Изобретение относится к соединениям, модулирующим рецепторы глюкокортикоидов, и к их применению в терапии.
Внутриклеточные рецепторы - это класс структурно родственных белков, вовлеченных в регуляцию генов белков. Стероидные рецепторы являются подклассом таких рецепторов, включающим рецептор глюкокортикоидов (GR), рецептор прогестерона (PR), рецептор андрогена (AR), рецептор эстрогена (ER) и рецептор минералокортикоида (MR). Для регуляции генов такими рецепторами или факторами требуются внутриклеточный рецептор и соответствующий лиганд, который способен селективно связываться с рецептором таким образом, чтобы это влияло на транскрипцию генов.
Существующие модуляторы глюкокортикоидных рецепторов (глюкокортикоиды), такие как преднизолон и другие, являются очень эффективными противовоспалительными агентами, используемыми при более чем 100 синдромах в области ревматологии, гематологии, пульмонологии, дерматологии, гастроэнтерологии, эндокринологии, неврологии, нефрологии. Подвергающиеся лечению заболевания включают ревматоидный артрит (RA), воспалительные заболевания кишечника (IBD), волчанку, аллергию, астму, псориаз и множество других (J.D Baxter, Advances in Internal Medicine 45; 317-349; 2000). Предполагают, что противовоспалительные эффекты этих соединений опосредованы ингибированием экспрессии провоспалительных медиаторов, таких как молекулы адгезии, цитокины, хемокины и ферменты, согласно механизму, включающему взаимодействие лиганд-связанного GR с транскрипционными факторами. Этот механизм известен как трансрепрессия (M. Karin, Cell 93; 487-490; 1998).
Использование имеющихся стероидных глюкокортикоидов сопровождается метаболическими и прочими побочными эффектами (например, диабетом, повышенным кровяным давлением, остеопорозом, атрофией мышечной ткани и т.д.). Предполагается, что часть этих побочных эффектов опосредована прямым взаимодействием лиганд-связанного GR с глюкокортикоидзависимыми элементами (GRE) в ДНК таргетных генов и последующей индукцией экспрессии генов (J.D Baxter, Advances in Internal Medicine 45; 317-349; 2000; M. Karin, Cell 93; 487-490; 1998). Другая часть этих побочных эффектов может быть обусловлена кросс-реактивностью к другим стероидным рецепторам, таким как рецептор минералокортикоида (MR) или прогестерона (PR).
Нестероидные глюкокортикоиды не имеют сходства в молекулярной структуре со стероидами, и, таким образом, можно предположить также различия в физико-химических свойствах, параметрах фармакокинетики (PK), распределении в тканях (например, ЦНС по сравнению с периферической нервной системой) и, важнее, нестероидные глюкокортикоиды могут не иметь/иметь низкую кросс-реактивность с другими стероидными рецепторами или могут не иметь/иметь меньшие метаболические и другие побочные эффекты.
Настоящее изобретение представляет нестероидные соединения, модулирующие активность рецепторов глюкокортикоидов. Особенно, настоящее изобретение представляет высокоаффинные нестероидные соединения для взаимодействия с GR, демонстрирующие противовоспалительные эффекты in vitro и in vivo. В соответствии с настоящим изобретением предоставляются соединения, имеющие общую формулу I, или их пролекарственные формы, или их фармацевтически приемлемые соли.
Настоящее изобретение представляет нестероидные соединения, модулирующие активность рецепторов глюкокортикоидов. Особенно, настоящее изобретение представляет высокоаффинные соединения, которые являются агонистами, частичными агонистами или антагонистами рецепторов глюкокортикоидов. В соответствии с настоящим изобретением представляются соединения, имеющие общую формулу I
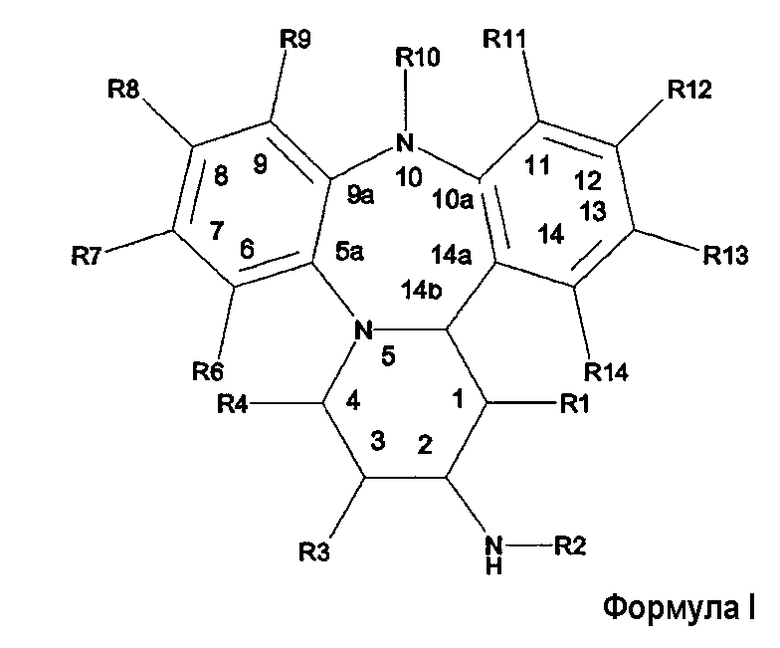
или их фармацевтически приемлемые соли.
В этой формуле R-группы имеют следующие значения:
R1 представляет собой Н или (1-4С)алкил;
R2 представляет собой -С(О)R15 или -S(O)2R15;
R3 представляет собой H, (1-4C)алкил или -OR16;
R4 представляет собой H, (1-4C)алкил или -OR16;
R6 представляет собой H или -C(H)NOR16;
R7 представляет собой H или галоген, циано-;
(1-6С)алкил, (2-6С)алкенил или (2-6С)алкинил, причем все три указанные группы необязательно замещены OH, галогеном или NH2;
-С(Н)NOR16, -OR16, -C(O)R16 или -C(O)OR16;
R8 представляет собой H, циано-, галоген, нитро-;
(1-6С)алкил, (2-6С)алкенил, (2-6С)алкинил или -О(1-6С)алкил, причем все указанные группы необязательно замещены амино, гидрокси или галогеном;
(гетеро)арил, необязательно замещенный циано-, галогеном, (1-4С)алкилом, (1-4С)алкокси, (1-4С)алкокси(1-4С)алкилом;
-C(H)NOR16, -C(O)NHR17, -C(O)R18, -C(O)OR19, -NHC(O)R20, -NHS(O)2R21 или -C(1-4C)алкилNOR21;
R9 представляет собой Н, галоген, циано- или (1-4С)алкил, необязательно замещенный галогеном;
R10 представляет собой Н или (1-4С)алкил;
R11 представляет собой H;
R12 представляет собой Н, циано или (1-4С)алкил;
R13 представляет собой Н, (1-4С)алкил, галоген или формил;
R14 представляет собой Н, галоген, циано-, (1-4С)алкил, (2-6С)алкенил, C(O)R21 или (гетеро)арил;
R15 представляет собой H;
(1-6С)алкил, (2-6С)алкенил, (2-6С)алкинил, -O(2-6C)алкил, -О(2-6С)алкенил или О(2-6С)алкинил, причем все указанные группы необязательно замещены одним или более ОН, галогеном, циано- или (гетеро)арилом;
(гетеро)арил, необязательно замещенный (1-4С)алкилом, галогеном, циано-, нитро- или амино-; NH2, (ди)(1-4С)алкиламино, (1-4С)алкил(1-4С)алкоксиамин, (1-4С)алкилтио(1-4С)алкил или (1-4С)алкокси(1-4С)алкил;
R16 представляет собой Н, (1-6С)алкил, (2-6С)алкенил или (2-6С)алкинил;
R17 представляет собой H,
(1-6С)алкил, необязательно замещенный галогеном, (1-4С)алкокси или (гетеро)арилом, необязательно замещенным галогеном, (1-4С)алкилом или (1-4С)алкокси;
(3-6С)циклоалкил или
(гетеро)арил, необязательно замещенный галогеном, (1-4С)алкилом или (1-4С)алкокси;
R18 представляет собой H, NH2, -C(O)R21 или (1-4С)алкил, необязательно замещенный ОН, галогеном, циано- или -S(1-4C)алкилом;
R19 представляет собой Н или (1-6С)алкил, необязательно замещенный ОН или галогеном;
R20 представляет собой H,
(1-6С)алкил или (2-6С)алкенил, которые необязательно замещены галогеном, О(1-6С) алкилом, (гетеро)арилом, необязательно замещенным (1-4С)алкилом или галогеном;
(3-6С)циклоалкил (1-6С)алкокси; (1-6С)алкенилокси; или (гетеро)арил, необязательно замещенный (1-4С)алкилом, NH2, -NH(1-6С)алкилом или -NH(гетеро)арилом;
и
R21 представляет собой Н или (1-6С)алкил.
Так, было обнаружено, что вышеупомянутый класс соединений, соответствующих формуле I, или их фармацевтически приемлемые соли имеют активность модуляторов рецепторов глюкокортикоидов.
Термин «(1-6С)алкил», как использовано в определении изобретения, означает разветвленную или неразветвленную алкильную группу, состоящую из 1-6 атомов углерода, например, метил, этил, пропил, изопропил, бутил, втор-бутил и трет-бутил, пентил и гексил. Предпочтителен (1-4С)алкил.
Термин «(1-4С)алкил», как использовано в определении изобретения, означает разветвленную или неразветвленную алкильную группу, состоящую из 1-4 атомов углерода, например, метил, этил, пропил, изопропил, бутил, втор-бутил и трет-бутил. Предпочтительны метил и этил. Наиболее предпочтителен метил.
Термин «(3-6С)циклоалкил» означает циклическую алкильную группу, состоящую из 3-6 атомов углерода.
Термин «галоген» означает фтор, хлор, бром или йод.
Термин «(2-6С)алкенил» означает разветвленную или неразветвленную алкенильную группу, состоящую из 2-6 атомов углерода, такую как этенил, 2-бутенил, пентенил и гексенил. Предпочтителен (2-4C)циклоалкенил.
Термин «(2-4C)алкенил» означает разветвленную или неразветвленную алкенильную группу, состоящую из 2-4 атомов углерода, такую как этенил и 2-бутенил.
Термин «(2-6С)алкинил» означает разветвленную или неразветвленную алкинильную группу, состоящую из 2-6 атомов углерода, такую как этинил, пропинил, бутилил, пентинил и гексинил. Предпочтителен (2-4С)алкинил.
Термин «(2-4С)алкинил» означает разветвленную или неразветвленную алкинильную группу, состоящую из 2-4 атомов углерода, такую как этинил и пропинил.
Термин «-О(1-6С)алкил» означает (1-6С)алкокси, где (1-6С)алкил имеет вышеуказанное значение.
Термин «-О(2-6С)алкенил» означает (2-6С)алкенилокси, где (2-6С)алкенил имеет вышеуказанное значение.
Термин «-О(2-6С)алкинил» означает (2-6С)алкинилокси, где (2-6С)алкинил имеет вышеуказанное значение.
Термин «(1-4С)алкилокси» означает алкилоксигруппу, состоящую из 1-4 атомов углерода, в которой алкильная часть имеет вышеуказанное значение; (1-2С)алкилоксигруппы предпочтительны.
Термин «(1-4С)алкокси(1-4)алкил» означает (1-4С)алкокси, присоединенный к (1-4С)алкильной группе; обе группы имеют ранее определенные значения.
Термин «(ди)(1-4С)алкиламино» означает аминокомпонент с по меньшей мере одним, необязательно двумя, атомами водорода, замещенным на (1-4С)алкильную группу, как определено ранее.
Термин «-S(1-4C)алкил» означает (1-4С)алкилтиогруппу, где (1-4С)алкильная группа имеет ранее определенное значение.
Термин «(1-4С)алкилтио(1-4С)алкил» означает (1-4С)алкилтиогруппу, присоединенную к (1-4С)алкильной группе; обе группы имеют ранее определенные значения.
Термин «арил» означает 6-членную кольцевую ароматическую систему.
Термин «гетероарил» означает 5- или 6-членную кольцевую ароматическую систему, содержащую по меньшей мере один гетероатом, выбираемый из группы, состоящей из N, O и S в 5-членном кольце и N - в 6-членном кольце, таком как пиридил, пиримидил, тетразол или тиадиазол.
Термин «(гетеро)арил» означает арил или гетероарил, как ранее обозначено.
Термин «фармацевтически приемлемая соль» представляет те соли, которые являются, с точки зрения специалиста в области медицины, подходящими для использования в контакте с тканями человека или низших животных без излишней токсичности, раздражения, аллергических реакций и подобного, и соответствуют разумному отношению «польза/риск». Фармацевтически приемлемые соли хорошо известны в соответствующей области. Они могут быть получены в процессе окончательного выделения и очистки соединений изобретения, или отдельно в реакции свободной основной функции с подходящими неорганическими кислотами, такими как соляная кислота, фосфорная кислота или серная кислота, или органическими кислотами, такими как, например, аскорбиновая кислота, лимонная кислота, винная кислота, молочная кислота, малеиновая кислота, малоновая кислота, фумаровая кислота, гликолевая кислота, янтарная кислота, пропионовая кислота, уксусная кислота, метансульфоновая кислота и им подобные. Кислотная функция может использоваться в реакции с органическими или неорганическими основаниями, такими как гидроксид натрия, гидроксид калия или гидроксид лития.
Соединения настоящего изобретения обладают по меньшей мере двумя хиральными атомами и могут, таким образом, быть получены в виде чистых энантиомеров или смеси энантиомеров, или смеси диастереомеров. Методы получения чистых энантиомеров известны в данной области, например, кристаллизация солей, полученных из оптически активных кислот и рацемических смесей, или хроматография на хиральных колонках. Для разделения диастереомеров могут быть использованы обычные или обращеннофазные колонки.
Изобретение также касается соединений, соответствующих формуле I, где R7 представляет собой H, галоген или -OR16.
Изобретение также касается соединений, соответствующих формуле I, где R2 представляет собой С(O)R15.
В еще другом аспекте изобретение касается соединений, соответствующих формуле I, где R7 представляет собой H.
Изобретение далее касается тех соединений, где R10 в формуле I является метилом.
Изобретение также касается тех соединений, где R4 в формуле I представляет собой Н или (1-4С)алкил.
Другой аспект изобретения касается соединений, соответствующих формуле I, где R16 представляет собой Н или (1-6С)алкил.
В другом аспекте изобретение касается соединений, где R14 представляет собой (1-4С)алкил.
В еще другом аспекте изобретение касается соединений, где R15 представляет собой (1-4)алкил, необязательно замещенный галогеном, циано-, нитро- или амино.
В еще другом аспекте изобретение касается соединений, где R15 представляет собой (1-4)алкил, необязательно замещенный галогеном.
В еще другом аспекте изобретение касается соединений формулы I, где R15 представляет собой трифторметил или (гетеро)арил, необязательно замещенный (1-4)алкилом.
Изобретение также касается соединений формулы I, где R15 представляет собой (гетеро)арил, необязательно замещенный (1-4С)алкилом.
В дальнейшем аспекте R21 в соединениях, соответствующих формуле I, представляет собой (1-4С)алкил.
Изобретение также касается соединений, соответствующих формуле I, где R8 представляет собой Н, галоген, циано-, нитро, -C(O)R18, -NHC(O)R20 или (гетеро)арил, необязательно замещенный циано-, (1-4С)алкилом, (1-4С)алкокси, (1-4С)алкокси(1-4С)алкилом или (гетеро)арилом.
Изобретение также касается соединений, соответствующих формуле I, где R8 представляет собой H, галоген, циано-, нитро, -C(O)R18, -NHC(O)R20 или (гетеро)арил, необязательно замещенный циано-, (1-4С)алкилом, (1-4С)алкокси, (1-4С)алкокси(1-4С)алкилом.
Изобретение, более того, касается тех соединений, где R8 представляет собой H, циано-, пиридил- или нитро.
В другом аспекте изобретение касается соединений, где R8 представляет собой циано-, пиридил- или нитро. Изобретение также касается соединений, где R8 представляет собой циано.
В еще одном аспекте изобретение касается соединений, соответствующих формуле I, где R1, R3, R4, R6, R7, R9, R11, R12, R13 и R14 представляют собой Н.
Еще один аспект изобретения касается соединений, где все специфические определения групп R1-R21, определенные здесь выше, объединены в соединении формулы I.
Изобретение также предоставляет соединения, соответствующие формуле I, высоко специфичные к рецепторам глюкокортикоидов. Специфичность может быть определена тестированием соединений, как описано далее для рецепторов глюкокортикоидов, для других хорошо известных рецепторов, таких как рецептор прогестерона, рецептор андрогена, рецептор минералкортикоидов или рецептор эстрогена.
Синтез
Последовательность стадий путем синтеза соединений данного изобретения описана на схеме 1.
Соединения настоящего изобретения могут быть получены путем, во-первых, сочетания 2-галогеннитроарилов общей структуры 1, где Х имеет ранее определенное для галогенов значение, с производными (N-алкил)анилина.
Вышеупомянутую реакцию обычно проводят при повышенной температуре в присутствии карбоната калия и в присутствии или в отсутствие органического растворителя. 2-галогеннитроарилы общей структуры 1 либо являются коммерчески доступными или легко получаются синтетическим путем, хорошо известным из литературы. Эта реакция описана в литературе G.W. Rewcastle, et al. J. Med. Chem., 30, 1987, 843. Альтернативно эти реакции могут быть проведены в присутствии карбоната цезия, ацетата палладия и BINAP для получения аналогичных продуктов.
Схема 1

Соединения общей структуры 2 могут затем быть восстановлены для получения соединений общей структуры 3.
Вышеупомянутую реакцию обычно проводят при температуре окружающей среды в присутствии хлорида олова(II) в органических растворителях с последующей обработкой гидроксидом.
Соединения общей структуры 3 затем могут быть N-формилированы для получения соединений общей структуры 4.
Вышеупомянутую реакцию обычно проводят при кипячении в муравьиной кислоте с обратным холодильником без использования какого-либо органического растворителя.
Соединения общей формулы 4 могут затем подвергаться реакции замыкания цикла для образования кольца С и получения трициклических соединений общей структуры 5.
Вышеупомянутую реакцию обычно проводят при температуре окружающей среды в присутствии пентахлорида фосфора, с использованием органических растворителей. Альтернативно эти реакции могут быть проведены в присутствии полифосфорной кислоты и трихлорида оксида фосфора (V) без использования каких-либо органических растворителей для получения желаемого продукта.
Соединения общей структуры 5 могут затем подвергаться реакции типа гетеро-Дильса-Альдера для формирования пиперинового кольца и получения тетрациклических соединений. Эти соединения могут быть затем восстановлены in situ для получения тетрациклических спиртов общей структуры 6, получаемых в основном в транс-конфигурации.
Реакцию Дильса-Альдера обычно проводят при пониженной температуре в присутствии либо триметил[(1-метилен-2-пропенил)окси]силана, триметил[(1-метилен-2-бутенил)окси]силана, либо [(3-метокси-1-метилен-2-пропенил)окси]триметилсилана (диена Данишевского) и трифторметансульфоната иттербия при использовании органического растворителя. Эти сырые продукты могут затем быть восстановлены при температуре окружающей среды в присутствии борогидрида натрия с использованием органического растворителя.
Соединения общей структуры 6 могут затем подвергаться реакции при условиях Мицунобу для получения азидных соединений общей структуры 7.
Вышеупомянутую реакцию обычно проводят при температуре окружающей среды в присутствии трифенилфосфина, диизопропилазодикарбоксилата и дифенилфосфорилазида с использованием органического растворителя.
Соединения общей структуры 7 могут быть затем восстановлены для получения свободных аминосоединений общей структуры 8. Эту реакцию обычно проводят при температуре окружающей среды в присутствии трифенилфосфина и воды с использованием органического растворителя. Эти соединения могут быть затем конденсированы посредством обычных процедур с производными карбоновых кислот (кислотами, хлорангидридами или эфирами кислот) для получения амидных продуктов 9.
Соединения 6, 8 и 9 являются ключевыми промежуточными соединениями при образовании всех остальных описанных здесь соединений. Функционализация этих ключевых промежуточных соединений может быть осуществлена путем выбора подходящих исходных веществ или галогенированием, нитрованием, формилованием и т.д. и модифицирована далее при помощи описываемых методов (например, методов Бухвальда, Сузуки, Стилле, ароматического замещения и т.д.) для получения желаемого соединения 9 с желаемой цис-стехиометрией.
Соединения настоящего изобретения имеют по меньшей мере два стереогенных атомов углерода и могут, таким образом, быть получены как чистые энантиомеры, смесь энантиомеров или смесь диастереоизомеров. Как правило, их выделяют в виде смеси энантиомеров. Диастереоизомеры могут быть разделены при использовании обычной или обращеннофазовой хроматографии. Методы получения чистых энантиомеров хорошо известны в соответствующей области, например, в хроматографии с использованием хиральных колонок.
Биологическая активность
Методы определения связывания с рецепторами, так же как и исследования биологической активности соединений in vitro и in vivo, хорошо известны. В целом экспрессируемый рецептор обрабатывают тестируемым соединением, после чего измеряют связывание, стимулирование или ингибирование функционального ответа.
Для измерения связывания может быть использован изолированный цитозоль, содержащий экспрессированный GR. Также могут быть использованы радиоактивно или флуоресцентно меченные соединения. В качестве контрольного соединения может быть использован природный гормон или иные связывающиеся с рецептором соединения. В качестве альтернативы может также применяться метод конкурентного связывания. Эти подходы для определения связывания могут быть либо разработаны на месте, либо заказаны в виде коммерчески доступных исследований (китов). Экспериментальные методики определения способности к связыванию (аффинности) хорошо известны в области техники.
Для отбора модуляторов GR способность связывания соединений с рецептором должна быть ниже 10-5 М. Более предпочтительна аффинность <10-7 М и, наиболее предпочтительна, аффинность <10-8 М.
Для измерения функционального ответа изолированная ДНК, содержащая кодирующий рецептор глюкокортикоидов ген, предпочтительно рецептор человека, экспрессируется в подходящей линии клеток, например, линии клеток человеческой остеобластомы U2OS.
Методы получения клеточной линии, экспрессирующей рекомбинантный рецептор глюкокортикоидов, хорошо известны в области техники. Экспрессия рецептора осуществляется при экспрессии ДНК, кодирующей желаемый белок. На данный момент все методики сайт-направленного мутагенеза, лигирования дополнительных последовательностей, ПЦР и конструирования подходящей экспрессионной системы хорошо известны в области техники. Полноразмерная ДНК или ее фрагменты, кодирующие желаемый белок, могут быть сконструированы синтетически при использовании стандартных твердофазных методов, предпочтительно для включения рестрикционых сайтов для облегчения лигирования. Подходящие контрольные элементы для транскрипции и трансляции включенных кодирующих последовательностей могут быть вставлены в кодирующие последовательности ДНК. Как хорошо известно, на данный момент доступны экспрессионные системы, сочетающиеся с широким спектром организмов-хозяев: прокариотических систем, таких как бактерии, эукариотических систем, таких как дрожжи, растительные клетки, клетки насекомых, клетки млекопитающих, клетки птиц и им подобные.
In vitro, для линии человеческих клеток, стабильно трансфицированных ДНК GR человека, может быть моделировано воспаление, что стимулирует секрецию цитокинов, хемокинов и других воспалительных медиаторов. Противовоспалительный эффект соединений может быть оценен количественно при измерении воспалительного ответа в этих линиях клеток. EC50 может быть рассчитано и для тестируемого соединения, и для контрольного соединения, такого как преднизолон, при исследовании кривой зависимости ответа от полной дозы вещества. Значение EC50 можно сравнить со значением EC50 для преднизолона, полученным в том же клеточном анализе. Предпочтительно, соединения имеют значения EC50, сопоставимые со значением EC50, полученным для преднизолона. Более предпочтительно, значения EC50 ниже полученных для преднизолона.
Специалист в данной области поймет, что желаемое значение EC50 зависит от тестируемого соединения. Например, соединение со значением EC50, меньшим, чем 10-5 М, в целом, рассматривается в качестве кандидата для селекции лекарства. Предпочтительно, это значение меньше, чем 10-7 М. Однако соединение, которое имеет более высокое значение EC50, но селективно для определенного типа рецепторов, может быть даже лучшим кандидатом.
In vivo, противовоспалительный эффект соединений может быть исследован на мышах, которым вводили липополисахарид (LPS). Соединения могут быть введены системно во время или после введения LPS. Противовоспалительный эффект может быть количественно оценен как ингибирование в сыворотке крови мышей LPS-индуцированного TNFα или любого другого воспалительного цитокина или хемокина (S.R. Hyde & R.E. McCallum, Infection and Immunity, 60; 976-982 (1992)). Способность ингибировать развитие артрита может быть исследована на мышиной модели артрита, индуцированного коллагеном 2-го типа - (CIA) как способность ингибирования набухания конечностей (D.E. Trentham et al. J. exp. Med. 146; 857-868 (1997)) или на других моделях артрита.
Изобретение также относится к фармацевтическим композициям, содержащим соединения или их соли, имеющие общую формулу I. Так, соединения, соответствующие формуле I, могут быть использованы в терапии.
Подходящие пути введения соединений формулы I или их фармацевтически приемлемых солей, также обозначенные здесь как активные компоненты, представляют собой внутримышечные инъекции, подкожные инъекции, внутривенные инъекции или внутрибрюшинные инъекции, пероральное или интраназальное введение. Предпочтительно, соединения могут быть введены перорально. Точная доза и режим применения активного компонента или фармацевтических композиций на его основе будут обязательно зависеть от желаемого эффекта, которого требуется достичь (например, лечение астмы, R.A, I.B.D), и могут варьировать для индивидуальных соединений, пути введения и возраста, и состояния индивидуального субъекта, которому должно быть введено лекарственное средство. В целом терапевтически эффективная ежедневная доза составляет от примерно 0,001 мг до примерно 15 мг/кг массы тела в день для каждого соединения изобретения; предпочтительно, от примерно 0,01 мг до примерно 10 мг/кг массы тела в день; и, более предпочтительно, от примерно 0,1 до примерно 1,5 мг/кг массы тела в день. Определенная степень обычной оптимизации дозы может требоваться для определения оптимального уровня и режима дозирования.
Дальнейший аспект изобретения относится к применению соединений, соответствующих формуле I или их фармакологически приемлемых солей, или их сольватов для изготовления лекарственного средства для всех заболеваний, где требуется модуляция рецепторов глюкокортикоидов, то есть в области ревматологии, гематологии, пульмонологии, дерматологии, гастроэнтерологии, эндокринологии, неврологии или нефрологии, которые на данный момент поддаются лечению стероидными глюкокортикоидами, такими как преднизолон. Наиболее предпочтительна область ревматологии, в особенности, например, ревматоидный артрит.
Так, соединения настоящего изобретения модулируют активность рецептора глюкокортикоидов, и они могут быть использованы при лечении иммунологических и воспалительных заболеваний. В особенности, соединения могут быть использованы для лечения ревматических заболеваний, таких как ревматоидный артрит, юношеский артрит и анкилозирующий спондилит, дерматологические заболевания, включая псориаз и пузырчатку, аллергические заболевания, включая аллергический ринит, атопический дерматит, контактный дерматит, легочные состояния, включая астму и хроническое обструктивное заболевание легких, и другие иммунные и воспалительные заболевания, включая болезнь Крона, неспецифический язвенный колит, системную красную волчанку, аутоиммунный хронический активный гепатит, остеоартрит, тендинит и бурсит. Кроме того, соединения могут быть использованы для предотвращения отторжения органов после трансплантации.
Более конкретно соединения могут быть использованы для лечения ревматоидного артрита, псориаза, астмы и хронического обструктивного заболевания легких, болезни Крона или неспецифического язвенного колита, и соединения могут быть использованы для предотвращения отторжения органов послетрансплантации.
Так, соединения по изобретению могут быть использованы при лечении этих заболеваний, то есть всех заболеваний, где пациенту требуется модуляция рецептора глюкокортикоидов.
Примеры
Пример 1
NB: Нумерация в примерах относится к схеме 1, где R1, R3, R4, R6-R9=Н, R10=Me, R11-R14=H, если не обозначено иначе.
Пример 1
Цис-2,2,2-трифтор-N-(8-формил-1,2,3,4,10,14b-гексагидро-10-метилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)ацетамид (9:R2-CF3-C(O)-; R8=CH(O)-)
Перемешиваемый раствор 2-бромнитробензола (1) (301 г, 1,5 моль) и N-метиланилина (176,5 г, 1,65 моль) в толуоле (2,5 л) дегазировали пропусканием N2 в течение 15 минут. Затем добавляли при перемешивании карбонат цезия (537 г, 1,65 моль), Pd(OAC)2 (973 мг, 4,3 ммоль) и рац-BINAP (15,1 г, 24,3 ммоль), реакционную смесь нагревали до 85°С и выдерживали в течение 20 часов. Реакцию останавливали H2O и промывали органический слой с 6М НСl, затем H2O и, наконец, насыщенным рассолом, после чего обезвоживали (Na2SO4). Органическую фазу концентрировали при пониженном давлении, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле для получения (2) (341 г, 99%). Данные (m/z)=229 (M+H)+.
К перемешиваемому раствору соединения (2) (100 г, 0,5 моль) в этаноле (1 л) добавляли SnCl2·2H2O (451 г, 2,0 моль) и смесь перемешивали при температуре окружающей среды в течение ночи. Спиртовой раствор разделяли на 4 части и каждую выливали в 6М NaOH в H2O (1 л). Смесь перемешивали до обесцвечивания раствора, после чего продукт экстрагировали EtOAc. Органические фазы объединяли и промывали насыщенным рассолом, после чего обезвоживали (Na2SO4). Органическую фазу концентрировали при пониженном давлении и сырой продукт очищали с помощью колоночной хроматографии на силикагеле для получения (3) (78 г, 79%). Данные: (m/z)=199 (M+H)+.
Перемешиваемый раствор соединения (3) (78 г, 0,39 моль) в муравьиной кислоте (500 мл) кипятили с обратным холодильником и выдерживали в течение 20 часов. Муравьиную кислоту удаляли при пониженном давлении, полученное масло растворяли в EtOAC. Органический слой промывали раствором NаНСО3, затем Н2О и, наконец, насыщенным рассолом, после чего обезвоживали (Na2SO4). Органическую фазу концентрировали при пониженном давлении и сырой продукт очищали с помощью колоночной хроматографии на силикагеле для получения (4) (61 г, 69%). Данные: (m/z)=227 (M+H)+.
К перемешиваемому раствору соединения (4) (61 г, 270 ммоль) в DCM (500 мл) частями добавляли PCl5 (56,3 г, 270 ммоль). Реакционную смесь перемешивали в течение 1 часа при температуре окружающей среды, после чего выливали в раствор МаНСО3 в Н2О (1 л). рН реакционной смеси доводили твердым NaHСО3 до основных значений по лакмусовой бумаге. Органическую фазу отделяли и концентрировали при пониженном давлении. Полученное масло растворяли в Et2O. Затем добавляли 6М раствор НСl в H2O, после чего смесь перемешивали в течение 30 минут. Водную фазу отделяли, органическую фазу дважды промывали 6М раствором НСl в Н2О. Водные фракции объединяли, промывали Et2O, после чего нейтрализовали. Продукт экстрагировали EtOAc и промывали насыщенным рассолом, обезвоживали (Na2SO4) и концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией на силикагеле для получения (5) (30 г, 54%). Данные: (m/z)=209 (М+Н)+.
Перемешиваемый раствор вещества (5) (30 г, 144 ммоль) в толуоле (350 мл) охлаждали до 0°С, после чего добавляли диен Данишевского (28,4 г, 144 ммоль) и Yb(OTf)3 (4,47 г, 7,2 ммоль). Раствор оставляли нагреваться до комнатной температуры и перемешивали в течение 2 часов. Реакцию останавливали 0,1 М НСl в Н2О. Добавляли воду и экстрагировали продукт толуолом. Органическую фазу промывали насыщенным рассолом, обезвоживали (Na2SO4), затем концентрировали при пониженном давлении для получения 10,14b-дигидро-10-метил-дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2(1Н)она (47 г, сырой). Это соединение суспендировали в этаноле (1 л), затем добавляли NaBH4 (21,8 г, 576 ммоль) и реакционную смесь перемешивали в течение 8 часов при температуре окружающей среды. Органическую фазу частично выпаривали при пониженном давлении, полученную суспензию выливали в насыщенный раствор NH4Cl в Н2О, после чего экстрагировали EtOAc. Органическую фазу промывали насыщенным раствором, обезвоживали (Na2SO4) и концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией на силикагеле для получения (6) (23,4 г, 58%). Данные: (m/z)=281 (М+Н)+.
Перемешиваемый раствор вещества (6) (10,0 г, 35,7 ммоль) и трифенилфосфина (12,2 г, 46,4 ммоль) в безводном ТГФ (150 мл) охлаждали до 0°С и по каплям добавляли диизопропилазодикарбоксилат (9,2 мл, 46,4 ммоль). По каплям добавляли дифенилфосфорилазид (10,0 мл, 46,4 ммоль), после чего снимали охлаждение. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении и сырой продукт очищали колоночной хроматографией на силикагеле для получения (7) (13,1 г, 100%). Данные: (m/z)=306 (M+H)+.
К перемешиваемому раствору вещества (7) (10,9 г, 35,7 ммоль) и трифенилфосфина (13,4 г, 51,1 ммоль) в ТГФ (150 мл)добавляли Н2О (2 мл). Реакционную смесь затем перемешивали в течение 24 часов при температуре окружающей среды, после чего концентрировали при пониженном давлении для получения (8) (35 г, сырое). Данные: (m/z)=280 (M+H)+.
Неочищенное вещество (8) растворяли в МеОН (400 мл), перемешивали, затем добавляли триэтиламин (19,4 мл, 140 ммоль) и этилтрифторацетат (20,9 мл, 175 ммоль) и нагревали реакционную смесь до 50°С в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении, затем очищали колоночной хроматографией на силикагеле для получения цис-2,2,2-трифтор-N-(1,2,3,4,10,14b-гексагидро-10-метилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)ацетамида (9:R2=СF3-С(О)-) (5,7 г, 58% для двух стадий). Данные: (m/z)=376 (M+H)+.
Оксалил хлорид (436 мкл, 5 ммоль) осторожно добавляли по каплям, при перемешивании, к ДМФ (0,5 мл) при 0°С и выдерживали в течение 25 минут. Раствор цис-2,2,2-трифтор-N-(1,2,3,4,10,14b-гексагидро-10-метилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)ацетамида (9:R2=СF3-С(О)-) (400 мг, 1,07 ммоль) в ДМФ (2 мл) добавляли по каплям к полученной суспензии, реакционную смесь нагревали до 80° и выдерживали в течение 1,5 часов. Реакцию охлаждали до температуры окружающей среды и останавливали добавлением по каплям раствора NаНСО3 в H2O. Продукт экстрагировали EtOAc, обезвоживали (Na2SO4) и концентрировали при пониженном давлении. Затем сырой продукт очищали колоночной хроматографией на силикагеле для получения цис-2,2,2-трифтор-N-(8-формил-1,2,3,4,10,14b-гексагидро-10-метилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)ацетамида (9:R2=СF3-С(O)-; R8=CHO) (130 мг, 30%). Данные: (m/z)=404 (М+Н)+. [При хроматографии также получается цис-2,2,2-трифтор-N-(6-формил-1,2,3,4,10,14b-гексагидро-10-метилдибензо[b, f]пиридо[1,2-d][1,4]диазепин-2-ил)ацетамид (9:R2=СF3-С(О)-; R6=CH(O)-) (45 мг, 10%). Данные: (m/z)=404 (М+Н)+.]
Пример 2
Цис-2,2,2-трифтор-N-(1,2,3,4,10,14b-гексагидро-10-метил 8-нитродибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)ацетамид (9:R2=СF3-C(O)-; R8=NO2-)
Перемешиваемый раствор TFAA (188 мкл, 1,33 ммоль) и TBAN (406 мг, 1,33 ммоль) в DCM (5 мл) охлаждали до 0°С и перемешивали в течение 20 минут, после чего по каплям добавляли к раствору вещества (9:R2=СF3-С(О)-) (250 мг, 0,67 ммоль) в DCM (5 мл). Реакционную смесь перемешивали в течение 1,5 часов при 0°С, затем останавливали (при 0°С) раствором NaHСО3 в H2O. Органическую фазу отделяли и промывали Н2О, затем насыщенным рассолом, затем обезвоживали (РЕ-фильтр) и концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией на силикагеле для получения цис-2,2,2-трифтор-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-нитродибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)ацетамида (9: R2=CF3-C(О)-; R8=NO2-) (110 мг, 42%). Данные: (m/z)=421 (М+Н)+.
Пример 3
Цис-N-(8-циано-1,2,3,4,10,14b-гексагидро-10-метилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2,2,2-трифторацетамид (9:R2=CF3-C(O)-; R8=CN)
Перемешиваемый раствор (6) (12,7 г, 45,4 ммоль) в ацетоне (175 мл) охлаждали до 0°С и по частям добавляли N-бромсукцинимид (10,4 г, 58,4 ммоль). Раствор оставляли нагреваться до температуры окружающей среды и перемешивали в течение 3 часов. Затем реакцию останавливали раствором NaHCO3 в Н2О и экстрагировали продукт EtOAc. Органическую фазу промывали насыщенным рассолом, обезвоживали (Na2SO4) и концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией на силикагеле для получения вещества (6:R8=Br) (11,0 г, 67%). Данные: (m/z)=360 (M+H)+.
(7:R8=Br). Это соединение было получено аналогично способу, описанному в примере 1 (см. ниже). Продукт не очищали и использовали на следующей стадии как таковой. Данные: (m/z)=385 (M+H)+.
(9:R2=CF3-C(О)-; R8=Br). Это соединение было получено аналогично способу, описанному в примере 1 (35% для трех стадий). Данные: (m/z)=455 (M+H)+.
Раствор вещества (9:R2=CF3-С(О)-; R8=Br) (2 г, 4,4 ммоль) и CuCN (1 г, 11 ммоль) был дегазован при пропускании азота в течение 0,5 часов. Реакционную смесь затем нагревали до 200°С и поддерживали при этой температуре в течение 4 часов при перемешивании. Реакцию останавливали раствором NH4OH в воде, затем фильтровали. Продукт экстрагировали EtOAc и органическую фазу промывали Н2O, затем обезвоживали (Na2SO4). Органическую фазу концентрировали при пониженном давлении и сырой продукт очищали колоночной хроматографией на силикагеле для получения цис-N-(8-циано-1,2,3,4,10,14b-гексагидро-10-метилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2,2,2-трифторацетамида (9:R2=CF3-С(О)-; R8=CN) (1,1 г, 62%). Данные: (m/z)=401 (M+H)+.
Пример 4
Цис-1,2,3,4,10,14b-гексагидро-10-метил-N-фенил-2-(2,2,2-трифторацетиламино)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-8-карбоксамид (9: R2=CF3-C(О)-; R8=Ph-NH-C(О)-)
К перемешиваемому раствору цис-N-(8-циано-1,2,3,4,10,14b-гексагидро-10-метилдибензо[b,f]пиридо[1,2-d][1,4] диазепин-2-ил)-2,2,2-трифторацетамида (9:R2=CF3-С(О)-; R8=CN) (900 мг, 2,25 ммоль) в EtOH (50 мл) добавляли 6N КОН (20 мл). Реакционную смесь нагревали до 120°С и выдерживали в течение 1,5 часов, используя микроволновую печь (120 Вт). Реакционную смесь охлаждали до температуры окружающей среды, нейтрализовали с 2М НСl в H2O и удаляли растворитель при сушке сублимацией. Сырой продукт растворяли в МеОН (50 мл), после чего добавляли триэтиламин (1,5 мл, 10,8 ммоль) и этилтрифторацетат (1,4 мл, 11,7 ммоль). Реакционную смесь нагревали до 50°С и поддерживали при этой температуре в течение 3 часов, после чего охлаждали до температуры окружающей среды. Продукт выделяли кислотно-щелочной экстракцией и органическую фазу промывали Н2О, затем насыщенным рассолом, затем обезвоживали (Na2SO4) и концентрировали при пониженном давлении для получения сырого вещества (9:R2=CF3-С(О)-; R8=CO2H) (737 мг, 78% для двух стадий). Данные: (m/z)=420 (М+Н)+.
К перемешиваемому раствору вещества (9: R2=CF3-С(О)-; R8=CO2H) (30 мг, 0,72 ммоль) в ДМФ (1 мл)добавляли TBTU (34,5 мг, 0,108 ммоль) и DIPEA (68,3 мкл, 0,360 ммоль) и перемешивали смесь в течение 10 минут. Затем добавляли анилин (7,2 мкл, 0,792 ммоль), перемешивали реакционную смесь при температуре окружающей среды в течение 70 часов. Реакцию затем останавливали раствором Na2CO3 в H2O и экстрагировали продукт с DCM. Органическую фазу обезвоживали (Na2SO4) и концентрировали при пониженном давлении, после чего сырой продукт очищали колоночной хроматографией на силикагеле для получения цис-1,2,3,4,10,14b-гексагидро-10-метил-N-фенил-2-(2,2,2-трифторацетиламино)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-8-карбоксамида (9:R2=CF3-С(О)-; R8=Ph-NH-C(О)-) (25 мг, 70%). Данные: (m/z)=495 (M+H)+.
Пример 5
Цис-2,2,-дихлор-N-(8-циано-1,2,3,4,10,14b-гексагидро-10-метилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)ацетамид (9:R2-CHCl2-C(O)-; R8=CN)
К перемешиваемому раствору цис-N-(8-циано-1,2,3,4,10,14b-гексагидро-10-метилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2,2,2-трифторацетамида (9:R2=CF3-С(О)-; R8=CN) (900 мг, 2,25 ммоль) в EtOH (50 мл) добавляли 2N NaOH (12 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 3 часов, после чего продукт выделяли кислотно-щелочной экстракцией и органическую фазу, промывали Н2О, затем насыщенным рассолом, обезвоживали (Na2SO4) и концентрировали при пониженном давлении для получения сырого вещества (8:R8=CN) (687 мг, 100%). Данные: (m/z)=305 (M+H)+.
Перемешиваемый раствор вещества (8:R8=CN) (20 мг, 0,066 ммоль) и триэтиламина (5,75 мкл, 0,069 ммоль) в DCM (0,5 мл) охлаждали до 0°С, после чего по каплям добавляли раствор дихлорацетилхлорида (6 мкл, 0,069 ммоль). Реакцию перемешивали в течение 2 часов, затем останавливали раствором Na2СО3 в Н2О и экстрагировали продукт DCM. Органическую фазу обезвоживали (Na2SO4) и концентрировали при пониженном давлении, затем сырой продукт очищали колоночной хроматографией на силикагеле для получения цис-2,2-дихлор-N-(8-циано-1,2,3,4,10,14b-гексагидро-10-метилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)ацетамида (9:R2=CHCl2-C(O)-; R8=CN) (20 мг, 81%). Данные: (m/z)=415 (M+H)+.
Пример 6
Цис-N-[1,2,3,4,10,14b-гексагидро-10-метил-N-(2,2,2-трифторацетиламино)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-8-ил]-2-метилпропанамид (9:R2=CF3-С(О)-;
R8=CH3 (CH3) CH-C(О)-NH-)
К перемешиваемому раствору вещества (9:R2=CF3-С(О)-; R8=Br) (5,16 г, 11,36 ммоль), Pd2(dba)3 (0,1 г), 2-(ди-трет-бутилфосфино)бифенила (0,2 г) и NaOBut (2,18 г, 22,7 ммоль) в DME (150 мл) добавляли бензиламин (2,48 мл, 22,7 ммоль), реакционную смесь нагревали до 75°С и поддерживали при этой температуре в течение 20 часов. Реакционную смесь охлаждали до температуры окружающей среды и останавливали реакцию добавлением EtOAc и раствора NaHCO3 в H2O.
Органическую фазу отделяли и промывали H2O, затем насыщенным рассолом, затем обезвоживали (Na2SO4) и концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией на силикагеле для получения (9:R2=CF3-С(О)-, R8=Ph-CH2-NH-) (4,51 г, 83%). Данные: (m/z)=481 (M+H)+.
К перемешиваемому раствору вещества (9:R2=CF3-С(О)-; R8=Ph-CH2-NH-) (2,00 г, 4,16 ммоль) в EtOH (35 мл) добавляли 10% Pd/C (0,2 мл, 22,7 ммоль) и НСl в диоксане (1 мл) реакционную смесь очищали три раза с азотом. Реакционную смесь перемешивали в течение 20 часов в атмосфере водорода (2 бар). Реакцию останавливали добавлением EtOAc и раствора NaHCO3 в H2O и затем фильтровали (целит). Органическую фазу отделяли и промывали H2O, затем насыщенным рассолом, затем обезвоживали (Na2SO4) и концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией на силикагеле для получения (9:R2=CF3-С(O)-; R8=NH2) (1,39 г, 86%). Данные: (m/z)=391 (M+H)+.
Перемешиваемый раствор вещества (9:R2=CF3-С(О)-, R8=NH2) (250 мг, 0,643 ммоль) и триэтиламина (95 мкл, 0,675 ммоль) в DCM (7,5 мл) охлаждали до 0°С и по каплям добавляли раствор изобутирилхлорида (70 мкл, 0,675 ммоль). Реакцию перемешивали при 0°С в течение 1 часа, затем останавливали добавлением EtOAc и раствора NаНСО3 в H2O. Органическую фазу отделяли и промывали H2O, затем насыщенным рассолом, обезвоживали (Na2SO4) и концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией на силикагеле для получения цис-N-[1,2,3,4,10,14b-гексагидро-10-метил-10-2,2,2-трифторацетиламино)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-8-ил]-2-метилпропанамида (9:R2=CF3-С(О)-;
R8=CH3 (СН3) СН-С(О)-NH-) (208 мг, 70%). Данные: (m/z)=461 (M+H)+.
Пример 7
Цис-N-[1,2,3,4,10,14b-гексагидро-10-метил-N-фенил-2-(2,2,2-трифторацетиламино)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-8-ил]-
4-метил-1,2,3-тиадиазол-5-карбоксамид (9:R2=CF3-C(O)-; R8=4-метил-1,2,3-тиадиазолил-С(O)-NH-)
Это соединение было получено аналогично способу, описанному в примере 6, из вещества (9:R2=CF3-С(О)-, R8=NH2) для получения цис-N- [1,2,3,4,10,14b-гексагидро-10-метил-N-фенил-2-(2,2,2-трифторацетиламино)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-8-ил]-4-метил-1,2,3-тиадиазол-5-карбоксамида (9:R2=CF3-С(О)-; R8=4-метил-1,2,3-тиадиазолил-С(О)-NH-) (43%). Данные: (m/z)=517 (М+Н)+.
Пример 8
Этил цис-1,2,3,4,10,14b-гексагидро-10-метил-2-(2,2,2-трифторацетиламино)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-8-карбоксилат (9:R2=CF3-С(О)-; R8=этил-O-С(О)-)
Перемешиваемый раствор вещества (9:R2=CF3-С(О)-; R8=Br) (500 мг, 1,10 ммоль) в безводном ТГФ (10 мл) охлаждали до -75°С и по каплям добавляли н-BuLi (1,44 мл, 2,10 ммоль). Через 5 минут по каплям добавляли этилхлорформиат (525 мкл, 5,50 ммоль) и перемешивали реакционную смесь при -75°С в течение 1,5 часов. Реакцию останавливали добавлением по каплям H2O и продукт экстрагировали EtOAc. Органическую фазу обезвоживали (Na2SO4) и концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией на силикагеле для получения этил цис-1,2,3,4,10,14b-гексагидро-10-метил-2-(2,2,2-трифторацетиламино)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-8-карбоксилат (9:R2=CF3-С(О)-; R8=этил-O-С(О)-) (75 мг, 15%). Данные: (m/z)=448 (M+H)+.
Пример 9
Цис-N-(7-хлор-8-циано-1,2,3,4,10,14b-гексагидро-10-метил-дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2,2,2-трифторацетамид (9:R2=CF3-C(O)-; R7=Cl; R8=CN)
Это соединение было получено аналогично способу, описанному в примере 3, из вещества (8:R7=Cl; R8=Br) для получения цис-N-(7-хлор-8-циано-1,2,3,4,10,14b-гексагидро-10-метил-дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2,2,2-трифторацетамид (9:R2=CF3-С(О)-; R7=Cl; R8=CN) (40%). Данные: (m/z)=435 (M+H)+.
Пример 10
Цис-2,2,2-тpифтop-N-(1,2,3,4,10,14b-гексагидро-10,14-диметил-8-нитро-дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)ацетамид (9:R2=CF3-C(O)-; R8=NO2; R14=СН3)
Это соединение было получено аналогично способу, описанному в примере 2, из вещества (9:R2=CF3-С(О)-; R14=СН3) для получения цис-2,2,2-трифтор-N-(1,2,3,4,10,14b-гексагидро-10,14-диметил-8-нитро-дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)ацетамид (9:R2=CF3-С(O)-; R8=NO2; R14=СН3) (59%). Данные: (m/z)=435 (М+Н)4.
Пример 11
Цис-N-(14-бром-1,2,3,4,10,14b-гексагидро-10-метил-8-нитро-дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2,2,2-трифторацетамид (9:R2=CF3-С(О)-; R8=NO2; R14=Br)
Это соединение было получено аналогично способу, описанному в примере 2, из вещества (9:R2=CF3-С(О)-; R14=Br) для получения цис-N-(14-бром-1,2,3,4,10,14b-гексагидро-10-метил-8-нитро-дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2,2,2-трифторацетамида (9:R2=CF3-С(О)-; R8=NO2; R14=Br) (45%). Данные: (m/z)=500 (M+H)+.
Пример 12
Цис-2,2,2-трифтор-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-нитро-14-(пиридин-2-ил)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)ацетамид (9:R2=CF3-C(О)-; R8=NO2; R14=2-пиридил-)
К перемешиваемому раствору вещества (9:R2=CF3-С(О)-; R8=NO2; R14=Br) (40 мг, 0,080 ммоль) в толуоле (3 мл) добавляли РdСl2(PPh3)2 (3,5 мг, 0,005 ммоль), йодид меди (8,0 мг, 0,005 ммоль), фторид цезия (24 мг, 0,160 ммоль) и 2-(трибутилстаннил)пиридин (44 мг, 0,120 ммоль). Реакционную смесь нагревали до 120°С и выдерживали в течение 24 часов. Реакцию останавливали раствором NаНСО3 в H2O и продукт экстрагировали EtOAc. Органическую фазу промывали Н2О, затем насыщенным рассолом, затем обезвоживали (Na2SO4) и концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией на силикагеле для получения цис-2,2,2-трифтор-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-нитро-14-(пиридин-2-ил)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)ацетамид (9:R2=CF3-С(О)-; R8=NO2; R14=2-пиридил-) (8 мг, 20%). Данные: (m/z)=498 (М+Н)+.
Пример 13
Цис-N-(8,14-дициано-1,2,3,4,10,14b-гексагидро-10-метилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2,2,2-трифторацетамид (9:R2=CF3-С(О)-; R8=CN; R14=CN)
Это соединение было получено аналогично способу, описанному в примере 3, из вещества (9:R2=CF3-C(О)-; R8=Br; R14=Br) для получения цис-N-(8,14-дициано-1,2,3,4,10,14b-гексагидро-10-метилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2,2,2-трифторацетамид (9:R2=CF3-С(О)-; R8=CN; R14=CN) (74%). Данные:
(m/z)=426 (M+H)+.
Пример 14
(2α,4α,14bα)-2,2,2-трифтор-N-[1,2,3,4,10,14b-гексагидро-4,10-диметил-8-(пиридин-4-ил)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил]ацетамид (9:R2=CF3-С(О)-; R4=СН3; R8=4-пиридил-)
Перемешиваемый раствор вещества (9:R2=CF3-С(О)-; R4=СН3; R8=Br) (405 мг, 0,865 ммоль), РdСl2(РРh3)2 (18,2 мг, 0,026 ммоль), К3РO4·7H2O (342,6 мг, 0,101 ммоль), АdРb3 (17,7 мг, 0,058 ммоль) и 2,2-диметилпропандиол циклический эфир пиридин-4-боровой кислоты (456 мг, 2,38 ммоль) в смеси вода:диоксан 1:6 (3,5 мл) нагревали при 30 ватт до 15°С в микроволновой печи в течение 15 минут. Реакцию останавливали раствором NaHСО3 в H2O и продукт экстрагировали DCM. Органическую фазу промывали водой и концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией на силикагеле для получения (2α,4α,14bα)-2,2,2-трифтор-]-N-[1,2,3,4,10,14b-гексагидро-4,10-диметил-8-(пиридин-4-ил)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил]ацетамида (9:R2=CF3-С(О)-; R4=СН3; R8=4-пиридил-) (342 мг, 85%). Данные: (m/z)=467 (M+H)+.
Пример 15
Цис-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-нитродибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2-тиофенэтанамид (9:R2=тиофен-метилен-С(О)-; R8=NO2)
Это соединение было получено аналогично способу, описанному в примере 4, из вещества (8: R8=NO2) для получения цис-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-нитродибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2-тиофенэтанамид (9:R2=тиофен-метилен-С(О)-; R8=NO2) (75%). Данные: (m/z)=449 (M+H)+.
Пример 16
Цис-N-(7-хлор-1,2,3,4,10,14b-гексагидро-4,10-диметилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2,2-дифторацетамид (9:R2=CHF2-C (O)-; R4=СН3; R7=Cl; R8=H)
Это соединение было получено аналогично способу, описанному в примере 4, из вещества (8:R4=СН3; R7=Cl) для получения цис-N-(7-хлор-1,2,3,4,10,14b-гексагидро-4,10-диметилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2,2-дифторацетамид (9:R2=CHF2-C(О)-; R4=СН3; R7=Cl; R8=H) (71%). Данные: (m/z)=406 (M+H)+.
Пример 17
Цис-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-нитро-дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-N',N'-диметилмочевина (9:R2=диметиламино-С(О)-; R8=NO2)
Это соединение было получено аналогично способу, описанному в примере 5, из вещества (8:R8=NO2) для получения цис-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-нитро-дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-N',N'-диметилмочевина (9:R2=диметиламино-С(О)-; R8=NO2) (54%). Данные: (m/z)=396 (M+H)+.
Пример 18
Цис-2,2,2-трифтор-N-(1,2,3,4,10,14b-гексагидро-8-[1-(гидроксиимино)этил)-10,14-диметилдибензо[b,f]пиридо[1,2-d][1,4] диазепин-2-ил)ацетамид (9:R2=CF3-С(О)-; R8=HO-N=C(СН3)-; R14=СН3)
Раствор вещества (9:R2=CF3-С(О)-; R8=CH3-C(О)-; R14=CH3, пример 21) (50 мг, 0,116 ммоль), гидроксиламина·НСl (12 мг, 0,174 ммоль) и триэтиламина (1 капля) в ТГФ (1 мл) нагревали до 50°С и выдерживали в течение 20 часов. Раствор NаНСО3 в H2O добавляли и продукт экстрагировали EtOAc. Органическую фазу промывали Н2О, затем насыщенным раствором, затем обезвоживали (Na2SO4). Органическую фазу концентрировали при пониженном давлении и сырой продукт очищали колоночной хроматографией на силикагеле для получения цис-2,2,2-трифтор-N-(1,2,3,4,10,14b-гексагидро-8-[1-(гидроксиимино)этил)-10,14-диметилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)ацетамида (9:R2=CF3-С(O)-; R8=НО-N=С(СН3)-; R14=СН3) (49 мг, 94%). Данные: (m/z)=447 (М+Н)+.
Пример 19
Цис-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-нитро-дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)фуран-3-карбоксамид (9:R2=фуранил-С(O)-; R8=NO2)
Это соединение было получено аналогично способу, описанному в примере 5, из вещества (8:R8=NO2) для получения цис-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-нитро-дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)фуран-3-карбоксамида (9:R2=фуранил-С(O)-; R8=NO2) (45%). Данные: (m/z)=419 (M+H)+.
Пример 20
Цис-2,2,2-трифтор-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-(пиридин-4-ил)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил) ацетамид (9:R2=CF3-C(О)-; R8=4-пиридил-)
Это соединение было получено аналогично способу, описанному в примере 5, из вещества (8:R8=4-C5H4N) для получения цис-2,2,2-трифтор-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-(пиридин-4-ил)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)ацетамида (9:R2=CF3-С(О)-; R8=4-пиридил-) (31%). Данные: (m/z)=481 (M+H)+.
Пример 21
Цис-N-(8-ацетил-1,2,3,4,10,14b-гексагидро-10,14-диметилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2,2,2-трифторацетамид (9:R8=CF3-C(O)-; R8=CH3-C(O)-; R14=СН3)
Это соединение было получено аналогично способу, описанному в примере 12, из вещества (9:R2=CF3-C(О)-; R8=Br; R14=СН3) для получения цис-N-(8-ацетил-1,2,3,4,10,14b-гексагидро-10,14-диметилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2,2,2-трифторацетамид (9:R2=CF3-C(О)-; R8=СН3-С(O)-; R14=CH3) (22%). Данные: (m/z)=432 (M+H)+.
Пример 22
Цис-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-нитродибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2(метилтио)ацетамид (9:R2=CH3-S-CH2-C(O)-; R8=NO2)
Это соединение было получено аналогично способу, описанному в примере 4, из вещества (8:R8=NO2) для получения цис-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-нитродибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2-(метилтио) ацетамид (9:R2=СН3-3-СН2-С(О)-; R8=NO2) (25%). Данные: (m/z)=413 (M+H)+.
Пример 23
Цис-2,2,2-трифтор-N-[1,2,3,4,10,14b-гексагидро-10,14-диметил-8-(пиримидин-2-ил)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил] ацетамид (9:R2=CF3-С(О)-; R8=2-пиримидинил-; R14=CH3)
Это соединение было получено аналогично способу, описанному в примере 12, из вещества (9:R2=CF3-С(О)-; R8=Br; R14=CH3) для получения цис-2,2,2-трифтор-N-[1,2,3,4,10,14b-гексагидро-10,14-диметил-8-(пиримидин-2-ил]дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)ацетамида (9:R2=CF3-С(О)-; R8=2-пиримидинил-; R14=СН3) (14%). Данные: (m/z)=468 (M+H)+.
Пример 24
Цис-N-(1,2,3,4,10,14b-гeкcaгидpo-10-8-нитродибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)пропионамид (9:R2=этил-С(O)-; R8=NO2)
Это соединение было получено аналогично способу, описанному в примере 5, из вещества (8:R8=NO2) для получения цис-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-нитродибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)пропионамида (9:R2=этил-С(О)-; R8=NO2) (55%). Данные: (m/z)=381 (M+H)+.
Пример 25
(2α,4α,14bα)-2,2,2-трифтор-N-(8-фтор-1,2,3,4,10,14b-гексагидро-4,10-диметилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)ацетамид (9:R2=CF3-C(O)-; R4=CH3; R8=F)
Это соединение было получено аналогично способу, описанному в примере 1, из вещества (8:R4=СН3; R8=F) для получения (2α,4α,14bα)-2,2,2-трифтор-N-[8-фтор-1,2,3,4,10,14b-гексагидро-4,10-диметилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил]ацетамида (9:R2=CF3-С(O)-; R4=СН3; R8=F) (62%). Данные: (m/z)=408 (M+H)+.
Пример 26
Цис-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-нитродибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)метоксиацетамид (9:R2=метоксиметил-С(О)-; R8=NO2)
Это соединение было получено аналогично способу, описанному в примере 5, из вещества (8:R8=NO2) для получения (цис-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-нитродибензо[b,f]пиридо[1,2-d][1,4] диазепин-2-ил)метоксиацетамида (9:R2=мeтoкcимeтил-C(О)-; R8=NO2) (46%). Данные: (m/z)=397 (M+H)+.
Пример 27
Цис-2,2,2-трифтор-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-(2-метил-2Н-тетразол-5-ил)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)ацетамид (9:R2=CF3-C(О)-; R8=2-метил-5-тетразолил-)
К перемешиваемому раствору вещества (9:R2=CF3-C(О)-; R8=CN, Пример 3) (100 мг, 0,250 ммоль) в ДМФ (2,5 мл) добавляли азид натрия (195 мг, 3,00 ммоль) и хлорид аммония (160 мг, 3,00 ммоль). Реакционную смесь нагревали при 20 ватт до 150°С в микроволновой печи и выдерживали в течение 5 минут. Проводили кислотно-щелочную экстракцию и продукт экстрагировали EtOAc. Органическую фазу промывали водой, затем обезвоживали (Na2SO3) и концентрировали при пониженном давлении для получения твердого вещества (48 мг, 43%). Сырой продукт (39 мг, 0,088 ммоль) растворяли в смеси ДМФ:ацетон 1:1 (5 мл), затем добавляли бикарбонат натрия (11,1 мг, 0,132 ммоль) и метилйодид (54,7 мг, 0,880 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 24 часов. Затем добавляли EtOAc и промывали реакционную смесь водой, обезвоживали (Na2SO4), затем концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией на силикагеле для получения цис-2,2,2-трифтор-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-(2-метил-2Н-тетразол-5-ил)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)ацетамида (9:R2=CF3-C(О)-; R8=2-метил-5-тетразолил-) (7 мг, 17%). Данные: (m/z)=458 (M+H)+.
Пример 28
Цис-2,2,2-трифтор-N-(1,2,3,4,10,14b-гексагидро-8-[(гидроксиимино)этил]-10-метилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)ацетамид (9:R2=CF3-С(О)-; R8=HO-N=C(СН3)-)
Это соединение было получено аналогично способу, описанному в примере 18, из вещества (9:R2=CF3-C(О)-; R8=СОСН3) для получения цис-2,2,2-трифтор-N-(1,2,3,4,10,14b-гексагидро-8-[(гидроксиимино)этил]-10-метилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)ацетамида (9:R2=CF3-С(О)-; R8=HO-N=C (СН3)-) (55%). Данные: (m/z)=433 (M+H)+.
Пример 29
(2α,4α,14bα)-N-(7-хлор-1,2,3,4,10,14b-гексагидро-4,10-диметил-дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-3,5-диметилизоксазол-4-карбоксамид (9:R8=3,5-диметил-4-изоксазолил-C(O)-; R4=CH3; R7=Cl; R8=H)
Это соединение было получено аналогично способу, описанному в примере 5, из вещества (8:R4=СН3; R7=Cl) для получения (2α,4α,14bα)-N-(7-хлор-1,2,3,4,10,14b-гексагидро-4,10-диметил-дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-3,5-диметилизоксазол-4-карбоксамида (9:R2=3,5-диметил-4-изоксазолил-C(O)-; R4=СН3; R7=Cl) (15%). Данные: (m/z)=451 (M+H)+.
Пример 30
Цис-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-нитродибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-5-метилизоксазол-4-карбоксамид (9:R2=5-метил-4-изоксазолил-С(О)-; R8=NO2)
Это соединение было получено аналогично способу, описанному в примере 4, из вещества (8:R8=NO2) для получения цис-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-нитродибензо[b,f] пиридо[1,2-d][1,4]диазепин-2-ил)-5-метилизоксазол-4-карбоксамида (9:R2=5-метил-4-изоксазолил-С(О)-; R8=NO2) (27%). Данные: (m/z)=434 (M+H)+.
Пример 31
Цис-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-нитродибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-4-метил-1,2,3-тиадиазол-5-карбоксамид (9:R2=4-метил-5-тиадиазолил-С(О)-; R8=NO2)
Это соединение было получено аналогично способу, описанному в примере 5, из вещества (8:R8=NO2) для получения цис-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-нитродибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-4-метил-1,2,3-тиадиазол-5-карбоксамида (9:R2=4-метил-5-тиадиазолил-С(О)-; R8=NO2) (72%). Данные: (m/z)=451 (M+H)+.
Пример 32
Цис-N-(8-(6-цианопиридин-2-ил)-1,2,3,4,10,14b-гексагидро-10-метилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2,2,2-трифторацетамид (9: R2=CF3-С(О)-; R8=CN-пиридинил-)
Это соединение было получено аналогично способу, описанному в примере 12, из вещества (9:R2=CF3-C(О)-; R8=Br) для получения цис-N-(8-(6-цианопиридин-2-ил)-1,2,3,4,10,14b-гексагидро-10-метил-дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2,2,2-трифторацетамида (9:R2=CF3-С(О)-; R8=N-пиридинил-) (58%). Данные: (m/z)=478 (M+H)+.
Пример 33
Цис-N-[8-(5-этил-1,2,4-оксадиазол-3-ил)-1,2,3,4,10,14b-гексагидро-10-метилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2,2,2-трифторацетамид (9:R2=CF3-С(O)-; R8=5-этил-(3-оксадиазолил)-)
К перемешиваемому раствору вещества (9:R2=CF3-С(О)-; R8=CN) (391 мг, 0,980 ммоль) и триэтиламина (212 мкл, 1,51 ммоль) в этаноле (5 мл) добавляли гидроксиламин гидрохлорид (102 мг, 1,47 ммоль), реакционную смесь нагревали до 80°С и выдерживали в течение 24 часов. Реакционную смесь восстанавливали на роторном испарителе для получения масла, которое было растворено в DCM и промыто H2O. Органическую фазу обезвоживали (Na2SO4) и концентрировали при пониженном давлении для получения масла (420 мг, 100%). Сырой продукт (31 мг, 0,072 ммоль) растворяли в толуоле (1 мл), затем добавляли пиридин (23 мкл, 0,280 ммоль) и пропионил хлорид (12,5 мкл, 0,140 ммоль) и реакционную смесь нагревали до 120°С в течение 2 часов. Реакционную смесь промывали водой, обезвоживали (Na2SO4), затем концентрировали при пониженном давлении. Сырой продукт очищали обращеннофазной ВЭЖХ для получения цис-N-[8-(5-этил-1,2,4-оксадиазол-3-ил)-1,2,3,4,10,14b-гексагидро-10-метилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2,2,2-трифторацетамид (9:R2=CF3-С(О)-; R8=5-этил-(3-оксадиазолил)-) (6 мг, 18%). Данные: (m/z)=472 (М+Н)+.
Пример 34
Цис-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-нитродибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2-гидроксипропанамид (9:R2=НО-этил-С(О)-; R8=NO2)
Это соединение было получено аналогично способу, описанному в примере 5, из вещества (8:R8=NO2). Изолированное соединение затем растворяли в EtOH и добавляли 8% раствор NaOH. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Большую часть EtOH удаляли при пониженном давлении. Затем добавляли H2O и экстрагировали продукт DCM. Органическую фазу промывали насыщенным рассолом, затем обезвоживали (Na2SO4). Органическую фазу концентрировали при пониженном давлении и сырой продукт очищали колоночной хроматографией на силикагеле для получения цис-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-нитродибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2-гидроксипропанамида (9:R2=НО-этил-С(О)-; R8=NO2) (49%). Данные: (m/z)=397 (M+H)+.
Пример 35
Цис-N-(9-хлор-8-циано-1,2,3,4,10,14b-гексагидро-10-метилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2,2,2-трифторацетамид (9:R2=CF3-C(O)-; R8=CN; R9=Cl)
Это соединение было получено аналогично способу, описанному в примере 3, из вещества (9:R2=CF3-С(О)-; R8=Br; R9=Cl) для получения цис-N-(9-хлор-8-циано-1,2,3,4,10,14b-гексагидро-10-метилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2,2,2-трифторацетамида (9: R2=CF3-С(О)-; R8=CN; R9=Cl) (60%). Данные: (m/z)=435 (M+H)+.
Пример 36
Цис-N-(5-метоксипиридин-3-ил)-1,2,3,4,10,14b-гексагидро-10-метил-2-(2,2,2-трифторацетиламино)дибензо[b,f]пиридо[1,2-d][1,4] диазепин-8-карбоксамид (9:R2=CF3-С(О)-; R8=Ме-O-пиридин-NН-С(О)-)
К перемешиваемому раствору вещества (9:R2=CF3-С(О)-; R8=CHO2, пример 4) (20 мг, 0,048 ммоль) в DCM (0,5 мл) и ДМФ (2 капли) добавляли раствор оксалил хлорида (6,8 мкл, 0,078 ммоль) в DCM (0,5 мл). Реакционную смесь перемешивали в течение 1 часа при комнатной температуре. Растворитель затем удаляли при пониженном давлении и полученное масло растворяли в ТГФ (0,5 мл). Затем добавляли триэтиламин (7,3 мкл, 0,052 ммоль) и реакционную смесь охлаждали до 0°С. Затем добавляли раствор 5-амино-2-метоксипиридина (6,5 мг, 0,052 ммоль) в ТГФ (0,5 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 20 часов. Реакцию останавливали добавлением раствора NаНСО3 в H2O и продукт экстрагировали EtOAc, обезвоживали (Na2SO4) и концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией на силикагеле для получения цис-N-(5-метоксипиридин-3-ил)-1,2,3,4,10,14b-гексагидро-10-метил-2-(2,2,2-трифторацетиламино)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-8-карбоксамида (9:R2=CF3-C(О)-; R8=Ме-O-пиридин-NH-C(O)-) (39%). Данные: (m/z)=526 (M+H)+.
Пример 37
Цис-1,2,3,4,10,14b-гексагидро-10,14-диметил-2-(2,2,2-трифторацетиламино)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-8-карбоксамид (9:R2=CF3-С(О)-; R8=NH2-C(О)-; R14=СН3)
Это соединение было получено аналогично способу, описанному в примере 1, из вещества (9:R8=NH2-C(О) -; R14=СН3) для получения цис-1,2,3,4,10,14b-гексагидро-10,14-диметил-2-(2,2,2-трифторацетиламино)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-8-карбоксамида (9:R2=CF3-С(О)-; R8=NH2-C(О)-; R14=СН3) (19%). Данные: (m/z)=433 (M+H)+.
Пример 38
Цис-2,2,2-трифтор-N-[1,2,3,4,10,14b-гексагидро-8-(2-гидроксиацетил)-10,14-диметилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил]ацетамид (9:R2=CF3-С(О)-; R8=OH-CH2-C(О)-)
К перемешиваемому раствору вещества (9:2-CF3; 8-С(O)СН3) (660 мг, 1,58 ммоль) в диоксане (20 мл) по каплям добавляли раствор брома (68,4 мкл, 1,58 ммоль) в Et2O (5 мл). Реакционную смесь нагревали до 40°С в течение 30 минут. Реакцию останавливали добавлением раствора NaHCO3 в Н2О и продукт экстрагировали EtOAc, обезвоживали (Na2SO4) и концентрировали при пониженном давлении. Сырой продукт очищали колоночной хроматографией на силикагеле для получения (9:R2=CF3-С(О)-; R8=BrCH2-C(О)-) (350 мг, 45%). Продукт (9:R2=CF3-С(O)-; R8=BrCH2-C(О)-) (170 мг, 0,343 ммоль) растворяли в EtOH/H2O (85:15) (20 мл) и частями добавляли формиат натрия (140 мг, 2,058 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 20 часов. Реакцию останавливали добавлением раствора NaHCO3 в H2O и продукт экстрагировали EtOAc, обезвоживали (Na2SO4) и концентрировали при пониженном давлении. Сырой продукт очищали обращеннофазовой ВЭЖХ для получения цис-2,2,2-трифтор-N-[1,2,3,4,10,14b-гексагидро-8-(2-гидроксиацетил)-10,14-диметилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил]ацетамида (9:R2=CF3-С(О)-; R8=OH-CH2-C(О)-) (46 мг, 31%). Данные: (m/z)=434 (M+H)+.
Пример 39
Цис-1,2,3,4,10,14b-гексагидро-10-метил-N-пропил-2-(2,2,2-трифторацетиламино)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-8-карбоксамид (9:R2=CF3-С(О)-; R8=пропил-NH-C(O)-)
Это соединение было получено аналогично способу, описанному в примере 36, из вещества (9:R2=CF3-С(О)-; R8=CHO2, пример 4) для получения цис-1,2,3,4,10,14b-гексагидро-10-метил-N-пропил-2-(2,2,2-трифторацетиламино)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-8-карбоксамид (9:R2=CF3-С(О)-; R8=пропил-NH-С(O)-) (27%). Данные: (m/z)=461 (M+H)+.
Пример 40
Цис-2,2,2-трифтор-N-[1,2,3,4,10,14b-гексагидро-10-метил-8-(5-метоксиметил-1,2,4-оксадиазол-3-ил)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил]ацетамид (9:R2=CF3-С(O)-; R8=5-(СН3-O-СН2)-(3-оксадиазолил)-)
К перемешиваемому раствору вещества (9:R2=CF3-С(О)-; R8=CN) (391 мг, 0,980 ммоль) и триэтиламина (212 мкл, 1,51 ммоль) в этаноле (5 мл) добавляли гидроксиламин гидрохлорид (102 мг, 1,47 ммоль), реакционную смесь нагревали до 80°С и выдерживали в течение 24 часов. Реакционную смесь упаривали на роторном испарителе для получения масла, которое растворяли в DCM и промывали Н2О. Органическую фазу обезвоживали (Na2SO4) и концентрировали при пониженном давлении для получения масла (420 мг, 100%). Сырой продукт (60 мг, 0,140 ммоль) растворяли в пиридине (1 мл) и добавляли метоксиацетилхлорид (25,5 мкл, 0,280 ммоль), реакционную смесь нагревали с обратным холодильником в течение 3 часов. Реакционную смесь промывали водой, обезвоживали (Na2SO4) и концентрировали при пониженном давлении. Сырой продукт очищали обращеннофазной ВЭЖХ для получения цис-2,2,2-трифтор-N-[1,2,3,4,10,14b-гексагидро-10-метил-8-(5-метоксиметил-1,2,4-оксадиазол-3-ил)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил]ацетамид (9:R2=CF3-С(О)-; R8=5-(СН3-O-СН2)-(3-оксадиазолил)-) (12 мг, 18%). Данные: (m/z)=488 (M+H)+.
Пример 41
Цис-2,2,2-трифтор-N-[1,2,3,4,10,14b-гексагидро-10-метил-8-(3-метокси-пиридин-5-ил)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил]ацетамид (9:R2=CF3-C(O)-; R8=СН3-О-пиридинил-)
Это соединение было получено аналогично способу, описанному для соединения 12, из вещества (9:R2=CF3-С(О)-; R8=Br)для получения цис-2,2,2-трифтор-N-[1,2,3,4,10,14b-гексагидро-10-метил-8-(3-метокси-пиридин-5-ил)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил]ацетамид (9:R2=CF3-С(О)-; R8=СН3-O-пиридинил-) (54%). Данные: (m/z)=483 (M+H)+.
Пример 42
Цис-N-(12-циано-1,2,3,4,10,14b-гексагидро-10-метил-8-нитро-дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2,2,2-трифторацетамид (9:R8=CF3-C(О)-; R8=NO2; R12=CN)
Это соединение было получено аналогично способу, описанному в примере 2, из вещества (9:R2=CF3-С(О)-; R12=CN) для получения цис-N-(12-циано-1,2,3,4,10,14b-гексагидро-10-метил-8-нитро-дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2,2,2-трифторацетамид (9:R2=CF3-С(О)-; R8=NO2; R12=CN) (10%). Данные: (m/z)=446 (M+H)+.
Пример 43
Цис-N-[8,13-дибром-1,2,3,4,10,14b-гексагидро-10,14-диметилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил]-2,2,2-трифторацетамид (9:R2=CF3-C(O)-; R8=Br; R13=Br; R14=CN)
Это соединение было получено аналогично способу, описанному в примере 3, из вещества (9:R2=CF3-С(О)-; R14=CN) для получения цис-N-[8,13-дибром-1,2,3,4,10,14b-гексагидро-10,14-диметилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил]-2,2,2-трифторацетамид (9:R2=CF3-С(О)-; R8=Br; R13=Br; R14=CN) (33%). Данные: (m/z)=548 (M+H)+.
Пример 44
Цис-N-[1,2,3,4,10,14b-гексагидро-10-метил-8-(пиридин-4-ил)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил]метансульфонамид (9:R2=СН3-3(O)2-; R8=4-пиридил-)
К перемешиваемому раствору вещества (8:R8=4-C5H4N) (200 мг, 0,54 ммоль) в дихлорметане (10 мл) добавляли триэтиламин (81 мкл) и метансульфонилхлорид (45 мкл), поддерживая во время введения температуру в 0°С.Затем реакционную смесь перемешивали в течение 2 часов при температуре окружающей среды. Реакцию останавливали водой, промывали насыщенным водным раствором гидрокарбоната натрия и обезвоживали над сульфатом магния. Реакционную смесь концентрировали при пониженном давлении и сырой продукт очищали колоночной хроматографией на силикагеле для получения цис-N-[1,2,3,4,10,14b-гексагидро-10-метил-8-(пиридин-4-ил)дибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил] метансульфонамида (9:R2=CH3-S(О)2-; R8=4-пиридил-) (160 мг, 66%). Данные: (m/z)=449 (M+H)+.
Пример 45
Цис-N-(1,2,3,4,10,14b-гексагидро-10-метил-8-цианодибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-N'-метил-N'-метоксимочевина (9:R2=Me(MeO)N-C(О)-; R8=CN)
К перемешиваемому раствору вещества цис-N-(8-циано-1,2,3,4,10,14b-гексагидро-10-метилдибензо[b,f]пиридо[1,2-d][1,4]диазепин-2-ил)-2,2,2-трифторацетамида (9:R2=CF3-С(О)-; R8=CN) (4,36 г, 10,9 ммоль) в EtOH (72 мл) добавляли 2N NaOH (19,2 мл). Реакционную смесь перемешивали в течение ночи при температуре окружающей среды, затем выливали в воду и экстрагировали EtOAc. Органическую фазу промывали водой, затем насыщенным рассолом, затем обезвоживали (Na2SO4) и концентрировали при пониженном давлении для получения сырого (8:R8=CN) (3,04 г, 92%). Данные: (m/z)=305 (M+H)+.
К перемешиваемому раствору вещества (8:R8=CN) (200 мг, 0,658 ммоль) в EtOAc (8 мл) добавляли каталитическое количество активированного древесного угля и трихлорметилхлорформиата (94,8 мкл, 0,197 ммоль). Реакционную смесь перемешивали с обратным холодильником в течение 2 часов. Реакционную смесь фильтровали через дикалит и концентрировали при пониженном давлении для получения сырого продукта (8:изоцианат в положении 2 (NCO), R8=CN) (217 мг, 100%). Данные: (m/z)=331 (M+H)+.
К перемешиваемому раствору вещества (8:(NHR2=NCO; R8=CN) (54 мг, 0,164 ммоль) в EtOAc (10 мл) добавляли раствор N,O-диметилгидроксиламингидрохлорида (80 мг, 0,197 ммоль) с триэтиламином (23,7 мкл, 0,197 ммоль) в EtOAc (5 мл). Реакционную смесь перемешивали при 50°С в течение 2 дней, а затем выливали в воду и экстрагировали EtOAc. Органический слой промывали Н2О и насыщенным рассолом, затем высушивали (Na2SO4) и концентрировали при пониженном давлении для получения после очистки (9:R2=Me(MeO)N-C(O)-, R8=CN) (5,9 мг, 9,2%). Данные: (m/z)=392 (M+H)+.
Пример 46
Активность связывания с рецептором глюкокортикоидов
Афинность соединений тестировали, используя Glucocorticoid. Receptor Competitor Assay kit (PanVera®). Компоненты набора размораживали с -80°С на льду (Fluormone GS1, рекомбинантный GR человека (GR)) или при комнатной температуре (GR буфер для скрининга, стабилизирующий пептид и DTT). 10 мМ тестируемого соединения вручную разводили в 20 мкл, затем ступенчато разводили до концентраций от 10 мкМ до 0,1 нМ, используя BioMek 2000 (Beckman-Coulter) в темно-стенных 384-луночных планшетах (Matrix technologies). В следующем порядке: fluormone GS1 (1 нМ итоговая концентрация) добавляли во все лунки, исключая лунку контроля буфера; GR (4 нМ итоговая концентрация) добавляли во все лунки, кроме лунок минимального контроля и контроля буфера, кортизол (10 мкМ итоговая концентрация) добавляли только в лунку контроля fluormone GS1, буфер добавляли во все лунки до итогового объема 40 мкл. Планшет закрывали и выдерживали при комнатной температуре при перемешивании в течение 90 минут. Считывание значений производили с помощью Analyst (LJL) во флуоресцентно-поляризационном режиме. MilliP соотношение рассчитывали из значений «счет в секунду», полученных в параллельном и перпендикулярном режимах. Процент эффективности связывания лиганда рассчитывали для каждой концентрации и строили кривые дозо-ответа для рассчета ECso. Ее сравнивали со значениями для известного стандарта (11β,17β)-11-(1,3-бензодиоксол-5-ил)-17-гидрокси-17-1-пропинил)эстра-4,9-диен-3-она (CAS №189035-07-2, ЕС50=10-8 М). Все соединения, приведенные в примерах, имели активность связывания <2×10-8 М.
Пример 47
Функциональные ответы in vitro
Для количественной оценки способности соединения ингибировать экспрессию воспалительных генов in vitro оценивали действие соединений на линию человеческих клеток U20S, стабильно трансфецированную ДНК рекомбинантного GR человека. U20S клетки стимулировали с TNFα и IFNγ, что приводило к секреции МСР-1 в супернатант. Секрецию МСР-1 количественно оценивали, используя два антитела против МСР-1 человека: одно - меченное с флуоресцентным донором Европием, другое - с флуоресцентным акцептором Аллофикоцианином (АРС). Секреция МСР-1 в супернатанте количественно оценивается путем регистрации флуоресценции при длине волны эмиссии АРС (665 нм), тогда как возбуждение проводится при длине волны Европия (340 нм). Способность соединений (преднизолона или соединений, соответствующих формуле I) ингибировать экспрессию МСР-1 количественно оценивали и рассчитывали значение EC50. Соединения согласно примерам 1,2, 10, 11 и 14-21 имели EC50 в интервале 0,2-2 нМ, тогда как значение для преднизолона составляет 2 нМ.
Пример 48
Противовоспалительная активность in vivo
Способность соединений подавлять воспаление количественно оценивали на модели введения липополисахарида (LPS) мышам. Противовоспалительный эффект оценивали как подавление LPS, вызванного TNFα (S.R.Hyde & R.E.McCallum, Infection & Immunity, 60; 976-982 (1992)). Мышам внутрибрюшинно вводили 0,5 мг/кг LPS. Соединения (преднизолон или соединения, соответствующие формуле I) применяли системно посредством либо перорального, либо подкожного введения за 1 час до индукции LPS. Через 1,5 часа после LPS-индукции собирали сыворотку и мышей убивали. Уровень TNFα в сыворотке оценивали количественно, используя коммерчески доступный Elisa-kit, в соответствии с описанием производителя. Как преднизолон, так и соединения примеров 2-7, 9 и 31 дозозависимо ингибировали TNFα (ED50: 0,5-20 мг/кг по сравнению с 0,5 для преднизолона).
Пример 49
Антиартритная активность in vivo
Способность соединений подавлять развитие артрита тестировали на мышиной модели коллаген 2 типа индуцированного артрита (D.E.Trentham et al., J Exp.Med. 146; 8570868 (1997)). В этой модели Dba/1 мыши были иммунизированы и поддерживающе иммунизированы (через 3 недели) коллагеном. Артрит оценивали по распуханию конечностей. Мышам, у которых развивался артрит, в течение 3 недель вводили либо преднизолон, либо соединения, соответствующие формуле I, либо перорально, либо подкожно (терапевтическая модель). Альтернативно преднизолон или соединения формулы I вводили перорально или подкожно до начала развития артрита (полутерапевтическая модель). Как в терапевтической, так и в полутерапевтической моделях дальнейшее развитие артрита оценивали три раза в неделю. Через 3 недели мышей убивали. Способность соединений подавлять артрит оценивали как способность ингибировать набухание конечностей. Как преднизолон, так и тестированные соединения примеров (примеры 2-5) (в дозе 10 или 20 мг/кг) проявляли способность значительно подавлять развитие артрита.
| название | год | авторы | номер документа |
|---|---|---|---|
| НЕСТЕРОИДНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРОВ ГЛЮКОКОРТИКОИДОВ | 2006 |
|
RU2420528C2 |
| НЕСТЕРОИДНЫЕ МОДУЛЯТОРЫ РЕЦЕПТОРА ПРОГЕСТЕРОНА | 2003 |
|
RU2309155C2 |
| НОВЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ МОНОАЦИЛГЛИЦЕРИНЛИПАЗЫ | 2019 |
|
RU2769507C1 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ ФАКТОРА Ха | 2004 |
|
RU2346944C2 |
| ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СПОСОБ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ ВАЗОПРЕССИНА | 1994 |
|
RU2149160C1 |
| ИНГИБИТОРЫ ОКСАЗИНМОНОАЦИЛГЛИЦЕРИНЛИПАЗЫ (MAGL) | 2019 |
|
RU2794334C2 |
| НОВЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ МОНОАЦИЛГЛИЦЕРИНЛИПАЗЫ | 2019 |
|
RU2801190C2 |
| НОВЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2019 |
|
RU2809257C2 |
| АЗАИНДОЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФАКТОРА Xa | 2004 |
|
RU2330853C2 |
| ПРОИЗВОДНЫЕ ГИДРИРОВАННЫХ ПИРИДО(4,3-B)ИНДОЛОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1995 |
|
RU2140417C1 |
Описываются новые производные дибензо[b,f]пиридо[1,2-d][1,4] диазепинила общей формулы I:

или их фармацевтически приемлемые соли (значения радикалов приведены в формуле изобретения), которые являются модуляторами рецепторов глюкокортикоидов и могут найти применение при лечении иммунологических и воспалительных заболеваний. 4 н. и 7 з.п. ф-лы.
1. Соединение, соответствующее общей формуле I:
или его фармацевтически приемлемая соль,
где R1 представляет собой Н;
R2 представляет собой -C(O)R15,
где R15 представляет собой (1-4С)алкил(1-4С)алкоксиамин или (1-6С)алкил, необязательно замещенный одним или более ОН галогеном или тиофенилом, или R15 представляет собой (ди)(1-4С)алкиламино, (1-4С)алкил(1-4С)алкоксиамин, (1-4С)алкилтио(1-4С)алкил, (1-4С)алкокси(1-4С)алкил или гетероарил, выбранный из группы, содержащей фуранил, изоксазолил, необязательно замещенный (1-4С)алкилом, или тиадиазолил, необязательно замещенный(1-4С)алкилом; или
R2 представляет собой -S(O)2R15,
где R15 представляет собой (1-6С)алкил;
R3 представляет собой Н;
R4 представляет собой Н или (1-4С)алкил;
R6 представляет собой Н;
R7 представляет собой Н или галоген;
R8 представляет собой Н, циано, галоген или нитро;
или R8 представляет собой пиридил, необязательно замещенный циано или (1-4С)алкокси, пиримидил, тетразолил, необязательно замещенный (1-4С)алкилом, оксадиазолил, необязательно замещенный (1-4С) алкилом
или (1-4С)алкокси(1-4С)алкилом;
или R8 представляет собой -C(O)NHR17, -C(O)R18, -C(O)OR19, -NHC(O)R20- или -С(1-4С)алкилNOR21;
R9 представляет собой Н или галоген;
R10 представляет собой (1-4С)алкил;
R11 представляет собой Н;
R12 представляет собой Н или циано;
R13 представляет собой Н или галоген;
R14 представляет собой Н, галоген, циано, (1-4С)алкил или пиридил;
R17 представляет собой Н или (1-6С)алкил, фенил или пиридил, необязательно замещенный (1-4С)алкокси;
R18 представляет собой Н, NH2 или (1-4С)алкил, необязательно замещенный ОН;
R19 представляет собой (1-6С)алкил;
R20 представляет собой (1-6С)алкил или тиазолил, необязательно замещенный (1-4С)алкилом;
каждый R21 независимо представляет собой Н.
2. Соединение по п.1, где R7 представляет собой Н.
3. Соединение по п.1 или 2, где R10 представляет собой метил.
4. Соединение по п.1, где R2 представляет собой C(O)R15.
5. Соединение по п.1, где R15 представляет собой (1-4С)алкил, необязательно замещенный галогеном.
6. Соединение по п.5, где R15 представляет собой трифторметил.
7. Соединение по п.1, где R8 представляет собой Н, галоген, циано, нитро, C(O)R18 или -NHC(O)R20:
или R8 представляет собой пиридил, необязательно замещенный циано, пиримидил, тетразолил, необязательно замещенный (1-4С)алкилом, оксадиазолил, необязательно замещенный (1-4С)алкилом или (1-4С)алкокси(1-4С)алкилом.
8. Соединение по п.7, где R8 представляет собой Н, циано, пиридил или нитро.
9. Применение соединения по пп.1-8 или его фармацевтически приемлемой соли для производства лекарственного средства для пациентов, нуждающихся в модуляции рецептора глюкокортикоидов.
10. Применение соединения по пп.1-8 или его фармацевтически приемлемой соли для производства лекарственного средства для лечения иммунологических и воспалительных заболеваний.
11. Применение соединения по пп.1-8 или его фармацевтически приемлемой соли для производства лекарственного средства в области ревматологии.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| US 4054572 А, 18.10.1977 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| RU 2004132215/04 А, 20.04.2005. | |||

