Настоящее изобретение относится к лекарственному средству для лечения хронического ревматоидного артрита.
Хронический ревматоидный артрит является трудноизлечимым аутоиммунным заболеванием, которое ассоциируется с набуханием, воспалением, ригидностью (неэластичностью) и болью сустава, и демонстрирует клиническую картину генерализованного полиартрита. То есть, это - генерализованная болезнь, при которой живой организм распознает "свое" как "не свое (чужое)", исходя из дефекта распознавания "свое" и "не свое", и воздействует на собственную (аутологичную) ткань, вызывая аномальный иммунный ответ, что приводит к воспалению соединительной ткани.
Основным белком, составляющим соединительную ткань, является коллаген, и, по крайней мере, пять разновидностей (типов I, II, III, IV и V) коллагенов обнаружено у теплокровных (с постоянной температурой тела). Сустав содержит суставной хрящ, который содержит в качестве основного компонента коллаген типа II. Основным местом поражения хроническим ревматоидным артритом является синовиальная ткань, где появляется лимфоцитарная инфильтрация (инфильтрат), состоящая в основном из Т-клеток. Кроме того, традиционно считали, что клон, который специфически взаимодействует с коллагеном типа II, существует в клетках CD4+T, которые проникли в синовиальную мембрану. В качестве антигена, способного продуцировать аутореактивные Т-клетки, известны, помимо коллагена типа II, протеогликан, аденовирус, вирус EB и белок теплового шока. Среди них коллаген типа II, как полагают, является одним из наиболее известных аутоантигенов, так как он является основным компонентом хряща и имеется в суставе. Хронический ревматоидный артрит вызывает повреждение хряща и кости при повторной ремиссии и обострении (болезни), вызывая структурную деформацию периферического сустава.
В качестве фармакотерапии для хронического ревматоидного артрита используют противовоспалительный стероид (например, преднизолон), нестероидные противовоспалительные лекарственные средства (например, индометацин, аспирин), иммунодепрессивное средство (например, циклоспорин А, такролимус (FK506), метотрексат, циклофосфамид, азатиоприн) и болезнь-корректирующее противоревматическое лекарственное средство (например, препарат соли золота). Хотя противовоспалительное средство может ослабить боль и набухание, регулируя воспаление, подавить развитие этой болезни трудно. Считают, что среди вышеуказанных иммунодепрессивных средств циклоспорин А и такролимус оказывают иммунодепрессивное действие путем подавления продуцирования IL-2 из T-клеток. Хотя механизм действия болезнь-корректирующего противоревматического лекарственного средства никогда не был объяснен, вышеуказанное лекарственное средство обладает седативным воздействием и оказывает влияние на ремиссию этого заболевания.
Как было изложено выше, любые вышеупомянутые лекарственные средства только облегчают симптом (заболевания) и замедляют развитие болезни и, кроме того, вызывают побочные действия, связанные с применением препарата на протяжении длительного периода времени, и поэтому указанные лекарственные средства являются неудовлетворительными лекарственными средствами. Поэтому существует значительная потребность в разработке нового лекарственного средства для лечения хронического ревматоидного артрита.
Известно, что производное аминостилбазола настоящего изобретения имеет высокую противораковую активность и очень низкую токсичность (Международная публикация W095/27699).
Целью настоящего изобретения является разработка нового лекарственного средства для хронического ревматоидного артрита.
Исходя из вышеизложенной точки зрения, изобретателями были проведены исследования на различных соединениях с целью обнаружения соединения, которое подавляет хронический ревматоидный артрит. В результате ими было установлено ингибирующее действие производных аминостилбазола (в дальнейшем иногда называемых соединениями настоящего изобретения), представленных нижеследующей общей формулой [1], на индуцируемый коллагеном типа II артрит. Таким образом, настоящее изобретение было завершено.
Настоящее изобретение относится к лекарственному средству для хронического ревматоидного артрита, содержащему в качестве активного компонента соединение, представленное общей формулой [1], или его соль

[где A представляет необязательно замещенный гетероарил или его оксид, B представляет необязательно замещенный этенилен, D представляет необязательно замещенный фенилен, E представляет нижеследующую формулу

(где G представляет необязательно замещенный фенил, R представляет (1) водород, (2) необязательно замещенный алкил, (3) необязательно замещенный алкенил, (4) алкинил, или (5) -COR°, R° представляет алкил, алкокси, арилокси, необязательно замещенный гетероарил, необязательно замещенный гетероарилметил или имеет нижеследующую формулу:

где n представляет целое число от 1 до 5, R5 и R6 являются одинаковыми или отличными и каждый представляет водород, алкил, гидроксиалкил или аминоалкил, или R5 и R6 взяты вместе с соседними атомами азота как -NR5(R6), образуя необязательно замещенный циклический амино, и циклический амино может иметь один атом кислорода, серы или азота, в качестве кольцевого атома, в добавление к атому азота)].
Предпочтительно, настоящее изобретение относится к лекарственному средству для хронического ревматоидного артрита, содержащему в качестве активного компонента соединения настоящего изобретения или соль их, где A представляет необязательно замещенный пиридил или 1-оксид пиридила, B представляет незамещенный этенилен, D представляет незамещенный или аминоалкокси-замещенный 1,2-фенилен, R в -N(R)-SO2-G, представляющем E, означает алкил, который может быть замещен заместителем, выбранным из группы, состоящей из галогена, амино, моноалкиламино, диалкиламино, морфолино, алкокси, гидрокси, циано, формулы -CONR1R2 (где R1 и R2 являются одинаковыми или отличными и каждый представляет водород, гидрокси, алкил, алкокси, циклоалкил, циклоалкилокси, арил, арилокси, аралкил, аралкилокси, циклоалкилалкил, циклоалкилалкилокси или тетрагидрофуранилокси), и формулы -SO2NR3R4 (где R3 и R4 являются одинаковыми или отличными и каждый представляет водород или алкил), алкенил, который может быть замещен галогеном, или -COR°, R° представляет алкил, циклический аминоалкил или диалкиламиноалкил, и G представляет 4-алкоксифенил.
Более предпочтительно, настоящее изобретение относится к лекарственному средству для хронического ревматоидного артрита, содержащему в качестве активного компонента соединения настоящего изобретения или их соль, где A представляет незамещенный 4-пиридил или 1-оксид-4-пиридил, B представляет этенилен незамещенной транс-формы, D представляет незамещенный или аминоалкокси-замещенный 1,2-фенилен, R в -N(R)-SO2-G, представляющем E, означает водород, гидроксиалкил или -COR°, R° представляет алкил, морфолиноалкил или диалкиламиноалкил, и G представляет 4-метоксифенил.
Особенность настоящего изобретения заключается в том, что соединение формулы [1] оказывает ингибирующее действие на хронический ревматоидный артрит и является полезным для лечения хронического ревматоидного артрита. Это ни разу не было описано ни в одном документе и, кроме того, никогда не было известно, что вышеупомянутое соединение обладает таким действием.
Ниже настоящее изобретение описывается детально.
Термины, используемые в настоящем описании и определениях соответствующих заместителей, являются таким, как следует ниже.
Используемый здесь термин "лекарственное средство для хронического ревматоидного артрита" относится к лекарственному средству, которое облегчает боль сустава, уменьшая воспаление сустава, или лекарственному средству, которое поддерживает или восстанавливает функцию сустава, предотвращая повреждение кости и хряща.
Примеры "гетероарила" могут включать (5-6)-членный гетероарил, имеющий от 1 до 2 атомов азота в кольце. Вышеуказанный гетероарил может иметь от 1 до 2 заместителей в любых положениях, и примеры заместителя могут включать галоген, алкил, алкокси, гидрокси, аминоалкил и т.п. Примеры "гетероарила", представляющего A, могут включать 6-членный гетероарил, например 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидинил, 4-пиримидинил, 5-пиримидинил, 3-пиридазинил, 4-пиридазинил и пиразинил. Среди них предпочтительным является незамещенный 4-пиридил. Примеры гетероарильной части "гетероарила" и "гетероарилметила", как R°, могут включать (5-6)-членный гетероарил, например 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидинил, 4-пиримидинил, 5-пиримидинил, 3-пиридазинил, 4-пиридазинил, 1-имидазолил, 2-имидазолил и 4-имидазолил. Среди них предпочтительным является пиридил.
"Этенилен" может иметь заместитель на соответствующих углеродных атомах, и примеры вышеуказанного заместителя могут включать циано, бром, алкил и т.п. Среди них предпочтительным является незамещенный этенилен.
Примеры "фенилена" могут включать 1,2-фенилен, 1,3-фенилен и 1,4-фенилен. Такая фениленовая группа может иметь от 1 до 2 заместителей в любых положениях, и примеры заместителя могут включать гидрокси, галоген, амино, алкил, алкокси, аминоалкокси и т.п. Предпочтительным является незамещенный или аминоалкокси-замещенный 1,2-фенилен.
"Фенил" может иметь от 1 до 2 заместителей, и примеры вышеуказанного заместителя могут включать гидрокси, алкокси и т.п. Среди них предпочтительным является алкокси-замещенный фенил и, в частности, предпочтительным является 4-метоксифенил.
Примеры "галогена" могут включать фтор, хлор, бром, иод и т.п.
Примеры "алкила" могут включать алкил с линейной или разветвленной цепью, имеющий от 1 до 6 атомов углерода, например метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, н-гексил, изогексил и т.п. Среди них алкил, имеющий от 1 до 4 атомов углерода, является предпочтительным, и, в частности, предпочтителен метил. Алкил, представляющий R, может быть замещен заместителем, выбранным из группы, состоящей из галогена, амино, моноалкиламино, диалкиламино, морфолино, алкокси, гидрокси, циано, формулы -CONR1R2 (где R1 и R2 являются одинаковыми или отличными и каждый представляет водород, гидрокси, алкил, алкокси, циклоалкил, циклоалкилокси, арил, арилокси, аралкил, аралкилокси, циклоалкилалкил, циклоалкилалкилокси или тетрагидрофуранилокси) и формулы -SO2NR3R4 (где R3 и R4 являются одинаковыми или отличными и каждый представляет водород или алкил).
Примеры алкильной части "гидроксиалкила", "моноалкиламино", "диалкиламино", "аминоалкила", "циклоалкилалкила" и "циклоалкилалкилокси" могут включать примеры "алкила", описанные выше.
Примеры "алкокси" могут включать алкокси с прямой или разветвленной цепью, имеющую от 1 до 6 атомов углерода, например метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси, н-пентилокси, изопентилокси, н-гексилокси и изогексилокси. Среди них алкокси, имеющая от 1 до 4 атомов углерода, является предпочтительной, и, в частности, метокси является более предпочтительным.
Примеры алкокси части "аминоалкокси" могут включать обозначения "алкокси", описанной выше.
Примеры "арилокси" могут включать необязательно замещенную арилокси, имеющую от 6 до 10 атомов углерода, например фенокси, 1-нафтилокси и 2-нафтилокси. Среди них предпочтительным является фенокси. Примеры заместителя могут включать алкил, галоген, гидрокси, алкокси и т.п.
Примеры "алкенила" могут включать алкенил с линейной или разветвленной цепью, имеющий от 2 до 6 атомов углерода, например винил, 1-пропенил, 2-пропенил, изопропенил, 2-бутенил, 3-бутенил, изобутенил, металлил, пренил, изопренил и 1,1-диметилаллил. Среди них алкенил, имеющий от 2 до 4 атомов углерода, является предпочтительным. "Алкенил", представляющий R, может быть замещен галогеном.
Примеры "алкинила" могут включать алкинил с линейной или разветвленной цепью, имеющий от 2 до 6 атомов углерода, например этинил, 1-пропинил, 2-пропинил, 2-бутинил, 3-бутинил и 3-метил-2-бутинил. В частности, алкинил, имеющий от 2 до 4 атомов углерода, является предпочтительным.
В алкилене, представленном "-(CH2)n-", атом водорода может быть замещен одной аминогруппой или алкилом в любых положениях.
Примеры "циклического амина" могут включать (5-8)-членные циклические амины, например пирролидин-1-ил, пиперидино, гексаметиленимино, тетрагидропиридин-1-ил, октагидроазоцин-1-ил, пиперазин-1-ил, гомопиперазин-1-ил, морфолино и тиоморфолино. Вышеуказанный циклический амино может иметь от 1 до 2 заместителей, выбранных из группы, состоящей из алкильной, алкенильной, алкинильной, арильной, аралкильной и гетероциклической группы, имеющей атом азота в любых положениях. Среди них (5-6)-членный циклический амин является предпочтительным, и пиперазин-1-ил, замещенный пиридилом, незамещенным пирролидин-1-илом, пиперидино или морфолино является предпочтительным.
Примеры "арила" могут включать арил, имеющий от 6 до 10 атомов углерода, например фенил, 1-нафтил и 2-нафтил.
Примеры "аралкила" могут включать аралкил, имеющий от 7 до 8 атомов углерода, например бензил и фенэтил.
Примеры аралкильной части "аралкилокси" могут включать аралкилы, описанные выше.
Примеры "гетероцикла, имеющего атом азота", могут включать циклический амин и гетероарил, описанные выше. Вышеуказанный гетероцикл может иметь от 1 до 2 заместителей, выбранных из группы, состоящей из алкила, амино, гидрокси и оксо.
Примеры "циклоалкила" могут включать циклоалкил, имеющий от 3 до 8 атомов углерода, например циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил.
Примеры циклоалкильной части "циклоалкилокси", "циклоалкилалкила" и "циклоалкилалкилокси" могут включать циклоалкилы, описанные выше.
Примеры "соли" соединений настоящего изобретения могут включать фармакологически приемлемые соли, например соли с неорганическими кислотами, такими как хлористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, фтористоводородная кислота и бромистоводородная кислота; и соли с органическими кислотами, такими как уксусная кислота, винная кислота, молочная кислота, лимонная кислота, фумаровая кислота, малеиновая кислота, янтарная кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, толуолсульфоновая кислота, нафталинсульфоновая кислота и камфорсульфоновая кислота.
Соединения настоящего изобретения могут существовать в форме цис (Z форма) и транс (E форма) изомеров, и каждый изомер и смесь их также входит в объем настоящего изобретения. В частности, Е форма является предпочтительной.
Примеры соединений настоящего изобретения могут включать соединения формулы [1]. Среди них предпочтительными являются
(E)-4-[2-{2-[N-ацетил-N-(4-метоксибензолсульфонил)амино]фенил}-этенил]пиридин 1-оксид,
(E)-4-[2-{2-[N-ацетил-N-(4-метоксибензолсульфонил)амино]фенил}-этенил]пиридин,
(E)-4-[2-{2-[N-(4-метоксибензолсульфонил)амино]фенил}-этенил]пиридин 1-оксид,
(E)-4-[2-{2-[N-(4-метоксибензолсульфонил)амино]фенил}-этенил]пиридин,
(E)-4-[2-{2-[N-(2-гидроксиэтил)-N-(4-метоксибензолсульфонил)-амино]фенил}этенил]пиридин 1-оксид,
(E)-4-[2-{2-[N-(2-гидроксиэтил)-N-(4-метоксибензолсульфонил)-амино]фенил}этенил]пиридин,
(E)-4-[2-{3-(2-аминоэтилокси)-2-[N-(4-метоксибензолсульфонил)-амино]фенил}этенил]пиридин и
(E)-4-[2-{3-(2-аминоэтилокси)-2-[N-(4-метоксибензолсульфонил)-амино]фенил}этенил]пиридин 1-оксид.
Соединения настоящего изобретения можно получить способом, описанным в международной публикации W095/27699, международной публикации W001/44195 или в не прошедшей экспертизу заявке на патент Японии № 2001-261649, или его модификацией.
В тех случаях, когда соединения настоящего изобретения применяют в качестве лекарственного средства для хронического ревматоидного артрита, они могут вводиться млекопитающему, включая человека, как таковые или в смеси с фармацевтически приемлемым нетоксичным инертным носителем, например, в виде фармацевтической композиции, содержащей соединение при уровне от 0,1 до 99,5 мас.%, предпочтительно от 0,5 до 90 мас.%.
Носитель, который может быть использован, включает твердый, полутвердый и жидкий разбавитель, наполнитель и другие вспомогательные средства для формулирования, и, по крайней мере, один из них используют селективно. Фармацевтическую композицию предпочтительно вводят в дозированной лекарственной форме. Фармацевтическую композицию настоящего изобретения можно вводить пероральным путем или парентерально (например, при помощи инъекции или ректально). Несомненно, должна использоваться лекарственная форма, подходящая для каждого конкретного способа введения. Например, пероральное введение является особенно предпочтительным.
Доза соединения, используемого в качестве лекарственного средства для хронического ревматоидного артрита, может предпочтительно регулироваться, принимая во внимание показатели, свойственные конкретному пациенту, такие как возраст, масса тела и т.д., характер и тяжесть заболевания и т.д., а также способ введения; суточная доза, как активного компонента, для взрослого, при введении перорально, обычно составляет от 0,1 мг до 300 мг на взрослого, предпочтительно от 1 мг до 100 мг на взрослого. В некоторых случаях, может быть достаточна более низкая доза или может потребоваться более высокая доза. Вышеупомянутую суточную дозу предпочтительно вводят один раз или ее вводят несколько раз в виде поделенной на части.
Пероральное введение можно осуществить, используя твердую или жидкую дозированную лекарственную форму, такую как частица, порошок, таблетка, покрытая сахарной оболочкой таблетка, капсула, гранула, суспензия, жидкость, сироп, капли, подъязычная таблетка, или другие лекарственные формы.
Частицу получают измельчением соединения (вещества) настоящего изобретения до частиц подходящего размера. Порошок можно получить измельчением соединения настоящего изобретения до частиц подходящего размера с последующим смешением с фармацевтическим носителем, таким как годный в пищу углевод, включая крахмалы или маннит, который также может быть измельчен до частиц подходящего размера. Вспомогательными веществами, которые могут быть добавлены при необходимости, являются придающие вкус добавки, консерванты, диспергирующие средства, красители, придающие аромат добавки и т.п.
Капсулу можно получить путем загрузки частиц или порошка, который был измельчен, как описано выше, или гранул, полученных, как описано выше в разделе таблетки, например в капсулу, такую как желатиновая капсула. Кроме того, до операции наполнения капсулы можно смешать измельченное (активное) вещество с добавкой, такой как смазка, псевдоожижающее средство, такое как коллоидный диоксид кремния, тальк, стеарат магния, стеарат кальция или твердый полиэтиленгликоль. Эффективность лекарственного средства после приема внутрь капсулы может быть повышена путем добавления дезинтегратора или солюбилизатора, такого как карбоксиметилцеллюлоза, кальций-карбоксиметилцеллюлоза, гидроксипропилцеллюлоза с низкой степенью замещения, натрий-кроскармеллоза, натрий-карбоксиметилкрахмал, карбонат кальция или карбонат натрия.
Тонкоизмельченный порошок может быть суспензирован, и диспергирован в растительном масле, полиэтиленгликоле, глицерине и поверхностно-активном веществе, и затем инкапсулирован в желатиновом слое, в результате чего получают мягкую капсулу.
Таблетку можно получить путем приготовления порошкообразной композиции, добавляя наполнитель, ее грануляции или комкования зерен сыпучего материала из нее (зернение), добавления дезинтегратора или смазки в полученный гранулят и затем прессования образовавшейся гранулированной смеси в таблетку.
Порошкообразную композицию можно получить путем смешения надлежащим образом измельченного вещества с разбавителем или основой, описанной выше, при необходимости вместе со связующим веществом (например, натрий-карбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, желатин, поливинилпиррролидон и поливиниловый спирт), замедлителем растворения (например, парафин), активатором резорбции (например, четвертичная соль) или адсорбентом (например, бентонит, каолин и дифосфат кальция). Порошкообразную композицию можно гранулировать путем увлажнения связующим, таким как сироп, крахмальный клей, аравийская камедь, раствор целлюлозы или раствор полимера, с последующим смешением при перемешивании, сушкой и измельчением. Вместо операции для гранулирования порошка, описанной выше, можно использовать другую операцию, в которой смесь сначала подвергают (обработке) в таблетировочной машине, получая морфологически дефектный (несовершенную) заготовку, которую затем измельчают. Полученная таким образом гранула может содержать, в качестве смазки, стеариновую кислоту, стеараты, тальк, минеральное масло и т.п. для предотвращения какой-либо адгезии друг с другом. Смазанную таким образом смесь затем прессуют в таблетки. Полученная таким образом плоская таблетка может быть покрыта пленкой или покрыта сахарной оболочкой.
Соединения настоящего изобретения можно смешать с псевдоожиженным инертным носителем и затем спрессовать непосредственно в таблетки, не подвергая операции гранулирования или зернения, описанной выше. Кроме того, может быть использована прозрачная или полупрозрачная защитная пленка в форме герметизирующей пленки из шеллака, пленки из сахара или полимерного вещества и глянцевой пленки из воска. Другие пероральные лекарственные формы, такие как раствор, сироп и эликсир, могут быть формулированы в виде дозированной лекарственной формы, определенное количество которой содержит определенное количество лекарственного средства. Сироп получают растворением соединений настоящего изобретения в ароматизированном водном растворе, в то время как эликсир получают, используя нетоксичный спиртовой носитель. Суспензию формулируют, диспергируя соединение в нетоксичном носителе. Кроме того, по необходимости, могут быть также добавлены добавки, такие как солюбилизирующее средство, эмульгатор (например, этоксилированные изостеариловые спирты, сложные эфиры полиоксиэтилированного сорбита, консервант и ароматизатор (например, эфирное масло из перечной мяты, сахарин).
При желании, состав стандартной дозы, предназначенный для перорального введения, может представлять микрокапсулу. Такой состав может быть покрыт или встроен в полимер, воск или т.п. с тем, чтобы получить пролонгированную активность или замедленное высвобождение активного компонента.
Для парентерального введения можно использовать инъекцию и суппозиторий. Парентеральное введение может быть осуществлено, используя жидкую дозированную лекарственную форму, такую как раствор или суспензия, для подкожного, внутримышечного или внутривенного введения. Такую дозированную лекарственную форму можно получить путем суспендирования или растворения заранее установленного количества соединения настоящего изобретения в нетоксичном жидком носителе, таком как водная или маслянистая среда, совместимым с целью инъекции, с последующей стерилизацией суспензии или раствора. Кроме того, возможно добавление нетоксичной соли или раствора соли с целью получения изотонического раствора для инъекции. Возможно также использование стабилизатора, консерванта, эмульгатора и т.п.
Ректальное введение можно осуществить, используя суппозиторий или т.п., полученный путем суспендирования или растворения соединений настоящего изобретения в водорастворимом или водонерастворимом твердом веществе, имеющем низкую точку плавления, таком как полиэтиленгликоль, масло какао, полусинтезированные жиры и жирные масла (например, Witepsol (R)), высшие сложные эфиры (например, миристилпальмитат), а также их смеси.
В лекарственном средстве для хронического ревматоидного артрита, заявляемого в настоящем изобретении, возможно использование в комбинации с другими компонентами, например противовоспалительного стероида, нестероидного противовоспалительного средства, иммунодепрессивного средства, болезнь-изменяющего противоревматического лекарственного средства и т.п.
Настоящее изобретение будет изложено ниже более подробно, ссылаясь на Примеры получения типичных исходных веществ (Ссылочные примеры), Примеры получения соединений настоящего изобретения (Примеры), Примеры приготовления фармацевтических составов и Примеры испытаний, однако настоящее изобретение ими не ограничивается. Удельное вращение измеряют при 20°C.
Ссылочный пример 1
Получение (E)-2-(2-трет-бутоксикарбониламиноэтокси)-6-[2-(4-пиридил)этенил]анилина
(Стадия 1) Синтез 3-гидрокси-2-нитробензальдегида
3,62 г 3-метокси-2-нитробензальдегида растворяют в 80 мл метиленхлорида и по каплям при охлаждении льдом добавляют раствор трибромида бора в метиленхлориде (15,03 г трибромида бора, 40 мл метиленхлорида), с последующим перемешиванием при 0°C в течение часа. Реакционный раствор выливают в лед, экстрагируют хлороформом, сушат и затем концентрируют, получая 3,32 г требуемого соединения.
(Стадия 2) Получение (E)-2-ацетокси-6-[2-(4-пиридил)этенил]-нитробензола
К 3,17 г соединения, полученного на стадии 1, добавляют 1,94 г 4-пиколина и 4,79 г уксусного ангидрида и затем смесь кипятят с обратным холодильником при перемешивании в течение 12 часов. Реакционный раствор выливают в лед, нейтрализуют карбонатом калия, экстрагируют хлороформом и затем сушат над сульфатом магния. После дистилляции растворителя при пониженном давлении остаток подвергают колоночной хроматографии на силикагеле (CHCl3), получая 4,78 г требуемого соединения.
(Стадия 3) Получение (E)-2-гидрокси-6-[2-(4-пиридил)этенил]-нитробензола
4,54 г соединения, полученного на стадии 2, растворяют в 160 мл метанола и добавляют 4,42 г карбоната калия с последующим перемешиванием при комнатной температуре в течение 2 часов. После концентрирования реакционного раствора остаток объединяют с водой со льдом и нейтрализуют 2N хлористоводородной кислотой и затем осажденный порошок собирают фильтрацией, получая 3,47 г требуемого соединения. Полученное соединение используют в качестве вещества без дополнительной очистки.
(Стадия 4) Получение (E)-2-(2-трет-бутоксикарбониламиноэтокси)-6-[2-(4-пиридил)этенил]нитробензола
2,18 г соединения, полученного на стадии 3, растворяют в 9 мл ДМСО (DMCO) и в потоке газа-аргона добавляют 0,54 г 60% гидрида натрия с последующим перемешиванием при комнатной температуре в течение одного часа. Добавляют 4,63 г 2-бромэтил-трет-бутоксикарбониламина с последующим перемешиванием при нагревании при 120°C в течение 3 часов. Реакционный раствор выливают в воду со льдом, объединяют с этилацетатом, промывают водой (3 раза), промывают насыщенным раствором соли и затем сушат над сульфатом магния. После дистилляции растворителя при пониженном давлении остаток подвергают колоночной хроматографии на силикагеле (CHCl3:MeOH=50:1), получая 1,90 г требуемого соединения.
(Стадия 5) Получение (E)-2-(2-трет-бутоксикарбониламиноэтокси)-6-[2-(4-пиридил)этенил]анилина
1,90 г соединения, полученного на стадии 4, растворяют в 50 мл 70% водного метанола и добавляют 4,90 г восстановленного железа и 0,30 г хлорида кальция и затем смесь кипятят с обратным холодильником при перемешивании в течение 4 часов. После фильтрования реакционного раствора через целит фильтрат концентрируют и остаток подвергают колоночной хроматографии на силикагеле (CHCl3:MeOH=50:1), получая 1,45 г требуемого соединения.
Пример 1
(E)-4-[2-{2-[N-феноксикарбонил-N-(4-метоксибензолсульфонил)амино]-фенил}этенил]пиридин
1,00 г (E)-4-[2-{2-[N-(4-метоксибензолсульфонил)амино]-фенил}этенил]пиридина суспендируют в 40 мл хлороформа и после добавления 1,82 г фенилхлоркарбоната медленно при охлаждении льдом добавляют 1,20 г триэтиламина. Затем смесь перемешивают при комнатной температуре в течение 5 минут. Растворитель отгоняют при пониженном давлении и требуемый продукт очищают на колонке с силикагелем (носитель: Wako Gel C200, проявляющий растворитель-хлороформ), получая требуемое соединение. Требуемое соединение обрабатывают этанолом, получая 0,71 г белых гранулированных кристаллов.
Элементный анализ (для С27Н22N2O5S)
Рассчитано (%): H, 4,55; C, 66,65; N, 5,75
Найдено (%): H, 4,51; C, 66,44; N, 5,67
Таким же способом синтезируют нижеследующие требуемые соединения.
Пример 2
(E)-4-[2-{2-[N-метоксикарбонил-N-(4-метоксибензолсульфонил)-амино]фенил}этенил]пиридин
Элементный анализ (для С22Н20N2O5S)
Рассчитано (%): H, 4,75; C, 62,25; N, 6,60
Найдено (%): H, 4,83; C, 61,97; N, 6,51
Пример 3
(E}-4-[2-{2-[N-этоксикарбонил-N-(4-метоксибензолсульфонил)-амино]фенил}этенил]пиридин
Элементный анализ (для С23Н22N2O5S·1/5Н2О)
Рассчитано (%): H, 5,11; C, 62,49; N, 6,34
Найдено (%): H, 5,09; C, 62,39; N, 6,34
Пример 4
(E)-4-[2-{2-[N-н-пропоксикарбонил-N-(4-метоксибензолсульфонил)-амино]фенил}этенил]пиридин гидрохлорид
Элементный анализ (для С24Н24N2O5S·HCl·1/2Н2О)
Рассчитано (%): H, 5,26; C, 57,88; N, 5,63
Найдено (%): H, 5,23; C, 58,12; N, 5,72
Пример 5
(E)-4-[2-{2-[N-н-бутоксикарбонил-N-(4-метоксибензолсульфонил)-амино]фенил}этенил]пиридин гидрохлорид
Элементный анализ (для С25Н26N2O5S·HCl·1/2Н2О))
Рассчитано (%): H, 5,51; C, 58,64; N, 5,47
Найдено (%): H, 5,47; C, 58,44; N, 5,49
Пример 6
(E)-4-[2-{3-(2-аминоэтокси)-2-[N-(4-метоксибензолсульфонил)-амино]фенил}этенил]пиридин дигидрохлорид
(Стадия 1) Получение (E)-4-[2-{3-(2-трет-бутоксикарбониламино-этокси)-2-[N-(4-метоксибензолсульфонил)амино]фенил}этенил]пиридина
1,42 г (E)-2-(2-трет-бутоксикарбониламиноэтокси)-6-[2-(4-пиридил)этенил]анилина, полученного на стадии 5 Ссылочного примера 3, растворяют в 14 мл пиридина и добавляют 0,99 г 4-метоксибензолсульфонилхлорида с последующим перемешиванием при комнатной температуре на протяжении ночи. Реакционный раствор концентрируют и остаток объединяют с водой со льдом, экстрагируют хлороформом и затем сушат над сульфатом магния. После удаления растворителя дистилляцией при пониженном давлении остаток подвергают колоночной хроматографии на силикагеле (CHCl3:MeOH=30:1), получая 2,19 г требуемого соединения.
(Стадия 2) Получение (E)-4-[2-{3-(2-трет-бутоксикарбониламиноэтокси)-2-[N-(4-метоксибензолсульфонил)амино]фенил}этенил]пиридин 1-оксида
0,56 г соединения, полученного на стадии 1, растворяют в 5 мл хлороформа и добавляют м-хлорпербензойную кислоту с последующим перемешиванием при комнатной температуре в течение одного часа. Реакционный раствор промывают водой три раза и затем сушат над сульфатом магния. После удаления растворителя дистилляцией при пониженном давлении остаток подвергают колоночной хроматографии на силикагеле (CHCl3: MeOH=20:1), получая 0,46 г требуемого соединения.
(Стадия 3) 2,10 г соединения, полученного на стадии 2, растворяют в 10 мл метиленхлорида и по каплям при охлаждении льдом добавляют 10,5 мл трифторуксусной кислоты с последующим перемешиванием при комнатной температуре в течение 2 часов. Реакционный раствор объединяют с водой со льдом, слабо подщелачивают путем добавления карбоната калия, экстрагируют хлороформом и затем сушат над сульфатом магния. После удаления растворителя дистилляцией при пониженном давлении остаток подвергают колоночной хроматографии на силикагеле (CHCl3:MeOH=20:1), получая 1,33 г свободной формы.
Свободную форму растворяют в 20 мл метанола и при охлаждении льдом добавляют избыточное количество раствора 20% хлористоводородная кислота-простой эфир с последующим перемешиванием в течение одного часа. Реакционный раствор концентрируют и обрабатывают этанолом, получая 1,30 г требуемого соединения (бледно-желтые кристаллы).
Элементный анализ (для С22Н23N3O4S·2HCl·H2O)
Рассчитано (%): C, 51,17; H, 5,27; N, 8,14
Найдено (%): C, 51,04; H, 4,99; N, 8,02
Пример 7
(E)-4-[2-{3-(2-аминоэтокси)-2-[N-(4-метоксибензолсульфонил)амино]-фенил}этенил]пиридин 1-оксид дигидрохлорид
2,38 г соединения, полученного на стадии 2 Примера 6, растворяют в 23 мл метиленхлорида и добавляют по каплям при охлаждении льдом 23 мл трифторуксусной кислоты с последующим перемешиванием при комнатной температуре в течение 2 часов. Реакционный раствор объединяют с водой со льдом, слабо подщелачивают путем добавления карбоната калия, экстрагируют хлороформом и затем сушат над сульфатом магния. После удаления растворителя дистилляцией при пониженном давлении остаток подвергают колоночной хроматографии на силикагеле (CHCl3:MeOH:28%NH3(водн.)=90:10:1), получая 1,48 г свободной формы.
Свободную форму растворяют в 50 мл метанола и при охлаждении льдом добавляют избыточное количество раствора 20% хлористоводородная кислота-простой эфир с последующим перемешиванием в течение одного часа. Реакционный раствор концентрируют и обрабатывают этанолом, получая 1,50 г требуемого соединения (бледно-желтые кристаллы).
Элементный анализ (для С22Н23N3O5S·2HCl·0,3H2O)
Рассчит. (%): C, 50,83; H, 4,96; N, 8,08
Найдено (%): C, 50,86; H, 4,88; N, 7,99
Пример испытания 1: Ингибирующее действие на артрит
Как описано ниже, ингибирующее действие на артрит соединений настоящего изобретения может быть подтверждено, используя в качестве модели, близкой к человеческому ревматоидному артриту, индуцированный коллагеном типа II артрит и используя в качестве испытываемого соединения (E)-4-{2-[2-{N-ацетил-N-[(4-метоксифенил)сульфонил]амино}фенил]этенил}пиридин 1-оксид (в дальнейшем называемое соединением 1).
Использовали мышей-самцов линии DBA/1J возраста 8 недель (от 7 до 10 мышей). 0,1 мл эмульсии, полученной смешением раствора коллагена типа II (2 мг/мл), полученного из бычьего суставного хряща, в 0,1M растворе уксусной кислоты в физиологическом растворе и полного адъюванта Фрейнда при отношении концентраций компонентов смеси 1:1 инъецируют внутрикожно мышам в основание хвоста (первичная сенсибилизация). Спустя 21 день 0,1 мл эмульсии, полученной таким же способом, как и в случае первичной сенсибилизации, снова инъецируют подкожно мышам в спинку (вторичная сенсибилизация). Испытываемое соединение суспендируют в 0,5% водном растворе метилцеллюлозы и вводят перорально мышам один раз в день, который начинался с первичной или вторичной сенсибилизации. Спустя 35 дней от первичной сенсибилизации визуально наблюдали симптомы артрита и результаты использовали как оценку артрита.
Результаты представлены в табл.1 и 2.
Ингибирующее действие на артрит в случае, если введение испытываемого соединения начинали после первичной сенсибилизации
Ингибирующее действие на артрит в случае, если введение испытываемого соединения начинали после вторичной сенсибилизации
Как следует из вышеупомянутых таблиц, соединение 1 оказывает значительное ингибирующее действие при дозе 5 мг/кг в случае, если введение испытываемого соединения начинали после проведения первичной сенсибилизации, или при дозах 5 и 10 мг/кг в случае, если введение испытываемого соединения начинали после проведения вторичной сенсибилизации. То есть соединение 1 вызывает ингибирующее действие на индуцируемый коллагеном артрит даже в схеме введения испытываемого соединения после вторичной сенсибилизации и также является полезным в качестве лекарственного средства для лечения хронического ревматоидного артрита.
Пример испытания 2: Острая токсичность
Использовали мышей-самцов линии CDF1 возраста 7 недель (6 мышей). Соединение 1 суспендировали в 0,5% метилцеллюлозе и полученную суспензию вводили перорально один раз при помощи перорального зонда. Спустя 2 недели определяли коэффициент смертности и значение LD50 рассчитывали пробит-анализом. В результате было установлено, что значение LD50 соединения 1 составляет 620,5 мг/кг и что соединение 1 имеет очень незначительную токсичность.
Вышеприведенные результаты четко указывают на то, что соединения настоящего изобретения имеют очень низкую токсичность и являются высокобезопасными. Результаты примеров испытания 1 и 2 четко указывают на то, что соединения настоящего изобретения оказывают превосходное ингибирующее действие на артрит, индуцируемый коллагеном типа II, и имеют низкую токсичность.
Примеры фармацевтических составов
Пример 1. (180 мг на таблетку для внутреннего использования)
Вышеупомянутые компоненты взвешивают согласно составу. Компоненты, включая поливиниловый спирт и стеарат магния, смешивают равномерно и получают гранулы для таблетирования (гранулят) методом влажной грануляции, используя раствор поливинилового спирта в качестве связующего. После смешения гранулята со стеаратом магния смесь подвергают прессованию в таблетированной машине с получением таблеток, при этом каждая таблетка диаметром 8 мм весит 180 мг, получая таким образом таблетку для внутреннего использования.
Пример 2 (220 мг на твердую капсулу)
Вышеупомянутые компоненты взвешивают в соответствии с составом и после смешения до однородного состояния 220 мг смеси заполняют в капсулу #2, используя машину для наполнения капсул, получая таким образом твердую капсулу.
Пример 3 (1 г на гранулу)
Вышеупомянутые компоненты взвешивают согласно составу и после проведения операции смешения и замеса с образованием однородной смеси полученную смесь подвергают формованию в гранулирующей машине (гранулятор) с получением гранул, каждая диаметром 0,7 мм, получая таким образом гранулы.
Пример 4 (1 мл на инъекцию)
(Соединение 2 представляет (E)-4-{2-[2-{N-(4-метоксибензол-сульфонил)-N-[4-(2-пиридил)пиперадино]ацетиламино}фенил]этенил}-пиридин 1-оксид дигидрохлорид)
Способ получения
После растворения соединений настоящего изобретения и маннита в воде для инъекции полученный раствор асептически фильтруют через мембранный фильтр (размер пор: 0,22 мкм). После заполнения в ампулу, ампулу подвергают сушке вымораживанием, получая состав для инъекции, подлежащий растворению перед использованием.
Пример 5 (1 мл на инъекцию)
Способ получения
После растворения соединений настоящего изобретения и мальтозы в воде для инъекции полученный раствор асептически фильтруют через мембранный фильтр (размер пор: 0,22 мкм). После заполнения в ампулу, ампулу подвергают сушке вымораживанием, получая состав для инъекции, который подлежит растворению перед использованием.
Поскольку соединения настоящего изобретения оказывают сильное ингибирующее действие на артрит при дозе, которая намного ниже, чем доза, достаточная для проявления противораковой активности, и могут вводиться пероральным путем, они могут быть безопасно использованы в качестве лекарственного средства для лечения ювенильного ревматоидного артрита (болезнь Стилла), синдрома Рейтера и SLE, синдрома Бенкета, в добавление к хроническому ревматоидному артриту.
| название | год | авторы | номер документа |
|---|---|---|---|
| АЗОТСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРОТИВОРАКОВЫЙ АГЕНТ | 2000 |
|
RU2248969C2 |
| СПОСОБ ПРИМЕНЕНИЯ КОМБИНАЦИИ ПРОИЗВОДНОГО БЕНЗОФЕНОНА ИЛИ ЕГО СОЛИ И ИММУНОСУПРЕССОРА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ДАННЫЕ КОМПОНЕНТЫ | 2009 |
|
RU2496490C2 |
| ПРОИЗВОДНЫЕ АМИНОСТИЛЬБАЗОЛА ИЛИ ИХ ГИДРАТЫ, ИЛИ ИХ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1995 |
|
RU2138482C1 |
| ФЕНИЛАЛАНИНОЛОВЫЕ ПРОИЗВОДНЫЕ | 2000 |
|
RU2238265C2 |
| ОКТАГИДРОПЕНТАЛЕНОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АНТАГОНИСТОВ ХЕМОКИНОВЫХ РЕЦЕПТОРОВ | 2008 |
|
RU2481332C2 |
| ПИРАЗОЛОПИРИДИНОВЫЕ ПРОИЗВОДНЫЕ КАК ИНГИБИТОРЫ НАДФН-ОКСИДАЗЫ | 2009 |
|
RU2569303C2 |
| НЕСТЕРОИДНЫЕ МОДУЛЯТОРЫ ГЛЮКОКОРТИКОИДНЫХ РЕЦЕПТОРОВ | 2006 |
|
RU2409577C2 |
| ПРОИЗВОДНОЕ КУМАРИНА | 2013 |
|
RU2646756C2 |
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗЫ БРУТОНА | 2014 |
|
RU2634723C2 |
| ЗАМЕЩЕННЫЕ 2-АРИЛ-3-(ГЕТЕРОАРИЛ) ИМИДАЗО [1,2-А] ПИРИМИДИНЫ, СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СВЯЗАННЫЕ С НИМИ СПОСОБЫ | 2000 |
|
RU2264403C2 |
Лекарственное средство содержит в качестве активного компонента производное аминостилбазола формулы [А] или его соль. Предпочтительным соединением является (Е)-4-{2-[2-{N-ацетил-Н-[(4-метоксифенил)сульфонил]амино}фенил]этенил}пиридин 1-оксид. Технический результат заключается в реализации указанного назначения: средство предназначено для лечения хронического суставного ревматизма. 3 н. и 4 з.п. ф-лы, 2 табл.

(где G представляет фенил, необязательно замещенный С1-6алкокси, R представляет (1) водород, (2) C1-6гидроксиалкил, или (3) -COR°, R° представляет C1-6алкил, C1-6алкокси, фенокси).

где G представляет фенил, необязательно замещенный C1-6алкокси, R представляет (1) водород, (2) C1-6 гидроксиалкил, или (3) -COR°, R° представляет C1-6алкил, C1-6алкокси, фенокси.
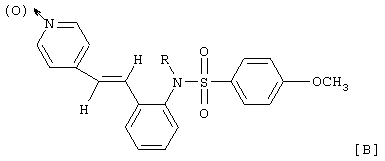
где R означает водород, C1-6гидроксиалкил или -COR°, R° представляет C1-6алкил, C1-6алкокси, фенокси.
(Е)-4-[2-{2-[N-ацетил-N-(4-метоксибензолсульфонил)амино]-фенил}этенил]пиридин 1-оксида,
(Е)-4-[2-{2-[N-ацетил-N-(4-метоксибензолсульфонил)амино]-фенил}этенил]пиридина,
(Е)-4-[2-{2-[N-(4-метоксибензолсульфонил)амино]фенил}этенил]-пиридин 1-оксида,
(Е)-4-[2-{2-[N-(4-метоксибензолсульфонил)амино]фенил}-этенил]пиридина,
(Е)-4-[2-{2-р[N-(2-гидроксиэтил)-N-(4-метоксибензолсульфонил}-амино]фенил}этенил]пиридин 1-оксида и
(Е)-4-[2-{2-[N-(2-гидроксиэтил)-N-(4-метоксибензолсульфонил)-амино]фенил}этенил]пиридина.
| МАШКОВСКИЙ М.Д., Лекарственные средства, Москва, ООО «Новая Волна», т.1, 2001, с.168-177 | |||
| RU 94030471 A1, 20.04.1996 | |||
| ПРОИЗВОДНЫЕ АМИНОСТИЛЬБАЗОЛА ИЛИ ИХ ГИДРАТЫ, ИЛИ ИХ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1995 |
|
RU2138482C1 |
| ЕР 754682 A1, 22.01.1997 | |||
| WO 00/64875 A1, 02.11.2000 | |||
| Перекатываемый затвор для водоемов | 1922 |
|
SU2001A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| ХАРКЕВИЧ Д.А., ФАРМАКОЛОГИЯ, Москва, Медицина, 1987, с.47-48. | |||

