Область техники, к которой относится изобретение
Данное изобретение относится к ряду замещенных имидазопиримидинов и содержащим их фармацевтическим композициям. Соединения изобретения ингибируют продуцирование ряда цитокинов воспаления, в частности - TNF-α и IL-1β. Соединения данного изобретения полезны при лечении заболеваний, опосредуемых р38, таких как ревматоидный артрит, воспалительное заболевание кишечника, септический шок, остеопороз, остеоартрит, нейродегенеративные заболевания и заболевания, связанные со СПИДом.
Предпосылки создания изобретения
Цитокины воспаления TNF-α и IL-1β играют важную роль при ряде воспалительных заболеваний, таких как ревматоидный артрит (Dinarello et al., Curr. Opin. Immunol., 1991, 3: 941-8). Артрит является воспалительным заболеванием, поражающим миллионы людей, и может разрушить любой сустав человека. Его симптомы колеблются от легкой боли и слабого воспаления в порженных суставах до тяжелой и изнуряющей боли и сильного воспаления. Хотя заболевание ассоциируется, главным образом, с пожилыми людьми, оно поражает и людей другого возраста.
Наиболее распространенное лечение артрита включает применение нестероидных противовоспалительных средств ("NSAID") для облегчения симптомов. Однако несмотря на широкое применение NSAID, многие индивидуумы не могут переносить дозы, необходимые для лечения в течение длительного времени. Кроме того, NSAID лечит только симптомы заболевания, не воздействуя на причины, лежащие в его основе. Когда пациент плохо реагирует на NSAID, часто применяют другие лекарственные средства, такие как метотрексат, D-пенцилламин, соли золота и фреднион. Такие лекарственные средства также имеют значительную токсичность, и механизм их действия пока неизвестен.
При клинических испытаниях в небольшом масштабе показано, что антагонисты рецепторов к IL-1β и моноклональные антитела к TNF-α ослабляют симптомы ревматоидного артрита. Кроме терапии, основанной на применении белков, существуют средства с небольшими молекулами, ингибирующие продуцирование указанных цитокинов, и их активность показана на животных моделях артрита (Boehm et al., J. Med. Chem., 1996, 39: 3929-37). Доказано, что средство из числа таких средств с небольшими молекулами, SB 203580, эффективно снижает продуцирование TNF-α и IL-1β в LPS-стимулированных клеточных линиях моноцитов человека со значениями IC50 50-100 нМ (Adams et а1., WO 93/14081, 23 июля 1993). В дополнение к такому испытанию in vitro показано, что SB 203580 ингибирует продуцирование цитокинов воспаления у крыс и мышей при величинах IC50 15-25 мг/кг (Badger et al., J. Pharm. Exp.Therap., 1996, 279: 1453-61). Хотя на сегодняшний день сведений о действии SB 203580 на человеческий организм пока нет, доказано, что моноклональные антитела к TNF-α эффективны при лечении ревматоидного артрита (Elliot et al., Arthritis Rheum., 1993, 36: 1681-90). На основании пероральной активности SB 203580 и его эффективности на животных моделях исследователи предполагают, что соединение с таким профилем имеет практический потенциал для лечения ревматоидного артрита (Badger et al., J. Pharm. Exp.Therap., 1996, 279: 1453-61).
SB 203580 и другие средства с небольшими молекулами снижают продуцирование цитокинов воспаления, ингибируя активность серин/треонинкиназы р38, которую иногда называют CSBP, при IC50 200 нМ (Griswold et al., Pharm. Commun., 1996, 7: 323-9). Хотя точная роль данной киназы неизвестна, она вовлекается как в продуцирование TNF-α, так и в сигнальные реакции, связанные с рецептором к TNF-α.
В WO 91/00092 описывается способ ингибирования продуцирования интерлейкина-1 моноцитами и/или макрофагами у людей путем введения диарилзамещенного имидазола, сконденсированного с другим гетероциклическим кольцом, содержащим атом азота у мостиковой связи, где указанное другое кольцо также может содержать атом серы, кислорода или дополнительный атом азота, и может содержать дополнительную ненасыщенность.
В WO 90/15534 и ЕР 0403251 описывается лечение людей, пораженных Т-клеточно-вирусной (TIV) инфекцией, включающее введение эффективного количества средства, снижающего активность монокинов.
В WO 91/19497 описывается диарилзамещенный имидазол, полезный для двойного ингибирования - при заболеваниях, опосредуемых путем метаболизма 5-липоксигеназы, и при заболеваниях, опосредуемых путем метаболизма циклооксигеназы. Данное соединение конденсируется с другим ненасыщенным 5- или 6-членным гетероциклическим кольцом, содержащим атом азота у мостиковой связи, где указанное другое 5-членное кольцо также содержит атом серы или атом кислорода, а указанное другое 6-членное кольцо может также содержать дополнительный атом азота.
Несмотря на существование указанных известных соединений и способов, в данной области остается потребность в усовершенствованных способах снижения продуцирования цитокинов воспаления посредством ингибирования активности серин/треонинкиназы р38, и в соответствующих способах лечения и предупреждения артрита и других воспалительных расстройств.
Краткое изложение сущности изобретения
Данное изобретение относится к новым соединениям, ингибирующим in vitro активность р38 в интервале наномолярных концентраций, а также к способам их получения. Кроме того, соединения настоящего изобретения ингибируют in vitro секрецию TNF-α и IL-1β в интервале наномолярных концентраций.
Животные модели демонстрируют ингибирование продуцирования TNF-α, индуцированного LPS. Соединения, биологическая активность которых демонстрируется in vitro и in vivo с помощью описанных здесь испытаний, являются соединениями настоящего изобретения, представленными формулой I

Данное изобретение также относится к фармацевтической композиции, содержащей указанное соединение и фармацевтически приемлемый носитель, а также к соответствующим способам синтеза.
Данное изобретение также относится к способу лечения субъекта, страдающего от состояния, облегчение которого достигается снижением цитокинов воспаления, действие которых вносит вклад в указанное состояние, причем указанный способ включает введение субъекту терапевтически эффективной дозы фармацевтической композиции по настоящему изобретению.
Данное изобретение также относится к способу ингибирования возникновения у субъекта состояния, облегчение которого достигается снижением цитокинов воспаления, действие которых вносит вклад в указанное состояние, причем указанный способ включает введение субъекту профилактически эффективной дозы фармацевтической композиции по настоящему изобретению.
Подробное описание изобретения
Данное изобретение относится к соединению формулы I
или его фармацевтически приемлемой соли, где
(a) R1 выбирают из группы, состоящей из NH2, C1-5алкиламино, ди-C1-5алкиламино, гидрокси, С1-5алкокси, фенилметиламино, гетероциклилметила, С1-5алкилкарбониламино и замещенного фенилкарбониламино, где
указанные фенилметиламино и гетероциклилметил могут быть замещены в своей фенильной группе одним или несколькими заместителями, выбранными из группы, состоящей из галогена, C1-5алкила, С1-5алкокси, арил-С1-3алкиламино, R'R"NCH=N- и OR"', причем R', R' и R"' выбирают, независимо, из Н, С1-5алкила, фенилметила, замещенного фенилметила, α-алкилфенилметила, замещенного α-алкилфенилметила, гетероциклилметила и замещенного гетероциклилметила;
(b) Y выбирают из группы, состоящей из Н, галогена, гетероцикла, OR4, SR4, NHR4 и NR4R5, где
R4 и R5 выбирают, независимо, из Н, гетероциклила, С3-5карбоцикла, фенила, α-алкилфенилС1-5алкила, прямого или разветвленного алкила, необязательно замещенного R, NHR, N(R)2, С3-5карбоциклом, фенилом или замещенным фенилом, где (i) R представляет Н, галоген, C1-5алкил, фенилметил, замещенный фенилметил, SO2Ph, пиридил или пиридилметил, и (ii) указанные фенил, гетероциклил и α-алкилфенилС1-5алкил могут быть замещены одним или несколькими заместителями, выбранными из группы, состоящей из галогена, С1-5алкила, С1-5алкокси, арил-C1-3алкиламино, фенилметила, замещенного фенилметила, R'R"NCH=N- и OR"', как указано в (а);
(c) R2 представляет от одного до пяти членов, независимо выбранных из группы, включающей галоген, трифторметил, -NHCH2PH, С1-5алкила и С1-5алкокси;
(d) R3 представляет Н, или R3, взятые вместе, образуют ароматическое кольцо; и
(e) Х представляет N или СН.
В одном из вариантов соединений настоящего изобретения R1 представляет NH2. В другом варианте R2 является членом, выбранным из группы, состоящей из галогена, трифторметила, -NHCH2PH и С1-5алкокси. Еще в одном из вариантов Y представляет NHR4, и R4 представляет фенилметил. Еще в одном варианте Х представляет СН.
Если не указано иначе, термин "алкил" относится к прямому, разветвленному или циклическому заместителю, состоящему только из углерода и Н в отсутствии ненасыщенности. Термин "алкокси" относится к O-алкилу, где алкил имеет значение, указанное выше. Термин "ароматическое кольцо" относится к 5-6-членному кольцу, содержащему систему делокализованных конъюгированных пи-связей из 6 электронов, такому как фенил, фуранил и пирролил. Термин "арил" включает моно- и конденсированные ароматические кольца, такие как фенил, нафтил, дифенил, фторфенил, дифторфенил, бензил, бензоилоксифенил, карбоэтоксифенил, ацетилфенил, этоксифенил, феноксифенил, гидроксифенил, карбоксифенил, трифторметилфенил, метоксиэтилфенил, ацетамидофенил, толил, ксилил, диметилкарбамилфенил и т.п. Термин "галоген" обозначает фтор, хлор, бром и иод. Символ "Ph" относится к фенилу. Термин "гетероциклил", "гетероцикл" или "гетероциклический остаток" представляет отдельное кольцо или конденсированные кольца, имеющие в качестве кольцевого атома по меньшей мере один атом, иной чем атом углерода, например, пиридин, пиримидин, оксазолин, пиррол, имидазол, морфолин, фуран, индол, бензофуран, пиразол, пирролидин, пиперидин и бензимидазол.
Замещенный гетероциклилметил и замещенный фенилметил содержат заместители такие, как галоген, С1-5алкил, C1-5алкокси, арил-С1-3алкиламино, R'R"NCH=N- и OR"', где R', R" и R"' выбирают, независимо, из Н, С1-5алкила, фенилметила, замещенного фенилметила, α-алкилфенилметила и замещенного α-алкилфенилметила, гетероциклилметила и замещенного гетероциклилметила.
Термин "FCS" представляет фетальную телячью сыворотку, "ТФК" обозначает трифторуксусную кислоту, и "RPMI" обозначает среду от Roswell Park Memorial Inst. (Sigma, cat. № R0833).
"Независимо" обозначает, что когда заместителей несколько, они могут быть различными. "DME" обозначает этиленгликольдиметил. Термин "NaHMDS" относится к гексаметилдисилазиду натрия.
Выражение "фармацевтически приемлемая соль" обозначает соли свободного основания, обладающие требуемой фармакологической активностью свободного основания, не являющиеся нежелательными с биологической или иной точки зрения. Такие соли могут быть образованы неорганическими или органическими кислотами. Примерами неорганических кислот являются хлористоводородная кислота, бромистоводородная кислота, иодистоводородная кислота, перхлорная кислота, азотная кислота, серная кислота и фосфорная кислота. Примерами органических кислот являются уксусная кислота, пропионовая кислота, гликолевая кислота, молочная кислота, виноградная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, щавелевая кислота, памовая кислота, сахарная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, метилсульфоновая кислота, салициловая кислота, гидроэтансульфоновая кислота, бензолсульфоновая кислота, 2-нафталинсульфоновая кислота, п-толуолсульфоновая кислота, циклогексансульфаминовая кислота и т.п.
Когда в соединениях данного изобретения существует один или несколько центров стереоизомерии, следует иметь в виду, что все возможные оптические изомеры, антиподы, энантиомеры и диастереомеры, появляющиеся вследствие наличия таких центров стереоизомерии, которые могут существовать в оптических изомерах, антиподах, их рацематы и рацемические смеси также являются частью данного изобретения. Антиподы можно разделить способами, известными специалистам в данной области техники, такими как, например, дробная перекристаллизация диастереомерных солей энантимерно чистых кислот. С другой стороны, антиподы можно разделить методом хроматографии на колонке типа Pirkle.
Примерами соединений настоящего изобретения являются следующие соединения:
соединение 1: 2-(4-фторфенил)-3-(4-пиридинил)имидазо[1,2-а] пиримидин-7-амин;


соединение 2: 2-(3-фторфенил)-3-(4-пиридинил)имидазо[1,2-а]пиримидин-7-амин;
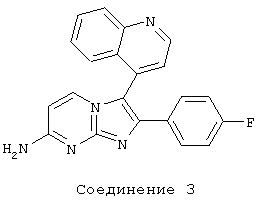
соединение 3: 2-(4-фторфенил)-3-(4-хинолинил)имидазо[1,2-а]пиримидин-7-амин;
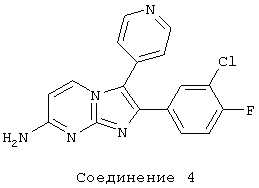
соединение 4: 2-(3-хлор-4-фторфенил)-3-(4-пиридинил)имидазо [1,2-а]пиримидин-7-амин;
соединение 5: 2-фенил-3-(4-пиридинил)имидазо[1,2-а]пиримидин-7-амин;


соединение 6: 2-(4-фторфенил)-3-[2-[(фенилметил)амино]-4-пиридинил]имидазо[1,2-а]пиримидин-7-амин;
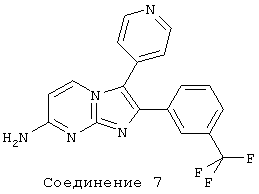
соединение 7: 3-(4-пиридинил)-2-[3-(трифторметил)фенил]имидазо[1,2-а]пиримидин-7-амин;

соединение 8: 3-[2-[(фенилметил)амино]-4-пиридинил]-2-[3-(трифторметил)фенил]имидазо[1,2-а]пиримидин-7-амин;
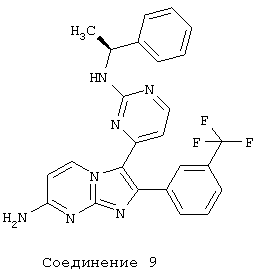
соединение 9: 3-[2-[[(1S)-l-фенилэтил]амино]-4-пиримидинил]-2-[3-(трифторметил)фенил]имидазо[1,2-а]пиримидин-7-амин;

соединение 10: 2-(4-фторфенил)-3-[2-(метилтио)-4-пиримидинил]имидазо[1,2-а]пиримидин-7-амин;
соединение 11: 3-[2-(метилтио)-4-пиримидинил]-2-[3-(трифторметил)фенил]имидазо[1,2-а]пиримидин-7-амин;


соединение 12: 2-(3-фторфенил)-3-[2-(метилтио)-4-пиримидинил]имидазо[1,2-а]пиримидин-7-амин;
соединение 13: 3-(4-пиримидинил)-2-[3-(трифторметил)фенил]имидазо[1,2-а]пиримидин-7-амин;


соединение 14: 2-фенил-3-[2-(1-пиперидинил)-4-пиримидинил]имидазо[1,2-а]пиримидин-7-амин;
соединение 15: 3-[2-(метилтио)-4-пиримидинил]-2-фенилимидазо[1,2-а]пиримидин-7-амин;


соединение 16: 2-фенил-3-(4-пиримидинил)имидазо[1,2-а]пиримидин-7-амин;
соединение 17: 3-[2-(метилсульфонил)-4-пиримидинил]-2-фенилимидазо[1,2-а]пиримидин-7-амин;
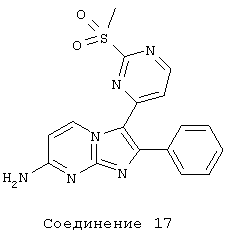

соединение 18: 3-[2-(метилсульфонил)-4-пиримидинил]-2-[3-(трифторметил)фенил]имидазо[1,2-а]пиримидин-7-амин;
соединение 19: 2-фенил-3-[2-[[(1S)-1-фенилэтил]амино]-4-пиримидинил]имидазо[1,2-а]пиримидин-7-амин;


соединение 20: 3-[[[(4-метоксифенил)метил]амино]-4-пиримидинил]-2-фенилимидазо[1,2-а]пиримидин-7-амин;
соединение 21: 2-(4-фторфенил)-3-[3-[[(1S)-1-фенилэтил]амино]-4-пиридинил]имидазо[1,2-а]пиримидин-7-амин;

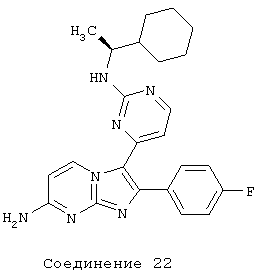
соединение 22: 3-[2-[[(1S)-1-циклогексилэтил]амино]-4-пиримидинил]-2-(4-фторфенил)имидазо[1,2-а]пиримидин-7-амин;
соединение 23: 3-(2-метокси-4-пиримидинил)-2-фенилимидазо [1,2-а]пиримидин-7-амин;


соединение 24: 2-(4-фторфенил)-3-(4-пиримидинил)имидазо[1,2-а]пиримидин-7-амин;
соединение 25: 2-(3-хлорфенил)-3-(4-пиридинил)имидазо[1,2-а]пиримидин-7-амин;


соединение 26: 3-(2-бром-4-пиридинил)-2-(4-фторфенил)имидазо [1,2-а]пиримидин-7-амин и
соединение 27: 3-(2-бром-4-пиридинил)-2-[3-(трифторметил) фенил]имидазо[1,2-а]пиримидин-7-амин

Данное изобретение также относится к фармацевтической композиции, содержащей указанное соединение и фармацевтически приемлемый носитель.
Фармацевтические композиции, содержащие соединение настоящего изобретения в качестве активного ингредиента в однородной смеси с фармацевтическим носителем, можно получить обычными методами, принятыми в фармации. Носитель может принимать самые разные формы, в зависимости от формы препарата, требуемого для введения, в том числе, такого как местное введение и системное введение, включая, но не ограничиваясь ими, внутривенную инфузию, пероральное, назальное или парентеральное введение. При получении композиций в лекарственной форме для перорального введения можно использовать любые обычные известные фармацевтические носители, такие как вода, глицерин, гликоли, масла, спирты, корригенты, консерванты, красители, сироп и подобные вещества в случае жидких пероральных препаратов (например, суспензий, эликсиров и растворов); или носители, такие как крахмалы, сахара, метилцеллюлоза, стеарат магния, дикальцийфосфат, маннит и подобные вещества в случае твердых пероральных препаратов (например, порошков, капсул и таблеток). Все эксципиенты можно смешивать, по необходимости, с веществами, способствующими рассыпанию, разбавителями, веществами, способствующими грануляции, смазывающими веществами, связующими веществами и т.п., с использованием обычных методов, известных специалистам в области получения лекарственных форм.
Предпочтительным способом введения является пероральное введение. В силу простоты введения таблетки и капсулы представляют выгодную стандартную лекарственную форму для перорального введения, и в таком случае, очевидно, используются твердые фармацевтические носители. При необходимости, на таблетки обычными способами можно нанести сахарное покрытие или энтеросолюбильное покрытие. В случае парентеральных форм носитель, как правило, будет содержать стерильную воду, хотя можно добавить другие ингредиенты, например, способствующие растворению или для целей сохранения. Также можно получить суспензии для инъекций, и в таком случае можно использовать соответствующие жидкие носители, суспендирующие средства и т.п.
Используемый здесь термин "цитокин" относится к белкам TNF-α и IL-1β. Расстройствами, связанными с цитокинами, являются заболевания людей и других млекопитающих, где сверхпродуцирование цитокинов вызывает симптомы заболевания. Сверхпродуцирование цитокинов TNF-α и IL-1β связано с рядом заболеваний.
Соединения настоящего изобретения ингибируют продуцирование TNF-α и IL-1β.Таким образом, настоящее изобретение также относится к способу лечения субъекта, страдающего от состояния, облегчение которого достигается снижением цитокинов воспаления, действие которых вносит вклад в указанное состояние, включающему введение указанному субъекту терапевтически эффективной дозы фармацевтической композиции по настоящему изобретению. Используемый здесь термин "субъект" включает, но не ограничивается ими, любое животное или искусственно измененное животное. В предпочтительном варианте воплощения изобретения субъектом является человек.
Данное изобретение также относится еще к способу ингибирования возникновения у субъекта состояния, облегчение которого достигается снижением цитокинов воспаления, действие которых вносит вклад в указанное состояние, включающему введение указанному субъекту профилактически эффективной дозы фармацевтической композиции по настоящему изобретению.
В одном из вариантов воплощения изобретения такое состояние выбрано из группы, в которую входят артрит, воспалительное заболевание пищеварительного тракта, септический шок, остеопороз, остеоартрит, невропатическая боль, репликация ВИЧ, деменция, вызванная ВИЧ, вирусный миокардит, инсулинзависимый диабет, инсулиннезависимый диабет, периодонтальная болезнь, рестеноз, гнездная алопеция, падение числа Т-лимфоцитов при ВИЧ-инфекции или СПИДе, псориаз, острый панкреатит, отторжение аллотрансплантата, аллергическое воспаление легких, атеросклероз, рассеянный склероз, кахексия, болезнь Альцгеймера, удар, болезнь Крона, ишемия, застойная сердечная недостаточность, фиброз легких, гепатит, глиобластома, синдром Гийена-Барре и системная красная волчанка. В предпочтительном варианте воплощения изобретения таким состоянием является ревматоидный артрит.
Используемый здесь термин "лечение" обозначает устранение или, иначе, ослабление причины и/или ее действий. "Ингибирование" возникновения расстройства означает профилактику, сдерживание или уменьшение вероятности такого возникновения. Подобным образом, "терапевтически эффективные" и "профилактически эффективные" дозы являются дозами, создающими возможность, соответственно, лечения и ингибирования расстройства. Способы определения терапевтически и профилактически эффективных доз для фармацевтической композиции настоящего изобретения являются способами, известными в данной области. Эффективную дозу для введения фармацевтической композиции человеку, например, можно определить математически из результатов исследований на животных.
В одном варианте воплощения изобретения пероральные дозы соединений настоящего изобретения находятся в интервале от примерно 0,05 до примерно 100 мг/кг в сутки. В другом варианте пероральные дозы находятся в интервале от примерно 0,05 до примерно 50 мг/кг в сутки, и в еще одном варианте - от примерно 0,05 до примерно 20 мг/кг в сутки. Инфузионные дозы могут составлять, например, от примерно 1,0 до 1,0×104 мкг/кг/мин соединения настоящего изобретения, смешанного с фармацевтически приемлемым носителем, в течение времени в интервале от нескольких минут до нескольких дней. В случае местного введения соединение настоящего изобретения можно смешивать с фармацевтически приемлемым носителем в концентрации, например, от примерно 0,1 до примерно 10% лекарственного средства относительно наполнителя.
Наконец, данное изобретение относится к способам получения соединений настоящего изобретения. Указанные соединения можно получить так, как показано ниже, из легко доступных исходных веществ и/или промежуточных соединений согласно способам, известным в данной области.
Данное изобретение будет более понятно при обращении к экспериментальным подробностям, описанным далее, но специалисты в данной области техники легко представят, что они являются только иллюстративными для изобретения, которое более полно описано в формуле изобретения, приведенной ниже.
Кроме того, в данной заявке цитируются различные публикации. Все указанные публикации включены в настоящее описание в качестве ссылок для более полного описания уровня техники, к которой относится данное изобретение.
ЭКСПЕРИМЕНТАЛЬНЫЕ ПОДРОБНОСТИ
А. Схемы и синтезы
Соединения формулы I, где R1 представляет NH2, и R3 и Y представляют Н, можно получить по схеме I. Исходное соединение типа 1а такое, как 4-метилпиридин или 4-метилхинолин, можно смешать с эфиром бензойной кислоты типа 1b и двумя эквивалентами подходящего основания, имеющего пространственные затруднения, такого как гексаметилдисилазид натрия, в подходящем растворителе таком, как ТГФ, при комнатной температуре и получить енолят 1 с, который затем бромируют до соединения типа Id. Затем промежуточное соединение типа Id можно ввести во взаимодействие с 2,6-диаминопиримидином и получить соединение формулы I, где R1 представляет NH2, и Y представляет Н.
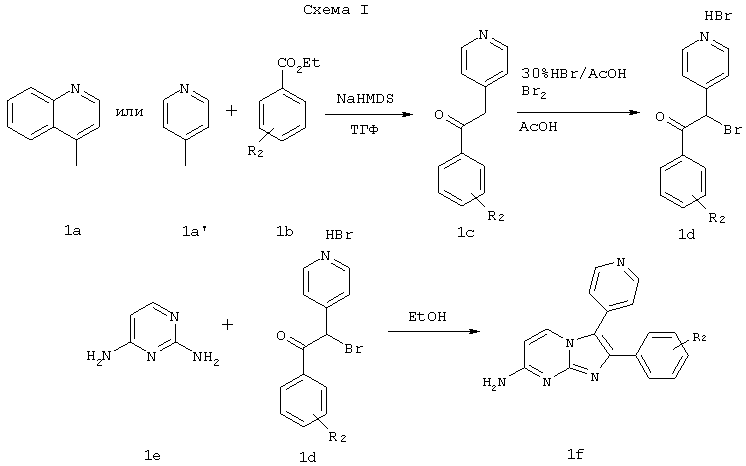
Хотя способом, показанным на схеме, получают соединение формулы I, где R1 представляет NH2, X представляет СН, и Y представляет Н, указанной схемой можно также воспользоваться для получения других соединений изобретения.
Схема II показывает, как можно получить соединения формулы I, где Х представляет N, и Y, R2 и R3 имеют значения, указанные выше.

Схема III показывает, как можно получить соединения формулы I, где Х представляет СН, Z' представляет F, Cl или Br, и Y, R2 и R3 имеют значения, указанные выше.

Схема IV показывает, как можно получить соединения формулы I, где Х представляет СН, Y представляет NHR4, и R4 имеет значения, указанные выше.

В примерах, приведенных ниже, описывается более конкретно химический синтез характерных соединений настоящего изобретения. Остальные соединения, описываемые здесь, можно получить подобным образом в соответствии с одним или несколькими из указанных способов. Попытка оптимизации выходов в указанных реакциях не предпринималась, и для специалиста в данной области техники должно быть ясно, что изменения во времени, температурах, растворителях и/или реагентах реакций могут повысить такие выходы.
Пример 1
3-[2-[(Фенилметил)амино]-4-пиридинил]-2-[3-(трифторметил) фенил]имидазо[1,2-а]пиримидин-7-амин

К 5,44 г (27,44 ммоль) 2-бензиламино-4-метилпиридина в 40 мл трет-бутанола добавляют 6,59 г (31,18 ммоль) ди-трет-бутилдикарбоната. Через 18 часов растворитель удаляют в вакууме. Остаток растирают с гексаном и фильтруют.Фильтрат концентрируют в вакууме и получают 4,25 г амина с защитной группой. 1H ЯМР (300 МГц, ДМСО-d6) δ 8,22 (1Н, д, J=5,1 Гц), 6,99 (1Н, д, J=5,1 Гц), 5,10 (2Н, с), 2,31 (3H, с), 1,38 (9Н, с).

К раствору 8,97 г (30,07 ммоль) H-Вос-2-бензиламино-4-метилпиридина и 6,58 г (30,07 ммоль) этил 3-трифторметилбензоата в 60 мл тетрагидрофурана с помощью капельной воронки в атмосфере азота добавляют по каплям 61 мл (61 ммоль) 1,0 М раствора бис(триметилсилил)амида натрия в тетрагидрофуране. Через восемнадцать часов реакцию гасят насыщенным раствором хлорида аммония и удаляют растворитель в вакууме. Остаток экстрагируют 300 мл этилацетата и экстракт промывают 2×200 мл воды, 1×100 мл рассола, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, получая коричневое масло. Колоночная хроматография с использованием смеси гексан/этилацетат, 5:1, дает 10,92 г продукта реакции в виде густого желтого масла. 1H ЯМР (300 МГц, ДМСО-d6) δ 7,59 (1Н, с), 5,11 (2Н, с), 4,62 (2Н, с), 1,33 (9Н, с).

Амин с защитной группой (10,92 г, 23,21 ммоль) кипятят с обратным холодильником в течение 1 часа в 100 мл тетрагидрофурана, содержащего 20 мл 6 М HCl, смесь охлаждают, разбавляют 220 мл воды и экстрагируют 2×250 мл этилацетата. Органические слои отделяют, объединяют, промывают 200 мл воды, 2×100 мл рассола, сушат над сульфатом магния, фильтруют и концентрируют в вакууме, получая 7,91 г вязкого красного масла. МН+=371.

К 2,33 г (6,29 ммоль) кетона в 10 мл ледяной уксусной кислоты добавляют 1,30 мл (6,61 ммоль) 30% раствора бромистого водорода в уксусной кислоте. Добавляют по каплям раствор 0,35 мл (6,79 ммоль) брома в 1,65 мл ледяной уксусной кислоты и реакционную смесь нагревают при 60°С в течение одного часа, охлаждают до комнатной температуры и разбавляют эфиром. Образовавшийся маслянистый остаток промывают эфиром и получают 2,27 г (4,28 ммоль) сырого бромида. МН+=450.

Раствор 1,89 г (17,13 ммоль) 2,4-диаминопиримидина в 20 мл этанола нагревают до 80°С. С помощью капельной воронки добавляют по каплям раствор 2,27 г (4,28 ммоль) неочищенного бромида в 50 мл этанола. Реакционную смесь перемешивают при 80°С в течение одного часа и затем охлаждают до комнатной температуры. Удаляют в вакууме приблизительно половину объема растворителя. После охлаждения до комнатной температуры реакционную смесь фильтруют. Фильтрат концентрируют в вакууме, остаток разбавляют 250 мл этилацетата и промывают 2х100 мл 0,5 М раствора гидроксида натрия, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, получая красно-коричневое масло. Колоночная хроматография с использованием 2% раствор метанола в этилацетате дает 0,4161 г соединения 8 (3-[2-[(фенилметил)амино]-4-пиридинил]-2-[3-(трифторметил)фенил]имидазо[1,2-а]пиримидин-7-амин) в виде не совсем белого твердого вещества. МН+=461.
Пример 2
1-Фенил-2-(4-пиридинил)этанон и 1-фенил-2-бром-2-(4-пиридинил)этанон
К раствору 1,8 г (0,02 моль) 4-пиколина и 3,0 г (0,02 моль) этилбензоата в 60 мл тетрагидрофурана с помощью капельной воронки в атмосфере азота добавляют 1,0 М раствор бис(триметилсилил)амида натрия (40 мл, 0,04 моль) в тетрагидрофуране. Через восемнадцать часов реакцию гасят насыщенным раствором хлорида аммония и удаляют растворитель в вакууме. Остаток экстрагируют 100 мл этилацетата и экстракт промывают 2×200 мл воды, 1×100 мл рассола, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, получая масло. Растирание с эфиром дает 1,6 г продукта реакции 1-Фенил-2-(4-пиридинил)этанона. МН+ 198.
К 1,6 г (8,1 ммоль) кетона в 10 мл ледяной уксусной кислоты добавляют 1,8 мл (8,9 ммоль) 35% раствора бромистого водорода в уксусной кислоте. Добавляют по каплям раствор 0,46 мл (8,9 ммоль) брома в 1,65 мл ледяной уксусной кислоты и реакционную смесь нагревают при 60°С в течение одного часа, охлаждают до комнатной температуры и разбавляют эфиром. Образовавшееся твердое вещество промывают эфиром и получают 2,5 г бромида в виде соли HBr 1-фенил-2-бром-2-(4-пиридинил)этанона. МН+=276.
Пример 3
2-Фенил-3-(4-пиридинил)имидазо[1,2-а]пиримидин-7-амин
Раствор 1,2 г (11 ммоль) 2,4-диаминопиримидина в 10 мл этанола нагревают до 80°С. С помощью капельной воронки добавляют по каплям раствор 1,0 г (2,8 ммоль) бромида в 20 мл этанола. Реакционную смесь перемешивают при 80°С в течение 3 часов и затем охлаждают до комнатной температуры. Удаляют в вакууме приблизительно половину объема растворителя. После охлаждения до комнатной температуры реакционную смесь фильтруют. Фильтрат концентрируют в вакууме, остаток разбавляют 250 мл этилацетата и промывают 2×100 мл 0,5 М раствора гидроксида натрия, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, получая красно-коричневое масло. Растирание остатка с EtOAc с последующей фильтрацией дают 0,108 г соединения 5 в виде не совсем белого твердого вещества. МН+=288.
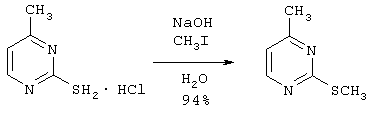
К раствору 13,38 г (84,73 ммоль) гидрохлорида 2-меркапто-4-метилпиримидина и 7,46 г (186,4 ммоль) гидроксида натрия в 120 мл воды при помощи шприца добавляют по каплям 13,23 г (93,2 ммоль) иодметана. Через 2 часа реакционную смесь экстрагируют 2×125 мл дихлорметана. Органические слои отделяют, объединяют, сушат над Na2SO4, фильтруют и концентрируют в вакууме, получая 11,14 г (79,45 ммоль) 4-метил-2-(метилтио) пиримидина в виде красного масла. МН+=140,9.
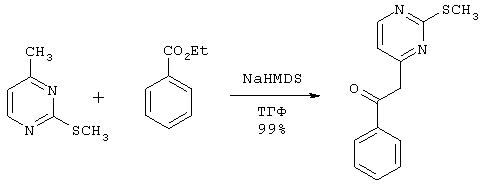
К раствору 6,03 г (43 ммоль) 4-метил-2-(метилтио) пиримидина и 6,46 г (43 ммоль) этилбензоата в 86 мл тетрагидрофурана в атмосфере азота при помощи капельной воронки добавляют по каплям 86 мл (86 ммоль) 1,0 М раствора бис(триметилсилил)амида натрия в тетрагидрофуране. Через 2 часа реакцию гасят насыщенным раствором хлорида аммония. Большую часть тетрагидрофурана удаляют в вакууме. Остаток разбавляют 400 мл этилацетата и 200 мл воды. Органический слой отделяют и промывают 2×100 мл насыщенного раствора хлорида натрия, отделяют, сушат над Na2SO4, фильтруют и концентрируют в вакууме, получая 10,45 г (42,77 ммоль) 2-[2-(метилтио)пиримидин-4-ил]-1-фенилэтанона в виде вязкого красно-коричневого масла, которое отверждается при стоянии. MH+=244,9.
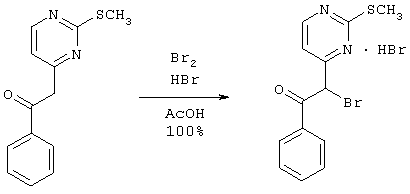
К 10,45 г (42,77 ммоль) кетона в 80 мл ледяной уксусной кислоты добавляют 9 мл (44,91 ммоль) 30% раствора бромистого водорода в уксусной кислоте. Добавляют по каплям раствор 2,40 мл (46,19 ммоль) брома в 2,60 мл ледяной уксусной кислоты, и реакционную смесь нагревают при 60°С в течение 45 минут, охлаждают до комнатной температуры и разбавляют эфиром. Образовавшуюся суспензию фильтруют, промывают эфиром и сушат в вакууме, получая 18,06 г (44,69 ммоль) сырого бромида. МН+=324,9.

Раствор 18,83 г (171,08 ммоль) 2,4-диаминопиримидина в 150 мл этанола нагревают до 80°С. С помощью капельной воронки добавляют по каплям раствор 18,06 г (42,77 ммоль) неочищенного бромида в 350 мл этанола. Реакционную смесь перемешивают при кипячении с обратным холодильником в течение двух часов. После охлаждения до комнатной температуры реакционную смесь фильтруют. Осадок перемешивают со 150 мл 0,5 М раствора гидроксида натрия. Осадок собирают фильтрацией, промывают водой, эфиром и гексаном, получая 6,72 г (21,1 моль) продукта реакции в виде светло-желтого твердого вещества. МН+=334,9.

Смесь 0,60 г (1,79 ммоль) тиометилпиримидина, приблизительно 4 мл 50% никеля Ренея в водном растворе, 40 мл этанола и 20 мл воды кипятят с обратным холодильником в течение восемнадцати часов в атмосфере азота. Реакционную смесь охлаждают до комнатной температуры и фильтруют через целит. Целит промывают этанолом. Объединенные фильтраты концентрируют в вакууме. Остаток растирают с этанолом, собирают фильтрацией и промывают эфиром, получая 0,2310 г пиримидина в виде желтого твердого вещества (соединение 16). MH+=289,0.

Раствор 8,28 г (13,46 ммоль) оксона в 75 мл воды с помощью капельной воронки добавляют по каплям к 1,50 г (4,49 ммоль) тиометилпиримидина в 77 мл метанола. Полученную суспензию перемешивают при комнатной температуре в течение восемнадцати часов, фильтруют и фильтрат концентрируют в вакууме, удаляя метанол. Остаток разбавляют 100 мл воды и нейтрализуют твердым бикарбонатом натрия. Полученную суспензию фильтруют, а осадок промывают водой, эфиром и сушат, получая 1,27 г (3,46 ммоль) метилсульфонпиримидина в виде желтого твердого вещества (соединение 17). МН+=367,0.

Смесь 0,55 г (1,5 ммоль) метилсульфонпиримидина и 1,82 г (15 ммоль) (S)-(-)-α-метилбензиламина нагревают при 140°С в течение 30 минут, охлаждают до комнатной температуры, разбавляют 100 мл этилацетата и промывают 3×50 мл воды и 1×50 мл насыщенного раствора хлорида натрия, сушат над Na2SO4, фильтруют и концентрируют в вакууме, получая желтое масло. Колоночная хроматография с использованием 100% этилацетата в качестве элюента дает 0,3977 г (0,98 ммоль) продукта реакции в виде светло-желтого твердого вещества (соединение 19). МН+=408,1.
В. АНАЛИЗЫ
Пример 4
Анализы на ингибирование р38
Биологическую активность некоторых соединений изобретения демонстрируют с помощью анализов in vitro и in vivo. Как указано выше, средства, ингибирующие активность фермента р38, ингибируют продуцирование цитокинов воспаления TNF-α и IL-1β.
Перечень выбранных соединений изобретения приводится в табл.1, в которой также приводятся данные масс-спектрального анализа, а также данные, показывающие способность каждого соединения ингибировать р38, как показано ингибированием продуцирования TNF-α. Анализы, в которых получены такие данные, описаны ниже.
Соединения, испытанные на их способность ингибировать р38, показанную через ингибирование продуцирования TNF-α
Анализ целых клеток РВМС
Характерные соединения настоящего изобретения испытывают в анализе in vitro целых клеток с использованием мононуклеарных клеток периферической крови ("РВМС"), которые получают из крови человека следующим образом. В свежевзятую венозную кровь добавляют противосвертывающее средство гепарин, разбавляют ее равным объемом забуференного фосфатом физиологического раствора ("PBS") и помещают в стерильную пробирку или другую емкость. Аликвоты (30 мл) такой смеси переносят в центрифужные пробирки, в которые вводят подслой фиколла-гипака (15 мл). Подготовленные пробирки центрифугируют при 400×g без торможения в течение 30 мин при комнатной температуре. Осторожно удаляют пипеткой приблизительно 1/2-2/3 слоя осадка над прослойкой мононуклеарных клеток. Осторожно с использованием пипетки удаляют большую часть слоя мононуклеарных клеток, и эти РВМС разводят PBS и центрифугируют при 600×g в течение 15 мин. Полученные РВМС промывают другой порцией PBS и центрифугируют при 400×g в течение 10 мин при комнатной температуре. Извлеченные осадки разводят в культуральной среде RPMI/1% FCS с низким содержанием эндотоксинов и получают концентрацию клеток 0,5-2,0×106 РВМС/мл. Извлекают небольшой объем суспензии для подсчета на гемоцитометре, а остальной препарат центрифугируют при 200×g в течение 15 мин при комнатной температуре. Извлеченные осажденные РВМС ресуспендируют в RPMI/1% FCS до концентрации 1,67×106/мл.
Для того чтобы осуществить анализ, суспензию РВМС переносят в сдвоенные ячейки 96-луночного титрационного микропланшета с плоскодонными лунками и инкубируют в течение 1 часа при 37°С.В каждую лунку добавляют раствор испытываемого соединения (10 мкл, полученный как 20× требуемая конечная концентрация) и планшет инкубируют в течение 1 часа при 37°С. Добавляют раствор (10 мкл) LPS в RPMI/1% FCS (200 нг/мл) и планшет инкубируют в течение ночи при 37°С. Извлекают супернатант (100 мкл) из каждой лунки и разводят RPMI/1% FCS (400 мкл). Образцы анализируют на TNF-α с использованием коммерческого набора ELISA (Genzyme). Результаты приводятся выше в табл.1.
Анализ in vivo на грызунах
Способность соединений формулы I ингибировать продуцирование TNF-α, индуцированное LPS, показывают в описанных далее анализах in vivo на грызунах. Мышей (самки BALB/cJ, Jackson Laboratories) не кормят за 30 мин до перорального введения дозы в 5-10 мл/кг с 5-50 мг/кг испытываемого соединения. Через тридцать минут после введения дозы животным инъецируют интраперитонеально LPS в количестве 1 мг/кг и возвращают их в клетки на 1 час. Животным дают наркоз CO2, выпускают кровь посредством кардиальной пункции, и собирают цельную кровь (0,1-0,7 мл). Крови дают возможность свертываться и сыворотку переносят в центрифужную пробирку. Полученный образец центрифугируют и сыворотку собирают, делят на аликвоты и замораживают при -80°С. Образцы испытывают с помощью коммерческого набора ELISA на TNF-α (эндоген для мышиного TNF-α). Вычисляют % ингибирования испытываемых соединений с помощью формулы
% инигибирования=[1-(образец-BKG)/(СТRL-BKG)]×100.
Результаты приводятся выше в табл.1.
Анализ рекомбинантного р38
Соединения изобретения проверяют на их способность ингибировать активность р38 с помощью описанного далее анализа in vitro. Раствор (38 мкл) очищенного рекомбинантного р38 (где количество фермента определяют эмпирически, причем рассматривается линейная область анализа и приемлемое соотношение сигнала и шума; 6×His-p38 экспрессирован в E.coli), основного белкового субстрата миелина (также определяют эмпирически) и буфера рН 7,5 (Hepes - 25 мМ; MgCl2 - 10 мМ; MnCl2 - 10 мМ) добавляют в 92 лунки 96-луночного пропиленового планшета с круглодонными лунками. Остальные лунки используют для контроля ("CTRL") и фона ("BKG"). CTRL получают с ферментом, буфером для субстрата и 21% ДМСО, a BKG получают с буфером для субстрата и 2% ДМСО. В лунки для испытаний добавляют раствор (12 мкл) испытываемого соединения в ДМСО (соединения разводят до 125 мкМ смесью 10% ДМСО/Н2О и анализируют при 25 мкМ, где конечная концентрация ДМСО составляет 2%). Во все лунки добавляют раствор АТР/33Р-АТР (10 мкл, содержащих 50 мкМ немеченного АТР и 1 мкКи 33Р-АТР), и в заполненных планшетах все перемешивают и инкубируют при 30°С в течение 30 мин. В каждую лунку добавляют охлажденный на льду раствор 50% ТСА/10 мМ фосфата натрия (60 мкл) и планшеты выдерживают на льду в течение 15 мин. Содержимое каждой лунки переносят в лунки 96-луночного фильтрационного планшета (Millipore, MultiScreen-DP) и помещают фильтрационный планшет в вакуумную установку, снабженную поддоном для сбора отходов. Лунки промывают в вакууме пять раз смесью 10% ТСА/10 мМ фосфата натрия (200 мкл). Добавляют сцинтиллятор MicroScint-20, планшеты герметизируют с использованием листов Topseal-S и проводят подсчет в сцинтилляционном счетчике Packard TopCount с использованием программы для жидкости с 33P с коррекцией тушения цвета, где результат выражается в ч.им./мин с исправлением на тушение цвета. Хотя соединения испытывают сначала при 10 мкМ, если есть основания, то соединения испытывают при 4-х кратном увеличении и снижении указанной концентрации. Кроме того, для некоторых соединений вычисляют IC50 с использованием программы подбора 4-параметрических кривых Дельтаграф. Результаты не приводятся.
Анализ на IL-1β in vitro
Способность соединений изобретения ингибировать продуцирование IL-1β можно определить с помощью следующего анализа in vitro. Из РВМС получают клетки, прилипающие к пластику. РВМС добавляют в лунки 96-луночного планшета, как описано выше, инкубируют в течение 1 часа при 37°С и получают прилипающие клетки, осторожно ресуспендируя неприлипающие клетки пипеттором, удаляя и отбрасывая их, и осторожно промывая лунки 3 раза 200 мкл культуральной среды. После последней промывки в лунки снова добавляют культуральную среду (180 мкл). Добавление соединения, стимуляцию LPS, инкубирование и сбор супернатанта осуществляют так же, как для TNF-α. Супернатанты анализируют на интерлейкин-1β, используя коммерческий набор ELISA (Genzyme). Результаты не приводятся.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ КИНАЗЫ | 2005 |
|
RU2348635C2 |
| ПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ В ОТНОШЕНИИ МСН | 2005 |
|
RU2373197C2 |
| СПИРО-6,7-ДИГИДРО-5Н-ПИРАЗОЛО[1,2а]ПИРАЗОЛ-1-ОНЫ, КОТОРЫЕ РЕГУЛИРУЮТ ВОСПАЛИТЕЛЬНЫЕ ЦИТОКИНЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ | 2002 |
|
RU2272040C2 |
| НОВЫЕ ГЕТЕРОЦИКЛЫ | 2009 |
|
RU2572616C2 |
| ЗАМЕЩЕННЫЕ 3-ПИРИДИЛ-4-АРИЛПИРРОЛЫ И СВЯЗАННЫЕ С НИМИ ТЕРАПЕВТИЧЕСКИЕ И ПРОФИЛАКТИЧЕСКИЕ СПОСОБЫ | 2000 |
|
RU2238940C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2005 |
|
RU2390522C2 |
| СОЕДИНЕНИЯ, КОТОРЫЕ ИНГИБИРУЮТ ВЫСВОБОЖДЕНИЕ ВОСПАЛИТЕЛЬНЫХ ЦИТОКИНОВ | 2002 |
|
RU2278864C2 |
| 6,7-ДИГИДРО-5H-ПИРАЗОЛО[1,2-А]ПИРАЗОЛ-1-ОНЫ, РЕГУЛИРУЮЩИЕ ВОСПАЛИТЕЛЬНЫЕ ЦИТОКИНЫ (ВАРИАНТЫ), И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2003 |
|
RU2289584C2 |
| 6,7-ДИГИДРО-5H-ПИРАЗОЛО[1,2-a]ПИРАЗОЛ-1-ОНЫ (ВАРИАНТЫ), ИНГИБИРУЮЩИЕ ВЫСВОБОЖДЕНИЕ ВОСПАЛИТЕЛЬНЫХ ЦИТОКИНОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ | 2002 |
|
RU2299885C2 |
| ЗАМЕЩЕННЫЕ ИМИДАЗОЛЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБЫ ЛЕЧЕНИЯ ЦИТОКИН-ОПОСРЕДОВАННЫХ ЗАБОЛЕВАНИЙ | 1998 |
|
RU2222534C2 |
Изобретение относится к новым замещенным 2-арил-3-(гетероарил)имидазо[1,2-а]пиримидинам формулы I

или к их фармацевтически приемлемым солям, где (a) R1 выбирают из группы, состоящей из NH2, С1-5алкиламино, ди-С1-5алкиламино, фенилметиламино; (b) Y выбирают из группы, состоящей из Н, галогена, пиперидина, OR4, SR4, -SO2СН3, NHR4 и NR4R5, где R4 и R5 выбирают, независимо, из Н, α-алкилфенил-С1-5алкила, прямого или разветвленного алкила, необязательно замещенного С3-5карбоциклом, фенилом или замещенным фенилом, где указанный фенил может быть замещен одним или несколькими заместителями, выбранными из С1-5алкокси; (с) R2 представляет от одного до пяти членов, независимо выбранных из группы, включающей Н, галоген, трифторметил; (d) R3 представляет Н, или радикалы R3, взятые вместе, образуют ароматическое кольцо; и (е) Х представляет N или СН. Изобретение также относится к способам получения указанных соединений и к способу лечения на основе этих соединений. Технический результат - получение новых соединений, которые могут применяться для состояний, облегчение которых достигается снижением цитокинов воспаления, например, указанное состояние представляет собой ревматоидный артрит. 5 н. и 35 з.п. ф-лы, 1 табл.

или их фармацевтически приемлемые соли, где
(a) R1 выбирают из группы, состоящей из NH2, С1-5алкиламино, ди-С1-5алкиламино, фенилметиламино,
(b) Y выбирают из группы, состоящей из Н, галогена, пиперидина, OR4, SR4, -SO2СН3, NHR4 и NR4R5, где
R4 и R5 выбирают, независимо, из Н, α-алкилфенил-C1-5алкила, прямого или разветвленного алкила, необязательно замещенного С3-5карбоциклом, фенилом или замещенным фенилом, где указанный фенил может быть замещен одним или несколькими заместителями, выбранными из С1-5алкокси;
(c) R2 представляет от одного до пяти членов, независимо выбранных из группы, включающей Н, галоген, трифторметил;
(d) R3 представляет Н, или радикалы R3, взятые вместе, образуют ароматическое кольцо;
(e) Х представляет N или СН.
включающий
а) превращение соединения 2а в присутствии NaOH и СН3I в соединение 2b




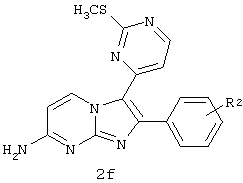
b) взаимодействие соединения 2b с соединением 2c в присутствии NaHMDS и ТГФ с образованием соединения 2d;
c) превращение соединения 2d в соединение 2е в присутствии НВг, Br2 и АсОН;

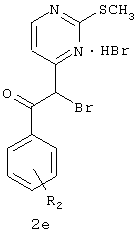
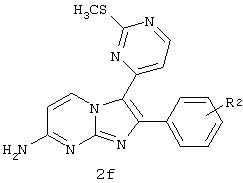
d) взаимодействие соединения 2е с соединением 1e в присутствии EtOH с образованием соединения 2f.

а) превращение соединения 2f в соединение 2h в присутствии оксона и МеОН;
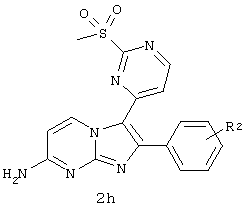

b) взаимодействие соединения формулы 2h с соединением Y,
где Y представляет галоген, пиперидин, OR4, SR4, NHR4 или NR4R5, с образованием соединения формулы 2i.
а) превращение соединения формулы 4а в соединение формулы 4b в присутствии (ВОС)2O и трет-BuOH;



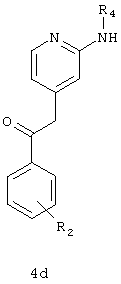
b) взаимодействие соединения формулы 4b с соединением формулы 4с в присутствии NaHMDS и HCl с образованием соединения формулы 4d;
c) превращение соединения формулы 4а в соединение формулы 4е в присутствии 30% HBr/АсОН, Br2 и АсОН; и



d) взаимодействие соединения формулы 4e с соединением формулы 1е в присутствии EtOH с образованием соединения формулы 4f.
| WO 9100092 A1, 10.01.1991 | |||
| WO 9119497 A1, 26.12.1991 | |||
| WO 9807425 A1, 26.02.1998 | |||
| 1,2,3,4-ТЕТРАГИДРОПИРАЗОЛО [5,1-c](1,2,4)ТРИАЗИН ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИНГИБИРУЮЩАЯ ВЫРАБОТКУ ИНТЕРЛЕЙКИНА-1 И ФАКТОРА НЕКРОЗА ОПУХОЛИ, СПОСОБ ПРОФИЛАКТИЧЕСКОГО ИЛИ ТЕРАПЕВТИЧЕСКОГО ЛЕЧЕНИЯ БОЛЕЗНЕЙ, ОПОСРЕДОВАННЫХ ИНТЕРЛЕЙКИНОМ-1 И ФАКТОРОМ НЕКРОЗА ОПУХОЛИ | 1994 |
|
RU2124517C1 |

