольные посевы из ячеек, проведенные через 1-4 сутки воздействия соединениями общей формулы I, показали практически фунгицидный эффект соединения XVIII в концентрации около 5·10-5 М.
Таким образом, соединения общей формулы 1 проявляют фунгистатический и/или фунгицидный эффекты, величина которых зависит от строения и проявляется в интервале концентраций от 10-4 до 10-6 М.
Пример 20
Композиции настоящего изобретения могут быть использованы в виде дезинфицирующих, антисептических и фармацевтических препаратов (например, в твердой, полутвердой или жидкой формах), содержащих предлагаемые в настоящем изобретении соединения в качестве активных ингредиентов в смеси с органическим или неорганическим носителем или наполнителем, приемлемым для внутримышечного, внутривенного, интраназального, перорального, сублингвального, ингаляционного и интраректального введения. Активный ингредиент может быть включен в композицию вместе с обычно используемыми нетоксичными, фармацевтически приемлемыми носителями, пригодными для изготовления растворов, таблеток, пилюль, капсул, суппозиториев, эмульсий, суспензий, спреев, ингаляторов, капель, мазей и любых других лекарственных форм. В качестве носителей могут быть использованы вода, глюкоза, лактоза, аравийская камедь, желатин, крахмал, триксилит магния, тальк, кукурузный крахмал, мочевина, полиэтиленгликоль и другие носители, пригодные для изготовления твердых, мягких или жидких препаратов. При этом в качестве добавок могут быть использованы стабилизаторы, загустители, красители и отдушки.
Активное действующее соединение общей формулы I вводят в композицию в количестве, достаточном для получения нужного вирулицидного эффекта.
При изготовлении разовой лекарственной формы количество активного ингредиента, используемого в комбинации с носителем, может варьировать в зависимости от реципиента, подвергающегося лечению, от конкретного способа введения лекарственного средства.
Так, например, при использовании соединений настоящего изобретения в виде растворов для инъекций содержание действующего начала в них составляет 0.001-1%. В качестве разбавителя вещества могут быть использованы 0,9% раствор натрия хлорида, дистиллированная вода, раствор новокаина для инъекций, раствор Рингера, раствор глюкозы. При использовании соединения общей формулы (I) в виде таблеток и суппозиториев количество вещества составляет 1,0-100,0 мг на единичную дозированную форму. Для таблеток и суппозиториев в качестве фармацевтического наполнителя используют любую фармацевтически пригодную основу.
Примеры лекарственных форм
A. Желатиновые капсулы
Состав вводимого в капсулу порошка:
соединение, соответствующее общей формуле (I) - 1-50 мг,
оксид магния - 50 мг,
крахмал - 100-200 мг.
Указанные выше ингредиенты смешивают и смесь вводят в твердые желатиновые капсулы в количестве 151-285 мг.
Б. Таблетированная форма
Таблетированную форму получают, используя приведенные ниже ингредиенты, мг:
Компоненты смешивают и прессуют для образования таблеток весом 200 мг каждая.
B. Аэрозольная форма
Состав аэрозольной смеси, рассчитанной на 10 приемов, мг:
Соединение смешивают с наполнителями и помещают в специальное устройство для распыления.
Г. Суппозитории
В качестве суппозиторной основы могут быть использованы:
основы, нерастворимые в воде - масло какао;
основы, растворимые в воде или смешиваемые с водой - желатино-глицериновые или полиэтиленоксидные;
комбинированные основы - мыльно-глицериновые.
Пример состава суппозитория:
Соединение, соответствующее общей формуле (I), - 1-50 мг,
Масло какао - количество, необходимое для получения суппозитория.
При необходимости возможно изготовление ректальных, вагинальных и уретральных суппозиториев с соответствующими наполнителями.
Д. Мази
В качестве мазевой основы могут быть использованы:
углеводородные мазевые основы - вазелин белый и желтый (Vaselinum album, Vaselinum flavum), вазелиновое масло (Oleum Vaselini), мазь белая и жидкая (Unguentum album, Unguentum flavum), а в качестве добавок для придания более плотной консистенции твердый парафин и воск;
абсорбтивные мазевые основы - гидрофильный вазелин (Vaselinum hydrophylicum), ланолин (Lanolinum), кольдкрем (Unguentum leniens);
мазевые основы, смываемые водой - гидрофильная мазь (Unguentum hydrophylum); водорастворимые мазевые основы - полиэтиленгликолевая мазь (Unguentum Glycolis Polyaethyleni), бентонитовые основы и другие.
Пример состава мази, г:
Мази изготавливают по соответствующей технологии.
Е. Раствор для инъекций
В качестве растворителя при приготовлении раствора для инъекций могут быть использованы 0,9% раствор натрия хлорида, дистиллированная вода, раствор новокаина. Форма выпуска - ампулы, флаконы, шприц-тюбики.
Состав раствора для инъекций:
Возможно изготовление различных лекарственных форм для инъекций - стерильных растворов, стерильных порошков и таблеток.
Примеры композиций для дезинфицирующих и антисептических средств.
D. Поверхностно-активное соединение (ПАВ) катионного, анионного или амфотерного типа
Таким образом, заявленные производные гемина общей формулы I обладают антибактериальной и противогрибковой активностью, в том числе в отношении патогенного для человека S. aureus; соединения общей формулы I по п.1 - вирулицидной активностью в низких дозах и способны инактивировать вирусы различного строения, в том числе гриппа и герпеса. Кроме того, практически все заявленные производные гемина в различной степени обладают способностью увеличивать проницаемость модельных липидных мембран, что, как известно из литературных данных, представляет собой важную часть механизма антимикробного (антибактериального, противогрибкового, вирулицидного) действия антимикробных агентов. Эффективность отдельных представителей соединений, соответствующих общей формуле I, подтверждает их пригодность для индустриального применения в составе дезинфицирующих, антисептических и терапевтических средств с противогрибковым, антибактериальным и вирулицидным действием.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ГЕМИНА И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ, ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2002 |
|
RU2238950C2 |
| Катионный пептид, проявляющий антибактериальные свойства | 2021 |
|
RU2778856C1 |
| АНТИМИКРОБНОЕ ВЕЩЕСТВО | 2010 |
|
RU2447896C1 |
| ПРОИЗВОДНЫЕ НОНАПЕПТИДОВ ИЛИ ИХ СОЛЕЙ С ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫМИ КИСЛОТАМИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2115659C1 |
| ПЕПТИД, ОБЛАДАЮЩИЙ ЛЕЧЕБНЫМ ДЕЙСТВИЕМ ПРОТИВ БОЛЕЗНИ АЛЬЦГЕЙМЕРА | 2014 |
|
RU2558242C1 |
| ГЕМИНПЕПТИДЫ, ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ФАРМКОМПОЗИЦИЯ И ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОТИВООПУХОЛЕВЫХ АГЕНТОВ | 2004 |
|
RU2280649C1 |
| ПРОИЗВОДНОЕ АНАЛОГА GLP-1 ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ИХ ПРИМЕНЕНИЕ | 2010 |
|
RU2565536C2 |
| Пептид, обладающий лечебным действием против болезни Альцгеймера | 2016 |
|
RU2626972C2 |
| ПОЛИПЕПТИДЫ ЛИЗИНА, АКТИВНЫЕ ПРОТИВ ГРАМОТРИЦАТЕЛЬНЫХ БАКТЕРИЙ | 2016 |
|
RU2724545C2 |
| Пептид, обладающий лечебным действием против болезни Альцгеймера | 2018 |
|
RU2677296C2 |
Изобретение относится к биоорганической химии, а именно к новым производным гемина общей формулы (I), их фармацевтически приемлемым солям, способу получения, фармацевтическим и дезинфицирующим композициям. Соединения обладают деструктивным действием в отношении липидной мембраны, антимикробными свойствами, в том числе антибактериальными и противогрибковыми, а также одновременно вирулицидными и антимикробными свойствами. 8 н. и 7 з.п. ф-лы, 2 ил., 11 табл.
1. Производные гемина или их фармацевтически приемлемые соли, представляющие собой соединения общей формулы I:
 ,
,
где R1 представляет собой ОН и
R2=-Val-Gly-Ala-(D-Leu)-Ala-(D-Val)-Val-(D-Val)-Trp-(D-Leu)-X-(D-Leu)-Trp-(D-Leu)-Trp-NHCH2CH2OH, где Х=Trp, или Phe, или Tyr (грамицидин D),
 , -Arg-Gly-Asp-OH, или
, -Arg-Gly-Asp-OH, или
 ,
,  ,
,  ,
,
 ,
, 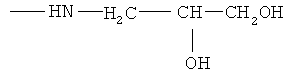 ,
,
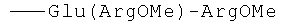
Y- представляет собой Cl-;
Men+ представляет собой Fe2+ или Fe3+.
2. Производные гемина общей формулы I по п.1, обладающие деструктирующим действием в отношении липидной мембраны.
3. Производные гемина общей формулы I по п.1, обладающие антибактериальными и противогрибковыми свойствами.
4. Производные гемина общей формулы I по п.1, обладающие одновременно вирулицидными и антимикробными свойствами.
5. Способ получения производных гемина или их фармацевтически приемлемых солей общей формулы I:
где R1 представляет собой ОН и
R2=-Val-Gly-Ala-(D-Leu)-Ala-(D-Val)-Val-(D-Val)-Trp-(D-Leu)-X-(D-Leu)-Trp-(D-Leu)-Trp-NHCH2CH2OH, где Х=Trp, или Phe, или Tyr (грамицидин D),
, -Arg-Gly-Asp-OH,
или
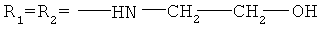 ,
, 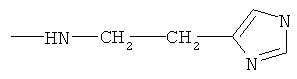 , ,
, ,
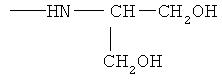 , ,
, ,
Y- представляет собой Cl-; Men+ представляет собой Fe2+ или Fe3+; заключающийся в том, что активированное по карбоксильной/ым группе/ам производное гемина вводят во взаимодействие с аминокомпонентом, процесс ведут в органическом растворителе при перемешивании.
6. Способ по п.5, где в качестве активированного производного гемина использован 6,7-бис-N-оксисукцинимидный эфир гемина или 6(7)-моно-N-окси-5-норборнен-2,3-дикарбоксиимидный эфир гемина, в качестве растворителя использован N,N-диметилформамид.
7. Способ по п.5, где для активирования по одной из карбоксильных групп гемина используют ди-трет-бутилпирокарбонат, процесс ведут в N,N-диметилформамиде в присутствии пиридина.
8. Способ по п.5, где в качестве аминокомпонента используют трипептид H-Arg-Gly-AspOH в силилированной форме, в качестве активированного по карбоксильным группам гемина используют 6,7-бис-N-оксисукцинимидный эфир; полученный при этом конъюгат обрабатывают водным метанолом для снятия силильных групп.
9. Способ по п.8, где трипептид H-Arg-Gly-Asp-OH в силилированной форме получают исходя из глицина, который силилируют N,O-бис-(триметилсилил)-ацетамидом в хлористом метилене при кипячении, образующийся при этом бистриметилсилилглицин ацилируют трикарбобензоксиаргинином методом смешанных ангидридов, удаляют триметилсилильную защиту карбоксильной группы дипептида и α бутиловым эфиром аспарагиновой кислоты методом смешанных ангидридов; обработкой полученного производного трипептида 90-100% трифторуксусной кислотой отщепляют трет-бутильные группы, и затем удаляют карбобензокси-группы гидрированием полученного продукта над 10-20% палладием на активированном угле; трипептид силилируют действием N,О-бис-(триметисилил)ацетамида в хлористом метелене.
10. Фармацевтическая композиция, обладающая антимикробной активностью, содержащая эффективное количество производного гемина или его фармацевтически приемлемой соли общей формулы I:
где R1=OH, R2=-ArgArgTrpHisArgLeuLysGlu(OMe)OH,
ArgTrpHisArgLeuLysGlu(OMe)OH;
R1=ОН,
R2=-Val-Gly-Ala-(D-Leu)-Ala-(D-Val)-Val-(D-Val)-Trp-(D-Leu)-X-(D-Leu)-Trp-(D-Leu)-Trp-NHCH2CH2OH, где Х=Trp, или Phe, или Tyr (грамицидин D),
R1=OH,  , -Arg-Gly-Asp-OH,
, -Arg-Gly-Asp-OH,
или
, , ,
, ,
или R1=R2=-ArgOMe, -SerOMe, -βAlaHA,
Y- представляет собой Cl-;
Men+ представляет собой Fe2+ или Fe3+;
и фармацевтически приемлемый носитель.
11. Фармацевтическая композиция, обладающая вирулицидными, антибактериальными и противогрибковыми свойствами, содержащая эффективное количество производного гемина или его фармацевтически приемлемой соли общей формулы I:
где R1=R2=-GlyOMe, -Glu(ArgOMe)ArgOMe;
R1=OH,R2=-=-Val-Gly-Ala-(D-Leu)-Ala-(D-Val)-Val-(D-Val)-Trp-(D-Leu)-X-(D-Leu)-Trp-(D-Leu)-Trp-NHCH2CH2OH, где X=Trp, Phe, Tyr (грамицидин D),
R1=OH, 
Y- представляет собой Cl-;
Men+ представляет собой Fe2+ или Fe3+;
и фармацевтически приемлемый носитель.
12. Дезинфицирующая и/или антисептическая композиция, обладающая антибактериальной и противогрибковой активностью, содержащая эффективное количество производного гемина или его фармацевтически приемлемой соли общей формулы I:
где R1=OH, R2=-ArgArgTrpHisArgLeuLysGlu(OMe)OH,
ArgTrpHisArgLeuLysGlu(OMe)OH;
R1=OH и
R2=-Val-Gly-Ala-(D-Leu)-Ala-(D-Val)-Val-(D-Val)-Trp-(D-Leu)-X-(D-Leu)-Trp-(D-Leu)-Trp-NHCH2CH2OH, где Х=Trp, или Phe, или Tyr (грамицидин D),
или
R1=OH,  , -Arg-Gly-Asp-OH,
, -Arg-Gly-Asp-OH,
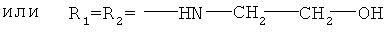 , , ,
, , ,
, ,
или R1=R2=-ArgOMe, -SerOMe, -βAlaHA,
Y- представляет собой Cl-;
Men+ представляет собой Fe2+ или Fe3+;
и приемлемый носитель.
13. Дезинфицирующая, антисептическая композиция, обладающая вирулицидным, антибактериальным и противогрибковым действием, содержащая эффективное количество производного гемина или его фармацевтически приемлемой соли общей формулы I:
где R1=R2=-OMe, -βAlaHis, -GlyOMe, -Glu(ArgOMe)ArgOMe;
R1=OH, R2=-Val-Gly-Ala-(D-Leu)-Ala-(D-Val)-Val-(D-Val)-Trp-(D-Leu)-X-(D-Leu)-Trp-(D-Leu)-TrpNHCH2CH2OH, где X=Trp, Phe, Tyr (грамицидин D),
R1=OH, 
Y- представляет собой Cl-;
Меn+ представляет собой Fe2+ или Fe3+;
и приемлемый носитель.
14. Средство, обладающее антибактериальной, вирулицидной и противогрибковой активностью, представляющее собой соединение общей формулы I:
где
R1=OH, R2=-ArgArgTrpHisArgLeuLysGlu(OMe)OH,
ArgTrpHisArgLeuLysGlu(OMe)OH;
R1=OH,
R2=-Val-Gly-Ala-(D-Leu)-Ala-(D-Val)-Val-(D-Val)-Trp-(D-Leu)-X-(D-Leu)-Trp-(D-Leu)-Trp-NHCH2CH2OH, где Х=Trp, или Phe, или Tyr (грамицидин D),
R1=OH,  , -Arg-Gly-Asp-OH,
, -Arg-Gly-Asp-OH,
или
, , ,
, ,
или R1=R2=-ArgOMe, -SerOMe, -βAlaHA,
Y- представляет собой Cl-;
Меn+ представляет собой Fe2+ или Fe3+
или его фармацевтически приемлемые соли.
15. Средство, обладающее вирулицидной, противогрибковой и антибактериальной активностью, в том числе против резистентных бактерий, представляющее собой соединение общей формулы I:
где R1=R2=-OMe, -GlyOMe, -Glu(ArgOMe)ArgOMe;
R1=OH, R2=-Val-Gly-Ala-(D-Leu)-Ala-(D-Val)-Val-(D-Val)-Trp-(D-Leu)-X-(D-Leu)-Trp-(D-Leu)-TrpNHCH2CH2OH, где X=Trp, Phe, Tyr (грамицидин D),
R1=OH, 
Y- представляет собой Cl-;
Men+ представляет собой Fe2+ или Fe3+
или его фармацевтически приемлемые соли.
| ГЕМИНПЕПТИДЫ, ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ФАРМКОМПОЗИЦИЯ И ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОТИВООПУХОЛЕВЫХ АГЕНТОВ | 2004 |
|
RU2280649C1 |
| ПРОИЗВОДНЫЕ ГЕМИНА И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ, ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2002 |
|
RU2238950C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ГЕМИНПЕПТИДОВ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ НУКЛЕОЛИТИЧЕСКИХ АГЕНТОВ | 2003 |
|
RU2250906C2 |
| ГЕМИНПЕПТИД, ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОТИВОВИРУСНОГО И ВИРУЛЕЦИДНОГО АГЕНТА | 2004 |
|
RU2296131C2 |
| Способ получения несимметричных 6,7-пептидил-протогеминов 1х | 1978 |
|
SU717039A1 |
| RU 2007125604 А, 20.01.2009 | |||
| Г.А.Желтухина и др | |||
| «Синтез и структурно-функциональные исследования искусственных нуклеаз на основе конъюгатов гемина с пептидными фрагментами фактора дифференцировки клеток HLDF», Биоорганическая химия, 2006, 32(2), с.198-210. | |||

